
Thalassaemia-like carriers not linked to the beta-globin gene cluster
11
Thalassaemia-like carriers not linked to the b-globin gene cluster The b-thalassaemias are a very heterogeneous group of autosomal recessive disorders characterised by reduced (b + ) or absent (bŶ) production of b-globin chains (Weatherall et al, 2001). To date, approximately 200 different molecular defects affecting the b-globin gene and leading to the b-thalassaemia phenotype have been reported (Huisman et al, 1997). Homo- zygosity or compound heterozygosity for b-thalassaemia usually result in the severe transfusion-dependent thalassaemia major phenotype or, more rarely, in the milder non-transfu- sion dependent variety referred to as thalassaemia intermedia. These milder forms are produced from homozygosity or compound heterozygosity for mild or silent b-thalassaemia mutations or from the coinheritance of homozygosity for a typical severe b-thalassaemia mutation with some ameliorating genetic determinants, such as a-thalassaemia or hereditary persistence of fetal Hb. Even more rarely, thalassaemia intermedia is caused by double heterozygosity for typical b-thalassaemia and the triple a globin gene rearrangements or heterozygosity for hyperunstable Hb molecules. The large majority of the b-thalassaemia heterozygotes show the typical phenotype of the b-thalassaemia carrier state, namely low mean corpuscular volume (MCV) and mean corpuscular haemoglobin (MCH), high HbA2 levels, and unbalanced globin chain synthesis. A minor group, dubbed silent b- thalassaemia, display normal red blood cell indices and normal/border-line HbA2 levels, and are defined solely by unbalanced globin chain synthesis (Gonzalez-Redondo et al, 1989; Ristaldi et al, 1990; Rosatelli et al, 1995; Moi et al, 2004). A few subjects of Italian, English, and Portuguese origin who present a b-thalassaemia-like carrier phenotype (b-thalas- saemia like determinant) with completely normal b-globin gene sequences have been reported (Murru et al, 1992; Thein et al, 1993; Pacheco et al, 1995). Furthermore, the co-inher- itance of such a b-thalassaemia-like determinant with typical b-thalassaemia has been postulated to explain the thalassaemia intermedia phenotype manifested by otherwise typical hetero- Valeria Faa `, 1 Alessandra Meloni, 1 Loredana Moi, 2 Giuseppe Ibba, 2 Maurizio Travi, 3 Antonio Vitucci, 4 Antonio Cao 1 and Maria Cristina Rosatelli 2 1 Istituto di Neurogenetica e Neurofarmacologia, CNR, Cagliari, 2 Dipartimento di Scienze Biomediche e Biotecnologie, Universita ` degli Studi, Cagliari, 3 IRCCS Ospedale Maggiore Policlinico Mangiagalli e Regina Elena, Milano, and 4 Divisione di Ematologia II, Universita ` di Bari, Bari, Italy Received 29 August 2005; accepted for publication 14 November 2005 Correspondence: Maria Cristina Rosatelli, Dipartimento di Scienze Biomediche e Biotecnologie, Universita ` di Cagliari, via Jenner s/n, 09121, Cagliari, Italy. E-mail: [email protected] Summary This study describes the largest series reported to date, of individuals belonging to unrelated families carrying a b-thalassaemia-like phenotype in whom the b-globin gene was found to be structurally intact by sequence analysis. This genetic determinant appears haematologically heterogeneous, displaying either a silent b-thalassaemia-like phenotype or a typical b-thalassaemia carrier-like phenotype in different families. Compound heterozygosity for both b-thalassaemia-like determinant and typical b-thalassaemia allele resulted either in thalassaemia intermedia or thalassaemia major. By linkage analysis both the silent and the typical b-like determinants were found not to be linked to the b-globin cluster. Sequence analysis of the hypersensitive site cores of locus control region and of the genes coding for the transcription factors erythroid Kruppel-like factor and nuclear factor (erythroid-derived 2) were normal. b-globin mRNA levels determined by real-time polymerase chain reaction were reduced in both types of b-like carriers. These results indicate the existence of causative genetic determinants not yet molecularly defined, but most likely, resulting from either the reduction or loss of function of a gene coding for unknown transcriptional regulator(s) of the b-globin gene. The knowledge of these rare b-thalassaemia-like determinants have implications for clinical and, especially, prenatal diagnosis of b-thalassaemia. Keywords: b-thalassaemia, gene expression, globin genes, quantitative polymerase chain reaction, transcription factors. research paper doi:10.1111/j.1365-2141.2005.05915.x ª 2006 Blackwell Publishing Ltd, British Journal of Haematology, 132, 640–650
-
Upload
independent -
Category
Documents
-
view
3 -
download
0
Transcript of Thalassaemia-like carriers not linked to the beta-globin gene cluster

Thalassaemia-like carriers not linked to the b-globin gene cluster
The b-thalassaemias are a very heterogeneous group of
autosomal recessive disorders characterised by reduced (b+)or absent (b�) production of b-globin chains (Weatherall et al,
2001). To date, approximately 200 different molecular defects
affecting the b-globin gene and leading to the b-thalassaemia
phenotype have been reported (Huisman et al, 1997). Homo-
zygosity or compound heterozygosity for b-thalassaemia
usually result in the severe transfusion-dependent thalassaemia
major phenotype or, more rarely, in the milder non-transfu-
sion dependent variety referred to as thalassaemia intermedia.
These milder forms are produced from homozygosity or
compound heterozygosity for mild or silent b-thalassaemia
mutations or from the coinheritance of homozygosity for a
typical severe b-thalassaemia mutation with some ameliorating
genetic determinants, such as a-thalassaemia or hereditary
persistence of fetal Hb. Even more rarely, thalassaemia
intermedia is caused by double heterozygosity for typical
b-thalassaemia and the triple a globin gene rearrangements or
heterozygosity for hyperunstable Hb molecules. The large
majority of the b-thalassaemia heterozygotes show the typical
phenotype of the b-thalassaemia carrier state, namely low
mean corpuscular volume (MCV) and mean corpuscular
haemoglobin (MCH), high HbA2 levels, and unbalanced
globin chain synthesis. A minor group, dubbed silent b-thalassaemia, display normal red blood cell indices and
normal/border-line HbA2 levels, and are defined solely by
unbalanced globin chain synthesis (Gonzalez-Redondo et al,
1989; Ristaldi et al, 1990; Rosatelli et al, 1995; Moi et al, 2004).
A few subjects of Italian, English, and Portuguese origin who
present a b-thalassaemia-like carrier phenotype (b-thalas-saemia like determinant) with completely normal b-globingene sequences have been reported (Murru et al, 1992; Thein
et al, 1993; Pacheco et al, 1995). Furthermore, the co-inher-
itance of such a b-thalassaemia-like determinant with typical
b-thalassaemia has been postulated to explain the thalassaemia
intermedia phenotype manifested by otherwise typical hetero-
Valeria Faa,1 Alessandra Meloni,1
Loredana Moi,2 Giuseppe Ibba,2
Maurizio Travi,3 Antonio Vitucci,4
Antonio Cao1 and Maria Cristina
Rosatelli2
1Istituto di Neurogenetica e Neurofarmacologia,
CNR, Cagliari, 2Dipartimento di Scienze
Biomediche e Biotecnologie, Universita degli Studi,
Cagliari, 3IRCCS Ospedale Maggiore Policlinico
Mangiagalli e Regina Elena, Milano, and4Divisione di Ematologia II, Universita di Bari,
Bari, Italy
Received 29 August 2005; accepted for
publication 14 November 2005
Correspondence: Maria Cristina Rosatelli,
Dipartimento di Scienze Biomediche e
Biotecnologie, Universita di Cagliari, via
Jenner s/n, 09121, Cagliari, Italy.
E-mail: [email protected]
Summary
This study describes the largest series reported to date, of individuals
belonging to unrelated families carrying a b-thalassaemia-like phenotype in
whom the b-globin gene was found to be structurally intact by sequence
analysis. This genetic determinant appears haematologically heterogeneous,
displaying either a silent b-thalassaemia-like phenotype or a typical
b-thalassaemia carrier-like phenotype in different families. Compound
heterozygosity for both b-thalassaemia-like determinant and typical
b-thalassaemia allele resulted either in thalassaemia intermedia or
thalassaemia major. By linkage analysis both the silent and the typical
b-like determinants were found not to be linked to the b-globin cluster.
