
THE GENETICS OF MANIC DEPRESSIVE ILLNESS: A PREDIGREE ...
197
THE GENETICS OF MANIC DEPRESSIVE ILLNESS: A PREDIGREE AND LINKAGE STUDY by Kathleen Donovan Bucher Department of Biostatistics University of North Carolina at Chapel Hill Institute of Statistics Mimeo Series No. 1141 August 1977
-
Upload
khangminh22 -
Category
Documents
-
view
2 -
download
0
Transcript of THE GENETICS OF MANIC DEPRESSIVE ILLNESS: A PREDIGREE ...

THE GENETICS OF MANIC DEPRESSIVE ILLNESS:A PREDIGREE AND LINKAGE STUDY
by
Kathleen Donovan Bucher
Department of BiostatisticsUniversity of North Carolina at Chapel Hill
Institute of Statistics Mimeo Series No. 1141
August 1977

KATHLEEN DONOVAN BUCHER. The Genetics of Manic Depressive Illness:A Pedigree and Linkage Study. (Under the direction of R.C. ELSTON.)
A likelihood method of pedigree analysis is applied to four
previously reported sets of data on manic depressive illness, and
linkage analysis is performed on a large family with manic depressive
illness, colorblindness and the Xg blood group. Some of the problems
inherent in the study of mental illnesses are alleviated in this
analysis: all the information in the family structure is used, the
variable age of onset is taken into account, the fact that families are
ascertained via one or more probands is allowed for, and one- and two-
gene hypotheses are tested versus a general alternative by a likelihood
ratio test.
Methods are given to correct for a two-stage ascertainment which is
sometimes used in an attempt to reduce genetic heterogeneity in a set of
families. In this type of sample selection families are first ascer-
tained through a proband in the usual way, then a subset of these
families is chosen for analysis on the basis of some criterion of family
history. Corrections are explicitly given for the cases in which the
second stage criterion is that there are at least two affected people in
the family, and that there is no father-to-son transmission of the
disease.
The analysis is consistent over all four sets of data, and indicates
that neither a single X-linked gene nor a single autosomal gene can ac-
count for the transmission of manic depressive illness. None of the two-
gene models examined were significantly more likely than the single-gene
models. However, linkage analysis on a single large family gives some

suggestion that there may be a locus on. the X-chromosome closely
linked to the Xg blood group which has a rare allele that can cause
manic depressive illness in.an occasional family with the disease.
\, \

ACKNOWLEDGEMENTS
I would like to express my appreciation to my advisor, Dr. R. C.
Elston, who suggested the topic of this research and provided valuable
guidance. Thanks also go to the members of my committee, Drs. M.E.
Francis, C. H. Langley, W. S. Pollitzer, and C. D. Turnbull.
Ellen Kaplan did the computer programming and provided endless
assistance with running the programs. Her help is gratefully
acknowledged.
Drs. R. Green, J. Helzer, and G. Winokur shared their data and
were very cooperative and helpful.
Thanks go to Carolyn Vaughan who typed this dissertation.
Finally, I want to thank my husband John for his help and patience
and my son Tom for his distractions during the past eight months.
, I1 '

TABLE OF CONTENTS
Page
ACKNOWLEDGMENTS
LIST OF TABLES
LIST OF FIGURES
Chapter
I. INTRODUCTION AND REVIEW OF THE LITERATURE
iii
v
ix
1
1.1 Introduction. . . . . . . . . • . . . 11.2 The genetic basis of manic depressive psychosis. 21.3 Statistical methods used to detect major
genes in human family data . . . . . . 28
II. METHODS AND MODELS FOR AFFECTIVE ILLNESS DATA 46
2.1 Specific models for affective illness data . 462.1.1 The phenotypic model. . . .. .... 462.1.2 The ascertainment function. . . . .. 482.1.3 The sampling correction. . . 492.1.4 Genetic models. . . . . . . 562.1.5 Summary of the model and its parameters 63
2.2 Extensions of the likelihood method: Twostage ascertainment. . . . . . . . 65
2.2.1 The selection procedure, ascertainmentfunction, and general form of thesampling correction . . . . . . . • • .. 65
2.2.2 Families with two or more affect~d
people. . . . . . • • . . . • . • • • •. 712.2.3 Exclusion of families with father to
son transmission. . . . . . . . 73
III. ANALYSIS OF DATA FROM HELZER AND WINOKUR (1974)
3.1 Description of the data ..3.2 Segregation Analysis ...
3.2.1 Single gene models.3.2.2 Two gene models.
3.3 Conclusions .
78
7886869091

v
Chapter Page eIV. ANALYSIS OF DATA FROM WINOKUR. CLAYTON AND
REICH (1969) . · · · · 100
4.1 Description of data · 1004.2 Segregation Analysis. · 105
4.2.1 Single gene models · · 1064.2.2 Two gene models. · · · · 108
4.3 Conclusions · · · · · · · · · · 110
V. ANALYSIS OF DATA FROM GOETZL ET. AL. (1974) 119
5.1 Description of the data · · . · · 1195.2 Segregation Analysis. · 123
5.2.1 Single gene models · 1235.2.2 Two gene models. 126
5.3 Conclusions · · · · · · · 127
VI. ANALYSIS OF DATA FROM STENSTEDT (1952) · · · · · 136
6.1 Description of the data · 1366.2 Segregation Analysis. · · · 141
6.2.1 Single gene models 1416.2.2 Two gene models. · · . . 143
6.3 Conclusions · . · · · 144
VII. LINKAGE ANALYSIS · · · · · · 151
7.1 Description of the family · , · · · · · 1517.2 Segregation Analysis. · · · · · · 1537.3 Linkage analysis. 1557.4 Conclusions · · · · 156
VIII. SUMMARY AND CONCLUSIONS. 165
BIBLIOGRAPHY. . . . . . · · · · . · · · · · 175

LIST OF TABLES
Table
1.1 Classification of mental illnesses.
1.2 Pairwise concordance rates.
1.3 Morbid risks to relatives of affectively illprobands. . . . . . • . • . . .
1.4 "Etiologic" groups of psychiatric illnesses.
2.1 Genetic models for affective illness.
2.2 Parameters of the model and their interpretation.
3.1 Sex ratios among affectively ill people.Helzer and Winokur (1974) • . . .
3.2 Age of onset. Helzer and Winokur (1974)
Page
4
11
14
21
59
64
93
94
3.3 First degree family history. Helzer and Winokur (1974). 84
3.4 Preliminary autosomal models. Helzer and Winokur (1974) 95
3.5 Preliminary X-linked models. Helzer and Winokur (1974). .. 96
3.6 Autosomal models. Helzer and Winokur (1974) 97
3.7 X-linked models. Helzer and Winokur (1974) .. 98
3.8 Two gene models. Helzer and Winokur (1974). 99
4.1 Sex ratios among affectively ill people.Clayton and Reich (1969). ..••
Winokur,112
4.2 Age of onset. Winokur, Clayton and Reich (1969) 113
4.3 First degree family history.and Reich (1969) • • • .
Winokur, Clayton. . . . . . . . . . . 114
4.4 Morbidity risks, Str~mgren age corrected.Winokur, Clayton and Reich (1969) .... 115

Table
vii
Page
4.5
4.6
Preliminary single gene autosomal models.Winokur, Clayton and· Reich (1969) . • •
Preliminary single gene X-linked models.Winokur, Clayton and Reich (1969) .••
115
116
4.7 Single dominant gene models, autosomal and X-linked.Winokur, Clayton and Reich (1969) . . • . . . . . • . 117
4.8 Two-gene models. Winokur, Clayton and Reich (1969) . 118
5.1 Sex ratios among affectively ill people.Goetzl et. al (1974). . . . . . · . . . 120
5.2 Age of onset. Goetzl et. al. (1974). . 129
5.3 First degree family history. Goetzl et. al. (1974) · 129
5.4 Weinberg age corrected morbidity risks.Goetzl et. al. (1974) . . . . . . . . . . · 123
5.5 Preliminary autosomal models. Goetzl et. al. (1974) 130
5.6 Preliminary X-linked models. Goetzl et. al. (1974) · . 131
5.7 Single gene dominant models: Autosomal andX-linked. Goetzl et. al. (1974)
5.8 Single gene dominant models with ~ fixed at 7.0:Autosomal and X-linked. Goetzl ~l. al. (1974) .•
132
133
5.9 Two gene models. Goetzl et. al. (1974) • 134
6.1 Sex ratios among affectively ill people.Stenstedt (1952) .
6.2 Age of onset. Stenstedt (1952) .
6.3 First degree family history. Stenstedt (1952) ..
6.4 Preliminary autosomal models. Stenstedt (1952)
145
145
146
147
6.5 Preliminary single gene models:Normal. Stenstedt (1952) .
X-linked, Alcoholics =148
6.6 Single gene dominant models:Stenstedt (1952) ....••
Autosomal and X-linked.. . . " . 149
6.7 Two locus models. Stenstedt (1952) 150

Table
7.1 Segregation analysis under an autosomal model.Lava family . . . . • • . • • . • . .
7.2 Segregation analysis under an X-linked model.Lava family . . . . . • . •
7.3 Lod scores for colorblindness and Xg. Lava family
viii
Page
160
161
162
7.4
8.1
Phenotypes of proband's brothers on whom completeinformation is available. Lava family
Summary of unrestricted autosomal models over alldata sets. • .....••....•...
162
167

LIST OF FIGURES
Figure
1.1 Age of onset of affective illness. . ....Page
7
1.2 Age of onset in unipolar and bipolar disease.
2.1 Range of onset for which u(2,0,a) is greater than zero.
2.2 Map of the X-chromosome.
16
53
62
31. Age of onset with best-fitting normal and lognormaldistributions. Helzer and Winokur (1974) .... 83
3.2 Age of onset with best-fitting exponential distribution.Helzer and Winokur (1974) . . • • . • . . • . . . . . 83
3.3 Proportion of affectively ill people who are probands,by decade of onset. Helzer and Winokur (1974). . 85
4.1 Age of onset with best-fitting normal and lognormaldistributions. Winokur, Clayton and Reich (1969) 104
4.2 Age of onset with best-fitting exponential distribution.Winokur, Clayton and Reich (1969) . • • . . . . . . . .. 105
4.3 Proportion of affectively ill people who are probands,by decade of onset. Winokur, Clayton and Reich (1969).. 106
5.1 Age of onset with best-fitting normal and lognormaldistributions. Goetz1 et. al. (1974) ....•. 122
5.2 Age of onset with best-fitting exponential distribution.Goetzl et. al.· (1974). .•....••..•.• 122
5.3 Proportion of affectively ill people who are probands,by decade of onset. Goetzl et. a1. (1974) •.••• 124
6.1 Age of onset with best-fitting nofmal and lognormaldistributions. Stenstedt (1952) ...•.•.. 139
6.2 Age of onset with best-fitting exponentialdistributions. Stenstedt (1952) ..•. • • • 41 • • • •
" ") '~
139

Figure
x
Page
6.3 Proportion of affectively ill people who are probands,by decade of onset.' Stenstedt (1952) . . . . . . • 140
7.1 Lava family:relatives .
Pedigree of the proband's first-degree
, ,
152

CHAPTER I
INTRODUCTION AND REVIEW OF THE LITERATURE
The study of the genetics of manic depressive illness has been
hampered by many problems. As with the study of any mental disease,
there are problems with the definition and diagnosis of manic depressive
illness, and with enlisting the cooperation of the families of manic
depressive probands. These difficulties aggravate complications such
as incomplete penetrance, variable age of onset, and possible genetic
heterogeneity, that make the detection of genes in human populations
difficult. Previous family studies have, for the most part, been
concerned with calculating age corrected risks to various classes of
relatives of manic depressive probands. These risks are then compared
to the segregation ratios expected on various genetic hypotheses. This
method wastes information present in the family structures and does
not have the power to discriminate among genetic hypotheses in the
face of the problems present in the analysis of manic depressive
illness. Genetic hypotheses that have been suggested include a single
X-linked dominant gene, a single autosomal dominant gene, and two-gene
models including one X-linked and one autosomal dominant. Linkage
with X-linked markers has also been tested. While it is quite
generally acknowledged that there is a genetic component in the
etiology of manic depressive illness, no single hypothesis has
achieved widespread acceptance.

2
This thesis will be concerned with the analysis of four sets of
data, all previously reported in the literature, to examine these
hypotheses using Elston's approach (Elston & Stewart 1971, Elston
1973, Elston & Yelverton 1975) to pedigree analysis, which helps
overcome the difficulties present in the analysLs of mental illness.
Previously suggested modes of inherit~ce will be tested, and one
large family with data on manic depressive illness and the X-linked
markers of colorblindness and the Xg blood group will be used for
linkage analysis. Sections 2 and 3 of this chapter will review the
literature on the genetics of manic depressive illness and statistical
methods used to detect genes in human populations, respectively.
Chapter II presents a detailed description of the models to be used
and some extensions to the likelihood method of pedigree analysis.
Chapter III - VI present segregation analyses of the four sets of data,
and Chapter VII presents linkage analysis of a large family.
1.2 The genetic basis of manic depressive psychosis.
Manic depressive psychosis as a disease entity separate from other
forms of mental illness was first described by Kraeplin in the late
1880's (Kraeplin 1907, 1917). Kraeplin's nosological scheme classified
non-organic mental illness into several broad categories: melancholia,
maniacal-depressive conditions, dementia praecox (schizophrenia),
katatonia, and paranoia (Kraeplin 1917). He described manic depressive
psychosis as marked by recurrent attacks of mania and depression
separated by lucid intervals. Recovery from episodes is spontaneous
and there is no progressive mental degeneration as in schizophrenia.
Kraeplin says the prognosis is good in that recovery from individual

3
episodes is almost certain, but poor in that more attacks are likely
in the future (Kraeplin 1907).
Mania is characterized by psychomotor restlessness, flight of
ideas, great impulsiveness, irritability, insomnia, grandiosity, short
attention span, and sometimes delusions or hallucinations. Depression
is characterized by psychomotor retardation, inability to concentrate,
dearth of ideas, despondent view of life, loss of appetite, insomnia,
social withdrawl, and sometimes feelings of sinfulness, worthlessness
or hypochondria. These symptoms, described by Kraeplin, are almost
exactly the same diagnostic criteria used today by Winokur's group
(eg., Winokur, Clayton, and Reich 1969), Mend1ewicz's group
(Mend1ewicz and Fleiss 1974), and others (Abrams and Taylor 1974,
Goetz1 et. al. 1974, Hopkinson and Ley 1969, Gershon et. al. 1973).
Kraeplin's scheme is also quite similar to that of the American
Psychiatric Association (1968) (see Table 1.1), which divides non
organic psychoses into schizophrenia, major affective disorders,
paranoid states, and other psychoses. The category of major affective
disorders is further subdivided into involutional melancholia (a
form of depression with onset in late middle age); manic depressive
illness, depressive type (unipolar depression); manic depressive
illness, manic type (unipolar mania); manic depressive illness,
circular type (bipolar illness); and other (including "mixed"
disorders with mania and depression almost simultaneously). These
states are all characterized by, among other ~hings, an onset that
is independent of any obvious environmental trigger such as death
of a spouse. A depression occurring after such an event and severe
enough to be called psychotic is a psychotic depressive reaction and

4
Table 1.1 Classification of mental illnesses.
Classification of the AmericanPsychiatric Association (1968)
Kraeplin's classification(taken from Kraeplin (1917)
Melancholia
Delirium, delusions, hysteria
Maniacal-depressive conditions
Melancholia
Imbecility, idiocyCoarse brain lesions, epilepsy
ParanoiaAlcoholic, moral, drug psychoses
Dementia praecox, katatonia
J
I.II.
III.
Mental retardation }Organic brain syndromesPsychoses not attributed to
physical conditions295. Schizophrenia (& subtypes)296. Major affective disorders
.0 Involutional melancholia
.1 Manic depressive illness,manic type
.2 Manic depressive illness,depressive type
.3 Manic depressive illness,circular type
.8 Other major affectivedisorders
297. Paranoid states298. Other psychoses
.0 Psychotic depressivereaction
IV - X. Neuroses, childhooddisorders, etc.
is classified under "other psychoses" (American Psychiatric
Association (1968).
Reports of the frequency of manic depressive illness in the
population vary quite widely. Book (1953) found a lifetime morbid
risk of 0.0007 in Northern Sweden, while Tomasson (1938) found a
lifetime risk of 0.07 in Iceland. The most commonly reported figures,
however, average about 0.005 - 0.01 (Kallman 1954, Rosenthal 1970,
Winokur and Pitts 1965, Stenstedt 1952, Baldwin 1971). Part of the
variation can be explained by differences in diagnostic criteria used
by different investigators, and different methods of study (for example,

5
a study based on hospitalized patients will find lower prevalence than
one based on a population survey that includes milder cases).
Differing genetic, ethnic, and social environments may also contribute
to the differences. It is noteworthy, however, that in nearly all
studies where sex-specific risks were determined women tend to be
affected about twice as often as men, regardless of the overall
population prevalence (Helgason 1964, Kraep1in 1907, Stenstedt 1952,
Kallman 1954, Rosenthal 1970, Baldwin 1971).
Figures on population frequency are generally given in terms of
lifetime morbid risk, which is defined as the probability that a person
in the population under study will develop the disease sometime during
his life if he lives through the risk period (eg., Winokur, Clayton,
and Reich 1969). This implies that the risk period for the disease is
known. For affective disorders the risk period is usually taken as
15 - 60 (Winokur, Clayton, and Reich 1969; Winokur et. al. 1972),
20 - 50 (Stenstedt 1952, Slater 1935), 20 - 60 (Amark 1951, Stenstedt
1959), or 15 - 80 years (Perris 1974). There are two common methods
of estimating the morbid risk in a population where the members are
of variable ages and not all members have passed through the risk
period (Stenstedt (1952) gives a discussion). The simplest is the
Weinberg abridged method. It is assumed that the risk is uniformly
distributed over the entire period of risk. An estimate of the morbid
a
individuals not yet in the risk period, and b = number of individualsm
in the risk period. This is equivalent to giving a weight of 1 to all

6
people who have passed the risk period, a weight of 0.5 to all
people in the risk period, and a weight of 0 to all people not yet
in the risk period. The denominator is then a weighted average of
the number of people in the population.
The other method, devised by Stromgren, is based on the same
principle with a more complicated weighting scheme. The estimate is
aEb.c. ' where a = number of affected individuals, bi = number of
~ ~
1 in the ~th age d 1 ti . k t th iddlpeop e ~ group, an c. = cumu a ve r~s up oem e~
thof the i age group. Usually five- or ten-year age groups are used
and the weights are determined either from previous population surveys
or from the sample.
These methods are both approximations and there are some
objections to their use when the "population" is a specified group
of relatives of probands. For both methods the age of onset distribu-
tion in the population used to calculate weights (or risk period), and
that in the relatives must be similar or else the risk may be
considerably biased (Rosenthal 1970). Also, for the Weinberg method
to be valid the population distribution of age of onset must be
symmetric and there should be no correlation in age of onset between
relatives.
Age of onset distributions (Stenstedt 1952, Slater 1935) for
affective illness have been published for several populations. Within
each study, the distribution tends to be quite similar for men and
women (see Figure 1.1). These distributions, however, are based on
rather broadly defined affective disorders, including bipolar manic

)C..'1
7
Figure 1.1 Age of onset of affective illness •
..-r---------------------~~"'*-_jlf__--_..,oo
.,.oo
L~g
!~N!ioo~~ao-+---lp:::;;;.---r----.,-----.r-----t----t-----..,..---;
. 10.0 20.0 30.0 '0.0 50.0 60.0 10.0 80
)( Males}
y Females Stenstedt (1952)
• Males
() Females} Slater (1935)

8
depression, unipolar depression, and possibly involutional depressions
and thus may reflect the heterogeneity in the data.
The fact that there is a genetic component in the etiology of
manic depressive illness was recognized by workers as early as
Kraeplin, who said that 70% to 80% of the cases had an "hereditary
taint" and recommended that people who "seemed to be predisposed" to
the disease not marry (Kraeplin 1907). The earliest family studies
were done by Vogt (1910) and Hoffman (1921). Vogt examined 108
probands and their first degree relatives and found about 10% of the
parents had manic depressive psychoses. Hoffman examined the relatives
of people with endogenous depressions and found a very high incidence
of manic depressive illness in the children. This study was widely
criticized (for example by Slater (1935) and Stenstedt (1952)) for
use of very broad definitions of manic depressive illness (cycloid
personalities and "quiet humorists" were included as affected), and
for inadequate statistical treatment. Luxenburger (1932) collected
the data of several other investigators, including Hoffman, and found
incidences of manic depressive illness of 30.2% in children of pro
bands, 12.6% in sibs, 10.6% in parents, 2.6% in nephews, nieces and
cousins, and 4.8% in aunts and uncles.
Some of the earlier investigators constructed quite elaborate
genetic theories. Hoffman hypothesized three independent factors
carrying different weights with a certain total weight necessary for
psychosis and lesser weights causing cycloid personalities. Rudin
(1923) proposed that one autosomal dominant and two autosomal recessive
factors were necessary, and Luxenburger (1932) suggested one dominant

9
and one recessive factor. Slater (1935) criticized the methodology
and conclusions of all earlier studies and collected his own series
from Kraeplin's clinic. He considered as manic depressive illness all
cases with one clear manic and one clear depressive episode, or three
clear depressions where onset was younger than 50. Using StrBmgren
age correction with five-year intervals and a risk period of 20 to
50, he found the morbid risk for manic depressive illness to parents
of probands was 15.4% ± a standard error of 1.6% and to children was
15.8 ± 2.5%. He also criticized the earlier genetic theories, saying
they were premature, improbable, and supported by no real evidence.
He considered manic depressive illness to be transmitted by an
autosomal dominant gene with reduced penetrance. Part of the reduced
penetrance was explained by problems of diagnosis and ascertainment,
and part by the influence of the "genetic milieu." Slater noted the
increased incidence in women but said that X-linkage was unlikely,
and accounted for it by endocrine balance and social factors.
Several other investigators, however, have considered·the possi
bility of an X-linked factor. Rosanoff, Handy and P1esset (1935)
suggest a rather common autosomal dominant "cyclothymic" factor and
a less common X-linked dominant activating factor. Burch (1964),
drawing parallels between manic depressive illness and autoimmune
diseases, shows the data fit a model of a fairly common X-linked
dominant gene and a less common autosomal dominant. More recently,
Winokur and Tanna (1969), Mendlewicz et. ale (1972), and Taylor and
Abrams (1973) have suggested that a single X-linked dominant may be
responsible for bipolar manic depressive illness in some families.

10
Large twin studies have been done by Rosanoff, Handy and Plesset
(1935), Kallman (1950, 1954), Harvald and Hauge (1965), Kringlen
(1967), Allen et. al. (1974), and others. The special problems of
dealing with twins seem even more troublesome when mental disorders
are considered. In the early literature there is almost certainly an
over-reporting of concordant monozygotic twins (Rosanoff, Handy and
Plesset 1935). The special shared environment of twins may tend to
produce an excess of concordant pairs. Also, there has been some
suggestion that just being a twin may increase the likelihood of
mental disorders; for instance, Munro (1965) found 11 of 153 of his
probands with primary affective illness were one of a set of twins,
while only 3 of 163 controls had a twin. Tienari (1963) found a
similar situation for schizophrenia. However, several large
population surveys found no increased incidence of mental disorders
in twins over the general population (Harvald and Hauge 1965,
Kringlen 1967). In general, the studies have shown higher pairwise
concordance rates for monozygotic than dizygotic twins, and for
bipolar than unipolar disease (see Table 1.2). Rosanoff, Handy and
Plesset (1935) separated their sets by sex and found higher
concordance in female than male pairs, although the numbers were
quite small.
The likelihood that affective illness is not a genetically
homogeneous entity has been recognized for a long time. Slater (1935)
says, "We have little ground for supposing the clinical entity, which
is a matter of conception and convenience, and the underlying genetic
entity correspond." The major division made in order to obtain a

11
Table 1.2 Pairwise concordance rates.
Reference
Monozygotic probandBipolar Unipolar
II % II %
Dizygotic probandBipolar Unipolar
II % II %
Allen et. al. 1974 5 20 10 40 15 0 10 0Harvald & Hauge 1965 15 67 40 5Kring1en 1967 6 33 21 0Kallman 1950 23 96* ? 61* 52 26* ? 7*Luxenburger 1932 4 75 13 0Rosanoff et. al. 1935 ~ 14 71 ~ 27 22
0' 9 67 d'8 25opposite sex 32 9
*age-corrected rates
more homogeneous group of probands is between unipolar and bipolar
illness. Bipolar illness is defined, quite uniformly by various
investigators, as an affective illness with at least one clear-cut
episode of mania and one of depression (Slater 1935; American
Psychiatric Association 1968; Perris 1968a; Winokur, Clayton andG
Reich 1969; Mendlewicz and Fleiss 1972; Goetzl et. al. 1974).
Unipolar mania is usually reported to be quite rare: Perris (1968a)
reports 4.5% of patients exhibiting mania have unipolar mania, and
Bratfos and Haug (1968) find 2%. An exception is Abrams and Taylor
(1974) who find 28%. Unipolar manics, when found, are usually
considered to have bipolar illness.
Unipolar depression is generally diagnosed in one of two ways:
either by at least one clear cut depressive episode with no mania
(Winokur, Clayton and Reich 1969; Winokur 1970, Goetzl et. al. 1974,
~lendlewicz and Fleiss 1974), or by at least three depressions with no

12
mania (Slater 1935; Perris 1968a, 1969). Perris (1968a, 1968b) argues
in favor of using three depressive episodes to define unipolar
illness on the grounds that manic depressive illness is defined as a
recurrent disease and one or two isolated depressions are not suf-
ficient. Also, in about two-thirds of bipolar cases the first episode
is a depression (Kraeplin 1907, Stenstedt 1952, Perris 1968b), so a
person who had had only one depression may well change polarity with
the next episode. Perris (1968b) finds, however, that only 15% of
patients who have had three consecutive depressions and no mania have
a manic episode at some later date. Thus, he says requiring three
depressions provides a reasonable cut-off point for diagnosing
unipolar illness.
However, this criterion eliminates many (mostly young) people who
have had one or two depressions and have not yet passed enough of the
risk period to have had three depressions, thus exaggerating the age
of onset problem. Also, bipolar illness needs only two episodes to
•be defined while unipolar needs three. The average number of
depressions per unipolar patient may be less than three (Stenstedt
1952, Helzer and Winokur 1974; Perris 1968b quotes others' results).
Even Perris (1968b, 1969), using a minimum of three depressions to
define unipolar illness, finds the average number of episodes to be
only 4.2. However, it seems that for purposes of finding morbid
risks to relatives of unipolar probands, the definition makes very
little difference. Perris (1968b), using three depressions, finds
the morbid risk to first degree relatives is about 11%. Winokur et. a1.
(1972) using one depression find a risk of about 14%, and Mendlewicz

13
and Rainer (1974) find 22.4%. The risk is substantial, regardless
of the definition.
Bipolar illness is rarer than unipolar, with the proportion
of all affective disorders that are bipolar varying from about 8%
(Clayton, Pitts and Winokur 1965) to about 35% (Perris 1969). Bipolar
patients tend to have more episodes than unipolar (Perris 1968b, 1969;
Helzer and Winokur 1974), and to have their first episodes at a
younger age (Clayton, Pitts and Winokur 1965; Perris 1968b; Brodie
and Leff 1971; Gershon et. al. 1971; Taylor and Abrams 1973; Winokur
1975). Risks to first degree relatives of bipolar probands tend to
be higher than to first degree relatives of unipolar probands
(Perris 1968a; Cadoret, Clayton and Winokur 1970; Brodie and Leff
1971) (See Table 1.3). Studies which pool together all affective
disorders tend to find intermediate risks (Slater 1935, Stenstedt
1952). Thus, there seems to be good reason for separating probands
by diagnosis of unipolar or bipolar illness.
Another division that is sometimes made is "early" vs. "late"
onset. "Early" onset can mean before 30, 40, or 50 years. Winokur
and Clayton (1967b) separated a group of probands with affective
disorder into those with onset before 50 and those with onset after
50 and found the morbid risks for mothers, fathers, and brothers of
late onset probands all about 12% and for sisters 19%. In the early
onset probands, the risks were 30% for mothers, 22% for fathers, 11%
for brothers, and 21% for sisters. The probands were not split by
diagnosis of unipolar or bipolar illness. In a group of bipolar
probands, however, Winokur (1975) finds that risks to parents and sibs

14
+Table 1.3 Morbid risks to relatives (%) .
Reference
Parents
Mo. Fa. All
Sibs
Sis. Bro. All
Children
Dau. Son All
I. Bipolar Probands
57.1** 28.6**76.9** 57.1**
12.0
27.0 22.09.1 10.0
46.0 23.039.0 30.044.0 27.0 35.0
18.2 9.4
10.3
18.812.5
1. 9 11. 2
50.0 23.063.0 0.056.0 13.0 34.0
Mend1ewicz & Rainer 33.7 39.2 59.0(1974) :..:c..::;..__--"-"'-'-' ..:c.:- --"-..;;....;...;'-.-
Winokur et. a1(1972)
Perris (1968a)Winokur, Clayton
& Reich (1969)!t probandt proband
overallGoetz1 et. a1.
proband ~ 24. 2(1974)probandg 18.2
II. Unipolar Probands
Winokur et. a1.(1972)
Cadoret, Winokur,& Clayton (1971)~ probandi5' proband
Perris (1968a)
12.8
16.1 7.114.1 18.2
5.4 4.5
15.3
25.86.6
11.4
8.84.9
6.4
9.7
9.40
6.04
24.1
13.815.8
2.53
5.06
9.0
6.04 10.0
12.7
5.48 6.62
10.27 14.29 12.26 14.29
3.4
10.412.0
15.49.79 4.81 7.35
7.22 3.21 5.25
Slater (1935)*Hoffman (1921)*Banse (1929)*Ro11 & Entres
(1936)*Luxenberger (1942)
III. Endogenous depression, or all affective1y ill probands
Stenstedt (1952)"certain""certain"+"uncertain"
**These risks are based onvery small numbers
*Taken from Rosenthal (1970) + Risks areWeinberg or
age corrected by either theStrtlmgren method.

