
Developing of Micellar Drug Carriers for the Treatment of ...
235
University of Nebraska Medical Center University of Nebraska Medical Center DigitalCommons@UNMC DigitalCommons@UNMC Theses & Dissertations Graduate Studies Fall 12-14-2018 Developing of Micellar Drug Carriers for the Treatment of Breast Developing of Micellar Drug Carriers for the Treatment of Breast Cancer Bone Metastasis Cancer Bone Metastasis Tong Liu University of Nebraska Medical Center Follow this and additional works at: https://digitalcommons.unmc.edu/etd Part of the Pharmacy and Pharmaceutical Sciences Commons Recommended Citation Recommended Citation Liu, Tong, "Developing of Micellar Drug Carriers for the Treatment of Breast Cancer Bone Metastasis" (2018). Theses & Dissertations. 335. https://digitalcommons.unmc.edu/etd/335 This Dissertation is brought to you for free and open access by the Graduate Studies at DigitalCommons@UNMC. It has been accepted for inclusion in Theses & Dissertations by an authorized administrator of DigitalCommons@UNMC. For more information, please contact [email protected].
-
Upload
khangminh22 -
Category
Documents
-
view
2 -
download
0
Transcript of Developing of Micellar Drug Carriers for the Treatment of ...

University of Nebraska Medical Center University of Nebraska Medical Center
DigitalCommons@UNMC DigitalCommons@UNMC
Theses & Dissertations Graduate Studies
Fall 12-14-2018
Developing of Micellar Drug Carriers for the Treatment of Breast Developing of Micellar Drug Carriers for the Treatment of Breast
Cancer Bone Metastasis Cancer Bone Metastasis
Tong Liu University of Nebraska Medical Center
Follow this and additional works at: https://digitalcommons.unmc.edu/etd
Part of the Pharmacy and Pharmaceutical Sciences Commons
Recommended Citation Recommended Citation Liu, Tong, "Developing of Micellar Drug Carriers for the Treatment of Breast Cancer Bone Metastasis" (2018). Theses & Dissertations. 335. https://digitalcommons.unmc.edu/etd/335
This Dissertation is brought to you for free and open access by the Graduate Studies at DigitalCommons@UNMC. It has been accepted for inclusion in Theses & Dissertations by an authorized administrator of DigitalCommons@UNMC. For more information, please contact [email protected].

DEVELOPING OF MICELLAR DRUG CARRIERS FOR THE TREATMENT OF
BREAST CANCER BONE METASTASIS
by
Tong Liu
A DISSERTATION
Presented to the Faculty of
The Graduate College in the University of Nebraska
In Partial Fulfillment of the Requirements
For the Degree of Doctor of Philosophy
Pharmaceutical Sciences Graduate Program
Under the Supervision of Professor Tatiana K. Bronich
University of Nebraska Medical
Center Omaha, Nebraska
November 2018

II
ACKNOWLEDGMENT
The journey in the past five years was an unforgettable experience for me to
explore the scientific researches. Many ups and downs, joys and sorrows challenged
every aspect of my personal and professional to extreme. I'm glad that I went through
finally and now look forward to moving to the next step for a new career. It could never
be possible without the contribution of people who lend their hands to the shapes the
research projects and my graduation. Please allow me at this special moment to express
the sincere appreciation and the gratitude for the invaluable support.
My heartfelt appreciation goes to my mentor, Dr. Bronich, who is a brilliant and
dedicated scientist and has been guiding me constantly throughout the Ph.D. training. It
could never be imagined for me to finish the researches without her countless support
and patience. She's been critical and inspiring during each discussion, through which I
grew my profession steadily. Many researching opportunities would be missed if she did
not provide innovative and sharp comments and suggestion. From her, I learned to
conduct researches with the attention to details and to present data concisely. She also
granted me plenty of opportunities and flexibilities to explore and evaluate my ideas. It
has been a great pleasure to be her student. Dr. Rakesh Singh has been enormously
helpful in the biological study in the bone metastases, and I was able to gain the skill of
intracardiac injection under his instruction. He was so kind that no matter how busy the
schedule was, he always found the time to sit down with me and shared his insight. I feel
lucky to have him in the committee. I would extend my thankfulness to other committee
members, Dr. Band, Dr. Li, and Dr. Vetro. They have been effective co-mentors and

III
shaped the achievement with valuable advice and constructive criticism. Special thanks
to Dr. Li for his kindness and suggestion on my life as a student. I would also like to
thank my current and previous lab members. Dr. Svetlana Romanova made the efforts in
the synthesis of the polymers and helped to prepare the material in the animal studies.
Dr. Hangting Hu brought me aboard of the cell-based experiments and transferred the
essential techniques, which was the foundation of many of the researches.
I thank Dr. Swapnil Desale, Dr. Jinjin Zhang, Dr. Krtui Soni, Dr. Fan Lei and Dr.
Shaheen Ahmed, Xinyuan Xi, Nan Zhao, Fei Wang and Zi Wang for their kind help,
friendship, and the opportunity to work with them. The Ph.D. study is focused on a
pinpoint and dives deeply. When reaching an interdisciplinary field like pharmaceutical
sciences, one’s knowledge is always limited. Thanks to the colleagues in UNMC, who
are willing to help at every moment. They are Dr. Ying Xie, Michelle Varney, Linyun Wu,
Dr. Xiaobei Wang, Dr. Xin Wei, Zhifeng Zhao, Dr. Kumar Virender, Yang Peng, Hongjun
Wang, Dr. Di Wen, Lin Feng, Zhiyi Lin, Hang Su, Dr. Dongwei Guo, Zhihao Mao,
Nicholas Wojtynek, Bowen Qi, Dr. Beth Clymer, Yu Hang, Weiming Tang. Also, I would
like to acknowledge the assistance from UNMC core facilities, IACUC, comparative
medicine and financial support from NIH, UNMC Program of Excellence Graduate
Assistantship, and the China Scholar Council. The acknowledgment would be
incomplete without mentioning my friends on and off the campus, Dr. Chaojun Wang, Dr.
Xiaoyan Yang, Dr. Miaorong Yu, Chenjun Shi, Dr. Shengqi Hou, Dr. Jiaqiang Wang, Dr.
Nimin Wu, Yike Wang, Yuxiang Zhu, Chunyi Zhou, Qingfeng He, Jian Zhang, Yuning
Zhang and Junming Zhou.
In the end, I'm so grateful for my parents and their unconditioned love over the
past three decades. They are the backbones and ready to be supportive whenever it is

IV
needed. Their encouragement and willingness to share any burden make me brave on
the road chasing my dream. I feel extremely blessed to be their child.
Tong Liu
November 2018

V
DEVELOPMENT OF MICELLAR DRUG CARRIERS FOR THE
TREATMENT OF BREAST CANCER BONE METASTASIS
Tong Liu, Ph.D.
University of Nebraska Medical Center, 2018
Advisor: Tatiana K. Bronich, Ph.D.
Breast cancer (BC) remains one of the most frequently diagnosed cancer
worldwide. Bone is one of the most common sites and often the first clinical indication of
metastatic progression of BC. Current treatments including radiation, surgery, and
chemotherapy are rarely curative with limited effect in overall survival. The challenge is
true especially for patients with triple-negative BC that are not responsive to receptor-
targeted therapy, or patients who had exposure to chemotherapy before and might
already gain the resistance to some of the drugs.
Here we took advantage of the aminobisphosphonate, alendronate (ALN), that
can bind to the bone mineral efficiently and engineered ALN-decorated polymeric
micelles to target Docetaxel (DTX) to bone metastasis. DTX/ALN-m showed high affinity
to hydroxyapatite in vitro, exhibited similar cytotoxic activity as free drug and
demonstrated the potential to intervene the tumor microenvironment by inhibiting bone
resorption and macrophage recruitment. Systemic treatment with the DTX/ALN-m led to

VI
significant attenuation of bone metastatic tumor burden and improved survival time in the
immunocompetent mouse model of BC dissemination to the bone.
To further improve the efficacy of the metastasis treatment, we developed a
micellar formulation of DTX and Dasatinib (Das), an inhibitor of multiple tyrosine kinases.
Dual drug-loaded micelles inhibited the osteoclast differentiation, migration of cancer
cells, exhibited strong synergy in metastatic BC cell lines and retained the comparable
efficacy even in DTX-tolerant cells. Mechanistic studies revealed the connection of drug
tolerance to the activation of AMP-activated protein kinase and the role of both drugs in
mitigating adaptive resistance. We also demonstrated that this micellar drug
combination exerted enhanced antitumor activity delaying the progression of metastatic
disease. Overall, the micellar drug carriers provide an effective platform for the
treatment of breast cancer bone metastasis by targeting tumor and their
microenvironment.

VII
TABLE OF CONTENT
ACKNOWLEDGMENT ..................................................................................................... II
LIST OF FIGURES ........................................................................................................ IX
LIST OF TABLES ....................................................................................................... XIII
LIST OF ABBREVIATION ........................................................................................... XIV
LIST OF CONTRIBUTORS ....................................................................................... XXIII
CHAPTER I INTRODUCTION ........................................................................................ 1
1.1 CANCER AND METASTASES......................................................................................... 2
1.2 BONE METASTASES ...................................................................................................... 4
1.3 BONE-TUMOR MICROENVIRONMENT ......................................................................... 8
1.4 CURRENT TREATMENT OPTIONS IN THE CLINIC FOR BONE METASTASES ........ 14
1.5 CHEMO DRUG RESISTANCE....................................................................................... 20
1.6 NANOCARRIERS-BASED DRUG DELIVERY SYSTEM FOR CANCER THERAPY .... 29
1.7 CONCLUSION ............................................................................................................... 40
1.8 REFERENCES ............................................................................................................... 41
CHAPTER II TARGETED POLYMERIC MICELLES FOR THE TREATMENT OF
BREAST CANCER BONE METASTASIS ....................................................................55
2.1 INTRODUCTION ............................................................................................................ 56
2.2 MATERIALS AND METHODS........................................................................................ 59

VIII
2.3 RESULTS AND DISCUSSION ....................................................................................... 74
2.4 CONCLUSION ............................................................................................................. 111
2.5 REFERENCE ............................................................................................................... 112
CHAPTER III COMBINATION THERAPEUTIC PLATFORM FOR BREAST CANCER
BONE METASTASIS TREATMENT ........................................................................... 116
3.1 INTRODUCTION .......................................................................................................... 117
3.2 MATERIALS AND METHODS...................................................................................... 120
3.3 RESULT AND DISCUSSION ....................................................................................... 135
3.4 CONCLUSION ............................................................................................................. 197
CHAPTER IV SUMMARY AND FUTURE STUDY....................................................... 204
4.1 SUMMARY ................................................................................................................... 205
4.2 FUTURE STUDY .......................................................................................................... 208
4.3 REFERENCE ............................................................................................................... 210

IX
LIST OF FIGURES
Scheme 2.1: Synthesis of the bone-targeted copolymer. ...............................................76
Figure S2.1: 1H-NMR spectra (400 MHz, DMSO-d6) of synthesized block-copolymers.77
Figure S2.2: GPC traces................................................................................................79
Figure S2.3: The plot of fluorescence intensities ratio of I3/I1.. ......................................80
Figure 2.1: In vitro characterization of ALN-m and ALN-m/DTX micelles. ......................86
Figure S2.4: Binding of the ALN(37%)-m to the bone mineral HA in DI water and in the
presence of 3.4 mM CaCl2 in the solution. ....................................................................89
Figure S2.5: In vitro toxicity of free ALN in 4T1 cancer breast cells as determined by
MTT assay. ...................................................................................................................93
Figure 2.2: Effects of ALN-m on osteoclastogenesis. .....................................................96
Figure 2.3: ALN-m inhibits the resorption activity of osteoclasts. ...................................98
Figure 2.4: Inhibition of tumor-induced macrophage migration by ALN-m. ................... 100
Figure 2.5:. Anti-metastatic efficacy of ALN-m/DTX in syngeneic 4T1 breast cancer bone
metastasis model. ....................................................................................................... 105
Figure S2.6: BLI captures and individual mouse respose. ........................................... 106
Figure 2.6: Treatment efficiencies of sequential chemotherapy with ALN-m/DTX and
radiotherapy (RT). ....................................................................................................... 109

X
Figure S2.7: Quantification of bone metastasis burden in individual mice treated ........ 110
Figure 3.1: Representative image of the morphology change in DTX and Das treated
4T1 cells. ..................................................................................................................... 136
Figure 3.2: Dose-responsive profile of DTX to 20 nM 36 h pre-treated 4T1. ................ 138
Figure:3.3: Adapted response to DTX treatment. ......................................................... 140
Figure 3.4: Adaptive response to Das. ......................................................................... 141
Figure 3.5: Recovery of DTX adapted 4T1 cells. ......................................................... 142
Figure 3.6: The regimen of different drug treatments for the analysis of the corresponded
adaptive responses. .................................................................................................... 145
Figure 3.7: Adaptive response of p-AMPK level to the drug treatments. ...................... 146
Figure 3.8: Parent 4T1 and drug adapted 4T1 in 96-well scanned by Licor Odyssey. .. 148
Figure 3.9: Representative fluorescent images parent 4T1 and drug adapted 4T1. ..... 149
Figure 3.10: Quantification of p-AMPK level/total AMPK change in DTX and Das adapted
cells by In-Cell ELISA. ................................................................................................. 150
Figure 3.11: 1H-NMR (400 MHz, DMSO-d6) of synthesized block-copolymers. ............ 155
Figure 3.12: 1H-NMR (400 MHz, TFA-d1) of synthesized block-copolymers. ................ 156
Figure 3.13: In vitro release profile of DTX/m, Das/m and (DTX+Das)/m. .................... 163
Figure 3.14: Relative sensitivity to Das. ....................................................................... 167

XI
Figure 3.15: Growth inhibition of AICAR and Compound C. ......................................... 171
Figure 3.16: Cell viability after 24 h treatment of Das/m. .............................................. 176
Figure 3.17: 4T1 migration assay. ............................................................................... 177
Figure 3.18: 4T1 migrated through the transwell membrane. ....................................... 178
Figure 3.19: Migrated MDA-MB-231 using preOB-CM. ................................................ 179
Figure 3.20: Das inhibited osteoclast at nanomolar concentration. .............................. 182
Figure 3.21: Representative image of the differentiated osteoclast under different
treatment groups. ........................................................................................................ 183
Figure 3.22: Tumor growth in each mouse. ................................................................. 185
Figure 3.23: Average tumor growth of mice with different treatment. ........................... 186
Figure 3.24: Body weight loss along with tumor growth. .............................................. 188
Figure 3.25: Acute toxicity study based on H&E staining of the major organs collected 24
h after the final treatment. ............................................................................................ 189
Figure 3.26: Kaplan-Meier survival curves of tumor-bearing mice following the
treatments. .................................................................................................................. 191
Figure 3.27: Correlation between initial tumor burden (bioluminescence) and the survival
time within each treatment groups. .............................................................................. 193
Figure 3.28: Blood count 24 h and five days after the final treatment. .......................... 195

XII
Figure 3.29: Representative image of the blood smear from the tumor-bearing mice. . 196

XIII
LIST OF TABLES
Table S2.1: Polymer characteristics determined by 1H NMR and GPC ..........................78
Table 2.1: Physicochemical characteristics of DTX-loaded micelles ..............................84
Table 2.2: IC50 values of free DTX and DTX-loaded micelles in 4T1 breast cancer cell
line. ...............................................................................................................................92
Table S2.2: PK parameters of DTX in plasma in tumor-free nude mice. ...................... 104
Table 3.1: Polydispersity of synthesized polymers by gel permeation chromatography.
.................................................................................................................................... 157
Table 3.2: Yield of preparing drug-loaded micelles with DTX, Das and the combinations.
.................................................................................................................................... 159
Table 3.3: Size, PDI and distribution modal of the drug-loaded micelles. ..................... 161
Table 3.4: In vitro tumor growth inhibition of drug-loaded micelles. .............................. 166
Table 3.5: Growth inhibition in MDA-MB-231 and PC-3 IC50 (µM). ............................... 169
Table 3.6: Growth inhibition of the combination with AICAR and Compound C, IC50 µM.
.................................................................................................................................... 173

XIV
LIST OF ABBREVIATION
%ID Percentage of injected dose
1H-NMR Proton nuclear magnetic resonance
4T1/Luc Luciferase-expressing 4T1
ABC ATP-binding cassette
Abs Absorbance
ACC Acetyl-CoA carboxylase
ACN Acetonitrile
AICAR 5-Aminoimidazole-4-carboxamide
ribonucleotide
AKT v-Akt Murine thymoma viral oncogene
ALK Alkynyl
ALK-m Non-conjugated micelles
ALN Alendronate
ALN-m Alendronate-conjugated micelles

XV
ALK-PEG-NH2 α-Amino-ω-ALK polyethylene glycol
AMPK 5’ AMP-activated protein kinase
ANOVA Analysis of variance
ATCC American type culture collection
AUC Area under the curve
BCA Bicinchoninic acid assay
BCRP Breast cancer resistance protein
CI Combination index
CMC Critical micelle concentration
Das Dasatinib
Das/m Das-loaded micelles
Deff Hydrodynamic diameters
DLS Dynamic light scattering
DMEM Dulbecco's modified Eagle medium
DMF N, N-dimethyl formamide
DMSO-d6 Deuterated Methyl sulfoxide

XVI
DTX Docetaxel
ALK-m/DTX DTX-loaded non-conjugated micelles
ALN-m/DTX DTX-loaded alendronate-conjugated micelles
DTX/m DTX-loaded micelles
DTX/m+Das/m Drug loaded micellar cocktail
DTX+Das/m Combinational drug-loaded micelles
EC50 Half-maximal effective concentration
EDC 1-3-dimethylaminopropyl-3-ethylcarbodiimide
hydrochloride
EDTA Ethylenediaminetetraacetic acid
EGFR Epidermal growth factor receptor
EGTA Ethylene glycol-bisβ-aminoethyl ether-
N,N,N’,N’-tetraacetic acid
ELISA Enzyme-linked immunosorbent assay
Em Emission
EPR Enhanced permeability and retention
Ex Excitation

XVII
F-12K Ham's F-12K Kaighn's Medium
FBS Fetal bovine serum
GAS Growth arrest-specific
VCAM Cell adhesion molecule
Glu L-glutamic acid γ-benzyl ester
GPC Gel permeation chromatography
H&E Hematoxylin and eosin
HAP Hydroxyapatite
HCl Hydrochloride
HPLC High-performance liquid chromatography
HPMA N-2-hydroxypropylmethacrylamide
HSC Hemopoietic stem cells
i.p. Intraperitoneal
i.v. Intravenous
IACUC Animal care and use committee
IC50 Half-maximal inhibitory concentration

XVIII
IL Interleukin
IR Irradiation
IVIS In vivo imaging system
L.C. Loading capacity
M-CSF Macrophage colony-stimulating factor
MDR Multidrug resistance protein
MEM Minimum essential medium eagle
mPEG Methoxy polyethylene glycol5k
mPEG-PLE Methoxy polyethylene glycol5k-block-polyL-
glutamic acid
mPEG-PLE-PLF Methoxy polyethylene glycol5k-block-polyL-
glutamic acid-block-polyL-phenylalanine
MRM Multiple reaction monitoring
MRP Multidrug resistance-associated protein
MRT Main drug residual time
MTD maximum tolerant doses
MTT 3-4,5-Dimethylthiazol-2-yl-2,5-

XIX
diphenyltetrazolium bromide
MW Molecular weight
MWCO Molecular weight cut off
MWw Weight-averaged molecular weight
Na2SO4 Sodium sulfate anhydrous
Na3VO4 Sodium orthovanadate
NaF Sodium fluoride
NaOH Sodium hydroxide
NBF Neutral buffered formaldehyde
NCA N-carboxy anhydride
NCA Non-compartmental analysis
NHS N-hydroxysuccinimide
OPN Osteopontin
PAGE Polyacrylamide gel electrophoresis
PBS Phosphate buffered saline
PDI Polydispersity index

XX
PE Plating efficient
PEG Polyethyleneglycol
PhA L-phenylalanine
PI Propidium iodide
PI3K Phosphoinositide 3-kinase
PLE PolyL-glutamic acid
PLF PolyL-phenylalanine
PMSF Phenylmethane sulfonyl fluoride
preOB-CM MC3T3-conditioned medium
QTRAP Triple quadrupole linear ion traps
RANK Receptor activator of NF Kappa B
RANKL Receptor activator of NF Kappa B ligand
Raptor Regulatory associated protein of mTOR
TFA-d1 Deuterated trifluoroacetic acid
RBC Red blood cells
RIPA Radioimmunoprecipitation assay

XXI
RPMI Roswell park memorial institute medium
r.t. Room temperature
Rt/R0 Post-treatment radiance over initial radiance
RTK Receptor tyrosine kinase
SD Standard deviation
SDS Sodium dodecyl sulfate
SF Surviving fraction
STAT Signal transducers and activators of
transcription
T1/2 Half-life time
TAM Tumor-associated macrophages
THPTA Tris3-hydroxypropyltriazolylmethylamine
TRAP Tartrate-resistant acid phosphatase
Tris Trishydroxymethylaminomethane
UV Ultraviolet–visible spectroscopy
v/v% Volume percentage

XXII
VD Volume distribution
WBC White blood cells

XXIII
LIST OF CONTRIBUTORS
1. Chapter II – Dr. Svetlana Romanova, Dr. YuXiang Dong, Dr. Xinming Liu assisted in the
polymer synthesis and conjugation. Dr. Rakesh guided the osteoclast differentiation
study and the establishment of the mouse model. Ms. Michelle Varney helped in TRAP
assay and cell maintenance. Mr. Hongjun Wang provided suggestions in the left-
ventricle injection. Dr. Larisa Poluektova supported the bone decalcification. Mr. Hang
Su helped in blood collection in the pharmacokinetic study. Dr. Samuel M Cohen
examined the tissue samples. Michelle Varney provides technique support for osteoclast
staining. Dr. Chi Zhang, Dr. Wang Shuo and Dr. Megan Hyun conduct IR treatment.
2. Chapter III – Dr. Svetlana Romanova performed the polymerization reaction and GPC
characterization. Dr. Beth K Clymer ran the western blot for AMPK/pAMPK analysis. Dr.
Michael A. Hollingsworth and Ms. Jenea Sweeter helped in-cell ELISA quantification.
Nicholas Wojtynek performed the fluorescent microscopic imaging. Dr. Xin Wei provided
the suggestion on bone tissue fixing and decalcification. Dr. Samuel M Cohen examined
the tissue samples and assisted in animal study design.
3. Tong Liu made major contributions in all chapters. The overall project was designed
under the guidance of Dr. Tatiana K. Bronich. Dr. Rakesh Singh provided valuable
direction to the projects.

XXIV
4. The work was financially supported by National Institutes of Health (U01 CA198910),
Nanomaterials Core Facility of the Center for Biomedical Research Excellence (CoBRE).
Award from the National Institute of General Medical Sciences of the National Institutes
of Health under grant number P20GM103480. The stipend support for Tong Liu was
provided by research assistantship from Nebraska Center for Nanomedicine (2013-
2014), Nanomaterials Core Facility of the Center of Biomedical Research Excellence
(CoBRE) (2014-2018), and China Scholar Council (2013-2017).

1
CHAPTER I
INTRODUCTION

2
1.1 CANCER AND METASTASES
1.1.1 Improved clinical care in cancer
Since its first documentation, cancer has a long history for more than three
thousand years[1]. Projected by the American cancer society, over 1.7M people are
newly diagnosed for cancer in the US, and 600 thousand patients will die in 2018.
Among all cases, breast cancer, lung cancer, and prostate cancer were forecasted to be
the most common ones[2]. Over the last few decades, the fundamental of the disease
was explored extensively with advanced molecular biological technologies. Researches
identified numerous pathways, genetic regulation, and mutations that associate with the
tumor cells and novel drugs were designed to target the discovered mechanism.
Diagnostic also evolved drastically, allowing the detection of the tumor cells at very early
stages[3] as well as personally profiles phenotypes that could be sensitive to specific
drug molecules[4]. Deep learning and artificial intelligence facilitated tools that emerge
since 2014 made the way into the clinics successfully, helping to build the most effective
treatment regimen for individual patients based on their bassline characterization[5].
With all the efforts above, people have achieved substantial improvement of the overall
therapeutic outcome, longer survival time, and better qualities of life.

3
1.1.2 The deadly challenge by cancer metastases
Even with so many achievements, cancer is still considered incurable. The
disease is found refractive even after complete remission and tumor-free for several
years. Once comes back, tumors are frequently found in distant organs other than the
primary location (metastases), for example in lung, liver, and bone[6]. Also, quite a few
populations were found to have the metastases at initial presentation due to the
relatively late diagnose. The forming of the bone metastases generally takes the steps of
disseminating from the primary site, gaining access to the systemic circulation, homing
to a distant organ and developing into metastatic lesions[7]. The metastases are usually
not dissectible by surgery as they disperse widely to multiple sites and diffuse deep into
the healthy tissue. If from recurrence, metastases are generally less sensitive to the
same drug, because the tumor that survives from the previous treatment has been
positively selected for the "right" gene or drug-resistant phenotypes. Tumor
microenvironment also supports the colonization of the metastases, making it extremely
challenging to combat[8].

4
1.2 BONE METASTASES
1.2.1 Preferential metastasis to bone
The selection of organs for circulating tumor cells to reside does not appear to be
random. Tumors with different origins have shown their preference. Among the cases,
bone tends to be one of the most popular places to settle, especially in patients with
breast and prostate cancer[9,10]. Both tumors will likely metastasize to the bone at the
late stage, and the bone undergoes pathological destruction and results in severe pain
and spinal paralysis if in the vertebrae. The reason why bone is attractive to the
disseminated tumor cells were not fully clarified. The widely acknowledged hypothesis is
the “seed and soil” theory[11]. Tumor cells were believed to seed itself to the bone due
to the favorable environment that has the stem cell niches to support the initial
colonization and host cells that can remodel the bone further by interacting with the
tumor cell. When looking into the details of how metastases were distributed in the
skeletons, it is interesting that a large portion of the lesion forms in the axial skeletons
which are consist of head bones and trunk of a vertebrate[12]. Both hematopoietic
condition and the way blood is drained into the skeletons are accredited for the frequent
incidence of metastases in the axial skeletons and the girdles as suggested by
Batson[13].

5
1.2.2 Prognosis of the bone metastases
The prognosis of the bone metastases varies, depending, on the origin of the
diseases as well as the existence of other metastases. For breast cancer bone
metastases at the first relapse, the median survival time from the diagnose is close to
two years[14]. It could be more than four years, in the bone metastases from prostate
cancer with only axial skeleton affected. However, lung cancer metastasized to bone has
a much shorter survival period that is counted by months[10]. Other metastases taking
places at the same time (e.g., liver, lung) can drastically deteriorate the prognosis and
shorten the survival time from years to only a few months. Besides, patients with
exclusive metastases in bones at the initial relapse might also be followed with the
secondary lesions in visceral organs, which could also substantially differentiate the
prognosis from those who do not. In a study of 17,251 patients with bone metastases
from multiple types of cancers, Elisabeth found the prognosis depends on both the
synchronous metastases as well as the types of cancer. However, it is interesting that
the report was suspected to have selection bias due to misclassification of the patients
with bone metastases of mild symptoms or lesser extent than other metastases to be
recorded. Hence, the modality risk could be potentially higher than estimated[15].

6
1.2.3 Major Clinical symptoms
Bone fracture is the symptom commonly occurs in bone metastases, especially
for breast cancer bone metastases. More than half of the patients with pathological
fracture were the one with breast cancer[16]. The symptom commonly happens in the
long bone, femur, rib and vertebral and lead to disability (broken long bone) or lung
diseases (vertebral collapse)[17,18]. Tumor-induced osteolytic lesion by the excessive
osteoclast resorptive activity contributes most to fractures. The process is associated
with unlocalized pain that does not relieve during sleep or by laying down and
significantly affect the quality of life[19]. Part of the pain could also be attributed to the
movement of the fractured bones. Pain in return is considered as an indication of
developing bone metastases[18].
Hypercalcemia is another frequently observed symptom. The increase calcium
level in serum is contributed mostly by the enhanced calcium reabsorption. The PTHrP
secreted from tumor cells can promote the process in Henle ascending limb and distal
convoluted tubule[20]. Pathological bone resorption by osteoclast also releases
additional soluble calcium to the serum. The factors like PTHrP, IL-6, IL-11, and VEGF
produced by tumor cells are known to affect the osteoblast cell and lead to elevated
expression of the RANKL, which is the differentiating stimuli for osteoclast maturation.
Some cell lines were also reported to produce the RANKL directly or secrete proteases
that cleave the anchored RNAKL into the soluble form[21]. Hypercalcemia leads to pains,
cognition difficulties, vomiting, etc., and requires immediate control. Hypercalcemia can

7
be severe and a sign of the disease progression. It generally comes with the poorer
prognosis, and about half of the patients with hypercalcemia died within one month[18].
The spinal cord compression is also frequently diagnosed in the clinic that
reduces the survival time substantially[22]. The affection of the spinal cord can result in
severe pain, vertebral collapse motility issues, bladder, bowels dysfunction. About one
out of five patients with bone metastases will end up with the spinal cord compression,
and it is common in breast, prostate and lung cancer[23]. Multiple evolvements at the
different location are frequently reported: thoracic spine accounts for 60-80% of all the
cases whereas the cervical and lumbar share the rest[24].

8
1.3 BONE-TUMOR MICROENVIRONMENT
The microenvironment of bone is a complicated and well-balanced biological
condition maintained by the extracellular matrix, hematopoietic cells, mesenchymal stem
cells bone marrow stromal cells and infiltrated immune cells. In 1889, Stephen Paget
highlighted the importance of the bone microenvironment and believed it to be the
proper "soil" that is used by cancer "seed." The follow-up studies have revealed more
details about how cancer cell turn the bone to its favor.

9
1.3.1 Hemopoietic stem cells (HSC) niche
The hemopoietic stem cells (HSC) niche was suggested to be the first
component in the bone that attracts circulating tumor cells to home, establish the initial
colony and protect it from chemotherapy. In the physiological condition, the CXCR4 is
produced by osteoblast and bone marrow stromal cells to direct hemopoietic cells to the
niches. The receptor of CXCR4, CXCR12 was also found on tumor cells. Studies in cells
and animals have validated that the CXCR12 is overexpressed in the highly metastatic
lesion and CXCR4/CXCR12 is one of the critical factors that regulate the tumor
migration and help the tumor cells to penetrate the basement membrane[25]. Once
arrived at the bone, the adhesive receptor Anxa2-R expressed on tumor cells can be
used for their retention and survival in the bone marrow, just as HSCs. Other ligands,
growth arrest-specific 6 (GAS-6), vascular cell adhesion molecule 1 (VCAM-1) and
matrix protein osteopontin (OPN) were also reported to be associated with the first stage
invasion. Just as mentioned above, tumor cells share a lot in similar to the HSCs, who
are regulated to go to bone niches via the physiological mechanisms[26]. Another way of
how tumor cells “hijack” the biological system is to compete with HSCs to occupy the
niches. It is reported that HSCs alter their expression of adhesive ligand Notch-1 and
Tie-2 and exit the niches, vacating the space for tumor cells to reside[27]. As a result of
such invasion, more unmatured hemopoietic cells were found in the circulation in a
metastatic prostate cancer model in bone.

10
1.3.2 Bone remodeling and the "vicious cycle."
Bone is a living organ and constantly under remodeling. In healthy adults,
osteoclast and osteoblast derived from monocyte linage work in conjunction to remove
the “outdated” mineral composition and replace it with the fresh one immediately. The
balance of both resorption and formation is critical for maintaining the bone mass and its
normal function[28,29]. One of the widely acknowledged mechanisms that bone
remodeling process relies on is the receptor activator of NF Kappa B (RANK) ligand
(RANKL) system, which is further regulated by osteoprotegerin (OPG). In vitro and in
vivo studies confirmed that RANKL is essential for differentiating macrophage into
matured osteoclast and maintain its resorptive function. While OPG serves as a
neutralizer and prevents the binding between RANK and RANKL, the relative
OPG/RANKL is suggested to control the bone remodeling process.
Tumor cells are found to secrete various cytokines that directly or indirectly affect
the RANKL/OPG level. These include parathyroid hormone, interleukins, and tumor
necrosis factors and can promote the production of RANKL in osteoblast while
suppressing the OPG, moving the balance toward bone resorption[30]. Research
observed higher expression level of PTHrP in metastases in bone than other visceral
organs. The finding fits into the tumor-induced bone resorption theory and hints the
association was not solely intrinsic but evolved along the time[31]. Other factors from
tumor cells, like colony stimulating factor (M-CSF), can also facilitate the osteoclast
formation[30].

11
The increased level of osteoclast and bone degradation will create the physical
space for the expansion of metastatic colony and more importantly release the
"nutrients" that was embedded inside the matrix when the bone was modeled. Bone is
known to be rich in growth factors like TGF-beta, IGFs, FGFs, PDGF and BMPs[32].
They are believed to influence the tumor growth from the following aspects: directly
promoting tumor cells survival and proliferation, stimulating the secretion of osteolytic
mediates by tumor cells, and even changing the tumor cells’ phenotype. In animal
studies, it has been uncovered that the Smad mediated pathway was involved in the
progression of the bone metastases. Mice inoculated with tumor cells that have Smad4
knockdown or express inhibitory Smad7 showed decreased bone metastases[33]. TGF-
beta also induces the production of osteolytic factors, e.g., PTHrP. Previous research
uncovered that high-level of expression in breast cancer bone metastases is not intrinsic
but rather the result of the stimulation by TGF-beta released from the matrix, or in other
words, a phenotypical change happened due to the interaction between tumor and
bone[34]. The PTHrP subsequently promote the expression of the RANKL in bone host
cells and consequently lead to enhanced osteoclast resorption. More interestingly, it is
not the only factor that represents the alteration of the tumor phenotype to utilize the
bone microenvironment. The interleukin-8 (IL-8) releasing level was found to be greater
in cells derived from metastatic lesion than in the primary tumor and suggested the
correlation between the metastatic potential and IL-8[35].
Tumor-associated macrophages (TAM) has been intensely investigated for
shaping the tumor microenvironment. The macrophage is generally classified into two

12
main phenotypes, M1 and M2: M1 is the phenotype driven by IFNγ or bacterial products
and is considered as "cytotoxic" phenotype that suppresses tumor growth; M2 is the one
induced by IL-4 or IL-13 and is believed to serve as a support role for tumor
progression[36]. The tumor-associated macrophages are thought to originate from
chemotactic recruitment from both peripheral blood circulation (monocyte or monocyte-
related cells) and residential macrophages[37,38]. It has been discovered that various
factors could act as the chemoattractant for the recruitment, including CSF‑ 1, VEGF,
IL‑ 34, CCL2, CCL5, C5a. Interestingly, these molecules were found not only produced
by tumor cells, but also by the TAM that already infiltrated inside the tumor tissue[39],
suggesting that tumor-TAM works as a whole entity in maintaining the stable population
of TAM in the tumor lesion as the tumor grows[40]. M2-like subtype has attracted
substantial attention and been studied as it promoted some of the key processes in
tumor development, including angiogenesis, immunosuppression, metastasis and
genetic instability[41]. When it comes to the effect on chemo drug treatment, mixed
results were reported with both improved efficacy and the rise of resistance[36], which
correlated with the researches that suggested the relationship of M1/M2 ratio to the
disease prognosis[42]. Evidence also indicated that the TAM is not the same as
standard format as seen in non-neoplastic tissue but has rather board heterogenicity (a
subpopulation of skewed M2), shaped by the metabolic re-programming in different
tumors[43,44]. Most of the studies in TAM focused on the primary tumors and soft organ
metastases while few have been explored in cancer bone metastases. One important
role that TAM involves in the metastatic cancer development is to follow the “scene” set
by the tumor cells and provide the trail that leads the circulating tumor cell to settle down
in the bone[45].

13

14
1.4 CURRENT TREATMENT OPTIONS IN THE CLINIC FOR BONE METASTASES
The overall paradigm in skeleton metastases treatment aims to target three
aspects of the disease: kill tumor cells (or suppress their proliferation), prevent bone loss
and symptom control. The tumor development is the cause and key of the complication,
thus controlling the tumor growth and even eliminate them is the essential and most
demanding need. The purpose could be achieved to some extent by the treatment of
antiproliferative chemo drug, targeted kinase inhibitors and androgen deprivation agents.
As discussed in the previous section, the tumor does progress alone, but also "hijack"
host cells into helpers that remodel the microenvironment in favor of tumor growth. Anti-
resorptive drugs are used in conjunction with the chemotherapy to interfere one of the
most crucial pathological process, the bone destruction, and showed beneficial in
prolonging patient survival time. Pain is one of the symptoms that are frequently seen in
cancer, especially in bone metastases where pain could be severe. Palliative therapies
are adopted to relief the symptom and improve patients’ quality of life.

15
1.4.1 Treatment directly against tumor growth:
Taxane (e.g., Docetaxel) and anthracyclines (e.g., Doxorubicin) remains the first-
line therapy in the chemotherapy system for patients with bone metastases. These small
cytotoxic molecules were used as a single agent or in combination or sequentially. For
breast cancer metastases, the median time to progression after chemotherapy ranges
from 3 months to a year and the median survival time ranges from 8 months to two
years[46]. Patients who had prior exposure to anthracycline tended to respond in a lower
rate to taxane, suggesting the emergence a possible multidrug resistance. Both classes
of drugs were limited by their toxicities: bone marrow suppression in taxane-based
treatment[47] and cardiotoxicity in anthracycline[48]. In metastatic breast cancer,
another commonly used therapy is androgen deprivation (or suppression), as more than
seventy percent of the patients are hormone receptor-positive[49]. This type of drugs
showed the generally good response the first time however the disease relapsed after
several cycles of the treatments. Hormonotherapy is also used in combination of with
chemotherapy. In a study of prostate bone metastases, 17-month improvement in
median survival time was observed when adding the docetaxel to the androgen
deprivation therapy[50]. For the subpopulation with HER2 positive tumors, a monoclonal
antibody that targets HER2 can be used in combination with chemotherapy. In a study of
186 patients with metastatic breast cancer, the median survival time increased from 6
months (Docetaxel alone) to 31months (Docetaxel and Trastuzumab)[51]. By 2018, over
30 receptor tyrosine kinase (RTK) inhibitors have been approved in the clinic for various
cancers[52]. This class of drugs is considered as the targeted therapy to intervene the
many aberrantly regulated signal transduction[53] and has proved the efficacy not only

16
as a single agent but also as the adjuvant to overcome the drug resistance[54]. Several
RTKs were also tested in patients with bone metastases[55]. For instance, sunitinib was
shown to prolong the survival in patients with bone metastases from cancer of unknown
primary[56].

17
1.4.2 Treatments for bone resorption:
Bisphosphonates were mainly used to treat osteoporosis and to prevent the bone
loss[57]. The chemical structure of bisphosphonate is compromised by two phosphorus
groups that covalently bound to the one carbon atom. This conformation grants it high
binding affinity to the hydroxyapatite which is the mineral compartment of the bone[58].
Systemically administered bisphosphonate will "shoot" to the bone, and upon bone
resorption, the drug will be uptake by osteoclast cells. The accumulation of
bisphosphonate in osteoclast cell will inhibit farnesyl pyrophosphate synthase and finally
cause the osteoclast cell detaching from the resorption surface. Depending on the
concentration and the efficacy of specific bisphosphonate, the osteoclast could also
become apoptotic after drug exposure[59]. The ability of bisphosphonates to target bone
and to limit the resorptive activity of osteoclast makes them good candidates to intervene
the bone remodeling in metastatic lesion. Indeed, zoledronate was found to delay the
occurrence of skeleton-related symptom in patients with prostate cancer bone
metastases. However the drug did not prolong the overall survival time[60], nor did they
decrease the risk of developing metastases when used as a prevention medicine[61].
Apart from directly acting on osteoclast cellular function, another approach is to
control the differentiation and activation of the osteoclast. As discussed in the previous
section RANKL is the key to regulate osteoclast-mediated bone resorption. Denosumab
is a human monoclonal IgG2 antibody that binds to RANKL with high specificity and
further prevents the contact between RANKL and RANK, cutting off the activation

18
signal[62]. A clinical study showed Denosumab had an even better response and could
delay the skeleton related events even more than what zoledronate did[63].

19
1.4.3 Treatment for pain control:
Non-invasive beam radiotherapy and radiopharmaceuticals are two major
approaches used in the clinic for palliating skeletal metastases. External beam
radiotherapy has been established to relieve the bone pain within 4–6 weeks. American
Society of Radiation Oncology suggests single fraction radiotherapy using a dose of 8Gy
to provide the palliation[64] and the repeated dosing is possible if pain recurs[65].
Radiopharmaceuticals like strontium-89, samarium-153, and radium-223 have been
approved by FDA as bone-targeted agents for pain control. These emitters serve as
substitutes, which will be uptake by osteoblast cells when they construct new bones in
prostate cancer (osteoblastic metastases)[66]. The radiopharmaceuticals were also
found to work well in combination of chemotherapy (taxane), and the toxicity was well
tolerated[67]. However, radiotherapy in general (except one study[68]) did not improve
the overall survival time in bone metastases[69].

