
Targeting Metabolic Enzymes in Cancer – Clinical Trials Update
29
1 Targeting Metabolic Enzymes in Cancer – Clinical Trials Update Julie O’Neal and Jason Chesney ## Division of Medical Oncology and Hematology, Department of Medicine, University of Louisville, Molecular Targets Program, James Graham Brown Cancer Center, Louisville, Kentucky ## Communicating Author, 505 South Hancock Street, Clinical and Translational Research Building Room 424, University of Louisville, Louisville, Kentucky, 40202 Key Words: Glycolysis, Chemotherapy, Glucose Financial Support: DOD CDMRP BC112204 (JO) and NCI R01-CA149438 (JC)
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Targeting Metabolic Enzymes in Cancer – Clinical Trials Update

1
Targeting Metabolic Enzymes in Cancer – Clinical Trials Update
Julie O’Neal and Jason Chesney##
Division of Medical Oncology and Hematology, Department of Medicine, University of Louisville,
Molecular Targets Program, James Graham Brown Cancer Center, Louisville, Kentucky
## Communicating Author, 505 South Hancock Street, Clinical and Translational Research Building
Room 424, University of Louisville, Louisville, Kentucky, 40202
Key Words: Glycolysis, Chemotherapy, Glucose
Financial Support: DOD CDMRP BC112204 (JO) and NCI R01-CA149438 (JC)

2
Abstract
The uptake and utilization of glucose and glutamine by cancer cells is markedly higher than by
most non-transformed, normal epithelial and mesenchymal cells. This metabolic shift enables the
production of ATP and anabolic precursors necessary for the synthesis of proteins, lipids and nucleotides
required for survival, proliferation and invasiveness. The observations that certain oncogenic proteins
(Ras, c-Myc and HIF-1α) and tumor suppressors (P53, PTEN, Rb and VHL) regulate the expression and
activity of several metabolic enzymes has supported their potential as molecular targets for the
development of anti-neoplastic agents. Indeed, recent pre-clinical studies have shown that several
established and novel inhibitors of metabolic enzymes exhibit reasonable therapeutic indices when tested
in xenograft models of tumorigenesis. In this review, we will discuss the rationale of targeting metabolic
enzymes for the treatment of cancer and then will describe published pre-clinical and clinical data for
several inhibitors of metabolism in cancer.

3
Introduction
Death due to cancer remains the second highest cause of mortality in the United States behind
heart disease (www.cdc.gov) indicating that new approaches to identifying and implementing effective
anti-cancer treatments may improve the overall survival and quality of life of our population. The
relatively recent shift from generalized cytotoxic chemotherapies to treatments targeted at genetic
alterations of cancer has dramatically improved clinical outcomes in certain types of cancer. For example,
Imatinib (Gleevec), which inhibits the fusion BCR/ABL protein caused by a t(9;22)(q34;q11)
translocation in patients with chronic myelogenous leukemia (CML), was developed in the 1990s and
now has essentially eradicated CML as a cause of serious morbidity and mortality in the world [1].
Today, approximately 37 targeted agents in the form of small molecule antagonists or blocking antibodies
are FDA-approved for the treatment of cancer. Such therapies are designed to inhibit cancer-specific
signaling pathways (e.g. downstream of oncogenic Ras or the EGF receptor) or processes (e.g. tumor
angiogenesis induced by VEGF) with the goal of preferentially killing tumor cells without affecting the
viability of normal cells. Although an old concept, metabolic reprogramming recently has been
recognized as an additional essential characteristic of human cancers [2] and several agents targeted
against signaling pathways have been found to modulate the metabolism of cancer cells (Table I). These
observations have encouraged the development of small molecules designed to take advantage of the
metabolic differences of normal versus tumor cells with the goal of generating improved anti-cancer
drugs with unique mechanisms of action. In this review, we will discuss the rationale for metabolic
reprogramming in cancer cells and describe several drugs that target metabolism and show promise in
preclinical studies and/or clinical trials.
Rationale for Increased Metabolism In Cancer
Tumor cells consume a relative excess of glucose via passive glucose transporters and the
quantitation of 18F-2-fluoro-2-deoxy-glucose (FDG) uptake by tumors using positron emission

4
tomography (PET) is a commonly used diagnostic measure that correlates directly with tumor
aggressiveness and patient prognosis [3, 4]. The increased metabolism of glucose to lactate by cancer
cells occurs even in the presence of oxygen (i.e. aerobic glycolysis or the “Warburg Effect”) [5].
Although currently an area of active research, it is generally accepted that rapidly dividing cancer cells
increase glycolytic flux for anabolic and energetic purposes enabling them to thrive in a low nutrient,
oxygen poor environment. Paradoxically, glycolysis only yields two ATP per glucose as opposed to 38
ATP via oxidative phosphorylation. However, the rate of ATP production via glycolysis is faster than via
oxidative phosphorylation with less free radical formation [6]. In addition to producing ATP, multiple
enzymes in glycolysis generate products that can be shunted to alternative pathways allowing for nucleic
acid, amino acid, and lipid synthesis essential for cell division. For example, glucose-6-phosphate,
fructose-6-phosphate and glyceraldehyde-3-phosphate can all be metabolized via the oxidative or non-
oxidative pentose phosphate pathways allowing for ribose-5-phosphate production and ultimately
nucleotide biosynthesis. Additionally, 3-phosphoglycerate (3PG) and pyruvate feed into pathways that
generate amino acids and 3PG and dihydroxyacetone are precursors for lipid biogenesis. High glycolytic
flux also lowers the extracellular pH causing p53 dependent apoptosis of normal cells facilitating
invasiveness [7] and the glycolytic end-product lactate also provides an essential carbon source for
supporting fibroblasts [8]. Increased glycolytic flux thus permits a ready supply of ATP and anabolic
precursors essential for cancer cell proliferation while simultaneously promoting invasiveness and support
from host cells.
Regulation of Metabolism by Oncogenes and Tumor Suppressors
Although the altered metabolism of cancer cells was discovered almost a century ago by Otto
Warburg, more recent research has demonstrated that changes in tumor metabolism may be causally
related to the selection of oncogenic/tumor suppressor mutations that directly induce and/or activate
glycolytic and mitochondrial proteins which, in turn, are required for survival, proliferation and
invasiveness. Expression of mutated Ras oncogenes causes increased glucose uptake [9, 10] and

