
(PDF) The Aquilegia genome provides insight into adaptive ...
Upload
independentCategory
view
0download
0
The molecular analysis of Trypanosoma cruzimetallocarboxypeptidase 1 provides insight into fold andsubstrate specificity
Gabriela Niemirowicz,1 Daniel Fernández,2
Maria Solà,3 Juan J. Cazzulo,1 Francesc X. Avilés2
and F. Xavier Gomis-Rüth3*1Instituto de Investigaciones Biotecnológicas – InstitutoTecnológico de Chascomús (IIB-INTECH), UniversidadNacional de General San Martín-CONICET, AvenidaGeneral Paz 5445, AR-1650 San Martín, Buenos Aires,Argentina.2Institut de Biotecnologia i de Biomedicina andDepartament de Bioquímica i Biologia Molecular,Facultat de Ciències, Universitat Autònoma deBarcelona, E-08193 Bellaterra, Spain.3Department of Structural Biology, Molecular BiologyInstitute of Barcelona (CSIC), Barcelona Science Park,c/Baldiri Reixac, 10–12 and 15–21, E-08028 Barcelona,Spain.
Summary
Trypanosoma cruzi is the aetiological agent ofChagas’ disease, a chronic infection that affects mil-lions in Central and South America. Proteolyticenzymes are involved in the development and pro-gression of this disease and two metallocarboxypep-tidases, isolated from T. cruzi CL Brener clone, haverecently been characterized: TcMCP-1 and TcMCP-2.Although both are cytosolic and closely related insequence, they display different temporary expres-sion patterns and substrate preferences. TcMCP-1removes basic C-terminal residues, whereasTcMCP-2 prefers hydrophobic/aromatic residues.Here we report the three-dimensional structure ofTcMCP-1. It resembles an elongated cowry, with along, deep, narrow active-site cleft mimicking theaperture. It has an N-terminal dimerization subdo-main, involved in a homodimeric catalytically activequaternary structure arrangement, and a proteolyticsubdomain partitioned by the cleft into an upper anda lower moiety. The cleft accommodates a catalyticmetal ion, most likely a cobalt, which is co-ordinated
by residues included in a characteristic zinc-bindingsequence, HEXXH and a downstream glutamate.The structure of TcMCP-1 shows strong topologicalsimilarity with archaeal, bacterial and mammalianmetallopeptidases including angiotensin-convertingenzyme, neurolysin and thimet oligopeptidase. Acrucial residue for shaping the S1� pocket in TcMCP-1,Met-304, was mutated to the respective residue inTcMCP-2, an arginine, leading to a TcMCP-1 variantwith TcMCP-2 specificity. The present studies pavethe way for a better understanding of a potentialtarget in Chagas’ disease at the molecular level andprovide a template for the design of novel therapeuticapproaches.
Introduction
Neglected diseases that do not affect developed countriesare burdens for poor populations (O’Connell, 2007). Theyare largely ignored by medical science, first-world publicopinion and pharmaceutical companies (Gutteridge,1997; Beyrer et al., 2007). Such diseases include para-sitic infections like malaria, leishmaniasis, sleeping sick-ness and Chagas’ disease. The latter, also known asAmerican trypanosomiasis, is caused by the flagellateprotozoan Trypanosoma cruzi and it affects an estimated16–18 million people mainly in Central and SouthAmerica, causing up to 45 000 deaths each year(Docampo, 2001; Barrett et al., 2003; Sealey-Cardonaet al., 2007). The intracellular lifestyle of T. cruzi enablesthe parasite to evade host defences and results in achronic disease stage. Thus, after years of asymptomaticinfection, 15–30% of patients develop signs of organdamage, which produce characteristic cardiac, digestiveor nervous forms of chronic Chagas’ disease; death is afrequent outcome. Currently available medication, suchas nitrofurans (nifurtimox) and nitroimidazoles (benznida-zole), are effective during the acute phase of the infectionbut are little active in the chronic stage of the disease(Cazzulo, 2002). Moreover, in addition to serious side-effects, resistance against both compounds leads toincreasing treatment failures (Urbina, 2002). These prob-lems, together with the current lack of vaccines, make the
Accepted 2 September, 2008. *For correspondence. E-mail [email protected]; Tel. (+34) 934020186; Fax (+34) 934034979.
Molecular Microbiology (2008) 70(4), 853–866 � doi:10.1111/j.1365-2958.2008.06444.xFirst published online 3 October 2008
© 2008 The AuthorsJournal compilation © 2008 Blackwell Publishing Ltd

development of novel therapeutic strategies againstChagas’ disease necessary (Urbina, 2001; Barrett et al.,2003). For this purpose, the elucidation of the moleculardeterminants of pathogen physiology and virulence on thebasis of structural information is essential (Greer et al.,1994).
In this context, the study of peptidases in protozoanparasites has acquired considerable importance over thelast decade. They play central roles in diverse processessuch as cell invasion, differentiation, cell cycle progres-sion, catabolism of host proteins and evasion of the hostimmune response (Klemba and Goldberg, 2002). There-fore, they are promising targets for the design of antimi-crobials (Potempa and Pike, 2005; Mittl and Grütter,2006). In particular, T. cruzi has been shown to haveseveral proteolytic activities (Cazzulo, 2002). Amongthem, the most abundant and best characterized is due tothe cysteine-protease cruzipain, which is an immun-odominant antigen in chronic disease. Its inhibition hasbeen shown to kill the parasite and to cure infected mice(Engel et al., 1998) and it is the only T. cruzi protease thathas been structurally analysed to date (McGrath et al.,1995).
Recently, we identified and biochemically studied twometallocarboxypeptidases (MCPs) from the T. cruziCL Brener clone (Zingales et al., 1997), TcMCP-1 andTcMCP-2 (Niemirowicz et al., 2007). These peptidasesare cytosolic enzymes that differ in their pattern ofexpression: while TcMCP-1 is present in all life stages ofT. cruzi, TcMCP-2 is mainly restricted to the stages thatoccur in the invertebrate host. Despite high sequenceidentity (64%), TcMCPs display remarkable biochemicaldifferences. Whereas TcMCP-1 cleaves off basic C-terminal residues, TcMCP-2 prefers aromatic and ali-phatic residues. Accordingly, they match, respectively, thespecificity of B-type and A-type pancreatic mammalianMCPs of the metallopeptidase (MP) tribe of relatives ofbovine carboxypeptidase A (Niemirowicz et al., 2007).However, neither TcMCP is sequentially related to theseclassic secreted MCPs, which belong to MEROPS data-base family M14 (Rawlings et al., 2006; Arolas et al.,2007). Instead, they were found to show significantsequence similarity to bacterial and archaeal MCPs of theM32 family: Thermus aquaticus carboxypeptidase (TaqC-P)(Lee et al., 1992; 1994; 1996) and Pyrococcus furiosuscarboxypeptidase (PfuCP)(Cheng et al., 1999). In addi-tion, orthologous genes to TcMCPs corresponding to hith-erto uncharacterized proteins have been retrieved fromthe genomes of related protozoa (Trypanosoma bruceiand Leishmania spp.). These parasites are the only groupof eukaryotic organisms that harbour peptidases of theM32 family so far. Relatives are also found in bacterialhuman pathogens such as Bacillus anthracis, Listeriamonocytogenes, Francisella tularensis, Vibrio cholerae,
Legionella pneumophila and several Serratia, Rickettsiaand Yersinia species. Most organisms that harbour apotential M32 orthologue also contain M14 sequences(Arndt et al., 2002).
To examine the mechanism of TcMCPs and to providea scaffold for rational drug design to advance in the treat-ment of American trypanosomiasis, we analysed thethree-dimensional structure of TcMCP-1 and the bio-chemical properties of TcMCP active-site point-mutants.