Sequence analysis of the hypersensitive site cores of locus control region and
of the genes coding for the transcription factors erythroid Kruppel-like factor
and nuclear factor (erythroid-derived 2) were normal. b-globin mRNA levels
determined by real-time polymerase chain reaction were reduced in both
types of b-like carriers. These results indicate the existence of causative
genetic determinants not yet molecularly defined, but most likely, resulting
from either the reduction or loss of function of a gene coding for unknown
transcriptional regulator(s) of the b-globin gene. The knowledge of these
rare b-thalassaemia-like determinants have implications for clinical and,
especially, prenatal diagnosis of b-thalassaemia.
Keywords: b-thalassaemia, gene expression, globin genes, quantitative
polymerase chain reaction, transcription factors.
research paper
doi:10.1111/j.1365-2141.2005.05915.x ª 2006 Blackwell Publishing Ltd, British Journal of Haematology, 132, 640–650

zygotes for b-thalassaemia (Rund et al, 1997; Gasperini et al,
1998; Ho et al, 1998a,b). In these cases, however, the severe
phenotype may be related to the presence of a somatic mosaic
of the stem cell pool for an acquired deletion involving the b-globin gene cluster and making a clone of erythroid precursor
de facto hemizygous for the b-thalassaemia mutation, as
recently reported in three families (Badens et al, 2002;
Galanello et al, 2004). The postulated unknown b-thalas-saemia-like determinant(s) may lie either within the locus
control region (LCR) or in the 3¢ b-globin enhancer (Grosveld,
1999; Li et al, 1999). However, in the best characterised cases
reported so far, the b-like determinants appeared unlinked to
the b-globin gene cluster. Therefore, this thalassaemia-like
phenotype most likely depends on the defective function of a
trans-acting erythroid-specific transcription factor, of which
possible candidates are erythroid Kruppel-like factor (EKLF)
(Miller & Bieker, 1993), nuclear factor (erythroid-derived 2)
(NF-E2 (Andrews et al, 1993), or GATA-1 (Weiss & Orkin,
1995). The first molecularly characterised condition so far
described that shows a thalassaemia phenotype not caused by
the defective function of any gene of the a or b-globin gene
cluster is the ATR-X syndrome [Online Mendelian Inheritance
in Man (OMIM) number 301040], which is characterised by
syndromic mental retardation and the a-thalassaemia pheno-
type resulting from a molecular defect in an X-linked gene
coding for a trans-acting chromatin-remodelling protein called
ATR-X (Gibbons et al, 1995). Quite recently a b-thalassaemia-
like phenotype was found to result from a defect of either a
general transcription factor, i.e. transcription factor IIH
(TFIIH), or of an erythroid-specific factor, GATA-1 with
mutations lying in its N-finger. However, in both cases the
b-thalassaemia phenotype is associated with other features,
namely trichothiodystrophy in the mutation of the TFIIH gene
and macrothrombocytopenia in the GATA-1 mutations
(Nichols et al, 2000).
In the past few years, while carrying out the molecular
characterisation of over 8000 subjects with the phenotype of
high HbA2 b-thalassaemia or silent b-thalassaemia, we detec-
ted several b-thalassaemia-like carriers, in whom the b-globingene was found to be structurally intact by sequencing analysis.
Herein, we report studies that include linkage analysis to the
b-globin gene cluster, sequence analysis of hypersensitive site
(HS) cores of LCR, NF-E2, and EKLF, and quantitative
b-globin mRNA analysis on these subjects with the aim of
characterising molecularly these atypical b-thalassaemia-like
phenotypes.
Patients and methods
Patients
This study includes 11 unrelated families of Italian origin with
several members displaying b-thalassaemia-like haematological
features. The pedigrees of these families are shown in
Figures 1–3.
The propositi in families A (II-1) and B (III-1 and III-2),
were affected by transfusion-dependent thalassaemia major,
while in families C (II-9) and K (II-1 and II-2) they presented
the phenotype of thalassaemia intermedia. The remaining
families included only subjects with the heterozygous
b-thalassaemia-like condition. Prenatal diagnoses by fetal
globin chain synthesis was carried out in families F, H, and I.
Methods
DNA analysis DNA extraction was performed according to
standard procedures (Miller et al, 1998). Informed consent
was obtained in all cases before the collection of blood samples.
Screening for common b-thalassaemia mutations in the
Mediterranean population was carried out on amplified DNA
by reverse dot blot hybridisation (Saiki et al, 1989). The
presence of unknown mutations was investigated by denatur-
ing gradient gel electrophoresis (DGGE) (Myers et al, 1985;
Cai & Kan, 1990; Rosatelli et al, 1992). Because these
techniques may fail to detect the molecular defects causing
b-thalassaemia, the b-globin gene was completely sequenced
from position )158 5¢ to the Cap site to position +60 3¢ fromthe polyadenilation signal, according to the dideoxy-chain
termination method of Sanger et al (1977) using the Big Dye
Terminator cycle sequencing kit (Applera, Norwalk, CT, USA).
Sequencing reactions were run on ABI PRISM 3100 (Applera).
b-Globin haplotypes were determined by studying 10
polymorphic sites along the b-globin cluster by restriction
enzyme digestion of amplified DNA fragments. The polymor-
phic loci analysed were HincII e, XmnI 5¢ Gc, HindIII Gc,HindIII Ac, HincII 5¢wb, HincII 3¢wb, HinfI 5¢b, RsaI 5¢b,HinfI 3¢b, and HpaI 3¢b(Varawalla et al, 1992). Five more
polymorphic sites (C fi T at codon 2 of exon 1, C fi G at
nt 16, G fi T at nt 74, C fi T at nt 81, and T fi C at nt
666 of the second introne) were detected within the b-globingene by DGGE or sequencing analysis. In order to detect large
deletions of the b-globin cluster, Southern Blot analysis was
performed according to standard procedures (Southern, 1975).
Genomic DNA was digested with the following restriction
endonucleases: HindIII, EcoRI, BamHI, HpaI, and BglII and
hybridised with d, wb, Ac, Gc, and e probes, as well as the
minilocus SK-l LAR probe containing HS 1, 2, 3, 4 and the Gcpromoter (Antonarakis et al, 1982; Orkin et al, 1982).
The HS-1, HS-2, HS-3, and HS-4 core regions of the locus
control region, as well as the EKLF and the NF-E2 genes were
analysed by direct sequencing. The sequencing primers are
listed in Table I.
Determination of a-globin gene number was assessed as
described previously (Gossens et al, 1980). Presence of the
aaaanti)4.2 allele was excluded by Southern blot analysis. The
aaaanti-3.7 allele was excluded by polymerase chain reaction
(PCR) as previously described (Dode’ et al, 1992). Two non-
deletional forms of a-thalassaemia were screened for using
NcoI digestion of PCR products from the a2-and a1-globingenes (Pirastu et al, 1984; Moi et al, 1987).
b-Thalassaemia not Linked to the b-Globin Gene
ª 2006 Blackwell Publishing Ltd, British Journal of Haematology, 132, 640–650 641
RNA analysis Erythrocytes and reticulocytes were separated
from 10 ml of peripheral blood using Sigmacell Type 50 (Sigma,
St Louis, MO, USA) and alpha cellulose (Sigma) resins (Beutler
& Gelbart, 1986). Reticulocyte enrichment was performed using
Percoll and phosphate-buffered saline gradients. Cytoplasmatic
RNA was extracted from reticulocytes using TRIzol LS (Life
Technologies, Carlsbad, CA, USA) (Chomczynski & Sacchi,
1987). cDNA synthesis was performed with the High Capacity
cDNA Archive kit (Applera) used according to the
manufacturer’s instructions. As we found a strongly reduced
expression of several commonly used housekeeping transcripts
(b-actin, 18SRNA, b2 microglobulin, and GHPDH) in
reticulocytes and a-globin genes in our patients were
structurally intact, we used the cDNA of alpha globin genes as
an internal control. Primers and probes were designed to span
exon junctions in the fully processed message in order to
prevent reporting of amplification of any possible
contaminating genomic DNA. The design of primers and
probes was performed by the Primer Express v.2 (Applera)
software. The following primers and probes were used: b-globinforward primer, 5¢-CACCTTTGCCACACTGAGT-GA-3¢;
b-globin reverse primer, 5¢-GTGATGGGCCAGCACACA-3¢;b-globin TaqMan probe, 5¢-FAM- ATCCTGAGAACTTCAGG-
CTCCTGGGC-TAMRA-3¢; a-globin forward primer, 5¢-GC-ACGCTGGCGAGTATGG-3¢; a-globin reverse primer, 5¢-TCGAAGTGCGGGAAGTAGGT-3¢; a-globin TaqMan probe,
5¢-VIC- AGGCCCTGGAGAGGATGTTCCTGTC-TAMRA-3¢.cDNA was amplified both for the target gene (beta) and for
the control gene (alpha). For each gene we calculated the cycle
threshold (Ct). Quantitative real time-PCR assay was
performed in an ABI Prism 7000 Sequence Detection Systems
(Applied Biosystems, Foster City, CA, USA). Relative
quantification of beta transcripts was calculated by a
comparative method 2^-DDCt (Livak & Schmittgen, 2001).