20
1.5 CHEMO DRUG RESISTANCE
Drug resistance was a concept initially in anti-bacterial treatment, where the
bacteria become tolerant to antibiotics. The similar phenomenon is observed in cancer
treatment. Despite the various therapeutic agents based on the different mechanisms of
action, the tumor cells could always develop resistance during the therapy or after
relapsing. Due to the toxicity of anti-cancer drugs, the dose is limited, and thus the
development of resistance significantly hinders the therapeutic effect. With the advanced
analytical tools in genomic and proteomic researches, several mechanisms of the drug
resistance have been discovered, and strategies were explored to overcome the barriers.
Studies revealed that different tumors with characterization are sensitive/resistant to
different treatments. The concept of precision medicine was also proposed in this
context, that the optimal drug candidates could be identified based on the unique
biological fingerprint of different tumors. However, the question that how the cells
establish or acquire such resistance remains controversial and the approach to avoid
inducing the resistance is still unknown.

21
1.5.1 Mechanism of the drug resistance
A variety of molecular mechanisms have been revealed in drug resistance.
These could be summarized and classified into the following aspects: the accumulation
of the drug inside cancer cells, upstream action of the drugs, and downstream cellular
response.
The drug efflux is considered the main barrier in drug uptake and one of the well-
studies mechanisms of drug resistance. Tumor cells actively pump out the cytotoxic drug
to keep the intracellular concentration at a lower level and survive through the regular
dose. The ATP-binding cassette (ABC) transporter family proteins play a significant role
in this kind of resistance. ABCs are essential to maintaining the physiology condition.
Epithelia cells in liver and intestine express the transporters at a high level to avoid over
accumulation of toxins. In tumor cells, three transporters including multidrug resistance
protein 1 (MDR1), multidrug resistance-associated protein 1 (MRP1), and breast cancer
resistance protein (BCRP) are accredited for resistance driven by drug efflux. These
transporters share a board range of substitutes specificity that overlaps with the
commonly used chemo drugs (taxane, anthracyclines and kinase inhibitors). Many of the
tumor (liver, kidney, and colon) were found to have higher expression of the transporter
proteins compared to the corresponding healthy tissue, whereas some of the tumors
were able to increase the expression upon chemo drug exposure. In the clinic, the
overexpression of MRP1 has been positively associated with chemoresistance and poor
outcome, in prostate, lung and breast cancer and the level of BCRP were used as a
predictive marker for potential drug response and survival rates in patients with small cell

22
lung cancer. The inherited resistance found in tumor stem cells was also suggested to
correlate with the expression of MDR and BCRP.
Another important upstream drug resistance is due to the drug inactivation.
Platinum-based drugs can be inactivated by thiol glutathione[70,71]. In other cases, the
prodrug cannot be metabolized into the active form, for example, capecitabine cannot be
converted into 5-FU by thymidine phosphorylase28 when the gene encoding thymidine
phosphorylase is inactivated by methylation[72]. Besides the alteration of
pharmacokinetics and pharmacodynamics, the drug’s target can also change to reduce
the efficacy. The change can be reflected as the different expression level or
composition alteration of targeted proteins. When using androgen deprivation agent, the
expression of androgen receptor was found to increase in the patients who showed
resistance to the therapy[73]. The increased quantities of the pharmacological targets
require more active inhibitors to show the treatment effect.
Even when the tumor cells are under stress by sufficient functional drugs that
successfully act on the target, the downstream survival response can still save the tumor
cells from dying. One typical mechanism is the DNA repairing, also known as DNA
damage response. The process is intrinsic in the normal cells and contributes to the
genome stability. The specific signal transduction pathways vary in the different type of
damage as reviewed by Alberto[74]. In the tumor, the nucleotide excision or mismatch
repair were recognized as the dominant mechanism that involved in resistance to
platinum-based DNA damaging agents[75]. However, tumors are frequently to have

23
mutations that cause the deficiency in one of the repairing pathways, and the actual
resistance relies on the redundant repairing pathways[76].
Apart from the repairing mechanism, the inhibition of apoptosis at the same time
plays an important role to allow tumor time to overcome the treatment effect. The
inhibition in cell death does not exclusively happen in tumors with DNA damage but also
in those treated with targeted inhibitors as well. Studies discovered that although
different chemo drugs may have distinguished mechanism of action, the intrinsic
apoptotic process is disrupted by the overexpression of BCL-2[77]. Extensive
researches have been done and identify a series of factors that dysregulate the
apoptotic function, including the BCL-2 family protein, p53, and inhibitors of apoptosis
proteins[78].
Related to the apoptosis inhibition, autophagy acts as a double-edged sword in
cancer resistance. Autophagy is considered a recycling process to increase the
threshold of the metabolic stress required for cellular apoptosis by eliminating the
malfunction organelles and pro-apoptotic signal transductors. Both apoptotic and
autophagic pathways share many signaling mediates in common, like p53 and BH3[79].
Evidence has shown that the pro-apoptotic factors can activate the autophagy, which is
a pro-survival activity[80]; while autophagy can end up with the completion of apoptotic
signaling transduction, which is the initiation of the cell death[81]. Thus, both inhibitors
and activators of autophagy have been proposed to overcome cancer resistance[82].

24
Epidermal growth factor receptor (EGFR) is another pro-survival factor that has
been intensely investigated. Activation of EGFR will subsequently trigger a series of
downstream signaling cascades, including the KRAS-BRAF-MEK-ERK pathway,
phosphoinositide 3-kinase (PI3K), phospholipase C gamma protein pathway, the anti-
apoptotic AKT kinase pathway, and the STAT signaling pathway, which tunes the
cellular activities like proliferation and survival[83]. Overactivation of EGFR was
commonly observed in many types of cancer and was considered as part of the
chemoresistance[84]. Small molecule inhibitors and monoclonal antibodies were
successfully developed and available for lung and colorectal cancer but yet failed to
prove the efficacy in patients with breast cancer[85].

25
1.5.2 The ways to acquire drug resistance
It has been widely acknowledged that the drug resistance can be driven by the
gene mutation. For example, cancer cells that resist to DNA damage were found to have
the mutation in p53[86], MLH1 and MSH2[87]; those resist to EGFR targeted inhibitors
showed gatekeeper residue mutations in EGFR[88] or mutations in the downstream
kinase, like ALK tyrosine kinase[89]. Indeed, the emerging of drug resistance has been
described as an evolution followed by Darwin's law. The genomic instability is one of the
hallmarks of cancer and contributes to a board spectrum of the tumor heterogeneity[90].
Chemotherapy holds the pressure of natural selection and allows the enrichment of the
subclonal populations that happen to have the resistant mutation[91] while increasing
the genomic instability to maintain the intratumoral heterogeneity[92]. A recent model
also supports the theory of evolution, where tumor cells were observed to gain the
amplification of an oncogene under the continuous treatment of ERK inhibitors[93].
However, the other study that focuses on the same oncogene but followed a different
route. The researchers observed profound variability in cellular dynamics with a small
population that had transient tolerance to the treatment, which ultimately converted to
the inheritable resistance via epigenetic reprogramming[94], suggesting the non-genetic
part of drug resistance. Another study about the multidrug resistance in HL60 leukemic
carried out the experiments to eliminate the selective effect and explained the
appearance of the resistance by Lamarckian induction, where tumor cells responded to
the stress with transcriptome changes and switched between resistant and non-resistant
phenotypes[95]. Similar phenotypical was found in breast cancer cells, and the

26
stochastic gene expression of CD24/CD44 was modeled to explain the chemotherapy-
induced resistance[96].
The nongenetic transition can be further interpreted under the concept of cell
attractors. Cells with the same genome can have multiple distinct phenotypes that are
considered to be stable, dictated by the "attractors." The relative stability of those cells
defines the popularity of each phenotype within the attractors, commonly creating a bell-
shaped cell distribution based on the gene expression level. Those who fall on the edge
of an attractor would have distinctive gene expression to the rest population. Cell
attractor can be redefined upon the exposure to chemo drugs, in other words, the
stability of the initial cell states can also change due to the treatment. If the state on the
edge of the attractor become more stable in the new condition, the subpopulation of
those cells will increase accordingly while the rest will decrease. Such phenotypical
transition shares the similar macrophenomenon as the natural selection but is based on
different mechanism. Qin analyzed the dynamics in a single cell resolution and proved
the transition continually happened within and between the attractors. Interestingly, the
study also found the cells tend to restore the original distribution after the loss a
subpopulation, suggesting that targeted elimination of drug-resistant or stem phenotypes
may not be a practical approach to overcome the resistance[97].

27
1.5.3 Overcome drug resistance
Many efforts have been made to overcome the drug resistance by developing
specific molecules that target the critical element on which the resistance relies. The
results were a mix of encouraging and disappointing.
Inhibiting the function of the overexpressed transporters was believed to enhance
the efficacy of cytotoxic molecules. Researches identified a few kinase inhibitors (e.g.,
Gefitinib) that can block the drug efflux and specific inhibitors with high affinity were also
developed into clinical stages (zosuquidar and tariquidar). However, the results of the
trial were negative, showing no improvement in overall survival or progression-free time.
The reason was suggested as "the high degree of functional redundancy” in transporter
family. Given the fact that chemo drug induces high expression of MDR, it could also be
speculated that the inhibition leads to "feedback response" in cells and results in more
production of the transporter to compensate the inhibitory effect.
For resistance to DNA damaging drugs, inhibition of the DNA repairing is a clear
approach. As the tumor cells commonly lack in one or more repairing pathway and rely
on the machinery that is redundant in normal cells, the synthetic lethality was
proposed[98]. The inhibitor of DNA damage response showed efficacy initially however
extra mutant was found to compensate the inhibition[99].

28
EFGR inhibitors are designed to cut off the pro-survival signaling and sensitize
the tumor cells to chemotherapy. However, the generation of secondary resistance is
almost inevitable. In an example of resistance against tyrosine kinase inhibitor, the
gatekeeper residue of BCR–ABL1 was mutated causing the inhibitor to lose the binding
affinity while maintaining the ontogenetical function[100]. Besides the alteration of drug
target, adaptive signaling bypass was discovered in the tumor that escaped from HER-
family tyrosine kinase inhibitors[101].
Such non-genetic adaptation is even more challenging to combat, especially
considering the complexity of the gene regulation network and the possible redundancy
in metabolic machinery. In the treatment against bacteria, people have found that the
resistance to one antibiotic may confer the susceptibility to another drug[102]. A similar
concept was proposed in cancer treatment that the resistance could be useful and
create the opportunity for “collateral sensitivity” as suggested by Mathew[103]. A
sequenced dosing regime was reported to overcome the adaptive resistance by inducing
the cell into the drug-tolerant state which is vulnerable to the secondary treatment[96].
Given the fact that the adaptive response could be universal and diverse to the stress
posed by the treatment, it is difficult to predict or avoid the actual resistance. Drug
combinations that affect different pathways at the same time is an option, as it could
diminish the pathway cross-talk[104] and decrease the possibility that tumor cells find an
effective way to transit into adaptation states.

29
1.6 NANOCARRIERS-BASED DRUG DELIVERY SYSTEM FOR CANCER THERAPY
Nanocarriers have been a versatile drug delivery platform for cancer therapy.
Various types of nanocarriers have been developed to encapsulate small molecules[105],
peptides[106], nucleic acids[107] and the combination of them. The chemical
composition and physical properties can be tuned to serve specific purposes, e.g.,
sustained release[108], stimuli-responsive behavior[109], tumor targeting[110],
prolonged circulation[111] etc. Systemically administration of the nanomedicine has
shown improved therapeutic outcome in the animal models as well as in the clinic[46].

30
1.6.1 Common types of nanocarriers and their general characteristics
Liposome:
The liposome is a sphere vesicle that consists of at least one lipid bilayer on as
an out shell and an aqueous phase in the middle. The lipid bilayer is usually
compromised by cholesterol and phospholipid. Depend on the number of the bilayers,
the liposome can be classified into multilamellar vesicles (multiple layers) and
unilamellar (single layers) vesicles. Two methods are frequently used to prepare drug-
loaded liposome, lipid film rehydration and reverse phase evaporation methods[112].
The former method produces small sized particles and the latter provide high
aqueous/lipid ratio and loading capacity. Due to the presence of aqueous phase and the
lipid bilayer, the liposome can encapsulate both hydrophilic and hydrophobic drugs
together and achieve better anticancer efficacy[113]. Even the carrier has the membrane
structure, it is still found to be treated as the foreigner when administered systematically
and is cleared by the mononuclear phagocyte system. PEGylation was reported to
effectively shield the liposome from being uptake and prolong the circulation time[114].
Liposome formulated chemo drugs have been successfully approved, and more clinical
candidates are under investigation[115].

31
Polymer-drug conjugate:
Polymer-drug conjugate is a macromolecule in which active pharmaceutical
ingredient is covalently attached to a water-soluble polymer via a labile linked in between.
The linker is supposed to undergo hydrolysis or be chopped off under the specific
condition, and the payload will then be released as active form. Various linker has been
designed to grant the conjugate with environmental responsive behavior to
pH(hydrazone), redox-condition(disulfide) and enzyme(cathepsin-sensitive
dipeptide)[116-118]. These linkers are chemically stable during the circulation and can
release the drug when being internalized into the tumor cells. N-(2-
hydroxypropyl)methacrylamide (HPMA) copolymers are one of the most widely used
polymers in drug conjugates. The hydroxy group in the side chain is ready to be modified
into suitable functional groups that can further attach the linkers and the drugs. Due to
the numerous side chain, it is accessible to conjugate drug combinations and to control
the ratio of individual drug in the same HPMA copolymer[119]. The dendrimer is another
popular format of the drug conjugates, a branched start-like polymer. Compared to
single strand copolymer (like HPMA), dendrimer had better-defined structure and well-
controlled molecular weight. Unlike HMPA-based conjugates, the dendrimer has the
drugs only attached to one end of the polymer, providing the ability to form secondary
amphiphilic micellar structures[120], but on the other reduce the loading capacity. Both
types of conjugates convert the small drug molecules into macromolecules which
prolongs the circulation time by avoiding renal filtration and limits the drug exposure to
healthy tissues[121]. Several polymer-drug conjugates have demonstrated the improved
antitumoral efficacy in animal models with different types of cancer. However no HPMA
or dendrimer based candidate so far had successfully clear the way to clinical
practice[122-124].

32

33
Antibody-drug conjugates:
The monoclonal antibody has a strong binding affinity and selectivity to the target
protein and is used to guide the small molecule drugs (warhead) to the tumor cells. Due
to the high specificity, the biodistribution of the warhead can be drastically improved,
which makes the drug candidate that is generally limited by off-target toxicity feasible
and well-tolerated in practice[125]. Each antibody typically comes with 2-3 warheads.
Similar to polymer-drug conjugates, the linker between antibody-drug conjugates and the
warhead is crucial in the successful design. The conjugation chemistry has evolved for
several generations. The attaching position has improved from random lysine residues
to the engineered cysteine group in the heavy chain, allowing better control of the
chemistry and quality. The labile linkers also changed from hydrazone-based to
protease-cleavable[126,127]. Thanks to the advanced understanding the fundamental
biology, the biparatopic antibody was adopted to enhance the receptor clustering and
facilitate the internalization process[128]. Antibody-drug conjugates have achieved great
success since their launch in the clinic[126] and many more candidates are entering the
clinical trials[129]. However, challenges remain in penetrating biological barriers and the
risk to cause fatal toxicity[130].

34
Polymeric micelles:
Micelles are self-assembling of the amphiphilic molecules (e.g., detergents) in
colloidal system. Similarly, polymers with both hydrophilic and hydrophobic residues can
form the micellar structure in the aqueous media. The polymeric micelles feature with of
high molecular weight, improved stability, and the potential to solubilize substantial
quantities of drugs. Most of the polymeric micelles reported were based on the block
copolymer and grafted polymers that comprised of a PEG shell and a hydrophobic core.
The length and the chemical composition of the polymers can be engineered to optimize
the micelles’ physicochemical properties, including size, surface charge, critic micellar
concentration, stability, drug loading capacity, stealth properties, targeting ability and
stimuli-responsive dissociation. The introduction of chemical functional groups (like
carboxy and primary amine) provided additional flexibility in loading drug with distinct
properties (hydrophilic drugs, nucleic acids drugs)[131] and in attaching tracers for
imaging purposes[132]. In particular, Swapnil used the carboxy group to crosslink the
micelles and further enhance the stability[131].
Moreover, the crosslinker can be replaced by labile ones to achieve stimuli-
responsive characteristic[133]. Drug-loaded polymeric micelles can be prepared by
several methods, film rehydration, dialysis, and nanoprecipitation[134]. A series of
studies have demonstrated the potential of polymeric micelles to improve the overall
survival in a mouse model of breast cancer, ovarian cancer, pancreatic cancer, and lung
cancer[131,135-137].

35
1.6.2 Passive tumor accumulation and active tumor targeting
One of the most important goals of formulating drugs into nanocarriers is to
improve the biodistribution and achieve accumulation selectively in tumor tissues.
Chemo drugs are generally toxic to the healthy tissue and may cause a severe adverse
effect, which limits the total dose in the treatment. As the on-site drug concentration is
directly related with the treatment efficacy, ideally the targeted delivery to the tumor can
increase the dose limit or enhance the effectiveness with the same dose, while keeping
the side effects at a minimum level.
Three main approaches were used to grant the nanocarriers with the tumor
targeting ability, either passively or actively. Tumor cells grow rapidly and commonly
have newly formed blood vessels that are formed by defective endothelial cells with
higher permeability than normal ones, while the lymphatic drainage is impaired. It is
believed that the leakage-retention would facilitate the diffusion of the small particles to
the tumor tissue during the circulation over the normal organs and lead to the
preferential drug accumulation(EPR effect)[138]. Since the born of the concept in 1986,
numerous studies have shown the evidence that supported the hypothesis and explored
the characteristics that determined whether the nanocarrier could take advantage of
EPR effect or not[139,140]. Size appeared to be one the most important factors and
particles with 50-100 nm diameter are generally considered to be effective[141-143].
Much less has been understood in the metastatic cancers that whether the EPR effect

36
exists or not. Two studies suggested the EPR effect may still be valid in lung and bone
metastases[144,145].
Besides the passive targeting ability based on EPR effect, decorating
nanocarriers with the binding ligands or antibodies has been extensively studied to
provide more specific targeting efficacy. Tumor cells were found to overexpress various
cell markers (e.g., CD19 CD44), EGFR, HER-2, folic receptor and ανβ3-Integrins,
providing the useful anchors for the targeting moieties. The nanocarriers, on the other
hand, have versatile reaction residues which can be chemically conjugated with the
targeting ligands, typically through click reaction, amide formation, ester formation, thiol-
maleimide reaction, etc [146,147]. The potential of different ligands or antibodies has
been explored, among which hyaluronic acid, folic acid, RGD peptides, monoclonal
antibodies to EGFR and HER-2 are those commonly used and the nanocarriers with the
targeting moiety exhibit enrichment in tumor and decreased accumulation in the liver or
other visceral organs[148-153].
For bone metastases, the current options are the targeting ligands that bind to
the mineral compartment of the bone or the antibody for the RANK ligand[154].
Bisphosphonate is one of the most commonly used ligands in the bone-targeted delivery
system, e.g., zoledronate and alendronate. It showed high binding affinity to
hydroxyapatite (HAP) on both bone formation and bone resorptive surface. Acidic
oligopeptides like Asp8 also exhibit strong binding to the HAP preferentially on the bone

37
resorption surface, on the contrary (AspSerSer)6 demonstrate selective accumulation on
the bone formation surface[155]. Beside of focusing on the HAP binding, Denosumab is
a monoclonal antibody that selectively neutralizes RANK ligand in the bone
microenvironment and has been approved for bone metastases treatment, offering an
alternative to the current targeting strategies.

38
1.6.3 Combinational therapy
The efficacy of monotherapy is generally limited due to many pro-survival
mechanisms and drug resistance in cancer. Combination of two or more drugs has been
proved to be an effective way to improve the therapeutic outcome and has been widely
adopted in the clinic. For patients with bone metastases, combination therapy prolonged
the survival time for 1-4 month when compared to monotherapy, and the improvement
tended to be more remarkable when the patients were not chemo-naieve[156]. The
conventional modalities contain two first-line chemotherapy or one with the addition of
targeted therapy that blocks the ontogenetic signaling pathways, suppressing the
survival response from multiple nodes[157,158]. Challenges remain to select the proper
drug unit for the combination. Exerting synergistic effect is an attracting principle and
serves as the fundamental that underlines the identification of optimal drug combination.
Synergy describes a scenario of the combination is more effective than the additive
result of the corresponding monotherapies. The concept allows fast screening of
possible combinations in vitro and determination of the optimal drug ratio. However, the
result may not be translated successfully in clinical trials. One important reason is the
different pharmacokinetic and biodistribution of two drugs could not ensure the tumor
cells are exposed to both drugs at the same time[159], whereas in vitro cell-based test
does not reflect the hindrance. Careful planning with computation modeling on the dose
regimes is necessary to compensate such difference[160-162] and the accuracy is
limited. With the help of nanocarrier, it is possible to load drug combinations inside the
particles and alter the PK profile of each drug to the same. Upon the uptake of drug-
loaded carriers, tumor cells are exposed to individual drugs at the predetermined ratio.

39
Another challenge of the combinational therapy is the additive toxicity. Therefore, each
drug component is applied under the optimal dosage. The increased risk of adverse
effect in the conventional treatment could also be offset by the change of biodistribution
in the nanoformulation. The nano strategy has been reported in a study by Keren Miller
that use bisphosphonate bearing HPMA-paclitaxel conjugate to treat breast cancer bone
metastases. The HPMA polymer did not only serve as a carrier but also the
antiangiogenetic agent that exerted the antitumoral efficacy in the combination of
paclitaxel[163,164].

40
1.7 CONCLUSION
Despite the remarkable advance in cancer therapy, metastatic malignancy remains
the leading cause of cancer death. Metastasis to bone is unique and yet dissertating.
The bone serves as fertile soil, and the host cells cooperate with tumor cells through
ligand-receptor crosstalk, supporting their growth and survival. Such interaction leads to
bone degradation, causing skeleton complications of pain, fracture, spinal compression
and hypercalcemia that drastically decrease the quality of life. However, the current
clinic treatments have limited efficacy other than pain control. The expected life span can
be only a few months or up to 3 years. The challenge is due to the bone-tumor
microenvironment as well as the "resistant" nature of the tumor itself. The studies in
adaptive resistant phenotype transition revealed another fact that needs to be taken into
consideration when developing new treatment modalities. Nanocarriers have been
developed as an attracting platform for cancer treatment. With the ability of tumor
targeting, combinational delivering and reducing toxicity, the nanocarrier-based delivery
system has demonstrated the potential to improve the therapeutic outcome and emerges
as one of the most useful tools in the treatment for bone metastases.

41
1.8 REFERENCES
1. Sudhakar A. History of cancer, ancient and modern treatment methods. J Cancer Sci Ther. 2009;1: 1–4. doi:10.4172/1948-5956.100000e2
2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 3rd ed. 2018;68: 7–30. doi:10.3322/caac.21442
3. Schiffman JD, Fisher PG, Gibbs P. Early detection of cancer: past, present, and future. Am Soc Clin Oncol Educ Book. 2015;35: 57–65. doi:10.14694/EdBook_AM.2015.35.57
4. Senft D, Leiserson MDM, Ruppin E, Ronai ZA. Precision oncology: the road ahead. Trends Mol Med. 2017;23: 874–898. doi:10.1016/j.molmed.2017.08.003
5. Bychkov D, Linder N, Turkki R, Nordling S, Kovanen PE, Verrill C, et al. Deep learning based tissue analysis predicts outcome in colorectal cancer. Sci Rep. 2018;8: 3395. doi:10.1038/s41598-018-21758-3
6. Hernandez RK, Wade SW, Reich A, Pirolli M, Liede A, Lyman GH. Incidence of bone metastases in patients with solid tumors: analysis of oncology electronic medical records in the United States. BMC Cancer. 2018;18: 44–54. doi:10.1186/s12885-017-3922-0
7. Valastyan S, Weinberg RA. Tumor metastasis: molecular insights and evolving paradigms. Cell. 2011;147: 275–292. doi:10.1016/j.cell.2011.09.024
8. Steeg PS. Tumor metastasis: mechanistic insights and clinical challenges. Nature Medicine. 2006;12: 895–904. doi:10.1038/nm1469
9. Obenauf AC, Massagué J. Surviving at a distance: organ specific metastasis. Trends Cancer. 2015;1: 76–91. doi:10.1016/j.trecan.2015.07.009
10. Coleman RE. Clinical Features of Metastatic Bone Disease and Risk of Skeletal Morbidity. Clinical Cancer Research. American Association for Cancer Research; 2006;12: 6243s–6249s. doi:10.1158/1078-0432.CCR-06-0931
11. Fidler IJ, Poste G. The “seed and soil” hypothesis revisited. Lancet Oncology. 2008;9: 808. doi:10.1016/S1470-2045(08)70201-8
12. Macedo F, Ladeira K, Pinho F, Saraiva N, Bonito N, Pinto L, et al. Bone Metastases: An Overview. Oncol Rev. 2017;11: 321. doi:10.4081/oncol.2017.321
13. Batson OV. The role of the vertebral veins in metastatic processes. Annals of internal medicine. Am Coll Physicians; 1942;16: 38–45.

42
14. Coleman RE, Rubens RD. The clinical course of bone metastases from breast cancer. Br J Cancer. 1987;55: 61–66.
15. Svensson E, Christiansen CF, Ulrichsen SP, Rørth MR, Sørensen HT. Survival after bone metastasis by primary cancer type: a Danish population-based cohort study. BMJ Open. 2017;7: e016022. doi:10.1136/bmjopen-2017-016022
16. Domchek SM, Younger J, Finkelstein DM, Seiden MV. Predictors of skeletal complications in patients with metastatic breast carcinoma. Cancer. 2000;89: 363–368.
17. Higinbotham NL, Marcove RC. The management of pathological fractures. Journal of Trauma - Injury, Infection and Critical Care. 1965;5: 792–798. doi:10.1097/00005373-196511000-00015
18. Selvaggi G, Scagliotti GV. Management of bone metastases in cancer: A review. Critical Reviews in Oncology/Hematology. 2005;56: 365–378. doi:10.1016/j.critrevonc.2005.03.011
19. Clohisy DR, Mantyh PW. Bone cancer pain. Cancer. 2003;97: 866–873. doi:10.1002/cncr.11144
20. Mundy GR. Metastasis: Metastasis to bone: causes, consequences and therapeutic opportunities. Nature Reviews Cancer , Nature Publishing Group; 2002;2: 584–593. doi:10.1038/nrc867
21. Lynch CC, Hikosaka A, Acuff HB, Martin MD, Kawai N, Singh RK, et al. MMP-7 promotes prostate cancer-induced osteolysis via the solubilization of RANKL. Cancer Cell. Cell Press; 2005;7: 485–496. doi:10.1016/j.ccr.2005.04.013
22. Rajer M, Kovač V. Malignant spinal cord compression. Radiology and Oncology. 2008;42: 23–31. doi:10.2478/v10019-007-0035-4
23. Cole JS, Patchell RA. Metastatic epidural spinal cord compression. The Lancet Neurology. 2008;7: 459–466. doi:10.1016/S1474-4422(08)70089-9
24. Tsuzuki S, Park SH, Eber MR, Peters CM, Shiozawa Y. Skeletal complications in cancer patients with bone metastases. International Journal of Urology. 2016;23: 825–832. doi:10.1111/iju.13170
25. Shiozawa Y, Pedersen EA, Havens AM, Jung Y, Mishra A, Joseph J, et al. Human prostate cancer metastases target the hematopoietic stem cell niche to establish footholds in mouse bone marrow. J Clin Invest. 2011;121: 1298–1312. doi:10.1172/JCI43414
26. Decker AM, Jung Y, Cackowski F, Taichman RS. The role of hematopoietic stem cell niche in prostate cancer bone metastasis. J Bone Oncol. 2016;5: 117–120. doi:10.1016/j.jbo.2016.02.005

43
27. Shiozawa Y, Pienta KJ, Taichman RS. Hematopoietic stem cell niche is a potential therapeutic target for bone metastatic tumors. Clinical Cancer Research. 2011;17: 5553–5558. doi:10.1158/1078-0432.CCR-10-2505
28. Hadjidakis DJ, Androulakis II. Bone remodeling. 2006. pp. 385–396. doi:10.1196/annals.1365.035
29. Zhou H, Lu SS, Dempster DW. Bone Remodeling: Cellular Activities in Bone. Osteoporosis in Men. Elsevier; 2010. pp. 15–24. doi:10.1016/B978-0-12-374602-3.00002-X
30. Zheng Y, Zhou H, Dunstan CR, Sutherland RL, Seibel MJ. The role of the bone microenvironment in skeletal metastasis. J Bone Oncol. 2013;2: 47–57. doi:10.1016/j.jbo.2012.11.002
31. Kohno N, Kitazawa S, Fukase M, Sakoda Y, Kanbara Y, Furuya Y, et al. The expression of parathyroid hormone-related protein in human breast cancer with skeletal metastases. Surgery Today. 1994;24: 215–220. doi:10.1007/BF02032890
32. Linkhart TA, Mohan S, Baylink DJ. Growth factors for bone growth and repair: IGF, TGFβ and BMP. 1996. pp. S1–S12. doi:10.1016/S8756-3282(96)00138-X
33. Kang Y, He W, Tulley S, Gupta GP, Serganova I, Chen C-R, et al. Breast cancer bone metastasis mediated by the Smad tumor suppressor pathway. Proc Natl Acad Sci USA. 2005;102: 13909–13914. doi:10.1073/pnas.0506517102
34. Chiechi A, Waning DL, Stayrook KR, Buijs JT, Guise TA, Mohammad KS. Role of TGF-β in breast cancer bone metastases. Adv Biosci Biotechnol. 2013;4: 15–30. doi:10.4236/abb.2013.410A4003
35. Bendre MS, Gaddy-Kurten D, Mon-Foote T, Akel NS, Skinner RA, Nicholas RW, et al. Expression of interleukin 8 and not parathyroid hormone-related protein by human breast cancer cells correlates with bone metastasis in vivo. Cancer Res. 2002;62: 5571–5579.
36. Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nature Reviews Clinical Oncology. 2017;14: 399–416. doi:10.1038/nrclinonc.2016.217
37. Feng X, Szulzewsky F, Yerevanian A, Chen Z, Heinzmann D, Rasmussen RD, et al. Loss of CX3CR1 increases accumulation of inflammatory monocytes and promotes gliomagenesis. Oncotarget. 2015;6: 15077–15094. doi:10.18632/oncotarget.3730
38. Qian B-Z, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, et al. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. 2011;475: 222–225. doi:10.1038/nature10138
39. Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454: 436–444. doi:10.1038/nature07205

44
40. Franklin RA, Liao W, Sarkar A, Kim MV, Bivona MR, Liu K, et al. The Cellular and Molecular Origin of Tumor-Associated Macrophages. Science. American Association for the Advancement of Science; 2014;344: 921–925. doi:10.1126/science.1252510
41. De Palma M, Lewis CE. Macrophage Regulation of Tumor Responses to Anticancer Therapies. Cancer Cell. Cell Press; 2013;23: 277–286. doi:10.1016/j.ccr.2013.02.013
42. Bingle L, Brown NJ, Lewis CE. The role of tumour-associated macrophages in tumour progression: implications for new anticancer therapies. J Pathol. 2002;196: 254–265. doi:10.1002/path.1027
43. Netea-Maier RT, Smit JWA, Netea MG. Metabolic changes in tumor cells and tumor-associated macrophages: A mutual relationship. Cancer Lett. 2018;413: 102–109. doi:10.1016/j.canlet.2017.10.037
44. Ruffell B, Affara NI, Coussens LM. Differential macrophage programming in the tumor microenvironment. Trends in Immunology. Elsevier Current Trends; 2012;33: 119–126. doi:10.1016/j.it.2011.12.001
45. Sousa S, Määttä J. The role of tumour-associated macrophages in bone metastasis. J Bone Oncol. 2016;5: 135–138. doi:10.1016/j.jbo.2016.03.004
46. Tran S, DeGiovanni P-J, Piel B, Rai P. Cancer nanomedicine: a review of recent success in drug delivery. Clin Transl Med. 2017;6: 44. doi:10.1186/s40169-017-0175-0
47. Guastalla JP, Diéras V. The taxanes: Toxicity and quality of life considerations in advanced ovarian cancer. Br J Cancer. 2003;89: S16–22. doi:10.1038/sj.bjc.6601496
48. Moazeni S, Cadeiras M, Yang EH, Deng MC, Nguyen K-L. Anthracycline induced cardiotoxicity: biomarkers and “Omics” technology in the era of patient specific care. Clin Transl Med. SpringerOpen; 2017;6: 17. doi:10.1186/s40169-017-0148-3
49. Xiong Z, Deng G, Huang X, Li X, Xie X, Wang J, et al. Bone metastasis pattern in initial metastatic breast cancer: a population-based study. Cancer Management and Research. Dove Press; 2018;10: 287–295. doi:10.2147/CMAR.S155524
50. Sweeney C, Chen Y-H, Carducci MA, Liu G, Jarrard DF, Eisenberger MA, et al. Impact on overall survival (OS) with chemohormonal therapy versus hormonal therapy for hormone-sensitive newly metastatic prostate cancer (mPrCa): An ECOG-led phase III randomized trial. J Clin Oncol. American Society of Clinical Oncology; 2014;32: LBA2–LBA2. doi:10.1200/jco.2014.32.18_suppl.lba2
51. Marty M, Cognetti F, Maraninchi D, Snyder R, Mauriac L, Tubiana-Hulin M, et al. Randomized phase II trial of the efficacy and safety of trastuzumab combined with docetaxel in patients with human epidermal growth factor receptor 2-

45
positive metastatic breast cancer administered as first-line treatment: the M77001 study group. J Clin Oncol. 2005;23: 4265–4274. doi:10.1200/JCO.2005.04.173
52. Ferguson FM, Gray NS. Kinase inhibitors: the road ahead. Nat Rev Drug Discov. 2018;17: 353–377. doi:10.1038/nrd.2018.21
53. Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nature Reviews Cancer. 2009;9: 28–39. doi:10.1038/nrc2559
54. Gross S, Rahal R, Stransky N, Lengauer C, Hoeflich KP. Targeting cancer with kinase inhibitors. J Clin Invest. 2015;125: 1780–1789. doi:10.1172/JCI76094
55. Gallick GE, Corn PG, Zurita AJ, Lin SH. Small-molecule protein tyrosine kinase inhibitors for the treatment of metastatic prostate cancer. Future Med Chem. 2012;4: 107–119. doi:10.4155/fmc.11.161
56. Ma Y, Zhou W, He S, Xu W, Xiao J. Tyrosine kinase inhibitor sunitinib therapy is effective in the treatment of bone metastasis from cancer of unknown primary: Identification of clinical and immunohistochemical biomarkers predicting survival. Int J Cancer. 8 ed. 2016;139: 1423–1430. doi:10.1002/ijc.30176
57. Papapoulos SE. Bone diseases: Bisphosphonates in osteoporosis - Beyond 5 years. Nature Reviews Rheumatology. 2013;9: 263–264. doi:10.1038/nrrheum.2013.57
58. Puljula E, Turhanen P, Vepsäläinen J, Monteil M, LECOUVEY M, Weisell J. Structural requirements for bisphosphonate binding on hydroxyapatite: NMR study of bisphosphonate partial esters. ACS Medicinal Chemistry Letters. 2015;6: 397–401. doi:10.1021/ml5004603
59. Drake MT, Clarke BL, Khosla S. Bisphosphonates: Mechanism of action and role in clinical practice. Mayo Clinic Proceedings. 2008;83: 1032–1045. doi:10.4065/83.9.1032
60. Saad F, Gleason DM, Murray R, Tchekmedyian S, Venner P, Lacombe L, et al. Long-term efficacy of zoledronic acid for the prevention of skeletal complications in patients with metastatic hormone-refractory prostate cancer. J Natl Cancer Inst. 2004;96: 879–882. doi:10.1093/jnci/djh141
61. Wirth M, Tammela T, Cicalese V, Gomez Veiga F, Delaere K, Miller K, et al. Prevention of bone metastases in patients with high-risk nonmetastatic prostate cancer treated with zoledronic acid: Efficacy and safety results of the zometa european study (ZEUS). European Urology. 2015;67: 482–491. doi:10.1016/j.eururo.2014.02.014
62. Kostenuik PJ, Nguyen HQ, McCabe J, Warmington KS, Kurahara C, Sun N, et al. Denosumab, a fully human monoclonal antibody to RANKL, inhibits bone resorption and increases BMD in knock-in mice that express chimeric (murine/human) RANKL. 2009. pp. 182–195. doi:10.1359/jbmr.081112

46
63. Fizazi K, Carducci M, Smith M, Damião R, Brown J, Karsh L, et al. Denosumab versus zoledronic acid for treatment of bone metastases in men with castration-resistant prostate cancer: A randomised, double-blind study. The Lancet. 2011;377: 813–822. doi:10.1016/S0140-6736(10)62344-6
64. Lutz S, Berk L, Chang E, Chow E, Hahn C, Hoskin P, et al. Palliative radiotherapy for bone metastases: an ASTRO evidence-based guideline. International journal of radiation oncology, biology, physics. 2011. pp. 965–976. doi:10.1016/j.ijrobp.2010.11.026
65. Hoskin PJ. Bisphosphonates and radiation therapy for palliation of metastatic bone disease. Cancer Treatment Reviews. 2003;29: 321–327.
66. Wong M, Pavlakis N. Optimal management of bone metastases in breast cancer patients. Breast Cancer (Dove Med Press). 2011;3: 35–60. doi:10.2147/BCTT.S6655
67. Morris MJ, Pandit-Taskar N, Carrasquillo J, Divgi CR, Slovin S, Kelly WK, et al. Phase I study of samarium-153 lexidronam with docetaxel in castration-resistant metastatic prostate cancer. J Clin Oncol. 2009;27: 2436–2442. doi:10.1200/JCO.2008.20.4164
68. Tu SM, Millikan RE, Mengistu B, Delpassand ES, Amato RJ, Pagliaro LC, et al. Bone-targeted therapy for advanced androgen-independent carcinoma of the prostate: a randomised phase II trial. Lancet. 2001;357: 336–341. doi:10.1016/S0140-6736(00)03639-4
69. Autio KA, Scher HI, Morris MJ. Therapeutic strategies for bone metastases and their clinical sequelae in prostate cancer. Curr Treat Options Oncol. 2012;13: 174–188. doi:10.1007/s11864-012-0190-8
70. Meijer C, Sluiter WJ, Mulder NH, Timmer-Bosscha H, Jan Meersma G, de Vries EGE. Relationship of Cellular Glutathione to the Cytotoxicity and Resistance of Seven Platinum Compounds. Cancer Res. 1992;52: 6885–6889.
71. Mehta K, Fok JY. Targeting transglutaminase-2 to overcome chemoresistance in cancer cells. Drug Resistance in Cancer Cells. 2009. pp. 95–114. doi:10.1007/978-0-387-89445-4_5
72. Kosuri KV, Wu X, Wang L, Villalona-Calero MA, Otterson GA. An epigenetic mechanism for capecitabine resistance in mesothelioma. Biochem Biophys Res Commun. 2010;391: 1465–1470. doi:10.1016/j.bbrc.2009.12.095
73. Palmberg C, Koivisto P, Hyytinen E, Isola J, Visakorpi T, Kallioniemi OP, et al. Androgen receptor gene amplification in a recurrent prostate cancer after monotherapy with the nonsteroidal potent antiandrogen Casodex (bicalutamide) with a subsequent favorable response to maximal androgen blockade. European Urology. 1997;31: 216–219. doi:10.1159/000474453
74. Ciccia A, Elledge SJ. The DNA Damage Response: Making It Safe to Play with Knives. Molecular Cell. 2010;40: 179–204. doi:10.1016/j.molcel.2010.09.019

47
75. Martin LP, Hamilton TC, Schilder RJ. Platinum resistance: The role of DNA repair pathways. Clinical Cancer Research. 2008;14: 1291–1295. doi:10.1158/1078-0432.CCR-07-2238
76. Bouwman P, Jonkers J. The effects of deregulated DNA damage signalling on cancer chemotherapy response and resistance. Nature Reviews Cancer. 2012;12: 587–598. doi:10.1038/nrc3342
77. Sentman CL, Shutter JR, Hockenbery D, Kanagawa O, Korsmeyer SJ. bcl-2 inhibits multiple forms of apoptosis but not negative selection in thymocytes. Cell. 1991;67: 879–888. doi:10.1016/0092-8674(91)90361-2
78. Wong RSY. Apoptosis in cancer: From pathogenesis to treatment. Journal of Experimental & Clinical Cancer Research 2017 36:1. 2011;30: 87. doi:10.1186/1756-9966-30-87
79. Mariño G, Niso-Santano M, Baehrecke EH, Kroemer G. Self-consumption: The interplay of autophagy and apoptosis. Nature Reviews Molecular Cell Biology. 2014;15: 81–94. doi:10.1038/nrm3735
80. Das CK, Mandal M, Kögel D. Pro-survival autophagy and cancer cell resistance to therapy. Cancer Metastasis Rev. 2018;: 1–18. doi:10.1007/s10555-018-9727-z
81. Gump JM, Thorburn A. Autophagy and apoptosis: What is the connection? Trends in Cell Biology. 2011;21: 387–392. doi:10.1016/j.tcb.2011.03.007
82. Levy JMM, Towers CG, Thorburn A. Targeting autophagy in cancer. Nature Reviews Cancer. 2017;17: 528–542. doi:10.1038/nrc.2017.53
83. Seshacharyulu P, Ponnusamy MP, Haridas D, Jain M, Ganti AK, Batra SK. Targeting the EGFR signaling pathway in cancer therapy. Expert Opinion on Therapeutic Targets. 2012;16: 15–31. doi:10.1517/14728222.2011.648617
84. Yarden Y, Pines G. The ERBB network: At last, cancer therapy meets systems biology. Nature Reviews Cancer. 2012;12: 553–563. doi:10.1038/nrc3309
85. Nakai K, Hung MC, Yamaguchi H. A perspective on anti-EGFR therapies targeting triple-negative breast cancer. Am J Cancer Res. 2016;6: 1609–1623.
86. Fan S, Bhatia K, El-Deiry WS, Bae I, Magrath I, Freeman J, et al. p53 Gene Mutations Are Associated with Decreased Sensitivity of Human Lymphoma Cells to DNA Damaging Agents. Cancer Res. 1994;54: 5824–5830.
87. Fink D, Aebi S, Howell SB. The role of DNA mismatch repair in drug resistance. Clinical Cancer Research. 1998;4: 1–6.
88. Sequist LV, Martins RG, Spigel D, Grunberg SM, Spira A, Jänne PA, et al. First-line gefitinib in patients with advanced non-small-cell lung cancer harboring somatic EGFR mutations. J Clin Oncol. 2008;26: 2442–2449. doi:10.1200/JCO.2007.14.8494

