
Synthesis and computation of diastereomeric phenanthrolineequinine ligands and their application in...
9
Synthesis and computation of diastereomeric phenanthrolineequinine ligands and their application in asymmetric Henry reaction Lili Zhang a , Hao Wu b , Zhongyue Yang b , Xiufang Xu b , Haitao Zhao c , Yaodong Huang a, * , Yongmei Wang b, * a Key Laboratory of Systems Bioengineering, Ministry of Education, School of Chemical Engineering and Technology, Tianjin University, Tianjin 300072, China b State Key Laboratory of Elemento-Organic Chemistry, Department of Chemistry, Nankai University, Tianjin 300071, China c Department of Chemistry, Tianjin University, Tianjin 300072, China article info Article history: Received 18 May 2013 Received in revised form 3 October 2013 Accepted 5 October 2013 Available online 21 October 2013 Keywords: Chiral phenanthroline Quinine Henry reaction Copper Mechanistic study abstract A class of chiral ligands has been developed by combining phenanthroline with quinine in a one-step method that does not require resolution. The synthesized three ligands were then coordinated with Cu (II) and the performance of the resultant chiral catalysts in the asymmetric Henry reaction was eval- uated. Moderate to good yields (up to 86%) with high enantioselectivities (up to 99% ee) were observed in the reactions catalyzed by one of the three catalysts. Theoretical calculations were performed to analyze the catalytic activities of the different Cu (II) -ligand catalysts. Three different ligands were investigated and one ligand was found to adopt an unexpected five-coordinated mode; the second coordinated with two nitrogen atoms of phenanthroline to give a complex, which activated both substrates of Henry re- action; the third was unable to form a complex with Cu (II) . Ó 2013 Elsevier Ltd. All rights reserved. 1. Introduction The asymmetric Henry reaction is an atom economical and powerful method for stereoselective carbonecarbon bond forma- tion. The resulting chiral adducts, b-nitro alcohols, can be conve- niently converted into b-amino alcohols, a-hydroxy carboxylic acids, aziridines, and other complex target molecules, which are then used as highly versatile building blocks for the synthesis of biological natural products and pharmaceutical agents. 1,2 Since the pioneering work of Shibasaki et al. in 1992, 3a remarkable efforts have been devoted toward the design of novel chiral ligands for asymmetric metal catalyzed Henry reactions. 3e8 Most of the chiral ligands have focused on 1-(2-hydroxynaphthalen-1-yl) naph- thalen-2-ols (BINOLs), 3 bisoxazolines (BOXs), 4 amino alcohols, 5 bis- amines, 6 N,N 0 -dioxides, 7 amino/imino-pyridines, 8 and Schiff bases (Salen-types). 9 Ligands based on 1,10-phenanthrolines can chelate a wide va- riety of different metals. Over the last two decades, considerable attention has been paid to CeH bond activation catalyzed by phe- nanthrolines with metals. 10 However, only a few examples of this basic scaffold with chiral groups have been reported for asym- metric catalysis. These include cyclopropanation, 11 allylic alkyl- ation, 12 hydrosilylation, 11b,13 transfer hydrogenation, 14 and allylic oxidation. 15 To our surprise, there has not been a single example of the asymmetric Henry reaction catalyzed by this class of ligands. Moreover, most of the chiral phenanthroline ligands reported in the literature were synthesized via long routes that required multiple steps and the chiral elements generally required a final resolution step by preparative chiral HPLC. With this in mind, the design of simple and easily accessible chiral phenanthroline ligands for asymmetric catalysis is very important. Cinchona alkaloids are regarded as one of the most important chirality inducers and they have found applications in chiral ligands and asymmetric organocatalysis. 16 These alkaloids have some po- tential for asymmetric catalysis owning to their chiral recognition capabilities, their structural diversity and their conformational characteristics. Considering the features of quinine and phenanthroline, it is interesting to see what will happen after the combination of the two compounds. Thus some phenanthrolineecinchona hybrid * Corresponding authors. E-mail addresses: [email protected] (Y. Huang), [email protected] (Y. Wang). Contents lists available at ScienceDirect Tetrahedron journal homepage: www.elsevier.com/locate/tet 0040-4020/$ e see front matter Ó 2013 Elsevier Ltd. All rights reserved. http://dx.doi.org/10.1016/j.tet.2013.10.010 Tetrahedron 69 (2013) 10644e10652
Transcript of Synthesis and computation of diastereomeric phenanthrolineequinine ligands and their application in...

lable at ScienceDirect
Tetrahedron 69 (2013) 10644e10652
Contents lists avai
Tetrahedron
journal homepage: www.elsevier .com/locate/ tet
Synthesis and computation of diastereomericphenanthrolineequinine ligands and their application inasymmetric Henry reaction
Lili Zhang a, Hao Wub, Zhongyue Yang b, Xiufang Xu b, Haitao Zhao c, Yaodong Huang a,*,Yongmei Wang b,*
aKey Laboratory of Systems Bioengineering, Ministry of Education, School of Chemical Engineering and Technology, Tianjin University, Tianjin 300072,Chinab State Key Laboratory of Elemento-Organic Chemistry, Department of Chemistry, Nankai University, Tianjin 300071, ChinacDepartment of Chemistry, Tianjin University, Tianjin 300072, China
a r t i c l e i n f o
Article history:Received 18 May 2013Received in revised form 3 October 2013Accepted 5 October 2013Available online 21 October 2013
Keywords:Chiral phenanthrolineQuinineHenry reactionCopperMechanistic study
* Corresponding authors. E-mail addresses:(Y. Huang), [email protected] (Y. Wang).
0040-4020/$ e see front matter � 2013 Elsevier Ltd.http://dx.doi.org/10.1016/j.tet.2013.10.010
a b s t r a c t
A class of chiral ligands has been developed by combining phenanthroline with quinine in a one-stepmethod that does not require resolution. The synthesized three ligands were then coordinated withCu(II) and the performance of the resultant chiral catalysts in the asymmetric Henry reaction was eval-uated. Moderate to good yields (up to 86%) with high enantioselectivities (up to 99% ee) were observed inthe reactions catalyzed by one of the three catalysts. Theoretical calculations were performed to analyzethe catalytic activities of the different Cu(II)-ligand catalysts. Three different ligands were investigatedand one ligand was found to adopt an unexpected five-coordinated mode; the second coordinated withtwo nitrogen atoms of phenanthroline to give a complex, which activated both substrates of Henry re-action; the third was unable to form a complex with Cu(II).
� 2013 Elsevier Ltd. All rights reserved.
1. Introduction
The asymmetric Henry reaction is an atom economical andpowerful method for stereoselective carbonecarbon bond forma-tion. The resulting chiral adducts, b-nitro alcohols, can be conve-niently converted into b-amino alcohols, a-hydroxy carboxylicacids, aziridines, and other complex target molecules, which arethen used as highly versatile building blocks for the synthesis ofbiological natural products and pharmaceutical agents.1,2 Since thepioneering work of Shibasaki et al. in 1992,3a remarkable effortshave been devoted toward the design of novel chiral ligands forasymmetric metal catalyzed Henry reactions.3e8 Most of the chiralligands have focused on 1-(2-hydroxynaphthalen-1-yl) naph-thalen-2-ols (BINOLs),3 bisoxazolines (BOXs),4 amino alcohols,5 bis-amines,6 N,N0-dioxides,7 amino/imino-pyridines,8 and Schiff bases(Salen-types).9
Ligands based on 1,10-phenanthrolines can chelate a wide va-riety of different metals. Over the last two decades, considerable
All rights reserved.
attention has been paid to CeH bond activation catalyzed by phe-nanthrolines with metals.10 However, only a few examples of thisbasic scaffold with chiral groups have been reported for asym-metric catalysis. These include cyclopropanation,11 allylic alkyl-ation,12 hydrosilylation,11b,13 transfer hydrogenation,14 and allylicoxidation.15 To our surprise, there has not been a single example ofthe asymmetric Henry reaction catalyzed by this class of ligands.Moreover, most of the chiral phenanthroline ligands reported in theliterature were synthesized via long routes that required multiplesteps and the chiral elements generally required a final resolutionstep by preparative chiral HPLC. With this in mind, the design ofsimple and easily accessible chiral phenanthroline ligands forasymmetric catalysis is very important.
Cinchona alkaloids are regarded as one of the most importantchirality inducers and they have found applications in chiral ligandsand asymmetric organocatalysis.16 These alkaloids have some po-tential for asymmetric catalysis owning to their chiral recognitioncapabilities, their structural diversity and their conformationalcharacteristics.
Considering the features of quinine and phenanthroline, it isinteresting to see what will happen after the combination of thetwo compounds. Thus some phenanthrolineecinchona hybrid

Fig. 1. X-ray crystal structure of the complex (R)-L1eCuCl2.
L. Zhang et al. / Tetrahedron 69 (2013) 10644e10652 10645
chiral catalysts for the asymmetric Henry reaction were designed,synthesized, and tested. Herein, a one-stepmethod for a novel classof chiral phenanthrolineequinine ligands was developed and ap-plied to asymmetric copper(II)-catalyzed Henry reaction, the cat-alytic activities of the catalysts in the reaction were alsoinvestigated through theoretical calculations in order to explain theunusually high enantioselectivity and great difference of theorganocatalysts.
2. Results and discussion
The chiral phenanthrolineecinchona ligands were constructedby combining 2-bromo-1,10-phenanthroline with cinchona alka-loids with a chiral element being provided by the cinchona alka-loids. Several advantages are associated with this strategy: 1) aneasily accessed catalytic system with only one-step; 2) both of thestarting materials are commercially available; 3) a readily modifiedstructure, which is useful for diverse catalytic applications; and 4)selective production of either enantiomeric product without theneed for a resolution process. Initially, chiral phenanthroline li-gands were synthesized by nucleophilic substitution between 2-bromo-1,10-phenanthroline 1 and quinine in DMSO. The mixturewas treatedwith NaH at 80e90 �C for 5 h to give the diastereomericproducts L1 and L2, which were easily separated by flash chro-matography in moderate yields (Scheme 1). Treatment of 2,9-dibromo-1,10-phenanthroline 2 with quinine in the same pro-cedure led to the only one product L3, which is probably due to theeffect of the bromine atom (Scheme 1). The absolute configurationsof L1 and L3 were unambiguously determined by X-ray crystal-lography to be 9-(R) (Figs. 1 and 2). The configuration of L2 wasdeduced to be 9-(S) according to the reported literature.17
Scheme 1. Synthetic route to chiral phenanthroline ligands derived from quinine.
