
Synthesis of Some Novel Heterocyclic and Schiff Base Derivatives as Antimicrobial Agents
Upload
independentCategory
view
6download
0
www.elsevier.com/locate/poly
Polyhedron 26 (2007) 5139–5149
Synthesis, characterisation and crystal structures of threetrinuclear cadmium(II) complexes with multidentate Schiff base ligands
Joy Chakraborty a, Santarupa Thakurta a, Brajagopal Samanta a, Aurkie Ray a,G. Pilet b, Stuart R. Batten c, Paul Jensen c, Samiran Mitra a,*
a Department of Chemistry, Jadavpur University, Raja S.C. Mullick Road, Kolkata 700 032, Indiab Groupe de Cristallographie et Ingenierie Moleculaire, Laboratoire des Multimateriaux et Interfaces UMR 5615 CNRS –
Universite Claude Bernard Lyon 1, Bat. Jules Raulin, 43 bd du 11 Novembre, 1918 69622 Villeurbanne, Cedex, Francec School of Chemistry, Monash University, P.O. Box 23, 3800 Victoria, Australia
Received 30 March 2007; accepted 16 July 2007Available online 4 September 2007
Abstract
Three novel Schiff base Cd(II) trimeric complexes, [Cd3(L1)2(SCN)2(CF3COO)2] (1), [Cd3(L1)2(SCN)2(HCONMe2)] (2) and[Cd3(L2)2{N(CN)2}2] (3) have been prepared from two different symmetrical Schiff bases H2L1 and H2L2 (where H2L1 = N1,N3-bis(sal-icylideneimino)diethylenetriamine, a potentially pentadentate Schiff base with a N3O2 donor set, and H2L2 = N1,N3-bis(3-methoxysali-cylideneimino)diethylenetriamine, a potentially heptadentate Schiff base with a N3O4 donor set). All the complexes have been synthesisedunder similar synthetic procedures and their crystal structures have been established by single crystal X-ray diffraction methods. Theligands and their metal complexes have been characterised by analytical and spectroscopic techniques. Among the three complexes, 1
and 3 are linear whereas 2 is a cyclic trimer. In 1 and 3, all the doubly phenoxo bridged Cd(II) metal centres are in a distorted octahedralenvironment. In complex 2, two of the three Cd(II) centres reside in a distorted octahedral environment and the remaining one enjoys amonocapped octahedral geometry. Altogether the variety in the bridging mode of two new salen-type ligands has been establishedthrough these complexes.� 2007 Elsevier Ltd. All rights reserved.
Keywords: Cadmium(II); Trimer; Schiff base; X-ray structure; Luminescence
1. Introduction
Metallo-organic coordination networks with variedcomplex molecular and crystalline architectures have beenextensively studied for their interesting chemical and phys-ical properties. Schiff base ligands have played an integraland important role in this area since the late 19th century.That metal complexes with this type of ligands are ubiqui-tous is a clear reflection of their facile synthesis, easily tun-able steric and electronic properties, good solubility incommon organic solvents, wide application and overall
0277-5387/$ - see front matter � 2007 Elsevier Ltd. All rights reserved.
doi:10.1016/j.poly.2007.07.038
* Corresponding author. Tel.: +91(033)2414 6666x2505; fax:+91(033)2414 6414/6210.
E-mail address: [email protected] (Samiran Mitra).
accessibility of diverse structural modifications. So, exten-sive research on these Schiff base metal complexes hasexpanded enormously nowadays, and has embraced verywide and diversified subjects comprising vast areas of orga-nometallic compounds and various aspects of bioinorganicchemistry [1,2]. Transition metal complexes, particularlywith oxygen and nitrogen donor Schiff bases, are of partic-ular interest [3] because of their ability to possess unusualconfigurations, structural ability and their sensitivity tomolecular environments [4]. Many excellent works havebeen devoted to the exploration of new synthetic pathwaysand structural aspects of the resulting systems, highlightingthe role of the ligands, suitable combinations of bridginggroups and of the different metal ions to design a chemicalpathway capable of tuning the synthesis from mononuclear

5140 J. Chakraborty et al. / Polyhedron 26 (2007) 5139–5149
to multinuclear systems [5–7]. Tridentate and tetradentateligands such as salen-type Schiff bases are quite capableof forming complexes with certain metal ions which canexhibit unusual coordination, high thermodynamic stabil-ity and kinetic inertness. The coordination geometry ofthese complexes depends upon the electronic configurationand size of the metal ions, repulsions between non-bondedatoms in different ligand arms, and the inherent rigidity dueto the presence of aromatic rings, etc. [8]. Such topologiesare also based on strong metal–ligand covalent bonds [9]and multiple weak non-covalent forces like hydrogenbonding [10] and p–p interactions [11]. The role of differenttransition and non-transition metal ions as templates,Schiff bases as blocking units and pseudohalides or carbox-ylates as bridges are very much noteworthy in this regard[12–15]. Pseudohalides, especially azide, thiocyanate ordicyanamide, have long been known for their versatilecoordination modes to exploit the desired architectures.Cadmium(II), well suited with its d10 configuration, permitsa wide range of symmetries and coordination numbers andhence reports on hexacoordinated cadmium(II) complexesare well documented [14,15]. However, heptacoordinatedcadmium(II) complexes are not very common [12,16]. Allthese features have prompted us to develop new coordina-tion complexes with increased dimensionality using the var-iation of donor sites. Both the Schiff bases used to preparethe reported complexes are symmetric in nature. The mostinteresting fact is that although the ligands are quite sym-metric in nature, their coordination behaviour to the metalatoms is not at all similar. The geometry of the coordina-tion around the metal centers is mostly distorted octahe-dral, but one of the complexes shows a unique presenceof both hexa- and heptacoordinated metal centers. Fromthe report, it is very much clear that the coordination envi-ronment around the metal centre is largely influenced bythe steric demand of the long chain multidentate ligand.
We have successfully prepared three trinuclear Cd(II)complexes and characterised them on the basis of micro-analytical, spectroscopic and other physicochemical prop-erties. X-ray crystallographic studies have been made toestablish their structures. The present paper reports asystematic study of three new trimeric Cd(II) complexeswith two multidentate Schiff bases. In addition, theinvestigation of the coordination properties of cadmiumis of interest because of the environmental aspects andbiological activity of Cd(II). The details of syntheses,structures and other spectroscopic behaviours aredescribed below.
2. Experimental
2.1. Materials
All the chemicals and solvents used for the syntheseswere of analytical grade. Cadmium nitrate, tetrahydrate;salicylaldehyde; 3-methoxysalicylaldehyde; sodium thiocy-anate; sodium dicyanamide and diethylenetriamine were
purchased from Aldrich Chemical Co. Inc. and were usedwithout any further purification. Methanol and N,N-dimethylformamide (HCONMe2) were used as solvents.Both the solvents were super dried before use. AR grademethanol was distilled with a good fractionating columnfor the exclusion of moisture. In this method, the watercontent was reduced to ca. 100 ppm. The solvent dimethylformamide (AR) was distilled over di-phosphorus pentox-ide before use in order to minimise the moisture contentin the solvents. Hydrated cadmium (II) trifluoroacetate[Cd(CF3COO)2 Æ xH2O] was prepared by the treatment ofcadmium(II) carbonate, CdCO3, (AR grade, E. Merck,India) with 60% trifluoroacetic acid (AR grade, E. Merck,India) followed by slow evaporation on a steam bath. Itwas then filtered through a fine glass-frit and preserved ina CaCl2 desiccator.