48
89. Kim DW, Mehra R, Tan DSW, Felip E, Chow LQM, Camidge DR, et al. Activity and safety of ceritinib in patients with ALK-rearranged non-small-cell lung cancer (ASCEND-1): updated results from the multicentre, open-label, phase 1 trial. The Lancet Oncology. 2016;17: 452–463. doi:10.1016/S1470-2045(15)00614-2
90. Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell. 2011;144: 646–674. doi:10.1016/j.cell.2011.02.013
91. Dagogo-Jack I, Shaw AT. Tumour heterogeneity and resistance to cancer therapies. Nature Reviews Clinical Oncology 2018;15: 81–94. doi:10.1038/nrclinonc.2017.166
92. Findlay JM, Castro-Giner F, Makino S, Rayner E, Kartsonaki C, Cross W, et al. Differential clonal evolution in oesophageal cancers in response to neo-adjuvant chemotherapy. Nat Commun. 2016;7: 11111. doi:10.1038/ncomms11111
93. Xue Y, Martelotto L, Baslan T, Vides A, Solomon M, Mai TT, et al. An approach to suppress the evolution of resistance in BRAF V600E-mutant cancer. Nature Medicine. 2017;23: 929–937. doi:10.1038/nm.4369
94. Shaffer SM, Dunagin MC, Torborg SR, Torre EA, Emert B, Krepler C, et al. Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature. 2017;546: 431–435. doi:10.1038/nature22794
95. Pisco AO, Brock A, Zhou J, Moor A, Mojtahedi M, Jackson D, et al. Non-Darwinian dynamics in therapy-induced cancer drug resistance. Nat Commun. Nature Publishing Group; 2013;4: 2467. doi:10.1038/ncomms3467
96. Goldman A, Majumder B, Dhawan A, Ravi S, Goldman D, Kohandel M, et al. Temporally sequenced anticancer drugs overcome adaptive resistance by targeting a vulnerable chemotherapy-induced phenotypic transition. Nat Commun. 2015;6: 6139. doi:10.1038/ncomms7139
97. Li Q, Wennborg A, Aurell E, Dekel E, Zou J-Z, Xu Y, et al. Dynamics inside the cancer cell attractor reveal cell heterogeneity, limits of stability, and escape. Proc Natl Acad Sci USA. 2016;113: 2672–2677. doi:10.1073/pnas.1519210113
98. Kaelin WG. The concept of synthetic lethality in the context of anticancer therapy. Nature Reviews Cancer. 2005;5: 689–698. doi:10.1038/nrc1691
99. Edwards SL, Brough R, Lord CJ, Natrajan R, Vatcheva R, Levine DA, et al. Resistance to therapy caused by intragenic deletion in BRCA2. Nature. 2008;451: 1111–1115. doi:10.1038/nature06548
100. Gorre ME, Mohammed M, Ellwood K, Hsu N, Paquette R, Rao PN, et al. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 2001;293: 876–880. doi:10.1126/science.1062538
101. Sergina NV, Rausch M, Wang D, Blair J, Hann B, Shokat KM, et al. Escape from HER-family tyrosine kinase inhibitor therapy by the kinase-inactive HER3. Nature. 2007;445: 437–441. doi:10.1038/nature05474

49
102. Ng CL, Siciliano G, Lee MCS, de Almeida MJ, Corey VC, Bopp SE, et al. CRISPR-Cas9-modified pfmdr1 protects Plasmodium falciparum asexual blood stages and gametocytes against a class of piperazine-containing compounds but potentiates artemisinin-based combination therapy partner drugs. Molecular Microbiology. 2016;101: 381–393. doi:10.1111/mmi.13397
103. Hall MD, Handley MD, Gottesman MM. Is resistance useless? Multidrug resistance and collateral sensitivity. Trends in Pharmacological Sciences. Elsevier Current Trends; 2009;30: 546–556. doi:10.1016/j.tips.2009.07.003
104. Jaeger S, Igea A, Arroyo R, Alcalde V, Canovas B, Orozco M, et al. Quantification of pathway cross-talk reveals novel synergistic drug combinations for breast cancer. Cancer Res. American Association for Cancer Research; 2017;77: 459–469. doi:10.1158/0008-5472.CAN-16-0097
105. Chen ZG. Small-molecule delivery by nanoparticles for anticancer therapy. Trends Mol Med. 2010;16: 594–602. doi:10.1016/j.molmed.2010.08.001
106. Patel A, Patel M, Yang X, Mitra AK. Recent advances in protein and peptide drug delivery: A special emphasis on polymeric nanoparticles. Protein and Peptide Letters. 2014;21: 1102–1120. doi:10.2174/0929866521666140807114240
107. Iyer AK, Ganesh S, Amiji MM. Nano-Platforms for Tumor-Targeted Delivery of Nucleic Acid Therapies. In: Alonso MJ, Garcia-Fuentes M, editors. Nano-Oncologicals: New Targeting and Delivery Approaches. Cham: Springer International Publishing; 2014. pp. 269–291.
108. Leroux JC, Allémann E, De Jaeghere F, Doelker E, Gurny R. Biodegradable nanoparticles - From sustained release formulations to improved site specific drug delivery. 1996. pp. 339–350. doi:10.1016/0168-3659(95)00164-6
109. Ganta S, Devalapally H, Shahiwala A, Amiji M. A review of stimuli-responsive nanocarriers for drug and gene delivery. Journal of Controlled Release. 2008;126: 187–204. doi:10.1016/j.jconrel.2007.12.017
110. Bahrami B, Hojjat-Farsangi M, Mohammadi H, Anvari E, Ghalamfarsa G, Yousefi M, et al. Nanoparticles and targeted drug delivery in cancer therapy. Immunology Letters. 2017;190: 64–83. doi:10.1016/j.imlet.2017.07.015
111. Amoozgar Z, Yeo Y. Recent advances in stealth coating of nanoparticle drug delivery systems. Wiley Interdisciplinary Reviews: Nanomedicine and Nanobiotechnology. 2012;4: 219–233. doi:10.1002/wnan.1157
112. Zununi Vahed S, Salehi R, Davaran S, Sharifi S. Liposome-based drug co-delivery systems in cancer cells. Mater Sci Eng C Mater Biol Appl. 2017;71: 1327–1341. doi:10.1016/j.msec.2016.11.073
113. Naderinezhad S, Amoabediny G, Haghiralsadat F. Co-delivery of hydrophilic and hydrophobic anticancer drugs using biocompatible pH-sensitive lipid-based

50
nano-carriers for multidrug-resistant cancers. RSC Advances. 2017;7: 30008–30019. doi:10.1039/c7ra01736g
114. Milla P, Dosio F, Cattel L. PEGylation of Proteins and Liposomes: a Powerful and Flexible Strategy to Improve the Drug Delivery. CDM. Bentham Science Publishers; 2012;13: 105–119. doi:10.2174/138920012798356934
115. Bulbake U, Doppalapudi S, Kommineni N, Khan W. Liposomal Formulations in Clinical Use: An Updated Review. Pharmaceutics. 2017;9. doi:10.3390/pharmaceutics9020012
116. Chang M, Zhang F, Wei T, Zuo T, Guan Y, Lin G, et al. Smart linkers in polymer-drug conjugates for tumor-targeted delivery. J Drug Target. 2016;24: 475–491. doi:10.3109/1061186X.2015.1108324
117. Chytil P, Koziolová E, Etrych T, Ulbrich K. HPMA Copolymer–Drug Conjugates with Controlled Tumor-Specific Drug Release. Macromol Biosci. 2018;18. doi:10.1002/mabi.201700209
118. Feng Q, Tong R. Anticancer nanoparticulate polymer-drug conjugate. Bioeng Transl Med. 2016;1: 277–296. doi:10.1002/btm2.10033
119. Krakovicová H, Etrych T, Ulbrich K. HPMA-based polymer conjugates with drug combination. Eur J Pharm Sci. 2009;37: 405–412. doi:10.1016/j.ejps.2009.03.011
120. Ambade AV, Savariar EN, Thayumanavan S. Dendrimeric micelles for controlled drug release and targeted delivery. Molecular Pharmaceutics. 2005;2: 264–272. doi:10.1021/mp050020d
121. Larson N, Ghandehari H. Polymeric conjugates for drug delivery. Chemistry of Materials. 2012;24: 840–853. doi:10.1021/cm2031569
122. Singh J, Desai S, Yadav S, Narasimhan B, Kaur H. Polymer Drug Conjugates:
Recent Advancements in Various Diseases. Curr Pharm Des 2016;22: 2821–
2843.
123. Yang J, Kopeček J. The Light at the End of the Tunnel-Second Generation HPMA Conjugates for Cancer Treatment. Curr Opin Colloid Interface Sci. 2017;31: 30–42. doi:10.1016/j.cocis.2017.07.003
124. Khan J, Alexander A, Ajazuddin, Saraf S, Saraf S. Exploring the role of polymeric conjugates toward anti-cancer drug delivery: Current trends and future projections. Int J Pharm. 2018;548: 500–514. doi:10.1016/j.ijpharm.2018.06.060
125. Panowski S, Bhakta S, Raab H, Polakis P, Junutula JR. Site-specific antibody drug conjugates for cancer therapy. MAbs. 2014;6: 34–45. doi:10.4161/mabs.27022

51
126. Beck A, Goetsch L, Dumontet C, Corvaïa N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat Rev Drug Discov. 2017;16: 315–337. doi:10.1038/nrd.2016.268
127. Sussman D, Westendorf L, Meyer DW, Leiske CI, Anderson M, Okeley NM, et al. Engineered cysteine antibodies: an improved antibody-drug conjugate platform with a novel mechanism of drug-linker stability. Protein Eng Des Sel. 2nd ed. 2018;31: 47–54. doi:10.1093/protein/gzx067
128. Li JY, Perry SR, Muniz-Medina V, Wang X, Wetzel LK, Rebelatto MC, et al. A Biparatopic HER2-Targeting Antibody-Drug Conjugate Induces Tumor Regression in Primary Models Refractory to or Ineligible for HER2-Targeted Therapy. Cancer Cell. 2016;29: 117–129. doi:10.1016/j.ccell.2015.12.008
129. Lambert JM, Berkenblit A. Antibody-Drug Conjugates for Cancer Treatment. Annual Review of Medicine. 2018;69: 191–207. doi:10.1146/annurev-med-061516-121357
130. Sau S, Alsaab HO, Kashaw SK, Tatiparti K, Iyer AK. Advances in antibody-drug conjugates: A new era of targeted cancer therapy. Drug Discov Today. 2017;22: 1547–1556. doi:10.1016/j.drudis.2017.05.011
131. Desale SS, Cohen SM, Zhao Y, Kabanov AV, Bronich TK. Biodegradable hybrid polymer micelles for combination drug therapy in ovarian cancer. J Control Release. 2013;171: 339–348. doi:10.1016/j.jconrel.2013.04.026
132. Movassaghian S, Merkel OM, Torchilin VP. Applications of polymer micelles for imaging and drug delivery. Wiley Interdisciplinary Reviews: Nanomedicine and Nanobiotechnology. 2015;7: 691–707. doi:10.1002/wnan.1332
133. Greco F, Vicent MJ. Combination therapy: opportunities and challenges for polymer-drug conjugates as anticancer nanomedicines. Adv Drug Deliv Rev. 2009;61: 1203–1213. doi:10.1016/j.addr.2009.05.006
134. Sezgin Z, Yüksel N, Baykara T. Preparation and characterization of polymeric micelles for solubilization of poorly soluble anticancer drugs. Eur J Pharm Biopharm. 2006;64: 261–268. doi:10.1016/j.ejpb.2006.06.003
135. Desale SS, Raja SM, Kim JO, Mohapatra B, Soni KS, Luan H, et al. Polypeptide-based nanogels co-encapsulating a synergistic combination of doxorubicin with 17-AAG show potent anti-tumor activity in ErbB2-driven breast cancer models. J Control Release. 2015;208: 59–66. doi:10.1016/j.jconrel.2015.02.001
136. Soni KS, Lei F, Desale SS, Marky LA, Cohen SM, Bronich TK. Tuning polypeptide-based micellar carrier for efficient combination therapy of ErbB2-positive breast cancer. Journal of Controlled Release. 2017;264: 276–287. doi:10.1016/j.jconrel.2017.08.038
137. Wan X, Min Y, Bludau H, Keith A, Sheiko SS, Jordan R, et al. Drug Combination Synergy in Worm-like Polymeric Micelles Improves Treatment Outcome for

52
Small Cell and Non-Small Cell Lung Cancer. ACS Nano. 2018;12: 2426–2439. doi:10.1021/acsnano.7b07878
138. Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: A review. Journal of Controlled Release. 2000;65: 271–284. doi:10.1016/S0168-3659(99)00248-5
139. Sykes EA, Dai Q, Sarsons CD, Chen J, Rocheleau JV, Hwang DM, et al. Tailoring nanoparticle designs to target cancer based on tumor pathophysiology. Proc Natl Acad Sci USA. 2016;113: E1142–51. doi:10.1073/pnas.1521265113
140. Fang J, Sawa T, Maeda H. Factors and mechanism of “EPR” effect and the enhanced antitumor effects of macromolecular drugs including SMANCS. Adv Exp Med Biol. Boston: Kluwer Academic Publishers; 2003;519: 29–49. doi:10.1007/0-306-47932-X_2
141. Prabhakar U, Maeda H, Jain RK, Sevick-Muraca EM, Zamboni W, Farokhzad OC, et al. Challenges and key considerations of the enhanced permeability and retention effect for nanomedicine drug delivery in oncology. Cancer Res. 2013;73: 2412–2417. doi:10.1158/0008-5472.CAN-12-4561
142. Lee H, Fonge H, Hoang B, Reilly RM, Allen C. The effects of particle size and molecular targeting on the intratumoral and subcellular distribution of polymeric nanoparticles. Molecular Pharmaceutics. 2010;7: 1195–1208. doi:10.1021/mp100038h
143. Perrault SD, Walkey C, Jennings T, Fischer HC, Chan WCW. Mediating tumor targeting efficiency of nanoparticles through design. Nano Lett. 2009;9: 1909–1915. doi:10.1021/nl900031y
144. Dozono H, Yanazume S, Nakamura H, Etrych T, Chytil P, Ulbrich K, et al. HPMA Copolymer-Conjugated Pirarubicin in Multimodal Treatment of a Patient with Stage IV Prostate Cancer and Extensive Lung and Bone Metastases. Target Oncol. Springer International Publishing; 2016;11: 101–106. doi:10.1007/s11523-015-0379-4
145. Maeda H. Toward a full understanding of the EPR effect in primary and metastatic tumors as well as issues related to its heterogeneity. Adv Drug Deliv Rev. 2015;91: 3–6. doi:10.1016/j.addr.2015.01.002
146. Friedman AD, Claypool SE, Liu R. The smart targeting of nanoparticles. Curr Pharm Des 2013;19: 6315–6329. doi:10.2174/13816128113199990375
147. Srinivasarao M, Low PS. Ligand-Targeted Drug Delivery. Chem Rev. 2017;117: 12133–12164. doi:10.1021/acs.chemrev.7b00013
148. Fahmy TM, Fong PM, Goyal A, Saltzman WM. Targeted for drug delivery. Materials Today. 2005;8: 18–26. doi:10.1016/S1369-7021(05)71033-6

53
149. Sadat SMA, Saeidnia S, Nazarali AJ, Haddadi A. Nano-pharmaceutical formulations for targeted drug delivery against HER2 in breast cancer. Curr Cancer Drug Targets. 2015;15: 71–86.
150. Akers WJ, Zhang Z, Berezin M, Ye Y, Agee A, Guo K, et al. Targeting of alpha(nu)beta(3)-integrins expressed on tumor tissue and neovasculature using fluorescent small molecules and nanoparticles. Nanomedicine (Lond). 2010;5: 715–726. doi:10.2217/nnm.10.38
151. Cheung A, Bax HJ, Josephs DH, Ilieva KM, Pellizzari G, Opzoomer J, et al. Targeting folate receptor alpha for cancer treatment. Oncotarget. 2016;7: 52553–52574. doi:10.18632/oncotarget.9651
152. Master AM, Gupta Sen A. EGF receptor-targeted nanocarriers for enhanced cancer treatment. Nanomedicine (Lond). 2012;7: 1895–1906. doi:10.2217/nnm.12.160
153. Wickens JM, Alsaab HO, Kesharwani P, Bhise K, Amin MCIM, Tekade RK, et al. Recent advances in hyaluronic acid-decorated nanocarriers for targeted cancer therapy. Drug Discov Today. 2017;22: 665–680. doi:10.1016/j.drudis.2016.12.009
154. Vinay R, KusumDevi V. Potential of targeted drug delivery system for the treatment of bone metastasis. Drug Deliv. 2016;23: 21–29. doi:10.3109/10717544.2014.913325
155. Zhang G, Guo B, Wu H, Tang T, Zhang B-T, Zheng L, et al. A delivery system targeting bone formation surfaces to facilitate RNAi-based anabolic therapy. Nature Medicine. Nature Research; 2012;18: 307–314. doi:10.1038/nm.2617
156. O’Shaughnessy J. Extending survival with chemotherapy in metastatic breast cancer. The Oncologist. AlphaMed Press; 2005;10 Suppl 3: 20–29. doi:10.1634/theoncologist.10-90003-20
157. Mokhtari RB, Homayouni TS, Baluch N, Morgatskaya E, Kumar S, Das B, et al. Combination therapy in combating cancer. Oncotarget. 2017;8: 38022–38043. doi:10.18632/oncotarget.16723
158. Lee JH, Nan A. Combination drug delivery approaches in metastatic breast cancer. J Drug Deliv. 2012;2012: 915375. doi:10.1155/2012/915375
159. Lopez JS, Banerji U. Combine and conquer: challenges for targeted therapy combinations in early phase trials. Nature Reviews Clinical Oncology 2017 14:7. 2017;14: 57–66. doi:10.1038/nrclinonc.2016.96
160. Chakrabarti S, Michor F. Pharmacokinetics and Drug Interactions Determine Optimum Combination Strategies in Computational Models of Cancer Evolution. Cancer Res. 2017;77: 3908–3921. doi:10.1158/0008-5472.CAN-16-2871

54
161. Curtis LT, van Berkel VH, Frieboes HB. Pharmacokinetic/pharmacodynamic modeling of combination-chemotherapy for lung cancer. J Theor Biol. 2018;448: 38–52. doi:10.1016/j.jtbi.2018.03.035
162. Fuso Nerini I, Cesca M, Bizzaro F, Giavazzi R. Combination therapy in cancer: effects of angiogenesis inhibitors on drug pharmacokinetics and pharmacodynamics. Chinese Journal of Cancer 2017 36:1. 2016;35: 61. doi:10.1186/s40880-016-0123-1
163. Miller K, Erez R, Segal E, Shabat D, Satchi-Fainaro R. Targeting bone metastases with a bispecific anticancer and antiangiogenic polymer-alendronate-taxane conjugate. Angew Chem Int Ed Engl. 2009;48: 2949–2954. doi:10.1002/anie.200805133
164. Miller K, Eldar-Boock A, Polyak D, Segal E, Benayoun L, Shaked Y, et al. Antiangiogenic antitumor activity of HPMA copolymer-paclitaxel-alendronate conjugate on breast cancer bone metastasis mouse model. Molecular Pharmaceutics. 2011;8: 1052–1062. doi:10.1021/mp200083n

55
CHAPTER II
TARGETED POLYMERIC MICELLES FOR THE TREATMENT OF
BREAST CANCER BONE METASTASIS

56
2.1 INTRODUCTION
Tumor metastases remain the primary cause of cancer-related mortality. Bone
has been widely recognized not only as one of the frequent destinations of metastatic
spread of multiple solid tumors, but also as an organ that can provide a favorable
microenvironment for cancer cell survival and further growth or dormancy of metastases.
The complex mechanism of bone metastasis involves a coordinating interplay of bone-
homing cancer cells, osteoclasts, osteoblasts and the immune cells in the bone
marrow[1]. Tumor cells secrete factors which stimulate osteoclast-mediated bone
destruction and negatively affect osteoblasts. The consequent release of numerous
factors immobilized within the bone matrix that act on cancer cells, promotes a more
aggressive tumor phenotype and potentiating cancer spread and bone destruction. In
addition, physical properties of bone, such as hypoxia, acidic pH, and a high
extracellular calcium concentration, also support tumor growth. In patients with breast
cancer, the skeleton is the most frequent site for metastases. It is estimated that up to
75% of late-stage breast cancer patients will eventually develop bone metastases[2].
These lesions lead to numerous skeletal complications, including osteolysis, bone pain,
hypercalcemia, pathologic fractures, and spinal cord and nerve compression
syndromes[3]. Such complications increase morbidity and diminish the quality of life in
these patients as well as often account for the poor prognosis[4]. While early diagnosis
and effective chemo- and radiotherapies have significantly reduced the mortality rate
from primary breast cancer, the current therapeutic modalities for advanced metastatic
breast cancer patients remain palliative. Radiation therapy continues to be a gold
standard for palliative care of painful bone metastases and suppression of local disease.
Osteoclast inhibitors, bisphosphonates (pamidronate, zoledronic acid) and denosumab,

57
an antibody targeting RANKL, are commonly prescribed therapeutics for management
the progression of the established disease and reducing the risk of occurrence of
skeletal-related events. However, the benefits of these agents on improved survival was
observed in only a small population of patients[5,6]. Another challenge in the treatment
of metastatic cancer is effective delivery of therapeutics to the tumor lesion due to poor
penetration of most drugs in the bone tissue. Moreover, these agents also affect non-
cancerous cells within the bone microenvironment, inhibiting normal bone healing
processes and potentially leading to serious adverse events. Thus, despite significant
advances in the care of these patients there is a need for more effective treatment
options to prolong the survival of patients with metastatic bone diseases.
During the last decades, substantial efforts have been made in targeting bone
metastases using various delivery systems such as polymeric nanoparticles, liposomes,
micelles, and polymer-drug conjugates. These systems aim to improve the
biodistribution and target site accumulation of drugs thus reducing side effects. To
further enhance targeting of nanoparticles to bone lesions, the bone-binding moieties,
such as bisphosphonates, were utilized as targeting ligands due to their affinity to the
mineral compartment in bone tissue[7,8]. Indeed, hydroxyapatite is exposed as bone
metastases progress providing a target for bisphosphonate attachment. In line with this,
detection of metastatic involvement of the skeleton using 99mTc - radiolabeled
bisphosphonates has been in clinical practice for many years[9]. A number of in vitro and
in vivo studies substantiated preferential and efficient accumulation of bone-targeted
drug carriers within the bone lesions. Interestingly, despite demonstrated improved drug
efficacy, decreased toxicity and delay of tumor progression in mouse models, no
significant survival improvement between targeted and non-targeted nanocarriers has

58
been reported[10-17]. Nonetheless, combining the benefits of drug delivery systems,
bisphosphonate bone targeting to manipulate the tumor cells and their microenvironment
may further enhance the capabilities of drug delivery to eliminate or ameliorate bone
metastases.
Herein, we engineered bone-homing polypeptide-based polymeric micelles
carrying the chemotherapeutic docetaxel for the targeted treatment of breast cancer
bone metastases. To achieve this, an amphiphilic block copolymer composed of
polyethylene glycol, polyglutamic acid and phenylalanine (PEG-PLE-PLF) was
functionalized with a bisphosphonate, alendronate (ALN). The phenylalanine moieties
facilitate self-assembly of block copolymers and formation of micelles and are essential
for solubilization of hydrophobic compounds. Using an immunocompetent mouse model
of breast cancer dissemination to the bone, this study demonstrates that targeted
micelle-based therapy significantly attenuated tumor burden and extended animal
survival, proving the effectiveness of this targeting approach for the delivery of
chemotherapeutics to the bone.

59
2.2 MATERIALS AND METHODS
2.2.1 MATERIALS
α-Amino-ω-alkyne-poly(ethylene glycol) (ALK-PEG-NH2 , MW = 5,000 g mol-1, Mw / Mn
= 1.02) was purchased from JenKem Technology USA Inc. L-glutamic acid γ-benzyl
ester (BLE), L-phenylalanine (LF), 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide
hydrochloride (EDC), N-hydroxysuccinimide (NHS), tris(3-
hydroxypropyltriazolylmethyl)amine (THPTA), hydroxyapatite (HA, powder, 10μm, ≥100
m2/g), ethylenediaminetetraacetic acid (EDTA), 3-bromopropionic acid, sodium
ascorbate, copper (II) sulfate pentahydrate, Phosphate Colorimetric Kit (MAK030), Acid
phosphatase Leukocyte (TRAP) Kit, and other chemicals were purchased from Sigma-
Aldrich. Receptor activator of nuclear factor kappa-Β ligand (RANKL) was obtained from
R&D System. Docetaxel (DTX) was purchased from Selleck Chemicals LLC.
Alendronate sodium trihydrate (ALN) was purchased from Alfa Aesar. CF488A amine
was obtained from Biotium. Fetal bovine serum (FBS), RPMI 1640 medium, DMEM
medium, penicillin/streptomycin, and other chemicals were purchased from Invitrogen.
XenoLight D-Luciferin - K+ Salt Bioluminescent Substrate was purchased from
PerkinElmer.

60
2.2.2 METHODS
Synthesis of ALK-PEG-PLE10-PLF10 block copolymers
The BLE-NCA (γ-benzyl L-glutamate-N- carboxyanhydride) and LF-NCA (L-
phenylalanine -N- carboxyanhydride) monomers were prepared as described
previously[18]. First, ALK-PEG-PLE diblock copolymer was synthesized via ring –
opening polymerization of BLE-NCA using ALK-PEG-NH2 as a macroinitiator. ALK-
PEG-NH2 (150 mg, 0.03 mmol) was dissolved in 10 mL of anhydrous DMF in an oven-
dried flask under nitrogen atmosphere followed by addition of BLE-NCA (79.6 mg, 0.302
mmol) dissolved in 5 mL of DMF. The feed molar ratio of ALK-PEG-NH2 to BLE-NCA
was 1:10. The reaction was allowed to proceed at 40°C for 72 h. An aliquot of the
reaction mixture was precipitated using an excess of diethyl ether, purified by repeated
precipitation from DMF/diethyl ether and dried under vacuum. The composition of ALK-
PEG–PLE copolymer was determined by 1H NMR from the peak intensity ratios of the
methylene protons of PEG and -benzyl groups (400 MHz, DMSO-d6, ppm: 3.52 (s,
4H, -OCH2CH2-), 5.14 (m, 2H, -CH2C6H5)). After the completion of the reaction, LF-NCA
(145 mg, 0.26 mmol), previously dissolved in 5 mL of anhydrous 1,4-dioxane, was then
added and let to react for an additional 120 h. The final product was isolated by
precipitation in diethyl ether, purified by repeated precipitation from DMF/diethyl ether
and dried under vacuum. The residue was dissolved in DMF and added dropwise to 0.5
N NaOH to deprotect glutamate residues. The mixture was stirred overnight and then
dialyzed using a dialysis membrane (MWCO 2 kDa) against distilled water for 48 h. After
dialysis, the product was filtered (0.8 μm filter) and lyophilized.

61

62
Synthesis of azido-ALN
3-azidopropionic acid was synthesized as previously described[19]. To 3-
azidopropionic acid (40.8 mg, 0.3 mmol) dissolved in 1 mL of anhydrous CH2Cl2, EDC
(66 mg,0.3 mmol) and NHS (48.6 mg,0.4 mmol) were added and the reaction was stirred
at room temperature for 6 h. The reaction mixture was washed with brine and an organic
layer was evaporated to remove the solvent. ALN (12.3 mg, 0.04 mmol) alendronate was
dissolved in 0.5 mL of water (pH 8.0). The NHS activated 3-azidopropionic acid (14.6 mg)
dissolved in 0.2 mL of acetonitrile/water mixture (1:4 v/v) was then added dropwise in
ALN solution while adjusting pH to 8,0 after each drop. The reaction was continued
overnight at r.t., then precipitated in ethanol three times to give the pure product and
dried. The structure was confirmed by 1H NMR.

63
Synthesis of ALN-PEG-PLE-PLF
The azide-ALN (6.64 mg, 0.02 mmol) was reacted with ALK-PEG-PLE10-PLF10
(13.4 mg, 0.002 mmol) dissolved in 10 mL of water and methanol mixture (1:1 v/v) in the
presence of copper (II) sulfate pentahydrate (0.5 mg, 0.002 mmol), THPTA (1.06 mg,
0.002 mmol) and ascorbic acid (3.52 mg, 0.02 mmol) for two days at r.t under argon
atmosphere. Then, EDTA (6.7 mg, 0.02 mmol) was added to the solution followed by
dialysis against deionized water (MWCO 2 kDa) for another 48 h to remove unreacted
small molecules. The dialyzed solution was then acidified and lyophilized to produce
ALN-PEG-PLE-PLF.

64
Polymer characterization
The 1H NMR spectra for the monomers and copolymers were acquired in DMSO-
d6 using a Bruker 400 MHz spectrometer. Gel permeation chromatography (GPC)
measurements to determine the molecular weights and polydispersity of the copolymers
were carried out at 40°C using a Shimadzu liquid chromatography system equipped with
TSK-GEL® column (G4000HHR) connected to Shimadzu RI and UV/vis detectors.
DMF/LiBr (10 mmol) was used as mobile phase at a flow rate of 0.6 mL/ min.
Poly(ethylene glycol) standards (Agilent Technologies, USA) with a molecular weight
range of 282 – 34,890 were used to generate the standard curve. Self-assembly
behavior of ALK-PEG-PLE-PLF and ALN-PEG-PLE-PLF copolymer was examined
using pyrene as a hydrophobic fluorescence probe[20]. The ALN conjugation efficiency
was determined by phosphate assay as previously reported[21]. Briefly, 20 µL of ALN-
PEG-PLE-PLF solution (5 mg/mL) was mixed with 20 µL of H2O2 and 120 µL H2SO4 in
the ampule and the mixture was heated for 15 min at 200 0C. 1.38 mL of Na2S2O5 (3
mg/mL) was added to the ampule and samples were incubated at 100 0C for another 15
min. Finally, the sample was filtered through a 0.2 µm filter and 200 µL of the solution
were collected into a 96 well plate. A series of standards was prepared in the same plate
according to the manufacture’s instruction ranging from 2.5 to100 µM. Finally, 30 µL of
the phosphorus reagent was added to each well and incubated with mild shaking
overnight at r.t. The absorbance at 650 nm was measured using by Spectramax M5
plate reader.

65
Polymeric micelles preparation and characterization
The empty and drug-loaded micelles were prepared by nanoprecipitation method.
Briefly, the polymers without or with DTX (polymer/drug molar ratio was 4:1) were
dissolved in minimal amount of 80% ethanol and dispersed into an excessive amount of
ice-cold water or PBS solution (water: ethanol = 20: 1 v/v). The solutions were stirred for
an additional 5 min and ethanol was evaporated under vacuum. The unbound drug was
removed by centrifugation (1000 g, 5 min). The drug content in micellar formulations was
determined by high-performance liquid chromatography (HPLC) analysis under isocratic
conditions using an Agilent 1200 HPLC system and a diode array detector set at 227 nm.
A Nucleosil C18 column was used as stationary phase (250 mm × 4.6 mm), and mobile
phase comprised of acetonitrile/water mixture (55/45, v/v) at a flow rate of 1 mL/min. The
loading capacity (LC) was expressed as LC (%) = 𝑊𝐷𝑇𝑋
𝑊𝐷𝑇𝑋+𝑊𝑝𝑜𝑙𝑦𝑚𝑒𝑟× 100%, where wDTX
and wpolymer are the weight amount of solubilized drug and polymer excipients in solution.
Intensity-mean z-averaged particle diameter (Deff), polydispersity index (PDI) and ζ-
potential of the drug-loaded micelles were determined by dynamic light scattering (DLS)
using Nano ZS Zetasizer (Malvern Instruments, UK). All measurements were performed
in automatic mode at 25°C at a fixed 173° scattering angle. Software provided by
manufacturer was used to calculate the Deff, PDI, and ζ-potential. Data are presented as
mean values ± SD (n = 3).

66
Drug release studies
The drug release from the micelles was examined in PBS (pH 7.4) or 70% FBS
in PBS at 37 0C by dialysis method. Briefly, 1 mL of drug-loaded micelle formulation in
PBS (approximately 0.1 mg/mL of DTX) was transferred to a floatable dialysis tube
(Slide-A-Lyzer G2, MWCO 3.5 kDa) and suspended in 45 mL PBS solution. At
predetermined time points, samples of 100 µL were withdrawn from the dialysis device
and analyzed quantified by HPLC using the same conditions reported above.

67
HA binding assay
To investigate the biomineral-binding ability of ALN-functionalized micelles, CF-
488 labeled copolymers were synthesized by EDC coupling (at a molar ratio of
[COOH]/[dye] = 10] and purified by dialysis to remove the uncoupled dye. The CF-488
labeled micelle solution in PBS (1.1 mg/mL, 1.5 mL) were incubated with HA powder (10
mg) for 4 hours at r.t. upon gentle shaking. HA was removed by centrifugation (2200 g,
for 1 min) and the fluorescence intensity of the supernatant (Isupernatant) was determined at
515 nm (ex=490 nm) and compared with the initial micelle solution (Io). The relative
binding affinity was calculated as (1 −𝐼𝑠𝑢𝑝𝑒𝑟𝑛𝑎𝑡𝑎𝑛𝑡
𝐼𝑜) × 100%. CF-488-labeled ALK-m were
used as controls. The binding test was performed in triplicate.

68
Cell culture and cytotoxicity studies
The murine 4T1 mammary adenocarcinoma cell line was purchased from the
American Type Culture Collection (ATCC). Cells were maintained in RPMI 1640
supplemented with 10% FBS, and antibiotics (streptomycin 100 U/mL and penicillin 100
U/mL) at 37 0C, 5% CO2. Cells were seeded in the 96-well plates at a density of 3500
cells per well and incubated for 12 h. The cells were then exposed to free DTX
(formulated as 10 mg DTX, 80 mg Tween 80, 648 mg PEG300, 275.9 mg ethanol 96%,
4 mg citric acid), DTX/ALK-m and DTX/ALN-m at serial concentrations for 36 h at 37 0C.
The cell viability was be evaluated by standard MTT assay[22] and the IC50 values were
calculated using GraphPad Prism software. To confirm the anticancer activity, 3D
clonogenic assay, cells were seeded at a density of 95 cells/cm2 in 24-well plates that
were pre-coated with Matrigel. Cells were treated with free DTX or DTX-loaded micelles
(0.1 µg/mL on DTX basis) for 36 h and allowed to recover in fresh medium for another
72 h. Colonies were stained with crystal violets and counted. The surviving fraction (SF)
was calculated as:
𝑆𝐹 =𝑁𝑜. 𝑜𝑓 𝑐𝑜𝑙𝑜𝑛𝑖𝑒𝑠 𝑎𝑡 𝑡ℎ𝑒 𝑒𝑛𝑑 𝑝𝑜𝑖𝑛𝑡 𝑜𝑓 𝑡ℎ𝑒 𝑠𝑡𝑢𝑑𝑦
𝑁𝑜. 𝑜𝑓 𝑠𝑒𝑒𝑑𝑒𝑑 𝑐𝑒𝑙𝑙𝑠 × 𝑃𝐸
where plating efficiency (PE) is calculated as:
𝑃𝐸 =𝑁𝑜. 𝑜𝑓 𝑐𝑜𝑙𝑜𝑛𝑖𝑒𝑠 𝑎𝑓𝑡𝑒𝑟 𝑠𝑒𝑒𝑑𝑖𝑛𝑔
𝑁𝑜. 𝑜𝑓 𝑠𝑒𝑒𝑑𝑒𝑑 𝑐𝑒𝑙𝑙𝑠

69
Inhibition of osteoclast differentiation and absorptive activity
The Raw264.7 monocyte/macrophage-like cells (ATCC, TIB-71) were cultured in
DMEM supplemented with10% FBS and 1% penicillin/streptomycin, in an atmosphere of
5% CO2 and 95% humidity at 37°C. Cells were harvested after reaching about 75%
confluency, seeded on 48-well plates (1.25×104 cells/well) and supplemented with
RANKL at the final concentration of 50 µg/mL. Cells were allowed to attach and then
were treated with empty ALN-decorated micelles or non-decorated micelles at serial
concentrations (0-100 µM on ALN basis) for one week. The culture medium containing
RANKL and corresponding treatment were exchanged every three days. Cells were then
fixed with 10% formaldehyde/PBS solution and stained with TRAP. The stain-positive
cells (osteoclasts) in each well were numerated and the IC50 were calculated using
GraphPad Prism. The bone resorption activity of osteoclasts derived from Raw264.7
cells was evaluated in 24-well Osteo Surface plates. Raw264.7 cells were continuously
cultured with RANKL (50 µg/mL) in the presence or absence of ALN-decorated micelles
or non-decorated micelles at two different concentrations (0.81 µM or 9 µM on ALN
basis) for 7 days. In another setting, Raw264.7 cells were cultured in the presence of
RANKL for 4 days and then exposed to the micelles for another 3 days. On day 8, cells
were bleached and the area/numbers of the pits (resulting from the absorption activity of
osteoclasts) appeared on the plate surface were quantified by using Image Pro software.

70
Macrophage migration assay
Cell migration assay was performed using Transwell permeable supports with 0.8
m pore membranes. Conditioned medium (CM) was collected from 4T1 culture
following 12 h of incubation in serum-free DMEM at 37°C and used as the
chemoattractant. 4T1 cells were seeded on 24-well plates (10,000 cells/well) and
cultured in serum-free DMEM for 12 h followed by replacement with CM and incubation
for an additional 8 h. Raw264.7 cells (1,000 per insert) were allowed to attach to the top
inserts and then were placed into the wells. The macrophages were allowed to migrate
to the underside of the top chamber for 24 hours in the presence of 4T1 cells and CM in
the bottom chamber. Where indicated, ALN-decorated or non-decorated micelles were
present in both top and bottom chambers throughout the assay. The wells without 4T1
cells and chemoattractant were used as negative controls. The nonmigratory cells on the
upper membrane surface were removed with cotton swabs and the migrated cells
attached to the bottom side of the membrane were fixed in 10% neutral buffered formalin
and then stained with 0.25% crystal violet aqueous solution. The numbers of migratory
cells were counted with an inverted microscope using a 20× objective. Each
determination represents the average of five random spots for each well (n = 3).

71
Pharmacokinetics studies in mice
All animal studies were performed in accordance with the guidelines of the
University of Nebraska Medical Center Institutional Animal Care and Use Committee
(IACUC). Pharmacokinetics of DTX and DTX-loaded micelles were determined in 8-
weeks old female Balb/C mice (Charles River Laboratories). The 18 mice were
randomly divided into three treatment groups of 6 mice and given either DTX (in Tween
80/ethanol/PEG300) or DTX-loaded ALN-micelles or DTX-loaded non-decorated
micelles at an equivalent dose of 10 mg DTX/kg body weight through intravenous tail
vein injection. At predetermined times, blood samples (100-150 μL) were withdrawn from
a submandibular vein into heparinized tubes, and then centrifuged at 10,000 g for 3 min.
DTX was extracted from plasma samples (10 μL) using excess (40 μL) of ice-cold
acetonitrile. All samples were sonicated for 1 min and then centrifuged at 10,000 g for 3
min. The clear supernatants were separated and used for analysis. Paclitaxel was used
as an internal standard at a final concentration of 625 ng/mL. Calibration curves were
built by spiking plasma samples from control animals to achieve the final concentration
of 5–2000 ng/mL DTX. These samples were analyzed on LC-MS/MS system using a
Nucleosil C18 column (250 mm × 4.6 mm) and a mobile phase of 80% acetonitrile in
water was applied under isocratic conditions at a flow rate of 0.5 mL/min. An LC-MS/MS
system equipped with triple quadrupole (QTRAP® 6500, Sciex) coupled to HPLC system
(Nexera × 2, Shimadzu) was used in multiple reaction monitoring (MRM) mode as
follows; m/z 853.995/876.25 → 286/308.25 for paclitaxel and 808.08/830.26 →
527/549.24 for DTX.