Results
This study reports the largest series to date, of subjects from 11
unrelated families of Italian origin carrying ab-thalassaemia-like
determinant not linked to the b-cluster. The clinical and
haematological characteristics show marked heterogeneity,
which led to the identification of at least two different
maF Aβ 1SVI 1- 10
tneliS β ht- a essal im a t tiar
maF B
maF C
VCM 58bH ·21 5
2AbH ·3 0α/β ·2 60
FbH 1<
M VC 77bH ·9 6
Hb 2A ·4 3α/β+γ ·3 69
FbH ·8 9
VCM 77bH ·21 9
2AbH ·2 6α/β ·4 1
FbH 1<
VCM 87 77bH ·41 8 ·21 2
2AbH ·2 6 2·2α/β ·1 68 ·2 8HbF 1< 1<
hT la ma oj r
β 93
lahT ma roj
M VC 38 ·68 5bH ·8 1 ·01 3
Hb 2A ·2 7 2·9
21
1
I
II
III
1 2
1 2 3
1 2 T lah m roja
I
II
VCM 87 08
bH 61 ·61 2
2AbH ·2 66 ·2 92
α/β ·1 71 ·1 64
FbH 1< 1<
VCM 75 78bH 13·5 12·4
2AbH 2·29 2·91α/β 2·2 1·74HbF 1< 1<
VCM 67bH ·61 2
2AbH ·2 96α/β ·2 1
FbH 1<
β 93
lahT mretni de ai
lahT mretni de ai
I
II
1 2 3
4 5 6
1 2 3
7 8 9
paH l epytoB/A D/C
D/A D/B D/E
D/B D/D
paH l epyto/F G H/I
I/F I/F
wonK n β itatumeneg- no
VCM 62 71 73bH 11·8 13·1 7·7
2AbH 4·46 2·52 2·8α/β 2·54 1·99 α/β+γ ·3 90
FbH 1< 01
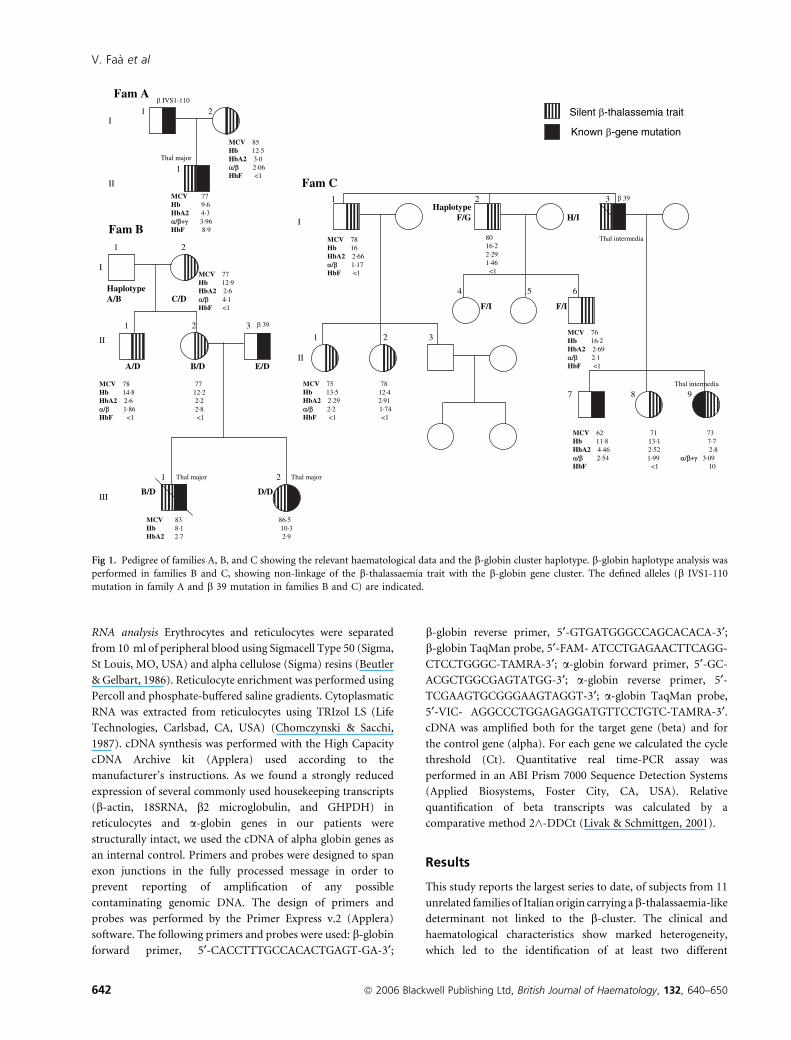
Fig 1. Pedigree of families A, B, and C showing the relevant haematological data and the b-globin cluster haplotype. b-globin haplotype analysis was
performed in families B and C, showing non-linkage of the b-thalassaemia trait with the b-globin gene cluster. The defined alleles (b IVS1-110
mutation in family A and b 39 mutation in families B and C) are indicated.
V. Faa et al
642 ª 2006 Blackwell Publishing Ltd, British Journal of Haematology, 132, 640–650

phenotypes of this b-like determinant, namely a silent b-thalas-saemia-like phenotype with low/normal MCV, normal HbA2
levels and markedly unbalanced globin-chain synthesis (type 1)
(families A, B and C) and a typical b-thalassaemia-like pheno-
type with low MCV–MCH, high HbA2 levels and unbalanced
globin chain synthesis (type 2) (families D, E, F, G, H, I, J and K).
In all members of these families, the silent as well the
typical b-like determinant showed a completely normal b-globin gene sequence from position )158 to the CAP site to
position +60 3¢ to the polyadenilation signal. Furthermore
the compound heterozygous state for the silent determinant
and a typical b-thalassaemia allele resulted either in the
phenotype of thalassaemia major (families A and B) or
intermedia (family C). On the contrary, compound hetero-
zygosity for the typical b-thalassaemia-like determinant and
typical b-thalassaemia allele produced either a fetus affected
by severe b-thalassaemia according to the very low b/c globin
chain synthesis ratio (family I) or patients with thalassaemia
intermedia (family K).
Type 1 silent b-thalassaemia-like determinant
The type 1 b-thalassaemia-like determinant was detected in
three families (A, B and C).
Family A In this family, the proband II-1 was affected by
transfusion-dependent thalassaemia major in spite of having a
b-globin gene affected by a typical b-thalassaemia mutation
(b+ IVS I-110) inherited from his father and a type 1 silent
b-thalassaemia-like determinant transmitted from his mother
(Fig 1). His mother (I-2) showed a silent b-thalassaemia-like
phenotype with MCV values of 85 fl, a HbA2 level of 3% and
an a/b ratio of 2Æ06.
