
Enhanced amphiphilic profile of a short β-stranded peptide improves Its antimicrobial activity

The Synthesis and Self-Assembly of ABA Amphiphilic BlockCopolymers Containing Styrene and Oligo(ethylene glycol)Methyl Ether Methacrylate in Dilute Aqueous Solutions:Elevated Cloud Point Temperatures forThermoresponsive Micelles
SIMON J. HOLDER,1 GERALDINE G. DURAND,1 CHERT-TSUN YEOH,1 ELODIE ILLI,1* NICHOLAS J. HARDY,2
TIM H. RICHARDSON2
1Functional Materials Group, School of Physical Sciences, University of Kent, Canterbury, Kent CT2 7NH, United Kingdom
2Department of Physics and Astronomy, Nanomaterials Engineering Group, The University of Sheffield, Hicks Building,Hounsfield Road, Sheffield S3 7RH, United Kingdom
Received 5 August 2008; accepted 9 September 2008DOI: 10.1002/pola.23077Published online in Wiley InterScience (www.interscience.wiley.com).
ABSTRACT: A series of ABA amphiphilic triblock copolymers possessing polystyrene(PS) central hydrophobic blocks, one group with ‘‘short’’ PS blocks (DP ¼ 54–86) andone with ‘‘long’’ PS blocks (DP ¼ 183–204) were synthesized by atom transfer radicalpolymerization. The outer hydrophilic blocks were various lengths of poly(oligoethy-lene glycol methyl ether) methacrylate, a comb-like polymer. The critical aggregationconcentrations were recorded for certain block copolymer samples and were found tobe in the range circa 10�9 mol L�1 for short PS blocks and circa 10�12 mol L�1 forlong PS blocks. Dilute aqueous solutions were analyzed by transmission electron mi-croscopy (TEM) and demonstrated that the short PS block copolymers formed spheri-cal micelles and the long PS block copolymers formed predominantly sphericalmicelles with smaller proportions of cylindrical and Y-branched cylindrical micelles.Dynamic light scattering analysis results agreed with the TEM observations demon-strating variations in micelle size with PS and POEGMA chain length: the hydro-dynamic diameters (DH) of the shorter PS block copolymer micelles increased withincreasing POEGMA block lengths while maintaining similar PS micellar core dia-meters (DC); in contrast the values of DH and DC for the longer PS block copolymermicelles decreased. Surface-pressure isotherms were recorded for two of the samplesand these indicated close packing of a short PS block copolymer at the air–waterinterface. The aggregate solutions were demonstrated to be stable over a 38-day pe-riod with no change in aggregate size or noticeable precipitation. The cloud pointtemperatures of certain block copolymer aggregate solutions were measured andfound to be in the range 76–93 �C; significantly these were �11 �C higher in temper-
Journal of Polymer Science: Part A: Polymer Chemistry, Vol. 46, 7739–7756 (2008)VVC 2008 Wiley Periodicals, Inc.
Additional Supporting Information may be found in theonline version of this article.
*Present address: Erasmus student visiting from l’EcolePolytechnique Universitaire de Montpellier, Place EugeneBataillon, 34095 Montpellier Cedex 5-France.
Correspondence to: S. J. Holder (E-mail: [email protected])
7739

ature than those of POEGMA homopolymer samples with similar chain lengths.VVC 2008 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem 46: 7739–7756, 2008
Keywords: amphiphilic block copolymer; atom transfer radical polymerization(ATRP); block copolymers; cloud point; colloids; micelles; oligo(ethylene glycol)methyl ether methacrylate; self-assembly; stimuli-sensitive polymers;thermoresponsive
INTRODUCTION
Amphiphilic block copolymers are capable of self-assembly in solvents where there is preferentialsolvation of one block.1 In such solvents they canform various aggregates, the most commonlyencountered being spherical micelles, cylindrical(or ‘‘worm-like’’) micelles, and vesicles.2–12 Themajority of such block copolymers possess blockswhere the glass transition temperatures of thehydrophobic blocks are lower than the ambienttemperature of self-assembly and hence in princi-ple the aggregates exit in a dynamic equilibriumwith individual solvated macromolecules. Conse-quently the aggregate structures are true equilib-rium structures. The most widely used and stud-ied commercially available examples of ‘‘soft’’amphiphilic block copolymers are the Pluronicsconsisting of poly(ethylene oxide) (PEO) andpoly(propylene oxide) (PPO) blocks.13,14
Block copolymers possessing hydrophobic blockssuch as poly(methyl methacrylate) and poly-styrene, whose Tgs are substantially higher thanthe ambient temperature,8–10 can be induced toform aggregates in aqueous dispersions by theslow addition of water to a solution in a water-miscible organic solvent, followed by removal ofthe solvent. Such block copolymers form sphericaland cylindrical micelles with ‘‘glassy’’ cores andvesicles with glassy wall interiors. Potentiallysuch aggregates are stable in aqueous solutionsince molecular motion in the hydrophobic part ofthe aggregate is frozen and no interaggregate co-polymer migration can occur. Such aggregates areof potential interest for solubilizing hydrophobicmolecules in aqueous solutions where stable andmechanically robust aggregates are desirable. Inthis article, we will report results from our studyof the synthesis and self-assembly into aggregatesin aqueous solutions of block copolymers contain-ing comb-like hydrophilic blocks of poly[oligo-(ethylene glycol) methyl ether methacrylate], athermoresponsive polymer.
The bulk of studies carried out on amphiphilicblock copolymer aggregates has been performed
on materials synthesized by living polymerizationmethods.15 Following its discovery in 1995,16–18
ATRP has been used extensively in the productionof functional polymers with a wide range of copol-ymer architectures, including blocks, grafts, stars,and hyperbranched polymers. Since the propagat-ing chain ends are radicals, ATRP is much moretolerant of monomer functionality compared toliving ionic polymerizations.19,20 Over the pastfew years, ATRP has been shown to be a versatiletechnique for the efficient polymerizations ofhydrophilic monomers21–27 and amphiphilic blockcopolymers using many of these monomers havealso been successfully synthesized.11
Polyethylene oxide (PEO, or poly(ethylene gly-col) (PEG)) has long been a major component ofblock copolymers studied for aqueous applica-tions. Current interest in its applications resultsfrom the fact that PEO is a biocompatible non-toxic polymer28 with potential applications as bio-materials for a variety of applications, such ascontrolled release of drugs.29 The biomedicalapplication of PEG copolymers has been of greatinterest due to their highly effective exclusionproperties such as low protein absorption and lowcell adhesion in aqueous solution.30 Linear blockcopolymers containing PEO can self-assemble inaqueous solution to give aggregates includingspherical and cylindrical micelles and vesicles.31
Graft copolymers exhibit many particular physi-cal-chemical characteristics which are differentfrom their corresponding linear homologues.32
Graft copolymers of (oligoethylene glycol) methylmethacrylate, having methacrylate backbonesand poly(ethylene glycol) branches, have beenstudied for some time33–37 as a consequence ofapparently similar biocompatibility to PEO.38–40
Armes and coworkers have described the synthe-sis of well-defined POEGMA22,23 and a number ofsyntheses and studies of block copolymers incorpo-rating POEGMA have been reported,41–46 includ-ing block copolymers with polypropylene oxide,47
polydimethylsiloxane,48 polymethylphenylsi-lane,49 and poly(octadecyl acrylate)50 hydrophobiccomponents, many of which form aggregates in
7740 HOLDER ET AL.
Journal of Polymer Science: Part A: Polymer ChemistryDOI 10.1002/pola

dilute aqueous solutions. An increasing number ofamphiphilic block copolymers with novel or hith-erto inaccessible macromolecular architecturesare being synthesized by controlled radical poly-merization techniques such as ATRP and RAFT.The study of the self-assembly of such systems invarious media is of importance since in contrast tomaterials prepared by classical living techniquessuch as ionic or ring-opening polymerizations,these often have relatively high polydispersitiesand possible ‘‘contamination’’ from terminationevents in the polymerization process (e.g., leadingto the presence of diblocks in triblock samples).
We have previously reported the selectivedelamination of thin films of triblock POEGMA-PS-POEGMA amphiphilic copolymers preparedby ATRP as a consequence of the microphase sep-arated morphologies formed in the solid state andtheir application in patterned cell-growth.51,52 Inthis publication we report on studies of theiraggregation behavior in dilute aqueous solutionsincluding critical aggregation constants (CAC),aggregate morphologies, behavior at the air–water interface, and demonstrate their anomalousbehavior with relation to their cloud point temper-atures compared to that of homopolymers ofPOEGMA.
EXPERIMENTAL
Materials
Copper (I) bromide (Cu(I)Br) (99%) (Aldrich),diethyl-meso-2,5-dibromoadipate (DMDA) (98%)(Aldrich), 2,20-dipryidyl (99þ%) (Acros), alumi-num oxide (activated neutral) (alumina) ([98%)(Acros), hexane (99%, analytical grade) (FischerChemicals), and methanol (99%, analytical grade)(Fischer Chemicals) were used as received. Tolu-ene (HPLC grade) (Fischer Chemicals) was driedover magnesium sulfate (MgSO4) then dried oversodium wire and then distilled over sodium wireand benzophenone under N2(g) immediately priorto use. Oligo(ethylene glycol) methyl ether meth-acrylate (OEGMA) (99%, Aldrich, Mn ¼ 300) andstyrene (99%, Acros Organics) were passedthrough dry alumina columns to remove theinhibitors and were then stored at 4 �C underN2(g). Poly(oligoethylene glycol methyl ethermethacrylate) (POEGMA) samples used for cloudpoint temperature measurements were synthe-sized according to literature procedures.22,23 Themolecular weight (MW) parameters of the
POEGMA samples used are given in the Support-ing Information.
Analysis
Nuclear magnetic resonance (NMR) spectra wererecorded using a JEOL GX-270 spectrometer fromsolutions in deuterated chloroform. Both 1H NMRand 13C NMR analysis were performed at 30 �C.NMR spectra were recorded and analyzed usingthe SpecNMR version 1.2.0 software.
Molecular weights of the polymers were esti-mated relative to polystyrene standards by size-exclusion chromatography (SEC) using equipmentsupplied by Polymer Laboratories Ltd. All deter-minations were carried out at 40 �C using a GPCsystem consisting of a Polymer Lab LC 1120HPLC pump, a Polymer Labs Shodex RefractiveIndex detector, and two 300 mm � 7.5 mm PLGel5 lm mixed-C columns. THF (HPLC grade) wasused as an eluent and was set to a flow rate of1 mL min�1. All GPC measurements were inter-preted using the Cirrus GPC online computersoftware version 2.0.
Transition electron microscopy was carried outusing a JEOL JEM-1230 operating at 80 kV. Mi-cellar solutions (copolymer concentration of 1 gL�1) were deposited onto copper EM grids (200mesh, Formar and C covered) and after 30 s waterwas removed by suction. The sample grids werethen stained with a solution of uranyl acetatedihydrate (C4H6U H2O) (1% w/v in water) by add-ing a drop of the staining solution onto the gridand removing after 5–15 s by suction.
