
Testing K d models of Cs + in the near field of a HLW repository in granite with a reactive...
6
Testing K d models of Cs + in the near field of a HLW repository in granite with a reactive transport model Javier Samper a, * , Hongyun Ma a , José Luis Cormenzana b , Chuanhe Lu a , Luis Montenegro a , Miguel Ángel Cuñado b a University of La Coruña, La Coruña 15071, Spain b ENRESA, C Emilio Vargas 7, Madrid 28043, Spain article info Article history: Received 26 October 2009 Received in revised form 11 March 2010 Accepted 10 April 2010 Available online 14 May 2010 Keywords: Apparent K d Caesium Reactive transport model HLW repository Bentonite Thermodynamic sorption model abstract Performance assessment models of high-level radioactive waste (HLW) repositories commonly rely on the simplifying assumption of a constant K d for radionuclide sorption. Testing the validity of this assump- tion has been prevented by the lack of nuclide thermodynamic sorption data and the unavailability of models for handling nuclide migration and sorption together with the geochemical evolution of the near field. The validity of the constant K d assumption has been tested with a multicomponent reactive trans- port model (RTM) for Cs + in the near field of a HLW repository in granite. Model results show that the apparent K d of Cs + , K a d , increases with time due to the decrease of the ionic strength, I, of the bentonite porewater caused by the out-diffusion of the aqueous species from the bentonite into the granite. Com- puted values of I are related to those of K a d through the following polynomial expression: I ¼ 253:5 1 K a d 2 þ 8:77 1 K a d 0:005. A thermodynamic justification for such an expression has been derived from the cation exchange reactions. A constant-K d model fails to reproduce the release rate of Cs + from the near field computed with the RTM. A variable-K d model which incorporates the dependence of K a d on I reproduces adequately the Cs + release rate, thus providing a surrogate for the constant-K d model. The results of the sensitivity runs to changes in model parameters and boundary conditions show that the water flux at the bentonite–granite interface affects strongly the K a d through changes in I while the effective diffusion coefficient of the bentonite plays a minor role on K a d . An increase in the cation exchange capacity leads to an increase of K a d , but it does not affect the time evolution of I. Competing cat- ions such as Ni 2+ and iron corrosion products decrease slightly the K a d of Cs + by competing for exchange sites and by increasing the I. Ó 2010 Elsevier Ltd. All rights reserved. 1. Introduction Current performance assessment (PA) models for radionuclide migration through the near field of a high-level radioactive waste (HLW) repository usually rely on simplifying assumptions such as the use of a constant K d for nuclide sorption and ‘‘the limited sol- ubility” for nuclide precipitation. Testing the validity of these assumptions has been limited by: (1) the lack of nuclide surface complexation and cation exchange data; and (2) the unavailability of computer codes which could handle simultaneously the migra- tion, sorption and precipitation of radionuclides and the geochem- ical evolution of the near field. Laboratory experiments performed in recent years have provided substantial data and understanding on the mechanisms of nuclide sorption. On the other hand, sophis- ticated research-oriented process-based models and computer codes have been developed which allow for the simultaneous mod- elling of migration, sorption, nuclide precipitation and multicom- ponent geochemical evolution of the near field. In this paper we test the validity of the constant-K d model for nuclide sorption and precipitation for the 0.75 m thick compacted bentonite barrier of a spent-fuel repository in granite designed according to the Spanish reference concept (ENRESA, 2005). Such testing has been performed by comparing the results of a PA model with those obtained with a reactive transport model performed with CORE (Samper et al., 2009a). Here we present the results for Cs + . The testing for other nuclides is reported by Samper et al. (2009b). Cs + sorption onto bentonite occurs mainly via cation exchange. Cs + sorbs rapidly onto bentonite and achieves equilibrium almost instantaneously (Tsai et al., 2001; Murali and Mathur, 2002; Khan, 2003; Montavon et al., 2006). No important changes in Cs + sorption are observed with pH because Cs + speciation and cation exchange are not pH-dependent. The dependence of Cs + sorption on ionic strength is normally due to an increase of the competition from other cations that exchange preferentially. Therefore, Cs + sorption 1474-7065/$ - see front matter Ó 2010 Elsevier Ltd. All rights reserved. doi:10.1016/j.pce.2010.04.002 * Corresponding author. E-mail address: [email protected] (J. Samper). Physics and Chemistry of the Earth 35 (2010) 278–283 Contents lists available at ScienceDirect Physics and Chemistry of the Earth journal homepage: www.elsevier.com/locate/pce
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Testing K d models of Cs + in the near field of a HLW repository in granite with a reactive...

Physics and Chemistry of the Earth 35 (2010) 278–283
Contents lists available at ScienceDirect
Physics and Chemistry of the Earth
journal homepage: www.elsevier .com/locate /pce
Testing Kd models of Cs+ in the near field of a HLW repository in granitewith a reactive transport model
Javier Samper a,*, Hongyun Ma a, José Luis Cormenzana b, Chuanhe Lu a, Luis Montenegro a,Miguel Ángel Cuñado b
a University of La Coruña, La Coruña 15071, Spainb ENRESA, C Emilio Vargas 7, Madrid 28043, Spain
a r t i c l e i n f o
Article history:Received 26 October 2009Received in revised form 11 March 2010Accepted 10 April 2010Available online 14 May 2010
Keywords:Apparent Kd
CaesiumReactive transport modelHLW repositoryBentoniteThermodynamic sorption model
1474-7065/$ - see front matter � 2010 Elsevier Ltd. Adoi:10.1016/j.pce.2010.04.002
* Corresponding author.E-mail address: [email protected] (J. Samper).