5
glycolytic flux [11] likely as a result of the ability of Ras to increase transcription and translation of
hypoxia-inducible factor 1-α (HIF1α) which promotes the transcription of multiple metabolic genes
involved in glycolysis including the glucose transporter-1 (GLUT1), hexokinase-2 (HK2), 6-
phosphofructo-1-kinase (PFK1) and lactate dehydrogenase-A (LDHA) [12]. Additionally, ectopic
expression of oncogenic Ras or HIF-1α increases protein levels of 6-phosphofructo-2-kinase/fructose-2,6-
biphosphatase-3 (PFKFB3), which, by producing fructose-2,6-bisphosphate (F26BP), activates 6-
phosphofructo-1-kinase (PFK1), a key regulated, irreversible and committed step of glycolysis [9, 13].
Like Ras, c-Myc alters tumor cell metabolism specifically through regulation of glycolytic genes
including hexokinase-2, PFK-1, LDH-A, enolase and pyruvate kinase M2 [14]. Congruently, the loss of
tumor suppressor function also has been found to activate glycolysis. For example, loss of functional p53
stimulates the expression of glucose transporters (GLUT1 and GLUT4) [15] and the glycolytic enzyme
phosphoglycerate mutase [16]. Additionally, the loss of PTEN has recently been found to reduce
APC/Cdh1-mediated degradation of PFKFB3 which activates glycolytic flux through PFK-1 [17]. Tumor
oxidative phosphorylation is also linked to oncogenes as expression of oncogenic Ras (H-RASV12) in
immortalized cells increases flux through the tricarboxylic acid cycle (TCA), oxygen consumption, and
sensitivity to electron transport (Complex I) inhibition [9] and cytochrome c oxidase Vb is a translational
target of H-RASV12 [18]. Additionally, multiple mitochondrial genes are c-Myc targets including
cytochrome c oxidase 5b and cytochrome c and c-Myc-deficient cells display reduced oxygen
consumption relative to Myc expressing cells [19]. The relatively recent observation that some tumor
cells die if glutamine is withdrawn led to the idea that some tumors are “glutamine addicted” [20]. The
glutamine requirement of cancer cells is at least in part due to c-Myc, which leads to mitochondrial
glutaminolysis [21]. As with glycolysis, the loss of a tumor suppressor, in this case Rb, also has been
found to increase glutamine metabolism [22, 23].
Basis for Targeting Metabolism as a Therapeutic Strategy in Cancer

6
Efforts to directly target the proteins that result from genetic changes that activate oncogenes (e.g.
Ras, c-Myc, etc.) have largely proven ineffective. Two notable exceptions are the previously mentioned
BCR/ABL inhibitor Imatinib which has had a great impact on the lives of CML patients and a new B-
RafV600E inhibitor, Vemurafenib, that is yielding dramatic albeit relatively transient partial responses in
stage IV melanoma patients [1, 24]. Unfortunately, the vast majority of these novel signaling inhibitors
have had only limited success in the clinic as a result of the acquisition of resistance mutations and hyper-
activation of alternative signaling pathways [25]. Small molecule antagonists of metabolic proteins have
a high potential for being effective and druggable targets since: (i) many metabolic regulators are
differentially expressed in normal versus tumor cells; (ii) cancer cells are frequently addicted to certain
metabolites, for example glutamine; (iii) tracing metabolites provides a feasible biomarker for clinical
responses (e.g. FDG-PET imaging); (iv) testing for genetic deletion of upstream oncogenes or tumor
suppressor genes should be predictive of clinical effectiveness (e.g. PTEN status and PFKFB3 inhibitors);
and (v) there is prior success targeting metabolism (i.e. inhibitors of nucleotide synthesis such as anti-
folates in acute lymphoblastic leukemia and 5-fluorouracil in gastrointestinal cancers). Below, we will
discuss a subset of promising small molecules that target tumor-specific metabolic proteins and that are
currently in preclinical and/or clinical development for the treatment of cancer.
Targeting LDA-A: AT-101 and FX11
The lactate dehydrogenase enzyme is a tetrameric complex comprised of LDH-A (LDH-M,
muscle) and/or LDH-B (LDH-H, heart) subunits encoded by two separate genes (LDHA and LDHB,
respectively) that combine to generate five separate isozymes (LDH1-5) which contain differing ratios of A
and B subunits. A separate gene, LDH-C encodes LDH-C that is expressed in sperm and testes only. The
LDH enzyme complex catalyzes the reversible conversion of pyruvate to lactate. Generation of lactate
from pyruvate replenishes NAD+ required for enhanced flux through the glyceraldehyde-3-phosphate
dehydrogenase step of glycolysis and can provide a carbon source to adjacent cells. Increased expression
of LDH-A has been observed in several tumor types [26]. Additionally, reduction of LDH-A expression

7
in tumor cells with siRNA molecules causes a dramatic decrease in tumor size and increase life span in a
murine model of breast cancer [27] as well as reduced tumor growth in a separate lung tumor xenograft
model [26] suggesting that inhibition of LDH-A activity in tumor cells may be an effective anti-tumor
therapy.
Gossypol is naturally found in the cotton plant and was originally tested in China as a male
contraceptive. AT-101 (Ascenta Therapeutics) is an orally bioavailable form of the R-(-) enantiomer of
gossypol that inhibits LDH activity. It also is reported to antagonize the anti-apoptotic BCL2 family of
proteins by acting as a BH3 domain mimetic [28]. Clinical efficacy results of AT-101 were mixed.
Reduced PSA levels were observed in some patients when AT-101 was given as a single agent to
castrate-resistant prostate cancer (CRPC) patients [29]. Preliminary results in a phase II study
demonstrated that 26% of newly diagnosed, metastatic, androgen dependent prostate cancer patients
treated with AT-101 and androgen deprivation therapy had undetectable PSA at seven months [30].
However, in a phase II, placebo controlled trial in metastatic CRPC patients, AT-101 given with
docetaxel and prednisone resulted in no statistical improvements in survival or progression free survival
[31]. Treatment of advanced small cell lung cancer (SCLC) patients with AT-101 and topotecan enabled
limited partial responses (PR) and stable disease (SD); however the study did not meet criteria for further
enrollment into the expansion Phase II of this Phase I/II study [32]. No improvement in PFS or response
rate (RR) were seen in patients with advanced non-small cell lung cancer (NSCLC) treated with AT-101
plus docetaxel, although an increase of 1.9 months in median survival was observed [33].
Disappointingly, no responses were seen in a trial of chemotherapy sensitive recurrent NSCLC patients
treated with AT-101 as a single agent [34]. A phase I study conducted in 24 patients with a variety of
refractory solid tumors treated with AT-101, paclitaxel, and carboplatin resulted in four CRPC patients
with SD and multiple PRs (two CRPC, one NSCLC, and one esophageal adenocarcinoma) [35].
Preliminary results from a Phase I trial conducted in previously untreated CLL patients with high risk
features treated with AT-101 resulted in multiple patients achieving decreases in lymphocyte counts,
lymphadenopathy and spleen size [36]. There are currently two ongoing Phase II clinical trials combining