Family B Three individuals (I-2, II-1 and II-2) were carriers of
the type 1 silent b-thalassaemia-like determinant (Fig 1). They
had MCV values of 77–78 fl and an a/b ratio of 4Æ1 in I-2, 1Æ86in II-1 and 2Æ8 in II-2. The husband of II-2 is heterozygous for
the b�39 mutation. Their two children III-1 and III-2, one of
maF D lacipyT β laht- ssa e im a t tiarmaF E
VCM 39bH ·01 9
2AbH ·5 5HbF 1<
VCM 67bH ·41 3
2AbH ·4 8α/β ·1 2HbF 1<
VCM 97 87bH ·31 2 ·31 8
2AbH ·5 3 5·4α/β ·1 2
FbH 1< 1<
I
II
III
1 2
1 2
1 2
VCM ·55 7bH ·11 5
2AbH ·6 1α/β ·3 1
VCM ·17 4bH 31
2AbH ·5 5α/β ·1 34
FbH 1<
1 2
1 2
1
β 93
VCM ·87 9bH ·31 4
2AbH ·5 7FbH 1<
I
II
III
maF F
VCM 64 5 1bH 10·6 1 0·8
2AbH 4·4 4 ·5
VCM ·97 6bH ·51 5
2AbH ·5 2α/β ·1 32
FbH 1<
I
II
1 2
1 2
β 93
maF G
VCM 82 7 9bH 11·9 1 3·2
2AbH 5·3 4 ·2α/β ·0 29 ·1 30
FbH 1< 1<
VCM 79bH 41
2AbH 8FbH 1<
I
II
1 2
1 2
paH l epytoM/L N /O
/L O /C C
C/L O /C
wonK n β itatum eneg- no
Fig 2. Pedigree of families D, E, F, and G showing the relevant haematological data and the b-globin cluster haplotype. b-globin haplotype analysis
was performed in family D, showing non-linkage of the b-thalassaemia trait with the b-globin gene cluster. The defined alleles (b 39 mutation in
families E and F) are indicated.
b-Thalassaemia not Linked to the b-Globin Gene
ª 2006 Blackwell Publishing Ltd, British Journal of Haematology, 132, 640–650 643

which died last year, were affected by transfusion-dependent
thalassaemia major. Both inherited the b�39 allele from their
father and the type 1 silent b-thalassaemia-like determinant
from their mother. Unexpectedly, this genetic combination led
to the production of a thalassaemia major phenotype.
Family C Six individuals from this family, i.e. I-1, I-2, II-1, II-2,
II-6 and II-8 were carriers of the type 1 silent b-thalassaemia-
like determinant (Fig 1). Their MCV varied from 71 to 80 fl,
and the a/b ratio from 1Æ17 to 2Æ2. HbA2 levels were within the
normal range. One second generation individual (II-9) had the
phenotype of thalassaemia intermedia (moderate spleen and
liver enlargement, Hb 7Æ7 g/dl; HbF 10%, a/b+c ratio 3.09).
She carried the codon 39 non-sense mutation, which was
probably inherited from her father (I-3), who was dead at the
time of examination and was reported to have been affected by
thalassaemia intermedia. The in trans b-globin gene was normal
and the triple a-globin gene arrangement was not detected. Her
brother (II-7) inherited the b�39 allele and showed the typical
b-thalassaemia carrier state phenotype. Her mother had low
MCV (69 fl), normal HbA2, and a balanced a/b ratio. Based on
these findings we postulate that, besides the b�39 allele, II-9
inherited the type 1 silent b-thalassaemia-like determinant
from her father. This genetic combination may be the reason
for the thalassaemia intermedia phenotype.
Type 2 typical b-thalassaemia-like carriers
The type 2 typical b-thalassaemia-like determinant was
detected in eight families (D, E, F, G, H, I, J and K).
Family D Four members of this family (I-2, II-1, III-1 and III-
2) in three generations carried the type 2 typical
b-thalassaemia-like determinant (Fig 2). The determinant
segregating in this family was characterised by a HbA2 level
of between 4Æ8% and 5Æ5%, borderline MCV-MCH, and
slightly unbalanced b-globin chain synthesis.
Family E Two individuals of this family, I-1 and II-1, were
carriers of type 2 typical b-thalassaemia-like determinant with
MCV values of 78Æ9 and 71Æ4 fl, and HbA2 levels of 5Æ7 and
5Æ5%, respectively (Fig 2). II-1 married a carrier of the b�39
maF H
VCM ·97 3bH ·31 2
2AbH ·4 8α/β ·1 23
FbH 1<
VCM 77 ·57 3bH ·51 3 ·31 1
2AbH ·4 9 4·7α/β ·1 54 ·1 43
FbH 1< 1<
1 2lacipyT β laht- ssa e im a
tiartmaF II
II
III
1 2 3 4 5
paH l /P epyto B Q /S
/B Q /P S /P Q /B S /B Q
VCM 67bH ·41 6
2AbH ·3 7FbH 1<
VCM 27bH ·41 3
2AbH ·3 9α/β ·1 97
FbH 1<
I
II
III
1 2
1 2
1
β/γ ·0 010
β 1SVI 1- 10
maF J
VCM 07bH 41
2AbH ·6 8FbH 1<
I
II
21
1
1 2
1 2 3
VCM 86bH 51
2AbH 6α/β ·2 30
FbH 1<
maF K
VCM ·77 5bH ·11 2
2AbH ·4 3
VCM 57·6 5 5·6 6 2bH 9·8 9 ·6 11·5
2AbH 8·4 7 4· 4 ·3
I
II
paH l epyto R/S /U T
β 1SVI 1-
lahT mretni de ai
R/U /T R /T R
β 93
wonK n β eneg-itatum no
VCM ·65 8bH ·9 1
2AbH ·4 6
Fig 3. Pedigree of families H, I, J, and K showing the relevant haematological data and the b-globin cluster haplotype. b-globin haplotype analysis was
performed in families H and K, showing non-linkage of the b -thalassaemia trait with the b-globin gene cluster. The defined alleles (b 39 mutation in
family H, b IVS1-110 mutation in family I, and b IVS1-1 mutation in family K) are indicated.
V. Faa et al
644 ª 2006 Blackwell Publishing Ltd, British Journal of Haematology, 132, 640–650
mutation. Their daughter (III-1), showed an MCV value of
55Æ4 fl, HbA2 level of 5Æ8%, and an a/b ratio of 3Æ1 at the age of22 months. She inherited the b�39 nonsense mutation from
her mother, and at last follow-up, at 9 years of age, her Hb was
11Æ5 g/dl, MCV 55Æ7 fl, and HbA2 level 6Æ1%.
Family F In this family subject I-1 was a carrier of the type 2
typical b-thalassaemia-like determinant with MCV values of
79Æ6 fl, HbA2 levels of 5Æ5%, and a/b ratio of 1Æ23 (Fig 2). He
married a carrier of the b�39 mutation (I-2) who showed the
typical features of the b-thalassaemia trait. The couple had two
pregnancies in which prenatal diagnosis was carried out by
both fetal DNA analysis and globin chain synthesis. In both
fetuses, DNA analysis showed heterozygosity for the b�39nonsense mutation. Fetal globin chain synthesis analysis
showed b/c ratios of 0Æ040 and 0Æ038. The fetuses were
considered heterozygotes for the b�39 nonsense mutation. The
haematological evaluation of the two children shown in
the figure was performed at 1 year of age and confirmed the
prenatal test.
Family G The three members of this family (I-1, II-1 and II-2)
displayed the type 2 typical b-thalassaemia-like determinant
(Fig 2). The determinant segregating in this family is
characterised by a HbA2 level of between 4Æ2 and 8%,
borderline MCV–MCH, and a normal b-globin chain
synthesis.