2.2. Syntheses
Caution! Although no problems were encountered in thiswork, trifluoroacetic acid and its vapour are potentiallycorrosive to human skin. It should be handled with everycare.
2.2.1. Synthesis of the Schiff base ligands
2.2.1.1. Synthesis of (OH)C6H4CH@N(CH2)2NH(CH2)2
N@CHC6H4(OH) (H2L1). The Schiff base chelator H2L1
was prepared following a reported [17] procedure with aslight modification. Salicylaldehyde (0.244 g, 2 mmol) wasrefluxed with diethylenetriamine (0.103 g, 1 mmol) in dehy-drated methanol. After 1 h, a deep orange yellow solutionwas obtained. It was subjected to TLC which revealed thepresence of some unreacted starting materials along withthe Schiff base product. The Schiff base product was iso-lated by column chromatography in order to get it in thepurified form. The purified ligand was then evaporatedunder reduced pressure to yield a gummy mass, whichwas dried and stored in vacuo over CaCl2 for subsequentuse. Yield: 0.264 g (85%). Anal. Calc. for C18H21N3O2: C,69.43; H, 6.80; N, 13.49. Found: C, 69.40; H, 6.82; N,13.50%. Mass spectrum (EI): m/z 311(M+ = L+). FT-IR(KBr, cm�1): m(C@N) 1640. UV–Vis (k, nm): 242, 369.
2.2.1.2. Synthesis of (OH)(OCH3)C6H3CH@N(CH2)2
NH(CH2)2N@CHC6H3(OH)(OCH3) (H2L2). The Schiffbase chelator H2L2 was prepared following the same proce-dure as adopted for H2L1 except 3-methoxysalicylaldehyde(0.305 g, 2 mmol) was refluxed with diethylenetriamine(0.103 g, 1 mmol) in dehydrated methanol. Yield: 0.304 g(82%). Anal. Calc. for C20H25N3O4: C, 64.67; H, 6.78; N,11.31. Found: C, 64.65; H, 6.80; N, 11.30%. Mass spectrum(EI): m/z 371 (M+ = L+). FT-IR (KBr, cm�1): m(C@N)1641. UV–Vis (k, nm): 240, 372.
2.2.2. Syntheses of the Schiff base complexes
2.2.2.1. Synthesis of [Cd3(L1)2(SCN)2(CF3COO)2] (1).
The appropriate quantity of solid Schiff base ligand H2L1

J. Chakraborty et al. / Polyhedron 26 (2007) 5139–5149 5141
(0.622 g, 2 mmol) was dissolved in dry methanol (20 ml). Asolution of Cd(CF3COO)2 Æ 2H2O (1.123 g, 3 mmol) in drymethanol (10 ml) was added to this solution (3:2 molarratio, M:L) followed by the addition of a 5 ml methanolicsolution of 0.162 g (2 mmol) NaSCN. The mixture wasrefluxed for 2 h at 80 �C. The resulting yellow colouredsolution was then filtered off and the filtrate was left undis-turbed. After 5 days fine colourless needle shaped X-raydiffraction quality single crystals separated out. They werefiltered, washed with methanol and dried in vacuo overCaCl2. Yield: 85%. Anal. Calc. for C42H40Cd3F6N8O8S2:C, 38.80; H, 3.10; N, 8.62. Found: C, 38.82; H, 3.07; N,8.65%.
2.2.2.2. Synthesis of [Cd3(L1)2(SCN)2 (HCONMe2)] (2).
0.622 g (2 mmol) of the solid Schiff base ligand H2L1
was dissolved in dry methanol (20 ml). A gently warmsolution of Cd(NO3)2 Æ 4H2O (0.930 g, 3 mmol) in drymethanol (10 ml) was added to this solution (3:2 molarratio, M:L) followed by the addition of 5 ml NaSCNsolution (0.162 g, 2 mmol). The solution turned turbid.N,N-Dimethylformamide was added dropwise till theturbidity just disappeared. The mixture was refluxedfor 3 h at 80 �C. The resulting yellow coloured solutionwas then filtered off and the filtrate was left undis-turbed. After 2 h colourless plate shaped X-ray diffrac-tion quality single crystals separated out. They werefiltered, washed with methanol and dried in vacuo overCaCl2. Yield: 65%. Anal. Calc. for C41H43Cd3N9O5S2:C, 43.08; H, 3.79; N, 11.03. Found: C, 43.05; H, 3.80;N, 11.00%.
2.2.2.3. Synthesis of [Cd3(L2)2{N(CN)2}2] (3). 0.742 g(2 mmol) of the solid Schiff base ligand H2L2 was dissolvedin 10 ml methanol. The solution was then added to a 10 mlgently warm solution of Cd(NO3)2 Æ 4H2O (0.930 g,3 mmol) followed by the slow dropwise addition of 5 mlsodium dicyanamide solution (0.178 g, 2 mmol). The mix-ture was then refluxed for 45 min at 85 �C. The resultingyellow coloured solution was filtered off and the filtratewas left undisturbed. After 5 days, yellow block shapedX-ray diffraction quality single crystals separated out fromthe medium. They were filtered, washed with methanol anddried in vacuo over CaCl2. Yield: 87%. Anal. Calc. forC44H46Cd3N12O8: C, 43.74; H, 3.84; N, 13.91. Found: C,43.76; H, 3.80; N, 13.94%.
2.3. Physical measurements
The Fourier Transform infrared spectra of the ligandsand the complexes were recorded on a Perkin–Elmer RX-I FT-IR spectrophotometer with a KBr disc in the range4000–200 cm�1. Elemental analyses (C, H, N) were car-ried out using a Perkin–Elmer 2400 II elemental analyser.The electronic spectra of the ligands were recorded on aPerkin–Elmer Lambda 40 (UV/Vis) spectrometer in meth-anol. Steady state fluorescence measurements were per-
formed using a Spex Fluorolog-II spectrofluorimeter.For spectroscopic measurements, the concentration ofthe solutions were ca. 2 · 10�5 M. Mass spectra wereobtained with a MAT 8200 (electron ionization, EIMS)instrument.