8
AT-101 with chemotherapy. In the first, AT-101 and docetaxel are being studied in patients with
squamous cell carcinoma of the head and neck (NCT01285635) and the second will assess AT-101 in
combination with chemotherapy (docetaxol and cisplatin or carboplatin) in laryngeal cancer patients
(NCT01633541).
FX-11 was originally identified in a study that sought to synthesize a specific inhibitor of malarial
LDH [37, 38]. It is a small molecule derivative of the LDH inhibitor, gossypol and is competitive to the
LDH substrate NADH. It has a Ki for LDH-A that is twenty times that of LDH-B (LDH-A: 0.05µM;
LDH-B: 1µM) [37, 38] and almost 40 times better than gossypol for LDH-A (1.9µM) [38]. Treatment of
cells with FX11 (or siRNA mediated knockdown of LDH-A) increases oxygen consumption and
generation of reactive oxygen species through elevated mitochondrial glucose oxidation [39] [27]. FX11
has been found to be effective in reducing tumor burden in three mouse models: (i) a P4398 B cell
xenograft model where treatment was initiated just after tumors were palpable; (ii) a human lymphoma
xenograft model; and (iii) a P198 pancreatic adenocarcinoma model where treatment was initiated after
tumors were well established (200 mm3). Mice treated with FX11 displayed no weight loss, normal
hematology and blood chemistry that included multiple markers of kidney (blood urea nitrogen, creatine)
and liver (aspartate aminotransferase, alanine aminotransferase or alkaline phosphatase) function,
suggesting that FX11 is not toxic [39]. Humans deficient for LDHA develop normally although they
suffer from exertional myopathy [40], suggesting that inhibition of LDH-A will be well-tolerated under
non-exertional circumstances. Although there are no current clinical trials involving FX11, a recent study
reports the synthesis of new gossypol derivatives that show toxicity to tumor cell lines suggesting that
these agents will continue to be developed for possible clinical trial testing [41].
Targeting PDK: CPI-613 and Dichloroacetate (DCA)
The pyruvate dehydrogenase complex (PDH) is composed of three enzymes: pyruvate
dehydrogenase (E1; comprised of two α and two β subunits with the active site in the α subunit),

9
dihydrolipoamine acetyltransferase (E2) and lipoamide dehydrogenase (E3). Together, they catalyze the
conversion of pyruvate to Acetyl CoA for further oxidation in the TCA cycle. PDH is positively regulated
in situations of low energy by pyruvate dehydrogenase phosphatase (PDP) and negatively regulated by
phosphorylation of the E1α subunit by the serine/threonine pyruvate dehydrogenase kinase (PDK). When
PDH is active, it becomes saturated with cofactors, including covalently bound lipoate (lipoamide) that,
when sensed by PDK, results in phosphorylation of the E1α subunit of the PDH complex leading to its
inactivation and reduced conversion of pyruvate into Acetyl CoA. Glycolysis and the TCA cycle are
linked by PDH which, when activated, directs carbons away from lactate production and into the TCA
cycle. Since the TCA cycle is important for the production of anabolic precursors, inhibition of PDH is
potentially an effective anti-tumor therapy. There are two promising drugs that target PDH: CPI-613 that
inhibits PDH and DCA that activates PDH (discussed below). At first glance it seems counterintuitive
that both inactivating and activating PDH would be toxic to tumor cells. However, since tumor cells
require an activated glycolytic pathway, TCA cycle and electron transport chain, maintaining a controlled
integration between these pathways via PDH is thought to be essential for cancer cell survival [42].
A liopamide mimic, CPI-613 activates PDK, leading to phosphorylation and inactivation of PDH
[43]. Preclinical studies have revealed that CPI-613 reduced PDH activity and effectively killed tumor
cells but was less toxic to primary normal cell counterparts. Knockdown of all four PDK isoforms
resulted in resistance to CPI-613 treatment, indicating a requirement of PDK expression for CPI-613-
mediated cell death. Significantly, reduced tumor volumes were seen in pancreatic and lung xenograft
models with median survival in the pancreatic model extended to 192.5 days with CPI-613 treatment
versus 48 days with vehicle [43]. CPI-613, the lead clinical candidate in Cornerstone Pharmaceuticals
Altered Energy Metabolism Directed Platform, was granted orphan drug status for pancreatic cancer by
the FDA, and is currently being tested in clinical trials. A phase I trial to determine the safety,
tolerability, MTD, efficacy and pharmacokinetic profile of CPI-613 given IV twice a week for three
weeks in patients with advanced hematologic malignancies (NCT01034475) and a phase I/II trial

10
(NCT00741403) to assess the same criteria in patients with advanced solid malignancies and lymphoma
are currently ongoing. Promising results from the hematologic study showed that CPI-613 was well
tolerated in the thirteen patients (dose ranged from 420-1386mg/M2) with no bone marrow suppression or
dose-limiting-toxicities (DLT). Seven of the thirteen patients had SD or better with an overall response
rate of 54%, suggesting CPI-613 may be an effective treatment for hematologic malignancies [44]. A
Phase I/II study (NCT00907166) will assess the safety of CPI-613 and gemcitabine in patients with solid
tumors (phase I) with the phase II part of the trial comparing CPI-613 and gemcitabine treatment to
gemcitabine alone in pancreatic cancer patients. Results from the phase I part of the trial revealed that
CPI-613 was well tolerated (no DLTs), and four of the eight patients with breast and colon cancer treated
had SD (4-16 weeks) with reductions in glucose uptake (4-42%) by FDG-PET imaging [45]. Initial
results from the phase II study showed that administration of CPI-613 and gemcitabine was well tolerated
and that prolonged survival correlated with increased CPI-613 dose [46]. The lack of toxicity and initial
positive patient outcomes suggests that targeting PDH with CPI-613 may hold promise as an anti-cancer
therapeutic agent in patients with solid tumors including pancreatic cancer.
As opposed to CPI-613, which activates PDK, and therefore inhibits PDH, DCA inhibits PDK,
leading to activation of PDH [47, 48]. This results in a decrease in glycolytic flux to lactate and increase
in oxidative metabolism which may starve adjacent cancer cells and supporting host cells of oxygen.
Additionally, as mentioned above, since both activating and inhibiting PDH activity can be toxic to
cancer cells, cancer cells may be especially sensitive to disruption of the normal metabolic integration of
the glycolytic pathway and the TCA cycle. The validation of DCA as an anti-cancer agent stems from an
initial report that demonstrated dramatic reductions in tumor size after DCA treatment in a nude rat A549
xenograft tumor model [48]. DCA is appealing as a therapeutic agent for widespread testing as a
monotherapy and in combination with standard agents since it is orally bioavailable and has little if any
patent restrictions. However, DCA was not found to be effective in reducing tumor size or improving
survival in a xenograft mouse model of breast cancer [49] suggesting that DCA may be effective in only
certain types of tumors. Since glioblastoma multiforme (GBM) tumors are very glycolytic, and DCA can

11
cross the blood brain barrier, it was tested in a small clinical trial that included five GBM patients. Three
patients had failed prior standard therapy (debulking surgery, radiation therapy (RT), temozolomide
(TMZ)) and subsequent chemotherapies. These three patients were treated with DCA as a single agent.
One patient had a large tumor with brain edema at the start of treatment and died three months later from
complications related to the edema. However, after fifteen months of DCA treatment, the remaining four
patients had stable disease by CT imaging and all were still alive at eighteen months [50]. Although a
small trial, the results of this study provide hope that DCA may inhibit tumor growth and prolong survival
in GBM patients. There now are three ongoing cancer clinical trials involving DCA. The first is a Phase I
safety and efficacy study in patients with recurrent brain tumors (NCT0111097). The second is a Phase I
trial that is examining the safety of DCA in patients with recurrent or metastatic solid tumors
(NCT00566410) and the third is a placebo controlled Phase II study that will determine safety and
efficacy of DCA treatment in combination with cisplatin and radiation in patients with head and neck
carcinomas (NCT01386632).
Targeting Glucose Transporter 1 (GLUT1): STF-31
Von Hippel Lindau (VHL) is a tumor suppressor inactivated in most (~80%) spontaneous renal
cell carcinomas (RCC). A synthetic lethal screen designed to find agents that specifically kill VHL null
cells identified STF-31, a compound that is toxic to VHL negative RCCs in a HIF-1α−dependent manner
[51]. Since HIF-1α regulates expression of multiple glycolytic enzyme genes including the glucose
transporter GLUT1, STF-31 was tested for its ability to inhibit GLUT1 functions. STF-31 blocked
glucose uptake and reduced tumor size in renal cell carcinoma xenograft models with no reported
toxicities. STF-31 is thought to inhibit GLUT1 and the prediction thus is that it will be effective against
multiple tumor types that express GLUT1, and not just VHL negative RCCs, although this has yet to be
tested. STF-31 is currently licensed to Ruga Incorporated and is in preclinical testing with Phase I trials
predicted for 2013/2014 (Rugacorp.com).