a b s t r a c t
Performance assessment models of high-level radioactive waste (HLW) repositories commonly rely onthe simplifying assumption of a constant Kd for radionuclide sorption. Testing the validity of this assump-tion has been prevented by the lack of nuclide thermodynamic sorption data and the unavailability ofmodels for handling nuclide migration and sorption together with the geochemical evolution of the nearfield. The validity of the constant Kd assumption has been tested with a multicomponent reactive trans-port model (RTM) for Cs+ in the near field of a HLW repository in granite. Model results show that theapparent Kd of Cs+, Ka
d, increases with time due to the decrease of the ionic strength, I, of the bentoniteporewater caused by the out-diffusion of the aqueous species from the bentonite into the granite. Com-puted values of I are related to those of Ka
d through the following polynomial expression:I ¼ 253:5 1
Kad
� �2þ 8:77 1
Kad
� �� 0:005. A thermodynamic justification for such an expression has been
derived from the cation exchange reactions. A constant-Kd model fails to reproduce the release rate ofCs+ from the near field computed with the RTM. A variable-Kd model which incorporates the dependenceof Ka
d on I reproduces adequately the Cs+ release rate, thus providing a surrogate for the constant-Kd
model. The results of the sensitivity runs to changes in model parameters and boundary conditions showthat the water flux at the bentonite–granite interface affects strongly the Ka
d through changes in I whilethe effective diffusion coefficient of the bentonite plays a minor role on Ka
d. An increase in the cationexchange capacity leads to an increase of Ka
d, but it does not affect the time evolution of I. Competing cat-ions such as Ni2+ and iron corrosion products decrease slightly the Ka
d of Cs+ by competing for exchangesites and by increasing the I.
� 2010 Elsevier Ltd. All rights reserved.
1. Introduction
Current performance assessment (PA) models for radionuclidemigration through the near field of a high-level radioactive waste(HLW) repository usually rely on simplifying assumptions suchas the use of a constant Kd for nuclide sorption and ‘‘the limited sol-ubility” for nuclide precipitation. Testing the validity of theseassumptions has been limited by: (1) the lack of nuclide surfacecomplexation and cation exchange data; and (2) the unavailabilityof computer codes which could handle simultaneously the migra-tion, sorption and precipitation of radionuclides and the geochem-ical evolution of the near field. Laboratory experiments performedin recent years have provided substantial data and understandingon the mechanisms of nuclide sorption. On the other hand, sophis-ticated research-oriented process-based models and computercodes have been developed which allow for the simultaneous mod-
ll rights reserved.
elling of migration, sorption, nuclide precipitation and multicom-ponent geochemical evolution of the near field.
In this paper we test the validity of the constant-Kd model fornuclide sorption and precipitation for the 0.75 m thick compactedbentonite barrier of a spent-fuel repository in granite designedaccording to the Spanish reference concept (ENRESA, 2005). Suchtesting has been performed by comparing the results of a PA modelwith those obtained with a reactive transport model performedwith CORE (Samper et al., 2009a). Here we present the results forCs+. The testing for other nuclides is reported by Samper et al.(2009b).
Cs+ sorption onto bentonite occurs mainly via cation exchange.Cs+ sorbs rapidly onto bentonite and achieves equilibrium almostinstantaneously (Tsai et al., 2001; Murali and Mathur, 2002; Khan,2003; Montavon et al., 2006). No important changes in Cs+ sorptionare observed with pH because Cs+ speciation and cation exchangeare not pH-dependent. The dependence of Cs+ sorption on ionicstrength is normally due to an increase of the competition fromother cations that exchange preferentially. Therefore, Cs+ sorption

J. Samper et al. / Physics and Chemistry of the Earth 35 (2010) 278–283 279
decreases when the ionic strength of the solution increases due tothe effect of other cations which compete for exchange positions(Murali and Mathur, 2002). Groundwater composition can affectthe sorption of a cation onto montmorillonite because it deter-mines the aqueous form in which the element is found in solution.Dissolved Cs is usually found as Cs+, except for high Cl� concentra-tions when the concentration of CsCl(aq) may be relevant also. Themost important anion affecting Cs+ sorption is Cl�. Large Cl� con-centrations may decrease Cs+ exchange. The experimental resultsobtained in different studies show that the Kd of Cs+ decreasesfor large concentrations of dissolved Cs+ (Murali and Mathur,2002; Khan, 2003). However, the concentration of Cs+ does not af-fect the Kd when the concentration of exchanged Cs+ is much smal-ler than the cation exchange capacity, CEC.
The paper starts by describing the reference concept and theconceptual and numerical models Then, model results are pre-sented for the geochemical evolution, the base run and the sensi-tivity runs. Competition effects are then evaluated. Finally, theresults of the reactive transport and Kd models are compared.The paper ends with the main conclusions.
