
Tesis DTP Leticia Melo Lopez Nov 25 2016.pdf
165
-
Upload
khangminh22 -
Category
Documents
-
view
3 -
download
0
Transcript of Tesis DTP Leticia Melo Lopez Nov 25 2016.pdf



CENTRO DE INVESTIGACIÓN EN QUÍMICA APLICADA
TESIS
“Desarrollo de Membranas poliméricas sulfonadas de poli(estireno-co-ácido acrílico) y su
modificación con partículas de platino para su aplicación en celdas de combustible”
Presentado por:
Leticia Melo López
Para obtener el Grado de:
DOCTOR EN TECNOLOGÍA DE POLÍMEROS
Asesor: Dr. Roberto Benavides Cantú
Co-asesor: Dr. Juan Guillermo Martínez Colunga
Saltillo, Coahuila 30 de Junio del 2016



Agradecimientos Agradezco a mi mami y mi hermana Susy que siempre hayan estado al pendiente de mí, que
me han demostrado cada día que me tienen en su pensamiento y en su corazón a pesar de la
distancia. A Cris por brindarme su amistad incondicional desde hace 17 años sin que el tiempo
la diluya. A los compañeros y amigos que hicieron más agradable mi estadía en el CIQA y en
Saltillo.
Gracias al CONACYT y al CIQA por haberme dado la oportunidad de continuar con mi
formación académica en el área de investigación científica, pero que a su vez permitieron mi
crecimiento en el aspecto personal. Agradezco al Dr. Roberto Benavides Cantú y al Dr. Juan
Guillermo Martínez Colunga por su asesoría brindada durante la realización de la tesis. De
igual manera doy las gracias a la Dra. Diana Morales Acosta por su disponibilidad para
orientarme en el vasto conocimiento del área electroquímica y por su amistad. Gracias a los
sinodales; Dr. Mario Humberto Gutiérrez Villarreal, Dra. Leticia Larios López, Dr. Héctor
Iván Meléndez Ortiz, Dra. Tatiana Romero Castañón y Dr. Edgar Jocsan Ruiz Ruiz, por sus
observaciones y sugerencias.
Durante estos años también recibí el apoyo y ayuda para la caracterización de los materiales
obtenidos, a cada uno de ellos le agradezco; Ing. Adolfo López Ángeles, I. Q. Adán Herrera
Guerrero, Q. F. B. Bertha Alicia Puente Urbina, M. C. Blanca Margarita Huerta Martínez,
L. C. Q. Jesús Alejandro Espinosa Muñoz, Q. F. B. Jesús Ángel Cepeda Garza, Tec. Juan
Francisco Zendejo Rodríguez, L. C. Q. Luis Enrique Reyes Vielma, T. L. Q. Marcelina
Sánchez Adame, L. Q. Marcelo Israel Ulloa Pérez, M. C. María del Rosario Rangel Ramírez,
Lic. María Guadalupe Méndez Padilla, M. C. María Luisa López Quintanilla, Lic. Maricela
García Zamora, Q. F. B. Myriam Lozano Estrada, M. C. Silvia Torres Rincón, C. P. Irma
Imelda Vargas García, Lic. Nancy Guadalupe Espinosa Pinales y a la M. C. María Concepción
González Cantú que además de su ayuda en el laboratorio me brindó su amistad.

RESUMEN
i
RESUMEN
Se sintetizaron copolímeros al azar de estireno-co-ácido acrílico mediante reacciones
radicálicas en solución, con una relación molar de 94:6 (St:AA). Se experimentó adicionando
y variando la concentración de un agente entrecruzante: divinil benceno (DVB) o
trimetacrilato de trimetilol propano (TMPTMA), para obtener copolímeros parcialmente
reticulados. Los copolímeros entrecruzados con las mejores propiedades para formar película
se sulfonaron empleando por separado tres agentes sulfonantes: ácido sulfúrico, acetil sulfato
y ácido sulfúrico con sulfato de plata. Los mejores copolímeros sulfonados se utilizaron para
preparar membranas mediante disolución-evaporación con diferentes disolventes para obtener
membranas optimizadas en sus propiedades fisicoquímicas. Una vez estandarizado el proceso
de disolución-evaporación, las membranas fueron sometidas al proceso de reducción
transmembrana para depositar platino en una de sus caras, con la intención de uso posterior
como catalizador del combustible en una celda. Los copolímeros fueron caracterizados por
mediante TGA, DSC, GPC, DMA, IR y RMN 1H, y los cambios en los copolímeros debidos al
proceso de sulfonación fueron corroborados mediante la determinación de insolubles por
extracción soxhlet, GPC, IR, DSC, TGA y grado de sulfonación. Las membranas preparadas
fueron caracterizadas determinando su IEC, absorción de agua, relación de hinchamiento,
permeación de metanol y morfología mediante SEM. El proceso de formación del platino
mediante la reducción transmembrana en su superficie se evaluó con espectrofotometría
UV-VIS, DRX, cuantificación de platino por espectrometría de emisión de plasma, IEC,
absorción de agua, relación de hinchamiento, permeación de combustible y SEM. Los
copolímeros parcialmente entrecruzados sintetizados mostraron pesos moleculares de hasta
260,000 g/mol cuando se usa DVB y hasta de 303,000 g/mol con el TMPTMA, con
temperaturas de descomposición (Td) y transición vítrea (Tg) mejoradas. Los resultados de IR
y de RMN 1H muestran que los dos comonómeros están presentes en el copolímero y que la
relación molar de 94:6 (St:AA) alimentada en el reactor varía dependiendo del agente
entrecruzante empleado, debido al cambio en sus reactividades ocasionadas por la naturaleza
del medio de reacción. Los espectros de IR muestran la incorporación de grupos sulfónicos en
los copolímeros tratados con los 3 agentes sulfonantes, sin embargo, utilizando solo ácido
sulfúrico se generan grupos sulfonas que entrecruzan el material, volviéndolo insoluble y
aumentando su peso molecular. Por otro lado, con sulfato de acetilo el peso molecular de los

RESUMEN
ii
copolímeros tratados se disminuye y aumenta su solubilidad en agua. La opción del ácido
sulfúrico con sulfato de plata permite obtener membranas sulfonadas de propiedades
mecánicas convenientes para formar membranas. La mezcla de disolventes THF/DMSO
permitió obtener membranas mediante disolución-evaporación con propiedades
electroquímicas mejoradas, obteniéndose altos niveles de absorción de agua y valores de
capacidad de intercambio iónico (IEC) del orden de los que presenta una membrana de
referencia (Nafion), lo cual se adjudica a la porosidad observada por microscopía electrónica
de barrido, pero sin presentarse permeación de metanol (combustible). El método de reducción
transmembrana fue efectivo para formar partículas de platino metálico en una de las caras de
las membranas, con efectos pequeños en las propiedades fisicoquímicas evaluadas antes de
este procedimiento. De manera general, es factible preparar membranas de copolímeros
sulfonados económicos parcialmente entrecruzados mediante copolimerización radicálica en
solución. Estas membranas, debidamente preparadas mediante disolución-evaporación con
mezcla de disolventes, consiguen propiedades de intercambio iónico similar al Nafion y
además factibles de ser incorporadas con un catalizador metálico para su uso en una celda de
combustible.

INTRODUCCIÓN
iii
INTRODUCCIÓN
Las celdas de combustible son dispositivos electroquímicos que transforman directamente la
energía química de una reacción en energía eléctrica. Para que esto suceda, la celda de
combustible debe contar con un electrolito entre los electrodos, el ánodo y el cátodo. Los
compartimentos anódico y catódico (electrodos negativo y positivo) de la celda se alimentan
continuamente con el combustible y el oxidante respectivamente, lo que origina que las
reacciones electroquímicas se lleven a cabo en los electrodos para producir la corriente
eléctrica y una especie química con carga iónica que se difunde por el electrolito. La especie
iónica y su dirección de transporte varía dependiendo del tipo de celda de combustible, el ion
puede ser positivo o negativo, y también el sitio donde se genera el agua (subproducto) puede
variar de celda a celda. El punto donde convergen los reactivos (combustible u oxidante), el
electrolito y el catalizador es la frontera triple de fases, y la naturaleza de ésta interface es
primordial en el desempeño electroquímico en la celda de combustible. La función catalítica
de los electrodos es más importante en celdas de combustible de baja temperatura en
comparación con las celdas de alta temperatura porque las velocidades de las reacciones de
ionización incrementan con la temperatura.
Las celdas de combustible pueden ser clasificadas por la combinación del tipo de combustible
y oxidante, por el tipo de electrolito, por la temperatura de operación, etc. La clasificación más
común es por el tipo de electrolito: 1) celdas de combustible de membrana polimérica
intercambiadora de protones (proton exchange membrane fuel cell –PEMFC–), 2) celda de
combustible alcalina (alkaline fuel cell –AFC–), 3) celda de combustible de ácido fosfórico
(phosphoric acid fuel cell –PAFC–), 4) celda de combustible de carbonato fundido (molten
carbonate fuel cell –MCFC–) y 5) celda de combustible de óxido sólido (solid oxide fuel cell –
SOFC–). Las celdas mencionadas estan enlistadas en orden de temperatura de operación,
siendo de ~80 °C para PEMFC, ~100 °C para AFC, ~200 °C para PAFC, ~650 °C para MCFC
y de 800 °C a 1000 °C para SOFC. De acuerdo con la temperatura de operación, se selecciona
el tipo de combustible que se puede utilizar en ellas.
En las PEMFC, el electrolito es una membrana sólida de polímero con capacidad de
intercambiar protones, que por su estado físico tiene la ventaja de facilitar el cierre herméico
del dispositivo electroquímico, reducuendo los problemas de corrosión causados por el
electrolito y prolongando el tiempo de vida útil de la celda. La baja temperatura de operación

INTRODUCCIÓN
iv
requerida permite que el encendido de este tipo de celdas sea rápido y de respuesta inmediata a
los cambios de demanda energética. Sin embargo, éste tipo de celdas también presenta puntos
débiles, entre ellos la distribución del agua en la membranas para el buen desempeño de la
PEMFC. Las condiciones de operación deben permitir que el agua generada como
subproducto se evapore a menor velocidad a la que se forma, para que la membrana se
mantenga hidratada, así que para evitar problemas con el balance del agua la temperatura de
operación debe ser menor a 100 °C [1].
Una membrana polímérica conductora de protones puede ser empleada en celdas que operen
con hidrógeno o metanol, por lo cual los requerimientos de la membrana dependerán de la
aplicación a la cual sea destinada. Actualmente se encuentran comercialmente disponibles las
membranas de Nafion y membranas similares de ácido perfluorosulfónico, estas membranas
son durables y de buen desempeño, pero sus inconvenientes son su precio, su inhabilidad para
operar a temperaturas alrededor de los 120 °C por baja humedad [2] y la elevada permeación a
metanol debida a la diferencia de concentración que existe entre los compartimentos anódico y
catódico [3]. Debido a estos inconvenientes, existen otros materiales conductores de iones bajo
investigación, como los copolímeros de poliestireno sulfonado en di y tribloque, derivados de
estireno como el α-metilestireno, el α,β,β-trifluoroestireno, poli(p-fenilenos) sulfonados,
poliimidas sulfonadas, polifosfacenos sulfonados, polibenzimidazoles sulfonados, poli(éter
éter cetona) sulfonada, la poli(arileno éter sulfona), etc. [4-10].
En éste trabajo de tesis las alternativas a estudiar como electrolito polimérico son los
copolímeros de poliestireno-co-poliácido acrílico entrecruzados (con divinilbenceno o
trimetacrilato de timetilol propano) y posteriormente sulfonados. El poliácido acrílico
comúnmente es utilizado como agente espesante, adhesivo, dispersante en pinturas, floculante
de residuos para clarificación de aguas, pero también se utiliza en resinas de intercambio
iónico reticuladas [11]. Además de que los polímeros iónicos que contienen segmentos de
ácido acrílico y un monómero no polar combinan características atractivas como la formación
de regiones hidrófobas e hidrófilas [12]. Por otro lado el grupo sulfónico es un grupo ácido
fuerte, que puede proporcionarle conductividad protónica al copolímero.
En este trabajo se describe la obtención de membranas intercambiadoras de protones de
poliestireno-co-ácido acrílico entrecruzado con DVB o TMPTMA y sulfonados, para su uso

INTRODUCCIÓN
v
en celdas de combustible, procurando que sean térmicamente estables, impermeables a
metanol, con capacidad de absorción de agua y capacidad de intercambio iónico.
En el primer capítulo se describe la polimerización radicálica en solución de los copolímeros
de PS-PAA, realizada en dietilbenceno como medio de reacción para mantener un mejor
control de la temperatura y viscosidad de la reacción. Se describe el proceso de selección de
concentraciones de peróxido de benzoílo y de agentes entrecruzantes para obtener copolímeros
de peso molecular que permita formar películas mediante disolución-evaporación (casting).
En el capítulo dos se describe el proceso de sulfonación de los copolímeros de PS-PAA a
diferentes tiempos de reacción y variando la concentracion de tres diferentes sistemas
sulfonantes: H2SO4, CH3COOSO3H o H2SO4-Ag2SO4. En donde se encontró que el mejor
sistema sulfonante es el H2SO4 con la sal de plata porque se reduce el entrecruzamiento de las
cadenas poliméricas por formación de sulfonas.
El tercer capítulo describe el proceso para la preparación de las membranas con los
copolímeros sulfonados y la evaluación de su capacidad de intercambio iónico, absorción de
agua, relación de hinchamiento, grado de sulfonación y permeación de metanol.
El proceso de reducción transmembrana es descrito en el cuarto capítulo, con el cual se
modificaron algunas membranas con la formación de platino metálico directamente sobre su
superficie, comprobando la factibilidad de incorporar el catalizador sin la necesidad de
emplear un soporte adicional para su preparación previa, así como tampoco fue necesaria la
preparación de una tinta catalítica y la optimización de un método para su aplicación.
Se llegó a la obtención de membranas intercambiadoras de protones y su modificación
mediante la incorporación de platino metálico en su superficie, sin embargo son necesarias
evaluaciones adicionales de conducción protónica, permeación de H2(g) y O2(g), resistencia
mecánica ante la preparación del ensamble de la membrana en una celda, etc., para conocer su
competitividad total frente a las membranas de Nafion, las cuales son las líderes en el
mercado.

HIPÓTESIS
vi
HIPÓTESIS
La síntesis de copolímeros al azar de estireno con ácido acrílico puede prepararse mediante
reacciones de tipo radicálico los cuales pueden ser entrecruzados para mejorar su estabilidad
mecánica, química y térmica. Además pueden ser sulfonados para incorporar grupos ácidos
fácilmente disociables.
Por lo anterior es posible obtener electrolitos poliméricos de estireno con ácido acrílico
entrecruzados y sulfonados para preparar membranas intercambiadoras de protones para
celdas de combustible (metanol).

OBJETIVOS
vii
OBJETIVOS
OBJETIVO GENERAL
Sintetizar copolímeros de poli(estireno-co-ácido acrílico) entrecruzados y sulfonados, para la
preparación de membranas para celdas de combustible (metanol), así como su modificación
superficial con un catalizador de platino.
OBJETIVOS PARTICULARES
1) Evaluar el efecto del iniciador BPO y los entrecruzantes DVB y TMPTMA en la
síntesis de copolímeros de St-AA para formar película mediante disolución-evaporación.
2) Determinar el agente sulfonante y las condiciones de reacción para la sulfonación de
los copolímeros St-AA. Que permita obtener membranas insolubles en agua y solubles en
THF, con capacidad de intercambio iónico y retención de agua; además de baja permeación de
metanol.
3) Sintetizar partículas de platino metálico directamente sobre la membrana sulfonada, las
cuales actúen como catalizadores de las reaccioens electroquímicas.

ÍNDICE
viii
ÍNDICE
Resumen…...…………………………………………………………………………………..i Introducción…..……………………………………………………………………………...iii Hipótesis…….…..………………………………………………………………………........vi Objetivo General…………..………………………………….……………………………..vii Objetivos Particulares……………………………………….……………………………...vii CAPÍTULO 1. SÍNTESIS DE COPOLÍMEROS PS-PAA ENTRECRUZADOS 1.1 Antecedentes………………………………………………………………………………1 1.2 Experimental………………………………………………………………………............3 1.2.1 Materiales………………………………………………………………………............3 1.2.2 Metodología……………………………………………………………………............4 1.2.2.1 Preparación y purificación de reactivos…………………………………………….4 1.2.2.2 Reacciones de copolimerización……………….…………………………………...4 1.2.2.3 Elección del tiempo de reacción de copolimerización………………………...……5 1.2.2.4 Preparación de películas…………………………………………………………....6 1.2.3 Caracterización………………………………………………………………...............6 1.2.3.1 Análisis termogravimétrico …………………………………………………...6 1.2.3.2 Calorimetría diferencial de barrido………………………………………………....7 1.2.3.3 Cromatografía de permeación en gel…………………………………………….…7 1.2.3.4 Análisis dinámico mecánico…………………………………………………….….7 1.2.3.5 Espectroscopía infrarrojo………………………………………………………...…7 1.2.3.6 Resonancia magnética nuclear de protón…………………………………………...7 1.3 Resultados y discusión…………………………………………………………………….8 1.3.1 Elección del tiempo de reacción de copolimerización y
escalamiento…………………………………………………………………………..8 1.3.2 Efecto del peso molecular en el desempeño fisicoqímico de los
copolímeros……………………..……………………………………………………..9 1.3.3 Estabilidad térmica de los copolímeros……………………...……………………….12 1.3.4 Calorimetría diferencial de barrido…………………………………………...………15 1.3.5 Análisis dinámico mecánico………………………………………………………….19 1.3.6 Espectroscopía infrarrojo……………………………………………………………..19 1.3.7 Resonancia magnética nuclear de protón………………………………………….….20 1.4 Conclusiones del capítulo 1…………………………………………………………..….27 CAPÍTULO 2. SULFONACIÓN DE COPOLÍMEROS DE PS-PAA ENTRECRUZADOS 2.1. Antecedentes…………………………………………..………………………………...28 2.2. Experimental……………………………………………….……………………………29 2.2.1 Materiales……………………………………………………………………………..29 2.2.2 Preparación de reactivos…………..………………………………………………….29 2.2.2.1 Sulfato de acetilo………………...………………………………………………...30 2.2.2.2 Sulfato de plata………………………….………………………………………...31 2.2.3 Sulfonación del copolímero…………………………...……………………………...31 2.2.4 Caracterización……………………………………………………………………….32 2.2.4.1 Determinación de la capacidad de intercambio iónico………………………...….32

ÍNDICE
ix
2.2.4.2 Determinación de material insoluble mediante extracción soxhlet……………….33 2.2.4.3 Cromatografía de permeación en gel..…………………………………………….33 2.2.4.4 Espectroscopía infrarrojo……………..…………………………………………...33 2.2.4.5 Calorimetría diferencial de barrido………………………………………………..34 2.2.4.6 Análisis termogravimétrico………………………………………………………..34 2.3 Resultados y discusión…………………………………………………………………...34 2.3.1 Solubilidad y desempeño físico y iónico de los copolímeros sulfonados …………..35 2.3.2 Determinación de insolubles mediante extracción soxhlet…………………………...41 2.3.3 Efecto del peso molecular en el desempeño ficicoquímico de los copolímeros
sulfonados……………………………………………………………………………..43 2.3.4 Espectroscopía infrarrojo…………………………………………………………….49 2.3.5 Calorimetría diferencial de barrido…………………………………………………..55 2.3.6 Estabilidad térmica de los copolímeros sulfonados………………………………….57 2.4 Conclusiones del capítulo 2……………………………………………………………..60 CAPÍTULO 3. MEMBRANAS 3.1 Antecedentes…………………………………………………………………………….61 3.2. Experimental…………………………………………………………………………….63 3.2.1 Materiales…………………………………………………………………………….63 3.2.2 Preparación de las membranas……………………………………………………….63 3.2.3 Activación de las membranas………………………………………………………...64 3.2.4 Caracterización……………………………………………………………………….64 3.2.4.1 Determinación de disolvente residual en las membranas…………………………64 3.2.4.2 Capacidad de intercambio iónico de las membranas……………………………..64 3.2.4.3 Absorción de agua de las membranas…………………………………………….65 3.2.4.4 Relación de hinchamiento de las membranas…………………………………….65 3.2.4.5 Caracterización electroquímica de las membranas……………………………….65 3.2.4.5.1 Fabricación de la celda de difusión……………………………………………65 3.2.4.5.2 Limpieza del electrodo de trabajo…………………………………………….66 3.2.4.5.2.1 Pulido mecánico del electrodo de trabajo………………………………….66 3.2.4.5.2.2 Pulido electroquímico del electrodo de trabajo…………………………….67 3.2.4.5.3 Curva de calibración para la determinación de permeación de metanol a través
de las membranas………………………………………………………………67 3.2.4.5.4 Determinación de la permeación de combustible a través de las membranas…74 3.2.4.6 Microscopía electrónica de barrido de las membranas……………………………75 3.2.4.7 Análisis termomecánico de las membranas……………………………………….76 3.2.4.8 Grado de sulfonación de las membranas………………………………………….76 3.3 Resultados y discusión…………………………………………………………………..77 3.3.1 Determinación del disolvente residual en las membranas……………………………77 3.3.2 Capacidad de intercambio iónico de las membranas…………………………………78 3.3.3 Absorción de agua de las membranas………………………………………………...85 3.3.4 Relación de hinchamiento de las membranas………………………………………..88 3.3.5 Permeación de combustible a través de las membranas……………………………...91 3.3.5.1 Determinación del coeficiente de difusión de la membrana de nafion 117………91 3.3.5.2 Determinación del coeficiente de difusión de las membranas preparadas con los
copolímeros sulfonados…………………………………………… …………….93

ÍNDICE
x
3.3.6 Microscopía electrónica de barrido de las membranas……………………………...100 3.3.7 Análisis termomecánico de las membranas…………………………………………105 3.3.8 Grado de sulfonación de las membranas……………………………………………110 3.4 Conclusiones del capítulo 3…………………………………………………………….112 CAPÍTULO 4. MODIFICACIÓN DE MEMBRANAS CON PLATINO 4.1 Antecedentes……………………………………………………………………………113 4.2 Experimental……………………………………………………………………………115 4.2.1 Materiales……………………………………………………………………………115 4.2.2 Incorporación de platino en las membranas…………………………………………116 4.2.3 Espectrofotometría UV-Vis…………………………………………………………117 4.2.4 Difracción de rayos-X……………………………………………………………….117 4.2.5 Digestión ácida y cuantificación del platino en las membranas por espectrometría de
emisión de plasma………………………………………………………………….117 4.3 Resultados y discusión…………………………………………………………………119 4.3.1 Espectrofotometría UV-Vis…………………………………………………………119 4.3.2 Difracción de rayos-X……………………………………………………………….120 4.3.3 Cuantificación del platino en las membranas por espectrometría de emisión de
plasma……………………………………………………………………………….121 4.3.4 Capacidad de intercambio iónico……………………………………………………123 4.3.5 Absorción de agua…………………………………………………………………..124 4.3.6 Relación de hinchamiento…………………………………………………………..124 4.3.7 Permeación de combustible…………………………………………………………125 4.3.8 Microscopía electrónica de barrido…………………………………………………126 4.4 Conclusiones del capítulo 4……………………………………………………………132 Conclusiones finales………………………………………………………………………..133 Referencias………………………………………………………………………………….135 Lista de abreviaciones……………………………………………………………………..148

CAPÍTULO 1
1
CAPÍTULO 1. SÍNTESIS DE COPOLÍMEROS PS-PAA ENTRECRUZADOS
1.1 ANTECEDENTES
El empleo de copolímeros estirénicos sulfonados como polielectrolitos fue visualizado en
2009 por Chee y Gan. Ellos analizaron copolímeros sulfonados de poliestireno (PS) y
poliácido acrílico (PAA), encontrando que a mayor cantidad de grupos carboxílicos en el
copolímero, mayor cantidad de agua absorbe el material aumentando su hinchamiento, debido
a que el poliácido acrílico es más hidrófilo que el poliestireno sulfonado [13]. La absorción de
agua es una característica favorable, tomando en cuenta que una de las desventajas a superar
del Nafion es su deshidratación debido a la naturaleza química hidrófoba de su cadena
principal, a pesar de contener grupos sulfónicos en las cadenas laterales.
Es necesario que las membranas poliméricas a utilizar en celdas de combustible sean afines al
agua y que mantengan la humedad, sin embargo el agua absorbida merma las propiedades
mecánicas de la membrana porque actúa como plastificante, disminuyendo la temperatura de
transición vítrea (Tg) y el módulo (la razón de un componente de esfuerzo a un componente de
deformación) de la membrana [10]. Además, el carácter hidrófilo no es exclusivo del poliácido
acrílico, sino que los grupos sulfónicos también lo presentan, inclusive conforme aumenta el
grado de sulfonación de un polímero, éste se vuelve cada vez más soluble en agua [14].
Cuando se tiene un 100 % de los anillos aromáticos sulfonados, el polímero es totalmente
soluble [15], lo cual no es una característica deseable para un material que se desea utilizar
como membrana que estará en contacto constante con agua. El cambio de las propiedades
mecánicas por la presencia de los grupos hidrófilos puede ser compensado reduciendo la
movilidad molecular del material mediante el entrecruzamiento de las cadenas poliméricas con
la formación de redes [16]. Pudiéndose lograr con la inclusión de un agente entrecruzante,
además de que para membranas poliméricas, el entrecruzamiento evita el cambio del área
superficial y el espesor de las membranas en estado hidratado [17].
Cuando se utiliza un agente entrecruzante en la síntesis de polímeros, cuando el grado de
entrecruzamiento es bajo, su efecto se refleja como un incremento del peso molecular de las
cadenas de polímero. Si se emplea una cantidad de agente entrecruzante elevada, las cadenas
poliméricas formarán una red entrecruzada insoluble que únicamente se hincharía al estar en

CAPÍTULO 1
2
contacto con un disolvente [18]. Los agregados de cadenas poliméricas entrecruzadas se
denominan geles e influencian en el comportamiento del polímero durante su procesado [19].
Por ello, durante el diseño y síntesis de un polímero iónico para formar una membrana que
incluye entrecruzamientos químicos, debe existir un balance entre la cantidad de grupos
iónicos y el grado de entrecruzamiento, para tener las mejores propiedades químicas y
mecánicas de la membrana [20].
Para la síntesis de los copolímeros de poliestireno-co-poliácido acrílico de este trabajo se
utilizaron dos agentes entrecruzantes, con diferente número de enlaces activos: el divinil
benceno (DVB) que es un entrecruzante bifuncional y el trimetacrilato de trimetilolpropano
(TMPTMA) que es trifuncional. Estos agentes entrecruzantes le otorgan diferentes
características a las membranas poliméricas, por ejemplo, un copolímero de estireno
sintetizado con un 5 % de TMPTMA es más flexible que uno obtenido con el mismo
porcentaje de DVB [21].
A la fecha se utilizan membranas de estireno-divinilbenceno para la electrodiálisis, para su
obtención se polimeriza estireno (St) con divinilbenceno y posteriormetne el polímero se
sulfona o amina para formar las membranas de intercambio catiónico o aniónico [22]. Las
membranas de intercambio catiónico de copolímeros de St y DVB sulfonados son útiles en la
separación de iones en solución acuosa [23], pudiendo ser selectivas ante diferentes iones de la
misma carga variando el contenido de entrecruzamiento de la membrana [22].
Chee y Gan [13] han realizado reacciones de copolimerización de estireno con ácido acrílico,
a los cuales les incorporaron DVB para obtener copolímeros entrecruzados, que
posteriormente sometieron a reacciones de sulfonación. A partir de estas reacciones
obtuvieron materiales que absorben agua con una característica en común, a mayor grado de
entrecruzamiento menor capacidad de absorción de agua.
El DVB posee en su estructura química dos dobles enlaces polimerizables, cuando cada uno
de ellos se incorpora en dos cadenas poliméricas diferentes se genera su entrecruzamiento.
Cuando los polímeros tienen baja densidad de entrecruzamiento, mayor capacidad de
hinchamiento tienen, sin embargo en partículas de polímero sinteizadas por polimerización en
emulsión el hinchamiento también está en función de la densidad del polímero y el área
superficial de las partículas [13].

CAPÍTULO 1
3
Las resinas poliméricas entrecruzadas más comunes y comercialmente disponibles son de
carácter aromático y de St-DVB, pero cuando se requieren polímeros entrecruzados de tamaño
de poro grande, se utiliza el TMPTMA como agente entrecruzante. Por ello los polímeros
porosos de TMPTMA son útiles como absorbentes en medios acuosos y no acuosos, además
cuando se les incorporan grupos funcionales iónicos en su estructura polimérica, sirven como
resinas intercambiadoras de iones. No obstante, la porosidad del polímero también depende
del tipo de disolvente empleado [24].
Las polimerizaciones radicálicas pueden llevarse a cabo en medio heterogéneo u homogéneo,
entre las últimas se encuentran las reacciones en masa y en solución. Las polimerizaciones en
masa son exotérmicas y la energía de activación en combinación con la presencia del efecto
gel dificulta la disipación del calor. Para poder controlarlas se requieren equipos robustos de
agitación que soporten el rápido incremento de la viscosidad del medio de reacción a
conversiones relativamente bajas para poder disipar la temperatura de la reacción. De lo
contrario, la viscosidad y el efecto exotérmico pueden generar regiones localizadas de mayor
temperatura dentro del medio de reacción, que originen la degradación del polímero. El
incremento de la temperatura aumenta la concentración de radicales libres y la velocidad de la
reacción, además de disminuir el peso molecular promedio. En casos extremos, la rápida
aceleración de la velocidad de polimerización puede resultar en reacciones exotérmicas
descontroladas que ocasionan explosiones [11, 25].
Las dificultades mencionadas en las polimerizaciones en masa se pueden evitar empleando un
disolvente como medio de reacción, lo cual se conoce como polimerización en solución. El
disolvente transfiere el calor de la polimerización y disminuye la viscosidad de la mezcla de
reacción, permitiendo emplear equipos de agitación menos robustos. Además de que se
obtienen pesos moleculares mayores, debido a que la velocidad de terminación bimolecular
está controlada por la difusión, por lo que la constante de terminación es inversamente
proporcional a la viscosidad del medio (disolvente + monómeros) [26].
1.2 EXPERIMENTAL
1.2.1 MATERIALES
Acetona (CTR Scientific), ácido acrílico (99 %, Aldrich), cloroformo deuterado 99.8 %
átomos de D (Sigma Aldrich), cloruro de calcio (J.T. Baker), dietilbenceno (Aldrich),

CAPÍTULO 1
4
divinilbenceno (Aldrich), estireno (99 %, Aldrich), fenotiazina (Sigma aldrich), hidróxido de
sodio (Aldrich), metanol (CTR Scientific), peróxido de benzoílo (Aldrich), tetrahidrofurano
libre de inhibidor ≥ 99.9 % (Aldrich), triclorometano (Sigma Aldrich), trimetacrilato de
trimetil propano (Aldrich).
1.2.2 METODOLOGÍA
1.2.2.1 PREPARACIÓN Y PURIFICACIÓN DE REACTIVOS
El estireno se purificó lavándolo con una solución acuosa de hidróxido de sodio (NaOH) al
20 % en peso y posteriormente con agua destilada. Se dejó en contacto con cloruro de calcio
durante toda la noche y al día siguiente se destiló a presión reducida [27]. El ácido acrílico se
puso en agitación con fenotiazina y se destiló a presión reducida [28]. El iniciador peróxido de
benzoílo (BPO) se disolvió en triclorometano a temperatura ambiente, se precipitó
adicionando a la solución un volumen igual de metanol, los cristales formados se filtraron y se
secaron con vacío a temperatura ambiente durante 24 horas [27]. El St, AA y BPO, una vez
purificados se almacenaron en frío hasta su uso. El metanol se destiló de forma convencional.
1.2.2.2 REACCIONES DE COPOLIMERIZACIÓN
Los copolímeros de PS-PAA se prepararon haciendo reaccionar 94 unidades molares de St y
6 unidades de AA, tomando como base estudios previos de reacciones de copolimerización de
estos monómeros con diferentes relaciones molares (90:10, 92:08 y 94:06), siendo el
copolímero sintetizado con la relación entre monómeros 94:06 el de estabilidad química
superior durante la reacción de sulfonación con ácido sulfúrico concentrado [29, 30].
Las reacciones se llevaron a cabo mediante polimerización radicálica en solución, utilizando
dietilbenceno (DEB) como disolvente (empleando un volumen de disolvente por cada
volumen de monómeros), BPO como iniciador y DVB o TMPTMA como agente
entrecruzante. Las reacciones se llevaron a cabo en un reactor de vidrio, con agitación
mecánica a 200 rpm, atmósfera de nitrógeno y temperatura de reacción de 90 °C, para
descartar la iniciación térmica de la polimerización de los monómeros [31, 32]. Se utilizó una
corriente de nitrógeno para desplazar al oxígeno del interior del reactor, y evitar la activación
o desactivación el iniciador y/o la degradación oxidativa de las cadenas poliméricas por la
presencia de oxígeno [33]. Se armó el reactor, se conectó el baño de recirculación, se

CAPÍTULO 1
5
adicionó el disolvente y se mantuvo flujo de N2 durante todo el procedimiento. Una vez que
el DEB alcanzó los 90 °C, se adicionó la mezcla de reacción (monómeros, entrecruzante e
iniciador disuelto) e inmediantamente después se inició la agitación. Una vez que la mezcla
de reacción recuperó los 90 °C se empezó a contar el tiempo de reacción. Para detener la
reacción y precipitar el copolímero se adicionó metanol al reactor [34, 35]. El proceso de
purificación de cada copolímero se realizó disoviendolo en acetona, precipitándolo en
metanol, redisolviéndolo en tetrahidrofurano (THF) y precipitándolo nuevamente en metanol,
asi como colocándolo a vacio por 48 h a 65-70 °C para secarlo.
1.2.2.3 ELECCIÓN DEL TIEMPO DE REACCIÓN DE COPOLIMERIZACIÓN
La determinación del tiempo de reacción para obtener los copolímeros entrecruzados con
DVB o TMPTMA se llevó a cabo a partir de una reacción de 5 horas empleando St, AA,
0.175 % mol de BPO y 0.15 % mol de DVB. Durante la reacción se tomaron alícuotas de
aproximadamente 1 mL a diferentes intervalos de tiempo, cada una se pesó inmediatamente
después de ser extraída y se le adicionó metanol, se purificó disolviéndolo en acetona y THF
por separado y precipitándolo en metanol [35, 36]. El polímero se filtró, se dejó a temperatura
y presión ambiente durante toda la noche, se secó a 80 °C y presión reducida por 6 horas y se
pesó. Los cálculos de la conversión de cada muestra se hicieron tomando en cuenta que la
alimentación del reactor fue de 94 moles de St por cada 6 moles de AA, por lo que la relación
entre monómeros puede indicarse de la siguiente manera:
94 mol St = 10805.8499 mL = 9790.1 g
6 mol AA = 411.428571 mL = 432 g
Monómeros totales = 11217.278471 mL = 10222.1 g
Además por cada volumen de monómeros se adicionó un volumen de disolvente
DEB = 11217.278471 mL = 10095.5506 g
Mezcla de reacción = 22434.556942 mL = 20317.6506 g
En la mezcla de reacción existe disolvente, monómeros y/o polímero. Por tratarse de una
polimerización por adición, si se tuviera un 100 % de conversión, todos los átomos de los
monómeros estarían incorporados en el polímero formado, sin generación de
subproductos [11]. Si el peso gravimétrico del polímero equivale al peso gravimétrico de los
monómeros, la conversión se puede calcular relacionando el peso del polímero con la cantidad

CAPÍTULO 1
6
de monómeros iniciales presentes en cada muestra (ecuación 1.1).
%푐표푛푣푒푟푠푖ó푛 = ( í )( )( ó )
(ec.1.1)
Para conocer el peso de los monómeros en cada muestra, se multiplicó el peso de la alícuota
por el factor (ecuación 1.2) que involucra la relación gravimétrica existente entre los
monómeros iniciales y el total de la mezcla de reacción (ecuación 1.3).
푓푎푐푡표푟 = . ó . ó
(ec.1.2)
%푐표푛푣푒푟푠푖ó푛 = ( í )( )( í )( )
(ec.1.3)
De esa manera se llevó a cabo el procedimiento y cálculo de la conversión en cada muestra
para determinar el tiempo máximo de reacción para las reacciones subsecuentes.
1.2.2.4 PREPARACIÓN DE PELÍCULAS
La selección de los copolímeros de PS-PAA a sulfonar posteriormente se basó tomando en
cuenta que éstos deben formar membranas, las cuales permitan el paso del ión móvil y que
además sen impermeables y resistentes a la presión del combustible y del oxidante (líquido y/o
gas) para evitar su combinación. La preparación de las películas poliméricas depende del tipo
de polímero y disolvente a emplear y del arreglo morfológico que se quiera obtener [37]. La
obtención de membranas para celdas de combustible comúnmente se realiza mediante el
proceso de evaporación de disolvente, o el llamado casting [37-40]. Este proceso consiste en
formar una solución homogénea con el polímero y un disovente que se vierte en un molde, en
el cual se forma la película. Las películas de los copolímeros de PS-PAA se prepararon
mezclando 0.4 g de material con 2 mL de THF, la solución se vertió en un molde de 16 cm2 y
el disolvente se dejó evaporar a temperatura y presión ambiente.
1.2.3 CARACTERIZACIÓN
1.2.3.1 ANÁLISIS TERMOGRAVIMÉTRICO
Para estudiar la estabilidad térmica de los copolímeros, un aproximado de 4.5 mg de cada
muestra fue evaluada en un Analizador Termogravimétrico modelo Q500 de TA Instruments,

CAPÍTULO 1
7
a una velocidad de calentamietno de 10 °C/min desde temperatura ambiente hasta 600 °C bajo
atmósfera de gas nitrógeno (N2).
1.2.3.2 CALORIMETRÍA DIFERENCIAL DE BARRIDO
La temperatura de transición vítrea de los copolímeros se determinó mediante calorimetría
diferencial de barrido, utilizando un DSC modelo 2920 (TA Instruments) empleando una
velocidad de calentamiento de 10 °C/min.
1.2.3.3 CROMATOGRAFÍA DE PERMEACIÓN EN GEL
El peso molecular de los copolímeros se determinó por Cromatografía de Permeación en Gel
(GPC). Se utilizó un cromatógrafo Hewlett Packard con detector de Índice de Refracción,
equipado con 3 columnas de Ultrastyragel (105, 104 y 103 Å), calibrado con estándares de
poliestireno, la velocidad de elución utilizada fue de 1 mL/min a 40 °C. Las soluciones de los
copolímeros se prepararon a una concentración de 1 mg/mL en THF y se pasaron por filtros
de 0.2 µm.
1.2.3.4 ANÁLISIS DINÁMICO MECÁNICO
El comportamiento mecánico de las películas de los copolímeros se determinó en un
analizador dinámico mecánico modelo Q800 de TA Instruments, utilizando el modo de
tensión, una rampa de calentamiento de 2 °C/min y 1 Hz de frecuencia.
1.2.3.5 ESPECTROSCOPÍA INFRARROJO
Para corroborar la incorporación de las unidades de AA en los copolímeros en forma de
película se empleó un espectroscopio de infrarrojo Nicolet modelo Avatar 316, empleando
32 scan y resolución de 4 cm-1. Con el software OMNIC 5.2 se hizo una corrección
automática de la línea base y se normalizaron los espectros tomando como base la señal que
se encuentra en 1602 cm-1, la cual corresponde a la tensión C=C del anillo aromático [41],
para la posterior comparación cualitativa y semicuantitativa de las bandas.
1.2.3.6 RESONANCIA MAGNÉTICA NUCLEAR DE PROTÓN
La relación molar entre las unidades estirénicas y las de ácido acrílico en los copolímeros se

CAPÍTULO 1
8
determinó por resonancia magnética nuclear de protón (RMN 1H) con un equipo Jeol operado
a 300 MHz a partir de soluciones preparadas con 5 mg de copolímero y 0.7 ml de cloroformo
deuterado (CDCl3).
1.3 RESULTADOS Y DISCUSIÓN
1.3.1 ELECCIÓN DEL TIEMPO DE REACCIÓN DE COPOLIMERIZACIÓN Y
ESCALAMIENTO
A partir de la copolimerización de St con AA (DVB 0.15 % mol y BPO 0.175 % mol) llevada
a cabo durante 5 horas, se calculó la conversión a diferentes tiempos (Figura 1.1).
Figura 1.1. Gráfica de conversión contra tiempo de reacción para determinar el intervalo óptimo de tiempo a emplear en los demás experimentos.
En la Figura 1.1 se observa que la conversión aumenta durante los primeros 90 min de
reacción y a partir de los 125 min se vuelve asintótica, por lo que decidió que las siguientes
reacciones de copolimerización se llevaron a cabo durante 120 minutos.
Las copolimerizaciones realizadas para la optimización de la concentración de iniciador y de
DVB se llevaron a cabo en un reactor con capacidad de 100 mL, sus productos se
caracterizaron mediante DSC, TGA, GPC y DMA y una vez seleccionado el mejor
copolímero, se preparó un lote mayor. Mientras que para la optimización de la concentración
de TMPTMA todas las reacciones se llevaron a cabo en cantidad suficiente para ser utilizado
en las reacciones de sulfonación sin necesidad de preparar más copolímero.
0 50 100 150 200 250 3000
10
20
30
40
Con
vers
ión
(%)
Tiempo de reacción (min)