Dynamic light scattering measurements werecarried out on a Malvern High Performance Parti-cle Sizer (HPPS HPP5001) at 25 �C with a He-Ne,3.0 mW,633 nm laser using a noninvasive back-scattering technique. Four runs with an averageof 14 scans each were performed. Data was ana-lyzed with HPPS Malvern software version 1.10and hydrodynamic diameters quoted are from in-tensity measurements and calculations.
Fluorescence measurements were carried outon a Perkin–Elmer LS50B fluorescence spectrom-eter at room temperature.
Techniques
Preparation of Block Copolymer AggregateSolutions
The typical procedure for the preparation of themicellar solution was as follows: a sample of copoly-mer (10 mg) was dissolved in filtered THF
SYNTHESIS OF ABA AMPHIPHILIC BLOCK COPOLYMERS 7741
Journal of Polymer Science: Part A: Polymer ChemistryDOI 10.1002/pola

(1 mL). Pure water (9 mL) was added dropwiseover a 1-h period with vigorous stirring. Sampleswere transferred to dialysis tubing, sealed (tomaintain a constant volume), and dialysedagainst distilled water for 24 h to remove theTHF and to obtain a final concentration of co-polymer in water of �1 g L�1. The water (3 L)surrounding the bags was changed three timesduring this procedure. After dialysis, the sampleswere transferred to jars, sealed, and stored in thedark at room temperature. For TEM and DLSanalysis, samples were allowed to equilibrate for3 h before any measurements.
Cloud Point Temperature Determinations
The cloud point temperatures (TCP) of selectedcopolymers were determined at fixed concentra-tions of copolymers (1 g L�1). A typical procedurefor the determination of the cloud point tempera-ture described by Bahadur was followed.7 A Pyrexcuvette containing �2 mL of the micellar solutionunder study was immersed in an oil bath. Thetemperature was raised at a rate of 1–2 �C min�1
and the first appearance of cloudiness in solutionwas recorded and was taken as the cloud point.The measurements were repeated twice and theaverage value of temperature upon the onset ofturbidity on heating was taken as the cloud point.
Critical Aggregation Concentration Determination
Deionized water was added to a solution of pyrenein acetone (6 � 10�2 M) in such a quantity that af-ter the acetone was removed under reduced pres-sure at 25 �C, an aqueous solution of pyrene witha final concentration of 6 � 10�7 M was obtained.The standard procedure for the determination ofthe critical aggregation concentration (CAC)using pyrene was as follows. An aqueous solutionof the pyrene in deionized water (9 mL, 6 � 10�7
mol L�1) was added dropwise over 1 h to a stirredTHF solution of a copolymer (1 mL, 1 g L�1). Sub-sequently the solution was diluted with furtheraqueous pyrene solution to obtain a series of solu-tions with copolymer concentrations ranging from10�10 to 1 g L�1 while maintaining a constant py-rene concentration of 6 � 10�7 M. The solutionswere then dialysed against the stock pyrene solu-tion over 24 h. The emission spectrum of eachsample was recorded (kexc ¼ 339 nm) at room tem-perature and the CAC values were obtained fromplots of the I1/I3 emission band ratios (I1 ¼ inten-sity at 373 nm and I3 ¼ intensity at 383 nm) ver-
sus copolymer concentration according to pub-lished procedures (see Supp. Info.).
Aggregation Numbers
CS Chem3D Pro (Version 5.0, CambridgeSoft,1999) was used to determine the Connelly-SolventExcluded Volume for an energy minimized,extended 44 unit PS chain which was divided by44 to obtain the volume for a single styrene unit(Vsty). Aggregation numbers (Nagg) were then cal-culated by dividing the PS core volume deter-mined from TEM measurements of core radius(rcore) with the total core volume dependent uponthe chain length (DPPS) of the PS.
Nagg ¼ 4
3pr3core
� ��Vsty �DPPS
� �
No measurements or analyses of packing den-sities were carried out so these values are only in-dicative.
Surface Pressure–Area Isotherms
Surface pressure–area isotherms were recordedusing a NIMA Model 112D Langmuir trough,whose subphase surface area could be continu-ously varied from 80 to 20 cm2 during the mea-surement of an isotherm. The trough was cleanedthoroughly prior to use with chloroform and awater pump suction device was used to removeany contamination from the pure water ([15 MXcm) subphase surface before spreading of the poly-mer solution. Chloroform was also used as the sol-vent for the spreading solutions. The two poly-mers ABAB 11 and ABA 13 were made up to con-centrations of 0.40 and 0.44 mg mL�1,respectively. The quantities of solution spreadwere 50 and 30 lL (for the two ABA 11 isotherms)and 40 lL for the ABA 13 isotherm. A Wilhelmyplate incorporating a filter paper plate was usedto measure the surface pressure.
Syntheses
a,x-Dibromopolystyrene Macroinitiators (PS1-PS6)
A typical synthesis was as follows: A Schlenk tubecontaining Cu(I)Br (0.208 g, 1.45 mmol), 2,20-dipyridyl (0.453 g, 2.9 mmol), DMDA (0.261 g,0.725 mmol), and styrene (5 mL, 43.63 mmol) wassealed and subjected to three freeze-vacuum-thawcycles. The vessel was then immersed in an oil
7742 HOLDER ET AL.
Journal of Polymer Science: Part A: Polymer ChemistryDOI 10.1002/pola
bath set at a temperature of 108 �C and stirred atthis temperature until an hour after the onset ofgelation (indicated by a cessation of stirring). Themixture was diluted with THF and then passedthrough a column of alumina with additionalTHF as the eluent. Finally the reaction mixturewas precipitated into an excess of methanol priorto analysis by SEC and NMR. The product wasthen collected and dried in vacuum for 24 h at50 �C.
1H NMR (CDCl3) d: 6.0–7.5 ppm (C-Ph), 4.4–4.5 ppm (CH-Br), 0.8–2.5 ppm (CH2 and CH ofpolystyrene backbone).
13C NMR (CDCl3) d: 127–129 ppm and 146ppm (CHAC6H5), 40–49 ppm (CH2ACH)
A range of initiator:monomer concentrationmolar ratios were employed to synthesize a num-ber of polystyrene samples of various molecularweights. The relevant experimental parametersfor each sample synthesized are given in Table 1.
Poly[oligo(ethylene glycol) MethylEther Methacrylate]-block-polystyrene-block-poly[oligo(ethylene glycol) Methyl EtherMethacrylate] (POEGMA-PS-POEGMA) (ABA 1–15)
A typical synthesis was as follows: A Schlenk tubecontaining a,x-dibromopolystyrene (PS5, Mn ¼8700, Mw/Mn ¼ 1.27, 1.00 g, 0.12 mmol), Cu(I)Br(0.017 g, 0.12 mmol), 2,20-dipyridyl (0.036 g, 0.23mmol), and toluene (5 mL) was sealed and sub-jected to three freeze-vacuum-thaw cycles.OEGMA (0.692 g, 2.31 mmol) was then added viaa degassed syringe and the mixture in theSchlenk was then placed in an oil bath set to atemperature of 108 �C and stirred until at least 1h after the reaction reached gel-point (indicatedby cessation of stirring). The mixture was exposed
to air and was then diluted with THF (30 mL)(whereupon the reaction mixture turned fromdark brown to green) and passed through an alu-mina column to remove the copper catalyst withadditional THF as the eluent. The combined solu-tion was then added slowly (dropwise) to anexcess of stirred hexane. The product precipitatedas a clear gel-like solid and was isolated and driedovernight under vacuum at 50 �C prior to SECand NMR analysis.
1H NMR (CDCl3) d: 6.0–7.5 ppm (C-Ph), 4.4–4.5 ppm (CH-Br), 4.1 ppm (CO2CH2CH2OA), 3.5–3.8 ppm (AOCH2CH2A), 3.35 ppm (OCH3), 1.65–2.1 ppm (ACH2C(CH3)A), 0.8–1.3 ppm(ACH2C(CH3)A) 13CNMR (CDCl3) d: 179 ppm(C¼¼O), 127–129 and 146 ppm (CHAC6H5), 60–72ppm (AOCH2CH2A), 59 ppm (OCH3), 40 ppm(ACH2C(CH3)A), 25 ppm (ACH2C(CH3)A), 17ppm (ACH2C(CH3)A).
A range of initiator:monomer concentrationmolar ratios were employed to synthesize 15POEGMA-PS-POEGMA samples of various mo-lecular weights. The relevant experimental pa-rameters for each sample synthesized are given inTable 2.
Kinetic Analysis
Monomer conversions during the syntheses ofpolystyrene and block copolymers were made bytaking small aliquots from the reaction Schlenkat suitable time intervals and immediately dilut-ing the samples in analytical grade THF. Thesamples were then subsequently filtered througha short alumina column to remove the catalystand then analyzed by 1H NMR spectroscopy andSEC. Monomer conversions were calculated from
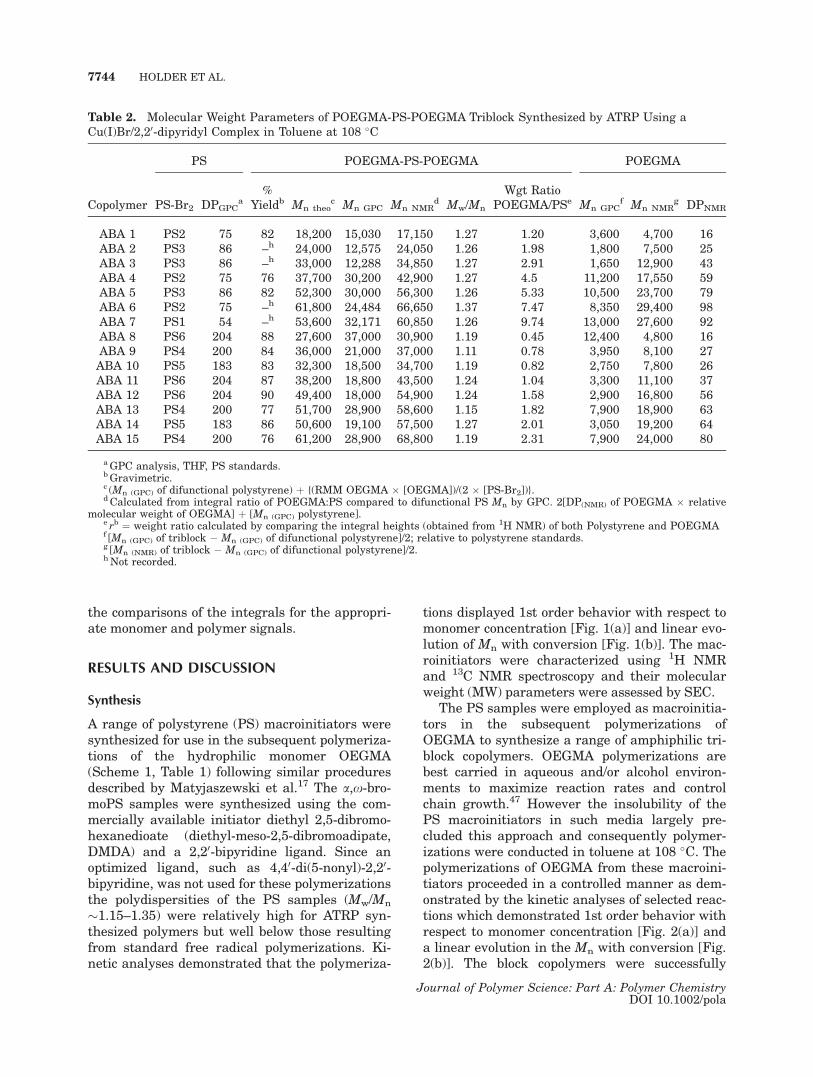
Table 1. Reaction Conditions for the ATRP of Styrene Using a Cu(I)Br/2,20-dipyridyl Complex in Bulk at 108 �Cand Molecular Weight Parameters of the Resultant a,x-Dibromopolystyrene Samples
No Sty:Cu(I):Bipy:Ia DPtheob Mn theo
c Mn GPCd DPGPC
d Mw/Mnd Yielde
PS1 30:1:2:0.5 60 6,250 5,600 54 1.27 80PS2 30:1:2:0.5 60 6,250 7,800 75 1.15 88PS3 30:1:2:0.5 60 6,250 9,000 86 1.24 89PS4 100:1:2:0.5 200 20,800 20,800 200 1.26 85PS5 100:1:2:0.5 200 20,800 19,100 183 1.20 85PS6 100:1:2:0.5 200 20,800 21,290 204 1.25 82
aMolar ratio styrene: Cu(I)Br: 2,20-bipyridine: diethyl-meso-2,5-dibromoadipate.bDP of styrene calculated according to [M0]/[I].c [M0]/[I] � relative molecular mass of styrene, initiator mass not included.dGPC analysis, THF, PS standards.eGravimetrically determined.