2. Conceptual and numerical model
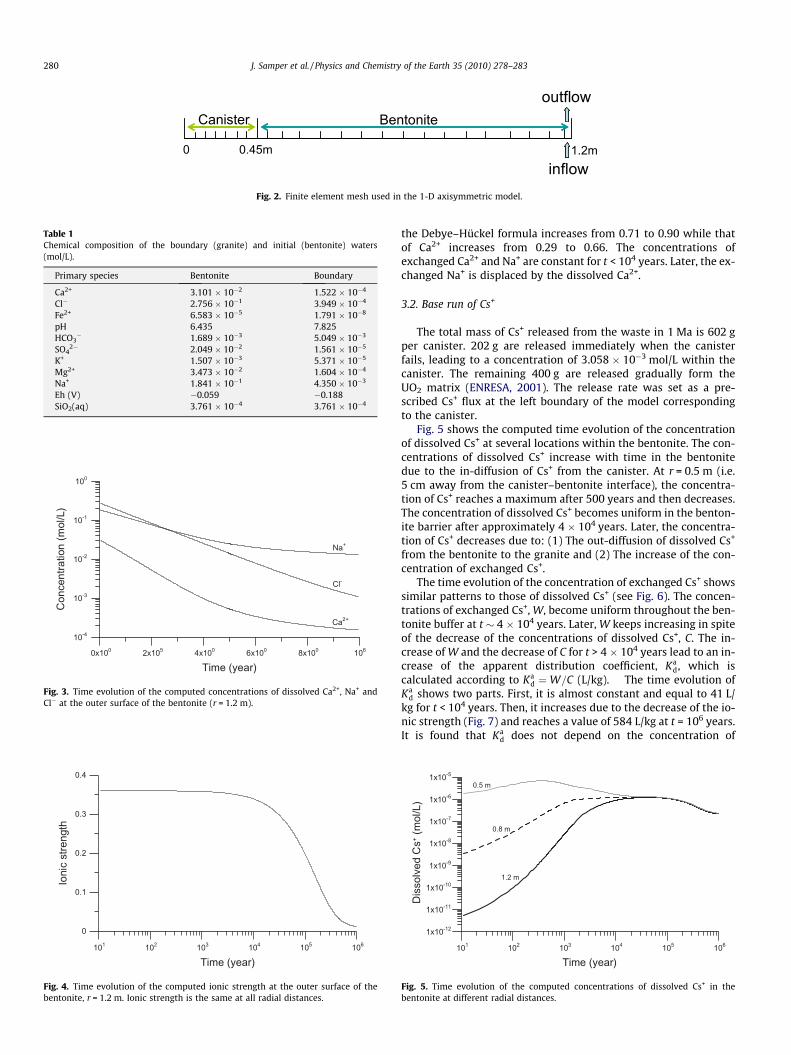
The Spanish reference concept for the disposal of spent-fuel in arepository in a granite formation envisages carbon-steel canistersin 500 m long horizontal drifts excavated at a depth of 500 m inthe granite formation by tunnel boring. Canisters are disposed incylindrical disposal cells built with blocks of compacted bentonitehaving a dry density of 1700 kg/m3 to achieve a final dry density of1600 kg/m3. The blocks are initially unsaturated with a gravimetricwater content of 14%. The disposal drifts have a diameter of 2.4 m.The dimensions of an individual ‘‘cell” corresponding to a singlecanister are shown in Fig. 1 (ENRESA, 2001).
A numerical model has been used to simulate radionuclidemigration after the canister has failed so that water can fill allthe voids within the canister. At this stage of the repository evolu-tion, the engineered barrier is fully saturated and the thermal pulsehas dissipated. Therefore, calculations are performed by assumingisothermal and fully water-saturated conditions. Simulations arerun for 1 Ma with a 1-D axisymmetric finite element grid (seeFig. 2). Selectivity coefficients for exchanged cations and protolysisconstants for surface complexation were taken from Bradbury andBaeyens (2005), Fernández et al. (2001) and Samper et al. (2008a).Porewaters in the canister and bentonite are assumed to have ini-tially the same chemical composition (Samper et al., 2008a) whichis controlled by mineral dissolution/precipitation at chemical equi-librium (calcite and quartz), proton surface complexation and cat-ion exchange. Initial volume fractions of calcite and quartz inbentonite are both equal to 0.01 (Samper et al., 2008a). Gypsum,magnetite, siderite and goethite are minerals which are not ini-tially present in the system, but are allowed to precipitate. Cation
Canister
Bentonite
4.54 m
5.94 m
2.40 m 0.90 m
Fig. 1. Dimensions of an individual disposal cell.
exchange capacity (CEC) in bentonite is 87 meq/100 g. The surfacecomplexation site densities of strong sites and two weak sites inbentonite are equal to 2 � 10�6, 4 � 10�5 and 4 � 10�5 mol/g,respectively. An ‘equivalent flux’ of 0.06 L/year-canister is usedfor the boundary condition at the bentonite/granite interface (seeFig. 2). The chemical composition of inflow water coincides withthat of the initial granite porewater. The initial chemical composi-tions of bentonite and granite porewaters are listed in Table 1. Theconcentrations of most of dissolved species in the bentonite arelarger than those in the granite except for bicarbonate.
Given the low hydraulic conductivity of bentonite, diffusion isthe dominant transport process in bentonite. All dissolved chemi-cal species are assumed to have the same effective diffusion coef-ficient which is equal to 4.07 � 10�11 m2/s. Bentonite porosity isequal to 0.407.
Simulations using the Kd-approach have been performed withGoldSim (Golder Associates, 2007). The reactive transport model(RTM) has been performed with CORE2D, a 2-D Galerkin finite ele-ment multicomponent reactive transport code which solves forgroundwater flow, heat transport and multicomponent reactivesolute transport in saturated/unsaturated steady or transientgroundwater flow under general boundary conditions in 1-, 2- or3-D axisymmetric conditions (Xu et al., 1999). It accounts foracid–base, redox, aqueous complexation, surface adsorption viasurface complexation, cation exchange, mineral dissolution/pre-cipitation, and gas dissolution/ex-solution. Parameters for surfacecomplexation include protolysis constants and site concentrationsfor each type of sorption site. The coupled transport and chemicalequations are solved by the sequential iterative approach (Samperet al., 2009a). CORE2D has been widely used to model laboratoryand in situ experiments performed for HLW disposal (Molineroand Samper, 2006; Samper et al., 2008b; Zheng and Samper,2008; Zheng et al., 2008), to evaluate the long-term geochemicalevolution of radioactive waste repositories in clay (Yang et al.,2008), and to model the transport of corrosion products and theirgeochemical interactions with bentonite (Samper et al., 2008a).