2.4. Single crystal X-ray structure determination
A good diffraction quality air stable single crystal of 1
(dim 0.01 · 0.01 · 0.01 mm) was mounted in a NoniusKappaCCD diffractometer while that of 2 (dim0.12 · 0.12 · 0.03 mm) and 3 (dim 0.40 · 0.50 · 0.50 mm)in Bruker X8 APEXII CCD and Bruker AXS P4 diffrac-tometers, respectively. All the diffractometers wereequipped with graphite monochromated Mo Ka radiation(k = 0.71069 A for 1 and 0.71073 A for 2 and 3). Crystaldata were collected using the software COLLECT [18a] at atemperature of 293 K for 1 and using Bruker SMART [18b]and Bruker XCANS [18c] softwares at a temperature 123 Kfor 2 and 3, respectively. Cell refinements were carriedout using DENZO-SCALEPACK [18a], Bruker SAINT and BrukerXCANS for 1, 2 and 3, respectively. No significant intensityvariation was observed for each of them. A total of 8876reflections (6368 unique reflections, Rint = 0.021), 16582reflections (8470 unique reflections, Rint = 0.087) and27073 reflections (6801 unique reflections, Rint = 0.049)for 1, 2 and 3 were collected applying the boundary condi-tion I > 3r(I) for 1 and I P 2r(I) for 2 and 3. No absorp-tion correction was applied to the data sets for 1 and 3.Multiscan absorption correction was applied to the inten-sity values (Tmax = 0.9533, Tmin = 0.8302) empiricallyusing SADABS for 2 [18d]. Data reduction was performedusing DENZO-SCALEPACK for 1 [18a], while Bruker SAINT
[18e] and Bruker SHELXTL [18f] softwares were used for 2
and 3, respectively. According to the observed systematicextinctions, the structure of 1 was solved by direct methodsusing the SIR97 [18g] program combined with Fourier dif-ference syntheses and refined against F using reflectionsmaintaining the boundary condition I/r(I) > 3 (CRYSTALS
program [18(h)]). For 2 and 3, the final structures weresolved by direct methods using the program SHELXS-97and refined with full-matrix least-squares based on F2 usingSHELXL-97 [18i]. The molecular graphics and crystallo-graphic illustrations were prepared using the CAMERON
[18j], ORTEP-III [18k] and CRYSTALMAKER [18l], Bruker SHEL-
XTL [18f] programs for 1, 2 and 3, respectively. For all non-hydrogen atoms, the anisotropic displacement parametershave been refined successfully. Hydrogen atoms of the aro-matic rings and the imino groups were placed geometricallyand refined as a riding model taken from a difference Fou-rier map and refined with isotropic thermal parameters.Maximum and minimum peaks (e A�3) in the final differ-ence Fourier synthesis were found as 1.96 and �1.99,1.363 and �1.273, 1.12 and �0.84 for 1, 2 and 3, respec-tively. Relevant crystallographic data and structure refine-ment parameters for all the three complexes aresummarised in Table 1.

Table 1Crystallographic parameters for complexes 1, 2 and 3
Complex 1 2 3
Formula C42H40Cd3F6N8O8S2 C41H43Cd3N9O5S2 C44H46Cd3N12O8
Formula weight 1300.17 1143.16 1208.16Crystal system triclinic triclinic monoclinicSpace group P�1(no. 2) P�1(no. 2) C2/c(no. 15)a (A) 10.506(5) 10.148(15) 10.142(10)b (A) 10.832(5) 11.456(11) 24.684(3)c (A) 12.362(5) 21.100(3) 18.492(3)a (�) 68.288(5) 75.374(6) 90b (�) 67.608(5) 88.362(6) 99.924 (10)c (�) 72.489(5) 66.317(6) 90V (A3) 1187.3(9) 2166.1(5) 4560.2(10)Z 1 2 4T (K) 293 123 123kMo Ka (A) 0.71069 0.71073 0.71073Dcalc (g cm�3) 1.818 1.753 1.760l (mm�1) 1.502 1.610 1.453F(000) 642 1136 2408Crystal dimension (mm) 0.01 · 0.01 · 0.01 0.12 · 0.12 · 0.03 0.40 · 0.50 · 0.50h (�) 1.9, 29.5 2.3, 26.0 3.4, 30.4Total, unique data, Rint 8876, 6368, 0.021 16582, 8470, 0.087 27073, 6801, 0.049Observed data 4853 [I > 3r(I)] 5259 [I P 2r (I)] 5728 [I P 2r(I)]Ra, Rw
b 0.0526, 0.0742 0.0812, 0.1556 0.0282, 0.0679Goodness-of-fit 1.125 1.082 1.033Dqmax, min (e A�3) 1.96, �1.99 1.36, �1.27 1.12, �0.84
a R ¼PðjF o � F cjÞ=
PjF oj.
b Rw ¼ fP½wðjF o � F cjÞ2�=
P½wjF oj2�g1=2.
5142 J. Chakraborty et al. / Polyhedron 26 (2007) 5139–5149
3. Results and discussion
3.1. Fourier Transform infrared spectra
The solid state Fourier Transform infrared spectra of allthe complexes, recorded on a FT-IR spectrophotometer inthe range 4000–200 cm�1 are fully consistent with their sin-gle crystal structures. The samples were studied as powdersdispersed in KBr pellets. The spectral region for all thecomplexes is more or less similar due to the similarity incoordination modes of the ligands with the metal centres.Sharp peaks were found at 1640 cm�1 and 1641 cm�1 forH2L1 and H2L2, respectively, which can be correlated tothe stretching frequency of the azomethine (CH@N) groupin the Schiff base ligands. This stretching frequency of theazomethine (CH@N) group observed near about1640 cm�1, is shifted to lower frequency values upon com-plexation with the metal by 20–40 cm�1 [19,20], suggestingcoordination via the azomethine nitrogen in all the com-plexes. m(N–H) at 3435 cm�1 in the IR spectrum ofcomplex 1, suggests the presence of a protonated nitrogenatom in the ligand [21]. Several sharp weak peaks observedfor the complexes in the range 3180–2855 cm�1 are likely tobe due to the aromatic stretches [22]. Sharp bandsappearing at 455 and 375 cm�1 correspond to m(Cd–N),and m(Cd–O), respectively in all the complexes. Them(C–O) mode is present as a very strong band at about1240 cm�1. Complex 1 also shows asymmetric andsymmetric stretching vibrations of the trifluoroacetategroups at 1600 and 1450 cm�1, respectively. The difference
between masym(COO) and msym(COO) (Dm = 150 cm�1),which is smaller than 164 cm�1 observed in ionic trifluoro-acetate, reflects the bidentate bridging coordination mode[21]. In the mid energy range, characteristic absorptionpeaks for terminal SCN� groups were found at2105 cm�1and 2109 cm�1, respectively for 1 and 2, alongwith the distinctive peaks in the low energy region near1215 cm�1 attributable to dCN vibrations [22]. In the caseof complex 3, sharp characteristic peaks are found at2300, 2245 and 2185 cm�1, which may be assigned tomsym + masym(CN), masym(CN) and msym(CN) stretching fre-quencies, respectively. The characteristic peaks invariablyprove the presence of a terminal dicyanamide group incomplex 3 [12a].