SYNTHESIS OF ABA AMPHIPHILIC BLOCK COPOLYMERS 7743
Journal of Polymer Science: Part A: Polymer ChemistryDOI 10.1002/pola
the comparisons of the integrals for the appropri-ate monomer and polymer signals.
RESULTS AND DISCUSSION
Synthesis
A range of polystyrene (PS) macroinitiators weresynthesized for use in the subsequent polymeriza-tions of the hydrophilic monomer OEGMA(Scheme 1, Table 1) following similar proceduresdescribed by Matyjaszewski et al.17 The a,x-bro-moPS samples were synthesized using the com-mercially available initiator diethyl 2,5-dibromo-hexanedioate (diethyl-meso-2,5-dibromoadipate,DMDA) and a 2,20-bipyridine ligand. Since anoptimized ligand, such as 4,40-di(5-nonyl)-2,20-bipyridine, was not used for these polymerizationsthe polydispersities of the PS samples (Mw/Mn
�1.15–1.35) were relatively high for ATRP syn-thesized polymers but well below those resultingfrom standard free radical polymerizations. Ki-netic analyses demonstrated that the polymeriza-
tions displayed 1st order behavior with respect tomonomer concentration [Fig. 1(a)] and linear evo-lution of Mn with conversion [Fig. 1(b)]. The mac-roinitiators were characterized using 1H NMRand 13C NMR spectroscopy and their molecularweight (MW) parameters were assessed by SEC.
The PS samples were employed as macroinitia-tors in the subsequent polymerizations ofOEGMA to synthesize a range of amphiphilic tri-block copolymers. OEGMA polymerizations arebest carried in aqueous and/or alcohol environ-ments to maximize reaction rates and controlchain growth.47 However the insolubility of thePS macroinitiators in such media largely pre-cluded this approach and consequently polymer-izations were conducted in toluene at 108 �C. Thepolymerizations of OEGMA from these macroini-tiators proceeded in a controlled manner as dem-onstrated by the kinetic analyses of selected reac-tions which demonstrated 1st order behavior withrespect to monomer concentration [Fig. 2(a)] anda linear evolution in the Mn with conversion [Fig.2(b)]. The block copolymers were successfully
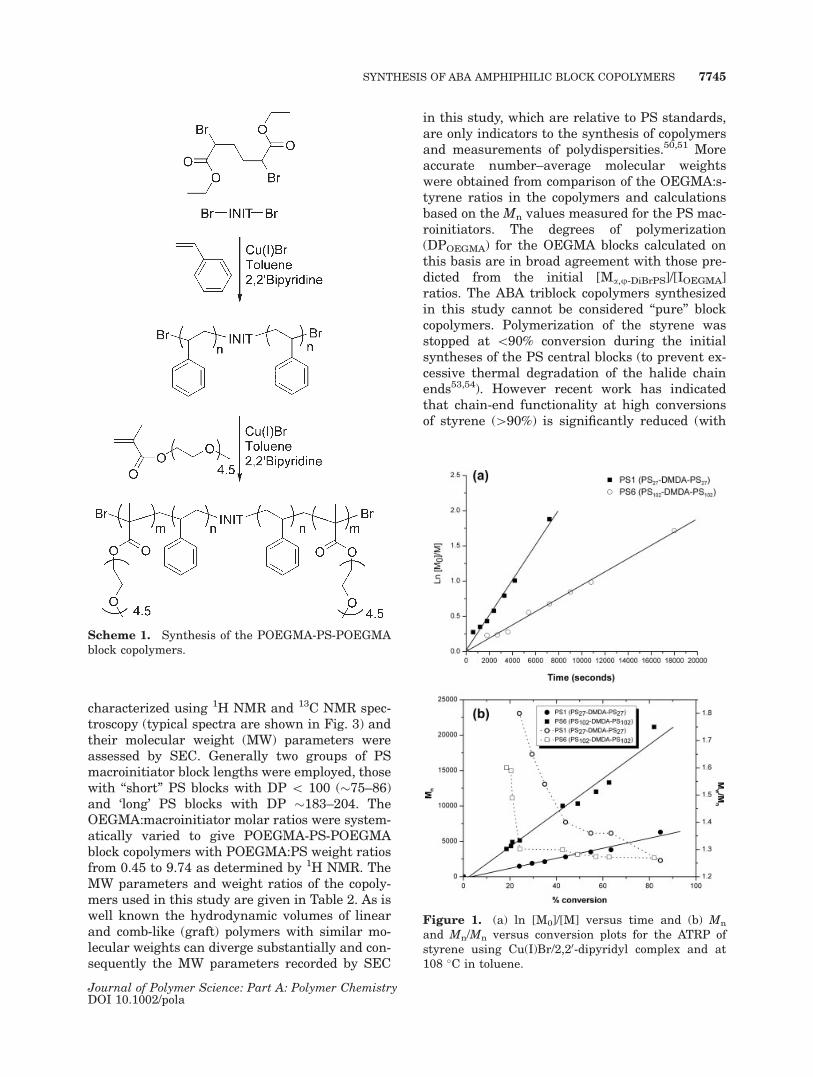
Table 2. Molecular Weight Parameters of POEGMA-PS-POEGMA Triblock Synthesized by ATRP Using aCu(I)Br/2,20-dipyridyl Complex in Toluene at 108 �C
Copolymer
PS POEGMA-PS-POEGMA POEGMA
PS-Br2 DPGPCa
%Yieldb Mn theo
c Mn GPC Mn NMRd Mw/Mn
Wgt RatioPOEGMA/PSe Mn GPC
f Mn NMRg DPNMR
ABA 1 PS2 75 82 18,200 15,030 17,150 1.27 1.20 3,600 4,700 16ABA 2 PS3 86 –h 24,000 12,575 24,050 1.26 1.98 1,800 7,500 25ABA 3 PS3 86 –h 33,000 12,288 34,850 1.27 2.91 1,650 12,900 43ABA 4 PS2 75 76 37,700 30,200 42,900 1.27 4.5 11,200 17,550 59ABA 5 PS3 86 82 52,300 30,000 56,300 1.26 5.33 10,500 23,700 79ABA 6 PS2 75 –h 61,800 24,484 66,650 1.37 7.47 8,350 29,400 98ABA 7 PS1 54 –h 53,600 32,171 60,850 1.26 9.74 13,000 27,600 92ABA 8 PS6 204 88 27,600 37,000 30,900 1.19 0.45 12,400 4,800 16ABA 9 PS4 200 84 36,000 21,000 37,000 1.11 0.78 3,950 8,100 27ABA 10 PS5 183 83 32,300 18,500 34,700 1.19 0.82 2,750 7,800 26ABA 11 PS6 204 87 38,200 18,800 43,500 1.24 1.04 3,300 11,100 37ABA 12 PS6 204 90 49,400 18,000 54,900 1.24 1.58 2,900 16,800 56ABA 13 PS4 200 77 51,700 28,900 58,600 1.15 1.82 7,900 18,900 63ABA 14 PS5 183 86 50,600 19,100 57,500 1.27 2.01 3,050 19,200 64ABA 15 PS4 200 76 61,200 28,900 68,800 1.19 2.31 7,900 24,000 80
aGPC analysis, THF, PS standards.bGravimetric.c (Mn (GPC) of difunctional polystyrene) þ {(RMM OEGMA � [OEGMA])/(2 � [PS-Br2])}.d Calculated from integral ratio of POEGMA:PS compared to difunctional PS Mn by GPC. 2[DP(NMR) of POEGMA � relative
molecular weight of OEGMA] þ [Mn (GPC) polystyrene].e rb ¼ weight ratio calculated by comparing the integral heights (obtained from 1H NMR) of both Polystyrene and POEGMAf [Mn (GPC) of triblock � Mn (GPC) of difunctional polystyrene]/2; relative to polystyrene standards.g [Mn (NMR) of triblock � Mn (GPC) of difunctional polystyrene]/2.hNot recorded.
7744 HOLDER ET AL.
Journal of Polymer Science: Part A: Polymer ChemistryDOI 10.1002/pola
characterized using 1H NMR and 13C NMR spec-troscopy (typical spectra are shown in Fig. 3) andtheir molecular weight (MW) parameters wereassessed by SEC. Generally two groups of PSmacroinitiator block lengths were employed, thosewith ‘‘short’’ PS blocks with DP \ 100 (�75–86)and ‘long’ PS blocks with DP �183–204. TheOEGMA:macroinitiator molar ratios were system-atically varied to give POEGMA-PS-POEGMAblock copolymers with POEGMA:PS weight ratiosfrom 0.45 to 9.74 as determined by 1H NMR. TheMW parameters and weight ratios of the copoly-mers used in this study are given in Table 2. As iswell known the hydrodynamic volumes of linearand comb-like (graft) polymers with similar mo-lecular weights can diverge substantially and con-sequently the MW parameters recorded by SEC
in this study, which are relative to PS standards,are only indicators to the synthesis of copolymersand measurements of polydispersities.50,51 Moreaccurate number–average molecular weightswere obtained from comparison of the OEGMA:s-tyrene ratios in the copolymers and calculationsbased on the Mn values measured for the PS mac-roinitiators. The degrees of polymerization(DPOEGMA) for the OEGMA blocks calculated onthis basis are in broad agreement with those pre-dicted from the initial [Ma,u-DiBrPS]/[IOEGMA]ratios. The ABA triblock copolymers synthesizedin this study cannot be considered ‘‘pure’’ blockcopolymers. Polymerization of the styrene wasstopped at \90% conversion during the initialsyntheses of the PS central blocks (to prevent ex-cessive thermal degradation of the halide chainends53,54). However recent work has indicatedthat chain-end functionality at high conversionsof styrene ([90%) is significantly reduced (with
Scheme 1. Synthesis of the POEGMA-PS-POEGMAblock copolymers.