15
are the same in the early (before 30) and late onset groups.
Hopkinson and Ley (1969), using probands with bipolar manic depres-
sive psychosis or unipolar depressions find that in both men and
women onset later than 40 is associated with significantly lower
risks to first degree relatives than onset before 40.
Reports of the sex ratios in early and late onset groups are
conflicting. Winokur (1975) finds more women in the early onset
group but concedes that the literature is divided on this point,
while Hopkinson and Ley (1969) and Taylor and Abrams (1973) find
no excess of women in the early onset group. Thus, while there is
some evidence that familial cases tend to have earlier onset, dividing
probands by "early" or "late" onset does not seem to give homogeneous
groups. It seems likely that whatever differences there are between
early and late onset groups arise from the earlier onset of bipolar
patients as compared to unipolar patients (see Figure 1.2). While
bipolar patients tend to be younger at onset, the age of onset
distributions of unipolar and bipolar patients are sufficiently
overlapping to make early vs. late onset not a useful criterion.
A more useful criterion for reducing heterogeneity within a
sample of families may be to analyze separately families with twoI
or more cases of affective illness, thus eliminating families of
probands who have a "sporadic" or environmentally caused case. This
has been done by Winokur's group (Winokur and Clayton 1967a,b,
Winokur and Pitts 1965) and Mendlewicz's group (Mendlewicz et. ale
1972, Mendlewicz et. ale 1973), by selecting families with one or
more additional cases among the first degree relatives of probands.

Figure 1.2 Age of onset in unipolar and bipolar disease.
16
x-«i
10.0 20.0 30.0 50.0 60.0 70.0 80
Y Males
)( Females} Bipolar (Winokur, Clayton and Reich 1969)
() Males }• Females
Unipolar (Winokur et ~. 1971)

17
Mendlewicz's group found that the affectively ill people in the
positive family history group had earlier onset with manic and
depressive episodes more nearly equal in frequency than the negative
family history group, and that the positive family history group more
nearly fit the model of transmission of a single dominant gene.
Winokur's group split families on the basis of whether or not the
proband had an affected parent, and found significantly more mania
and more affected sibs in the group of probands with an affected
parent. Ho~ever, none of these studies took into account the manner
in which the sample was selected.
An interesting finding, and possibly a more significant one from
a genetic standpoint, is that it may be possible to form meaningful
groups of probands on the basis of response to drug therapy.
Biochemical theories of mania and depression center around two related
concepts. It has been found that levels of brain neurotransmitters
(norepinephrine and dopamine) are functionally decreased in depression
and increased in mania. Tricyclic compounds (eg., imipramines) and
monoamine oxidase inhibitors (MAOI) are used to treat depressions,
since their effect is to slow the breakdown of the neurotransmitters
(Bunney et. ale 1972, Janowsky 1972). There may also be an upset of
the ionic balance in the nerve endings, and one theory is that the
basic defect may be in the membrane, interfering with the neuronal
uptake process, and associated with ionic balance maintained by the
membrane (Bunney et. ale 1972). Lithium salts are used to treat
both depression and mania (Bunney et. ale 1972; Mendlewicz, Fieve
and Stallon 1973). Studies of drug response in manic depressive
patients and their relatives have been small, but they are suggestive.

18
Angst (1961) found that 66% of 105 people improved when treated
with imipramine for endogenous depression. Of these, there were 9
who had a first or second degree relative who had also been treated
with imipramine. Eight of the pairs had concordant responses; only
one pair was discordant.
Pare et. al. (1962), using imipramines or MAOI on a heterogenous
group of outpatients, found that reactive depressions tend to respond
to MAOI, and endogenous depressions tend to respond to imipramines,
and hypothesized different genetic mechanisms for the two types of
depression. They were also able to find seven proband-first degree
relative pairs treated with the same drug, and all pairs showed
concordant responses. In a later study, Pare and Mack (1971) reported
12 proband-relative pairs (presumably different from the 7 above)
treated with the same drug, and 10 pairs were concordant.
Mendlewicz, Fieve and Stallon (1973), using a more homogeneous
group of 73 bipolar probands in a study of lithium vs. placebo,
found that there was a significant association between positive
response to lithium and the presence of bipolar illness in first
degree relatives.
These studies all ~ggest that affective illness within a family
tends to have the same biochemical (ie., genetic) basis and that
response to drugs may provide a way of discriminating between genetic
and environmental forms of affective illness.
A problem has been raised which is the opposite of trying to
divide affective illnesses in order to get a homogeneous group of
probands: whether there is any reason to subdivide mental illness
into affective disorders, schizophrenia, and so forth. There have

19
been several studies published dealing with the relationship of
manic depressive illness to schizophrenia, involutional psychoses,
and the so-called "mixed" or schizoaffective disorders.
Most studies have found that there is very little, if any,
increased risk of schizophrenia in relatives of manic depressive
probands. Slater (1935), Stenstedt (1942), Kallman (1950, 1954), and
Winokur et. al. (1972) all find morbid risks for schizophrenia
in parents and sibs of manic depressive probands of about 0.5% to
2%. The risk for schizophrenia to the general population is about
0.8% to 1% (Kallman 1954, Rosenthal 1970), so these risks are not
excessively high. The risks to children, however, tend to be
somewhat higher: 1.6% to 3.1% (Slater 1935, Kallman 1954, Rosenthal
1970). This finding has never been satisfactorily explained, but
it seems the simplest explanation is that being raised by a manic
depressive parent can lower the threshold for schizophrenia and
cause a child to show the schizophrenic phenotype when he ordinarily
would not (Rosenthal 1970).
The relationship of manic depressive illness to involutional
melancholia is somewhat hazy. One problem is that unless age periods
are carefully defined involutional melancholia can be confused with
unipolar depression with rather late onset. Stenstedt (1959) using
a possibly heterogeneous group of probands (307 with endogenous
depression including manic depressive and involutional psychoses)
finds risks to sibs of 3.0% for involutional depression and 1.7% for
manic depression; the risks to parents were 1.6% and 0.6% respec
tively. These figures are not above those for the general

20
population. Rosenthal (1970) finds that the morbid risks for manic
depressive illness are a little higher, on the order of 5%, and
concludes that involutional melancholia is genetically associated
with other affective disorders.
The "mixed" (schizoaffective, schizophreniform, or cycloid)
psychoses are those that can be diagnosed neither as schizophrenia
nor as affective disorders but have some of the characteristics of
each. Whether or not the schizoaffective disorders are a distinct
entity is not agreed upon. Perris (1974) and Mitsuda (1965) consider
schizoaffective disorders as a separate disease. Perris, in a study
of 60 probands, finds morbid risks for schizoaffective disorders in
parents and sibs considerably higher than risks for schizophrenia
or manic depressive illness (20% in parents and 9% in sibs for schizo
affective disorders; 0% and 1.4% for schizophrenia; 3.1% and 0.68%
for manic depression). Mitsuada (1965), using twin and family
studies, finds no twin pair (out of 16) in which one twin has
schizophrenia and the other a schizoaffective disorder. He also
finds that risks for schizoaffective disorder in families of
schizophrenia or manic depressive probands are small. These studies
support the view that schizoaffective disorders are not generally
related to schizophrenia or affective disorders, but must be
considered as independent.
Clayton, Rodin and Winokur (1968) and Stromgren (1965), on the
other hand, consider the schizoaffective disorders as a symptomological
concept, not a distinct entity. Clayton, Rodin and Winokur, using a
group of 39 probands with schizoaffective disorder, find 11 of their
parents had an affective disorder, and that the risks to first degree

21
relatives for affective disorder is higher than for schizoaffective
disorder or schizophrenia. They conclude that schizoaffective dis-
order is a variant of affective disorder. Stromgren (1965) says
that the schizoaffective psychoses are a heterogeneous group, made
up partly of atypical schizophrenias and partly of atypical affective
disorders.
The schizoaffective disorders are relatively rare, however.
Clayton, Rodin and Winokur (1968) in their series of 426 cases of
primary affective disorder find 39 patients with schizoaffective
disorder. For the most part manic depressive psychoses tend to
"breed true." Gershon et. al. (1971), after an extensive review
of the literature, construct three groups of psychiatric illnesses on
the basis of "common etiologic genetic factors" (see Table 1.4).
Table 1. 4 "Etiologic" groups of psychiatric illnesses.(After Gershon et. al. 1971)
Affective & related
Unipolar depressionPsychotic depressive
reactionBipolar manic depressionInvolutional melan
choliaAlcoholism (?)Antisocial personality (?)
Schizophrenia & related
Chronic schizophreniaSchizoidParanoidInadequate personality
Mixed
SchizoaffectiveAntisocial (?)Alcoholism (?)Some mental
retardation (?)
It is generally agreed that at least a portion of affective illnesses
are caused to a large degree by genetic factors. The nature of the
genetic factors, however, is still open to question. A major point

22
of disagreement is whether bipolar manic depressive disease is caused
by an X-linked gene, at least in some families. The old observation
that about twice as many women as men are affected is suggestive of
a rare X-linked dominant gene. However, there may be other explana
tions for the sex ratio. Kallman (1954) says that social factors may
be causing the excess: men have more of an advantage in concealing
the disease, and women have less control over their lives and are
therefore more likely to be hospitalized. Ther~ may also be a slight
excess of completed suicide in men (Kallman 1954, Stenstedt 1952,
Gershon et. al. 1971), although if there is any sex difference it
is not large (Pitts and Winokur 1964, Perris 1969). Slater (1935)
says the sex difference may be due to "different endocrine balance"
in women, and Stenstedt (1952) says manifestation is greater in women
due to "constitution." There has been some suggestion (Brown et. al.
1973) that the overall sex ratio in families with affective illness
is aberrant, with a preponderance of women. This has not been
found in other studies (Winokur and Reich 1970; Cadoret and Winokur
1975 give other references).
A more stringent requirement of the X-linkage hypothesis is
that there be no father to son transmission of the disease. A
number of groups refute the X-linked hypothesis on this basis
(eg., Goetzl et. al. 1974; Perris 1968a, 1969; Von Grieff et. al.
1975). Perris (1968a, 1969) finds morbid risks to sons and daughters
of bipolar men about equal, and that the male to female ratio for
bipolar patients is about one to one. He concludes that bipolar
illness cannot be due to an X-linked gene. Goetzl et. al. (1974)

23
find four instances of male to male transmission in their series of
39 manic probands, as well as an equal number of men and women
affected, and conclude that a locus on the X chromosome "is not itself
a necessary or sufficient condition for the transmission of manic
depressive illness."
Perris (1968a, 1969) finds a preponderance of women among his
unipolar patients, and that risks to daughters of unipolar men are
much higher than risks to sons (although there are a few cases of
father to son transmission). He quotes Angst (1966) as obtaining
results very similar to these, and concludes that unipolar depression,
but not bipolar illness, may be due to an X-linked dominant gene.
Other groups (eg., Taylor and Abrams 1973, Mendlewicz and
Rainer 1974, Reich, Clayton and Winokur 1969) reach exactly opposite
conclusion: that bipolar illness may be due to an X-linked gene but
not unipolar illness. In some studies with bipolar probands there is
no male to male transmission found (Winokur, Clayton and Reich 1969,
Taylor and Abrams 1973, Helzer and Winokur 1974); in others there is
some (Reich, Clayton and Winokur 1969, Mendlewicz and Rainer 1974),
but these people generally conclude that there is heterogeneity of
the disease rather than reject the X-linkage hypothesis.
In further support of the hypothesis, Winokur, Clayton and Reich
(1969) find that the risk bo mothers of female probands is about twice
that to fathers, and the risk to sisters is about twice that to
brothers. For male probands the risk to fathers is 0 and is 63% to
mothers, while the risk to brothers and sisters is about equal (see
Table 1.3). This is apparently the only study (of this group or

24
others) where the risks turned out to support the X-linkage hypothesis
so strongly. Usually, at least a few cases of father to son
transmission are observed, or there is some other abberation in the
sex ratios of observed risks. However, in most studies father to son
transmission of mania is very rare (Dunner and Fieve 1975).
Cadoret, Winokur and Clayton (1971), using unipolar probands from
their Family History Studies series and four other series in the
literature find that sons and brothers of female probands have smaller
morbid risks than daughters and sisters, and that all children of
male probands have about the same risk. They rule out X-linkage for
unipolar depression and hypothesize that there are two subgroups of
depressive illness: one that is sex limited to women and one that is
expressed equally in men and women. There seems to be no simple
explanation for the discrepancy in results of these groups, and the
results of Perris, Goetzl et. al. and others. Gershon et. al. (1976)
conclude, after an extensive review of the literature, that bipolar
illness may be transmitted by an X-linked gene in some families but
that family studies do not suggest that this is the usual mode of
inheritance.
Another hypothesis that has been suggested is that transmission
is polygenic. Slater (1966) used a very simple approach to determine
that if transmission of a disease is polygenic then pairs of affected
relatives should be in the ratio one bilateral (ie., a maternal and
a paternal relative) to two unilateral (ie., two maternal or two
paternal). If transmission is via a single dominant gene then there
should be an excess of unilateral pairs (assuming mating is at random).

25
This model has been applied by Perris (1971) and Slater et. al.
(1971) who find that the polygenic model fits for both unipolar and
bipolar diseases, and by Baker et. al. (1972) who find it fits
unipolar disease. Mendlewicz et. al. (1973) divide their bipolar
patients by those who have at least one first degree relative with
affective illness and those who do not. They find the polygenic model
fits the negative family history group but not the positive family
history group. However, Slater et. al. (1971) point out biases that
can arise in this type of analysis, including an excess of bilateral
pairs caused by assortative mating or the possibility that knowledge
of mental illness on one side of the family increases the likelihood
of its being discovered on the other side.
Another observation of interest is that alcoholism seems to be
more frequent in families with affective illness than would be expected
by chance. Winokur's group, particularly, has looked at this in some
detail (Helzer and Winokur 1974; Winokur, Rimmer and Reich 1971;
Pitts and Winokur 1966; Winokur and Clayton 1967a; Winokur and Reich
1970). They find, in general, that in families with affective
illness alcoholism tends to be increased in men, but not in women.
Winokur and Reich (1970) find that 16% of the fathers of 61 manic
probands were alcoholic, and that 25 probands had alcoholism in the
family, with no difference in incidence in maternal and paternal sides.
Winokur and Clayton (1967a) find the risks for alcoholism in brothers
of affectively ill probands greater than in sisters, and that the risk
for affective illness to sisters of female probands (21.6%) was much
greater than the risk to brothers (7.4%). If it is assumed that
alcoholism is an affective equivalent, risks to brothers and sisters

26
of male and female probands are about equal. However, alcoholism is
not rare enough to assume all alcoholism in the families is affective
disorder. Also the risk for alcoholism in brothers of male and female
probands is about equal, but the risk for affective illness is not.
The increased risk for alcoholism in families with affective
illness apparently does not go the other way, however. Amark (1951),
in a study of alcoholic probands, found increased risk of alcoholism
to fathers and brothers but not to mothers and sisters. The risk to
parents and sibs for manic depressive illness and schizophrenia was
not increased over the general population. It has also been found
(Pitts and Winokur 1966, Rosenthal 1970, Gershon et. al. 1971) that
alcoholism tends to be increased in the families of people with other
mental diseases such as schizophrenia and various neuroses, and thus
the association of alcoholism with mental disease may be rather
nonspecific.
Winokur and Reich (1970), and Morrison (1975), however, suggest
a two gene hypothesis for bipolar manic depression that involves
alcoholism. The risk to first degree relatives of bipolar patients
tends to be less than the 50% expected for a dominant gene. Thus,
the gene either has reduced penetrance or there is more than one
gene involved. Winokur (1970) observes that about 43% of the affec
tively ill sibs and children of manic probands showed mania, and
points out that this is about what would be expected if there were
another dominant gene that caused mania in the presence of the gene
causing affective illness. In later papers (Winokur and Reich 1970,
Morrison 1975) it is suggested that the second gene may be expressed

27
as alcoholism in the absence of the primary factor. They hypothesize
also that the primary factor causing affective illness is an X-linked
dominant and the second factor is an autosomal dominant. This is
quite an elaborate hypothesis and while it fits much of the data it
has not been rigorously tested.
The hypothesis that bipolar disease is due to an X-linked gene
has recently been tested by linkage analysis (Fieve, Mendlewicz and
Fleiss 1969; Winokur and Tanna 1969; Winokur, Clayton and Reich 1969;
Mendlewicz, Fleiss and Fieve 1972; Mendlewicz and Fleiss 1974).
Winokur and Tanna (1969), using ten two-generational families with
unipolar or bipolar disease and tested for nine blood groups, found
no evidence of linkage between unipolar depression and any of the
markers. However, they found significant evidence of linkage between
bipolar disease and the Xg blood group. The sample was very small
(only two families were informative), and although there was no
information on the mothers' phases they assumed both were in coupling
and counted "recombinants." Winokur's group also has two large
families in which both bipolar illness and colorblindness are present
(Winokur, Clayton and Reich 1969; Reich, Clayton and Winokur 1969).
In these families they find that there are 11 people with the
colorblindness gene and the hypothesized manic depression gene, and
only (possibly) one person with one gene and not the other (one young
man is colorblind but not yet affectively ill). They say the
probability this is due to chance is (.5)11 = .0005, and that this is
strong evidence for linkage.
Mendlewicz and Fleiss (1974) take all the informative families of
Winokur's group, plus 15 of their own, and using lod scores as

28
tabulated by Edwards (1971), find maximum likelihood estimates of
recombination fractions of 0.07 for bipolar-deuteranopia, 0.10 for
bipolar-protanopia, and 0.19 for bipolar-Xg. The maximum lod scores
are 4.47 for linkage with deuteranopia, 3.73 with protanopia, and
2.95 with Xg. If valid, this would be strong evidence for linkage
with both the colorblindness loci and Xg. However, Gershon and Bunney
(1976) have pointed out some of the serious methodological problems
with these studies, and it would be premature to conclude that
linkage between a gene for manic depressive illness and an X-linked
marker has been demonstrated.
1.3 Statistical methods used to detect major genes in human family
data.
There are many problems that arise in trying to detect genes that
cause disease in human populations. Small sample sizes and non-random
sampling are among the more serious statistical problems; genetic
problems include the probable genetic heterogeneity of many diseases,
incomplete penetrance and sporadic cases, and a variable age of onset.
As a result the classical Mendelian segregat~on ratios are virtually
never observed in human families for any traits of interest. Two
major approaches used to detect major genes are segregation analysis
and its variants, and linkage analysis.
Classical segregation analysis calculates the expected segregation
ratios of affected children conditional on parental phenotypes,
assuming a specific one gene model (eg, Morton 1969, Morton et. al.
1971). It can take into account incomplete penetrance, non-random
sampling, and sporadic cases. It must be used on families of parents

29
and their children only, however, and a simulation study (Smith 1971)
shows that classical segregation analysis is not very powerful for
detecting major genes in the face of complications like incomplete
penetrance and a variable age of onset.
Other approaches to the problem of incomplete penetrance have
been made by Trankell (1955, 1956); O'Brien, Li and Taylor (1965);
Defrise-Gussenhoven (1962), Wilson (1974) and others. The basic
model taken by Trankell and by O'Brien et. al. is that of a single
locus with two alleles, resulting in two phenotypes. O'Brien et. al.
consider a recessive gene with incomplete penetrance and calculate
the frequencies of sib-pair phenotypes as a function of the penetrance
parameters and the gene frequency. They derive maximum likelihood
estimates of these parameters. Trankell takes a more general approach.
Each genotype is allowed a different probability of affectation, and
expected proportions of affected children from different mating types
are calculated. These proportions are expressed as a function of the
probability of affectation for each genotype and of the population
gene frequency.
The approach of Defrise-Gussenhoven is to try to discriminate
a two-gene hypothesis from that of a single gene with incomplete
penetrance. The model is of two genes, each with two alleles,
resulting in two phenotypes. Biologically reasonable modes of
interaction of the two genes are considered, and the frequencies of
the phenotypes in children of different mating types are calculated
as a function of the gene frequencies. Similar calculations are made
on the hypothesis of one incompletely penetrant gene, and charts of

30
the distribution of phenotypes as a function of gene frequency are
given to aid in the rejection of unreasonable hypotheses.
Two approaches to analysis of traits with a variable age at onset
have been made by Batschelet (1962, 1963) and Elandt-Johnson (1973).
Batschelet's model is of one locus, two alleles, and two phenotypes.
Assuming Hardy-Weinberg equilibrium and no correlation in age of onset
among relatives, he defines a penetrance function p(x)=Pr(age at
onset~x) and allows for an upper age cutoff beyond which it is assumed
no one acquires the disease. The basic calculation is Pr(k parents
and r sibs [out of s] are affected I ages, genetic hypothesis, and
parental mating type), and is a function of the p(x) for the ages of
everyone in the family. However, Batschelet makes the approximation
of replacing the parents' ages by their average, and the childrens'
ages by their average, which converts the probabilities to products of
binomials. This introduces a bias which gets larger as the sample
size increases. The approximation is not necessary, but only makes
the probabilities easier to write down in a closed form. Batschelet
also considers corrections for ascertainment in the form of single
selection or truncate selection.
Elandt-Johnson (1973) uses a life-table approach to estimate the
probability that a child will be genetically susceptible to a disease
given the parental mating type, when onset of the disease is variable.
Her model assumes that onset is variable but early (so that onset is
complete in the parents), that all susceptible genotypes develop the
disease eventually, and that all parents of a given mating type have
the same segregation pattern. Actuarial-type techniques are used

31
to estimate probabilities of a child developing the disease at age x
and the overall proportion of children susceptible to the disease
from parents of given phenotypes.
Recently, more powerful and general models for segregation
analysis have been devised (MacLean et. al. 1975, Morton and MacLean
1974, Elston and Stewart 1971, Elston 1973, Elston and Yelverton
1975). These methods are based on calculating the likelihood of a
family's phenotypes based on a genetic hypothesis. The method leads
to maximum likelihood estimates of parameters, and hypotheses are
tested by likelihood ratio tests.
An important assumption underlying these models is that there is
no non-genetic correlation in phenotype between parent and offspring.
Violation of this assumption might lead to the detection of a
spurious major gene. Morton's model (Morton & MacLean 1974)
specifically allows for an environmental correlation among sibs.
Elston's model assumes that there is no sibling environmental correla-
tion, but simulation studies (Go et. al. 1977) suggest that the method
is robust against violations of this assumption.
Morton's model is currently programed for the mixed model of a
major gene (with two alleles) plus polygenic and environmental
variation for families of parents and their children. The likelihood
is expressed as
00 3 3 s 3
L(f)=Looj~l k~l ~jku n~l i~lPi,jkPr(~nlgi,u)du

32
where
phenotype of jth person; there are 2 parents and s children
. 1 f .thmaJor ocus genotype 0 J person
(midParent breeding value + ) I ]
Pr[gj,gk'u = environment common to sibship ~f'~m
= Pr[child's genotype is g.lhis parents' genotypes are1
and
Pr(~ Ig.,u) is the phenotype distribution, conditional onn 1
genotype and u.
These are expressed as functions of the mean and variance of the
trait in question, the degree of dominance'of the major locus, the
displacement of major locus (i.e., the distance between homozygotes),
the gene frequency of major locus, polygenic heritability, and
relative variance due to common environment.
The phenotype may be a quantitative measure, a dichotomy (normal
vs. affected) or trichotomy (normal, intermediate, affected), and
the model specifically allows for an environmental correlation among
sibs (but not between parents and offspring). The null hypothesis
of no major locus is tested by setting the gene frequency equal to
zero. Morton and MacLean (1974) have performed simulation experiments
to test the power and robustness of the model, and found that the
method has good power to recognize a major gene when applied to '
quantitative data on samples of several hundred families. It is
reasonably robust against parent-child environmental correlations,

33
assortative mating, and heterogeneity among families if and only if
the model includes both polygenic variation and sibling environmental
correlation, and tests of heterogeneity are performed.I
Elston's model can, at least in theory, encompass a wide
variety of genetic models, including one- and two-major gene models
(with linkage analysis as a special case), polygenic, and the mixed
model of one major gene plus polygenic background. The likelihood
is formulated for pedigrees of nearly any size and structure, and
can take into account incomplete penetrance, variable age of onset,
and nonrandom sampling.
The likelihood of observing the phenotypes of a family under a
genetic model can be expressed in terms of:
(i)~u' the population frequency of genotype u;
(ii)p ,the probability that a child has genotype u, givenstu
that his parents have genotypes sand t; and
(iii) 8hu (z), the probability (or likelihood) that an individual
has phenotype z, given he has genotype u and sex h (Elston
and Stewart 1971).
The genetic model can be one gene, two linked genes, two unlinked
genes, polygenic, or the mixed model of one major gene with polygenic
background. The type of genetic transmission is expressed in the
transition matrix of probabilities, p . For the one- or two-genestu
models, the p t are discrete probabilities; for the polygenic modelss u
they are expressed in terms of normal densities. The segregation
analysis considered here is concerned with detection of major genes;
therefore models of one or two genes will be of interest. The pstu

34
can then be expressed in terms of transmission probabilities:
'ss'u = Pr(genotype sst transmits allele u). For one autosomal
locus with two alleles, the matrix P = (p ) is 3x3x3, and under- stu
Mendelian inheritance, 'AAA = 1.0, 'AaA = 0.5, and 'aaA = 0.0.
The phenotypic model is expressed in the distributions g (z).u
Phenotype may be a continuous measure, or categorical with two states
(as in a disease) or multiple states (as in some blood groups, for
example). The distributions may be formulated to take into account
such things as reduced penetrance, sporadic cases of a disease, or
variable age of onset. For example, in the simple case of a partially
penetrant or incompletely dominant gene, where zi 1 if the i th
person is affected, and 0 if he is normal, the phenotypic model might
be
1
f
o
l-f
g (0) = 0aa
g (0) = 1aa
The likelihood of a nuclear family, assuming that it is a random
sample from the population and that there is no environmental
correlation in phenotype among family members, is
where zf = father's phenotype, Z = mother's phenotype, z. = children'sm 1
phenotypes (Elston and Stewart 1971). The likelihood calculations are
easily extended (in theory) to complex families of more than two
generations (Lange and Elston 1975), and to include half sibs and
twins (Elston and Yelverton 197&).

35
The correction for ascertainment is necessary when families
are ascertained through one or more probands. The likelihood of a
family is expressed as L(flassertained through one or more probands)=
L(f)L(one or more probandsl~) (Elston 1973).L(one or more probands)
L(one or more probandslf) is called the ascertainment function.
If we assume that probands are ascertained independently from only
one source, then the ascertainment function for the family is the
product of the individuals' ascertainment functions.
The ascertainment function for an individual is a(zi,bi,ai
)=
thPr(i person has proband status b.1 disease status z , and age of
1 i
onset a.)1
where 1·f h .th . b d dt e 1 person 1S a pro an , an
if he is not.
The ascertainment function for the whole family is
a(~,E.,.9:.) = J}a(z. ,b., a.).1. 111.
where i indexes all individuals in the family. If we let TI(z.,a.,) =1. 1
Pr(a person with phenotype z. and age of onset a. is a proband) then1. 1
(Elston and Yelverton 1975). Various models for TI(zi,ai
) have been
proposed. The simplest is to assume that the probability that an
affected person is a proband is the same for all affected people:
TI(l,a.) = TI, and TI(O,a.) = O.1. 1.

36
The denominator of the conditional likelihood is referred to as
the sampling correction. Define B = Pr(no probands in family). Then
the sampling correction Pr(~ 1 probands in family) = I-B. B is condi-
tional on the size, structure, and ages at examination observed in the
family but not on the phenotypes (disease status, age of onset) of
the family members. It is equal to the multiple intrega1 over ages
of onset and multiple ?ummation over disease statuses of the numerator,
with all proband statuses, b. set equal to 0 (Elston 1973). The1
calculation of B is somewhat simplified by the fact that in the
expression for L(f) there is just one factor for each person that
depends on his phenotype. Therefore, the order in which operations
are carried out may be rearranged and the numerator (for a two-genera-
tiona1 family) is
, , nE~ a(zf,b f ,af )gl (zf,af,af)L~ a(z ,b ,a )gZt(z a ,a ).TI1Epssstt m m m m, m m 1= u stu
a ( z . , b . ,a. ) gh u (z . ,a ~ , a . ) ,111.111
1 . __....-_
where h = I if a person is male and h = 2 if female.
The integration and summation can be performed separately for each
person, so that B depends on functions
r;h u =i
fa. Z ,
1 E a (z ., 0 , a . ) gl ( Z. ,a . , a . ) d_('0 Zj =0 1 1 Ii lJ 1 1 1 a i
(Elston and Yelverton 1975). r;h may be interpreted as the prob.u1
ability of not being a proband by age a~, conditional of sex hi and
genotype u. B can be calculated using the same algorithm as L(f),,
with r;h ~eplacing gh (z.,a.,a.). It depends on the partieular.u .u 1 1 11 1
forms of the ascertainment and phenotypic models.