Results and discussion
Structure of TcMCP-1
The protein monomer is ellipsoidal with maximum dimen-sions ~75 (width) ¥ ~55 (height) ¥ ~60 Å (depth). Theshape is reminiscent of a cowry, with the active-site cleftlooking like the cowry aperture (Fig. 1A). According to thestandard orientation (Gomis-Rüth et al., 1993), MPs arebest displayed by looking into the active-site cleft runninghorizontally from left (non-primed side) to right (primedside) (Fig. 1A–C). This view reveals that the protein con-sists of an N-terminal dimerization subdomain (DS; Ser-0–Leu-94), which participates in dimerization and cleftdelimitation (see below), and a proteolytic subdomain,which is divided into an upper (UM; Glu-195–Arg-341 + Tyr-417–Asp-500) and a lower moiety (LM; Leu-95–Leu-194 + Val-342–Gly-416) (Fig. 1C and D). DSconstitutes the upper left of the molecule and includes thefirst four helices (a1–a4), which are arranged in two setsof roughly parallel a-hairpins (a1a2 and a3a4), whoseaxes are rotated ~130° relative to each other. DS is con-nected to LM, which is fully helical and forms the lowerhalf of the molecule. It starts with an a-hairpin (a5a6),which shapes the lower front of the molecule. Subse-quently, the loop connecting helices a6 and a7 (La6a7)leads to a7, whose axis is approximately perpendicular tothe previous a-hairpin. This helix ends at the bottom of theactive-site cleft on its non-primed side. Thereafter, loopLa7a8 disembogues into a long helix, a8, which runshorizontally on the back surface and reaches the rightflank of the molecule. Approximately half way up, aproline-induced (Pro-181) kink accounts for a change inthe helix direction of ~30° and partial unwinding of helix a8from Ser-178 to Pro-181. After a8, the polypeptide passesthrough the first cross-point between LM and UM at Leu-194–Gly-195. In contrast to DS and LM, UM shows a/btopology and starts with a segment in extended confor-mation (Gly-195–Gly-203), which runs along the rightfront and reaches the top of the molecule. Subsequently,a turn leads to a9, which crests the molecule from front toback. The following loop (La9b1) leads to the uppermoststrand (b1) of a three-stranded mixed b-sheet of connec-tivity +2, -1 and with the strands linked by same-end
854 G. Niemirowicz et al. �
© 2008 The AuthorsJournal compilation © 2008 Blackwell Publishing Ltd, Molecular Microbiology, 70, 853–866

Fig. 1. Molecular structure and topology of TcMCP-1.A. Protein monomer of TcMCP-1 in standard orientation shown with its electrostatic-potential surface (colour coding: red, < -10 kB T; blue,>10 kB T, where kB is the Boltzmann constant and T is the temperature in Kelvin). The magenta sphere pinpoints the active-site metal ion.B. Cross-sectional view of (A) after a vertical ~90° rotation to illustrate the depth and width of the channel.C. Richardson diagram (Richardson, 1985) of TcMCP-1 oriented almost as in (A) depicting helices as ribbons and b-strands as arrows, whichare labelled a1-a21 and b1-b3 respectively. The magenta sphere denotes the catalytic metal ion. Colour coding: dark blue, dimerizationsubdomain; pale green, upper moiety of the proteolytic subdomain; sky blue, lower moiety of the proteolytic subdomain.D. Topology scheme illustrating the secondary structure elements and subdomain structure and extension as shown in (C) in the sameorientation. The amino acid positions at the beginning and the end of each element are indicated (for the amino acid sequence, see fig. 2a inNiemirowicz et al., 2007 and UniProt Q6ZXC0).E. Functionally relevant quaternary structure of TcMCP-1 viewed perpendicular to the twofold axis relating the two monomers to each another.The left monomer is superimposed with its Connolly surface. The dimerization domains are coloured in orange and magenta respectively.
Structure of Trypanosoma cruzi metallocarboxypeptidase 855
© 2008 The AuthorsJournal compilation © 2008 Blackwell Publishing Ltd, Molecular Microbiology, 70, 853–866

connections. This causes the lowermost strand, b2, to runantiparallel to b1 and b3 and to a bound substrate (seebelow) (Fig. 1C and D). Strand b3 is connected with a10,the ‘active site helix’, which traverses the molecule almosthorizontally from right to left. Thereafter, the polypeptidechain enters a11, the ‘glutamate helix’, which runs directlybelow a10 from front to back. A rotation in the direction ofthe polypeptide chain of ~70° provokes that the followinghelix a12 is nearly vertical and ends at the top of themolecule. A subsequent downstream turn leads to a13,which progresses from back to front. Thereafter, thepolypeptide chain leaves UM at Arg-341 to enter ana-hairpin (a14a15) within LM, which forms the bottom ofthe molecule. After a15, a ~180° rotation in the direction ofthe polypeptide leads to the 310-helix a16, which backsa-hairpin a5a6 and leads to a17, the ‘tyrosine helix’. Thebeginning of the latter constitutes the second subdomaincross-point to re-enter UM. The helix is partially unwoundfrom Tyr-417 to Pro-419 and adopts a 310-helix con-formation. The tyrosine helix crosses the molecule fromfront to back and reaches the surface at Leu-439. Aftera17, the chain extends vertically along the back surfaceand enters a18 and a19. The latter helix opens up afterIle-464 and leads to a20. Thereafter, a turn leads to theC-terminal helix a21, which backs a8 within LM.
Quaternary structure of TcMCP-1
The quaternary structure of TcMCP-1 is a homodimer inwhich two essentially identical monomers (see Experi-mental procedures) associate through a vertical non-crystallographic twofold axis (Fig. 1E). The superstructureshows approximate dimensions ~95 (width) ¥ ~60(depth) ¥ ~75 Å (height) and confirms size-exclusionchromatography studies, in which the catalytically activeenzyme eluted with an apparent molecular mass of128 kDa, i.e. roughly twice the value of a monomer
(58 kDa) (Niemirowicz et al., 2007). This symmetric asso-ciation occurs through the upper-left frontal surface ofeach monomer and is reminiscent of the two-headedmythical god Janus, with the fully accessible active-siteclefts resembling a mouth on each of the opposite faces ofthe dimer. The axes of the clefts intersect at an angle of~55°. Dimerization occurs through an interface of 1413 Å2
(6.5% of the total monomer surface) and is shaped by theDSs via a1, La1a2 and a2 (segment Lys-13–His-50), aswell as by the concave face of the b-sheet of each UM.Further participating segments include La9b1, the begin-ning of b1 (segment Asp-224–Arg-229), Lb2b3 and thebeginning of b3 (segment Glu-243–Arg-246). Dimeriza-tion is based on 67 close contacts (< 4 Å), of which 58 aresymmetric, and includes 15 hydrophobic interactions,seven hydrogen bonds and five salt bridges (see Table 1).
Related structures – the cowrin family of MPs
As expected from previous sequence similarity searches(Niemirowicz et al., 2007), two MCPs from P. furiosus[Protein Data Bank access code (PDB) 1K9X; Arndt et al.,2002] and Thermus thermophilus (PDB 1WGZ; no publi-cation to date), ascribed to the MEROPS family M32,were identified as the closest structural homologues (seeFig. 2A and B). These proteins displayed, respectively,29% and 31% sequence identity with TcMCP-1 and couldbe superimposed on the protozoan enzyme with rms-deviations of 2.2 Å and 2.3 Å respectively. A detailedinspection of the superposed structures (see Fig. 3D)revealed that the prokaryotic and the protozoan enzymesevince a closely related chain trace throughout the entiremolecule. They all possess an N-terminal DS consisting offour a-helices and a similar relative arrangement of LMand UM. The former comprises six a-helices and the latter10 helices and a three-stranded b-sheet of identical con-nectivity in all three cases. Main insertions and deletions,
Table 1. Dimerization interactions in TcMCP-1 (distances in Å).
Hydrophobic interactions Hydrogen bonds
Arg-16A Cz – Ala-42B Cb 3.31 His-19A Ne2 – Arg-40B Ne 2.98Arg-16A Cb – Glu-16B Cb 3.65 Met-31A O – Arg-229B Nh1 2.90Tyr-17A Ce1 – Glu-46B Cg 3.87 Lys-35A N – Asp-226B O 2.82His-19A Cd2 – Ala-43B Cb 3.46 Arg-40A Ne – His-19B Ne2 2.94Met-20A Ce – Ala-24B Cb 3.68 Asp-226A O – Cys-34B N 3.41Leu-23A Cd2 – Ala-24B Ca 3.75 Asp-226A O – Lys-35B N 2.86Ala-24A Cb – Met-20B Ce 3.77 Arg-229A Nh1 – Met-31B O 2.83Ala-24A Ca – Leu-23B Cd2 3.72Ala-39A Cb – Glu-243B Cd 3.49 Salt bridgesAla-42A Cb – Arg-16B Cz 3.39 Asp-27A Od1 – Arg-246B Nh1 2.93Ala-43A Cb – His-19B Cd2 3.22 Lys-35A Nz – Asp-224B Od2 2.80Met-44A Ca – Met-20B Ce 3.49 Asp-224A Od2 – Lys-35B Nz 2.76Glu-46A Cb – Arg-16B Cb 3.72 Arg-246A Nh1 – Asp-27B Od1 2.76His-50A Ce1 – His-50B Ce1 3.25 Asp-27A Od1 – solvent – Asp-27B Od1 2.75, 2.84Glu-243A Cd – Ala-39B Cb 3.52
856 G. Niemirowicz et al. �
© 2008 The AuthorsJournal compilation © 2008 Blackwell Publishing Ltd, Molecular Microbiology, 70, 853–866

which lead to distinct local chain traces, are observed atthe N-terminus and at La6a7, La8a9, Lb2b3, Lb3a10 andwithin all the connecting loops between helices a10 anda18. The most significant deviation occurs in the segmentbetween a8 and a9, which connects LM and UM, with amaximal displacement of 10 Å (for the pair TcMCP-1 Leu-194 and PfuCP Pro-197; see Fig. 3D). These differenceswere also highlighted by seven insertion or deletionsobserved in a structure-based sequence alignment(Fig. 2B).