The enantiopure phenanthroline ligands L1eL3 were thenscreened to evaluate their abilities to promote the copper(II)-catalyzed enantioselective Henry reaction between benzaldehyde(3a) and nitromethane (4a) in the presence of CuCl2$2H2O (5mol %)in EtOH at room temperature. Gratifyingly, (S)-L2 promoted thereaction smoothly, giving the desired product (5a) with moderateyield and good enantioselectivity (Table 1, entry 2, 68% yield, 75%ee). In sharp contrast, only traces of the product could be detectedwhen (R)-L1 and (R)-L3 were used, even when a higher tempera-ture or a longer reaction time was used (Table 1, entries 1 and 3).
Different copper salts were then tested to evaluate the effect ofthe counterions on the reaction. No improvements in enantiose-lectivity were obtained (Table 1, entries 4e6). Then smaller catalyst
loading was tested and this resulted in lower yields althoughcomparable enantioselectivities were achieved (Table 1, entries 7and 8). Further investigations showed that the reaction is highlysensitive to the nature of the solvent (Table 1, entries 9e15). Amixed solvent of THF and i-PrOH (1:1) was found to be the bestsolvent, affording a moderate yield and high enantioselectivity(Table 1, entry 17, 57% yield, 85% ee).
Because the Henry reaction is thought to generate a nitronateintermediate, a base is usually added to the reaction system to in-crease the reactivity of the catalyst.6d,p Therefore a series of baseswere tested in the reaction and the results are summarized inTable 2. As expected, the activity of the catalyst and the yield of the
product (5a) were significantly improved (Table 2, entries 1e5). N-Meemorpholine was the optimal additive in terms of yield andenantioselectivity (Table 2, entry 5, 83% yield, 80% ee). Lowering thetemperature of the reaction led to an impressive improvement inthe enantioselectivity, albeit in a lower yield (entry 6, 70% yield,90% ee). No significant improvements in yields and ee values wereachieved by changing the amount of the base, although comparableenantioselectivities were obtained (entries 7e9).
Having optimized the reaction conditions, the generality of thecatalytic system for aldehydes bearing different substituents wasthen investigated (Table 3). A wide range of aldehydes bearingelectron-withdrawing and electron-donating groups all reactedunder the optimized reaction conditions with moderate to good

Fig. 2. X-ray crystal structure of (R)-L3.
Table 1Optimization of reaction conditionsa
Entry L Metal salts Solvents Time (h) Yieldb (%) eec (%)
1 L1 CuCl2$2H2O EtOH 120 Trace ndd
2 L2 CuCl2$2H2O EtOH 72 68 753 L3 CuCl2$2H2O EtOH 120 Trace ndd
4 L2 Cu(OTf)2 EtOH 72 43 655 L2 Cu(OAc)2$H2O EtOH 72 77 576 L2 Cu(NO3)2$3H2O EtOH 72 46 667e L2 CuCl2$2H2O EtOH 72 58 768f L2 CuCl2$2H2O EtOH 72 36 739 L2 CuCl2$2H2O THF 72 42 8510 L2 CuCl2$2H2O CH2Cl2 68 33 8111 L2 CuCl2$2H2O CH3CN 92 38 7912 L2 CuCl2$2H2O CHCl3 68 30 7613 L2 CuCl2$2H2O Toluene 120 52 7814 L2 CuCl2$2H2O MeOH 44 60 7715 L2 CuCl2$2H2O i-PrOH 40 73 7516 L2 CuCl2$2H2O THF/EtOH¼1:1 21 56 8117 L2 CuCl2$2H2O THF/iPrOH¼1:1 21 57 85
a Unless otherwise stated, all reactions were carried out with 3a (0.25 mmol) and4a (2.5 mmol) in the solvent (1 mL) in the presence of Cu(II) salts (5 mol %) and Li-gands (6 mol %) at room temperature.
b Isolated yield.c Determined by HPLC analysis (Chiralcel OD-H column).d Not determined.e 3 mol % CuCl2$2H2O and 3.3 mol % ligand were used.f 1 mol % CuCl2$2H2O and 1.1 mol % ligand were used.
Table 2Effect of bases on the Henry reactiona
Entry Bases Loading (%)b Time (h) Yieldc (%) eed (%)
1 Et3N 5 20 78 712 DIPEA 5 20 75 763 DABCO 5 20 70 774 Morpholine 5 20 62 795 N-Meemorpholine 5 20 83 806e N-Meemorpholine 5 38 70 907e N-Meemorpholine 1 34 44 898e N-Meemorpholine 3 34 52 869e N-Meemorpholine 10 34 75 87
a Unless otherwise stated, all reactions were carried out with 3a (0.25 mmol) and4a (2.5 mmol) in THF/iPrOH (1:1, 1 mL) in the presence of CuCl2$2H2O (5 mol %) andL2 (6 mol %) at room temperature.
b Equivalent based on benzaldehyde 3a.c Isolated yield.d Determined by HPLC analysis (Chiralcel OD-H column).e Reaction was carried out at 0 �C.
Table 3Enantioselective Henry reaction of aldehydes with nitroalkanesa
∗ ∗
Entry R1 R2 Time (h) Yieldb (%) eec (%)
1 Ph (5a) H 38 68 902 3-ClC6H4 (5b) H 36 50 813 2,4-ClC6H4 (5c) H 36 68 774 4-FC6H4 (5d) H 38 47 765 4-NO2C6H4 (5e) H 14 83 716 2-MeOC6H4 (5f) H 15 79 667 3-MeOC6H4 (5g) H 38 73 888 4-MeOC6H4 (5h) H 36 60 929 2,4-MeOC6H4 (5i) H 17 62 9010 2-MeC6H4 (5j) H 36 65 8411 4-MeC6H4 (5k) H 36 58 8412 4-HOC6H4 (5l) H 41 67 8913 2-Thiophenyl (5m) H 38 69 9014 2-Furyl (5n) H 38 65 8115 3-Furyl (5o) H 41 50 8616 PhCH]CH (5p) H 17 86 8617 n-Pr (5q) H 41 52 8518 i-Pr (5r) H 41 49 8219 Cyclohexyl (5s) H 21 55 8620d Ph (5t) Et 24 62 79/8921d 4-MeO (5u) Et 21 68 99/8822d 2-Thiophenyl (5y) Et 21 52 59/89
a Reactions were performed on a 0.25 mmol scale: CuCl2$2H2O (5 mol %), L2(6 mol %) were stirred in THF/i-PrOH (1:1, 1 mL) at room temperature for 1 h, thenthe aldehyde 3 (1 equiv), nitropropane 4 (10 equiv) and N-Meemorpholine(5 mol %) were added and the reaction mixture was stirred for intend time at 0 �C.
b Isolated yield.c Determined by HPLC analysis.d 5t, dr¼35/65; 5u, dr¼52:48; 5y, dr¼46:51.
L. Zhang et al. / Tetrahedron 69 (2013) 10644e1065210646
yields and moderate to high enantioselectivities (Table 3, entries1e12, 47e83% yields, 66e92% ee). Generally, the benzaldehydeswith electron-donating groups gave higher enantioselectivitiesthan those with electron-withdrawing groups (Table 3, entries 2e5vs 7e12). A substitution at the ortho-position of R1 had a detri-mental effect on the ee value (Table 3, entries 6e8, 14, 15). Heter-oaromatic aldehydes (Table 3, entries 13e15) and cinnamaldehyde(Table 3, entry 16) were also suitable for this catalytic system,
affording the desired products in 50e86% yields and 86e90% ee.Notably, there appears to be a significant tolerance for aliphaticaldehydes, including those with alicyclic and cyclic substitutions(Table 3, entries 17e19). Finally, nitropropane was investigated andthe corresponding adducts were obtained in high to excellentenantioselectivities, albeit with moderate diastereoselectivities(Table 3, entries 20e22).
To explain the significantly different catalytic activities of thethree ligands in Henry reaction, theoretical calculation is an

L. Zhang et al. / Tetrahedron 69 (2013) 10644e10652 10647
essential and efficient method. For this purpose, great endeavorwas devoted to cultivate single crystals from the ligands and theircomplexes. However, apart from L3 and L1eCuCl2, all attempts toobtain single crystals suitable for X-ray analysis failed. From the X-ray crystal structure, (R)-L1 was found to coordinate with Cu(II) togive a Cu(II)eL1 complex, which is an inactive catalyst for the Henryreaction. The X-ray crystallography structure suggested that threenitrogen atoms including the bridge-head nitrogen atom of thequinine coordinate to the copper, giving an unexpected five-coordinate complex (Fig. 1). To the best of our knowledge, therehas been no report about this novel complexation of phenanthro-line ligands with Cu(II). The absolute configuration of the complexwas determined to be 9-(R), and thus L1 was also deduced to be 9-(R), which is the same as quinine.
Notably, L2 also formed a Cu(II)eL2 complex. However, thiscomplex is an efficient enantioselective catalyst in the Henry re-action. Even though a single crystal of the Cu(II)eL2 complex couldnot be obtained, the different catalytic activity indicates that theremust be crucial differences between the two structures of thecomplexes that significantly affect their catalytic activities.
Computational study has been performed to help understandingthe different catalytic activities of these copper complexes. Allpossible isomers of the species under computation have beenconsidered, and the most stable ones are screened out by confor-mation search and further geometry optimization. Geometry op-timization and frequency analysis on the most stable species havebeen performed using the B3LYP functional and a basis set of 6-31G* for all the atoms. All calculations were carried out withGaussian 09.18 Table 4 listed the binding energies, dipole momentsand dihedral angle changes after the two ligands L1 and L2 co-ordinated to Cu(II) salts. The optimized geometry of the Cu(II)eL1complex is consistent with the X-ray crystallography structure, inwhich two chlorine atoms bind to copper while three nitrogenatoms coordinate with copper to form a five-dentate pattern (Table4, h3-L1). As well known, Cu(II) could have up to six coordinate sites.Thus, in the Cu(II)eL1 complex not enough coordinate sites areremained to coordinate simultaneously to the substrates 3a and 4a.This is the reasonwhy the reaction between 3a and 4a cannot occurwith Cu(II)eL1 complex.