37
If there are N families (assumed independent) in the sample
then the likelihood of the sample is L = WL(fi)L(ascertainmentl~l.i=l l-8
i
Maximum likelihood estimates of the parameters of the model are
obtained via a search algorithm (Kaplan and Elston 1973). Hypotheses
are tested by likelihood ratio tests. An hypothesis corresponds to
placing restrictions on one or more parameters, and the likelihood
is maximized both in the unrestricted case and under the hypothesis.
If L denotes the unrestricted maximum likelihood and L k denotesr r-
the maximum likelihood with k independent restrictions on the r"-
parameters, then 2 (lnL - I.nL k' is asymptotically distributed as ar r-
X2 with k degrees of freedom under the null hypothesis (see, ego
Kendall and Stuart 1973).
When the principal question of interest is whether or not there
is a major gene, hypotheses about the transmission probabilities
T I are tested. The null hypothesis that there is a major geness u
corresponds to fixing the T'S at their Mendelian values. For an
autosomal locus the hypothesis is Ho
: TAAA
= 1.0, TAaA = 0.5,
2TaaA
= 0.0, and the X has 3 degrees of freedom. The "environmental"
hypothesis of no vertical transmission is Ho
: TAAA
= TAaA = TaaA
,
2and the X has 2 deg~ees of freedom.
Linkage analysis attempts to find an association of phenotypes
within families that is not present in the general population. Two
genetic loci are said to be syntenic if they are on the same
chromosome, and are linked if they are on the same chromosome close
enough together so that there is an association of phenotypes
within families. If it is possible to demonstrate linkage between

38
an hypothesized locus for a disease and a well-known marker locus
(such as a blood type), then this is convincing evidence that there
is a major gene that causes the disease, since it is difficult to
construct situations where an environmentally-caused disease could
mimic linkage with a marker locus.
The problem of detecting linkage would be simple were it not
for the process of recombination, or crossing-over. At a certain
stage in the formation of gametes, homologous chromosomes are paired
and genetic material can be exchanged within a pair. This means
that it is not particular alleles that are linked, but only the loci:
in an equilibrium population there is no association of traits due
to linkage. The recombination fraction, A, is defined as the
proportion of offspring that have genotypes different from the
parental genotypes. For example, in a mating DdMrn x ddmm the off
spring are of four types: DdMrn, ddrnrn (the parental types), ddMrn, and
Ddrnrn (the recombinant types). With no linkage, the expected
"recombination" fraction is .5, since all types are equally likely.
Values of the recombination fraction less than .5 indicate some
association of loci. Only certain mating types are capable of giving
information on linkage; one parent must be heterozygous at both loci,
and neither parent can be homozygous for a dominant at either locus.
The most informative mating is a double backcross DdMrn x ddmm. X
linkage is the easiest to deal with, since if it, is known that both
traits are X-linked then they are on the same chromosome, and all
informative matings are essentially double back-cross (ie.,
DdMrn S x dm t- ). Problems in detecting linkages in human data include

39
fairly small family sizes, inability to control matings, and the
small prior probability that two random loci are linked.
Early methods of detecting linkage in human data included the
Fisher-Finney u-scores, Penrose's sib-pair methods, and the
probability ratio test suggested by Haldane and Smith. Fisher
(1934, 1935) suggested modifications of Bernstein's scores that
resulted in a maximum-likelihood scoring procedure, appropriate for
detecting linkage from two-generational data. The method was further
developed by Finney (1940 - 1943). Fisher (1935) showed that the
method is fully efficient in the limit for loose linkage, but the
efficiency approaches zero as the recombination fraction approaches
zero.
Haldane and Smith (1947) described a probability ratio test to
detect linkage from families of any size, without the assumption of
normality required by maximum likelihood scoring. Smith (1953b) shows
that the method of u-scores and the sib-pair method are also forms of
the probability ratio test.
Morton (1955) was the first to suggest using the sequential
probability ratio test. This test has the advantage of having the
smallest expected sample size on the null or alternative hypothesis,
and Morton (1955) shows that the mean expected sample size (averaged
over a "reasonable" range of alternate hypothesis recombination
fractions) of the sequential test is about one-third that required
by a u-score test with the same error probabilities.
The assumptions required for the test are: (1) parental genotypes
are known with certainty except for phase, (2) segregation ratios

40
are not disturbed by incomplete penetrance or differential
viability, (3) the method of ascertainment and selection of families
in properly allowed for (Morton 1955). Alternatives to the null
hypothesis of no linkage (A = .5) include the negation of any of
these assumptions, nonrandom segregation of nonhomologous chromosomes,
or linkage. The test procedure is based on lod scores, defined as
Zi = 10glO (~~ ~~~-I ~5n, where Pr (f i IA) is the probability of observing
the i th family if the recombination fraction is A. Values of the lod
scores have been extensively tabulated by Morton (1955, 1957,
Steinberg and Morton 1956, Smith 1968, 1969, Maynard-Smith et. al.
1961, Edwards 1971), and others. The total score for the sample is
2 Lzi
. Morton suggests using a small type I error probability,
a = 0.001, in order to exclude as many false linkages as possible.
Since the a priori probability that two random autosomal loci are
linked is on the order of 1/22 = 0.045, about 4 in every 100 pairs
of loci are on the same chromosome. If the usual significance level
of 0.05 is used, about 5 of every 100 pairs will be detected as
"linked" when they are not, and thus such a test would detect more
false than true linkages (Smith 1953b). Using a = 0.001 helps correct
this. For this a, and power of the test of 0.90, the stopping rule
is to accept linkage (at the nominal value Qf AI) if Z~3 and reject
linkage (ie., accept~= .5) if 2<-2.
Morton's original paper derived in detail the lod scores for
autosomal loci with two alleles. Ascertainment corrections were
derived for selection in either or both factors. Later papers
extended the method to multiple alleles at the marker locus

41
(Steinberg and Morton 1956), multiple alleles at both loci (Morton
1957), and considered more mating types.
The sequential test approach to detecting linkage has been
criticized (Smith 1959), and a Bayesian approach suggested (Smith
1959, 1968; Renwick 1971, 1969; Renwick and Schultze 1964; Denniston
1969).
Smith's (1959) objections to the sequential procedure are mainly
philosophic. He argues that sequential procedures were developed for
applications such as industrial quality control, and the stopping
rule has no significance in linkage studies. He appeals to the
"principle of self-sufficiency", saying that if a random sample from
a population is available, all information on the population is
contained in the sample, with the order in which the observations
occurred giving no information. Renwick (1971) argues in favor of
Bayesian analysis on the grounds that a reasonable prior distribution
of A is available, and that the prior probability that two loci are
linked is so small that a large posterior likelihood of linkage is
necessary before it can be believed. Also, the Bayesian approach
gives a value for the relative odds of linkage to no linkage without
the necessity of setting significance levels or accepting or
rejecting hypotheses (Smith 1968).
The form of Bayes' theorem used in linkage analysis can be
described as follows: Consider the hypotheses H : A = .5 andI a
H1
: A = A1
, and denote by E the sample that has been observed. Then
the relative final probability is Pr(Hl~) = Pr(Hll x L(~U!11Pr(H~) Pr(H) L(~ H )000

42
(Smith 1968). The prior distribution of the recombination fraction
is usually taken as uniform on the interval (0,0.5), with
Pr (A = 0.5) = 0.95.
When the alternate hypothesis is not the simple one A = some
specified value A1
, but H1
: the two loci are linked, the relative
final probabilities can be calculated as
kJ;pr(A) L(IIA)dA.
Pr(H ) L(I IDo 0
The numerator represents the average value of the posterior proba-
bility on the alternate hypothesis. This can be approximated by
assuming A takes on only discrete valueg such as 0.00,0.01, ••• ,0.49
and replacing integration by summation (Smith 1959, 1963; Denniston
1969). The Bayesian method has been extended to more than two loci
(Renwick 1971, Renwick and Bolling 1971), but human applications are
scarce and prior distributions of map intervals become difficult.
Regardless of whether a Bayesian or sequential approach is used,
the basis of the analysis lies in the 10d scores or their antilogs.
The basic calculations and the ascertainment corrections are the
same in either case.
Some of the problems involved in linkage analysis are minimzed
when X-linked loci are involved. Recombination is then observed
only in women, so every informative mating is essentially a double
backcross with respect to sons. Phase in a doubly hetero~gous
woman can be determined from the phenotype of the father. If both
loci are known to be X-linked, they are obviously syntenic and the
only question is whether linkage is close enough to be demonstrated.

43
Lod scores and ascertainment corrections have been tabulated
for two- and three-generational families (Smith 1968, 1969; Edwards
1971). Although usually only sons are informative for linkage,
daughters can be used if the father has recessive (or codominant)
alleles at both loci (Smith 1968). Ascertainment of families is
usually assumed to be through sons with a recessive disease, and a
"family" consists of sons (including daughters if they are informa-
tive), mother, and the mother's father. Fraser (1968) shows that
ascertainment corrections are sometimes needed even when three
generations are used. Situations where this is necessary include
truncate selection* at the disease locus and any selection at the
marker locus, and cases where the _mother's mother is used to determine
the phase or genotype of the mother. Fraser shows, however, that no
bias arises when there is single ascertainment at the disease locus
and truncate selection at the marker locus.
In the linkage studies so far performed on manic depressive
illness data (Fieve, Mendlewicz and Fleiss 1969; Mendlewicz, Fleiss
and Fleiss 1972; Mendlewicz and Fleiss 1974) tables of lod scores
as tabulated by Edwards (1971) have been used. These tables were
constructed under the assumption that selection at the marker locus
is independent of disease locus phenotypes. Gershon and Matthysse
(1977) point out that this may not be the case for some of the
affective illness data since the probands are usually colorblind,
manic depressive males. They derive the proper ascertainment cor-
rections when selection of families is through "doubly ill" probands.
*If A = Pr(an affected person is a proband, then truncateselection corresponds to A = 1 and single selection to A~.

44
Comparisons of results obtained with two generation and three
generation families were made by Fraser and Mayo (1968). They noted
discrepancies in estimates of A between Xg and G6PD: using two
generations significant evidence of linkage was found (A = 0.26), but
no evidence of linkage was found when data from three generations were
used. Using simulation studies and large samples (1000 two-son
families) they found that the three generational data had larger
maximum lod scores and that values of Al up to about 0.3 could be
detected. If there was no linkage, this could be detected when
Al = 0.33. In two generational data, linkage could reliably be
detected only up to A = 0.10-0.20, and true H were accepted onlyo
against Al = 0.17 or tighter. They conclude that three generational
data carries far more information than two generations, and "linkages"
detected from two generational data with A as large as 0.25 may not be
linkages at all, even in large samples.
Morton (1955) briefly considers incomplete penetrance or natural
selection in the disease trait. It is assumed that the marker is
fully penetrant, viable, and subject to complete or truncate se1ec-
tion. Then, if the disease is fully penetrant but subvital, linkage
analysis can still be done and the Type I and Type II error prob-
abilities are unchanged. If the disease is not fully penetrant, the
method is still valid but the power will be low if the penetrance is
low.
Ott (1974), using the method of Elston and Stewart (1971) to ca1-
culate the likelihood, takes account of reduced penetrance in the

matrix of probabilities Pr(phenotypelgenotype). The penetrance
parameters can be estimated along with any other unknown parameters
in the likelihood.
45

CHAPTER II
METHODS AND MODELS FOR AFFECTIVE ILLNESS DATA
This Chapter will outline models to be used on the manic
depressive illness data, and present some extensions of Elston's
pedigree likelihood method.
The likelihood of observing a family's phenotypes conditional
on the family being 'ascertained through one or more probands is
L(flmethod of ascertainment) = L(f)L(ascertainmentLtLL(ascertainment)
(Elston, 1973). L(f) is the likelihood assuming the family is a
random one from the population, and is derived under fairly general
conditions by Elston and Stewart (1971). L(ascertainmentlf), the
ascertainment function, can be expressed as the product, over all
individuals in the pedigree, of the individuals' ascertainment
functions, if it can be assumed that probands are ascertained inde-
pendently. The denominator, L(ascertainment) is the same as
L(one or more probands in the family) = l-L(no probands in the
family).
2.1 Specific models for affective illness data.
2.1.1 The phenotypic model
The variables observed include z, the disease status; a, the age
of onset of affective illness; and a', the age at examination.

47
We will assign phenotypes in two ways:
(i) i th person has bipolar manic depressive diseasei th person has unipolar depressioni th person is normal or alcoholic
(ii) Z. = ti2 if i th person has bipolar disease1 1 if i th person has unipolar depression
o if i th person is normalor alcoholism
The age of examination is observed or estimated for all individuals
on whom z. is observed; age of onset is observed for a subsample of1
those with depression and/or mania, but there are no data available
on age of onset of alcoholism. It is assumed that only individuals
with phenotype z = 2 can possibly be probands.
The phenotypic model to be considered is a trichotomy with age
of onset, model I (Yelverton, Namboodiri, and Elston 1975). For
this model the,
gh (zi,a. ,a.).u 1 11
= pr(ith
person has phenotype zilgenotype u and sex hJwith age of onset a.
1
are as follows:
,gh (2 ,a. , a .) = Y2f h ' (a.)
.u 1 1 .u 11 1,
gh (2,a.,?) = YZ' Fh
(a~).u 1 .u 11 1
,~ (l,a.,a.) = ylfh (a.)-hiU 1 1 iU 1
,= ylFh (a.)
iU 1
g' ,hiu(~,ai'-) = 1-(Yl+Y2)Fh.u(ai)
1
where y. = Pr(a person will develop disease form i if he lives long1
enough); fh.u(a) is the density of age of onset, dependent on sex and1

48
a'genotype; Eh (a') = f f h (a) da.u _00 u
i i
The usual form of the model assumes that there is some trans-
formation of age of onset that makes the distribution approximately
normal. This is not a necessary restriction, however, and sampling
corrections will be derived below for the case where f is an
exponential distribution.
In this model it is assumed that the parameters of the age of
onset distribution may depend on sex and/or genotype, while the
susceptibilities (y,) depend only on the form of the diseaseJ.
(unipolar or bipolar).
2.1.2 The ascertainment function
In the case of mental illness data, it may be that the prob-
ability that an affected person becomes a proband depends on his
age of onset. There is evidence for this in schizophrenia (Spence
et. ale 1976). One might expect that the probability of becoming a
proband would decrease with age of onset. An ascertainment model
that allows this is an age dependent exponential function (Elston
and Yelverton (1975):
r.(Ko+Kl a+K2a2
)~(z,a) = mJ.n e ,1 ,
o ,if z = 2.
if z 0,1
2 2where either K2=0, or K2<0 and Kl~4KOK2' If K2<0 and Kl~4K2KO then
2Ko
+Kl
a+K2
ae ~l always, and ~(z,a) can increase to a maximum then
decrease again. If K2=0 then ~(z,a) either increases'to a maximum
of 1 and then remains constant, or decreases with age from a maximum

49
of 1. Elston et. al. (1976) found a quadratic function was necessary
for schizophrenia; a linear or constant function may be adequate for
affective illness.
2.1.3 The sampling correction: l-Pr(O probands in the family).
B = Pr(O probands in the family) can be expressed in terms of
functions ~hu (see section 1.3), which depend on the sex, genotype,
and age at examination for each person, as well as the phenotypic
and ascertainment models. For g and ~ as above,
~hu
ghu (2,a' ,a) da
= 1 - (yl +y2)Fhu (a') + ylFhu(a') + yZ Fhu(a')
= 1
The ~hu for the case were fhu(a) is a normal distribution have
been worked out by Yelverton et. a1. (1975). It has been suggested
(Crowe and Smouse 1976) that age of onset in affective illness may
fit an exponential distribution; therefore 'hu for this distribution
will be derived below.
In this case we have f(a) =(ee-e(a~L) ,a~L. The parametersto ,a~L
are e, a "rate" of onset and L, the lowest age of onset. In theory,
both e and L could be sex and genotype dependent. For simplicity,
we will let 6hu depend on sex and/or genotype, but assume there is

if a~L
50
There are two major subcases: K2<0 and K2=0.
1.
Here
always, so
l;hu
[2.1]
The exponent equals
Therefore,
[2.1]
1 - Y26hu/_~ exp\ 2
K +6h Lo u
x
[K -6 J-12iC (L+ 1 hU)
2 2K2 [2.2]

Where
1 2
Jx ! -202 (t-~)
;-;;::::z edt.-<Xl 21T0
That is,
51
!;hu= {[2.2l for a'~L
1 for a'<L
The !;hu may be readily derived for cases which do not fit the
2restrictions K2<O and Kl~4KoK20 These restrictions imply that the
probability of being a proband increases with age of onset up to a
certain point then decreases. This model is reasonable for many
cases in practice and in particular may be appropriate for mental
illness data.
Thus the cases Kl>O and Kl<O must be considered separately.
If Kl>O, then
a(z,O,a)Ko+Kla
if 2 and Ko/Kl= e z = a<
if z = 2 and a~ Ko/Kl
if z = 0 or 1
There are several subcases depending on the relationships among
a' ,L, and -Ko/Klo If a'~L then fhu(a') = Fhu(a') = 0, and
ghu(2,a' ,a) = ghu(l,a' ,a) = 0 and ghu(O,a' ,-) = 1, so that !;hu = 1.

52
In the cases where a'>L,
1;; =hu
[2.3]
Where the limits of the integral (q and r) differ according to the
relationships among a' ,L, and -Ko/Kl
: the integration is performed
over values of a for which a(2,O,a) is not zero.
[2.3] reduces to
-6 (a-L)hue
[2.4]
In the present case, a(2,O,a»O if a<-K /K and the major subcaseso 1
are L<-Ko/K1 and L~-Ko/Kl (see Figure 2.1 (A))
If L<a' < - Ko/Kl then q = Land r = a', so
((K -8 )a' (K -8 )L1
1 hu 1 hu Je -e

53
Figure 2.1 Range of age of onset for which a(2,O,a) isgreater than zero.
KA.
0a<- _.K •1
t I I a or _00 , ( a
0 L-Ko -Ko L
K1 Xl
B.Koa>- K1
_00 J I I ~a or aK L 0 L K
0 0--K1K1

54
KIf - KO
~L<a, then a(2,0,a) = ° for all a>L, so ~2 = 0.1
Summarizing and simplifying these results,
~u = 1 ,if a'~L
-Ko- S:L<a'Kl -
1-Y2[
_e(K1-ShU)L ])
-6 (a'-L)hu
+ Y2e
If Kl
<0, then
K +6h L [6 0 .uhue 1
Kl -6hu
Ko
, if L<- K s:a' .1
-Kif L<a' < -.2.
Kl
a(z,O,a) =
°1 z = °or 1
This case is similar to the previous one, and ~hu is of the same form.
Let ~l' ~2' q, and r be defined as in [2.4]. The major subclasses

here are L>-K /Kl
and L< -K /K (See Figure 2.1).- 0 0 1
[K ] K +6h L
-6 - --.Q - L -6 (a' -L) 6 e 0 ul: = hu Kl + hu + -:...h_u _"'2 -e e K -6 x
1 hu
If -Ko/Kl~L<a', then q = Land r = a' so
55
K +Sh L-S (a'-L) Shue 0 u
e hu -1 + --::.::.~---Kl-Shu
Simplifying and summarizing these results,
~u: 1, a'.$L
1 -[
-Shu (~+L)Y l-e 1-
Z
YZShu e(Ko+6hUL+(Kl-ShU)a')
K1-Shu
KL<- ~ <a'
Kl
KL<a'~- .-Q., K
1

56
In each case of the age dependent threshold model there is one
term that depends on age and one that depends only on sex and
genotype.
2.1.4 Genetic models
The genetic models to be used are those suggested by previous
studies on the genetics of manic depressive illness. Among the
simple models suggested are a single X-linked dominant gene, a single
autosomal dominant gene, and two gene models with one autDsomal and
one X-linked dominant.
A model of a simple autosomal dominant has been proposed, but
this cannot explain the excess of affected women, or the deficiency
of affected sons of affected men. It is possible that environmental
or social factors, or genetic factors unrelated to the primary
affective illness gene are responsible for these observations.
Allowing sex and genotype dependencies in the age of onset dis-
tribution parameters may explain sex differences without necessarily
requiring an X-linked gene. A model of a single autosomal gene is
the simplest possible. The matrix of transition probabilities,
P t ' has dimensions 3x3x3 and is independent of sex.s u
The other single gene model that has been proposed is that of
an X-linked dominant. Support for the model comes from the observa-
tions that prevalence of affective disorder in women is about twice
that in men, and that affected men have a deficiency of affected sons
and fathers. There are, however, exceptions to the requirement for
the X-linked model that there be no father to son transmission of
the disease. In nearly every study there are at least a few

57
father-son pairs where both are affectively ill. If there is an
X-linked gene for manic depressive illness, then these pairs might
be explained as incomplete penetrance (ie., the mother has the
defective gene but does not show the disease), or as sporadic cases
(ie., either the father or the son is genotypically normal, but has
the disease anyway). Pedigree analysis should be able to account
for these situations.
For an X-linked model the matrix of transition probabilities
must be modified. There are now only two possible genotypes for
men, so the dimensions are 2x3x2 for sons (if "s" is the father's
genotype) and 2x3x3 for daughters. In the likelihood of the family
(L(f» and the sampling correction (S) the summations over p ts u
depend on the sex of the child.
Several two gene models for the inheritance of manic depressive
illness have been proposed (Winokur 1970, Rosanoff et. al. 1935).
These models both involve one autosomal dominant and one X-linked
dominant gene. In Winokur's model, primary affective illness is
caused by an X-linked gene, with a second autosomal dominant gene
necessary for mania. Alcholism may be one way in which the secondary
(autosomal) factor is expressed in the absence of the primary factor.
This model therefore has three or four phenotypes, depending on
whether alcoholics are counted as a separate class.
Rosanoff et. al. (1935) proposed a model wherein both a
cyclothymic autosomal dominant gene and an activating X-linked gene
are necessary for mania and/or depression. Only two phenotypes,
affectively ill and normal, are considered in this model.

58
Another two gene model may be used in an attempt to detect
heterogeneity. Here either an X-linked or an autosomal gene
segregating within a family may cause manic depressive illness while
in the population both genes are present. Allowing both genes in the
model may split the sample into two parts: one in which an autosomal
gene is segregating and one in which an X-linked gene is segregating.
There are, of course, many other ways in which genes at two
loci may interact. For two autosomal loci, there are 50 distinct
ways in which the 9 genotypes can be split into two phenotypes
(Hartl and Maruyama 1968). If one of the loci is X-linked there
will be more since the sexes must be considered separately. The
models outlined here seem most reasonable, based on examination of
the data and previously suggested genetic mechanisms. See Table 2.1
for a summary of the genotype-phenotype correspondences in the
models considered.
For two gene models the genetic transition matrices may be
generated from the corresponding single gene matrices as follows.
Let P and P be the genetic transition matrices appropriate for the-x -a
X-linked and autosomal loci, respectively. Then the genetic
transition matrix for the loci together is
P = P lIP P-a -x
P 4P P'-a -x
if P is 3x2x3 or 3x2x2-x
if P is 2x3x3 or 2x3x2-x
(Elston and Stewart 1971). The Kronecker product of two three-
dimensional matrices is given by Elston and Stewart (1971). ! may

Table 2.1 Genetic models for affective illness.
Females Males
59
One gene models: AA Aa aa Aa Aa aa
Autosomal GItlBJ GIiIiJBB Bb bb B. b.
X-linked CiIiJLI CLGJ
M M N
M M N
D D N
I I N
I I N
N N N
I I I
I I I
I I N
Two Gene models:
(1) Winokur 1970
(2) Double dominant
(Rosanoff et.al.1935)
(3) Heterogenity
AA
Aa
aa
AA
Aa
aa
AA
Aa
aa
BB
BB
BB
Bb
Bb
Bb
bb
bb
bb
B. b.
:~aa~
B. b.
AA~W1j
B. b.
:~aa tttti
I = Affectively ill, M= mania, D= depression, N= normal.

60
also be expressed in terms of .'s. Since an autosomal and an X-
linked gene must be unlinked, • = • T for femalesss'xx'uy ss'u xx'y
and. = • • for males.ss'x.uy ss'u x.y
A subset of the data may be used to test for linkage between a
gene for manic depressive illness and an X-linked marker. It has
been suggested that there is such a linkage with the Xg blood group
(Fieve et. al. 1973, Winokur and Tanna 1969), colorblindness
(Winokur, Clayton and Reich 1969), or both (Mend1ewicz and Fleiss
1974). A map of the X-chromosome (Berg and Bern 1968, Levitan and
Montagu 1971) indicates that it is unlikely that linkage between a
locus for manic depressive illness and both Xg and co10rb1indness
would be detected (see Figure 2.2). The methodological problems in
the studies suggesting these linkages may be sufficient to cause the
detection of nonexistant linkages. An analysis including a variable
age of onset for manic depressive illness and proper ascertainment
corrections should help clarify the situation.
Linkage analysis may also be put in the framework of the like-
lihood method. This is similar to, but not identical with the lad
score approach of Morton (1955). The lads are conditional on the
parental mating type, and consider only the estimation of the
recombination fraction A. The likelihood is not conditional on
parental mating types, and other parameters (gene frequencies,
penetrances, etc.) may be estimated simultaneously with A.
Biological assumptions for linkage analysis include (i) non-
homologous chromosomes segregate at random, (ii) there is no
epistatic or pleiotropic interaction of phenotypes between the

Figure 2.2 Map of the X-Chromosome.
61
Xg
IchthyosisOcu1ara1binism
Fabry's disease
XmDeutanG6PDProtan
Hemophi1a A
Hemophila B
Distances are in centimorgans: 1 centimorgan corresponds to 1%recombination. Thus a distance of 50 centimorgans representsfree recombination. (After Levitan and Montagu (1971».

62
two loci under consideration, and (iii) there is random mating with
respect to the two loci. In order for linkage analysis to be valid, it
is necessary for the population to be in linkage equilibrium. This
implies that the frequency of genotypes in coupling (DM/dm; ie. D and M
are on the same chromosome) equals the frequency of genotypes in repul-
sion (Dm/dM), where D, d are the alleles at the disease locus and M,
m are the alleles at the marker locus. If the population is not in
linkage equilibruim then there is an association of phenotypes in the
population. Linkage disequilibrium can arise when there is admixture of
two populations with different genotypic frequencies, and can persist
for thousands of years if there is tight linkage, or if the loci are
linked to a gene which is subject to selection (Thompson 1977). Before
a linkage analysis is undertaken, it is important to test for population
association of the traits under consideration.
Let ~ = (zd,zm) = (phenotype at disease locus, phenotype at
marker locus). If the biological assumptions hold then
g (z) = g (zd)g (z). The genetic transition matrix may be generatedu - ud
um
m
as for two unlinked loci, with the modification that the transmission
probabilities L are dependent on the recombination fraction. The
question of interest is whether or not the two loci are linked. Testing
the null hypothesis that the loci are not linked corresponds to
H : A = 0.5.o
For each of the other models summarized in Table 2.1, the
parameters and hypotheses are similar. Parameters include the
gene frequencies, transmission probabilities (L's), means and
variances of the age of onset distributions, susceptibilities

63
(Yl
,Y2), and parameters of the ascertainment function (Ko ,Kl ,K2)·
There is quite a large number of parameters, and some assumptions
will be made. A common variance for all age of onset distributions
will be assumed, and for the two gene models not all genotypes will
be allowed a separate mean.
Hypotheses to be tested include (i) a constant ascertainment
function is adequate (Kl
=K2=0), (ii) the hypothesized gene is a
dominant, (iii) there is Mendelian segregation at one or both loci
(TAAA
=1.0,TAaA=.5, TaaA=O), (iv) there is no vertical transmission
(T -T =T )AAA- AaA aaA'
2.1.5 Summary of the model and its parameters
The phenotypic model to be applied to the affective illness data
is a trichotomy with the variable age of onset taken into account.
Phenotypes will be assigned in two ways: (1) bipolar - unipolar -
unaffected and alcoholic, and (2) bipolar - unipolar and alcoholic -
unaffected. This is to be done because of the suggestion (eg.,
Winokur 1970) that alcoholism may be an "affective equivalent," at
least in some individuals. Genetic models to be specifically tested
include a single autosomal gene, a single X-linked gene, and the
heterogeneity and double dominant two gene models (see Table 2.1).
Table 2.2 gives a list of the parameters and their interpreta-
tions. In the general single gene model with no restrictions there
are twelve parameters. The two gene models have these same
parameters, plus one more gene frequency. It may be possible to
make some additional assumptions such as VAA=VAa
or KI=O in order to

64
Table 2.2 Parameters of the model and their interpretations
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
TAaA
TaaA
q
o
The probability that an individual of genotype AAtransmits allele A.
The probability that an individual of genotype Aatransmits alleJe A.
The probability that an individual of genotype aatransmits allele A.
Gene frequency of the abnormal allel, A. For thetwo locus models there will be two gene frequencies,qA and qB·
The probability that a random member of the populationwill develop unipolar depression if he lives longenough.
The probability that a random member of the population will develop bipolar manic depression 1f helives long enough.
The mean of the natural log of age of onset forpeople with genotype AA.
The mean of the natural log of age of onset forpeople with genotype Aa.
The mean of the natural log of age of onset forpeople with genotype aa.
The standard deviation of the natural log of ageof onset.
:} Ascertainment parameters.