CAPÍTULO 1
9
1.3.2 EFECTO DEL PESO MOLECULAR EN EL DESEMPEÑO FISICOQÍMICO DE
LOS COPOLÍMEROS
Para relacionar el comportamietno físico de los copolímeros sintetizados con su estructura
química, se hizo el seguimiento de sus pesos moleculares mediante GPC, en la Tabla 1.1 se
indica la cantidad de iniciador y de agente entrecruzante empleado en la síntesis de cada uno
de ellos y su peso molecular. El primer copolímero se sintetizó con 0.3 % mol de BPO y no
contenía agente entrecruzante (copolímero A), al preparar su película y una vez que el
disolvente se evaporó, la película se fracturó. Su resultado de GPC indicó que su peso
molecular promedio en peso (Mw) es de 44,923 g/mol, valor que corresponde a un polímero
quebradizo, según lo reportado en la literatura para poliestirenos con Mw menor a
150,000 g/mol [42].
Tabla 1.1. Copolímeros de PS-PAA sintetizados con diferente concentración de reactivos, comportamiento físico y peso molecular
Reacción BPO % mol DVB % mol
Película Mw (g/mol) Rendimientos (%)
A 0.300 0 NO 44,923 33.1 B 0.300 0.05 NO 41,509 30.8 C 0.300 0.10 NO 47,658 33.9 D 0.300 0.30 Frágil 138,606 37.2 L 0.300 0.35 NO (insoluble) - 40.0 E 0.175 0.15 Frágil 100,371 33.4 F 0.050 0 Frágil 93,662 35.1 G 0.050 0.05 Frágil 87,155 35.9 H 0.050 0.20 SI 156,048 44.8 I 0.045 0.25 SI 182,799 43.6 J 0.045 0.30 SI 245,590 57.4 K 0.050 0.30 SI 248,100 58.3
La fragilidad de un polímero se puede reducir incrementando su peso molecular [43] y para
lograrlo se pueden emplear agentes entrecruzantes procurando evitar la formación de redes de
cadenas poliméricas insolubles [18]. Para incrementar el peso molecular y la resistencia
mecánica del copolímero A (Tabla 1.1), se utilizaron diferentes concentraciones de agente
entrecruzante (DVB): 0.05, 0.10, 0.30 y 0.35 % molar (copolímeros B, C, D y L de la
Tabla 1.1). Después de su purificación y secado, los copolímeros se caracterizaron por GPC y
se siguió el procedimiento para formar película con ellos. Emplear 0.05 y 0.10 % molar de

CAPÍTULO 1
10
DVB generó materiales que no formaron película y sus Mw fueron de 41,509 y 47,658 g/mol,
valores que también se encuentran por debajo e los 150,000 g/mol, lo cual explica su
fragilidad. Inclusive está reportado que el PS comercial que se emplea en moldeo y extrusión
debe tener un Mw cercano a los 180,000 g/mol [42].
El copolímero sintetizado con 0.30 % molar de DVB y de BPO cuyo Mw es de 138,606 g/mol
(copolímero D de la Tabla 1.1) formó una película mediante disolución-evaporación, pero se
fracturó al desmoldarla, a pesar de que su valor de peso molecular duplica al de los
copolímeros previamente sintetizados (copolímeros A, B y C). Utilizar 0.35 % mol de DVB
(reación L de la Tabla 1.1) generó un polímero insoluble, inapropiado tanto para llevar a cabo
el proceso de disolución-evaporación como para la determinación de su peso molecular. No
obstante, la síntesis del copolímero L sirvió para determinar que el nivel máximo utilizable de
agente entrecruzante en conjunto con 0.3 % mol de BPO, es 0.3 % mol de DVB. Los
copolímeros de las reacciones A, B, C, D y L se descartaron por no formar película, pero su
caracterización por GPC (excepto L) se utilizó para evaluar el efecto del DVB en el peso
molecular de los materiales.
Otra estrategia para obtener un copolímero de mayor Mw en una polimerización radicálica
consiste en disminuir la cantidad de radicales del medio de reacción para aumentar la relación
monómeros/radicales activos (monómeros/iniciador) [11, 44].
Los copolímeros F y G mostraron un incremento del peso molecular; 93,662 y 87,155 g/mol
respecto a sus análogos preparados con más iniciador (copolímeros A y B), no obstante
tampoco formaron película. El efecto de incremento de peso molecular sucede debido a que
en las reacciones de polimerización radicálica, en teroría la reacción de propagación podría
proceder indefinidamente hasta agotar todo el monómero en el medio de reacción; siempre y
cuando no ocurrieran las reacciones de terminación. Las constantes típicas de velocidad de las
reacciones de terminación (kt) se encuentran en el intervalo de 106 – 108 L mol-1 s-1, que son
varios órdenes de magnitud mayor a las constantes de velocidad de las reacciones de
propagación (kp = 102–104 L mol-1 s-1). A pesar de que el valor de la kt es mucho mayor que
la kp, la propagación y el crecimiento de las cadenas sucede porque las especies radicálicas se
encuentran en bajas concentraciones y estadísticamente es más probable que encuentren un
monómero para seguir creciendo, en comparación a la probabilidad de que se encuentren
entre ellos para dar lugar a una terminación bimolecular. Además, la velocidad de

CAPÍTULO 1
11
polimerización es directamente proporcional a la kp, e inversamente proporcional a la kt pero
ésta última se encuentra elevada a la ½ [11].
Al emplear las dos estrategias para incrementar el peso molecular fue posible lograrlo, sin
embargo los copolímeros obtenidos no formaron película. Razón por la que se sintetizaron los
copolímeros E, H, I, J y K de la Tabla 1.1 con menor concentración de BPO y mayor cantidad
de DVB. En la Figura 1.2 se muestra el efecto de la concentración del BPO y del DVB
empleados en las síntesis respecto al peso molecular de los copolímeros obtenidos. Los
copolímeros H, I, J y K formaron películas que no se fracturaban, comprobando lo reportado
en la literatura; que el incremento del peso molecular de los copolímeros aumenta su
estabilidad mecánica [45].
Además se puede mencionar que en las reacciones con mayor concentración de BPO
(radicales activos) se llega a un límite máximo utilizable de DVB antes de que se obtenga un
copolímero insoluble. Esto se puede deber a que el DVB reacciona 3.54 veces más rápido que
el St al utilizar BPO a la temperatura de 90 ± 0.3 °C [46]. Este consumo acelerado de DVB
daría lugar al incremento de entrecruzamientos entre cadenas poliméricas generando
materilese insolubles, como sucede con la reacción L.
De las películas preparadas con los copolímeros H, I, J y K se analizó su comportamiento
mecánico para seleccionar el mejor copolímero a utilizar para las reacciones de sulfonación
en la siguiente etapa, la sulfonación.
Figura 1.2. Efecto de la concentración de BPO y DVB en el peso molecular de los copolímeros. Los círculos grises indican los copolímeros que formaron película, los círculos blancos indican los copolímeros que no formaron película o es muy frágil, la línea gris indica las concentraciones empleadas para el copolímero L.

CAPÍTULO 1
12
Del análisis mecánico (DMA) se determinó que emplear 0.045 % mol de BPO y 0.25 % mol
de DVB generó el mejor copolímero (reacción I) cuyo peso molecular fue de 182,799 g/ mol.
Utilizando las mismas condiciones de síntesis se repitió la reacción pero en mayor cantidad,
obteniéndose un copolímero de peso molecular Mw = 259,095 g/mol. El cambio en el Mw,
en sus Tg y en el inicio de la Td de los copolímeros obtenidos al emplear volúmenes y
reactores diferentes (100 mL y 3 L), fue el resultado de la variación en el tiempo de contacto
entre reactivos, la rampa de calentamiento y el incremento de la temperatura que alcanzó la
mezcla de reacción.
Para evaluar el efecto del agente entrecruzante trifuncional TMPTMA se emplearon seis
porcentajes molares: 0.250, 0.270, 0.300, 0.3125, 0.375 y 0.500 en conjunto con
0.045 % molar de BPO. Cinco copolímeros resultaron entrecruzados e insolubles y un
copolímero soluble con Mw de 302,607 g/mol, el cual formó película que no se fracturó. Este
copolímeros se sintetizó con 0.045 % molar de BPO y 0.25 % molar de TMPTMA y fue el
que se seleccionó para las reacciones de sulfonación.
1.3.3 ESTABILIDAD TÉRMICA DE LOS COPOLÍMEROS
Una celda de combustible de membrana polimérica opera a una temperatura menor a los
100 °C, utilizando hidrógeno gaseoso (H2) o metanol como combustible y catalizadores de
platino [47- 49]. Esta temperatura de operación es una ventaja porque la celda puede empezar
a funcionar rápidamente en condiciones de ambiente, siempre y cuando el H2 sea puro y no
exista permeación de metanol. Si el H2 contiene impurezas como el monóxido de carbono o
cuando el metanol alcanza el cátodo, los electrodos de platino se envenenán debido a que el
carbón (del CO o del CH3OH) se une a los sitios catalíticos reduciendo el desempeño de la
celda. Para disminuir el envenenamiento ocasionado por la quimisorción y electrooxidación de
dichos compuestos sobre el platino se puede operar la celda de combustible a una temperatura
mayor a los 120 °C [48], sin embargo eso afectaría el contendo de agua además de que el
polímero de la membrana debe ser estable a esa temperatura. Para conocer la estabilidad de los
copolímeros de PS-PAA entrecruzados con DVB y TMPTMA, se realizó su análisis
termogravimétrico (TGA). En la Figura 1.3a se encuentran los termogramas correspondientes,
en ellos se observa que existe una primera pérdida de masa que inicia alrededor de los 100 °C,
la cual puede ser a causa de la volatilización de humedad y/o disolvente residual [37, 50-52].

CAPÍTULO 1
13
A esa temperatura el copolímero entrecruzado con TMPTMA pierde mayor cantidad de masa
en comparación a los copolímeros entrecruzados con DVB sintetizados con las mismas
condicones. Una posible explicación a esa diferencia es el acomodo molecular y estructura de
las cadenas poliméricas de esos materiales. Se sabe que los polímeros entrecruzados con
TMPTMA forman poros y/o canales de mayor dimensión en comparación con los de los
materiales entrecruzados con DVB, inclusive los polímeros porosos de TMPTMA son útiles
como absorbentes [24]. La siguiente pérdida de masa que se observa en la Figura 1.3a
corresponde a la temperatura de descomposición (Td) de las muestras, la cual comienza cerca
de los 315 °C como consecuencia de la vaporización inmediata de los compuestos que se
forman por la degradación térmica [53]. La presencia de una sola pérdida de peso mayoritaria
indica que las unidades de ácido acrílico se encuentran dispersas homogéneamente en todo el
copolímero y no en forma de estructuras tipo bloques. Este resultado concuerda con el
esperado tomando en cuenta que en una copolimerización los monómeros tienden a la
alternación si el producto de sus constantes de reactividad se acerca al valor de cero [11], tal y
como sucede con el producto de las relaciones de reactividad para St y el AA reportado en la
literatura (r1r2 = 0.01 y 0.15) [25, 54-56].
Figura 1.3. Td de los copolímeros PS-PAA obtenidas de TGA. a) termogramas sobrepuestos, b) termogramas presentados en forma escalonada.
En la Figura 1.3b, se muestra los termogramas graficados de forma escalonada (no
sobrepuestos) y en orden ascendente para evitar que las curvas se solapen, y de esta forma
mostrar con mayor claridad la temperatura en la cual existe un cambio abrupto en su pendiente
debido a la pérdida mayoritaria de masa de los materiales. La temperatura de degradación se
100 200 300 4000
50
60
70
80
90
100a)
Copolímeros PS-PAA entrecruzados con: DVB: A - K (excepto I) DVB: I DVB: I-escalado TMPTMA
Pérd
ida
de m
asa
(%)
Temperatura (° C) 240 280 320 360 400
Pé
rdid
a de
mas
a (%
) DVB-Iescalado 323.2° C
DVB-
Temperatura (°C)
DVB-DVB-DVB-DVB-DVB-DVB-DVB-DVB-DVB-DVB-
TMPTMA 328.10° C
A 315.56° CB 316.51° CC 318.35° CD 318.69° CE 317.67° CF 314.20° CG 316.72° CH 317.87° CI 319.30° CJ 324.75° CK 327.96° C
DVB-
10%
b)

CAPÍTULO 1
14
encuentra en el intervalo de 314 a 329 °C, la cual es intermedia entre las temperaturas de
degradación del poliestireno puro y del poliácido acrílico. La degradación térmica del PS puro
puede variar entre los 350 °C a los 450 °C dependiendo de la técnica empleada para su
síntesis [57], mientras que el poliácido acrílico se degrada a una temperatura de 300 °C [58].
La degradación térmica involucra rompimiento de enlaces carbono-carbono de la cadena
principal, rompimiento de enlaces carbono-hidrógeno, recombinación radicálica y reacciones
de desproporcionación entre radicales. Los productos de descomposición del PS son los
trímeros, dímeros, monómeros de estireno y productos de bajo peso molecular como el
tolueno, alilbenceno, difenil metano, difenil etano, 1,2-difenil propano, 1,3-difenil propano,
1,4-difenil butadieno y 1,4-difenil butileno [59, 60]. Mientras que la degradación térmica de
los grupos carboxílicos del PAA inicia con su deshidratación y posteriormente se forman
anhídridos entre grupos carboxílicos cercanos [58, 61].
Comparando el inicio de la Td de los tres copolímeros sintetizados con 0.045 % mol de BPO y
0.25 % mol de agente entrecruzante: copolímero I (319.30 °C), copolímero I escalado
(323.2 °C) y el entrecruzado con TMPTMA (328.10º C), se observa que el último es
térmicamene más estable. Este resultado puede deberse a que el TMPTMA es un agente
trifuncional mientras que el DVB es bifuncional, por lo que a pesar de utilizar la misma
cantidad molar de cada uno de ellos en las síntesis, la cantidad de cadenas que puede
entrecruzar el TMPTMA es 50 % más que al usar DVB en la misma cantidad molar. Esta
mayor cantidad de cadenas restringidas en movimiento dificulta la propagación de los
radicales que se generan durante la descoposición térmica, generando mayor estabilidad
térmica.
La diferencia de temperatura de inicio de descomposición entre el copolímero I escalado con
respecto al original puede deberse a sus diferencias en peso molecular
(259,095 vs 182,799 g/mol), tomando en cuenta que los copolímeros consisten principalmente
de unidades de estireno. La descomposición térmica del PS inicia con la formación de
radicales libres en los enlaces que se encuentran al final de las cadenas, los cuales se propagan
hasta afectar la totalidad de la matriz polimérica [62]. El copolímero I escalado (de mayor
peso molecular), será térmicamente mas estable porque necesitará más energía para que los
radicales libres afecten la totalidad de las cadenas, en comparación de lo que sucede con las
cadenas del copolímero I original, cuyas cadenas de menor tamaño se ven afectadas a menor

CAPÍTULO 1
15
temperatura.
En la Figura 1.4 se aprecia la relación de variación de los valores de inicio de la Td de los
copolímeros respecto a la cantidad de reactivos empleados para su síntesis.
Figura 1.4. Comparación de la tendencia que existe entre las Td de los copolímeros de PS-PAA entrecruzados con DVB.
A mayor cantidad de agente entrecruzante, la Td se desplaza a temperaturas mayores,
probablemente como consecuencia de la movilidad reducida de las cadenas poliméricas,
tendencia que concuerda con lo reportado en la literatura [62].
Cabe destacar que en la Figura 1.4 no se incorporó el dato de la temperatura de
descomposición del material sintetizado a escala con las condiciones del copolímero I, debido
a que existieron diferencias en el desarrollo de la reacción de copolimerización, y su valor no
sería representativo dentro de este gráfico.
1.3.4 CALORIMETRÍA DIFERENCIAL DE BARRIDO
Los resultados del análisis de calorimetría diferencial de barrido (DSC) de los copolímeros de
PS-PAA entrecruzados con DVB se muestran en la Figura 1.5. Los copolímeros A y F,
sintetizados sin entrecruzante (Figuras 1.5a y 1.5b) presentan las Tg de menor valor,
probablemente porque el movimiento de cadenas no está restringido por el entrecruzamiento.
Inclusive sus Tg están por debajo de la temperatura de trabajo óptima de una celda de
combustible, por lo que en caso de que hubieran formado película, ésta podría perder su
estabilidad dimensional al estar dentro de una celda en funcionamiento.

CAPÍTULO 1
16
Figura 1.5. Termogramas de DSC de copolímeros de PS-PAA con diferente cantidad de DVB. a) nivel alto de BPO (0.30 % mol), b) nivel bajo de BPO (0.050 y 0.045 % mol*).
En la figura 15 se compararan los termogramas de los copolímeros sintetizados con con la
misma cantidad de iniciador (0.30, 0.045 o 0.050 % molar) y variando la cantidad de DVB.
Se aprecia que la Tg se desplaza a temperaturas mayores con el incremento de la
concentración de agente entrecruzante. Este efecto ya ha sido reportado en la literatura por
Li y col. [63] quienes indicaron que para poliestirenos polimerizados radicálicamente con
BPO y entrecruzados con DVB la Tg incrementa con el entrecruzamiento. Sin embargo esto
se cumple siempre y cuando la cantidad de DVB empleada sea menor al 30 % en peso
(60 % mol de DVB respecto al St), donde las uniones entre las cadenas aún permiten su
flexión y reacomodo con el incremento de la temperatura.
Ahora bien, cuando se hace una variación de la cantidad de BPO, manteniendo constante la
concentración de DVB (Figura 1.6), también existen cambios en la Tg. A mayor cantidad de
BPO empleada, la Tg de los materiales se desplaza a temperaturas menores; aunque el
desplazamiento no es tan drástico como el observado al incrementar la concentración de
DVB.
40 60 80 100 120 140 160
DVB
D 0.30%
C 0.10%
B 0.05%
A 0.00%
a
107.8 °C
112.4 °C
113.10 °C
Fluj
o de
cal
or (W
/g)
Temperatura (°C)
93.59 °C
BPO = 0.3% mol ex
o
40 60 80 100 120 140 160
BPO = 0.050% mol 0.045% mol *
F 0.00%
G 0.05%
**
b
119.2 °C
119.4 °C
Fluj
o de
cal
or (W
/g)
Temperatura (°C)
111.9°C 120.4 °C
118.8 °C
91.8°C
H 0.20%I 0.25%
J 0.30%
K 0.30%
**
DVB
exo

CAPÍTULO 1
17
Figura 1.6. Termogramas de DSC de copolímeros de PS-PAA con diferente cantidad de BPO. Concentración de DVB: a) 0.05 % mol y b) 0.30 % mol.
Una vez discutido el efecto de la variación de la concetración de iniciador y de agente
entrecruzante por separado, se visualiza mejor la tendencia general que existe en la variación
de las Tg de los copolímeros y que se muestra en la Figura 1.7. En dicha Figura también
seincluye la reacción E (BPO = 0.175 % mol y DVB = 0.150 % mol) con Tg de 116.9 °C, que
se realizó para corroborar la tendencia descrita.
Figura 1.7. Efecto del BPO y DVB en la Tg de los copolímeros de PS-PAA.
Después de encontrar las condiciones de síntesis para el mejor copolímero entrecruzado con
DVB, se llevó a cabo la búsqueda de las condiciones de síntesis de un copolímero
entrecruzado con TMPTMA con capacidad de formar película. Las condiciones de síntesis
del mejor copolímero entrecruzado con TMPTMA son las mismas que las empleadas para
40 60 80 100 120 140 160
DVB = 0.05% mol
a
Fluj
o de
cal
or (W
/g)
Temperatura (°C)
107.8 °C
111.9 °CB 0.300%
G 0.050%
BPO
exo
40 60 80 100 120 140 160
DVB = 0.30% mol
D 0.300%
b
Fluj
o de
cal
or (W
/g)
Temperatura (°C)
K 118.8 °C
J 119.2 °C
D 113.1 °C
K 0.050%
J 0.045%
BPO
exo

CAPÍTULO 1
18
obtener el copolímero I entrecruzado con DVB (0.045 % mol de BPO y 0.25 % mol de agente
entrecruzante). En la Figura 1.8 se muestra el comparativo entre los termogramas de los tres
copolímeros mencionados: copolímero I, copolímero I-escalado y el entrecruzado con
TMPTMA.
Figura 1.8. Termogramas obtenidas por DSC de los copolímeros PS-PAA.
El copolímero entrecruzado con TMPTMA presenta su Tg a mayor temperatura, lo cual
puede estar relacionado con la cantidad de entrecruzamientos químicos que el TMPTMA
genera en comparación con la misma cantidad molar del DVB, a consecuencia de su
trifuncionalidad. Las cadenas de los copolímeros entrecruzados con DVB tienene mayor
libertad de movimiento por lo que necesitan menos temperatura para acomodarse. Además, la
Tg del copolímero I-escalado (121.9 °C) es mayor a la del copolímero I inicial (119.4 °C), lo
cual puede relacionarse a sus pesos moleculares (259,095 vs 182,799 g/mol) tomando en
cuenta que cuando se suministrar energía térmica a un polímero, sus cadenas empiezan a
moverse al final de cada una de ellas. Y si un polímero conformado de cadenas largas
(Mw elevado) contiene pocos finales de cadena por unidades totales en comparación a un
polímero de bajo Mw, esto explicaría la variación de su Tg. Sin embargo la diferencia de la
Tg de los copolímeros I es pequeña (Figura 1.8), a pesar de que la variación de peso
molecular es considerable, probablemente porque para poliestirenos la Tg es independiente
del peso molecular a partir de los 25 000 g/mol [64], y estos copolímeros están compuestos
mayoritariamente de PS, por lo que su comportaminto debe ser similar.
40 60 80 100 120 140 160
0.045% mol BPO0.25% mol agente entrecruzante
I-escalado 121.9° C
Fluj
o de
cal
or (W
/g)
Temperatura (°C)
TMPTMA 129.3 °C
I (DVB) 119.4 °C
exo

CAPÍTULO 1
19
1.3.5 ANÁLISIS DINÁMICO MECÁNICO
Para seleccionar entre los copolímeros entrecruzados con DVB (copolímeros H, I, J y K) con
cual se seguiría el proceso de escalamiento y posterior sulfonación, se prepararon sus
respectivas películas y se les realizó un análisis dinámico mecánico. En la Figura 1.9 se
muestra los termogramas correspondientes a la variación del módulo de almacenamiento
respecto a la variación de la temperatura, mientras que en la Tabla 1.2 se reportan los valores
correspondientes de cada uno de ellos a 30 ºC. A esa temperatura, el copolímero I sintetizado
con 0.045 % mol de BPO y 0.25 % mol de DVB, tiene el mayor módulo elástico, razón por la
cual se seleccionó para las reacciones posteriores.
Figura 1.9. Módulo de almacenamiento de los copolímeros entrecruzados con DVB que dieron lugar a la formación de películas.
Tabla 1.2. Módulos de almacenamiento de los copolímeros sintetizados con DVB que forman película.
Copolímero entrecruzado con DVB E’ (MPa) I 2922 K 2569 J 2061 H 1482
1.3.6 ESPECTROSCOPÍA INFRARROJO
Como primer análisis, mediante espectroscopía de infrarrojo se corroboró la incorporación de
unidades de ácido acrílico en los copolímeros sintetizados. En la Figura 1.10 se presenta el
comparativo de los espectros de un PS puro (de la base de datos) y de los copolímeros de
PS-PAA entrecruzados con 0.045 % mol de BPO y 0.25 % mol de DVB o TMPTMA. En
comparación con el espectro del PS puro, los copolímeros muestran la señal de carbonilos
30 45 60 75 90 105 1200
500
1000
1500
2000
2500
3000
H
J
K
E' (M
pa)
Temperatura (°C)
I

CAPÍTULO 1
20
ubicada en la región de ~1700 cm-1 [65], indicando la incorporación de unidades de AA en
los copolímeros. Además de que se enuentra su confirmación con el ensanchamiento de las
bandas en la región entre 3300 a 2500 cm-1 debido al estiramiento del enlace O-H, las señales
correspondientes al estiramiento del enlace carbono-oxígeno en 1240 cm-1 [66] perteneniente
a los ácidos carboxílicos, y las señales correspondientes a la flexión en el plano del enlace
oxígeno-hidrógeno en 1430 cm-1 [66].
Figura 1.10. Espectros infrarrojos de: a) PS puro de la base de datos, b) copolímero PS-PAA entrecruzado con DVB y c) copolímero PS-PAA entrecruzado con TMPTMA. 1.3.7 RESONANCIA MAGNÉTICA NUCLEAR DE PROTÓN
La caracterización por RMN de 1H se utilizó para cuantificar los anillos estirénicos en los
copolímeros que se utilizarán para incorporarles los grupos sulfónicos (-SO3H) mediante
reacciones de sulfonación [10]. Para controlar el grado de sulfonación se puede variar la
cantidad de agente sulfonante que se va a adicionar al medio de reacción respecto al número
de anillos estirénicos presentes en la muestra. Por ello es necesario conocer la relación real
entre unidades de estireno y de ácido acrílico presentes en los copolímeros, lo cual se hizo
mediante RMN de 1H. Esto se debe a que a pesar de que en la alimentación del reactor en
todas las síntesis de los copolímeros mantuvo la relación molar St:AA de 94:6, existen
factores que pueden cambiar esa relación en el copolímero resultante. Entre los factores se
encuentra la viscosidad del medio de reacción, la afinidad química de los monómeros con el
3500 2800 1800 1600 1400 1200 1000 800 600
3500 2800 1800 1600 1400 1200 1000 800 600
a
c
b
Tran
smita
ncia
Número de onda (cm-1)

CAPÍTULO 1
21
medio de reacción debido a su polaridad, la variación en la partición de los monómeros entre
el medio de reacción y la cadena en crecimiento por diferencia de composición química, la
variación de la reactividad de los monómeros, etc [11, 25, 67].
En la Figura 1.11 se encuentra el espectro de RMN de 1H para el copolímero PS-PAA
entrecruzado con DVB (I-escalado) y la asignación de las señales respecto a una estructura
representativa del copolímero; PS-PAA/DVB 1H RMN (CDCl3, ppm): = 11.42 (Hm de
COO-H) [68, 69], = 7.07 (Hb meta,Hb para de St y Hg de DVB) [70-74], = 6.6 (Hc orto de
aromático) [68, 70-74], = 3.74 (agua involucrada por la presencia de los grupos
carboxílicos) [71, 75, 76], = 2.4 (Hd unido a C-C=O) [68, 77, 78], = 1.85 (Hf, unido a C
cuaternario de cadena principal) [79], = 1.47(He de CH2 de cadena principal) [74, 77].
Figura 1.11. Asignación de las señales de RMN de 1H para el copolímero PS-PAA entrecruzado con DVB.
La Figura 1.12 corresponde al espectro de RMN de 1H del copolímero PS-PAA entrecruzado
con TMPTMA. La asignación de los desplazamientos químicos de las señales de 1H es la
siguiente: PS-PAA/TMPTMA 1H RMN (CDCl3, ppm): = 11.41 (Hm de COO-H) [68, 69],
12 11 10 9 8 7 6 5 4 3 2 1 ppm
OOH
HH
HH
H
H
H
H
H
HH H HH
HHHH
H HH
b g
c
d
e
f
g g
d
c c
bbb
f ee f e
m
mg g

CAPÍTULO 1
22
= 7.07 (Hb meta y Hb para de aromático) [70-74], = 6.59 (Hc orto de aromático) [68, 70-
74], = 4.15 (Hh unidos a C cuaternario y O de ester) [80], = 3.74 (agua involucrada por la
presencia de los grupos carboxílicos) [71, 75, 76], = 2.48 (Hd unido a C-C=O) [68, 77, 78],
= 1.85 (Hf, unido a C cuaternario de la cadena principal) [79], = 1.47(He de CH2 de cadena
polimérica; y Hi de CH2-CH3 del TMPTMA) [74, 77, 80], = 1.17 (Hg correspondietnes al
metilo α) [81], = 0.87.(Hk de CH3-CH2 de TMPTMA) [80].
Figura 1.12. Asignación de las señales de RMN de 1H para el copolímero PA-PAA entrecruzado con TMPTMA.
La cuantificación de unidades de St y de AA en los copolímeros se realizó utilizando las
señales de los protones aromáticos que se encuentran en 7.07 ppm y 6.6 ppm, así como con la
señal del protón alfa del grupo carboxílico etiquetado como Hd y cuyo desplazamiento se
encuentra en 2.4 ppm.
Se determinó que la suma de las integrales de las señales en 7.07 ppm y 6.6 ppm corresponde
al área que representa todos los protones aromáticos presentes en la muestra, además que la
integral del protón Hd (en 2.4 ppm) equivale a los protones de ácido acrílico presente. En este
paso, cuando se determina la integral de los protones aromáticos, se indicó con el software
12 11 10 9 8 7 6 5 4 3 2 1 ppm
OOH
O
O
O
O
O
O
H
HH
HH
H
CH3
H HH
H HHH
HH
CH3
HHH
HH
HH H
g
k
d
c c
bbb
f ee e
i
h
b
c
d
e
f
g
i
k hm
m

CAPÍTULO 1
23
empleado que los límites de una sola integral sean de 7.6 ppm hasta 6.2 ppm, debido a que
estas señales no están bien resueltas. Posteriormente, se normalizan los valores de las áreas de
tal forma que la integral del protón Hd (o Hα) tenga el valor de 1 y los protones aromáticos
adopten el valor proporcional correspondiente. Lo cual se logra empleando la ecuaciones
1.4 y 1.5.
Área Hα AA normalizada = á á
=1 (ec.1.4)
ÁreaH’saromáticosnormalizada = á ′ á á α
(ec.1.5)
Se tomó en cuenta que en una unidad de ácido acrílico existe un solo Hα, por lo que se utilizó
el valor de 1 para el área (ecuación 1.6), además también se consideró que en una unidad de
estireno existen 5 protones aromáticos, por lo que el área de los H’s aromáticos normalizada se
debe dividir ente 5, tal y como se muestra en la ecuación 1.7.
Número de monómeros de AA = Área Hα AA normalizada = 1 (ec.1.6)
Número de monómeros de St = Á ’ á
(ec.1.7)
Se sumaron el númro de monómeros para obtenemos su cantida total (ecuación 1.8):
Monómeros totales = Número de monómeros de AA+ Número de monómeros de St
Es decir
Número de monómeros totales = 1+ Número de monómeros de St (ec.1.8)
Para obtener la relación porcentual molar de cada monómero en el copolímero se emplean las
ecuaciones 1.9 y 1.10.
%푚표푙푎푟푑푒푆푡 = ú ∗ %ú ó
(ec.1.9)
y
%푚표푙푎푟퐴퐴 = ú ∗ %ú ó
= ∗ %ú ó
(ec.1.10)
Con esta metodología se calcularon las relaciones porcentuales molares reales de cada
copolímero, las cuales se reportan en la Tabla 1.3.

CAPÍTULO 1
24
Tabla 1.3. Determinación de la relación molar real existente en los copolímeros PA-PAA
Entrecruzante empleado en el
copolímero PS-PAA
Área de la integral Área normalizada No. monómeros de: % molar
Hα AA
H's aromáticos
Hα AA
H’s aromáticos AA St St AA
DVB 2989 390900 1 130.7795 1 26.1559 96 4
TMPMA 5561 407500 1 73.2781 1 14.6556 94 6
En la Tabla 1.3 se puede ver que la relación molar del copolímero PS-PAA entrecruzado con
TMPTMA se mantuvo (St:AA = 94:6), mientras que en el copolímero entrecruzado con DVB
la relación cambia, se incorpora menos cantidad de ácido acrílico a la esperada
(St:AA = 96:4).
La diferencia de la relación de unidades repetitivas incoporadas en los copolímeros
sintetizados radicálicamente ya ha sido reportada con anterioridad, sobre todo en reacciones
donde el copolímero formado es poco soluble en el medio de reacción. Bajo esas condiciones,
la composición del copolímero se altera y uno de los monómeros es adsorbido
preferencialmente por el copolímero. Por ejemplo, para la copolimerización del metil
metacrilato(M1)-N-vinilcarbazol(M2), las relaciones de reactividad en benceno son r1 = 1.80,
r2 = 0.06, mientras que en metanol son r1 = 0.57 y r2 = 0.75. La viscosidad también afecta a
los valores de r. La copolimerización en masa de estireno(M1)-metil metacrilato(M2) genera
un copolímero que contiene menos estireno que cuando la reacción se lleva a cabo en solución
utilizando benceno como disolvente. Esto se debe a que en las reacciones en masa, el efecto
gel disminuye la movilidad del estireno que a su vez reduce el valor de r1 e incrementa el valor
de r2. Un efecto similar sucede cuando la copolimerización involucra la combinación de un
monómero polar (M1) y un monómero no polar (M2), el comportamiento dependerá de la
polaridad del medio de reacción. La composición del copolímero será más rico en el
monómero menos polar cuando la reacción se lleve a cabo en un disolvente polar o al menos
más polar que el monómero. Muchos mecanismos se han propuesto para describir este efecto
para monómeros no ionizables: lo cual dio lugar al estudio del efecto “bootstrap” o partición
del monómero [11].

CAPÍTULO 1
25
En el modelo bootstrap, el efecto del disolvente sobre la velocidad de propagación es atribuida
a la partición y con ello a la diferencia resultante entre la concentración localizada del
monómero en comparación con la concentración del total de la mezcla de reacción. Un
disolvente puede afectar el coeficiente de la velocidad de propagación sin cambiar la
reactividad del paso de propagación. El efecto bootstrap puede ser consecuencia de diversos
factores, uno de ellos consiste en la formación de complejos radical-disolvente o
monómero-disolvente que una vez que se han formado, no se propagan, pudiendo llegar a
cambiar la concentración efectiva del radical o del monómero [67]. En nuestro caso, el
disolvente empleado (DEB), no cuenta con grupos químicos polarizables, que por lo general
son los que pueden dar lugar a la formación de complejos. Sin embargo el efecto bootstrap
también puede generarse en reacciones de copolimerización como consecuencia de la
absorción preferencial de uno de los comonómeros alrededor de la cadena polimérica en
crecimiento; generalmente sucede cuando uno de los monómeros no es buen disolvente del
copolímero resultante [67]. Un mecanismo parecido a éste último descrito podría ser la causa
de la diferencia en composición encontrada en el copolímero entrecruzado con DVB.
Cuando se llevaron a cabo las reacciones de copolimerización empleando DVB, una vez
transcurrido el tiempo de reacción, la mezcla dentro del reactor era homogénea y después de
apagar el calentamiento, su viscosidad no mostraba cambio de viscosidad visible conforme
disminuía un poco su temperatura durante la apertura del reactor y justo antes de verter
elmedio de reacción en metanol. Sin embargo, cuando se utilizó TMPTMA, la mezcla de
reacción mantenía una apariencia homogénea y sin cambio de viscosidad visible siempre y
cuando la mezcla de reacción se mantuviera a 90 °C, pero una vez que se apagaba el
calentamiento y empezaba a disminuír la temperatura del medio de reacción, se podía apreciar
un cambio de densidad en la mezcla de reacción. La parte superior la mezcla se tornaba menos
concentrada, mientras que al fondo se formaba un gel que aumentaba de viscosidad conforme
la temperatura disminuía. Debido a este efecto, una vez transcurrido el tiempo de reacción, el
medio de reacción del reactor se vertía lo más rápido posible en metanol para precipitar y
dispersar el polímero, para evitar con ello que el polímero se enfriara con el DEB formando
una masa hinchada en lugar de estar disuelto. Este comportamiento de precipitación y/o
sedimentación del copolímero entrecruzado con TMPTMA indica que la miscibilidad del
material disminuye conforme va procediendo la reacción de copolimerización; lo cual es

CAPÍTULO 1
26
totalmente diferente a lo observado durante la reacción del copolímero PS-PAA entrecruzado
con DVB, que permanece disuelto homogéneamente incluso al disminuir la temperatura y
hasta el momento previo en que se vertió en el metanol. En estas reacciones de
copolimerización de PS-PAA parece ser que el efecto bootstrap se debe al efecto de partición
del monómero, que da como resultado una diferencia en la concentración efectiva del
monómero, que a su vez genera una discrepancia entre la velocidad de propagación
pronosticada a la real [67].

CAPÍTULO 1
27
1.4 CONCLUSIONES DEL CAPÍTULO 1
Se encontró que 0.045 % mol de BPO y 0.25 % mol de agente entrecruzante son las
concetraciones adecuadas para preparar copolímeros de PS-PAA útiles para formar
películas.
El peso molecular de los copolímeros de PS-PAA incrementa proporcionalmente a la
concnetrción de entrecruzante e inversamente al disminuir la concentración de
iniciador. Pero la variación del agente entrecruzante tiene mayor efecto en el Mw.
La Td y la Tg de los copolímeros sintetizados se desplaza a temperaturas mayores
cuando se utiliza mayor cantidad de agente entrecruzante y/o menor cantidad de BPO.
Debido al incremento de los entrecruzamientos químicos y el aumento de tamaño de
cadenas, limitando la movilidad de las cadenas.
La caracterización por espectroscopía de IR y por RMN de 1H comprobó la presencia
de ambos monómeros (St y AA) en los copolímeros. El copolímero entrecruzado con
DVB tiene una relación molar St:AA = 96:4 y copolímero entrecruzado con TMPTMA
tiene una relación molar St:AA = 94:6. Las relaciones entre monómeros incorporados
es similar e igual a la alimentada en el proceso de síntesis (St:AA= 94:6), por lo cual se
puede decir que la polimerización radicálica en solución (empleando DEB) es buen
método para copolimerizar St con AA.