Table 4Binding energies, dipole moments and dihedral angle changes after the two ligandsL1 and L2 coordinated to Cu(II) salts
Entry Complex Binding energy(kcal/mol)d
Dihedral anglechange (�)e
Dipole moment(Debye)
1a h3-L1 �69.9 11.2 7.18722b h3-L2 �64.5 61.4 6.79573c h2-L2 �67.1 d 10.6340
a The five-dentate Cu(II)eL1 complex was indicated as h3-L1.b The hyposized five-dentate Cu(II)eL2 complex, which was similar as Cu(II)eL1
complex was indicated as h3-L2 for computational study.c The four-dentate Cu(II)eL2 complex was indicated as h2-L2.d Binding energy ¼ GM � GCuCl2 � GM�noCu where G is Gibbs free energy.e Dihedral angle change is the difference between the dihedral angles
C1eN2eN3eN4 and C10eN2
0eN30eN4
0 in ligands and that in Cu(II)eL complexes,respectively.
In comparison, it is proposed that L2 coordinates to Cu(II)
through the two nitrogen atoms of phenanthroline to form a four-dentate copper complex (Table 4, h2-L2), which can coordinatesimultaneously to the substrates 3a and 4a and thus catalyzes thereaction. The evidence for this structural proposal of h2-L2 is sup-ported by three facts. First, the Cu(II)eL2 complex was locatedmuchlower on the same thin layer chromatography (TLC) plate than theCu(II)eL1 complex, indicating the two complexes have differentstructures. The computational study showed that if the Cu(II)eL2was five dentate (h3-L2) then the dipole moment of the h3-L2complex would be 6.7957 Debye, which is lower than that of the
h3-L1 complex (7.1872 Debye, Table 4, entries 1 and 2). In this case,the polarity of the Cu(II)eL2 complex (h3-L2) would be lower thanthat of the Cu(II)eL1 complex (h3-L1) and it is inconsistent with theTLC results. However, if the Cu(II)eL2 complex is four-dentate (h2-L2), the dipole moment of h2-L2 is higher than that of h3-L1 (Table4, entries 1 and 3), which is in agreement with the TLC results.Therefore, Cu(II) is more likely to coordinate to the two nitrogenatoms of the phenanthroline. Second, the changes of the dihedralangles in two ligands before and after they coordinate to copper(Fig. 3) also support a four-dentate structure. The dihedral anglesC1eN2eN3eN4 and C1
0eN20eN3
0eN40, as shown in Fig. 3a, were
149.0� and 129.9�, respectively. The change of the dihedral angle(C1eN2eN3eN4) between L2 and h3-L2was almost six times largerthan the change of the dihedral angle (C1
0eN20eN3
0eN40) between
L1 and h3-L1 (Table 4, entries 1 and 2, Fig. 3b and c). This suggeststhat the h3-L2 complex is much more distorted than the h3-L1complex. A more deformed structure will lead to higher energy andless stability of it. Accordingly, it should be difficult to form the 3-nitrogen-coordinate h3-L2 complex. Finally, the binding free en-ergy of h2-L2 is higher than that of h3-L2 (Table 4, entries 2 and 3),indicating that the transformation from h2-L2 to h3-L2 is not fa-vorable. Therefore, CuCl2 binds to the two nitrogen atoms of thephenanthroline to form a four-dentate h2-L2 complex, which isable to activate the reaction between substrates 3a and 4a.
To obtain insight into the mechanism of the reaction of L3 withCuCl2, its product was checked by mass spectrometry using elec-trospray ionization (ESI) model. The spectrum only gave m/z 583.1([Mþ3H]þ, 100%) and 584.1 ([Mþ4H]þ, 28%) while no peaks re-lating to complex product were observed, implying L3 did not co-ordinate with cupric salt. However, under the same reaction andexamination conditions, the one of L2 gave m/z 503.2 ([MþH]þ,100%), 504.2 ([Mþ2H]þ, 33%), 600.2 ([MþCuCl]þ, 50%), 602.2([MþCuClþ2H]þ, 39%), 603.3 ([MþCuClþ3H]þ, 15%), 604.2([MþCuClþ4H]þ, 9%), 605.2 ([MþCuClþ5H]þ, 4%). The observationof m/z 503.2 and 504.2 is expected since some complexes maydecompose during the ionization. The peak cluster of 600.2e605.2indicates unambiguously L2 forms complex product with CuCl2. L3can not form a complex with Cu(II) and thus is inactive for the Henryreaction. The geometries of L1 and L3with their van derWaals radiiare shown in Fig. 4 and a comparison shows that the Br atomprobably sterically blocks the attack of the copper ion, which couldbe the reason that the complex did not form.
Based on the above theoretical calculations, a possible transitionstate, which explains the stereochemical outcome of the reaction isproposed in Scheme 2. According to the model proposed by Evanset al.,4d the aldehyde and nitromethane coordinate to the coppercenter in such a way that there is a maximum activation for thereactive partners. Thus, the oxygen atom of the nitromethane ap-proaches the metal center from the axial side and the carbonyloxygen atom comes in from the equatorial side. The nucleophiliccarbon of the nitronate ion forms in situ by the deprotonation of thenitromethane and then attack the aldehyde from the Re-face to givethe R isomer.
3. Conclusion
A novel class of chiral phenanthroline ligands has been de-veloped by combining 1,10-phenanthroline with quinine. Thesechiral ligands can be easily prepared through a one-step procedurewith commercially available starting materials and do not requireany resolution. The ligand L2was coordinated to Cu(II) and thenwasemployed in the asymmetric Henry reaction with good substrategenerality, moderate to good yields and high enantioselectivities.Theoretical calculations showed that L2 coordinated with two ni-trogen atoms of phenanthroline to give a Cu(II)eL2 complex, whichwas able to activate both aldehyde and nitroalkane in the reaction.
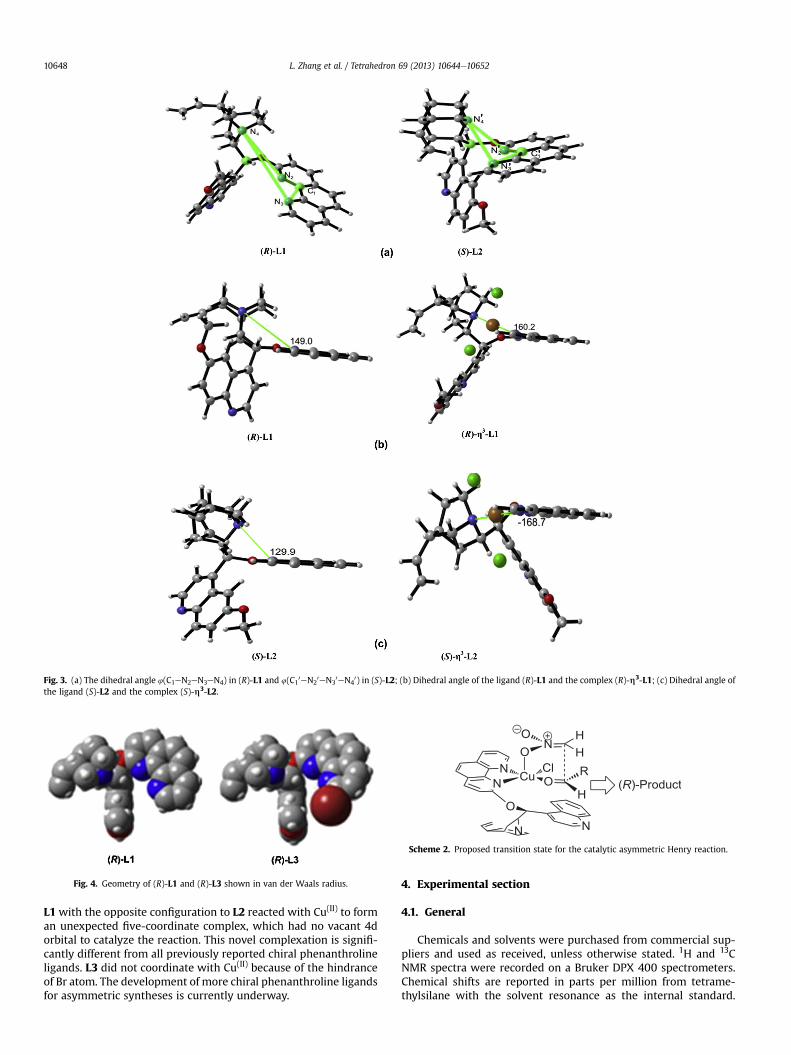
Fig. 3. (a) The dihedral angle 4(C1eN2eN3eN4) in (R)-L1 and 4(C10eN2
0eN30eN4
0) in (S)-L2; (b) Dihedral angle of the ligand (R)-L1 and the complex (R)-h3-L1; (c) Dihedral angle ofthe ligand (S)-L2 and the complex (S)-h3-L2.
Fig. 4. Geometry of (R)-L1 and (R)-L3 shown in van der Waals radius.
Scheme 2. Proposed transition state for the catalytic asymmetric Henry reaction.
L. Zhang et al. / Tetrahedron 69 (2013) 10644e1065210648
L1 with the opposite configuration to L2 reacted with Cu(II) to forman unexpected five-coordinate complex, which had no vacant 4dorbital to catalyze the reaction. This novel complexation is signifi-cantly different from all previously reported chiral phenanthrolineligands. L3 did not coordinate with Cu(II) because of the hindranceof Br atom. The development of more chiral phenanthroline ligandsfor asymmetric syntheses is currently underway.
4. Experimental section
4.1. General
Chemicals and solvents were purchased from commercial sup-pliers and used as received, unless otherwise stated. 1H and 13CNMR spectra were recorded on a Bruker DPX 400 spectrometers.Chemical shifts are reported in parts per million from tetrame-thylsilane with the solvent resonance as the internal standard.