3.2. UV/Vis spectra
Electronic spectra of all the complexes have been mea-sured in HPLC grade methanol. In a like manner, thebands in the 465–335 nm range can be assigned to the n–p* transition from the azomethine (CH@N) group [4]. Inthe spectra of the complexes, these n–p* transitions, areshifted to lower frequencies indicating that the imine nitro-gen atom is involved in coordination to the Cd(II) metalion. The bands at higher energies (300–210 nm range) areassociated with benzene p–p* transitions. The spectra ofthe complexes show intense bands in the high-energyregion at 305–375 nm which can be attributed to L!Mcharge transfer bands [23]. Although the precise nature ofthis transition involving the phenoxide group is not fully
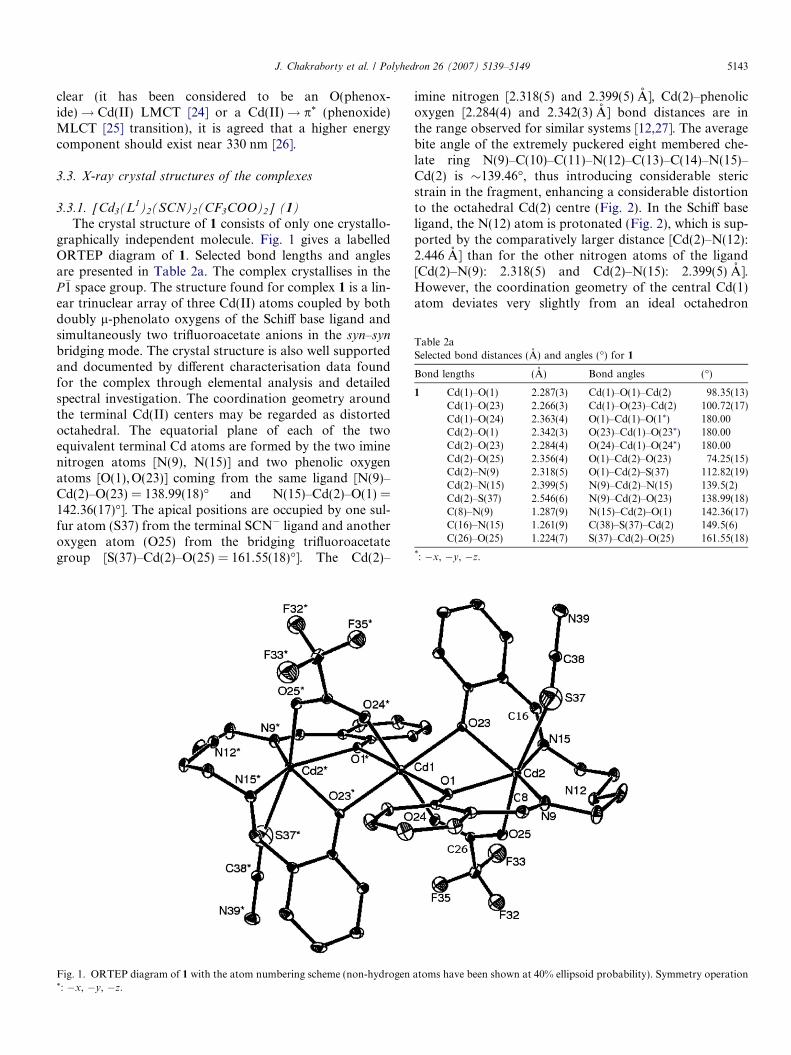
Table 2aSelected bond distances (A) and angles (�) for 1
Bond lengths (A) Bond angles (�)
1 Cd(1)–O(1) 2.287(3) Cd(1)–O(1)–Cd(2) 98.35(13)Cd(1)–O(23) 2.266(3) Cd(1)–O(23)–Cd(2) 100.72(17)Cd(1)–O(24) 2.363(4) O(1)–Cd(1)–O(1*) 180.00Cd(2)–O(1) 2.342(3) O(23)–Cd(1)–O(23*) 180.00Cd(2)–O(23) 2.284(4) O(24)–Cd(1)–O(24*) 180.00Cd(2)–O(25) 2.356(4) O(1)–Cd(2)–O(23) 74.25(15)Cd(2)–N(9) 2.318(5) O(1)–Cd(2)–S(37) 112.82(19)Cd(2)–N(15) 2.399(5) N(9)–Cd(2)–N(15) 139.5(2)Cd(2)–S(37) 2.546(6) N(9)–Cd(2)–O(23) 138.99(18)C(8)–N(9) 1.287(9) N(15)–Cd(2)–O(1) 142.36(17)C(16)–N(15) 1.261(9) C(38)–S(37)–Cd(2) 149.5(6)C(26)–O(25) 1.224(7) S(37)–Cd(2)–O(25) 161.55(18)
*: �x, �y, �z.
J. Chakraborty et al. / Polyhedron 26 (2007) 5139–5149 5143
clear (it has been considered to be an O(phenox-ide)! Cd(II) LMCT [24] or a Cd(II)! p* (phenoxide)MLCT [25] transition), it is agreed that a higher energycomponent should exist near 330 nm [26].
3.3. X-ray crystal structures of the complexes
3.3.1. [Cd3(L1)2(SCN)2(CF3COO)2] (1)
The crystal structure of 1 consists of only one crystallo-graphically independent molecule. Fig. 1 gives a labelledORTEP diagram of 1. Selected bond lengths and anglesare presented in Table 2a. The complex crystallises in theP�1 space group. The structure found for complex 1 is a lin-ear trinuclear array of three Cd(II) atoms coupled by bothdoubly l-phenolato oxygens of the Schiff base ligand andsimultaneously two trifluoroacetate anions in the syn–syn
bridging mode. The crystal structure is also well supportedand documented by different characterisation data foundfor the complex through elemental analysis and detailedspectral investigation. The coordination geometry aroundthe terminal Cd(II) centers may be regarded as distortedoctahedral. The equatorial plane of each of the twoequivalent terminal Cd atoms are formed by the two iminenitrogen atoms [N(9), N(15)] and two phenolic oxygenatoms [O(1),O(23)] coming from the same ligand [N(9)–Cd(2)–O(23) = 138.99(18)� and N(15)–Cd(2)–O(1) =142.36(17)�]. The apical positions are occupied by one sul-fur atom (S37) from the terminal SCN� ligand and anotheroxygen atom (O25) from the bridging trifluoroacetategroup [S(37)–Cd(2)–O(25) = 161.55(18)�]. The Cd(2)–
Fig. 1. ORTEP diagram of 1 with the atom numbering scheme (non-hydrogen*: �x, �y, �z.