Figure 1. (a) ln [M0]/[M] versus time and (b) Mn
and Mn/Mn versus conversion plots for the ATRP ofstyrene using Cu(I)Br/2,20-dipyridyl complex and at108 �C in toluene.
SYNTHESIS OF ABA AMPHIPHILIC BLOCK COPOLYMERS 7745
Journal of Polymer Science: Part A: Polymer ChemistryDOI 10.1002/pola

respect to active C-Br chain ends) and may leadto in the region of only 80% end-group functions.55
Taking 80% as a guide to active Br chain endsthis would lead to 4% (20/100 � 20/100) of chainswithout any Br end-groups and up to 16% (20/100� 80/100) of chains with one end-group. Fractio-nation by precipitation using selective solventsduring workup for the block copolymers wouldremove any small quantities of homopolymer andconsequently contamination of the triblock sam-ples with significant amounts of homopolymer isnot likely. However the possibility of contamina-tion by a significant degree of AB diblock materi-als cannot be excluded without extensive analysis(e.g., MALDI-TOFF) and/or fractionation of theproducts. Despite this it is noticeable that allPOEGMA-PS-POEGMA samples showed mono-modal distributions and no significant deviationsfrom expected molecular weights or highly broadpolydispersity indices were observed.
Critical Aggregation Concentrations
The critical aggregation concentrations (CAC) ofsix copolymer samples (ABA 1, 3, 5, 13, 14, and15) were determined by a fluorescent probe (py-rene) method.7,56–58 Pyrene is a hydrophobic fluo-rescent molecule that exhibits different fluores-cence behavior depending upon the properties ofthe solubilizing medium (its fluorescence tends tobe quenched in polar media) and consequentlybetween the micelle (aggregate) interiors and thesurrounding aqueous medium. Emission spectrawere recorded for different concentrations of eachcopolymer colloidal solution prepared in the pres-ence of pyrene. Typical overlaid fluorescence spec-tra (for ABA 3) of pyrene (kexc ¼ 339 nm) in vari-ous concentrations are shown in Figure 4(a). TheCAC values were obtained from plots of the I1/I3emission band ratios (I1 ¼ intensity at 373 nmand I3 ¼ intensity at 383 nm) versus copolymerconcentration (see also Supp. Info. for detailed ex-planation).56 The CAC values were identified asthe midpoint of the transition from high to lowvalues of the I1/I3 ratio [a typical plot is shown inFig. 4(b)] and are given in Table 3. The copoly-mers studied were three copolymers with a shortPS segment and varying POEGMA block lengths,ABA 1, 3, and 5, and three copolymers with a con-stant long PS segment and varying POEGMAblock lengths, ABA 13, 14, and 15. All of thecopolymers displayed very low CACs with the val-ues for the short PS groups in the range circa
Figure 2. Typical (a) ln [M0]/[M] versus time and(b) Mn and Mn/Mn versus conversion plots for theATRP of OEGMA using Cu(I)Br/2,20-dipyridyl com-plex, PS5 macroinitiator at 108 �C in toluene.
Figure 3. 1H NMR and 13C NMR spectra ofPOEGMA-PS-POEGMA triblock copolymer (reaction 5).
7746 HOLDER ET AL.
Journal of Polymer Science: Part A: Polymer ChemistryDOI 10.1002/pola
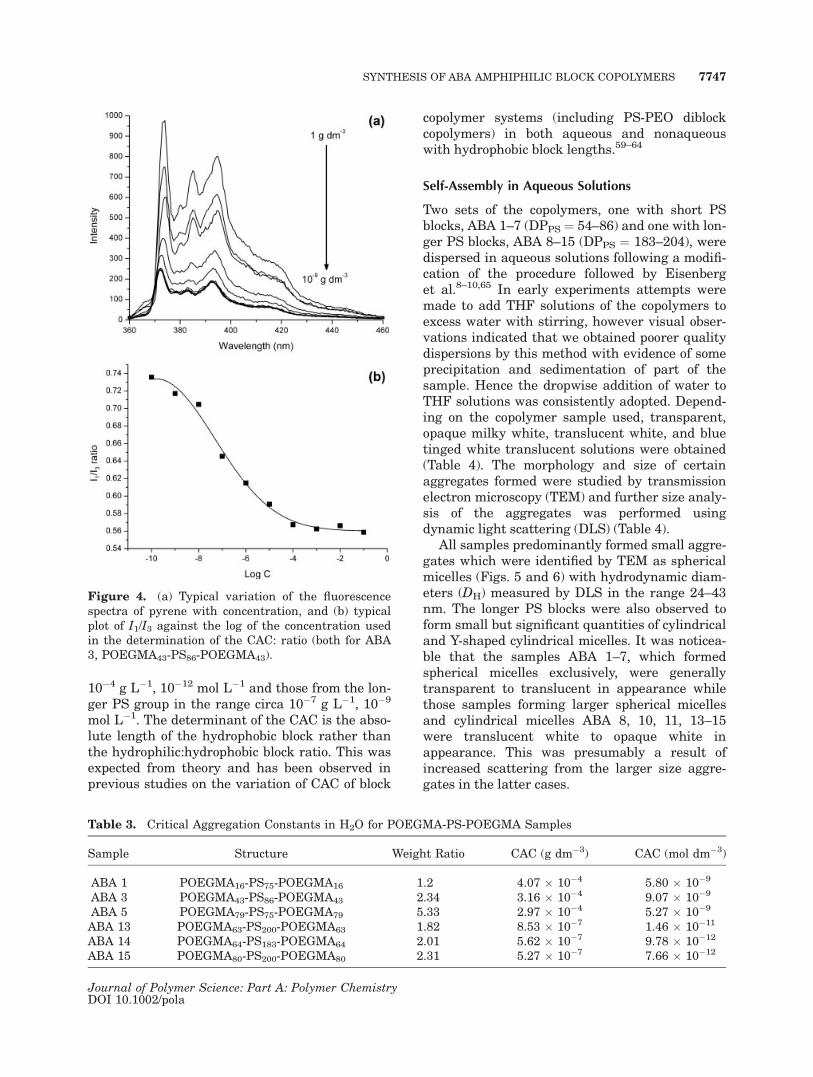
10�4 g L�1, 10�12 mol L�1 and those from the lon-ger PS group in the range circa 10�7 g L�1, 10�9
mol L�1. The determinant of the CAC is the abso-lute length of the hydrophobic block rather thanthe hydrophilic:hydrophobic block ratio. This wasexpected from theory and has been observed inprevious studies on the variation of CAC of block
copolymer systems (including PS-PEO diblockcopolymers) in both aqueous and nonaqueouswith hydrophobic block lengths.59–64
Self-Assembly in Aqueous Solutions
Two sets of the copolymers, one with short PSblocks, ABA 1–7 (DPPS ¼ 54–86) and one with lon-ger PS blocks, ABA 8–15 (DPPS ¼ 183–204), weredispersed in aqueous solutions following a modifi-cation of the procedure followed by Eisenberget al.8–10,65 In early experiments attempts weremade to add THF solutions of the copolymers toexcess water with stirring, however visual obser-vations indicated that we obtained poorer qualitydispersions by this method with evidence of someprecipitation and sedimentation of part of thesample. Hence the dropwise addition of water toTHF solutions was consistently adopted. Depend-ing on the copolymer sample used, transparent,opaque milky white, translucent white, and bluetinged white translucent solutions were obtained(Table 4). The morphology and size of certainaggregates formed were studied by transmissionelectron microscopy (TEM) and further size analy-sis of the aggregates was performed usingdynamic light scattering (DLS) (Table 4).
All samples predominantly formed small aggre-gates which were identified by TEM as sphericalmicelles (Figs. 5 and 6) with hydrodynamic diam-eters (DH) measured by DLS in the range 24–43nm. The longer PS blocks were also observed toform small but significant quantities of cylindricaland Y-shaped cylindrical micelles. It was noticea-ble that the samples ABA 1–7, which formedspherical micelles exclusively, were generallytransparent to translucent in appearance whilethose samples forming larger spherical micellesand cylindrical micelles ABA 8, 10, 11, 13–15were translucent white to opaque white inappearance. This was presumably a result ofincreased scattering from the larger size aggre-gates in the latter cases.
Figure 4. (a) Typical variation of the fluorescencespectra of pyrene with concentration, and (b) typicalplot of I1/I3 against the log of the concentration usedin the determination of the CAC: ratio (both for ABA3, POEGMA43-PS86-POEGMA43).
Table 3. Critical Aggregation Constants in H2O for POEGMA-PS-POEGMA Samples
Sample Structure Weight Ratio CAC (g dm�3) CAC (mol dm�3)
ABA 1 POEGMA16-PS75-POEGMA16 1.2 4.07 � 10�4 5.80 � 10�9
ABA 3 POEGMA43-PS86-POEGMA43 2.34 3.16 � 10�4 9.07 � 10�9
ABA 5 POEGMA79-PS75-POEGMA79 5.33 2.97 � 10�4 5.27 � 10�9
ABA 13 POEGMA63-PS200-POEGMA63 1.82 8.53 � 10�7 1.46 � 10�11
ABA 14 POEGMA64-PS183-POEGMA64 2.01 5.62 � 10�7 9.78 � 10�12
ABA 15 POEGMA80-PS200-POEGMA80 2.31 5.27 � 10�7 7.66 � 10�12
SYNTHESIS OF ABA AMPHIPHILIC BLOCK COPOLYMERS 7747
Journal of Polymer Science: Part A: Polymer ChemistryDOI 10.1002/pola
Table
4.