6S
reduce the number of parameters. These hypotheses will be tested.
The susceptibility parameters, Yl and Y2 , express the probability
that a random individual from the population will become ill if he
lives long enough, regardless of his genotype. These parameters
must be interpreted in conjunction with the gene frequency and the
means and variance of the age of onset distribution. Y1+Y2 can be
very large without implying that nearly everyone in the population
is affectively ill if, for example, the gene frequency of the
abnormal allele (A) is small and ~ is very large. This wmuldaa
mean that virtually no one with genotype aa lives long enough to
develop the disease and that a large proportion of the population
has genotype aa.
The probability that a bipolar individual is a proband is
assumed to be a function of the form exp{Ko+Kla}, where a is the
natural logarithm of age of onset. This form of the ascertainment
function allows as a special case the usual model where the prob-
ability that an affected person is a proband is constant for all
affected people. This is equivalent to assuming Kl=O.
For the preliminary analyses, the transmission probabilities
will be set at their Mendelian values of TAAA=l.O, TAaA=O.5 and
TaaA=O.O. Under the X-linked models it is assumed that TA.A=TAAA
and T A=T aA' Maximum likelihood estimates will be obtained undera. a
the Mendelian models for the other parameters. The hypotheses that
the gene is a dominant (Ho:~AA=~Aa) and that a constant ascertainment
function is adequate (Ho :K1=O) will be tested. Based on the results
of these tests, an adequate model with the fewest possible parameters

66
will then be used to test the Mendelian hypothesis (Ho :LAAA=1.0,
LAaA
=O.5, LaaA=O.O) and the environmental hypothesis
(HO:LAAA
= LAaA=LaaA). The results of these analyses for the indivd
ua1 sets of data will be presented in Chapters III through VI, and
overall conclusions and comparisons among datasets will be presented
in Chapter VIII.
2.2 Extensions of the likelihood: Two stage ascertainment.
2.2.1 The selection procedure, ascertainment function, andgeneral form of the sampling correction
One of the most serious problems in analyzing human genetic
diseases is that the sample is virtually never a random one from the
population. Families are generally selected through one or more
probands who are ascertained because they have the disease in
question. Corrections for this type of sample selection have been
worked out for classical segregation analysis (see ego Morton 1969),
and for pedigree analysis using the likelihood methods (Elston 1973,
Morton and MacLean 1974). The problem of nonrandom sampling has
been almost entirely ignored in the analysis of the genetics of
mental diseases, however.
In this section, ascertainment corrections for a two-stage
sampling procedure are derived. This type of sampling is often
used in linkage analysis and may also be used in an attempt to reduce
genetic heterogeneity within a sample of families.
A major problem in the genetic analysis of human disease is
the probable genetic heterogeneity of many diseases. There are
several approaches that can be taken in order to obtain a sample

67
in which the heterogeneity is reduced. A single large pedigree may
be analyzed, since within anyone family there is likely to be only
one genetic mechanism operating. However, it is difficult to get
good quality data on distant or dead relatives of the proband, and
this approach is more likely to be useful for genetic counselling
rather than population inferences. Another possibility is to use a
model of two major genes. If, within any family, the disease is
caused by a single gene, but there are two or more genes in the
population, then a model of this kind may be appropriate ..
The approach to be considered in more detail here is a two
stage sampling procedure. A sample of families is first ascertained
in the usual way, then a subset of these families is chosen for
analysis on the basis of some criterion of family history. This
approach has been used, for example, to select families with two or
more cases of a disease to exclude sporadic, environmentally caused
cases (Winokur and Clayton 1967 a,b; Mendlewicz et. al. 1972). In
order to avoid examining the entire family, which may be time
consuming and expensive, it may be required that there be a secondary
case in some specified set of relatives. For example, one could
choose for analysis only those families in which a parent or a sib
of the proband is affected. A two-stage selection procedure could
also be used to exclude families in which there is father to son
transmission in order to obtain a sample more likely to be
segregating for an X-linked gene.
The selection procedure is as follows:

68
(1) A family is ascertained via one or more probands in the
usual way. Let P represent the event that there is a proband in
the family.
(2) The family is examined to determine whether it meets the
secondary selection criterion. If it does, it is retained for
further analysis; if not, it is rejected. Let S represent the
event that the secondary selection criterion is met. The likelihood
of the family conditional on the method of ascertainment is
L(f).L(family is ana1yzed'f)- ~ . L(f) is the unconditional likelihood,L(fami1y is analyzed)
as before. The ascertainment function, L(fami1y is analyzed I f),
is the same as for the usual method of ascertainment via one or more
probands; this will be shown below. The denominator,
Pr(fami1y is analyzed) Pr(P) x Pr(Slp)
Pr(SOP) = 1 - Pr(SUP)
1 - (Pr(P) + Pr(S) - Pr(SOP))
1 - Pr(P) - Pr(pnS) = 1 -eo -81 .
8 is the sampling correction as before. The correction for twoo
stage sampling, then, consists of subtracting an extra term from
the denominator of the conditional likelihood. The corrections for
the case where the secondary selection criterion is that there are
at least two affected people in the family, and for the case of
eliminating families with father-to-son transmission will be derived
below.

69
The ascertainment function
It will be assumed that the two stages of selection are
independent, which is likely to be the case in practice. It will be
necessary to temporarily distinguish between two types of "proband."
The first type is the index case (ie., the first proband in the
family) from the initial ascertainment of the family. The other type
is a "proband" for the second· stage of sampling. A person will be
called a second stage proband if he satisfies the secondary
selection criterion, 5. For example, if we are selecting families
with two or more cases, then every affected person other than the
index case is a second stage proband; if we select families without
father to son transmission then every man without an affected father
first stage of sampling
otherwise
if the i th person satisfies S
otherwise
Define
or son is a second stage proband.
if the ith
person is the index case of the
and
Let TIl(zi,ai
) = Pr(ith
person is the index case I zi,ai
)
TIZ(zi,ai ) = Pr(ith
person satisfies 51 zi,ai )
-.:. (1 if zi satisfies 5
10 otherwise
The ascertainment function for the ith
person is

a([b 1 ·,b 2 ·J,z.,a.)1 1 1 1
b1i
1-b1i
= ['IT1(z.,a.)] [l-'IT
I(z.,a.)]1 1 1 1
b2i 1-b .x ['IT
2(z.,a.)] [l-'IT
2(z.,a.)] 21
1 1 1 1
whether b2i
= 0 or 1.
70
Hence the ascertainment function is the same as when ascertainment is
via one proband, and there is no need to define b2i or 'IT 2 . As
before,
and
·f h . th . b d1 tel person 1S a pro an
otherwise,
a(z. ,b. ,a.) = ['IT(Z. ,a.)11111
1-'IT(z.,a.) if1 1
if b. = 11
b. = O.1
The denominator: Pr(fami1y is analyzed)
The denominator is 1 - Pr(no probands) - Pr(at least 1 proband
and S is not satisfied) = 1 - 80-8
1. 8
0is the multiple integral (or
sum) of the numerator over phenotypes and ages of onset of all
people in the family with all b i set equal to zero. This is the same
as in the case of ascertainment via one proband. 81
depends on
what the secondary selection criterion is. The case of selection of
families with two or more affected people will be considered first.