12
Targeting 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase (PFKFB3): 3PO/PFK158
The PFKFB3 kinase domain phosphorylates fructose 6-phosphate (F6P) to generate fructose 2,6-
bisphosphate (F26BP), a potent allosteric activator of PFK-1. Since activation of PFK-1 is a key
regulatory step in glycolysis, modulation of PFKFB3 activity directly affects flux through the entire
glycolytic pathway [52]. PFKFB3 is a member of a family of dual kinase:bisphosphatases (PFKFB1-4)
that phosphorylate and dephosphorylate F6P. Unlike the other family members, PFKFB3 functions
essentially solely as a kinase with a kinase:bisphosphatase ratio of ~740:1 [53]. PFKFB3 is expressed at
low levels in most normal human tissues, and is not expressed in neurons [54], but is highly expressed in
many human cancers including lung, breast, prostate and colon tumors when compared to matched
normal samples [55]. Importantly, PFKFB3 is upregulated by multiple oncoproteins including HIF-1α
and Ras [9, 56]. Additionally, recent data suggests that PFKFB3 is negatively regulated by the PTEN
tumor suppressor gene [57] which promotes the APC/CDH1 degradation complex that post-
translationally negatively regulates PFKFB3 [58]. PTEN null cells therefore are predicted to have higher
PFKFB3 expression and potentially higher reliance on the activity PFKFB3. Accordingly, PTEN status in
human tumors may be a predictive biomarker for sensitivity to PFKFB3 inhibitors. Last, the requirement
of PFKFB3 for neoplastic transformation was recently demonstrated by the observations that
heterozygous genomic deletion of the Pfkfb3 gene reduced the concentration of F2,6BP, glucose uptake,
glycolytic flux to lactate and anchorage-independent growth of the LT/H-RasV12-transformed fibroblasts
as xenograft tumors in syngeneic mice [52, 59].
In silico screening identified an inhibitor of PFKFB3, 3-(3-pyridinyl)-1-(4-pyridinyl)-2-propen-1-
one (3PO), that inhibits glucose uptake, cellular F26BP production, glycolytic flux to lactate, growth of
cancer cell lines, and importantly, glucose uptake and tumor growth in multiple mouse tumor models
[60]. Development of 3PO derivatives by the biotechnology company, Advanced Cancer Therapeutics,
has resulted in the identification of an initial lead compound, PFK15 with improved (~100X) potency for

13
inhibition of recombinant enzyme activity over 3PO (2011 American Association for Cancer Research
Annual Meeting, Abstract #2825). PFK15 provided a synthetic platform for the synthesis of third
generation derivatives, which led to the pharmaceutical-grade agent, PFK158, that exhibits increased
potency and improved pharmacokinetic properties. PFK158 has undergone IND-enabling rat and beagle
toxicity testing with Phase I clinical trials slated to begin in 2013
(www.advancedcancertherapeutics.com). Kancera AB (Sweden) is also testing PFKFB3 inhibitors that
are in preclinical lead optimization phase with the plan to identify a candidate for clinical trial testing by
the end of 2012 (www.kancera.com).
Targeting Complex I: Metformin
For millennia, the herb Galega officinalis (French Lilac, Italian Fitch or Goat’s Rue) has been
used to produce tea to relieve frequent urination and sweet-‐smelling breath. This herbal remedy
for what was eventually found to be caused by the hyperglycemia of Diabetes Mellitus (DM) led
several investigators during the 20th century to purify the active components of the herb,
biguanides, including Phenformin, Buformin and Metformin (i.e. N’,N’-‐dimethylbiguanide).
Although Phenformin and Buformin were limited by toxicity related to lactic acidosis, Metformin is
currently FDA-‐approved and widely used for the treatment of DM. The precise mechanism of action
of metformin is not well defined but the agent does inhibit complex I of the electron transport
chain, oxygen consumption and ATP in hepatocytes [61, 62]. Such a decrease in the intracellular
concentration of ATP will cause an allosteric activation of 6-‐phosphofructo-‐1-‐kinase and resultant
elevation in glucose uptake and glycolytic flux [63] as well as activation of AMP kinase (AMPK)
which increases glucose transporter (GLUT4) expression and translocation in myocytes [64] which
may in part explain the anti-‐diabetic effects of Metformin. Several retrospective epidemiological
studies of diabetic patients who were treated with Metformin have found that these patients had a
lower risk of developing all types of cancer and of cancer-‐related deaths relative to diabetic patients

14
who received other oral glucose-‐lowering agents [65-‐68]. Additionally, diabetic breast cancer
patients on Metformin were found after resection to experience a higher rate of microscopic
complete responses after neoadjuvant chemotherapy than diabetic patients not being treated with
Metformin or non-‐diabetic patients [69]. The mechanism for these statistically significant effects is
an area of active investigation and several pre-‐clinical and clinical investigators are now attempting
to improve outcomes of standard anti-‐neoplastic agents with Metformin. One potential hypothesis
is that inhibition of electron transport chain activity will limit the availability of NAD+ that is
required for TCA cycling which, in turn, is required for the production of anabolic precursors.
Importantly, preclinical studies in multiple non-‐diabetic mouse tumor models in vivo have
demonstrated that metformin may be effective in patients with normal metabolism (i.e. non-‐
diabetics). For example, treatment of mice with metformin prior to development of breast tumors
(MMTV-‐HER-‐2/neu), resulted in delayed tumor onset, reduced number of tumors, and improved
overall survival [70]. Metformin also has been found to reduce tumorigenicity in mouse models of
liver cancer [71], lung cancer [72] and intestinal cancer [73]. Several clinical trials are now underway
to assess whether metformin as a single agent or in combination with other chemotherapies can improve
patient outcomes specifically in non-diabetic patients. A completed Phase I trial combining metformin
and temsirolimus conducted in patients with solid tumors evaluated eight patients for disease outcomes.
Two patients had disease progression, five had SD, and one had a PR. One of the patients with SD was a
melanoma patient that had radiologic progression on chemotherapy prior to the study who had SD for 22
months. Although a small study, these clinical results demonstrated that some solid cancers may be
sensitive to combined temsirolimus and metformin treatment [74].
Final Comments and Future Directions
There is and always will be concern that targeting metabolic pathways required for both normal
and tumor cells will yield unacceptable therapeutic indices in cancer patients. However, given the lack of