imine nitrogen [2.318(5) and 2.399(5) A], Cd(2)–phenolicoxygen [2.284(4) and 2.342(3) A] bond distances are inthe range observed for similar systems [12,27]. The averagebite angle of the extremely puckered eight membered che-late ring N(9)–C(10)–C(11)–N(12)–C(13)–C(14)–N(15)–Cd(2) is �139.46�, thus introducing considerable stericstrain in the fragment, enhancing a considerable distortionto the octahedral Cd(2) centre (Fig. 2). In the Schiff baseligand, the N(12) atom is protonated (Fig. 2), which is sup-ported by the comparatively larger distance [Cd(2)–N(12):2.446 A] than for the other nitrogen atoms of the ligand[Cd(2)–N(9): 2.318(5) and Cd(2)–N(15): 2.399(5) A].However, the coordination geometry of the central Cd(1)atom deviates very slightly from an ideal octahedron
atoms have been shown at 40% ellipsoid probability). Symmetry operation
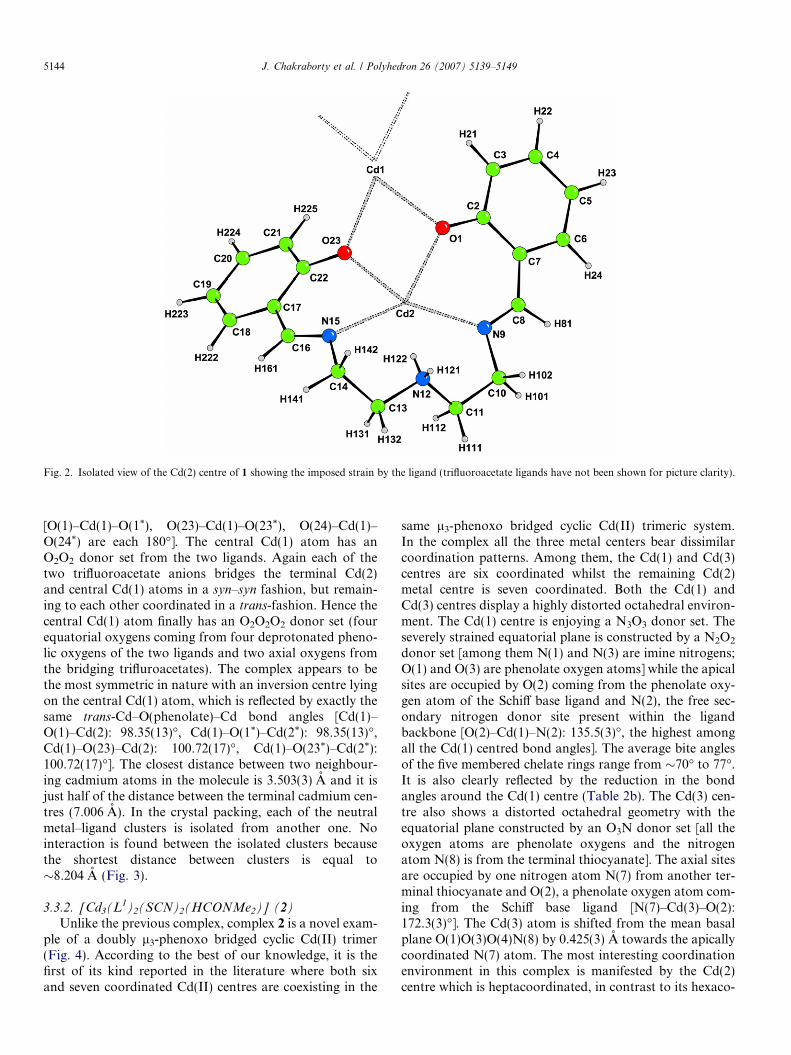
Fig. 2. Isolated view of the Cd(2) centre of 1 showing the imposed strain by the ligand (trifluoroacetate ligands have not been shown for picture clarity).
5144 J. Chakraborty et al. / Polyhedron 26 (2007) 5139–5149
[O(1)–Cd(1)–O(1*), O(23)–Cd(1)–O(23*), O(24)–Cd(1)–O(24*) are each 180�]. The central Cd(1) atom has anO2O2 donor set from the two ligands. Again each of thetwo trifluoroacetate anions bridges the terminal Cd(2)and central Cd(1) atoms in a syn–syn fashion, but remain-ing to each other coordinated in a trans-fashion. Hence thecentral Cd(1) atom finally has an O2O2O2 donor set (fourequatorial oxygens coming from four deprotonated pheno-lic oxygens of the two ligands and two axial oxygens fromthe bridging trifluroacetates). The complex appears to bethe most symmetric in nature with an inversion centre lyingon the central Cd(1) atom, which is reflected by exactly thesame trans-Cd–O(phenolate)–Cd bond angles [Cd(1)–O(1)–Cd(2): 98.35(13)�, Cd(1)–O(1*)–Cd(2*): 98.35(13)�,Cd(1)–O(23)–Cd(2): 100.72(17)�, Cd(1)–O(23*)–Cd(2*):100.72(17)�]. The closest distance between two neighbour-ing cadmium atoms in the molecule is 3.503(3) A and it isjust half of the distance between the terminal cadmium cen-tres (7.006 A). In the crystal packing, each of the neutralmetal–ligand clusters is isolated from another one. Nointeraction is found between the isolated clusters becausethe shortest distance between clusters is equal to�8.204 A (Fig. 3).
3.3.2. [Cd3(L1)2(SCN)2(HCONMe2)] (2)
Unlike the previous complex, complex 2 is a novel exam-ple of a doubly l3-phenoxo bridged cyclic Cd(II) trimer(Fig. 4). According to the best of our knowledge, it is thefirst of its kind reported in the literature where both sixand seven coordinated Cd(II) centres are coexisting in the
same l3-phenoxo bridged cyclic Cd(II) trimeric system.In the complex all the three metal centers bear dissimilarcoordination patterns. Among them, the Cd(1) and Cd(3)centres are six coordinated whilst the remaining Cd(2)metal centre is seven coordinated. Both the Cd(1) andCd(3) centres display a highly distorted octahedral environ-ment. The Cd(1) centre is enjoying a N3O3 donor set. Theseverely strained equatorial plane is constructed by a N2O2
donor set [among them N(1) and N(3) are imine nitrogens;O(1) and O(3) are phenolate oxygen atoms] while the apicalsites are occupied by O(2) coming from the phenolate oxy-gen atom of the Schiff base ligand and N(2), the free sec-ondary nitrogen donor site present within the ligandbackbone [O(2)–Cd(1)–N(2): 135.5(3)�, the highest amongall the Cd(1) centred bond angles]. The average bite anglesof the five membered chelate rings range from �70� to 77�.It is also clearly reflected by the reduction in the bondangles around the Cd(1) centre (Table 2b). The Cd(3) cen-tre also shows a distorted octahedral geometry with theequatorial plane constructed by an O3N donor set [all theoxygen atoms are phenolate oxygens and the nitrogenatom N(8) is from the terminal thiocyanate]. The axial sitesare occupied by one nitrogen atom N(7) from another ter-minal thiocyanate and O(2), a phenolate oxygen atom com-ing from the Schiff base ligand [N(7)–Cd(3)–O(2):172.3(3)�]. The Cd(3) atom is shifted from the mean basalplane O(1)O(3)O(4)N(8) by 0.425(3) A towards the apicallycoordinated N(7) atom. The most interesting coordinationenvironment in this complex is manifested by the Cd(2)centre which is heptacoordinated, in contrast to its hexaco-
Fig. 3. Packing diagram of 1 along the ac plane.
Fig. 4. ORTEP diagram of 2 with the atom numbering scheme (non-hydrogen atoms have been shown at 40% ellipsoid probability).