L. Zhang et al. / Tetrahedron 69 (2013) 10644e10652 10649
Multiplicity was indicated as follows: s (singlet), d (doublet), t(triplet), q (quartet), m (multiplet), dd (doublet of doublet), br s(broad singlet). Coupling constants were reported in Hertz (Hz).Accurate mass data and routine mass data were obtained with anAgilent Technologies 6520 Accurate-Mass Q-TOF LC/MS instrumentand a LCQ Advantage MAX instrument, respectively, under ESImodel. Optical rotations were measured with a PerkineElmer 341polarimeter at 25 �C. HPLC analysis was performed with a Shi-madzu CTO-10 AS instrument and a Chiralpak AD-H column pur-chased from Daicel Chemical Industries, LTD.
4.2. Synthesis of ligands L1eL3
To a solution of NaH (5 equiv, 20 mmol) in 10 mL DMSO wasadded quinine (4mmol) and themixturewas stirred for 1 h at roomtemperature. Then 2-bromo-1,10-phenanthroline 1 (6 mmol) or2,9-dibromo-1,10-phenanthroline 2 (6 mmol) was added and thereaction was kept for 5 h at 80e90 �C under stirring. The mixturewas cooled to room temperature and 20 mL H2O was added care-fully, following extracted with CH2Cl2 (3�20 mL). The organicphases were combined, washed with brine, dried over anhydrousNa2SO4, and evaporated in vacuo. The residue was purified by flashchromatography to afford L1eL3.
L1: White powder, mp 101e103 �C; [a]D24 þ3.0 (c 1.4, CH2Cl2);IR (KBr) nmax: 2927, 1618, 1504, 1454, 1349, 1260, 1076, 1027, 993,850 cm�1. 1H NMR (400 MHz, CDCl3): d 9.06 (d, J¼3.1 Hz, 1H), 8.73(d, J¼4.5 Hz, 1H), 8.36 (s, 1H), 8.13 (m, J¼7.1 Hz, 2H), 8.04e7.92 (m,2H), 7.73 (d, J¼4.4 Hz, 1H), 7.65 (d, J¼8.7 Hz, 1H), 7.61e7.51 (m,2H), 7.36 (dd, J¼9.1, 2.0 Hz, 1H), 7.28 (d, J¼8.5 Hz, 1H), 5.81e5.68(m, 1H), 5.06e4.88 (m, 2H), 4.21 (s, 3H), 3.50 (t, J¼8.3 Hz, 1H),3.40e3.22 (m, 2H), 2.87 (d, J¼13.3 Hz, 1H), 2.77e2.66 (m, 1H),2.37 (s, 1H), 2.17e2.07 (m, 1H), 1.91 (s, 1H), 1.85e1.66 (m, 2H), 1.55(s, 1H). 13C NMR (101 MHz, CDCl3): d 161.35, 158.54, 149.48, 147.16,144.48, 143.89, 140.96, 139.86, 136.27, 131.33, 129.12, 126.86,125.97, 125.18, 124.23, 123.00, 122.60, 118.52, 115.00, 113.69,101.67, 73.20, 59.68, 56.72, 42.85, 39.26, 29.70, 27.60, 26.99, 21.34.HRMS: calculated for [MþH]þ C32H30N4O2 503.2442, found503.2443.
L2: White yellow powder, mp 205e206 �C; [a]D24 þ178.2 (c0.7, CH2Cl2); IR (KBr) nmax: 3389, 2926, 1618, 1504, 1454, 1354,1251, 1134, 1026, 918, 848 cm�1. 1H NMR (400 MHz, CDCl3):d 9.05 (s, 1H), 8.77 (d, J¼4.2 Hz, 1H), 8.13 (dd, J¼28.8, 8.3 Hz, 3H),7.95 (m, J¼10.2 Hz, 2H), 7.76e7.48 (m, 5H), 7.37 (d, J¼8.8 Hz, 1H),5.74 (dd, J¼17.1, 6.7 Hz, 1H), 5.24e5.03 (m, 2H), 4.13 (d,J¼26.2 Hz, 3H), 3.88e3.75 (m, 1H), 3.27 (s, 2H), 2.72 (s, 1H), 2.02(d, J¼17.1 Hz, 2H), 1.71 (d, J¼11.0 Hz, 1H), 1.53 (s, 1H), 1.35e1.17(m, 3H). 13C NMR (101 MHz, CDCl3): d 160.57, 158.40, 149.23,147.36, 144.93, 143.13, 139.60, 137.70, 136.31, 131.63, 128.98,127.71, 126.32, 125.52, 124.07, 122.65, 122.31, 116.90, 115.15,102.63, 58.02, 56.35, 54.12, 41.70, 37.03, 30.92, 29.68, 26.76,24.79. HRMS: calculated for [MþH]þ C32H30N4O2 503.2442,found 503.2443.
L3: White yellow powder, mp 195e197 �C; [a]D24 �103.7 (c 0.5,CH2Cl2); IR (KBr) nmax: 2927, 1619, 1579, 1490, 1449, 1364, 1267,1232, 1109, 1026, 970, 852 cm�1. 1H NMR (400 MHz, CDCl3): d 8.75(d, J¼4.4 Hz, 1H), 8.08 (d, J¼8.7 Hz, 1H), 8.00e7.84 (m, 4H), 7.73 (d,J¼4.3 Hz, 1H), 7.64 (dd, J¼18.4, 8.5 Hz, 2H), 7.53 (d, J¼8.6 Hz, 1H),7.34 (dd, J¼9.1, 2.1 Hz,1H), 7.20 (d, J¼8.6 Hz,1H), 5.90e5.78 (m,1H),5.00 (m, J¼12.7 Hz, 2H), 4.04 (s, 3H), 3.66e3.55 (m, 1H), 3.44 (d,J¼11.6 Hz, 1H), 3.23e3.10 (m, 1H), 2.74 (d, J¼12.6 Hz, 2H), 2.34 (s,1H), 1.91 (d, J¼31.7 Hz, 4H), 1.60 (s, 1H). 13C NMR (101 MHz, CDCl3):d 161.61,158.14,147.36,145.39,144.71,143.02,141.80,139.50,138.04,131.45, 127.85, 127.39, 126.45, 125.37, 123.38, 122.22, 119.83, 114.57,114.01, 101.76, 74.49, 59.41, 58.31, 56.77, 56.34, 42.77, 39.69, 30.94,27.78. HRMS: calculated for [MþH]þ C32H29BrN4O2 581.1547, found581.1548.
4.3. General experimental procedure for Henry reaction
Themixture of L2 (7.5 mg, 0.015mmol) and CuCl2$2H2O (2.1mg,0.0125 mmol) was stirred in THF/i-PrOH (1:1, 1 mL) at room tem-perature under N2 protection for 1 h to form the complex. Theresulting mixture was cooled to 0 �C, then nitroalkanes (2.5 mmol),N-Meemorpholine (1.4 mL) and aldehydes (0.25 mmol) were addedand stirring continued for intend time at 0 �C. The solvent wasremoved under reduced pressure and the resulting mixture wasdirectly purified by column chromatography to afford the Henryreaction product.
(R)-2-Nitro-1-phenylethanol (5a, Table 4, entry 1).4d The titlecompound was prepared according to the general procedure andpurified by column chromatography (petroleum ether/ethylacetate¼6:1). Enantiomeric excess (90% ee, Table 4, entry 1) wasdetermined by HPLC (Chiralcel OD-H), hexane/i-PrOH¼90:10,1.0 mL/min, 254 nm, major enantiomer (R) tr¼12.847 min, minorenantiomer (S) tr¼16.136 min; 1H NMR (400 MHz, CDCl3):d 7.43e7.29 (m, 5H), 5.39 (m, 1H), 4.56 (dd, J¼13.2, 9.6 Hz, 1H), 4.46(dd, J¼13.2, 3.1 Hz, 1H), 3.27 (d, J¼3.6 Hz, 1H). 13C NMR (101 MHz,CDCl3): d 138.28, 129.04, 128.96, 126.01, 81.28, 71.01.
(R)-1-(3-Chlorophenyl)-2-nitroethanol (5b, Table 4, entry 2).19
The title compound was prepared according to the general pro-cedure and purified by column chromatography (petroleum ether/ethyl acetate¼6:1). Enantiomeric excess (73% ee, Table 4, entry 2)was determined by HPLC (Chiralcel OD-H), hexane/i-PrOH¼90:10,1.0 mL/min, 215 nm, major enantiomer (R) tr¼12.731 min, minorenantiomer (S) tr¼16.514 min; 1H NMR (400 MHz, CDCl3):d 7.50e7.12 (m, 4H), 5.34 (dd, J¼9.1, 3.5 Hz, 1H), 4.46 (m, J¼13.3,6.3 Hz, 2H), 3.65 (s, 1H). 13C NMR (101MHz, CDCl3): d 140.31,134.77,130.37, 129.01, 126.23, 124.23, 80.95, 70.28.
(R)-1-(2, 4-Dichlorophenyl)-2-nitroethanol (5c, Table 4, entry3).6n The title compound was prepared according to the generalprocedure and purified by column chromatography (petroleumether/ethyl acetate¼6:1). Enantiomeric excess (77% ee, Table 4,entry 3) was determined by HPLC (Chiralcel AD-H), hexane/i-PrOH¼90:10, 0.5 mL/min, 254 nm, major enantiomer (R)tr¼14.592 min, minor enantiomer (S) tr¼17.432 min; 1H NMR(400 MHz, CDCl3): d 7.61 (d, J¼8.4 Hz, 1H), 7.40 (d, J¼2.0 Hz, 1H),7.34 (dd, J¼8.4, 2.0 Hz, 1H), 5.79 (m, J¼9.4, 3.9, 2.4 Hz, 1H), 4.64 (dd,J¼13.7, 2.3 Hz, 1H), 4.42 (dd, J¼13.7, 9.5 Hz, 1H), 3.20 (d, J¼4.2 Hz,1H). 13C NMR (101 MHz, CDCl3): d 135.24, 134.16, 132.09, 129.51,128.60, 127.99, 79.09, 67.44.