15
effective therapies against the majority of human cancers that have metastasized and the abundance of
pre-clinical data demonstrating efficacy without overt clinical toxicity, the clinical testing of metabolic
inhibitors seems warranted. Importantly, AT-101 has thus far been well tolerated in humans, as has been
the BCR/ABL inhibitor Imatinib, which is a potent inhibitor of glycolysis in CML cells [75].
Additionally, unanticipated mechanisms of action may be appreciated in clinical trials of metabolic
inhibitors which may in turn explain positive pre-clinical in vivo data. For example, rapidly dividing T
cells utilize glycolysis [76] and the PFKFB3 inhibitor, 3PO, has been found to suppress T cell activation
in vitro and in vivo [77]. Given the immunosuppressive effects of regulatory T cells in cancer patients
[78], PFKFB3 inhibitors may have positive anti-tumor effects on the immune system which may
contribute to their anti-tumor properties. Last, since metabolic pathways are increased in tumor versus
normal cells, a therapeutic window for metabolic modulators may be reached whereby treatment is below
the threshold for causing toxicity, but sufficient to kill tumor cells. Since metabolic reprogramming is
universal to tumor cells, modulating this phenotype with drugs has the potential to be useful for the
treatment of a wide range of tumor types. This does not imply however that the same metabolic inhibitors
will be effective in all tumor types. For example, as discussed above, PTEN status may predict tumor
sensitivity to inhibition of PFKFB3 but perhaps not to inhibition of other metabolic proteins. Given the
distinct mechanisms of action of the aforementioned metabolic inhibitors, we expect the development of
multiple phase I/II trials in which these inhibitors are combined with FDA-approved agents that are the
standard of care for distinct cancer types (e.g. vemurafenib in melanoma) as well as the rational
combination of glycolytic inhibitors such as PFK158 with agents that suppress angiogenesis, electron
transport chain activity, glutamine metabolism or alternative pathways for energy and anabolic precursor
production such as autophagy.

16
Figure Legend
Figure 1. Metabolic Inhibitors In or Entering Clinical Trials. Ras, HIF-1α and c-Myc increase the
expression of metabolic transporters and enzymes which in turn cause a reprogramming of metabolic
utilization that supports the enhanced energetic and anabolic requirements of cancer cells. Several
metabolic inhibitors are under study in clinical trials, including the following inhibitors (targets in
parantheses): (i) STF-31 (Glut1); (ii) 3PO/PFK158 (PFKFB3); (iii) FX11/AT-101 (LDH-A); (iv)
DCA/CPI-613 (PDK); and (v) Metformin (Met; Complex I). Black lines indicate suppression and red
lines indicate stimulation of activity and/or expression. Key regulators, transporters and enzymes are
highlighted in red. GLUT1: Glucose transporter 1; HK2: Hexokinase 2; PFK1: 6-phosphofructo-1-kinase;
PK-M2: Pyruvate kinase M2; PDH: Pyruvate dehydrogenase; LDH-A: Lactate dehydrogenase A;
PFKFB3: 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3.

17
Acknowledgements
We would like to thank Brian Clem, Yoannis Imbert-Fernandez, Sucheta Telang, Alden Klarer and John
Eaton for their critical reviews of this article. We also acknowledge the essential support of the National
Cancer Institute (R01-CA149438; JC) and the Congressionally Directed Medical Research Program
(BC112204; JO) for the completion of this review article.

18
References
1. Cortes, J., et al., Front-‐line and salvage therapies with tyrosine kinase inhibitors and
other treatments in chronic myeloid leukemia. J Clin Oncol. 29(5): p. 524-‐31.
2. Hanahan, D. and R.A. Weinberg, Hallmarks of cancer: the next generation. Cell, 2011.
144(5): p. 646-‐74.
3. Kunkel, M., et al., Overexpression of Glut-‐1 and increased glucose metabolism in
tumors are associated with a poor prognosis in patients with oral squamous cell
carcinoma. Cancer, 2003. 97(4): p. 1015-‐24.
4. Mochiki, E., et al., Evaluation of 18F-‐2-‐deoxy-‐2-‐fluoro-‐D-‐glucose positron emission
tomography for gastric cancer. World J Surg, 2004. 28(3): p. 247-‐53.
5. Warburg, O., On the origin of cancer cells. Science, 1956. 123(3191): p. 309-‐14.
6. Guppy, M., E. Greiner, and K. Brand, The role of the Crabtree effect and an endogenous
fuel in the energy metabolism of resting and proliferating thymocytes. Eur J Biochem,
1993. 212(1): p. 95-‐9.
7. Gatenby, R.A. and R.J. Gillies, Why do cancers have high aerobic glycolysis? Nat Rev
Cancer, 2004. 4(11): p. 891-‐9.
8. Koukourakis, M.I., et al., Comparison of metabolic pathways between cancer cells and
stromal cells in colorectal carcinomas: a metabolic survival role for tumor-‐associated
stroma. Cancer Res, 2006. 66(2): p. 632-‐7.
9. Telang, S., et al., The oncoprotein H-‐RasV12 increases mitochondrial metabolism. Mol
Cancer, 2007. 6: p. 77.

19
10. Ramanathan, A., C. Wang, and S.L. Schreiber, Perturbational profiling of a cell-‐line
model of tumorigenesis by using metabolic measurements. Proc Natl Acad Sci U S A,
2005. 102(17): p. 5992-‐7.
11. Racker, E., R.J. Resnick, and R. Feldman, Glycolysis and methylaminoisobutyrate
uptake in rat-‐1 cells transfected with ras or myc oncogenes. Proc Natl Acad Sci U S A,
1985. 82(11): p. 3535-‐8.
12. Pylayeva-‐Gupta, Y., E. Grabocka, and D. Bar-‐Sagi, RAS oncogenes: weaving a
tumorigenic web. Nat Rev Cancer, 2011. 11(11): p. 761-‐74.
13. Obach, M., et al., 6-‐Phosphofructo-‐2-‐kinase (pfkfb3) gene promoter contains hypoxia-‐
inducible factor-‐1 binding sites necessary for transactivation in response to hypoxia. J
Biol Chem, 2004. 279(51): p. 53562-‐70.
14. Dang, C.V., A. Le, and P. Gao, MYC-‐induced cancer cell energy metabolism and
therapeutic opportunities. Clin Cancer Res, 2009. 15(21): p. 6479-‐83.
15. Schwartzenberg-‐Bar-‐Yoseph, F., M. Armoni, and E. Karnieli, The tumor suppressor
p53 down-‐regulates glucose transporters GLUT1 and GLUT4 gene expression. Cancer
Res, 2004. 64(7): p. 2627-‐33.
16. Kondoh, H., et al., Glycolytic enzymes can modulate cellular life span. Cancer Res,
2005. 65(1): p. 177-‐85.
17. Garcia-‐Cao, I., et al., Systemic elevation of PTEN induces a tumor-‐suppressive
metabolic state. Cell. 149(1): p. 49-‐62.
18. Telang, S., et al., Cytochrome c oxidase is activated by the oncoprotein Ras and is
required for A549 lung adenocarcinoma growth. Mol Cancer. 11(1): p. 60.