J. Chakraborty et al. / Polyhedron 26 (2007) 5139–5149 5145
ordinated neighbours Cd(1) and Cd(3). The geometryaround the metal centre can be best described as a severelydistorted monocapped octahedron. The equatorial plane is
constructed by a O3N donor set [among them N(4) is animine nitrogen; O(4) is a l-phenolato oxygen bridging theCd(2) and Cd(3) centres; O(2) is a l3-phenolato oxygen

Table 2bSelected bond distances (A) and angles (�) for 2
Bond lengths (A) Bond angles (�)
2 Cd(1)–O(1) 2.325(8) O(1)–Cd(1)–O(2) 76.9(3)Cd(1)–O(2) 2.348(7) O(2)–Cd(1)–O(3) 68.8(2)Cd(1)–O(3) 2.382(7) O(1)–Cd(1)–N(1) 77.9(3)Cd(1)–N(1) 2.281(10) O(2)–Cd(1)–N(2) 135.5(3)Cd(1)–N(2) 2.585(11) O(3)–Cd(2)–N(4) 65.3(3)Cd(1)–N(3) 2.255(8) O(2)–Cd(2)–O(5) 157.1(3)Cd(2)–O(2) 2.277(6) O(4)–Cd(2)–O(5) 91.4(3)Cd(2)–O(3) 2.398(7) N(4)–Cd(2)–O(3) 65.3(3)Cd(2)–O(4) 2.331(9) N(8)–Cd(3)–O(3) 157.0(3)Cd(2)–O(5) 2.316(6) N(7)–Cd(3)–O(4) 102.8(3)Cd(2)–N(4) 2.594(10) N(7)–Cd(3)–O(2) 172.3(3)Cd(2)–N(5) 2.380(11) Cd(1)–O(1)–Cd(3) 98.5(3)Cd(2)–N(6) 2.343(9) Cd(1)–O(2)–Cd(2) 97.5(3)Cd(3)–O(1) 2.223(8) Cd(2)–O(2)–Cd(3) 88.4(2)Cd(3)–O(2) 2.549(8) Cd(1)–O(2)–Cd(3) 89.3(2)Cd(3)–O(3) 2.393(8) Cd(1)–O(3)–Cd(2) 93.3(2)Cd(3)–O(4) 2.266(8) Cd(2)–O(3)–Cd(3) 89.4(2)Cd(3)–N(7) 2.285(10) Cd(1)–O(3)–Cd(3) 92.4(2)Cd(3)–N(8) 2.211(10) Cd(2)–O(4)–Cd(3) 94.3(3)
5146 J. Chakraborty et al. / Polyhedron 26 (2007) 5139–5149
which simultaneously bridges all the three Cd centres andO(5) comes from the coordinated HCONMe2 molecule].The trans-axial sites are occupied by one nitrogen atomN(5) from the secondary nitrogen donor site of the diaminefragment and O(3), a l3-phenolato oxygen. Another bond,Cd(2)–N(6), is directed through the centre of the trigonalface furnished by N(5), O(4) and O(5). It fulfils the sevencoordinated geometry around the metal centre [theCd(2)–N(4), Cd(2)–O(2), Cd(2)–O(4) and Cd(2)–O(5) bondlengths are 2.594(10), 2.277(6), 2.331(9) and 2.316(3) A,respectively in the equatorial plane; Cd(2)–N(5) andCd(2)–O(3) are 2.380(11) and 2.398(7) A, respectively inthe apical zone and Cd(2)–N(6) is 2.343(9) A]. The appar-
Fig. 5. ORTEP diagram of 3 with the atom numbering scheme (non-hydrogen*: 1 � x, y, 1/2 � z.
ent anomaly in the corresponding bond lengths around theCd(2) centre is probably due to the severe strain imposedby the ligand system to the metal atom [the Cd(2) centreis deviated from the mean plane constructed by N(4),O(2), O(4) and O(5) by 0.6561(2) A towards the apicalN(5) atom]. The most noteworthy structural novelty ofcomplex 2 lies in the interesting variation of the coordina-tion behaviour of the ‘‘salen’’ moieties present in the twofag ends of the completely symmetric ligand H2L1. In thedoubly deprotonated ligand (L2�), one phenoxo oxygen[O(1) or O(4)] is l-bridged between two adjacent metal cen-tres [Cd(1)–Cd(3) or Cd(2)–Cd(3)] while its immediateneighbour on the other end [O(2) or O(3)] bridges threeCd(II) centers simultaneously, which is a very rare phe-nomenon observed in the chemical literature in the caseof such salen-type Schiff base complexes. Thus there areactually two different kinds of phenolato oxygen atomspresent in the ligand: one of them doubly coordinated tothe adjacent metal centres and the other kind shows a l3
bridging mode which helps to afford a cyclic trimer ofCd(II). The average M–O(phenolate)–M bond angles arearound 93�. All other relevant bond lengths and bondangles are given in Table 2b. The nearest Cd–Cd distancesare: Cd(1)–Cd(2), 3.477(3); Cd(2)–Cd(3), 3.370(5); Cd(1)–Cd(3), 3.445(2) A, thereby showing the least chance ofinteractions among the adjacent metal centres. Again fromthe view of crystal packing, any sort of non-covalent orweak interaction is absent amongst the neutral metal-Schiffbase clusters, with them being isolated at large distances.
3.3.3. [Cd3(L2)2{N(CN)2}2] (3)
A perspective view of the trimeric asymmetric unit ofcomplex 3 has been represented in Fig. 5 as an ORTEPplot and a packing diagram is shown in Fig. 6. Coordi-
atoms have been shown at 40% ellipsoid probability). Symmetry operation

Fig. 6. Packing diagram of 3.
J. Chakraborty et al. / Polyhedron 26 (2007) 5139–5149 5147
nates of the non-hydrogen atoms, and selected bond dis-tances and bond angles are given in Table 2c. The struc-ture is a linear trimeric one, bearing two equivalentterminal Cd(II) centres. Complex 3 crystallises in theC2/c (monoclinic) space group. Adjacent Cd(II) atomsin the linear trinuclear array are coupled to each otherdoubly with l-phenoxo bridges. The crystal structure isalso well supported and documented by different charac-
Table 2cSelected bond distances (A) and angles (�) for 3
Bond lengths (A) Bond angles (�)
3 Cd(1)–O(2) 2.3702(17) O(9)–Cd(1)–O(9*) 145.12(5)Cd(1)–O(2*) 2.3702(17) O(2)–Cd(1)–O(25) 142.84(5)Cd(1)–O(9) 2.2601(15) O(2*)–Cd(1)–O(25*) 142.84(5)Cd(1)–O(9*) 2.2601(15) O(25)–Cd(1)–O(25*) 95.97(6)Cd(1)–O(25) 2.2134(15) N(11)–Cd(2)–O(25) 149.63(6)Cd(1)–O(25*) 2.2134(15) N(14)–Cd(2)–O(9) 147.90(6)Cd(2)–O(9) 2.3256(15) N(17)–Cd(2)–N(31) 115.55(8)Cd(2)–O(25) 2.3077(15) O(9)–Cd(2)–O(25) 71.99(5)Cd(2)–N(11) 2.2932(18) N(11)–Cd(2)–N(14) 71.96(6)Cd(2)–N(14) 2.560(2) O(9)–Cd(2)–N(11) 77.80(6)Cd(2)–N(17) 2.290(2) Cd(1)–O(9)–Cd(2) 105.36(6)Cd(2)–N(31) 2.246(2) Cd(1)–O(25)–Cd(2) 107.52(6)
*: 1 � x, y, 1/2 � z.