3. Model results
3.1. Geochemical evolution
A reference run without nuclides was performed to evaluate thelong-term time evolution of the chemical conditions in the nearfield. The solute mass flux from the bentonite into the granite orfrom the granite to the bentonite takes place until the concentra-tions attain steady values. Except for HCO3
�, dissolved anionsand cations decrease due to solute diffusion from the bentoniteinto the granite. The concentration of HCO3
� in granite porewateris larger than that of bentonite and therefore it increases with timein the bentonite. pH increases from 6.4 to 7.4 because the pH ofgranite porewater is higher than that of bentonite. Calcite dissolu-tion in the bentonite also contributes to increase the pH. Eh de-creases in the near field due to the equilibration of the redoxpotentials of the bentonite and granite porewaters.
The ionic strength, I, decreases because most ions diffuse outfrom the bentonite into the granite (Fig. 3). At the end of the sim-ulation it reaches a value similar to that of the boundary granitewater (see Fig. 4). The diffusive mass flux from the bentonite intothe granite can be much larger than the advective solute flux atthe boundary. The gradients of the concentrations of dissolved spe-cies in the bentonite are negligible except for Cs+. Therefore, thetime evolution of dissolved concentrations and ionic strength isthe same everywhere in the bentonite barrier.
The activities of aqueous species increase because the ionicstrength decreases. The activity coefficient of Na+ calculated with
Bentonite
1.2m
Canister
0 0.45m
inflow
outflow
Fig. 2. Finite element mesh used in the 1-D axisymmetric model.
Table 1Chemical composition of the boundary (granite) and initial (bentonite) waters(mol/L).
Primary species Bentonite Boundary
Ca2+ 3.101 � 10�2 1.522 � 10�4
Cl� 2.756 � 10�1 3.949 � 10�4
Fe2+ 6.583 � 10�5 1.791 � 10�8
pH 6.435 7.825HCO3
� 1.689 � 10�3 5.049 � 10�3
SO42� 2.049 � 10�2 1.561 � 10�5
K+ 1.507 � 10�3 5.371 � 10�5
Mg2+ 3.473 � 10�2 1.604 � 10�4
Na+ 1.841 � 10�1 4.350 � 10�3
Eh (V) �0.059 �0.188SiO2(aq) 3.761 � 10�4 3.761 � 10�4
0x100 2x105 4x100 6x100 8x100 106
Time (year)
10-4
10-3
10-2
10-1
100
Con
cent
ratio
n (m
ol/L
)
Na+
Cl-
Ca2+
Fig. 3. Time evolution of the computed concentrations of dissolved Ca2+, Na+ andCl� at the outer surface of the bentonite (r = 1.2 m).
101 102 103 104 105 106
Time (year)
0
0.1
0.2
0.3
0.4
Ioni
c st
reng
th
Fig. 4. Time evolution of the computed ionic strength at the outer surface of thebentonite, r = 1.2 m. Ionic strength is the same at all radial distances.
280 J. Samper et al. / Physics and Chemistry of the Earth 35 (2010) 278–283
the Debye–Hückel formula increases from 0.71 to 0.90 while thatof Ca2+ increases from 0.29 to 0.66. The concentrations ofexchanged Ca2+ and Na+ are constant for t < 104 years. Later, the ex-changed Na+ is displaced by the dissolved Ca2+.
3.2. Base run of Cs+
The total mass of Cs+ released from the waste in 1 Ma is 602 gper canister. 202 g are released immediately when the canisterfails, leading to a concentration of 3.058 � 10�3 mol/L within thecanister. The remaining 400 g are released gradually form theUO2 matrix (ENRESA, 2001). The release rate was set as a pre-scribed Cs+ flux at the left boundary of the model correspondingto the canister.
Fig. 5 shows the computed time evolution of the concentrationof dissolved Cs+ at several locations within the bentonite. The con-centrations of dissolved Cs+ increase with time in the bentonitedue to the in-diffusion of Cs+ from the canister. At r = 0.5 m (i.e.5 cm away from the canister–bentonite interface), the concentra-tion of Cs+ reaches a maximum after 500 years and then decreases.The concentration of dissolved Cs+ becomes uniform in the benton-ite barrier after approximately 4 � 104 years. Later, the concentra-tion of Cs+ decreases due to: (1) The out-diffusion of dissolved Cs+
from the bentonite to the granite and (2) The increase of the con-centration of exchanged Cs+.