More distantly though still significantly related toTcMCP-1 were mammalian MCPs, dipeptidyl carboxy-peptidases and even endopeptidases, namely humanC-terminal and N-terminal domains of angiotensin-converting enzyme [PDB 1O8A (Natesh et al., 2003);PDB 2C6F (Corradi et al., 2006)], human angiotensin-converting enzyme-like carboxypeptidase (PDB1R42; Towler et al., 2004), Drosophila melanogasterangiotensin-converting enzyme-like MP (PDB 1J38; Kimet al., 2003), rat neurolysin (PDB 1I1I; Brown et al., 2001)and human thimet oligopeptidase (PDB 1S4B; Ray et al.,2004) (Fig. 2A). Structural similarity of TcMCP-1 wasalso found with the bacterial proteins dipeptidyl carbox-ypeptidase Dcp from Escherichia coli (PDB 1Y79;Comellas-Bigler et al., 2005), Bacillus stearothermophilusoligopeptidase F (PDB 2H1J; Gerdts et al., 2006),Enterocuccus faecium oligoendopeptidase F (PDB 2QR4;no publication to date) and a putative peptidase fromChlamydophila abortus (PDB 3CE2; no publication todate). These MPs belong to MEROPS families M2, M3Aand M3B. They all align in sequence with TcMCP-1 withE-values in the range of 1E-30–1E-25, but sequence simi-larity (6–7%) and rms-deviation scores (3.8–4.0 Å) aremuch lower. In any case, superposition of these structuresreveals clear homology (Fig. 3D and E). They all share acommon structural core composed of a total of three ofthe four helices found in TcMCP-1 DS, six out of the sevenhelices of LM, as well as eight of the 10 helices and thethree b-strands of UM. In addition, the catalytic metalbinding sites are equivalent (see below). Accordingly,these peptidases can be grouped in one single family.Given the shape of TcMCP-1 and the related members,which recalls a cowry shell, the name cowrins is proposedto refer to this family (Gomis-Rüth, 2008). Among thecowrin structures reported, only angiotensin-convertingenzyme-like carboxypeptidase has been analysed both inan unambiguously unbound state and in an inhibitorcomplex (Towler et al., 2004). These studies revealed alarge hinge-bending motion, similar to the action of aclosing clam shell, which narrows the active-site cleftupon substrate or inhibitor binding. Similarly, structuralanalysis of E. coli Dcp provided additional evidence for ahinge movement upon substrate binding (Comellas-Bigleret al., 2005). In addition, the latter study provided the only
structure of the cowrin family in a product complex, with acleaved tetrapeptide, and it was used for modellingstudies with TcMCP-1 (see below).
Active-site cleft of TcMCP-1
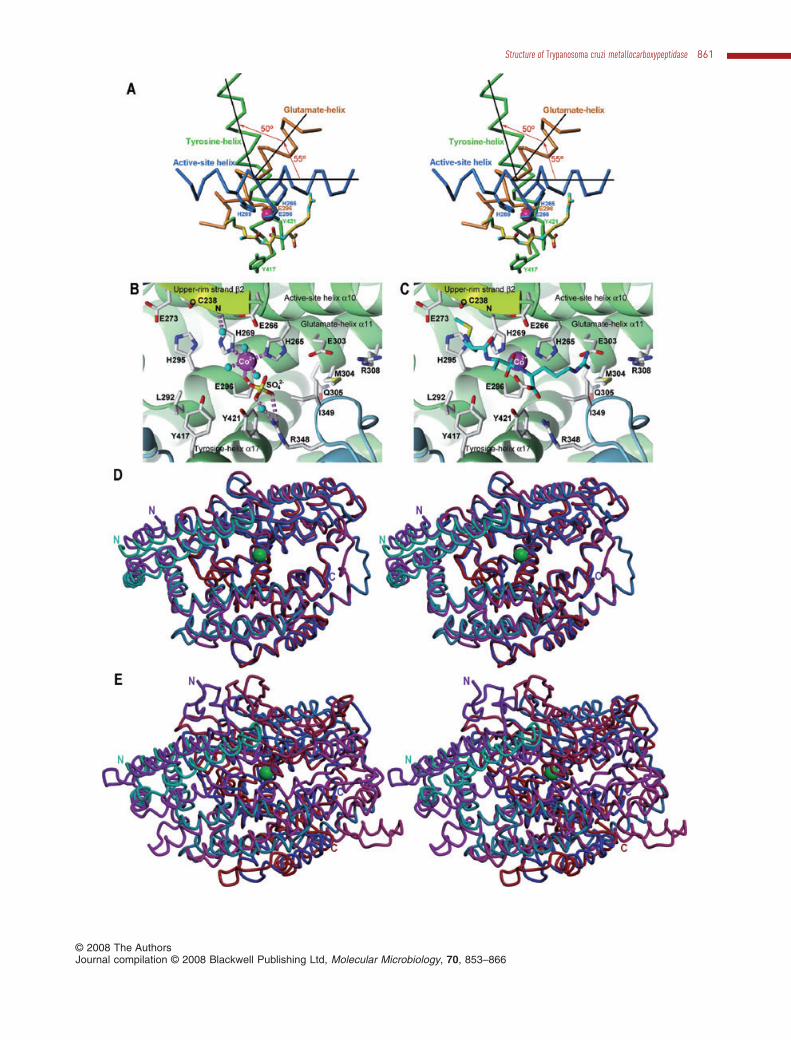
The active site cleft completely traverses the moleculefrom left to right and features a deep, narrow canyon witha minimal width of 10 Å (Fig. 1A and B). Such narrowclefts have been implicated within cowrins in the cleavageof oligopeptides or partially unfolded substrates only(Brown et al., 2001). The cleft is mostly negativelycharged and delimited by six elements (Fig. 1A and D):DS, which creates a protrusion above the cleft on itsnon-primed side; strand b2 of the three-stranded b-sheetin UM, which gives rise to an ‘upper rim’ above the cleft;the beginning of the tyrosine helix a17, the active-sitehelix a10 and the glutamate helix a11 of UM; and helix a5of the LM. The active-site helix, the glutamate helix andthe tyrosine helix constitute the core of the TcMCP-1molecule. They are placed directly below each other andtheir axes intersect on a vertical axis approximately tra-versing the Ca positions of Glu-266 (active-site helix),Ser-299 (glutamate helix) and Ala-428 (tyrosine helix) at~55° (helices a10 and a11) and ~50° (helices a11 anda17) (see Fig. 3A). The helices are tightly packed withtheir axes spaced ~7.5 Å. This is allowed for by the pres-ence of short side-chains at the two cross-point inter-faces, namely, Gly-268 of the active-site helix; Gly-297,Ser-299 and Ala-302 of the glutamate helix; and Gly-424,Ala-425 and Ala-428 of the tyrosine helix.