For simplicity, in the derivation below it will
71
2.2.2 Families with two or more affected people
In this case ~l = Pr(family has 1 proband and no other affected
people). It is the sum of N terms, where N is the number of people
in the family. Each term is the multiple integral of the numerator
over ages of onset with one b. = 1 and z. = 2, and the rest1. 1.
bj~i=O,Zj~i=O.
If, instead of selecting families with a secondary case in any
relative, the procedure is to select for analysis families with
two or more affected people in a specified set of relatives of the
proband (eg., families of probands with an affected sib), ~l is
slightly different. Let Ri
denote the set of relatives examined
'f h .th . b d d N b h b fl'1. t e 1. person 1.S a pro an ,an . e t e num er 0 peop e 1.n1.
Ri . Then ~l is the sum of N terms, with the i th term the multiple
integral of the numerator over all ages of onset and summation over
z.=O,l,2 for all relatives not in R., with b =1 zi=O, andJ 1. i'
bkERi=O,ZkERi=O.
be assumed that R. is the set of all available relatives.1.
As in the calculation of ~ , the integration in the terms ofa
~l can be performed for each person separately. Thus, each term
of ~l depends on two functions:
~~~) = J~: z~O a(l,z,a)ghu(z,a' ,a)da (1 of these)
~(2) =hu
a'J_oo
a(O,O,a) ghu(O,a' ,a)da (N-l of these)
Each term of al can thus be calculated using the same algorithm used
to calculate the unconditional likelihood of the family, L(f). In

72
this case, each term is of the same form as L(f) with ~~~) replacing
one of the g-functions and ~~~) replacing the other N-1 g-functions.
fa' 2 (0)So' as before, depends on _00 z~oa(z,o,z)ghu(z,a',a)da= ~hu .
The ~'s can be interpreted as
(0)~hu
(1)~hu =
~(2)hu
Pr(not being a proband by age a' I sex, genotype)
Pr(being a proband by age a' i sex, genotype)
Pr(not being affected by age a' I sex, genotype).
The calculations are simplified by the fact that ~~~)
This is shown as follows:
1_ r(O)
"'hu .
2EOTI(z,a)gh (z,a' ,a)daz= u
The age of onset density function is denoted by f(a), and may depend
on sex and/or genotype. The ~(O) have already been calculated for ahu
trichotomy with age of onset Model I, where the age of onset
distribution is normal (Yelverton et. a1. 1975) or exponential
(see section 2.1.3), with age dependent threshold model for
ascertainment.
The probability of not being affected by age a' conditional on
(2)sex and genotype, ~hu ' may be calculated as follows:

73
fa'= gh (O,a',a)da-~ u
since (O,O,a) = 1
~u(O,a' ,-) = 1 - (Yl+Y2)Fhu (a').
Thus, each term in 81
is of the same form as L(f) with (l-~~~»
replacing one of the g-functions and 1-(Yl +Y2)Fhu (a') replacing
the other N-l g-functions.
2.2.3 Exclusion of families with father to son transmission
Consider the case of a tricholomy (or dichotomy). We are
concerned with the case in which there may be two modes of inheritance
for the same phenotype, one of which is an X-linked gene, and we
wish to choose a subset of families which is more likely to be
segregating for the X-linked gene. We will therefore exclude all
families in which there is a father-son pair both of whom have z10.
The ascertainment function a(~,~,~), is the same as with one-
stage ascertainment. The denominator, Pr(family is analyzed) =
l-(Pr(O probands) + Pr(~l proband and father-son transmission»
81
= Pr(~l proband, and father-son transmission)
1 Pr(O probands, or no father-son transmission)
1 Pr(O probands)- Pr(no father-son transmission) +
Pr(O probands and no father-son transmission)
Thus, Pr(family is analyzed)
= 1 - (80+l-80-Pr(no father-son transmission) +
Pr(O probands and no father-son transmission»

74
= Pr(no father-son transmission) -Pr(O probands and no
father-son transmission)
The first term is the numerator integrated over all ages of onset,
summed over b's, and summed over z's with the restriction that when
a father's z~O all his sons must have z=O. The second term is the
numerator with all b's set equal to 0, integrated over all ages of
onset, and summed over z's again with the restriction that when a
father's z~O all his sons have z set equal to O.
The form of each term will be basically the same as that of
L(f) with functions s(j) (to be defined below) replacing thehu
First term:
For each female in the family there is only one factor in the
numerator that depends on her b, a, and z. Thus the integration
and summation can be performed separately for each female, analagous
to the calculation of So' Let ~~~) = J~~ z~O bfou(z,b,a)g2U(z,a' ,a)da.
(1)Then ~2u replaces g2u(z,a' ,a) for each female.
The values of z to be summed over for a male depend on the z-
value of his father at the current stage of summation. There is
thus a dependence between generations and the integration and
summation cannot be performed separately for each male. The z's for
all males in the family are restricted by the z of the original
father. For example, when the original father has z=l or 2, then
all his sons must have z=O; the grandsons have z summed over 0,1,2;
the greatgrandsons have z=O when the grandson's z=l or 2 and z

75
summed over 0,1,2 when the grandson's z=O; and so forth. The men
who marry into the family have z summed over 0,1,2 and then the
z-values of their male descendants are restricted. Since there is
this dependence between generations there will be extra summations
in this term that are not present in L(f).
Define
~(l) fa' ~ thu = -00 z=O b=Oa(z,b,a)ghu(z,a' ,a)da
c (3) fa' 1~lu = -00 b~Oa(O,b,a)glu(O,a',a)da
~ (2)lu
c (3)"'lu .
Note that
as ~(2) +lu
+ ~(3) = ~(l) so summing over z=0,l,2 is the samelu lu
Then, for males, when z is restricted to 0, ~~~)
is used to replace glu(z,a' ,a) and when z is to be summed over
0,l,2'q ~2 ~(qn) is used to allow for dependence between generations.n
That is, in the calculation of this term, the same basic structure
as L(f) is used, but with ghu(z,a' ,a) replaced by.~~~)in the
thn generation, where
,
~ (1) if h:::2 (ie. , for females)2u
3 ~(qll)* (n)
L if h=l and his father is not in the pedigreeq =2 iu~hu n
3~~~A)L if h=l and q 1=3 (his father is in pedigree)q =2 n-n
~ (3) if h=l and g 1=2 (his father is in the pedigree)lu n-

76
Se~ond term:
This term is similar to the first, except that here all b's
are set equal to 0 instead of summing over b=O,l. Define
a' 2~2u f_
ooz~Oa(z,0,a)g2u(z,a' ,a)da (as in the calculation
for S )o
(2) fa' 2 ,~lu _00 z~la(z,O,a) glu(z,a ,a)da
This term is also of the same basic form as L(f) with ghu(z,a',z)
1 d *(n). h th . hrep ace by ~hu l.n ten generat l.on, were
el
~2u if h=2
q 22 si~n) if h=l and his father is not in the pedigreen
if h=l and q 1=3 (his father is in the pedigree)n-
if h=l and q 1=2 (his father is in the pedigree)n-
Calculations for s~~) and s~~) for the trichotomy with age of onset
Model I are as follows
~ (1) a' ~ 1hu J_oo z=O b~Oa(z,b,a)ghu(z,a',a)da
, .
Ja [gh (O,a' ,a)+gh (l,a' ,a)+(l-7T(a»gh (2,a' ,a)+-00 u u u

77
fa'[gh (O,a' ,a)+gh (l,a',a)+~ (2,a' ,a)]da_00 u . u . -nu
the cumulative distribution of the age of onset
= 1.
(2) fa' ~ 1 ,~lU = _00 z~l b~Oa(z,b,a)glu(z,a ,a)da
a'f [gl (l,a',a)+(l-~(a»gl (2,a',a)+~(a)gl (2,a' ,a)]da_00 u u u
,fa [gl (l,a',a)+gl (2,a' ,a)]da
-00 u u
~(3)lu
fa'= gl (O,a' ,a)da
-00 u
~hu has already been calculated for age of onset distributed normally
or exponentially.
r(2) fa' 2 ( 0) ( , ) and r(3)~h = Ila z, ,a gh z,a ,a , ~huu _00 z- u
a'= f a(O,O,a)gh (O,a' ,a)da
-00 u
Therefore I;hu = ~(2)+1;(3) and 1;(2) can be calculated as~~' ~
r _r(3) r(3) fa'~hu ~hu ' where ~hu = _ooghu(O,a' ,a)da =

CHAPTER III
ANALYSIS OF THE DATA OF HELZER AND WINOKUR (1974)
3.1 Description of the data.
The data used in this Chapter have been reported by Helzer
and Winokur (1974) and Helzer (1975). The probands are from a
series of consecutive admissions to a psychiatric hospital from
July 1970 to July 1971. Those selected as probands were all males
who were manic at admission; there are a total of 30 probands in
the final sample. Personal interviews were conducted with the
proband and all available first-degree relatives who were over 15
years old and lived within 150 miles of the hospital. Ninety-
four percent of all relatives meeting these criteria were interviewed;
these represented 56% of all first-degree relatives. Diagnostic
criteria for all mental illnesses were those of Feighner et. ale
(1972) and the same standards were used for probands and relatives.
Information on relatives more distant than first degree was
obtained from various family members, and is therefore of variable
reliability. In general, information was included in the final
data set if it was obtained from someone no further than one
generation away, unless it was noted on the chart that "information
seems unreliable" or some similar comment. In most cases this caused
people in the generation of the grandparents of the proband to be
included, although information on the sibs of the grandparents is

79
probably incomplete. Information on first cousins of the proband
is extremely scanty and therefore all cousins were excluded.
Information on children of sibs (usually obtained from the sib) is
included. Thus the data consist of, for the most part, the proband,
his sibs and their children if any, his parents and their sibs,
and his grandparents. In some families there is enough information
on the sibs of the grandparents to include them also. Information
on wives of the probands was generally very incomplete, and unless
some definite information was available they were not included
in the pedigrees.
Age at examination or death was available for about 85-90%
of all individuals. Most of the missing data is from aunts and
uncles, grandparents, and sibs of grandparents. Estimates of the
unknown ages were obtained with priorities as follows:
(1) If the ages of some individuals in the sibship were known, the
average of known ages was used as an estimate of unknown ages.
(2) If the ages of some of the children were known, the estimate
was age of oldest child plus 25 for males and age of oldest child
child plus 20 for females; or if more precise information was
available (eg., "mother died when proband was lO"), that was used
instead of the child's current age.
All the unknown ages could be estimated in this way.
There are 6 sets of twins in the 30 families: 3 female-female,
1 male-male, and 2 male-female. None of the probands is a twin,

80
3 of the sets are sibs of the proband, and 2 sets are the mother of
a proband and her twin. For purposes of further analysis, one
of each pair of twins was discarded at random, except in the two
cases where the proband's mother was a twin, where the mother was
retained.
Other information in the data includes the Xg blood type and
colorblindness tests on all interviewed people. There were 13
families in which at least one person had an Xg-phenotype and two
families with colorblindness; some of these families may be infor
mative for linkage analysis. Information was also recorded on the
course of the illness for nearly all affectively ill people. The
number and type of episodes, symptoms observed, length of episodes,
and whether the person was hospitalized and/or treated as an out
patient was recorded.
The probands ranged in age from 18 to 69 at the time of index
admission. For 5 probands the index episode was the first episode
of affective illness. Nineteen of the probands were white and 11
were black. Helzer (1975) found no significant differences between
blacks and whites in the course or severity of the illness, number
of symptoms reported, or family history, except for an excess of
blacks with alcoholism on the paternal side.
There is information on a total of 518 individuals in the 30
families. Excluding the (all male) probands, there were 488 people:
238(48.8%) females and 250 (51.2%) males. The probands have infor
mation on an average of 16.2 relatives each, of which 7.1 are first
degree relatives. There was no significant differences between

81
the number of brothers and sisters (46 and 50, respectively) the
probands have. The average size of the proband's sibship is 4.2.
Of the twenty probands who had children the average number of
children is 2.8.
There are 79 affectively ill people. Among the affectively
ill (counting uncertain cases as affected), the sex ratios are as
shown in Table 3.1. Since all probands were male the categories
including probands are considerably biased. When probands are
excluded, there is an excess of women among all affectively ill
people. This excess is due" to those with unipolar depression;
among bipolars, the sex ratio is one to one. The number of bipolars
is very small, however. When alcoholics are counted as affectively
ill, the excess of women disappears; in fact, there is a slight
excess of men. It has been suggested (eg., Winokur and Clayton
1967a) that alcoholism may be an alternative expression of a gene
for manic depressive illness. However, since alcoholism is fairly
common, it may not be valid to count all alcoholics as affectively
ill.
The age of onset distributions show no significant differences
between males and females (Kolmogorov-Smirnov 2 sided test, p >0.20),
probands and all affectively ill (Kolmogorov-Smirnov 2-sided test
p >0.20), or people with mania and those with depression only
(Kolmogorov-Smirnov 2-sided test, 0.10<p<0.20) (see also Table 3.2).
Some of the non-significance may be due to small sample sizes. In
particular, manics seem to have an earlier onset than depressives.

82
The age of onset was recorded for 66 of 79 affectively ill
people. The range is 15-62 years with a mean of 30.2 and standard
deviation of 12.4. The skewness is significantly greater than
O. Plots were made of the empirical cumulative distribution
along with the best-fitting exponential, normal, lognormal,
-1squareroot normal, and (age of onset) normal distributions. Visual
inspection of the plots indicates that either an exponential or
lognormal distribution provides an adequate fit, with little
difference between these distributions (see Figures 3~1, 3.2). In
subsequent analyses, it will be assumed that age of onset is
approximately lognormally distributed, since this is computationally
simpler.
The number of affected first-degree relatives and their diagnoses
are shown in Table 3.3. There are only two fathers of probands
with affective illness, one with unipolar and one with bipolar
disea~e. One son of a proband had depression, but the proband's
wife was also depressed. Helzer and Winokur (1974) give morbid risks
(age-corrected using the Weinberg abridged method with risk period
15-60) as:
mothers: 40%
fathers: 5%
sisters: 19%
brothers: 8%
daughters:
sons:
40% )
18% jnumbers are too small for reliable conclu-
sions (based on 15 daughters and 11 sons
over 15 years old)

o~::"""i!L.---;t-----t----,...----r----r---....,.----1. 10.0 20.0 SO.O 'l0.0 50.0 60.0 70.0 80.0
Il8E II' ONSET
Figure 3.1 Age of onset with best fitting normal andlognormal distributions. Helzer and Winokur(1974).
Figure 3.2 Age of onset with best fitting exponentialdistribution. "Helzer and Winokur (1974).
83
~•:.
~
I
~
I
~~I~•~,~
10.0 20.0 30.0 'l0.0 50.0flOE OF ONSET
10.0 70.0 80.0

84
Table 3.3 First degree family history. (Data of Helzer andWinokur 1974)
Mo Fa Sis Bro Dau Son
Depression 6 1 3 0 3 1
Uncertain depression 2 0 2 1 0 0
Mania ± depression 1 1 2 3 0 0
Hypomania ± depression 0 0 1 1 0 0----------------
Total affectively ill 9 2 8 5 3 1
Total number older than 15 30 30 38 44 15 11
Some of the deficiency of affectively ill male relatives is
made up by alcoholism: 5 fathers and 6 brothers were alcoholic.
Other psychiatric illnesses in these families include sociopathy
(in 6 families), compulsive gambling (in 4 families), and some
cases in which a relative (usually a second or third degree relative)
was hospitalized for a long time with an unknown mental illness.
There was no well-documented case of schizophrenia. Six of the
probands had a totally negative family history of affective disorder,
and 6 more had illness in a second or third degree relative but
none in first degree relatives.
The proportion of affectively ill people who are probands, as a
function of age of onset, is shown in Figure 3.3. Also plotted are
the best-fitting curves of the form
In(p)
and In(p) KO+ Ki In(a).

Figure 3.3 Proportion of affective1y ill people who areprobands by decade of onset. Helzer andWinokur (1974).
85
1
P
o
-I- ,\ .. "", /, /
\ -~,/
-~, ,, ""-I' _ .. J:---- ....
'" /....- -- ....~ ~
---- -~ " ~L.~ .... -~
.~
..a
Age of onset, a
____ 1n (p) = 20.6 - 13.2 (In a) + 2.0 (In a)2
---- 1n (p) = -1.3 + 0.2 (In a)
Vertical lines indicate ± standard error

86
These functions are used since the ascertainment function is
2assumed to be n exp{K
O+K
l(lna)+K
2(Ina) }.The proportions are
2not significantly heterogeneous among age classes (X4=7.0l7,p>O.10).
However, P = number of ill people who are probands is a biasedtotal number of ill people
estimate of n, and the true porportions are smaller. In subsequent
analyses, tests for the significance of maximum likelihood estimates
of Kl
will be performed.
3.2 Segregation Analyses.
3.2.1 Single gene models
Several hypotheses under both an X-linked and an autosomal
single gene model are examined. For the preliminary analyses,
it is assumed that there is Mendelian segregation at a single locus.
Maximum likelihood estimates are obtained assuming a codominant
model (~AA'~Aa'~aa all distinct) or a dominant model (~AA=~Aa)'
and assuming the probability of ascertainment of bipolar individuals
is a constant or linear exponential function of age of onset
(n exp{KO
+K1
(lna)}; for the constant function Kl
is set equal to 0).
These analyses are done first counting alcoholics as normal with
respect to affective illness (z=O), and then considering alcoholism
as equivalent to unipolar depression (z=l). The hypotheses to be
tested on these models are HI: Kl
= O(ie., the probability that a
bipolar person is a proband is constant for all bipolar people),
and H2 : ~=~Aa (ie., the hypothesized gene is a dominant).
Table 3.4 shows the results of the preliminary analyses
assuming an autosomal gene. A comparison of corresponding columns

87
from parts (A) and (B) shows that the likelihood is much higher
when alcoholics are considered normal than when they are considered
affected. There is no information on age of onset of alcoholism
so that the joint likelihood of affective illness (z=l or 2) and
age of onset when alcoholics are considered affected is comparable
to the likelihood when they are considered normal. Therefore all
further analyses assume that alcoholics are normal with respect to
affective illness.
The hypothesis that Kl = 0 is rejected with p = 0.01 under
both the dominant and codominant models. Subsequent models therefore
assume that the probability of ascertainment is a linear exponential
function of age of onset.
The hypothesis that ~AA = ~Aa is also rejected (xi=6.896,
p=O.Ol). Thus the model in which the age of onset distribution is
a mixture of three lognormal distributions fits the data significantly
better than one in which age of onset is a mixture of two lognormal
distributions. The three means correspond to an early onset group
. h 3.108 22 4 1 h 3.952W1t mean e =. , a ate onset group wit mean e = 52.0,
and an "unaffected" group with mean e 5 •295 = 199.3. Three standard
deviations below the highest mean is e(5.295 - 3(0.230» = 100 years,
so that virtually no one with genotype aa lives long enough to
develop manic depressive illness. In the two-mean (dominant) model,
there is an early onset group with genotypes AA or Aa that has mean
3.643 4.728e = 38.2 and a group with genotype aa that has mean e =
113.1. Two standard deviations below this mean corresponds to
4.728-2(0.425) 47 5 h fe = • years, so t at a air proportion of those

88
with genotype aa live long enough to manifest the disease. In an
attempt to force a dichotomy of affected people with genotypes AA
or Aa and unaffected people with genotype aa the log mean of the
upper distribution was set at 7.0. The results are shown in Table
3.4. Except for the ascertainment parameters the estimates do not
differ greatly from the model with ~ free; however, the model withaa
~aa = 7.0 is significantly less likely (xi = 7.]80, p<O.Ol). It
appears that, at least under a Mendelian autosomal model, there is
significant heterogeneity in the age of onset in these data.
The maximum likelihood estimates of gene frequency are fairly
sensitive to the choice of a dominant or codominant model: the
gene frequency is much higher under a codominant model. Suscepti-
bilities under this model are correspondingly lower than under a
dominant model.2
Under the codominant model, about 49% «0.286) +
2(0.286)(1-0.286» of the population carries the gene for manic
depressive illness and about 30.3% (0.216+0.087) of these individuals
will develop the disease if they live long enough (virtually no one
with genotype aa lives long enough to develop the disease). Thus,
the codominant model is one of a fairly common gene with low
penetrance. Under the dominant model, about 9% «0.046)2+2(0.046)
(1-0.046» of the population carries the defective gene and 50.4%
(0.369+0.135) of these individuals are susceptible. However, a
fair proportion of people with genotype aa live long enough to
develop manic depressive illness, so that the dominant model is of
a relatively rare gene with incomplete penetrance and some sporadic
cases among "normal" homozygotes. More detailed analyses are
reported later on both the codominant and dominant models.

89
Table 3.5 shows the results of analysis on the preliminary
X-linked models. As with the autosomal models, the likelihood is
much higher when alcoholics are considered normal, and this will be
assumed in all further analyses. The hypothesis
rejected under the X-linked models (for dominant
that Kl
=2
model, Xl
o is not
1.094,
2p>0.25; for the codominant model Xl = 2.150, p>0.25). It will be
assumed that Kl
= O. Under this model, three means are significantly
2more likely than two (Xl = 7.748, p<O.Ol), indicating that an X-linked
codominant is more likely than an X-linked dominant. However, a
significant proportion of people with genotype aa live long enough
to become affected (2 standard deviations below ~ correspondsaa
to 42.9 years in the dominant model). To force a dichotomy ~aa
is fixed at 7.0. This results in little change in the likelihood
or estimates of the parameters.
Under the X-linked models, the maximum likelihood estimates are
quite similar whether a dominant or codominant gene is hypothesized.
In both cases the model is of a fairly common gene (frequency about
0.35) with incomplete penetrance and some people with the "normal"
genotype being affected. Further analyses will be performed on the
codominant model, and the dominant model with ~ set equal to 7.0.aa
A comparison of corresponding models under the autosomal and
X-linked hypotheses shows that the autosomal models are more likely.
The smallest difference in log likelihoods is 7.17, indicating the
autosomal model is about 1300 times more likely than the X-linked
model. Estimates of the gene frequency and susceptibility also
differ between the X-linked and autosomal models. The X-linked

90
models tend to have higher gene frequencies and lower suscepti-
bilities than the autosomal. The age of onset distribution and
ascertainment parameters are not greatly different.
Table 3.6 shows the results of analyses on the autosomal models.
Two hypotheses are tested: HI: there is Mendelian segregation at a
single locus (TAAA= 1.0, TAaA=0.5, Taaa=O.O) and H2 : there is no
vertical transmission (TAAA=TAaA=Taaa). Regardless of whether a
dominant or codominant model is assumed, both of these hypotheses
are easily rejected (p<O.OOl). In this setting the environmental
hypothesis is somewhat less likely than the Mendelian hypothesis.
Table 3.7 shows similar results under the X-linked model. The
Mendelian hypothesis is rejected (p<O.OOl) whether or not a dichotomy
is forced by setting ~ equal to 7.0. It doesnQt appear that a,aa
single gene, either autosomal or X-linked, can account for tpe
familial illness patterns in these data.
3.2.2 Two gene models
The two-gene models analyzed are the heterogeneity model and the
double dominant model (see Table 2.1 for the genotype-phenotype
correspondences). For the heterogeneity model, the presence of
either an autosomal dominant gene A or an X-linked dominant B leads
to manic depressive illness. For the double dominant model, both an
autosomal dominant gene A and an X-linked dominant gene Bare neces-
sary for an individual to develop manic depressive illness. The
parameters of this model are similar to those of the single gene
models above, with the addition of an extra gene frequency. It is

91
assumed that there are two means for the age of onset distribution,
one for affected people and one (set equal to 7.0 on the log scale)
for unaffected. See Table 3.8 for the genotypes which correspond
to each mean for the two models. Either of the single gene models
may be obtained from the two-gene model by fixing the other gene
frequency at zero. The single gene models can thus be considered
as a two gene model in which the frequency of one of the genes is
zero, so the likelihood of the two gene model must necessarily be
larger than the likelihood of a corresponding single gene model.
Table 3.8 shows the results when the transmission probabilities
are fixed at their Mendelian values for both loci. In each model
the frequency of one of the X-linked alleles converges to 1.0, so
that these models are essentially single-gene autosomal models. The
likelihood is virtually the same as the autosomal dominant model
(Column 5 of Table 3.4). Thus a two gene model provides no better
fit to these data than a single gene model. Although the models
examined are only two of many possible two-gene models including
one autosomal and one X-linked gene, they are the ones that seemed
most reasonable for affective illness data and it is unlikely that
an adequate description of the data could be found by examining more
two-gene models.
3.3 Conclusions.
Helzer and Winokur (1974) conclude that these data support the
hypothesis that manic depressive illness is an X-linked disease. In
the thirty families there is only one father-son pair in which both
were affectively ill with the mother well, while there were 9 probands

92
with an ill mother, and 3 ill daughters of probands. There are also
about twice as many ill females as males. Some of the sex ratios
were not as expected. The probands (all male) had significantly
more ill sisters than brothers, while a simple X-linked dominant
model wo~ld predict an equal number. Helzer and Winokur suggest
that alcoholism may be an affective equivalent, since in these data
the sex ratios are more compatible with a two locus, autosomal plus
X-linked hypothesis when alcoholics are considered affected.
The current analysis does not support these conclusions. An
X-linked model is much less likely than an autosomal model, and two
different two-gene models do not fit the data significantly better
than the one gene models. None of the models provides an adequate
explanation for the familial illness patterns, and it is probably
safe to conclude that the transmission mechanism in these families
is not as simple as segregation at one or two Mendelian loci. The
analyses in this chapter should be more powerful than that of
Helzer and Winokur (1974) since information in the whole family
structure is used instead of that on first degree relatives only,
the variable age of onset is taken into account in a more exact way,
and the method of ascertainment is taken into account. It is
therefore not surprising that the relatively simple genetic models
examined were rejected. A more complicated genetic model may
explain the data. It is also possible that, within certain families,
manic depressive illness is caused by a single gene, but that these
families are genetically heterogeneous so that the genetic mechanism
in individual families is obscured.

Table 3.1 Sex ratios among affectively ill people.(Data of Helzer and Winokur 1974).
Females Males Total
All affectively ill 34 (0.430) 45 (0.570) 79
All affectively ill + alcoholics 35 (0.340) 68 (0.660) 103
All affectively ill, excludingprobands 34 (0.694)* 15 (0.306) 49
All affectively ill, excluding 35 (0.479) 38 (0.521) 73probands, +. alcoholics
Bipo1ars 7 35 42
Bipo1ars, excu1ding probands 7 (0.583) 5 (0.417) 12
Unipo1ars 27 (0.730)* 10 (0.270) 37
Unipo1ars + alcoholics 28 (0.459) 33 (0.541) 61
Only "certain" alcoholics included in table.Percentages in parentheses
**Significant1y different from one-to-0ne sex ratio,p<O.Ol; *p<0.05.
93

Table 3.2 Age of onset. (Data of Helzer and Winokur 1974)
94
N Minimum Maximum Mean Std. Dev. Skewness
All data 66 15 62 30.2 12.4 0.76 **
males 46 15 58 29.1 12.9 0.87 **
females 20 17 62 32.7 11.3 0.65
Probands 30 15 58 29.8 13.6 0.84 *(males)
All bipolars 38 15 58 28.3 12.8 LOS **
All unipolar 22 15 62 32.7 12.6 0.54 x(depression)
Significantly different from 0*p<.05, **p<.Ol
x sample too small to judge significance

e e e
Table 3.4 Preliminary autosomal single gene models (Data of Helzer and Winokur 1974)
A. Alcoholics = Normal B. Alcoholics = Affected
Linear Ascertainment Constant Ascertainment ~AA=~Aa Linear Ascertainment Constant AscertainmentCodominant Dominant Codominant Dominant ~aa
=7.0 Codominant Dominant Codominant Dominant
q 0.286 0.046 0.371 0.077 0.072 0.219 0.228 0.216 0.228
Yl 0.216 0.369 0.197 0.295 0.292 0.401 0.399 0.405 0.397
Y2 0.087 0.135 0.082 0.106 0.106 0.077 0.076 0.077 0.076
~AA 3.108}3.643
3.088p.43l }3.496
3.292p.465
3.287 p.465~Aa 3.952 3.948 3.499 3.505
~aa 5.295 4.728 5.511 4.999 7.0 5.734 5.740 5.922 5.894
a 0.230 0.434 0.237 0.438 0.459 0.423 0.423 0.426 0.423
KO 1.393 -0.666 -17.141 -17.027 -16.125 -16.994 -16.915 16.550 -16.956
Kl -1.113 -1. 847 0 0 0.020 0.014 0.014 0 0
1n L -234.960 -238.408 -238.154 -241.817 -241. 998 -296.620 -296.852 -296.619 -296.857
For all models, TAAA=1.0, TAaA=0.5, TaaA=O.O
The hypotheses are (1) HO: Kl=O - for codominant
for dominant
2xl =6.388
2Xl=6.818
2(2) HO: ~AA=~a - when Kl=O Xl =7.326
when Kl~O xI=6.896\0\J1

Table 3.5 Preliminary single gene X-linked models (Data of Helzer and Winokur 1974)
A. Alcoholics = Normal B. Alcoholics = Affected• a
Linear Ascertainment Constant Ascertainment 11M =l1AaCodominant Dominant Codominant Dominant 11aa=7.0 Dominant, constant ascertainment
q 0.237 0.314 0.326 0.400 0.479 0.532
"'(1 0.210 0.180 0.204 0.178 0.177 0.230,
'(2 0.076 0.065 0.074 0.065 D.064 0.044
11M 3.296 }3.345 3.297}3.438 h.477 h .616
11Aa 3.714 3.763
11aa 4.462 4.495 4.639 4.652 7.0 3.233
cr 0.357 0.394 0.408 0.447 0.453 0.381
KO -10.682 -17.071 -17.131 -17 .153 -17.154 -17.680
K1 -1.159 -0.137 0 0 0 0
In L -244.483 -248.885 -245.558 -249.432 -250.102 -301.344
For all models 'AAA=l.O, 'AaA=0.5,
HO: K1=0 dominant - xi=1.094
codominant - xi=2.150
e
'aaA=O.O HO: llM=llAa when KI=O,
when K 40IT ,
e
2XI =7.748
2xl =8.804
e
1.00'\

e e e
Table 3.6 Single gene autosomal models. (Data of Helzer and Winokur 1974)
A. Codominant Model B. Dominant Model
Unrestricted Mendelian Environmental Unrestricted Mendelian - Environmental
TAAA 1.000 1.0 1.000 1.0
TAaA 0.942 0.5 } 0.633 1.000 0.5 } 0.278
TaaA 0.134 0.0 0.046 0.0
q 0.038 0.286 0.212 0.014 0.046 0.096
Y1 0.324 0.216 0.161 0.473 0.369 0.307
Y2 0.119 0.087 0.058 0.172 0.135 0.112
~AA 3.088 3.108 3.058 } 3.623 } 3.643 } 3.502~ 3.824 3.952 3.781Aa
~aa 5.296 5.295 5.251 5.882 4.728 5.817
0 0.211 0.230 0.211 0.452 0.434 0.462Ke -4.661 1.393 -16.095 -11.466 0.666 -14.758K1 -1.326 -1.113 -0.035 -0.112 -1.847 -0.153
In L -214.802 -234.960 -237.163 -219.074 -238.408 -243.8882
40.316 44.722 38.668 49.628X --- ---For the Mendelian models TAAA=1.0, TAaA=0.5, TaaA=O.O For the environmental models, TAAA=TAaA=T
aaA.
\0'-J

Table 3.7 Single gene X-linked models. (Data of Helzer and Winokur 1974)
A. ~=0, "VAA=llAa B. K1=0, llAA=llAa' llaa=7.0
Unrestricted Mendelian Unrestricted Mendelian
lAM 0.999 1.0 0.999 1.0
lAaA 1.000 0.5 1.000 0.5
laaA 0.124 0.0 0.105 0.0
q 0.058 0.400 0.032 0.479
Yl 0.293 0.178 0.386 0.177
Y2 0.106 0.065 0.140 0.064
llA_ 3.380 3.438 3.510 3.477
llaa 4.809 4.652 7.0 7.0L
0 0.373 0.447 0.439 0.453
KO -16.352 -17 .153 -16.261 -17.154
K1 0 0 0 0
In L -227.348 -249.432 -224.761 -250.1022 44.168 50.682X --- ---
For Mendelian models, l AM=l. 0, l AaA=0 •.5, l aaA=0.0.\0ex>
e e e

99
Table 3.8 Two-gene models. (Data of Helzer and Winokur 1974)
Double dominant Heterogeneity
Y2
III
JJ 2
cr
In L
0.999
0.067
0.299
0.109
3.510
7.0
0.469
-16.613
0.028
-241.981
0.000
0.075
0.302
0.110
3.544
7.0
0.470
-4.402
-0.176
-242.215
Transmission probabilitiesare fixed at their Mendelianvalues for both loci, and112 is fixed at 7.0. qB is
the frequency of the Xlinked gene and qA is the
frequency of the autosomalgene.
JJAab · ,JJAAbb
JJAab' , IIAabb
Double dominant:
llMAB" JJAABB
JJAaB" JJAaBB
llAaBb
JJAaBb
JJ JJAab" AAbb
JJAab" JJAabb
JJaab" JJaabb
JJaaB" JJaaBB
, llaa Bb
JJ2
Heterogeneity:

CHAPTER IV
ANALYSIS OF THE DATA OF WINOKUR, CLAYTON AND REICH (1969)
4.1 Description of data.
The 62 families analyzed in this Chapter were reported by
Winokur, Clayton and Reich (1969) and Winokur and Tanna (1969). The
probands are from two groups of consecutive admissions to a
psychiatric hospital. Group A consists of admissions from July 1964
through June 1965. Data were collected on all patients admitted
during this period (a total of 1075), and patients who were manic at
the time of admission were used for this study. There are 32 manic
probands in this group. Group B consists of all patients who were
manic at admission from January through May, 1967. This group has
29 probands. The families reported in Winokur and Tanna (1969) were
selected for having affective disorder in two generations, living
parents, and at least three sibs, and were used for linkage analysis.
Two of the families informative for Xg were among the 61 from groups
A and B, and we have information on the other family informative
for linkage with Xg. These three families were selected on the basis
of Xg phenotypes, but apparently not through the proband.
There are thus a total of 62 probands. Thirty-five are female
and 27 male; 57 are white and 5 are black. Personal interviews were
conducted with all available first degree relatives. Forty-nine per
cent of the first degree relatives of group A probands were
\'

101
interviewed, as were 83% of group B first degree relatives.
Di~gnostic criteria for probands were very similar to those of
Feighner et. al. (1972). For relatives the diagnosis was based on
course of illness, symptomology, and quality of recovery. Affective
illness was diagnosed if a relative suffered from an illness with
"prominent affective symptoms from which he recovered without
personality defect." Schizophrenia was diagnosed if the illness
