
Synthesis, an experimental and quantum chemical computational study: proton sharing in...
10
Synthesis, an experimental and quantum chemical computational study of a new nonlinear optical material: 2-Picolinium hydrogensquarate Ufuk Korkmaz ⇑ , Ahmet Bulut Department of Physics, Faculty of Arts and Sciences, Ondokuzmayıs University, Kurupelit 55139, Samsun, Turkey highlights 2-Picolinium hydrogensquarate. Non-linear optical material. Supramolecular architectures and a-dimer. IR, UV spectroscopy and single crystal XRD. Ab initio calculations. graphical abstract The X-ray analysis clearly indicated that the crystal packing has shown the hydrogen bonding ring pat- tern of D 2 2 ð10Þ (a-dimer) through NAHO interactions. The hydrogensquarate anions form a-dimer, while 2-Picolinium molecule interacts through NAHO and CAHO with the hydrogensquarate anion. article info Article history: Received 24 January 2014 Received in revised form 4 April 2014 Accepted 7 April 2014 Available online 20 April 2014 Keywords: Squaric acid X-ray diffraction Strong hydrogen bonding Vibrational spectra Quantum chemical calculations Non-linear optical materials abstract The experimental and theoretical investigation results of a novel organic non-linear optical (NLO) organic squarate salt of 2-Picolinium hydrogensquarate (1), C 6 H 8 N þ C 4 HO 4 , were reported in this study. The space group of the title compound was found in the monoclinic C2/c space group. It was found that the asymmetric unit consists of one monohydrogen squarate anion together with mono protonated 2-Pic- olinium, forming the (1) salt. The X-ray analysis clearly indicated that the crystal packing has shown the hydrogen bonding ring pattern of D 2 2 ð10Þ (a-dimer) through NAHO interactions. The hydrogensquarate anions form a-dimer, while 2-Picolinium molecule interacts through NAHO and CAHO with the hydrogensquarate anion. The structural and vibrational properties of the compound were also studied by computational methods of ab initio performed on the compound at DFT/B3LYP/6-31++G(d,p) (2) and HF/6-31++G(d,p) (3) level of theory. The calculation results on the basis of two models for both the optimized molecular structure and vibrational properties for the 1 obtained are presented and com- pared with the X-ray analysis result. On the other the molecular electrostatic potential (MEP), electronic absorption spectra, frontier molecular orbitals (FMOs), conformational flexibility and non-linear optical properties (NLO) of the title compound were also studied at the 2 level and the results are reported. In order to evaluate the suitability for NLO applications thermal analysis (TG, DTA and DTG) data of 1 were also obtained. Ó 2014 Elsevier B.V. All rights reserved. http://dx.doi.org/10.1016/j.saa.2014.04.038 1386-1425/Ó 2014 Elsevier B.V. All rights reserved. ⇑ Corresponding author. Tel.: +90 5365291998; fax: +90 362 4576600. E-mail address: [email protected] (U. Korkmaz). Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 130 (2014) 376–385 Contents lists available at ScienceDirect Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy journal homepage: www.elsevier.com/locate/saa
Transcript of Synthesis, an experimental and quantum chemical computational study: proton sharing in...

Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 130 (2014) 376–385
Contents lists available at ScienceDirect
Spectrochimica Acta Part A: Molecular andBiomolecular Spectroscopy
journal homepage: www.elsevier .com/locate /saa
Synthesis, an experimental and quantum chemical computational studyof a new nonlinear optical material: 2-Picolinium hydrogensquarate
http://dx.doi.org/10.1016/j.saa.2014.04.0381386-1425/� 2014 Elsevier B.V. All rights reserved.
⇑ Corresponding author. Tel.: +90 5365291998; fax: +90 362 4576600.E-mail address: [email protected] (U. Korkmaz).