CAPÍTULO 2
28
CAPÍTULO 2. SULFONACIÓN DE COPOLÍMEROS DE PS-PAA
ENTRECRUZADOS
2.1. ANTECEDENTES
Las propiedades de las membranas intercambiadoras de iones están determinadas por el
polímero base y el tipo y concentración de grupos iónicos que contenga. El polímero base
determina la estabilidad química, mecánica y térmica de la membrana, y suelen ser polímeros
hidrófobos como el poliestireno, polietileno, politetrafluoroetileno, las polisulfonas, etc. Estos
polímeros son insolubles en agua y tienen baja o nula relación de hinchamiento, pero al
incorporarles grupos iónicos pueden volverse completamente compatibles con el agua, siendo
necesario el entrecruzamiento de la matriz polimérica para evitar la disolución del polímero
iónico en el agua. En el caso de membranas, el entrecruzamiento limita su hinchamiento, le
proporcionar estabilidad térmica, y selectividad. El tipo y concentración de cargas iónicas en
una membrana determina su selectividad, su resistencia eléctrica, sus propiedades mecánicas y
su hinchamiento. Los grupos ácidos que comúnmente se incorporan en las matrices
poliméricas son –SO3-, -COO-, -PO3
2- y -AsO32-, cuya diferencia radica en su fuerza de
acidez [82].
En este capítulo se describen las reacciones de sulfonación de los copolímeros entrecruzados
sintetizados previamente, empleando tres sistemas sulfonantes. La sulfonación de los
copolímeros de PS-PAA darán como resultado materiales poliméricos con dos tipos de grupos
ácidos: carboxílicos y sulfónicos, cada grupos con una función específica. Los grupos
carboxílicos poseen un pKa entre 4.7 y 4.9 [83, 84], su fuerza ácida es media, por lo que sus
protones no se disocian fácilmente, no obstante este tipo de grupos químicos forman puentes
de hidrógeno [85] que le imparten hidrofilidad al material, que favoreceran la hidratación de
las membranas cuando estén en funcionamiento en una celda de combustible. Respecto a la
conductividad protónica de la membrana, ésta se consigue incorporando grupos ácidos con
valor de pKa bajo, como los grupos sulfónicos cuyo pKa es igual o menor a 1 [86]. La fuerza
ácida de éstos grupos garantizando la desprotonación, y volviéndolos una excelente opción en
copolímeros para membranas intercambiadoras de protones [87].
La obtención de polímeros para membranas con grupos -SO3H se puede lograr polimerizando
monómeros que contengan el grupo ácido, o polimerizando monómeros que no lo contengan y

CAPÍTULO 2
29
realizando una reacción de sulfonación al polímero obtenido. Esta última metodología es la
más empleada, entre los agentes sulfonantes que se han utilizado se encuentran el ácido
sulfúrico, concentrado, ácido clorosulfónico, trióxido de azufre, sulfato de acetilo, ácido
trimetilsililclorosulfonico, mezcla de ácido sulfúrico con ácido clorosulfónico [15, 88-92].
Las condiciones y reactivos empleados para la sulfonación de polímeros pueden propiciar
reacciones secundarias además de la incorporación de grupos sulfónicos. Es recomendado
emplear disolventes que no reaccionen durante la sulfonación; como hidrocarburos alquílicos,
diclorometano, dicloroetano, éteres clorados o mezclas de ellos [93, 94]. Un ejemplo de
reaccion secundaria es el entrecruzamiento de grupos sulfónicos, que origina sulfonas [14, 95]
por reacciones intramoleculares o intermolecuares [96, 97]. Para copolímeros con grupos
carboxílicos, existe la posibilidad de la reducción de éstos grupos por reacciones de acilación
seguida de reacciones de deshidratación, tal y como ha sido reportada en copolímeros de
estireno/metilacrilato [98, 99]. Por otro lado, también puede suceder el caso contrario, los
agentes sulfonantes sean muy suaves, como el trimetilsililclorosulfonato, se genera baja
eficiencia de sulfonación, incluso utilizándolo en exceso [100].
2.2. EXPERIMENTAL
2.2.1 MATERIALES
Ácido acético (99.7 % Sigma Aldrich), ácido clorhídrico (36.5-38 %, Sigma Aldrich),
ácido sulfúrico (98 % J.T.Baker), anhídrido acético (Sigma Aldrich), cloruro de sodio
(J. T. Baker), copolímero de PS-PAA entrecruzado con DVB (copolímero I-escalado) de
Mw = 259,095 g/mol, copolímero de PS-PAA entrecruzado con TMPTMA de
Mw = 302,607 g/mol, diclorometano anhidro (≥ 99.8 %, Aldrich), hidróxido de sodio
(Aldrich), nitrato de plata (≥ 99.0 %, CTR Scientific), tetrahidrofurano libre de
inhibidor ≥ 99.9 % (Aldrich).
2.2.2 PREPARACIÓN DE REACTIVOS
Los tres sistemas sulfonantes fueron: ácido sulfúrico (H2SO4), sulfato de acetilo
(CH3COOSO3H) y ácido sulfúrico con sulfato de plata (Ag2SO4). La cantidad molar de agente
sulfonante corresponde directamente a la cantidad de H2SO4, que se calculó respecto a la
cantidad de anillos aromáticos presentes en la muestra a sulfonar (ecuación 2.1). Para los

CAPÍTULO 2
30
cálculos se consideraron las relaciones molares reales de St:AA determinadas previamente por
RMN de 1H para los copolímeros entrecruzados con DVB y TMPTMA, 96:4 y 94:6
respectivamente.
A continuación se muestran los cálculos realizados para el copolímero entrecruzado con DVB
(St:AA = 96:4), sin embargo se realizaron de de ifual forma para el copolímero entrecruzado
con TMPTMA, empleando la relación molar St:AA = 94:6.
Cantidad de H2SO4 a utilizar para el copolímero DVB (St:AA = 96:4)
96 mol St = 11035.76 mL = 9998.4 g
4 mol AA = 274.285 mL = 288 g
Monómeros totales teóricos = 11310.045 mL = 10286.4 g
Masa de copolímero total = 10286.4 g
푚표푙푒푠푑푒퐻 푆푂 = ( )( . )(% )( . í )( . / ) %
(ec.2.1)
en donde
g muestra: peso de la muestra de copolímero que se va a sulfonar
% sulfonante: % de agente sulfonante a utilizar en el experimento
La cantidad de moles de H2SO4 se interpretan a mL tomando en cuenta la pureza, densidad y
peso molecular del reactivo.
2.2.2.1 SULFATO DE ACETILO
El sulfato de acetilo se preparó antes de ser utilizado y la reacción que rige su química de
preparación es la siguiente:
1 H2SO4 + 1 (CH3CO)2O 1 CH3COOSO3H + 1 CH3COOH
Estequiométricamente se requiere un mol de H2SO4 y un mol de anhídrido acético
((CH3CO)2O) para obtener un mol de CH3COOSO3H y un mol de ácido acético (CH3COOH),
no obstante por cada mol de H2SO4 se adicionaron 1.2 moles de anhídrido acético. El exceso
de anhídrido acético se adicionó para que reaccione con el agua que pudiera existir en el medio
de reacción y que afecta la relación estequiométrica H2SO4:anhídrido acético.
El procedimiento reportado para la preparación del CH3COOSO3H [101-104], consiste en
mezclar durante 10 min en baño de hielo con flujo constante de nitrógeno, un volumen de
anhídrido acético con un volumen de diclorometano, y posteriormente se adiciona el H2SO4 y
se deja reaccionar por 10 min.

CAPÍTULO 2
31
2.2.2.2 SULFATO DE PLATA
La preparación del Ag2SO4 se llevó a cabo protegiendo de la luz la mezcla de reacción, para
llevarla a cabo se mezclaron 5 g de nitrato de plata (AgNO3) con 5 mL de H2SO4 al 98 %, se
calentó hasta la ebullición del ácido y la emisión de humo negro. Una vez cesada la emisión
de humo, el medio de reacción aún caliente se vertió en 500 mL de agua desionizada fría para
formar la sal Ag2SO4, los cristales se filtraron y lavaron con agua desionizada fría hasta
eliminar el ácido residual (pH neutro del agua de los lavados). Los cristales se secaron a
110 °C y se dejaron enfriar en un desecador [27, 105]. El proceso de purificación del Ag2SO4
se seleccionó considerando que el AgNO3 es totalmente soluble en agua, mientras que el
Ag2SO4 es prácticamente insoluble en la misma (0.8 % a 17 °C) [106].
La cantidad de Ag2SO4 a utilizar se calculó empleando la ecuación 2.2, tomando como base
los moles de H2SO4 a emplear y considerando que la cantidad de la sal de plata no superara el
1 % respecto a la cantidad de ácido, según lo indicado en la bibliografía [22, 107-112].
푔푑푒퐴푔 푆푂 = ( )( . )(% )(% )( . / )( . í )( . / )
(ec.2.2)
en donde
g muestra: peso de la muestra de copolímero (entrecruzado con DVB) que se va a sulfonar
% sulfonante: % de agente sulfonante a utilizar en el experimento
% Ag2SO4: % de sulfato de plata a utilizar en el experimento
2.2.3 SULFONACIÓN DEL COPOLÍMERO
Las reacciones de sulfonación se realizaron en un reactor con eje de vidrio ambos, con una
propela de teflón. El copolímero se mezcló con el diclorometano
(relación peso:volumen = 1:3) a 40 °C bajo atmósfera de nitrógeno a 200 rpm durante 40 min
y posteriormente se adicionó el agente sulfonante. Transcurrido el tiempo de sulfonación, el
medio de reacción se vertió en un recipiente amplio y se le aplicó una corriente de aire para
eliminar el diclorometano y enfriar la reacción. Los copolímeros sulfonados se lavaron
consecutivamente hasta que el agua empleada alcanzó pH neutro, a continuación los
copolímeros sulfonados se secaron por 48 h con una corriente de aire constante a temperatura
ambiente y posteriormente en estufa por 72 h a 40 °C a presión reducida.

CAPÍTULO 2
32
Al emplear H2SO4 con Ag2SO4 se hicieron dos modificaciones:
a) Al reactor se adicionó CH3COOH para compatibilizar al copolímero con el medio de
reacción durante la sulfonación. La cantida de CH3COOH adicionado equivale a la
mitad del volumen de H2SO4 a utilizar en la reacción.
b) La mezcla sulfonante se preparó previa a su adición mezclando el Ag2SO4, el H2SO4 y
otro volumen igual de CH3COOH que el adicionado al reactor.
A continuación, cuando se habla del porcentaje molar o cantidad de agente sulfonante, es
referente a los moles de agente sulfonante respecto a los moles de anillos de estireno del
copolímero utilizado.
2.2.4 CARACTERIZACIÓN
2.2.4.1 DETERMINACIÓN DE LA CAPACIDAD DE INTERCAMBIO IÓNICO
La capacidad de intercambio iónico (ionic exchange capacity -IEC-) de las membranas se
midió con el método de titulación indirecta. La membrana se colocó en ácido clorhídrico
(HCl) 1 M durante 24 h, posteriormente se enjuagó y se sumergió en una solución de cloruro
de sodio (NaCl) 1 M durante 24 h. A continuación la membrana se retiró de la solución de
NaCl y se enjuagó con agua desionizada, procurando que el agua del enjuague cayera dentro
del vaso que contenía la solución de NaCl. El número de protones desplazados de la
membrana se determinó utilizando un pH-metro para detectar el punto de equivalencia durante
la titulación de la solución de NaCl con una solución de NaOH 0.005M previamente valorada.
La membrana se regeneró sumergiéndola en HCl 1M durante 24 h, se enjuagó con agua
desionizada, se le secó el agua superficial con un papel absorbente, e inmediatamente se pesó
(푊 ). La membrana se dejó a temperatura ambiente durante 24 h, después en la estufa
a 44 °C con vacío constante durante 24 h, se dejó enfriar en un desecador y se volvió a pesar
(푤 ).
La capacidad de intercambio iónico se calculó mediante la ecuación 2.3 [113, 114]:
퐼퐸퐶 = (ec.2.3)
Donde 푎 es la concentración de NaOH usada (mmol/mL ~ meq/mL), 푏 es el volumen de la
solución de NaOH empleado (mL) y 푊 es el peso (g) de la membrana seca.

CAPÍTULO 2
33
2.2.4.2 DETERMINACIÓN DE MATERIAL INSOLUBLE MEDIANTE
EXTRACCIÓN SOXHLET
El contenido de material insoluble de las muestras, el cual está relacionado con formación de
geles por entrecruzamiento, se determinó por extracciones en sistema soxhlet. Se utilizó una
muestra de aproximadamente 0.5 g de copolímero (w1) que se colocó dentro de un cartucho
de celulosa previamente tarado a peso constante (w2); el cartucho se sometió a reflujo dentro
del equipo soxhlet con THF por 12 horas. Posteriormente el dedal se secó a 80 °C y presión
reducida por 12 h, obteniendo el peso nuevamente (w3). El contenido de gel (fracción en
peso) se calculó utilizando la ecuación 2.4:
퐶표푛푡푒푛푖푑표푑푒푔푒푙(%) = 푥100 (ec.2.4)
el cual indica indirectamente el grado de entrecruzamiento [115].
2.2.4.3 CROMATOGRAFÍA DE PERMEACIÓN EN GEL
El peso molecular de los copolímeros de determinó por Cromatografía de Permeación en Gel
(GPC) empleando un cromatógrafo Hewlett Packard con detector de índice de refracción,
equipado con 3 columnas de Ultrastyragel (105, 104 y103 Å), calibrado con estándares de
poliestireno. Las soluciones de los copolímeros se prepararon con concentración de 1mg/mL
en THF anhidro y se filtraron por tamices de 0.2 µm. Para su análisis de utilizó una velocidad
de elución de 1 mL/min y una temperatura de 40 °C. Posiblemente la concentración de las
soluciones analizadas es menor a la concentración preparada, debido a la presencia de
material insoluble en la muestra, por lo que además el peso molecular obtenido no es
inequívoco pero permitió conocer la tendencia general de los pesos moleculares al cambiar
las condiciones de reacción de sulfonación.
2.2.4.4 ESPECTROSCOPÍA INFRARROJO
Para verificar que la sulfoanación de los copolímeros se haya llevado a cabo se empleó un
espectrofotómetro infrarrojo Nicolet modelo Avatar 316. Las muestras se evaluaron en forma
de película empleando 32 scans y una resolución de 4 cm-1. A los espectros se les aplicó una
corrección automática de la línea base y una normalización con el software OMNIC 5.2,
tomando como base la señal de 1602 cm-1 correspondiente a la tensión C=C del anillo

CAPÍTULO 2
34
aromático [41], considerando que ésta estructura química es estable y no se genere su
rompimiento durante las reacciones de sulfonación.
2.2.4.5 CALORIMETRÍA DIFERENCIAL DE BARRIDO
La temperatura de transición vítrea de los copolímeros obtenidos se evaluó mediante
calorimetría diferencial de barrido, utilizando un DSC modelo 2920 (TA Instruments)
empleando una velocidad de calentamiento de 10 °C/min.
2.2.4.6 ANÁLISIS TERMOGRAVIMÉTRICO
Para estudiar la estabilidad térmica de los copolímeros, un aproximado de 4.5 mg de cada
muestra fue evaluada en un Analizador Termogravimétrico (TGA) modelo Q500 de TA
Instruments, a una velocidad de 10 °C/min desde temperatura ambiente hasta 600 °C bajo
atmósfera de N2.
2.3 RESULTADOS Y DISCUSIÓN
La búsqueda de condiciones de sulfonación y mejor agente sulfonante se hizo para obtener
materiales sulfonados que cumplieran con las siguientes características: insolubles en agua,
solubles en THF, carentes de geles que les imposiblilite su procesamiento, utiles para formar
película mediante disolución-evaporación, resistencia a su manipulación y activación,
capacidad de absorber agua, capacidad de intercambio iónico, baja permeabilidad de
combustible competitiva al de la membrana de Nafion. Para obtener los materiales sulfonados
que cumplan con estas características se llevaron a cabo reacciones de sulfonación a dos
diferentes copolímeros empleando diferentes sistema sulfonante, porcentaje de agente
sulfonante, tiempos de sulfonación, condiciones de preparación de películas, etc. A partir de
cada cambio realizado fue necesario evaluar y modificar el procedimiento para mejorar o
lograr las características deseadas sin afectar otras.
La búsqueda de las mejores condiciones de sulfonación se comenzó con el mejor copolímero
entrecruzado con DVB (al cual se nombrará copolímero D a partir de esta parte del
documento), y conforme se fueron encontrando, esas mismas condiciones se reprodujeron con
el mejor copolímero entrecruzado con TMPTMA (copolímero T). Los primeros agentes
sulfonantes utilizados fueron el sulfato de acetilo (CH3COOSO3H) y el ácido sulfúrico

CAPÍTULO 2
35
(H2SO4), su utilización se hizo por separado pero a la par para encontrar las condiciones donde
convergen los experimentos que se compararían entre sí. La sulfonación de los copolímeros
con CH3COOSO3H o H2SO4 originó materiales de diferentes índoles: no procesables
(entrecruzados), solubles en agua, procesables pero sin capacidad de formar película y capaces
de formar película pero que no intercambiaban iones. Estos inconvenientes llevaron al uso de
otro sistema sulfonante: ácido sulfúrico en conjunto con sulfato de plata (H2SO4-Ag2SO4), con
el cual se obtuvieron materiales procesables que cumplen las características buscadas.
El significado de la nomencalrura de los copolímeros que se empleará a partir de este capítulo
es la siguiente. La primera letra corresponde al copolímero D o T, referente al agente
entrecruzante utilizado para su síntesis, DVB o TMPTMA. Las letras siguientes (H, C o Ag)
se asocian al nombre del sistema sulfonante: H para H2SO4, C para CH3COOSO3H, Ag para el
sistema acido sulfúrico-Ag2SO4. El primer número corresponde al tiempo de sulfonación en
minutos y va seguido del símbolo min. El segundo número indica el porcentaje molar de
agente sulfonante (H2SO4) y va acompañado del símbolo %. En caso de haber un tercer
número, éste corresponde al porcentaje molar de Ag2SO4 y va acompañado del símbolo %. Por
ejemplo, la etiqueta TAg-120min170%0.220% corresponde a una muestra preparada con el
copolímero de PS-PAA entrecruzado con TMPTMA, sometido a sulfonación con el sistema
acido sulfúrico-Ag2SO4, la reacción se llevó a cabo durante 120 minutos empleando
170 % mol de ácido sulfúrico y 0.220 % mol de Ag2SO4.
2.3.1 SOLUBILIDAD Y DESEMPEÑO FÍSICO Y IÓNICO DE LOS COPOLÍMEROS
SULFONADOS
En una celda de combustible de PEM debe haber humedad para que los protones de los grupos
ácidos se disocien y se genere la conducción protónica [116]. Debido a la permanentemente
humedad en la celda de combustible, se presupone que el material de la membrana debería ser
insoluble en agua, para evitar que se reblandezca y/o rompa afectando otros componentes de la
celda. Además la membrana debe ser mecánicamente resistente para soportar los cambios
dimensionales ocasionados durante el ciclo de operación de la celda por la variación de la
temperatura y humedad relativa [117].
Las membranas se prepararon mediante disolución-evaporación, y los copolímeros sulfonados
deben formar película, ser insolubles en agua y solubles en un disolvente volátil o de fácil

CAPÍTULO 2
36
extracción. La técnica de disolución-evaporación se escogió porque genera membranas
isótropas [118] y se requiere poco material para prepararlas, a diferencia de la extrusión o uso
de placas que además involucrarían más variables.
En la Tabla 2.1 se reportan el rendimiento de las reacciones de sulfonación de los copolímeros
D y T, su solubilidad en agua y su capacidad de formar película, observándose que la
solubilidad en agua y el rendimiento de la reacción están relacionados directamente. Esto se
debe a que una vez que termina el tiempo de sulfonación, al medio de reacción se adiciona
agua para detener la reacción y eliminar los remanentes de sistema sulfonante. Es entonces
cuando las cadenas poliméricas sulfonadas sin entrecruzar, se solubilizan en el agua
integrándose a la fase acuosa, generando rendimientos menores al 100% (cantidad de material
insoluble y recuperado).
Las primeras reacciones de sulfonación se llevaron a cabo por 1 y 2 horas con 100 % mol de
agente sulfonante (serie I de experimentos en Tabla 2.1); condiciones de sulfonación ya
utilizadas por el grupo de trabajo para copolímeros de PS-PAA pero sintetizados en masa,
encontrándose buenos resultados [119]. Sin embargo, al emplear esas concentraciones y
tiempos de reacción con los copolímeros de PS-PAA entrecruzados con DVB y TMPTMA
sintetizados en solución se observó un comportamiento diferente. Al emplear sulfato de acetilo
se generaron materiales solubles en agua, mientras que con H2SO4 se generaron materiales
insolubles en agua u otro disolvente orgánico, incluyendo THF, imposibilitando su
procesamiento y utilización, por lo que fue necesario buscar alternativas.

CAPÍTULO 2
37
Tabla 2.1. Comportamiento de los copolímeros D y T sulfonados en diferentes disolventes y despeño físico para la preparación de membranas mediante disolución-evaporación
Serie Copolímero sulfonado Rendimiento (%) Hidrosoluble Película
I
DC-120min100% 0 Si N.D. DH-120min100% ~100 No N.D. DC- 60min100% 0 Si N.D. DH- 60min100% ~100 No N.D.
II
DC-120min90% 0 Si N.D. DC- 60min90% 0 Si N.D. DC-120min80% 0 Si N.D. DC- 60min80% 0 Si N.D. DC-120min70% 0 Si N.D. DC- 60min70% 0 Si N.D. DC-120min60% 0 Si N.D.
III
DC- 60min60% 10.1 Si No (fractura al secar) DC-120min50% 4.0 Si No (fractura al secar) DH-120min60% ~100 No Si (incompleta y geles) DH- 60min60% ~100 No Si (incompleta y geles)
IV
DC-120min40% 80.1 Si Si (fractura al desmoldar) DC- 60min40% 85.3 Si Si (fractura al desmoldar) DH-120min40% ~100 No Si (geles mínimos) DH- 60min40% ~100 No Si (geles mínimos) DC-120min20% 92.4 Si Si (fractura al desmoldar) DC- 60min20% 97.0 Si Si (fractura al desmoldar) DH-120min20% ~100 No Si (geles mínimos) DH- 60min20% ~100 No Si (geles mínimos)
V
DC-30min20% 98.2 Si Si DC-10min20% ~100 No Si DC- 2min20% ~100 No Si DH-30min20% ~100 No Si DH-10min20% ~100 No Si DH- 2min20% ~100 No Si TC-30min20% ~100 No Si TC-10min20% ~100 No Si TC- 2min20% ~100 No Si TH-30min20% ~100 No Si TH-10min20% ~100 No Si TH- 2min20% ~100 No Si
VI
DAg-120min 85%0.110% ~100 No Si DAg-240min 85%0.110% ~100 No Si DAg-240min170%0.110% ~100 No Si DAg-360min 85%0.110% ~100 No Si DAg-240min225%0.110% ~100 No Si DAg-240min170%0.220% ~100 No Si DAg-240min170%0.055% ~100 No Si TAg-120min 85%0.110% ~100 No Si TAg-240min 85%0.110% ~100 No Si TAg-120min170%0.110% ~100 No Si TAg-120min170%0.220% ~100 No Si TAg-360min 85%0.110% ~100 No Si TAg-120min225%0.110% ~100 No Si TAg-120min170%0.055% ~100 No Si

CAPÍTULO 2
38
Durante la sulfonación de unidades de estireno con H2SO4 la reacción química principal es la
incorporación de grupos sulfónicos en los anillos aromáticos, pero también ocurren en baja
proporción reacciones secundarias que pasan desapercibidas [120]. Dichas reacciones son la
formación de sulfonas, que actúan como entrecruzantes entre cadenas de polímero [121]. La
importancia de la generación de sulfonas está documentada para poliestirenos sulfonados
totalmente solubles en agua, donde la viscosidad de sus soluciones cambia dependiendo de la
cantidad de sulfonas presentes [120, 122, 123]. Además se sabe que la cantidad de sulfonas
aumenta con el incremento de la temperatura de sulfonación [103], y empiezan a formarse a
diferentes temperaturas de reacción dependiendo del agentes sulfonantes empleado, a 40 °C
con sulfato de acetilo [14], a 55 °C con H2SO4 [124] y entre 30 y 60 °C con sílica-ácido
sulfúrico [125]. Además la formación de sulfonas no está limitada a anillos estirénicos,
también sucede con el poliéter éter cetona (PEEK) al ser sulfonado con ácido clorosulfónico a
50 °C [126]. Y la temperatura a la que se forman las sulfonas es baja, considerando que las
reacciones de sulfonación pueden llevarse a cabo en un amplio intervalo de temperaturas,
desde -20 hasta 300 °C [103]. Una vez expuesto el efecto de la formación y presencia de
grupos sulfona en los polímeros, se puede relacionar su presencia con la insolubilidad del
copolímero D sulfonado con H2SO4 por 1 y 2 h. Debido a que los entrecruzamiento del DVB
podrían sumarse al los de tipo sulfona, sobrepasando el límite en el cual el polímero es
soluble.
Respecto a la solubilidad en agua del copolímero D sulfonado con CH3COOSO3H por 1 y 2 h,
está relacionado con la cantidad de grupos sulfónicos, en donde a mayor grado de sulfonación
mayor solubilidad del copolímerpo en agua como se verá a continuación. Está reportado que
polímeros de estireno con grados de sulfonación del 40 % mol son sumamente afines al agua;
inclusive, después de purificarlos se han obtenido rendimientos del 42 % de su masa
inicial [14]. De forma similar, los polímeros tipo poliéter-eter-cetona sulfonados (sPEEK), con
grado de sulfonación de 60 % se solubilizan totalmente en mezclas de agua-metanol, y con
grados de sulfonación de 100 % son solubles completamente en agua caliente [127].
Ambos problemas encontrados al utilizar 100 % mol de agente sulfonante por 1 y 2 h podrían
reducirse disminuyendo el tiempo de reacción y/o la cantidad de agente sulfonante [14]. La
primer alternativa que se empleó fue disminuir la cantidad de agente sulfonante,
de 100 % a 90, 80, 70, 60 y 50 %, manteniendo los mismos tiempos de reacción (series II y III

CAPÍTULO 2
39
en la Tabla 2.1). Emplear 60 % mol por 1 h y 50 % mol por 2 h con de sulfato de acetilo
fueron las condiciones más extremas a utilizar con el copolímero D para obtener copolímeros
sulfonados insolubles en agua y solubles en THF, con los que se llevó el proceso de
disolución-evaporación pero las películas se fracturaron conforme se evaporó el THF. Este
comportamiento descalificó a los copolímeros sulfonados de las series II y III, por lo que se
redujo a 20 y 40 % la cantidad de agente sulfonante (serie IV en Tabla 2.1) obteniéndose
copolímeros sulfonados que formaron películas que se fracturaron al desmoldarlas o que
contenían geles.
La siguiente alternativa fue la reducción del tiempo de sulfonación, a 30, 10 y 2 minutos,
manteniendo 20 % mol de agente sulfonante (serie V en Tabla 2.1). Con estas condiciones se
obtuvieron materiales sulfonados insolubles en agua, solubles en THF, útiles para formar
película que no se fracturaron durnate su secado, desmolde o purificación y activación. Dichas
características indicaron la conveniencia de utilizar condiciones suaves de sulfonación, por lo
que también se utilizaron con el copolímero T, además de que en la literatura se menciona que
el uso de H2SO4 concentrado con polímeros de estireno-TMPTMA disminuye la estabilidad
física y/o química de polímero por el rompimiento de los grupos químicos del TMPTMA,
originando polímeros sulfonados hidrosolubles [128].
A pesar de la estabilidad mecánica de las membranas de los copolímeros sulfonados por 2, 10
y 30 minutos con 20 % mol de agentes sulfonantes, la evaluación de la capacidad de
intercambio iónico (IEC) indicó que no poseían propiedades competitivas con la membrana
Nafion 117, como se muestra en la Tabla 2.2.

CAPÍTULO 2
40
Tabla 2.2. Valores de IEC de los materiales (membranas) sulfonados con H2SO4 o CH3COOSO3H. IEC (meq/g)
Etiqueta de la muestra IEC (meq/g) Nafion 117 0.87
DC-30min20% 0.0007 DC-10min20% 0.0004 DC- 2min20% 0.00004 DH-30min20% 0.0006 DH-10min20% 0.0006 DH- 2min20% 0.0005 TC-30min20% 0.0007 TC-10min20% 0.0007 TC- 2min20% 0.0006 TH-30min20% 0.0006 TH-10min20% 0.0006 TH- 2min20% 0.0006
DAg-240min170%0.110% 0.5524 TAg-120min170%0.110% 0.9030
Nafion 117: Membrana comercial, no se preparó en el laboratorio
La importancia de la evaluacion de la IEC de las membranas radica en la estrecha relación que
existe entre esta propiedad y la conductividad protónica de una membrana para celda de
combustible [129] como se verá a continuación. La IEC es el número de miliequivalentes de
cargas intercambiables en 1.0 g de matriz polimérica seca [130], dichas cargas corresponden a
los grupos ácidos, por lo que el IEC es un indicativo del grado de sulfonación de un material, y
se puede determinar mediante titulación. A su vez, debido a que los grupos ácidos son los
responsables de la conducción de protones, la IEC da un aproximado indirecto de la
conductividad protónica [131], inclusive está reportado que la conductividad iónica se
incrementa con el aumento de la capacidad de intercambio iónico del polímero [9].
Debido a que el la IEC da un aproximado indirecto de la conductividad protónica, una vez que
se determinó que los copolímetos D y T sulfonados que generaban membranas tenían una IEC
que tiende a cero, se buscaron alternativas para sulfonar los copolímeros que a su vez evitaran
la formación de reacciones secundarias.
Para poder evitar la formación de grupos sulfona es necesario conocer el mecanismo por el
cual se forman. Estos grupos químicos surgen de la reacción entre dos grupos sulfónicos de
diferentes anillos, generando entrecruzamientos intermacromoleculares o

CAPÍTULO 2
41
intramacromoleculares, siendo más probable el que sucede entre grupos sulfónicos de
diferente cadenas, además conforme aumenta la cantidad de agente entrecruzante incrementa
la cantidad de grupos sulfona [14, 132]. En este punto de la experimentación, la opción de
utilizar bajas cantidades de agente entrecruzante ya se llevó a cabo con resultado desfavorable,
así que se opto por otra opción menos utilizada, que consiste en adicionar sulfato de plata. En
la literatura se menciona que ésta sal actúa como catalizador para acelerar la reacción de
sulfonación con H2SO4, así como prevenir la formación de grupos sulfonas [133], sin embargo
la función concreta de los iones plata (Ag+) del Ag2SO4 es formar un complejo metálico con
los grupos sulfónicos, para disminuir la probabilidad de que un anillo estirénico que contiene
éste grupo se encuentre con otro, evitando que reaccionen entre si y se forme un grupo
sulfona. La cantidad de Ag2SO4 a utilizar en conjunto con H2SO4 no está establecida en la
literatura, se menciona su uso en cantidades catalíticas o que la cantidad a utilizar no debe
supera el 1 % respecto al H2SO4 [22, 107-112]. Para los experimentos llevados a cabo se
utilizaron tres concentraciones de Ag2SO4: 0.055, 0.110 y 0.220 % mol, 3 tiempos de
sulfonación (2, 4 y 6 horas) y los 3 porcentajes molares de H2SO4 (85, 170 y 225 % mol).
2.3.2 DETERMINACIÓN DE INSOLUBLES MEDIANTE EXTRACCIÓN SOXHLET
La determinación de material insoluble en los copolímeros se realizó para averiguar si existía
una tendencia respecto a las condiciones de sulfonación empleadas y la cantidad de geles
formados en los copolímeros debido a la reacción de sulfonación.
Las primeras reacciones de sulfonación del copolímero D se llevaron a cabo por 1 y 2 horas
utilizando 100 % mol de agente sulfonante. Los copolímeros sulfonados con H2SO4 estuvieron
sumergidos en THF y con agitación durante 2 días, pero no se disolvieron para formar una
solución, solo se hincharon. En cambio los copolímeros sulfonados con CH3COOSO3H
utilizando las mismas condiciones, fueron totalmente solubles en agua, razón por la que se
descartaron.
Debido a estos inconvenientes se utilizaron menores cantidades de agente sulfonante y mismos
tiempos de reacción. Con H2SO4 nuevamente se obtuvieron materiales con geles que se
caracterizaron mediante extracción soxhlet con THF durante 12 horas. En la Figura 2.2 se
muestran los resultados. Cuando se sulfonó durante 1 hora, no se obtuvo una tendencia, pero al

CAPÍTULO 2
42
llevar a cabo la reacción por 120 min, se encontró que a mayor cantidad de H2SO4 utilizado,
myor cantidad de material insoluble se genera, llegando a alcanzar 48.2 % en peso. La
presencia de geles en los copolímeros afecta su disolución en THF generando soluciones
heterogéneas, y al evaporarse el THF se forman películas incompletas y/o con partículas que
sobresalen de la superficie de la película. Los geles insolubles son el resultado de
entrecruzamientos químicos en el copolímero después de la sulfonación con H2SO4, debido a
la reacción entre dos grupos sulfónicos que dan lugar a una sulfona que entrecruza cadenas. La
formación de geles fue un resultado es inesperado, debido a que las reacciones de sulfonación
se llevaron a cabo a 40 °C, mientras que en la literatura, estudios de degradación térmica,
reportan la ocurrencia de este tipo de entrecruzamientos pero a temperaturas mayores [134].
Figura 2.2. Insolubilidad de los copolímeros DH sulfonados durante 1 y 2 horas con diferente % mol de ácido sulfúrico, determinado como contenido de gel en 12 horas de extracción.
Al utilizar 60, 40 y 20 % mol de CH3COOSO3H por 1 y 2 horas de reacción (Figura 2.3),
también se observan cambios en la cantidad de material insoluble, sin embargo en ningún caso
supera el 10 % peso. Además las soluciones preparadas con ellos en THF son homogéneas,
ideales para el proceso de disolución-evaporación.
Figura 2.3. Insolubilidad de los copolímeros DC sulfonados durante 1 y 2 horas con diferente % mol de sulfato de acetilo, determinado como contenido de gel en 12 horas de extracción.
0
10
20
30
40
50
H2SO4 (mol %)
Gel
(%) 29.3%
47.6%
36.7%
1.6%0 20 40 60
60 min
0
10
20
30
40
50 120 min
1.6%
34.8%41.0%
48.2%
0 20 40 60
Gel
(%)
H2SO4 (mol %)
0
10
20
30
40
50
CH3COOSO3H (mol %)
1.6% 2.1%9.9%
2.2%
0 20 40 60
Gel
(%)
60 min
0
10
20
30
40
50
1.6% 1.3% 1.6% 2.9%
0 20 40 50
Gel
(%)
120 min
CH3COOSO3H (mol %)

CAPÍTULO 2
43
Una vez comprobado que al utilizar concentraciones menores al 100 % de agente sulfonante se
obtuvieron bajos niveles de geles, se continuó con la disminución de los porcentajes de
sulfonante y tiempo de sulfonación, buscando mejorar la respuesta mecánica de los materiales
sulfonados. No obstante, con las condiciones suaves de sulfonación se detectó baja capacidad
de intercambio iónico en los copolímeros, por lo que se procedió a cambiar de sistema
sulfonante. El sistema sulfonante alternativo fue H2SO4 en conjunto con Ag2SO4, con el cual
se obtuvieron copolímeros sulfonados insolubles en agua y solubles en THF, capaces de
formar película, con capacidad de intercambio iónico competitiva al Nafion 117;
específicamente los copolímeros DAg-240min170%0.110% y TAg-120min170%0.110% son
los que cumplen con dichas características. En la Figura 2.4 se encuentran los porcentajes de
material insoluble formado en estos copolímeros sulfonados y de los copolímeros D y T sin
sulfonar. Se observa que utilizar Ag2SO4 no suprime la presencia de geles pero restringe su
generación, formándose 10.4 y 8.3 % de material insoluble respectivamente. Este efecto es
evidente al comparar los copolímeros sulfonados con 100 % mol de H2SO4 únicamente, los
cuales eran insolubles, y los copolímeros sulfonados con 20, 40 y 60 % mol de H2SO4
(Figura 2.2) presentan 29.3 a 48.2 % de material insoluble.
Figura 2.4. Grado de insolubilidad de los copolímeros sulfonados D, T y sus análogos sulfonados con H2SO4-Ag2SO4, determinado como contenido de gel en 12 horas de extracción.
2.3.3 EFECTO DEL PESO MOLECULAR EN EL DESEMPEÑO FICICOQUÍMICO
DE LOS COPOLÍMEROS SULFONADOS
Los resultados de contenido de gel comprobaron que utilizar H2SO4 para sulfonar genera
material insoluble, pero no explican la hidorsolubilidad e incapacidad de formar membranas
del copolímero D sulfonado con CH3COOSO3H. Tomando en cuenta que un polímero es
quebradizo y sin resistencia mecánica cuando su peso molecular es bajo [135], se determinó la
0
10
20
30
40
50
1.6%
10.4%
2.1%8.3%
Gel
(%)
D DAg T TAg

CAPÍTULO 2
44
variación del peso molecular de la fracción soluble de los copolímeros sulfonados con
CH3COOSO3H y de otros sulfonados con H2SO4.
En la Tabla 2.3 se encuentran los valores del peso molecular promedio en peso (Mw), el peso
molecular promedio en número (Mn), el peso molecular del máximo del pico (Mp) y la
dispersidad (Ð) de los copolímeros D y T antes y después de las reacciones de sulfonación.
Comparando el peso molecular del copolímero D y sus análogos sulfonados por 1 y 2 horas
con CH3COOSO3H, se observan valores de Mw entre 10,028 g/mol y 3,413 g/mol, por debajo
del copolímero D antes de ser sulfonado (259,095 g/mol). En la literatura se encuentra
reportado que cuando un poliestireno posee un Mw menor a 150,000 es frágil, por lo que los
poliestirenos comerciales para moldeo y extrusión tienen Mw cercanos a 180,000 g/mol [136].
Tabla 2.3. Mn, Mw, Mp y Ð de los copolímeros D y T antes y después de ser sulfonados con H2SO4, CH3COOSO3H y H2SO4-Ag2SO4.
Reacciones de
sulfoanción Etiqueta de la muestra Mn
(g/mol) Mw
(g/mol) Mp
(g/mol) Ð
- D 68,012 259,095 103,405 3.8 T 54,068 302,607 88,826 5.6
Grupo I
DC- 60min60% 1,903 5,498 6,834 2.9 DC-120min50% 903 3,413 4,258 3.8 DC-120min40% 4,415 7,122 7,394 1.6 DC- 60min40% 1,825 4,902 5,708 2.7 DC-120min20% 2,337 10,028 13,061 4.3 DC- 60min20% 3,092 9,092 12,566 2.9
Grupo II
DC-30min20% 45,427 174,256 79,310 3.8 DC-10min20% 22,685 77,992 51,228 3.4 DC- 2min20% 51,276 247,495 105,221 4.8 DH-30min20% 44,867 195,426 64,734 4.4 DH-10min20% 45,315 224,963 64,086 5.0 DH- 2min20% 60,183 244,377 98,836 4.1 TC-30min20% 5,057 12,812 8,528 2.5 TC-10min20% 7,597 18,629 13,280 2.5 TC- 2min20% 27,667 210,429 50,000 7.6 TH-30min20% 57,059 527,475 91,226 9.2 TH-10min20% 45,820 504,790 93,860 11.0 TH- 2min20% 50,177 524,232 91,979 10.4
Grupo III DAg-240min170%0.110% 69,546 250,366 93,021 3.6 TAg-120min170%0.110% 53,174 377,535 92,873 7.1
En la Figura 2.5 se encuentran los cromatogramas de los copolímeros D sulfonados por 1 y 2 h
con CH3COOSO3H, (Grupo I de experimentos en Tabla 2.3). Se observa que la población de

CAPÍTULO 2
45
cadenas poliméricas sulfonadas disminuyó de peso molecular en comparación con el
copolímero D. Las cadenas poliméricas de mayor tamaño de los copolímeros sulfonados
empiezan en 40,000 g/mol y las más pequeñas se detectaron cerca de los 100 g/mol. Con estos
resultados se confirmó que los bajos pesos moleculares son los responsables de la ineficiencia
de estos materiales para formar películas mediante disolución-evaporación, ya sea porque las
películas se fracturan desde que el disolvente empezó a evaporarse, o al desmoldarlas.
Figura 2.5. Distribución de peso molecular del copolímero D sulfonado a diferentes grados y tiempos con CH3COOSO3H.
En la Figura 2.5 se observa que el perfil tipo campana de Gauss se va perdiendo por la
aparición de hombros e incluso hay regiones que llegan a parecer mesetas (DC-60min60%), lo
cual difiere del perfil del cromatograma del copolímero D. Estos cambios pueden ser el
resultado de reacciones secundarias de la sulfonación con CH3COOSO3H relacionadas con
procesos de degradación, a pesar de que las reacciones se llevaron a cabo a 40 °C.
El empleo de copolímeros de PS-PAA para usos diferentes a celdas de combustible se ha
reportado en la literatura, en donde se indica que durante la purificación de este tipo de
copolímeros, la temperatura no debe exceder los 90 °C para evitar reacciones de
deshidratación y la formación de grupos anhídridos [137]. La degradación térmica de grupos
carboxílicos comienza con la eliminación de agua unida a los grupos ácidos, seguida de la
deshidratación de los grupos –COOH vecinos para dar lugar a la formación de grupos
anhídridos, a lo cual prosiguen las reacciones de descarboxilación [58, 134, 136, 138, 139] y la
formación de grupos insaturados hasta llegar a la destrucción total de la matriz polimérica por
la depolimerización de la cadena principal [140].
1E7 1,000,000 100,000 10,000 1,000 1000.0
0.5
1.0
1.5
2.0
2.5
UA
Peso Molecular (g/mol)
DC-120min50% DC- 60min60% DC-120min40% DC- 60min40% DC-120min20% DC- 60min20% D

CAPÍTULO 2
46
Los estudios de degradación térmica de grupos carboxílicos que se encuentran en la literatura
se han llevado a cabo a altas temperaturas, sin embargo Arthur Ferris [141] en su patente
"Carboxysulphonic cation-exchange resins" menciona la presencia de reacciones de
degradación a temperaturas menores. Ferris indicó que durante el proceso de sulfonación de
copolímeros de estireno y viniltolueno con AA, ácido metacrílico o ésteres, sucede la pérdida
de grupos carboxílicos y posterior formación de estructuras cíclicas en las cadenas de
polímero, debido a la presencia del agente sulfonante en el medio de reacción. También
menciona que las reacciones de descarboxilación son más frecuentes en copolímeros con AA,
en comparación a copolímeros con éster. Ferris también mencionó que cuando la temperatura
de reacción es cercana a los 45 °C, la pérdida de grupos carboxílicos incrementa rápidamente,
alcanzando una pérdida de hasta el 50 % o más al llegar a los 60 °C.
Con fundamento en la información existente, se considera que cuando se utiliza
CH3COOSO3H para sulfonar el copolímero D, posiblemente ocurren reacciones de
descarboxilación catalizadas por el ambiente ácido, seguidas de la destrucción de la matriz
polimérica, ocasionando la reducción del peso molecular del copolímero. Proceso similar al
reportado por Ferris, con la diferencia de que las reacciones de descomposición están
sucediendo 5 °C por debajo de la temperatura que el reportó [141].
En las Figuras 2.6 y 2.7 se encuentran los cromatogramas de los copolímeros D y T sulfonados
con CH3COOSO3H durante 2, 10 y 30 min, se observa que los materiales obtenidos son de
peso molecular menor al del copolímero de partida (D o T).
Figura 2.6. Efecto del tiempo de reacción en el peso molecular del copolímero D al utilizar 20 % mol de CH3COOSO3H como agente sulfonante.
1E7 1,000,000 100,000 10,000 1,000 1000.00
0.25
0.50
0.75
1.00
DC-30min20% DC-10min20% DC- 2min20% D
UA
Peso molecular (g/mol)6.6E6 - 3.2E6