Aggregate
Morpholog
iesandSizeDistribution
sfrom
TEM
andDLSMea
suremen
tsfrom
Dilute
Solution
sof
POEGMA-PS-POEGMA
Dilute
AqueousSolution
s
Sample
Structure
Weight
Ratioa
Mw/M
nb
Con
c.(g
dm
�3)
DcTEM
(nm)c
Morpholog
yD
HDLS
(nm)d
Solution
Appea
rance
Nagge
ABA
1POEGMA16-PS75-POEGMA16
1.20
1.27
120�
3Spherical
27
Translucentwhite
494
ABA
2POEGMA25-PS86-POEGMA25
1.98
1.26
1–
–30
Translucentwhite
–ABA
3POEGMA43-PS86-POEGMA43
2.91
1.27
124�
4–
31
Translucentblue/white
745
ABA
4POEGMA59-PS75-POEGMA59
4.5
1.27
122�
3Spherical
27
Cleartransp
arent
658
ABA
5POEGMA79-PS75-POEGMA79
5.33
1.26
120�
4Spherical
39
Cleartransp
arent
494
ABA
6POEGMA98-PS75-POEGMA98
7.47
1.37
1–
–35
Cleartransp
arent
–ABA
7POEGMA92-PS54-POEGMA92
9.74
1.26
1–
–33
Cleartransp
arent
–ABA
8POEGMA16-PS204-POEGMA16
0.45
1.19
132�
3SphericalCylindrical
43
Opaquewhite
745
ABA
8POEGMA16-PS204-POEGMA16
0.45
1.19
0.1
34�
3Spherical
(46.2)
Translucentwhite
893
ABA
8POEGMA16-PS204-POEGMA16
0.45
1.19
0.01
33�
4Spherical
(56.6)
Translucentwhite
816
ABA
10
POEGMA26-PS183-POEGMA26
0.82
1.19
137�
5SphericalCylindrical
45
Opaquewhite
1,283
ABA
11POEGMA37-PS204-POEGMA37
1.04
1.24
137�
5SphericalCylindrical
39
Opaquewhite
1,940
ABA
13
POEGMA63-PS200-POEGMA63
1.82
1.15
1–
–39
Translucentblue/white
–ABA
14
POEGMA64-PS183-POEGMA64
2.01
1.27
120�
3Spherical
31
Translucentblue/white
203
ABA
15
POEGMA80-PS200-POEGMA80
2.31
1.19
126�
4SphericalCylindrical
32
Translucentblue/white
407
aWeightratioof
hydrophilic
(POEGMA)to
hydrophob
ic(PS)block
s.bPolydispersity
index
ofcopolymer
sample
from
SEC.
cDiameter
ofmicelle
PScore
from
TEM.
dHydrodynamic
diameter
from
DLS.
eAggregation
number
calculatedfrom
Dc.
7748 HOLDER ET AL.
Journal of Polymer Science: Part A: Polymer ChemistryDOI 10.1002/pola

In all cases the sizes of the spherical micellesmeasured by DLS was larger than that recordedby TEM. TEM measures the diameters of the non-stained cores for such micelles and is thereforereflective only of the dimensions of the phase-sep-arated polystyrene core component.66 The calcu-lated lengths of the PS chains for the PS75 and PS
86 samples (fully extended) are 17.4 and 20.0 nm,respectively, and these values correspond well thetypical diameters measured by TEM of 20–24 nmand suggest a significant degree of stretching forthe PS chains. The calculated lengths of the PSchains for the PS183-PS204 samples (fullyextended) are 42.5–47.4 nm which are signifi-cantly longer than the diameters measured by
TEM of 32–37 nm for copolymers 8, 10, and 11.This suggests that a higher degree of coiling forthe PS chains may be occurring in these micellesthan in the shorter PS chain aggregates. Howevermore data is required before any firm judgmentsabout degree of coiling in the cores can be made.The diameters measured for the PS cores by TEMfor ABA 14 and 15 of 20 and 26 nm are signifi-cantly smaller than expected, however, and thesesmall cores are apparently reflected in the DLSdiameters of 31 and 32 nm and low aggregationnumbers.
While no immediate trend is visible uponinspection of the data in Table 4, a plot of totalchain ‘‘length’’ (the total number of carbon–carbon
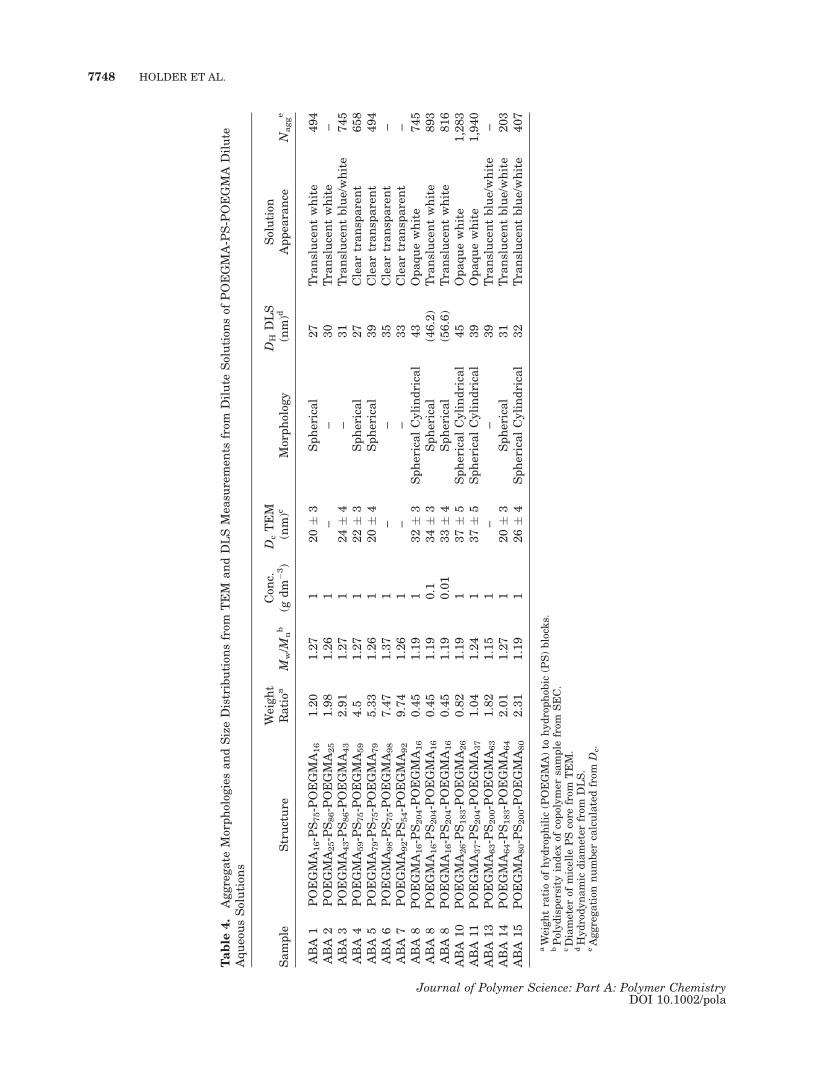
Figure 5. TEM pictures for the aggregates in solution of POEGMA-PS-POEGMAfrom the long PS group: (a) ABA 10; (b) ABA 8; (c) ABA 11.
SYNTHESIS OF ABA AMPHIPHILIC BLOCK COPOLYMERS 7749
Journal of Polymer Science: Part A: Polymer ChemistryDOI 10.1002/pola
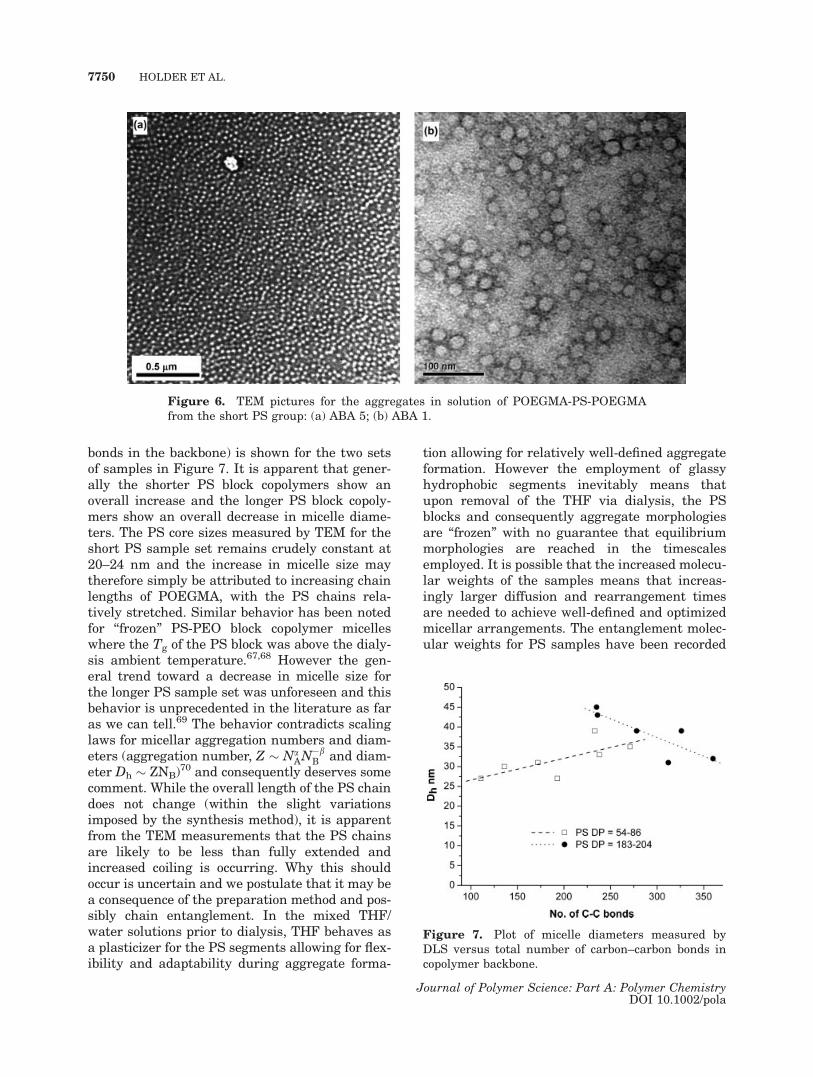
bonds in the backbone) is shown for the two setsof samples in Figure 7. It is apparent that gener-ally the shorter PS block copolymers show anoverall increase and the longer PS block copoly-mers show an overall decrease in micelle diame-ters. The PS core sizes measured by TEM for theshort PS sample set remains crudely constant at20–24 nm and the increase in micelle size maytherefore simply be attributed to increasing chainlengths of POEGMA, with the PS chains rela-tively stretched. Similar behavior has been notedfor ‘‘frozen’’ PS-PEO block copolymer micelleswhere the Tg of the PS block was above the dialy-sis ambient temperature.67,68 However the gen-eral trend toward a decrease in micelle size forthe longer PS sample set was unforeseen and thisbehavior is unprecedented in the literature as faras we can tell.69 The behavior contradicts scalinglaws for micellar aggregation numbers and diam-eters (aggregation number, Z � Na
AN�bB and diam-
eter Dh � ZNB)70 and consequently deserves some
comment. While the overall length of the PS chaindoes not change (within the slight variationsimposed by the synthesis method), it is apparentfrom the TEM measurements that the PS chainsare likely to be less than fully extended andincreased coiling is occurring. Why this shouldoccur is uncertain and we postulate that it may bea consequence of the preparation method and pos-sibly chain entanglement. In the mixed THF/water solutions prior to dialysis, THF behaves asa plasticizer for the PS segments allowing for flex-ibility and adaptability during aggregate forma-
tion allowing for relatively well-defined aggregateformation. However the employment of glassyhydrophobic segments inevitably means thatupon removal of the THF via dialysis, the PSblocks and consequently aggregate morphologiesare ‘‘frozen’’ with no guarantee that equilibriummorphologies are reached in the timescalesemployed. It is possible that the increased molecu-lar weights of the samples means that increas-ingly larger diffusion and rearrangement timesare needed to achieve well-defined and optimizedmicellar arrangements. The entanglement molec-ular weights for PS samples have been recorded
Figure 6. TEM pictures for the aggregates in solution of POEGMA-PS-POEGMAfrom the short PS group: (a) ABA 5; (b) ABA 1.