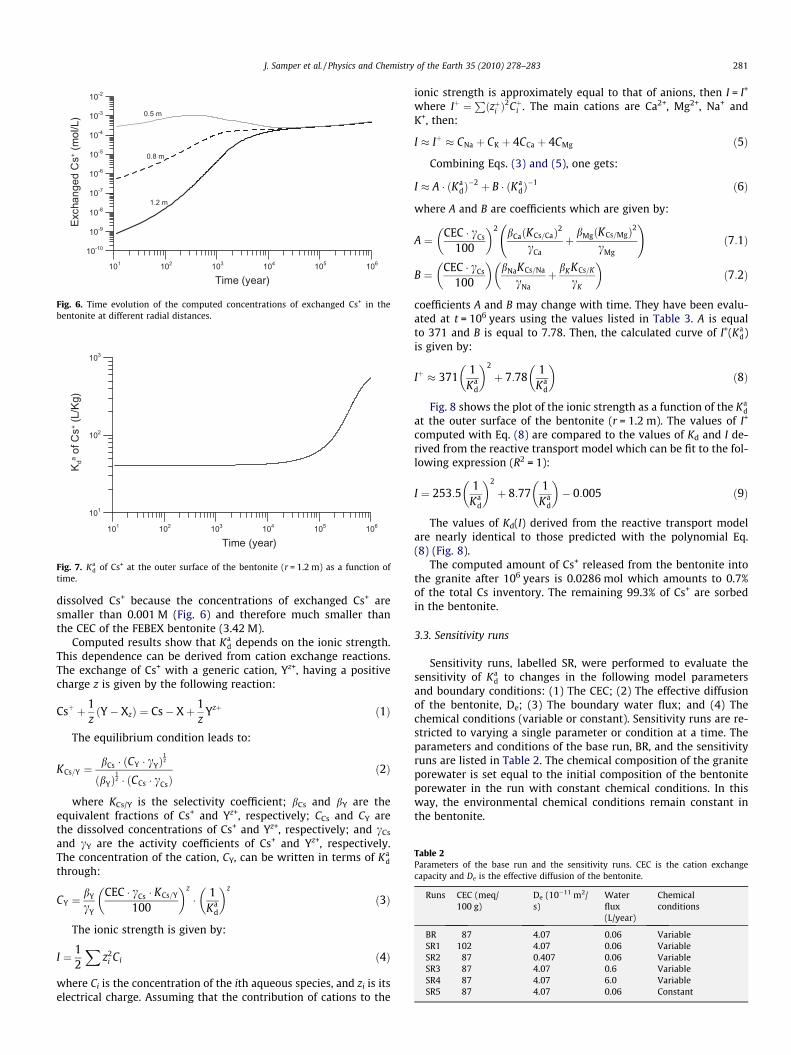
The time evolution of the concentration of exchanged Cs+ showssimilar patterns to those of dissolved Cs+ (see Fig. 6). The concen-trations of exchanged Cs+, W, become uniform throughout the ben-tonite buffer at t � 4 � 104 years. Later, W keeps increasing in spiteof the decrease of the concentrations of dissolved Cs+, C. The in-crease of W and the decrease of C for t > 4 � 104 years lead to an in-crease of the apparent distribution coefficient, Ka
d, which iscalculated according to Ka
d ¼W=C (L/kg). The time evolution ofKa
d shows two parts. First, it is almost constant and equal to 41 L/kg for t < 104 years. Then, it increases due to the decrease of the io-nic strength (Fig. 7) and reaches a value of 584 L/kg at t = 106 years.It is found that Ka
d does not depend on the concentration of
101 102 103 104 105 106
Time (year)
1x10-12
1x10-11
1x10-10
1x10-9
1x10-8
1x10-7
1x10-6
1x10-5
Dis
solv
ed C
s+ (m
ol/L
)
1.2 m
0.8 m
0.5 m
Fig. 5. Time evolution of the computed concentrations of dissolved Cs+ in thebentonite at different radial distances.
101 102 103 104 105 106
Time (year)
10-10
10-9
10-8
10-7
10-6
10-5
10-4
10-3
10-2
Exch
ange
d C
s+ (m
ol/L
)
1.2 m
0.8 m
0.5 m
Fig. 6. Time evolution of the computed concentrations of exchanged Cs+ in thebentonite at different radial distances.
101 102 103 104 105 106
Time (year)
101
102
103
Kda o
f Cs+ (
L/Kg
)
Fig. 7. Kad of Cs+ at the outer surface of the bentonite (r = 1.2 m) as a function of
time.
Table 2Parameters of the base run and the sensitivity runs. CEC is the cation exchangecapacity and De is the effective diffusion of the bentonite.
Runs CEC (meq/100 g)
De (10�11 m2/s)
Waterflux(L/year)
Chemicalconditions
BR 87 4.07 0.06 VariableSR1 102 4.07 0.06 VariableSR2 87 0.407 0.06 VariableSR3 87 4.07 0.6 VariableSR4 87 4.07 6.0 VariableSR5 87 4.07 0.06 Constant
J. Samper et al. / Physics and Chemistry of the Earth 35 (2010) 278–283 281
dissolved Cs+ because the concentrations of exchanged Cs+ aresmaller than 0.001 M (Fig. 6) and therefore much smaller thanthe CEC of the FEBEX bentonite (3.42 M).
Computed results show that Kad depends on the ionic strength.