CAPÍTULO 2
47
Figura 2.7. Efecto del tiempo de reacción en el peso molecular del copolímero T al utilizar 20 % mol de CH3COOSO3H como agente sulfonante. En la Figura 2.8 se muestran los cromatogramas del copolímero D sulfonado con H2SO4, se
observa que la dispersidad y tamaño de las cadenas poliméricas de los materiales sulfonados
no cambian respecto al del copolímeros D sin sulfonar; inclusive el cromatograma del
copolímero DH-2min20% se sobrepone al del copolímero D. En cambio, para los materiales
sulfonados por 2, 10 y 30 minutos utilizando 20 % mol de H2SO4, provenientes del
copolímero T (Figura 2.9) la población se vuelve más dispersa y con un Mw entre 527,475 y
504,709 g/mol, siendo mayor que el del copolímero T sin sulfonar (Mw = 302,607 g/mol). Lo
anterior indica que los entrecruzamientos ocasionados por el H2SO4 forman geles insolubles a
nivel macroscópico, pero a nivel nanométrico aumenta el peso molecular de las cadenas, lo
cual se deduce tomando en cuenta que las soluciones analizadas se pasaron a través de filtros
de 0.2 micras.
Figura 2.8. Efecto del tiempo de reacción en el peso molecular del copolímero D al utilizar 20 % mol de H2SO4 como agente sulfonante.
1E7 1,000,000 100,000 10,000 1,000 1000.00
0.25
0.50
0.75
1.00
TC-30min20% TC-10min20% TC- 2min20% T
UA
Peso Molecular (g/mol)
1E7 1,000,000 100,000 10,000 1,000 1000.00
0.25
0.50
0.75
1.00
DH-30min20% DH-10min20% DH- 2min20% D
UA
Peso molecular (g/mol)6.6E6 - 3.2E6

CAPÍTULO 2
48
Figura 2.9. Efecto del tiempo de reacción en el peso molecular del copolímero T al utilizar 20 % mol de H2SO4 como agente sulfonante.
Al emplear H2SO4 en conjunto con Ag2SO4, para evitar las reacciones de entrecruzamiento de
los copolímeros D y T durante la sulfonación, para ambos copolímeros las mejores
concentraciones son 170 % mol de H2SO4 y 0.110 % mol de Ag2SO4, sin embargo el tiempo
de reacción para el copolímero D fue de 4 horas y de 2 horas para el copolímero T. Los
cromatogramas de estos materiales sulfonados se encuentran en la Figura 2.10.
Figura 2.10. Distribución de peso molecular de los copolímeros sulfonados con 170 % mol de H2SO4 y 0.011 % mol de Ag2SO4 a tiempos de: a) 4 h para el copolímero D, b) 2 h para el copolímero T.
En la Figura 2.10 se observa que el cromatograma del copolímero DAg-240min170%0.110%
mantiene el mismo perfil que el del copolímero D con la diferencia del hombro ubicado entre
2 y 6x106 g/mol. El perfil del cromatograma del copolímero TAg-120min170%0.110%, a
diferencia del correspondiente al copolímero T, presenta un hombro en el intervalo de peso
molecular de 1.77 a 12x105 g/mol, por lo que la dispersidad aumenta de 5.6 a 7.1, y su
1E7 1,000,000 100,000 10,000 1,000 1000.00
0.25
0.50
0.75
1.00
TH-30min20% TH-10min20% TH- 2min20% T
UA
Peso Molecular (g/mol)6.6E6 - 3.4E5
1E7 1,000,000 100,000 10,000 1,000 1000.00
0.25
0.50
0.75
1.00
UA
Peso molecular (g/mol)
DAg-240min170%0.110% D
1E7 1,000,000 100,000 10,000 1,000 1000.00
0.25
0.50
0.75
1.00
UA
Peso Molecular (g/mol)
TAg-120min170%0.110% T

CAPÍTULO 2
49
Mw cambia de 302,607 a 377,535 g/mol, respecto al del copolímero T sin sulfonar. El proceso
de sulfonación con los tres sistemas sulfonantes genera cambios en el tamaño de las cadenas
poliméricas, lo cual indica que además de la incorporación de grupos sulfónicos están
sucediendo reacciones secundarias que afectan a la cadena principal.
El uso de CH3COOSO3H disminuye el tamaño de las cadenas, incluso a bajos tiempos de
reacción y porcentajes de agente sulfonante. Cuando se utiliza H2SO4, la fracción soluble de
los copolímeros sulfonados aumenta de peso molecular, afectando más al copolímero T en
comparación al copolímero D. Y al usar los porcentajes molares mayores de H2SO4 en
conjunto con la sal de plata, se reduce el entrecruzamiento de cadenas, a diferencia de lo que
sucede en ausencia del Ag2SO4.
2.3.4 ESPECTROSCOPÍA INFRARROJO
La espectroscopía de infrarrojo se utilizó como primer recurso para comprobar la
incorporación de grupos sulfónicos en los copolímeros, mediante el seguimiento de la
variación de intensidad y/o de la ubicación de las señales de los copolímeros D y T, así como
de la aparición de señales cuando son sulfonados. Al sulfonar los copolímeros se observa un
incremento de las señales correspondientes a los compuestos de azufre, de 1390 a 1290 cm-1
para los estiramientos asimétricos del grupo O=S=O y de 1190 a 1120 cm-1 para los
estiramientos simétricos [66]. La señal ubicada de 1042 a 1020 cm-1 corresponde al grupo
sulfónico [14, 96, 98, 142] y la banda de la flexión de este mismo grupo (flexión S=O del
grupo sulfónico) aparece cerca de los 900 cm-1 [143]. En la mayoría de los espectros de los
materiales sulfonados disminuye la intensidad de las señales ubicadas en 702, 760, 1453 y
1498 cm-1, y la ubicada entre 3200 y 3000 cm-1 correspondientes a vibraciones C-H del anillo
aromático [144] debido a la sustitución de sus protones por otros grupos químicos. Además,
empleando los mismos tiempos de reacción y mismo porcentaje de agente sulfonante, la
disminución de la intensidad de las señales correspondientes a las vibraciones C-H del anillo
es más pronunciada al utilizar CH3COOSO3H en comparación al uso de H2SO4. Estas
variaciones de señales se observan en las Figuras 2.11-2.13.

CAPÍTULO 2
50
Figura 2.11. Comparación entre espectros con tiempo de sulfonación constante y porcentaje molar de agente sulfonante diferente
3500 2800 1800 1600 1400 1200 1000 800 600
30
40
50
60
70
80
90
3500 2800 1800 1600 1400 1200 1000 800 600
a)
*
Tran
smita
ncia
(%)
Número de onda (cm-1)
D DC-120min20% DC-120min40% DC-120min60%
cm-1
3450
~1700cm-1
1315 - 1050 cm-1
1683cm-1
3500 2800 1800 1600 1400 1200 1000 800 600
30
40
50
60
70
80
90
3500 2800 1800 1600 1400 1200 1000 800 600
b)
*cm-1
Tran
smita
ncia
(%)
Número de onda (cm-1)
D DC-60min20% DC-60min40% DC-60min60%
3450
~1700cm-1
1315 - 1050 cm-1
1683cm-1
3500 2800 1800 1600 1400 1200 1000 800 600
30
40
50
60
70
80
90
3500 2800 1800 1600 1400 1200 1000 800 600
c)
*cm-1
Tran
smita
ncia
(%)
Número de onda (cm-1)
D DH-120min20% DH-120min40% DH-120min60%
3450
~1700cm-1
1315 - 1050 cm-1
3500 2800 1800 1600 1400 1200 1000 800 600
30
40
50
60
70
80
90
3500 2800 1800 1600 1400 1200 1000 800 600
d)
cm-1
3450
Tran
smita
ncia
(%)
Número de onda (cm-1)
D DH-60min20% DH-60min40% DH-60min60%
~1700cm-1
1315 - 1050 cm-1
*

CAPÍTULO 2
51
Figura 2.12. Comparación entre espectros con porcentaje molar de agente sulfonante constante y tiempo de sulfonación diferente.
3500 2800 1800 1600 1400 1200 1000 800 600
30
40
50
60
70
80
90
3500 2800 1800 1600 1400 1200 1000 800 600
a)
*
cm-1
Tran
smita
ncia
(%)
Número de onda (cm-1)
D DC- 2min20% DC- 10min20% DC- 30min20% DC- 60min20% DC-120min20%
16833450 cm-1
~1700cm-1
1315 - 1050 cm-1
3500 2800 1800 1600 1400 1200 1000 800 600
30
40
50
60
70
80
90
3500 2800 1800 1600 1400 1200 1000 800 600
b)
*
Tran
smita
ncia
(%)
Número de onda (cm-1)
D DH- 2min20% DH- 10min20% DH- 30min20% DH- 60min20% DH-120min20%
3450 cm-1
~1700cm-1
1315 - 1050 cm-1
3500 2800 1800 1600 1400 1200 1000 800 600
30
40
50
60
70
80
90
3500 2800 1800 1600 1400 1200 1000 800 600
c)
*
cm-1
Tran
smita
ncia
(%)
Número de onda (cm-1)
T TC- 2min20% TC-10min20% TC-30min20%
1730
cm-11683
3450 cm-1
~1700cm-1
1315 - 1050 cm-1
3500 2800 1800 1600 1400 1200 1000 800 600
30
40
50
60
70
80
90
3500 2800 1800 1600 1400 1200 1000 800 600
d)
*
Tran
smita
ncia
(%)
Número de onda (cm-1)
T TH- 2min20% TH-10min20% TH-30min20%
cm-1
3450
~1700cm-1
1315 - 1050 cm-1

CAPÍTULO 2
52
Figura 2.13. Comparación entre espectros de copolímeros D y T sulfonados con H2SO4-Ag2SO4 con variación de: tiempo de sulfonación y/o porcentajes de ácido sulfúrico y/o porcentaje de Ag2SO4.
3500 2800 1800 1600 1400 1200 1000 800 600
30
40
50
60
70
80
90
3500 2800 1800 1600 1400 1200 1000 800 600
*
a)
Tran
smita
ncia
(%)
Número de onda (cm-1)
D DAg-120min85%0.110% DAg-240min85%0.110% DAg-360min85%0.110%
cm-1
3450
~1700cm-1
1492cm-1
1457cm-1
1315 - 1050 cm-1
cm-1760
702cm-1
3500 2800 1800 1600 1400 1200 1000 800 60030
40
50
60
70
80
90
3500 2800 1800 1600 1400 1200 1000 800 600
1050 cm-1
1315 -
d)
*
Tran
smita
ncia
(%)
Número de onda (cm-1)
T TAg-120min85%0.110% TAg-240min85%0.110% TAg-360min85%0.110%
1492cm-1
1457cm-1
702cm-1
760cm-1
cm-1
3450
~1700cm-1
3500 2800 1800 1600 1400 1200 1000 800 60030
40
50
60
70
80
90
3500 2800 1800 1600 1400 1200 1000 800 600
*
b)
Tran
smita
ncia
(%)
Número de onda (cm-1)
D DAg-240min 85%0.110% DAg-240min170%0.110% DAg-240min255%0.110%
cm-1
3450
~1700cm-1
1492cm-1
1457cm-1
1050 cm-1
1315 -
cm-1760 702cm-1
3500 2800 1800 1600 1400 1200 1000 800 60030
40
50
60
70
80
90
3500 2800 1800 1600 1400 1200 1000 800 600
*
e)
Tran
smita
ncia
(%)
Número de onda (cm-1)
T TAg-120min 85%0.110% TAg-120min170%0.110% TAg-120min225%0.110%
cm-1
3450
~1700cm-1
1492cm-1
1457cm-1
1315 - 1050 cm-1
cm-1760
702cm-1
3500 2800 1800 1600 1400 1200 1000 800 600
30
40
50
60
70
80
90
3500 2800 1800 1600 1400 1200 1000 800 600
*
c)
Tran
smita
ncia
(%)
Número de onda (cm-1)
D DAg-240min170%0.055% DAg-240min170%0.110% DAg-240min170%0.220%
cm-1
3450
~1700cm-1
1492cm-1
1457cm-1
1315 - 1050 cm-1
cm-1760 702cm-1
3500 2800 1800 1600 1400 1200 1000 800 600
30
40
50
60
70
80
90
3500 2800 1800 1600 1400 1200 1000 800 600
Tran
smita
ncia
(%)
Número de onda (cm-1)
T TAg-120min170%0.055% TAg-120min170%0.110% TAg-120min170%0.220%
f)
*
cm-1
3450 ~1700cm-1
1492cm-1
1457cm-1
1315 - 1050 cm-1
cm-1760
702cm-1

CAPÍTULO 2
53
En las Figuras 2.11c, 2.11d y 2.12b la intensidad de la señal en 702 cm-1 aumenta
probablemente debido a los estiramientos simétrico y asimétrico de C-S-C que se presentan en
esa longitud de onda [145-149], los cuales se relacionan con la formación de grupos sulfona.
El incremento de esta señal es congruente a lo esperado, ya que estos materiales se sulfonaron
con H2SO4, generando geles insolubles por el incremento de entrecruzamientos.
Otra diferencia importante entre los espectros de los copolímeros sulfonados con las mismas
condiciones de reacción y variando entre H2SO4 y CH3COOSO3H es la intensidad de la señal
en 3450 cm-1, la cual indica la presencia de grupos O-H provenientes del grupo –SO3H [150].
Cuando se utiliza H2SO4 la señal de 3450 cm-1 es menor que cuando se utiliza CH3COOSO3H,
análogo a lo que sucede con la señal de 615 cm-1 correspondiente al estiramiento del enlace
simple S-O del grupo sulfónico [151]. Esta diferencia podría corresponder a una mayor
incorporación de grupos –SO3H al emplear CH3COOSO3H en comparación al uso de H2SO4.
Además de las señales características debidas a las reacciones de sulfonación, se observa que
la señal del grupo carbonilo (1704 cm-1) cambia de intensidad y/o posición, dependiendo del
tipo de agente sulfonante empleado. El enlace C=O de cetonas, aldehídos, ácidos carboxílicos,
esteres, lactonas, haluros de acilo, anhídridos, amidas y lactamas muestran una banda de
absorción por el estiramientos C=O en la región de 1870-1540 cm-1. Dentro de este intervalo
la posición del la banda del grupo carbonilo está determinada por factores como el estado
físico de la muestra, efecto electrónico de los sustituyentes vecinos, los puentes de hidrógeno
(intermolecular e intramolecular) y la presencia de anillos aromáticos [65].
Cuando se utiliza CH3COOSO3H (Figuras 2.11a y b, 2.12c y d), la señal de carbonilo
(1704 cm-1) disminuye de intensidad, inclusive desaparece en algunos de los materiales
sulfonados, además de que aparece otra señal de carbonilos ubicada en 1683 cm-1. Esa región
del espectro de infrarrojo puede corresponder al estiramiento de un enlace C=O de cetonas
conjugadas con un doble enlace C=C, lo cual genera una deslocalización de los electrones π de
ambos grupos insaturados. La deslocalización de los electrones π del grupo C=O reduce el
carácter de doble enlace entre C-O, causando una absorción a menores números de onda
(longitud de onda mayores), tal y como sucede con la conjugación con un grupo fenilo, que
causa una absorción en la región de entre 1685-1666 cm-1 [152]. De modo que la presencia de
las dos señales ubicadas en 1683 y 1704 cm-1 pueden corresponder respectivamente al

CAPÍTULO 2
54
estiramiento del enlace C=O de una cetona α,β conjugada formada durante la
sulfonación [153], y al de un grupo carboxílico [140] propio del copolímero base D o T.
Al cotejar los espectros de las reacciones donde se utilizó únicamente H2SO4
(Figuras 2.11c, 2.11d, 2.12c, 2.12d) en comparación al uso del ácido sulfúrico con Ag2SO4
(Figura 2.13), se observa que la banda correspondiente al estiramiento del enlace C=O del
grupo carboxílico (1704 cm-1) se mantiene en todos los casos a pesar de que en algunos de
ellos disminuye de intensidad. Sin embargo también aparece una segunda señal a números de
onda mayores (1730 cm-1), que también puede corresponder a un enlace C=O pero de tipo
éster α,β insaturado en conjunto con una señal múltiple de 1300 a 1160 cm-1 correspondiente
al estiramiento C-C(=O)-O [65]. La señal entre 1300 a 1160 cm-1 es difícil de corroborar
porque en esa región también se encuentran las señales correspondientes a los compuestos
azufrados.
Otra explicación a la aparición de la señal en 1730 cm-1 es un una interacción entre unidades
de ácido acrílico o con el ambiente químico que las rodea. Ostrowska reportó [154] que un
poliácido acrílico, además de mostrar una señal intensa y predominante en ~1700 cm-1,
también puede presentar hombros en las cercanías de 1695, 1723 y 1737 cm-1. La señal
principal de ~1700 cm-1 corresponde a la vibración de los grupos carbonilo cuando se
encuentran formando “ciclos diméricos”, que consisten en la interacción de dos grupos
carboxílicos mediante dos puentes de hidrógeno. Las señales tipo hombro corresponden a
estructuras con diferentes puentes de hidrógeno, donde un oxígeno de un grupo carbonilo
interacciona con un átomo de hidrógeno de otro ácido carboxílico, y donde ese segundo grupo
carboxílico interacciona con otro diferente, y así sucesivamente, las cuales son llamadas
“dímero abierto”. Las señales tipo hombro también pueden deberse a la existencia de puentes
de hidrógeno entre grupos carboxílicos y moléculas de agua cercanas. Aunado a esto, el ancho
de estas señales depende de la superposición de las bandas de grupos carbonilos que se
desplazan unas de las otras debido a una distribución no-homogénea de los mismos a lo largo
de la cadena polimérica [154, 155].
En todas las reacciones donde se utilizó ácido sulfúrico con Ag2SO4 se obtuvieron materiales
insolubles en agua, solubles en THF y aptos para formar películas mediante
disolución-evaporación, además de que se obtuvo una mayor incorporación de grupos
sulfónicos, lo cual se ve reflejado cualitativamente en señales de mayor intensidad
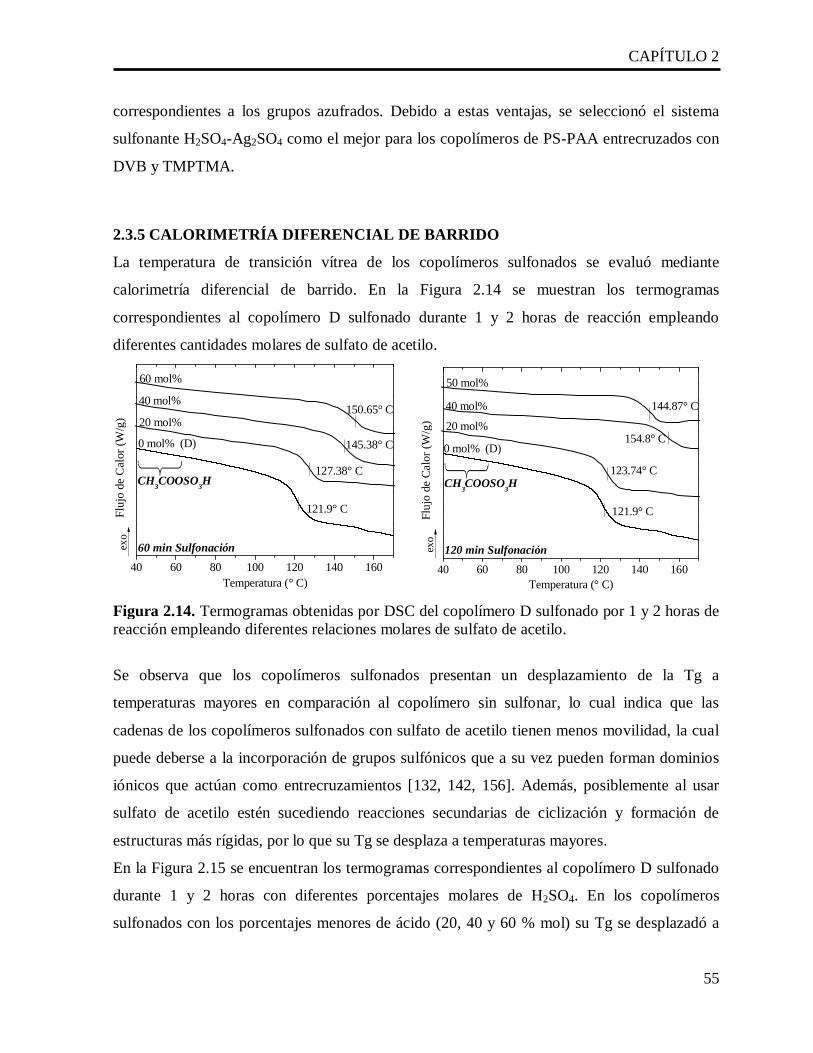
CAPÍTULO 2
55
correspondientes a los grupos azufrados. Debido a estas ventajas, se seleccionó el sistema
sulfonante H2SO4-Ag2SO4 como el mejor para los copolímeros de PS-PAA entrecruzados con
DVB y TMPTMA.
2.3.5 CALORIMETRÍA DIFERENCIAL DE BARRIDO
La temperatura de transición vítrea de los copolímeros sulfonados se evaluó mediante
calorimetría diferencial de barrido. En la Figura 2.14 se muestran los termogramas
correspondientes al copolímero D sulfonado durante 1 y 2 horas de reacción empleando
diferentes cantidades molares de sulfato de acetilo.
Figura 2.14. Termogramas obtenidas por DSC del copolímero D sulfonado por 1 y 2 horas de reacción empleando diferentes relaciones molares de sulfato de acetilo.
Se observa que los copolímeros sulfonados presentan un desplazamiento de la Tg a
temperaturas mayores en comparación al copolímero sin sulfonar, lo cual indica que las
cadenas de los copolímeros sulfonados con sulfato de acetilo tienen menos movilidad, la cual
puede deberse a la incorporación de grupos sulfónicos que a su vez pueden forman dominios
iónicos que actúan como entrecruzamientos [132, 142, 156]. Además, posiblemente al usar
sulfato de acetilo estén sucediendo reacciones secundarias de ciclización y formación de
estructuras más rígidas, por lo que su Tg se desplaza a temperaturas mayores.
En la Figura 2.15 se encuentran los termogramas correspondientes al copolímero D sulfonado
durante 1 y 2 horas con diferentes porcentajes molares de H2SO4. En los copolímeros
sulfonados con los porcentajes menores de ácido (20, 40 y 60 % mol) su Tg se desplazadó a
40 60 80 100 120 140 160
Fluj
o de
Cal
or (W
/g)
Temperatura (° C)
60 min Sulfonación
CH3COOSO3H
121.9° C
127.38° C
145.38° C
150.65° C
60 mol%
20 mol%
40 mol%
0 mol% (D)
exo
40 60 80 100 120 140 160
123.74° C
Fluj
o de
Cal
or (W
/g)
Temperatura (° C)
154.8° C
144.87° C
CH3COOSO3H
120 min Sulfonación
0 mol% (D)
20 mol%
40 mol%
50 mol%
121.9° C
exo

CAPÍTULO 2
56
temperaturas menores en comparación al copolímero D. En cambio, cuando se utiliza
100 % mol de H2SO4, la temperatura a la cual se presenta la transición vítrea es mayor.
Figura 2.15. Termogramas obtenidas por DSC del copolímero D sulfonado por 1 y 2 horas de reacción empleando diferentes relaciones molares de H2SO4.
La disminución de la Tg al emplear los porcentajes molares de H2SO4 menores al 100 %
puede deberse al incremento del espacio entre cadenas poliméricas cuando se incorpora el
grupo químico sulfónico, el cual genera mayor impedimento estérico que el protón existente
inicialmente en el polímero, al cual sustituye [157]. El espacio adicional entre cadenas les
otorga libertad de movimiento, por lo que su Tg se aprecia a temperaturas menores. En
cambio, cuando se utiliza 100 % mol de H2SO4, la Tg del copolímero sulfonado es mayor a la
Tg del copolímero sin sulfonar, este efecto contrario con un mismo agente sulfonante puede
deberse a que al utilizar concentraciones altas de H2SO4, el copolímero se sulfona pero
también se entrecruza, lo cual se comprobó mediante la determinación de material insoluble
mediante extracción soxhlet. Los entrecruzamientos restringen el movimiento de las cadenas
disminuyendo su flexibilidad, por lo cual los copolímeros sulfonados por 1 y 2 horas con
100 % mol de H2SO4 presentan su Tg a temperaturas mayores [158].
Los termogramas de los copolímeros sulfonados con el sistema H2SO4-Ag2SO4 se muestran en
la Figura 2.16.
40 60 80 100 120 140 160
60 min Sulfonación
Fluj
o de
Cal
or (W
/g)
Temperatura (° C)
121.9° C
113.74° C
114.91° C
115.25° C
130.40° C
100 mol%
H2SO4
0 mol% (D)
20 mol%
40 mol%
60 mol%
exo
40 60 80 100 120 140 160
120 min Sulfonación
121.9° C
113.68° C
116.24° C
112.11° C
138.9° C100 mol%
0 mol% (D)
60 mol%
40 mol%
20 mol%
Fluj
o de
Cal
or (W
/g)
Temperatura (° C)
H2SO
4
exo

CAPÍTULO 2
57
Figura 2.16. Termogramas obtenidas por DSC de los copolímeros D y T sulfonado con ácido sulfúrico-Ag2SO4.
En éstos termogramas se puede ver que a pesar de llevar a cabo las reacciones de sulfonación
por 2 y 4 horas con 170 % mol de H2SO4, la Tg de los materiales sulfonados es menor que la
Tg de los materiales de partida (copolímeros D y T), como efecto de la utilización de la sal de
plata. El desplazamiento de la Tg de los copolímeros sulfonados a temperaturas menores
puede deberse a la incorporación de grupos sulfónicos (comprobada mediante espectroscopía
de infrarrojo), sin la formación de material de entrecruzamientos químicos, comprobado
mediante extracción por soxhlet y GPC. Por lo tanto los termogramas de estos materiales
sulfonados indican que el incremento del espacio entre cadenas que genera la presencia de
grupos sulfónicos en los anillos estirénicos tiene un efecto superior a la restricción generada
por los entrecruzamientos formados entre ellas.
2.3.6 ESTABILIDAD TÉRMICA DE LOS COPOLÍMEROS SULFONADOS
Para determinar si las membranas preparadas con los mejores copolímeros sulfonados serían
térmicamente estables a la temperatura de trabajo de una celda de combustible de hidrógeno o
metanol (80 – 100 °C) [88], se llevó a cabo su caracterización mediante TGA. En las
Figuras 2.17 y 2.18 se presentan los termogramas corresponden a los copolímeros D y T
sulfonados con ácido sulfúrico-Ag2SO4.
40 60 80 100 120 140 160
D
T
DAg-240min170%0.110%
TAg-120min170%0.110%
Fluj
o de
cal
or (W
/g)
Temperatura (° C)
121.9° C
129.3° C
110.0° C
84.7° C
exo

CAPÍTULO 2
58
Figura 2.17. Análisis termogravimétrico del copolímero D sulfonado con diferentes tiempos y concentraciones de ácido sulfúrico y sulfato de plata.
Figura 2.18. Análisis termogravimétrico del copolímero T sulfonado con diferentes tiempos y concentraciones de ácido sulfúrico y sulfato de plata.
Puede observarse que desde el inicio de la prueba hasta el primer cambio abrupto de pendiente
(aproximadamente a 150 °C), los copolímeros sulfonados pierden masa, lo cual puede deberse
a la degradación de los grupos carboxílicos de los copolímeros D y T sulfonados catalizada
por el ambiente ácido de los grupos sulfónicos, originando anhídridos y estructuras cíclicas,
cuya ocurrencia ha sido reportado desde los 25 hasta los 150 °C [159].
La primera pérdida drástica de masa observada cerca de los 150 °C puede deberse a la pérdida
de agua o disolvente resudual. A pesar de que 150° C es una temperatura por arriba de la
prevista para la evaporación de agua, en la literatura sido reportado la pérdida de agua
adsorbida en copolímeros de alcohol polivinilico/poliestireno sulfonado a temperatura de
100 200 300 400 500
Pérd
ida
en m
asa
(%)
Temperatura (°C)
288.66° C
318.91° C
337.96° C
295.80° C
310.10° C281.51° C
298.42° C
323.2° Cabcd
efg
325.10° C
10%
100 200 300 400 5000
80
85
90
95
100
Pérd
ida
en p
eso
(%)
Temperatura (°C)
Nafion 117 D
a DAg120min- 85%0.110% b DAg240min- 85%0.110% c DAg240min-170%0.110% d DAg360min- 85%0.110% e DAg240min-225%0.110% f DAg240min-170%0.220% g DAg240min-170%0.055%
d
b
e
g
f
c
a
100 200 300 400 500
288.66° C
Temperatura (°C)
286.75° C315.57° C
331.29° C
310.57° C
288.90° C313.43° C284.40° C
328.10° Chiklm
np
100 200 300 400 5000
80
85
90
95
100
Pérd
ida
de m
asa
(%)
Temperatura (°C)
m
k l
pi
nh Nafion 117 T
h TAg120min- 85%0.110% i TAg240min- 85%0.110% k TAg120min-170%0.110% l TAg120min-170%0.220% m TAg360min- 85%0.110% n TAg120min-225%0.110% p TAg120min-170%0.055%

CAPÍTULO 2
59
100 a 150 °C [51], y hasta 200 °C cuando el agua se encuentra adsorbida en muestras porosas
de copolímero de poliestireno sulfonado-poliácido acrílico; además esta pérdida de agua
conlleva formación de estructuras cíclicas anhidridas [134]. Otra explicación a la pérdida de
masa en 150 °C concuerda a lo observado por McGaugh en un estudio de degradación térmica
de poliácido acrílico, donde la pérdida de grupos carboxílicos y formación de anhídridos
sucede entre 150 y 275 °C [159]. Por otro lado, a partir de los 300 °C se pueden formar grupos
cetona y éster, y a los 350 °C estructuras cíclicas. Rhim encontró en el 2004 mediante análisis
termogravimétricos de copolímeros de alcohol polivinilico/poliestireno sulfonado, que se
presenta una disminución de masa entre 250–400 °C debida a la pérdida de grupos sulfónicos
y rompimiento de grupos éster [51].
También se puede observar que los copolímeros T sulfonados presentan mayor pérdida de
masa en comparación a los copolímeros D sulfonados en la temperatura de 150 a 300 °C, esta
pérdida puede estar relacionada a que el entrecruzante del copolímero T contiene enlaces tipo
éster, los cuales se degradan entre 250-400 °C y cuyo efecto se suma al de degradación de
grupos sulfónicos, que se presenta en el mismo intervalo de
temperatura (250-400 °C) [51, 160, 161].
Otra observación general de los termogramas de los copolímeros sulfonados, es que su pérdida
mayoritaria de peso ocurre a temperaturas menores que la del copolímero sin sulfonar
(cerca de 300 ºC), lo cual probablemente esté relacionado a la presencia de grupos sulfónicos,
que aceleran la degradación del resto de la estructura polimérica.
A pesar de esta disminución de la temperatura de degradación por la presencia de los grupos
sulfónicos, todos ellos se degradan a una temperatura superior a los 280 °C, siendo un valor de
temperatura mayor a la de operación en una PEMFC en funcionamiento (80–100 °C) [88].

CAPÍTULO 2
60
CONCLUSIONES DEL CAPÍTULO 2
El proceso de sulfonación con los tres sistemas sulfonantes genera cambios en el
tamaño de las cadenas poliméricas, indicando la ocurrencia de reacciones secundarias
además de la incorporación de grupos sulfónicos
La diferencias primordiales entre los tres sistemas sulfonantes son: El uso de
CH3COOSO3H disminuye el tamaño de las cadenas, incluso empleando bajos tiempos
de reacción y porcentajes de agente sulfonante. Cuando se utiliza únicamente H2SO4,
se forman entrecruzamientos, incrementando la fracción insoluble, mientras que la
fracción soluble aumenta de peso molecular, además de que éste efecto es mayor en el
copolímero T en comparación al copolímero D. Al emplear H2SO4-Ag2SO4 se reduce
el entrecruzamiento de cadenas, a diferencia de lo que sucede en ausencia de la sal de
plata.
Emplear tiempos menores o iguales a 30 minutos de sulfonación con 20% de agente
sulfonante genera materiales sulfonados insolubles en agua, solubles en THF, útiles
para formar película que no se fracturaron al secar, desmoldar o durante su purificación
y activación, pero sin IEC competitiva a la de Nafion 117.
El mejor sistema sulfonante para los copolímeros de PS-PAA entrecruzados con DVB
y TMPTMA fue el H2SO4 - Ag2SO4, con el cual se generaron copolímeros sulfonados
insolubles en agua y solubles en THF, capaces de formar película y con mayor
incorporación de grupos sulfónicos (comprobado cualitativamente con la intensidad de
las señales de IR correspondientes a los grupos azufrados).
Los dos mejores materiales sulfonados con H2SO4-Ag2SO4 fueron los copolímeros
DAg-240min170%0.110% y TAg-120min170%0.110%, debido a su capacidad de
intercambio iónico competitiva al Nafion 117; 0.5524, 0.9030 y 0.87 meq/g
respectivamente.

CAPÍTULO 3
61
CAPÍTULO 3. MEMBRANAS
3.1 ANTECEDENTES
En una celda de combustible de membrana intercambiadora de protones, entre los combustible
que se pueden utilizar son hidrógeno o un alcohol, y los iones móviles de carga positiva que
deben pasar a través de la membrana para cerrar el circuito químico son protones [82, 162]. La
primera membrana de intercambio protónico se preparó en 1950, la cual consistía en una
mezcla de resina de intercambio iónico en polvo con un polímero que lo mantenía unido [162].
Posteriormente se lograron mejoras para la obtención de membranas iónicas, iniciando por el
cambio del mezclado mecánico por la síntesis química de polímeros iónicos para formar con
ellos la membrana. En 1962 se obtuvo el primer electrolito polimérico intercambiador de
protones con base en poliestireno sulfonado que se utilizó en la celda de combustible del
satélite espacial Gemini. A pesar de este logro, se continuó la búsqueda de electrolitos
poliméricos de mayor tiempo de vida en las celdas de combustible, y fue en 1963 cuando
Walther Grot, trabajador de la compañía Du Pont sintetizó un polímero de ácido
perfluorosulfónico el cual es químicamente más estable que la resina de poliestireno sulfonado
y que se nombró Nafion [163].
Actualmente el Nafion se utiliza en la mayoría de los estudios científicos como estándar de
referencia, el cual posee grupos de ácido fuerte, lo cual le confiere conductividad protónica
elevada, además de poseer estabilidad química, buen desempeño mecánico bajo condiciones
normales de operación (menos de 80 °C) y porque se encuentran comercialmente disponible
aunque su precio es elevado [164]. A pesar de sus características favorables, las membranas de
Nafion presentan limitaciones; permeación de metanol desde el ánodo hacia el cátodo, lo que
reduce el voltaje y de la eficiencia de las celdas de éste combustible [9, 164, 165]. Además, en
una PEMFC que utiliza Nafion, cuando la temperatura de trabajo es cercana a los 100 °C la
conductividad de protones disminuye o se interrumpe por la deshidratación de la membrana.
Las membranas de Nafion tienen baja velocidad de hidratación, por lo que genera problemas
con el balance del agua en el total de la membrana y deshidratación del lado del ánodo [95].
Otro problema del Nafion es debido a los ciclos de hidratación y deshidratación que van
acompañados de incremento y reducción de dimensiones de la membrana, lo cual genera
discontinuidad de la capa catalítica en la membrana, incrementando la resistencia entre el

CAPÍTULO 3
62
electrodo y ésta, además de generar su agrietamiento o perforación, pudiendo producir un
corto circuito químico que culmine en una reacción exotérmica violenta [95].
Aunado a lo anterior, la presencia de un disolvente polar (agua o metanol) hincha las
membranas de Nafion, reduciéndose las fuerzas intermoleculares, e incrementando su
elongación, además de que la membrana se vuelve más dúctil. El disolvente genera un impacto
directo en la relación esfuerzo-deformación, lo que genera que su módulo de Young
disminuya, haciendo que la membrana se vuelve susceptible a la deformación permanente con
debilitamiento gradual, ocasionando su falla en la celda de combustible cuando existen
gradientes de presión en ella [166, 167]. Las membranas de Nafion son difíciles de reciclar,
sintetizar, procesar, disolver, fundir o extruir en su forma iónica, lo cual restringe el diseño de
celdas novedosas cuyas geometrías no sean planas. Y debido a su alto costo, se limita su uso
en aplicaciones masivas [9, 164, 165].
Debido a estos inconvenientes, actualmente hay muchos polímeros conductores alternativos al
Nafion para preparar membranas bajo investigación, los cuales incluyen poliarilenos,
poliimidas, polifosfacenos, polibenzimidazoles, compositos orgánicos, inorgánicos e híbridos,
copolímeros de PS en di y tribloque, etc. [5-9, 168]; o como en este trabajo, copolímeros
sulfonados de PS-PAA entrecruzados con DVB o TMPTMA. Estas investigaciones tienen
como fin conseguir materiales con las siguientes propiedades:
(1) Estabilidad química y electroquímica (durabilidad). Deben ser químicamente estables en
las condiciones de operación, debido a que la naturaleza ácida de la membrana y el ambiente
oxidante de ánodo producen condiciones muy agresivas, que en conjunto culminen en la
degradación de las membranas y la descomposición de los electrocatalizadores [169].
(2) Elevada conductividad protónica. Conducir especies iónicas (iones hidronio y/o protones)
entre los electrodos con la mínima caída óhmica [170].
(3) Impermeabilidad de combustible a través de la membrana. Funcionar como barrera física
(o baja permeabilidad) para evitar la mezcla directa del combustible y el oxidante [171, 172].
(4) Retención de agua. Deben ser capaces de retener moléculas de agua en las condiciones de
trabajo, debido a que la transferencia de protones ocurre cuando el electrolito polimérico se
encuentra hidratado [163].
(5) Bajo costo. El electrolito polimérico debe ser económicamente accesible para que su uso
en una FC sea viable [163].

CAPÍTULO 3
63
(6) Estabilidad mecánica. El material utilizado para la membrana debe poseer resistencia
mecánica en estado hidratado para soportar los procesos a los que se expondrán [163, 172].
Las propiedades mecánicas de una membrana polimérica están gobernadas por su naturaleza
química; peso molecular del polímero, rigidez de la estructura química, ramificaciones en el
polímero, grado de sulfonación, absorción de agua, etc. Cada uno de estos factores genera un
efecto en su desempeño, que está relacionado con la estabilidad de la membrana en la celda de
combustible [173]. El deterioro de las propiedades mecánicas de las membranas poliméricas
se refleja como perforaciones casi imperceptibles, hoyos, fisuras e incluso desgarros, para
evitar que esto suceda, es necesario que las membranas sean uniformes para que la presión a la
que son sometidas durante la preparación de ensambles membrana-electrodo y entre los platos
bipolares sea igualmente uniforme al momento en que la celda se encuentre en operación [88].
Los procesos a los que son sometidas las membranas incluyen su preparación (extrusión,
disolución-evaporación, etc.), incorporación de capas catalíticas, compresión aplicada durante
la preparación del ensamble membrana-electrodo, presión de ensamble y cerrado de la
PEMFC para probar su desempeño, cambio dimensional durante la hidratación y
deshidratación de la membrana, así como la presión generada por el oxidante y el combustible
(gaseoso o líquido). De aquí la importancia de la evaluación de las propiedades químicas y
mecánicas de las membranas preparadas con los copolímeros sulfonados obtenidos en este
trabajo.
3.2. EXPERIMENTAL
3.2.1 MATERIALES
Ácido clorhídrico (36.5-38 %, Sigma Aldrich), ácido nítrico (70 %, CTR Scientific), ácido
sulfúrico (98 % J.T.Baker), cloruro de sodio (J.T. Baker), copolímero de PS-PAA
entrecruzado con DVB sulfonado , copolímero de PS-PAA entrecruzado con TMPTMA
sulfonado,dimetilsulfóxido (≥ 99.5 %, Sigma Aldrich), hidróxido de sodio (Aldrich),
membrana de Nafion 117 (Sigma Aldrich), peróxido de hidrógeno (solución al 30 % en agua,
Sigma Aldrich), tetrahidrofurano libre de inhibidor ≥ 99.9 % (Aldrich).
3.2.2 PREPARACIÓN DE LAS MEMBRANAS
Las películas se obtuvieron mediante disolución-evaporación, para lo cual se disolvió el
copolímero en THF o mezcla de THF con dimetilsulfóxido (DMSO). La solución polimérica

CAPÍTULO 3
64
se vertió en un molde de área conocida y se dejó evaporar el disolvente. Se utilizó una relación
de 2.4 mL de disolvente o mezcla de disolventes por cada 0.4 g de copolímero, y un molde de
area igual a 16 cm2. Una vez volatilizado el disolvene a temperatura y presión ambiente, la
película se desmoldó, se le extrajo el DMSO y los iones plata, y posteriormente se activó la
membrana.
Se utilizaron dos porcentajes de DMSO, 55 y 110 %, que equivale a emplear 0.4 g de
copolímero con 0.22 o 0.44 mL de DMSO (55 y 110 % respectivamente), y se completó el
volumen a 2.4 mL con THF. La extracción líquido-líquido del dimetilsulfóxido presente en las
membranas se realizó con agua durante 2 días. Mientras que los iones plata de las películas
preparadas con los copolímeros sulfonados con H2SO4-Ag2SO4, se extrajeron sumergiéndolas
en una solución de ácido nítrico (HNO3) 0.5 M por 24 h.
3.2.3 ACTIVACIÓN DE LAS MEMBRANAS
La activación de las películas de PS-PAA sulfonadas se llevó a cabo colocándolas en peróxido
de hidrógeno al 5 % y 80°C durante 1 h, en H2SO4 0.5 M a 80 °C por una hora, y en agua
desionizada a 80 °C por una hora. Este procedimiento es similar al reportado para las
membranas de Nafion [174, 175].
3.2.4 CARACTERIZACIÓN
3.2.4.1 DETERMINACIÓN DE DISOLVENTE RESIDUAL EN LAS MEMBRANAS
La verificación de la eliminación de DMSO de las membranas se llevó a cabo mediante
Cromatografía de Gases con detector de Masas, para lo cual se utilizó un cromatógrafo de
gases AGILENT Instrument con una columna HP-5M5 de 30 m x 0.25 mm, corriente de helio
como gas portador a velocidad de 1.5 mL/min y temperatura del horno e inyector de 200 °C.
La membrana se colocó en estufa a 100 °C por 1 h, aún caliente se transfirió a un vial
hermético, el cual se calentó a 200 °C durante 1 h para y a continuación se tomó la muestra de
la parte gaseosa del vial.
3.2.4.2 CAPACIDAD DE INTERCAMBIO IÓNICO DE LAS MEMBRANAS
La IEC de las membranas se midió indirectamente mediante titulación de la solución de NaCl
en la cual se intercambiaron sus protones por iones sodio, método descrito en el capítulo 2.