(R)-1-(4-Fluorophenyl)-2-nitroethanol (5d, Table 4, entry 4).4d
The title compound was prepared according to the general pro-cedure and purified by column chromatography (petroleum ether/ethyl acetate¼6:1). Enantiomeric excess (76% ee, Table 4, entry 4)was determined by HPLC (Chiralcel OD-H), hexane/i-PrOH¼90:10,0.8 mL/min, 215 nm, major enantiomer (R) tr¼13.513 min, minorenantiomer (S) tr¼115.837 min; 1H NMR (400 MHz, CDCl3):d 7.43e7.29 (m, 2H), 7.14e7.00 (m, 2H), 5.41 (dd, J¼9.5, 3.2 Hz, 1H),4.56 (dd, J¼13.2, 9.5 Hz, 1H), 4.47 (dd, J¼13.2, 3.2 Hz, 1H), 3.27 (d,J¼66.4 Hz, 1H). 13C NMR (101 MHz, CDCl3): d 164.09, 134.03, 127.84,116.07, 81.15, 70.34.
(R)-2-Nitro-1-(4-nitrophenyl)ethanol (5e, Table 4, entry 5).4d
The title compound was prepared according to the general pro-cedure and purified by column chromatography (petroleum ether/ethyl acetate¼5:1). Enantiomeric excess (71% ee, Table 4, entry 5)was determined by HPLC (Chiralcel OD-H), hexane/i-PrOH¼85:15,1.0 mL/min, 215 nm, major enantiomer (R) tr¼15.571 min, minorenantiomer (S) tr¼19.195 min; 1H NMR (400 MHz, CDCl3): d 8.26 (d,J¼8.7 Hz, 2H), 7.63 (d, J¼8.7 Hz, 2H), 5.61 (d, J¼4.3 Hz, 1H),4.69e4.50 (m, 2H), 3.45 (s, 1H). 13C NMR (101MHz, CDCl3): d 148.10,145.14, 126.98, 124.19, 80.68, 69.98.
(R)-1-(2-Methoxyphenyl)-2-nitroethanol (5f, Table 4, entry 6).4d
The title compound was prepared according to the general

L. Zhang et al. / Tetrahedron 69 (2013) 10644e1065210650
procedure and purified by column chromatography (petroleumether/ethyl acetate¼6:1). Enantiomeric excess (66% ee, Table 4,entry 6) was determined by HPLC (Chiralcel OD-H), hexane/i-PrOH¼90:10, 0.8 mL/min, 215 nm, major enantiomer (R)tr¼13.740 min, minor enantiomer (S) tr¼16.487 min; 1H NMR(400MHz, CDCl3): d 7.44e7.28 (m, 2H), 6.96 (m, J¼24.4, 15.7, 4.4 Hz,2H), 5.60 (dd, J¼9.3, 3.1 Hz, 1H), 4.59 (dd, J¼12.8, 3.2 Hz, 1H), 4.49(dd, J¼12.8, 9.3 Hz,1H), 3.84 (d, J¼6.6 Hz, 3H), 3.66 (d, J¼6.4 Hz,1H).13C NMR (101 MHz, CDCl3): d 156.07, 129.76, 127.06, 126.35, 121.00,110.66, 79.90, 67.56, 55.42.
(R)-1-(3-Methoxyphenyl)-2-nitroethanol (5g, Table 4, entry7).20 The title compound was prepared according to the generalprocedure and purified by column chromatography (petroleumether/ethyl acetate¼6:1). Enantiomeric excess (88% ee, Table 4,entry 7) was determined by HPLC (Chiralcel OD-H), hexane/i-PrOH¼90:10, 1.0 mL/min, 215 nm, major enantiomer (R)tr¼21.053 min, minor enantiomer (S) tr¼27.262 min; 1H NMR(400 MHz, CDCl3): d 7.30 (m, J¼8.1 Hz, 1H), 6.90 (m, J¼14.2, 8.4,4.5 Hz, 3H), 5.44e5.35 (m, 1H), 4.58 (dd, J¼13.1, 9.6 Hz, 1H), 4.48(dd, J¼13.1, 3.1 Hz, 1H), 3.81 (s, 3H), 3.42 (d, J¼3.7 Hz, 1H). 13C NMR(101 MHz, CDCl3): d 159.98, 139.98, 130.11, 118.16, 114.35, 111.55,81.27, 70.90, 55.35.
(R)-1-(4-Methoxyphenyl)-2-nitroethanol (5h, Table 4, entry8).6n The title compound was prepared according to the generalprocedure and purified by column chromatography (petroleumether/ethyl acetate¼6:1). Enantiomeric excess (92% ee, Table 4,entry 8) was determined by HPLC (Chiralcel OD-H), hexane/i-PrOH¼85:15, 0.8 mL/min, 215 nm, major enantiomer (R)tr¼14.708 min, minor enantiomer (S) tr¼17.596 min; 1H NMR(400 MHz, CDCl3): d 7.30 (d, J¼8.6 Hz, 2H), 6.91 (d, J¼8.7 Hz, 2H),5.38 (d, J¼7.9 Hz, 1H), 4.59 (dd, J¼13.1, 9.6 Hz, 1H), 4.46 (dd, J¼13.1,3.1 Hz, 1H), 3.79 (d, J¼9.2 Hz, 3H), 3.02 (s, 1H). 13C NMR (101 MHz,CDCl3): d 160.01, 130.32, 127.32, 114.40, 81.30, 70.68, 55.38.
(R)-1-(2,4-Dimethoxyphenyl)-2-nitroethanol (5i, Table 4, entry9). The title compound was prepared according to the generalprocedure and purified by column chromatography (petroleumether/ethyl acetate¼6:1). It was a colorless oily compound. Enan-tiomeric excess (90% ee, Table 4, entry 9) was determined by HPLC(Chiralcel OD-H), hexane/i-PrOH¼90:10,1.0 mL/min, 215 nm, majorenantiomer (R) tr¼14.768 min, minor enantiomer (S)tr¼22.478 min; 1H NMR (400 MHz, CDCl3): d 7.29 (d, J¼8.4 Hz, 1H),6.47 (m, J¼12.1, 9.0, 4.9 Hz, 2H), 4.80 (d, J¼6.9 Hz, 1H), 4.56 (m,J¼6.6 Hz, 2H), 3.79 (dd, J¼19.9, 8.0 Hz, 6H), 3.50 (d, J¼5.9 Hz, 1H).13C NMR (101 MHz, CDCl3): d 161.10, 157.20, 130.61, 127.95, 118.72,104.63, 80.12, 75.60, 67.54, 55.42. HRMS: calculated for [MþH]þ
C10H13NO5 228.0867, found 228.0865.(R)-1-(2-Methyphenyl)-2-nitroethanol (5j, Table 4, entry 10).4d
The title compound was prepared according to the general pro-cedure and purified by column chromatography (petroleum ether/ethyl acetate¼6:1). Enantiomeric excess (84% ee, Table 4, entry 10)was determined by HPLC (Chiralcel OD-H), hexane/i-PrOH¼85:15,0.8 mL/min, 215 nm, major enantiomer (R) tr¼9.507 min, minorenantiomer (R) tr¼13.965 min; 1H NMR (400 MHz, CDCl3):d 7.51e7.12 (m, 4H), 5.60 (m, J¼9.6, 3.1 Hz, 1H), 4.47 (dd, J¼13.2,9.7 Hz, 1H), 4.37 (dd, J¼13.2, 2.7 Hz, 1H), 3.28 (d, J¼3.6 Hz, 1H), 2.35(s, 3H). 13C NMR (101 MHz, CDCl3): d 136.44, 134.61, 130.87, 128.67,126.76, 125.67, 80.30, 67.94, 18.85.
(R)-1-(4-Methyphenyl)-2-nitroethanol (5k, Table 4, entry 11).6n
The title compound was prepared according to the general pro-cedure and purified by column chromatography (petroleum ether/ethyl acetate¼6:1). Enantiomeric excess (84% ee, Table 4, entry 11)was determined by HPLC (Chiralcel OD-H), hexane/i-PrOH¼95:5,0.5 mL/min, 215 nm, major enantiomer (R) tr¼17.917 min, minorenantiomer (S) tr¼21.810min; 1H NMR (400MHz, CDCl3): d 7.21 (m,J¼22.3, 8.1 Hz, 4H), 5.33 (d, J¼9.6 Hz, 1H), 4.53 (dd, J¼13.1, 9.6 Hz,1H), 4.42 (dd, J¼13.1, 3.2 Hz, 1H), 3.36 (d, J¼3.6 Hz, 1H), 2.33 (d,
J¼11.0 Hz, 3H). 13C NMR (101 MHz, CDCl3): d 138.83, 135.41, 129.67,125.97, 81.29, 70.89, 21.17.
(R)-1-(4-Hydroxyphenyl)-2-nitroethanol (5l, Table 4, entry12).6o The title compound was prepared according to the generalprocedure and purified by column chromatography (petroleumether/ethyl acetate¼3:1). Enantiomeric excess (89% ee, Table 4,entry 12) was determined by HPLC (Chiralcel OJ-H), hexane/i-PrOH¼70:30, 0.8 mL/min, 215 nm, major enantiomer (R)tr¼14.101 min, minor enantiomer (S) tr¼16.844 min; 1H NMR(400MHz, DMSO): d 9.49 (d, J¼23.4 Hz,1H), 7.49e7.10 (m, 2H), 6.80(m, J¼20.0, 17.5 Hz, 2H), 5.91 (s, 1H), 5.28e4.43 (m, 3H). 13C NMR(101 MHz, DMSO): d 157.10, 128.96, 127.45, 114.93, 82.04, 69.81.
(S)-2-Nitro-1-(thiophen-2-yl)ethanol (5m, Table 4, entry 13).6n
The title compound was prepared according to the general pro-cedure and purified by column chromatography (petroleum ether/ethyl acetate¼6:1). Enantiomeric excess (90% ee, Table 4, entry 13)was determined by HPLC (Chiralcel OD-H), hexane/i-PrOH¼90:10,0.5 mL/min, 215 nm, major enantiomer (R) tr¼27.999 min, minorenantiomer (S) tr¼30.711 min; 1H NMR (400 MHz, CDCl3):d 7.43e7.26 (m, 1H), 7.18e6.94 (m, 2H), 5.69 (dd, J¼9.3, 3.3 Hz, 1H),4.65 (m, J¼16.7, 13.3, 6.4 Hz, 2H), 3.60 (s, 1H). 13C NMR (101 MHz,CDCl3): d 141.43, 127.28, 126.15, 125.13, 80.84, 67.05.