Figure 7. Plot of micelle diameters measured byDLS versus total number of carbon–carbon bonds incopolymer backbone.
7750 HOLDER ET AL.
Journal of Polymer Science: Part A: Polymer ChemistryDOI 10.1002/pola
as 13,00056,57 and consequently the shorter PSsamples are not entangled and the longer PSchains are entangled in the solid state. Withincreasing overall molecular weights (throughincrease in POEGMA length), the time necessaryfor disentanglement (to adopt more extended PSchains) is increased for the long set (DP ¼183–204) of block copolymers. It is noticeable thatthe longer PS block copolymers formed cylindricalmicelles in significant quantities, including Y-branched cylindrical micelles [Fig. 5(b)].71,72 Thebranching of cylindrical micelles, that is, both theformation of networks and individual Y-junctions,is associated with defect formation and has beenattributed to a frustrated packing of the polymersegments inside the aggregate.73,74 Significantdifferences in molecular packing behaviorbetween a long and short PS sample at the air–water interface support this interpretation (seesubsequent section).
However, this is presently speculated as a sub-ject area of possible interest for future study. It isencouraging that well-dispersed colloidal solu-tions were prepared with no significant insolublefraction remaining. Where solutions were repeat-edly prepared near identical aggregate distribu-tions and morphologies were obtained. Further-more no changes in aggregate morphology wereobserved by decreasing the concentration of thepreparative solution as observed for ABA 8 (Table4), though no cylindrical micelles were observedfor the lower concentrations and the micelle sizeincreased (by DLS).
Self-Organization at the Air–Water Interface
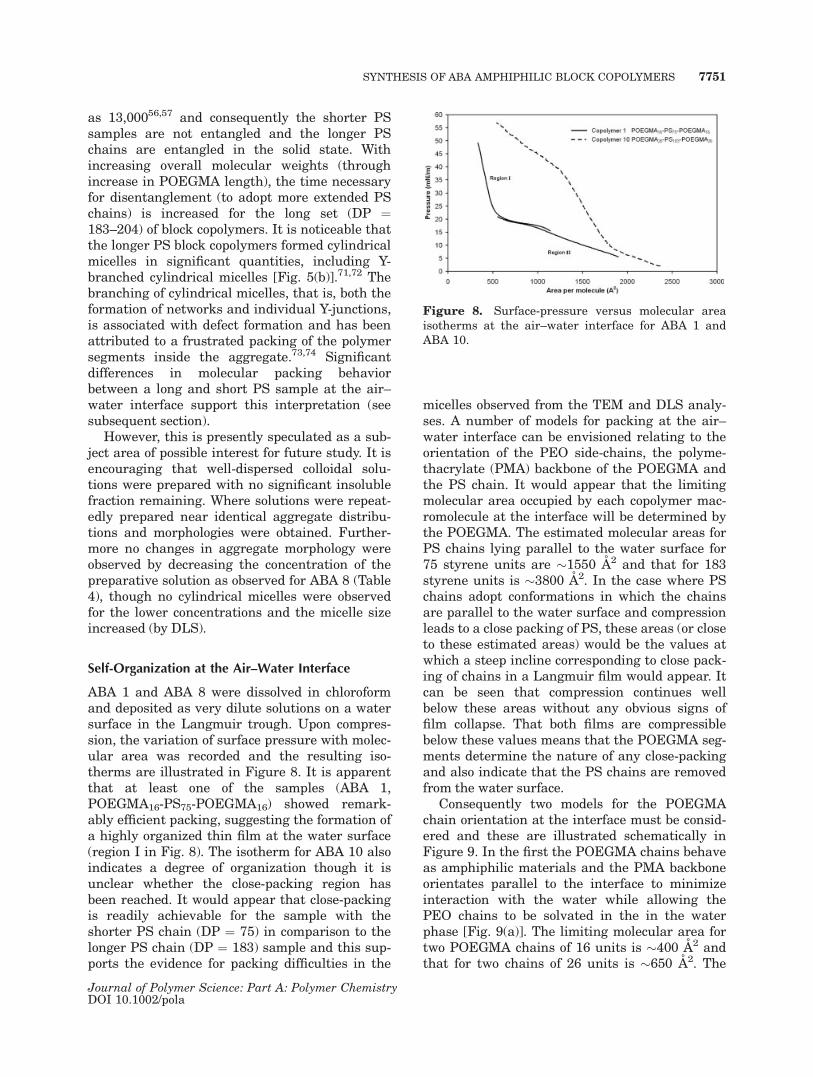
ABA 1 and ABA 8 were dissolved in chloroformand deposited as very dilute solutions on a watersurface in the Langmuir trough. Upon compres-sion, the variation of surface pressure with molec-ular area was recorded and the resulting iso-therms are illustrated in Figure 8. It is apparentthat at least one of the samples (ABA 1,POEGMA16-PS75-POEGMA16) showed remark-ably efficient packing, suggesting the formation ofa highly organized thin film at the water surface(region I in Fig. 8). The isotherm for ABA 10 alsoindicates a degree of organization though it isunclear whether the close-packing region hasbeen reached. It would appear that close-packingis readily achievable for the sample with theshorter PS chain (DP ¼ 75) in comparison to thelonger PS chain (DP ¼ 183) sample and this sup-ports the evidence for packing difficulties in the
micelles observed from the TEM and DLS analy-ses. A number of models for packing at the air–water interface can be envisioned relating to theorientation of the PEO side-chains, the polyme-thacrylate (PMA) backbone of the POEGMA andthe PS chain. It would appear that the limitingmolecular area occupied by each copolymer mac-romolecule at the interface will be determined bythe POEGMA. The estimated molecular areas forPS chains lying parallel to the water surface for75 styrene units are �1550 A2 and that for 183styrene units is �3800 A2. In the case where PSchains adopt conformations in which the chainsare parallel to the water surface and compressionleads to a close packing of PS, these areas (or closeto these estimated areas) would be the values atwhich a steep incline corresponding to close pack-ing of chains in a Langmuir film would appear. Itcan be seen that compression continues wellbelow these areas without any obvious signs offilm collapse. That both films are compressiblebelow these values means that the POEGMA seg-ments determine the nature of any close-packingand also indicate that the PS chains are removedfrom the water surface.
Consequently two models for the POEGMAchain orientation at the interface must be consid-ered and these are illustrated schematically inFigure 9. In the first the POEGMA chains behaveas amphiphilic materials and the PMA backboneorientates parallel to the interface to minimizeinteraction with the water while allowing thePEO chains to be solvated in the in the waterphase [Fig. 9(a)]. The limiting molecular area fortwo POEGMA chains of 16 units is �400 A2 andthat for two chains of 26 units is �650 A2. The
Figure 8. Surface-pressure versus molecular areaisotherms at the air–water interface for ABA 1 andABA 10.
SYNTHESIS OF ABA AMPHIPHILIC BLOCK COPOLYMERS 7751
Journal of Polymer Science: Part A: Polymer ChemistryDOI 10.1002/pola

other model would involve the full solvation of thePEOGMA blocks as illustrated schematically inFigure 9(b) in which case the limiting area wouldcorrespond to the cross-sectional area of aPOEGMA ‘‘cylinder’’ and be �380 A2 for one chainand 760 A2 for two chains (presuming no interdi-gitation). The cross-sectional area of an atactic PSchain is �92 A2, and consequently is far smallerthan the POEGMA head groups (either model).Space considerations would not allow for the closepacking and hence perpendicular orientation ofthe PS chains away from the water, and thereforeit can be presumed they would adopt coiled con-formations as illustrated in Figure 9.
A linear extrapolation to the X-axis of region Iof the copolymer 1 isotherm gives an intercept of647 A2. This is larger than the limiting moleculararea for two POEGMA chains with PMA back-bones parallel to the surface (�400 A2) and it issmaller than calculated for the fully solvatedPOEGMA model (760 A2). However, in the lattercase the crude model does not take into accountinterdigitation that is likely to occur to someextent between PEO chains. The model with PMAorientated parallel to the water surface seems theless likely postulate on the basis of other observa-tions such as the complete solubility of POEGMAin water (below the LCST, see below) and no evi-dence of intermolecular aggregate formation hasbeen found by us or other groups.75,76 Conse-quently the packing model illustrated in Figure9(b) is the most likely arrangement.
Cloud Point Temperatures of Micelle Solutions
Many hydrophilic polymers possess a lower criti-cal solution temperature (LCST), below this tem-perature a single phase molecular solution exists,and above it the polymer becomes hydrophobicand precipitates from solution. The polymer loseswater solubility by the breaking of its hydrogenbonds with water and a cloudy dispersion resultsat the cloud point temperature (TCP).
7,77–81 Poly-mers and copolymers that display this property insolution are consequently thermoresponsivematerials and are of considerable current inter-est.82–89
Since the outer corona of amphiphilic block co-polymer aggregates consist of a hydrophilic block,such materials display an LCST as a result.Numerous studies of poly(N-isopropylacrylamide)(PNIPAM) block copolymers have demonstratedthis effect.90–93 PNIPAM displays a decrease inthe LCST with increasing chain length, becomingmore hydrophobic with increasing MW despitethe nominal hydrophobic part to hydrophilic partsremaining constant.94,95 This has been most con-vincingly related to higher values of the Flory-Huggins interaction parameter (vc) of the lowerMW polymer compared to the higher MW poly-mer, and higher values of DH and DS associatedwith the phase separation of low MW chains.96,97
In contrast, studies on polyoxyalkylene triblockcopolymers (containing linear PEO chains) inaqueous solution by Chu et al. have demonstratedthat an increase in the hydrophobic block lengthof the amphiphile leads to an exponentialdecrease in TCP and an increase in the hydrophilicblock length leads to a linear increase in TCP.