CAPÍTULO 3
65
3.2.4.3 ABSORCIÓN DE AGUA DE LAS MEMBRANAS
La determinación de la absorción de agua de las membranas se hizo a partir de la diferencia de
su peso en estado hidratado y seco, utilizando la ecuación 3.1 [37, 176]:
푎푏푠표푟푐푖ó푛푑푒푎푔푢푎(%푝푒푠표) = ( ) 푥100 (ec.3.1)
Los valores de 푊 푦푊 de la ecuación (3.1) se obtuvieron durante la determinación
de la IEC.
3.2.4.4 RELACIÓN DE HINCHAMIENTO DE LAS MEMBRANAS
La relación de hinchamiento de las membranas se determinó a partir de su diferencia de
espesor en estado hidratado y seco mediante la ecuación 3.2. [177]. El proceso de hidratación
es igual el descrito para la determinación de absorción de agua.
푟푒푙푎푐푖ó푛푑푒ℎ푖푛푐ℎ푎푚푖푒푛푡표(%푒푠푝푒푠표푟) = ( ) 푥100 (ec. 3.2)
Los valores de 푑 푦푑 corresponden respectivamente a los espesores de las
membranas en estado hidratado y seco.
3.2.4.5 CARACTERIZACIÓN ELECTROQUÍMICA DE LAS MEMBRANAS
Las mediciones electroquímicas se realizaron con un Potenciostato Galvanostato BioLogic
Science Instruments SP-300, se emplearon 3 electrodos: uno estándar de trabajo de platino con
cuerpo aislante de PEEK, diámetro exterior de 6 mm y diámetro del electrodo de 1.6 mm; un
electrodo de referencia de plata/cloruro de plata con electrolito de NaCl 3 M
(potencial = 0.2 V) y un contraelectrodo de platino de 50 mm de longitud, 0.5 mm de diámetro
y ~ 0.7 cm2 de área superficial. Para el mantenimiento del electrodo de trabajo se emplearon:
solución de diamante de 1 μm, solución de alúmina de 0.05 μm, almohadilla pulidora para
diamante y almohadilla pulidora para alúmina.
3.2.4.5.1 FABRICACIÓN DE LA CELDA DE DIFUSIÓN
La permeación de metanol a través de las membranas se determinó mediante el siguimiento
del gradiente de concentración originado por el flujo de metanol de un compartimento al otro
en una celda de difusión, generando la oxidación electroquímica del alcohol mediante
voltamperometría cíclica. La celda de difusión utilizada (Figura 3.1a) está formada por dos

CAPÍTULO 3
66
compartimentos con capacidad de 50 mL cada una (3.7 x 3.7 x 5 cm) interconectados
lateralmente mediante una abertura circular de 2 cm diámetro (d) (Figura 3.1b) por lo que el
área efectiva de difusión la membrana utilizada en cada experimento fue de 3.14 cm2.
Figura 3.1. a) Celda de dos compartimentos para permeación de metanol, b) Interconexión lateral de los compartimentos.
3.2.4.5.2 LIMPIEZA DEL ELECTRODO DE TRABAJO
Para la deterincación de la concetración de metanol permeado a través de la membrana se
obtuvo el perfil del voltamperograma cíclico del electrodo de trabajo de platino, empleando
como electrolito una solución de H2SO4 0.5 M. Para minimizar los cambios en el perfil del
voltamperograma inicial se estableció una metodología para su limpieza y se determinó la
rugosidad del electrodo, así como la disminución del área catalítica activa por los subtroductos
generados de la oxidación del metanol.
3.2.4.5.2.1 PULIDO MECÁNICO DEL ELECTRODO DE TRABAJO
Antes del inicio de las mediciones electroquímicas, el electrodo de trabajo se pulió de la
siguiente manera. Se humedeció la almohadilla pulidora para la solución de diamante, se
adicionaron 3 gotas de la solución de diamante de 1 μm y con movimientos circulares se pulió
durante 10 minutos. Posteriormente se humedeció la almohadilla pulidora para alúmina, se
adicionaron 3 gotas de la solución de alúmina de 0.05 μm y se pulió durante 5 minutos.
Posteriormente se enjuagó el cuerpo aislante del electrodo y el área del platino con agua
destilada.

CAPÍTULO 3
67
3.2.4.5.2.2 PULIDO ELECTROQUÍMICO DEL ELECTRODO DE TRABAJO
Previo a la realización de la curva de calibración para la determinación de permeación de
metanol a través de la membrana, y posterior al pulido mecánico del electrodo de trabajo, se
llevó a cabo el pulido electroquímico de éste. Los tres electrodos; el electrodo de trabajo, el
electrodo de referencia y el contra electrodo se colocaron en la tapa de uno de los
compartimentos de la celda de difusión (Figura 3.2), y en ese compartimento se adicionaron
50 mL de H2SO4 0.5 M, además se agitó y burbujeó con N2 durante 30 minutos para eliminar
el O2 disuelto.
Figura 3.2. Configuración de los electrodos empleados en la celda de difusión: A = electrodo de trabajo, B = Electrodo de referencia y C = contra electrodo.
Una vez transcurrido los 30 min se detuvo la agitación y se mantuvo el burbujeo de N2 durante
5 min. Se retiró el N2 de la solución y se realizaron 40 ciclos desde -0.175 V a +0.975 V
versus el electrodo de referencia de plata/cloruro de plata (Ag/AgCl) a una velocidad de
50 mV/s. Posteriormente se realizaron 3 ciclos a 20 mV/s, y se usó el tercero como el perfil
del electrodo de platino.
3.2.4.5.3 CURVA DE CALIBRACIÓN PARA LA DETERMINACIÓN DE
PERMEACIÓN DE METANOL A TRAVÉS DE LAS MEMBRANAS
Para construir la curva de calibración fue necesario solo uno de los compartimentos de la celda
de difusión, por lo que se colocó una placa de vidrio entre los dos compartimentos de la celda
(Figura 3.3). En el compartimento I se colocaron los electrodos, una solución de H2SO4 0.5 M
y se hicieron adiciones consecutivas de metanol para tener concentraciones de 10, 25, 50, 75,
100, 125, 150, 175, 200 y 225 mM. Después de cada adición de metanol se agitó y burbujeó

CAPÍTULO 3
68
con N2 la solución por 3 minutos, y posteriormente se registraron 3 ciclos de
oxidación-reducción a 20 mV/s.
Figura 3.3. Configuración de la celda de difusión para obtener la curva de calibración. Compartimento I: electrodos, H2SO4 0.5 M y adición consecutiva de metanol. Compartimento II: vacío.
La presencia de metanol en una solución se detecta por la generación de una corriente de pico
que empieza en el potencial de 0.55 V [178]. En la Figura 3.4 se muestra el intervalo de
potencial de 0.025 hasta 1 V correspondiente el tercer ciclo de la corriente generada para cada
concentración de metanol empleada para construir la curva de calibración.
Figura 3.4. Secuencias de voltamperogramas I vs. V correspondientes al aumento de concentración de metanol en la solución de H2SO4 0.5 M. Electrodo de trabajo de platino y velocidad de barrido = 20 mV/s.
0.0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1.00.0
0.2
0.4
0.6
0.8
1.0
1.2
1.4
g
Potencial (V) vs ENH
k: 225 mM MeOHj: 200 mM MeOHi: 175 mM MeOHh: 150 mM MeOHg: 125 mM MeOHf: 100 mM MeOHe: 75 mM MeOHd: 50 mM MeOHc: 25 mM MeOHb: 10 mM MeOHa: 0 mM MeOH
h
Cor
rient
e (µ
A)
ab
c
de
f
kj
i

CAPÍTULO 3
69
En los voltamperogramas se aprecia lo siguiente: 1) conforme incrementa la concentración de
metanol en el sistema, la corriente de pico correspondiente a la oxidación del alcohol aumenta,
hasta que alcanza un máximo en el potencial cercano a 0.83 V, 2) el incremento de la corriente
generada no es lineal respecto al aumento de la concentración del alchohol, 3) conforme
aumenta la concentración de metanol en el sistema, el área bajo la curva de oxidación
(0.025 V a 0.3 V) va disminuyendo, indicando el envenenamiento del electrodo de trabajo. El
envenenamiento del electrodo de Pt se debe a que los sitios catalíticos se obstruyen por la
formación y adsorción de especies como el monóxido de carbono (CO) y el óxido de platino
(Pt-OH) [179].
Para conocer la correlación entre la corriente del pico (máximo) y la concentración de metanol
que le dio origen tomando en cuenta el envenenamiento del electrodo, se requiere:
1) determinar el área superficial real o área electrocatalítica (ECA) del electrodo en cada
adición de metanol, 2) determinar la densidad de corriente (j) correspondiente a la oxidación
del metanol en cada adición, 3) relacionar la densidad de corriente con la concentración de
metanol que le dio origen.
El área superficial real de un electrodo se puede representar con la ecuación 3.3 [180]:
푆 = 푛푁푎 (ec.3.3)
Donde:
푆 = área superfical real (u2)
푛 = moles de gas hidrógeno adsorbido en una monocapa (mol)
푁 = Número de Avogadro (1/mol)
푎 = área ocupada por partícula adsorbida (u2)
Cada átomo o molécula de gas está unido por un enlace covalente a cada sitio de adsorción,
por lo tanto necesitamos conocer la cantidad de gas adsorbido y la densidad atómica de
superficie del electrodo. Éste último dato se puede conocer a partir de datos cristalográficos,
considerando una superficie teóricamente plana. De esa manera el área superficial real puede
ser calculada de la siguiente manera (ecuación 3.4).
푆 = (ec.3.4)
Donde 푑 es la densidad atómica de superficie del platino

CAPÍTULO 3
70
El valor de 푑 es calculado tomando en cuenta que para el platino, los planos predominantes
son aquellos cuyos índices son (100), (110) y (111), y cada uno de ellos se encuentran en una
proporción de 33 % [181]. Es decir, la superficie del platino está compuesta por tres caras
cristalinas predominantes ((100): 1.3x1015 átomos/cm2, (110): 0.92x1015 átomos/cm2 y (111):
1.56x1015 átomos/cm2), por lo cual el promedio de la densidad atómica de superficie es de
1.3x1015 átomos/cm2 [49].
Tomando en cuenta que éste es un proceso de transferencia de carga en el cual la adsorción de
un átomo en un sitio de platino involucra un electrón, los moles de átomos de hidrógeno
adsorbidos se pueden calcular como
푛 = (ec.3.5)
푄 = carga asociada con la formación de una monocapa
퐹 = constante de Faraday
Y a su vez la constante de Faraday es la cantidad de carga eléctrica en un mol de electrones
퐹 = 푁푒 (ec.3.6)
푒 = carga eléctrica elemental = 1.602x10-19 C
de la ecuación 3.6 despejamos el número de Avogadro y sustituimos en la ecuación 3.4
푁 = 푆 =
푆 = (ec.3.7)
Ahora sustituimos la ecuación (3.5) en (3.7)
푆 =푄퐹 퐹푒푑
푆 = (ec.3.8)
En este punto calcular 푄 es la incógnita principal
Tomando en cuenta que
퐼 = (ec.3.9)
Entonces
푄 = ∫ 퐼푑푡 (ec.3.10)

CAPÍTULO 3
71
Donde 푡 es el momento en el que la adsorción de hidrógeno comienza y 푡 es el momento en
el que una monocapa se completa. Para una voltamperometría cíclica, el potencial aplicado al
electrodo en un barrido varía como
푉 = 푉 − 푣푡 (ec.3.11)
푉 = potencial inicial aplicado
푣 = velocidad de barrido del potencial (푣 = 푑푉 푑푡⁄ )
푡 = es el tiempo
despejando el tiempo de la ecuación (3.11) tenemos
푡 = 푡 = (ec.3.12)
y
푑푡 = (ec.3.13)
la ecuación 3.10 se puede reescribir como:
푄 = 퐼푑푉1푣
la 푣 es constante durante el experimento así que puede salir de la integral
푄 = ∫ 퐼푑푉 (ec.3.14)
La ecuación 3.14 nos indica que 푄 puede calcularse del voltamperograma de la Figura 3.5,
integrando la curva correspondiente a la desorción de hidrógeno (área cuadriculada). Sin
embargo la corriente capacitiva (área gris) no se restringe a la región de la doble capa, sino
que se extiende a todo el intervalo del potencial del electrodo.

CAPÍTULO 3
72
Figura 3.5. Voltamperograma cíclico típico obtenido con un electrodo de platino, usando una solución de H2SO4 0.5 M como electrolito (Imagen adaptada de Rodríguez y col. [180]).
Por esa razón 푄 debe ser calculada conforme a la ecuación 3.15.
푄 = ∫ 퐼푑푉 −푄 (ec.3.15)
Donde 푄 es la carga asociada a la doble capa capacitiva de la región de desorción de
hidrógeno (zona donde coincide el área gris y el área cuadriculada en la Figura 3.5)
Conociendo cómo calcular el valor de 푄 a partir de cada uno de los voltamperogramas
cíclicos obtenidos, se regresa a la ecuación 3.8 para determinar el área superficial real presente
en el electrodo de trabajo en cada una de las mediciones hechas con el incremento de
concentración de metanol para la curva de calibración.
A continuación se muestra el análisis dimensional que nos indica que unidades las unidades
que se deben usar al sustituir valores en las ecuaciones 3.8 y 3.15.
푄 =1푣 퐼푑푉 −푄 [=]
1 ∗ (퐴푉 − 퐴푉)[=]
푠푉
(퐴푉)[=](퐴푠)[=]퐶
푆 =푄푒푑
[=]퐶
퐶 ∗ [=] [=]
퐶 ∗ 푐푚퐶
[=]푐푚
Después de calcular el ECA de cada voltamperograma, se obtiene la densidad de corriente de
cada una de ellas mediante la ecuación 3.16
Desorción de Hidrógeno Región de
la doble capa
Región de la doble capa
Adsorción de Hidrógeno
Corriente capacitiva debido a la doble capa
Potencial (V)
Cor
rient
e (A
)
0

CAPÍTULO 3
73
푗 = (ec.3.16)
푗 = densidad de corriente (A/cm2)
퐼 = Intensidad de corriente máxima en el pico de la curva de oxidación (A)
푆 = área electrocatalítica o real del electrodo de trabajo (cm2)
Una vez que se conoce la densidad de corriente de cada voltamperograma, se puede realizar la
curva de calibración graficando la densidad de corriente (j) de la oxidación de metanol versus
concentración de metanol presente en la solución de H2SO4 0.5 M (Figura 3.6).
Figura 3.6. Curva de calibración entre densidad de corriente (j) de la oxidación de metanol versus concentración de metanol en la solución de H2SO4 0.5 M empleada.
La relación de variables de la curva de calibración de la Figura 3.6 no sigue una tendencia
lineal, sino que se ajusta a una ecuación polinomial de segundo grado (ecuación 3.17):
j = -0.0017C2 + 0.7417C + 0.8493 (ec.3.17)
La ecuación 3.17 se resolvió con una de las raíces de la ecuación cuadrática (ecuación 3.18)
퐶표푛푐.푑푒푚푒푡푎푛표푙 = . ( . ) ( . ∗( . )).
(ec.3.18)
Donde
j = densidad de corriente en μA/cm2
A partir de la ecuación 3.18 es posible conocer la concentración correspondiente a
determinada densidad de corriente obtenida durante la experimentación de permeación de
metanol de las membranas.
0 40 80 120 160 200 2400
15
30
45
60
75
90
j (µA
/cm
2 )
Concentración de metanol (mmol/L)

CAPÍTULO 3
74
3.2.4.5.4 DETERMINACIÓN DE LA PERMEACIÓN DE COMBUSTIBLE A TRAVÉS
DE LAS MEMBRANAS
La permeación de metanol a través de la membrana en un determinado tiempo se expresa
como un coeficiente de difusión D (cm2/s), el cual se calcula a partir de la ecuación 3.19. ∆∆
= (ec.3.19)
Donde ∆퐶 es la diferencia de concentración de metanol (mmol/L) entre los dos
compartimentos de la celda de difusión al principio del experimento, ∆푋 es el espesor de la
membrana (cm), k es la pendiente (mM/min) de la línea de tendencia de la variación de la
concentración de metanol versus tiempo, V es el volumen existente en el compartimento
(cm3), y A es el área de la membrana (cm2).
De la ecuación 3.19, para calcular D, la incógnita que no se conoce directamente es la
pendiente de la línea de tendencia de la variación de la concentración de metanol versus
tiempo, por lo cual es necesario construir el gráfico correspondiente con el siguiente
procedimiento.
Se coloca la membrana entre los compartimentos de la celda de difusión (Figura 3.7) y en cada
compartimento se coloca un agitador magnético, una manguera con flujo de N2 y 50 mL de
H2SO4 0.5 M. En el compartimento I se colocan los electrodos y en el compartimento II se
adiciona el metanol para tener una concentración 1 M (3.2 % en peso), que correspondería a la
alimentación en una celda de combustible de metanol directo (direct methanol fuel cell -
DMFC-) (1 - 6 % en peso) [182]. Después de la adición del metanol se agitó por 2 min y se
burbujeó N2 por 5 min, se detuvo la agitación y el burbujeo y se registraron
3 voltamperometrías cíclicas a 20 mV/s (tiempo = 0). A continuación se repitió el proceso
cada 30 minutos durante 5 horas: 30 min burbujeo//90segundos agitación//detener
agitación//sacar mangueras//registrar 3 ciclos a 20mV/s [183].

CAPÍTULO 3
75
Figura 3.7. Configuración de la celda de difusión para la determinación de permeabilidad de metanol a través de una membrana. Compartimento I: electrodos y H2SO4 0.5 M. Compartimento II: Metanol 1 M + H2SO4 0.5 M.
De los voltamperogramas correspondientes a la permeación de metanol a diferentes tiempos,
se obtienen los valores de la corriente generada por la presencia de metanol en el
compartimento I. Dichos valores se sustituyen en la ecuación 3.16 y con las ecuaciones 3.8 y
3.15 se obtiene la densidad de corriente. Los valores de densidad de corriente se sustituyen en
la ecuación de la curva de calibración (ecuación 3.18), y con ello se conoce directamente la
concentración de metanol (C (mmol/L) existente en el compartimento I de la celda de difusión
a diferentes tiempos (t (min)) en el lapso de 5 horas, como consecuencia de la permeación
demetanol a través de la membrana.
Posteriormente se grafica de variación de concentración de metanol contra el tiempo para
obtener una línea recta y = mx + b (ecuación 3.20).
C = kt + b (ec.3.20)
De esta línea se obtiene el valor de k para calcular el coeficiente de difusión D de la membrana
con la ecuación 3.19.
3.2.4.6 MICROSCOPÍA ELECTRÓNICA DE BARRIDO DE LAS MEMBRANAS
La topografía superficial y transversal de las membranas se obtuvo mediante microscopía
electrónica de barrido (SEM) empleando un equipo JEOL JSM-7401F.

CAPÍTULO 3
76
3.2.4.7 ANÁLISIS TERMOMECÁNICO DE LAS MEMBRANAS
El comportamiento mecánico de las membrans se determinó empleando la prueba de flexión
en tres puntos, utilizando muestras rectangulares de las membranas, para calcular su módulo
de elasticidad. El cual se determina a partir de la relación entre el esfuerzo aplicado para
generar una deformación elástica del material, y se calcula trazando una tangente a la parte
lineal de mayor pendiente de la curva de esfuerzo-deformación y aplicando la
ecuación 3.21 [184]. En la prueba realizada la fuerza máxima aplicada fue de 1 N a una
velocidad de 0.15 N/min.
퐸∗ = 퐿 푚 4푏푑⁄ (ec.3.21)
Donde E* es el módulo de elasticidad (MPa), L es la separación de los dos puntos que soportan
la muestra durante la prueba, b es el ancho de la muestra, d es el espesor de la membrana,
y m es la pendiente de la tangente de la parte lineal (N/m) de la curva de
esfuerzo-deformación.
3.2.4.8 GRADO DE SULFONACIÓN DE LAS MEMBRANAS
Para la cuantificación de los grupos sulfónicos de los copolímeros sulfonados se empleó la
relación entre unidades repetitivas (St:AA) de los copolímeros sin sulfonar determinadas por
RMN de 1H en conjunto con los resultados de la titulación ácido-base directa de soluciones
preparadas con los copolímeros sulfonados. El procedimiento consistió en pesar 0.5 g
(aproximadamente, pero registrando el peso exacto) del copolímero sulfonado y disolverlo en
50 mL de THF, la solución se acidificó con 5 mL de HCl 0.06 M (en agua) para generar la
desprotonación de los grupos ácidos del copolímero. La solución polimérica se tituló
empleando un pH-metro con electrodo de vidrio y una solución acuosa de NaOH 0.03 M. Este
procedimiento de titulación potenciométrica es una variación de la metodología empleada para
construir diagramas de distribución de especies de copolímeros de PS-PAA sulfonados [29] y
para la cuantificación de grupos ácidos en materiales solubles en disolventes
no acuosos [185-187].
Para calcular el grado de sulfonación generalmente se utiliza la ecuación 3.22 [186-189], sin
embargo esta ecuación se emplea para polímeros conformado únicamente por unidades
repetitivas sulfonables.

CAPÍTULO 3
77
퐺푟푎푑표푑푒푠푢푙푓표푛푎푐푖ó푛(%) =
∗ 100 (ec.3.22)
Para poder emplear la ecuación 3.22 fue necesario hacer algunos cálculos previos para
involucrar la presencia de unidades de ácido acrílico en los copolímeros de PS-PAA, las
cuales no se sulfonan, pero que poseen masa y sus grupos -COOH consumen titulante durante
el procedimiento. Para el copolímero D la relación molar es de 96 unidades de St por cada 4
unidades de AA (determinada por RMN de 1H), por lo tanto la relación gravimétrica entre
monómeros puede indicarse de la siguiente manera: 96 mol St = 9998.4 g,
4 mol AA = 288.24 g y monómeros totales en el copolímero = 10286.64 g.
Si la masa de copolímero sulfonado es de 0.48 g y se gastaron 9.1 mL de NaOH 0.03M
(0.000273 moles de NaOH) para neutralizar los grupos –SO3H y –COOH, los cálculos
correspondientes son:
푚표푙푑푒 − 퐶푂푂퐻 = 0.48푔푚푢푒푠푡푟푎 ∗288.24푔퐴퐴
10286.64푔푃푆 ∗1푚표푙퐴퐴
72.06푔퐴퐴 ∗1푚표푙퐶푂푂퐻
1푚표푙퐴퐴 = 1.8665푥10 푚표푙푒푠
푚표푙푒푠푑푒−푆푂 퐻 = 0.000273푚표푙푁푎푂퐻푔푎푠푡푎푑표푠 − 1.8665푥10 푚표푙푑푒퐶푂푂퐻 = 8.635푥10 푚표푙푒푠
푚푎푠푎푑푒푙표푠푔푟푢푝표푠푆푂 퐻푖푛푐표푟푝표푟푎푑표푠(푔) = 8.635푥10 푚표푙푑푒푆푂 퐻 ∗81푔푆푂 퐻
1푚표푙푆푂 퐻 = 6.9943푥10 푔
푔푃푆푞푢푒푠푒푠푢푙푓표푛푎푟표푛 = 8.635푥10 푚표푙푆푂 퐻 ∗1푚표푙푃푆푠푢푙푓1푚표푙푆푂 퐻 ∗
1푚표푙푃푆1푚표푙푃푆푠푢푙푓 ∗
104.15푔푃푆1푚표푙푃푆
= 8.9933푥10 푔
푔푃푆푒푛푙푎푚푢푒푠푡푟푎 = 0.48푔푚푢푒푠푡푟푎 ∗9998.4푔푃푆
10286.64푔푚푢푒푠푡푟푎 − 6.9943푥10 푔푔푟푢푝표푠푆푂 퐻 = 0.4595푔
퐺푟푎푑표푑푒푠푢푙푓표푛푎푐푖ó푛(%) = 푔푃푆푞푢푒푠푒푠푢푙푓표푛푎푟표푛푔푃푆푒푛푙푎푚푢푒푠푡푟푎 ∗ 100 =
8.9933푥100.4595 ∗ 100 = 1.9%
3.3 RESULTADOS Y DISCUSIÓN
3.3.1 DETERMINACIÓN DEL DISOLVENTE RESIDUAL EN LAS MEMBRANAS
Las membranas preparadas mediante disolución-evaporación utilizando la mezcla
THF-DMSO generaron valores de IEC y de absorción de agua más altos que al prepararlas
únicamente con THF. Para descartar o confirmar que la presencia de DMSO en las membranas

CAPÍTULO 3
78
genera alguna interacción que favorece las dos propiedades mencionadas, se llevó a cabo la
determinación de disolvente residual (DMSO) mediante Cromatografía de Gases acoplada a
Espectrometría de Masas. En la Figura 3.8 se muestra el cromatograma de la membrana que se
analizó, siendo ésta la que presentó el valor de IEC más alto y la que se preparó con 110 % de
DMSO (TAg-120min170%0.110%).
Figura 3.8. Cromatograma de gases para determinación de disolvente residual.
Los resultados mostrados en la Figura 3.8 se observa que la membrana contenía dietilbenceno,
proveniente del medio de polimerización, así como THF, que es el disolvente mayoritario
empleado en la preparación de las membranas. Noobstante no se observa la presencia de
DMSO, posiblemente debido a la baja concentración empleada en la preparación de las
membranas. Sin embargo se observó que es necesario mejorar los procesos de purificación.
3.3.2 CAPACIDAD DE INTERCAMBIO IÓNICO DE LAS MEMBRANAS
La preparación de membranas con los copolímeros sulfonados mediante el proceso de
disolución-evaporación inicialmente se llevó a cabo utilizando THF como disolvente,
generando valores de capacidad de intercambio cercanos a cero a pesar de que los resultados
de espectroscopía IR y titulación directa de grupos ácidos indicaban mayor cantidad de grupos
sulfónicos en los copolímeros. En la búsqueda de información que pudiera ayudar a
comprender la discrepancia de resultados, se encontró que en el 2012 Jun y col. publicaron
que para membranas de SPEEK, dependiendo del tipo de disolventes utilizado durante su
proceso de preparación, su capacidad de conducción protónica se modifica [37]. Por otro lado,
Silva y col. observaron que la conductividad protónica y la absorción de agua de las
0 1 2 3 4 5 6 7 8 90
20
40
60
80
100
11.6 %THF
31.2 %
24.4 %Abu
ndan
cia
(%)
Tiempo de retención (min)
32.7 %
(DEB = 88.3 %) o,p & m-dietilbenceno

CAPÍTULO 3
79
membranas de Nafion difieren dependiendo del disolvente que se utilice durante su proceso de
preparación [39]. Además, dependiendo del espesor de la película, tipo de disolvente utilizado
y velocidad de evaporación del disolvente se pueden obtener películas con diferentes
morfologías, así como también se obtienen películas con ordenamiento de las partes polares y
no polares del copolímero por el uso de mezcla de disolventes [190].
Tomando en cuenta la información encontrada, para mejorar los resultados de IEC, e
indirectamente la conducción protónica, se procedió a hacer un cambio en el disolvente
empleado (THF hasta este punto) durante el proceso de disolución-evaporación. Los
disolventes alternativos deberían tener diferente velocidad de evaporación, ser miscibles entre
ellos, que el disolvente de menor velocidad de evaporación pudiera ser removido mediante
presión reducida o extracción con un disolvente volátil que no disolviera o dañara la película
formada, y que el copolímero sulfonado fuera miscible en la mezcla de disolventes. La
primera opción fue THF-H2O pero el polímero no fue soluble en la mezcla, la siguiente opción
fue la mezcla THF-DMSO que cumple con los requerimeintos mencionados. Una vez
seleccionado la mezcla THF-DMSO, se utilizaron dos relaciones respecto a la masa del
copolímero utilizado: 55 % y 110 % en peso. En la Tabla 3.1 se encuentran los valores de IEC
de las membranas preparadas con THF y con la mezcla de THF-DMSO.

CAPÍTULO 3
80
Tabla 3.1. Valores de IEC de los materiales con los se obtuvieron membranas mediante el proceso de disolución-evaporación utilizando diferentes relaciones de THF-DMSO.
Etiqueta de la muestra IEC (meq/g) determinada para películas
preparadas con diferente relación THF-DMSO
0% DMSO 55% DMSO 110% DMSO
Nafion 117 0.87 N.D. N.D. DC-30min20% 0.0007 N.D. N.D. DC-10min20% 0.0004 N.D. N.D. DC- 2min20% 0.00004 N.D. N.D. DH-30min20% 0.0006 N.D. N.D. DH-10min20% 0.0006 N.D. N.D. DH- 2min20% 0.0005 N.D. N.D. TC-30min20% 0.0007 N.D. N.D. TC-10min20% 0.0007 N.D. N.D. TC- 2min20% 0.0006 N.D. N.D. TH-30min20% 0.0006 N.D. N.D. TH-10min20% 0.0006 N.D. N.D. TH- 2min20% 0.0006 N.D. N.D.
DAg-120min 85%0.110% 0.0089 0.0541 0.4607 DAg-240min 85%0.110% 0.0303 0.4260 0.4580 DAg-240min170%0.110% 0.1436 0.4231 0.5524
DAg-360min 85%0.110%* 0.0286 - - DAg-240min225%0.110% 0.0872 0.1746 0.2230
DAg-240min170%0.220%** 0.1210 0.1603 0.1800 DAg-240min170%0.055% 0.0310 0.0960 0.2255 TAg-120min 85%0.110% 0.1501 0.3853 0.6468 TAg-240min 85%0.110% 0.0608 0.4304 0.5552 TAg-120min170%0.110% 0.0783 0.6525 0.9030 TAg-120min170%0.220% 0.0044 0.3764 0.9233 TAg-360min 85%0.110% 0.1436 0.3542 0.5100
TAg-120min225%0.110%** 0.1546 0.1910 0.2316 TAg-120min170%0.055% 0.0503 0.3905 0.7605
* El copolímero DAg-360min 85%0.110%no fue apto para formar película utilizando DMSO en la solución para el proceso de disolución-evaporación. ** Los copolímero sulfonados DAg-240min170%0.220% y TAg-120min225%0.110% presenta una cantidad baja de geles que son imperceptibles durante la preparación de las soluciones para el proceso de disolución-evaporación, sin embargo, una vez que se ha evaporado el THF se hacen evidentes los geles presentes en las mismas, los cuales se observan como pequeños gránulos. Nafion 117: Membrana comercial, no se preparó en el laboratorio
Se observa que las membranas de los copolímeros sulfonados con 20 % mol de H2SO4 o
CH3COOSO3H por 2, 10 o 30 minutos preparadas con THF tienen valores de IEC que tineden
a cero. Con el sistema ácido sulfúrico-Ag2SO4, la selección de las mejores condiciones de

CAPÍTULO 3
81
sulfonación se obtuvo comparando el valor de IEC entre conjuntos de tres reacciones,
manteniendo constantes dos condiciones y variando una. En la Figura 3.9 se indican las
reacciones que conformaban cada conjunto de experimentos, y la secuencia que se siguió para
seleccionar las mejores condiciones de reacción tomando como base los resultados de
capacidad de intercambio iónico.
Figura 3.9. Conjunto de experimentos realizados y comparados para encontrar las mejores condiciones de sulfonación del copolímero D con ácido sulfúrico-Ag2SO4.
A partir de la comparación de los valores de IEC0%DMSO de las membranas de los
copolímeros D sulfonados con ácido sulfúrico-Ag2SO4, se observó que alincrementar la
concentración de H2SO4 de 85 a 170 % mol, se genera un aumento en la IEC, es decir, a
mayor cantidad de H2SO4 utilizado, más grupos sulfónicos en el copolímero. Pero al aumentar
de 170 a 225 % mol, la IEC disminuye, probablemente porque la concentración de los iones
Ag+ es de 0.110 % mol respecto a los moles de H2SO4, pudiéndose formar entrecruzamientos
entre grupos SO3-, y como consecuencia generar la disminución de la IEC.
DAg-120min-85%0.110% IEC0%DMSO = 0.0089DAg-240min-85%0.110% IEC0%DMSO = 0.0303 ←DAg-360min-85%0.110% IEC0%DMSO = 0.0286
DAg-240min- 85%0.110% IEC0%DMSO = 0.0303DAg-240min-170%0.110% IEC0%DMSO = 0.1436 ←DAg-240min-225%0.110% IEC0%DMSO = 0.0872
DAg-240min-170%0.055% IEC0%DMSO = 0.0310DAg-240min-170%0.110% IEC0%DMSO = 0.1436 ←DAg-240min-170%0.220% IEC0%DMSO = 0.1210
240 min de reacción, 170 mol% de H2SO4 y 0.110 mol% de Ag2SO4
Diferente tiempo de sulfonación
Diferente concentración de H2SO4
Diferente concentración de Ag2SO4
Mejores condiciones de sulfonación para el copolímero D con H2SO4- Ag2SO4:
Primer conjunto de experimentos
Segundo conjunto de experimentos
Tercer conjunto de experimentos
mejores condiciones del conjunto
mejores condiciones del conjunto
mejores condiciones del conjunto

CAPÍTULO 3
82
Una vez comparados los valores de IEC de los tres conjuntos de experimentos se determinó
que el copolímero sulfonado con 170 % mol de H2SO4 y 0.110 % mol de Ag2SO4 durante
240 min de reacción, genera el material sulfonado con el IEC más alto de todos
(0.1436 meq/g). Sin embargo este valor aun es menor en comparación al de la membrana de
Nafion 117 (0.87 meq/g).
Con los valores de IEC de las tres membranas preparadas con cada copolímero sulfonado
(Tabla 3.1) se construyó la gráfica de la Figura 3.10.
Figura 3.10. Valores de IEC de las membranas del copolímero D sulfonado con ácido sulfúrico-Ag2SO4, preparadas mediante disolución-evaporación utilizando diferente composición de disolventes.
En la Figura 3.10 se observa que las membranas preparadas con THF generan el valor más
bajo de IEC, el valor intermedio corresponde al uso de 55 % en peso de DMSO y el valor más
alto corresponde a las membranas preparadas con 110 % en peso de DMSO.
El material DAg240min-170%0.110% genera la membrana con mayor IEC; 0.5524 meq/g, lo
que significa que al emplear Ag2SO4 en conjunto con H2SO4 fué posible obtener una
membrana mejor que al emplear únicamente H2SO4 o sulfato de acetilo
(IEC mayor = 0.007meq/g). Sin embargo el valor de IEC (0.5524 meq/g) aún es menor que el
de la membrana de Nafion 117 (0.87 meq/g). De forma análoga se hizo con el copolímero T,
con el siguiente orden de comparación de variables: tiempo de reacción, concentración de
H2SO4 y concentración de Ag2SO4. Posteriormente con cada material se prepararon tres
membranas empleando diferentes relaciones de THF-DMSO, de las cuales se utilizaron las
0.00.10.20.30.40.50.60.70.80.91.0
DA
g240
min
-170
%0.
055%
DA
g240
min
-170
%0.
220%
**
DA
g240
min
-225
%0.
110%
DA
g360
min
-85%
0.11
0%*
DA
g240
min
-170
%0.
110%
DA
g240
min
-85%
0.11
0%
DA
g120
min
-85%
0.11
0%
Naf
ion
IEC
(meq
/g)
Disolvente:THF-110%DMSOTHF- 55%DMSOTHF- 0%DMSO

CAPÍTULO 3
83
preparadas con 110 % de DMSO para comparar entre ellas y seleccionar las mejores
condiciones de sulfonación con ácido sulfúrico-Ag2SO4 (Figura 3.11).
Figura 3.11. Experimentos realizados y comparados para encontrar las mejores condiciones de sulfonación del copolímero T con ácido sulfúrico-Ag2SO4.
En la Figura 3.12 se pude ver que las membranas de los copolímeros
TAg120min-170%0.220% y TAg120min-170%0.110% preparadas con 110 % en peso de
DMSO presentan los valores de IEC más altos (0.9233 meq/g y 0.9030 meq/g
respectivamente) que el de Nafion 117 (0.87 meq/g), por lo que estos copolímeros pudieran
ser competitivos respecto al de la membrana de Nafion 117.
TAg-120min-85%0.110% IEC110%DMSO = 0.6468TAg-240min-85%0.110% IEC110%DMSO = 0.5552TAg-360min-85%0.110% IEC110%DMSO = 0.5100
TAg-120min- 85%0.110% IEC110%DMSO = 0.6468TAg-120min-170%0.110% IEC110%DMSO = 0.9030TAg-120min-225%0.110% IEC110%DMSO = 0.2316
TAg-120min-170%0.055% IEC110%DMSO = 0.7605 TAg-120min-170%0.110% IEC110%DMSO = 0.9030TAg-120min-170%0.220% IEC110%DMSO = 0.9233
120 min de reacción, 170 mol% de H2SO4 y 0.110 mol% o 0.220 mol% de Ag2SO4
Diferente tiempo de sulfonación
Diferente concentración de H2SO4
Diferente concentración de Ag2SO4
Mejores condiciones de sulfonación para el copolímero T con H2SO4- Ag2SO4:
Primer conjunto de experimentos
Segundo conjunto de experimentos
Tercer conjunto de experimentos
mejores condiciones del conjunto
mejores condiciones del conjunto
mejores condiciones del conjunto
←
←

CAPÍTULO 3
84
Figura 3.12. Valores de IEC de las membranas del copolímero T sulfonado con ácido sulfúrico-Ag2SO4, preparadas mediante disolución-evaporación utilizando diferente relación de disolventes.
La capacidad de intercambiar iones y de absorber agua son dos propiedades relacionadas con
la conductividad protónica en una PEM, por esa razón se ha hecho énfasis en obtener
membranas cuyos valores de IEC sean lo más cercanos a los valores de la membrana de
estándar de Nafion 117. Además es importante conocer el mecanismo por el cual está
sucediendo la accesibilidad de los grupos ácidos de os copolímeros sulfonados.
La morfología de las membranas poliméricas depende de la naturaleza química del polímero y
de factores externos como el método de preparación de la membrana, el tipo de disolvente
empleado para su preparación, los esfuerzos aplicados durante la extrusión, el moldeo por
compresión, etc. [9]. En este trabajo las membranas fueron preparadas mediante
disolución-evaporación, por lo que la variación del comportamento de las membranas
preparadas con un mismo copolímero sulfonado probablemente proviene del efecto generado
por el disolvente empleado.
Existen diversos reportes [39, 190, 191] sobre cambios morfológicos en membranas
poliméricas dependiendo del método por el cual se preparen, sin embargo existen pocos
reportes sobre la influencia de la morfología respecto a propiedades vinculadas a membranas
para celdas de combustible. Uno de éstos es el de Peckham [191] quien observó que en el
proceso de disolución-evaporación, a mayor afinidad del disolvente con el copolímero, se
genera mayor ordenamiento morfológico en la membrana, a pesar de no encontrar una
0.00.10.20.30.40.50.60.70.80.91.0
IEC
(meq
/g)
TAg1
20m
in-1
70%
0.05
5%
TAg1
20m
in-2
25%
0.11
0%
TAg3
60m
in-8
5%0.
110%
TAg1
20m
in-1
70%
0.22
0%
TAg1
20m
in-1
70%
0.11
0%
TAg2
40m
in-8
5%0.
110%
TAg1
20m
in-8
5%0.
110%
Naf
ion
Disolvente:THF-110%DMSOTHF- 55%DMSOTHF- 0%DMSO

CAPÍTULO 3
85
correlación obvia de las propiedades del disolvente empleado y la morfología adoptada por el
copolímero.
Los resultados de IEC de los copolímeros D y T sulfonados con ácido sulfúrico-Ag2SO4,
indican que se genera un incremento de la IEC cuando se utiliza como disolvente una mezcla
de THF-DMSO, lo cual podría deberse a un mejor ordenamiento de las partes polares y no
polares del copolímero y que a su vez está relacionado con la velocidad de volatización de los
disolventes empleados [190], lo cual genera materiales sulfonados con IEC competitivo al de
Nafion 117. Una posible explicación al efecto que ocasiona la mezcla de disolvente puede
deberse a que la presión de vapor saturado de los disolventes empleados es diferente, 23.5 kPa
y 0.077 kPa para el THF y DMSO respectivamente [192], lo cual ocasiona que el THF se
evapore mientras que el DMSO permanece con el copolímero, por lo que es necesaria su
extracción posterior. Además, probablemente existe un reacomodo del copolímero sulfonado
en las membranas, que incrementa la accesibilidad de los grupos funcionales generando el
incremento en su capacidad de intercambio iónico. En la literatura está reportado el efecto
relacionado a la evaporación y la adición de un no-disolvente en el proceso de preparación de
membranas mediante disolución-evaporación, indicando que su presencia modifica su
morfología por la separación de fases que se presenta [193].
3.3.3 ABSORCIÓN DE AGUA DE LAS MEMBRANAS
En la Tabla 3.2 se presentan los valores de absorción de agua de las membranas de los
copolímeros D y T sulfonados, mismas a las que anteriormente se les determinó su capacidad
de intercambio iónico.