(S)-1-(Furan-2-yl)-2-nitroethanol (5n, Table 4, entry 14).6n Thetitle compound was prepared according to the general procedureand purified by column chromatography (petroleum ether/ethylacetate¼6:1). Enantiomeric excess (81% ee, Table 4, entry 14) wasdetermined byHPLC (Chiralcel AD-H), hexane/i-PrOH¼97:3, 0.5mL/min, 254 nm, major enantiomer (R) tr¼51.923 min, minor enantio-mer (S) tr¼56.810 min; 1H NMR (400 MHz, CDCl3): d 7.42 (d,J¼0.8 Hz, 1H), 6.53e6.33 (m, 2H), 5.47 (dd, J¼9.0, 3.2 Hz, 1H), 4.78(dd, J¼13.4, 9.0 Hz,1H), 4.67 (dd, J¼13.4, 3.5 Hz,1H), 3.16 (s, 1H). 13CNMR (101MHz, CDCl3): d 150.79,143.19,110.68,108.19, 78.44, 64.85.
(S)-1-(Furan-3-yl)-2-nitroethanol (5o, Table 4, entry 15).21 Thetitle compoundwaspreparedaccording to thegeneral procedure andpurified by column chromatography (petroleum ether/ethylacetate¼6:1). Enantiomeric excess (86% ee, Table 4, entry 15) wasdeterminedbyHPLC (ChiralcelAD-H), hexane/i-PrOH¼90:10,1.0mL/min, 215 nm, major enantiomer (R) tr¼12.052 min, minor enantio-mer (S) tr¼16.430 min; 1H NMR (400 MHz, CDCl3): d 7.63e7.18 (m,2H), 6.37 (d, J¼0.9 Hz, 1H), 5.35 (dd, J¼9.2, 3.4 Hz, 1H), 4.58 (dd,J¼13.1, 9.2 Hz, 1H), 4.50 (dd, J¼13.1, 3.5 Hz,1H), 3.39 (s,1H). 13C NMR(101 MHz, CDCl3): d 144.04, 139.99, 123.55, 108.10, 80.29, 64.13.
(R,E)-1-Nitro-4-phenylbut-3-en-2-ol (5p, Table 4, entry 16).22
The title compound was prepared according to the general pro-cedure and purified by column chromatography (petroleum ether/ethyl acetate¼6:1). Enantiomeric excess (86% ee, Table 4, entry 16)was determined by HPLC (Chiralcel OD-H), hexane/i-PrOH¼85:15,0.8 mL/min, 215 nm, major enantiomer (R) tr¼31.843 min, minorenantiomer (S) tr¼28.015 min; 1H NMR (400 MHz, CDCl3):d 7.34e7.24 (m, 5H), 6.67 (dd, J¼15.7, 9.5 Hz, 1H), 6.12e5.97 (m,1H),4.59e4.45 (m,1H), 4.39 (dd, J¼6.7, 3.6 Hz, 2H), 3.32 (s, 1H). 13C NMR(101 MHz, CDCl3): d 133.41, 128.83, 128.54, 126.85, 125.37, 121.32,80.06, 69.70.
(R)-1-Nitropentyl-2-ol (5q, Table 4, entry 17).23 The title com-pound was prepared according to the general procedure and pu-rified by column chromatography (petroleum ether/ethylacetate¼6:1). Enantiomeric excess (85% ee, Table 4, entry 17) wasdetermined by HPLC (Chiralcel AD-H), hexane/i-PrOH¼95:5,1.0 mL/min, 215 nm, major enantiomer (R) tr¼14.555 min, minorenantiomer (S) tr¼23.733 min; 1H NMR (400 MHz, CDCl3):d 4.43e4.29 (m, 2H), 4.30e4.20 (m, 1H), 3.65 (d, J¼5.4 Hz, 1H),1.51e1.42 (m, 2H),1.41e1.30 (m, 2H), 0.90 (dd, J¼9.9, 4.2 Hz, 3H). 13CNMR (101 MHz, CDCl3): d 80.84, 68.53, 35.79, 18.34, 13.61.
(R)-3-Methyl-1-nitro-butyl-2-ol (5r, Table 4, entry 18).4d Thetitle compound was prepared according to the general procedureand purified by column chromatography (petroleum ether/ethyl

L. Zhang et al. / Tetrahedron 69 (2013) 10644e10652 10651
acetate¼6:1). Enantiomeric excess (82% ee, Table 4, entry 18) wasdetermined by HPLC (Chiralcel OD-H), hexane/i-PrOH¼98:2,0.5 mL/min, 215 nm, major enantiomer (R) tr¼30.822 min, minorenantiomer (S) tr¼33.880 min; 1H NMR (400 MHz, CDCl3): d 4.40(m, J¼22.0, 12.7, 6.1 Hz, 2H), 4.12e3.98 (m, 1H), 3.43 (d, J¼5.6 Hz,1H), 1.73 (d, J¼6.3 Hz, 1H), 0.92 (m, J¼6.5 Hz, 6H). 13C NMR(101 MHz, CDCl3): d 79.43, 73.51, 31.78, 18.29, 17.40.
(R)-1-Cyclohexyl-2-nitroethanol (5s, Table 4, entry 19).4d Thetitle compound was prepared according to the general procedureand purified by column chromatography (petroleum ether/ethylacetate¼6:1). Enantiomeric excess (86% ee, Table 4, entry 19) wasdetermined by HPLC (Chiralcel AD-H), hexane/i-PrOH¼95:5,0.7 mL/min, 215 nm, major enantiomer (R) tr¼18.313 min, minorenantiomer (S) tr¼19.588 min; 1H NMR (400 MHz, CDCl3): d 4.46(m, J¼21.9, 12.7, 6.1 Hz, 2H), 4.07 (s, 1H), 3.48 (s, 1H), 1.89e1.72 (m,3H), 1.67 (dd, J¼14.9, 7.3 Hz, 2H), 1.45 (m, J¼12.4, 6.2, 3.1 Hz, 1H),1.32e1.15 (m, 3H), 1.15e1.03 (m, 2H). 13C NMR (101 MHz, CDCl3):d 79.45, 72.95, 41.45, 28.69, 27.92, 25.85.
(1R,2R)-2-nitro-1-phenylbutan-1-ol (5t, Table 4, entry 20).6n
The title compound was prepared according to the general pro-cedure and purified by column chromatography (petroleum ether/ethyl acetate¼6:1). Enantiomeric excess (86% ee, Table 1, entry 8)was determined by HPLC (Chiralcel AS-H), hexane/i-PrOH¼95:5,0.8 mL/min, 215 nm, anti: major enantiomer tr¼16.295 min, minorenantiomer tr¼14.373 min, syn: major enantiomer tr¼22.682 min,minor enantiomer tr¼18.061 min; 1H NMR (400 MHz, CDCl3):d 7.58e7.15 (m, 5H), 4.98 (dd, J¼32.8, 7.5 Hz,1H), 4.64e4.48 (m,1H),3.52 (s, 1H), 1.77 (m, J¼14.5, 10.9, 7.3 Hz, 1H), 1.32 (m, J¼14.8, 7.5,3.4 Hz, 1H), 0.83 (m, J¼29.8, 7.4 Hz, 3H). 13C NMR (101 MHz, CDCl3):d 138.78, 129.07, 127.06, 126.44, 95.31, 23.84, 9.85.
(1S,2R)-1-(4-Methoxyphenyl)-2-nitrobutan-1-ol (5u, Table 4,entry 21).8d The title compound was prepared according to thegeneral procedure and purified by column chromatography (pe-troleum ether/ethyl acetate¼6:1) to give colorless oil. Enantiomericexcess (86% ee, Table 1, entry 8) was determined by HPLC (ChiralcelAD-H), hexane/i-PrOH¼95:5, 1.0 mL/min, 215 nm, anti: major en-antiomer tr¼17.237 min, minor enantiomer tr¼18.603 min, syn:major enantiomer tr¼23.030 min, minor enantiomertr¼24.785 min; 1H NMR (400 MHz, CDCl3): d 7.26 (m, J¼5.7 Hz, 2H),6.90 (m, J¼7.5 Hz, 2H), 4.98 (dd, J¼30.5, 7.6 Hz, 1H), 4.66e4.47 (m,1H), 3.79 (d, J¼9.2 Hz, 3H), 2.92 (s, 1H), 1.80 (m, J¼14.5, 10.9, 7.3 Hz,1H), 1.38 (m, J¼14.6, 7.4, 3.4 Hz, 1H), 0.87 (dd, J¼24.3, 16.9 Hz, 3H).13C NMR (101 MHz, CDCl3): d 160.03, 130.87, 128.22, 114.35, 95.47,75.14, 55.35, 23.95, 10.07.
(1S,2R)-2-Nitro-1-(thiophen-2-yl)butan-1-ol (5y, Table 4, entry22).24 The title compound was prepared according to the generalprocedure and purified by column chromatography (petroleumether/ethyl acetate¼6:1). Enantiomeric excess (86% ee, Table 1,entry 8) was determined by HPLC (Chiralcel AD-H), hexane/i-PrOH¼95:5, 0.8 mL/min, 215 nm, anti: major enantiomertr¼15.912 min, minor enantiomer tr¼15.248 min, syn: major en-antiomer tr¼22.375 min, minor enantiomer tr¼21.640 min; 1HNMR (400 MHz, CDCl3): d 7.35 (d, J¼5.0 Hz, 1H), 7.10e6.97 (m, 2H),5.30 (d, J¼8.9 Hz, 1H), 4.69e4.59 (m, 1H), 2.97 (s, 1H), 1.85 (m,J¼14.6, 10.6, 7.3 Hz, 1H), 1.56 (m, J¼14.8, 7.5, 3.9 Hz, 1H), 0.91 (m,J¼7.4 Hz, 3H). 13C NMR (101 MHz, CDCl3): d 141.74, 127.18, 126.60,126.25, 95.30, 71.15, 23.84, 9.89.