This dependence can be derived from cation exchange reactions.The exchange of Cs+ with a generic cation, Yz+, having a positivecharge z is given by the following reaction:
Csþ þ 1zðY � XzÞ ¼ Cs� Xþ 1
zYzþ ð1Þ
The equilibrium condition leads to:
KCs=Y ¼bCs � ðCY � cYÞ
1z
ðbYÞ1z � ðCCs � cCsÞ
ð2Þ
where KCs/Y is the selectivity coefficient; bCs and bY are theequivalent fractions of Cs+ and Yz+, respectively; CCs and CY arethe dissolved concentrations of Cs+ and Yz+, respectively; and cCs
and cY are the activity coefficients of Cs+ and Yz+, respectively.The concentration of the cation, CY, can be written in terms of Ka
d
through:
CY ¼bY
cY
CEC � cCs � KCs=Y
100
� �z
� 1Ka
d
� �z
ð3Þ
The ionic strength is given by:
I ¼ 12
Xz2
i Ci ð4Þ
where Ci is the concentration of the ith aqueous species, and zi is itselectrical charge. Assuming that the contribution of cations to the
ionic strength is approximately equal to that of anions, then I = I+
where Iþ ¼Pðzþi Þ
2Cþi . The main cations are Ca2+, Mg2+, Na+ andK+, then:
I � Iþ � CNa þ CK þ 4CCa þ 4CMg ð5Þ
Combining Eqs. (3) and (5), one gets:
I � A � ðKadÞ�2 þ B � ðKa
dÞ�1 ð6Þ
where A and B are coefficients which are given by:
A ¼ CEC � cCs
100
� �2 bCaðKCs=CaÞ2
cCaþ
bMgðKCs=MgÞ2
cMg
!ð7:1Þ
B ¼ CEC � cCs
100
� �bNaKCs=Na
cNaþ bK KCs=K
cK
� �ð7:2Þ
coefficients A and B may change with time. They have been evalu-ated at t = 106 years using the values listed in Table 3. A is equalto 371 and B is equal to 7.78. Then, the calculated curve of I+(Ka
d)is given by:
Iþ � 3711
Kad
� �2
þ 7:781
Kad
� �ð8Þ
Fig. 8 shows the plot of the ionic strength as a function of the Kad
at the outer surface of the bentonite (r = 1.2 m). The values of I+
computed with Eq. (8) are compared to the values of Kd and I de-rived from the reactive transport model which can be fit to the fol-lowing expression (R2 = 1):
I ¼ 253:51
Kad
� �2
þ 8:771
Kad
� �� 0:005 ð9Þ
The values of Kd(I) derived from the reactive transport modelare nearly identical to those predicted with the polynomial Eq.(8) (Fig. 8).
The computed amount of Cs+ released from the bentonite intothe granite after 106 years is 0.0286 mol which amounts to 0.7%of the total Cs inventory. The remaining 99.3% of Cs+ are sorbedin the bentonite.
3.3. Sensitivity runs
Sensitivity runs, labelled SR, were performed to evaluate thesensitivity of Ka
d to changes in the following model parametersand boundary conditions: (1) The CEC; (2) The effective diffusionof the bentonite, De; (3) The boundary water flux; and (4) Thechemical conditions (variable or constant). Sensitivity runs are re-stricted to varying a single parameter or condition at a time. Theparameters and conditions of the base run, BR, and the sensitivityruns are listed in Table 2. The chemical composition of the graniteporewater is set equal to the initial composition of the bentoniteporewater in the run with constant chemical conditions. In thisway, the environmental chemical conditions remain constant inthe bentonite.
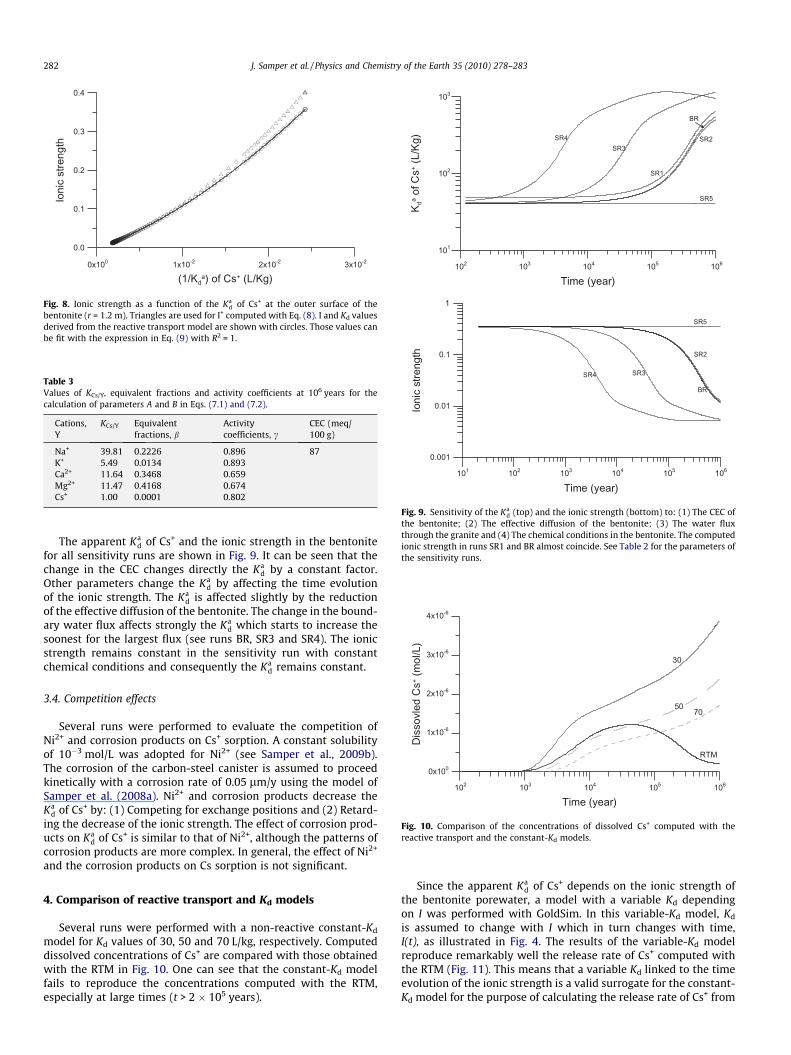
Table 3Values of KCs/Y, equivalent fractions and activity coefficients at 106 years for thecalculation of parameters A and B in Eqs. (7.1) and (7.2).