CAPÍTULO 3
86
Tabla 3.2. Absorción de agua de membranas obtenidas con varias relaciones de THF-DMSO.
Etiqueta de la muestra
Absorción de agua (% peso) determinada para películas preparadas con diferente relación THF-DMSO
0% DMSO 55% DMSO 110% DMSO
Nafion 117 23.2 N.D. N.D. DC-30min20% 0.03 N.D. N.D. DC-10min20% 0.01 N.D. N.D. DC- 2min20% 0.01 N.D. N.D. DH-30min20% 0.03 N.D. N.D. DH-10min20% 0.00 N.D. N.D. DH- 2min20% 0.00 N.D. N.D. TC-30min20% 0.02 N.D. N.D. TC-10min20% 0.01 N.D. N.D. TC- 2min20% 0.00 N.D. N.D. TH-30min20% 0.07 N.D. N.D. TH-10min20% 0.04 N.D. N.D. TH- 2min20% 0.01 N.D. N.D.
DAg-120min 85%0.110% 7.0461 41.1529 99.0174 DAg-240min 85%0.110% 15.4858 84.6131 89.7400 DAg-240min170%0.110% 15.2210 64.9186 105.1197 DAg-360min 85%0.110%* 19.1119 - - DAg-240min225%0.110% 9.3430 50.5221 104.8327 DAg-240min170%0.220%* 11.8003 54.4654 102.5821 DAg-240min170%0.055% 10.6000 56.2099 104.7677 TAg-120min 85%0.110% 22.0057 61.6535 101.8309 TAg-240min 85%0.110% 13.6876 76.8808 117.9192 TAg-120min170%0.110% 14.9514 83.5415 139.8289 TAg-120min170%0.220%* 14.8453 42.6589 114.0819 TAg-360min 85%0.110% 9.4713 55.9745 100.4200 TAg-120min225%0.110% 10.2635 61.9496 99.8587 TAg-120min170%0.055% 13.6132 64.7000 108.9560
* El copolímero DAg-360min 85%0.110%no fue apto para formar película utilizando DMSO en la solución para el proceso de disolución-evaporación. ** Los copolímero sulfonados DAg-240min170%0.220% y TAg-120min225%0.110% presenta geles imperceptibles durante la preparación de las soluciones para el proceso de disolución-evaporación, pero evidentes una vez que se ha evaporado el THF. Nafion 117: Membrana comercial, no se preparó en el laboratorio
En las Figuras 3.13 y 3.14 se observa que al emplear DMSO para el proceso de
disolución-evaporación, las membranas presentan mayor absorción de agua, superando la
capacidad de absorción de la membrana de Nafion 117.

CAPÍTULO 3
87
Figura 3.13. Absorción de agua de las membranas del copolímero D sulfonadas con ácido sulfúrico-Ag2SO4, preparadas con diferente composición de disolventes.
Figura 3.14. Absorción de agua de las membranas del copolímero T sulfonadas con ácido sulfúrico-Ag2SO4, preparadas con diferente composición de disolventes.
En las Figuras 3.13 y3.14 se observa que las membrana DAg240min-170%-0.110% y
TAg120min-170%0.110% son las que resultaron con mayor capacidad de absorción de agua
así como mayor IEC, por lo que fueron seleccionadas para su posterior modificación mediante
incorporación de platino.
El uso de la mezcla THF-DMSO para la preparación de las membranas favorece su absorción
de agua y capacidad de intercambio iónico, lo cual concuerda con lo reportado en la
0
20
40
60
80
100
120
140
Abs
orci
ón d
e ag
ua (%
pes
o)
Naf
ion
DA
g120
min
-85%
0.11
0%
DA
g240
min
-85%
0.11
0%
DA
g240
min
-170
%0.
110%
DA
g360
min
-85%
0.11
0%*
DA
g240
min
-225
%0.
110%
DA
g240
min
-170
%0.
220%
**
DA
g240
min
-170
%0.
055%
Disolvente:THF-110%DMSOTHF- 55%DMSOTHF- 0%DMSO
0
20
40
60
80
100
120
140
Abs
orci
ón d
e ag
ua (%
pes
o)
Naf
ion
TAg1
20m
in-8
5%0.
110%
TAg2
40m
in-8
5%0.
110%
TAg1
20m
in-1
70%
0.11
0%
TAg1
20m
in-1
70%
0.22
0%
TAg3
60m
in-8
5%0.
110%
TAg1
20m
in-2
25%
0.11
0%
TAg1
20m
in-1
70%
0.05
5%
Disolvente:THF-110%DMSOTHF- 55%DMSOTHF- 0%DMSO

CAPÍTULO 3
88
literatura [194]. Esta mejoría probablemente esté relacionada con el acomodo morfológico del
polímero, lo cual podría ser similar a lo reportado por Silva [39], quien encontró que durante
la preparación de membranas con Nafion 117 líquido bajo un mismo procedimiento
(180 °C por 90 min) pero utilizando diferentes disolventes se obtienen diferentes valores de
conductividad (dimetilformamida > etilenglicol > DMSO) y absorción de agua
(dimetilformamida > DMSO > etilenglicol). La razón hipotética que propone Silva es que una
continuidad de aglomerados de iones absorbe más agua que iones separados. Los resultados de
éste trabajo muestran un comportamiento similar, sin embargo es necesario complementar su
caracterización mediante SEM para poder comprobar que sea un acomodo de dominios
hidrófilos e hidrófobos o una separación de fases del sistema debido a la presencia del
no-disolvente en la mezcla, que da lugar a las membranas.
3.3.4 RELACIÓN DE HINCHAMIENTO DE LAS MEMBRANAS
Dentro de una celda de combustible, las membranas intercambiadoras de protones están
expuestas a esfuerzos mecánicos, creep, deformación y degradación mecánica. Dicha
degradación es originada por los cambios dimensionales que resultan de la hidratación y
deshidratación (expansión y compresión) debido a los cambios de temperatura y humedad que
suceden durante el arranque y apagado de la FC [117]. Por lo anterior, la durabilidad e
integridad se han convertido en características a obtener en una membrana intercambiadora de
protones.
La presencia de agua en una membrana es necesaria para que suceda la disociación de
protones provenientes de los grupos ácidos, así como para su movilidad a través de la
membrana [177]. No obstante, la presencia de agua genera cambio dimensional en la
membrana, y cuando éste es referido al cambio de espesor, se conoce como relación de
hinchamiento.
En la Tabla 3.3 se encuentran los espesores en estado seco e hidratad y la relación de
hinchamiento de las membranas Nafion 117, DAg-240-170%0.110% y
TAg-120-170%0.110%.

CAPÍTULO 3
89
Tabla 3.3. Valores de absorción de agua y espesor de membrana Nafion 117 y membranas de copolímeros sulfonados D y T selecconadas, antes y después de ser hidratadas
Membrana Espesor Seca (mm)
Espesor Hidratada (mm)
Relación de hinchamiento
(%) DAg-240-170%0.110%DMSO0% 0.157±0.039 0.161±0.028 2.5
DAg-240-170%0.110%DMSO55% 0.207±0.063 0.228±0.067 10.1 DAg-240-170%0.110%DMSO110% 0.275±0.057 0.301±0.049 9.5
TAg-120-170%0.110%DMSO0% 0.196±0.024 0.208±0.031 6.1 TAg-120-170%0.110%DMSO55% 0.205±0.070 0.227±0.074 10.7
TAg-120-170%0.110%DMSO110% 0.238±0.080 0.264±0.071 10.9 Nafion 117 0.189±0.009 0.230±0.012 21.7
Nafion 117: Membrana comercial, no se preparó en el laboratorio
De los datos de relación de hinchamiento de las membranas (Tabla 3.3) se puede ver que las
membranas de los copolímeros sulfonados D y T, tienen bajo cambio dimensional al hidratarse
(entre 2.5 y 10.9 %) en comparación con el cambio dimensional de la membrana de
Nafion 117 (21.7 %). La baja relación de hinchamiento de una membrana es una característica
favorable, la cual generalmente está directamente relacionada con la capacidad de absorción
de agua.
Durante la evaluación de la capacidad de absorción de agua se determinó que las membranas
de los copolímeros sulfonados D y T, aquellas preparadas utilizando DMSO (Tabla 3.2)
absorben cantidades de agua entre ~41 y ~139%, porcentajes que superan al de la membrana
de Nafion 117 (32.2%). Estos valores indican que la relación de hinchamiento y la capacidad
de absorción de agua de las membranas de los copolímeros sintetizados es diferente a la
esperada, tal y como se muestra en la Figura 3.15.
Figura 3.15. Relación de hinchamiento contra Absorción de agua de las membranas de Nafion 117 y de los copolímeros D y T preparadas con diferente mezcla de disolventes.
0 20 40 60 80 100 120 1400
3
6
9
12
15
18
21
24
Rel
ació
n de
hin
cham
ient
o (%
)
Absorción de agua (%)
DAg240min-170%0.110% DMSO 0% DAg240min-170%0.110% DMSO 55% DAg240min-170%0.110% DMSO 110% TAg120min-170%0.110% DMSO 0% TAg120min-170%0.110% DMSO 55% TAg120min-170%0.110% DMSO 110% Nafion 117

CAPÍTULO 3
90
En la Figura 3.15 se observa que las membranas de los copolímeros sulfonados D y T
preparadas únicamente con THF presentan una relación de hinchamiento baja, de 2.5 y 6.1 %
respectivamente. Mientras que las membranas de éstos mismos copolímeros pero preparadas
con soluciones que contenían DMSO presentan valores de absorción de agua de 60 a 140% y
relación de hinchamiento de 9.5 a 21.7%, correlación de valores diferente a lo esperado.
Sin embargo, la absorción de agua y la relación de hinchamiento están en función de la
estructura e interacciones entre las cadenas poliméricas (así como los puentes de hidrógeno)
de la membrana [177], por lo que es necesario tomar en cuenta otros factores. En la
Figura 3.16 se se puede ver el cambio de espesor de las membranas en estado seco e hidratado.
Figura 3.16. Espesor de las membranas Nafion 117 y de los mejores copolímeros D y T sulfonados en estado seco e hidratado.
Se observa que cuando se utiliza únicamente THF durante el proceso de preparación de las
membranas se obtiene el menor espesor (1 unidad (1 u)), mientras que para las membranas
preparadas con THF-DMSO su espesor es mayor a la unidad, a pesar de haber utilizado la
misma cantidad de polímero para su preparación. La diferencia en dimensión de las
membranas ocasionada por el uso de la mezcla THF-DMSO podría contribuír a los elevados
porcentajes de absorción de agua, con poco cambio de dimensión. Tomando en cuenta los
0.000.050.100.150.200.250.300.35
0.000.050.100.150.200.250.300.35
TAg1
20m
in-1
70%
0.11
0%-D
MSO
55%
TAg1
20m
in-1
70%
0.11
0%-D
MSO
0%
DA
g240
min
-170
%0.
110%
-DM
SO11
0%
TAg1
20m
in-1
70%
0.11
0%-D
MSO
110%
Naf
ion
117
DA
g240
min
-170
%0.
110%
-DM
SO0%
Espe
sor (
mm
)
DA
g240
min
-170
%0.
110%
-DM
SO55
%
Membranas hidratadasMembranas secas
1.32
u
1u 1.75
u
1.05
u
1u 1.21
u

CAPÍTULO 3
91
siguientes puntos. (1) El DMSO en estado puro actúa como no-disolvente para los
copolímeros sulfonados, pero en mezcla con THF mantiene a los copolímeros en solución. (2)
Durante la preparación de las películas, una vez evaporado el THF, el DMSO permanece entre
las cadenas del copolímero sulfonado hasta su extracción. (3) Una vez extraído el DMSO de
las membranas, permanecen los espacios (ahora vacíos) que inicialmente ocupaba el DMSO.
Aunado a la variación dimensional de las membranas debido al proceso de preparación,
también está el ocasionado por la absorción de agua. Las películas preparadas con THF
únicamente, absorben agua y se hinchan pero en porcentajes bajos, que puede estar
relacionado con los entrecruzamientos entre cadenas que disminuyen el hinchamiento de la
membrana [195]. Sin embargo, en las membranas preparadas con DMSO, los espacios
vacantes que éste disolvente dejó, pudieran llenarse con agua, con lo cual la película
aumentaría de peso (absorción de agua), pero no de dimensión (relación de hinchamiento). Un
efecto similar se ha visto en copolímeros de estireno entrecruzados con DVB, los cuales están
formados por estructuras porosas llenas de aire, que no presentan hinchamiento al estar en
contacto con agua, debido a que ésta se acomoda dentro de los poros del material [196, 197].
3.3.5 PERMEACIÓN DE COMBUSTIBLE A TRAVÉS DE LAS MEMBRANAS
La permeación de metanol y determinación del coeficiente de difusión de metanol se llevaron
a cabo para la membrana de Nafion 117 y para las membranas de los copolímeros
DAg-240-170%0.110% y TAg-120-170%0.110% para poder comparar su desempeño.
3.3.5.1 DETERMINACIÓN DEL COEFICIENTE DE DIFUSIÓN DE LA MEMBRANA
DE NAFION 117
En la Figura 3.17 se muestran los voltamperogramas correspondientes a la permeación de
metanol a través de la membrana de Nafion 117

CAPÍTULO 3
92
Figura 3.17. Voltamperogramas a diferentes tiempos que reflejan la permeación de metanol a través de a membrana de Nafion 117en la celda de difusión.
En la figur 3.17 se observa que conforme transcurre el tiempo e incrementa la intensidad de la
corriente, indicando un aumento de la concentración de metanol debido a la permeación que
sucede a travez de la membrana de Nafion 117. Con las ecuaciones 3.8, 3.15, 3.16 y utilizando
los valores de corriente de la Figura 3.17 se obtuvieron sus correspondientes valores de
densidad de corriente (Figura 3.18).
Figura 3.17. Densidad de corriente generada por la permeación de metanol a través de la membrana de Nafion 117 a diferentes tiempos durante 5 horas.
Con los datos de la Figura 3.17 y la ecuación 3.18 se calculó la concentración de metanol que
permeó a través de la membrana de Nafion 117 a diferentes tiempos (Figura 3.19).
0.0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1.00.0
0.2
0.4
0.6
0.8
1.0
1.2
1.4
Cor
rient
e (µ
A)
Potencial (V) vs ENH
n: 300 minm:270 mink: 240 mini: 210 minh: 180 ming: 150 minf: 120 mine: 90 mind: 60 minc: 30 minb: 0 mina: Sin MeOH
nmkihg
fe
d
c
ba
0 60 120 180 240 3000
10
20
30
40
50
60
j (µA
/cm
2 )
Tiempo (min)

CAPÍTULO 3
93
Figura 3.19. Variación de la concentración de metanol respecto al tiempo debida a la permeación a través de la membrana de Nafion 117.
En la Figura 3.19 se observa la relación lineal que existe entre la concentración de metanol
(C (mmol/L)) y el tiempo (t (min)), la cual se puede expresar como una línea recta
(ecuación 3.23)
C = 0.3145t + 0.3081 (ec.3.23)
De la ecuación 3.23 se tomó el valor de la pendiente y se utilizó para calcular el coeficiente de
difusión de metanol de la membrana de Nafion 117 en H2SO4 0.5 M con la ecuación de la
curva de calibración, ecuación 3.19. El valor obtenido fue 1.48x10-3 cm2/s, el cual se
encuentra dentro del valor esperado tomando en cuenta que el área efectiva de difusión de la
celda es de 3.14 cm2
3.3.5.2 DETERMINACIÓN DEL COEFICIENTE DE DIFUSIÓN DE LAS
MEMBRANAS PREPARADAS CON LOS COPOLÍMEROS SULFONADOS
Para determinar el coeficiente de difusión de metanol de las membranas de los copolímeros
DAg-240-170%0.110% y TAg-120-170%0.110% se realizó el procedimiento descrito en la
sección 3.2.3.5.4 cuyos resultados de mustran en las Figuras 3.20 y 3.21.
0 60 120 180 240 3000
20
40
60
80
100
Con
cent
raci
ón d
e m
etan
ol (m
mol
/L)
Tiempo (min)

CAPÍTULO 3
94
Figura 3.20. Voltamperometría cíclica para la determinación de la permeación de metanol a través de las membranas DAg-240min170%0.110% preparadas con 0, 55 y 110 % en peso de DMSO (5 horas).

CAPÍTULO 3
95
Figura 3.21. Voltamperometría cíclica para la determinación de la permeación de metanol a través de las membranas TAg-120min170%0.110% preparadas con 0, 55 y 110 % en peso de DMSO (5 horas).

CAPÍTULO 3
96
La determinación de la permeación metanol a través de las membranas de los copolímeros
DAg-240-170%0.110% y TAg-120-170%0.110% se llevaron a cabo con la misma
metodología utilizada para la evaluación de la membrana de Nafion 117 (Figura 3.15),
obteniéndose resultados diferentes.
Cuando se evalúan las membranas de los copolímeros D y T sulfonados, el perfil de los
voltamperogramas correspondiente al electrodo de trabajo de platino no cambia durante las 5 h
(Figuras 3.20 y 3.21) indicando que las membranas no permiten su paso de metanol. Inclusive
al prolongar la duración de uno de los experimentos durante 10 días con la membrana
TAg-120-170%0.110%DMSO0% (Figura 3.22) se observó que la impermeabilidad al metanol
se mantuvo. Estos resultados son ideales tomando en cuenta que este comportamiento es el
requerido en las membranas a utilizar en DMFC, porque se evitaría que el alcohol se difunda
desde la región anódica hasta la catódica.
Figura 3.22. Voltamperogramas a diferentes tiempos en un intervalo de 10 días para la determinación de la permeación metanol por la membrana TAg-120min170%0.110%DMSO0%.
En la literatura está reportado que cuando una membrana posee elevada absorción de agua,
también presenta hinchamiento elevado, inestabilidad mecánica y dimensional, asi como
permeación de metanol [113, 194]. Sin embargo, las membranas de los copolímeros D y T
sulfonados no presentaron permeación de metanol a pesar de tener IEC y capacidad de
absorción de agua altos.
0.0 0.2 0.4 0.6 0.8 1.0
0.2
0.4
0.6
0.8
1.0
10 días 0 a 300 min Sin metanol
TAg-120min170%0.110%DMSO0%
Cor
rient
e (µ
A)
Potencial (V) vs ENH

CAPÍTULO 3
97
Las características de una PEM relacionadas entre sí son la conductividad protónica,
capacidad de intercambio iónico, cantidad de grupos ácidos, absorción de agua, permeación de
combustible y el cambio dimensional. Estas características, su tendencia y relación se reumen
en la Figura 3.23.
Figura 3.23. Relación existente entre las características que posee una membrana polimérica intercambiadora de protones. Ahora bien, para que una membrana intercambiadora de protones funcione como tal debe
poseer grupos ácidos [198], los cuales se pueden disociar o intercambiar su protón por un ión
sodio (Na+), proceso conocido como capacidad de intercambio iónico [113]. Además,
conforme aumenta la cantidad de grupos ácidos, la absorción de agua del material también
aumenta, debido a su naturaleza hidrófila [14].
De los mecanismos de movilidad de protones, los que suceden únicamente cuando la
membrana está hidratada son el de solvatación de protones por moléculas de agua y el
mecanismo de Grotthus, en el cual los protones saltan rápidamente de una molécula de agua a
otra formando momentáneamente estructuras iónicas tipo cation hidronio [199]. El agua que
absorbe una membrana se acomoda entre las cadenas de polímero separándolas, aumentando
las dimensiones de la membrana (conocido como relación de hinchamiento). Por lo que el
combustible puede permear a través de la membrana cuando el espacio entre cadenas
poliméricas incrementa en demasía [194].
Una vez que se ha mencionado como se relacionan las características de una PEM, queda más
claro porque con los resultados de IEC y absorción de agua determinados para las membranas
Con
duct
ivid
ad p
rotó
nica
Perm
eaci
ón d
e co
mbu
stib
leR
elac
ión
de h
inch
amie
nto
Absorción de aguaCapacidad de intercambio iónico
Cantidad de grupos ácidos

CAPÍTULO 3
98
DAg-240-170%0.110%DMSO110% y TAg-120-170%0.110%DMSO110% (Tablas 3.1 y 3.2)
se esperaba que la permeación de metanol fuera elevada. Para dar explicación a estos
resultados se buscó información en la literatura, encontrándose que en la síntesis y
modificación de polímeros para obtener membranas con baja permeación de metanol existe
una característica común: el entrecruzamiento.
Entre las alternativas para controlar la permeación de metanol en las membranas poliméricas
se encuentran la formación de membranas tipo compósito, formadas de polímeros conductores
de protones y compuestos inorgánicos [200], las membranas multicapa [177], la incorporación
de iones metálicos [201] o compuestos inorgánicos con cargas positivas al polímero
sulfonado [202], así como la formación de enlaces covalentes entre las cadenas
poliméricas [203-205]. A continuación se explican los procedimientos mencionados.
1) Membranas compósito preparadas con un polímero conductor de protones modificado
con compuestos inorgánicos. Las membranas resultantes presentan propiedades intermedias
entre las de los componentes que le les dieron origen. En estado hidratado el polímero
sulfonado y el compuesto inorgánico (SnO2) son buenos conductores de protones, por lo que
el compósito también lo es. La permeación de metanol del compósito está relacionada con la
absorción de los componentes por separado, los cuales tienen el siguiente orden:
polímero sulfonado (37 % en peso) > compósito (13 % peso) > SnO2 (3 % en peso) [200].
2) Membranas multicapa. Son membranas compactas compuestas por una capa de
nanotubos de carbono sulfonados hubicada entre dos capas de copolímero sulfonado. La capa
de nanotubos proporciona estabilidad dimensional, resistencia a la elongación y a la fractura, y
evita que la membrana se hinchen, disminuyendo la permeación de metanol respecto a la
membrana de Nafion 117 [177]
3) Polímero con grupos sulfónicos y iones metálicos. La incorporación de iones metálicos
al copolímero sulfonado genera un entrecruzamiento mediante interacción electrostática entre
las cargas negativas de dos grupos sulfónicos de diferentes cadenas poliméricas con un ion
divalente como el estroncio. El entrecruzamiento reduce la absorción de agua de la membrana
y la permeación de metanol, pero disminuye la conductividad protónica [201].
4) Polímero con grupos sulfónicos y compuestos inorgánicos con cargas positivas. Un
polímero sulfonado con cargas negativas y un componente inorgánico (sílica con grupos
amina) pueden formar entrecruzamientos iónicos por interacción electrostática entre cargas.

CAPÍTULO 3
99
Este enlace evita la pérdida de la sílica, controla el hinchamiento del polímero, disminuye la
absorción de agua y reduce la permeación de combustible incrementando la densidad de la
membrana. Sin embargo se disminuye la conductividad protónica [202].
5) Formación de enlaces covalentes entre las cadenas poliméricas. La disminución de la
permeabilidad en membranas de SPEEK entrecruzados covalentemente, se atribuye a que los
entrecruzamientos entre cadenas poliméricas reducen los dominios hidrófilos, con lo cual
disminuye la absorción de agua y el hinchamiento de la membrana; consecuentemente se
reduce el transporte de especies polares como el metanol [204] mediante la formación de una
estructura densa [203]. El transporte de metanol en la membrana requiere canales con buena
conectividad, la reducción de la permeación de metanol se debe a que los entrecruzamientos
obstaculizan la movilidad de las cadenas, suprimiendo el hinchamiento de la membrana, que a
su vez reduce el tamaño de los canales por los que pueden pasar las moléculas de
metanol [205].
De forma general, las alternativas mencionadas para disminuir la permeación de metanol en
las PEM consisten en la incorporación de entrecruzamientos ya sean electrostático, iónico o
covalente. De forma similar, para las membranas DAg240-170%0.110% y
TAg120-170%0.110% se comprobó mediante extracción soxhlet la presencia de
entrecruzamientos por unidades de DVB o TMPTMA, así como el incremento de material
insoluble debido a las reacciones de sulfonación. La presencia e incremento de
entrecruzamientos podrían ser los responsables de la ausencia de permeación de metanol a
través de estas membranas. Por otro lado, a pesar de que se comprobó mediante
espectroscopía IR y determinación de capacidad de intercambio iónico que las membranas de
los copolímeros D y T sulfonados cuentan con grupos ácidos tanto carboxílicos como
sulfónicos, probablemente la cantida de grupos ácidos es insuficiente para generar continuidad
entre grupos. En la literatura está reportado que si los grupos ácidos en una membrana se
encuentren aislados dentro de la propia matriz polimérica, éstos se encuentran inaccesibles e
imposibilitados para contribuir a la capacidad de intercambio iónico de la membrana [206]. A
valores por debajo del umbral de percolación de la IEC, la transición morfología de la
membrana consiste en una discontinuidad entre grupos iónicos, donde la conectividad entre
estos dominios iónicos no es suficiente para que los protones y el agua se transporten a través
de ellos, lo que conlleva a valores de conductividad protónica bajos [207]. De forma similar,

CAPÍTULO 3
100
con las membranas de los copolímeros D y T sulfonados, la falta de conectividad entre los
dominios iónicos (regiones hidrófilas) evitaría que hubiera regiones por las cuales pudiera
atravesar el metanol, y en caso de haberlas, los entrecruzamientos evitarían que la separación
entre cadenas fuera tan amplia para dejar pasar estas moléculas, traduciéndose en nula
permeación de metanol a través de las membranas.
3.3.6 MICROSCOPÍA ELECTRÓNICA DE BARRIDO DE LAS MEMBRANAS
Los resultados de capacidad de intercambio iónico, absorción de agua, relación de
hinchamiento y permeación de metanol han sido favorables para la aplicación de las
membranas para celdas de combustible de metanol. Para ampliar y complementar la
caracterización de las membranas, así como para poder relacionar la morfología de las
membranas con su desempeño, éstas se analizaron mediante microscopía electrónica de
barrido. Se obtuvieron las micrografías de la topografía superficial y de la fractura criogénica
de las membranas de los copolímeros D y T sin sulfonar, y de las preparadas mediante
disolución-evaporación utilizando THF o THF-DMSO con los copolímeros
DAg240-170%0.110% y TAg120-170%0.110%.
Las Figura 3.24 y 3.25 corresponden a las morfologías de las membranas de los copolímeros
D y T sin sulfonar, se observa que las superficies de las membranas son lisas, y su fractura
muestra que son densas y sin huecos. A pesar de que estos copolímeros no contienenen grupos
sulfónicos, se prepararon sus membranas (con THF) para relacionar su morfología con su
desempeño mecánico, y para compararlas respecto a las membranas preparadas con los
copolímeros sulfonados.
Figura 3.24. Morfología de la membrana preparada con THF y el copolímero D sin sulfonar: a) topografía superficial y b) fractura criogénica.

CAPÍTULO 3
101
Figura 3.25. Morfología de la membrana preparada con THF y el copolímero T sin sulfonar: a) topografía superficial y b) fractura criogénica.
En las Figuras 3.26 y 3.27 se presenta la topografía superficial y la fractura criogénica de las
membranas de los copolímeros DAg240-170%0.110% y TAg120-170%0.110% preparadas
con THF. El proceso de sulfonación de los copolímeros genera cambios en su interacción con
el disolvente, que resulta en membranas menos densas en comparación a las de los
copolímeros sin sulfonar y con poros tanto en el cuerpo como en el extremo más cercano a la
cara de la membrana que estuvo en contacto con el ambiente durante la evaporación del THF.
Figura 3.26. Morfología de la membrana preparada con THF y el copolímero DAg240-170%0.110%: a) y b) topografía superficial, c) y d) fractura criogénica.

CAPÍTULO 3
102
Figura 3.27. Morfología de la membrana preparada con THF y el copolímero TAg120-170%0.110%: a) y b) topografía superficial, b) y c) fractura criogénica.
La presencia de poros en el interior de las membranas sulfonadas ya ha sido reportada por
Mulijani y col. para membranas de poliestireno sulfonadas, atribuidos a la presencia de
grupos -SO3H, indicando que conforme aumenta el contenido de grupos ácidos la cantidad de
poros incrementa [15]. Respecto a los poros en la superficie de las membranas, son más
abundantes y homogéneos, su presencia probablemente se deba al fenómeno llamado Proceso
de rocío [208], cuyo mecanismo de formación ha sido descrito y estudiado por Escalé [209],
además ha sido utilizado para formar películas con superficies porosas tipo panal de abejas.
Para que el Proceso de rocío suceda deben controlarse el material polimérico a utilizar, el
disolvente y la humedad en el aire. El mecanismo consiste en la formación de poros o burbujas
de aire en la superficie de la película polimérica por la evaporación del disolvente, siguiendo
los pasos que se enlistan a continuación y que se ilustran en la Figura 3.28 [208, 210]:
1) El disolvente empieza a evaporarse
2) La superficie del líquido se enfría por la evaporación del disolvente
3) La humedad del aire condensa en la superficie fría del líquido y forma pequeñas gotas
4) Las gotas de agua crecen y la solución polimérica las cubre
5) Se evapora el disolvente y las gotas de agua y los poros permanecen en la película

CAPÍTULO 3
103
Figura 3.28. Proceso de rocío para la formación de superficie porosa en membranas.
Para las membranas preparadas en el laboratorio, el Proceso de rocío pudo haber ocurrido
debido a que la etapa de volatilización del disolvente se realizó en condiciones ambientales,
sin controlar de la humedad de la atmósfera. Además, probablemente el Proceso de rocío se
puede inducir debido a los grupos ácidos hidrófilos de los copolímeros favorece la
condensación de agua en la superficie de las membranas.
Las Figuras 3.29 y 3.30 corresponden a las morfologías de las membranas preparadas con la
mezcla THF-DMSO (55 % DMSO) y los copolímeros DAg240-170%0.110% y
TAg120-170%0.110%. En estas membranas también se observan poros en la superficie y en el
cuerpo de las membranas, posiblemente formados mediante el proceso descrito.
Figura 3.29. Morfología de la membrana preparada con THF-55%DMSO y el copolímero DAg240-170%0.110%: a) y b) topografía superficial, c) y d) fractura criogénica.
Película con superficie porosa
Molde con solución polimérica
Vapor de disolvente
Vapor de disolvente
Vapor de agua
Vapor de disolventeVapor
de aguaDisolvente frío y nucleación de la
humedad
Crecimiento y acomodo de las gotas de agua
Evaporación de disolvente y
gotas de agua
gotas de agua ordenadas
pequeñas gotas de agua
desordenadas

CAPÍTULO 3
104
Figura 3.30. Morfología de la membrana preparada con THF-55%DMSO y el copolímero TAg120-170%0.110%: a) y b) topografía superficial, c) y d) fractura criogénica. En las Figuras 3.31 y 3.21 se muestran las micrografías de las membranas preparadas con la
mezcla de THF y 110 % de DMSO, los poros de la superficie son heterogéneos en forma y
tamaño, en comparación con las membranas anteriores (55 % DMSO). Pudiendo estar
relacionado con una diferente interacción de la humedad que condensa y la superficie de la
membrana, debido a que existe mayor cantidad de DMSO en la solución pólimérica.
Figura 3.31. Morfología de la membrana preparada con THF-110%DMSO y el copolímero DAg240-170%0.110%: a) y b) topografía superficial, c) y d) fractura criogénica.

CAPÍTULO 3
105
Figura 3.32. Morfología de la membrana preparada con THF-110%DMSO y el copolímero TAg120-170%0.110%: a) y b) topografía superficial, c) y d) fractura criogénica.
En el interior de las membranas los poros son de mayor tamaño y el espesor de las paredes que
los forman es menor, por lo que existe menor cantidad de polímero entre cavidad y cavidad,
sobre todo en la membrana TAg120-170%0.110% (Figura 3.32). El origen de los poros en las
membranas preparadas con las mezclas de THF-DMSO posiblemente se debe a un efecto
combinado: la presencia de un no-disolvente (DMSO) en la mezcla de disolventes utilizada y
la velocidad de evaporación de la solución. Ambos factores influencían en la morfología de la
membrana; se ha reportado que la combinación de estos dos factores puede generar estructuras
tipo esponja, macrocavidades y superficie celular [193]. Las cavidades se forman debido a que
una vez que el THF se ha volatilizadom el DMSO permanece entre el copolímero sulfonado
de la membrana, y una vez que es extraído, el espacio que ocupaba el DMSO permanece en
forma de poro.
3.3.7 ANÁLISIS TERMOMECÁNICO DE LAS MEMBRANAS
La comparación de resistencia mecánica de las membranas fue realizada mediante el
procedimiento descrito en la norma ASTM D790 [184], la cual es comúnmente utilizada en la
industria del plástico por que tiene las ventajas de simplificar el maquinado de las probetas y
evitar los problemas asociados al empleo de mordazas. Este procedimiento se utiliza como
medida de la resistencia del material ante la carga aplicada y su rigidez, con lo cual se puede

CAPÍTULO 3
106
determinar la resistencia a la flexión y el módulo de elasticidad de plásticos reforzados y sin
reforzar. La resistencia a la flexión representa el máximo esfuerzo desarrollado en la superficie
de la probeta soportada cerca de sus extremos ante una carga aplicada en el centro hasta que
ocurra la falla. En cambio, el módulo de elasticidad es la relación existente dentro del límite
elástico del esfuerzo, correspondiente a la máxima deformación que se ocasiona en la muestra
(fuerza/área [=] Pascales).
En función del número de puntos de apoyo de la muestra pueden realizarse ensayos de flexión
en tres puntos y en cuatro puntos. Para el análisis mecánico de las membranas, se utilizó el
modo de deformación de flexión en tres puntos, donde la carga se aplicó al centro de la cara
superior de la probeta en forma de barra, soportada por sus extremos sobre un portamuestras
que cuenta con dos puntos de apoyo. A partir de éstos ensayos se obtuvieron los datos
necesarios para calcular el módulo elástico de cada membrana, en las Figuras 3.33 y 3.34 se
encuentran los gráficos que muestran la deformación generada por la fuerza aplicada. A partir
de los gráficos se calcularon los valores de módulo elástico (Tabla 3.4) de las membranas
secas e hidratadas de Nafion 117 comercial, de los copolímeros D y T sin sulfonar y de los
copolímeros sulfonados DAg240-170%0.110% y TAg120-170%0.110% preparadas con
diferentes relaciones de THF-DMSO. En las etiquetas solo se indica el porcentaje de DMSO
utilizado para acortar lo más posible el nombre de las muestras, sin embargo en todas ellas se
utilizó THF como disolvente.
Figura 3.33. Comportamiento mecánico en estado seco e hidratado de membranas de Nafion 117 y de copolímero D y DAg240-170%0.110%. Diferente relación entre THF y DMSO.
0 -100 -200 -300 -400 -5000.0
0.2
0.4
0.6
0.8
1.0D
D
D
D
D
Fuer
za (N
)
Deformación (µm)500
0800
160024003200
1991 1723 1355 14051096
1834
390 53
2685
Mód
ulo
de
elas
ticid
ad (M
Pa)
D
D D secaDAg-240min170%0.110% DMSO 0% secaDAg-240min170%0.110% DMSO 0% hidratadaDAg-240min170%0.110% DMSO 55% secaDAg-240min170%0.110% DMSO 55% hidratadaDAg-240min170%0.110% DMSO 110% secaDAg-240min170%0.110% DMSO 110% hidratadaNafion 117 secaNafion 117 hidratada

CAPÍTULO 3
107
Figura 3.34. Comportamiento mecánico en estado seco e hidratado de membranas de Nafion 117 y de copolímero T y TAg120-170%0.110%. Diferente relación entre THF y DMSO. Tabla 3.4. Módulo elástico de membranas obtenido mediante análisis termomecánico
Membrana A (MPa) B (MPa) C (%) D (%) Nafion 117 390 53 86 -
D 2685 - - - T 3113 - - -
DAg240-170%0.110% DMSO0% 1991 1834 8 0 DAg240-170%0.110% DMSO55% 1723 1355 21 13
DAg240-170%0.110% DMSO110% 1405 1096 22 29 TAg120-170%0.110% DMSO0% 2433 1063 56 0
TAg120-170%0.110% DMSO55% 2110 605 71 13 TAg120-170%0.110% DMSO110% 555 332 40 77
A = Membranas secas B = Membranas hidratadas C= Disminución porcentual del módulo elástico por hidratación de la membranas D = Disminución porcentual del módulo elástico en las membranas secas por presencia de DMSO en el proceso de preparación. Se tomó como membrana base la preparada con THF como único disolvente. Nafion 117: Membrana comercial, no se preparó en el laboratorio
A partir del ensayo de flexión se observan los siguientes puntos:
Comparación entre Nafion y los materiales preparados en este trabajo: La membrana de
Nafion 117 es más flexible que cualquiera de las membranas preparadas, lo cual puede
deberse a la naturaleza rígida de los copolímeros de partida: poliestireno y poliácido
acrílico.
Efecto de la presencia de grupos –SO3H: Los copolímeros sin sulfonar D y T son más
0 -100 -200 -300 -400 -5000.0
0.2
0.4
0.6
0.8
1.0
T
T
T
T
T
T
T
Fuer
za (N
)
Deformación (µm)500
0800
160024003200
2110
695 555 3321063
390 53
3113
T
Mód
ulo
de
elas
ticid
ad (M
Pa)
2433
T T seca TAg-120min170%0.110% DMSO 0% secaTAg-120min170%0.110% DMSO 0% hidratadaTAg-120min170%0.110% DMSO 55% secaTAg-120min170%0.110% DMSO 55% hidratadaTAg-120min170%0.110% DMSO 110% secaTAg-120min170%0.110% DMSO 110% hidratadaNafion 117 secaNafion 117 hidratada