Family H Three individuals from this family, i.e. I-2, II-1 and
II-3 were carriers of the type 2 typical b-thalassaemia-like
determinant (Fig 3). Their MCV varied from 75 to 79 fl, their
HbA2 levels were between 4Æ7% and 4Æ9%, and their a/b ratio
from 1Æ32 to 1Æ54. Individual II-1 married a carrier of the b�39mutation showing the typical features of the b-thalassaemia
trait. The couple had two pregnancies in which prenatal
diagnoses was carried out by both fetal DNA analysis and
globin chain synthesis analysis. In both fetuses DNA analysis
showed heterozygosity for the b�39 nonsense mutation. Fetal
globin chain synthesis analysis showed b/c ratios of 0Æ035 and
0Æ040. The fetuses were considered heterozygotes for the b�39
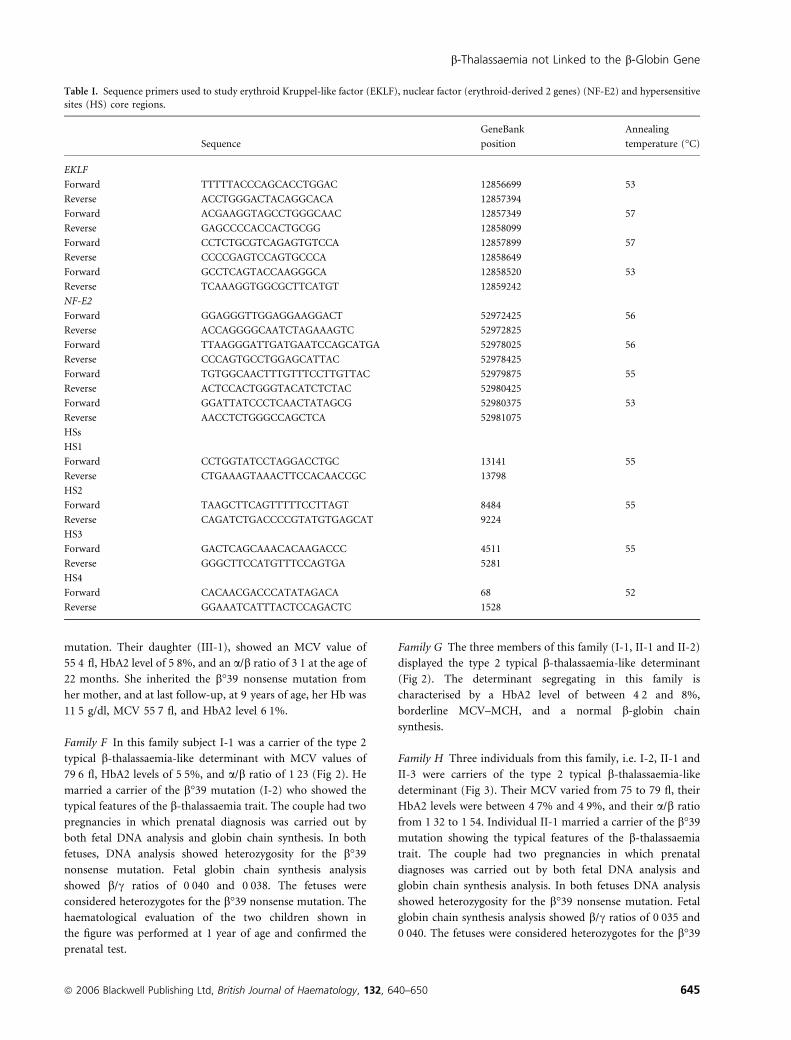
Table I. Sequence primers used to study erythroid Kruppel-like factor (EKLF), nuclear factor (erythroid-derived 2 genes) (NF-E2) and hypersensitive
sites (HS) core regions.
Sequence
GeneBank
position
Annealing
temperature (�C)
EKLF
Forward TTTTTACCCAGCACCTGGAC 12856699 53
Reverse ACCTGGGACTACAGGCACA 12857394
Forward ACGAAGGTAGCCTGGGCAAC 12857349 57
Reverse GAGCCCCACCACTGCGG 12858099
Forward CCTCTGCGTCAGAGTGTCCA 12857899 57
Reverse CCCCGAGTCCAGTGCCCA 12858649
Forward GCCTCAGTACCAAGGGCA 12858520 53
Reverse TCAAAGGTGGCGCTTCATGT 12859242
NF-E2
Forward GGAGGGTTGGAGGAAGGACT 52972425 56
Reverse ACCAGGGGCAATCTAGAAAGTC 52972825
Forward TTAAGGGATTGATGAATCCAGCATGA 52978025 56
Reverse CCCAGTGCCTGGAGCATTAC 52978425
Forward TGTGGCAACTTTGTTTCCTTGTTAC 52979875 55
Reverse ACTCCACTGGGTACATCTCTAC 52980425
Forward GGATTATCCCTCAACTATAGCG 52980375 53
Reverse AACCTCTGGGCCAGCTCA 52981075
HSs
HS1
Forward CCTGGTATCCTAGGACCTGC 13141 55
Reverse CTGAAAGTAAACTTCCACAACCGC 13798
HS2
Forward TAAGCTTCAGTTTTTCCTTAGT 8484 55
Reverse CAGATCTGACCCCGTATGTGAGCAT 9224
HS3
Forward GACTCAGCAAACACAAGACCC 4511 55
Reverse GGGCTTCCATGTTTCCAGTGA 5281
HS4
Forward CACAACGACCCATATAGACA 68 52
Reverse GGAAATCATTTACTCCAGACTC 1528
b-Thalassaemia not Linked to the b-Globin Gene
ª 2006 Blackwell Publishing Ltd, British Journal of Haematology, 132, 640–650 645

nonsense mutation. The haematological evaluation of one
child (III-1) was performed at 1 year of age and confirmed the
prenatal test.
Family I Subject II-2 from this family was a carrier of the
b+ IVS-I nt110 mutation and had the typical features of the
b-thalassaemia trait. Her husband (II-1) and her mother-in-law
(I-2) have the typical haematological features of heterozygous
b-thalassaemia, but the b-globin gene was completely intact
(Fig 3). The couple presented with an ongoing pregnancy and
decided to have prenatal diagnosis. This was carried out both by
fetal DNA analysis and by globin chain synthesis. DNA analysis
of the fetus showed heterozygosity for the b+ IVS-I nt110
mutation. Fetal globin chain synthesis analysis showed very low
b-globin chain production (b/c 0Æ010) indicative of an affected
fetus. Because the fetus (III-1) was considered affected by
thalassaemia major resulting from the presence of the b+ IVS-I
nt110 mutation and an unknown molecular defect, the
pregnancy was terminated by the parents’ request.
Family J In this family, father and son were carriers of the type
2 typical thalassaemia-like determinant (Fig 3). MCV values
varied between 68Æ2 and 70 fl, and HbA2 between 6 and 6Æ8%.
II-1 had a a/b globin chain ratio of 2Æ03.
Family K In this family, two subjects, II-1 and II-2, were
affected by a thalassaemia intermedia phenotype (Fig 3). They
inherited only one b-globin gene affected by a typical
b-thalassaemia mutation (b+ IVS I-1) from their father and
a type 2 typical b-thalassaemia-like determinant transmitted by
their mother. The mother (I-2) had MCV values of 77Æ5 fl and
a HbA2 level of 4Æ3%. One individual in the second generation
(II-3) inherited the b+ IVS I-1 allele from her father and
showed the typical b-thalassaemia carrier state phenotype.
Haplotype analysis
b-Haplotypes were constructed from 10 restriction fragment
length polymorphisms: HincII e, XmnI 5¢ Gc, HindIII Gc,HindIII Ac, HincII 5¢wb, HincII 3¢wb, HinfI 5¢b, RsaI 5¢b,HinfI 3¢b, and HpaI 3¢b (Table II). A further five polymorphic
sites, located in the b-globin gene, were detected by DGGE at
codon 2 of exon 1 (C fi T), at nt 16 (C fi G), at nt 74
(G fi T), at nt 81 (C fi T), and at nt 666 (T fi C) of the
second intron.
Haplotype analysis of the b-globin gene cluster was
performed in families B, C, D, H, and K (Figs 1, 2 and 3).
In family B, patient III-1 was affected by thalassaemia major.
Haplotype analysis indicated that the codon 39 nonsense
mutation was linked to haplotype D ()++)+++) and the type
1 silent b-thalassaemia-like determinant was linked to haplo-
type B ()))))++). When the mother became pregnant
prenatal diagnosis was carried out. The fetus (III-2) showed
haplotype D/F, which suggested that he inherited only the
codon 39 nonsense mutation. The infant was born at term and
in spite of the fact that she was a simple heterozygote for the
codon 39 mutation, she was found to be affected by
thalassaemia major. Re-evaluation and extension of b-globinhaplotype analysis indicated that, in this family, the type 1
silent b-thalassaemia like determinant was most probably not
linked to the b-globin cluster (Fig 1, Table II). However, we
cannot rule out the remote possibility of linkage resulting from
a recombination event in the db-globin region in III-1.
In family D, b-globin haplotype analysis indicates that the
type 2 typical b-thalassaemia like determinant was most
probably not linked to the b-globin cluster (Fig 2, Table II).
However, as in family B, we cannot rule out the possibility of a
recombination event in the db-globin region in subject III-1.