20
19. Morrish, F., et al., The oncogene c-‐Myc coordinates regulation of metabolic networks
to enable rapid cell cycle entry. Cell Cycle, 2008. 7(8): p. 1054-‐66.
20. Yuneva, M., et al., Deficiency in glutamine but not glucose induces MYC-‐dependent
apoptosis in human cells. J Cell Biol, 2007. 178(1): p. 93-‐105.
21. Wise, D.R., et al., Myc regulates a transcriptional program that stimulates
mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci U S
A, 2008. 105(48): p. 18782-‐7.
22. Clem, B.F. and J. Chesney, Regulation of Metabolism by Rb. Clinical Cancer Research,
2012. 18.(22): p. 4.
23. Reynolds, M., et al., Control of Glutamine Metabolism by the Tumor Suppressor Rb.
Oncogene, 2013. In Press.
24. Natarajan, N., et al., Novel immunotherapeutic agents and small molecule antagonists
of signalling kinases for the treatment of metastatic melanoma. Drugs. 71(10): p.
1233-‐50.
25. Chesney, J. and S. Telang, Regulation of Glycolytic and Mitochondrial Metabolism by
Ras. Curr Pharm Biotechnol, 2012.
26. Xie, H., et al., LDH-‐A inhibition, a therapeutic strategy for treatment of hereditary
leiomyomatosis and renal cell cancer. Mol Cancer Ther, 2009. 8(3): p. 626-‐35.
27. Fantin, V.R., J. St-‐Pierre, and P. Leder, Attenuation of LDH-‐A expression uncovers a link
between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell,
2006. 9(6): p. 425-‐34.

21
28. Kitada, S., et al., Discovery, characterization, and structure-‐activity relationships
studies of proapoptotic polyphenols targeting B-‐cell lymphocyte/leukemia-‐2 proteins. J
Med Chem, 2003. 46(20): p. 4259-‐64.
29. Liu, G., et al., An open-‐label, multicenter, phase I/II study of single-‐agent AT-‐101 in
men with castrate-‐resistant prostate cancer. Clin Cancer Res, 2009. 15(9): p. 3172-‐6.
30. M. N. Stein, I.K., M. Hussain, G. Liu, G. Wilding, E. M. Posadas, W. M. Stadler, C.
Jeyamohan, S. Eddy, R. S. DiPaola, Phase II study of AT-‐101 to abrogate Bcl-‐2-‐
mediated resistance to androgen-‐deprivation therapy (ADT) in patients (pts) with
newly diagnosed androgen-‐dependent metastatic prostate cancer (ADMPC). in Journal
of Clinical Oncology2011. p. abstracy 137.
31. Sonpavde, G., et al., Randomized phase II trial of docetaxel plus prednisone in
combination with placebo or AT-‐101, an oral small molecule Bcl-‐2 family antagonist,
as first-‐line therapy for metastatic castration-‐resistant prostate cancer. Ann Oncol,
2012. 23(7): p. 1803-‐8.
32. Heist, R.S., et al., Phase I/II study of AT-‐101 with topotecan in relapsed and refractory
small cell lung cancer. J Thorac Oncol, 2010. 5(10): p. 1637-‐43.
33. Ready, N., et al., Double-‐blind, placebo-‐controlled, randomized phase 2 study of the
proapoptotic agent AT-‐101 plus docetaxel, in second-‐line non-‐small cell lung cancer. J
Thorac Oncol, 2011. 6(4): p. 781-‐5.
34. Baggstrom, M.Q., et al., A phase II study of AT-‐101 (Gossypol) in chemotherapy-‐
sensitive recurrent extensive-‐stage small cell lung cancer. J Thorac Oncol, 2011. 6(10):
p. 1757-‐60.

22
35. Y. Zhao, M.G., H. Lin, K. Harris Addo, K. Taber Levinson, M. LaRosiliere, S. Goodin, R.
A. Moss, A. R. Tan, M. N. Stein, Phase I study of at-‐101 (R-‐(-‐)-‐gossypol) in combination
with paclitaxel (P) and carboplatin (C) in solid tumors including castrate-‐resistant
prostate cancer (CRPC). 2011: Journal of Clinical Oncology. p. abstract 169.
36. D. F. James, J.E.C., O. Loria, C. E. Prada, R. A. Aguillon, T. J. Kipps, AT-‐101, a small
molecule Bcl-‐2 antagonist, in treatment naïve CLL patients (pts) with high risk
features; Preliminary results from an ongoing phase I trial. Journal of Clinical
Oncology, 2006 ASCO Annual Meeting Proceedings Part I. , 2006. Vol 24( June 20
Supplement).
37. Deck, L.M., et al., Selective inhibitors of human lactate dehydrogenases and lactate
dehydrogenase from the malarial parasite Plasmodium falciparum. J Med Chem,
1998. 41(20): p. 3879-‐87.
38. Yu, Y., et al., Selective active site inhibitors of human lactate dehydrogenases A4, B4,
and C4. Biochem Pharmacol, 2001. 62(1): p. 81-‐9.
39. Le, A., et al., Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits
tumor progression. Proc Natl Acad Sci U S A, 2010. 107(5): p. 2037-‐42.
40. Kanno, T., et al., Lactate dehydrogenase M-‐subunit deficiency: a new type of hereditary
exertional myopathy. Clin Chim Acta, 1988. 173(1): p. 89-‐98.
41. Wei, J., et al., Synthesis and biological evaluation of Apogossypolone derivatives as
pan-‐active inhibitors of antiapoptotic B-‐cell lymphoma/leukemia-‐2 (Bcl-‐2) family
proteins. J Med Chem. 53(22): p. 8000-‐11.
42. DeBerardinis, R.J., et al., The biology of cancer: metabolic reprogramming fuels cell
growth and proliferation. Cell Metab, 2008. 7(1): p. 11-‐20.

23
43. Zachar, Z., et al., Non-‐redox-‐active lipoate derivates disrupt cancer cell mitochondrial
metabolism and are potent anticancer agents in vivo. J Mol Med (Berl), 2011. 89(11):
p. 1137-‐48.
44. Timothy S. Pardee, L.M.D.-‐W., Erica Peronto, Denise A. Levitan, David Duane Hurd,
Steven Kridel, Robin Harrelson, Megan Manuel, Susan Lyerly, Bayard L. Powell,
Evaluation of the first-‐in-‐class antimitochondrial metabolism agent CPI-‐613 in
hematologic malignancies. Journal of Clinical Oncolgy, 2012. 30: p. Abstract 6524.
45. A. S. Retter, R.S., R. Rodriguez, K. Hoffman, F. Volterra, A. D. Hoffman, N. Huppert, K.
Lee, Phase I trial of CPI-‐613, a lipoic acid analog, and gemcitabine in patients with
advanced solid tumors. Journal of Clinical Oncolgy , 2010. 28: p. Abstract e13136.
46. Retter, A.S., Translational assessment of the efficacy of CPI-‐613 against pancreatic
cancer in animal models versus patients with stage IV disease. Journal of Clinical
Oncolgy, 2012. 30: p. Abstract 3075.
47. Whitehouse, S., R.H. Cooper, and P.J. Randle, Mechanism of activation of pyruvate
dehydrogenase by dichloroacetate and other halogenated carboxylic acids. Biochem J,
1974. 141(3): p. 761-‐74.
48. Bonnet, S., et al., A mitochondria-‐K+ channel axis is suppressed in cancer and its
normalization promotes apoptosis and inhibits cancer growth. Cancer Cell, 2007.
11(1): p. 37-‐51.
49. Robey, I.F. and N.K. Martin, Bicarbonate and dichloroacetate: evaluating pH altering
therapies in a mouse model for metastatic breast cancer. BMC Cancer, 2011. 11: p.
235.