CAPÍTULO 3
108
rígidos que sus análogos sulfonados, lo cual se puede deber a que el acomodo entre cadenas
es diferente por la presencia de grupos -SO3H en los anillos aromáticos, los cuales ocupan
un volumen mayor que el átomo de hidrógeno que sustituyeron, así como por las cargas
iónicas de los grupos ácidos.
Efecto de la hidratación: Al comparar el comportamiento mecánico de las membranas en
estado seco y en estado hidratado, se observa que la hidratación disminuye el módulo. Este
efecto se presenta en las membrans de los copolímeros sulfonados D y T pero también en
las membranas de Nafion 117 debido a que el agua entra entre las cadenas poliméricas
generando un efecto plastificante en el material [10, 113]. La plastificación ocasiona que
con la misma fuerza aplicada se genera una mayor deformación en las membranas
hidratadas respecto a las secas. Este efecto ha sido observado en membranas de
SPEEK [176], donde el hinchamiento de las membranas por la presencia de agua está
inversamente relacionado con su resistencia mecánica.En la Tabla 3.4 se muestra que la
hidratación de las membranas afecta más a la membrana de Nafion 117 que a las de los
copolímeros sulfonados, ya que la disminución del módulo para la membrana de
Nafion 117 por la presencia de agua es de 86 %, mientras que para las membranas de los
copolímeros sulfonados la mayor disminución del módulo varía entre el 8 y el 71 %. Este
diferencia puede deberse a que los copolímeros D y T sulfonados contienen
entrecruzamientos que limitan el movimiento e hinchamiento de los materiales [176], a
diferencia de las membranas de Nafion 117 no entrecruzados (de acuerdo a las
especificación del fabricante).
Efecto de los entrecruzamientos: Cuando se compara el módulo de las membranas de los
copolímeros D y T sin sulfonar se observa que la membrana del copolímero T posee un
módulo mayor. Ésta diferencia está relacionado con el contenido de gel y por consiguiente
la cantidad de material entrecruzado presente en las membranas; la presencia de
entrecruzamientos en las películas incrementa su resistencia mecánica [176]. Al comparar
el efecto de la hidratación en las membranas de los copolímeros sulfonados, la disminución
del módulo para el copolímero DAg240-170%0.110% es menor (8, 21 y 22 %) que la
disminución del copolímero TAg120-170%0.110% (40, 56 y 71 %), sugiriendo una
plastificación mayor para éste último. Esta diferencia puede deberse a que con TMPTMA,
cada entrecruzamiento cuenta con tres ramificaciones, teniendo como centro de la molécula

CAPÍTULO 3
109
un carbono cuaternario y en cada funcionalización un enlace tipo éster que es móvil. En
cambio, en el copolímero entrecruzado con DVB, cada unidad de DVB genera
entrecruzamientos entre dos cadenas unidas por un anillo aromático, cuyos carbonos tienen
hibridación tipo sp2, cuya movilidad es menor en comparación a las de tipo éster del
TMPTMA.
Efecto del DMSO en la preparación de membranas: Utilizar la mezcla THF-DMSO en la
preparación de membranas genera membranas menos rígidas a partir de un mismo
copolímero sulfonado. Este efecto se ve reflejado en valores de módulo elástico menores,
que puede estar relacionado con la presencia de huecos que se comprimen a cargas bajas
durante la prueba de flexión, resistiendo con ello menos presión que una membrana densa.
Al utilizar DMSO con el copolímero DAg240-170%0.110% se genera una disminución del
módulo elástico de 13 y 29 % al emplear 55 y 110 % de DMSO respectivamente; mientras
que para el copolímero TAg120-170%0.110%, el módulo elástico disminuye 13 y 77 %
respectivamente, es decir, el DMSO se modifica más el comportamiento mecánico de las
membranas TAg120-170%0.110% en comparación a las DAg240-170%0.110%.
Comportamiento no lineal de la relación esfuerzo vs deformación. Las membranas de los
copolímeros sulfonados no presentan un comportamiento lineal similar al que presentan las
membranas de Nafion 117 y las membranas de los copolímeros D y T. Esta diferencia
puede deberse a que durante la prueba de flexión, en la cara superior de la membrana
existen fuerzas de compresión, mientras que en la cara opuesta se generan fuerzas de
tensión. Durante el proceso de compresión, las paredes de los huecos de los materiales
porosos pueden romperse con el incremento de la presión ejercida por la sonda, con lo cual
el material se vuelve más y más compacto. Sin embargo la presencia de poros cercanos a la
cara posterior de la probeta pueden actuar como puntos de iniciación de fractura. La
propagación de fracturas en materiales porosos que no son dúctiles cambia dependiendo de
la porosidad, pudiendo ser lineal o en trayectoria curva, según suceda el rompimiento de los
puentes entre poros. Por ésta razón, los materiales porosos suelen tener proceso de
deformación y compactación no homogénea y complejo [211, 212].
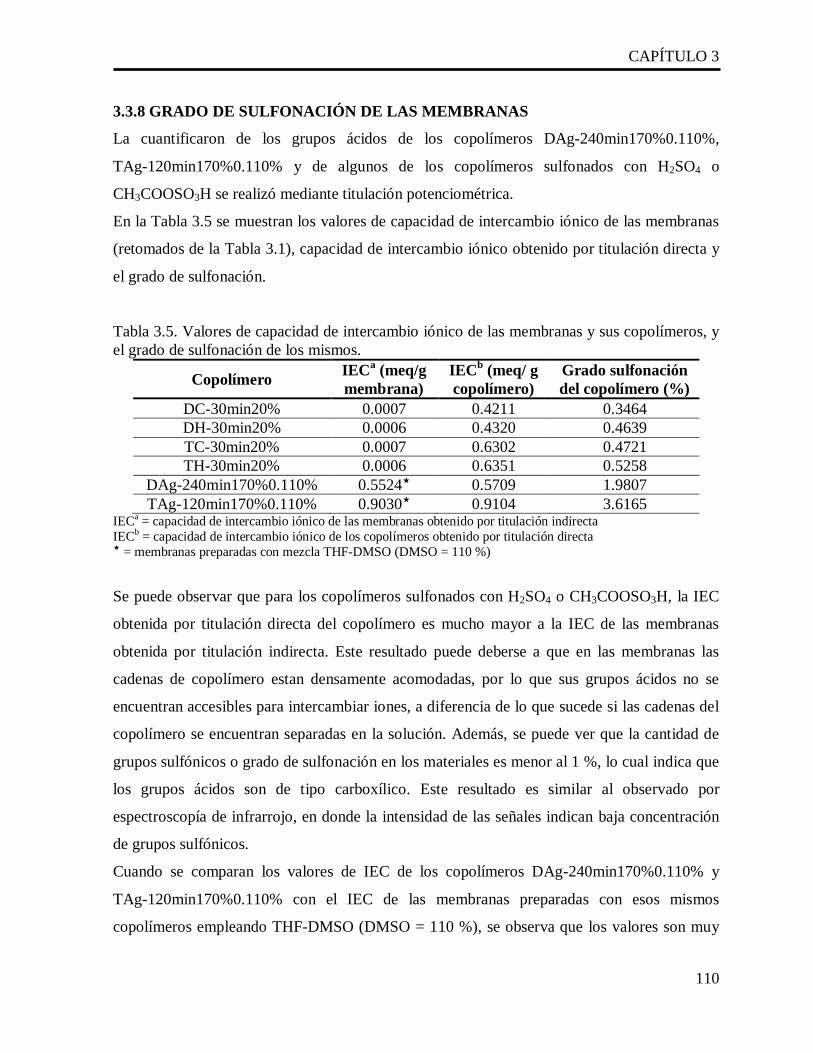
CAPÍTULO 3
110
3.3.8 GRADO DE SULFONACIÓN DE LAS MEMBRANAS
La cuantificaron de los grupos ácidos de los copolímeros DAg-240min170%0.110%,
TAg-120min170%0.110% y de algunos de los copolímeros sulfonados con H2SO4 o
CH3COOSO3H se realizó mediante titulación potenciométrica.
En la Tabla 3.5 se muestran los valores de capacidad de intercambio iónico de las membranas
(retomados de la Tabla 3.1), capacidad de intercambio iónico obtenido por titulación directa y
el grado de sulfonación.
Tabla 3.5. Valores de capacidad de intercambio iónico de las membranas y sus copolímeros, y el grado de sulfonación de los mismos.
Copolímero IECa (meq/g membrana)
IECb (meq/ g copolímero)
Grado sulfonación del copolímero (%)
DC-30min20% 0.0007 0.4211 0.3464 DH-30min20% 0.0006 0.4320 0.4639 TC-30min20% 0.0007 0.6302 0.4721 TH-30min20% 0.0006 0.6351 0.5258
DAg-240min170%0.110% 0.5524 0.5709 1.9807 TAg-120min170%0.110% 0.9030 0.9104 3.6165
IECa = capacidad de intercambio iónico de las membranas obtenido por titulación indirecta IECb = capacidad de intercambio iónico de los copolímeros obtenido por titulación directa = membranas preparadas con mezcla THF-DMSO (DMSO = 110 %)
Se puede observar que para los copolímeros sulfonados con H2SO4 o CH3COOSO3H, la IEC
obtenida por titulación directa del copolímero es mucho mayor a la IEC de las membranas
obtenida por titulación indirecta. Este resultado puede deberse a que en las membranas las
cadenas de copolímero estan densamente acomodadas, por lo que sus grupos ácidos no se
encuentran accesibles para intercambiar iones, a diferencia de lo que sucede si las cadenas del
copolímero se encuentran separadas en la solución. Además, se puede ver que la cantidad de
grupos sulfónicos o grado de sulfonación en los materiales es menor al 1 %, lo cual indica que
los grupos ácidos son de tipo carboxílico. Este resultado es similar al observado por
espectroscopía de infrarrojo, en donde la intensidad de las señales indican baja concentración
de grupos sulfónicos.
Cuando se comparan los valores de IEC de los copolímeros DAg-240min170%0.110% y
TAg-120min170%0.110% con el IEC de las membranas preparadas con esos mismos
copolímeros empleando THF-DMSO (DMSO = 110 %), se observa que los valores son muy

CAPÍTULO 3
111
similares entre sí. Esta similitud podría deberse a que las membranas cuentan con cavidades
formadas por paredes delgadas, que permiten su hidratación e hinchamiento por la presencia
de agua. Esto propicia que los grupos ácidos estén más accesibles para llevar a cabo su
intercambio al encontrarse en una solución básica. Además, la cantidad de grupos sulfónicos
en las membranas DAg-240min170%0.110% y TAg-120min170%0.110% es mayor al de las
primeras membranas evaluadas ~2 % y ~3.7 %, sin embargo el grado de sulfonación sigue
siendo bajo.
A pesar de que los valores de IEC de las membranas de los copolímeros
DAg-240min170%0.110% y TAg-120min170%0.110% son competitivos a los de la
membrana de Nafion 117, la IEC proviene de los grupos sulfónicos pero también de los
carboxílicos. Hay que tomar en cuenta que los grupos -COOH son tipo ácido débil, mientras
que los grupos -SO3H son de tipo fuerte pero se encuentran en poca cantidad. Además la
estructura de los copolímeros de PS-PAA sulfonados consiste en cadenas poliméricas con
grupos aromáticos voluminosos y sustituidos, lo cual les confiere rigidez, a diferencia de lo
que sucede con el Nafion 117 que está formado por una estructura lineal con grupos laterales
móviles tipo éter. Por ésta razón, a pesar de tener valores de IEC similares, el acomodo en las
cadenas poliméricas que conforman las diferentes membranas no es el mismo. En el caso de
que las membranas de los copolímeros DAg-240min170%0.110% y
TAg-120min170%0.110% la formación de regiones hidrófilas es baja.

CAPÍTULO 3
112
3.4 CONCLUSIONES DEL CAPÍTULO 3
Las membrans DAg240-170%0.110% y TAg120-170%0.110% presentaron nula
permeación de metanol debido al bajo grado de sulfonación (1.98 y 3.6 %
respectivamente), así como al entrecruzamiento de las cadenas de polímero que
restringe su movimiento, evitando que las cadenas se separen y con ello la difusión del
metanol.
Se comprobó mediante SEM que el uso de THF-DMSO para preparar las membranas
genera diferentes morfologías y porosidades. A mayor cantidad de DMSO empleada,
mayor cantidad de huecos, mayor cercanía entre ellos, y menor espesor de las paredes
que conforman los poros.
La disminución de espesor entre poros en las membranas incrementa la accesibilidad a
los grupos funcionales ácidos y con ello sus protones, alcanzando valores de IEC
cercanos a los esperados por titulación directa de los copolímeros.
La presencia de poros en las membranas explica porque las membranas preparadas con
THF-DMSO al hidratarse pueden llegar a duplicar su peso y a la vez tener relaciones
de hinchamiento menores al 12%. En donde la mayor cantidad de agua se aloja en los
poros y solo una parte entra entre cadenas que generan el hinchamiento.
Las membranas son mecánicamente estables para llevar a cabo su activación y
caracterización pero en comparación con la membrana de Nafion 117 son frágiles, la
presencia de poros disminuye el modulo elástico y ocasiona que se fracturen a cargas
bajas (< 1 N).

CAPÍTULO 4
113
CAPÍTULO 4. MODIFICACIÓN DE MEMBRANAS CON PLATINO
4.1 ANTECEDENTES
Los componentes básicos de una celda de combustible son los platos colectores de electrones
que poseen canales por los cuales fluye el combustible o el oxidante, y el ensamble
membrana-electrodo (membrane electrode assembly -MEA-). Ésta se compone de una
membrana intercambiadora de protones, el catalizador en cada costado de la membrana (donde
se lleva a cabo las reacciones electroquímicas), una capa difusora de gases y empaques que
sirven para contener herméticamente los reactivos entre los platos colectores y el ensamble
membrana-electrodo [213, 214].
Existen dos alternativas para formar el MEA, una consiste en incorporar el catalizador
(generalmente soportado en un material poroso) en forma de tinta catalítica en la capa difusora
de gases, que puede ser tela o papel de carbón. En la otra alternativa se incorpora el
catalizador directamente en la membrana con un pincel, un rodillo, mediante impresión o
pulverizando la tinta catalítica sobre la membrana [215, 216]. La tinta catalítica es una
suspensión uniforme que se prepara dispersando un catalizadoren en una mezcla de disolvente
con ionómero polimérico que sirve como adhesivo, siendo el Nafion comercial en
presentación de solución el ionómero generalmente empleado. El máximo aprovechamiento de
la tinta o capa catalítica se logra optimizando la frontera triple de fases
(triple phase boundary - TPB -), la cual es el punto donde coinciden las partículas de
catalizador, el polímero conductor de protones y la trayectoria de difusión del
reactivo [213, 217, 218]. La optimización de la TPB involucra utilizar la cantidad óptima de
Nafion, puesto que si se utiliza poco, la conductividad iónica de la capa catalítica puede ser
insuficiente (lo que genera catalizador inactivo), pero si se utiliza demasiado, éste puede cubrir
toda la superficie del catalizador y/o su soporte, evitando que el gas acceda a la superficie del
catalizador [215]. Una alternativa para maximizar el contacto del catalizador con el polímero
conductor de iones es la formación de las partículas de catalizador directamente sobre la
membrana y para lograrlo se puede utilizar el método Takenaka-Torikai [219]. Éste método
también es conocido como reducción transmembrana [220] y con él es posible la formación e
incorporación de partículas metálicas catalizadoras directamente sobre una membrana en un
solo paso. La cantidad de catalizador que se incorpora se puede controlar variando el tiempo y

CAPÍTULO 4
114
la temperatura de reacción, la concentración de la sal precursora del metal y la concentración
del agente reductor.
La incorporación de partículas catalíticas directamente sobre una membrana tiene su
aplicación en el campo de las celdas de combustible [221, 222], en la síntesis
electrocatalítica [223-225], en la generación de hidrógeno y oxígeno gaseosos [226], en
generadores de ozono [227], sistemas purificadores de agua [228] y sensores [229, 230]. Por
ello, dependiendo de la reacción que se desee catalizar y del ambiente químico al que estarán
expuestas las partículas catalíticas, será el tipo de sal precursora de metal o metales que se
utilicen para su síntesis.
Para las celdas de combustible de membrana intercambiadora de protones de tipo DMFC o
PEMFC, las partículas metálicas deben catalizar las reacciones de oxidación del combustible
(metanol o hidrógeno) y reducción de oxígeno. Para el caso de las PEMFC, varios
investigadores han hecho esfuerzos experimentales y teóricos para encontrar el mejor
catalizador para las reacciones que suceden en el ánodo y en el cátodo. Entre los reportes de
mayor importancia se encuentran aquellos realizados por Nørskov [231] y Greeley [232] que
involucran cálculos computacionales para estudiar metales puros (Ag, As, Au, Bi, Cd, Co, Cu,
Fe, Ir, Mo, Ni, Pd, Pt, Re, Rh, Ru, Sb, y W) y aleaciones de ellos, obteniendo modelos teóricos
que predicen la relación entre la velocidad de la reacción del catalizador y la energía de
adsorción del reactivo y desorción de productos.
Con dichos modelos se ha encontrado que cuando el reactivo se une con mucha fuerza a la
superficie del metal o aleación, su actividad catalítica es muy alta al inicio, pero debido a que
los productos de la reacción también se adsorben fuertemente, no se remueven de la superficie
del catalizador, por lo que rápidamente decrece su actividad. En el otro extremo, cuando la
energía de enlace del reactivo con el catalizador es muy débil, el reactivo puede acercarse y
adsorberse débilmente al catalizador, pero posteriormente se alejaría de él sin haber
reaccionado, por lo que la actividad del catalizador sería baja desde un inicio. Nørskov y
Greeley determinaron con sus modelos computacionales que el Pt es metal puro que mantiene
el mejor equilibrio entre la velocidad de la reacción del catalizador y la energía de adsorción
del reactivo, tanto para el oxígeno [231] como para el hidrógeno [232]. Sin embargo al utilizar
el mismo catalizador se presentan diferencia entre la reacción de reducción de oxígeno y la
reacción de oxidación del hidrógeno [215].

CAPÍTULO 4
115
En las DMFC, donde la reacción principal es la oxidación del metanol, el Pt es un buen
catalizador, sin embargo suele envenenarse rápidamente por la adsorción de CO en su
superficie. Una forma de subsanar dicho inconveniente consiste en utilizar un segundo metal
como co-catalizador, el cual sea capaz de formar especies metal-OH que actúen como fuente
de oxígeno, el cual es necesario para que el CO adsorbido se oxide a dióxido de carbono,
dando lugar a la liberación de los sitios activos del Pt en su superficie [233-235].
Como ya se mencinó, el platino es el metal ideal a seleccionar como catalizador para una
membrana en una celda de combustible de tipo PEMFC, más que para una tipo DMFC. Sin
embargo, debido a que la finalidad principal de ésta investigación es la preparación de
membranas, y como punto secundario su modificación con la incorporación de partículas
catalíticas, se seleccionó el Pt, esperando que los resultados abran el campo de visión de la
viabilidad de incorporación de nanopartículas catalíticas formadas in situ mediante el método
de reducción transmembrana.
4.2 EXPERIMENTAL
Las membranas preparadas con diferentes mezclas de disolventes y los copolímeros D y T
sulfonados con ácido sulfúrico-Ag2SO4, fueron modificadas con platino en una de sus
superficies. Su caracterización incluye difracción de rayos X, determinación de contenido de
platino mediante espectrometría de emisión de plasma, capacidad de intercambio iónico,
absorción de agua, relación de hinchamiento, permeación de combustible y microscopía
electrónica de barrido. Algunas de estas caracterizaciones ya se había llevado a cabo en estas
membrans pero sin platinino, sin embargo se presuponía que la presencia de platino sobre la
membrana podría ocasionar una obstrucción parcial o total de la superficie, que a su vez
ocasionaría cambios en sus propiedades, o incluso cambios generados por el proceso de
reducción de platino transmembrana.
4.2.1 MATERIALES
Membranas de los copolímeros DAg240-170%0.110% y TAg120-170%0.110%, ácido
clorhídrico (36.5-38 %, Sigma Aldrich), ácido hexacloroplatínico hexahidratado (≥ 37.50 % Pt
Sigma Aldrich), ácido nítrico (70 %, CTR Scientific), ácido sulfúrico (98 % J.T.Baker),
borohidruro de sodio 98 % (Sigma aldrich), cloruro de sodio (J.T. Baker), hidróxido de sodio

CAPÍTULO 4
116
(Aldrich), membrana de Nafion 117 (Sigma Aldrich).
4.2.2 INCORPORACIÓN DE PLATINO EN LAS MEMBRANAS
Las primeras reacciones de incorporaciónde platino mediante reducción transmembrana se
realizaron con la membrana de Nafion 117, empleando diferentes concentraciones de la sal
precursora del metal y del reactivo reductor. Las membranas de Nafion 117 cubiertas con
platino obtenidas se caracterización por SEM, y a partir de dicha caracterización se
seleccionaron las concentraciones de ácido hexacloroplatínico (H2PtCl6) y de borohidruro de
sodio (NaBH4) a utilizar con las membranas de los copolímeros DAg240min-170%0.110% y
TAg120min-170%0.110%.
El procedimiento de reducción transmembrana o Takenaka-Torikai [219] se llevó a cabo en
una celda con dos compartimentos separados por la membrana intercambiadora de iones, cada
compartimento tiene una capacidad de 50 mL y diámetro interno de 2.94 cm. Uno de los
compartimentos contiene la sal precursora del metal y el otro la solución del agente reductor.
El reductor se difunde a través de la membrana y reacciona con los iones metálicos que se
encuentran al otro lado de la membrana [220]. En la Figura 4.1 se muestra un esquema de la
celda empleada.
Figura 4.1. Sistema para llevar a cabo la incorporación de platino mediante reducción transmembrana.
En uno de los compartimentos se adicionó 15 mL de la solución de la sal precursora de platino
(3.33 o 12.2 mM) y se dejó reposar por 30 minutos. Una vez transcurrido el tiempo, en el otro
compartimento se adicionó 50 mL de la solución de NaBH4 (60 o 100 mM) y se dejó
reaccionar por 3 horas. El reductor se difundió a través de la membrana por un área efectiva de

CAPÍTULO 4
117
6.7886 cm2 y redujo los iones metálicos en la cara de la membrana que se encuentra en
contacto con la solución precursora del metal. Durante el transcurso de la reacción se
acumularon burbujas en la superficie de la membrana, razón por la cual el sistema se agitó
cada 15 min hasta terminar las 3 horas de reacción.
Una vez transcurrido el tiempo de reacción se retiraron las soluciones del sistema y la solución
de la sal precursora del metal se reservó para analizarla posteriormente. La membrana se
removió del sistema, se enjuagó con agua destilada, se sumergió por 2 horas en 75 mL de
H2SO4 0.5 M a temperatura ambiente, se transfirió a 75 mL de agua destilada por 1 hora y
finalmente se secó a 80 °C con vacío constante.
4.2.3 ESPECTROFOTOMETRÍA UV-VIS
Para corroborar la reducción de los iones metálicos durante el experimento, se hizo una
dilución 1:4 con agua desionizada y cada solución precursora del metal utilizada: la inicial y
las residuales, las cuales se midieron por separado en una celda de cuarzo con un
espectrofotómetro Shimadzu UV-2401PC.
4.2.4 DIFRACCIÓN DE RAYOS X
Las propiedades estructurales del platino formado sobre las membranas fue caracterizado por
difracción de rayos X, colocando directamente sobre el portamuestras del difractómetro la
membrana cubierta de platino. Para las mediciones se utilizó un difractómetro Siemens D5000
con una fuente de radicación de CuKα, operado a 25 mA y 35 Kv, y se realizó un barrido en la
escala de 2θ de 15 a 80 ° con pasos de 0.06 °.
4.2.5 DIGESTIÓN ÁCIDA Y CUANTIFICACIÓN DEL PLATINO EN LAS
MEMBRANAS POR ESPECTROMETRÍA DE EMISIÓN DE PLASMA
Para la cuantificación del platino se llevo a cabo la digestión ácida de cada membrana y con el
material resultante que contenía los iones metálicos se prepararon soluciones acuosas que s
eanalizaron en un Espectrofotómetro de Emisión por Plasma marca Thermo Jarrell Ash
modelo Iris Advantage.
Las membranas con platino se calentaron a 100 °C por 1 hora y se dejaron enfriar en un

CAPÍTULO 4
118
desecador, a continuación se cortaron círculos de las membranas en las zonas que contenían
platino y se calculó su área. Los círculos se colocaron en vasos de precipitado, se les agregó a
cada uno 2 mL de H2SO4 al 98%, se cubrieron con vidrios de reloj, se calentaron para generar
la degradación y posterior evaporación del contenido líquido y sólido, para conseguir que en
los vasos permanecieran únicamente los iones metálicos. Los vasos se dejaron enfriar y se
agregó a cada uno de ellos 1 mL de HNO3 al 70% y 3 mL de HCl al 36.5-38 %, se agitaron y
se calentaron hasta sequedad. Los vasos se dejaron enfriar y se les adicionó 1 mL de HCl al
36.5-38 % y 15 mg de NaCl, se procuró disolver la sal y limpiar las paredes de los vasos con
la solución. Los vasos se calentaron y se dejó evapora su contenido hasta sequedad. Se
agregaron 3 mL de HCl al 36.5-38 % y 15 mL de agua desionizada, se filtraron las soluciones
con papel Whatman numero 42 y las soluciones filtradas se aforaron a 50 mL con agua
desionizada [236, 237].
Para calcular la concentraciones de platino incorporado mediante la reducción transmembrana
se utilizó la ecuación 4.1.
푃푙푎푡푖푛표푒푛푙푎푠푚푒푚푏푟푎푛푎푠(푚푔 푐푚⁄ ) = ∗ . á ( )
(ec.4.1)
Donde C es la concentración de platino determinada por Espectrofotometría de Emisión por
Plasma en ppm (mg/L) y 0.05 L corresponde al volumen total de la solución que contenía el
platino de la muestra tratada por digestión ácida.
Si todo el platino presente en los 15 mL de la solución de H2PtCl6 3.33 mM utilizada en el
proceso de reducción transmembrana se hubiera reducido e incorporado en los 6.7886 cm2
correspondientes al área efectiva expuesta, se hubiera tenido el 100 % de incorporación del
metal. Dicha concentración se calculó empleando el peso molecular del platino (195.08 g/mol)
y la ecuación 4.2.
100%푑푒푖푛푐표푟푝표푟푎푐푖ó푛푑푒푝푙푎푡푖푛표(푚푔 푐푚⁄ ) = 0.00333 . .
195080 =
1.4353 푚푔 푐푚⁄ (ec.4.2)
Conociendo la concentración de platino (1.4352 mg/cm2) equivalente al 100 % de
incorporación de metal en la membrana, se relacionaron las concentraciones reales
incorporadas y se determinó su porcentaje correspondiente.

CAPÍTULO 4
119
4.3 RESULTADOS Y DISCUSIÓN
4.3.1 ESPECTROFOTOMETRÍA UV-VIS
Para corroborar que haya ocurrido la reducción de los iones platino mediatne el método
Takenaka-Torikai con las membranas de los copolímeros DAg240min-170%0.110% y
TAg120min-170%0.110%, se llevó a cabo el análisis por espectrofotometría UV-Vis de las
soluciones iniciales del precursor metálico y de las soluciones utilizadas en cada experimento
(Figura 4.2). Se observa que el espectro de la dilución de la solución inicial de H2PtCl6
presenta la banda característica del ion PtCl62– con máximo en 260 nm [238] y otra
correspondiente al ion Pt2+ en 220 nm [239].
Figura 4.2. Espectros UV-Vis de soluciones de la sal H2PtCl6, antes y después de ser empleadas en el proceso de Reducción transmembrana.
La intensidad de las señales de los iones metálicos de todas las soluciones empleadas en los
experimentos disminuyó. Con las membranas del copolímero DAg240min-170%0.110%, las
membranas preparadas con DMSO la diminución dué mayor que con la membrana preparada
únicamente con THF. Mientras que con las membranas del copolímero
TAg120min-170%0.110%, la ausencia de las bandas con máximos en 260 y 220 nm indica la
reducción total de los iones metálicos.
La cuantificación del platino reducido mediante la relación de la disminución de bandas en
260 y 220 nm y que correspondería a la cantidad de Pt0 incorporado en las membranas podría
haberse llevado a cabo [239, 240] si el platino reducido se hubiera formado únicamente sobre
ellas. Sin embargo esto no sucedió así, en todos los experimentos el platino metálico también
se formó y se adhirió en las paredes del compartimento que contenía la sal precursora del
metal. No obstante, la caracterización de las solucones mediante UV-Vis sirvió para
200 240 280 320 360 4000.0
0.5
1.0
1.5
2.0
2.5
Abs
orba
ncia
(u. a
.)
Longitud de onda (nm)
DAg240min-170%0.110%
200 240 280 320 360 4000.0
0.5
1.0
1.5
2.0
2.5TAg120min-170%0.110%
Abs
orba
ncia
(u. a
.)
Longitud de onda (nm)
Solución inicial de H2PtCl6
Solución remanente utilizada con las membranas preparadas con: 0% DMSO 55% DMSO 110% DMSO

CAPÍTULO 4
120
evidenciar que el método de reducción transmembrana puede ser utilizado con las membranas
de los copolímeros de PS-PAA sulfonados; a pesar de que necesita ser perfeccionado.
Además, a partir de los espectros de UV-Vis se deduce que la difusión del agente reductor a
través de las membranas del copolímero TAg120min-170%0.110% sucede con mayor
efectividad que en las membranas del copolímero DAg240min-170%0.110%, debido a que
para el primer copolímero T, las soluciones ya no contenían iones metálicos una vez finalizado
los experimentos.
4.3.2 DIFRACCIÓN DE RAYOS-X
Las membranas metalizadas mediante la reducción transmembrana se analizaron mediante
difracción de Rayos-X, generando los patrones de difracción que se muestran en la Figura 4.3.
Figura 4.3. Patrón de difracción de Rayos-X de las membranas de los copolímeros de PSS-PAA sometidas al proceso de reducción de platino transmembrana.
En la Figura 4.3 se observan señales en la escala 2θ en 40.1, 46.6 y 67.8 °, las cuales al
compararlas con la base de datos JCP2, se encontró que coinciden en posición e intensidad
(intensidad porcentual relativa de 100, 30 y 16) con las correspondientes al platino de
estructura cristalina tipo cúbica centrada en la cara, cuyos planos cristalográficos son (111),
(200) y (220) (PDF: 10-1194). No se observan otros picos de difracción, indicando la ausencia
de subproductos con estructura cristalina.
20 30 40 50 60 70 80 90
(220)(200)
TAg120min-170%0.110% DMSO 0%
TAg120min-170%0.110% DMSO 55%
TAg120min-170%0.110% DMSO 110%
Inte
nsid
ad (U
A)
2 (°)
DAg240min-170%0.110% DMSO 110%
DAg240min-170%0.110% DMSO 55%
DAg240min-170%0.110% DMSO 0%
(111)
JCP2 PDF: 10-1194 Pt FCC
40.0°46.5° 67.8°

CAPÍTULO 4
121
Debido a que el estireno es el componente principal de los copolímeros, y en menor
proporción el ácido acrílico, se esperaría que la interacción de los átomos en el copolímero
fuera similar al del poliestireno puro. Por ello, posiblemente la señal amplia en ~ 20 ° de la
escala 2θ que se observa en la Figura 4.3 corresponde a la interacción por fuerzas tipo Van der
Waals entre anillos estirénicos [241, 242].
Los resultados de DRX confirman la efectividad del método Takenaka-Torikai para la
formación de platino metálico cristalino sobre las membranas de PSS-PAA, de forma similar a
lo reportado para membranas de Nafion [174, 229, 243-247]. La utilización de éste método
reduce las variables a ajustar al obtener una membrana con catalizador, puestoque no fue
necesario un soporte adicional para la síntesis previa de las partículas de platino. En un solo
paso se formó e incorporó el platino metálico sobre las membranas, lo cual puede mejorar la
conductividad protónica como consecuencia del contacto efectivo entre la membrana
intercambiadora de protones y la capa catalítica [215]. Además, al no ser necesaria la
preparación de una tinta catalítica para incorporar las partículas metálicas sobre la membrana,
no se necesita un adhesivo (generalmente Nafion en solución), evitando la reducción de la
ocurrencia de la frontera triple de fases. Tampoco se presentaron problemas de
incompatibilidad entre la tinta catalítica y las membranas cuando estas están compuestas de
polímeros diferentes al Nafion.
4.3.3 CUANTIFICACIÓN DEL PLATINO EN LAS MEMBRANAS POR
ESPECTROMETRÍA DE EMISIÓN DE PLASMA
Después de comprobar la incorporación de platino metálico sobre la superficie de las
membranas DAg240-170%0.110% y TAg1240-170%0.110% mediante difracción de rayos X,
se llevó a cabo la cuantificación de paltino mediante espectrometría de emisión de plasma. En
la Figura 4.4 se comparan las membranas antes y después de incorporarles el metal, y se indica
con círculos de línea discontinua el área aproximada que se empleó para llevar a cabo la
digestión ácida.

CAPÍTULO 4
122
Figura 4.4. Membranas de los copolímeros DAg240-170%0.110% y TAg1240-170%0.110% antes y después de incorporarles el metal mediante reducción transmembrana.
La presencia de grupos carboxílicos y sulfónicos en las membranas de los copolímeros D y T
sulfonados pueden adquirir cargas negativas cuando las membrans se encuentran hidratadas y
sus protones se solvatan. Las cargas negativas podrían repeler a los iones complejos PtCl62-
presentes en la solución de la sal precursora del metal. Sin embargo, puede existir interacción
entre cargas los grupos –COO- y –SO3- y los iones Pt2+; los grupos cargados negativamente
podrían estar actuando como quelante hasta el instante en que el reductor se difunde a través
de la membrana y alcanca los iones metálicos para generar Pt0. La interacción entre iones
positivos de platino con grupos sulfónicos ya ha sido reportada con anterioridad por Waje y
col. [248] durante la deposición de nanopartículas de platino en nanotubos de carbono
funcionalizados con grupos sulfónicos, lo cual facilita precisamente la deposición uniforme de
Pt0 en la superficie de los nanotubos.
En la Tabla 4.1 se indican los porcentajes de platino incorporados en las membranas, los
cuales no superan el 25% respecto a la cantidad de platino presente en la solución empleada en
el proceso de reducción transmembrana. Sin embargo la concentración de platino (mg/cm2)
formada sobre la superficie de las membranas es superior a la concetración de platino con
actividad catalítica que se busta incorportar en fechas recientes.
0%DMSO 55%DMSO110%DMSO
Después
Antes
Reducción trans-
membrana de platino
Membranas del copolímero DAg240-170%0.110% empleando THT-DMSO para su preparación
0%DMSO 55%DMSO 110%DMSO
Membranas del copolímero TAg120-170%0.110% empleando THT-DMSO para su preparación

CAPÍTULO 4
123
Tabla 4.1. Platino incorporado mediante reducción transmembrana. Exp. Membrana Platino (mg/cm2) Platino (%)
1 DAg240min-170%0.110% DMSO 0% 0.1449 10.1 2 DAg240min-170%0.110% DMSO 55% 0.0243 1.7 3 DAg240min-170%0.110% DMSO 110% 0.0765 5.3 4 TAg120min-170%0.110% DMSO 0% 0.3561 24.8 5 TAg120min-170%0.110% DMSO 55% 0.1160 8.1 6 TAg120min-170%0.110% DMSO 110% 0.0914 6.4
En los inicios de los estudios de celdas de combustible, la cantidad de catalizador de platino
que se empleaba era de hasta 28 mg/cm2 [215], sin embargo, debido a los elevados costos de
este metal y/o de sus sales precursoras, se ha ido reduciendo la cantidad de platino a utilizar
llegando a emplear concentraciones de 0.1 mg/cm2 con resultados prometedores para
PEMFC [249].
4.3.4 CAPACIDAD DE INTERCAMBIO IÓNICO
En la Tabla 4.2 se encuentran los valores de capacidad de intercambio iónico de las
membranas de los copolímeros DAg240min-170%0.110% y TAg120min-170%0.110% antes
y después de ser sometidas al proceso de reducción transmembrana con H2PtCl6 y NaBH4.
Tabla 4.2. Capacidad de intercambio iónico de las membranas antes y después de ser modificadas por el proceso de reducción de platino transmembrana.
Membrana IEC (meq/g) Reducción de IEC por
presencia de platino (%) SIN platino CON platino
DAg240min-170%0.110% DMSO 0% 0.1436 0.1242 13.51 DAg240min-170%0.110% DMSO 55% 0.4231 0.4150 1.91 DAg120min-170%0.110% DMSO 110% 0.5524 0.5434 1.63 TAg120min-170%0.110% DMSO 0% 0.0783 0.0654 16.47 TAg120min-170%0.110% DMSO 55% 0.6525 0.6219 4.69 TAg120min-170%0.110% DMSO 110% 0.9030 0.8844 2.06
De los valores de la Tabla 4.2 se observa que la incorporación de platino sobre las membranas
disminuye su capacidad de intercambio iónico, particularmente en las membranas preparadas
únicamente con THF. Esta disminución puede deberse a que éstas membrans tienen un mayor
contenido de platino en su superficie y posiblemente están más obstruidas, disminuyendose la
accesibilidad de los protones.

CAPÍTULO 4
124
4.3.5 ABSORCIÓN DE AGUA
En la Tabla 4.3 se indican lo valores de absorción de agua de las membranas antes y después
de incorporarles platino en una de sus superficies. Al igual que lo sucedido con la capacidad
de intercambio iónico, la capacidad de absorción de agua de las membranas ha disminuido
después de haber sido exponerse al proceso de reducción de platino transmembrana,
probablemente porque el platino puede estar dificultando la entrada del agua a las regiones
hidrófilas de las membranas.
Tabla 4.3. Capacidad de absorción de agua de las membranas antes y después de ser modificadas por el proceso de reducción de platino transmembrana.
Membrana Absorción de agua (% peso) SIN platino CON platino
DAg240min-170%0.110% DMSO 0% 15.2210 15.0673 DAg240min-170%0.110% DMSO 55% 64.9186 64.3408 DAg120min-170%0.110% DMSO 110% 105.1197 104.4259 TAg120min-170%0.110% DMSO 0% 14.9514 14.2666 TAg120min-170%0.110% DMSO 55% 83.5415 81.4613 TAg120min-170%0.110% DMSO 110% 139.8289 137.0323
4.3.6 RELACIÓN DE HINCHAMIENTO
En la Tabla 4.4 se encuentran lo valores de relación de hinchamiento de las membranas al
hidratarse, antes y después de que se les incorporó platino sobre una de sus superficies. Se
observa que el cambio de dimensión las membranas con platino es menor en comparación a
las membranas que no lo contienen.
Tabla 4.4. Relación de hinchamiento de las membranas antes y después de ser modificadas por el proceso de reducción de platino transmembrana.
Membrana Relación de hinchamiento (%) SIN platino CON platino
DAg240min-170%0.110% DMSO 0% 2.5 2.0 DAg240min-170%0.110% DMSO 55% 10.1 9.6 DAg120min-170%0.110% DMSO 110% 9.5 8.9 TAg120min-170%0.110% DMSO 0% 6.1 5.8 TAg120min-170%0.110% DMSO 55% 10.7 9.7 TAg120min-170%0.110% DMSO 110% 10.9 9.4

CAPÍTULO 4
125
4.3.7 PERMEACIÓN DE COMBUSTIBLE
El platino es un metal catalíticamente activo que generalmente se utiliza para PEMFC cuano el
combustible es H2. Si se emplea en DMFC, los sitios activos del platino tienden a envenenarse
debido a la adsorción de moléculas de CO formadas durante la oxidación de metanol. No
obstante en este trabajo se llevó a cabo la determinación de permeación de metanol a través de
las membranas modificadas con platino, para determinar si la exposición de las membranas al
agente reductor (NaBH4) afectaba su permeabilidad ante el metanol. En las Figuras 4.5 y 4.6
se encuentran los voltamperogramas correspondientes a la medición de permeación de
metanol para las membranas de los copolímeros DAg-240-170%0.110% y
TAg-120-170%0.110% expuestas al proceso de reducción transmembrana.
Figura 4.5. Voltamperogramas de permeación metanol (5 h) en membranas del copolímero DAg-240min170%0.110% sometidas a reducción de platino transmembrana.

CAPÍTULO 4
126
Figura 4.6. Voltamperogramas de permeación metanol (5 h) en membranas del copolímero TAg-120min170%0.110% sometidas a reducción de platino transmembrana.
Los voltamperogramas cíclicos obtenidos para las membranas con platino no muestran
corriente generada por la oxidación de metanol, indicando la ausencia de permeación a través
de ellas y por consiguiente, que no hubo degradación en la membrana por estar en contacto
con el NaBH4 durante el proceso. Estos resultados comprueban que que es factible emplear
ésta metodología con las membranas de los copolímeros sulfonados de PS-PAA para
incorporles algun metal, en este caso platino.
4.3.8 MICROSCOPÍA ELECTRÓNICA DE BARRIDO
Los primeros experimentos de reducción de platino transmembrana se llevaron a cabo con la
membrana de Nafion 117, en la Tabla 4.5 se indican las cuatro combinaciones de
concentración de H2PtCl6 y de NaBH4 utilizadas, las cuales generaron el platino metálico cuya
morfología obtenida por SEM se muestra en la Figura 4.7.