In contrast to MCPs of the bovine carboxypeptidase AMP tribe, which display a hallmark cul-de-sac that trapsC-terminal tails of substrates (Arolas et al., 2007), theTcMCP-1 active site is midway along a cleft that is openbeyond S1′. This is more reminiscent of endopeptidasesthan exopeptidases and it was previously described forcarboxypeptidases such as PfuCP (Arndt et al., 2002) andhuman angiotensin-converting enzyme-like carboxypepti-dase (Towler et al., 2004). The centre of the cleft floor isoccupied by a metal ion. Atomic absorption studies did notenable to identify the nature of the ion (data not shown).Therefore, it was tentatively assigned as a divalent cobaltion (Co999), based on reasonable thermal displacementparameters and the absence of major negativesA-weighted (Fobs-Fcalc)-type electron density after crystal-lographic refinement of the final model. Additional evi-dence was provided by protein purification, which hadincluded cobalt-affinity chromatography, and by the studyof the effect of bivalent cations on activity, which hadrevealed that cobalt restores activity when added to apo-forms of TcMCP-1 and -2 (Niemirowicz et al., 2007). Fur-thermore, the apo-form of closely related PfuCP wasshown to be reconstituted by Co2+ but not by Zn2+ (Cheng
Structure of Trypanosoma cruzi metallocarboxypeptidase 857
© 2008 The AuthorsJournal compilation © 2008 Blackwell Publishing Ltd, Molecular Microbiology, 70, 853–866

et al., 1999). The TcMCP-1 metal co-ordination polyhe-dron is an octahedron, with two solvent molecules (atdistances ranging from 1.8 to 2.4 Å in the four proteinmolecules observed in the asymmetric unit), His-265 Ne2(2.2–2.3 Å) and Glu-296 Oe2 (2.2–2.3 Å) on a plane withthe metal, and a further solvent molecule (2.2–2.3 Å) andHis-269 Ne2 (2.2–2.4 Å) at the apical positions (Fig. 3B).In most MPs having a zinc cation at their active-sites, themetal is co-ordinated by three protein ligands and a singlesolvent molecule in distorted tetrahedral geometry(Matthews et al., 1972; Rees et al., 1983; Gomis-Rüthet al., 1994). In contrast, the putative cobalt cation isco-ordinated in TcMCP-1 by a total of three solvent mol-ecules in addition to the three protein ligands. This isconsistent with studies revealing a higher preference ofthe latter metal for an octahedral co-ordination sphere inprotein complexes when compared with zinc (Wray et al.,2000). The two TcMCP-1 histidines engaged in metalbinding, His-265 and His-269, are provided by the active-site helix and make up, together with the general base/acid glutamate Glu-266 (see below), the short zinc-binding consensus sequence, HEXXH (amino-acid one-letter code; X for any amino acid), which is characteristicfor the ‘zincin’ tribe of MPs (Bode et al., 1993; Hooper,1994). The third protein metal ligand is a downstreamglutamate, Glu-296, which comes from the glutamatehelix. All these elements are conserved among cowrinsand this, in turn, assigns this family to the ‘gluzincin’ clanof MPs (Hooper, 1994; Gomis-Rüth, 2003; 2008).
Modelling of substrate binding
All attempts to obtain complexes of TcMCP-1 with stan-dard MCP inhibitors proved unsuccessful. Therefore, inorder to gain further insight into the structural determi-nants of substrate binding and catalysis of TcMCP-1, atetrahedral reaction-intermediate of sequence Met–Ala–Arg was modelled on the basis of the co-ordinates of acleaved substrate found in the cleft of E. coli Dcp(Comellas-Bigler et al., 2005) after optimal superpositiononto TcMCP-1 (see Fig. 3C and E). Additional anchorpoints to superpose the substrate were a sulphate anionpotentially mimicking the C-terminus of a substrate oncebound, a solvent molecule bonded to Cys-238 N from the
upper-rim strand b2, and the metal-bound solvent mol-ecules, which coincided with the oxygen atoms of thereaction-intermediate gem-diolate (see below) (Fig. 3B).This model enabled us to assess that TcMCP-1 is likely toopen and close like a hinge upon substrate binding, asfound in E. coli Dcp and human angiotensin-convertingenzyme-like carboxypeptidase (Towler et al., 2004;Comellas-Bigler et al., 2005). This motion would enableGlu-266, above the cleft, and Tyr-421 and Tyr-417, belowthe cleft, to trap a substrate. In addition, the modelenabled us to identify the protein residues shaping distinctsubsites of the cleft, S1′, S1 and S2. The S1′ or specificitypocket would be shaped by the side-chains Thr-258, Leu-300, Glu-303, Met-304, Arg-308, Tyr-356, Ala-425 andIle-593 and would be ideally conceived to accommodate asubstrate side-chain that was both basic to interact withthe Glu-303 side-chain and hydrophobic to contact theother, mostly hydrophobic, residues. The S1 pocket wouldbe shaped by Val-408, His-409, Met-414 and Tyr-417, i.e.suitable to harbour large aromatic side-chains, and the S2
pocket would be framed by Cys-238, His-269, Glu-273and His-295 and could easily accommodate a methionine(Fig. 3C).
Proposed working mechanism
The present structure, together with previous data onother cowrins and HEXXH-proteases, indicates thatTcMCP-1 most likely acts on its substrates via apromoted-water pathway with a polarized solvent mol-ecule performing a nucleophilic attack on the scissilepeptide bond in a similar manner to bovine carboxypepti-dase A and thermolysin (Matthews, 1988; Arndt et al.,2002; Towler et al., 2004; Comellas-Bigler et al., 2005).Prior to catalysis, the substrate would be anchored to theactive-site cleft of TcMCP-1 through main-chain atoms ofthe upper-rim strand b2 of the enzyme, Pro-236–Cys-238.This interaction would give rise to an antiparallel b-sheet-like binding of the substrate main chain (Fig. 4, I). TheC-terminal carboxylate group of the substrate wouldco-ordinate to Arg-348 and Tyr-421. The latter tyrosineresidue may also bind the main-chain amide nitrogenpreceding the scissile bond. Tyr-356, in the vicinity of theC-terminal carboxylate group, may also participate in
Fig. 2. Phylogenetic analysis and sequence alignment.A. Rootless phylogenetic tree showing the relatedness of structurally analysed members of the cowrin family of metallopeptidases. The valuesobtained for each branch path are the absolute number of substitutions, i.e. not single amino-acid substitutions, as each position in asequence may go through a number of changes during evolution. Therefore, genetic distance reflects relatedness much more accurately thanmere sequence identity or similarity. The acronym coding is: DMACE, Drosophila melanogaster angiotensin-converting enzyme-relatedmetalloprptidase; ECDCP, E. coli peptidyl dipeptidase Dcp; HACE2, human angiotensin-converting enzyme-related carboxypeptidase; HACEC,human ACE testis or somatic C-terminal domain; HACEN, human ACE somatic N-terminal domain; HSTOP, human thimet oligopepeptidase;NEURO, rat neurolysin; OPEPF, B. stearothermophilus oligopeptidase F; PFUCP, P. furiosus carboxypeptidase; TCMCP, T. cruzimetallocarboxypeptidase 1; TTHCP, T. thermophilus carboxypeptidase. See also (Gomis-Rüth, 2008).B. Structure-based sequence alignment of TcMCP-1 and the closest structural relatives from P. furiosus and T. thermophilus. The regularsecondary structure elements depicted above each sequence block correspond to the protozoan enzyme (see also Fig. 1D).
858 G. Niemirowicz et al. �
© 2008 The AuthorsJournal compilation © 2008 Blackwell Publishing Ltd, Molecular Microbiology, 70, 853–866

Structure of Trypanosoma cruzi metallocarboxypeptidase 859
© 2008 The AuthorsJournal compilation © 2008 Blackwell Publishing Ltd, Molecular Microbiology, 70, 853–866

substrate-anchoring. Upon substrate binding, the scissilecarbonyl oxygen would enter the catalytic metal co-ordination sphere and replace two of the solvent mol-ecules found in the unbound structure. The remainingsolvent molecule would still bind to the cation, but prob-ably shift towards the carboxylate of Glu-266 of theHEXXH motif. The latter acidic residue would act as ageneral base and abstract a proton from the water (Fig. 4,I). The resulting incipient hydroxide would perform anucleophilic attack onto the scissile amide carbonylcarbon. This would lead to a negatively charged tetrahe-dral reaction-intermediate (Fig. 4, II) that could interact ina bidentate manner with, and be thus stabilized by, themetal ion, which would remain penta-co-ordinate (Kimand Lipscomb, 1991). Further stabilization of the interme-diate may be provided by other residues such as Tyr-417and His-409. Glu-266 would subsequently act as ageneral acid catalyst, delivering the proton captured fromthe solvent to the scissile amide nitrogen, which wouldbecome a secondary ammonium (Fig. 4, II) (Monzingoand Matthews, 1984). The gem-diolate proton would betransferred in a second step, possibly via Glu-266, to thesecondary ammonium and this would make the interme-diate collapse into two products (Fig. 4, III). Eventually,the cleavage products would leave and the metal-bindingsolvent molecules could be replenished, rendering theenzyme poised for a new round of catalysis.