24
50. Michelakis, E.D., et al., Metabolic modulation of glioblastoma with dichloroacetate. Sci
Transl Med, 2010. 2(31): p. 31ra34.
51. Chan, D.A., et al., Targeting GLUT1 and the Warburg effect in renal cell carcinoma by
chemical synthetic lethality. Sci Transl Med, 2011. 3(94): p. 94ra70.
52. Telang, S., et al., Ras transformation requires metabolic control by 6-‐phosphofructo-‐2-‐
kinase. Oncogene, 2006. 25(55): p. 7225-‐34.
53. Sakakibara, R., et al., Characterization of a human placental fructose-‐6-‐phosphate, 2-‐
kinase/fructose-‐2,6-‐bisphosphatase. J Biochem (Tokyo), 1997. 122(1): p. 122-‐8.
54. Herrero-‐Mendez, A., et al., The bioenergetic and antioxidant status of neurons is
controlled by continuous degradation of a key glycolytic enzyme by APC/C-‐Cdh1. Nat
Cell Biol, 2009. 11(6): p. 747-‐52.
55. Atsumi, T., et al., High expression of inducible 6-‐phosphofructo-‐2-‐kinase/fructose-‐2,6-‐
bisphosphatase (iPFK-‐2; PFKFB3) in human cancers. Cancer Res, 2002. 62(20): p.
5881-‐7.
56. Yalcin, A., et al., Regulation of glucose metabolism by 6-‐phosphofructo-‐2-‐
kinase/fructose-‐2,6-‐bisphosphatases in cancer. Exp Mol Pathol, 2009. 86(3): p. 174-‐9.
57. Garcia-‐Cao, I., et al., Systemic elevation of PTEN induces a tumor-‐suppressive
metabolic state. Cell, 2012. 149(1): p. 49-‐62.
58. Song, M.S., et al., Nuclear PTEN regulates the APC-‐CDH1 tumor-‐suppressive complex in
a phosphatase-‐independent manner. Cell, 2011. 144(2): p. 187-‐99.
59. Chesney, J., et al., Targeted disruption of inducible 6-‐phosphofructo-‐2-‐kinase results in
embryonic lethality. Biochem Biophys Res Commun, 2005. 331(1): p. 139-‐46.

25
60. Clem, B., et al., Small-‐molecule inhibition of 6-‐phosphofructo-‐2-‐kinase activity
suppresses glycolytic flux and tumor growth. Mol Cancer Ther, 2008. 7(1): p. 110-‐20.
61. El-‐Mir, M.Y., et al., Dimethylbiguanide inhibits cell respiration via an indirect effect
targeted on the respiratory chain complex I. J Biol Chem, 2000. 275(1): p. 223-‐8.
62. Owen, M.R., E. Doran, and A.P. Halestrap, Evidence that metformin exerts its anti-‐
diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain.
Biochem J, 2000. 348 Pt 3: p. 607-‐14.
63. Beatty, C.H., M.K. Young, and R.M. Bocek, Control of glycolysis in skeletal muscle from
fetal rhesus monkeys. Pediatr Res, 1976. 10(3): p. 149-‐53.
64. Towler, M.C. and D.G. Hardie, AMP-‐activated protein kinase in metabolic control and
insulin signaling. Circ Res, 2007. 100(3): p. 328-‐41.
65. Decensi, A., et al., Metformin and cancer risk in diabetic patients: a systematic review
and meta-‐analysis. Cancer Prev Res (Phila). 3(11): p. 1451-‐61.
66. Evans, J.M., et al., Metformin and reduced risk of cancer in diabetic patients. BMJ,
2005. 330(7503): p. 1304-‐5.
67. Landman, G.W., et al., Metformin associated with lower cancer mortality in type 2
diabetes: ZODIAC-‐16. Diabetes Care. 33(2): p. 322-‐6.
68. Libby, G., et al., New users of metformin are at low risk of incident cancer: a cohort
study among people with type 2 diabetes. Diabetes Care, 2009. 32(9): p. 1620-‐5.
69. Jiralerspong, S., et al., Metformin and pathologic complete responses to neoadjuvant
chemotherapy in diabetic patients with breast cancer. J Clin Oncol, 2009. 27(20): p.
3297-‐302.

26
70. Anisimov, V.N., et al., Effect of metformin on life span and on the development of
spontaneous mammary tumors in HER-‐2/neu transgenic mice. Exp Gerontol, 2005.
40(8-‐9): p. 685-‐93.
71. Bhalla, K., et al., Metformin prevents liver tumorigenesis by inhibiting pathways
driving hepatic lipogenesis. Cancer Prev Res (Phila), 2012. 5(4): p. 544-‐52.
72. Memmott, R.M., et al., Metformin prevents tobacco carcinogen-‐-‐induced lung
tumorigenesis. Cancer Prev Res (Phila), 2010. 3(9): p. 1066-‐76.
73. Tomimoto, A., et al., Metformin suppresses intestinal polyp growth in ApcMin/+ mice.
Cancer Sci, 2008. 99(11): p. 2136-‐41.
74. MacKenzie, M.J., et al., A phase I study of temsirolimus and metformin in advanced
solid tumours. Invest New Drugs, 2012. 30(2): p. 647-‐52.
75. Boren, J., et al., Gleevec (STI571) influences metabolic enzyme activities and glucose
carbon flow toward nucleic acid and fatty acid synthesis in myeloid tumor cells. J Biol
Chem, 2001. 276(41): p. 37747-‐53.
76. Greiner, E.F., M. Guppy, and K. Brand, Glucose is essential for proliferation and the
glycolytic enzyme induction that provokes a transition to glycolytic energy production.
J Biol Chem, 1994. 269(50): p. 31484-‐90.
77. Telang, S., et al., Small molecule inhibition of 6-‐phosphofructo-‐2-‐kinase suppresses t
cell activation. J Transl Med. 10: p. 95.
78. Jacobs, J.F., et al., Regulatory T cells in melanoma: the final hurdle towards effective
immunotherapy? Lancet Oncol. 13(1): p. e32-‐42.
79. Gottschalk, S., et al., Imatinib (STI571)-‐mediated changes in glucose metabolism in
human leukemia BCR-‐ABL-‐positive cells. Clin Cancer Res, 2004. 10(19): p. 6661-‐8.