72
Evaluation of therapeutic efficacy
To establish the animal model of breast cancer bone metastases, 1×104 4T1/Luc
cells, constitutively expressing firefly luciferase, were suspended in 50 μL of sterile PBS
and injected into the left cardiac ventricle[23] of 6-week old female Balb/C mice under
isoflurane anesthesia. Disease progression and dissemination to the bone were followed
by noninvasive bioluminescence imaging (BLI) using IVIS Imaging System (Xenogen).
Total photon flux (photons/sec) was measured from fixed regions of interest (ROI) over
the entire mouse. On day 12 post-intracardiac injection, mice were randomized (4
treatment groups, n = 8) and treated with saline (control), DTX (in Tween
80/ethanol/PEG300), DTX-loaded ALN-decorated micelles and DTX-loaded non-
decorated micelles at an equivalent dose of 10 mg DTX/kg body weight. Treatments
were administered via tail vein injections daily for three consecutive days. At 48 h after
the last treatment, 3 mice per each treatment group were euthanized, tissues (liver,
spleen, kidney, bones) were dissected and fixed in 10% neutral buffered formalin. Bones
were decalcified using Regular Cal Immuno (BBC Biochemical) according to the
manufacturer’s protocol. After fixation, tissues were processed for routine embedding in
paraffin, preparation of 5 micron thick sections, and stained with hematoxylin and eosin.
The slides were evaluated with the pathologist blinded as to which group was being
evaluated. The rest of the animals were sacrificed at signs of paralysis or low body
condition score. We also used two additional groups of animals to elucidate the efficacy
of targeted treatment of bone metastases in combination with radiotherapy. Metastatic-
bearing mice (n = 6) received 3 i.v. injections on the daily basis of either saline or DTX-
loaded ALN-decorated micelles (10 mg/kg on DTX basis). 24 h following the last dose of
micelles or vehicle mice were irradiated using the TrueBeam Linear accelerator (Varian

73
Medical Systems, Inc. Milpitas, CA) at a dose rate of 3 Gy/min with the total effective
dose of 5 Gy in one fraction with Anterio-posterior/posterior-anterior opposing beams.

74
2.3 RESULTS AND DISCUSSION
Synthesis of alendronate-conjugated polymers.
Scheme 1 shows the synthesis of the parent ALK-PEG-PLE10-PLF10
copolymer and the subsequent modification of the distal end of the PEG block
with ALN moieties using azide-alkyne “click” chemistry. The unreacted small
molecules were removed by dialysis against deionized water (MWCO 2 kDa).
The chemical composition, molecular weights and polydispersity of synthesized
copolymers were determined by 1H NMR and GPC analyses (Supporting
information, Figure. S2.1 and S2.2, Table S2.1). The ALN conjugation efficiency
was determined using a phosphate assay21 and was ∼75%. We have previously
demonstrated that the incorporation of hydrophobic PLF block into triblock
copolymers confer amphiphilic properties and facilitates self-assembly of block
copolymers in an aqueous medium, which was ascribed to hydrophobic and π–π
stacking interactions of the phenylalanine units, and was essential for
solubilization of various hydrophobic compounds[18]. An association behavior of
PEG-PLE-PLF block copolymers was confirmed by fluorescence measurements
using pyrene as a probe (Figure S2.3). Using this approach, we determined that
the critical micelle concentration (CMC) for ALK-PEG-PLE-PLF was low (0.4
mg/L). The onset of aggregation of functionalized ALN-PEG-PLE-PLF chains
was shifted to higher concentrations with determined CMC value of 8.2 mg/L.
The observed increased CMC value is consistent with the presence of distal

75
charged ALN groups on the polymer chains that are leading to increased
repulsion between the copolymer chains upon formation of micellar aggregates.
This CMC value is still extremely low and comparable with those reported for
PEG-poly(lactide) micelles [24-25], which suggests that micelles formed from
these polymers should be expected to exhibit good stability. As was previously
demonstrated, the incorporation of aromatic units in the polymer chains forming
the hydrophobic micellar core can considerably improve the stability, loading
capacity and drug retention of taxane-loaded polymeric micelles[26]. Therefore,
we believe that PLF-based cores of the micelles may provide a suitable
environment for encapsulation of DTX.

76
Scheme 2.1: Synthesis of the bone-targeted copolymer.
Conditions: A). DMF, 40 0C, N2, Glu(z)-NCA, 72 h. B). DMF / 1,4-dioxane, 40
0C, N2, PhA-NCA, 120 h. C).
THF / 0.5N NaOH, RT, 24 h. D). MeOH / H2O (1:1, v/v), CuSO4 x 5H2O, THPTA, ascorbic acid, Ar, RT. E).
ACN, NaN3, reflux, 6 h; (F) DCM, EDC/NHS, RT, 6 h. (G) ACN/H2O (1:4, v/v), pH 8.0, RT, 24 h.
HO Br
O
HO N3
O
O N3
N
O
O
O
OP
NH
OH
HO
OHO
OO
NH
HN H
OO
114 10O
ONH
HN
OO
114 10
HN
O
H
10
OO
NH
HN
OHO
114 10
HN
O
H
10
PO
OHHO
OO
NH
HN
OHO
114 10
HN
O
H
10
NN
HN
OP
NH
OH
HO
OHO
PO
OHHO
N3
B
G
D
OP
NH2
OH
HO
OH
PO
OHHO
HN
OO
NH2114
O O
O
A
C
+
E
F
O
HN
O
HN
O
HN
O
HN
O

77
Figure S2.1: 1H-NMR spectra (400 MHz, DMSO-d6) of synthesized block-copolymers. A). ALK-PEG-
PBLE10. B. ALK-PEG-PBLE10-PLF10.
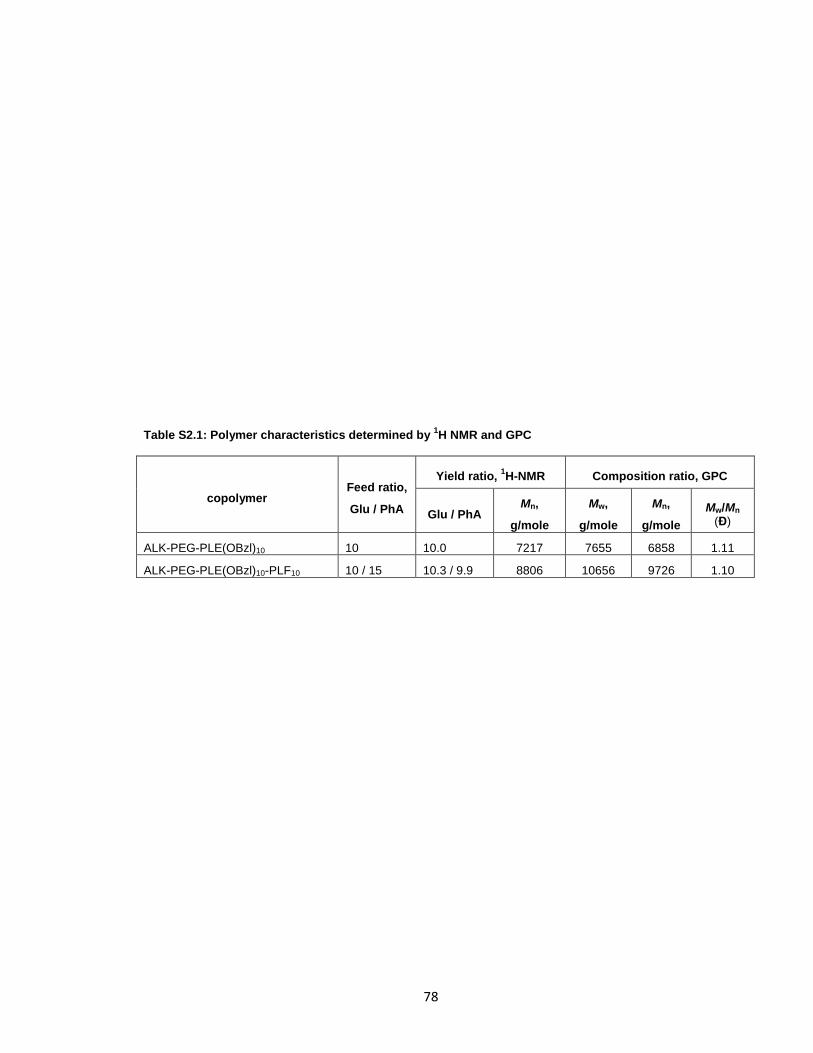
78
Table S2.1: Polymer characteristics determined by 1H NMR and GPC
copolymer Feed ratio,
Glu / PhA
Yield ratio, 1H-NMR Composition ratio, GPC
Glu / PhA Mn,
g/mole
Mw,
g/mole
Mn,
g/mole
Mw/Mn (Đ)
ALK-PEG-PLE(OBzl)10 10 10.0 7217 7655 6858 1.11
ALK-PEG-PLE(OBzl)10-PLF10 10 / 15 10.3 / 9.9 8806 10656 9726 1.10

79
Figure S2.2: GPC traces (DMF + 10 mM LiCl; 40 0C, 0.6 mL/min). A). ALK-PEG-NH2. B). ALK-PEG-
PBLE10. C). ALK-PEG-PBLE10-PLF10.
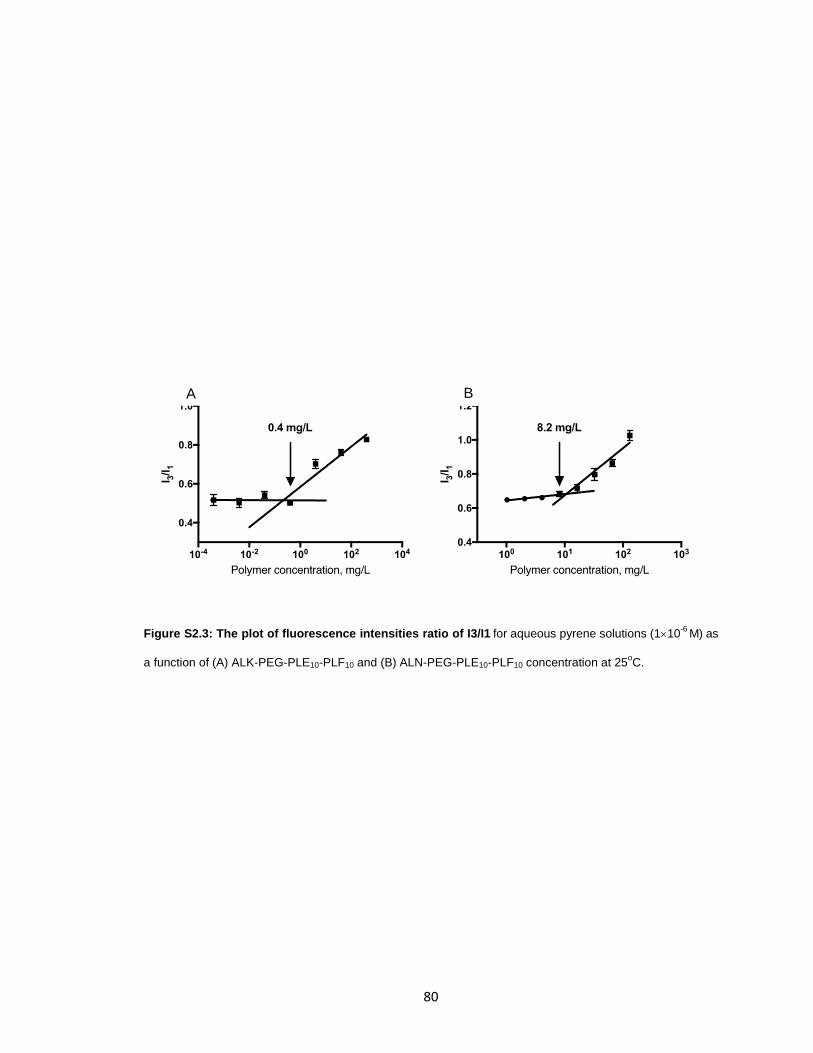
80
Figure S2.3: The plot of fluorescence intensities ratio of I3/I1 for aqueous pyrene solutions (110-6
M) as
a function of (A) ALK-PEG-PLE10-PLF10 and (B) ALN-PEG-PLE10-PLF10 concentration at 25oC.
A B

81
Preparation of docetaxel-loaded polymeric micelles and characterization of their
physicochemical properties
DTX, a taxane that inhibits cell replication by stabilizing the microtubule
cytoskeleton, is one of the chemotherapeutic agents approved for the first-line treatment
of metastatic breast cancer[27,28]. Despite its potent anticancer activity, DTX exhibits
serious dose-limiting toxicities partly due to the formulation excipients. Polyoxylated
surfactant, polysorbate 80, is utilized in Taxotere, a commercial formulation of DTX, and
has been implicated in observed hypersensitivity reactions, nonallergic anaphylaxis, and
injection- and infusion-site adverse events[29]. For these reasons, we selected DTX as
the model chemotherapeutic agent for solubilization into polymeric micelles. The micelle
formation and drug loading were accomplished in one single step using a method known
as nanoprecipitation. A polymer or a mixture of a polymer and drug were dissolved in a
minimal amount of ethanol and the resulting solution was injected into a 20 volume of
ice-cold DI water or PBS. Under these conditions we were able to obtain stable
dispersions of DTX-loaded micelles with a loading capacity of 22.4% as was determined
by HPLC and it was associated with high loading efficiency (about 97%). Targeted
micelles with varying ALN density were prepared by co-assembly of ALK-PEG-PLE-PLF
and ALN-PEG-PLE-PLF copolymers at various molar ratios. In this way, we prepared
ALN-m with the various proportion of ALN moieties ranging from 3.8% to 75%. Micelles
prepared from ALK-only copolymers (ALK-m) were used as a non-targeted control for
comparison. Of note, the modification with targeting ligand practically did not change the
size of the micelles: the average hydrodynamic diameters (Deff) of the ALN-only micelles
(ALN-m) and ALK-m in DI water at neutral pH were about 70 and 67 nm, respectively, as
measured by DLS. The DTX-loaded micelles had a greater size (82-84 nm) than that of

82
empty micelles and exhibited uniform size distribution (PDI < 0.2) (Table 2.1). Both types
of micelles displayed net negative charge which can be explained by the presence and
ionization of carboxylic acid groups of PLE chains in the shell of the micelles. As
expected, the ζ-potential of the ALN-m/DTX was further decreased to −32 ± 2 mV
compared to −17.0 ± 1 mV for the unmodified ALK-m/DTX (Table 2.1), which is
indicative of the presence of additional negatively charged ALN moieties on the surface
of ALN-m. One of the common metabolic complications of bone metastasized breast
cancer is hypercalcemia. It is characterized by elevated serum calcium level (about 3 -
3.5 mM for moderate cases) and indicates poor prognosis[30]. Therefore, we next
examine how the presence of calcium ions may affect the characteristics of DTX-loaded
micelle. When ALN-m/DTX were incubated in DI water in the presence of 3.4 mM Ca2+,
their size substantially decreased to ca. 57 nm and ζ-potential increased (Table 2.1).
Likewise, a decrease in size and the net negative charge was observed for ALK-m/DTX,
however, in the less pronounced manner. This behavior is consistent with effective
chelation of calcium ions by bisphosphonate groups as well as partial neutralization of
carboxylate groups of PLE chains leading to decrease of net charge, reduced repulsions
between the polymer chains in the shell of the micelles and collapse of the micelle.
Similarly, the size of DTX-loaded micelles was also affected by the ionic strength:
micelles prepared in PBS were substantially smaller compared to those in DI water
(Table 2.1) due to the masking of the electrostatic repulsions in the shell of the micelles
by the electrolyte (0.15 M NaCl). Interestingly, the addition of calcium ions to dispersions
of micelles in PBS did not lead to a further decrease in particle size. Based on these
results, we can expect that dimensions of ALN-m/DTX under physiological conditions will
be around 60 nm and thus will be suitable for bone targeting and extravasation since the
capillaries and sinusoids in the bone marrow have pores of approximately 80-100
nm[31]. Dispersions of DTX-loaded micelles remained stable in solution, and no

83
changes in size or PDI were detectable within at least 4 weeks of storage at ambient
conditions.
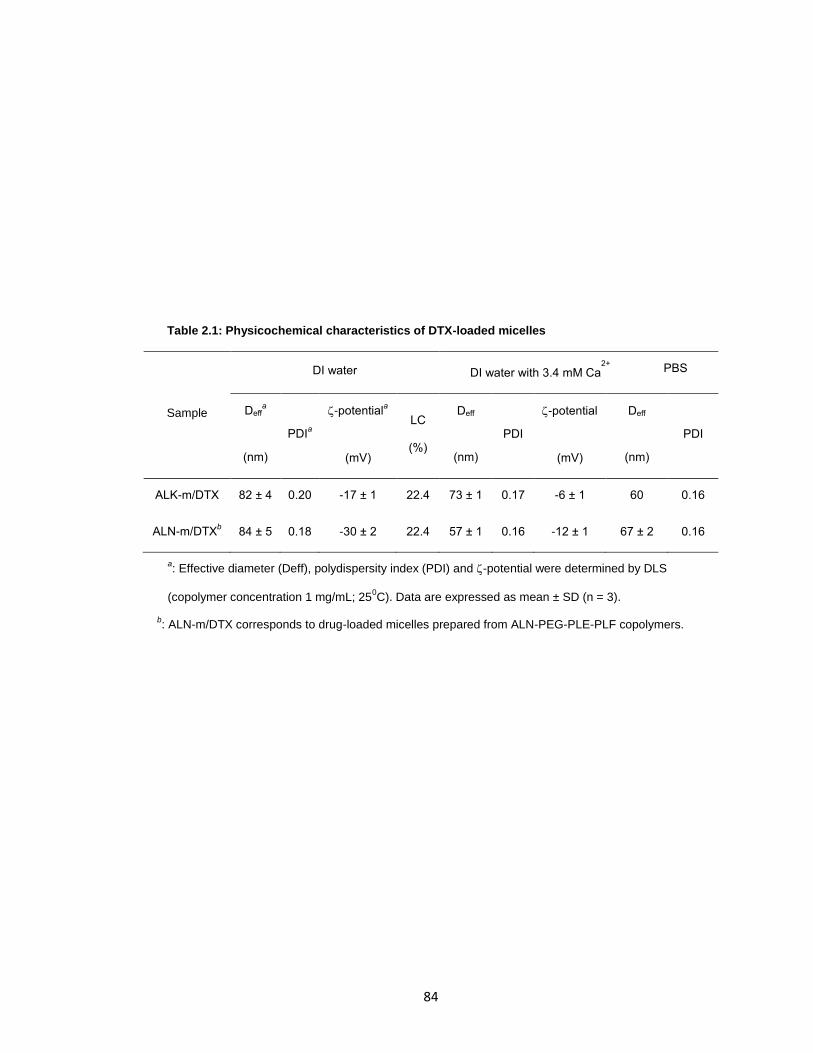
84
Table 2.1: Physicochemical characteristics of DTX-loaded micelles
Sample
DI water DI water with 3.4 mM Ca2+
PBS
Deffa
(nm)
PDIa
-potentiala
(mV)
LC
(%)
Deff
(nm)
PDI
-potential
(mV)
Deff
(nm)
PDI
ALK-m/DTX 82 ± 4 0.20 -17 ± 1 22.4 73 ± 1 0.17 -6 ± 1 60 0.16
ALN-m/DTXb 84 ± 5 0.18 -30 ± 2 22.4 57 ± 1 0.16 -12 ± 1 67 ± 2 0.16
a: Effective diameter (Deff), polydispersity index (PDI) and -potential were determined by DLS
(copolymer concentration 1 mg/mL; 250C). Data are expressed as mean ± SD (n = 3).
b: ALN-m/DTX corresponds to drug-loaded micelles prepared from ALN-PEG-PLE-PLF copolymers.

85
Both DTX-loaded formulations displayed similar sustained release profiles
without burst release (Figure 2.1A). Under normal physiological conditions (PBS, pH 7.4)
the cumulative release of DTX from both types of micelles was about 25% within 12 h.
The slightly slower release rates were observed for ALN-m/DTX as compared to ALK-
m/DTX at longer periods of times. Furthermore, the presence of serum albumin in the
medium (70% FBS) did not appear to have a significant impact on DTX release from
ALN-m/DTX suggesting that limited drug leakage from the micelles can be expected in
the circulation, which is the first step of the successful delivery.
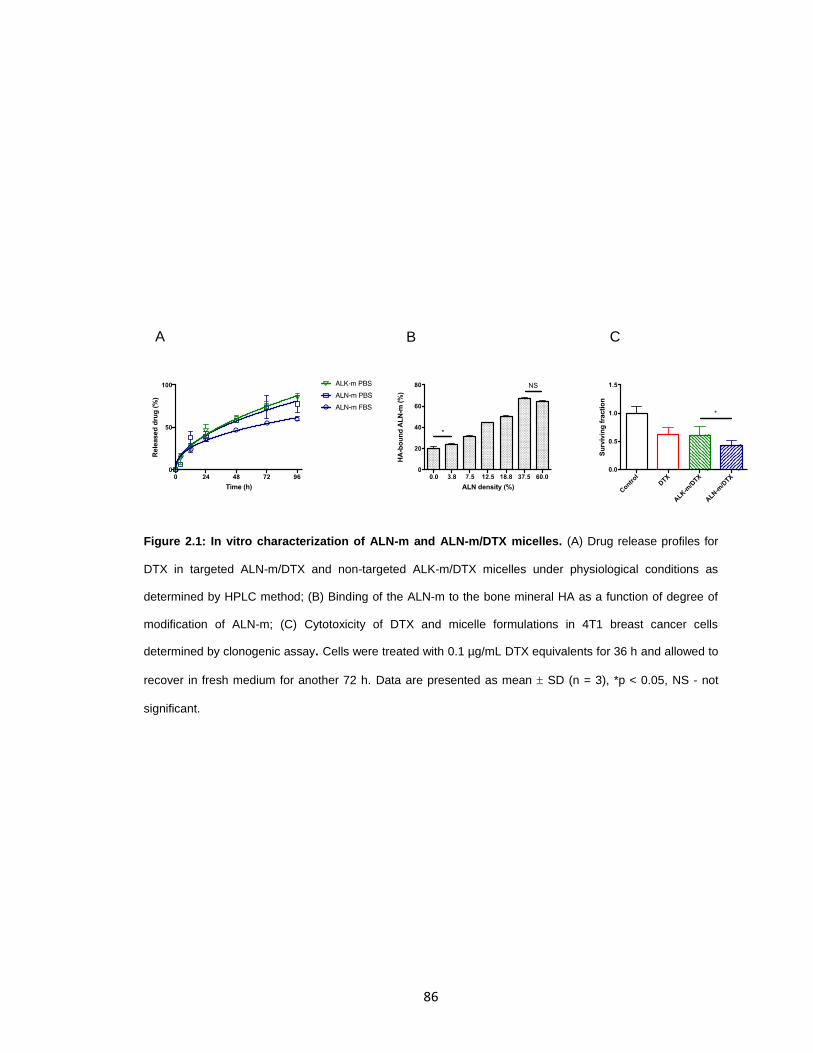
86
Figure 2.1: In vitro characterization of ALN-m and ALN-m/DTX micelles. (A) Drug release profiles for
DTX in targeted ALN-m/DTX and non-targeted ALK-m/DTX micelles under physiological conditions as
determined by HPLC method; (B) Binding of the ALN-m to the bone mineral HA as a function of degree of
modification of ALN-m; (C) Cytotoxicity of DTX and micelle formulations in 4T1 breast cancer cells
determined by clonogenic assay. Cells were treated with 0.1 µg/mL DTX equivalents for 36 h and allowed to
recover in fresh medium for another 72 h. Data are presented as mean SD (n = 3), *p < 0.05, NS - not
significant.
A B C

87
Bone mineral binding of ALN-decorated micelles.
Bone metastases cause structural damages in the skeleton, which ultimately lead
to exposure of bone mineral crystals within the metastatic lesions. These bone
resorption sites thus represent an ideal target for the design of osteotropic systems
using calcium chelating molecules such as bisphosphonates. The bone targeting and
retention capacity of ALN-m/DTX were evaluated using HA powder as a bone mineral
mimicking model[32]. To this end, we have screened a series of fluorescently-labeled
targeted micelles with various content of ALN moieties that was controlled by the molar
ratio of fluorescently labeled ALK-PEG-PLE-PLF and ALN-PEG-PLE-PLF copolymers.
The quantification of HA-associated fluorescence indicated that all bone-targeted
micelles displayed a good affinity to HA as a result of multivalent interactions and
anchoring via ALN moieties (Figure 2.1B). As expected, the binding of targeted ALN-m
exceeded that of the non-targeted ALK-m and became more efficient as the density of
targeting ligands increased. The bound fraction of ALN-m reached saturation level
(approximately 70%) for the ALN-m with 37% ALN moieties on their surfaces and a
further increase in ligand density did not provide statistically significant improvements in
HA binding. The observed ligand density effect can be related but not limited to improper
ligand presentation since ALN moieties attached to the flexible PEG chains may be
buried in the shell surrounding the core of the micelles or steric hindrance of the ligands
due to the size of the micelles. Importantly, the binding capacity of ALN-m was not
compromised by the presence of free calcium ions in the solution (Figure S2.4). This
would imply that binding of ALN-m to calcium ions present in the bloodstream upon
intravenous administration would not prevent their adsorption to the mineral matrix in
bone. In addition, since calcium-bound ALN-m had less negative ζ-potential (Table 2.1),

88
this observation also supports the argument that binding of ALN-m to HA is mainly
driven by specific interactions via ALN moieties rather than by the negative ζ-potential of
the micelles. In this context, it is also noteworthy that non-targeted ALK-m displayed
relatively high binding efficiency (ca. 20%, Figure 2.1B). It may be attributed to the
contribution of binding via PLE block in the outer shell of the micelles. Indeed, it is well
known that acidic oligopeptides of aspartic acid or glutamic acid have an affinity toward
HA. Their binding capacity increases when repeating sequences of amino acid residues
are present in the sequence such as in osteopontin and sialoprotein, non-collagenous
bone proteins[33]. Thus, it is likely that an incorporation of short hydrophilic PLE blocks
(10 units) into the shell of the ALN-m may be beneficial not only for stabilization of
micellar structures but also by enabling additional targeting capabilities. Overall, these
results suggest that ALN-m could be a promising drug delivery system to target
chemotherapeutics to the bone resorption sites. Besides being an excellent bone
targeting agent, ALN pharmacological mechanism of action also involves an inhibitory
effect on osteoclasts as well as beneficial effects on osteocytes and osteoblasts[32]. In
addition to anti-resorptive properties, experimental data suggest that bisphosphonates
may also have indirect effects on tumor cells[34]. To evaluate whether the design of our
delivery system can capitalize on the above-mentioned ALN properties, we choose ALN-
m with maximal content of conjugated ALN moieties (ca. 75%) for the further
experiments.

89
Figure S2.4: Binding of the ALN(37%)-m to the bone mineral HA in DI water and in the presence of
3.4 mM CaCl2 in the solution. The binding capacity of ALN-m was not compromised by the presence of
free calcium ions in the solution.
DI water DI water + 3.4mM CaCl2
0
20
40
60
80
HA
-bo
un
d A
LN
-m (
%)

90
In vitro cytotoxicity studies.
Murine 4T1 triple-negative breast cancer cell line is known to be highly invasive,
relatively resistant to chemotherapy and radiotherapy[35] and is widely used to establish
primary or metastatic tumor models in BALB/c mice. The cytotoxic effect of DTX loaded
into the polymeric micelles was first evaluated against 4T1 cells using MTT assay. As
summarized in Table 2.2, cytotoxicity of the DTX formulated into ALN-m was
substantially increased compared to ALK-m/DTX or conventional DTX formulation
(Tween 80/ethanol/PEG300): the IC50 value for ALN-m/DTX was 0.035 0.016 M, an
order of magnitude lower than those for other treatments. Interestingly, when the micelle
formulations were tested under the conditions mimicking hypercalcemia (3.4 mM Ca2+),
the IC50 values were further lowered. It could be explained by the neutralization of the
net surface charge of the micelles, which may facilitate their enhanced cellular uptake.
Of note, ALN alone at the concentration corresponding to the highest tested
concentration of ALN-m (about 30 M) was found not to be toxic to 4T1 cells (Figure
S2.5). To further validate these results, an additional study was conducted using a
clonogenic assay (Figure 2.1C). Reduction in the colony-forming potential of 4T1 cells
was observed following a 36 h exposure to all DTX formats. Consistent with MTT assay
results, ALN-m/DTX showed the most potent cytotoxicity and resulted in significantly
lower (p < 0.05) fraction of surviving cells. The mechanism explaining observed
enhanced cytotoxicity of ALN-m/DTX is not clear. One possibility is that ALN conjugation
may affect the cellular entry of the micelles and/or alter their cellular trafficking. The
Hammond group has previously reported that liposomes which were coated with
poly(acrylic acid) functionalized with ALN via a “layer-by-layer” method were more
effectively taken up by osteosarcoma cells compared to non-targeted liposomes[36].

91
These ALN-targeted liposomes loaded with doxorubicin also displayed higher levels of
cytotoxicity compared to the uncoated control. More studies are needed to understand
the observed cytotoxic effects of ALN-m/DTX.

92
Table 2.2: IC50 values of free DTX and DTX-loaded micelles in 4T1 breast cancer cell line.
Sample
IC50, µMa
RPMI RPMI (3.4 mM Ca2+
)
DTX 2.16 ± 0.82 -
ALK-m/DTX 1.39 ± 0.49 0.26 ± 0.05
ALN-m/DTX 0.035 ± 0.002 0.012 ± 0.020
a: The IC50 values represent the means SD (n = 3).
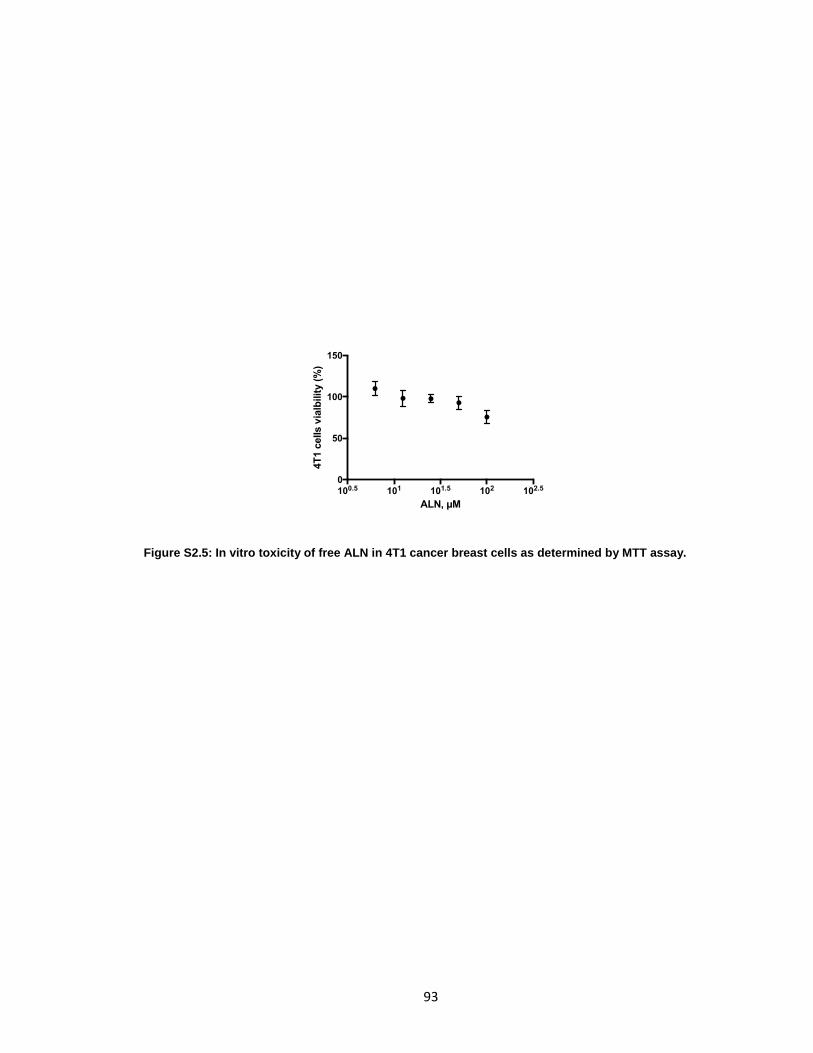
93
Figure S2.5: In vitro toxicity of free ALN in 4T1 cancer breast cells as determined by MTT assay.
100.5 101 101.5 102 102.50
50
100
150
ALN, µM
4T
1 c
ells v
ialb
ilit
y (
%)

94
Remodeling of the tumor-bone microenvironment.
As a therapeutic agent ALN was reported to inhibit osteoclast formation and limit
their resorptive activity. ALN was also reported to indirectly affects osteoclast precursors,
which are a source of resorption-stimulating cytokines[37]. We next investigated whether
ALN moieties tethered to the surface of the micelles could retain its inhibitory effect on
osteoclast formation in vitro. Murine monocyte/macrophage-like RAW264.7 cells that
maintain the capacity to differentiate into osteoclast-like cells in the presence of RANKL
were used in these studies. To this end, osteoclast precursor RAW264.7 cells were
stimulated by RANKL (50 ng/mL) with or without the presence of free ALN or ALN-m in
the culture medium. RAW-derived osteoclasts were confirmed through the staining of
TRAP enzyme, a key marker involved in osteoclastic bone resorption. As shown in
Figure 2.2A, treatment with ALN-m attenuated osteoclasts formation in a concentration-
dependent manner similarly to free ALN. The IC50 values for free ALN and ALN-m (6.4
2.5 M and 8.8 4.6 M, respectively) did not differ significantly from each other (p >
0.05). Importantly, treatment with ALK-m did not have any noticeable effect on
osteoclastogenesis (Figure 2.2B). The tumor cells are also known to play a critical role in
this cycle by cleaving anchored RANKL into soluble format via an elevated expression of
MMP-7[38]. Thus, we next performed osteoclastogenesis assay using the conditioned
medium (CM) from cultured 4T1 cells that contained secreted differentiation factors.
Consistent with the former results, ALN-m significantly repressed (p < 0.01) the
formation of mature RAW-derived osteoclasts at the ALN concentration corresponding to
its IC50 value (9 M) (Figure 2.2C). In contrast, multiple multinucleated TRAP-positive
osteoclasts were formed in 4T1-CM treated RAW264.7 cells in the presence of ALK-m at

95
the same polymer concentration. These results suggest that ALN-m provide substantial
inhibitory capacity against cancer-induced osteoclastogenesis.
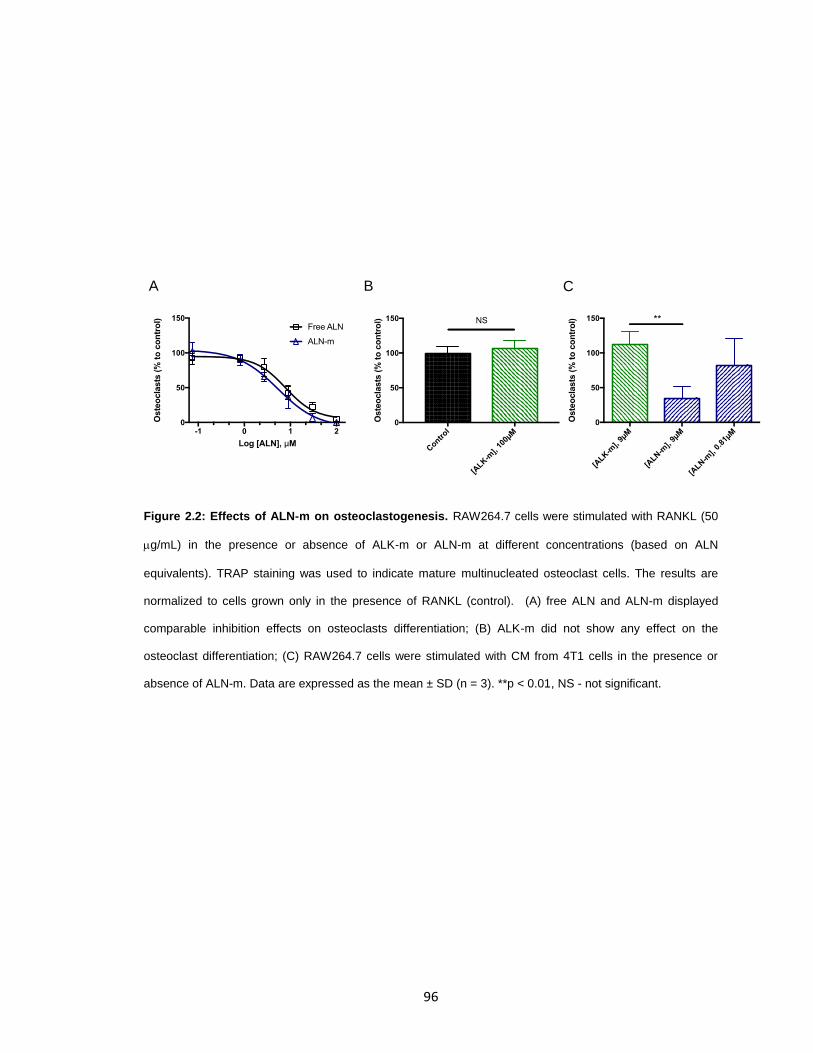
96
Figure 2.2: Effects of ALN-m on osteoclastogenesis. RAW264.7 cells were stimulated with RANKL (50
g/mL) in the presence or absence of ALK-m or ALN-m at different concentrations (based on ALN
equivalents). TRAP staining was used to indicate mature multinucleated osteoclast cells. The results are
normalized to cells grown only in the presence of RANKL (control). (A) free ALN and ALN-m displayed
comparable inhibition effects on osteoclasts differentiation; (B) ALK-m did not show any effect on the
osteoclast differentiation; (C) RAW264.7 cells were stimulated with CM from 4T1 cells in the presence or
absence of ALN-m. Data are expressed as the mean ± SD (n = 3). **p < 0.01, NS - not significant.
A B C

97
In breast cancer bone metastasis microenvironment bi-directional interactions
between tumor cells and osteoclasts lead to both osteolysis and tumor growth[39].
Hence, it is also crucial to suppress the resorptive activity of the osteoclasts. To evaluate
the effects of ALN-m on osteoclast bone lytic activity, RAW264.7 cells were cultured on
plates coated with microcrystalline calcium phosphate and stimulated with RANKL in the
presence or absence of ALN-m. We selected two concentrations of ALN-m, 9 M and
0.81 M (on ALN basis) that were determined to suppress or not affect osteoclast
differentiation, respectively. The resorption activity was quantified as a total resorptive
area of “pits” formed by osteoclasts (Figure 3A). RANKL-stimulated cells formed a
number of pits, suggesting that the bone resorption activity of RANKL-treated cells made
them into functionally active state-resembling osteoclasts. As expected, non-targeted
ALN-m did not affect their resorption activity while treatment with ALN-m suppressed
formation of resorption pits. Treatment with 0.81 M ALN-m, the concentration that
ineffectively inhibited osteoclast differentiation, significantly reduced (p <0.01) overall
area of resorption pits compared with treatment with RANKL alone. Osteoclastic bone
resorption was inhibited practically completely by the treatment with 9 M ALN-m which
is in line with our observation that ALN-m at this concentration also significantly inhibit
osteoclast formation (Figure 2.2C). To exclude the direct effect of ALN-m on
osteoclastogenesis, in parallel experiment RAW264.7 cells were first pre-differentiated
into osteoclasts for 4 days and then were treated with ALN-m. As it seen in Figure 2.3B,
osteoclasts treatment with ALN-m significantly inhibited their resorption activity in a
concentration-independent manner. Together these results implicate that the ALN-m
have an inhibitory activity on both osteoclast differentiation and function and thus could
provide additional benefit in combating bone metastases by intervening the disease-
induced bone loss.
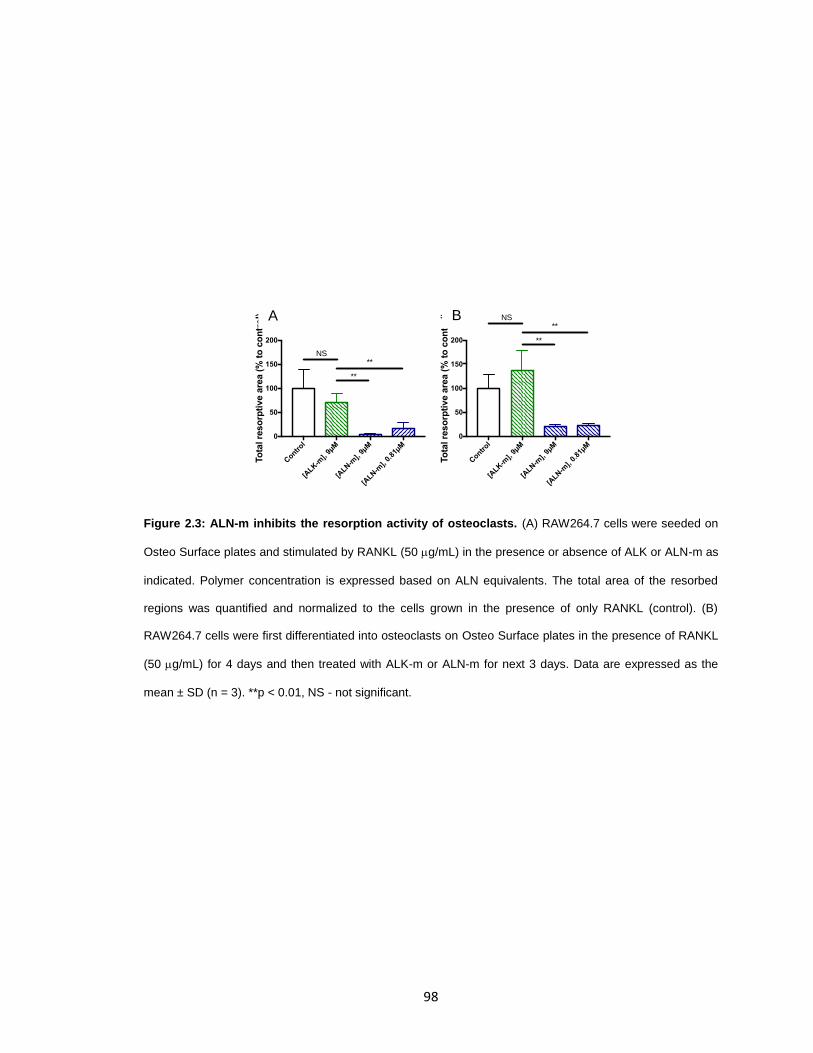
98
Figure 2.3: ALN-m inhibits the resorption activity of osteoclasts. (A) RAW264.7 cells were seeded on
Osteo Surface plates and stimulated by RANKL (50 g/mL) in the presence or absence of ALK or ALN-m as
indicated. Polymer concentration is expressed based on ALN equivalents. The total area of the resorbed
regions was quantified and normalized to the cells grown in the presence of only RANKL (control). (B)
RAW264.7 cells were first differentiated into osteoclasts on Osteo Surface plates in the presence of RANKL
(50 g/mL) for 4 days and then treated with ALK-m or ALN-m for next 3 days. Data are expressed as the
mean ± SD (n = 3). **p < 0.01, NS - not significant.
Contr
ol
[ALK
-m],
9µM
[ALN
-m],
9µM
[ALN
-m],
0.81µM
0
50
100
150
200
To
tal re
so
rpti
ve a
rea (
% t
o c
on
tro
l)
**
**
NS
Contr
ol
[ALK
-m],
9µM
[ALN
-m],
9µM
[ALN
-m],
0.81µM
0
50
100
150
200
To
tal re
so
rpti
ve a
rea (
% t
o c
on
tro
l)
**
**
NS
A B