Ufuk Korkmaz ⇑, Ahmet BulutDepartment of Physics, Faculty of Arts and Sciences, Ondokuzmayıs University, Kurupelit 55139, Samsun, Turkey
h i g h l i g h t s
� 2-Picolinium hydrogensquarate.� Non-linear optical material.� Supramolecular architectures anda-dimer.� IR, UV spectroscopy and single crystal
XRD.� Ab initio calculations.
g r a p h i c a l a b s t r a c t
The X-ray analysis clearly indicated that the crystal packing has shown the hydrogen bonding ring pat-tern of D2
2ð10Þ (a-dimer) through NAH� � �O interactions. The hydrogensquarate anions form a-dimer,while 2-Picolinium molecule interacts through NAH� � �O and CAH� � �O with the hydrogensquarate anion.
a r t i c l e i n f o
Article history:Received 24 January 2014Received in revised form 4 April 2014Accepted 7 April 2014Available online 20 April 2014
Keywords:Squaric acidX-ray diffractionStrong hydrogen bondingVibrational spectraQuantum chemical calculationsNon-linear optical materials
a b s t r a c t
The experimental and theoretical investigation results of a novel organic non-linear optical (NLO) organicsquarate salt of 2-Picolinium hydrogensquarate (1), C6H8Nþ � C4HO�4 , were reported in this study. Thespace group of the title compound was found in the monoclinic C2/c space group. It was found thatthe asymmetric unit consists of one monohydrogen squarate anion together with mono protonated 2-Pic-olinium, forming the (1) salt. The X-ray analysis clearly indicated that the crystal packing has shown thehydrogen bonding ring pattern of D2
2ð10Þ (a-dimer) through NAH� � �O interactions. The hydrogensquarateanions form a-dimer, while 2-Picolinium molecule interacts through NAH� � �O and CAH� � �O with thehydrogensquarate anion. The structural and vibrational properties of the compound were also studiedby computational methods of ab initio performed on the compound at DFT/B3LYP/6-31++G(d,p) (2)and HF/6-31++G(d,p) (3) level of theory. The calculation results on the basis of two models for boththe optimized molecular structure and vibrational properties for the 1 obtained are presented and com-pared with the X-ray analysis result. On the other the molecular electrostatic potential (MEP), electronicabsorption spectra, frontier molecular orbitals (FMOs), conformational flexibility and non-linear opticalproperties (NLO) of the title compound were also studied at the 2 level and the results are reported. Inorder to evaluate the suitability for NLO applications thermal analysis (TG, DTA and DTG) data of 1 werealso obtained.
� 2014 Elsevier B.V. All rights reserved.

Table 1Crystal data and structure refinement for compound.
Formula C6H8NþC4HO�4Formula weight (g) 207.18Temperature (K) 293
U. Korkmaz, A. Bulut / Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 130 (2014) 376–385 377
Introduction
The hydrogen bonding is one of the most important forcesamong the interactions in crystal engineering especially consider-ing the arrangement of molecules and ions as building blocks [1–4]. The stability as well as supramolecular structure in crystalengineering is determined by the type of the hydrogen bondinginteraction [5–7]. Choosing proper donor and acceptor site, onecan obtain a novel hydrogen bonding pattern that is unique andhas a desired physical and chemical properties [8–10]. The hydro-gen-bonded systems formed by organic cations and anions shouldalso be mentioned here due to their strong interactions compare toneutral molecules [11–15]. It should be added here that the pro-ton-transfer in the structure significantly affects the hydrogenbonding features in the crystal structure of a salt [15–17]. Thisstrong interaction also promotes the nonlinear optical propertyof the material. Therefore many scientists in this contextsynthesize the novel materials showing nonlinear optical behaviorand study their potential in optical applications such as opticalcomputing, optical information processing, optical disk data stor-age, and laser remote sensing [18–20].
Ongoing study on squaric acid and its anions because of theirpotential in forming supramolecular structure as well as their non-linear optical properties, the title compound has been synthesized.Squaric acid (H2SQ) can be found in three forms uncharged H2SQ,the HSQ� monoanion and SQ2� dianions on deprotonating byamines [13–17,21–23]. In the squarate anion form, the anions aregenerally linked to amines by NAH� � �O hydrogen bonds [21–27].In the supramolecular architectures the monoanion HSQ� displaysa number of polyanionic forms classified as a-chains, b-chains, a-dimers and a-tetramers [13,14,21,25,28]. This study reports thesynthesis, spectroscopic, structural and optical characterization of1 (C6H8N+�C4HO�4 ) molecule (Fig. 1). The aim of this research istwofold. The first one is to determine the supramolecular architec-tures of title structure together with spectroscopic properties. Thesecond is to obtain optical properties of 1 for possible use as opticalmaterial. In order to do further investigation on the properties ofthe title compound, ab initio calculations were carried out. A com-parison of the experimental and theoretical results has been givenwhich is useful in making correct assignments in physical andchemical properties of the molecular structure. In order to these,molecular electrostatic potential (MEP), electronic absorptionspectra, frontier molecular orbitals (FMOs) and conformationalflexibility of the title compound were studied at the 2 level. Thenonlinear optical properties together with physicochemicalstability of the title compound were also studied.