A single point mutation changes substrate specificity
In contrast to TcMCP-1, its paralogue TcMCP-2 is not ableto hydrolyse basic residues at P1′ and acts best on ali-phatic, neutral and aromatic amino acids (Niemirowiczet al., 2007). Homology models of TcMCP-2 based on thecrystal structure of TcMCP-1 were utilized to map thepositions of the potentially critical residues involved insubstrate specificity (data not shown). Two residueslocated within the active site of TcMCP-1, Met-304 andGln-305, are an arginine and a methionine, respectively,in TcMCP-2. To investigate whether these residues par-ticipated in specificity, we constructed the cross-mutants
TcMCP-1(M304R), TcMCP-1(Q305M), TcMCP-1(M304R/Q305R) and TcMCP-2(R304M). These forms wereexpressed and purified, and assayed for proteolytic activ-ity against A-type (with a C-terminal aromatic residue) andB-type (with a C-terminal basic residue) chromogenic sub-strates (see Table 2). The kinetic parameters of mutantTcMCP-1(Q305M) on the B-type substrate were similar tothose of the wild-type enzyme, suggesting that thisresidue is not indispensable for subsite S1′. Notably, themost marked effect was seen for TcMCP-1(M304R)and TcMCP-1(M304R/Q305R), which lacked the activitytowards the B-type substrate, but were able to cleave theA-type substrate with a similar efficiency than wild-typeTcMCP-2. The reciprocal substitution was made inTcMCP-2. TcMCP-2(R304M) could hydrolyse both theB-type substrate and the A-type substrate, although herekcat was significantly impaired. These data confirmed therole of the residue at position 304 in specificity. While amethionine here may interact with the aliphatic and/oraromatic part of an arginine or lysine of a B-type sub-strate, the arginine observed in TcMCP-2 [or introduced inTcMCP-1(M304R) and TcMCP-1(M304R/Q305R)] wouldgenerate electrostatic repulsion with an incoming posi-tively charged residue, thus hampering substrate binding.In contrast, this arginine could easily interact through p–pstacking with the aromatic ring of an A-type substrate, i.e.it could be easily accommodated and cleaved.
The role of position 304 in specificity is further high-lighted if the nature of the amino acids present is analysedthroughout all M32-peptidases annotated in the MEROPSdatabase. As more than three quarters of the 128sequences listed have either a methionine or an aspar-agine, we decided to analyse also the asparaginemutants. Interestingly, TcMCP-1(M304N) and TcMCP-2(R304N) displayed activity on both substrates, althoughwith lower efficiency (Table 2). This is in accordance withbiochemical data on three asparagine-containing M32-peptidases, PfuCP, TaqCP and Thermococcus sp. NA1carboxypeptidase, which display broad substrate speci-ficity at P1′ (Lee et al., 1994; 1996; 2006; Cheng et al.,1999). Therefore, an asparagine might account for
Fig. 3. Active-site cleft and related structures.A. Stereo-cartoon showing the Ca-traces of the active-site helix (dark blue), the glutamate helix (orange) and the tyrosine helix (green), asseen after a horizontal downwards ~90° rotation of the orientation shown in Fig. 1C. The active-site metal (magenta sphere) and some helixresidues key for metal binding and catalysis are shown as sticks, which are coloured according to helix they belong to. A modelled tetrahedralintermediate is also shown as a stick model colour-coded yellow for carbons, blue for nitrogens, green for sulphurs, and red for oxygens todelineate the active-site cleft [see also (C)].B. Close-up view of Fig. 1C focusing on the active-site cleft. Residues relevant for substrate binding and catalysis are shown by theirside-chains, which are colour-coded as in (A) except for carbons, shown in white. Some hydrogen bonds and the metal-binding interactionsare displayed as dashed lilac rods. Solvent molecules are shown as cyan spheres and the catalytic metal ion as a magenta sphere.C. Same as (B) but showing the modelled transition-state analogue as a stick model [same atom colour coding as in (A) except for thecarbons, shown in cyan].D. Superimposed Ca ribbon in standard orientation and stereo of TcMCP-1 (chain evolving from cyan to dark blue) and PfuCP (chain evolvingfrom magenta to red) and (E) of TcMCP-1 (cyan to dark blue) and E. coli Dcp (magenta to red). The N- and C-termini are labelled and thecatalytic metal ion of the protozoan enzyme is depicted as a green sphere in each case.
860 G. Niemirowicz et al. �
© 2008 The AuthorsJournal compilation © 2008 Blackwell Publishing Ltd, Molecular Microbiology, 70, 853–866
Structure of Trypanosoma cruzi metallocarboxypeptidase 861
© 2008 The AuthorsJournal compilation © 2008 Blackwell Publishing Ltd, Molecular Microbiology, 70, 853–866

substrate promiscuity in unicellular cowrins with carbox-ypeptidase activity.
Experimental procedures
Protein purification and activity assays
TcMCP-1 (503 residues; UniProt sequence database entryQ6ZXC0) was produced by recombinant overexpression withan N-terminal polyhistidine-tag and purified by affinity chroma-tography using Co2+-nitriloacetic acid resin, as reported(Niemirowicz et al., 2007). The cloning strategy led to theinclusion of additional N-terminal residues in the recombinant
protein (MGGS-H6-GMASMTGGQQMGRDLYDDDDKDRWGS). Site-directed mutations were introduced into TcMCP-1and TcMCP-2 genes cloned in plasmid pGEX2T using theQuickChangeTM site-directed mutagenesis kit (Stratagene).For biochemical studies, wild-type and mutated enzymes wereexpressed as fusion proteins with glutathione-S-transferase inE. coli BL21(Codon Plus) cells. Recombinant fusion proteinswere purified in one step using glutathione–agarose resin(Sigma) equilibrated with 50 mM Tris-HCl, 150 mM NaCl,pH 7.6. The column was washed with five column volumes ofequilibration buffer. Samples were eluted with equilibrationbuffer further 10 mM in reduced glutathione. Kinetic param-eters for wild-type TcMCP-1 and the mutants TcMCP-
Fig. 4. TcMCP-1 mediated peptide cleavage. Proposed hydrolytic mechanism, evolving from the Michaelis complex over a tetrahedralintermediate to a product complex. Potential key residues for catalysis are depicted and labelled. Hydrogen bonds are shown as dashed lines.
Table 2. Kinetic parameters of TcMCP-1 and -2 variants.
Enzyme variant/substrate Km (mM) kcat (s-1) kcat/Km (s-1/mM)
TcMCP-1FA–Ala–Lys 144 10 69FA–Phe–Phea ND ND ND
TcMCP-1(M304R/Q305M)FA–Ala–Lys ND ND NDFA–Phe–Phe 12 0.5 42
TcMCP-1(M304R)FA–Ala–Lys ND ND NDFA–Phe–Phe 11 0.3 27
TcMCP-1(Q305M)FA–Ala–Lys 154 8 52FA–Phe–Phe ND ND ND
TcMCP-1(M304N)FA–Ala–Lys 235 0.17 0.7FA–Phe–Phe 56 0.04 0.7
TcMCP-2FA–Ala–Lys ND ND NDFA–Phe–Phe 19 1.2 63
TcMCP-2(R304M)FA–Ala–Lys 182 0.13 0.7FA–Phe–Phe 53 0.02 0.4
TcMCP-2(R305N)FA–Ala–Lys 450 0.7 1.5FA–Phe–Phe 87 0.05 0.6
Kinetic parameters of recombinant TcMCPs and mutants were determined from Lineweaver–Burk plots, as described in the Experimentalprocedures section. ND, not detected under assay conditions.a. Activity on FA–Phe–Phe was detected only in the presence of 50 mM CoCl2. The values for Km and kcat measured under these conditions were74 mM and 0.5 s-1respectively.
862 G. Niemirowicz et al. �
© 2008 The AuthorsJournal compilation © 2008 Blackwell Publishing Ltd, Molecular Microbiology, 70, 853–866

1(M304R), TcMCP-1(Q305M), TcMCP-1(M304R/Q305R) andTcMCP-1(M304N) were determined using the standard chro-mogenic MCP substrates {N-(3-[2-furyl]acryloyl)}–Ala–Lys(FA–Ala–Lys; B-type MCP substrate) and {N-(3-[2-furyl]acryloyl)}–Phe–Phe (FA–Phe–Phe; A-type MCP sub-strate) in 100 mM MES, pH 6.2. Activity of wild-type TcMCP-2and mutants TcMCP-2(R304M) and TcMCP-2(R304N) wasassayed in 50 mM Tris-HCl, pH 7.6. Initial steady-state veloc-ity was monitorized by continuous assay for a range of sub-strate concentrations at 340 nm with a Beckman DU 650spectrophotometer. One unit of activity was defined as theamount of enzyme to process 1 mmol of substrate per minuteat 25°C.