99
In the osteolytic cycle in bone metastases, residential macrophages and
circulating monocytes stimulating by the cytokines secreted by the tumor can be
recruited and infiltrated into the tumor microenvironment[40,41]. The tumor-associated
macrophages are not only capable of osteoclast differentiation but also are considered
to play an important role in angiogenesis, immunosuppression and thus contribute to
disease progression and poor chemotherapy response. Here, we intended to determine
whether ALN- m were able to attenuate macrophage recruitment by cancer cells in vitro.
To this end, we used the Transwell method where the Raw264.7 cells were seeded on
the upper insert and allowed to migrate across the membrane to the lower chamber, on
which 4T1 cells were seeded (Figure 2.4A). In the 24 h period, a substantial number of
the macrophages were found to migrate across the membrane toward 4T1 cells, with
more than a 10-fold increase in a number of migrated cells compared to the negative
control (Figure 2.4, B and C). When treated with ALN-m, as low as 0.81 µM on ALN
base, migration ability of macrophages was suppressed to a practically negligible level
(p > 0.05 compared to negative control). Given that ALN-m at such concentration is non-
toxic to both types of cells, it could be assumed that the “cooperation” between tumor
and macrophage was inhibited.
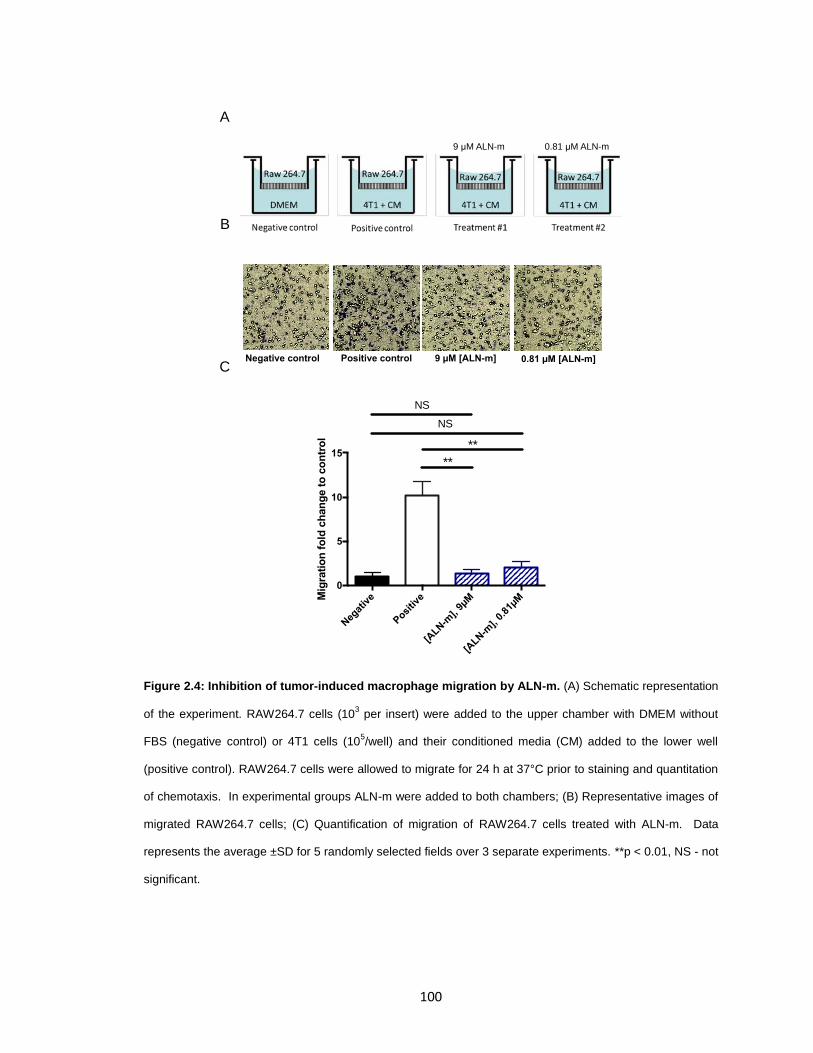
100
Figure 2.4: Inhibition of tumor-induced macrophage migration by ALN-m. (A) Schematic representation
of the experiment. RAW264.7 cells (103 per insert) were added to the upper chamber with DMEM without
FBS (negative control) or 4T1 cells (105/well) and their conditioned media (CM) added to the lower well
(positive control). RAW264.7 cells were allowed to migrate for 24 h at 37°C prior to staining and quantitation
of chemotaxis. In experimental groups ALN-m were added to both chambers; (B) Representative images of
migrated RAW264.7 cells; (C) Quantification of migration of RAW264.7 cells treated with ALN-m. Data
represents the average ±SD for 5 randomly selected fields over 3 separate experiments. **p < 0.01, NS - not
significant.
9 µM ALN-m 0.81 µM ALN-m
Negative control Positive control 9 µM [ALN-m] 0.81 µM [ALN-m]
Neg
ativ
e
Posi
tive
[ALN
-m],
9µM
[ALN
-m],
0.81µM
0
5
10
15
Mig
rati
on
fo
ld c
han
ge t
o c
on
tro
l
**
**
NS
NS
A
B
C

101
In vivo therapeutic efficacy.
Before assessing the therapeutic efficacy of DTX-loaded micelles, we evaluated
the pharmacokinetics of both ALN-m/DTX and ALK-m/DTX in healthy BABL/C mice and
compared to “free” DTX (in Tween 80/ethanol/PEG300) at an equivalent dose of 10
mg/kg DTX. The PK parameters calculated using a non-compartment model are
presented in Supplementary Table S2.2. DTX incorporation into micelles resulted in a
modest increase in plasma half-life (8.9 h for ALK-m/DTX and 7.7 h for ALK-m/DTX vs.
6.2 h for free DTX). Moreover, DTX plasma exposure as measured by the area-under-
the-curve (AUC) was ~1.5 times higher for ALN-m/DTX than that for ALK-m/DTX. Both
the volume of distribution and clearance were decreased by 1.3-fold and 1,5-fold,
respectively, in the ALN-m/DTX treatment group compared with the ALK-m/DTX
treatment group. These data suggest that ALN-m can prolong circulation time of DTX
through reducing its rapid elimination and contributing to more DTX distributed into bone
tissue.
To test the therapeutic effect of ALN-m/DTX on established bone metastases, we used
syngeneic 4T1 breast cancer model[42]. The 4T1/Luc cells were inoculated into a left
cardiac ventricle of Balb/C mice to establish disseminated metastatic disease. Bone
metastatic seeding and burden was monitored by whole body BLI. The disease was
found to be very aggressive and associated with substantial weight loss: tumor burden
exclusively inside the skeleton was successfully established in 1-2 weeks after cell
injection and often resulted in mortality within 3 weeks. Due to the aggressiveness of the
disease, the treatments were administered daily for the first three days to assure that all
individual experimental subjects received the same total dose of the drug. In this model
of late treatment of aggressive bone metastasis, we detected a trend of delayed

102
progression of bone metastasis by either ALN-m/DTX or ALK-m/DTX treatment
compared with the control group (p < 0.05, Figure 2.5A and Figure S2.6). In addition,
both treatments attenuated animal weight loss (the effect of ALN-m/DTX was statistically
significant, p < 0.02 vs. control group) and can also be indicative of their therapeutic
potential (Figure 2.5B). This is in contrast with the treatment of DTX, which had a
negligible effect on reducing bone metastasis (p ≈ 0.28 versus the control group), which
can be explained by the aggressiveness of the disease model and the drug-resistant
nature of the 4T1 cell line. Of note, neither of the micelles-based formulations reached
statistical significance compared to free DTX treatment at the endpoint of the BLI
monitoring (18 days) when the mice in the control group succumbed to metastatic
cancer. However, the promising efficacy of DTX micellar formulations translated into
improved survival (Figure 2.5C). In line with BLI studies, the treatment with free DTX did
not provide any improvement in animal survival over the saline control. The non-targeted
ALK-m/DTX slightly extended the mean survival time from 18 to 21 days, albeit all mice
reached the endpoint before day 24. Remarkably, the ALN-m/DTX demonstrated the
best therapeutic outcome and significantly improved the survival time (p < 0.05)
compared to ALK-m/DTX group with 2 out of 5 long-term survivors who reached the end
point at day 35 (Figure 2.5C). Such a therapeutic response to ALN-m/DTX is far superior
to either ALK-m/DTX or DTX alone suggesting a benefit of bone targeting along with
chemotherapy in late-stage bone metastasis. Notably, no visceral organ toxicities were
observed as a result of the treatments as representatively assessed for liver, spleen and
kidney by tissue histopathology analysis (Figure 2.5D). Extramedullary hematopoiesis is
often reported as a consequence of bone metastases in this model[43]. In the control
group, strong extramedullary hematopoiesis was observed in the liver, while in the
treatment groups it was significantly reduced. Marrow toxicity indicated by decreased
cellularity was occasionally present in the bone sections, but was similar between

103
groups. These observations suggest a favorable toxicity profile for the DTX micellar
formulations.
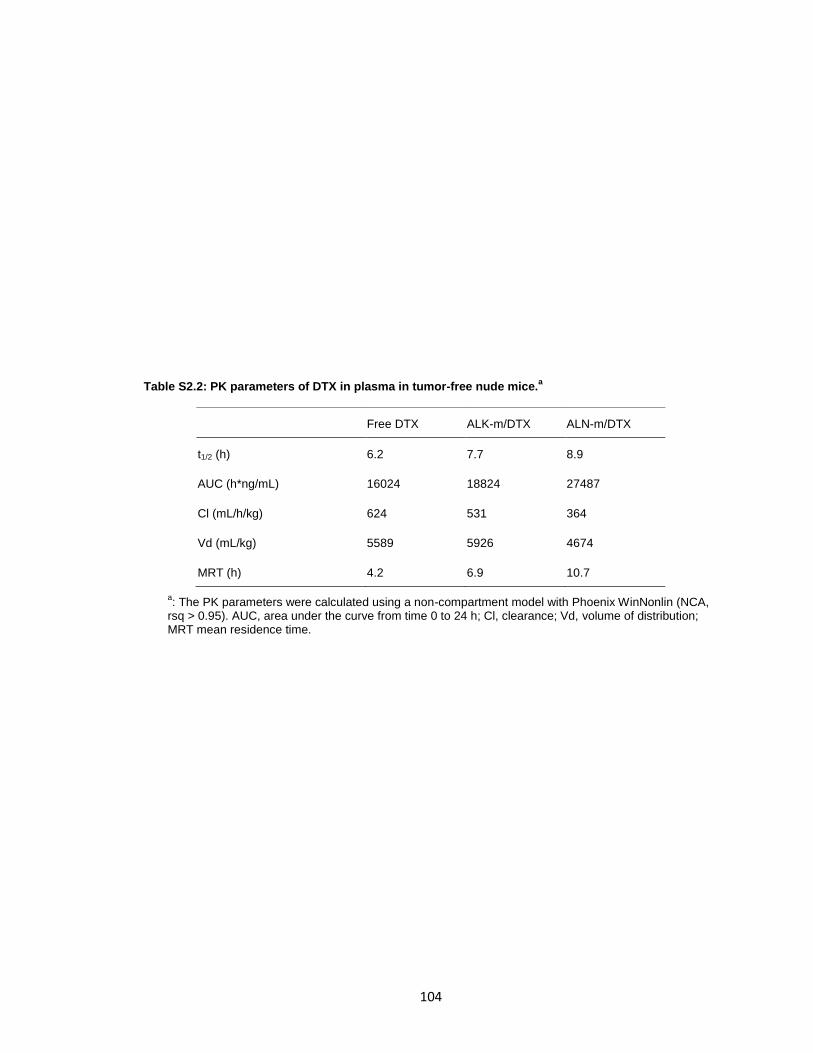
104
Table S2.2: PK parameters of DTX in plasma in tumor-free nude mice.a
Free DTX ALK-m/DTX ALN-m/DTX
t1/2 (h) 6.2 7.7 8.9
AUC (h*ng/mL) 16024 18824 27487
Cl (mL/h/kg) 624 531 364
Vd (mL/kg) 5589 5926 4674
MRT (h) 4.2 6.9 10.7
a: The PK parameters were calculated using a non-compartment model with Phoenix WinNonlin (NCA,
rsq > 0.95). AUC, area under the curve from time 0 to 24 h; Cl, clearance; Vd, volume of distribution; MRT mean residence time.
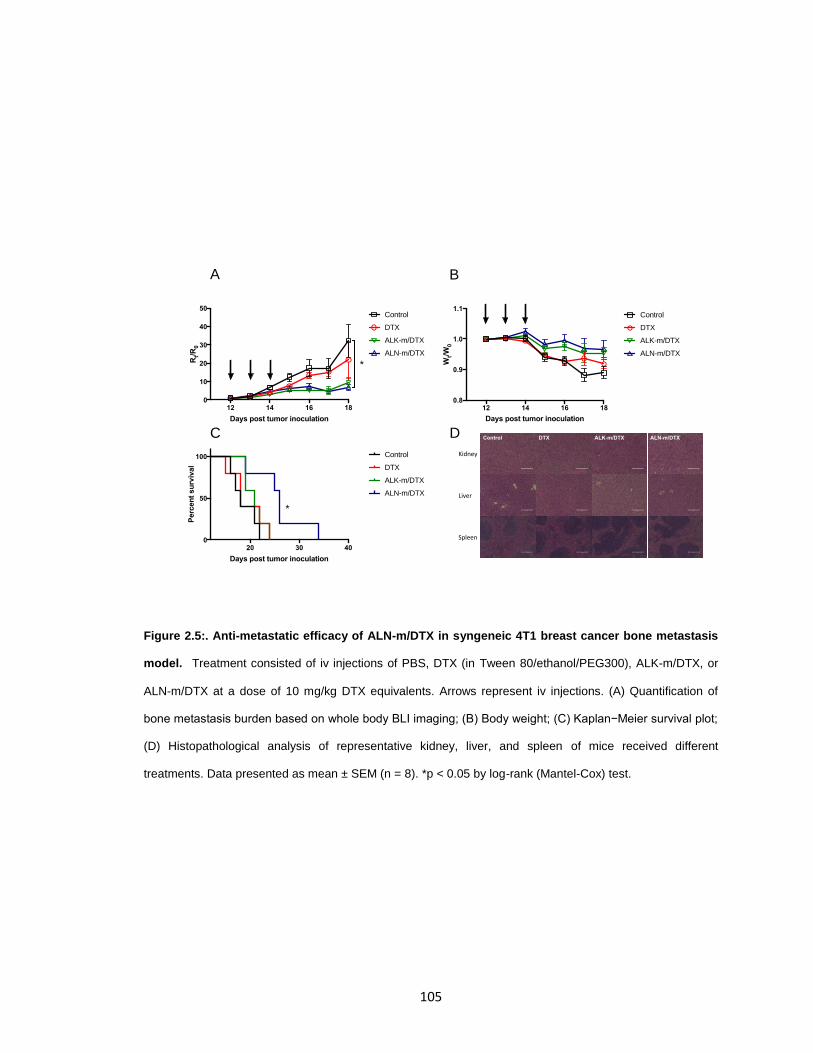
105
Figure 2.5:. Anti-metastatic efficacy of ALN-m/DTX in syngeneic 4T1 breast cancer bone metastasis
model. Treatment consisted of iv injections of PBS, DTX (in Tween 80/ethanol/PEG300), ALK-m/DTX, or
ALN-m/DTX at a dose of 10 mg/kg DTX equivalents. Arrows represent iv injections. (A) Quantification of
bone metastasis burden based on whole body BLI imaging; (B) Body weight; (C) Kaplan−Meier survival plot;
(D) Histopathological analysis of representative kidney, liver, and spleen of mice received different
treatments. Data presented as mean ± SEM (n = 8). *p < 0.05 by log-rank (Mantel-Cox) test.
12 14 16 180
10
20
30
40
50
Days post tumor inoculation
Rt/R
0
Control
DTX
ALK-m/DTX
ALN-m/DTX
*
20 30 400
50
100
Days post tumor inoculation
Perc
en
t su
rviv
al
Control
DTX
ALK-m/DTX
ALN-m/DTX
*
12 14 16 180.8
0.9
1.0
1.1
Days post tumor inoculation
Wt/W
0
Control
DTX
ALK-m/DTX
ALN-m/DTX
A
C
B
D Control DTX ALK-m/DTX ALN-m/DTX
Kidney
Liver
Spleen
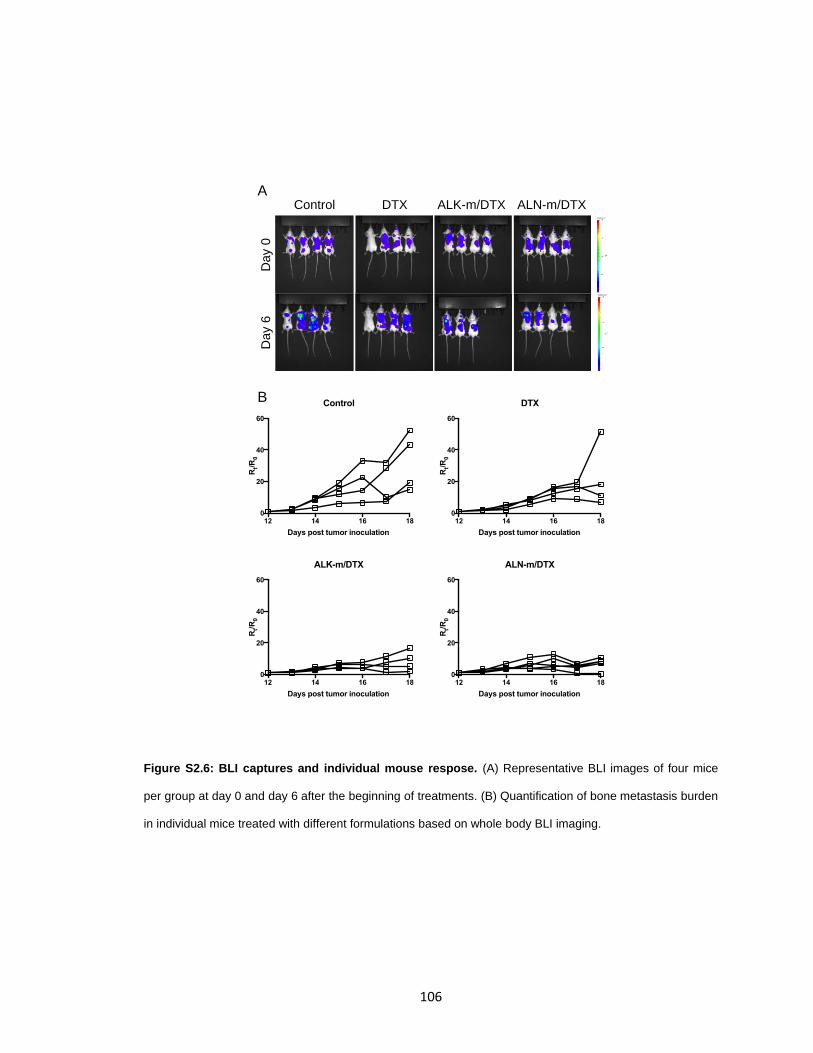
106
Figure S2.6: BLI captures and individual mouse respose. (A) Representative BLI images of four mice
per group at day 0 and day 6 after the beginning of treatments. (B) Quantification of bone metastasis burden
in individual mice treated with different formulations based on whole body BLI imaging.
Control ALK-m/DTXDTX ALN-m/DTX
Day 0
Day 6
12 14 16 180
20
40
60
Days post tumor inoculation
Rt/R
0
Control
12 14 16 180
20
40
60
Days post tumor inoculation
Rt/R
0
ALK-m/DTX
12 14 16 180
20
40
60
Days post tumor inoculation
Rt/R
0
DTX
12 14 16 180
20
40
60
Days post tumor inoculation
Rt/R
0
ALN-m/DTX
A
B

107
Among all options treating metastatic breast cancer including chemotherapy,
hormonal therapy and targeted therapy, radiation therapy (RT) remains an essential
modality particularly for the palliative purpose. The most common utilities of palliative
RT are for pain control, improving ambulation, preventing fractures and controlling spinal
cord compression/neurologic deficits and preserving the quality of life particularly for
patients with bone metastases. During the past few decades, multiple randomized
controlled clinical trials have demonstrated equivalent short-term pain relief between a
single 8 Gray (Gy) fraction and multiple fraction regimens, including most commonly 30
Gy in 10 fractions, 24 Gy in 6 fractions and 20 Gy in 5 fractions[44-48]. While single
fraction treatment optimized convenience of patients and their caregivers and was
related with lower acute toxicity, retreatment rates to the same anatomic site due to
recurrent pain were higher in those who received single fractions compared to
fractionated treatment courses. It is important for further studies to explore the
possibilities of combined therapy to enhance the efficacy of single fraction RT. To test
this notion, we conducted a proof-of-concept experiment utilizing sequential
chemotherapy with ALN-m/DTX and RT. Two groups of animals with developed
metastasis received three i.v. injections daily of either saline or ALN-m/DTX (10 mg/kg
on DTX basis) and then, 24 h after the last treatment, mice received a total of 5 Gy of
radiation at a dose rate of 3 Gy/min. 5Gy in one fraction is known clinically as an
ineffective dose for tumor control and is lower than the threshold dose causing
gastroenteral radiation syndrome as in human studies when large area or whole body is
irradiated. The treatment of late-stage metastasis in our mouse model with a single 5
Gy RT was not surprisingly ineffective and did not delay the progression of the disease
(Figure 2.6A). In contrast, the administration of ALN-m/DTX followed by RT delayed
tumor growth and extended the animals median survival from 19 days (RT) to 30 days
(Figure 2.6B). Due to the large variations within the RT group (Figure S2.7), the

108
differences in tumor growth or survival between RT and combination group was close to
but not statistically significant (p = 0.06 vs. the RT-only treatment group). We did not
observe any episodes of diarrhea or hematochezia in the mice treated with RT. Further
detailed studies are needed to confirm the benefits of this combined treatment regimen.
Particularly, it might be interesting to test this combination therapy in another model of
bone metastasis, when breast cancer cells are injected directly into bone (such as tibia
or femur). While it is technically not a model of metastasis, it can replicate tumor-induced
changes in bone[42] and will permit to apply RT locally. Nevertheless, these results
suggest that treating cancer patients with bone-targeted chemotherapy combined with
radiotherapy could potentially be more effective and suppress the development and
dissemination of bone metastasis.
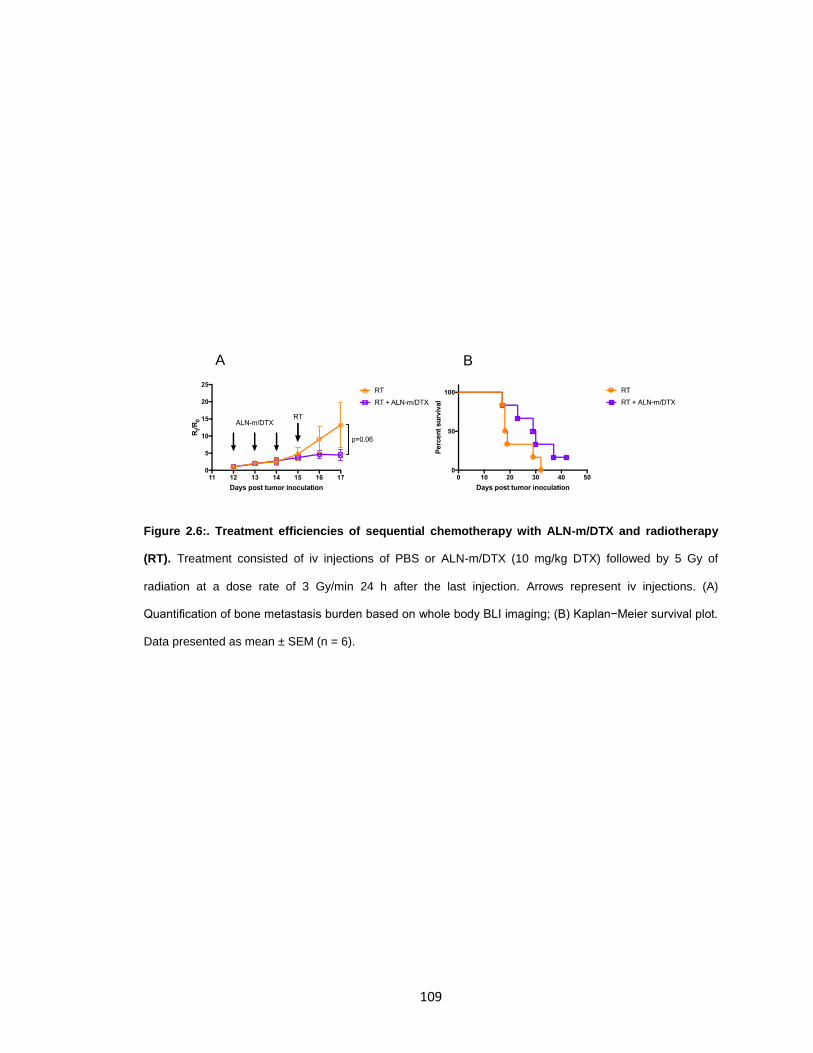
109
Figure 2.6:. Treatment efficiencies of sequential chemotherapy with ALN-m/DTX and radiotherapy
(RT). Treatment consisted of iv injections of PBS or ALN-m/DTX (10 mg/kg DTX) followed by 5 Gy of
radiation at a dose rate of 3 Gy/min 24 h after the last injection. Arrows represent iv injections. (A)
Quantification of bone metastasis burden based on whole body BLI imaging; (B) Kaplan−Meier survival plot.
Data presented as mean ± SEM (n = 6).
A B
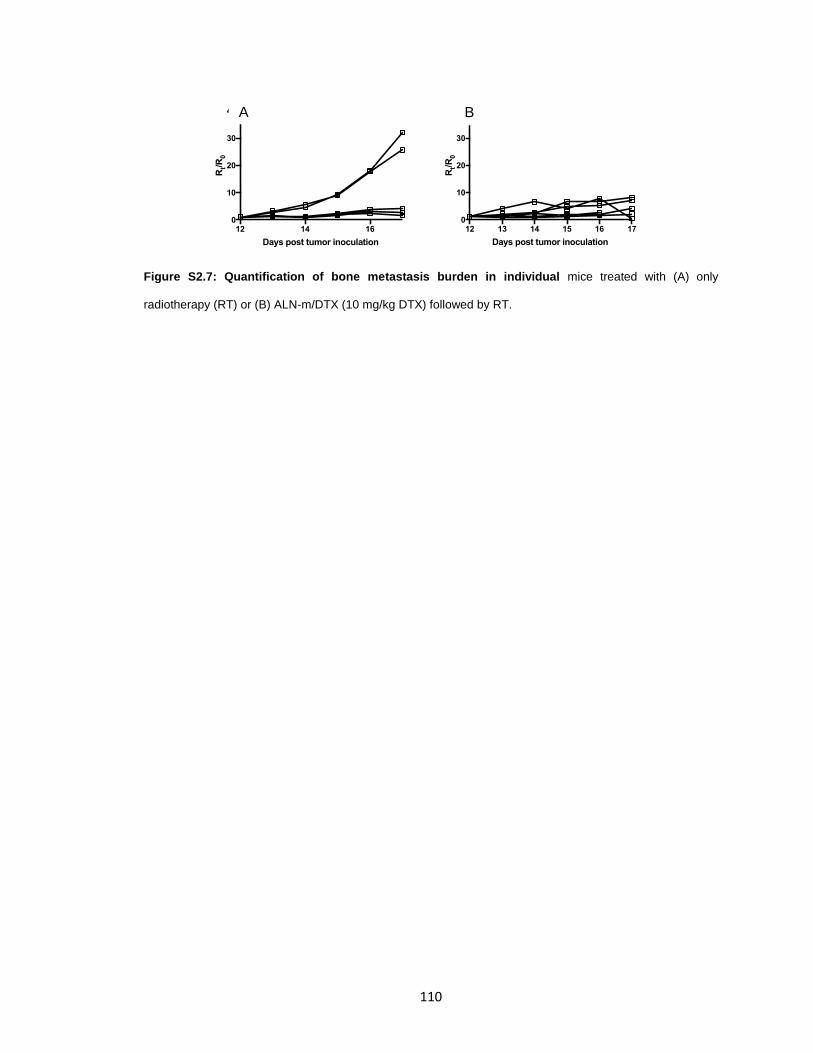
110
Figure S2.7: Quantification of bone metastasis burden in individual mice treated with (A) only
radiotherapy (RT) or (B) ALN-m/DTX (10 mg/kg DTX) followed by RT.
12 14 160
10
20
30
40
Days post tumor inoculation
Rt/R
012 13 14 15 16 17
0
10
20
30
40
Days post tumor inoculation
Rt/R
0
A B

111
2.4 CONCLUSION
We engineered a dual-functional bone-homing drug carrier based on
biodegradable polypeptidebased micelles that can deliver the drug to the bone and
shows a propensity for simultaneous remodeling the tumor-bone microenvironment. The
drug-loaded micelles were prepared via a robust procedure that allowed obtainment of a
high loading of docetaxel using an alendronate-modified copolymer with a relatively
short length of the hydrophobic segment. Docetaxel, when encapsulated into the ALN-m
was found to retain its cytotoxicity and to be even more effective in 4T1 breast cancer
cell killing in the conditions mimicking hypercalcemia in patients with metastases. These
ALN-m demonstrated enhanced binding to the bone mineral analog and were effective in
inhibiting osteoclast differentiation and their resorption activity as well as attenuate
tumor-induced migration of macrophages in vitro. In line with the in vitro results, ALN-
m/DTX delayed disease progression and improved survival in a syngeneic murine model
of breast cancer bone metastasis. This model represents a useful tool for examining the
later stages of breast cancer bone metastasis, however, the skeletal sites of metastatic
deposition and number and size of the metastases are extremely variable. Nevertheless,
our work provides new insight into the fusion of ALN moieties with drug-loaded micelles
to maximize the therapeutic efficiency of chemotherapy by targeting bone metastases
and their microenvironment.

112
2.5 REFERENCE
1. Chen YC, Sosnoski DM, Mastro AM. Breast cancer metastasis to the bone: mechanisms of bone loss. Breast Cancer Res. 7 ed. 2010;12: 215. doi:10.1186/bcr2781
2. Fang J, Xu Q. Differences of osteoblastic bone metastases and osteolytic bone metastases in clinical features and molecular characteristics. Clin Transl Oncol. 2015;17: 173–179. doi:10.1007/s12094-014-1247-x
3. Cleeland C, Moos von R, Walker MS, Wang Y, Gao J, Chavez-MacGregor M, et al. Burden of symptoms associated with development of metastatic bone disease in patients with breast cancer. Support Care Cancer. 2016;24: 3557–3565. doi:10.1007/s00520-016-3154-x
4. Kingsley LA, Fournier PGJ, Chirgwin JM, Guise TA. Molecular biology of bone metastasis. Mol Cancer Ther. 2007;6: 2609–2617. doi:10.1158/1535-7163.MCT-07-0234
5. O'Carrigan B, Wong MH, Willson ML, Stockler MR, Pavlakis N, Goodwin A. Bisphosphonates and other bone agents for breast cancer. Cochrane Database Syst Rev. 2017;10: CD003474. doi:10.1002/14651858.CD003474.pub4
6. Kuchuk I, Hutton B, Moretto P, Ng T, Addison CL, Clemons M. Incidence, consequences and treatment of bone metastases in breast cancer patients-Experience from a single cancer centre. J Bone Oncol. 2013;2: 137–144. doi:10.1016/j.jbo.2013.09.001
7. Wang D, MILLER S, Kopečková P, KOPECEK J. Bone-targeting macromolecular therapeutics. Adv Drug Deliv Rev. Elsevier; 2005;57: 1049–1076. doi:10.1016/j.addr.2004.12.011
8. Cole LE, Vargo-Gogola T, Roeder RK. Targeted delivery to bone and mineral deposits using bisphosphonate ligands. Adv Drug Deliv Rev. 2016;99: 12–27. doi:10.1016/j.addr.2015.10.005
9. Love C, Din AS, Tomas MB, Kalapparambath TP, Palestro CJ. Radionuclide Bone Imaging: An Illustrative Review. Radiographics. 2003;23: 341–358. doi:10.1148/rg.232025103
10. Ramanlal Chaudhari K, Kumar A, Megraj Khandelwal VK, Ukawala M, Manjappa AS, Mishra AK, et al. Bone metastasis targeting: a novel approach to reach bone using Zoledronate anchored PLGA nanoparticle as carrier system loaded with Docetaxel. J Control Release. 2012;158: 470–478. doi:10.1016/j.jconrel.2011.11.020
11. Wang X, Yang Y, Jia H, Jia W, Miller S, Bowman B, et al. Peptide Decoration of Nanovehicles to Achieve Active Targeting and Pathology-Responsive Cellular

113
Uptake for Bone Metastasis Chemotherapy. Biomater Sci. 2014;2: 961–971. doi:10.1039/C4BM00020J
12. Thamake SI, Raut SL, Gryczynski Z, Ranjan AP, Vishwanatha JK. Alendronate coated poly-lactic-co-glycolic acid (PLGA) nanoparticles for active targeting of metastatic breast cancer. Biomaterials. 2012;33: 7164–7173. doi:10.1016/j.biomaterials.2012.06.026
13. Fernandes C, Monteiro S, Belchior A, Marques F, Gano L, Correia JDG, et al. Novel (188)Re multi-functional bone-seeking compounds: Synthesis, biological and radiotoxic effects in metastatic breast cancer cells. Nucl Med Biol. 2016;43: 150–157. doi:10.1016/j.nucmedbio.2015.11.004
14. Ye W-L, Zhao Y-P, Li H-Q, Na R, Li F, Mei Q-B, et al. Doxorubicin-poly (ethylene glycol)-alendronate self-assembled micelles for targeted therapy of bone metastatic cancer. Sci Rep. 2015;5: 14614. doi:10.1038/srep14614
15. Miller K, Eldar-Boock A, Polyak D, Segal E, Benayoun L, Shaked Y, et al. Antiangiogenic antitumor activity of HPMA copolymer-paclitaxel-alendronate conjugate on breast cancer bone metastasis mouse model. Molecular Pharmaceutics. 2011;8: 1052–1062. doi:10.1021/mp200083n
16. Clementi C, Miller K, Mero A, Satchi-Fainaro R, Pasut G. Dendritic poly(ethylene glycol) bearing paclitaxel and alendronate for targeting bone neoplasms. Molecular Pharmaceutics. 2011;8: 1063–1072. doi:10.1021/mp2001445
17. Miller K, Clementi C, Polyak D, Eldar-Boock A, Benayoun L, Barshack I, et al. Poly(ethylene glycol)-paclitaxel-alendronate self-assembled micelles for the targeted treatment of breast cancer bone metastases. Biomaterials. 2013;34: 3795–3806. doi:10.1016/j.biomaterials.2013.01.052
18. Desale SS, Cohen SM, Zhao Y, Kabanov AV, Bronich TK. Biodegradable hybrid polymer micelles for combination drug therapy in ovarian cancer. J Control Release. 2013;171: 339–348. doi:10.1016/j.jconrel.2013.04.026
19. Srinivasan R, Tan LP, Wu H, Yang P-Y, Kalesh KA, Yao SQ. High-throughput synthesis of azide libraries suitable for direct “click” chemistry and in situ screening. Org Biomol Chem. 2009;7: 1821–1828. doi:10.1039/b902338k
20. Ananthapadmanabhan KP, Goddard ED, Turro NJ, Kuo PL. Fluorescence Probes for Critical Micelle Concentration. Langmuir. 1985;1: 352–355. doi:10.1021/la00063a015
21. Zhang S, Wright JEI, Bansal G, Cho P, Uludag H. Cleavage of disulfide-linked fetuin-bisphosphonate conjugates with three physiological thiols. Biomacromolecules. 2005;6: 2800–2808. doi:10.1021/bm050273s
22. Ferrari M, Fornasiero MC, Isetta AM. MTT colorimetric assay for testing macrophage cytotoxic activity in vitro. Journal of Immunological Methods. Elsevier; 1990;131: 165–172. doi:10.1016/0022-1759(90)90187-Z

114
23. Lelekakis M, Moseley JM, Martin TJ, Hards D, Williams E, Ho P, et al. A novel orthotopic model of breast cancer metastasis to bone. Clinical & experimental metastasis. Springer; 1999;17: 163–170.
24. Kim JO, Oberoi HS, Desale S, Kabanov AV, Bronich TK. Polypeptide nanogels with hydrophobic moieties in the cross-linked ionic cores: synthesis, characterization and implications for anticancer drug delivery. J Drug Target. 2nd ed. Taylor & Francis; 2013;21: 981–993. doi:10.3109/1061186X.2013.831421
25. Bertrand N, Grenier P, Mahmoudi M, Lima EM, Appel EA, Dormont F, et al. Mechanistic understanding of in vivo protein corona formation on polymeric nanoparticles and impact on pharmacokinetics. Nat Commun. Nature Publishing Group; 2017;8: 777. doi:10.1038/s41467-017-00600-w
26. Jeong B, Kibbey MR, Birnbaum JC, Won YY, Gutowska A. Thermogelling biodegradable polymers with hydrophilic backbones: PEG-g-PLGA. Macromolecules. 2000;33: 8317–8322. doi:10.1021/ma000638v
27. Pierri E, Avgoustakis K. Poly(lactide)-poly(ethylene glycol) micelles as a carrier for griseofulvin. J Biomed Mater Res A. 2005;75: 639–647. doi:10.1002/jbm.a.30490
28. Lyseng-Williamson KA, Fenton C. Docetaxel: a review of its use in metastatic breast cancer. Drugs. 2005;65: 2513–2531. doi:10.2165/00003495-200565170-00007
29. Jones SE, Erban J, Overmoyer B, Budd GT, Hutchins L, Lower E, et al. Randomized phase III study of docetaxel compared with paclitaxel in metastatic breast cancer. J Clin Oncol. 2005;23: 5542–5551. doi:10.1200/JCO.2005.02.027
30. Schwartzberg LS, Navari RM. Safety of Polysorbate 80 in the Oncology Setting. Adv Ther. 2018;35: 754–767. doi:10.1007/s12325-018-0707-z
31. Albanese A, Tang PS, Chan WCW. The Effect of Nanoparticle Size, Shape, and Surface Chemistry on Biological Systems. http://dxdoiorg/101146/annurev-bioeng-071811-150124. Annual Reviews; 2012;14: 1–16. doi:10.1146/annurev-bioeng-071811-150124
32. Zlatev HP, Auriola S, Monkkonen J, Määttä JA. Uptake of free, calcium-bound and liposomal encapsulated nitrogen containing bisphosphonates by breast cancer cells. Eur J Pharm Sci. 2016;86: 58–66. doi:10.1016/j.ejps.2016.02.016
33. Coleman RE. Clinical features of metastatic bone disease and risk of skeletal morbidity. Clinical Cancer Research. American Association for Cancer Research; 2006;12: 6243s–6249s. doi:10.1158/1078-0432.CCR-06-0931
34. Cabral H, Matsumoto Y, Mizuno K, Chen Q, Murakami M, Kimura M, et al. Accumulation of sub-100 nm polymeric micelles in poorly permeable tumours depends on size. Nature Nanotechnology. Nature Publishing Group; 2011;6: 815–823. doi:10.1038/nnano.2011.166

115
35. Papadopoulou V, Kosmidis K, Vlachou M, Macheras P. On the use of the Weibull function for the discernment of drug release mechanisms. Int J Pharm. 2006;309: 44–50. doi:10.1016/j.ijpharm.2005.10.044
36. Russell RGG, Watts NB, Ebetino FH, Rogers MJ. Mechanisms of action of bisphosphonates: similarities and differences and their potential influence on clinical efficacy. Osteoporos Int. 4 ed. Springer London; 2008;19: 733–759. doi:10.1007/s00198-007-0540-8
37. Journé F, Kheddoumi N, Chaboteaux C, Duvillier H, Laurent G, Body J-J. Extracellular calcium increases bisphosphonate-induced growth inhibition of breast cancer cells. Breast Cancer Res. 2008;10: R4. doi:10.1186/bcr1845
38. Lynch CC, Hikosaka A, Acuff HB, Martin MD, Kawai N, Singh RK, et al. MMP-7 promotes prostate cancer-induced osteolysis via the solubilization of RANKL. Cancer Cell. Cell Press; 2005;7: 485–496. doi:10.1016/j.ccr.2005.04.013
39. Mundy GR. Metastasis to bone: Causes, consequences and therapeutic opportunities. Nature Reviews Cancer. 2002;2: 584–593. doi:10.1038/nrc867
40. Vasiliadou I, Holen I. The role of macrophages in bone metastasis. J Bone Oncol. 2013;2: 158–166. doi:10.1016/j.jbo.2013.07.002
41. Chow E, Harris K, Fan G, Tsao M, Sze WM. Palliative radiotherapy trials for bone metastases: a systematic review. J Clin Oncol. 2007;25: 1423–1436. doi:10.1200/JCO.2006.09.5281

116
CHAPTER III
COMBINATION THERAPEUTIC PLATFORM FOR BREAST CANCER
BONE METASTASIS TREATMENT

117
3.1 INTRODUCTION
Chemo drugs based anticancer therapy remains dominant in the clinic. The
resistance to the toxic molecules, including the microtubing inhibitors, DNA damaging
agents and targeted kinase inhibitors, can drastically limit the treatment outcomes[1].
The general process of the generation of the resistance follows Darwin principle in which
the drug-tolerant cells (with right mutation) are positively selected by repeated drug
exposure[2,3]. Apart from that, researchers have also identified the acquired resistance
that emerges based on the cellular dynamics without genetic alteration[4,5]. The clinic
also showed evidence that such resistance occurred soon after the initial treatment[6]
and was found to be a drug-induced effect[7]. Its molecular mechanism was understood
as stochastic dynamics inside and between cell attractors, which are defined by the
microenvironment[8]. Such a phenomenon is clinically important, as it implies that even
the chemo drug naïve patients can develop something quickly that subsequently
reduces the drug efficacy.
The adaptive resistance unlike the intrinsic one, does not specify a certain class
of drugs. It could potentially arise against any environmental stress, including those
cytotoxic molecules. Several strategies were developed either to avoid or overcome the
adaptive[9-12]. However, the introduction of the new drug would likely to induce a
secondary adaptive resistance and how to resolve the puzzle is still uncleared. A recent
study[13] just prove the potential of what Matthew had suggested that the resistance
could create the new “Acais heel” and lead to ‘‘collateral sensitivity’’, where the resistant

118
might be useful[14]. By further extending the idea, a pair of drugs whose adaptive
statuses are sensitive to the counterparts would close the loop of generating secondary
adaptive resistance.
Described by the multiple cell attractor theory, a position that on the edge of an
attractor is unstable and the viability is low[15]. In the context of adaptive resistance in
cancer, a phenotype that falls outsides of the attractor defined by the chemo stress is
more sensitive. So, drug combinations that define different cell attractors (ideally without
any overlap) are critical to achieving the proposed paradigm. In other words, the drugs
need to induce different (ideally opposite) adaptive dynamics. In this study, we first
validated the existence of the adaptive resistance in drug-treated 4T1 cells and their
potential survival under high-concentration drug exposure. The level of AMPK activation
was evaluated as an indicator of two different adapted statuses. DTX and Das happened
to have the opposite effects on the adaptive dynamics. Therefore, we hypothesized that
the drug combinations of DTX and Das could have enhanced antitumor efficacy.
The paradigm was considered to work best when cells are exposed to both drugs
at the same time. The polypeptide-based micellar system has demonstrated the
capability to transport two or more drug simultaneously[16]. So, we aimed to engineer
the micelles with that could encapsulate both drugs efficiently and release them at a
similar pace. The size of the carriers would have to be small enough to harness the EPR
effect and passively targeted to the tumor area. After developing the formulation, breast
and prostate cancer cells were used to test the combinational treatment in vitro. Given

119
that DTX and Das are known to have their independent cytotoxic mechanisms[17,18],
and when applied together, it would be expected naturally to have some synergy effect
due to pathway crosstalk inhibition[19]. To separate the trait of the anticancer efficacy
from the sensitizing effect and validate the involvement of AMPK activation level in
establishing the drug-tolerant status, AMPK activator and inhibitor at nontoxic
concentrations were employed in conjunction with the chemo drugs.
Bone metastases remain the leading cause of cancer death, and no effective
treatment was available other than pain control[20]. One of the reasons why it is so
challenging might be associated with the adaptive resistance. Tumor in the bone
appears to be highly vascularized and could be expected to have sufficient nutrients
supply. In vitro, we observed that frequently refreshing the growth medium helped the
survival in Docetaxel and regrowth of the cells. Here, we used the murine breast cancer
bone metastases as established in Chapter II and evaluated the effectiveness of the
micellar drug combinations in vivo.