Fig. 1. The molecular structure of compound showing the atom-numberingscheme. Displacement ellipsoids are drawn at the 40% probability level and Hatoms are shown as small spheres of arbitrary radii. The hydrogen bonds are shownas the dotted lines.
Experimental and theoretical methods
General method
The IR spectra were recorded on a Vertex 80v Bruker FTIR spec-trometer using KBr pellets and operating in the 4000–400 cm�1
range. Electronic absorption spectra were obtained for aqueoussolutions of the compound (10�3 M) with an Agilent TechnologiesCary 60 UV–Vis spectrometer in the range 800–200 nm. Thermalanalysis curves (TG, DTA and DTG) were recorded simultaneouslyon a Shimadzu DTG-60 thermal analyzer in N2 atmosphere at aheating rate of 10 �C �min�1 in the temperature range 50–600 �C using platinum crucibles.
Synthesis
All chemical reagents were analytical grade commercial prod-ucts. Solvent was purified by conventional methods. Squaric acid(H2Sq; 0.23 g, 2 mmol) and 2-Picoline (0.19 g; 2 mmol) were dis-solved in water (25 cm3) mixture in the molar ratio 1:1 and thesolution was heated to 50 �C in a temperature-controlled bathand stirred for one hour. The reaction mixture was then slowlycooled to room temperature. The crystals formed were filteredand washed with 10 cm3 of water and methanol and dried in air.A few days later, well-formed crystals were selected for X-ray stud-ies. Elemental analysis for 1, (colorless, yield 43%) C10H9NO4: Calc.:C, 57.97; H, 4.38; N, 6.76%. Found: C, 57.83; H, 4.32; N, 6.38%.M.p.:214 �C.
X-ray crystallography
The diffraction data were collected on a STOE IPDSII image platedetector using Mo Ka radiation (k = 0.71073 Å, T = 293 K). Thetechnique used was w-2h scan mode with limits 1.9–27.6�. A sum-mary of the key crystallographic information is given in Table 1.Data collection: Stoe X-AREA [29]. Cell refinement: Stoe X-AREA[29]. Data reduction: Stoe X-RED [29]. The structure was solvedby direct-methods using SHELXS-97 [30] and anisotropic displace-ment parameters were applied to non-hydrogen atoms in a
Wavelength (Å) (Mo Ka) 0.71069Crystal system MonoclinicSpace group C2/c
Unit cell dimensionsa, b, c (Å) 11.715 (5), 13.684 (5), 13.200 (5)b (�) 115.061 (5)Volume (Å3) 1916.9 (13)Z 8Absorption coefficient (mm�1) 0.11Calculated density (g cm�3) 1.436Crystal size (mm) 0.52 � 0.36 � 0.13F(000) 864h range (�) 1.7–27.2Index ranges �14 6 h 6 13; �17 6 k 6 17;
�16 6 l 6 16Measured reflections 7461Independent reflections 2024Reflections observed [I > 2r(I)] 1005Absorption correction IntegrationRefinement method Full-matrix least-squares on F2
Final R indices [I > 2r(I)] R1 = 0.0397 wR2 = 0.098Goodness-of-fit on F2 0.87Largest diff. peak and hole (e �3) 0.11, �0.12

378 U. Korkmaz, A. Bulut / Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 130 (2014) 376–385
full-matrix least-squares refinement based on F2 using SHELXL-97[30]. All H atoms attached to carbon atoms were positioned geo-metrically and refined by a riding model with Uiso 1.2 times thatof attached atoms and remaining H atoms were located from theFourier difference map. Molecular drawings were obtained usingDIAMOND 3.0 (demonstrated version) [31] and Mercury 2.3 [32].