resulted in chronic hospitalization, did not begin after age 40, and
was not accompanied by marked affective symptoms. "Undiagnosed
psychiatric illness" was used if the symptomology and/or course was
unclear. A diagnosis of alcoholism or drug abuse was based on
chronic abuse along with trouble at home, work, or with the law.
The data consist of, for the most part, the proband, his sibs
and their children, his children, parents, aunts and uncles and
grandparents. It was not possible to tell from whom information on
relatives more distant than first degree was obtained. Information
on first cousins and sibs of grandparents was very sketchy and
therefore these relatives are not included in the final pedigrees.
There is little information on spouses of probands, and none on
spouses of the proband's sibs. Three male and no female probands
had a spouse with affective illness. Age at examination or death
was available for about 85% of all individuals, with most missing
data from aunts, uncles and grandparents. Unknown ages were estimated
as explained in Section 3.1.
There are 12 sets of twins in the data: 4 are female-female,
6 male-male, and 2 male-female. Five of the probands are one of a

102
set of twins, and one set consists of the mother of a proband and
her twin. About 2.3% of all people in these families are one of a
set of twins. One member of each set of twins was discarded at
random, except in the cases where a proband or his mother was a twin,
when the proband or mother was retained.
There are a total of 1028 people, 510 (49.6%) males and 518
(50.4%) females. Among probands the sex ratio is 27 (43.5%) male:
35 (56.5%) female. Neither of these ratios is significantly dif
ferent from 1:1.
The probands ranged in age from 16-73 years, with an average age
of 39 (38 for females, 40 for males). For eight probands (4 male,
4 female) the index admission was the first episode of affective
illness. The female probands have an average of 6.0 first degree
relatives each, the males an average of 5.5. The average size of a
proband's sibship is 3.4 (3.6 for female probands, 3.1 for male).
Of the 36 probands (20 female, 16 male) who had children the average
number was 2.2 (2.4 for females, 2.0 for males). Both female and
male probands have an approximately equal number of brothers and
sisters, and of sons and daughters.
Among affectively ill people, the sex ratios are as in Table 4.1.
There is an excess of women in all categories, though the excess
does not reach significance in the categories concerning manics only.
Including alcoholics as affectively ill makes the sex ratios much
closer to one to one.
Age of onset was recorded for 119 of 177 affectively ill people.
The onset distributions are not significantly different for males

103
and females (Kolmogorov-Smirnov 2-sided test, p>0.20), or probands\
and other affectively ill (Kolmogorov-Smirnov 2-sided test, p~0.20).
People with mania have a significantly earlier age of onset than
those with depression only (Kolmogorov-Smirnov 2-sided test,
0.01<p<0.025). For all affectively ill people, the average age of
onset was 32.1 with a standard deviation of 14.8. See Table 4.2.
One-third of all affectively ill people had onset by age 23,
half by 29, and two-thirds by 36. The minimum age was 14 and maximum
74.
The observed cumulative age of onset distribution was compared
to the best fitting exponential, normal, lognormal, squareroot normal,
d ( f ) -1 1 d· "b .an age 0 onset norma ~str~ ut~ons. The exponential dis-
tribution, F(x)=l-exp{-e(x-L)} gives a very good fit, with MLE'sA A
L = min (x.) = 14, and e= l/(x-L) = 0.06. The use of an exponential~
distribution to describe age of onset was suggested by Crowe and
Smouse (1976), using the probands from this series. A lognormal
distribution also fits fairly well (see Figures 4.1 and 4.2). The
parameters for the distribution are a mean of the log ages of 3.37
and variance of 0.03. Since a lognormal distribution fits almost as
well as an exponential distribution, and since it is computationally
simpler to use the lognormal, in subsequent analyses it will be
assumed that age of onset is approximately lognormally distributed.
The distribution and diagnosis of affectively ill first degree
relatives is shown in Table 4.3. There are no male probands with an
ill father or an ill son. Winokur, Clayton and Reich (1969) give
morbidity risks (StrBmgren age corrected)as shown in Table 4.4. Of

•g lognorma1----~~~
104
o.-t:=;......Jl~-_--......,...---_r__--__r_---"T"""--___t---_;
• 10.0 20.0 30.0 '0.0 50.0 60.0 70.0 80.0ftGE OF ONSET
Figure 4.1 Age of onset with best fitting normal and lognormaldistributions. Winokur, Clayton and Reich (1969).
Figure 4.2 Age of onset with best fitting exponentialdistributivn. Winokur, Clayton and Reich (1969).
~-r----------------------:-::-v"---X---'~o
o-+---"'<>--..,r-----,----r-----...,.-----r----__r_-----!• 10.0 20.0 30.0 '0.0 50.0 60.0 0.0 80.0
flOE OF ONSET

lOS
the 62 probands, 9 (6 female, 3 male) had totally negative family
histories. Six more had a history of affective illness in a second
or third degree relative but none in first degree (2 of the probands
were female, 4 male).
The proportion of affectively ill people who are probands as a
function of age of onse~ is plotted in Figure 4.3, along with the
2best fitting curves of the form ln p = K
O+ Klln a + K
2(ln a) and
In p = KO+ Ki(ln a). The proportions are not significantly heter
2eogeneous among age class~s (XS=3.368, p>O.S). A constant ascertain-
ment function is probably adequate for these data. This hypothesis
will be tested in Section 4.2.
4.2 Segregation analysis.
4.2.1 Single gene models
Maximum likelihood estimates for the preliminary autosomal model
assuming Mendelian transmission are shown in Table 4.S. The models
examined are a dominant (~AA=~Aa) and a condominant (~AA'~Aa' and ~aa
all distinct) model, with ascertainment either a constant or linear
exponential function of age of onset. The hypothesis that Kl
= 0
(that is, a constant ascertainment function is adequate) is not
rejected for either the dominant or codominant model; it will there-
fore be assumed that Kl = O. The hypothesis that ~AA = ~Aa is also
not rejected, which implies that a two-mean model provides an
adequate description of the data. Therefore, further analysis will
be performed on an autosomal dominant model with the probability of
ascertainment assumed to be constant for all affected persons. Since

100
Figure 4.3 Proportion of affectively ill people who are probands,by decade of onset. Winokur, Clayton and Reich (1969).
1.0
807060o ......._~----:~_:~._----;~---:L-_~=-..JL--JI.....--_
10 20 30 40 50Age of onset, a
P
--- ln (p) = -5.7 - 1.6 (In a) + 0.2 (In a)2
--- ln (p) = 0.9 - 0.5 (In a)
Vertical lines indicate ~ standard error.

107
this model is much more likely when alcoholics are considered normal
(and there is no information on age of onset of alcoholism), no
further runs will be done with alcoholics considered affected.
Under the autosomal dominant model, the means of the age of
di 'b ' d 3.533onset str1 ut10n correspon to e6.477
= 34.2 years and e = 650
years. Three standard deviations below the upper mean is 154 years,
so that virtually no one with genotype aa lives long enough to
become affected. About 17.2% «0.091)2+2 (0.091)(1-0.091» of the pop-
ulation carries the defective gene, and 47.5% (0.356+0.119) of these
individuals will develop the disease if they live long enough. Since
no one with genotype aa is likely to live long enough to become ill,
the dominant model is one of a gene with frequency about 9% with
incomplete penetrance and virtually no sporadic cases.
Table 4.6 shows the results of similar analyses assuming an
X-linked gene with Mendelian transmission. The conclusions are
similar to those for the autosomal locus: an adequate description
of the data is obtained with a dominant model with a constant
ascertainment function. The likelihood is much higher when alcoholics
are considered normal. Under this model, the mean age of onset is
34.1 years for people with genotype AA and Aa and is 377 years for
people with genotype aa. Three standard deviations below the upper
mean corresponds to 90 years so that very few people with genotype
aa would live long enough to be affected. The gene frequency is
slightly higher than under the autosomal model (13% instead of 9%).
The susceptibilities are about the same: about 45.6% of the popula-
tion would be expected to develop the disease if they live long enough.

108
Therefore the X-linked dominant model is of a fairly common gene with
incomplete penetrance and very few sporadic cases. A comparison of
Tables 4.5 and 4.6 shows that the X-linked model is more likely than
the autosomal model. The difference in log likelihoods is 13.6,
indicating the X-linked model is about 7.7xl05
times as likely as
the autosomal.
Table 4.7 shows the results of testing the Mendelian and
environmental hypotheses for the X-linked dominant and the autosomal
dominant models. The hypothesis of segregation at a single Mendelian
locus is tested by comparing the likelihood when L AAA =1.0,
L = 0.5, and L A = 0.0 with the likelihood without these restric-Aaa aa
tions. The environmental hypothesis of no vertical transmission is
tested by comparing the likelihood under the restrictions
TAAA
= LAaA = LaaA with the unrestricted likelihood. Both the
Mendelian and environmental hypotheses are rejected with p«O.OOl
under both the autosomal and X-linked models. The environmental
model has a somewhat lower likelihood than either the Mendelian
autosomal or X-linked models. The transmission of affective illness
in these families cannot be explained by segregation at a single
locus, either autosomal or X-linked. The environmental hypothesis
is also rejected, however, so it is possible that some more compli-
cated genetic mechanism is operating.
4.2.2 Two gene models
The double dominant and heterogeneity models are considered,
as with the other sets of data. In addition a model suggested by
Winokur and his colleagues (eg., Winokur 1970) will be considered.

109
Under this model, an X-linked dominant gene B causes affective
illness, with mania caused by an autosomal dominant gene A in the
presence of B. See Table 2.1 for the genotype-phenotype correspon
dences for the three models. Table 4.8 shows maximum likelihood
estimates when transmission probabilities are fixed at their
Mendelian values. The mean of one of the age of onset distributions
is set at 7.0 in order to reduce convergence problems. Doing this
made little difference to the estimates of the likelihood in the
single gene models (see column 5 of Tables 4.5 and 4.6). The
heterogeneity model gives essentially the same results as the single
X-linked dominant model: the frequency of the autosomal gene con
verges to 0 and the likelihood is nearly equal to that of the single
X-linked gene model. The double dominant model gives a somewhat
higher likelihood than the heterogeneity model, and neither gene
frequency converges to O. This model is of two genes each with
frequency about 17%, and quite high penetrances since the sum of the
susceptibilities is 0.844.
Winokur's (1970) model essentially reduces to the double
dominant model. The upper two means are so high that virtually no
one with those genotypes will live long enough to become affected.
Under this model, however, the autosomal gene frequency is higher
and the susceptibilities lower than under the double dominant model.
Although the double dominant model and Winokur's model have
higher likelihoods than the single gene Mendelian models, they are
far less likely than the unrestricted single gene models. There is
therefore no point in formally testing the Mendelian hypothesis on

110
the two-gene models since these hypotheses would be rejected.
Although there are many other two-gene models theoretically possi.ble,
the models considered here are probably the most reasonable for
affective i.llness and it seems unlikely that a simple two-gene
model that fits these data could be found.
4.3 Conclusions.
Winokur, Clayton and Reich (1969) conclude that transmission of
manic depressive illness in these families is due to a single
X-linked dominant gene. Their conclusion is based on morbidity
risks, age adjusted by the Stromgren method. There is no direct
father to son transmission of mania or depression, while the risk
to mothers of male probands is 63%. The risk to fathers of female
probands is 23% and to mothers, 50%. Female probands have twice
as many ill sisters as ill brothers, and male probands have about
an equal number of ill brothers and sisters. Overall there are
about twice as many ill women as men. All of these observations
are suggestive of a rare X-linked dominant gene.
The results presented in this Chapter do not support this
conclusion. Although an X-linked dominant model gives reasonable
estimates under a Mendelian hypothesis, this hypothesis is soundly
rejected when rigorously tested. An autosomal dominant gene is
also an untenable model. The two-gene models examined do not
fit substantially better than the single gene models. The hypothesis
of no vertical transmission is also rejected under the single gene
models, and is less likely than the Mendelian hypothesis. This

suggests that there may be a genetic mechanism operating in these
families that is too complex or too heterogeneous to be detected
by the models examined in this Chapter.
111

Table 4.1 Sex ratios among affectivelyill people(Data of Winokur, Clayton and Reich 1969)
112
Females Males Total
All affectively ill 119 (0.672)** 58 (0.328) 177
All affectively ill + 127 (0.567)* 97 (0.433) 224alcoholics
All ill, excluding probands 84 (0.730)** 31 (0.370) 115
All ill, excluding probands, 92 (0.568) 70 (0.432) 162+ alcoholics
Unipolar 66 (0.750)** 22 (0.250) 88
Unipolar + Alcoholics 74 (0.548) 61 (0.452) 135
Bipolar 53 (0.596) 36 (0.404) 89
Bipolar, excluding probands 18 (0.667) 9 (0.333) 27
Significantly different from 1:1*p<.05 **p<.Ol

Table 4.2 Age of onset (Data of Winokur, Clayton and Reich 1969)
113
/I obs min max mean s.d. skewness
A11affective1y 119 14 74 32.1 14.7 0.89**ill
Males 44 16 70 34.0 14.6 0.57*
Females 75 14 74 31.0 14.8 1.09**
Probands 61 16 66 29.0 13.0 1.05**
Manics 77 14 66 29.0 12.9 0.93**
Depressed 40 15 74 38.2 16.5 0.60*
* Significantly different from 0, p<0.05** Significantly different from 0, p<O.Ol

Table 4.3 First degree family history(data of Winokur, Clayton and Reich 1969)
114
Female Probands Male ProbandsMo Fa Sis Bro Dau Son Mo Fa Sis Bro Dau Son
Depression 14 5 8 3 1 1 12 0 4 4 2 0
Mania + 1 2 5 2 3 1 4 0 2 3 0 0Depression
Total affec-tively ill 15 7 13 5 4 2 16 0 6 7 2 0
Total older 35 34 48 37 11 10 27 26 21 28 5 13than 15
Table 4.4 Morbidity risks, Stromgren age corrected(Winokur, Clayton, Reich 1969)
Female Probands Male Probands Overall
Fathers 23 + 7.7% 0 13 + 4.6
Mothers 50 + 9.1 63 + 9.9 56 + 6.8
Brothers 23 + 9.0 30 + 9.5 27 + 6.6
Sisters 46 + 9.4 39 + 11.5 44 + 7.3
*Sons 40 + 21.9 0
*Daughters 73 + 18.9 80 + 25.3
*Weinberg age corrected with risk period 15-60 years.Very small numbers.

e
Table 4.5 Preliminary Single gene autosomal models(Data of Winokur, Clayton & Reich 1969)
A. Alcoholics = Normal
e
B. Alcoholics = Affected.
e
Linear Ascertainment Constant Ascertainment Dominant, Constant AscertainmentCodominant Dominant Codominant Dominant K
l=0 and).J =7. 0 Dominantaa
q 0.093 0.092 0.092 0.091 0.091 0.164
Yl0.359 0.371 0.354 0.356 0.356 0.447
Y2 0.125 0.124 0.118 0.119 0.119 0.099
).JAA 3.396 } 3.6013.428 } 3.533 } 3.533 } 3.501
~'Aa 3.626 3.533
).J 5.937 7.058 5.904 6.477 7.0 5.771aa(J 0.475 0.478 0.478 0.479 0.479 0.449
KO -14.232 -15.307 -16.850 -16.850 ~16.995 ! -15.384I
Kl -0.736 -0.673 0 0 0 0
1n L -554.817 -555.006 -556.414 -556.499 -556.500 -635.510
HO: Kl=O for dominant model, X~=2.986 0.05<p<0.10 2 p>0.50HO: ).JAA=).JAa when Kl=O, Xl =0.017 t-'t-'
for codominant model, X~=3.l94 when Kl:fO, xI=O.378 p>0.50 VI
0.05<p<0;10

Table 4.6 Preliminary Single gene X-linked Models (Data of Winokur, Clayton & Reich 1969)
A. Alcoholics=Normal B. Alcoholics=Affected
Linear AscertainmentCodominant Dominant
Constant AscertainmentCodominant Dominant
Dominant,Kl=O and u =7.0aa
Constant AscertainmentDominant
-541.523 -542.419 -542.945 -542.999
-16.980
} 3.531
HO: }JAA=}JAa when K1=0,
when K1,,0, ........0'
o
5.005
0.448
0.081
0.367
0.321
} 3.460
-15.673
-641.002
o
7.0
0.479
0.335
0.112
0.1310.130 0.132 0.129
0.355 0.339 0.342
0.118 0.113 0.114
} 3.5933.582
} 3.5313.479
5.885 5.875 5.932
0.474 0.475 0.479
-14.861 -16.993 -17.044
-0.601 0 0
for dominant model, xi=2.862 0.05<p<0.10
2for codominant model, xl =2.062 0.05<p<0.102
X~=1.052 p>0.25
Xl=0.027 p>0.50
q 0.131
'VI 0.350
'V2 0.117
~AA3.620
~Aa 3.547
~aa 5.875
a 0.466
KO -15.871--
K1 -0.625
In L -541. 388
H : K =0o 1
e e e
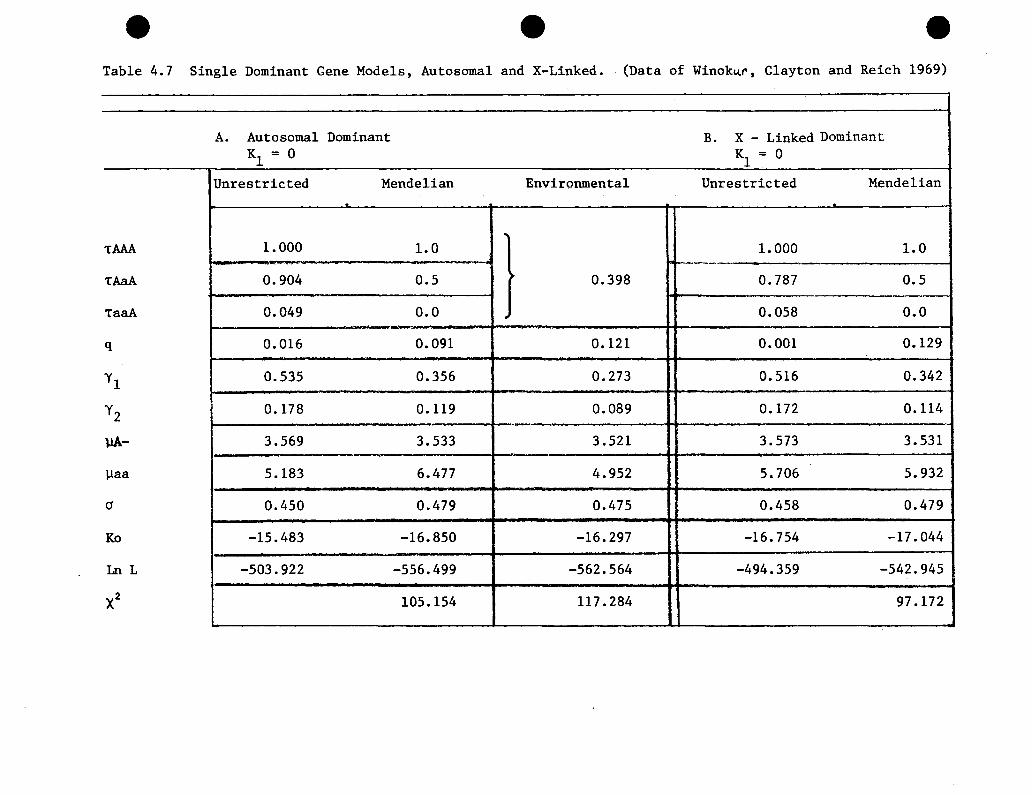
e e eTable 4.7 Single Dominant Gene Models, Autosomal and X-Linked. (Data of Winok~r, Clayton and Reich 1969)
A. Autosomal Dominant B. X - Linked DominantK = 0 K = 01 1
Unrestricted Mendelian Environmental Unrestricted Mendelian.
,TAAA 1.000 1.0 1.000 1.0- ,.......
TAaA 0.904 0.5 0.398 0.787 0.5
TaaA 0.049 0.0 . 0.058 0.0
q 0.016 0.091 0.121 0.001 0.129
Yl 0.535 0.356 0.273 0.516 0.342
Y2 0.178 0.119 0.089 0.172 0.114
llA- 3.569 3.533 3.521 3.573 3.531
llaa 5.183 6.477 4.952 5.706 5.932
(J 0.450 0.479 0.475 0.458 0.479
Ko -15.483 -16.850 -16.297 -16.754 -17.044
LnL -503.922 -556.499 -562.564 -494.359 -542.945
X2 105.154 117.284 97.172

118
Table 4.8 Two-gene models(Data of Winokur, Clayton and Reich 1969)
Double Dominant Heterogeneity Winokur 1970
qB 0.170 0.129 0.161 For all models,
qA 0.176 0.000 0.200transmission prob-abilities for both
Y1 0.633 0.339 0.585 loci are fixed at
Y2 0.211 0.113 0.190 their Mendelianvalues.
III 3.544 3.522 3.472The alleles A,a are
112 7.0 7.0 7.461 at autosomal locus
113 7.0 and the alleles B,bare at an X-linked
(J 0.484 0.463 0.461 locus.KO -16.223 -16.428 -16.217
1n L -533.707 -542.926 -534.448
Double dominant: Heterogeneity: Winokur (1970):
IlAAB· , IlAAB • , IlAABB IlAAB • ,
IlAaB· , IlAaB· , IlAaBB IlAaB • ,= III llaaB • , llaaBB = III
IlAABb IlAABb IlAABbIlAaBb = III Il
"aaB~} _aaB· ,IlAAb· , IlAAbb llaaBb llaaBb - 11 2PAab· , IlAabb IlAab· , IlAAbb IlAAb· , "MbjIlaab· , Ilaabb 112
IlAab·' IlAabb IlAab· , IlAabb =llaaB· , llaaBB
11 3Ilaab· , Ilaabb 11 2
Il aab· , IlaabllaaBb =

CHAPTER V
ANALYSIS OF THE DATA OF GOETZL ET. AL. (1974)
5.1 Description of the data.
The families analyzed in this Chapter were reported by Goetzl
et. ale (1974). The probands are from a series of patients admitted
consecutively to the Dartmouth Hitchcock Mental Health Center
between August 1968 and July 1971 who had a discharge diagnosis of
manic depressive illness, manic phase. There are 39 probands (20
female, 19 male) in 38 families; two of the probands are sisters.
Information on first degree relatives was obtained by sending a
questionnaire to all living relatives over 15 years old. Direct infor
mation was obtained on 119 of 155 living first degree relatives (76%).
Information on others, and on dead relatives, was obtained from the
proband and hospital records, if any. The families in most cases
consist of two or three generations. In a few families there is also
information on aunts, uncles, and grandparents. There is no information
on spouses. Age at examination is recorded for about 90% of the people
with unknown ages estimated as in section 3.1. Nearly all of those
with unknown ages are relatives more distant than first degree. Age of
onset is recorded for 55 of 71 affectively ill people.
There are a total of 321 individuals in the 38 families, 157
(48.9%) males and 164 (51.1%) females. The probands have an average of
6.3 first degree relatives (7.5 for female, 5.1 for males). The

120
average size of the proband's sibship is 2.0 (2.2 for female, 1.9
for male). Of the 19 probands (10 female, 9 male) who have children,
the average number is 2.6 (2.7 for females, 2.4 for males). Both male
and female probands have an approximately equal number of brothers
and sisters, and of sons and daughters.
Among the affectively ill people sex ratios are shown in Table 5.1.
Table 5.1 Sex ratios among affectively ill people.(Data of Goetzl et. al. 1974)
Female Male Total
All affectively ill 40 (0.563) 31 (0.437) 71
All ill, excluding probands 20 (0.625) 12 (0.375) 32
Manics 22 (0.524) 20 (0.476) 42
Manics, excluding probands 2 (0.667) 1 (0.333) 3
Depressives 17 (0.630) 10 (0.370) 27
All affectively ill plus alcoholics 42 (0.506) 41 (0.494) 83
All affectively ill plus alcoholics 22 (0.500) 22 (0.500) 44excluding probands
Depressives plus alcoholics 19 (0.487) 20 (0.513) 39
None of the ratios are significantly different from 0.50 at the
5% level, but the trends are the same as in other studies: there is
an excess of women among all affectively ill people, with the excess
due to those with depression only, and equal numbers of men and women
among those with mania. Counting alcoholics as affectively ill
results in approximately equal numbers of men and women in all
categories.

121
Age of onset was recorded for 55 affectively ill people and
some characteristics of the distribution are shown in Table 5.2.
Among all affectively ill people, the average age of onset is 32.7,
with a standard deviation of 13.1, and the distribution is signifi
cantly positively skewed. The minimum age of onset is 15 years, the
maximum 72 years. One-third of all ill people are affected by age
25, half by 32, and two-thirds by 40.
There are no significant differences between the age of onset
distributions of males and females, probands and other affectively
ill people with mania and those with depression only (Kolmogorov
Smirnov 2-sided p>0.25 in all cases). As in other studies, people
with mania tend to have earlier onset than those with depression only,
but in this study the difference is not significant.
The empirical age of onset distribution was compared with the
best fitting normal, lognormal, squareroot normal, (l/onset) normal,
and exponential distributions. The plots of the major distributions
considered, the normal, lognormal, and exponential, are shown in
Figures 5.1 and 5.2. Of the distributions considered, the lognormal
distribution provides the best fit for the age of onset distribution
in these data, with a mean of the log ages 3.4 and variance 0.029.
In all further analyses it will be assumed that age of onset is
approximately log normally distributed.
The distribution and diagnoses of affectively ill first degree
relatives is shown in Table 5.3. There is male to male transmission
in four of the families, while the general level of risk to relatives
is comparatively low (see Table 5.4). Sixteen of the probands

:T------------~::::;c:::::===~=,
lognormal
O-l""=-~-I--_,---_r---_r_---r_--__t---_l
· 10.0 20.0 30.0 'l0.0 50.0 60.0 70.0 80.0AGE OF ONSET
Figure 5.1 Age of onset with best fitting normal and lognormaldistributions. Goetzl et. ale (1974).
122
Figure 5.2 Age of onset with qest fitting exponential distribution.Goetzl et. ale (1974).
- x~•
!8co0
!"Z
I.~=
5......i~••~
~10.0 20.0 30.0 'l0.0 50.0 60.0 70.0 10.0
IlG£ Of ONSET

123
Table 5.4 Weinberg age-corrected morbidity risk~ foraffective illness. (Goetzl et. al. (1974)
Female Proband Male Proband
Mother 24.2% 18.5%
Father 18.8 12.5
Sister 27.0 9.1
Brother 2.2 10.0
Daughter 57.1} very small 76.9
{Son 28.6 numbers 57.1
(9 female and 7 male) have no affective illness in their first degree
relatives. Other illnesses in the family include alcoholism, two cases
of schizoaffective disorder, and one case of schizophrenia.
The proportion of all affectively ill who are probands, by
decade of onset, is shown in Figure 5.3. The best fitting curves of
form KO+Kl (In a) + K2 (ln a)2 = In p and K~+K~(ln a) = In p are also
plotted. The proportions are not significantly heterogeneous among
2age classes (X4 = 1.181, p>0.5). A constant ascertainment function
is probably adequate for these data. This hypothesis will be tested
in section 5.2.1.
5.2 Segregation Analyses.
5.2.1 Single gene models
As explained in section 2.1.4, the single gene models to be
examined are a single autosomal gene, and a single X-linked gene. The
data are analyzed first with alcoholics counted as normal, and then
with alcoholics counted as affected with z=l. For the preliminary,

Figure 5.3 Proportion of affectively ill people who are probandsby decade of onset. Goetzl et. al. (1974).
1
124
p
o
-!'" .. ....- ..- ..--'- ... --~ - ---,
".......
10 20 30 40Age of onset (a)
50 60
--- In p = -1.5 + 0.8 (In a)
--- In p = 0.2 - 0.2 (In a)
20.2 (In a)
Vertical lines indicate ± standard error.

125
exploratory runs, the transmission probabilities are fixed at their
Mendelian values. Under these conditions, the models examined under
each of the autosomal and X-linked hypotheses are a three mean model
(ll • II ,ll all distinct, corresponding to a " codominant" gene A)AA Aa aa
and a two mean model (llAA=llAa' corresponding to a "dominant" gene A),
with ascertainment either a linear or a constant (Kl=O) exponential
function of age of onset. Results of these runs are shown in Table
5.5 and 5.6.
Neither the hypothesis that Kl=O, nor the hypothesis that llAA=llAa
can be rejected under either the autosomal or the X-linked model,
whether or not alcoholics are counted as affected. Therefore, in all
further analyses it is assumed that llAA=llAa and Kl=O. Furthermore, in
both the autosomal and X-linked cases, the likelihoods are much higher
when alcoholics are considered normal. Since there is no information
on age of onset of alcoholism, subsequent analyses assume that
alcoholics are normal.
The X-linked and autosomal models with alcoholics considered
normal are strikingly similar. The likelihoods are very close: the
ratio of the likelihood of the autosomal to the X-linked model is
about 3.3. With the exception of the. gene frequency, which is higher
assuming an X-linked model, the estimates of the parameters are nearly
the same. Under a simple Mendelian hypothesis, these data do not
discriminate very well between the X-linked dominant and autosomal
models.
Table 5.7 shows the results of testing the Mendeliam
TAA A=l.O. TAa A=O.5. Taa A=O.O) and environmental

126
(H : T =T =T ) hypotheses under the X-linked and autosomalo AA A Aa A aa A
dominant models. Both hypotheses are rejected with p<O.OOl. The
environmental model is more likely than the Mendelian model for both
autosomal and X-linked genes, but the environmental model is also much
less likely than the unrestricted model.
Under the autosomal dominant Mendelian model, the means of the
f d ·"b" d 3.461 31 8 dage 0 onset 1str1 ut10n correspon to e =. years an
e5 . 035=153.7 years. Two standard deviations below the upper mean is
(5.035-2xO.494) 57 2 th t "f 1 "h te = . years, so a a port1on 0 peop e W1t geno ype
aa live long enough to develop manic depressive illness. A similar
situation exists under the X-linked dominant Mendelian model: the means
3.461 31 8 d 4.790 120 3 " h d d d "are e =. years an e = . years W1t two stan ar eV1a-
" b 1 h d" (4.790-2 x 0.538) 41 0tlons e ow t e upper mean correspon 1ng to e = .
years. If we want a model in which there are no such "sporadic" cases,
one approach is to fix the upper mean at some age large enough so that
no ones lives long enough to become affected. The results when ~aa
is set at 7.0 are shown in Table 5.8. Doing this makes very little
difference in the likelihoods or the estimates of the parameters, and
both the Mendelian and environmental hypotheses are rejected with
p<O.OOl, for both X-linked and autosomal models. Further manipulations
of single gene models seem unwarrapted, and it appears that neither
an autosomal nor an X-linked Mendelian model can account for transmis-
sion of affective illness in these data.
5.2.2 Two gene models
The two gene models examined in detail are the heterogeneity model,
and the double dominant model (see Table 2.1). Results of these

127
analyses are shown in Table 5.9. Transmission probabilities are fixed
at their Mendelian values for both loci and the upper mean is fixed
at 7.0. For the double dominant model, the gene frequencies are
similar to those in the single gene models (compare with columns 2 and
5 of Table 5.9), and all other estimates are similar. For the hetero
genity model, the frequency of the X-linked gene is much lower than
that of the single gene X-linked model but all other estimates are
similar. In both cases the likelihood of the two-gene models is only
slightly higher than the likelihood of either single-gene model.
There is no point in formally testing the Mendelian or environmental
hypotheses. Since the likelihoods under the two-gene model must be
larger than the likelihoods under the corresponding single gene models,
these hypotheses will necessarily be rejected.
These are just two of the many possible two-gene models. While
it is conceivable that there is a two-gene model that will fit these
data, it seems fruitless to search for it.
5.3 Conclusions.
In their analysis of these data, Goetzl et. al. (1974) conclude
that "any gene locus that exists on the X chromosome is not itself a
sufficient or necessary condition for the transmission of manic
depressive illness". They base this conclusion on their findings that
there is no significant difference in morbid risks to fathers and
mothers of either male or female probands, that the overall sex ratio
of affectively ill people is one to one, and that there are four ill
son-ill father pairs in the 38 families. The analysis presented in
this Chapter supports their conclusion that an X-linked gene cannot

128
explain the illness patterns in these families, and indicates that
a single autosomal gene is also an untenable model. There is no
reason to prefer either an X-linked or an autosomal model; when the
variable age of onset and ascertainment via probands is taken into
account both models fit equally poorly. Consideration of two genes
simultaneously does not help: the two-gene models examined also give
a very poor fit. It should be noted that the environmental models
of no vertical transmission of manic depressive illness also do not
fit. It may well be that there is some kind of genetic transmission
in these families, but it is too complex to be explained by the
models examined here.

129
Table 5.2 Age of onset. (Data of Goetz1 et. a1. 1974)
Number of StandardObservations Minimum Maximum .Mean Deviation Skewness
All affectively 55 15 72 32.7 13.1 0.72*ill
Males 26 15 58 32.1 12.4 0.48
Females 29 16 72 33.2 13.8 0.84*
Probands 39 15 57 31.4 11. 7 0.52
Manics 41 15 57 31.8 11.6 0.44
Depressives 10 19 72 38.7 17.5 0.64
*Significantly greater than 0, p<0.05
Table 5.3 First degree family hisotry. (Data of Goetz1 e1. a1. 1974)
Female Proband Male Proband
Mo Fa .Sis Bro Dau Son Mo Fa Sis Bra Dau Son
Depression 3 2 4 3 4 2 3 1 0 1 4 1
Mania 1 0 0 0 0 0 0 1 0 0 1 1
All affective 4 2 4 3 4 2 3** 2 0* 1 5 2ill
Totab15 years 19 19 2a 17 13 14 19 19 18 16 13 7
*1 sister was diagnosed as schizoaffective.
**1 mother was diagnosed as schizoaffective.

Table 5.5 Preliminary autosomal models. (Data of Goetzl et. al. 1974)
A. Alcoholics = Normal B. Alcoholics = Affected (z=l)
Linear Ascertainment Constant Ascertainment Linear Ascertainment Constant AscertainmentCodominant Dominant Codominant Dominant Codominant Dominant Codominant Dominant
q 0.151 0.145 0.134 0.140 0.233 0.227 0.209 0.182Y1 0.260 0.260 0.268 0.263 0.336 0.328 0.349 0.355YZ 0.072 0.072 0.074 0.073 0.067 0.066 0.071 0.071
IlAA 3.310 } 3.442 3.379 } 3.461 3.173 } 3.486 3.208 } 3.494IlAa 3.440 3.471 3.582 3.575Il aa 5.070 5.070 5.070 5.035 5.846 5.871 5.140 5.079
a 0.488 0.488 0.488 0.494 0.495 0.498 0.491 0.493K
O -2.494 -2.170 -1.489 -1.489 -2.505 -2.368 -1.430 -1.454Kl 0.285 0.203 0 0 0.293 0.255 0 0
In L -212.792 -212.838 -213.016 -213.038 -230.440 -230.907 -230.825 -231.154
Transmission probabilities are fixed at their Mendelian values. All hypotheses tested assumingalcoholics are normal.
(1) HO: Kl=O for dominant model xi = 0.400 } p>O. 50
codominant xi = 0.448
2}(2) HO: IlAA=IlAa when Kl=O, Xl = 0.044 p>0.50 ~
w2 0
when KIf0, Xl = 0.092
e e e

e e e
Table 5.6 Preliminary X-linked models. (Data of Goetzl et. al. 1974)
A. Alcoholics = Normal B. Alcoholics = Affected (z=l)
Linear Ascertainment Constant Ascertainment Linear Ascertainment Constant AscertainmentCodominant Dominant Codominant Dominant Codominant Dominant Codominant Dominant
I
q 0.322 0.333 0.276 0.250 0.325 0.326 0.296 0.357Yl 0.211 0.221 0.230 0.255 0.187 0.188 0.190 0.207Y2 0.062 0.062 0.065 0.074 0.037 0.037 0.038 0.042
~AA 3.340 } 3.419 3.349 } 3.461 3.076 } 3.068 3.098 } 3.230~Aa 3.364 3.451 3.052 3.068
~aa 4.571 4.480 4.626 4.790 3.791 3.791 3.813 3.944
0 0.490 0.497 0.489 0.538 0.260 0.260 0.260 0.345KO -2.353 -2.424 -1.430 -1. 423 -2.591 -2.587 -1.442 -1. 448K
l 0.253 0.269 0 0 0.320 0.318 0 0
In L -213.596 -213.898 -213.828 -214.222 -230.717 -230.733 -230.952 -232.642
Transmission probabilities are fixed at their Mendelian values. All hypotheses are tested assumingalcoholics are normal.
(1) HO: Kl=O when ~AA=~Aa2
Xl = 0.648
when ~AA:!:~Aa2Xl = 0.464
(2) HO: ~AA=lJAa when Kl=O xi = 0.788p>0.50
when Kl:!:O2
Xl = 0.604~w~

Table 5.7 Single gene dominant models: Autosomal and X-linked. (Data of Goetz1 et. al. 1974)
Autosomal X-linked
Unrestricted Mendelian Environmental Unrestricted MendelianTAM 1.000 1.0 1.000 1.0TAaA 0.996 0.5 l
0.639 0.999 0.5J
TaaA 0.044 0.0 0.101 0.0
q 0.006 0.140 0.023 0.015 0.250
Y1 0.589 0.263 0.237 0.534 0.255
Y2 0.164 0.073 0.066 0.151 0.074
]JA- 3.600 3.461 3.563 3.480 3.461]Jaa 5.476 5.035 4.801 5.179 4.790
0 0.467 0.494 0.492 0.436 0.538KO -1. 455 -1. 489 -1.446 -1.413 -1. 423
lnL -177.139 -213.038 -196.849 -186.079 -214.2222
71. 798 39.42 56.286X --- ---
- - e
~WN

e e e
Table 5.8 Single gene moce1s with ~ fixed at 7.0: Autosomal and X-linked.aa(Data of Goetz1 et. al. 1974)
A. Autosomal B. X-linked
Unrestricted Mendelian Environmental Unrestricted Mendelian
LAAA 1.000 1.0 1.000 1.0
LAaA 0.998 0.5 } 0.531 0.998 0.5
LaaA 0.038 0.0 0.103 0.0
q 0.006 0.149 0.005 0.169 0.422
'VI 0.705 0.264 0.302 0.733 0.230
'V2 0.181 0.073 0.084 0.209 0.060
~A- 3.762 3.548 3.679 3.787 3.525
~aa 7.0 7.0 7.0 7.0 7.0(J 0.536 0.518 0.558 0.549 0.512KO -1.439 -1.498 -1. 454 -1. 385 -1. 448
In L -177.372 -213.429 -199.567 -184.078 -215.0372
72.114 22.195 61. 918X --- ---
.....ww

134
'!';lblc 5.9 Two-locus models. (Data of Goetzl et. al. 1974)
---------------
Double dominant Heterogeneity
0.520
0.168
0.350
0.097
3.536
0.058
0.122
0.255
0.071
3.510
For both models, transmission probabilities arefixed at their Mendelianvalues for both loci,11
2=7.0, and K
1=K
2=0.
B is X-linked and A isautosomal.
7.0 7.0
() 0.519 0.508
-I. 483 -1. 489
III L -212.541 -213.152
Double dominant: Heterogeneity
IlAABB \IJAaBB i
IIl aaBB I11 AABb \IlAaBb
~JaaBb
IlAAbbIJ
Aahb
IlAaB ' ,
lIaaB · ,
~laaBB"-