Crystallization
Crystallization assays followed the sitting-drop vapor diffu-sion method. Reservoir solutions were prepared by aFreedom EVO robot (Tecan) and 200 nL sitting drops weredispensed on 96 ¥ 3-well CrystalQuick plates (Greiner) by aCartesian nanodrop robot (Genomic Solutions) at the jointIBMB-CSIC/PCB/IRB high-throughput crystallography plat-form (PAC) at Barcelona Science Park. Best crystals with theshape of square-based prisms were obtained with proteinsolution (3–5 mg ml-1 in 50 mM Tris-HCl, pH 7.6) and 2 M(NH4)2SO4, 0.1 M HEPES, pH 7.5 as reservoir solution afterabout 6 weeks in a steady-temperature Crystal Farm 400(Bruker) at 4°C. Crystals were harvested, cryo-protected with3M (NH4)2SO4, 30% glycerol, 0.1 M HEPES, pH 7.5, mountedon LithoLoops (Molecular Dimensions) and flash vitrified in
liquid N2. Complete diffraction data sets were collected at100K (Oxford Cryosystems) from a single crystal each on anADSC Quantum Q210 detector at beam line ID14-1 of theEuropean Synchrotron Radiation Facility (Grenoble, France)within the Block Allocation Group ‘BAG Barcelona’. Crystalswere monoclinic and contained four molecules per asymmet-ric unit [VM = 2.8 Å3/Da; 57% solvent; (Matthews, 1968)]. Dif-fraction data were integrated with program XDS (Kabsch,2001), corrected for radiation damage with program 0-DOSE(Diederichs et al., 2003) within program XSCALE of the XDS
package, and scaled, merged and reduced with programsCOMBAT and SCALA (Evans, 2006) within the CCP4 suite ofprograms (CCP4, 1994) (see Table 3). Crystals diffractedbeyond 2 Å resolution but displayed significant anisotropy, sothat a resolution cut-off of 2.1 Å was applied for the dataintended for final model refinement. A test set to monitor thefree Rfactor was created in 10 thin shells with program SFTOOLS
within CCP4 to account for the non-crystallographicsymmetry. The protein further crystallized in an orthorhombicspace group (P212121; a = 54.5–55.3 Å, b = 124.7–125.6 Å,c = 172.9–176.5 Å). These crystals contained one dimerper asymmetric unit and diffracted to lower resolution(2.4–2.6 Å). In addition, several crystals soaked in, orco-crystallized with, potential MCP inhibitors either did notdiffract or lacked the inhibitors in the active-site cleft.
Structure solution and refinement
The structure was solved by Patterson-search methods withprogram AMoRe (Navaza, 1994) employing all diffraction
Table 3. Crystallographic data.
Data set TcMCP-1Space group P21
Cell constants (a, b, c, in Å; b in °) 89.40, 136.25, 117.86, 103.06Wavelength (Å) 0.934No. of measurements/unique reflections 1,015,700/147,734Resolution range (Å) (outermost shell)a 46.23–2.10 (2.21–2.10)Completeness (%) 100.0 (100.0)Rr.i.m. = Rrmeas
b/Rp.i.m.b 0.084 (0.574)/0.033 (0.226)Average intensity (<[<I>/s(<I>)]>) 17.8 (4.1)B-Factor (Wilson) (Å2)/Average multiplicity 31.1/6.3 (6.3)Crystallographic Rfactor (free Rfactor)c 0.180 (0.221)Average multiplicityCorrelation coefficient Fobs - Fcalc (Fobs - Fcalc in test set) 0.957 (0.935)Estimated overall co-ordinate error based on free Rfactor (Å) 0.156No. of protein atoms/ions/ligands/solvent molecules 16,009/4 Co2+, 13 SO4
2-/11 glycerol, Gly-Ala dipeptide/1,120Rmsd from target values
bonds (Å)/angles (°) 0.016/1.48bonded B-factors (main chain/side-chain)(Å2) 1.08/2.28
Average B-factors for all atoms/within each of protein molecules A-D (Å2) 37.8/38.0, 36.8, 38.8, 39.4Main-chain conformational angle analysisd
Residues in favoured regions/outliers/all protein residues 1938/0/1975
a. Values in parentheses refer to the outermost resolution shell if not otherwise indicated.b. Rr.i.m. = Shkl(nhkl/[nhkl-1]1/2)Si |Ii(hkl) - <I(hkl)>|/ShklSiIi(hkl) and Rp.i.m. = Shkl(1/[nhkl-1]1/2)Si |Ii(hkl) - <I(hkl)>|/S hklS iIi(hkl), where Ii(hkl) is the i-th intensitymeasurement and nhkl the number of observations of reflection hkl, including symmetry-related reflections and <I(hkl)> its average intensity. Rr.i.m.
(alias Rmeas) and Rp.i.m. are improved multiplicity-weighted indicators of the quality of the data, the redundancy-independent merging R factor andthe precision-indicating merging R factor, the latter computed after averaging the multiple measurements (Weiss, 2001; Evans, 2006).c. Crystallographic Rfactor = Shkl ||Fobs| - k |Fcalc||/Shkl |Fobs|, with Fobs and Fcalc as the observed and calculated structure factor amplitudes; free Rfactor,same for a test set of reflections (> 500) not used during refinement.d. According to MOLPROBITY (Davis et al., 2007).
Structure of Trypanosoma cruzi metallocarboxypeptidase 863
© 2008 The AuthorsJournal compilation © 2008 Blackwell Publishing Ltd, Molecular Microbiology, 70, 853–866

data of the monoclinic crystal form between 15 and 4 Åresolution. The co-ordinates of a PfuCP dimer [PDB 1K9X;Arndt et al., 2002] were used as a searching model. Twosolutions were found at 62.8, 89.1, 266.4 and 52.8, 101.8,86.6 (a,b,g, in Eulerian angles) and 0.4783, 0.0001, 0.2474and 0.5554, 0.5100, 0.2240 (x,y,z, as fractional unit-cellco-ordinates), respectively, after rigid-body refinement. Thisdouble solution gave a combined correlation coefficient instructure factor amplitudes of 27.8% and a crystallographicRfactor of 53.4% (for definitions, see Table 3 and Navaza,1994). Subsequently, diffraction data were reprocessed to2.0 Å resolution and subjected to anisotropic scaling andellipsoidal truncation (Strong et al., 2006; http://www.doe-mbi.ucla.edu/~sawaya/anisoscale). A density modificationstep under fourfold non-crystallographic-symmetry-averagingwith program DM (Cowtan and Main, 1996) was performedwith these data using phases calculated from the four rotatedand translated protein monomers. These calculations ren-dered an improved electron density map, which was usedfor initial model building. Subsequently, manual modelcompletion on a Silicon-Graphics workstation using programTURBO-Frodo (Carranza et al., 1999) alternated with crystal-lographic refinement with program REFMAC5 (Murshudovet al., 1997) within CCP4 using the 2.1 Å resolution data set(see above and Table 3). The final model contained proteinresidues from Met-1 to Asp-500 (molecules A, C and D) andto Gly-503 (molecule B) plus one extra tag-residue precedingMet-1 (Ser-0) for molecules A, B and C, and two additionalresidues (Gly-1 and Ser-0) for molecule D. In each case, acobalt ion (Co999) was placed in the active site. The fourmolecules were fully defined by electron density and onlysegment Glu-97–Ala-145 comprising helices a5 and a6within each molecule was flexible, as denoted by above-average thermal displacement parameters. This flexibilitywas greater in molecule D, so that segment Thr-108–Phe-126 was not traced for this chain. In addition, sulphate ions,an isolated dipeptide of tentative sequence Gly–Ala, glycerolmolecules and solvent molecules were included in the finalmodel (see Table 3). Additional, discontinuous electrondensity peaks, which could not be unambiguously assigned,were found in the active-site cleft of all molecules. The fourmolecules (molecules A–D; pairwise rms-deviations ofequivalent Ca atoms equal 0.55–0.56 Å) and the two dimers(A–B and C–D; rms-deviation 0.60/0.62 Å) in the asymmetricunit are practically equivalent, so in the discussion only mol-ecule A and the A–B dimer were considered unless otherwisestated. The structure of the orthorhombic crystal was solvedusing a refined dimer from the monoclinic crystal form. In thiscase, the segment comprising helices a5 and a6 was com-pletely disordered within each of the two molecules in theasymmetric unit and the rest of the polypeptide chain wasindistinguishable from the monoclinic model. Therefore, theorthorhombic data were not further considered.