In families C, H, K, b-cluster haplotypes analysis indicatedthat both the type 1 (family C) and the type 2 (families H and
K) b-thalassaemia-like determinants were not linked to the
b-globin gene cluster (Figs 1 and 3, Table II).
d-Globin gene analysis
The b-thalassaemia-like determinant segregating in families D
and G was characterised by a HbA2 level of between 4Æ8% and
8Æ0%, low/normal MCV-MCH, and a slightly unbalanced/
normal b-globin chain synthesis. These characteristics could
result from a mutation in the d-globin gene, determining a
high output of d-globin chains. However, analysis of d-globingene from position )520 to the CAP site to position +140 from
the polyadenilation site in these subjects showed totally normal
sequences.
Locus control region analysis
The HS-1, HS-2, HS-3, HS-4 core regions of the LCR were
analysed by direct sequencing of amplified DNA in families B,
C, D, E, F, G, I, J, and no variations from the wild-type
sequences were detected. LCR analysis was not carried out in
family A because no DNA was available, or in families H and K
as there is strong evidence that there is no linkage to b-globingene cluster.
Southern blot analysis
Southern blot analysis carried out with d, wb, Ac, Gc, and eprobes, as well as with the minilocus SK-l LAR probe
containing HS 1, 2, 3, 4 and the Gc promoter, excluded the
presence of b globin cluster gross deletions. The finding of
heterozygosity for both intragenic and extragenic polymorphic
markers (Table II) further support this conclusion.
Transcription factors analysis
Sequence analysis of the genes encoding for NFE2 and EKLF
transcription factors was performed in families B, C, D, E, F,
H, I, and J. In families A, G, and K, the analysis was not carried
out as no patient DNA was available.
V. Faa et al
646 ª 2006 Blackwell Publishing Ltd, British Journal of Haematology, 132, 640–650

In subject II-6 of family C, sequence analysis of the EKLF
gene detected heterozygosity for a nucleotide change C fi T
at codon 102, which resulted in a proline to leucine
substitution. In family D, besides heterozygosity for the
C fi T substitution at position 102 subjects I-2, II-1, III-1,
and III-2 showed a T fi C change in position 182,
determining a phenylalanine to leucine aminoacid substitu-
tion. However, sequence analysis of the EKLF gene from 200
chromosomes of normal individuals of the same Sardinian
origin detected both the C fi T and the T fi C nucleotide
substitutions in 49% and 14% respectively of the chromo-
somes investigated, indicating that these sequence variations in
the EKLF gene are simple polymorphisms that do not affect its
function. No sequence variations of this gene were detected in
b-thalassaemia-like carriers from families B, E, F, H, I, and J.
Sequence analysis of the NF-E2 gene showed wild-type
sequences in all b-thalassaemia-like carriers from these fam-
ilies.
Analysis of the GATA-1 gene was not carried out in our
families because the b-thalassaemia-like determinants did not
show an X-linked transmission pattern and none of the
investigated b-thalassaemia carriers showed macrothrombo-
cytopenia. Likewise, analysis of the TFIIH gene was not carried
out as none of the subjects investigated showed the phenotype
of trichothiodystrophy.
RNA analysis
Quantitative analysis of reticulocyte mRNA was performed in
some members of the reported 11 families. In family B
quantitative analysis was performed in patients I-2, II-2, and
III-2, showing a 70–80% b-globin gene mRNA reduction. In
this family the proband III-2 showed a 99% beta globin gene
mRNA reduction, in agreement with the b-thalassaemia major
phenotype.
Patients II-1 and III-2 of family D, I-1 of family F, and II-1
and II-2 of family G, showed a 60–65% b-globin gene mRNA
reduction.
Patients I-1 and II-1 of family E, and I-2, II-1, and II-3 of
family H showed a 70–80% b-globin gene mRNA reduction.
On patient II-1 of family J the analysis showed a 47% beta
globin gene mRNA reduction (Fig 4).
a-Globin gene analysis
a-Globin gene analysis excluded the presence of a-globin gene
deletion as well as a-globin gene implication.
Discussion
Understanding the regulation of the b-globin gene could have
relevant therapeutic implications for the treatment of wide-
spread diseases, such as thalassaemia and sickle cell disease. It
is well established that the regulation of the b-globin gene is
mainly controlled at the level of transcription and is mediatedTable
II.b-G
lobin
genecluster
representedbytheb-haplotypes
denotedbythepresence
(+)orabsence
())ofthe15
polymorphic
sites.
HincIIe
XmnI5¢
Gc
HindIIIGc
HindIIIAc
HincII5¢wb
HincII3¢wb
HinfI5¢b
RsaI5¢b
cod2
Cfi
T
IVSII-16
Cfi
G
IVSII-74
Gfi
T
IVSII-81
Cfi
T
IVSII-666
Tfi
CHinfI3¢b
HpaI
3¢b
A)
)+
)+
++
+)
++
))
))
B+
))
))
)+
+)
+)
))
++
C+
))
))
)+
+)
++
))
)+
D)
)+
+)
++
+)
+)
))
++
E)
)+
)+
++
+)
+)
))
++
F+
)+
)+
++
++
)+
++
++
G)
))
))
)+
+)
++
))
))
H+
)+
)+
++
+)
+)
))
++
I)
))
))
)+
+)
+)
))
+)
L)
)+
+)
))
+)
++
))
)+
M+
))
))
++
++
)+
++
+)
N+
))
))
)+
++
)+
++
++
O)
)+
+)
++
++
)+
++
++
P)
++
)+
+)
++
+)
))
)+
Q)
))
))
)+
+)
+)
))
++
R+
))
))
)+
+)
))
))
++
S+
++
)+
++
))
+)
++
)+
T)
)+
)+
+)
+)
)+
))
)+
U+
+)
))
)+
++
)+
+)
)+
b-Thalassaemia not Linked to the b-Globin Gene
ª 2006 Blackwell Publishing Ltd, British Journal of Haematology, 132, 640–650 647

by tissue-specific and general transcription factors. Despite
important progress in the identification of b-globin gene
regulators only two general (ATR-X and TFIIH) and one
tissue-specific (GATA1) factors have been so far associated
with human thalassaemia disease. It is likely that several of
these regulators could be identified by reverse genetic analysis,
studying those thalassaemia patients that have mutations not
linked to the b-globin gene cluster. In attempt to identify these
factors, we have carried out an extensive molecular analysis of
the largest series reported to date, of b-thalassaemia hetero-
zygous individuals carrying a b-thalassaemia determinant not
linked to the b-cluster, which in compound heterozygosity
with the typical b-thalassaemia carrier state led to thalassaemia
major or intermedia. These results furthermore indicated the
existence of a markedly heterogeneous group of b-thalas-saemia-like disorders not yet molecularly characterised.
In all of our cases, sequence analysis of the b-globin gene
failed to detect a disease-causing mutation. Likewise, no
deletion that may inactivate the b-globin gene leaving its
sequences intact was found in the b-globin gene cluster by
Southern blot analysis. In addition, heterozygosity for intra-
genic as well extragenic polymorphisms indicated the presence
of two b-globin genes. Furthermore, sequence analysis of the
core region of HS1, HS2, HS3, and HS4 showed a wild-type
sequence thus excluding involvement of these control regions
in determining the b-thalassaemia-like phenotype. Based on
their phenotypic characteristics these b-thalassaemia-like car-
riers were classified into two groups. The first is the type 1
silent b-thalassaemia-like determinant, and is characterised by
normal haematological parameters, normal HbA2, and unbal-
anced b-globin chains, thus displaying the phenotype of silent
b-thalassaemia. The second group showed the typical charac-
teristics of heterozygosity for b-thalassaemia with low MCV-
MCH, high HbA2, and unbalanced globin chain synthesis, and
has been designated as a type 2 typical b-thalassaemia-like
determinant. Both the silent and typical b-thalassaemia-like
phenotypes have low frequency, as they have been detected
following the molecular analysis of >8000 chromosomes of
individuals with a b-thalassaemia carrier phenotype. In several
of these families some individuals are affected either by
thalassaemia intermedia (families C and K) or by thalassaemia
major (families A, B and I). This is quite surprising,
particularly when considering the cases in families A and B,
where the interaction of the type 1 silent b-thalassaemia-like
determinant with typical b-thalassaemia mutation determined
a severe unbalanced globin chain synthesis resulting in a
thalassaemia major phenotype, in spite of the fact that the
clinical phenotype resulting from compound heterozygosity
for a classical silent b-thalassaemia mutation (i.e. mutations in
the distal CACCC box of the b globin gene) and a typical
b-thalassaemia mutation usually results in a mild phenotype
referred to as thalassaemia intermedia.