\Il aaBb
I Il AAb · ,(1l<l,IBb I lJ 2 PAah · ,,IlAAbh III AAb · ,
Il Aah · , 11 Aabh
lIMI~B' ,II
A,I1L,

CHAPTER VI
ANALYSIS OF THE DATA OF STENSTEDT (1952)
6.1 Description of the data.
The data in this chapter were reported by Stenstedt (1952). The
probands are all cases of "manic depressive psychosis" who were
hospitalized in northwest Sweden during the years 1919-1948.
Stenstedt's definition of manic depressive illness includes both
cases of unipolar depression and bipolar manic depression. Probands
were ascertained using registers from state asylums and population
registers. Stenstedt personally interviewed the probands and many
of their first degree relatives. Probands and their families were
followed up until the end of the study in 1948. The original data
consist of 201 families. Of these, 51 have mania in a proband and
are used here. There are 54 probands in these 51 families, 30 female
and 24 male.
The diagnosis for the probands was the hospital diagnosis, and
was confirmed by Stenstedt by a chart review. The diagnosis of
relatives was based on symptomo10gy and social factors, and was
obtained from hospital records and/or personal examination when
possible, and information from relatives when records were unavailable
or personal examination impossible. Secondary cases (those other
than probands) were called "certain" if confirmatory hospital records

136
were available or the relative was personally diagnosed as affectively
ill by Stenstedt; they were called "uncertain" otherwise.
Diagnoses of mental illness were made on the basis of criteria
such as the following. Manic depressive disease was diagnosed if
the disease showed marked depressive or manic symptoms with poor
social functioning during episodes, but good functioning between
episodes. Schizophrenia was the diagnosis if the disease was
chronic and left "marked alteration of personality". A "cycloid
psychopath" had marked mood swings which were not severe enough to
be disabling. "Psychopathy" was a rather vague category applied to
a relative who was described as abnormal, but with no specific
details available, and who was not severely enough disabled to fit
into one of the other categories. Other diseases present in the
families include alcoholism, mental defects, senile and presenile
psychoses, and epilepsy.
There are a total of 549 individuals in 51 families. The data
consist of the proband, his parents, children and sibs. These are
thus simple, 2- and 3-generational pedigrees. The data include
the age of "disapperance from observation" (ie. age at the end of
the study, or death, or age at which a person's family lost track
of him) for nparly everyone, the number and type of episodes for
;It'fectively ill people, and description of "uncertain" cases. The
families are fairly large: the average size of the proband's
sibship is 6.8. Of the 33 probands who had children, the average
family size is 3.3.

137
The overall sex ratio is 267 female (48.6%): 282 male (51.4%).
Among probands it is 44.4% male: 55.6% female; this is not signif
2icantly different from 1:1 (X
l=0.677, p>0.25). Female probands have
significantly more sons than daughters (37 sons, 15 daughters,
2xl=9.3, p<O.Ol). but a similar difference is not present among
children of male probands. Both female and male probands have
approximately equal numbers of brothers and sisters.
There are a total of 76 affectively ill people (using a fairly
broad definition of "ill"--those who are designated "uncertain" cases
are included). The sex ratios are shown in Table 6.1. None of the
ratios differs significantly from a one-to-one sex ratio. There is,
however, an excess of women among all affectively ill, which is due
mainly to those who have unipolar depression only. When alcoholics
are included as affectively ill, the excess of women disappears.
The age of onset distribution is very similar in females and
males, and in probands and other affectively ill (see Table 6.2).
People with mania tend to have an earlier onset than those with
depression only (Kolmogorov-Smirnov statistic = 0.39; 2 sided p<O.Ol).
Age of onset was recorded for 74 of 76 affectively ill people. The
range was 14-72 years with mean 33 and standard deviation 13.4. The
median age of onset was 29, with one third affected by age 25, and
two thirds by 36. In general, the distributions for various break-
downs show positive skewness. The best fitting exponential, normal,
-1lognormal, squareroot normal, and (age of onset) normal were
compared to the empiric age of onset distribution. The lognormal
is the best fitting of the distributions, although there is not

138
much difference between the fits of the lognormal and exponential
distributions (Figures 6.1 and 6.2). For further analyses, it will
be assumed that age of onset is lognormally distributed.
In his original paper Stenstedt reported morbidity risks (age
corrected by the abridged Weisberg method using a risk period of
15-60 years) of
12.28% + 1.36 to sibs of probands
7.35% + 1.34 to parents, and
9.40% + 2.39 to children
(counting both "certain" and "uncertain" cases as affected). He
found no significant differences in risk between sexes, between
families of probands with "few attacks" (less than 3) and "several
attacks" (3 or more), or between families in which the proband had
mania and those in which the proband had depression only. Thirty-
three probands (19 female. 14 male) had no first degree relative
with affective illness. See Table 6.3 for a more complete breakdown
of first degree family history.
The proportion of affectively ill people who are probands as a
function of age of onset is shown in Figure 6.3, along with the best
2fitting curves of the form In p = K
O+ K
l(In a) + K
2(ln a) and
In p = KO+ Ki(ln a). The proportions are not significantly hetero-
2geneous among age classes (X
S=1.58, p>0.5). A constant ascertainment
function is probably adequate for these data. The hypothesis that
K = K = 0 will be tested in section 6.2.1 2

139
•g
10 50flOE OF ONSET
60 70 10
Figure 6.1 Age of onset and best fitting normal and lognormaldistributions. Stenstedt (1952).
Figure 6.2 Age of onset and best fitting exponential distribution.Stenstedt (1952) •
..,...--------------------:--;---'"'V"---,~o
·~-tlr-O-.o~..:.--r----r-----r-----r----r---..,..----420.0 ao.o '0.0 . 50.0 10.0 0.0 10.0
IIGE OF ONSET

140
Figure 6.3 Proportion of affectively ill people who are probands,by decade of onset. Stenstedt (1952).
1
..
..--------T
-~rtfl !I I I 11
I I I I '
oL __L. ~.L. __ .I __ .. J--J J L.-+-_10 20 30 40 50 60 70 80
Age of onset, a
p
--- In p -0.5 + 0.04 (In a) - 0.01 (In a )2
~- In p -0.6 + 0.1 (In a)
Vertical lines indicate ± standard error.

141
6.2 Segregation Analysis.
6.2.1 Single gene models
Preliminary analyses on these data were performed under an
autosomal and an X-linked model, assuming the transmission prob-
abilities are fixed at their Mendelian values of TAAA
= 1.0,
TAaA = 0.5 and TaaA = O. Results for the autosomal gene are shown
in Table 6.4. A constant ascertainment function is adequate since
HO
: Kl
= 0 cannot be rejected (p>0.5), and allowing the age of onset
distribution to be a mixture of three lognormal distributions is not
significantly better than a mixture of two (p>0.5). Therefore for
subsequent analyses under an autosomal model it will be assumed that
KI
= 0 and ~AA = ~Aa· A comparison of parts (A) and (B) of Table 6.4
shows that the likelihoods are much larger when alcoholics are
considered normal than when they are considered affected (with z-l)~
There is no information on age of onset of alcoholism, so no further
runs will be done considering alcoholics as affected.
Under the autosomal model chosen for further analysis (i.e.,
dominant with constant ascertainment and alcoholics considered normal),
the means of the age of onset distribution correspond to an "affected"
3.636 5 927mean of e =37.9 years and an "unaffected" mean of e· =375 years.
Three standard deviations below the upper mean corresponds to 92 years
so that very few people with genotype aa live long enough to become
affected. The model is thus of a dichotomy between genetically
susceptible people with genotypes AA and Aa, and genetically unsus-
ceptible people with genotype aa. The frequency of gene A is large
enough (0.126) so that homozygotes are not rare among affected

142
people. The susceptibilities are quite low, however. Only 0.113 +
0.050 = 16.3% of individuals are expected to develop the disease if
they live long enough, in spite of the high mean age of onset for
aa people. The model is thus of a fairly common dominant gene with
low penetrance.
Table 6.5 shows results of the preliminary analyses assuming
an X-linked gene. It is assumed that alcoholics are normal, since
under the autosomal model the likelihoods were so much higher when
this is assumed. Again, the hypothesis that Kl
o cannot be rejected
for either a codominant or a dominant model and the hypothesis that
~ = ~ is not reJ·ected.AA Aa
and ~AA= ~Aa·
Therefore it will be assumed that KI = 0
The most striking feature of the X-linked models is the high
gene frequency (0.740 for the model chosen). An attempt was made to
look for other local maxima with lower gene frequencies and higher
susceptibilities, but none with a larger likelihood was found. The
susceptibilities are very low: only 10.2% (0.070 + 0.032) of
individuals are expected to develop manic depressive illness if they
live long enough. The means of the age of onset distribution cor-
respond to an affected group with genotypes AA or Aa and mean
e3
. 564=35.3 and an unaffected group with genotype aa and mean
e 5 . 920=372. Three standard deviations helow the upper mean is 108
years, so virtually no one with genotype aa lives long enough to he
affected. The X-linked model is therefore of a very common dominant
gene with low penetrance.

143
A comparison of Tables 6.4 and 6.5 shows that these data do not
discriminate between the models of an X-linked and an autosomal gene.
The difference in log likelihoods is 0.859, indicating that an X-
linked model is about 2.4 times as likely as the autosomal. With
the exception of the gene frequency, estimates of the parameters
are also quite similar under the two models.
Table 6.6 shows the results of testing the Mendelian and
environmental hypotheses under the autosomal and X-linked models.
For both the X-linked and the autosomal models the Mendelian
hypothesis is rejected (p<O.OOOl). Since under the unrestricted
model LaaA converges to the bound 1.0, the degrees of freedom of
2the X 's are reduced to between 2 and 3 for the Mendelian hypothesis
and to between 1 and 2 for the environmental.
Under the autosomal model the environmental hypothesis is not
rejected (p>0.20). The segregation patterns in these families
cannot be explained by a single gene, either autosomal or X-linked;
it is more likely that transmission is entirely environmental.
6.2.2 Two gene models
Two different models assuming one X-linked dominant and one
autosomal dominant were examined. See Table 2.1 for the genotype-
phenotype correspondences. Table 6.7 shows the results of the
analysis. The double dominant model is quite similar to the single
gene autosomal and X-linked models. The X-linked gene in the
heterogeneity model has a much lower gene frequency but otherwise the
estimates are similar. The likelihoods of both two gene models,
the single gene autosomal, and the single gene X-linked models are

144
all quite similar. There is no point in testing the Mendelian
hypothesis for the two gene models since it would necessarily be
rejected. Thus these two gene models are also inadequate for
explaining the genetic transmission of manic depressive illness in
these data.
6.3 Conclusions.
Stenstedt's (1952) analysis of these data included all 201
families with both unipolar and bipolar probands. He found, however,
that there were no significant differences in morbid risks to first
degree relatives between unipolar and bipolar probands. Based on the
segregation patterns Stenstedt concludes that the mode of inheritance
cannot be exactly determined but that some kind of dominant trans
mission, probably an autosomal gene with incomplete penetrance, is
likely.
The analysis in this Chapter provides no support for a single
gene hypothesis, either autosomal or X-linked. The two gene models
examined are no more likely than the single gene models. If there
is a genetic mechanism operating in these families it is too complex
to be detected by the one-or two-locus models considered in this
Chapter. The environmental model provides a better explanation of
the illness pattern than any of the genetic models. It could be that
the phenotypic variability caused by genetic factors is too small in
comparison to the environmentally caused variability to be picked up
by these models. Another possibility is that there is a good deal of
gene.tic heterogeneity among families so that no single genetic
mechanism can be detected.

Table 6.1 Sex ratios among affectively ill people.(Data of Stenstedt 1952)
Females Males Total
All affectively ill 42 (0.553) 34 (0.447) 76
All affectively ill +42 (0.483) 45 (0.517) 87alcoholics
All affectively ill, 12 (0.545) 10 (0.455) 22excluding probands
All affectively ill, excluding 12 (0.387) 19 (0.613) 31probands,+ alcoholics
Unipolar depressives 11 (0.579) 8 (0.421) 19
Unipolar depressives 11 (0.324) 17 (0.676) 28+ alcoholics
Bipolar manic-depressives 31 (0.544) 26 (0.456) 57
Table 6.2 Age of onset.(Data of Stenstedt 1952)
N Min Max Mean Std. dev. Skewness
All affectively ill 74 14 72 33.0 13.4 0.88**
Males 34 16 72 32.8 14.3 1.10**
Females 40 14 65 33.3 12.8 0.66*
Probands 54 14 65 32.4 12 .5 0.83**
Bipolars 56 14 72 30.6 12.5 1.18**
Unipolars 18 18 65 39.4 13.7 0.38x
Significantly different from 0 *p<.05 **p<.Ol
xSample too small to detect significance.
145

Table 6.3 First degree family history.(Data of Stenstedt 1952)
146
Female Probands Male ProbandsMo Fa Sis Bro Dau Son Mo Fa Sis Bro Dau Son
Depression 2 1 1 2 0 0 1 0 1 0 0 0
Uncertain 1 1 0 1 0 0 0 1 2 2 0 0depression
Mania + 1 0 1 2 0 1 0 0 1 1 0 0depression
Total affective1y 4 2 2 5 0 1 1 1 4 3 0 0ill
Total number 15 36 32 80 78 18 42 24 24 56 51 9 41or older

e - e
Table 6.4 Preliminary autosomal models. (Data of Stenstedt 1952)
A. Alcoholics = Normal B. Alcoholics = Affected
Linear Ascertainment Constant Ascertainment ~A~~~Aa Linear Ascertainment Constant AscertainmentCodominant Dominant Codominant Dominant ~1 =7.0 Codominant Dominant Codominant Dominant
aa
q 0.221 0.219 0.237 0.219 0.191 0.380 0.373 0.373 0.339
Yl 0.096 0.094 0.093 0.094 0.097 0.112 0.112 0.112 0.115Y2 0.042 0.042 0.042 0.042 0.044 0.037 0.037 0.038 0.040
llAA 3.639 } 3.574 3.574 } 3.574 } 3.574 3.598 } 3.582 3.598 } 3.565llAa 3.555 3.574 3.565 3.565
~aa 5.820 5.830 5.786 5.830 7.0 5.748 5.748 5.748 5.718
(J 0.411 0.415 0.415 0.415 0.415 0.408 0.408 0.408 0.408KO -0.506 -0.433 -0.432 -0.433 -0.435 -0.420 -0.422 -0.422 -0.406Kl 0.006 -0.0002 0 0 0 -0.0003 -0.0005 0 0
In L -228.342 -228.404 -228.397 -228.406 -228.430 -247.304 -247.338 -247.305 -247.436
For all models, TAAA=I.O, TAaA=0.5, TaaA=O.O. All hypotheses tested assuming alcoholics are normal.
(1) HO: Kl=O for codominant xi=0.002
dominant xi=0.OI6
2(2) HO: ~AA=~Aa when KI=O X~=0.081
when KI~O Xl =0.067
I-'~".J

Table 6.5 Preliminary Single gene models: X-linked, Alcoholics = Normal.(Data of Stenstedt 1952)
7.0
o
0.070
0.032
0.743
0.410
-0.418
} 3.564
-228.232
Dominant-with constantascertainment and II =7.0aa
0.660 0.740
0.072 0.070
0.033 0.032
3.516 }. 3.5643.662
5.104 5.902
0.399 0.411
-0.419 -0.418
0 0
-227.947 -228.231
, A=O.Oaa.
p>.90
Constant AscertainmentCodominant Dominant
In all models 'AAA=l.O, 'AaA=0.5,
HO: Kl=O - for dominant X~=O.OlO}codominant Xi=0.002
2HO: llAA=uAa - for Kl=O Xl=0.568 } p>.40
Kl~O xr=0.597
Linear AscertainmentCodominant Dominant
q 0.652 0.743Yl 0.074 0.071Y2 0.033 0.032
llAA 3.527 } 3.564llAa 3.690
llaa 5.667 5.624(J 0.406 0.411K
O -0.349 -0.416K
l -0.024 -0.001
ln L -227.937 -228.226
f-'~(Xl
e e e

e e e
Table 6.6 Single gene dominant models: Autosomal and X-linked.(Data of Stenstedt 1952)
A. Autosomal B. X-linked
Unrestricted TIS Mendelian TIS equal Unrestricted . TIS MendelianT
AAA 0.00001 1.0 0.184 1.0T
AaA 0.995 0.5 } 0.454 0.428 0.5TaaA 0.535 0.0 1.000 0.0
q 0.064 0.126 0.055 0.096 0.740Y1 0.107 0.113 0.130 0.102 0.070Y2 0.050 0.050 0.061 0.048 0.032llA_ 3.649 3.636 3.649 3.633 3.564
llaa 5.868 5.927 5.920 5.780 5.902
a 0.436 0.468 0.436 0.427 0.411KO -0.414 -0.437 -0.413 -0.413 -0.418
1n L -213.355 -229.190 -213.931 -212.194 -228.2312
31.670X --- 1.152 --- 32.074
I-'-l::\0

e
Table 6.7 Two locus models (Data of Stenstedt 1952)
Double dominant Heterogeneity
qA 0.181 0.108
qB 0.741 0.008
Yl0.111 0.110
Y20.051 0.049
)Jl 3.593 3.610
)J2 7.0 7.0
a 0.429 0.432
KO -0.437 -0.442
ln L -227.991 -228.691
Transmission probabilities are fixed at their Mendelianvalues for both loci, and )J2 is fixed at 7.0. qA is thefrequency of the autosomal gene and qB is the frequencyof the X-linked gene.
e e
f-'V1o

CHAPTER VII
LINKAGE ANALYSIS
7.1 Description of the family.
This Chapter presents segregation and linkage analysis on a large
family, to be referred to as the Lava family. The family was reported
by Winokur (1971) but has not been extensively analyzed.
The proband is a woman with bipolar manic depressive disease.
She has six brothers and three sisters. Both colorblindness and the
Xg blood group are segregating in this sibship. Figure 7.1 shows the
pedigree of the proband's first degree relatives. There is also
information on manic depressive disease status available on her aunts
and uncles, grandparents, first cousins, and the children of her sibs,
for a total of 76 individuals. Affective illness is present on the
maternal side of the proband's family: her maternal grandmother and
several first cousins have had depressions, a maternal aunt has had
manias and depressions, and three first cousins and an uncle are
alcoholic. There is no affective illness or alcoholism on the
paternal side.
This family is well suited for linkage analysis for several
reasons. There is a large sibship, and its members are old enough
to have passed through a large part of the risk period for manic
depressive illness. Both Xg and colorblindness are segregating, but
the family was not selected on the basis of two traits through the

82' , 76
not CB
~36L ,
+CB
~81 J46W44
not CB
54, I+
not CB
56, ,+
not CB
I-'VIN
CB Colorblind
Bipolar manic depressive illiness
Unipolar depression
Legend:
0 Male
0 Female
~or ~
_ or •,. . Proband
Not CB not colorblind
+ Xg (+) phenotype
Xg (-) phenotype
? Unknown affective illness phenotype
Numbers refer to age at examination
Numbers in parentheses refer to age of onset ofaffective illness.
Figure 7.1 Lava family: Pedigree of the proband's first-degree relatives.
e e e

153
proband. Ascertainment was through a manic depressive proband, and
examination of her sibs revealed brothers with colorblindnessand
brothers with the Xg phenotype. This avoids the problem of ascertain-
ment through "doubly ill" probands that apparently has been present
in other linkage studies on manic depressive illness (Gershon and
Bunney 1976). In addition, ascertainment corrections to the lod
scores will not be necessary. The corrections cancel out when the
lod score is formed since selection of the marker traits (Xg and
colorblindness) was done without reference to affective illness pheno-
types, and the analysis is not conditional on parental phenotypes.
The remainder of this Chapter will be in two parts. First, seg-
regation analyses are performed in order to find the best-fitting
single gene model. The parameters of this model when the transmission
probabilities are set at their Mendelian values are then used as input
for the linkage analysis.
7.2 Segregation analysis.
Both X-linked and autosomal models are examined, since other sets
of data seem to contain insufficient information to discriminate
between these modes of inheritance. Alcoholics are considered normal
and it is assumed that KI =K2=O. Table 7.1 shows the results under an
autosomal model. The codominant model is not significantly better
2than the dominant model (X
l=O.OI6, p>O.5), so the Mendelian and
environmental hypotheses are tested under the assumption that ~AA=~Aa.
Under these conditions, neither the Mendelian nor the environmental
hypotheses can be rejected, although the Mendelian model fits somewhat
hetter than the environmental model. The parameters of the Mendelian

Three standard
154
and unrestricted models are quite similar, with the exception of the
gene frequency which is larger under the Mendelian model. Under the
environmental model the gene frequency is much higher than under the
unrestricted or Mendelian models, and the susceptibilities are higher
as well, with little difference in the likelihoods. This indicates
that the likelihood surface is quite flat so that a wide range of
models with different parameter values will have similar likelihoods.
Table 7.2 shows segregation analysis under an X-linked model.
2The hypothesis that ~AA=~Aa is not rejected at the 1% level (X
l=4.652,
0.025<p<0.05). Since. in addition, under the codominant model,
~AA<~Aa it seems reasonable to assume that a dominant model is adequate.
Under the assumption that ~AA=~Aa' the gene frequency is small (0.008)
and the susceptibilities moderate (yl=0.41l and y
2=0.103). The mean
age of onset for people with genotypes AA and Aa is e 3 . 208=24.7 and
f 1 h h · 5.660 287or peop e wit genotype aa t e mean 1S e =.
deviations below the upper mean is 112 years, so that virtually no
one with genotype aa lives long enough to become affected. Therefore,
the most reasonable Mendelian X-linked model is one of a rare dominant
gene with incomplete penetrance and virtually no sporadic cases.
The last two columns of Table 7.2 are used to test the Mendelian
hypothesis. As with the autosomal model, this hypothesis cannot be
rejected. The estimates of TAaA and TaaA in the unrestricted model
are close to their Mendelian values. The estimate of TAAA
(0.476)
is not close to the 1.0 expected under a Mendelian model; however,
individuals of genotype AA are extremely rare (expected frequency
(0.0002)2) so this parameter is not well estimated in these data. The

155
likelihood surface is again quite flat, with ~ fairly wide range of
gene frequency and susceptibility estimates resulting in similar
likelihoods. Although the Mendelian model fits these data, it is
not reasonable to conclude that there is an X-linked dominant gene
for manic depressive illness segregating in the family. The hypothesis
of an autosomal dominant gene is not rejected, and these data do not
discriminate between these two modes of inheritance. The difference
in log likelihoods between an autosomal and an X-linked gene is only
1.60, indicating the X-linked model is about five times as likely as
the autosomal model. The data are ambiguous as to the mode of
inheritance of manic depressive illness. Linkage analysis may shed
some light on this matter.
7.3 Linkage analysis.
Lad scores are calculated assuming that the estimates obtained
under an X-linked dominant model are the "true" values (column 2 of
Table 7.2). The gene frequency for the Xg(+) allele is taken as 0.53
and that of colorblindness as 0.02 (Levitan and Montagu 1971), values
which are often observed in American caucasian populations. Table
7.3 shows the lod scores when the recombination fraction is equal
to 0.0, 0.1, 0.2, 0.3, and 0.4.
For purposes of comparison, lod scores for Xg are shown both
taking the variable age of onset of affective illness into account,
and assuming that the gene for affective illness is fully penetrant
and completely dominant (as has been assumed in other linkage analyses,
e.g. Mendlewicz and Fleiss 1974, Tanna and Winokur 1968). There is no
qualitative difference between these two cases: the maximum lod score

156
is positive and occurs a~ A = 0.0 in both cases. However, the lod
scores are considerably lower when the variable age of onset is taken
into account. This is to be expected since under the more complex
model genotypes cannot be determined with certainty, so the possibility
of sporadic cases and incomplete penetrance is allowed. Under the
simple model, the genotype of all brothers of the proband is certain,
and "recombinants" can be counted. Table 7.4 shows the phenotypes
of all persons on whom there is complete information. There is
complete association of phenotypes: all brothers are either Xg(+)
and normal, or Xg(-) and depressed. No such association of phenotypes
has been found in the population (Gershon and Bunney 1976). There
is thus some evidence (far from conclusive) for close linkage
between Xg and a gene for manic depressive illness in this family.
Lod scores for colorblindness are shown in the third column of
Table 7.3. All lod scores are negative, and increase with increasing
A. There is thus no evidence for linkage of manic depressive illness
and colorblindness in this family. Any suggestion of linkage between
manic depressive illness and colorblindness must be regarded with
some suspicion, in view of the association between these traits in
the population suggested by Gershon et. al. (1976).
7.4 Conclusions.
The results of this analysis provide some support for the
hypothesis that there exists a gene closely linked to the Xg locus
that leads to the development of manic depressive illness in some
families. The evidence is far from conclusive, however, and more
data need to be collected before a confident statement can be made.

157
Segregation analyses presented in Chapters III ~ VI suggest that such
a gene (if it exists) is not the most common mode of transmission of
manic depressive illness.
There is no suggestion of linkage with co1orblindness in this
family, as has been reported by Mendelewicz and his co1legues
(Mendlewicz et. a1. 1972, Mendlewicz and Fleiss 1974, Fieve et. al.
1973). However. there are a number of methodological problems in
those analyses, as pointed out by Gershon and Bunney (1976). The
most serious of these difficulties are that ascertainment corrections
were improperly applied, the analysis was appropriate for nuclear
families but was applied to multigeneration pedigrees in some cases,
and the variable age of onset and incomplete penetrance were not
taken into account. The conclusion of Mendlewicz and Fleiss (1974)
that a gene for manic depressive illness is closely linked to both
Xg and the colorblindness loci (protanopia and deutanopia) is
difficult to accept in view of the large distance between Xg and
the colorblindness loci (e.g. Levitan and Montagu 1971; see Figure 2.2).
In addition, the suggested population association (Gershon et. al 1976)
between colorblindness and manic depressive illness makes it difficult
to conclude anything about linkage. It is probably best to disregard
data on linkage between manic depressive illness and co1orblindness
until the association is better understood.
The analysis of linkage with Xg is less difficult since no
population association has been suggested. Gershon and Bunney (1976)
present a more conservative analysis of the data of Mendlewicz's
group, and find a smaller maximum lod score (1.88 instead of 2.95,

158
both at a recombination fraction of 0.2). This is still suggestive
of linkage, though not as strongly as reported by Mendlewicz and
Fleiss (1974). Gershon and Bunney (1976) point out that there are
still problems with their analysis, probably the most serious of
which is the crude way in which variable age of onset is taken into
account (unaffected people younger than 25 years were not counted).
The conclusions of linkage analysis on the Lava family are similar
to those of Gershon and Bunney (1976): there is a slight suggestion
of tight linkage with Xg. The variable age of onset is taken into
account, which results in lower lod scores than when manic depressive
illness is considered to be caused by a fully penetrant gene. If
the data of Mendelwicz and Fleiss (1974) were subjected to an analysis
similar to the one presented in this chapter, we would expect to find
lod scores lower than those previously reported, but probably still
positive.
There are some difficulties in the present analysis. Ideally,
the gene frequencies, susceptibilities, and age of onset distribution
parameters should be estimated simultaneously with the recombination
fraction. However, if this is done the ascertainment corrections
no longer cancel out when the lod score is formed, and computational
problems arise. Therefore, the parameters as estimated by segregation
analyses are assumed to be the "true" or population values. This is
probably not a serious limitation. The age of onset distribution
parameters are fairly stable over a wide range of models (see Chapters
III-VI), and different gene frequency estimates do not have much
effect on lod scores or estimates of A (Ott 1974).

159
There is complete information on affective illness and Xg types
on only five men in one generation of the Lava family. It would
be desirable to have the Xg types of the parents and daughters as
well. The analysis of the available data does not exclude the
possibility that there is an X-linked gene for manic depressive
illness in this family.

Table 7.1 Segregation analysis under an autosomal model.Lava family.
UnrestrictedMendelian Models TransmissionCodominant Dominant Probabilities; ~AA=~Aa
'AAA 1.0* 1.0* 0.902
'AaA 0.5* 0.5* 0.564
'aaA 0.0* 0.0* 0.000
q 0.006 0.013 0.001
Y1 0.392 0.396 0.354Y2 0.098 0.101 0.089
~AA 3.075} 3.227 } 3.216
~Aa 3.207
~aa 5.476 5.519 5.422
a 0.323 0.312 0.323K
O -16.753 -17.174 -16.813
In L -34.629 -34.637 -34.567
X2 --- 0.140
*FixedFor all Models K
1=K
2=0.
e e
EnvironmentalModel;~AA=~Aa
} 0.166
0.211
0.471--0.118
} 3.226
5.537
0.316
-16.821--
-36.245
3.356
-.....0'o

- e
Table 7.2 Segregation analysis under an X-linked model.Lava family.
e
UnrestrictedMendelian Models Transmission
Codominant Dominant Probabilities; ~AA=~Aa
TAAA 1.0* 1.0* 0.476
'AaA 0.5* 0.5* 0.607TaaA 0.0* 0.0* 0.000
q 0.0008 0.008 0.0002
Y1 0.401 0.411 0.390Y2 0.100 0.103 0.096~AA 3.620 } 3.208 } 3.205llAa 3.011
~aa 5.494 5.660 5.386
(J 0.173 0.313 0.309
KO -16.099 -17.457 -16.651
1n L -30.715 -33.041 -32.3622
1.358X ---*Fixed
For all Models, K1
=K2
=0. .....0\.....

Table 7.3 Lod scores for colorblindness and Xg.
Marker LocusRecombination Xg Xg Colorblindnessfraction (taking age of (ignorillg age of (taking age of onset
J- on into account) onset) into account)
0.0 0.497 1.204 -0.321
0.1 0.368 0.975 -0.177
0.2 0.239 0.720 -0.091
0.3 0.121 0.436 -0.038
0.4 0.033 0.149 -0.009
Table 7.4 Phenotypes of proband's brothers on whomcomplete information is available.
Brother* Xg Affective illness Colorblindness
1 + Normal not CB
2 + Normal CB
5 + Normal not CB
9 + Normal CB
10 - Depressed not CB
......0\
*Numbering is from left to right on Figure 7.1. N
- - e

CHAPTER VIII
SUMMARY AND CONCLUSIONS
The sets of data analyzed in Chapters III - VI came from several
different investigators who reached different conclusions about the
genetic basis of manic depressive illness. Stenstedt's (1952)
families come from a mostly rural area of Sweden about a generation
ago, and represent an attempt to identify and follow up all manic
depressive patients in the area over a fairly long period of time.
Most individuals were personally interviewed and diagnosed by
Stenstedt, and in spite of the relatively subjective nature of the
diagnoses, the criteria on which the diagnoses were based were quite
similar to the research criteria proposed by Feighner et. al. (1972)
for most psychiatric illnesses. The other three sets of families
(Helzer and Winokur 1974; Winokur, Clayton and Reich 1969; and Goetzl
et. al. 1974) are from this country and were collected within the last
ten years. The probands were from series of consecutive admissions
of manic patients to mental hospitals, and diagnoses for probands
and their relatives were based on the Feighner criteria. Helzer
and Winokur (1974) and Winokur, Clayton and Reich (1969) attempted
to personally interview all first-degree relatives using a structured
interview schedule, while Goetzl et. aI, (1974) sent out a
questionnaire. Information on more distant relatives was obtained
from the proband or his first-degree relatives.

164
The method of analysis used by these investigators was the
calculation of age adjusted morbidity risks to fathers, mothers,
brothers, and sisters, and the comparison of these risks to
segregation ratios expected on specific genetic hypotheses. Helzer
and Winokur (1974), and Winokur, Clayton and Reich (1969) concluded
that an X-linked dominant gene was responsible for manic depressive
illness; Stenstedt (1952) and Goetzel et. al. (1974) concluded that
an autosomal dominant gene was a more reasonable genetic model.
In spite of the differences in data collection methodology, some
characteristics of the sets of data were quite similar. The sex
ratios of affectively ill people showed a consistent trend of an
excess of women among all affectively ill, while the sex ratio among
those who had had mania was about one to one. When alcoholics were
considered affectively ill the sex ratio was also close to one to
one. The age of onset distribution for all affectively ill people
was well approximated by a lognormal distribution in all studies.
Bipolar people tended to have earlier onset than unipolars, but
there were no significant differences in age of onset between men
and women, or probands and other affectively ill. The range of onset
was about 15 to 70 years, with a mean of 32 years and standard
deviation of 14, and the distribution was significantly positively
skewed. Age of onset was thus extremely v~riable and skewed towards
younger onset.
Affective illness was definitely familial in all sets of data,
but the overall level of risk to first degree relatives varied
somewhat among studies, ranging from about 10% in Stenstedt's (1952)

165
data to about 25% in that of Winokur, Clayton and Reich (1969).
These differences are not surprising in view of the span of time
and space over which the data were collected. The studies also
varied in the amount of observed father to son transmission of
affective illness. Helzer and Winokur (1974) found one case
and Winokur, Clayton and Reich (1969) found none, while Stenstedt
(1952) and Goetzl et. ale (1974) found that father to son trans
mission was not significantly rarer than father to daughter or mother
to son transmission. The different results obtained by different
investigators have so far been difficult to explain.
In view of these discrepancies, the segregation analyses
presented in Chapters III through VI are remarkably consistent
with regard to the conclusions drawn. A single gene model does not
fit, and the autosomal and X-linked models are equally poor. This
conclusion could be drawn only after a formal test of the Mendelian