Cations,Y
KCs/Y Equivalentfractions, b
Activitycoefficients, c
CEC (meq/100 g)
Na+ 39.81 0.2226 0.896 87K+ 5.49 0.0134 0.893Ca2+ 11.64 0.3468 0.659Mg2+ 11.47 0.4168 0.674Cs+ 1.00 0.0001 0.802
0x100 1x10-2 2x10-2 3x10-2
(1/Kda) of Cs+ (L/Kg)
0.0
0.1
0.2
0.3
0.4
Ioni
c st
reng
th
Fig. 8. Ionic strength as a function of the Kad of Cs+ at the outer surface of the
bentonite (r = 1.2 m). Triangles are used for I+ computed with Eq. (8). I and Kd valuesderived from the reactive transport model are shown with circles. Those values canbe fit with the expression in Eq. (9) with R2 = 1.
102 103 104 105 106
Time (year)
101
102
103
Kda o
f Cs+ (
L/Kg
) SR4SR3
SR1
SR2
BR
SR5
101 102 103 104 105 106
Time (year)
0.001
0.01
0.1
1
Ioni
c st
reng
th
SR4 SR3
BR
SR2
SR5
Fig. 9. Sensitivity of the Kad (top) and the ionic strength (bottom) to: (1) The CEC of
the bentonite; (2) The effective diffusion of the bentonite; (3) The water fluxthrough the granite and (4) The chemical conditions in the bentonite. The computedionic strength in runs SR1 and BR almost coincide. See Table 2 for the parameters ofthe sensitivity runs.
102 103 104 105 106
Time (year)
0x100
1x10-6
2x10-6
3x10-6
4x10-6
Dis
sovl
ed C
s+ (m
ol/ L
)
RTM
30
5070
Fig. 10. Comparison of the concentrations of dissolved Cs+ computed with thereactive transport and the constant-Kd models.
282 J. Samper et al. / Physics and Chemistry of the Earth 35 (2010) 278–283
The apparent Kad of Cs+ and the ionic strength in the bentonite
for all sensitivity runs are shown in Fig. 9. It can be seen that thechange in the CEC changes directly the Ka
d by a constant factor.Other parameters change the Ka
d by affecting the time evolutionof the ionic strength. The Ka
d is affected slightly by the reductionof the effective diffusion of the bentonite. The change in the bound-ary water flux affects strongly the Ka
d which starts to increase thesoonest for the largest flux (see runs BR, SR3 and SR4). The ionicstrength remains constant in the sensitivity run with constantchemical conditions and consequently the Ka
d remains constant.
3.4. Competition effects
Several runs were performed to evaluate the competition ofNi2+ and corrosion products on Cs+ sorption. A constant solubilityof 10�3 mol/L was adopted for Ni2+ (see Samper et al., 2009b).The corrosion of the carbon-steel canister is assumed to proceedkinetically with a corrosion rate of 0.05 lm/y using the model ofSamper et al. (2008a). Ni2+ and corrosion products decrease theKa
d of Cs+ by: (1) Competing for exchange positions and (2) Retard-ing the decrease of the ionic strength. The effect of corrosion prod-ucts on Ka
d of Cs+ is similar to that of Ni2+, although the patterns ofcorrosion products are more complex. In general, the effect of Ni2+
and the corrosion products on Cs sorption is not significant.
4. Comparison of reactive transport and Kd models
Several runs were performed with a non-reactive constant-Kd
model for Kd values of 30, 50 and 70 L/kg, respectively. Computeddissolved concentrations of Cs+ are compared with those obtainedwith the RTM in Fig. 10. One can see that the constant-Kd modelfails to reproduce the concentrations computed with the RTM,especially at large times (t > 2 � 105 years).
Since the apparent Kad of Cs+ depends on the ionic strength of
the bentonite porewater, a model with a variable Kd dependingon I was performed with GoldSim. In this variable-Kd model, Kd
is assumed to change with I which in turn changes with time,I(t), as illustrated in Fig. 4. The results of the variable-Kd modelreproduce remarkably well the release rate of Cs+ computed withthe RTM (Fig. 11). This means that a variable Kd linked to the timeevolution of the ionic strength is a valid surrogate for the constant-Kd model for the purpose of calculating the release rate of Cs+ from

0x100 2x105 4x105 6x105 8x105 106
Time (year)
0x100
2x10-8
4x10-8
6x10-8
8x10-8
Cs
rele
ase
rate
from
ben
toni
te (m
ol/y
ear)
Solid line: Variable Kd modelDash line: RTM
Fig. 11. Comparison of the release rates of Cs+ computed with the RTM and a time-variable-Kd model.
J. Samper et al. / Physics and Chemistry of the Earth 35 (2010) 278–283 283
the near field of a spent-fuel repository in granite designed accord-ing to the Spanish reference concept.
The release rate of Cs+ computed with the RTM for constantchemical conditions was compared with that obtained with anon-reactive constant-Kd model for Kd = 40.9 L/kg. The RTM solu-tion suffers from some numerical dispersion errors caused by thesequential iterative approach used by CORE to solve the coupledtransport and chemical equations. Several sensitivity runs wereperformed to evaluate possible ways to decrease such errors. Theydecrease at early times (t < 103 years) when the time incrementsare decreased by a factor of 10. For late times, however, decreasingthe convergence tolerance by a factor of 10 is more efficient inreducing dispersion errors than decreasing the time incrementsby a factor of 10.