CAPÍTULO 4
127
Tabla 4.5. Concentración de las soluciones de H2PtCl6 y NaBH4 empleadas para metalizar las membranas de Nafion 117.
Reacción Concentración mM de H2PtCl6 Concentración mM NaBH4 N1 12.2 100 N2 3.33 100 N3 12.2 60 N4 3.33 60
Figura 4.7. Micrografías de las superficies y fractura de las membranas de Nafion 117 sometidas al proceso de reducción de platino transmembrana.
De la Figura 4.7 se observa que las condiciones de reacción N1 generaron partículas grandes,
del orden de micras, las condiciones de reacción N3 no formaron partículas metálicas,
mientras que las condiciones de las reacciones N2 y N4 generaron partículas del tamaño
N3 b)
10 μm
N3 a)
200 nm
N3 d)
10 μm
N3 c)
200 nm
N2 b)
10 μm
N2 a)
200 nm
N2 d)
10 μm
N2 c)
200 nm
N1 b)
10 μm
N1 a)
200 nm
N1 d)
10 μm
N1 c)
200 nm
N4 b)
10 μm
N4 a)
200 nm
N4 d)
10 μm
N4 c)
200 nm
Superficie Fractura

CAPÍTULO 4
128
nanométrico. Sin embargo las partículas formadas en la reacción N2 están mejor delimitadas y
menos aglomeradas, generando con ello mayor área superficial y por consiguiente mayor
cantidad de sitios activos. Por lo tanto, las concentraciones utilizadas en la reacción N2,
H2PtCl6 3.33 mM y NaBH4 100 mM, se emplearon para llevar a cabo el proceso de reducción
transmembrana para las preparadas con los copolímeros DAg240-170%0.110% y
TAg120-170%0.110%. Con éstas concentraciones de reactivos se esperaba que las partículas
de platino que se formaran fueran de morfología similar a las observadas en la membrana de
Nafion 117 etiquetada como N2, sin embargo el resultado fue diferente.
En las Figuras 4.8 - 4.13 se muestran las imágenes comparatvas entre las membranas sin y con
platino.
Figura 4.8. Morfología de la membrana DAg240-170%0.110% DMSO 0% antes y después del proceso de reducción transmembrana para incorporarle platino en su superficie.

CAPÍTULO 4
129
Figura 4.9. Morfología de la membrana DAg240-170%0.110% DMSO 55 % antes y después del proceso de reducción transmembrana para incorporarle platino en su superficie.
Figura 4.10. Morfología de la membrana DAg240-170%0.110% DMSO 110% antes y después del proceso de reducción transmembrana para incorporarle platino en su superficie.

CAPÍTULO 4
130
Figura 4.11. Morfología de la membrana TAg120-170%0.110% DMSO 0% antes y después del proceso de reducción transmembrana para incorporarle platino en su superficie.
Figura 4.12. Morfología de la membrana TAg120-170%0.110% DMSO 55% antes y después del proceso de reducción transmembrana para incorporarle platino en su superficie.

CAPÍTULO 4
131
Figura 4.13. Morfología de la membrana TAg120-170%0.110% DMSO 110% antes y después del proceso de reducción transmembrana para incorporarle platino en su superficie.
Las membranas preparadas con el copolímero TAg120-170%0.110% (Figuras 4.11 – 4.13)
promueven la formación de partículas de menor tamaño y mejor dispersas, en comparación a
las partículas que se formaron en las membranas del copolímero DAg240-170%0.110%
(Figuras 4.8 – 4.10). Esto puede deberse a que el copolímero TAg120-170%0.110% posee
mayor cantidad de grupos ácidos en su estructura polimérica en comparación al copolímero
DAg240-170%0.110%, lo cual se refleja en una mayor capacidad de intercambio iónico y en
el grado de sulfonación. La interacción de los iones de platino con los grupos sulfónicos
permite la formación de partículas de platino metálico disminuyendo su agregación y
favoreciendo la homogeneidad de tamaños [248], además se sabe que los grupos carboxílicos
también interaccionan con iones metálicos antes de ser reducidos para formar nanopartículas
metálicas [250].
A pesar de haber empleado las mismas concentraciones de la sal precursora del metal y del
agente reductor en la reacción preliminar N2 realizada con Nafion 117, las morfologías
obtenidas en las membranas de los copolímeros de PSS-PAA difieren completamente. Esta
divergencia puede radicar en la naturaleza química y en el acomodo estructural de la cadena
principal y de la cantidad y fuerza ácida de los grupos presentes en el politetrafluoroetileno
sulfonado (Nafion) en comparación a los copolímeros de poliestireno sulfonado-poliácido
acrílico.

CAPÍTULO 4
132
4.4 CONCLUSIONES DEL CAPÍTULO 4
El método Takenaka-Torikai permite la formación, incorporación y permanencia de
partículas de platino metálico sobre las membranas de PS-PAA sulfonadas.
El platino metálico que se forma sobre las membranas mediante el método
Takenaka-Torikai es de estructura cristalina tipo FCC, la cual es adecuada para
catálisis.
A mayor grado de sulfonación en las membranas (cargas negativas fijas), mayor
incorporación (concentración) de Pt metálico en ellas.
La incorporación de platino metálico en las membranas disminuye su capacidad de intercambio iónico, absorción de agua y relación de hinchamiento, pero la ausencia de permeación de metanol se mantiene.

CONCLUSIONES FINALES
133
CONCLUSIONES FINALES
Fue posible la síntesis en solución de los copolímeros St-AA (94/6) entrecruzados con
DVB o TMPTMA.
El peso molecular de los copolímeros St-AA depende de la concentración de entrecruzante
(TMPTMA > DVB), e inversamente de la concentración de iniciador. El TMPTMA genera
Mw de hasta 302,607 g/mol, mientras que el DVB genera Mw de hasta 259,000 g/mol.
Donde Mw > 156,000 g/mol favorece la formación de películas.
En la mejor relación de iniciador:entrecruzante (0.045 % mol:0.25 % mol) se observa bajo
contenido de gel (1.6 y 2.1 %) donde la formación de películas no se ve afectada.
Durante la sulfonación de los copolímeros, el uso de H2SO4 incrementan los
entrecruzamientos entre cadenas (gel de 1.6 % a 48.2%). El CH3COOSO3H disminuye el
peso molecular debido a reacciones secundarias. Mientras el sistema H2SO4/Ag2SO4
minimiza la formación de material insoluble (gel de 1.6 % a 10.4%).
Las mejores condiciones de sulfonación encontradas para los copolímeros entrecruzados
son 170 % mol de H2SO4 y 0.110 % mol de Ag2SO4, durante 240 y 120 min para el
copolímero D y T respectivamente.
Las membranas DAg240-170%0.110% y TAg120-170%0.110% presentan una relación de
hinchamiento menor que la de Nafion 117 (9.5 y 10.9 % vs 21.7%) por el
entrecruzamiento.
Usar THF-DMSO promueve una mayor porosidad en las membranas,mayor retención de
agua (D, T y Nafion 117 = 105, 139 y 23 % respectivamente) e incrementa la accesibilidad
a los protones de los grupos ácidos, obteniéndose IEC de 0.55 y 0.90 meq/g
respectivamente, los cuales son competitivos al de Nafion 117 (0.87 meq/g).
La porosidad en las membranas sulfonadas disminuye su rigidez. Donde la membrana T
alcanzó valores de módulo elástico semejantes al del Nafion 117 (valores de 555 y
390 MPa respectivamente).
Se logró la síntesis de Pt en la superficie de las membranas mediante reducción
transmembrana. Obteniendo mayor concentración en las membranas
TAg120min-170%0.110% (0.35 mg/cm2). La presencia de Pt no afecta la absorción de

CONCLUSIONES FINALES
134
agua, relación de hinchamiento ni permeación de metanol, aunque reduce hasta un
16 % la IEC.

REFERENCIAS
135
REFERENCIAS
1 Hirschenhofer J. H., Stauffer D. B., Engleman R. R., Klett M.G., Fuel Cell Handbook, 4th ed. DOE/FETC-99/1076, Editado por U. S. Department of Energy, Federal Energy Technolog Center, 1998.
2 Gross M., Maier G. Fuller T., MacKinnon S., Gittleman, C. Design rules for the improvement of the performance of hydrocarbon-based membranes for proton exchange membrane fuel cells (PEMFC). Handbook of Fuel Cells. John Wiley & Sons 2010.
3 Barragán V. M., Ruiz-Bauzá C., Villaluenga J. P. G., Seoane B. Journal of Power Sources 2004, 130, 22.
4 Gao Y., Robertson G., Guiver M., Mikhailenko S., Li X., Kaliaguine S. Macromolecules 2004, 37, 6748.
5 Miyatake K., Shouji E., Yamamoto K., Tsuchida E. Macromolecules 1997, 30, 2941.
6 Miyatake K., Oyaizu K., Tsuchida E., Hay A. Macromolecules 2001, 34, 2065.
7 Allcock H., Hofmann M., Ambler C., Morford R. Macromolecules 2002, 35, 3484.
8 Serpico J., Ehrenberg S., Fontanella J., Jiao X., Perahia D., McGrady K., Sanders E., Kellogg G., Wnek G. Macromolecules 2002, 35, 5916.
9 Yang Y., Siu A., Peckham T., Holdcroft S. Advanced Polymer Science 2008, 215, 55.
10 Hickner M. A., Ghassemi H., Kim Y. S., Einsla B. R., McGrath J. E. Chem Rev 2004, 104, 4587.
11 Odian G. Principles of Polymerization. 4th Edition. John Wiley & Sons 2004.
12 Mori H., Müller A. Prog Polym Sci 2003 28, 1403
13 Chee S-Y., Gan S-N. J Appl Polym Sci 2009, 111, 1185
14 Martins C. R., Ruggeri G., De Paoli M. A. J Braz Chem Soc 2003, 14, 797.
15 Mulijani S., Dahlan K., Wulanawati A. International Journal of Materials, Mechanics and Manufacturing 2014, 2, 36.
16 Shina J-P., Changa B-J.; Kim J-H.; Lee S-B.; Suh D-H. Journal of Membrane Science2005, 251, 247.
17 Kabanov Y. High Energ Chem + 2004, 38, 57.
18 Van Der Goot A. J., Hettema R., Janssen L. P. B. M. Polymer Engineering and Science1997, 37, 511.

REFERENCIAS
136
19 Lange H. Colloid & Polymer Sci 1986, 264, 488.
20 Gubler L., Gürsel S., Youcef H., Wallasch F., Wokaun A., Scherer G. Fundamentals and Developments of Fuel Cells Conference. Nancy- France. 10 al 12 de Diciembre de 2008. Disponible en http://perso.ensem.inpl-nancy.fr/Olivier.Lottin/FDFC08/CD/Contributions/FCHGUBL12-09_76.pdf Consultado el 29 de junio de 2011.
21 Alexandratos S., Hussain L. Ind Eng Chem Res 1995, 34, 251.
22 Kariduraganavar M., Nagarale R., Kittur A., Kulkarni S. Desalination 2006, 197, 225
23 Ochoa J-R. Electrosíntesis y electrodiálisis: fundamentos, aplicaciones tecnológicas y tendencias. McGraw Hill 1996.
24 Albright R., U.S. Patent N°: 3,663,467 (1972)
25 Mishra M. K., Yagci Y. Handbook of Radical vinil polymerization. Marcel Dekker 1998.
26 Olaj O.F., Schnöll-Bitai I. Monatshefte für Chemie 1999, 130, 731.
27 Armarego W. L. F., Li C., Chai L. Purification of Laboratory Chemicals. Elsevier Inc. Sixth Edition, 2009.
28 Nakahara, S., Sakamoto, K., Matsumoto, Y., Sanada, K., Ueoka, M. U.S. PatentApplication 0005763 (2001)
29 Martins G., Fiori M. A., Benavides R., Szpoganicz B., Paula M. M. S., da Silva L. XI Congreso Internacional de la sociedad Mexicana del Hidrógeno 2011, Cuernavaca.
30 Da Silva L., Pedrosa R. P., Coelho F., Vieira L.F., Benavides R., Dal-Bó A. G., Frizon T. E., Paula M. M. S. Macromol Symp 2014, 343, 51–58
31 Johannes K., Werner P., Hans-Ulrich M. Macromol React Eng 2012, 6, 213.
32 Kotoulas C., Krallis A., Pladis P., Kiparissides C. Macromol Chem Phys 2003, 204, 1305.
33 Braun D., Cherdron H., Rehahn M., Ritter H., Voit B. Polymer Synthesis: Theory and Practice. Fundamentals, Methods, Experiments. Springer 2005.
34 Iatrou H., Hadjichristidis N. Macromolecules 1992, 25, 4649.
35 Ghosh P., Nandi D., Das T. J Chem Pharm Res 2010, 2, 122
36 Vollmert B. Polymer Chemistry. Springer Science & Business Media. 2012.
37 Jun M., Choi Y., Kim J. Journal of Membrane Science 2012, 396, 32.
38 Moore R. B., Marti C. R. Macromolecules 1988, 21, 1334.

REFERENCIAS
137
39 Silva R. F., De Francesco M., Pozio A. Electrochimica Acta 2004, 49, 3211.
40 Rafiq S., Man Z., Maulud A., Muhammad N., Maitra S. J Membr Sci 2011, 378, 444.
41 Duerto De Pérez Z., Pérez J., Gómez L., Vivas J., Orfila L. Revista Facultad de Farmacia 2003, 66, 67.
42 Herman, F. M. Encyclopedia of Polymer Science and Technology. Wiley-Blackwell, Vol. 4. 2003.
43 Billmeyer, F. W. Ciencia de los Polímeros, Ed. Reverté, 1975.
44 Chanda M. Introduction to Polymer Science and Chemistry. A Problem-Solving Approach. CRC Press 2013.
45 Haddad P. R., Jackson P. E. Ion Chromatography: Principles and applications. Journal of Chromatography Library. Vol 26. Elsevier 1990.
46 Storey B. T. Journal of Polymer Science Part A: General Papers 01, 1965, 3, 265.
47 Larminie J., Dicks A. Fuel Cell Systems Explained. John Wiley & Sons 2003.
48 Shekhawat D., Spivey J. J., Berry D. A. Fuel Cells: Technologies for Fuel Processing. Elsevier 2011
49 Srinivasan S. Fuel Cells: From Fundamentals to Applications. Springer Science & Business Media. 2006
50 Kim D. S., Guiver M. D., Nam S. Y., Yun T. I., Seo M. Y., Kim S. J., Hwang H. S., Rhim J. W. Journal of Membrane Science 2006, 281, 156.
51 Rhim J-W., Park H. B., Lee C-S., Jun J-H., Kim D. S., Lee Y. M. Journal of Membrane Science 2004, 238, 143.
52 Erdogan T., Erdal E. U. Inan T. Y., Birkan B. Journal of Membrane Science 2009, 344, 172.
53 Beyler C.L., Hirschler M. M. Society of Fire Protection Engineers Handbook of Fire Protection Engineering (3rd Edn)", National Fire Protection Association, Quincy, MA, 2001.
54 Braun D., Czerwinski W., Disselhoff G., Tudos F., Turcsányi B. Die Angewandte Makromolekulare Chemie 1984, 125, 161.
55 Wang S., Poehlein G. W. Journal of Applied Polymer Science 1993, 49, 991.
56 Brandrup J., Immergut E. H., Grulke E. A. Polymer Handbook. Fourth Edition Wiley-Interscience 1999.
57 Krishna S. V., Pugazhenthi G. Journal of Applied Polymer Science 2011, 120, 1322.

REFERENCIAS
138
58 Chambree D., Iditoiu C., Segal E., Cesaro A. J Therm Anal Calorim 2005, 82, 803.
59 Mathew A. P., Packirisamy S., Thomas S. Polymer Degradation and Stability 2001, 72, 423.
60 Zeng W. R., Chow W. K., Yao B. Presentado en Asia-Oceania Symposium on Fire Science & Technology 2007
61 Sadeghi S. M. T. Iran J Chem Chem Eng 2006, 25, 11.
62 Sreekumari N. P., Radhakrishnan T., Revaprasadu N., van Sittert C.G.C.E., Djokovic V., Luyt A. S. Materials Letters 2004, 58, 361.
63 Li Y., Fan Y., Ma J. Polymer Degradation and Stability 2001, 73 163.
64 Fox T. G., Flory P. J. Journal of Applied Physics 1950, 21, 581.
65 Silverstein R. M., Webster F. X., Kiemle D. Spectrometric Identification of Organic Compounds. 7th ed., Wiley, 2005
66 Stuart B. H. Infrared Spectroscopy: Fundamentals and Applications. Wiley 2004.
67 Matyjaszewski K., Davis T. P. Handbook of Radical Polymerization. Wiley Interscience 2002
68 Boyer C., Boutevin G., Robin J., Boutevin B. Journal of Polymer Science: Part A: Polymer Chemistry 2007, 45, 395.
69 Ran F., Nie S., Yin Z., Li J., Su B., Sun S., Zhao C. International Journal of Biological Macromolecules 2013, 55, 269.
70 Vargantwar P. H., Brannock M. C., Smith S. D., Spontak R. J. Journal of Materials Chemistry 2012, 22, 25262.
71 Yang J. C., Jablonsky M. J., Mays J. W. Polymer 2002, 43, 5125.
72 Müller F., Ferreira C. A., Franco L., Puiggalí J., Alemán C., Armelin E. J Phys Chem B, 2012, 116, 11767.
73 Baigl D., Seery T. A. P., Williams C. E. Macromolecules 2002, 35, 2318.
74 Aerdts A. M., de Haan J. W., German A. L. Macromolecules 1993, 26, 1965.
75 Robertson G. P., Mikhailenko S., Wang K., Xing P., Michael G., Kaliaguine S. Journal of Membrane Science 2003, 219, 113.
76 Amiinu I. S., Liang X., Tu Z., Zhang H., Feng J., Wan Z., Pan M. ACS Applied Materials & Interfaces 2013, 5, 11535.
77 Molnar R. M., Bodnar M., Hartmann J. F., Borbely J. Colloid Polym Sci 2009 287, 739.

REFERENCIAS
139
78 Feng Y., Billon L., Grassl B., Khoukh A., Francois J. Polymer 2002, 43, 2055.
79 Chijiwa, Sachiko. Tesis de Maestría 2001. Characterization of polystyrene-b-poly(acrylic acid) copolymer micelles by nuclear magnetic resonance spectroscopy. Disponible en http://digitool.Library.McGill.CA:80/R/-?func=dbin-jump-full&object_id=32983&silo_library=GEN01
80 Gode P., Hult A., Jannasch P., Johansson M., Karlsson L. E., Lindbergh G., Malmström E., Sandquist D. Solid State Ionics 2006, 177, 787.
81 Brar A. S., Hooda S., Kumar R. Journal of Polymer Science: Part A: Polymer Chemistry 2003, 41, 313.
82 Porter M. Handbook of Industrial Membrane Technology. Noyes Publications, 1990.
83 Bocourt M., Bada N., Argüelles-Monal W., Peniche C. Revista CENIC Ciencias Químicas 2009, 40, 81.
84 Rodríguez I. C., Cerezo A., Salem I. I. Ars Pharmaceutica 2000, 41, 115.
85 Baohui L., Lu X., Qiang W., Tiehong C., Pingchuan S., Qinghua J., Datong D., Xiaoliang W., Gi X., An-Chang S. Macromolecules 2007, 40, 5776
86 Bhowmik D., Kumar K. P. S. The Pharma Research 2010, 4, 138.
87 Park C. H., Lee C. H., Guiver M. D., Lee Y. M. Prog Polym Sci 2011, 36, 1443 .
88 Zaidi S. M. J., Matsuura T. Polymer Membranes for Fuel Cells. Springer 2009.
89 Peinemann K-V., Pereira N. S. Membranes for Energy Conversion. Vol 2. WILEY-VCH Verlag GmgH & Co. KGaA Weinheim. 2008.
90 Alkan G. S., Gubler L., Gupta B, Scherer G. G. Adv Polym Sci 2008, 215, 157.
91 Roziere J., Jones D. J. Annu Rev Mater Res 2003, 33, 503
92 Elabd Y. A., Napadensky E. Polymer 2004, 45, 3037.
93 Hodgdon R-B. U. S. Patent N°: 3,657,104 (1972)
94 Hodgdon R-B. U. S. Patent N°: 3,887,499 (1975)
95 Yen S-P., Narayanan S., Halpert G., Graham E., Yavrouian A. U. S. Patent N°: 5,795,496 (1998)
96 Phadnis S., Patri M., Hande V. R., Roychoudhury S., Deb P. C. J Appl Polym Sci 2004, 92, 2318.
97 Leng W., Zhou S., You B., Wu L. Langmuir 2010, 26, 17836.
98 Ratna D., Dalvi V., Chakraborty B. C., Deb P. C. J Appl Polym Sci 2003, 41, 2166.

REFERENCIAS
140
99 Ratna D., Dalvi V., Chakraborty B. C. J Appl Polym Sci 2007, 104, 1517.
100 Komber H., Chakraborty S., Voit B., Banerjee S. Polymer 2012, 53, 1624.
101 Zhang L., Katzenmeyer B. C., Cavicchi K. A., Weiss R. A., Wesdemiotis C. ACS Macro Lett 2013, 2, 217.
102 Li J., Xu Y., Wei H., Huo T., Wang E. Anal Chem 2007, 79, 5439.
103 Kučera F., Jančář J. Chem Papers 1996, 50, 224.
104 Lima A. F., Mansur C. R. E., Lucas E. F., González G. Energy Fuels 2010, 24, 2369.
105 Eastaugh N., Walsh V., Chaplin T., Siddall R. Pigment Compendium. Ed. Routledge 2008.
106 Jakubke H., Jeschkeit H. Concise Encyclopedia Chemistry. Walter de Gruyter. 1994.
107 Topp N. E., Pepper K. W. Journal of the Chemical Society 1949, 3299
108 Pope M. U.S. Patent 2 728 831 (1955).
109 Fujiwara H., Jawara K., Miyoshi K., Mukai K. U. S. Patent 4 025 401 (1977).
110 Tsukui T., Shimizu T., Doi R., Tsutsumi Y. U.S. Patent 4 537 840 (1985).
111 Tiang B., Yan C., Wang F. Chinese Journal of Polymer Science 2004, 22, 505.
112 Nitanan T., Akkaramongkolporn P., Rojanarata T., Ngawhirunpat T., Opanasopit P. Tropical Journal of Pharmaceutical Research 2014, 13, 191.
113 Zhou S., Hai S. D., Kim D. Fuel Cells 2012, 12, 589.
114 Klaysom C., Ladewig B. P., Lu G. Q. M., Wang L. Journal of Membrane Science 2011, 368, 48.
115 Tokuda S., Horikawa S., Negishi K., Uesugi K., Hirukawa H. Furukawa Review 2003, 23, 88.
116 Eikerling M., Kornyshev A., Spohr E. Adv Polym Sci 2008, 215, 15.
117 Sadeghi A. A., Goulet M.-A., Khorasany, R. M. H., Ghataurah J., Lim C., Lauritzen M., Kjeang E., Wang G. G., Rajapakse R. K. N. D. Fuel Cells 2015, 15, 204.
118 Basile A., Pereira S. Advanced membrane science and technology for sustainable energy and environmental applications. Woodhead Publishing Limited. 2011
119 Benavides R., Oenning L. W., Paula M. M. S., Da Silva L. Journal of New Materials for Electrochemical Systems 2014, 17, 1.
120 Roth H. H. Ind Eng Chem 1954, 46, 2435.

REFERENCIAS
141
121 Inagaki Y., Kuromiya M., Noguchi T., Watanabe H. Langmiur 1999, 15, 4171.
122 Turbak A. F. Ind Eng Chem Prod Res Dev 1962, 1, 275.
123 Inagaki Y., Kiuchi S. J Mater Cycles Waste Manag 2001, 3, 14.
124 Kumar A., Sandip R. Juvekar V. Polymer International 2007, 56, 167.
125 Sułkowski W. W., Nowak K., Sułkowska A., Wolinska A., Bajdur W. M., Pentak D., Mikuła B. Pure Appl Chem 2009, 81, 2417.
126 Di Vona M. L., Marani D., D’Ottavi C., Trombetta M., Traversa E., Beurroies I., Knauth P., Licoccia S. Chem Mater 2006, 18, 69.
127 Pu H. Polymers for PEM Fuel Cells. John Wiley & Sons 2014.
128 Clemens D. H., U.S. Patent 3 817 879 (1974).
129 Fuller T., Gasteiger H. A., Cleghorn S. Ramini V., Zhao T., Nguyen T. V., Haug A., Bock C., Lamy C., Ota K. Proton Exchange Membrane Fuel Cells. Vol 11 No. 1. ECS Transactions, 2007
130 Chakrabarty T., Rajesh A. M., Jasti A., Thakur A. K., Singh A. K., Prakash S., Kulshrestha V., Shahi V. K. Desalination 2011, 282, 2.
131 Sangeetha D. European Polymer Journal 2005, 41, 2644.
132 Martins C. R., Hallwass F., De Almeida Y. M. B., De Paoli M. A. Ann Magn Reson2007, 6, 46.
133 Carroll W.R., Eisenberg H. Journal of Polymer Science: Part A-2 1966, 4, 599.
134 Singare P. U., Lokhande R. S., Madyal R. S. Russian Journal of General Chemistry2010, 80, 527.
135 Bahadur P., Sastry N. V. Principles of Polymer Science. Alpha Science Int'l Ltd. 2nd ed. 2005.
135 Lee S.J., Mukerjee S., Ticianelli E.A., McBreen L. Electrochim Acta 1999, 44, 3283.
136 McGaugh M. C., Kottle S. J Appl Polym Sci 1968, 12, 1981.
137 Wang L. F., Pearce E. M., Kwei T. K. J Polym Sci Part C Polym Lett 1990, 28, 317.
138 Iditoiu C., Segal E., Chambrée D. J Therm Anal Calorim 1999, 56, 407.
139 Srivasta A., Mishra V., Singh P., Srivasta A., Kumar R. J Therm Anal Calorim 2012, 107, 211.
140 Suchocka-Gałas K., J Appl Polym Sci 2003, 89, 55.

REFERENCIAS
142
141 Ferris A. F., U.S. Patent 2 678 306 (1954).
142 Amarilla J. M., Rojas R. M., Rojo J. M., Cubillo M. J., Linares A., Acosta J. L. Solid State Ionics 2000, 127 133.
143 Detoni S., Hadzi D. Spectrochimica Acta 1957, 601-608. From The Colloquium Spectroscopium Internationale VI (Amsterdam 1956)
144 Mayo D. W., Miller F. A., Hannah R. W. Course notes on the Interpretation of Infrared and Raman Spectra. John Wiley & Sons Publications, 2003.
145 Sharma P., Khajuria D., Sachar N, Sachar R. Journal of Chemical and Pharmaceutical Research, 2014, 6, 2061.
146 Ramaprasad G. C., Kalluraya B., Kumar B. S., Mallya S. Der Pharma Chemica 2012, 4, 1026.
147 Salvi V. K., Sharma S., Sharma C., Bhambi D., Talesara G. L. Journal of Chemistry2009, 48B, 1583.
148 Azam F., Singh S., Khokhra S. L., Prakash O. Journal of Zhejiang University Science B2007,8, 446.
149 Ellis A., Zehentbauer F. M., Kiefer J. Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics 2013, 15, 1093.
150 Akkaramongkolporn P., Ngawhirunpat T., Opanasopit P. AAPS PharmSciTech 2009, 10, 641.
151 León R., Ramírez M. Rev Iberoamer Polím 2007, 8, 112.
152 Sharma B. K. Instrumental Methods of Chemical Analysis. Krishna Prakashan Media, 2000.
153 Casale J. F., Hays P. A. Microgram Journal 2004, 9, 33.
154 Ostrowska J., Narebska A. Colloid Polym Sci 1979, 257, 128.
155 Chapiro A. Pure & Appl Chem 1981, 53, 643.
156 Weiss R. A., Turner S. R., Lundberg R. D. Journal of Polymer Science: Part A Polymer Chemistry 1985, 23, 525.
157 Mehdipour-Ataei S., Bahri-Laleh N. Iranian Polymer Journal 2008, 17, 95.
158 Strong A. B. Fundamentals of Composites Manufacturing: Materials, Methods and Applications. Society of Manufacturing Engineers. 2008.
159 McGaugh M. C., Kottle S. Journal of Polymer Science Part B: Polymer Letters 1967, 5, 817.

REFERENCIAS
143
160 Watari T., Fang J., Tanaka K., Kita H., Okamoto K., Hirano T. Journal of Membrane Science 2004, 230, 111.
161 Abu-Thabit N. Y., Ali S. A., Zaidi S. M. J., Mezghani K. Journal of Materials Research2012, 27, 1958.
162 Sata T. Ion Exchange Membranes Preparation, Characterization, Modification and Application. RSC Advancing the Chemical Sciences, 2004
163 Ivanchev S. Russ J Appl Chem + 2008, 81, 569.
164 Elamathi S., Nithyakalyani G., Sangeetha D., Ravichandran S. Ionics 2008, 14, 377.
165 Fu L., Xiao G., Yan D. ACS Appl Mater Interfaces 2010, 2, 1601.
166 Kundu S., Simon L. C., Fowler M., Grot S. Polymer 2005, 46, 11707.
167 Kawano Y., Wang Y., Palmer R. A., Aubuchon S. R. Polímeros 2002, 12, 96.
168 Nicotera I., Kosma V., Simari C., Angioni S., Mustarelli P., Quartarone E. J Phys Chem C 2015, 119, 9745.
169 Kocha S., Yang J., Yi J. AIChE J 2006, 52, 1916
170 Haile S. Acta Mater 2003, 51, 5981.
171 Vaughn W. L., Wu M. L., U.S. Patent N°: 4,789,386 (1988).
172 Dorman L. C., Meyer V. E., Wu M. L., U.S. Patent N°: 4, 695,295 (1987).
173 Reyna-Valencia A., Kaliaguine S., Bousmina M. Journal of Applied Polymer Science2005, 98, 2380.
174 Wang C., Liu Z. X., Mao Z. Q., Xu J. M., Ge K.Y. Chem Eng J 2005, 112, 87.
175 Sundar P. S., Kal G. P., Ulaganathan M., Arunkumar J. Ionics 2011, 17, 361.
176 Mikhailenko S. D., Wang K., Kaliaguine S., Xing P., Robertson G. P., Guiver M. D.
Journal of Membrane Science 2004, 233, 93.
177 Feng M., You Y., Zheng P., Liu J., Jia K., Huang Y., Liu X. International Journal of Hydrogen Energy 2016, 41, 5113.
178 Oliveira A., Perez J., Napporn W. T., Ticianelli E. A., Gonzalez E. R. J Braz Chem Soc2000, 11, 39.
179 Seland F., Harrington D. A., Tunold R. Electrochemica Acta 2006, 52, 773.
180 Rodríguez J. M. D., Herrera J. A., Pérez J. Journal of Chemical Education 2000, 77, 1195.

REFERENCIAS
144
181 Wang C., Chen Y., Hu X., Guo R. Int J Electrochem Sci 2006, 1, 139.
182 Dai H., U.S. Patent N°: 7,402,351 B2 (2008)
183 Huang Q. M., Zhang Q. L., Huang H. L., Li W. S., Huang Y. J., Luo J. L. Journal of Power Sources 2008, 184, 338.
184 ASTM D790-07e1. Standard Test Methods for Flexural Properties of Unreinforced and Reinforced Plastics and Electrical Insulating Materials, ASTM International, West Conshohocken, 2007.
185 Seifriz I., Konzen M., Paula M. M. S., Goncalves N. S., Spoganickz B., Creczynski-Pasa T. B., Bonetti V. R., Beirith A., Calixto J. B., Franco C. V. Journal of Inorganic Biochemistry 1999, 76, 153.
186 Unnikrishnan L., Mohanty S., Nayak S. K. High Performance Polymers 2013, 25, 854.
187 Unnikrishnan L., Kumar N. S., Mohanty S., Sarkhel G. Polym Plastics Tech and Eng2010, 49, 1419.
188 Prasad M., Mohanty S., Nayak S. K. Royal Society of Chemistry Advances 2014, 4, 61178.
189 Yee R. S. L., Zhang K., Ladewig B. P. Membranes 2013, 3, 182.
190 Kim J., Kim B., Jung B., Kang Y. S., Ha H. Y., Oh I., Ihn K. I. Macromolecular Rapid Communications 2002, 23, 753.
191 Peckham T. J., Holdcroft S. Advanced Materials 2010, 22, 4667.
192 Yizhak M. The Properties of Solvents. Wiley 1998.
193 Lai J., Lin F., Wang C., Wang D. Journal of Membrane Science 1996, 118, 49.
194 Huang Y. J., Ye Y. S., Yen Y. C., Tsai L. D., Hwang B. J., Chang F, C. International Journal of Hydrogen Energy 2011, 36, 15333.
195 Neburchilov V., Martin J., Wang H., Zhang J. Journal of Power Sources 2007, 169, 221.
196 Millar J. R., Smith D. G., Marr W. E., Kressman T. R. E. J Chem. Soc 1963, 218.
197 Yan J., Wang X., Yang Y. Reactive & Functional Polymers 2000, 43, 227.
198 Smitha B., Sridhar S., Khan A. A. Journal of Membrane Science 2005, 259, 10.
199 Kreuer K. D., Paddison S. J., Spohr E., Schuster M. Chem Rev 2004, 104, 4637.
200 Mecheri B., D’Epifanio A., Traversa E., Licoccia S. Journal of Power Sources 2008, 178, 554.

REFERENCIAS
145
201 Dinh X. L., Dukjoon K. Solid State Ionics 2011, 192, 627.
202 Xue Y., Fu R., Wu C., Lee J. Y., Xu T. Journal of Membrane Science 2010, 350, 148.
203 Zhang Y., Wan Y., Zhang G., Shao K., Zhao C., Li H., Na H. Journal of Membrane Science 2010, 348, 353.
204 Abu-Thabit N. 1st International Electronic Conference on Materials 25 May-10 June 2014. Disponible en http://sciforum.net/conference/ecm-1
205 Feng S., Shang Y., Wang Y., Liu G., Xie X., Dong W., Xu J., Mathur V. K. Journal ofMembrane Science 2010, 352, 14.
206 Chen T., Leddy J. Langmuir 2000, 16, 2866.
207 Dubau L., Castanheira L., Maillard F., Chatenet M., Lottin O., Maranzana G., Dillet J., Lamibrac A., Perrin J., Moukheiber E., ElKaddouri A., De Moor G., Bas C., Flandin L., Caqué N. A review of PEM fuel cell durability: materials degradation, local heterogeneities of aging and possible mitigation strategies. WIREs Energy Environ 2014.
208 Zhang A., Bai H., Li L. Chemical Reviews 2015, 115, 9801.
209 Escalé P., Rubatat L., Billon L., Save M. European Polymer Journal 2012, 48, 1001.
210 Srinivasarao M., Collings D., Philips A., Patel S. Science 2001, 292, 79.
211 Borysovska K., Verbylo D., Podrezov Yu., Szafran M. Archives of Metallurgy and Materials 2009, 54, 875.
212 Zhang P., Chen G., Zheng X. F. Journal of Physics: Conference Series 2010, 240, 012152.
213 Snyder J. D., Elab Y. A. Journal of Power Sources 2009, 186, 385.
214 Stolten D., Samsun R. C., Garland N. Fuel Cells. Data, Facts and Figures. Wiley-VCH 2016.
215 Zhang J. PEM Fuel Cell Electrocatalysts and Catalyst Layers. Springer. 2008
216 Barbir F. PEM Fuel Cells: Theory and Practice. Academic Press, 2013
217 Brandon N. P., Brett D. J. Phil Trans R Soc A 2006, 364, 147.
218 Uchida M., Park Y., Kakinuma K., Yano H., Tryk D. A., Kamino T., Uchida H., Watanabe M. Phys Chem Chem Phys 2013, 15, 11236.
219 Takenaka H., Torikai E., Kawami Y., Wakabayash N. Int J Hydrogen Energy 1982, 7, 397.

REFERENCIAS
146
220 Barsukov V., Beck F. New Promising Electrochemical systems for Rechargeable Batteries. Kluwer Academic Publishers. 1995.
221 Lakeman J. B., Mepsted G. O., Adcock P. L., Mitchell P. J., Moore J. M. Journal of Power Sources 1997, 65, 179.
222 Huang L-M., Tang W-R., Wen T-C. J Power Sources 2007, 164, 519.
223 Ogumi Z., Nishio K., Yoshizana S. Electrochem Acta 1981, 26, 1779.
224 Jorissen J. Electrochem Acta 1996, 41, 553.
225 Fedkiw P. S., Potente J. M., Her W-H. The Elecrochem Soc 1990, 137, 1451.
226 Nuttall L. J. Int J Hydrogen Energy 1997, 2, 395.
227 Tatapudi P., Fenton J.M. J Electrochem Soc 1993, 140, 3527.
228 Grimm J. H., Bessarabov D. G., Simon U., Sanderson R. D. Journal of Applied Electrochemistry 2000, 30, 293.
229 Hwang B., Liu Y., Chen Y. Materials Chemistry and Physics 2001, 69, 267.
230 Liu Y., Hwang B., Hsu W. Sensors and Actuators B 2002, 87, 304.
231 Nørskov J. K., Rossmeisl J., Logadottir A., Lindqvist L., Kitchin J. R., Bligaard T., Jónsson H. J Phys Chem B 2004, 108, 17886
232 Greeley J., Jaramillo T. F., Bonde J., Chorkendorff I., Nørskov J. K. Nature Materials2006, 5, 909.
233 Lin W. F., Iwasita T., Vielstich W. J Phys Chem B 1999, 103, 3250.
234 Pozio A., Giorgi L., Antolini E., Passalacqua E. Electrochimica Acta 2000, 46, 555.
236 Clescerl L. S., Greenberg A. E., Eaton A. D. Standard Methods for Examination of Water & Wastewater. 20th Edition, 1999.
237 Analytical Methods for Atomic Absorption Spectroscopy. The PerkinElmer Inc. United States of America. 1996.
238 Georgieva M., Andonovski B. Anal Bioanal Chem 2003, 375, 836.
239 Zhou Z., Wang S., Zhou W., Jiang L., Wang G., Sun G., Zhou B, Xin Q. Phys Chem Chem Phys 2003, 5, 5485.
240 Gharibshahi E., Saion E. International Journal of Molecular Sciences 2012, 13, 14723.
241 Wu N., She X., Yang D., Wu X., Su F., Chen Y. Journal of Materials Chemistry 2012, 22, 17254.

REFERENCIAS
147
242 Zhu W., Wu Y., Yan C., Wang C., Zhang M., Wu Z. Materials 2013, 6, 5625.
243 Kita H., Fujikawa K., Nakajima H. Electrochimica Acta 1984, 29, 1721.
244 Nakajima H., Kita H. Electrochimica Acta 1988, 33, 521.
245 Liu R., Her W., Fedkiw P. S. J Electrochem Soc 1992, 139, 15.
246 Rajalakshmi N., Ryu H., Dhathathreyan K. S. Chemical Engineering Journal 2004, 102, 241.
247 Weissmann M., Coutanceau C., Brault P., Léger J.-M. Electrochemistry Communications 2007, 9, 1097.
248 Waje M. M., Wang X., Li W., Yan Y. Nanotechnology 2005, 16, S395.
249 Kongkanand A., Mathias M. F. J Phys Chem Lett 2016, 7, 1127.
250 Yu S., Lou Q., Han K., Wang Z., Zhu H. International Journal of Hydrogen Energy2012, 37, 13365.

LISTA DE ABREVIACIONES
148
LISTA DE ABREVIACIONES
(CH3CO)2O Anhídrido acético AA Ácido acrílico Ag Plata metálica Ag+ Iones plata Ag2SO4 Sulfato de plata AgCl Cloruro de plata AgNO3 Nitrato de plata BPO Peróxido de benzoílo CDCl3 Triclorometano deuterado CH3COOH Ácido acético CH3COOSO3H Sulfato de acetilo CO Monóxido de carbono –COOH Grupo funcional carboxílico D Coeficiente de difusión Ð Dispersidad DEB Dietilbenceno DMFC Celda de combustible de metanol DMSO Dimetilsulfóxido DSC Calorimetría diferencial de barrido DVB Divinil benceno ECA Área electrocatalítica FC Celda de combustible GPC Cromatografía de Permeación en Gel H2 Hidrógeno gaseoso H2PtCl6 Ácido hexacloroplatínico H2SO4 Ácido sulfúrico HCl Ácido clorhídrico HNO3 Ácido nítrico IEC Capacidad de intercambio iónico kp Constante de velocidad de propagación kt Constante de velocidad de terminación MEA Ensamble membrana-electrodo Mn Peso molecular promedio en número Mp Peso molecular del máximo del pico Mw Peso molecular promedio en peso N2 Gas nitrógeno Na+ Ión sodio NaBH4 Borohidruro de sodio NaCl Cloruro de sodio NaOH Hidróxido de sodio

LISTA DE ABREVIACIONES
149
O2 Gas oxígeno PAA Poliácido acrílico PEEK Poliéter-eter-cetona PEM Membrana intercambiadora de protones PEMFC Celda de combustible de membrana intercambiadora de protones PS Poliestireno PS-PAA Poliestireno-poliácido acrílico RMN 1H Resonancia Magnética Nuclear de Protón SEM Microscopía electrónica de barrido –SO3H Grupo sulfónico sPEEK Poliéter-eter-cetona sulfonado St Estireno Td Temperatura de descomposición Tg Temperatura de transición vítrea TGA Análisis termogravimétrico THF Tetrahidrofurano TMPTMA Trimetacrilato de trimetilolpropano TPB Frontera triple de fases Conductividad protónica




