98
Well-defined POEGMA samples with terminalmethyl ether groups with one ethylene oxide unitwere shown to be insoluble in water, those withtwo EO units displayed a TCP of 26 �C, and thosewith three EO units displayed a TCP of 52 �C.98
The TCP of POEGMAwith an average of 8/9 unitsof EO was observed to be �90 �C.75,76 We havesynthesized a range of POEGMA samples (withthe OEGMA monomer having an average Mn of300 corresponding to 4/5 EO units) with DPs from20 to 130 and their TCP values were determinedby simple visual turbidity observations. Thesesamples displayed TCP values from 60 to 64 �C;decreasing with increasing molecular weights inaccordance with previously reported results.98
These are in broad agreement for values thatwould be expected given the average of 4/5 unitsof EO in our samples.75,76,98,99 Concentration was
Figure 9. Schematic representations of possible mo-lecular packing arrangements for ABA copolymers atthe air–water interface.
7752 HOLDER ET AL.
Journal of Polymer Science: Part A: Polymer ChemistryDOI 10.1002/pola

observed to have little effect on these values (seeSupp. Info. for exact values).
The TCP values of solutions of the POEGMA-PS-POEGMA aggregate samples which weremilky white (such as ABA 8, 10, and 11) could notbe easily determined as a result of their high tur-bidity at room temperature. TCP values of eightcopolymer samples (six copolymers with PS DP of54–86 and 2 with a PS DP of 200) which werelargely transparent at room temperature weredetermined by simple visual turbidity observa-tions. The values for the low PS DPs (ABA 1–4, 6,and 7) were studied as a function of the length ofthe hydrophilic blocks and compared with the twosamples with long PS blocks (ABA 14 and 15).The average values taken from two measure-ments on heating are given in Table 5 and plots ofTCP against the DP of POEGMA are given inFigure 10. The TCP values fall in the range 93–76 �C upon increasing DPPOEGMA and are signifi-cantly higher than those of the POEGMA sam-ples. This was initially surprising given theexpected reduced mobility of the ends of thePOEGMA chains (at the PS-POEGMA interface).However, a similar phenomenon was recorded byLaschewsky et al. for a sample of poly[oligo(ethy-lene glycol methyl ether) methacrylate]-block-poly(n-butyl acrylate) (POEGMA-b-PBA).100 Inthis case the copolymer was observed to show aTCP of 93 �C compared to 83 �C for the POEGMAhomopolymer. They ascribed this behavior to ahindered collapse of the polymer chains in a con-fined geometry, similar to the behavior of tetheredpolymer chains on surfaces where increases inLCSTwere also observed.101
The decrease in TCP with increasing DPPOEGMA
contrasts with the behavior of the PEO-polyalky-lene block copolymers studied by Chu et al. andmimics that of the POEGMA samples. However,there is a significantly larger drop in TCP withincreasing MW (�11 �C) compared to POEGMA
over the same DPPOEGMA range (�2.5 �C). It isfurthermore notable that the aggregate solutionswith the long PS blocks displayed TCP values �5–6 �C higher than similar samples with short PSblocks. It is apparent that the PS component ofthese block copolymer aggregates has an effect onthe solution behavior of the POEGMA component(and hence of the aggregates themselves) thoughwhether aggregate size or morphology is causingthese effects remains uncertain and further studyis needed. If the cause of the increased TCP is theconfined geometry at the interface of PS andPOEGMA chains then determining the relation-ship between chain density and geometry at suchinterfaces for such aggregates is crucial for futureapplications. That the LCST behavior of the blockcopolymer aggregates is not simply an extrapola-tion of the linear analogues of the hydrophiliccomponent is of considerable importance in thepreparation and application of thermoresponsive
Table 5. Cloud Point Temperatures for POEGMA-PS-POEGMA Copolymers
Sample Structure Weight Ratio POEGMA:PS CP (�C)
ABA 1 POEGMA16-PS75-POEGMA16 1.20 94.9ABA 2 POEGMA25-PS86-POEGMA25 1.66 82.8ABA 3 POEGMA43-PS86-POEGMA43 2.86 81.2ABA 4 POEGMA59-PS75-POEGMA59 4.50 81.5ABA 6 POEGMA98-PS75-POEGMA98 7.47 78.1ABA 7 POEGMA92-PS54-POEGMA92 9.74 79.4ABA 14 POEGMA64-PS183-POEGMA64 2.01 84.9ABA 15 POEGMA80-PS200-POEGMA80 2.31 84.8
Figure 10. Plot of the cloud point temperaturesagainst the degree of polymerization of POEGMA for var-ious POEGMA-PS-POEGMA copolymer aggregate solu-tions and POEGMAsamples at three concentrations.
SYNTHESIS OF ABA AMPHIPHILIC BLOCK COPOLYMERS 7753
Journal of Polymer Science: Part A: Polymer ChemistryDOI 10.1002/pola

block copolymer aggregates based on OEGMAand related materials.76,102
Stability of the Colloidal Solutions
Control of the stability of colloidal suspensions iscrucial in their preparation and to their applica-tions. The stability of the POEGMA-PS-POEGMAABA 10 and ABA 11 aggregates in aqueous solu-tion was evaluated by measuring the evolutionand changes of the particles size with time byDLS techniques. The scattering intensity of thesolutions was recorded over a period of 918 h.Over this period no significant change in particlesizes, relative proportions, or sedimentation ofeither solution was observed. A plot of the particlesize of the micelles of ABA 10 and ABA 11 versustime is shown in Figure 11 (see Supp. Info. parti-cle size overlays for ABA 10). Thus, the colloidalsolutions of POEGMA-PS-POEGMA are stable atleast up to 38 days.
CONCLUSIONS
Novel amphiphilic block copolymers of polysty-rene with comb-like blocks of POEGMA havebeen synthesized by ATRP and their self-assem-bly in dilute aqueous solutions studied. Thecopolymers possessed low critical aggregation con-centrations and formed spherical micelles asexpected from the relative weight ratios of thehydrophilic (POEGMA) to hydrophobic (PS) com-ponents. DLS, TEM, and surface pressure–areaisotherm studies all support the conclusion that
there is significant difference in packing behaviorbetween samples with different lengths of PS.This difference was tentatively related to the criti-cal entanglement chain length of the polystyreneblock. Furthermore the cloud point temperaturesof the aggregates formed were significantly higherthan the simple homopolymer analogues which, ifdemonstrated to be a general phenomenon, willbe of considerable importance to potential app-lications of thermoresponsive block copolymersolutions.
The authors thank the EPSRC for support from grantnumber GR/R37463 and the loan of the Malvern HPPSfrom the EPSRCEngineering Instrument Loan Pool.
REFERENCES AND NOTES
1. Tuzar, Z.; Kratochvil, P. In Surface and ColloidScience, Matijevic, E., Ed.; Plenum Press: NewYork, 1993; Vol. 15, pp 1–35.
2. Discher, B. M.; Hammer, D. A.; Bates, F. S.; Dis-cher, D. E. Curr Opin Colloid Interface Sci 2000,5, 125–131.
3. Alexandridis, P.; Holzwarthf, J. F.; Hatton, T. A.Macromolecules 1994, 27, 2414–2425.
4. Alexandridis, P.; Hatton, T. A. Colloids Surf A1995, 96, 1–46.
5. Nakashima, K.; Bahadur, P. Adv Colloid Inter-face Sci 2006, 123, 75–96.
6. Jain, N. J.; Aswal, V. K.; Goyal, P. S.; Bahadur,P. J Phys Chem B 1998, 102, 8452–8458.
7. Bahadur, P.; Pandya, K. Langmuir 1992, 8,2666–2670.
8. Zhang, L.; Eisenberg, A. Polym Adv Technol1998, 9, 677–699.
9. Zhang, L.; Eisenberg, A. Science 1995, 268,1728–1731.
10. Zhang, L.; Eisenberg, A. J Am Chem Soc 1996,118, 3168–3181.
11. Lutz, J. F. Polym Int 2006, 55, 979–993.12. Gohy, J. F. Adv Polym Sci 2005, 190, 65–136.13. Schmolka, I. R. J Am Oil Chem Soc 1977, 54,
110–116.14. Kabanov, A. V.; Batrakova, E. V.; Alakov, V. Y. J
Controlled Release 2002, 82, 189–212.15. Quirk, R. P.; Kinning, D. J.; Fetters, L. J. In
Comprehensive Polymer Science; Allen, G.; Bev-ington, J. C.; Aggarwal, S. L., Eds.; PergamonPress: Oxford, 1989; Vol. 7, pp 1–26.
16. Kato, M.; Kamigaito, M.; Sawamoto, M.; Higa-shimura, T. Macromolecules 1995, 28, 1721–1723.
17. Wang, J. S.; Matyjaszewski, K. J Am Chem Soc1995, 117, 5614–5615.
Figure 11. Plot of POEGMA-PS-POEGMA micellediameters versus time demonstrating colloidal stability.
7754 HOLDER ET AL.
Journal of Polymer Science: Part A: Polymer ChemistryDOI 10.1002/pola