Miscellaneous
Figures were prepared with programs MOLMOL (Koradi et al.,1996), MOLSCRIPT (Kraulis, 1991) and TURBO-Frodo. Closecontacts and interaction surfaces, taking half the total surfaceburied at the interface obtained with a probe radius of 1.4 Å,were calculated with program CNS v. 1.2 (Brünger et al.,
1998). Structural similarity searches were undertaken withindatabase SCOP (Hubbard et al., 1997) and with programsDALI (http://www.ebi.ac.uk/msd) and DBAli v.2.0 (http://www.salilab.org/DBAli). A structure-based multiple sequence align-ment was performed with MAMMOTH-MULT (Lupyan et al.,2005). This alignment was used in a phylogenetic analysisbased on maximum-likelihood criteria with program PHYML
(Guindon and Gascuel, 2003). The resulting rootless phylo-genetic tree was represented with program NJPLOT (Perriereand Gouy, 1996). Pairwise sequence alignments in theabsence of structural information were calculated withprogram SIM(http://www.expasy.ch/tools/sim-prot.html). A tet-rahedral reaction intermediate of sequence Met–Ala–Arg wasmodelled and energy-minimized with the Dundee PRODRG-server at http://davapc1.bioch.dundee.ac.uk/prodrg/index.html. The final co-ordinates and structure factors ofTcMCP-1 have been deposited with the PDB at http://www.pdb.org (access code 3DWC).
Acknowledgements
This study was supported by the following grants: BIO2006-02668, BFU2006-09593, BIO2007-68046, PSE-010000-2007-1 and the CONSOLIDER-INGENIO 2010 Project‘La Factoría de Cristalización’ (CSD2006-00015) fromSpanish public agencies; EU FP6 Strep Project LSHG-2006–018830 ‘CAMP’; EU FP7 Collaborative Project 223101‘AntiPathoGN’; 2005SGR00280 and 2005SGR01027 fromthe National Catalan Government; and PICT2003 01–15042from the Agencia Nacional de Promoción Científica y Tec-nológica, Ministerio de Ciencia, Tecnología e Innovación Pro-ductiva, Argentina. We acknowledge the help provided byEuropean Synchrotron Radiation Facility local contacts.Funding for data collection was provided by the facility. Wefurther thank Robin Rycroft and, in particular, Josep Vendrellfor helpful contributions to the manuscript and TibisayGuevara for assistance during crystallization.
References
Arndt, J.W., Hao, B., Ramakrishnan, V., Cheng, T., Chan,S.I., and Chan, M.K. (2002) Crystal structure of a novelcarboxypeptidase from the hyperthermophilic archaeonPyrococcus furiosus. Structure 10: 215–224.
Arolas, J.L., Vendrell, J., Avilés, F.X., and Fricker, L.D. (2007)Metallocarboxypeptidases: emerging drug targets inbiomedicine. Curr Pharm Des 13: 349–366.
Barrett, M.P., Burchmore, R.J., Stich, A., Lazzari, J.O.,Frasch, A.C., Cazzulo, J.J., and Krishna, S. (2003) Thetrypanosomiases. Lancet 362: 1469–1480.
Beyrer, C., Villar, J.C., Suwanvanichkij, V., Singh, S., Baral,S.D., and Mills, E.J. (2007) Neglected diseases, civil con-flicts, and the right to health. Lancet 370: 619–627.
Bode, W., Gomis-Rüth, F.X., and Stöcker, W. (1993) Ast-acins, serralysins, snake venom and matrix metalloprotein-ases exhibit identical zinc-binding environments(HEXXHXXGXXH and Met-turn) and topologies andshould be grouped into a common family, the ‘metzincins’.FEBS Lett 331: 134–140.
Brown, C.K., Madauss, K., Lian, W., Beck, M.R., Tolbert,
864 G. Niemirowicz et al. �
© 2008 The AuthorsJournal compilation © 2008 Blackwell Publishing Ltd, Molecular Microbiology, 70, 853–866

W.D., and Rodgers, D.W. (2001) Structure of neurolysinreveals a deep channel that limits substrate access. ProcNatl Acad Sci USA 98: 3127–3132.
Brünger, A.T., Adams, P.D., Clore, G.M., DeLano, W.L.,Gros, P., Grosse-Kunstleve, R.W., et al. (1998) Crystallog-raphy & NMR System: a new software suite for macro-molecular structure determination. Acta Crystallogr D 54:905–921.
Carranza, C., Inisan, A.-G., Mouthuy-Knoops, E., Cambillau,C., and Roussel, A. (1999) Turbo-Frodo. In AFMB ActivityReport 1996–1999. Marseille: CNRS-UPR 9039, pp.89–90.
Cazzulo, J.J. (2002) Proteinases of Trypanosoma cruzi: pat-ential targets for the chemotherapy of Chagas’ desease.Curr Top Med Chem 2: 1261–1271.
CCP4 (1994) The CCP4 suite: programs for proteincrystallography. Acta Crystallogr D 50: 760–763.
Cheng, T.C., Ramakrishnan, V., and Chan, S.I. (1999) Puri-fication and characterization of a cobalt-activated carbox-ypeptidase from the hyperthermophilic archaeonPyrococcus furiosus. Protein Sci 8: 2474–2486.
Comellas-Bigler, M., Lang, R., Bode, W., and Maskos, K.(2005) Crystal structure of the E. coli dipeptidyl carbox-ypeptidase Dcp: further indication of a ligand-dependenthinge movement mechanism. J Mol Biol 349: 99–112.
Corradi, H.R., Schwager, S.L., Nchinda, A.T., Sturrock, E.D.,and Acharya, K.R. (2006) Crystal structure of the N domainof human somatic angiotensin I-converting enzyme pro-vides a structural basis for domain-specific inhibitor design.J Mol Biol 357: 964–974.
Cowtan, K.D., and Main, P. (1996) Phase combination andcross validation in iterated density-modification cal-culations. Acta Crystallogr D 52: 43–48.
Davis, I.W., Leaver-Fay, A., Chen, V.B., Block, J.N., Kapral,G.J., Wang, X., et al. (2007) MolProbity: all-atom contactsand structure validation for proteins and nucleic acids.Nucleic Acids Res 35 (Web Server issue): W375–W383.
Diederichs, K., McSweeney, S., and Ravelli, R.B. (2003)Zero-dose extrapolation as part of macromolecular syn-chrotron data reduction. Acta Crystallogr D 59: 903–909.
Docampo, R. (2001) Recent developments in the chemo-therapy of Chagas’ disease. Curr Pharm Des 7: 1157–1164.
Engel, J.C., Doyle, P.S., Hsieh, I., and McKerrow, J.H. (1998)Cysteine protease inhibitors cure an experimental Trypa-nosoma cruzi infection. J Exp Med 188: 725–734.
Evans, P. (2006) Scaling and assessment of data quality.Acta Crystallogr D 62: 72–82.
Gerdts, C.J., Tereshko, V., Yadav, M.K., Dementieva, I.,Collart, F., Joachimiak, A., et al. (2006) Time-controlledmicrofluidic seeding in nL-volume droplets to separatenucleation and growth stages of protein crystallization.Angew Chem Int Ed Engl 45: 8156–8160.
Gomis-Rüth, F.X. (2003) Structural aspects of the metzincinclan of metalloendopeptidases. Mol Biotechn 24: 157–202.
Gomis-Rüth, F.X. (2008) Structure and mechanism ofmetallocarboxypeptidases. Crit Rev Biochem Mol Biol 43:319–345.
Gomis-Rüth, F.X., Stöcker, W., Huber, R., Zwilling, R., andBode, W. (1993) Refined 1.8 Å X-ray crystal structure ofastacin, a zinc-endopeptidase from the crayfish Astacus
astacus L. Structure determination, refinement, molecularstructure and comparison with thermolysin. J Mol Biol 229:945–968.
Gomis-Rüth, F.X., Kress, L.F., Kellermann, J., Mayr, I., Lee,X., Huber, R., and Bode, W. (1994) Refined 2.0Å, X-raycrystal structure of the zinc-endopeptidase adamalysin II.Primary and tertiary structure determination, refinement,molecular structure and comparison with astacin, collage-nase and thermolysin. J Mol Biol 239: 513–544.
Greer, J., Erickson, J.W., Baldwin, J.J., and Varney, M.D.(1994) Application of the three-dimensional structures ofprotein target molecules in structure-based drug design.J Med Chem 37: 1035–1054.
Guindon, S., and Gascuel, O. (2003) A simple, fast, andaccurate algorithm to estimate large phylogenies bymaximum likelihood. Syst Biol 52: 696–704.