Theoretical methods
The crystal structure was used as an initial molecular geometry.Quantum chemical calculations are performed with GAUSSIAN 03program packages [33]. The output files are visualized by meansof GAUSSIAN VIEW 03 software [34]. The geometry of neutral formof title compound was optimized at two levels of theory: DensityFunctional Theory (DFT) and Hartree–Fock Theory using samebasis set of 6-31++G(d,p) [35,36], respectively. The DFT methodemployed is B3LYP, which combines Becke’s three-parameternon-local exchange function with the correlation function of Lee,Yang and Parr [37,38]. Molecular geometry of the studied structurewas fully optimized by the force gradient method using Berny’salgorithm. For the structure, the stationary points found on themolecule potential energy hyper surfaces were characterized usingstandard analytical harmonic vibrational analysis. The absence ofthe imaginary frequencies, as well as of negative eigenvalues ofthe second-derivative matrix, confirmed that the stationary pointscorrespond to minima of the potential energy hyper surfaces.
It is known that DFT is quite strong method to investigate non-linear optical properties of organic materials [39–43]. Therefore,DFT method was used total molecular dipole moment (ltot), linearpolarizability (atot) and first-order hyperpolarizability (btot) param-eters which were obtained from Gaussian result file [44].
Results and discussion
Description of crystal structures
The analysis of the XRD data indicated that the title compoundcrystalized in the monoclinic C2/c space group. Fig. 1 shows theasymmetric unit of compound that contains [2-Picolinium]+ cation,
Table 2Selected structural parameters by X-ray diffraction experimental (1) and calculated by th
Bond lengths (Å)
1 2 3
C1AO1 1.256 (2) 1.303 1.253 CC2AO2 1.314 (2) 1.340 1.343 CC2AC1 1.415 (3) 1.387 1.379 OC3AO3 1.246 (2) 1.215 1.198 OC3AC2 1.422 (2) 1.470 1.437 CC3AC4 1.487 (3) 1.553 1.530 CC4AO4 1.217 (2) 1.207 1.185 OC4AC1 1.483 (3) 1.504 1.504 OC6AN1 1.344 (2) 1.350 1.333 OC6AC5 1.477 (3) 1.506 1.502 OC7AC6 1.380 (3) 1.400 1.386 OC8AC7 1.362 (3) 1.394 1.384 OC9AC10 1.354 (3) 1.393 1.375 CC9AC8 1.378 (3) 1.395 1.389 C
Torsion angels (�)
1
O3AC3AC2AO2 0.8 (5)C4AC3AC2AO2 �179.4 (3)O3AC3AC2AC1 �179.4 (3)C4AC3AC2AC1 0.44 (18)C8AC9AC10AN1 �0.3 (4)O4AC4AC1AO1 2.4 (5)
and HSQ� anions. It is clearly seen in Fig. 1 that the nitrogen atomof pyridine ring of a 2-picoline molecule is protonated by theremoved hydrogen atom from the squaric acid molecule, formingthe salt 1.
Table 2 gives the selected structural parameters of the (1–3). Itcan be seen in the table three CACAC bond angles in the HSQ�
anion close to 90� whereas the last C1AC2AC3 bond angle is foundto be of 93.25� due to the hydroxyl group attached to C2 atom. Inthe planar squarate ring system of 1, the CAC distances reflect par-tial double-bond character for C2AC1, C3AC2, [1.415 (3) and 1.422(2) Å, respectively] and single-bond character for C3AC4 andC4AC1 [1.487 (3) and 1.483 (3) Å, respectively]. The HSQ� ionhas one CAO bond (C2AO2) that is longer than a normal singleCAO bond, and two intermediate CAO bonds (C1AO1 andC3AO3), while the C4AO4 bond is typical of a C@O double bond(Table 2). These lengths indicate a degree of delocalization in theHSQ� ion, as has been observed in previous studies [22–28,45,46].