120
3.2 MATERIALS AND METHODS
3.2.1 MATERIALS
Besides of the chemicals and reagents purchased in the 2.2.1, FxCycle
PI/RNase, RPMI 1640, FK-12, MEM-Alpha (without ascorbic acid) and FxCycle
PI/RNase Staining Solution were bought from Thermo Fisher Scientific. AMPK Alpha
Total and Phospho T172 In-Cell ELISA Kit were purchased from Abcam. Docetaxel
(DTX) and Dasatinib (Das) were purchased from the AdpoGen Life Science. The Cell
Counting Kit-8 was purchased from Dojindo Molecular technologies. Compound C and
AICAR were obtained by Dr. Robert Lewis’ group in the Eppley Institute at the University
of Nebraska Medical Center. MDA-MB-231 and PC-3 were originally purchased from
ATCC and were grown in the lab according to the ATCC’s instruction. MC3T3 were
obtained from Dr. Dong Wang in the Department of Pharmaceutical Sciences at the
University of Nebraska Medical Center.

121
3.2.2 METHODS
Preparation of adapted cells
4T1 and MDA-MB-231 cells were cultured in RPMI1640 that contained 10% fetal
bovine serum (FBS) and 100U/mL penicillin-streptomycin. PC-3 cells were cultured in F-
12 medium that also contained 10% FBS and 100U/mL penicillin-streptomycin. Adapted
cells were prepared freshly for all experiments. Cells were seeded on T75 flask with 10%
confluency, allowed to attach overnight and treated with free 10 µM DTX
(polysorbate/PEG300/95% ethanol) for three days. The complete growth medium that
contained the same concentration of the drugs was refreshed until the end point of the
adaptation whenever the phenol red became orange (the interval ranging from 8 to 24 h).
To demonstrate the generation of adaptive resistance at a clinically relevant
concentration, adapted 4T1 cells were also developed by treating with 20 nM DTX for 36
h.

122
Characterization of the polyploidy formation in the adapted cell population
The proportion of polyploidy population with different chromosome copies was
determined by flow cytometry, using propidium iodide (PI). Data were obtained on BD
FACSCalibur (BD Biosciences) (Ex: 488 nm, Em: 585/42 filter). The adapted cells and
the corresponded parent cells (used as the negative control) were harvested
(trypsinized), washed in ice-cold 1X PBS and fixed in 70% ethanol for 30 min at 4 0C.
Cells were then washed with deionized water and stained with FxCycle PI/RNase
Staining Solution for 30 min in the dark and analyzed by flow cytometry.

123
Quantification of AMPK protein concentration in parent and drug-adapted 4T1
cells
AMPK Alpha Total and Phospho T172 In-Cell ELISA Kit were used for measuring
the shift of p-AMPK level during the adaptation process. Cells 1) without treatment, 2)
treated with 10 µM / 20 nM DTX or 10 µM / 50 nM Das for 2 days, 3) treated with the first
drug (DTX or Das) for 2 days and then with the other one (Das or DTX) subsequently for
2 h (high dose) or 5 h (low dose), were washed with 1X PBS, fixed in situ with 4%
formaldehyde for 3 min and then permeabilized with methanol at -20 0C for 45 min. The
p-AMPK and total AMPK was measured according to the manufactory protocol.
Quantitative fluorescence intensity was read by LI-COR Odyssey CLx at both 700 nm
and 800 nm channels. The fluorescence intensity in each treated group was normalized
(dividing by the one in parent cells group). Change in the p-AMPK and total AMPK was
expressed as folds to control. Representative images were captured by Olympus IX73
Inverted Microscope with a xenon excitation source and an Olympus DP80 Digital
Camera and cellSens Dimension software.

124
Western Blot Analyses for p-AMPK and its downstream protein
Radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris-HCl, 1% NP-40, 0.5%
Na deoxycholate, 0.1% Na dodecyl sulfate, 150 mM NaCl, 0.5 mM Na3VO4, 2 mM EDTA,
2 mM EGTA, 10 mM NaF, 10μg/mL aprotinin, 20 mM leupeptin, 2 mM PMSF) was used
to prepare whole cell lysate from collected cells. Promega BCA protein assay was
utilized to evaluate protein concentration. SDS-PAGE gel electrophoresis was completed,
proteins were transferred to nitrocellulose membranes, membranes were blocked for 45
minutes in PBS-based blocking buffer (LI-COR Biosciences, 927-40000), and incubated
in primary antibody at 4°C overnight. Secondary antibodies (LICOR IR-Dye 680LT and
800CW) were diluted 1:10,000 in 0.1% TBS-Tween. The LI-COR Odyssey was used to
image the western blots. Primary antibodies were used in the indicated dilutions: pACC
(#3661, Cell Signaling) 1:1000, tACC (#3676, Cell Signaling) 1:1000, pRAPTOR (#2083,
Cell Signaling) 1:1000, tRAPTOR (#2280, Cell Signaling) 1:1000, pAMPKα1α2 (#2531,
Cell Signaling) 1:1000, AMPKα1α2 (#2532, Cell Signaling) 1:1000, and B-Actin (C-4,
47778, Santa Cruz) 1:2000. Anti-mouse and anti-rabbit secondary antibodies conjugated
to IRDye800 and IRDye680LT were used at 1:5000-1:10,000 dilutions.

125
Synthesis of triblock copolymer Methoxy polyethyleneglycol-b-polyglutamic acid-
b-polyphenylalanine (mPEG5K-PLE-PLF)
The same synthesis scheme in 2.2.2 was used to produce the triblock copolymer,
using methoxy-PEG-NH2 (M.W.=5,000 Da) as the macroinitiator. The length of the PLE
block was designed as five monomers and the polyphenylalanine as 15 monomers. The
chemical structure of the diblock copolymers, triblock copolymers, deprotected triblock
copolymers was confirmed by 1H-NMR. Deuterated dimethyl sulfoxide (DMSO-d6) was
used for the benzyl protected block copolymers and deuterated trifluoroacetic acid (TFA-
d1) for the deprotected block copolymers. The polydispersity of the benzyl protected
block copolymers was determined by gel permeation chromatography as described in
2.2.2.

126
Preparation and characterization of drug-loaded micelles DTX/m, Das/m,
(DTX+Das)/m
DTX, Das or both drugs were mixed with the synthesized triblock copolymers and
dissolved in the minimum amount of 80% ethanol. The solution was dispersed dropwise
into ice-cold 1X PBS (>20X volume) upon quick stirring. Then the ethanol was removed
under vacuum for 10 min, and the formulation was sterilized via a 0.45 µm syringe filter
(Millex, SLHV033RS).
Loaded drugs were confirmed by high-performance liquid chromatography
(HPLC) analysis under isocratic conditions using an Agilent 1200 HPLC system and a
diode array detector set at 227 nm for DTX and 325 nm for Das. A nucleosil C18 column
was used as stationary phase (250 mm × 4.6 mm), and mobile phase comprised of
acetonitrile/water mixture with 0.1% formic acid (60/40, v/v) at a flow rate of 1 mL/min.
The feed ratios were expressed as 𝑊𝐷𝑇𝑋+𝑊𝐷𝑎𝑠
𝑊𝐷𝑇𝑋+𝐷𝑎𝑠+𝑃𝑜𝑙𝑦𝑚𝑒𝑟𝑠%. The final yield after the filtration
was measured by the same HPLC method and calculated as
𝐷𝑟𝑢𝑔 𝑐𝑜𝑛𝑐𝑒𝑛𝑡𝑟𝑎𝑡𝑖𝑜𝑛𝑎𝑓𝑡𝑒𝑟 𝑓𝑖𝑙𝑡𝑟𝑎𝑡𝑖𝑜𝑛
𝐷𝑟𝑢𝑔 𝑐𝑜𝑛𝑐𝑒𝑛𝑡𝑟𝑎𝑡𝑖𝑜𝑛𝑏𝑒𝑓𝑜𝑟𝑒 𝑓𝑖𝑙𝑡𝑟𝑎𝑡𝑖𝑜𝑛% . All samples for HPLC analysis were prepared by
lyophilizing the 100 µl micelle solution and re-dissolving the solid residues in the same
amount of acetonitrile with 0.1% formic acid. The mixture was sonicated to extract the
drugs into the supernatant, and the rest insoluble contents were removed by
centrifugation.

127
Effective hydrodynamic diameters (Deff) of all micelles were determined at 25 0C
in 1X PBS solution by dynamic light scattering (DLS) using a Zetasizer Nano ZS
(Malvern Instruments Ltd., Malvern, UK). The measurements were performed per
manufacturer instruction to obtain the size distribution and polydispersity index (PDI).
The concentration of the polymers in measured samples was controlled in the range of
0.2-0.4 mg/mL. To test the stability of the drug-loaded micelles, (DTX+Das)/m was kept
at room temperature continuously for ten weeks and analyzed by DLS for potential
changes in size distribution. The experiments were repeated three times.
In vitro Drug release kinetics was studied by the dialysis method mentioned in
2.2.2 using 1X PBS as the releasing medium. 0.7 mL (DTX+Das)/m, DTX/m and Das/m
suspension in PBS that contained about 0.05 mg DTX and 0.03 mg Das were
transferred into the dialysis tubes (Slide-A-Lyzer G2, MWCO 3.5 kDa) and the tubes
were suspended in 50 mL PBS solution (sink condition). The drug release proceeded at
37 0C on the plate shaker. At predetermined time points (0, 4, 12, 24, 48, 72 h), an
aliquot of the 100 µL solution was sampled from each of the dialysis tubes. The
remaining drugs quantities in the aliquots were analyzed by the HPLC method in the
same manner as described above. The cumulative release was expressed as % of the
initial concentration. The experiments were repeated three times.

128
In vitro cell growth inhibition assays
Cells were seeded on 96-well plates, 10,000 cells per well, allowed to attach for 6
h (parent cells) or 12 h (DTX adapted cells) and treated with DTX, Das or the
combination of both drugs in the micellar formulation for 24 h. Viable cells were
quantified by using Cell Counting Kit-8 according to the manufactory’s protocol. The
absorbance (Abs) of the assay solution was read by SpectraMax M5 at 450 nm. All
reading was subtracted by the blank wells and normalized as % to the negative control.
The absolute IC50 was calculated in GraphPad Prism by fitting the dose-response data
with the non-linear function and interpolating the X values(concentration) when setting
Y(viability)=50%. Relative DTX sensitivity of adapted 4T1 to the parent cells was
calculated as 𝑉𝑖𝑎𝑙𝑏𝑖𝑙𝑖𝑡𝑦%𝑝𝑎𝑟𝑒𝑛𝑡 𝑐𝑒𝑙𝑙𝑠
𝑉𝑖𝑎𝑙𝑏𝑖𝑙𝑖𝑡𝑦%𝐷𝑇𝑋 𝑎𝑑𝑎𝑝𝑡𝑒𝑑 𝑟𝑒𝑛𝑡 𝑐𝑒𝑙𝑙𝑠. The experiments were repeated three times.

129
Sensitization of 4T1 cells to DTX or Das with 5-Aminoimidazole-4-carboxamide
ribonucleotide (AICAR) or Compound C
100 µM AICAR and 1 µM Compound C were first tested in 4T1 cell line for
potential growth inhibitory effect. Their sensitization effect was then evaluated in the 4T1
cell line. 4T1 cells were treated with two combinations DTX+100 µM AICAR and 1 µM
Compound C+Das for 24 h. Both tests used the same method in the growth inhibition
assay described above. The experiments were repeated three times.

130
Chemotaxis of tumor cells toward pre-osteoblast
MC3T3 cells were maintained in alpha-MEM (10% FBS and 100U/mL penicillin-
streptomycin, no ascorbic acid) at 80% confluency. MC3T3 conditioned medium (preOB-
CM) was prepared by incubating the cells with the same medium but without the FBS for
12 h. After incubation the medium was collected, sterilized by 0.2 µm syringe filter. 24-
well transwell permeable support (0.8 µm pore) was used for studying the migration. In
the experimental groups, MC3T3 cells with alpha MEM (10%FBS, no ascorbic acid)
were seeded on the receiver well and allowed to attach for 12 h. The complete growth
medium was then replaced by preOB-CM. MC3T3 with the PreOB-CM together served
as the chemoattractant. In the positive control group, the MEM medium (no ascorbic acid)
with 10% FBS was used as the chemoattractant. In the negative control group, only the
base MEM medium (no ascorbic acid) was used as a mock attractant. After setting up
the receiver, the inserts holding 15,000 cells (4T1/MDA-MB-231) with different
treatments were placed on the top of the receiver. Tumor cells were allowed to migrate
for 12 h, then fixed in 10% NBF and stained by crystal violet. The cells that did not travel
through (upper surface of the insert membrane) were wiped out gently by cotton swabs,
leaving the migrated cells on the lower surface. Migrated cells were counted under the
inverted light microscope (20X), in five random spots per well. The experiment was
repeated three times.
Osteoclast differentiation

131
10,000 Raw 264.7 cells were seeded in 24-well plates and incubated with the
complete growth medium (RMPI 1640 + 10%FBS + 100U/mL penicillin-streptomycin)
that contained additional 50 ng/mL of RANKL and Das/m treatment for 7 days. The
medium with exact RANKL and the drug were refreshed every three days. The cells
were fixed by 10% NBF for 60 sec, and the osteoclast was stained by the Acid
Phosphatase, Leukocyte (TRAP) Kit according to the manufactory protocol. The number
of multinucleated and positive stained cells in each treatment group was counted under
the inverted light microscope (20X). The experiment was repeated three times.

132
Establishment of murine bone metastatic model
Luciferase-expressing tumor cells (4T1/Luc) cells were cultured and selected in
the RPMI 1640 + 10%FBS as described in the in vitro experiment with the
supplementary of 15 µg/ mg of Blasticidin HCl for two days and without Blasticidin HCl
for another two days. Cells were sub-cultured when reaching 40% confluency. Cells
were trypsinized and washed thoroughly with ice-cold 1X DPBS. The suspension was
diluted to 200,000 cells/mL and was kept on ice for less than 20 min before being
injected. Eighty 5-week Balb/C mice were purchased from Charles River Laboratories.
For the convenience, the hair on the chest was removed before injection by Veet hair
removal gel. Mice were anesthetized by isoflurane and inoculated with the prepared cells
through the left ventricle injection, 10,000 cells/mouse in 50 µL DPBS (day0).

133
In vivo efficacy and survival study
Bioluminescence of the 4T1/Luc was measured by PerkinElmer In vivo Imaging
System (IVIS) starting from seven days post tumor injection to monitor the tumor growth.
Luciferin (15 mg/mL) was injected intraperitoneally (i.p.) for each mouse 10 min before
the imaging. On day9, mice were evenly distributed into five groups/10 mice per group
based on the bioluminescence intensity and randomly assigned to receive the
treatments 1) DPBS, 2) free DTX, 3) free DTX + free Das, 4) DTX/m + Das/m, 5)
(DTX+Das)/m respectively. The treatments that started on day10 were given via tail vein
injection with equiv. 10 mg/kg DTX, 6 mg/kg Das or both (based on the animal body
weight) on a daily basis and continuously for four days. The resulting cumulative dose
was 40 mg/kg DTX, 24 mg/kg Das or both. Body weight was monitored every day, and
euthanasia was performed when the body weight loss reached or exceeded more than
20% of the initial values or if the animals met other euthanasia criteria outlined by
(IACUC).
Twenty-four hours after the final treatments, three mice from each treatment
group were sacrificed. Whole blood was collected for blood cell counting and blood
chemistry analysis. Soft organs (the liver, kidney, spleen) and tibia/femur were collected
and fixed in 10% NBF for 24 h at room temperature. The tibias/femurs were further
decalcified in 14% EDTA-NH3H2O (pH=7.2) buffer for three weeks. Each piece of bone
tissue was incubated with the buffer (20-times volume) at room temperature with mild
shaking. The buffer was exchanged every day. The kidneys, livers, spleens and

134
decalcified tibias/femurs were processed subsequently in UNMC tissue facilities for
embedding, sectioning and H&E staining. The blood was again collected via maxillary
vein five days after the final treatment and counted for blood cell numbers. The blood
smear was also prepared to identify the unmatured neutrophils in the mice of the control
group (tumor-bearing).
The survival of the animals was recorded for 30 days from the first treatment.
The data was presented as Kaplan-Meier curves, and the Log-rank (Mantel-Cox) test
was employed to analyze the significance between the treatment groups. Animal death
was recorded when the mouse was found dead, the body weight dropped more than 20%
or suffered from stress that did not comply with IACUC.

135
3.3 RESULT AND DISCUSSION
Adaptive resistance in 4T1 cells
Cancer cells were reported previously to undergo phenotypical transition and
acquired adaptive resistance quickly when exposed to a variety of chemo drugs at
subtoxic concentration[6,21-23]. The adapted cancer cells are insensitive to the chemo
drugs and able to continue the proliferation after removal of the treatment[24]. In our
study, murine breast cancer cell line 4T1 was observed to transit into dormant status and
giant in size with the appearance of multiple nuclei. In both condition of 10 µM DTX and
10 µM Das, the morphology change started quickly after treatment for 24 h, however the
morphology was drastically different (Figure 3.1). Part of the cells was able to survive
through a 3-days treatment of 10 µM DTX with frequently refreshed culture medium
(containing the same drugs), which indicated the acquisition of the drug resistance. After
the removal of DTX, the dormant cells were found to produce daughter cells by budding,
similar to what was reported in work by Karuna[24].

136
Figure 3.1: Representative image of the morphology change in DTX and Das treated 4T1 cells. Upper
parent control, middle 10 µM DTX, lower 10 µM Das. Images were captured at 36 h of the treatment.
10µM Das
10µM DTX
Parent

137
To further confirm that the rise of such resistance was due to the adaptive
change rather than the selection of the subpopulation with intrinsic resistance (based on
genetic mutation), 4T1 cells were pre-treated with a lower dose of DTX (20 nM) for only
36 h. The treatment should not affect cell viability (Table 2.3). After the short drug
exposure, their sensitivity to DTX was further tested by growth inhibition assay for 24 h.
The IC50 increased more than three folds to ≈ 100 µM (Figure 3.2) compared to the
parent cells without the pre-treatment (Table 3.4). The drug exposure was also
considered as clinically relevant based on the pharmacokinetic data and the actual dose
regimen in pharmacy practice[25].

138
Figure 3.2: Dose-responsive profile of DTX to 20 nM 36 h pre-treated 4T1. Cells acquired the resistance
to DTX with the increased IC50 (≈100 µM).
12.5
0
25.0
0
50.0
0
100.
00
0
50
100
150
[DTX], µM
Via
lbilit
y %
to
co
ntr
ol

139
Polyploid formation in the adapted cells
Based on visual examination under the microscope, the adaptive cells were giant
(10 times larger than parent cells) and multinucleated. Flow cytometry was used to
analyze the DNA quantities to confirm the visual changes. Compare to the parent cells
which are diploid (2N), DTX adapted 4T1 showed a large population of polyploid cells
(4N-8N). Similar results were also found for human breast cancer cell line MDA-MB-231
and human prostate cancer cell line PC-3 (Figure 3.3). These cell lines are all known to
be metastatic, especially to the bone. Such kind of reaction could be explained as the
cell cycle was arrested due to the DTX’s inhibition in microtubing disassembly[26], while
the duplication of DNA and the synthesis of cellular protein continued. This observation
together with the fact of cell survival matched with the observation that breast cancer cell
could exit the cell cycle and stays in a sub G1 phase[24]. Also, chronic autophagy that
did not proceed to the apoptosis was considered to take place during this process and to
be part of the adaptive resistance acquisition[27,28]. On the contrary, the Das did not
show such an effect, even at a higher concentration of 100 µM (Figure 3.4). After
removing the DTX treatment, cells were recovered the proliferation gradually, and the
whole population trended to restore the initial diploidy condition (Figure 3.5).
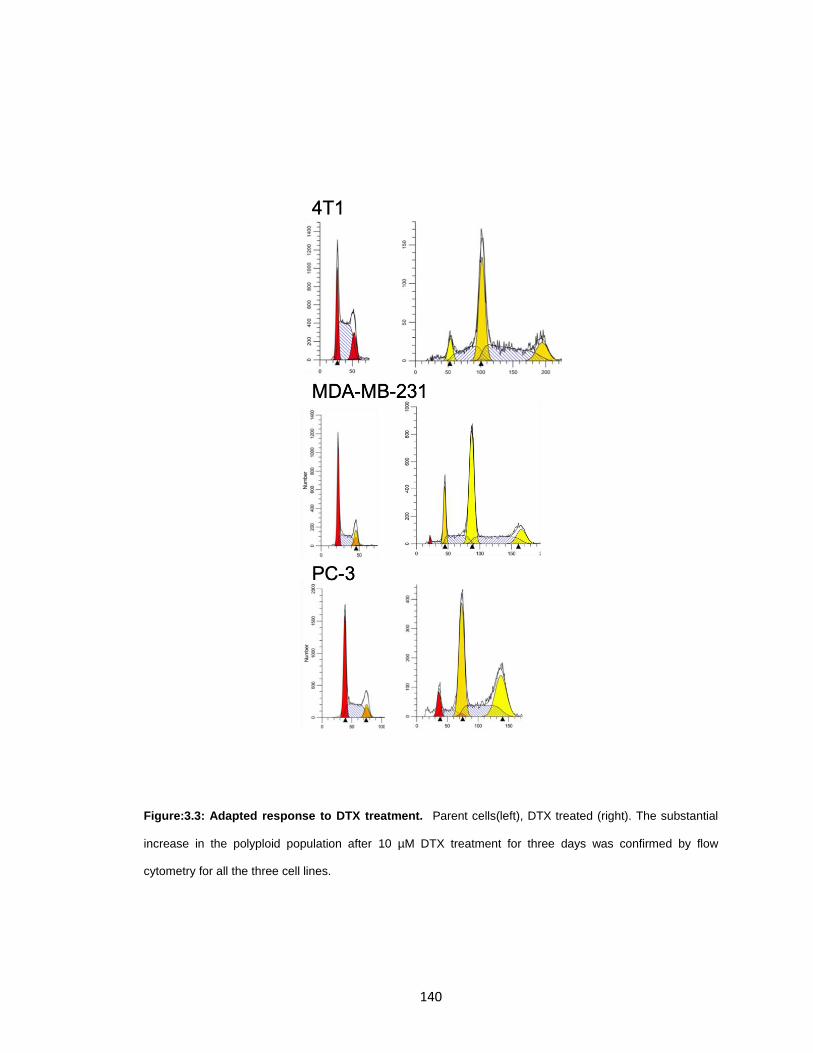
140
Figure:3.3: Adapted response to DTX treatment. Parent cells(left), DTX treated (right). The substantial
increase in the polyploid population after 10 µM DTX treatment for three days was confirmed by flow
cytometry for all the three cell lines.
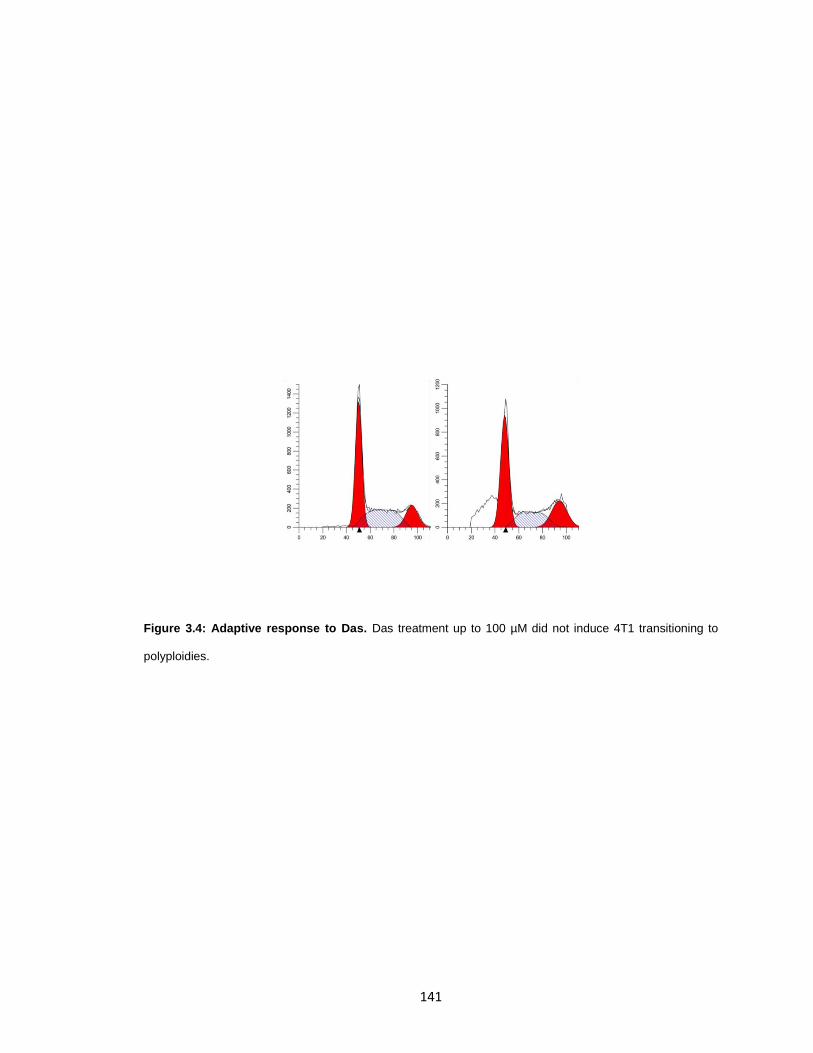
141
Figure 3.4: Adaptive response to Das. Das treatment up to 100 µM did not induce 4T1 transitioning to
polyploidies.
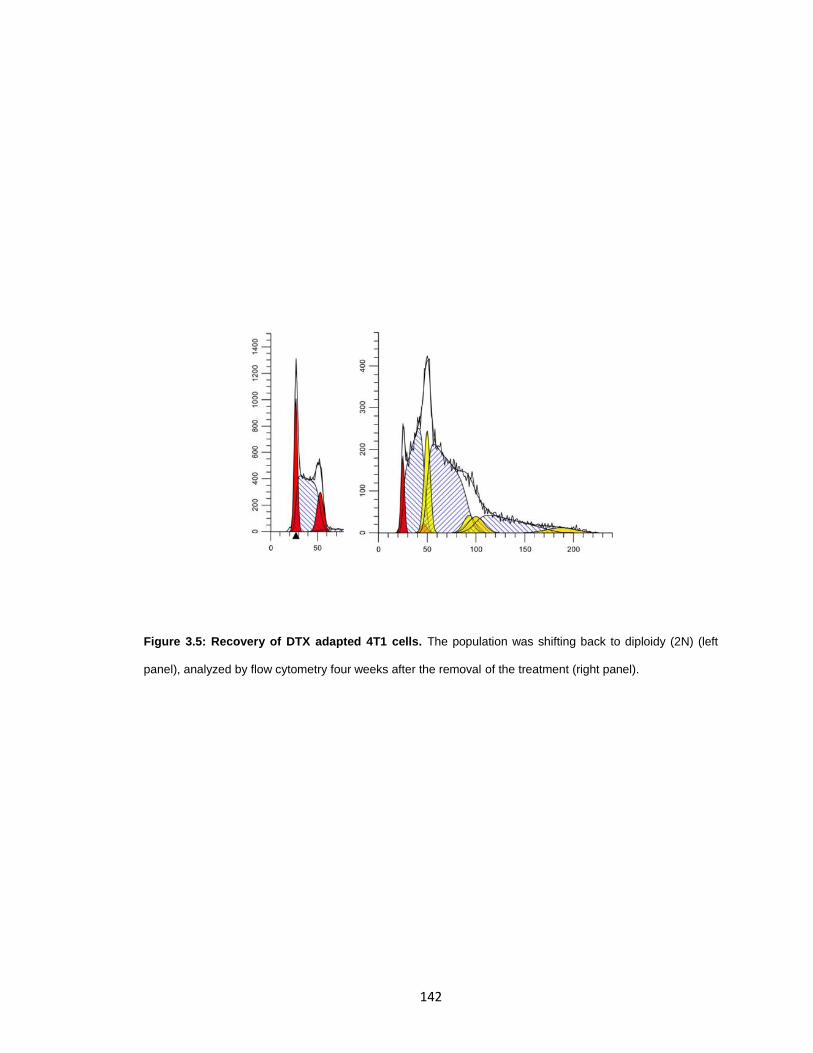
142
Figure 3.5: Recovery of DTX adapted 4T1 cells. The population was shifting back to diploidy (2N) (left
panel), analyzed by flow cytometry four weeks after the removal of the treatment (right panel).

143
p-AMPK protein level as a potential marker to determine the direction of adaptive
change in drug-treated cells
p-AMPK/AMPK known as the energy sensor is a key fundamental protein to
regulate the cell metabolic system[29,30]. Studies have revealed the complexity of its
functions, and some of them are closely related to cancer therapy, e.g., cell
proliferation/cell cycle regulation, autophagy, fatty acid synthesis, abnormal glutamic
metabolic, etc[31-33]. Inhibition on its downstream pathway (fatty acid synthesis) was
able to enhance the anticancer efficacy of the chemo drugs[34]. It is also reported that p-
AMPK level is crucial for the survival of the tetraploid cells which have the potential to
become cancer cells and the chemoprevention efficacy of aspirin was also attributed to
the capability of selective elimination of tetraploid cells[35].
These evidence make the p-AMPK an appealing target marker to classify the
adaptive response. Two studies were designed to determine the change in pAMPK level
after adapting to the DTX or Das. One was that 4T1 cells were exposed to 20 nM DTX
for 48 h and then to 50 nM Das for 5 h separately after removing the DTX; in contrast,
the other was that cells were exposed to Das for 48 h and then to DTX (Figure 3.6).
Because the doses were well below the IC50 level (Figure 3.4), few cells were observed
to be dead during the 48 h treatment. Therefore, the process did not select a
subpopulation cell that expressed a different level of p-AMPK. The parent control, the
cells with the primary treatment and with the addition of secondary treatment were
harvested for western blot analysis. The results showed decreased p-AMPK in DTX

144
adapted cells, but the increased level in Das group. After the subsequent treatment of
the counter drug, the trend of such changes reversed (Figure 3.7), which demonstrated
the different cellular responses to the two drugs separately. The results were also
implying that the change is dynamic, and cells can continuously answer to the chemo
stress even after the initial adaptation. To further clarify the p-AMPK level shift was the
part of the necessary rearrangement made by the cells to survive rather than the part of
the cytotoxic mechanism, the concentration of DTX and Das was both increased to 10
µM. In this case, since the dose was around IC50, a small proportion of the cells was
killed during the first treatment, and thus the cells collected for p-AMPK level analysis
were the survivors. The results (normalized to housekeeping protein) showed the same
trend of p-AMPK shifting as well as the reversal behavior just as what was observed in
lower dose sets. And the higher dose set seemed to lead to more considerable deviation
from the base level. Given the fact that either DTX or Das was not reported to have
direct interaction with p-AMPK/AMPK, it is reasonable to speculate the opposite p-AMPK
shift could be the consequences of distinguished adaptive response to the drugs.
Besides, the previous study already uncovered that the Das resistant CLL (chronic
lymphocytic leukemia) have a much higher base level of p-AMPK compared to the non-
resistant subtype[36], whereas DTX insensitive cell line (4T1) has a much lower base
level. The evidence in addition proved possible relation between p-AMPK level and the
adaptive resistance.
145
Figure 3.6: The regimen of different drug treatments for the analysis of the corresponded adaptive
responses. 10 µM DTX, 10 µM Das for high dose set; 20 nM DTX, 50 nM Das for low dose set. Cells at
each stage were analyzed for AMPK activation level.
Parent 4T1
DTX treatment2 days
Das treatment2 days
Parent 4T1
Das treatmentHigh dose (2h), low dose (5h)
DTX treatmentHigh dose (2h), low dose (5h)
DTX adapted
Das adapted
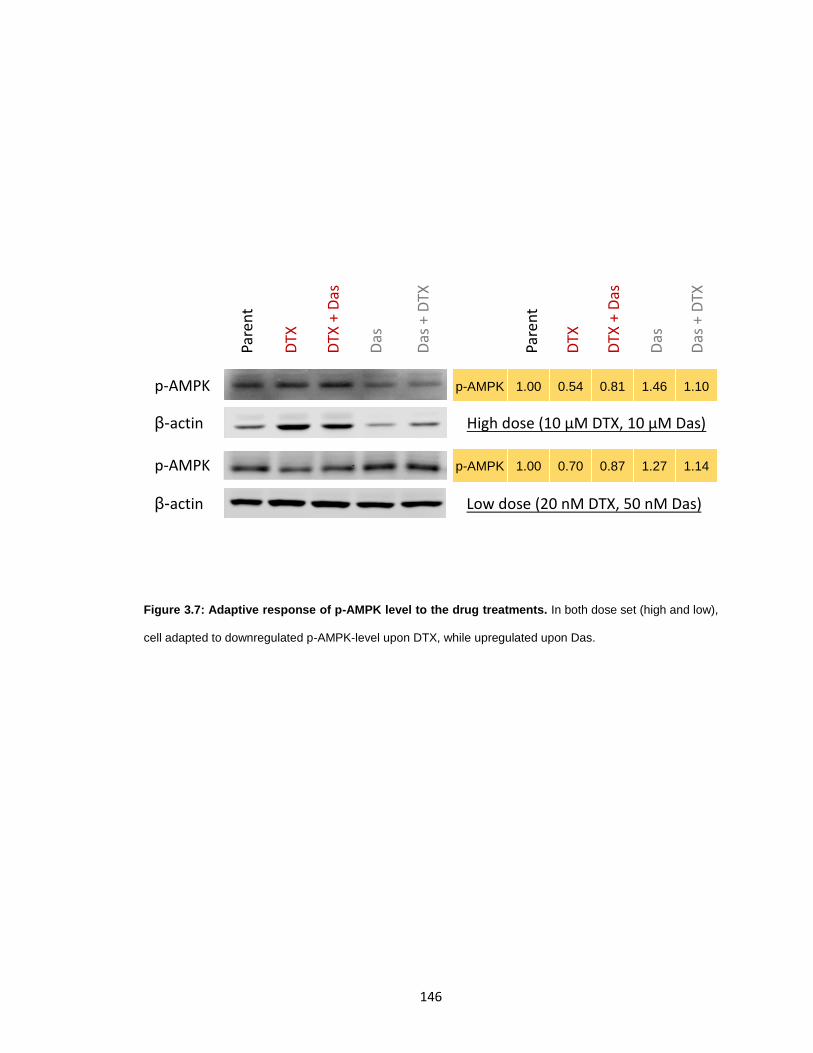
146
Figure 3.7: Adaptive response of p-AMPK level to the drug treatments. In both dose set (high and low),
cell adapted to downregulated p-AMPK-level upon DTX, while upregulated upon Das.
Pare
nt
DTX
DTX
+ D
as
Das
Das
+ D
TX
p-AMPK 1.00 0.54 0.81 1.46 1.10
p-AMPK 1.00 0.70 0.87 1.27 1.14
High dose (10 µM DTX, 10 µM Das)
Low dose (20 nM DTX, 50 nM Das)
Pare
nt
DTX
DTX
+ D
as
Das
Das
+ D
TX
p-AMPK
p-AMPK
β-actin
β-actin

147
Quantification of AMPK protein level shift
The adaptive change in p-AMPK level observed by western blot was double-
checked by a different method In-Cell ELISA. The similar design as used in western blot
studies was adopted, using the high-dose treatment set (10 µM DTX, 10 µM Das, 48 h
for the primary treatment, 2 h for the secondary Das treatment, but 12 h for DTX). Cells
at each of the three stages were fixed in situ and remained intact throughout the ELISA
assay. After incubating with the fluorophore label antibodies, the wells were scan in Licor
Odyssey at 680 nm for the total AMPK (showed in green) and 800 nm for p-AMPK
(showed in red). For both primary and secondary treatments, the color shifted toward
greenish (total AMPK) upon DTX exposure, and it turned to yellowish (total AMPK + p-
AMPK) upon Das exposure. The minimum amount of unspecified binding could be seen
in the negative control that omitted the primary antibody (Figure 3.8). Representative
fluorescent images were also captured to reflect the detailed change in the cellular level
(Figure 3.8). Quantified data were read out by Image Studio Software (Licor), then
normalized by the total quantities of the cells (Janus Green B) and expressed as folds to
the parent control (Figure 3.9). Similar to the western blot results, the DTX treatment
was confirmed to be associated with p-AMPK downregulation, Das with upregulation
instead; and the dynamic trend was also reversed upon secondary treatment. Compared
to DTX, Das had a quicker and more substantial effect on the cells that had been treated
with the counter drugs. In contrast to the activated AMPK, the total AMPK level remained
relatively stable upon all treatments.