Conflict of interest
None.
Acknowledgements
Generous financial support by the National Basic ResearchProgram of China (973 Program: 2010CB833300) and the State
Key Laboratory of Elemento-Organic Chemistry is gratefullyacknowledged.
References and notes
1. (a) Luzzio, F. A. Tetrahedron 2001, 57, 915e945; (b) Ono, N. The Nitro Group inOrganic Synthesis; Wiley-VCH: New York, NY, 2001.
2. For recent examples, see: (a) Uraguchi, D.; Nakamura, S.; Ooi, T. Angew. Chem.2010, 49, 7562e7565; (b) Metro, T. X.; Cochi, A.; Gomez Pardo, D.; Cossy, J. J.Org. Chem. 2011, 76, 2594e2602; (c) Liu, L.; Zhang, S.; Xue, F.; Lou, G.; Zhang, H.;Ma, S.; Duan, W.; Wang, W. Chem.dEur. J. 2011, 17, 7791e7795; (d) Lai, G.; Guo,F.; Zheng, Y.; Fang, Y.; Song, H.; Xu, K.; Wang, S.; Zha, Z.; Wang, Z. Chem.dEur. J.2011, 17, 1114e1117; (e) Kureshy, R. I.; Das, A.; Khan, N.-u. H.; Abdi, S. H. R.; Bajaj,H. C. ACS Catal. 2011, 1, 1529e1535; (f) Guo, Z.-L.; Deng, Y.-Q.; Zhong, S.; Lu, G.Tetrahedron: Asymmetry 2011, 22, 1395e1399; (g) Nitabaru, T.; Kumagai, N.;Shibasaki, M. Angew. Chem., Int. Ed. 2012, 51, 1644e1647.
3. For recent examples on asymmetric Henry reaction based on BINOLs-derivedligands, see: (a) Sasai, H.; Suzuki, T.; Arai, S.; Arai, T.; Shibasaki, M. J. Am.Chem. Soc. 1992, 114, 4418e4420; (b) Sasai, H.; Suzuki, T.; Itoh, N.; Shibasaki, M.Tetrahedron Lett. 1993, 34, 851e854; (c) Sasai, H.; Suzuki, T.; Itoh, N.; Tanaka, K.;Date, T.; Okamura, K.; Shibasaki, M. J. Am. Chem. Soc. 1993, 115, 10372e10373;(d) Sasai, H.; Tokunaga, T.; Watanabe, S.; Suzuki, T.; Itoh, N.; Shibasaki, M. J. Org.Chem. 1995, 60, 7388e7389; (e) Iseki, K.; Oishi, S.; Sasai, H.; Shibasaki, M.Tetrahedron Lett. 1996, 37, 9081e9084; (f) Shibasaki, M.; Yoshikawa, N. Chem.Rev. 2002, 102, 2187e2209; (g) Palomo, C.; Oiarbide, M.; Mielgo, A. Angew.Chem., Int. Ed. 2004, 43, 5442e5444; (h) Palomo, C.; Oiarbide, M.; Laso, A. Eur. J.Org. Chem. 2007, 2561e2574; (i) Blay, G.; Hernandez-Olmos, V.; Pedro, J. R.Synlett 2011, 1195e1211.
4. For recent examples on asymmetric Henry reaction based on BOX ligands, see:(a) Christensen, C.; Juhl, K.; Jørgensen, K. A. Chem. Commun. 2001, 2222e2223;(b) Knudsen, K. R.; Risgaard, T.; Nishiwaki, N.; Gothelf, K. V.; Jørgensen, K. A. J.Am. Chem. Soc. 2001, 123, 5843e5844; (c) Christensen, C.; Juhl, K.; Hazell, R. G.;Jørgensen, K. A. J. Org. Chem. 2002, 67, 4875e4881; (d) Evans, D. A.; Seidel, D.;Rueping, M.; Lam, H. W.; Shaw, J. T.; Downey, C. W. J. Am. Chem. Soc. 2003, 125,12692e12693; (e) Risgaard, T.; Gothelf, K. V.; Jorgensen, K. A. Org. Biomol. Chem.2003, 1, 153e156; (f) Lu, S.-F.; Du, D.-M.; Zhang, S.-W.; Xu, J. Tetrahedron:Asymmetry 2004, 15, 3433e3441; (g) Du, D.-M.; Lu, S.-F.; Fang, T.; Xu, J. J. Org.Chem. 2005, 70, 3712e3715; (h) Ginotra, S. K.; Singh, V. K. Org. Biomol. Chem.2007, 5, 3932e3937; (i) Toussaint, A.; Pfaltz, A. Eur. J. Org. Chem. 2008,4591e4597; (j) Gaab, M.; Bellemin-Laponnaz, S.; Gade, L. H. Chem.dEur. J. 2009,15, 5450e5462; (k) Spangler, K. Y.; Wolf, C. Org. Lett. 2009, 11, 4724e4727; (l)Xu, H.; Wolf, C. Chem. Commun. 2010, 8026e8028; (m) Didier, D.; Magnier-Bouvier, C.; Schulz, E. Adv. Synth. Catal. 2011, 353, 1087e1095; (n) Maggi, R.;Lanari, D.; Oro, C.; Sartori, G.; Vaccaro, L. Eur. J. Org. Chem. 2011, 2011,5551e5554; (o) Subba Reddy, B. V.; George, J. Tetrahedron: Asymmetry 2011, 22,1169e1175.
5. For recent examples on asymmetric Henry reaction based on amino alcoholsligands, see: (a) Kim, H. Y.; Oh, K. Org. Lett. 2009, 11, 5682e5685; (b) Reddy, B.V. S.; Reddy, S. M.; Manisha, S.; Madan, C. Tetrahedron: Asymmetry 2011, 22,530e535; (c) Guo, Z.-L.; Zhong, S.; Li, Y.-B.; Lu, G. Tetrahedron: Asymmetry 2011,22, 238e245; (d) Qin, D.-D.; Lai, W.-H.; Hu, D.; Chen, Z.; Wu, A.-A.; Ruan, Y.-P.;Zhou, Z.-H.; Chen, H.-B. Chem.dEur. J. 2012, 18, 10515e10518; (e) Xu, K.; Lai, G.;Zha, Z.; Pan, S.; Chen, H.; Wang, Z. Chem.dEur. J. 2012, 18, 12357e12362.
6. For recent examples on asymmetric Henry reaction based on bisamines ligands,see: (a) Arai, T.; Watanabe, M.; Fujiwara, A.; Yokoyama, N.; Yanagisawa, A.Angew. Chem. 2006, 45, 5978e5981; (b) Maheswaran, H.; Prasanth, K. L.;Krishna, G. G.; Ravikumar, K.; Sridhar, B.; Kantam, M. L. Chem. Commun. 2006,4066e4068; (c) Arai, T.; Watanabe, M.; Yanagisawa, A. Org. Lett. 2007, 9,3595e3597; (d) Arai, T.; Takashita, R.; Endo, Y.; Watanabe, M.; Yanagisawa, A. J.Org. Chem. 2008, 73, 4903e4906; (e) Kowalczyk, R.; Sidorowicz, q.; Skar _zewski,J. Tetrahedron: Asymmetry 2008, 19, 2310e2315; (f) Zhang, G.; Yashima, E.;Woggon, W.-D. Adv. Synth. Catal. 2009, 351, 1255e1262; (g) Breuning, M.; Hein,D.; Steiner, M.; Gessner, V. H.; Strohmann, C. Chem.dEur. J. 2009, 15,12764e12769; (h) Selvakumar, S.; Sivasankaran, D.; Singh, V. K. Org. Biomol.Chem. 2009, 7, 3156e3162; (i) Jin, W.; Li, X.; Huang, Y.; Wu, F.; Wan, B. Chem.dEur. J. 2010, 16, 8259e8261; (j) Steurer, M.; Bolm, C. J. Org. Chem. 2010, 75,3301e3310; (k) Noole, A.; Lippur, K.; Metsala, A.; Lopp, M.; Kanger, T. J. Org.Chem. 2010, 75, 1313e1316; (l) Chougnet, A.; Zhang, G.; Liu, K.; H€aussinger, D.;K€agi, A.; Allmendinger, T.; Woggon, W.-D. Adv. Synth. Catal. 2011, 353,1797e1806; (m) Lu, D.; Zhou, Y.; Li, Y.; Yan, S.; Gong, Y. J. Org. Chem. 2011, 76,8869e8878; (n) Zhou, Y.; Dong, J.; Zhang, F.; Gong, Y. J. Org. Chem. 2011, 76,588e600; (o) Gualandi, A.; Cerisoli, L.; Stoeckli-Evans, H.; Savoia, D. J. Org.Chem. 2011, 76, 3399e3408; (p) Jin, W.; Li, X.; Wan, B. J. Org. Chem. 2011, 76,484e491; (q) Yao, Q. J.; Judeh, Z. M. A. Tetrahedron 2011, 67, 4086e4092.
7. For recent examples on asymmetric Henry reaction based on N,N-dioxide li-gands, see: (a) Zhou, H.; Peng, D.; Qin, B.; Hou, Z. R.; Liu, X. H.; Feng, X. M. J. Org.Chem. 2007, 72, 10302e10304; (b) Qin, B.; Xiao, X.; Liu, X. H.; Huang, J. L.; Wen,Y. H.; Feng, X. M. J. Org. Chem. 2007, 72, 9323e9328; (c) Tan, C.; Liu, X. H.; Wang,L. W.; Wang, J.; Feng, X. M. Org. Lett. 2008, 10, 5305e5308.