hypothesis. In each set of data, however, for both the autosomal and
X-linked genes, a Mendelian model could be found that gave reasonable
estimates of the parameters. It is not surprising that investigators
assuming a single Mendelian locus, either autosomal or X-linked,
would conclude that the segregation patterns supported this hypothesis.
It is also not surprising that both X-linked and autosomal modes of
inheritance have been proposed. For the most part, if a Mendelian
model is assumed, the data do not discriminate very well between
these two modes of inheritance. In the data of Stenstedt (1952),
Helzer and Winokur (1974) and Goetzl et. ale (1974) the likelihoods of
the X-linked and autosomal Mendelian models are very similar. The

166
data of Winokur, Clayton and Reich (1969) is an exception: the
X-linked model has a much higher likelihood than the autosomal model
in these data.
The environmental model of no vertical transmission of manic
depressive illness was also rejected. This model sometimes had a
smaller likelihood than the corresponding Mendelian model, and some
times a larger likelihood. The only data in which the environmental
hypothesis was not easily rejected was that of Stenstedt. It is
difficult to draw any general conclusions in the face of these
variations, but it seems that these data cannot exclude the possi
bility of some kind of genetic transmission of manic depressive
disease. It is also possible that there is an environmentally caused
correlation in phenotype between parent and offspring that is
sufficient to result in the rejection of the hypothesis of no vertical
"transmission," but which does not simulate a major gene in this type
of analysis.
Table 8.1 gives a summary of the maximum likelihood estimates
under the unrestricted autosomal model over all sets of data. (A
similar table under the unrestricted X-linked model would look much
the same.) This table is interesting for several reasons. First,
except for Stenstedt's (1952) data, the estimates are very similar in
all sets of data. The ascertainment parameter (KO
) is larger in the
data of Goetzl et. al. (1974), but this is to be expected since two
of the probands are sisters in these data, while in Helzer and
Winokur (1974) and Winokur, Clayton and Reich (1969) there is only
one proband in each family. Stenstedt's (1952) data resulted in quite
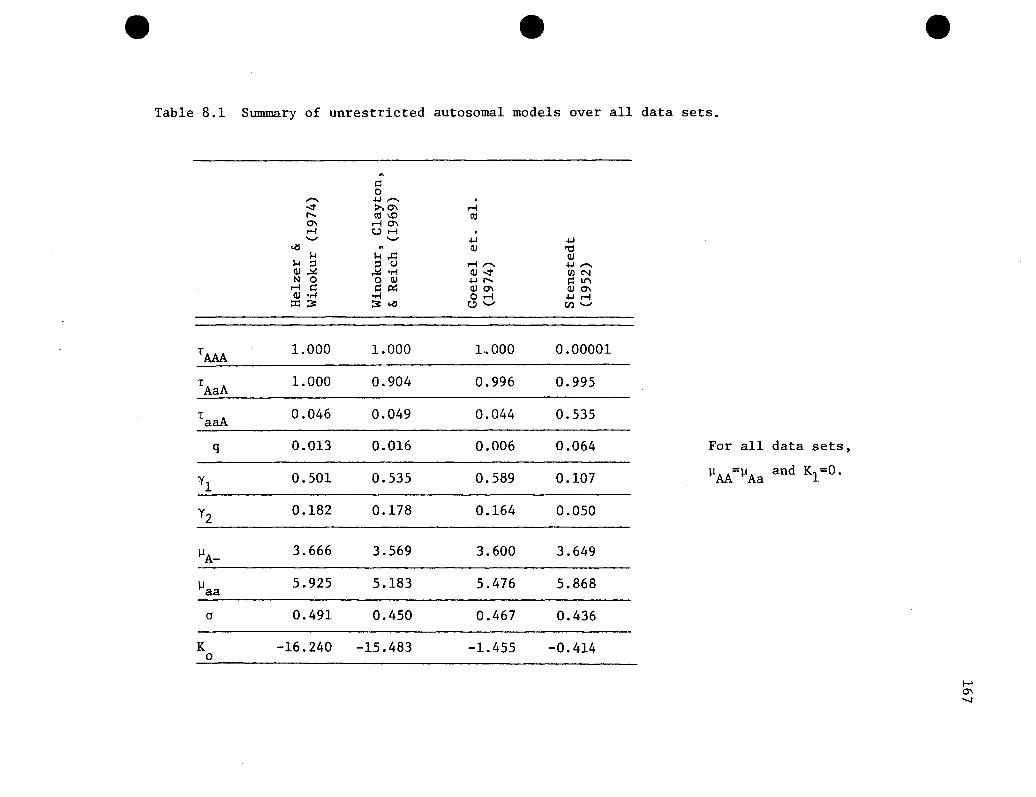
e e
Table 8.1 Summary of unrestricted autosomal models over all data sets.
e
~
~0...... +-I ...... .
..::t >-'0\ r-lt-. ell \0 ell0\ r-l0\r-l Ur-l .- - +-I +-I
~ ~ Qi 't:tI-l I-l,.c: Qi
I-l ::l ::l U r-l ...... +-I ......Qi~ ~ .,-j Qi..::t UlNN 0 o Qi +-It-. l=llt"l
r-l l=l l=lP:: QiO\ QiO\Qi .,-j .,-j Or-l +-Ir-l::C~ ~..., 0- V,l-
TAAA
1.000 1.000 LOOO 0.00001
TAaA 1.000 0.904 0.996 0.995
TaaA 0.046 0.049 0.044 0.535
q 0.013 0.016 0.006 0.064 For all data sets,
Yl0.501 0.535 0.589 0.107 ~AA=~Aa and K1=0.
Y2 0.182 0.178 0.164 0.050
~A-3.666 3.569 3.600 3.649
~aa 5.925 5.183 5.476 5.868
a 0.491 0.450 0.467 0.436
K -16.240 -15.483 -1.455 -0.4140
.....0\"-J

168
different estimates, particularly of the transmission probabilities
and susceptibilities. This is also the only set of data in which the
environmental hypothesis is not easily rejected, so the difference in
transmission probability estimates is not surprising.
The consistency of transmission probabilities in the other three
sets of data is noteworthy. In all cases these estimates are, approxi-
mately, TAM = T = 1 and T = O. In addition, the models are suchAaA aaA
that there are only two kinds of individuals: those susceptible to
affective illness and those not susceptible. Therefore, the transmis-
sion probabilities may be interpreted as follows: individuals who have
susceptible parents are susceptible, and individuals who have parents
who are not susceptible are themselves not susceptible. It appears
that there is a form of vertical transmission, though it does not con-
form to Mendelian segregation at a single locus.
Several two locus models were examined in the hope that a
slightly more complex model might prove to have a significantly better
fit. This. proved futile, perhaps not surprisingly in view of the
resounding rejection of the single locus models. None of the two gene
models resulted in a likelihood sufficiently larger than that of
the single gene models to even warrant formally testing the Mendelian
hypothesis. All of the models examined were of one autosomal and one
X-linked gene, and have been previously suggested to explain the trans-
mission of manic depressive illness. There are, of course, many
other possible two locus models. Other reasonable models might
include those with two autosomal or two X-linked genes, or models
involving more complicated types of interaction between the two loci.
However, there is absolutely nothing in any of the sets of data to

169
suggest that a different two locus model might be adequate to explain
the transmission of this disease. It is unlikely that an exhaustive
search of the possible two locus models would result in one that fit
the data well.
The results of segregation and linkage analysis of the single
large family (Chapter VII) are quite different from the results in
a group of families. In this family, both the X-linked and the
autosomal Mendelian models fit very well. However, the environmental
model also fits, although less well. An attempt was made to look
for other local maxima, and each set of initial estimates led to a
different set of maximum likelihood estimates, all with nearly equal
likelihoods. The likelihood surface must be extremely flat for such
a wide variety of parameter values to result in very similar likeli
hoods. As a result, nothing definite can be concluded from the
segregation analysis on this family.
Linkage analysis provides some support for the hypothesis that
there exists a gene for manic depressive illness that is closely
linked to that Xg locus. There is no suggestion of linkage with
colorblindness, which is compatible with the large distance between
colorblindness and Xg. The maximum lod score for xg is far too small
for linkage to be accepted, but the suggestion of a possible linkage
provides incentive to collect more data on families of this kind.
The results of Chapters III through VI indicate that an X-linked gene
is not the most common mechanism for transmission of manic depressive
illness, but is is possible that there are rare families in which
the disease is caused by an X-linked gene.

170
The method of analysis used in this dissertation alleviates
some of the difficulties encountered in analyzing the genetics of
manic depressive illness. All the information present in the
families is used, since there is no need to break the families into
groups of parents and their children, or to analyze only first degree
relatives of probands. The extremely variable age of onset is taken
into account in a manner specifically tailored for a lognormal
distribution, which is well approximated by the empiric distribution
observed among affectively ill individuals. Ascertainment of
families through a proband is allowed for, and the possibility
that the probability of a bipolar person being a proband varies
with age of onset is considered. Finally, the method allows the
rigorous testing of the hypothesis of a major gene against a general
alternative.
The increased power of this method over previously applied
methods is obvious. It was possible to reject an autosomal
dominant, an X-linked dominant, and three two-locus models as the
mode of inheritance of manic depressive illness. All of these models
had been suggested and had received some support from various
investigators, but none could be either confidently rejected or
strongly supported by previous methods of analysis.
It must be admitted that the models examined were quite simple,
and it is perhaps not surprising that they are untenable explana
tions for a disease as complex as manic depressive illness. Several
things can be done within the structure of the method to generalize
the model somewhat. It would be possible to allow sex differences

171
in the age of onset distribution parameters or the susceptibilities,
or both. Similarly, it would be possible to allow the suscepti
bilities and/or the variance of age of onset to depend on genotype
In particular, it is possible that allowing sex differences in
susceptibilities and the means of age of onset within the framework
of an autosomal model could account for the observed excess of ill
women and the segregation ratios in parents, sibs and children of
probands that look like something between an X-linked and an
autosomal gene. Further analyses of this kind are left for future
projects. However, it is unlikely (though possible) that such rela
tively minor generalizations could result in an acceptable model,
given the consistent and easy rejection of the simpler model.
An assumption underlying the genetic analysis is that there is
no non-genetic correlation in phenotype among family members. This
assumption is almost certainly violated for mental illnesses.
It is likely that there is a positive correlation among sibs, and
between parents and offspring. The method has been found to be
quite robust against sibling environmental correlation (Go et. al.
1977), at least for quantitative traits in nuclear families.
Obviously, any violation of this assumption did not lead to the
detection of a spurious major gene in these data.
Some potential difficulties with the analysis arise with regard
to the asymptotic maximum likelihood theory. If the sample size
is large enough the likelihood surface will have just one maximum
and the peak will be fairly sharp (eg., Kendall and Stuart 1973).
This is not the case with these data. The likelihood surface is

172
apparently quite flat, at least over a fairly wide region under the
Mendelian hypothesis. Several different sets of initial estimates
led to different sets of final estimates that had nearly equal
likelihoods. This effect seemed to be most pronounced in the Lava
family (see Chapter VII) and in the data of Helzer and Winokur
(1974) (Chapter III). There may well be more than one distinct
local maximum, and the standard errors of the estimates are almost
certainly large. It is also possible that twice the absolute
difference in log likelihoods between a restricted and an unre
stricted model does not have the asymptotic X2 distribution.
If the results of the analysis were borderline these
difficulties might be troublesome, but given the very large
X2 values for testing the Mendelian hypothesis and the consistency
of results over all data sets, the conclusions are not suspect
for these reasons.
Many of the problems involved in the analysis of mental illnesses
remain untouched by the methods applied in this dissertation. It
is possible that any genetic mechanism remains obscurred because
clinical diagnoses do not correspond to specific genotypes. More
work needs to be done on the definition and delineation of affective
illnesses, including investigation of biochemical differences
between well and ill people.
One of the difficulties in analyzing many families as one set
of data is that genetic heterogeneity of the disease is a possibility
and may cloud a genetic mechanism that is operating in some of the
families but not others. The two locus "heterogeneity" model was

173
an attempt at detecting any clear-cut heterogeneity in the data.
Analysis of a single large family (the Lava family) should reduce
heterogeneity to a minimum since there should be no more than one
genetic mechanism operating within a family. However. this approach
leads to other problems. such as getting reliable information on
distant relatives and having a large enough sample so that the
asympotic theory holds.
Another approach is to select families to be analyzed on
the basis of some criterion of family history. such as selection
of families with two or more affectively ill people or elimination
of families with father to son transmission. Sampling corrections
for these cases were worked out in Chapter II. but their application
awaits the development of computer programs. Approximate techniques
may be necessary since the amount of computer time and storage
required is apt to become very large.
More complex genetic models might also be examined. The mixed
model of a major gene plus polygenic and environmental variation
might prove to be more reasonable than the models investigated
here. This also must wait until appropriate computer programs are
developed.
The results presented in Chapters III through VI indicate that
the relatively simple genetic models thus far proposed for manic
depressive illness cannot account for the transmission of the
disease in the majority of families. while the linkage analysis
of Chapter VII does not discount the possibility that there is an
occasional family in which an X-linked gene causes the disease.

174
Table 8.1 suggests that there is some type of vertical transmission,
though it need not be genetic. Cultural inheritance, or even a
virus might be involved. If there is a genetic mechanism involved
in the transmission of manic depressive illness in a majority of
families with the disease, it must be much more complex than a single
major gene or the interaction of genes at two loci.

BIBLIOGRAPHY
Abrams, R. and M.A. Taylor (1974). Unipolar mania: A preliminaryreport. Arch Gen Psychiat 30: 441-443.
Allen, M.G., S. Cohen, W. Pollin, S.I. Greenspan (1974). Affectiveillness in veteran twins: A diagnostic review. Am J Psychiat131:1234-1239.
Amark, C. (1951). A study in alcoholism. Acta Psychiat et NeurolScand Supp! 70.
American Psychiatric Association (1968). Diagnostic and StatisticalManual of Mental Disorders, 2nd edition.
August, J. (1961). A clinical analysis of the effects of Tofranil indepression. Psychopharmacologica 2:381-407.
Angst, J. (1966). Zur Atiologie und Nosologie endogener DepressiverPsychosen. Springer-Verlag, Berlin. Cited in C. PerrisActa Psychiat Scand Suppl.203:45-52 (1968).
Baker, M., J. Dorzab, G. Winokur, R. Cadoret (1972). Depressiveillness: Evidence favoring polygenic inheritance based on ananalysis of ancestral cases. Arch Gen Psychiat 27:320-327.
Batschelet, E. (1962). Spurious correlation of the age of onset, withspecial reference to asthma bronchial and atopic rhinitis.Biometrische Zeitschrift 4:111-120.
Batschelet, E. (1963). Testing hypotheses and estimating parametersin human genetics if the age of onset is random. Biometrika50:265-279.
Baldwin, J.A. (1971). Five year incidence of reported psychiatricdisorder. In International Psychiatry Clinics, ed. J.A. Baldwin,Little, Brown & Co., Boston.
Book, J.A. (1953). A genetic and neuropsychiatric investigation of anorth Swedish population. Acta Genet et Statist Med 4:1-100.
Bratfos, o. and J.O. Haug (1968). The course of manic depressivepsychosis. Acta Psychiat Scan~ 44:89-111.

176
Brodie, H.K.H. and M.J. Leff (1971). Bipolar depression--Acomparative study of patient characteristics. Am J Psychiat127:1086-1090.
Brown, R.J., R.C. Elston, W.S. Pollitzer, A. Prange, I. Wilson(1973). Sex ratio in relatives of patients with affectivedisorder. Biol Psychiat 6:307-309.
Bunney, W.E., E.S. Gershon, D.L. Murphy, F.F. Goodwin (1972). Psychobiological and pharmacological studies of manic depressiveillness. J Psychiat Res 9:207-226.
Burch, P.R.J. (1964). Manic depressive psychosis: Some newetiological considerations. Brit J Psychiat 110:808-817.
Cadoret, R.J. and G. Winokur (1975). X-linkage in manic depressiveillness. Ann Rev Med 26:21-25.
Cadoret, R.J., G. Winokur, P. Clayton (1970). Family history studies:VII. Manic depressive disease vs. depressive disease. Brit JPsychiat 116:625-635.
Cadoret, R.J., G. Winokur, P. Clayton (1971). Family history studies:VI. Depressive disease types. Compr Psychiat 12:148-155.
Carter, T.C. and D.S. Falconer (1951). Stocks for detecting linkagein the mouse, and theory of their design. J Genet 50:307-323.
Clancy, J., M. Tsuang, B. Norton, G. Winokur (1974). The Iowa 500:A comprehensive study of mania, depression and schizophrenia.J Iowa Med Soc 64:394-398.
Clayton, P., F.N. Pitts, G. Winokur (1965). Affective disorder:IV. Mania. Compr Psychiat 6:313-322.
Clayton, P., L. Rodin, G. Winokur (1968). Family history studies:III. Schizoaffective disorder, clinical and genetic factorsincluding a one to two year followup. Compr Psychiat 9:31-49.
Cook, P.L., J. Noades, D.A. Hopkinson, E.B. Robson (1972). Demonstration of sex difference in recombination fraction in the looselinkage Rh and PGM1 • Ann Hum Gen 35:239-242.
Crowe, R.R. and P.E. Smouse (1976). Age dependent penetrance inmanic-depressive illness, and its implications. In press.
Defrise-Gussenhoven, E. (1962). Hypotheses de dimerie et de nonpen6trance. Acta Genet Basel 12:65-96.

177
Denniston, C. (1969). A note on the detection of linkage of apossible X-linked locus to a known X-linked locus. Appendixto K.N. McRae, I.A. Uchida, M. Lewis, Sex linked congenitaldeafness. Am J Hum Genet 21:415-422.
Dunner, D.L. and R.R. Fieve (1975). Psychiatric illness in fathersof men with primary affective illness. Arch Gen Pschiat32:1134-1137.
Edwards, J.H. (1960). The simulation of mende1ism. Acta Genet10:63-70.
Edwards, J.H. (1971). The analysis of X-linkage. Ann Hum Genet34:229-249.
E1andt-Johnson, R.C. (1973). Age at onset distribution in chronicdiseases. A life table approach to analyses of family data.J Chronic Dis 26:529-545.
Elston, R.C. (1973). Ascertainment and age of onset in pedigreeanalysis. Human Heredity 23:105-112.
Elston, R.C., K.K. Namboodiri, M.A. Spence, J.D. Rainer (1977). Agenetic study of schizophrenia pedigrees. II. One-locushypotheses. Neuropsychobio10gy, in press.
Elston, R.C. and J. Stewart (1971). A model for the genetic analysisof pedigree data. Human Heredity 21:523-542.
Elston, R.C. and K.C. Yelverton (1975). General models forsegregation analyses. Am J Hum Genet 27:31-45
Feighner, J.P., E. Robbins, S.B. Guze, R.A. Woodruff, G. Winokur,R. Munoz (1972). Diagnostic criteria for use in psychiatricresearch. Arch Gen Psychiat 26:57-63.
Fieve, R.R., J. Mende1wicz, F.L. F1eiss (1973). Manic depressiveillness: Linkage with Xg blood group. Am J Psychiat130:1355-1359.
Finney, D.J. (1940). The detection of linkage I. Ann Eugen10:171-214.
Finney, D.J. (1941). The detection of linkage II. Ann Eugen 11:10-30.
Finney, D.J. (1941). The detection of linkage III. Ann Eugen11J115-135.
Finney, D.J. (1942). The detection of linkage IV. Ann Eugen11:224-232.
Finney, D.J. (1942). The detection of linkage VI. Ann Eugen11:233-244.

178
Finney, D.J. (1943). The Detection of linkage VII. AnnEugen12:31-43.
Fisher, R.A. (1943).abnormalities.
Fisher, R.A. (1935).abnormalities.
The detection of linkage with "dominant"Ann Eugen 6:187-201.
The detection of linkage with recessiveAnn Eugen 6:339-350.
Fraser, G.R. (1968). Some complications of the use of the threegeneration method in the estimation of linkage relationshipson the X-chromosome in man. Ann Hum Genet 32:65-79.
Fraser, G.R. and O. Mayo (1968). A comparison of the two-generationand three-generation methods of estimating linkage values of theX-chromosome in man with special reference to the loci determining the Xg blood group and G6PD deficiency. Am J Hum Genet20:534-548.
Gershon, E.S., W.E. Bunney, J.F. Leckman, M. Van Eerdewegh, B.A.DeBauche, (1976). The inheritance of affective disorders: Areview of data and of hypotheses. Behaviour Genetics 6:227-261.
Gershon, E.S., D.L. Dunner, F.K. Goodwin (1971).affective disorders: Genetic contributions.25:1-15.
Toward a biology ofArch Gen Psychial
Gershon, E.S., and W.E. Bunney (1976). The question of X-linkage inmanic-depressive illness. J Psychiat Res 13:99-117.
Gershon, E.S., D.L. Dunner, L. Sturt, F.F. Goodwin (1973). Assortative mating in the affective disorders. BioI Psychiat 7:63-74.
Gershon, E.S. and S. Matthysse (1977). X-linkage: Ascertainmentthrough doubly ill probands. J Psychiat Res, in press.
Go, R.C.P., R.C. Elston, and E.B. Kaplan (1977).robustness of pedigree segregation analysis.in press.
Efficiency andAm J Hum Genet,
Goetzl, U., R. Green, P. Whybrow, R. Jackson (1974). X-linkagerevisited: A further family study of manic depressive illness.Arch Gen Psychiat 31:665-672.
Haldane, J.B.S. and C.A.B. Smith (1947). A new estimate of thelinkage between the genes for colorblindness and hemophiliain man. Ann Eugen 14:10-31.
Hartl, D.L. and T. Maruyama (1968). Phenogram enumeration: Thenumber of regular phenotype-genotype correspondences ingenetic systems. J Theoret BioI 20:129-163.

179
Harvald. B. and M. Hauge (1965). Hereditary factors elucidated bytwin studies. in Genetics and the Epidemiology of ChronicDiseases.
Helgason. T. (1964). Epidemiology of·mental disorders in Iceland.Acta Psychiat Scand Suppl. 1973.
Helzer. J.E. (1975). Bipolar affective disorder in black and whitemen. Arch Gen Psychiat 32:1140-1143.
Helzer. J. and G. Winokur (1974). Family interview study of malemanic depressives. Arch Gen Psychiat 31:73-77.
Hoffman. H. (1921). Die Nichkommenschaft be! endogen Psychosen.Berlin. cited in E. Slater. Proc Roy Soc Med 29:981-990. 1935.
Hopkinson. G. and P. Ley (1969). A genetic study of affectivedisorder. Brit J. Psychiat 115:917-922.
Jackson, C.E .• W.E. Symon, J.D. Mann (1964). X chromosome mapping ofgenes for red-green co1orb1indness and Xg. Am J Hum Genet16:403-409.
Janowsky. D.S .• J.M. Davis. M. Khaled E1-Yousef. H.J. Sekerke (1972).A cholinergic-adrenergic hypothesis of mania and depression.Lancet 2:632-635.
Kallman. F.J. (1950). The genetics of psychoses. Congres Internationale de Psychiatrie. vol. 6, Hermann & C1e • Paris.
Kallman. F.J. (1954). Genetic principles in manic depressivepsychosis. in Depression. ed. Hoch. P.H. and J. Zubin.Grune and Stratton. New York.
Kaplan. E.B. and R.C. Elston (1973).likelihood estimation (MAXLIK).Mimeo Series No. 823, UNC.
A subroutine for maximumInstitute of Statistics
Kendall. M.G. and A. Stuart (1973). The Advanced Theory of Statisticsvol. 2. Hafner Publishing Co .• New York.
Kraeplin. E. (1907). Lehrbuch der Psychiatrie. translated andabstracted from the 7th German edition by A.R. Diefendorf asClinical Psychiatry. MacMillian & Co. Ltd •• London.
Kraeplin, E. (1917). Lectures in Clinical Psychiatry. 3rd
Englished .• trans. and ed. by T. Johnstone. William Wood & Co •• NewYork.
Kringlen. E. (1967). Heredity and Environment in the FunctionalPsychoses. William Heinemann Medical Books. Ltd. London.

180
Kosambi, D.D. (1944). The estimation of map distances fromrecombination values. Ann Eugen 12:172-175.
Lange, K. and R.C. Elston (1975). Extensions to pedigree analysisI. Likelihood calculations for simple and complex pedigrees.Hum Hered. 25:95-105.
Lange, K., B.M. Page, R.C. Elston (1975). Age trends in humanchiasma frequencies and recombination fractions, I. Chiasmafrequencies. Am J Hum Genet 27:410-418.
Levitan, M. and A. Montagu (1971). Textbook of Human Genetics,Oxford University Press, New York.
Luxenburger, H. (1932). Nervenarzt 5:505, cited in A. Stenstedt,Acta Psychiat et Neuro1 Scand Suppl. 79 (1952).
MacLean, C.J., N.E. Morton and R. Lew (1975). Analysis of familyresemblence IV. Operational characteristics of segregationanalysis. Am J Hum Genet 27:365-384.
Maynard-Smith, S., L.S. Penrose, C.A.B. Smith (1961). MathematicalTables for Research Workers in Human Genetics, Churchill, London.
Mayo, O. (1970). The use of linkage in genetic counselling. HumanHeredity 20:473-485.
Mendlewicz, J., R.R. Feive, J.D. Rainer, M. Cataldo (1973). Affectivedisorder on paternal and maternal sides: Observations inbipolar patients with and without a family history. Brit JPsychiat 122:31-34.
Mend1ewicz, J., R.R. Fieve, J.D. Rainer, J.L. F1eiss (1972). Manicdepressive illness: A comparitive study of patients with andwithout a family history. Brit J Psychiat 120:523-530.
Mendlewicz, J., R.R. Fieve, F. Sta1lon (1973). Relationship betweenthe effectiveness of lithium therapy and family history. Am JPsychiat 130:1011-1013.
Mend1ewicz, J., and J.L. Fleiss (1974). Linkage studies with Xchromosome markers in bipolar (manic depressive) and unipolar(depressive) illnesses. BioI Psychiat 9:261-294.
Mendlewicz, J., J.L. F1eiss, M. Cataldo, J.D. Rainer (1975). Accuracyof the family history method in affective illness. Arch GenPsychia~ 32:309-314.
Mendlewicz, J., J.L. Fleiss, R.R. Fieve (1972). Evidence for Xlinkage in the transmission of manic depressive illness.JAmMed Assoc 222:1624-1627.

181
Mendlewicz, J. and J.D. Rainer (1974). Morbidity risk and genetictransmission in manic depressive illness. Am J Hum Genet26: 692-701.
Mitsuda, H. (1965). The concept of "atypical psychoses" from theaspect of clinical genetics. Acta Psychiat Scand41:372-377 .
Morrison, J.R. (1975). The family histories of manic depressivepatients with and without alcoholism. J Nerv Ment Dis160:227-229.
Morrison, J.R., J. Clancy, R. Crowe, G. Winokur (1972). The Iowa500: I. Diagnostic validity in mania, depression andschizophrenia. Arch Gen Psychiat 27:457-461.
Morton, N.E. (1955). Sequential tests for the detection of linkage.Am J Hum Genet 7:277-318.
Morton, N.E. (1956). The detection and estimation of linkage betweenthe genes for elliptocytosis and the Rh blood type. Am J HumGenet 8:80-96.
Morton, N.E. (1957). Further scoring types in sequential tests, witha critical review of autosomal and partial sex linkage in man.Am J Hum Genet 9:55-75.
Morton, N.E. (1969). Segregation analyses. In Computer Applicationsin Genetics, ed. N.E. Morton. University of Hawaii Press,Honolulu, Hawaii, pp. 129-137.
Morton, N.E., S. Yee, R. Lew (1971). Complex segregation analysis.Am J Hum Genetics 23:602-611.
Morton, N.E. and C.J. MacLean (1974). Analysis of family resemb1enceIII. Complex segregation of quant1tative traits. Am J HumGenet 26:489-503.
Munro, A. (1965). Depressive illness in twins. Acta Psychiat Scand41:111-116.
O'Brien, W.M., C.C. Li, F.H. Taylor (1965). Penetrance and thedistribution of sib-pair types, exemplified by taste abilityand rheumatoid arthritis. J. Chron Dis 18:675-680.
Ott, J. (1974).pedigrees.
Estimation of the recombination fraction in humanAm J Hum Genet 26:588-597.
Pare, C.M.B.', L. Rees, M.J. Sainsbury (1962). Differentiation oftwo genetically specific types of depression by the responseto antidepressants. Lancet 2:1340-1343.

182
Pare, C.M.B. and J.W. Mack (1971). Differentiation of twogenetically specific types of depression by the response to .antidepressant drugs. J Med Genet 8:306-309.
Penrose, L.S. (1935). The detection of autosomal linkage in datawhich consists of pairs of brothers and sisters of unspecifiedparentage. Ann Eugen 6:133-138.
Penrose, L.S. (1946). A further note on the sib-pair linkage method.Ann Eugen 13:25-29.
Penrose, L.S. (1953). The general purpose sib-pair linkage test.Ann Hum Genet 18:120-124.
Perris, C. (1968a). Genetic transmission of depressive psychoses.Acta Psychiat Scand Supp1. 203:45-52.
Perris, C. (1968b). The course of depressive psychoses. ActaPsychiat Scand 44:238-248.
Perris, C. (1969). The separation of bipolar from unipolar recurrentdepressive psychoses. Behavioral Neuropsychiatry 1:17-24.
Perris, C. (1971). Abnormality on paternal and maternal sides:Observations in unipolar and bipolar depressive psychoses.Brit J Psychiat 118:207-210.
Perris, C. (1974). A study of cycloid psychoses. Acta PsychiatScand Supp1. 253.
Pitts, F.N. and G. Winokur (1964). Affective disorder: III. Diagnostic correlates and incidence of suicide. J Nerv Ment Dis139:176-181.
Pitts, F.M. and G. Winokur (1966). Affective disorder: VII.Alcoholism and affective disorder. J Psychiat Res 4:37-50.
Porter, I.H., J. Schulze, V.A. McKusick (1962). Genetical linkagebetween the loci for G6PD deficiency and co1orb1indness inAmerican Negroes. Ann Hum Genet. 26:107-118.
Price, J. (1968). The genetics of depressive behavior. in RecentDevelopments in Affective Disorders, Coppen, H. and A. Walk,eds. Headley Brothers Ltd., London.
Reich, T.P. Clayton, G. Winokur (1969). Family history studies:V. The genetics of mania. Am J Psychiat 125:1358-1369.
Renwick, J.H. (1961). Elucidation of gene order. In Recent Advancesin Human Genetics, L.S. Penrose and H.L. Brown, eds. Little,Brown & Co. Boston.

183
Renwick, J.H. (1969). Progress in mapping human autosomes. Brit MedBull 25:65-73.
Renwick, J.H. (1971). The mapping of human chromosomes. Ann RevGenet 5:81-120.
Renwick, J.H. and D.R. Bolling (1971). An analysis procedure illustrated on a triple linkage of use for prenatal diagnosis ofmyotonic dystrophy. J Med Gen 8:399-406.
Renwick, J.H. and J. Schulze (1964). An analysis of some data onthe linkage between Xg and colorblindness in man. Am J HumGenet 16:410-418.
Rosanoff, A.J., L.M. Handy, l.R. Plesset (1935). The etiology ofmanic depressive syndromes with special reference to theiroccurence in twins. Am J Psychiat 91:725-762.
Rosenthal, D. (1970). Genetic Theory and Abnormal Behavior, McGrawHill Book Co., New York.
Rudin, E. (1923). Zeitschr. f. d. ges. Neural. u. Psychiat. 8:459.Cited in E. Slater Proc Roy Soc Med 29:981-990.
Slater, E. (1935). The inheritance of manic depressive insanity.Proc Roy Soc Med 29:981-990.
Slater, E. (1938). Z ges Neural Psychiat 163:1-47. Cited inT. Helgason, Acta Psychiat Scand Supp1 173 (1964).
Slater, E. (1966). Expectation of abnormality on paternal andmaternal sides: A computational model. J Med Genet 3:159-161.
Slater, E. J. Maxwell, J.S. Price (1971). Distribution of ancestralsecondary cases in bipolar affective disorders. Brit J Psychiat118:215-218.
Smith, C. (1971). Discriminating between different modes of inheritance in genetic disease. C1in Genet 2:303-314.
Smith, C.A.B. (1953). The separation of the sexes of parents in thedetection of linkage in man. Ann Hum Genet 18:278-301.
Smith, C.A.B. (1953b). The detection of linkage in human genetics.J Roy Statist Soc (B2. 15: 153-192.
Smith, C.A.B. (1959).linkage analysis.
Some comments on statistical methods used inAm J Hum Genet 11:289-304.
Smith, C.A.B. (1961). Statistical methods and theory. In RecentAdvances in Human Genetics, L.S. Penrose and H.L. Brown, eds.Little, Brown & Co., Boston.

184
Smith, C.A.B. (1963). Testing for heterogeneity of recombinationvalues in human genetics. Ann Hum Genet 27:175-182,.
Smith, C.A.B. (1968). Linkage scores in two- and three-generationfamilies. Ann Hum Genet 32:127-150.
Smith, C.A.B. (1969). Further linkage scores and corrections intwo- and three-generation families. Ann Hum Genet 33:207-223.
Spence, M.A., R.C. Elston, K.K. Namboodiri, and J.D. Rainer (1977).A genetic study of schizophrenia pedigrees. I. Demographicstudies: sample description, age of onset, ascertainment andclassification. Neuropsychobiology 2:328-340.
Steinberg, A.G. and N.E. Morton (1956). Sequential test for linkagebetween cystic fibrosis of the pancreas and the MNS locus.Am J Hum Genet 8:177-189.
Stenstedt, A. (1952). A study in manic depressive psychosis. ActaPsychiat et Neuro1 Scand Supp1. 79.
Stenstedt, A. (1959). Involutional melancholia. Acta Psychiat etNeuro1 Scand Supp1. 127.
Stromgren, E. (1965). Schizophreniform psychoses. Acta PsychiatScand 41:483-489.
Tanna, V.L. and G. Winokur (1968). A study of association and linkageof ABO blood types and primary affective disorder. Brit JPsychiat 114:1175-1181.
e-
Taylor, M. and R. Abrams (1973). Manic states:early and late onset affective disorders.28:656-8.
A genetic study ofArch Gen Psychiat
Tienari, P. (1963). Psychiatric illness in identical twins. ActaPsychiat Scand Supp1. 171.
Thompson, G. (1977). The effect of a selected locus on linkedneutral loci. Genetics 85:753-788.
Tomasson, H. (1938). Further investigations on manic depressivepsychosis. Acta Psychiat et Neuro1 Scand 13:517-523.
Tranke11, A. (1955). Aspects of genetics in psychology. Am J HumGenet 7:264-276.
Tranke11, A. (1956). Penetrance calculus in population genetics.Am J Hum Genet 8:44-48.
Vogt, R. (1910). Tiddsskrift for den norski 1aegeforening 30:417.Cited in A. Stenstedt Acta Psychiat et Neuro1 Scand Supp1 79(1952).

185
Von Grieff, H., P.R. McHugh, P.E. Stokes (1975). The familialhistory in 16 males with bipolar manic depressive illness.In Genetic Research in Psychiatry, eds. R.R. Fieve, D. Rosenthaland H. Brill. The Johns Hopkins University Press, Baltimore,pp. 219-232.
Weitkamp, L.R. (1972). Human autosomal linkage maps. In Proceedingsof the 4th International Congress of Human Genetics,J. deGrouchy, F.J.G. Ebling, and J.W. Henderson, eds. ExcerptaMedica, Amsterdam, pp. 445-460.
Wilson, S.R. (1971). Fitting model of incomplete penetrance tofamily data. Ann Hum Gen 35:99-108.
Wilson, S.R. (1974). Simulation of Mendelism by a non-genetic Markovchain model. Ann Hum Genet 38:225-229.
Winokur, G. (1970). Genetic findings and metholological considerations in manic depressive disease. Brit J Psychiat 117:267-274.
Winokur, G. (1971). The genetics of manic depressive illness. InDepression in the 70's R.R. Fieve, ed. Excerpta Medica, Amsterdam.pp. 47-53.
Winokur, G. (1975). The Iowa 500:depressive illness (bipolar).
Heterogeneity and course in manicCompr Psychiat 16:125-131.
Winokur. G., R. Cadoret, J. DorzaL, M~ Baker (1971). Depressivedisease: A genetic study. Arch Gen Psychiat 24:135-144.
Winokur, G. and P. Clayton (1967a). Family history studies: II. Sexdifferences and alcoholism in primary affective illness.Brit J. Psychiat 113:973-979.
Winokur, G. and P. Clayton (1967b).types of affective disorders.Psychiatry, vol. 9. J. Wortis,
Family history studies: I. TwoIn Recent Advances in Biologicaled. Plenum Press, New York.
Winokur, G., P. Clayton, T. Reich (1969). Manic Depressive Illness.The C.V. Mosley Co., St. Louis
Winokur, G., J. Morrison, J. Clancy, R. Crowe (1972).II. A blind family history comparison of mania,schizophrenia. Arch Gen Psychiat 27:462-464.
The Iowa 500:depression and
Winokur, G., and F.N. Pitts (1964).reactive depression an entity?
Affective disorder: I. IsJ Nerv Ment Dis 138:541-547.
Winokur, G., and F.N. Pitts (1965). Affective disorder: II. Afamily study of prevalences, sex differences and possiblegenetic factors. J Psychiat Res 3:113-123.

186
Winokur, G., and T. Reich (1970). Two genetic factors in manicdepressive disease. Compr Psychiat 11:93-99.
Winokur, G., J. Rimmer, T. Reich (1971). Alcoholism: IV. Is theremore than one type of alcoholism? Brit J Psychiat 118:125-131.
Winokur, G., and S. Ruangtrakoo1 (1966). Genetic vs. nongeneticaffective disorder. Am J Psychiat 123:703-705
Winokur, G., and V.L. Tanna (1969). Possible role of X-linkeddominant factor in manic depressive disease. Dis Nerv Sys30:89-94.
Woodruff, R., F.N. Pitts, G. Winokur (1964). Affective disorder:II. A comparison of patients with endogenous depressions withand without a family history of affective disorder. J NervMent Dis 139:49-52.
Yelverton, K.C., K.K. Naboodiri, R.C. Elston (1975). GeneralSegregation Analysis Package Writeup (GENSEG), unpublished.



