terisation data found for the complex through elementalanalysis and detailed spectroscopic investigation. Thecoordination geometry around the central Cd(1) can bebest described as a distorted octahedron. The centralCd(1) has an O2O2O2 donor set from the two ligands,among the four equatorial oxygens, two are from twodeprotonated phenolic oxygens [O(25) and O(25*)] whilethe other two are from two methoxy oxygens [O(2) andO(2*)] from the two different ligands. The two axial sitesare occupied by two phenolic oxygens [O(9) and O(9*)].The O(9)–Cd(1)–O(9*) angle of 145.12(5)� is the highestamong all the trans-bond angles. The other trans-equato-rial bond angles, O(2)–Cd(1)–O(25) and O(2*)–Cd(1)–O(25*), are same [142.84(5)�]. The coordination geometryaround each symmetrically equivalent terminal Cd(II) cen-tre may be regarded as a distorted octahedron. The termi-nal Cd(2) centre enjoys a N4O2 donor set. The severelystrained equatorial plane is constructed by a N3O donorset [among them N(17) is a imine nitrogen, N(14) is thefree secondary nitrogen donor site and O(9) is a phenolateoxygen atom of the same Schiff base ligand, while N(31)comes from the terminal dicyanamide ligand]. The apicalsites are occupied by O(25) coming from the phenolateoxygen atom and N(11), the imine nitrogen donor site

5148 J. Chakraborty et al. / Polyhedron 26 (2007) 5139–5149
present in the same Schiff base ligand. The trans-axialbond angle N(11)–Cd(2)–O(25) is 149.63(6)�. The averagebite angles of the two puckered five and six memberedchelate rings are �71.31� and 77.25�, respectively, whichinitiate a considerable distortion to the octahedral Cd(2)centre (Fig. 5). The comparatively larger distance[Cd(2)–N(14): 2.560(2) A] than for the other nitrogenatoms of the Schiff base ligand [Cd(2)–N(11): 2.2932(18)and Cd(2)–N(17): 2.290(2) A] may be attributed to theconsiderable steric strain present in the fragment. Themost interesting feature lies in the variation of the coordi-nation pattern of the o-vanilin moieties at the two ends ofthe Schiff base ligand (H2L2). In the doubly deprotonatedligand L2�, one –OMe group is coordinated to the centralCd(1) centre while its counterpart in the other end of thesame ligand remains free. The same behaviour is alsoobserved for the other ligand. The closest distancebetween two terminal Cd atoms in the molecule is7.050 A and it is approximately double that between theneighbouring Cd centres (3.647 A). In the packing dia-gram we did not find any trace of non-covalent or weakinteractions among the neutral metal-Schiff base clusters(Fig. 6).
3.4. Luminescence properties
Steady-state fluorescence studies have been performedto establish the metal Schiff base coordination for all thethree complexes. Fluorescence spectra for both the Schiffbase ligands and their respective Cd(II) complexes havebeen measured in methanol (Fig. 7). All of them show char-acteristic broad fluorescence emissions indicating a ligand-to-metal charge transfer (LMCT) nature of the bands. Sig-nificant differences in the positions of the emission maxi-mum of the complexes from that of the ligands may beconsidered as an evidence of metal–ligand complexation.All the complexes show intense bands at ca. 360 nm, whichmay be assigned to intraligand 1(p–p*) transitions. Theemission energy of all the complexes in methanol solutions
Fig. 7. Luminescence spectra for the ligands and the complexes H2L1:fluorescence spectra for the ligand H2L1, H2L2: fluorescence spectra forthe ligand H2L2, 1f, 2f, 3f: fluorescence spectra of 1, 2, 3 in MeOH solutionat 298 K, respectively. 1p, 2p, 3p: Phosphorescence spectra of 1, 2, 3 inMeOH glassy solution at 77 K, respectively.
at 298 K is almost same and is assignable to intraligandfluorescence [12,28–30]. The lifetimes of the complexesare in range 2.50–2.60 ns. In glassy solutions (methanolat 77 K) both fluorescence and 3(p–p*) phosphorescenceare observed at 525 nm.
4. Conclusion
The present study shows that the reaction of long chainmultidentate salen-type Schiff base ligands with variousmetal salts can afford a variety of fascinating metal-clusterframeworks. Both the ligands are flexible enough due totheir long chain lengths and hence they are able to adjusttheir conformations in order to fit with the geometries ofthe metal ion thereby reducing the steric strain within theligand backbone. We have been able to synthesise threenovel trinuclear coordination complexes with two analo-gous multidentate Schiff base ligands. The novelty of thework lies in the interesting variation of the coordinationpattern of the different donor sites due to the flexibility ofthe long chain ligand backbone.
Acknowledgements
Joy Chakraborty is especially grateful to the UniversityGrants Commission, New Delhi, Government of India forthe award of a Senior Research Fellowship to him. J.C. isalso thankful to Dr. Arnab Mukherjee, Department ofChemical Engineering, University Institute of ChemicalTechnology, Mumbai for the technical support to the lumi-nescence studies of the complexes. Santarupa Thakurta isgrateful to the CSIR, Govt. of India for a Junior ResearchFellowship.
Appendix A. Supplementary material
CCDC 636959, 636961, 636960 contain the supplemen-tary crystallographic data for 1, 2 and 3. These data canbe obtained free of charge via http://www.ccdc.ac.uk/con-ts/retrieving.html, or from the Cambridge CrystallographicData Centre, 12 Union Road, Cambridge CB2 1EZ, UK;fax: (+44) 1223-336-033; or e-mail: [email protected].
References
[1] Z.-F. Chen, Y.-Z. Tang, H. Liang, H.-K. Fun, K.-B. Yu, J. Coord.Chem. 59 (2006) 207.
[2] Q. Shi, L. Xu, J. Ji, Y. Li, R. Wang, Z. Zhou, R. Cao, M. Hong,A.S.C. Chan, Inorg. Chem. Commun. 7 (2004) 1254.
[3] (a) Z.-L. You, H.-L. Zhu, W.-S. Liu, Z. Anorg. Allg. Chem. 630(2004) 1617;(b) Z.-L. You, H.-L. Zhu, Z. Anorg. Allg. Chem. 630 (2004) 2754.
[4] A. Golcu, M. Tumer, H. Demirelli, R.A. Wheatley, Inorg. Chim.Acta 358 (2005) 1785.
[5] S. Brooker, Coord. Chem. Rev. 222 (2001) 33.[6] V. Amendola, L. Fabbrizzi, C. Mangano, P. Pallavicini, A. Poggi, A.
Taglietti, Coord. Chem. Rev. 219 (2001) 821.[7] P.A. Vigato, S. Tamburini, Coord. Chem. Rev. 248 (2004) 1717.