8. For recent examples on asymmetric Henry reaction based on imino(amino)-pyridine ligands, see: (a) Blay, G.; Climent, E.; Fern�andez, I.; Hern�andez-Ol-mos, V.; Pedro, J. R. Tetrahedron: Asymmetry 2006, 17, 2046e2049; (b) Blay, G.;Climent, E.; Fern�andez, I.; Hern�andez-Olmos, V.; Pedro, J. R. Tetrahedron:Asymmetry 2007, 18, 1603e1612; (c) Blay, G.; Hernandez-Olmos, V.; Pedro, J. R.Chem. Commun. 2008, 4840e4842; (d) Blay, G.; Domingo, L. R.; Hernandez-

L. Zhang et al. / Tetrahedron 69 (2013) 10644e1065210652
Olmos, V.; Pedro, J. R. Chem.dEur. J. 2008, 14, 4725e4730; (e) Blay, G.; Her-nandez-Olmos, V.; Pedro, J. R. Org. Biomol. Chem. 2008, 6, 468e476; (f) Blay, G.;Hernandez-Olmos, V.; Pedro, J. R. Org. Lett. 2010, 12, 3058e3061; (g) Blay, G.;Hernandez-Olmos, V.; Pedro, J. R. Chem.dEur. J. 2011, 17, 3768e3773.
9. For recent examples on asymmetric Henry reaction based on Schiff-bases li-gands, see: (a) Bandini, M.; Piccinelli, F.; Tommasi, S.; Umani-Ronchi, A.; Ventrici,C. Chem. Commun. 2007, 616e618; (b) Xiong, Y.; Wang, F.; Huang, X.; Wen, Y.;Feng, X. Chem.dEur. J. 2007,13, 829e833; (c) Bandini, M.; Benaglia, M.; Sinisi, R.;Tommasi, S.; Umani-Ronchi, A. Org. Lett. 2007, 9, 2151e2153; (d) Colak, M.; Aral,T.; Hosg€oren, H.; Demirel, N. Tetrahedron: Asymmetry 2007, 18, 1129e1133; (e)Jiang, J.-J.; Shi, M. Tetrahedron: Asymmetry 2007,18, 1376e1382; (f) Lai, G.; Wang,S.; Wang, Z. Tetrahedron: Asymmetry 2008, 19, 1813e1819; (g) Yang, W.; Liu, H.;Du, D. M. Org. Biomol. Chem. 2010, 8, 2956e2960; (h) Ingalsbe, M. L.; St. Denis, J.D.; Gleason, J. L.; Savage, G. P.; Priefer, R. Synthesis 2010, 98e102; (i) Yang,W.; Du,D.-M. Eur. J. Org. Chem. 2011, 2011, 1552e1556; (j) Wei, Y.; Yao, L.; Zhang, B.; He,W.; Zhang, S. Tetrahedron 2011, 67, 8552e8558; (k) Lang, K.; Park, J.; Hong, S.Angew. Chem., Int. Ed. 2012, 51, 1620e1624; (l) Boobalan, R.; Lee, G.-H.; Chen, C.Adv. Synth. Catal. 2012, 354, 2511e2520; (m) Cheng, H.-G.; Lu, L.-Q.; Wang, T.;Chen, J.-R.; Xiao, W.-J. Chem. Commun. 2012, 5596e5598.
10. For recent examples on CeH bond activation using phenanthrolines ligands,see: (a) Gao, K.; Yoshikai, N. Angew. Chem., Int. Ed. 2011, 50, 6888e6892; (b) Ye,M.; Gao, G. L.; Edmunds, A. J.; Worthington, P. A.; Morris, J. A.; Yu, J. Q. J. Am.Chem. Soc. 2011, 133, 19090e19093; (c) Uchiyama, N.; Shirakawa, E.; Nishikawa,R.; Hayashi, T. Chem. Commun. 2011, 11671e11673; (d) Ton, T. M.; Tejo, C.; Tiong,D. L.; Chan, P. W. J. Am. Chem. Soc. 2012, 134, 7344e7350.
11. Examples on asymmetric catalytic cyclopropanation by using chiral phenan-throlines ligands, see: (a) Chelucci, G.; Cabras, M. A.; Saba, A. J. Mol. Catal. A:Chem. 1995, 95, L7eL10; (b) Chelucci, G.; Gladiali, S.; Sanna, M. G.; Brunner, H.Tetrahedron: Asymmetry 2000, 11, 3419e3426; (c) Chelucci, G.; Thummel, R. P.Chem. Rev. 2002, 102, 3129e3170; (d) Puglisi, A.; Benaglia, M.; Annunziata, R.;Bologna, A. Tetrahedron Lett. 2003, 44, 2947e2951; (e) Kwong, H.-L.; Yeung, H.-L.; Yeung, C.-T.; Lee, W.-S.; Lee, C.-S.; Wong, W.-L. Coord. Chem. Rev. 2007, 251,2188e2222.
12. Examples on asymmetric catalytic allylic alkylation by using chiral phenan-throlines ligands, see: (a) Pe~na-Cabrera, E.; Norrby, P.-O.; Sj€ogren, M.; Vita-gliano, A.; De Felice, V.; Oslob, J.; Ishii, S.; O’Neill, D.;�Akermark, B.; Helquist, P. J.Am. Chem. Soc. 1996, 118, 4299e4313; (b) Chelucci, G.; Saba, A. Tetrahedron:Asymmetry 1998, 9, 2575e2578; (c) Chelucci, G.; Pinna, G. A.; Saba, A.; Sanna, G.J. Mol. Catal. A: Chem. 2000, 159, 423e427; (d) Chelucci, G.; Saba, A.; Sanna, G.;Soccolini, F. Tetrahedron: Asymmetry 2000, 11, 3427e3438; (e) Chelucci, G.Chem. Soc. Rev. 2006, 35, 1230e1243.
13. Examples on asymmetric catalytic hydrosilylation by using chiral phenan-throlines ligands, see: (a) Gladiali, S.; Pinna, L.; Delogu, G.; Graf, E.; Brunner, H.Tetrahedron: Asymmetry 1990, 1, 937e942.
14. Examples on asymmetric catalytic transfer hydrogenation by using chiralphenanthrolines ligands, see: (a) Gladiali, S.; Chelucci, G.; Chessa, G.; Delogu,
G.; Soccolini, F. J. Organomet. Chem. 1987, 327, C15eC17; (b) Gladiali, S.; Che-lucci, G.; Soccolini, F.; Delogu, G.; Chessa, G. J. Organmet. Chem. 1989, 370,285e294; (c) Gill, M.; Gim�enez, A.; Jhingran, A. G.; Palfreyman, A. R. Tetrahe-dron: Asymmetry 1990, 1, 621e634.
15. Examples on asymmetric catalytic allylic oxidation by using chiral phenan-throlines ligands, see: (a) Chelucci, G.; Loriga, G.; Murineddu, G.; Pinna, G. A.Tetrahedron Lett. 2002, 43, 3601e3604; (b) Chelucci, G.; Iuliano, A.; Muroni, D.;Saba, A. J. Mol. Catal. A: Chem. 2003, 191, 29e33.
16. For views on the use of Cinchona alkaloids and it’s derivatives in organicsynthesis and catalysis, see: (a) Tian, S.-K.; Chen, Y.; Hang, J.; Tang, L.; McDaid,P.; Deng, L. Acc. Chem. Res. 2004, 37, 621e631; (b) Song, C. E. Cinchona Alkaloidsin Synthesis and Catalysis; Wiley-VCH: New York, NY, 2009; (c) Jew, S.-s.; Park,H.-g. Chem. Commun. 2009, 7090e7130; (d) Marcelli, T.; Hiemstra, H. Synthesis2010, 1229e1279; (e) Zhou, Q.-L. Privileged Chiral Ligands and Catalysts; Wiley-VCH: New York, NY, 2011; pp 361e408; (f) Yeboah, E. M. O.; Yeboah, S. O.;Singh, G. S. Tetrahedron 2011, 67, 1725e1762; (g) Jiang, L.; Chen, Y.-C. Catal. Sci.Technol. 2011, 1, 354e365; (h) Melchiorre, P. Angew. Chem., Int. Ed. 2012, 51,9748e9770.
17. (a) Constable, E. C.; Kulke, T.; Neuburger, M.; Zehnder, M. New J. Chem. 1997, 21,633e646; (b) Constable, E. C.; Kulke, T.; Neuburger, T.; Zehnder, M. New J. Chem.1997, 21, 1091e1102.
18. Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.;Cheeseman, J. R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. A.; Na-katsuji, H.; Caricato, M.; Li, X.; Hratchian, H. P.; Izmaylov, A. F.; Bloino, J.; Zheng,G.; Sonnenberg, J. L.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.;Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Montgomery,J. A.; Peralta, J. E., Jr.; Ogliaro, F.; Bearpark, M.; Heyd, J. J.; Brothers, E.; Kudin, K.N.; Staroverov, V. N.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A.;Burant, J. C.; Iyengar, S. S.; Tomasi, J.; Cossi, M.; Rega, N.; Millam, J. M.; Klene,M.; Knox, J. E.; Cross, J. B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.;Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.;Martin, R. L.; Morokuma, K.; Zakrzewski, V. G.; Voth, G. A.; Salvador, P.; Dan-nenberg, J. J.; Dapprich, S.; Daniels, A. D.; Farkas, €O.; Foresman, J. B.; Ortiz, J. V.;Cioslowski, J.; Fox, D. J. Gaussian 09, Rev. B.01; Gaussian: Wallingford, CT, 2009.
19. Zheng, B.; Wang, M.; Li, Z.; Bian, Q.; Mao, J.; Li, S.; Liu, S.; Wang, M.; Zhong, J.;Guo, H. Tetrahedron: Asymmetry 2011, 22, 1156e1160.
20. Zhou, Z.-M.; Li, Z.-H.; Hao, X.-Y.; Zhang, J.; Dong, X.; Liu, Y.-Q.; Sun, W.-W.; Cao,D.; Wang, J.-L. Org. Biomol. Chem. 2012, 10, 2113e2118.
21. Gruber-Khadjawi, M.; Purkarthofer, T.; Skranc, W.; Griengl, H. Adv. Synth. Catal.2007, 349, 1445e1450.
22. Bulut, A.; Aslan, A.; Dogan, €O. J. Org. Chem. 2008, 73, 7373e7375.23. Yao, Q.; Gao, Q.; Zaher, M. A. J. Eur. J. Org. Chem. 2011, 4892e4898.24. Cheng, L.; Dong, J.-X.; You, J.-S.; Gao, G.; Lan, J.-B. Chem.dEur. J. 2010, 16,
6761e6765.




