5. Conclusions
The validity of the constant Kd assumption for Cs+ in the nearfield of a HLW repository in granite has been tested by using a mul-ticomponent reactive transport model (RTM). Model results showthat the apparent distribution coefficient of Cs+, Ka
d, remains con-stant for t < 104 years and later increases due to the decrease ofthe ionic strength, I, of the bentonite porewater caused by theout-diffusion of the aqueous species from the bentonite into thegranite. It has been found that I is related to Ka
d through:I ¼ 253:5 1
Kad
� �2þ 8:77 1
Kad
� �� 0:005. A thermodynamic justification
for such an expression has been presented.A run with constant chemistry leads to a constant Ka
d. Theapparent Kd does not depend on the concentration of dissolvedCs+ because the concentration of exchanged Cs+ is much smallerthan the concentrations of other exchanged cations in the benton-ite. A constant-Kd model fails to reproduce the release rate of Cs+
from the bentonite into the granite computed with a RTM. A modelwith a variable Kd which depends on the ionic strength of the ben-tonite porewater reproduces adequately the Cs+ release rate. Ka
d isnot sensitive to changes in the effective diffusion of the bentonite.It is slightly sensitive to the CEC. The Ka
d is strongly affected by thewater flux at the bentonite-granite interface because it affectsstrongly the time evolution of ionic strength. Competing cationssuch as Ni2+ and iron corrosion products affect slightly the Ka
d of
Cs+ by competing for exchange sites and increasing the ionicstrength.
Acknowledgements
This work was supported by ENRESA through PAMINA (contract#78000125), the European Union through the PAMINA Project(FP6-036404) and University of A Coruña through a research schol-arship awarded to the fourth author. Partial funding was providedalso by the Spanish Ministry of Science and Technology (ProjectCGL2006-09080). We thank Lara Duro & Cristina Domenech fromAMPHOS XXI (Spain) for providing support on thermodynamicdata and Wilfried Pfinsten from PSI for fruitful discussions on theuse of reactive transport models to quantify nuclide sorption forHLW performance assessment. We are grateful to the two anony-mous reviewers for their comments and recommendations whichimproved the paper.
References
Bradbury, M.H., Baeyens, B., 2005. Modelling the sorption of Mn(II), Co(II), Ni(II),Zn(II), Cd(II), Eu(III), Am(III), Sn(IV), Th(IV), Np(V) and U(VI) onmontmorillonite: linear free energy relationships and estimates of surfacebinding constants for some selected heavy metals and actinides. Geochimica etCosmochimica Acta 69 (4), 875–892.
ENRESA, 2001. ENRESA2000. Evaluación del Comportamiento y de la Seguridad deun Almacenamiento de Combustible Gastado en una Formación Granítica. 49-1PP-M-15-01 Rev.0. Diciembre 2001.
ENRESA, 2005. NF-PRO Project. Phenomenological Description. Reference Concept(Spent Fuel–Carbon Steel Canister–Bentonite–Granite). Deliverable D5.1.1. Part1. Final version.
Fernández, A.M., Cuevas, J., Rivas, P., 2001. Pore water chemistry of the FEBEXbentonite. Materials Research Society Symposium Proceedings 663, 573–588.
Golder Associates, 2007. GoldSim Users Guide. GoldSim Technology Group. <http://www.goldsim.com>.
Khan, S.A., 2003. Sorption of the long-lived radionuclides cesium-134, strotium-85and cobalt-60 on bentonite. Journal of Radioanalytical and Nuclear Chemistry258 (1), 3–6.
Molinero, J., Samper, J., 2006. Modeling of reactive solute transport in fracture zonesof granitic bedrocks. Journal of Contaminant Hydrology 82, 293–318.
Montavon, G., Alhajji, E., Grambow, B., 2006. Study of the interaction of Ni2+ and Cs+
on MX-80 bentonite: effect of compaction using the ‘‘capillary method”.Environmental Science and Technology 40, 4672–4679.
Murali, M.S., Mathur, J.N., 2002. Sorption characteristics of Am(III), Sr(II) and Cs(I)on bentonite and granite. Journal of Radioanalytical and Nuclear Chemistry 254(1), 129–136.
Samper, J., Lu, C., Montenegro, L., 2008a. Coupled hydrogeochemical calculations ofthe interactions of corrosion products and bentonite. Physics and Chemistry ofthe Earth 33 (Suppl. 1), S306–S316.
Samper, J., Zheng, L., Fernández, A.M., Montenegro, L., 2008b. Inverse modeling ofmulticomponent reactive transport through single and dual porosity media.Journal of Contaminant Hydrology 98 (3–4), 115–127.
Samper, J., Xu T., Yang, C., 2009a, A sequential partly iterative approach formulticomponent reactive transport with CORE2D, Computational Geosciences.Doi 10.1007/s10596-008-9119-5.
Samper, J., Lu, C., Cormenzana, J.L., Ma, H., Cuñado, M.A., 2009b, Benchmarkcalculations in granite, Milestone 4.1.2 of PAMINA Integrated Project.
Tsai, S.C., Ouyang, S., Hsu, C.N., 2001. Sorption and diffusion behaviour of Cs and Sron Jih-Hsing bentonite. Applied Radiation and Isotopes 54, 209–215.
Xu, T., Samper, J., Ayora, C., Manzano, M., Custodio, E., 1999. Modeling of non-isothermal multicomponent reactive transport in field scale porous media flowsystems. Journal of Hydrology 214, 144–164.
Yang, C., Samper, J., Montenegro, L., 2008. A coupled non-isothermal reactivetransport model for long-term geochemical evolution of a HLW repository inclay. Environmental Geology 53, 1627–1638. doi:10.1007/s00254-007-0770-2.
Zheng, L., Samper, J., 2008. Coupled THMC model of FEBEX mock-up test. Physicsand Chemistry of the Earth 33, S486–S498. doi:10.1016/j.pce.2008.10.023.
Zheng, L., Samper, J., Montenegro, L., Mayor, J.C., 2008. Flow and reactive transportmodel of a ventilation experiment in Opalinus clay. Physics and Chemistry ofthe Earth 33 (14–16), 1009–1018. doi:10.1016/j.pce.2008.05.012.




