148
Figure 3.8: Parent 4T1 and drug adapted 4T1 in 96-well scanned by Licor Odyssey. Greenish (680 nm
total AMPK) and yellowish (680 nm total AMPK and 800 nm p-AMPK) color shift was found in DTX treated
and Das treated respectively.
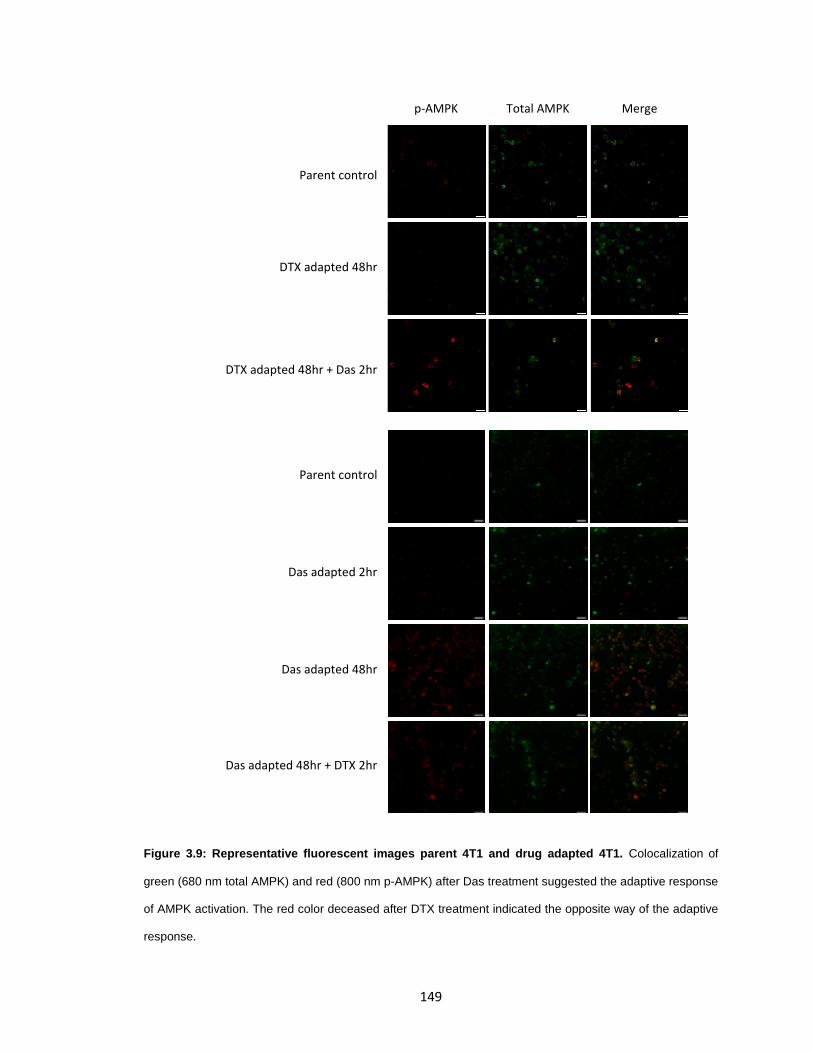
149
Figure 3.9: Representative fluorescent images parent 4T1 and drug adapted 4T1. Colocalization of
green (680 nm total AMPK) and red (800 nm p-AMPK) after Das treatment suggested the adaptive response
of AMPK activation. The red color deceased after DTX treatment indicated the opposite way of the adaptive
response.
p-AMPK Total AMPK Merge
Parent control
DTX adapted 48hr
DTX adapted 48hr + Das 2hr
Parent control
Das adapted 2hr
Das adapted 48hr
Das adapted 48hr + DTX 2hr
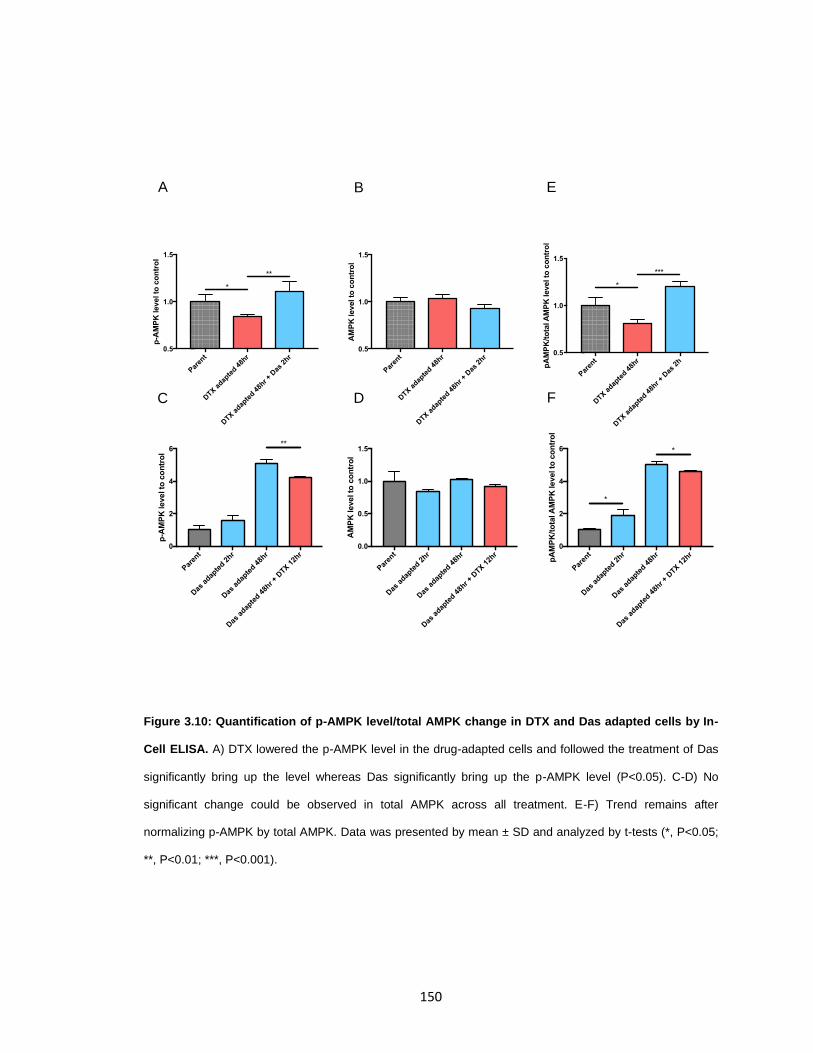
150
Figure 3.10: Quantification of p-AMPK level/total AMPK change in DTX and Das adapted cells by In-
Cell ELISA. A) DTX lowered the p-AMPK level in the drug-adapted cells and followed the treatment of Das
significantly bring up the level whereas Das significantly bring up the p-AMPK level (P<0.05). C-D) No
significant change could be observed in total AMPK across all treatment. E-F) Trend remains after
normalizing p-AMPK by total AMPK. Data was presented by mean ± SD and analyzed by t-tests (*, P<0.05;
**, P<0.01; ***, P<0.001).
Paren
t
DTX a
dapte
d 48h
r
DTX
adap
ted 4
8hr + D
as 2
hr0.5
1.0
1.5
p-A
MP
K level to
co
ntr
ol
**
*
Par
ent
Das
adap
ted 2
hr
Das
adap
ted 4
8hr
Das
adap
ted 4
8hr +
DTX
12h
r0
2
4
6
p-A
MP
K level to
co
ntr
ol
**
Paren
t
DTX a
dapte
d 48h
r
DTX
adap
ted 4
8hr +
Das
2hr
0.5
1.0
1.5
AM
PK
level to
co
ntr
ol
Par
ent
Das
adap
ted 2
hr
Das
adap
ted 4
8hr
Das
adap
ted 4
8hr + D
TX 1
2hr
0.0
0.5
1.0
1.5
AM
PK
level to
co
ntr
ol
Par
ent
DTX a
dapte
d 48h
r
DTX
adap
ted 4
8hr +
Das
2h
0.5
1.0
1.5
pA
MP
K/t
ota
l A
MP
K level to
co
ntr
ol
***
*
Paren
t
Das
adap
ted 2
hr
Das
adap
ted 4
8hr
Das
adap
ted 4
8hr + D
TX 1
2hr
0
2
4
6
pA
MP
K/t
ota
l A
MP
K level to
co
ntr
ol
*
*
A B
C D F
E

151
Rational of designing a micellar based formulation that co-delivers both DTX and
Das to overcome the adaptive resistance
Based on the results in western blot and In-Cell ELISA, it is clear that p-AMPK is
involved in the response of 4T1 cells to the DTX and Das treatment and the directions
were just opposite: one downregulated, and the other upregulated the p-AMPK. Also, the
mechanism of the drugs is known not directly connected the AMPK; the differences in
base levels of p-AMPK between drug sensitive and resistant cell lines are correlated with
the response trends observed in this study: it is reasonable to consider the response is
the adaptation for survival, belonging to “the way to resist to stress" rather than “the way
how cancer cells are killed".
Unlike the intrinsic resistance by drug selection or acquired resistance by a gene
mutation which was well defined by specific mechanism and takes time to develop, the
adaptive one is more dynamic, with the abilities to quickly enter a drug-tolerant status.
Given the variety of metabolic modes that cell could use potentially, it is difficult or nearly
impossible to inhibit one or two pathways to block the cell survival completely.
One strategy we proposed here is to use two drugs that can lead to the opposite
adaptive dynamics, like DTX and Das. The p-AMPK/AMPK serves as an “infrastructure”
element to support the proper cellular functions. And the same it is in drug adapted
cancer cells. An adaptive status (insensitive to the treatment) requires an appropriate p-

152
AMPK/AMPK level. To be specific, the survival of DTX treated cell relied on the low p-
AMPK level whereas Das adapted cells demanded the high p-AMPK level. Since there
would be only two conditions regarding the p-AMPK level (either low or high), cells would
not be able to locate at a proper status that can survive under both condition: any
adaptive effort that comes with a high level of p-AMPK would sensitize the cell to DTX,
while anyone ends with low level would be effectively killed by Das. Distinguished from
the classic approach that inhibits the pathways that cells use to survive, such a strategy
does not impede the adaption process directly, but making the drug adapted status more
sensitive to the counterpart treatment.

153
Synthesis of triblock copolymer methoxy-polyethyleneglycol-b-polyglutamic acid-
b-polyphenylalanine (mPEG5K-PLE5-PLF15)
The combination treatment of Docetaxel and Dasatinib has been explored and
tested for bone metastases in the clinical trials in 2013. However, apart from the well-
tolerated toxicity profile, the results were not encouraging. No significant improvement
was reported in term of the overall survival (OS) or bone associated biologic markers[37].
One reason for the inconsistency between in vitro and in vivo outcome might be the
discrepancy in pharmacokinetic profiles of both drugs[38], making it's challenging to
achieve the simultaneous drug exposure in the tumors.
In the proposed approach to enhance the anticancer efficacy, it is critical to
delivering both of the drugs simultaneously to achieve the best synergistic effect.
Polypeptide-based micellular nanoparticles have proved their capability to co-
encapsulate drug combination and transported them to the cancer cells at the same time.
To serve the purpose, mPEG5K-PLE5-PLF15 were synthesized. The polyphenylalanine
block was longer compared to the previous polymer described in Chapter II to
encapsulate DTX and Das together better, which are both hydrophobic and insoluble in
water. The chemical structures of the synthesized diblock copolymers, triblock
copolymers, and deprotected polymer were confirmed by 1H-NMR (Figure. 3.11, 3.12).
The polydispersity of the benzyl-protected polymers was characterized via GPC as
shown in (Table 3.1). The results suggested that the indicated polymers were
successfully synthesized with a low polydispersity within 1.05.

154
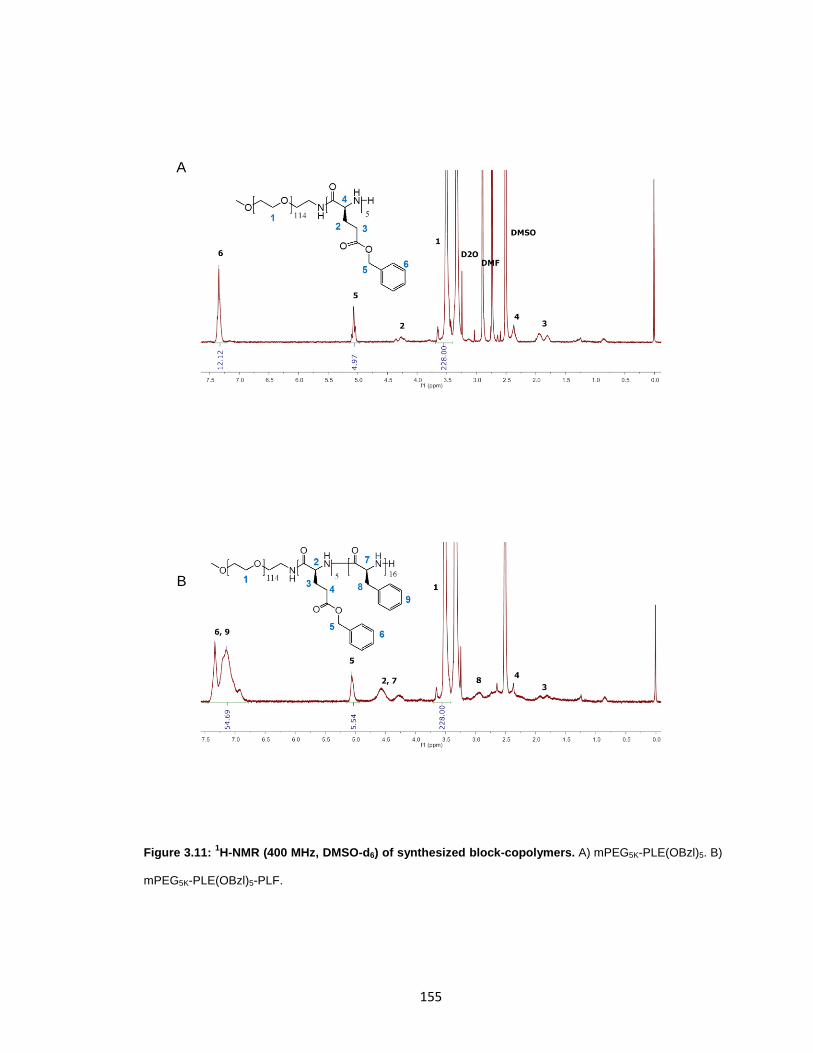
155
Figure 3.11: 1H-NMR (400 MHz, DMSO-d6) of synthesized block-copolymers. A) mPEG5K-PLE(OBzl)5. B)
mPEG5K-PLE(OBzl)5-PLF.
B
A
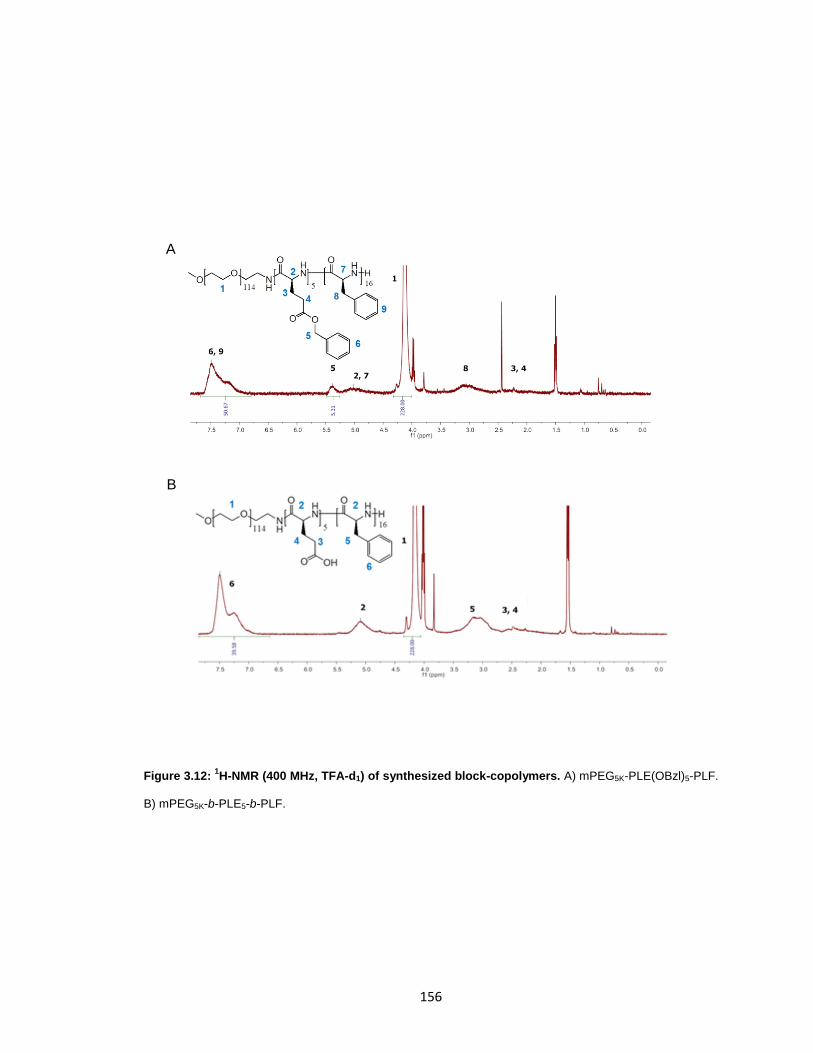
156
Figure 3.12: 1H-NMR (400 MHz, TFA-d1) of synthesized block-copolymers. A) mPEG5K-PLE(OBzl)5-PLF.
B) mPEG5K-b-PLE5-b-PLF.
B
A

157
MWw (Da) PDI
mPEG5K
-NH2 4839 1.03
mPEG5K
-PLE(OBzl) 5434 1.04
mPEG5K
-PLE(OBzl)-PLF 10542 1.05
Table 3.1: Polydispersity of synthesized polymers by gel permeation chromatography.

158
Preparation and characterization of drug-loaded micelles
The drug was loaded into the micelles through the nanoprecipitation method in
the ice-cold bath to achieve the best loading capacity as well as the narrow size
distribution. DTX-loaded micelles (DTX/m), Das-loaded micelles (Das/m), and two-in-one
combination (DTX+Das)/m were prepared separately by controlling the initial feed. The
micellar cocktail (DTX/m+Das/m) was obtained by mixing prepared DTX/m and Das/m
directly. No drug precipitation was observed during the loading procedure or after
centrifuging the formulation for 2 min at 1000g, which indicating an encapsulation
efficiency close to 100%. A small proportion (≈10% of the loaded drug) of the micelles
were found lost in the filtration through the 0.45 µm syringe filter (Table 3.2) by
comparing the quantity of drug concentration before and after the filtration. Hence the
total yield of the preparation was around 90%. Due to the high encapsulation efficiency,
the combination ratio between DTX and Das in the micelles can be precisely controlled.
Here, a 1:1 molar ratio was selected, and the total loading capacity of the (DTX+Das)/m
was 25%, consists of 15% DTX and 10% Das.
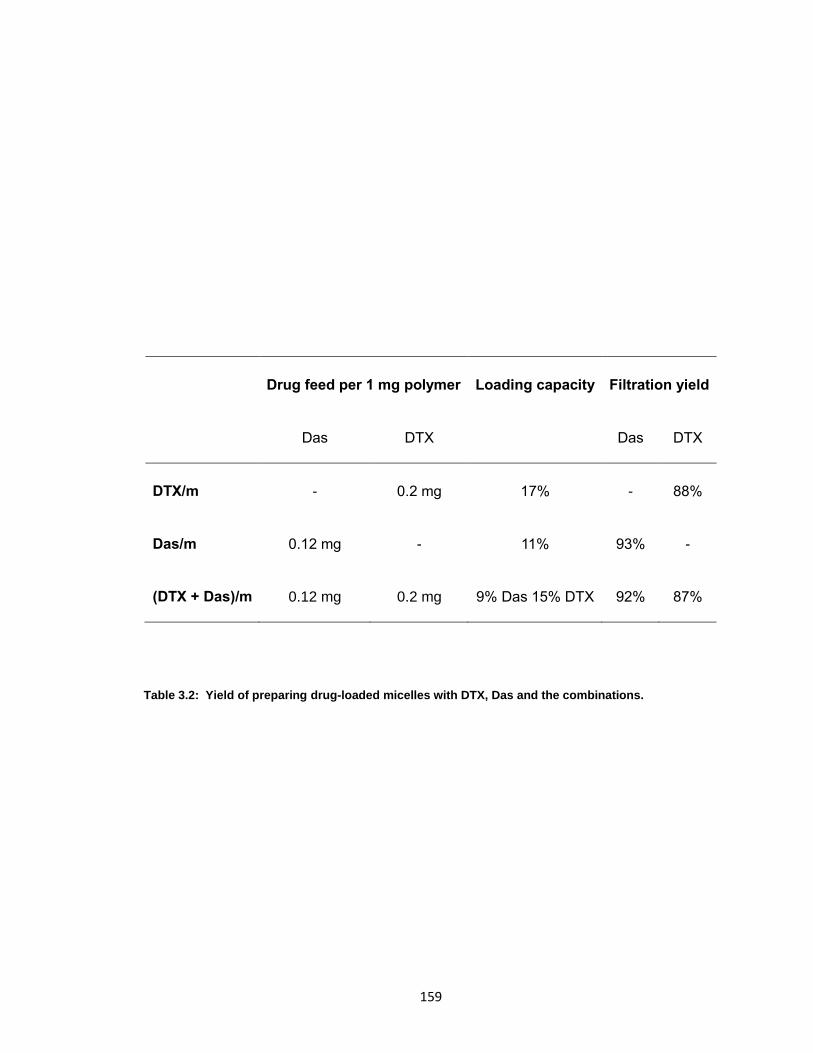
159
Drug feed per 1 mg polymer Loading capacity Filtration yield
Das DTX Das DTX
DTX/m - 0.2 mg 17% - 88%
Das/m 0.12 mg - 11% 93% -
(DTX + Das)/m 0.12 mg 0.2 mg 9% Das 15% DTX 92% 87%
Table 3.2: Yield of preparing drug-loaded micelles with DTX, Das and the combinations.

160
As summarized in Table 3.3, all drug-loaded micelles were mono-distributed with
the polydispersity index (PDI) below 0.2 and were around 35-40 nm on average. The
(DTX+Das)/m were found to be stable, and the size remained similar after being stored
at r.t. for ten weeks. Compared with the micelles synthesized in 2.2.2, the average sizes
were smaller, with the cutoff of the size distribution below 110 nm. The smaller size and
the lower cut off suggest most of the particles will be within the range that can harness
the EPR effect (discussed in 2.3).

161
Deff
mean ± SD nm PDI
Blank micelle 43 ± 1 0.19
DTX/m 34 ± 1 0.17
Das/m 42 ± 1 0.13
DTX/m + Das/m 36 ± 1 0.15
(DTX + Das)/m 35 ± 2 0.13
(DTX + Das)/m r.t. 10 weeks 42 0.16
Table 3.3: Size, PDI and distribution modal of the drug-loaded micelles.

162
As discussed previously, the simultaneous delivery of DTX and Das is the key to
success in the proposed approach. It requires a slow release during the circulation to
retain the drugs before reaching the cancer cells, and the similar release speed for both
drugs. The in vitro drug release studies in 1X PBS showed a moderate and steady
release profile that continued for more than three days, and the release rate of DTX and
Das were similar in single-drug-loaded micelles and double-drug-loaded micelles (Figure
3.13). Thus, it could be expected that the synthesized micelles have the potential to
serve our purpose of delivering both drugs at the same time.

163
Figure 3.13: In vitro release profile of DTX/m, Das/m and (DTX+Das)/m. The release continued steadily
for more than three days. Both drugs in single-drug-loaded micelles and double-drug-loaded micelles were
released at a similar rate. The cumulative release in % was calculated by normalizing the quantities of
released drugs to the total amount of the loaded drugs.
0 20 40 60 800%
50%
100%
Time (h)
Das, (DTX+Das)/m
DTX, (DTX+Das)/m
DTX, DTX/m
Das, Das/m

164
In vitro cytotoxicity of the drug-loaded micelles
The anticancer efficacy of drug-loaded micelles was evaluated in the cultured
murine breast cancer cells 4T1 in vitro. Both parent cell and DTX adapted one (pre-
treated with 10 µM DTX for three days) were used for the studies. The treatment
exposure time was limited to 24 h because it had been observed that cell might die after
exhausting the nutrients (refreshment of the medium with the same drug lead to cell
survival, 3.2.2). As shown in Table 3.4, the DTX and Das in the micelles had IC50 of 31
µM and 11 µM respectively; when given in combination, the drugs’ IC50 level of both
drugs decreased drastically to sub-micromolar concentration. The calculated
combination index (CI) was 0.107, indicating a strong synergistic effect. Not surprisingly,
the adapted cells that had been treated with 10 µM DTX developed the resistance, and
we could not observe any cell death even at 100 µM of DTX (data not shown). However,
the adapted cells became much more sensitive to the Das treatment, with one
magnitude lower IC50 (2.4 µM) in the single drug treatment group. Specifically, the
sensitivity to Das after the adaptation increased one-fold compared to the parent
counterpart (Figure 3.14). This was expected based on the studies of p-AMPK level shift
that the DTX treatment resulted in low p-AMPK response and such change made the
cell more vulnerable to Das. If we take a closer look and compare the IC50 between Das
and the combination in the adapted cells, it is noticeable that the addition of DTX did
provide an extra benefit, although in the single DTX treatment seemed already to lose all
the efficacy. It also reflects part of the hypothesis that DTX-adapted cell might be trying
to re-adapt to the secondary treatment (Das), and the existence of DTX made an
attempt unhelpful. Another point worthy to mention is that the IC50 of the combination

165
treatment remained comparable for both parent and adapted cells, which suggested the
combination might work very well not only for the patients who are chemo naïve but also
for the patients who have been treated with DTX.

166
DTX/m Das/m (DTX+Das)/m
Parent 4T1 31± 6 11 ± 0.3 0.87 ± 0.27
DTX adapted 4T1 >>100 2.4 ± 0.3 0.53 ± 0.04
Table 3.4: In vitro tumor growth inhibition of drug-loaded micelles. The absolute IC50 (µM) values were
the drug concentration to achieve 50% suppression of the tumor growth to the control. All treatment lasted
for 24 h.
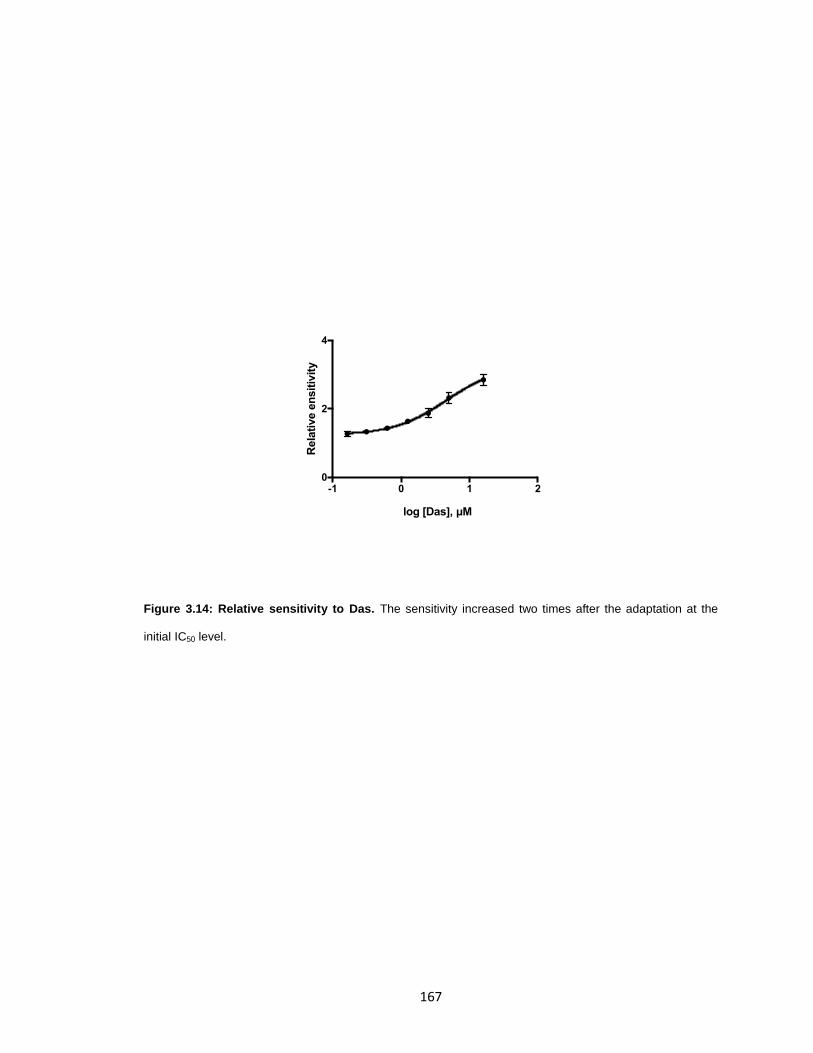
167
Figure 3.14: Relative sensitivity to Das. The sensitivity increased two times after the adaptation at the
initial IC50 level.
-1 0 1 20
2
4
log [Das], µM
Rela
tive e
nsit
ivit
y

168
To test if this approach was only limited in 4T1 or could apply to other cell lines or
different types of cancer. The same experiments were repeated on human breast cancer
cell line MDA-MB-231 and human prostate cancer cell line PC-3. The results
demonstrated the same trend as well as the relation between the IC50 of combination
and single drug-treated group (Table 3.5). Both cell lines showed clear evidence of
strong synergistic effects (CI<0.1) and the retained the similar efficacy in DTX adapted
cells.

169
DTX/m Das/m (DTX+Das)/m
Parent MDA-MB-231 6.2 ± 0.7 3.2 ± 3.0 0.11 ± 0.02
DTX adapted MDA-MB-231 >>100 0.47 ± 0.23 0.14 ± 0.12
Parent PC-3 1.7 ± 1.5 12 ± 8 0.12 ± 0.18
DTX adapted PC-3 >>100 3.3 ± 0.3 0.53 ± 0.28
Table 3.5: Growth inhibition in MDA-MB-231 and PC-3 IC50 (µM). Strong synergistic effect (CI<0.1) of the
combination was demonstrated, and the same level of efficacy was retained in DTX adapted cells.

170
Sensitization of 4T1 cells to DTX or Das with 5-Aminoimidazole-4-carboxamide
ribonucleotide (AICAR) or Compound C
To clarify that the strong synergy found between DTX and Das was indeed
associated with the adaptive dynamics represented by p-AMPK level shift and was not
only due to the cytotoxic effects via two independent targets by the combinations as
reported frequently in other drug combinations. However, in this case, the cytotoxic
stress was the foundation of the adaptive dynamics, and the adaptive dynamics was
responsive to the synergy. It would not be possible to lower the concentration of the drug
to a level that does not have a cytotoxic effect but retains the ability to induce the
adaptive response and deliver synergy based on that. Thus, to separate the trait of
drugs’ cytotoxicity from the trait of the sensitization via opposite adaptive dynamics, an
AMPK activator (AICAR) and an inhibitor (Compound C) was used as the analog to Das
and DTX respectively[39]. Neither of the activator or inhibitor could inhibit the growth of
4T1 in the working concentration of 100 µM and 1 µM respectively (Figure 3.15).

171
Figure 3.15: Growth inhibition of AICAR and Compound C. Neither 100 µM AICAR (left) nor 1 µM
Compound C (right) of the molecules inhibited 4T1 growth in 24 h treatment compared to non-treated control.
Contr
ol1µ
M Com
pound C
0
50
100
150
Via
lbilit
y%
Contr
ol
100µM
AIC
AR
0
50
100
150
Via
lbilit
y%

172
The AMPK activator AICAR upregulates the AMPK level and hence to provide
the similar effect as the Das could lead to in adapted cells, whereas the inhibitor
Compound C downregulate p-AMPK and accordingly to represent the DTX. Therefore,
the AICAR was used in combination with DTX while Compound C was with Das to test
their sensitization effect on 4T1 cells for 24 h. Just as expected, the reversal of the
adaptive dynamics (AMPK activation in DTX treated cells, AMPK inhibition in Das treat
cells) remarkably increase the cells sensitivity and brought down the IC50 to or even
exceeded the comparable levels that were achieved by the drug combinations (Table
3.6). So, it is clear that the p-AMPK level shift did play a critical role in the synergistic
effect between DTX and Das.
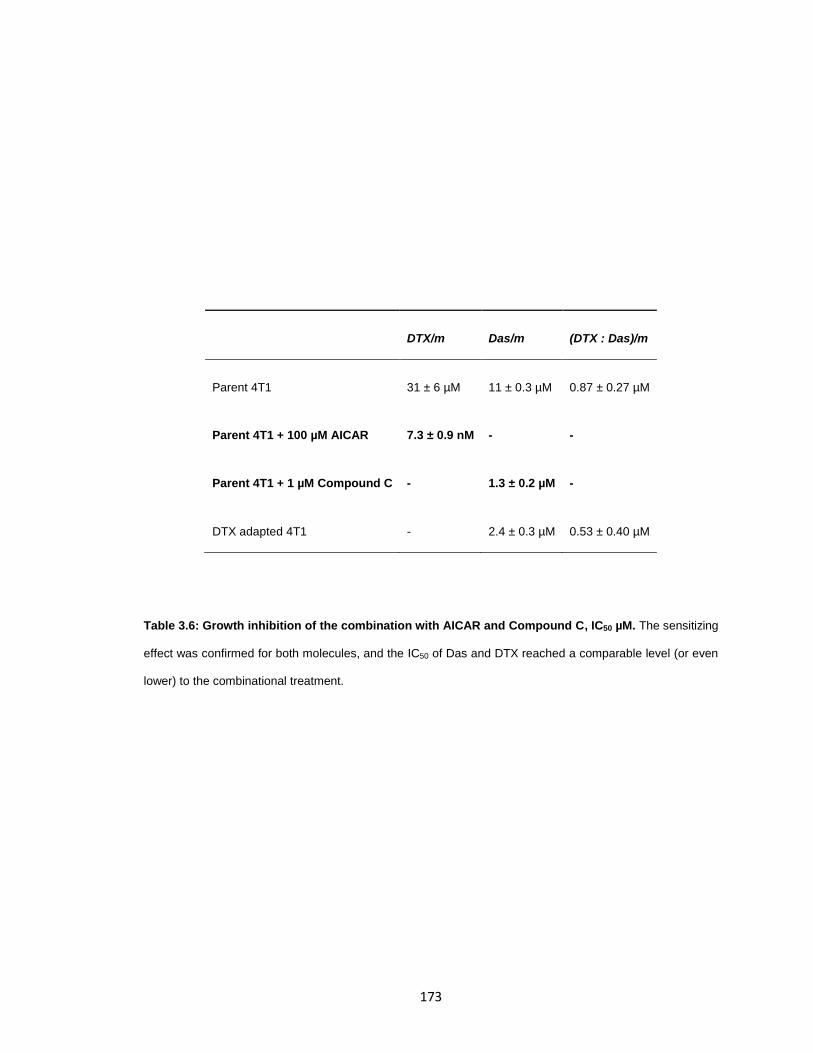
173
DTX/m Das/m (DTX : Das)/m
Parent 4T1 31 ± 6 µM 11 ± 0.3 µM 0.87 ± 0.27 µM
Parent 4T1 + 100 µM AICAR 7.3 ± 0.9 nM - -
Parent 4T1 + 1 µM Compound C - 1.3 ± 0.2 µM -
DTX adapted 4T1 - 2.4 ± 0.3 µM 0.53 ± 0.40 µM
Table 3.6: Growth inhibition of the combination with AICAR and Compound C, IC50 µM. The sensitizing
effect was confirmed for both molecules, and the IC50 of Das and DTX reached a comparable level (or even
lower) to the combinational treatment.

174
Chemotaxis of tumor cells toward pre-osteoblast
As reviewed by Maggague, Obenauf, and Weilbaecher, the preference of tumor
growth in bone may closely related to the stem-like niches that permit the homing of the
disseminated cancer cells and provides the support for their growth[40,41]. One of the
niches where cancer cells invade to has been hypothesized to be the endosteal one
which is filled up of osteoblasts. And evidence from in vivo studies also implies that the
niche could be one of the factors that determine if the cells will populate into the
metastatic lesion or remain sleepy[42]. Dasatinib has been reported to inhibit the
migration of human sarcoma cell lines[43]. In this study, we developed a simplified
model and aimed to demonstrate the inhibitory effect of the Dasatinib in the combination
could potentially stop the tumor's invasion into the osteoblast niches. The model
comprised by a trans-well system where the pre-osteoblast cells MC3T3 with their
conditioned medium (preOB-CM) were on the lower chamber to serve as the
chemoattractant and the tumor cells 4T1 were in the upper insert with the treatments.
The preOB-CM was found more effective than the 10% FBS which is typically
considered as a robust chemoattractant. However, MDA-MB-231 did not migrate under
the same condition (Figure 3.19). It might be some intercellular reaction that was species
specific.
When treated with just 10 nM or 20 nM Das/m, the migration was substantially
inhibited, and the number of migrated cells were close to the negative (Figure 3.17). As
the dose did not affect the cell viability (Figure 3.16), the inhibitory effect was not part of

175
the growth inhibition. Representative images of the migrated cells on the downside of the
membrane were shown in Figure 3.18. The results proved that the Das/m were potent in
inhibiting the tumor chemotaxis induced by the pre-osteoblast and therefore
demonstrated the potential to benefit the anticancer therapy by interfering the
association of osteoblast niches and tumors cells.

176
Figure 3.16: Cell viability after 24 h treatment of Das/m. The concentration of Das/m used in migration
study was 10 nM and 20 nM, and the duration of the drug exposure was 12 h. Hence, they would not affect
the viability of the cells throughout the study.
0.3125 0.6250
50
100
150
[Das], µM
Via
bilit
y %
to
co
ntr
ol
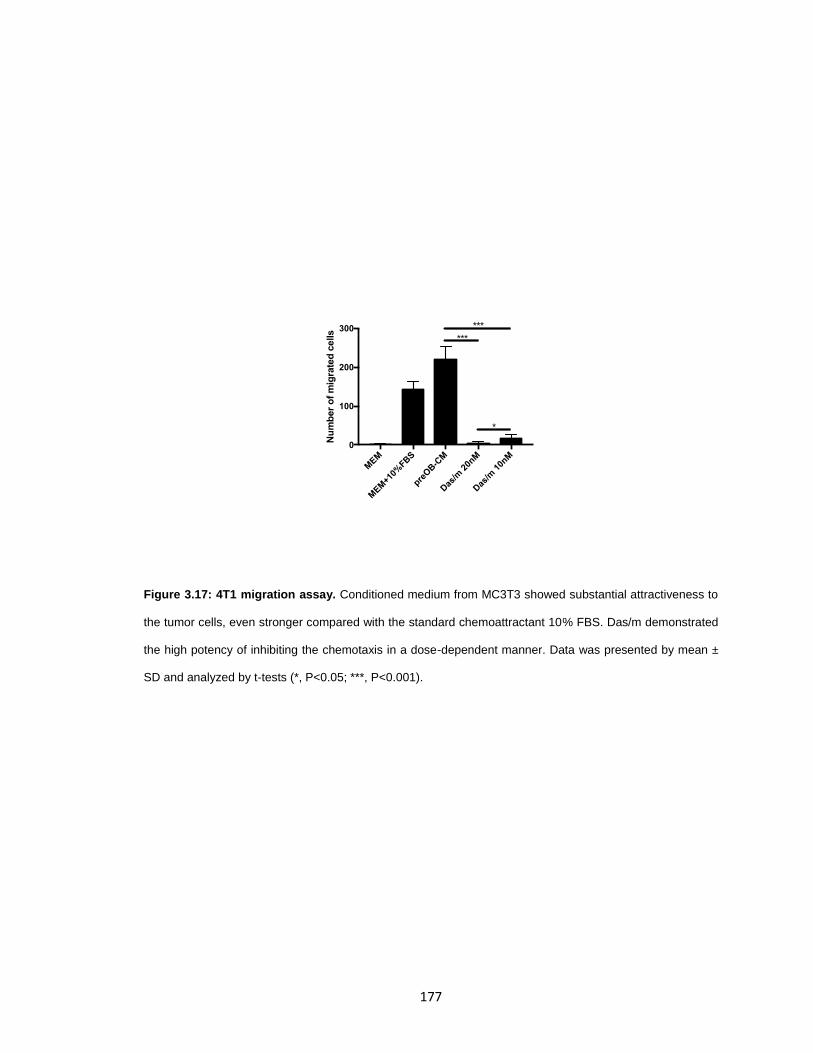
177
Figure 3.17: 4T1 migration assay. Conditioned medium from MC3T3 showed substantial attractiveness to
the tumor cells, even stronger compared with the standard chemoattractant 10% FBS. Das/m demonstrated
the high potency of inhibiting the chemotaxis in a dose-dependent manner. Data was presented by mean ±
SD and analyzed by t-tests (*, P<0.05; ***, P<0.001).
MEM
MEM
+10%
FBS
preOB-C
M
Das
/m 2
0nM
Das
/m 1
0nM
0
100
200
300N
um
ber
of
mig
rate
d c
ells
***
***
*
0.3125 0.6250
50
100
150
[Das], µM
Via
bilit
y %
to
co
ntr
ol
178
Figure 3.18: 4T1 migrated through the transwell membrane.
MEM MEM+10%FBS preOB-CM
Das/m 20nM Das/m 10nM

179
Figure 3.19: Migrated MDA-MB-231 using preOB-CM. No migrated cells could be seen on the membrane.
MDA-231 with preOB-CM

180
Inhibition of the osteoclast differentiation
In Chapter II, alendronate was attached to the micelles and served as not only
the targeting ligand but also a potential therapeutic agent who showed the efficacy of
amending the "vicious cycle" by inhibiting osteoclast differentiation and bone resorption
in vitro. However, the total dose of the alendronate administered in vivo was far below
the level used in the clinic, limiting the overall outcome. Dasatinib was also reported to
impede the osteoclast formation, and the potency was much higher compared to the
bisphosphonates[44,45].
Similar to the previous experiment in 2.2.2, the Raw cells were differentiated to
osteoclast cells with the RANKL. Polymer control, free Das (30 nM) and Das/m (30 nM,
10 nM) were given to the Raw cells from the beginning of the experiment. Compared to
the positive control, the number of differentiated osteoclasts was found to be remarkably
lower in Das treated groups but remain the same in polymer only group (Figure 3.20,
3.21). And loading Das into the micellar form seemed not to comprise the potency (no
significant difference was observed between free drug and micellar group). As expected,
the inhibitory effect was dose-dependent, and the IC50 was around the 10 µM level.
These results demonstrated that Das/m were more potent than alendronate to
suppress the osteoclast differentiation and thus could be a better option for treating the
pathological bone resorption in metastases.