J. Chakraborty et al. / Polyhedron 26 (2007) 5139–5149 5149
[8] S. Sen, P. Talukder, S.K. Dey, S. Mitra, G. Rosair, D.L. Hughes,G.P.A. Yap, G. Pilet, V. Gramlich, T. Matsushita, Dalton Trans.(2006) 1758.
[9] (a) S. Leininger, B. Olenyuk, P.J. Stang, Chem. Rev. 100 (2000) 853;(b) W. Meier, Chem. Soc. Rev. 29 (2000) 295.
[10] (a) T. Steiner, Angew. Chem., Int. Ed. Engl. 41 (2002) 48;(b) G.R. Desiraju, Acc. Chem. Res. 35 (2002) 565;(c) N.V. Belkova, E.S. Subina, L.M. Epstein, Acc. Chem. Res. 38(2005) 624.
[11] (a) C. Janiak, J. Chem. Soc., Dalton Trans. (2000) 3885;(b) M.J. Zaworotko, Chem. Commun. (2002) 199;(c) M.K. Milcic, V.B. Medakovic, D.N. Sredojevic, N.O. Juranic,S.D. Zaric, Inorg. Chem. 45 (2006) 4755.
[12] (a) J. Chakraborty, B. Samanta, G. Pilet, S. Mitra, Inorg. Chem.Commun. 10 (2007) 40;(b) A. Majumder, G. Rosair, A. Mallick, N. Chattopadhyay, S.Mitra, Polyhedron 25 (2006) 1753.
[13] (a) P. Comba, T.W. Hambley, Molecular Modelling of Inorganic andCoordination Compounds, second ed., VCH, Weinheim, 2000;(b) J. Wang, X. Qian, Chem. Commun. (2006) 109.
[14] (a) V. Alexander, Chem. Rev. 95 (1995) 273;(b) T.K. Karmakar, S.K. Chandra, J. Ribas, G. Mostafa, T.-H. Lu,B.K. Ghosh, Chem. Commun. (2002) 2364.
[15] (a) A.M. Golub, H. Kohler, V.V. Skopenko (Eds.), Chemistry ofPseudohalides, Elsevier, Amsterdam, 1986;(b) J. Ribas, A. Escuer, M. Monfort, R. Vicente, R. Cortes, L.Lezama, T. Rojo, Coord. Chem. Rev. 193 (1999) 1027.
[16] (a) H. Zhang, X. Wang, K. Zhang, B.K. Teo, Coord. Chem. Rev. 183(1999) 157;(b) S.H. Rahaman, R. Ghosh, G. Mostafa, B.K. Ghosh, Inorg.Chem. Commun. 8 (2005) 700, and refs. therein;(c) V. Haber, I. Cisaova, J. Fabry, Polyhedron 22 (2003) 3451.
[17] (a) C.-M. Liu, R.-G. Xiong, X.-Z. You, Y.-J. Liu, K.-K. Cheung,Polyhedron 15 (1996) 4565;(b) C.-M. Liu, R.-G. Xiong, X.-Z. You, H.-K. Fun, K. Sivakumar,Polyhedron 16 (1997) 119;(c) M.S. Ray, R. Bhattacharya, S. Chaudhuri, L. Righi, G. Bocelli, G.Mukhopadhyay, A. Ghosh, Polyhedron 22 (2003) 617.
[18] (a) Nonius COLLECT, DENZO, SCALEPACK, SORTAV: KAPPACCD ProgramB.V., Delft, The Netherlands 1999;(b) SMART: Data Collection Program for the CCD Area-DetectorSystem;(c) XCANS: Data Collection Program for the Bruker P4 system;(d) G.M. Sheldrick, SADABS, Programs for Area Detector AdsorptionCorrection, Institute for Inorganic Chemistry, University of Gottin-gen, Germany, 1996;
(e) SAINT: Data Reduction, Frame Integration Program for the CCDArea-Detector System, Bruker Analytical X-ray Systems Ltd., Mad-ison, Wisconsin, USA, 2005;(f) G.M. Sheldrick, SHELXTL version 5.1: Program for the Solution andRefinement of Crystal Structures, Bruker AXS P4, Inc., Madison,WI, USA, 1999;(g) G. Cascarano, A. Altomare, C. Giacovazzo, A. Guagliardi, A.G.Moliterni, D. Siliqi, M. Burla, G. Polidori, M. Camalli, ActaCrystallogr. A52 (1996) C-79;(h) D.J. Watkin, C.K. Prout, J.R. Carruthers, P.W. Betteridge,CRYSTALS Issue 11, Chemical Crystallography Laboratory, Oxford,UK, 1999;(i) G.M. Sheldrick, SHELX-97 (SHELXS and SHELXL) Program forSolution and Refinement of Crystal structures, University of Gottin-gen, Germany, 1997;(j) D.J. Watkin, C.K. Prout, L.J. Pearce, CAMERON, ChemicalCrystallography Laboratory, OXFORD, UK, 1996;(k) L.J. Farrugia, ORTEP-3 for Windows, J. Appl. Crystallogr. 30(1997) 565;(l) D.C. Palmer, CRYSTAL MAKER 7.04, Crystal Maker Software,Yarnton, UK, 2005.
[19] S.H. Rahaman, R. Ghosh, T.-H. Lu, B.K. Ghosh, Polyhedron 24(2005) 1525.
[20] M. Dolaz, M. Tumer, M. Dıgrak, Trans. Met. Chem. 29 (2004)528.
[21] M.T.H. Tarafder, A. Kasbollah, K.A. Crouse, A.M. Ali, B.M.Yamin, H.-K. Fun, Polyhedron 20 (2001) 236.
[22] (a) K. Nakamoto, Infrared and Raman Spectra of Inorganic andCoordination Compounds, fifth ed., John Wiley, New York, 1997,Part A and B;(b) R.T. Conley, Infrared Spectroscopy, Allyn & Bacon, Boston,1966.
[23] M. Tumer, Synth. React. Inorg. Met. Org. Chem. 30 (2000) 1139.[24] R. Atkins, G. Brewer, E. Kokot, G.M. Mockler, J.E. Sinn, Inorg.
Chem. 24 (1985) 127.[25] A.G. Suillerot, J.P. Albertini, A. Collet, L. Faury, J.M. Pastor, L.J.
Tosi, J. Chem. Soc., Dalton Trans. 44 (1981) 2544.[26] A.R. Amundsen, J. Whelan, B. Bosnich, J. Am. Chem. Soc. 99 (1977)
6730.[27] A.G. Blackman, Polyhedron 24 (2005) 1.[28] L.-Z. Cai, W.-T. Chen, M.-S. Wang, G.-C. Guo, J.-S. Huang, Inorg.
Chem. Commun. 7 (2004) 611.[29] J.-X. Chen, S.-X. Liu, E.-Q. Gao, Polyhedron 23 (2004) 1877.[30] D. Bose, J. Banerjee, Sk. H. Rahaman, G. Mostafa, H.-K. Fun, R.D.
Bailey Walsh, M.J. Zaworotko, B.K. Ghosh, Polyhedron 23 (2004)2045.
Copyright © 2022 FDOKUMEN




