The presence in some carriers (families D and G) of type 2
typical b-thalassaemia-like determinants characterised by high
HbA2 associated with minimal changes in red blood cell
indices suggested that this trait could be due to a mutation in
the d-globin region causing an increased output in d-globinchains from the affected locus. Nevertheless the d-globin chain
sequence was normal.
Linkage analysis using b globin gene cluster haplotypes
showed that both the type 1 and the type 2 b-thalassaemia-
like determinants segregated independently of the b globin
gene complex, indicating absence of linkage to this chromo-
somal region. However in two families (B and D), linkage
could not be ruled out as there is a remote possibility of a
recombination event in the db-globin region in one member
of each family.
The data produced in this study clearly indicate that both
b-thalassaemia-like determinants severely curtail b-globinchain production. Defective b-globin chain output could
result from a reduced transcription of the b-globin gene or
from a translation defect of the b-globin mRNA. The
reduction in steady state b-globin mRNA levels in both types
of b-thalassaemia-like heterozygotes in some of the examined
families indicated that these b-thalassaemia-like phenotypes
are caused by a defect in the transcription of the b-globin gene.
Defective transcription of the b-globin gene may result from a
defective promoter of the b-globin gene or from a lesion in a
gene coding for a transcription factor that regulates its
function, such as EKLF, GATA-1, or NF-E2. Nevertheless,
sequence analysis of the genes coding for EKLF and NF-E2
showed a wild-type sequence, thus probably ruling out a
defective function of these transcriptional factors.
The gene coding for GATA-1 was not examined because
neither b-thalassaemia-like determinant in these families
showed an X-linked transmission pattern and none of the
studied b-thalassaemia carriers displayed macrothrombo-
cytopenias, which are always detected in the b-thalassaemia-
like carrier resulting from a defect in the GATA-1 gene.
0
20
40
60
80
100
120
Nor
mal I-2
II-2
III-2 II-1
III-2 I-1
II-1
I-1
II-1
II-2
I-2
II-1
II-3
II-1
%
B D E F G H J
Fig 4. Quantitative analysis of reticulocyte b-mRNA performed on
several subjects from families B, D, E, F, G, H, and J. A normal indi-
vidual shows 100% overall b-globin gene expression In families B, E,
and H the analysis shows 20–30% overall b-globin gene expression.
Patient III-2 from family B is affected by thalassaemia major and shows
1% of b-globin gene expression In families D, F, and G quantitative
analysis shows 35–40% overall b-globin gene expression. In family J
patient II-1 shows about 50% overall b-globin gene expression.
V. Faa et al
648 ª 2006 Blackwell Publishing Ltd, British Journal of Haematology, 132, 640–650

Likewise, analysis of the TFIIH gene was not carried out as
none of the subjects investigated showed the phenotype of
trichothiodystrophy.
The results produced in this study indicate that the gene(s)
involved in the determining b-thalassaemia-like phenotypes
most probably code(s) for an unknown crucial transcriptional
regulator of the b-globin gene function. The description of
b-like determinants that may lead, in the compound hetero-
zygous state with typical heterozygous b-thalassaemia allele, to
thalassaemia major or intermedia, indicate the need for clinical
as well forprenatal diagnosis to carry out appropriate family
studies. In these families, prenatal diagnosis was carried out in
two steps, of which the first consisted of mutation analysis for
the paternal or maternal typical b-thalassaemia mutation in
the fetal DNA. In cases where the fetus was found to be
heterozygous for the b-thalassaemia mutation, globin chain
synthesis analysis on fetal blood was performed to detect the
compound heterozygote state for the thalassaemia-like deter-
minant and typical b-thalassaemia. The only possibility to
identify the molecular defects responsible for both the type 1
and the type 2 b-thalassaemia-like determinants is a genome-
wide screen for polymorphisms (microsatellites or single
nucleotide polymorphisms) associated with b-thalassaemia-
like determinants. However, simulation analyses indicate that
the number of families enrolled so far is not sufficient for such
an analysis, considering also the marked heterogeneity of the
phenotypes segregating in these families. We hope to be able to
collect more families with segregating b-thalassaemia-like
conditions in the future and establish a national and interna-
tional collaboration to finally determine the molecular pathol-
ogy of this interesting group of patients. The molecular
definition of these cases is also very important as it may lead to
the discovery of further novel transcription factors regulating
the function of the b-globin gene.
Acknowledgements
This work was supported by grants from Assessorato Igiene e
Sanita, Regione Sardegna (LR n 11, 1990: ‘Patologia Molecol-
are genetica e terapia genetico-somatica della b-thalassaemia’
and Progetto di Prevenzione e Educazione Sanitaria: ‘Malattie
genetiche nella popolazione sarda: Talassemia, Apeced, Mal-
attia di Wilson’).
The authors would like to thank: Prof. Renzo Galanello
(Diparimento di Scienze Biomediche e Biotecnologie, Ospe-
dale Microcitemico, Cagliari, Italy) for confirming haemato-
logical data, Prof Maurizio Longinotti (Istituto di Ematologia,
Universita’ di Sassari, Sassari, Italy), Dr Matteo Francese
(Dipartimento di Pediatria Seconda, Universita’ di Napoli,
Napoli, Italy), and Prof. Elisa Calzolari (Dipartimento di
Medicina Sperimentale e Diagnostica, Universita’ di Ferrara,
Ferrara, Italy) for collecting samples performing haematolog-
ical analysis and clinical evaluation, Maria Demurtas for
technical support and all the families included in this study for
their cooperation.
References
Andrews, N.C., Erdjument-Bromage, H., Davidson, M.B., Tempst, P.
& Orkin, SH. (1993) Erythroid transcription factor NF-E2 is he-
matopoietic-specific basic leucine zipper protein. Nature, 362, 722–
728.
Antonarakis, S.E., Boehm, C.D., Giardina, P.J.V. & Kazazian, H.H.
(1982) Nonrandom association of polymorphic restriction sites in
the b-globin gene cluster. Proceedings of the National Academy of
Sciences of the United States of America, 79, 137–141.
Badens, C., Mattei, M.G., Imbert, A.M., Lapoumeroulie, C., Martini,
N., Michel, G. & Lena-Russo, D. (2002) A novel mechanism for
thalassaemia intermedia. Lancet, 359, 132–133.
Beutler, E. & Gelbart, T. (1986) The mechanism of removal of leu-
kocytes by cellulose columns. Blood Cells, 12, 57–64.
Cai, S.P. & Kan, Y.W. (1990) Identification of the multiple b-tha-lassemia mutations by denaturing gradient gel electrophoresis.
Journal of Clinical Investigation, 85, 550–553.
Chomczynski, P. & Sacchi, N. (1987) Single-step method of RNA
isolation by acid guanidium thiocyanate-phenol-chloroform
extraction. Analytical Biochemistry, 162, 156–159.
Dode’, C., Krishnamoorthy, R., Lamb, J. & Rochette, J. (1992)
Rapid analysis of -a3.7 thalassemia and aaaanti3.7 triplication by
enzymatic amplification analysis. British Journal of Haematology, 82,
105–111.
Galanello, R., Perseu, L., Perra, C., Maccioni, L., Barella, S., Longinotti,
M., Cao, A. & Cazzola, M. (2004) Somatic deletion of the normal
beta-globin gene leading to thalassaemia intermedia in heterozygous
beta-thalassaemic patients. British Journal of Haematology, 127, 604–
606.
Gasperini, D., Perseu, L., Melis, M.A., Maccioni, L., Sollaino, C.,
Paglietti, E., Cao, A. & Galanello, R. (1998) Heterozygous beta-
thalassemia with thalassemia intermedia phenotype. American
Journal of Hematology, 57, 43–47.
Gibbons, R.J., Picketts, D.J., Villard, L. & Higgs, D.R. (1995) Mutations
in a putative global transcriptional regulator cause X-linked mental
retardation with alpha-thalassemia (ATR-X syndrome). Cell, 80,
837–845.