181

182
Figure 3.20: Das inhibited osteoclast at nanomolar concentration. Both free Das and Das loaded in
micelles significantly decreased the amount of the osteoclast. Data was presented by mean ± SD and
analyzed by t-test (*, P<0.05; **, P<0.01).
Neg
a Ctrl
Posi
Ctrl
Poly
mer
s
Fres
Das
30n
M
Das
/m 3
0nM
Das
/m 1
0nM
0
20
40
60
80
100N
um
ber
of
oste
ocla
st
*
**
**
NS
**
0.3125 0.6250
50
100
150
[Das], µM
Via
bilit
y %
to
co
ntr
ol

183
Figure 3.21: Representative image of the differentiated osteoclast under different treatment groups.
Free Das and Das/m 30 nM showed substantial cytotoxicity to the Raw cells over five days. Although Das/m
10 nM had no noticeable impact on the cell density compared to the two control groups (positive and
polymer), it still retained some inhibitory effect.

184
Anticancer efficacy in vivo
The combination micellar formulation (DTX+Das)/m and DTX/m+Das/m were
tested in the murine breast cancer bone metastases model, and their efficacy was
compared with free drug combinations as well as free DTX. As seen in the 2.3 the
disease model was aggressive. To ensure each mouse receive the same dosage of the
drugs, the treatments started as early as the bioluminescence appeared and was given
on a daily basis for four days in total. Starting from the day (day 0) before the first
treatment, the bioluminescence of the implanted tumor was monitor continuously for
seven days. Relative tumor growth (bioluminescent intensity normalized to the initial
read) of the individual mouse was showed in Figure 3.22, and the average for each
treatment group was summarized in Figure 3.23. Based on one-way ANOVA analysis
and multiple comparisons by Prism GraphPad, the free drug combinations did not
demonstrate any improvement over the single drug, however when encapsulated in
micelles, the combination significantly delayed the tumor progression. No statistical
difference could be observed between the DTX/m+Das/m and the (DTX+Das)/m groups.
Since both treatment groups shared a similar drug release profile and the particles size,
it could be reasonable to speculate the pharmacokinetics were close, which could
explain the similarity of the therapeutic outcome.
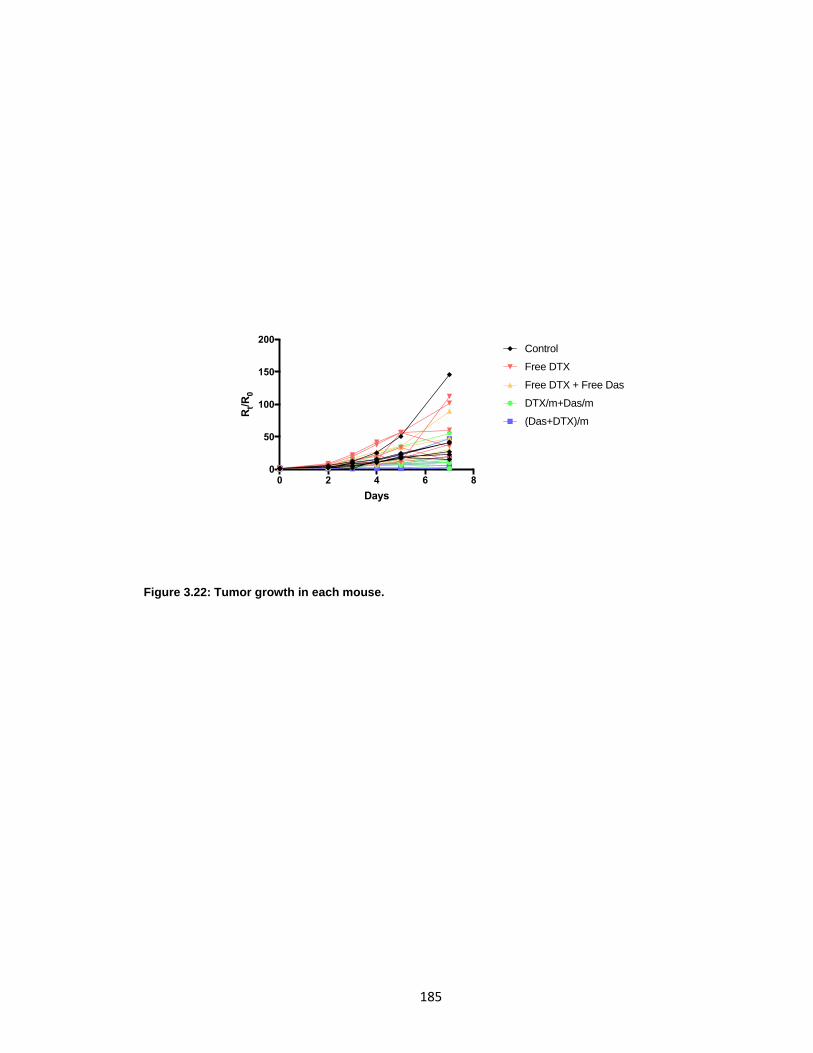
185
Figure 3.22: Tumor growth in each mouse.
0 2 4 6 80
50
100
150
200
Days
Rt/R
0
DTX/m+Das/m
(Das+DTX)/m
Free DTX + Free Das
Free DTX
Control
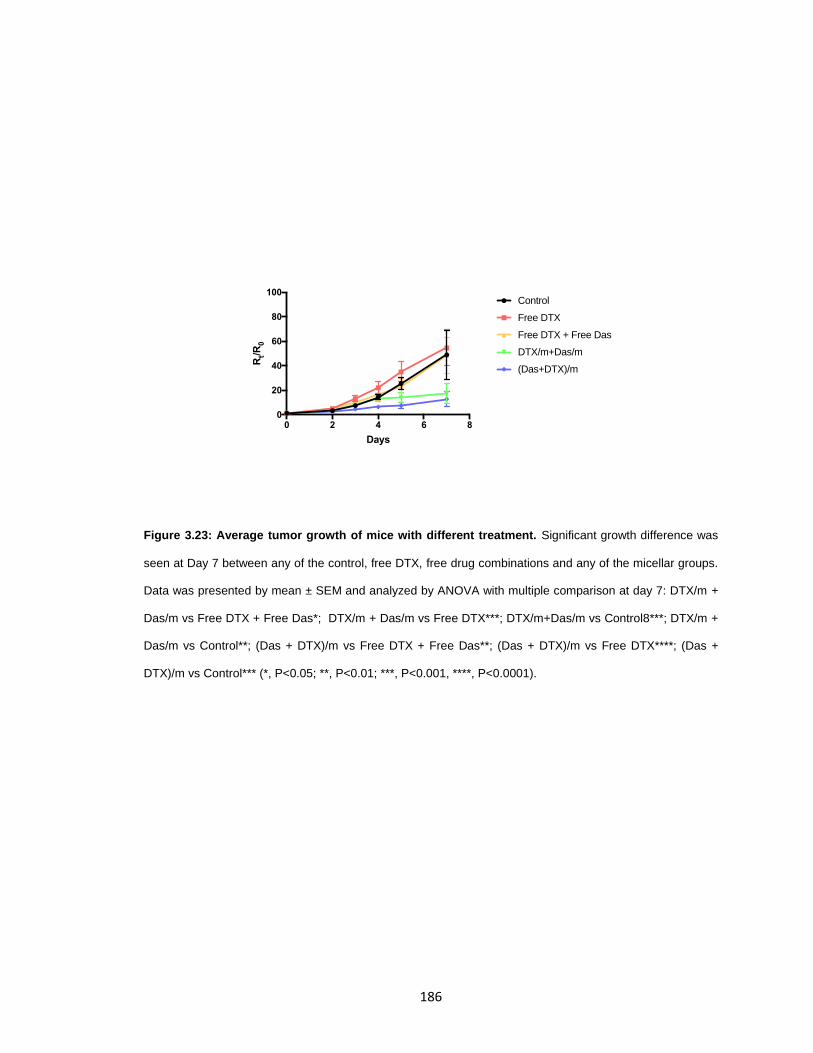
186
Figure 3.23: Average tumor growth of mice with different treatment. Significant growth difference was
seen at Day 7 between any of the control, free DTX, free drug combinations and any of the micellar groups.
Data was presented by mean ± SEM and analyzed by ANOVA with multiple comparison at day 7: DTX/m +
Das/m vs Free DTX + Free Das*; DTX/m + Das/m vs Free DTX***; DTX/m+Das/m vs Control8***; DTX/m +
Das/m vs Control**; (Das + DTX)/m vs Free DTX + Free Das**; (Das + DTX)/m vs Free DTX****; (Das +
DTX)/m vs Control*** (*, P<0.05; **, P<0.01; ***, P<0.001, ****, P<0.0001).
0 2 4 6 80
20
40
60
80
100
Days
Rt/R
0
Control
Free DTX
Free DTX + Free Das
DTX/m+Das/m
(Das+DTX)/m

187
As the disease progressed, substantial body weight loss happened for all tested
groups (Figure 3.24). Some of the mice were anesthetized to comply to IACUC protocol
and recorded as the death event. However, the weight loss seemed not associated with
the toxicity of the treatment. The cumulative dose of the DTX was 40 mg/kg, and Das
was 24 mg/kg. As the maximum tolerant doses (MTD) are 80 mg/kg for DTX and 70
mg/kg for Das, the toxicity was expected to be limited. One day after the final treatment,
three mice from each group were sacrificed, and the blood, spleen, kidney, liver and
femur/tibia were collected for histological analysis (Figure 3.25). No obvious damaged
were observed for any of the visceral organs. Apart from the toxicology, several
secondary metastases were found in the kidneys in the control group and both free drug
groups, while no metastases were found in any of the organs in the two micellar groups.
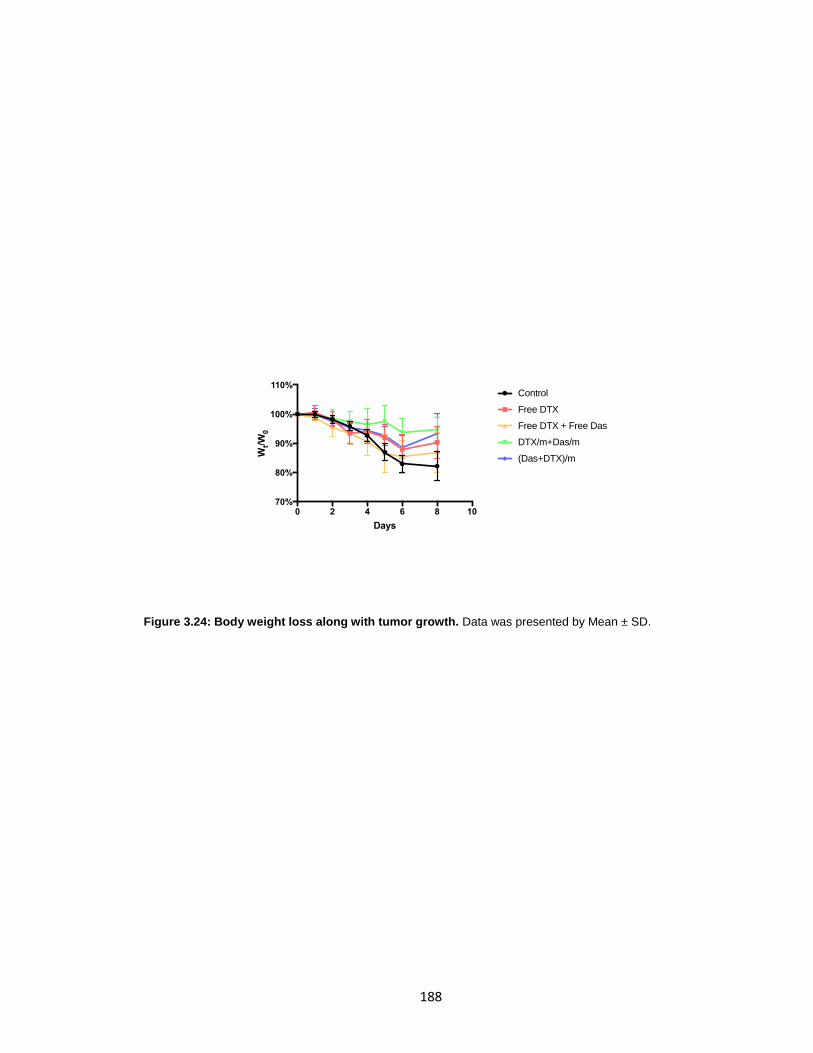
188
Figure 3.24: Body weight loss along with tumor growth. Data was presented by Mean ± SD.
0 2 4 6 8 1070%
80%
90%
100%
110%
Days
Wt/W
0
(Das+DTX)/m
DTX/m+Das/m
Free DTX + Free Das
Control
Free DTX

189
Figure 3.25: Acute toxicity study based on H&E staining of the major organs collected 24 h after the
final treatment. Pathologically, all the organs were in normal condition, indicating the treatments were
tolerated.
Kidney
Liver
Spleen
Control Free DTX DTX/m + Das/mFree DTX + Das (DTX + Das)/m

190
The survival of the mice was monitored up to thirty days. Apart from the
individuals that died naturally due to the tumor growth, some of the mice were
euthanized due to the weight loss > 20% or suffered from severe pain according to the
IACUC guidelines. Correlated with the tumor growth found previously, the survival of the
free drug-treated group barely showed any improvement over the control, while both
DTX/m+Das/m and (DTX+Das)/m demonstrate significantly prolonged survival time
compared to either control or the free drug combinations (Figure 3.26).
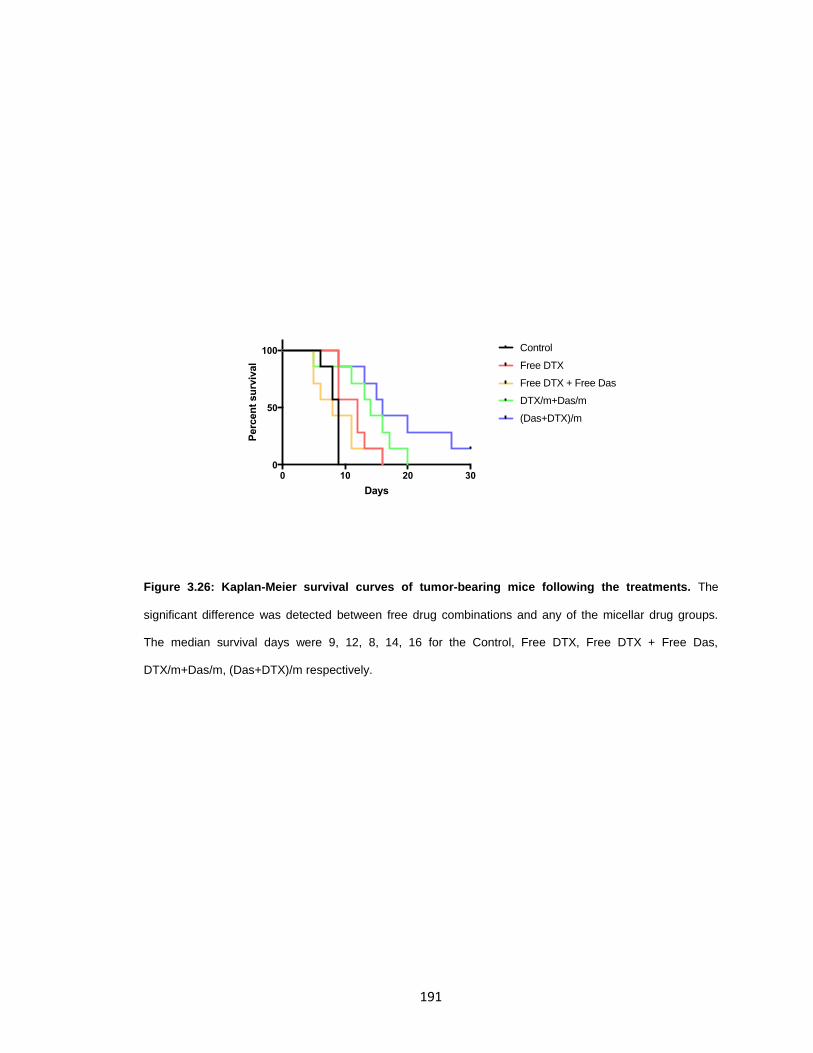
191
Figure 3.26: Kaplan-Meier survival curves of tumor-bearing mice following the treatments. The
significant difference was detected between free drug combinations and any of the micellar drug groups.
The median survival days were 9, 12, 8, 14, 16 for the Control, Free DTX, Free DTX + Free Das,
DTX/m+Das/m, (Das+DTX)/m respectively.
0 10 20 300
50
100
Days
Perc
en
t su
rviv
al
Control
Free DTX
Free DTX + Free Das
DTX/m+Das/m
(Das+DTX)/m

192
The bone metastases model we used, in general, has a broad distribution of the
initial tumor burden volume. The mice were evenly distributed into the ten groups and
randomly assigned to the five different treatments to mitigate any potential bias caused
by the initial tumor burden. The possible correlation between the survival time and the
initial tumor burden was also analyzed within each treatment groups. Data were fitted by
linear regression, and the slopes of the trend lines were not deviated (P>0.1, F test) from
zero and the correlation did not exist in this study (Figure 3.27). This result implies the
potential effect of uneven distribution of the animal with different initial tumor burden was
limited. After completing four-times treatment, the survival time was found to be
correlated with the tumor progression recorded at Day 5, explaining the consistency
between tumor growth and survival data. The mice with lower Rt/R0 ratio tended to
survive longer in general.

193
Figure 3.27: Correlation between initial tumor burden (bioluminescence) and the survival time within
each treatment groups. A) Trend lines were fitted by linear regression. However, none of the slopes
significantly deviated from zero. B) Data included all survived mice on Day 5. A linear correlation was
observed between survival time and the tumor growth rate (Rt/R0). The slope significantly deviates from zero
(P=0.01).
0 10 20 30 400.0
5.0×106
1.0×107
1.5×107
2.0×107
Survial time (Days)
Init
ial tu
mo
r in
ten
sit
y (
p/s
)
Control
Free DTX
Free DTX + Free Das
DTX/m+Das/m
(Das+DTX)/m
0 10 20 30 400
20
40
60
Survial time (Days)
Rt/R
0 a
t D
ay 5
A
B

194
Blood cells were counted one day and five days after the final treatment. As
listed in Figure 3.28, the tumor-bearing mice displayed a much higher level of white
blood cells (WBC) than normal healthy mice and spleen enlargement[46]. The blood
smear revealed that substantial amount (≈50%) of the unmatured neutrophils were in the
blood (Figure 3.29). Upon treatment of DTX or DTX+Das, the WBC seemed to be
eliminated drastically. However, after resting for five days without any treatment, the
number of WBCs recovered partially back to the control level. On the contrary to the
substantial change found in WBCs, the red blood cells (RBC) remained relatively stable
in all the treated groups.

195
Figure 3.28: Blood count 24 h and five days after the final treatment. WBC was considerably higher in
the tumor-bearing mice compared the standard control (STD). In drug-treated mice, the WBCs were
eliminated substantialy and partially recovered after five days.
WBC
LYM
MO
NNEU
0
5
10
40
80
Cell c
ou
nt
10
9/L
24 h after treatments
Control
Free DTX
Free DTX + Free Das
DTX/m + Das/m
(DTX+Das)/m
Healthy
WBC
LYM
MO
NNEU
0
20
40
60
80
100
Cell c
ou
nt
10
9/L
Recovery
Control
Free DTX
Free DTX + Free Das
DTX/m + Das/m
(DTX+Das)/m
Healthy

196
Figure 3.29: Representative image of the blood smear from the tumor-bearing mice. About half of the
neutrophils were unmatured.
Imm
ature
mat
ure
0
20
40
60
80
Fra
cti
on
% o
f to
tal W
BC

197
3.4 CONCLUSION
In this study, the 4T1 cells were observed to become drug-tolerant upon limited
exposure to Docetaxel. The adapted response was characterized by the exit of the cell
cycle and the formation of the polyploid cells. The adapted 4T1 showed the metabolic
dynamics of the p-AMPK that deviated substantially from the base level. Different drugs
were found to result in the different direction of the adaptive dynamics. Here, we
demonstrated that DTX-adapted cell was featured by low p-AMPK, whereas Das-
adapted by high p-AMPK. In other words, the new metabolic pattern that cells use to
survive under the chemo stress demands the p-AMPK to be low for DTX and to be high
for Das. The mechanism was further confirmed in the later study where the AMPK
activator and inhibitor restored the sensitivity to DTX and Das respectively.
Because of the opposite direction of cellular dynamics induced by DTX and Das,
the combinations of both could be a potential solution for overcoming the adaptive
resistance. The adaptation to DTX will lead to the sensitization to Das or vice versa.
And given the essentialness of the AMPK and only two possibilities of the activation level,
cells are not expected to develop anything outside of the box. The enhanced growth
inhibitory effect and strong synergy of the combination were validated in both parent and
DTX-adapted cell in vitro for all 4T1, MDA-MB-231 and PC-3.
Forty-nanometers sized micelles were synthesized to encapsulate both DTX and
Das with high loading capacity and sustained release profile. The micellar formulations

198
were tested in the model of murine breast cancer bone metastases, showing significantly
improved therapeutic compared to the free DTX and free drug combinations, while
keeping the toxicity at a tolerable level.
In the previous clinical trials, the combinations were considered no more effective
than the single DTX regimen, which might be due to the pharmacokinetic difference
between the two drugs. Our study reevaluated the potential of the combinational strategy
by developing a two-in-one micellar formulation that can deliver both drugs
simultaneously. The design achieved remarkable improvement and overcame the
adaptive resistance by inducing mirrored adaptive dynamics.

199
3.5 REFERENCE
1. Chabner BA, Roberts TG. Timeline: Chemotherapy and the war on cancer. Nature Reviews Cancer 2005;5: 65–72. doi:10.1038/nrc1529
2. Attolini CSO, Michor F. Evolutionary Theory of Cancer. Annals of the New York Academy of Sciences. Wiley/Blackwell (10.1111); 2009;1168: 23–51. doi:10.1111/j.1749-6632.2009.04880.x
3. Diaz LA Jr, Williams RT, Wu J, Kinde I, Hecht JR, Berlin J, et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature. Nature Publishing Group; 2012;486: 537–540. doi:10.1038/nature11219
4. Pisco AO, Huang S. Non-genetic cancer cell plasticity and therapy-induced stemness in tumour relapse: “What does not kill me strengthens me.” Br J Cancer. Nature Publishing Group; 2015;112: 1725–1732. doi:10.1038/bjc.2015.146
5. Salgia R, Kulkarni P. The genetic/non-genetic duality of drug “Resistance” in dancer. Trends Cancer. 2018;4: 110–118. doi:10.1016/j.trecan.2018.01.001
6. Abolhoda A, Wilson AE, Ross H, Danenberg PV, Burt M, Scotto KW. Rapid activation of MDR1 gene expression in human metastatic sarcoma after in vivo exposure to doxorubicin. Clinical Cancer Research. American Association for Cancer Research; 1999;5: 3352–3356.
7. Pisco AO, Brock A, Zhou J, Moor A, Mojtahedi M, Jackson D, et al. Non-Darwinian dynamics in therapy-induced cancer drug resistance. Nat Commun. Nature Publishing Group; 2013;4: 2467. doi:10.1038/ncomms3467
8. Gupta PB, Fillmore CM, Jiang G, Shapira SD, Tao K, Kuperwasser C, et al. Stochastic State Transitions Give Rise to Phenotypic Equilibrium in Populations of Cancer Cells. Cell. Cell Press; 2011;146: 633–644. doi:10.1016/j.cell.2011.07.026
9. Zheng Z, Fan S, Zheng J, Huang W, Gasparetto C, Chao NJ, et al. Inhibition of thioredoxin activates mitophagy and overcomes adaptive bortezomib resistance in multiple myeloma. Journal of Hematology & Oncology 2018 11:1. BioMed Central; 2018;11: 29. doi:10.1186/s13045-018-0575-7
10. Chen S, Zhang Y, Zhou L, Leng Y, Lin H, Kmieciak M, et al. A Bim-targeting strategy overcomes adaptive bortezomib resistance in myeloma through a novel link between autophagy and apoptosis. Blood. 2014;124: 2687–2697. doi:10.1182/blood-2014-03-564534
11. Wagner VP, Martins MAT, Martins MD, Warner KA, Webber LP, Squarize CH, et al. Overcoming adaptive resistance in mucoepidermoid carcinoma through inhibition of the IKK-β/IκBα/NFκB axis. Oncotarget. Impact Journals, LLC; 2016;7: 73032–73044. doi:10.18632/oncotarget.12195

200
12. Goldman A, Kulkarni A, Kohandel M, Pandey P, Rao P, Natarajan SK, et al. Rationally Designed 2-in-1 Nanoparticles Can Overcome Adaptive Resistance in Cancer. ACS Nano. 2016;10: 5823–5834. doi:10.1021/acsnano.6b00320
13. Goldman A, Majumder B, Dhawan A, Ravi S, Goldman D, Kohandel M, et al. Temporally sequenced anticancer drugs overcome adaptive resistance by targeting a vulnerable chemotherapy-induced phenotypic transition. Nat Commun. 2015;6: 6139. doi:10.1038/ncomms7139
14. Hall MD, Handley MD, Gottesman MM. Is resistance useless? Multidrug resistance and collateral sensitivity. Trends in Pharmacological Sciences. Elsevier Current Trends; 2009;30: 546–556. doi:10.1016/j.tips.2009.07.003
15. Li Q, Wennborg A, Aurell E, Dekel E, Zou J-Z, Xu Y, et al. Dynamics inside the cancer cell attractor reveal cell heterogeneity, limits of stability, and escape. Proc Natl Acad Sci USA. 2016;113: 2672–2677. doi:10.1073/pnas.1519210113
16. Soni KS, Lei F, Desale SS, Marky LA, Cohen SM, Bronich TK. Tuning polypeptide-based micellar carrier for efficient combination therapy of ErbB2-positive breast cancer. Journal of Controlled Release. 2017;264: 276–287. doi:10.1016/j.jconrel.2017.08.038
17. Kantarjian H, Jabbour E, Grimley J, Kirkpatrick P. Dasatinib. Nature Reviews Drug Discovery. 2006;5: 717–718. doi:10.1038/nrd2135
18. Montero A, Fossella F, Hortobagyi G, Valero V. Docetaxel for treatment of solid tumours: A systematic review of clinical data. Lancet Oncology. 2005;6: 229–239. doi:10.1016/S1470-2045(05)70094-2
19. Jaeger S, Igea A, Arroyo R, Alcalde V, Canovas B, Orozco M, et al. Quantification of Pathway Cross-talk Reveals Novel Synergistic Drug Combinations for Breast Cancer. Cancer Res. American Association for Cancer Research; 2017;77: 459–469. doi:10.1158/0008-5472.CAN-16-0097
20. Gdowski AS, Ranjan A, Vishwanatha JK. Current concepts in bone metastasis, contemporary therapeutic strategies and ongoing clinical trials. Journal of Experimental & Clinical Cancer Research 2017;36: 108. doi:10.1186/s13046-017-0578-1
21. Graham CH, Kobayashi H, Stankiewicz KS, Man S, Kapitain SJ, Kerbel RS. Rapid acquisition of multicellular drug resistance after a single exposure of mammary tumor cells to antitumor alkylating agents. J Natl Cancer Inst. 1994;86: 975–982.
22. Beck JF, Brügger D, Brischwein K, Liu C, Bader P, Niethammer D, et al. Anticancer drug-mediated induction of multidrug resistance-associated genes and protein kinase C isozymes in the T-lymphoblastoid cell line CCRF-CEM and in blasts from patients with acute lymphoblastic leukemias. Jpn J Cancer Res. Wiley-Blackwell; 2001;92: 896–903. doi:10.1111/j.1349-7006.2001.tb01178.x

201
23. Tomida A, Naito M, Tsuruo T. Acute induction of adriamycin-resistance in human colon carcinoma HT-29 cells exposed to a sublethal dose of adriamycin. Jpn J Cancer Res. 1995;86: 224–232. doi:10.1111/j.1349-7006.1995.tb03043.x
24. Mittal K, Donthamsetty S, Kaur R, Yang C, Gupta MV, Reid MD, et al. Multinucleated polyploidy drives resistance to Docetaxel chemotherapy in prostate cancer. Br J Cancer. 2017;116: 1186–1194. doi:10.1038/bjc.2017.78
25. Clarke SJ, Rivory LP. Clinical Pharmacokinetics of Docetaxel. Clin Pharmacokinet. Springer International Publishing; 1999;36: 99–114. doi:10.2165/00003088-199936020-00002
26. Ogden A, Rida PCG, Knudsen BS, Kucuk O, Aneja R. Docetaxel-induced polyploidization may underlie chemoresistance and disease relapse. Cancer Lett. 2015;367: 89–92. doi:10.1016/j.canlet.2015.06.025
27. Li Y-J, Lei Y-H, Yao N, Wang C-R, Hu N, Ye W-C, et al. Autophagy and multidrug resistance in cancer. Chinese Journal of Cancer 2017;36: 52. doi:10.1186/s40880-017-0219-2
28. Misirkic M, Janjetovic K, Vucicevic L, Tovilovic G, Ristic B, Vilimanovich U, et al. Inhibition of AMPK-dependent autophagy enhances in vitro antiglioma effect of simvastatin. Pharmacological Research. Academic Press; 2012;65: 111–119. doi:10.1016/j.p h.2011.08.003
29. Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nature Reviews Molecular Cell Biology 2012;13: 251–262. doi:10.1038/nrm3311
30. Herzig S, Shaw RJ. AMPK: guardian of metabolism and mitochondrial homeostasis. Nature Reviews Molecular Cell Biology. Nature Publishing Group; 2018;19: 121–135. doi:10.1038/nrm.2017.95
31. Zadra G, Batista JL, Loda M. Dissecting the dual role of AMPK in cancer: from experimental to human studies. Mol Cancer Res. American Association for Cancer Research; 2015;13: 1059–1072. doi:10.1158/1541-7786.MCR-15-0068
32. Li W, Saud SM, Young MR, Chen G, Hua B. Targeting AMPK for cancer prevention and treatment. Oncotarget. Impact Journals, LLC; 2015;6: 7365–7378. doi:10.18632/oncotarget.3629
33. Kuhajda FP. AMP-activated protein kinase and human cancer: cancer metabolism revisited. International Journal of Obesity (Lond) 2008 32:Suppl 4. S36–S41. doi:10.1038/ijo.2008.121
34. Menendez JA, Lupu R. Fatty acid synthase (FASN) as a therapeutic target in breast cancer. Expert Opinion on Therapeutic Targets. Taylor & Francis; 2017;21: 1001–1016. doi:10.1080/14728222.2017.1381087
35. Abd Al AM, Mahmoud AM, Sherbiny El GA, Moselhy El MA, Nofal SM, Latif El HA, et al. Resveratrol enhances the cytotoxic profile of docetaxel and doxorubicin in

202
solid tumour cell lines in vitro. Cell Proliferation. Wiley/Blackwell (10.1111); 2011;44: 591–601. doi:10.1111/j.1365-2184.2011.00783.x
36. Martinez Marignac VL, Smith S, Toban N, Bazile M, Aloyz R. Resistance to Dasatinib in primary chronic lymphocytic leukemia lymphocytes involves AMPK-mediated energetic re-programming. Oncotarget. 2013;4: 2550–2566. doi:10.18632/oncotarget.1508
37. Araujo JC, Trudel GC, Saad F, Armstrong AJ, Yu EY, Bellmunt J, et al. Docetaxel and dasatinib or placebo in men with metastatic castration-resistant prostate cancer (READY): a randomised, double-blind phase 3 trial. The Lancet Oncology. Elsevier; 2013;14: 1307–1316. doi:10.1016/S1470-2045(13)70479-0
38. Luo FR, Yang Z, Camuso A, Smykla R, McGlinchey K, Fager K, et al. Dasatinib (BMS-354825) pharmacokinetics and pharmacodynamic biomarkers in animal models predict optimal clinical exposure. Clinical Cancer Research. American Association for Cancer Research; 2006;12: 7180–7186. doi:10.1158/1078-0432.CCR-06-1112
39. Viollet B, Foretz M, Schlattner U. Bypassing AMPK phosphorylation. Chem Biol. 2014;21: 567–569. doi:10.1016/j.chembiol.2014.05.003
40. Massagué J, Obenauf AC. Metastatic colonization by circulating tumour cells. Nature. Nature Publishing Group; 2016;529: 298–306. doi:10.1038/nature17038
41. Weilbaecher KN, Guise TA, McCauley LK. Cancer to bone: a fatal attraction. Nature Reviews Cancer. Nature Publishing Group; 2011;11: 411–425. doi:10.1038/nrc3055
42. Ottewell PD. The role of osteoblasts in bone metastasis. J Bone Oncol. 2016;5: 124–127. doi:10.1016/j.jbo.2016.03.007
43. Shor AC, Keschman EA, Lee FY, Muro-Cacho C, Letson GD, Trent JC, et al. Dasatinib Inhibits Migration and Invasion in Diverse Human Sarcoma Cell Lines and Induces Apoptosis in Bone Sarcoma Cells Dependent on Src Kinase for Survival. Cancer Res. American Association for Cancer Research; 2007;67: 2800–2808. doi:10.1158/0008-5472.CAN-06-3469
44. Brownlow N, Mol C, Hayford C, Ghaem-Maghami S, Dibb NJ. Dasatinib is a potent inhibitor of tumour-associated macrophages, osteoclasts and the FMS receptor. Leukemia. 2009;23: 590–594. doi:10.1038/leu.2008.237
45. Li B, Ling Chau JF, Wang X, Leong WF. Bisphosphonates, specific inhibitors of osteoclast function and a class of drugs for osteoporosis therapy. Journal of Cellular Biochemistry. 2011;112: 1229–1242. doi:10.1002/jcb.23049
46. Beheshti A, Wage J, McDonald JT, Lamont C, Peluso M, Hahnfeldt P, et al. Tumor-host signaling interaction reveals a systemic, age-dependent splenic immune influence on tumor development. Oncotarget. 2015;6: 35419–35432. doi:10.18632/oncotarget.6214

203

204
CHAPTER IV
SUMMARY AND FUTURE STUDY

205
4.1 SUMMARY
Metastases in bone are the major obstacle in cancer therapy and decrease the 5-
years survival rate. No effective treatment is available in the clinic other than pain control.
The conventional monotherapy and combinational therapy only gained limited survival
advantage. As reviewed in Chapter I, the nanocarrier-based drug delivery system
provides an attracting opportunity in the treatment of bone metastases, with its versatility
in loading drug combination and tumor-targeting ability. Bisphosphonates have a long
history in osteoporosis treatments and are known to have the high binding affinity to the
skeleton, which makes it as the potential targeting ligand that can drive the chemo drug
to the bone-tumor site. Moreover, bisphosphonates exert the antiresorptive effect by
being cytotoxic to the osteoclasts and suppress their differentiation, which turns it into a
dual-functional molecule that can also strike on the tumor-bone microenvironment. As
the tumor-induced bone degradation serves a critical role in the propagation of the
"vicious cycle," the intervention of osteoclast activity would be beneficial.
In Chapter II, one of the most widely used bisphosphonate, alendronate, was
chemically linked on the surface of polypeptides-based micelles to shape the bone-
targeting carrier. DTX, one of the first-line chemo drugs in breast cancer, was
successfully loaded into the micelles with almost 100% efficiency by nanoprecipitation.
The preparation method was robust to yield high-concentrated formulation (2 mg/mL
DTX) without comprising the loading ability. The drug-loaded micelles had an average
size of 70 nm in PBS solution, small enough to be accumulated to the tumor tissue via

206
EPR effect. The formulation exhibited strong binding efficacy to the HAP which is the
mineral composition of the bone tissue. Comprehensive in vitro evaluation was made
demonstrating the stability, sustained drug release profile, retained anticancer efficacy
as well as the potential to reduce the formation of the osteoclasts and suppress the
macrophage recruitment by tumor cells. In mice, DTX showed prolonged circulation time
and increased AUC when encapsulated in the ALN-m. These characteristics were
translated into the treatment for the bone metastases murine model, where the targeted
micellar formulation was injected systematically through the tail vein and significantly
increased the animals' survival time compared without compromising the safety.
Das is a tyrosine kinase inhibitor that inhibits Src family proteins and was
reported to block the EGFR receptor that is frequently overexpressed in breast cancer
cells. As EGFR serves as the initiator of the pro-survival signaling in tumor cells, the
combination of Das and DTX could be an effective strategy to improve the therapeutic
outcome in bone metastases. However, clinical trials of such combination failed in
castration-resistance prostate bone metastases patients, which could be due to the
pharmacokinetic difference between two drugs. In Chapter III, we co-encapsulated the
Das and DTX at 1:1 molar ratio in a single micelle with a high loading capacity of 35% in
total. Both drugs were released at the same rate in vitro, suggesting the similar
pharmacokinetics profile. The combination showed substantial synergist effect (CI<0.1)
in breast and prostate cancer cell lines, with the IC50 that decreased in two magnitudes
compared to the monotherapy. Chemotherapies were discovered to induce the adaptive
drug tolerance in tumor cells rapidly and non-genetically, which is clinically relevant and
vital, as it may drastically impair the treatment efficacy. Notably, the chemo stress

207
imposed by Das and DTX resulted in opposite phenotype dynamics in tested cell lines.
Das-tolerant cells had a higher level of pAMPK, while DTX-tolerant cells had a lower
level. The appropriate level of pAMPK was demonstrated to be necessary for
maintaining the specific drug-tolerant status. The finding explained the observed synergy
between Das and DTX, as the tumor cells could not develop the resistance phenotype
that demands both up and downregulation of pAMPK when exposed to Das and DTX
simultaneously. Indeed, the DTX-adaptive cells which completely did not respond to the
continuous DTX exposure have the comparable sensitivity to the Das/DTX combination
as their parent cells. Besides the enhanced anticancer efficacy, Das also inhibited the
tumor migration and scavenged osteoclast in vitro. Subsequent in vivo studies further
revealed the potential of the drug combinational in micelles to delay the progression of
the disease and prolong the survival time, while keeping the regimen well tolerated.

208
4.2 FUTURE STUDY
1. The alendronate binds to the bone surface of formation and resorption without
preferential. In breast cancer bone metastases, tumor cells induce osteolytic lesion
that is mainly associated with bone resorption. Whereas prostate cancer originated
bone metastases are osteoblastic, and the bone formation is dominant. More specific
ligands can be chosen to target the surface of interest selectively. For example, Asp8
was reported to bind to resorption surface, and the (AspSerSer)6 showed specificity
to bone formation area[1]. Moreover, Denosumab[2] as a monoclonal antibody is
considered as a promising candidate that has not been investigated yet. It not only
shoots to RANKL with strong affinity but also cuts off the signaling of tumor-induced
osteoclast differentiation and activity, exhibiting both targeting and therapeutic
potential.
2. Using the two chemo agents that induce mirrored adaptive response is an intriguing
strategy to develop drug combination with high synergy. However, more detailed
investigation in the transcriptional or phosphorylated level of resistant phenotypical
transition is still needed. Due to the complication in the gene regulatory network, the
direction of the adaptive dynamic is described by a massive matrix of hyper
dimension. Proteomics and mRNA profile could be adopted to help identify individual
drug candidates that induce the distinctive adaptive response in tumor cells to the
maximum extent and thus to select the most effective combination.

209
3. In this project, the murine model of bone metastases was established by left-ventricle
injection. During the circulation, injected tumor cells are believed to seed them self to
the skeletons via tumor-bone interaction, mimicking the real situation. The model
allows the development of multiple-site bone metastases in the lower skeletons, skull,
ribs, spine). However, the model is highly aggressive, and the lifespan of the animals
was generally less than a month, making it difficult to arrange the dosing and
demonstrate the potential advantage of the treatments. Another model was recently
developed by Takahiro Kuchimaru[3]. Cancer cells were injected through caudal
arteries and trapped in the lower skeletons to form the bone metastases. The model
was more producible with a longer survival time of the animals and easier to handle
than the previous one achieved by intracardiac injection. Last but not least, the new
model opened up a new road to test the nanocarrier-based delivery system for bone
metastases.

210
4.3 REFERENCE
1. Zhang G, Guo B, Wu H, Tang T, Zhang B-T, Zheng L, et al. A delivery system targeting bone formation surfaces to facilitate RNAi-based anabolic therapy. Nature Medicine. 2012;18: 307–314. doi:10.1038/nm.2617
2. Vinay R, KusumDevi V. Potential of targeted drug delivery system for the treatment of bone metastasis. Drug Deliv. 2016;23: 21–29. doi:10.3109/10717544.2014.913325
3. Kuchimaru T, Kataoka N, Nakagawa K, Isozaki T, Miyabara H, Minegishi M, et al. A reliable murine model of bone metastasis by injecting cancer cells through caudal arteries. Nat Commun. 2018;9: 2981. doi:10.1038/s41467-018-05366-3




