27
80. Paszat, L., et al., Outcomes of surveillance mammography after treatment of primary
breast cancer: a population-‐based case series. Breast Cancer Res Treat, 2009. 114(1):
p. 169-‐78.
81. Zhao, Y., et al., Overcoming trastuzumab resistance in breast cancer by targeting
dysregulated glucose metabolism. Cancer Res. 71(13): p. 4585-‐97.
82. Sharma, R.I. and T.A. Smith, Colorectal tumor cells treated with 5-‐FU, oxaliplatin,
irinotecan, and cetuximab exhibit changes in 18F-‐FDG incorporation corresponding to
hexokinase activity and glucose transport. J Nucl Med, 2008. 49(8): p. 1386-‐94.
83. Zeng, Z., et al., Rapamycin derivatives reduce mTORC2 signaling and inhibit AKT
activation in AML. Blood, 2007. 109(8): p. 3509-‐12.
84. Ma, W.W., et al., [18F]fluorodeoxyglucose positron emission tomography correlates
with Akt pathway activity but is not predictive of clinical outcome during mTOR
inhibitor therapy. J Clin Oncol, 2009. 27(16): p. 2697-‐704.
85. Liu, L., et al., Glycolysis in Panc-‐1 human pancreatic cancer cells is inhibited by
everolimus. Exp Ther Med. 5(1): p. 338-‐342.
86. Walter, M.A., et al., Metabolic imaging allows early prediction of response to
vandetanib. J Nucl Med. 52(2): p. 231-‐40.
87. Keunen, O., et al., Anti-‐VEGF treatment reduces blood supply and increases tumor cell
invasion in glioblastoma. Proc Natl Acad Sci U S A. 108(9): p. 3749-‐54.
88. Fiume, L., et al., Effect of sorafenib on the energy metabolism of hepatocellular
carcinoma cells. Eur J Pharmacol. 670(1): p. 39-‐43.
28
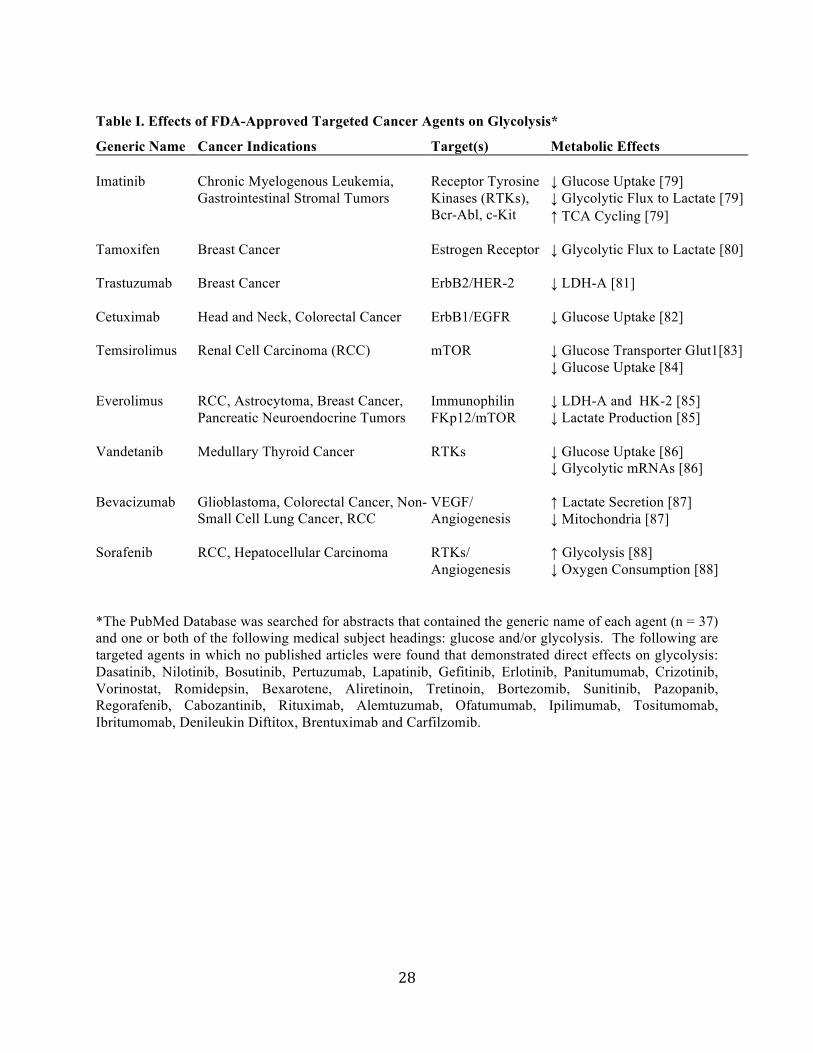
Table I. Effects of FDA-Approved Targeted Cancer Agents on Glycolysis*
*The PubMed Database was searched for abstracts that contained the generic name of each agent (n = 37) and one or both of the following medical subject headings: glucose and/or glycolysis. The following are targeted agents in which no published articles were found that demonstrated direct effects on glycolysis: Dasatinib, Nilotinib, Bosutinib, Pertuzumab, Lapatinib, Gefitinib, Erlotinib, Panitumumab, Crizotinib, Vorinostat, Romidepsin, Bexarotene, Aliretinoin, Tretinoin, Bortezomib, Sunitinib, Pazopanib, Regorafenib, Cabozantinib, Rituximab, Alemtuzumab, Ofatumumab, Ipilimumab, Tositumomab, Ibritumomab, Denileukin Diftitox, Brentuximab and Carfilzomib.
Generic Name Cancer Indications Target(s) Metabolic Effects Imatinib Chronic Myelogenous Leukemia,
Gastrointestinal Stromal Tumors Receptor Tyrosine Kinases (RTKs), Bcr-Abl, c-Kit
↓ Glucose Uptake [79] ↓ Glycolytic Flux to Lactate [79] ↑ TCA Cycling [79]
Tamoxifen Breast Cancer Estrogen Receptor ↓ Glycolytic Flux to Lactate [80]
Trastuzumab Breast Cancer ErbB2/HER-2 ↓ LDH-A [81]
Cetuximab Head and Neck, Colorectal Cancer ErbB1/EGFR ↓ Glucose Uptake [82]
Temsirolimus Renal Cell Carcinoma (RCC) mTOR ↓ Glucose Transporter Glut1[83] ↓ Glucose Uptake [84]
Everolimus RCC, Astrocytoma, Breast Cancer, Pancreatic Neuroendocrine Tumors
Immunophilin FKp12/mTOR
↓ LDH-A and HK-2 [85] ↓ Lactate Production [85]
Vandetanib Medullary Thyroid Cancer RTKs ↓ Glucose Uptake [86] ↓ Glycolytic mRNAs [86]
Bevacizumab Glioblastoma, Colorectal Cancer, Non-Small Cell Lung Cancer, RCC
VEGF/ Angiogenesis
↑ Lactate Secretion [87] ↓ Mitochondria [87]
Sorafenib RCC, Hepatocellular Carcinoma RTKs/ Angiogenesis
↑ Glycolysis [88] ↓ Oxygen Consumption [88]

Figure 1
Acetyl CoA
PDH
NADH
NAD+
2e-+2H+
H+
I
IIIII
IV
+½O2→H2O
TCA Cycle
Glucose
G6P
Glucose
HK2
F16P
F6P
PFK1
G3PDHA
Pyruvate
Ribose-5P
LDH-A
Lactate
X5P
PFKFB3
F26BP
PEP
PK-M2
GLUT1
+ + +
HIF-1a
Glutamine
Ras
Glutamine
PDKs
c-Myc
Glutamate
ATP-SMitochondria
STF-31
3PO
PFK158
FX11/AT-101
DCA
CPI-613+
Met




