Gutteridge, W.E. (1997) Designer drugs: pipe-dreams orrealities? Parasitology 114 (Suppl.): S145–S151.
Hooper, N.M. (1994) Families of zinc metalloproteases.FEBS Lett 354: 1–6.
Hubbard, T.J.P., Murzin, A.G., Brenner, S.E., and Chothia, C.(1997) SCOP: a structural classification of proteinsdatabase. Nucleic Acids Res 25: 236–239.
Kabsch, W. (2001) Chapter 25.2.9: XDS. In InternationalTables for Crystallography. Volume F: Crystallography ofBiological Macromolecules. Rossmann, M.G., and Arnold,E. (eds). Dordrecht: Kluwer Academic Publishers (for TheInternational Union of Crystallography), pp. 730–734.
Kim, H., and Lipscomb, W.N. (1991) Comparison of the struc-tures of three carboxypeptidase A – phosphonate com-plexes determined by X-ray crystallography. Biochemistry30: 8171–8180.
Kim, H.M., Shin, D.R., Yoo, O.J., Lee, H., and Lee, J.O.(2003) Crystal structure of Drosophila angiotensinI-converting enzyme bound to captopril and lisinopril.FEBS Lett 538: 65–70.
Klemba, M., and Goldberg, D.E. (2002) Biological roles ofproteases in parasitic protozoa. Annu Rev Biochem 71:275–305.
Koradi, R., Billeter, M., and Wüthrich, K. (1996) MOLMOL: aprogram for display and analysis of macromolecularstructures. J Mol Graphics 14: 51–55.
Kraulis, P.J. (1991) MOLSCRIPT: a program to produce bothdetailed and schematic plots of protein structures. J ApplCrystallogr 24: 946–950.
Lee, H.S., Kim, Y.J., Bae, S.S., Jeon, J.H., Lim, J.K., Kang,S.G., and Lee, J.H. (2006) Overexpression and character-ization of a carboxypeptidase from the hyperthermophilicarchaeon Thermococcus sp. NA1. Biosci BiotechnolBiochem 70: 1140–1147.
Lee, S.H., Minagawa, E., Taguchi, H., Matsuzawa, H., Ohta,T., Kaminogawa, S., and Yamauchi, K. (1992) Purificationand characterization of a thermostable carboxypeptidase(carboxypeptidase Taq) from Thermus aquaticus YT-1.Biosci Biotechnol Biochem 56: 1839–1844.
Lee, S.H., Taguchi, H., Yoshimura, E., Minagawa, E., Kami-nogawa, S., Ohta, T., and Matsuzawa, H. (1994) Carbox-ypeptidase Taq, a thermostable zinc enzyme, fromThermus aquaticus YT-1: molecular cloning, sequencing,and expression of the encoding gene in Escherichia coli.Biosci Biotechnol Biochem 58: 1490–1495.
Structure of Trypanosoma cruzi metallocarboxypeptidase 865
© 2008 The AuthorsJournal compilation © 2008 Blackwell Publishing Ltd, Molecular Microbiology, 70, 853–866

Lee, S.H., Taguchi, H., Yoshimura, E., Minagawa, E.,Kaminogawa, S., Ohta, T., and Matsuzawa, H. (1996)The active site of carboxypeptidase Taq possessesthe active-site motif His-Glu-X–X-His of zinc-dependentendopeptidases and aminopeptidases. Protein Eng 9:467–469.
Lupyan, D., Leo-Macias, A., and Ortiz, A.R. (2005) A newprogressive-iterative algorithm for multiple structurealignment. Bioinformatics 21: 3255–3263.
Matthews, B.W. (1968) Solvent content of protein crystals.J Mol Biol 33: 491–497.
Matthews, B.W. (1988) Structural basis of the action of ther-molysin and related zinc peptidases. Acc Chem Res 21:333–340.
Matthews, B.W., Jansonius, J.N., Colman, P.M., Schoen-born, B.P., and Dupourque, D. (1972) Three-dimensionalstructure of thermolysin. Nature 238: 37–41.
McGrath, M.E., Eakin, A.E., Engel, J.C., McKerrow, J.H.,Craik, C.S., and Fletterick, R.J. (1995) The crystal structureof cruzain: a therapeutic target for Chagas’ disease. J MolBiol 247: 251–259.
Mittl, P.R., and Grütter, M.G. (2006) Opportunities forstructure-based design of protease-directed drugs. CurrOpin Struct Biol 16: 769–775.
Monzingo, A.F., and Matthews, B.W. (1984) Binding ofN-carboxymethyl dipeptide inhibitors to thermolysin deter-mined by X-ray crystallography: a novel class of transition-state analogues for zinc peptidases. Biochemistry 23:5724–5729.
Murshudov, G.N., Vagin, A.A., and Dodson, E.J. (1997)Refinement of macromolecular structures by themaximum-likelihood method. Acta Crystallogr D 53: 240–255.
Natesh, R., Schwager, S.L.U., Sturrock, E.D., and Acharya,K.R. (2003) Crystal structure of the human angiotensin-converting enzyme-lisinopril complex. Nature 421: 551–554.
Navaza, J. (1994) AMoRe: an automated package formolecular replacement. Acta Crystallogr A 50: 157–163.
Niemirowicz, G., Parussini, F., Agüero, F., and Cazzulo, J.J.(2007) Two metallocarboxypeptidases from the protozoanTrypanosoma cruzi belong to the M32 family, found so faronly in prokaryotes. Biochem J 401: 399–410.
O’Connell, D. (2007) Neglected diseases. Nature 449: 157–157.
Perriere, G., and Gouy, M. (1996) WWW-query: an on-line
retrieval system for biological sequence banks. Biochimie78: 36436–36439.
Potempa, J., and Pike, R.N. (2005) Bacterial peptidases.Contrib Microbiol 12: 132–180.
Rawlings, N.D., Morton, F.R., and Barrett, A.J. (2006)MEROPS: the peptidase database. Nucleic Acids Res 34(Database issue): D270–D272.
Ray, K., Hines, C.S., Coll-Rodríguez, J., and Rodgers, D.W.(2004) Crystal structure of human thimet oligopeptidaseprovides insight into substrate recognition, regulation, andlocalization. J Biol Chem 279: 20480–20489.
Rees, D.C., Lewis, M., and Lipscomb, W.N. (1983) Refinedcrystal structure of carboxypeptidase A at 1.54 Åresolution. J Mol Biol 168: 367–387.
Richardson, J.S. (1985) Schematic drawings of proteinstructures. Methods Enzymol 115: 359–380.
Sealey-Cardona, M., Cammerer, S., Jones, S., Ruiz-Pérez,L.M., Brun, R., Gilbert, I.H., et al. (2007) Kinetic character-ization of squalene synthase from Trypanosoma cruzi:selective inhibition by quinuclidine derivatives. AntimicrobAgents Chemother 51: 2123–2129.
Strong, M., Sawaya, M.R., Wang, S., Phillips, M., Cascio, D.,and Eisenberg, D. (2006) Toward the structural genomicsof complexes: crystal structure of a PE/PPE proteincomplex from Mycobacterium tuberculosis. Proc Natl AcadSci USA 103: 8060–8065.
Towler, P., Staker, B., Prasad, S.G., Menon, S., Tang, J.,Parsons, T., et al. (2004) ACE2 X-ray structures reveal alarge hinge-bending motion important for inhibitor bindingand catalysis. J Biol Chem 279: 17996–18007.
Urbina, J.A. (2001) Specific treatment of Chagas disease:current status and new developments. Curr Opin Infect Dis14: 733–741.
Urbina, J.A. (2002) Chemotherapy of Chagas’ disease. CurrPharm Des 8: 287–295.
Weiss, M.S. (2001) Global indicators of X-ray quality. J ApplCryst 34: 130–135.
Wray, J.W., Baase, W.A., Ostheimer, G.J., Zhang, X.J., andMatthews, B.W. (2000) Use of a non-rigid region in T4lysozyme to design an adaptable metal-binding site.Protein Eng 13: 313–321.
Zingales, B., Pereira, M.E., Almeida, K.A., Umezawa, E.S.,Nehme, N.S., Oliveira, R.P., et al. (1997) Biological param-eters and molecular markers of clone CL Brener – thereference organism of the Trypanosoma cruzi genomeproject. Mem Inst Oswaldo Cruz 92: 811–814.
866 G. Niemirowicz et al. �
© 2008 The AuthorsJournal compilation © 2008 Blackwell Publishing Ltd, Molecular Microbiology, 70, 853–866
Copyright © 2022 FDOKUMEN




