Gonzalez-Redondo, J.M., Stoming, T.A., Kutlar, A., Kutlar, F., Lanclos,
K.D., Howard, E.F., Fei, Y.J., Aksoy, M., Altay, C., Gurgey, A., Basak,
A.N., Efremov, G.D., Petkov, G. & Huisman, T.H.J. (1989) A C-T
substitution at nt )101 in a conserved DNA sequence of the pro-
moter region of the b globin gene is associated with ‘silent’ b-tha-lassemia. Blood, 73, 1705–1711.
Gossens, M., Dozy, A.M., Embury, S.H., Zachariades, Z., Hadjiminas,
M.G., Stamatoyannopoulos, G. & Kan, Y.W. (1980) Triplicated
a-globin loci in humans. Proceedings of the National Academy of
Sciences of the United States of America, 77, 518–521.
Grosveld, F. (1999) Activation by locus control region? Current
Opinion in Genetics & Development, 9, 152–157.
Ho, P.J., Hall, G.W., Luo, L.Y., Weatherall, D.J. & Thein, S.L. (1998a)
Beta-thalassemia intermedia: is it possible consistently to
predict phenotype to genotype?. British Journal of Haematology, 100,
70–78.
Ho, P.J., Hall, G.W., Watt, S., West, N.C., Winperis, J.W., Wood, W.G.
& Thein, S.L. (1998b) Unusually severe heterozygous b-thalassemia:
evidence for an interacting gene affecting globin translation. Blood,
92, 3428–3435.
b-Thalassaemia not Linked to the b-Globin Gene
ª 2006 Blackwell Publishing Ltd, British Journal of Haematology, 132, 640–650 649

Huisman, T.H.J., Carver, M.F.H. & Baysal, E. (1997) A Syllabus of
Thalassemia Mutations. The Sickle Cell Anemia Foundation,
Augusta, GA, pp. 1–309.
Li, Q., Harju, S. & Peterson, KR. (1999) Locus control regions: coming
of age at a decade plus. Trends in Genetics, 15, 403–408.
Livak, K.J. & Schmittgen, T.D. (2001) Analysis of relative gene ex-
pression data using real-time quantitative PCR and the 2[-DD C(T)]
method. Methods, 25, 402–408.
Miller, I.J. & Bieker, J.J. (1993) A novel, erythroid b-thalassemia cell-
specific murine transcription factor that binds to the CACCC ele-
ment and is related to the Kruppel family of nuclear proteins.
Molecular Cellular Biology, 13, 2776–2786.
Miller, S.A., Dykes, D.D. & Polensky, H.F. (1998) A simple salting out
procedure for extracting DNA from human nucleated cells. Nucleic
Acids Research, 16, 1215.
Moi, P., Cash, F.E., Liebhaber, S.A., Cao, A. & Pirastu, M. (1987) An
initiation codonmutation (AUG fi GUG)of the humana1 – globingene: structural characterization and evidence for a mild thalassemic
phenotype. The Journal of Clinical Investigation, 80, 1416–1421.
Moi, P., Faa, V., Marini, M.G., Asunis, I., Ibba, G., Cao, A. & Rosatelli,
M.C. (2004) A novel silent b-thalassemia mutation in the distal
CACCC box affects the binding and responsiveness to EKLF. British
Journal of Haematology, 126, 881–884.
Murru, S., Loudianos, G., Porcu, S., Sciarratta, G.V., Agosti, S., Parodi,
M.I., Cao, A. & Pirastu, M. (1992) A b-thalassemia phenotype not
linked to the b-globin cluster in an Italian family. British Journal of
Haematology, 81, 283–287.
Myers, R.M., Fisher, S.G., Lerman, L.S. & Maniatis, T. (1985) Nearly all
single base substitutions in DNA fragments joined to a GC-clamp
can be detected by denaturing gradient gel electrophoresis. Nucleic
Acids Research, 13, 3131–3135.
Nichols, K.E., Crispino, J.D., Poncz, M., White, J.G., Orkin, S.H.,
Maris, J.M. & Weiss, M.J. (2000) Familial dyserythropoietic anaemia
and thrombocytopenia due to an inherited mutation in GATA-1.
Nature Genetics, 24, 266–270.
Orkin, S.H., Kazazian, H.H. Jr, Antonarakis, S.E., Goff, S.C., Boehm,
C.D., Sexton, J.P., Waber, P.G. & Giardina, P.J. (1982) Linkage of b-thalassemia mutations and b-globin gene polymorphisms with DNA
polymorphisms in human b-globin gene cluster. Nature, 296, 627–
631.
Pacheco, P., Peres, M.J., Faustino, P., Pischedda, C., Goncalves, J.,
Carvajales-Ramos, M., Seixas, T., Martins, M.C., Moi, P. & Lavinha,
J. (1995) Beta-thalassemia unlinked to the beta globin gene interacts
with sickle-cell trait in a Portuguese family. British Journal of Hae-
matology, 91, 85–89.
Pirastu, M., Saglio, G., Chang, J.C., Cao, A. & Kan, Y.W. (1984)
Initiation codon mutation as a cause of a-thalassemia. Journal of
Biological Chemistry, 259, 12315–12317.
Ristaldi, M.S., Murru, S., Loudianos, G., Casula, L., Porcu, S., Pigh-
eddu, D., Fanni, B., Sciarratta, G.V., Agosti, S., Parodi, M.I., Leone,
D., Camaschella, C., Serra, A., Pirastu, M. & Cao, A. (1990) The
C fi T substitution in the distal CACCC box of the b globin gene
promoter is a common cause of silent b-thalassemia in the Italian
polulation. British Journal of Haematology, 74, 480–486.
Rosatelli, M.C., Tuveri, T., Scalas, M.T., Leoni, G.B., Sardu, R., Faa, V.,
Meloni, A., Pischedda, M.A., Demurtas, M., Monni, G. & Cao, A.
(1992) Molecular screening and fetal diagnosis of b-thalassemia in
the Italian population. Human Genetics, 83, 590–592.
Rosatelli, M.C., Faa, V., Meloni, A., Fiorenza, F., Galanello, R.,
Gasperini, D., Amendola, G. & Cao, A. (1995) A promoter muta-
tion, C fi T at position )92, leading to a silent b-thalassemia.
British Journal of Haematology, 90, 483–485.
Rund, D., Oron-Karni, V., Filon, D., Rachmilewitz, E. & Oppenheim,
A. (1997) Genetic analysis of b-thalassemia intermedia in Israel:
diversity of mechanisms and unpredictability of phenotype. Ameri-
can Journal of Hematology, 54, 16–22.
Saiki, R.K., Walsh, P.S., Levenson, C.H. & Erlich, H.A. (1989) Genetic
analysis of amplified DNA with immobilized sequence-specific oli-
gonucleotide probes. Proceedings of the National Academy of Sciences
of the United States of America, 86, 6230–6234.
Sanger, F., Miicklen, S. & Coulson, A.R. (1977) DNA sequencing
with chain terminating inhibitors. Proceedings of the National
Academy of Sciences of the United States of America, 74, 5463–
5467.
Southern, E.M. (1975) Detection of specific sequences among DNA
fragments separated by gel electrophoresis. Journal of Molecular
Biology, 98, 503–517.
Thein, S.L., Wood, W.G., Wickramasinghe, S.N. & Galvin, M.C. (1993)
Beta-thalassemia unlinked to the beta-globin gene in an English
family. Blood, 81, 283–287.
Varawalla, N.Y., Fitches, A.C. & Old, J.M. (1992) Analysis of b-globinhaplotypes in Asian Indians: origin and spread of b-thalassemia on
the Indian subcontinent. Human Genetics, 90, 443–449.
Weatherall, D.J., Clegg, J.B., Higgs, D.R. & Wood, W.G. (2001) The
hemoglobinopathies. In: The metabolic and molecular bases of
inherited disease. (ed. by C.R. Scriver), pp. 4571–4636. McGraw-Hill,
New York.
Weiss, M.J. & Orkin, S.H. (1995) GATA transcription factors: key
regulators of hematopoiesis. European Journal of Biochemistry, 23,
99–107.
V. Faa et al
650 ª 2006 Blackwell Publishing Ltd, British Journal of Haematology, 132, 640–650




