18. Percec, V.; Barboiu, B. Macromolecules 1995, 28,7970–7972.
19. Matyjaszewski, K.; Gaynor, S. G. Macromole-cules 1997, 30, 7042–7049.
20. Matyjaszewski, K.; Miller, P. J.; Fossum, E. ApplOrg Chem 1998, 12, 667–673.
21. Wang, X. S.; Jackson, R. A.; Armes, S. P. Macro-molecules 2000, 33, 255–257.
22. Wang, X. S.; Lasceles, S. F.; Jackson, R. A.;Armes, S. P. Chem Commun 1999, 1817–1818.
23. Wang, X. S.; Armes, S. P. Macromolecules 2000,33, 6640–6647.
24. Robinson, K. L.; Khan, M. A.; Banez, M. V. D.Macromolecules 2001, 34, 3155–3158.
25. Ashford, E. J.; Naldi, V.; O’Dell, R.; Billingham,N. C.; Armes, S. P. Chem Commun 1999, 1285–1286.
26. Zeng, F.; Shen, Y.; Zhu, S.; Pelton, R. J PolymSci Part A: Polym Chem 2000, 38, 3821–3827.
27. Save, M.; Weaver, J. V. M.; Armes, S. P. Macro-molecules 2002, 35, 1152–1159.
28. Hui, S. W.; Kuhl, T. L.; Guo, Y. Q.; Israelachvili,J. Colloids Surf B 1999, 14, 213–222.
29. Yokoyama, M.; Kwon, G. S.; Okano, T.; Sakurai,Y.; Naito, M.; Kataoka, K. J Controlled Release1994, 28, 59–65.
30. Otsuka, H.; Nagasaki, Y.; Kataoka, K. Curr OpinColloid Interface Sci 2001, 6, 3–10.
31. Hamley, I. W. The Physics of Block Copolymers,1st ed.; Oxford Science Publication: Oxford,1998; Chapters 3 and 4.
32. Bhattacharya, A.; Misra, B. N. Prog Polym Sci2004, 29, 767–814.
33. Wesslen, B.; Wesslen, B. K. J Polym Sci Part A:Polym Chem 1992, 30, 355–362.
34. Bo, G.; Wesslen, B.; Wesslen, B. K. J Polym SciPart A: Polym Chem 1992, 30, 1799–1808.
35. Wesslen, B.; Wesslen, B. K. J Polym Sci Part A:Polym Chem 1989, 27, 3915–3926.
36. Ishizone, T.; Han, S.; Hagiwara, M.; Yokoyama,H. Macromolecules 2006, 39, 962–970.
37. Lutz, J. F. J Polym Sci Part A: Polym Chem2008, 46, 3459–3470.
38. Tao, L.; Mantovani, G.; Lecolley, F.; Haddleton,D. M. J Am Chem Soc 2004, 126, 13220–13221.
39. Ma, H. W.; Hyun, J. H.; Stiller, P.; Chilkoti, A.Adv Mater 2004, 16, 338–341.
40. Hyun, J. H.; Ma, H. W.; Zhang, Z. P.; Beebe, T.P.; Chilkoti, A. Adv Mater 2003, 15, 576–579.
41. Yi, Z.; Liu, X.; Jiao, Q.; Chen, E.; Chen, Y.;Xi F.J Polym Sci Part A: Polym Chem 2008, 46,4205–4217.
42. Gu, L.; Shen, Z.; Feng, C.; Li, Y.; Lu, G.; Huang,X. J Polym Sci Part A: Polym Chem 2008, 46,4056–4069.
43. Park, C. W.; Lee, S. J.; Kim, D.; Lee, D. S.; Kim,S. C. J Polym Sci Part A: Polym Chem 2008, 46,1954–1963.
44. Ganapathy, H. S.; Hwang, H. S.; Lee, M. Y.;Jeong, Y. T.; Gal, Y. S.; Lim, K. T. J Mater Sci2008, 43, 2300–2306.
45. Liu, H.; Jiang, X. Z.; Fan, J.; Wang, G. H.; Liu,S. Y. Macromolecules 2007, 40, 9074–9083.
46. Rao, J. Y.; Xu, J.; Luo, S. Z.; Liu, S. Y. Langmuir2007, 23, 11857–11865.
47. Robinson, K. L.; de Paz-Banez, M. V.; Wang, X.S.; Armes, S. P. Macromolecules 2001, 34, 5799–5805.
48. Kurjata, J.; Chojnowski, J.; Yeoh, C.-T.; Rossi, N.A. A.; Holder, S. J. Polymer 2004, 45, 6111–6121.
49. Holder, S. J.; Rossi, N. A. A.; Yeoh, C.-T.;Durand, G. G.; Boerakker, M. J.; Sommerdijk, N.A. J. M. J Mater Chem 2003, 13, 2771–2778.
50. Street, G.; Illsley, D.; Holder, S. J. J Polym SciPart A: Polym Chem 2005, 43, 1129–1143.
51. Popescu, D. C.; Rossi, N. A. A.; Yeoh, C.-T.;Durand, G. G.; Wouters, D.; Leclere, P. E. L. G.;Thune, P.; Holder, S. J.; Sommerdijk, N. A. J. M.Macromolecules 2004, 37, 3431–3437.
52. Popescu, D. C.; Lems, R.; Rossi, N. A. A.; Yeoh,C.-T.; Holder, S. J.; Bouten, C. V. C.; Sommer-dijk, N. A. J. M. Adv Mater 2005, 17, 2324–2329.
53. Lutz, J. F.; Matyjaszewski, K. Macromol ChemPhys 2002, 203, 1385–1395.
54. Matyjaszewski, K.; Davis, K.; Patten, T. E.; Wei,M.L. Tetrahedron 1997, 53, 15321–15329.
55. Lutz, J. F.; Matyjaszewski, K. J Polym Sci PartA: Polym Chem 2005, 43, 897–910.
56. Kalyanasundaram, K.; Thomas, J. K. J AmChem Soc 1977, 99, 2039–2044.
57. Zhao, C. L.; Winnik, M. A.; Riess, G.; Croucher,M. D. Langmuir 1990, 6, 514–516.
58. Narrainen, A. P.; Pascual, S.; Haddleton, D. M. JPolymSci Part A: PolymChem 2002, 40, 439–450.
59. Khougaz, K.; Zhong, X. F.; Eisenberg, A J PhysChem B 1997, 101, 4697–4708.
60. Shen, H.; Zhang, L.; Eisenberg, A. Macromole-cules 1996, 29, 3937–3949.
61. Nicholas, C. V.; Luo, Y.-Z.; Deng, N.-J.; Attwood,D.; Collett, J. H.; Price, C.; Booth, C. Polymer1993, 34, 138–144.
62. Gao, Z.; Eisenberg, A. Macromolecules 1993, 26,7353–7360.
63. Calderara, F.; Hruska, Z.; Hurtrez, G.; Lerch, J.P.; Nugay, T.; Riess, G. Macromolecules 1994, 27,1210–1215.
64. Booth, C.; Attwood, D. Macromol Rapid Commun2000, 21, 501–527.
65. Allen, C.; Maysinger, D.; Eisenberg, A. ColloidsSurf B 1999, 16, 3–27.
66. Zheng, Y.; Won, Y. Y.; Bates, F. S.; Davis, H. T.;Scriven, L. E.; Talmon, Y. J Phys Chem B 1999,103, 10331–10334.
67. Riess, G. Prog Polym Sci 2003, 28, 1107–1170.68. Jada, A.; Hurtrez, G.; Siffert, B.; Riess, G. Mac-
romol Chem Phys 1996, 197, 3697–3710.
SYNTHESIS OF ABA AMPHIPHILIC BLOCK COPOLYMERS 7755
Journal of Polymer Science: Part A: Polymer ChemistryDOI 10.1002/pola

69. Kaewsaiha, P.; Matsumoto, K.; Matsuoka, H.Langmuir 2005, 21, 9938–9945; this referencecontains an example of a reduction in hydrody-namic diameters upon an increase in hydro-phobic and hydrophilic chain lengths for poly(hydrogenated isoprene)-b-sodium poly(styrene-sulfonate), (Ip-h2)m-b-(SSNa)n, copolymers inwater. Generally micelle size was observed toincrease with increasing chain length (m ¼ 6–88) until a drop in micelle size was recordedfrom (Ip-h2)88-b-(SSNa)50, RH ¼ 38 nm, to (Ip-h2)131-b-(SSNa)54, RH ¼ 21 nm. The minimumentanglement length for polyisoprene is for a DP�90 (Mn � 6300).
70. Forster, S.; Zisenis, M.; Wenz, E.; Antonietti, M.J.; Chem Phys 1996, 104, 9956–9970; and refer-ences therein.
71. Jain, S.; Bates, F. S. Science 2003, 300, 460–464.72. Dan, N.; Safran, S. A. Adv Colloid Interface Sci
2006, 123–126, 323–331.73. Jain, S.; Bates, F. S. Macromolecules 2004, 37,
1511–1523.74. Safran, S. A. Surf Sci 2002, 500, 127–146.75. Lutz, J. F.; Hoth, A. Macromolecules 2006, 39,
893–896.76. Lutz, J. F.; Akdemir, O.; Hoth, A. J Am Chem
Soc 2006, 128, 13046–13047.77. Huibers, P. D. T.; Shah, D. O.; Katritzky, A. R. J
Colloid Interface Sci 1997, 193, 132–136.78. Almgren, M.; Bahadur, P.; Jansson, M.; Li, P.;
Brown, W.; Bahadur, A. J Colloid Interface Sci1992, 151, 157–162.
79. Desai, P. R.; Jain, N. J.; Bahadur, P. ColloidsSurf A 2002, 197, 19–26.
80. Brown, W.; Schillen, K.; Almgren, M.; Hvidt, S.;Bahadur, P. J Phys Chem 1991, 95, 1850–1858.
81. Pandya, K.; Bahadur, P.; Nagar, T. N.; Bahadur,A. Colloids Surf A 1993, 70, 219–227.
82. Bergbreiter, D. E.; Fu, H. J Polym Sci Part A:Polym Chem 2008, 46, 186–193.
83. Zhu, J.-L.; Zhang, X.-Z.; Cheng, H.; Li, Y.-Y.;Cheng, S.-X.; Zhuo, R.-X. J Polym Sci Part A:Polym Chem 2007, 45, 5354–5364.
84. Skrabania, K.; Li, W.; Laschewsky, A. MacromolChem Phys 2008, 209, 1389–1403.
85. Kayarna, M.; Okano, T. Macromolecules 2008,41, 504–507.
86. Yamamoto, S. I.; Pietrasik, J.; Matyjaszewski, K.J Polym Sci Part A: Polym Chem 2008, 46, 194–202.
87. Pasparakis, G.; Cockayne, A.; Alexander, C. JAm Chem Soc 2007, 129, 11014–11015.
88. Van Durme, K.; Van Assche, G.; Aseyev, V.;Raula, J.; Tenhu, H.; Van Mele, B. Macromole-cules 2007, 40, 3765–3772.
89. Zhou, J. F.; Wang, L.; Yang, Q.; Liu, Q. Q.; Yu,H. J.; Zhao, Z. R. J Phys Chem B 2007, 111,5573–5580.
90. Chiantore, O.; Guaita, M.; Trossarelli, L. Makro-mol Chemie Macromol Chem Phys 1979, 180,969–973.
91. Winnik, F. M. Macromolecules 1990, 23, 233–242.
92. Schild, H. G. Prog Polym Sci 1992, 17, 163–249.93. Rzaev, Z. M. O.; Dincer, S.; Piskin, E. Prog
Polym Sci 2007, 32, 534–595.94. Xia, Y.; Yin, X.; Burke, N. A. D.; Stover, H. D. H.
Macromolecules, 2005, 38, 5937–5934.95. Xia, Y.; Yin, X.; Burke, N. A. D.; Stover, H. D. H.
Macromolecules, 2006, 39, 2275–2283.96. Lessard, D. G.; Ousalem, M.; Zhu, X. X. Can J
Chem 2001, 79, 1870–1874.97. Patterson, D. Macromolecules 1969, 2, 672–
677.98. Liu, T.; Nace, V. M.; Chu, B. J Phys Chem B
1997, 101, 8074–8078.99. Han, S.; Hagiwara, M.; Ishizone, T. Macromole-
cules 2003, 36, 8312–8319.100. Mertoglu, M.; Garnier, S.; Laschewsky, A.; Skra-
bania, K.; Storsberg, J. Polymer 2005, 46, 7726–7740.
101. Wischerhoff, E.; Zacher, T.; Laschewsky, A.;Rekaı, E.-D. Angew Chem Int Ed 2000, 39,4602–4604.
102. Lutz, J. F.; Weichenhan, K.; Akdemir, O.; Hoth,A. Macromolecules 2007, 40, 2503–2508.
7756 HOLDER ET AL.
Journal of Polymer Science: Part A: Polymer ChemistryDOI 10.1002/pola
Copyright © 2022 FDOKUMEN




