The crystal packing is obtained by various forms of hydrogenbonding interaction. The network of the OAH� � �O hydrogen bondsforms D2
2ð10Þ type ring (Fig. 2) via HSQ� anions. According to theclassification of Gilli et al. [21,47,48], N� � �O distance should be inthe region of 2.72–2.78 Å, N� � �O distances in N1AH1� � �O1 (2.659(2) Å) (Table 3) which is called as [(+/�) CAHB] interaction. A dif-ference Fourier map (Fig. 3) [22] of the electron density clearlyindicated that the H atom is in between of the region of the N1and O1 atom, with the maximum lying closer to the N1 than theO1 atom, rather than being bound closely to the N1 atom to givea discrete neutral pair. In the interaction the behavior of the H1atom indicates a large degree of covalency. The (O� � �O) distancein the symmetry related O2AH2� � �O3i hydrogen bonding interac-tions are 2.542 (2) Å, given Table 3. Considering the ranges(2.38–2.50 Å) the values in the Gilli’s classification the interactionis also called as the negative charge assisted hydrogen bonding[(�) CAHB] [21,47,48] and is symbolized as [OAH� � �O]�. This is alsoobserved in the other squarate compounds formed by HSQ� anionsand is called ring a-dimer [16,17,21,25,28]. Looking at N� � �O dis-tances in the symmetry related hydrogen bonding between HSQ�
anions, it can be seen that the interaction is a bit shorter thanthe relevant interval values and is symbolized as either
e 2 and 3 for the compound.
Bond angels (�)
1 2 3
1AC4AC3 87.98 (14) 86.79 87.1901AC2AC3 93.25 (15) 94.57 95.9081AC1AC2 135.47 (18) 137.88 136.751AC1AC4 134.92 (17) 130.30 133.232AC3AC4 89.16 (14) 86.815 86.8852AC1AC4 89.60 (14) 91.82 90.0172AC2AC1 130.83 (15) 133.42 131.832AC2AC3 135.91 (17) 132.01 132.263AC3AC2 137.64 (19) 133.73 135.253AC3AC4 133.19 (17) 139.46 137.864AC4AC1 136.42 (17) 135.47 135.864AC4AC3 135.60 (17) 137.74 136.957AC8AC9 120.2 (2) 119.08 120.1610AC9AC8 118.6 (2) 118.15 117.86
2 3
�0.00652 �0.00363180.00000 179.99740�0.00652 �179.99741
0.00000 0.003630.00000 �0.000410.00577 0.00000

Fig. 2. The two-dimensional sheet assembly in the crystal structure of 1 viewed down the z-axes.
U. Korkmaz, A. Bulut / Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 130 (2014) 376–385 379
N+AH� � �O1/2� or N+AH� � �O� (+/�) CAHB. The oxygen atomsengaged with NAH� � �O or OAH� � �O hydrogen bonds are responsi-ble for forming the ribbons, the weak intermolecular interactionssuch as CAH� � �O (Table 3) and C@O� � �p [49] (Table 4) are respon-sible for connecting these ribbons [50] (Fig. 4) especially the cen-troid–centroid and interplanar distances of p-stacking interactionwhich are less than 4.00 Å as given in Table 4 [51].
Optimized geometry
The ab initio calculation methods were also applied to the com-pound at 2 and 3 level of theory (Fig. 5). Some optimized geometricparameters of the compound are listed in Table 2. The calculatedenergies and dipole moments for both 2 and 3 are�742.13878407 and �737.80592741 a.u. and 11.0516 and16.6375 D, respectively. A superposition of the molecular structureof the compound as established by quantum mechanical calcula-tions and X-ray study shows almost perfect matching (Fig. 6). Com-paring the theoretical data with the experimental results indicatethat optimized bond lengths and angle values are slightly different
from these of the experimental ones. This small difference origi-nate from that the theoretical calculations are performed for iso-lated molecule in gas-phase whereas the experimental databelong to solid-phase. It should be noted that the geometry ofthe solid state structure is subject to intra and intermolecularinteractions, such as hydrogen bonding and van der Waalsinteractions.
As seen in Table 2, the largest discrepancies between thecalculated and experimental geometrical parameters are observedfor OAH and NAH bond distances. These large deviations fromexperimental OAH and NAH bond distances may arise from thelow scattering factors of hydrogen atoms in the X-ray diffraction.Several authors have been reported that the HF methods underestimate some bond length and DFT/B3LYP methods predicts bondlengths which are systematically too long, especially CAH bondlengths [52–54].
The largest difference between experimental and calculated 2bond lengths is 0.066 Å (C3AC4). The calculation results of 3 leadsto geometric parameters much closer to experimental one. The big-gest difference between observed and calculated values of bond
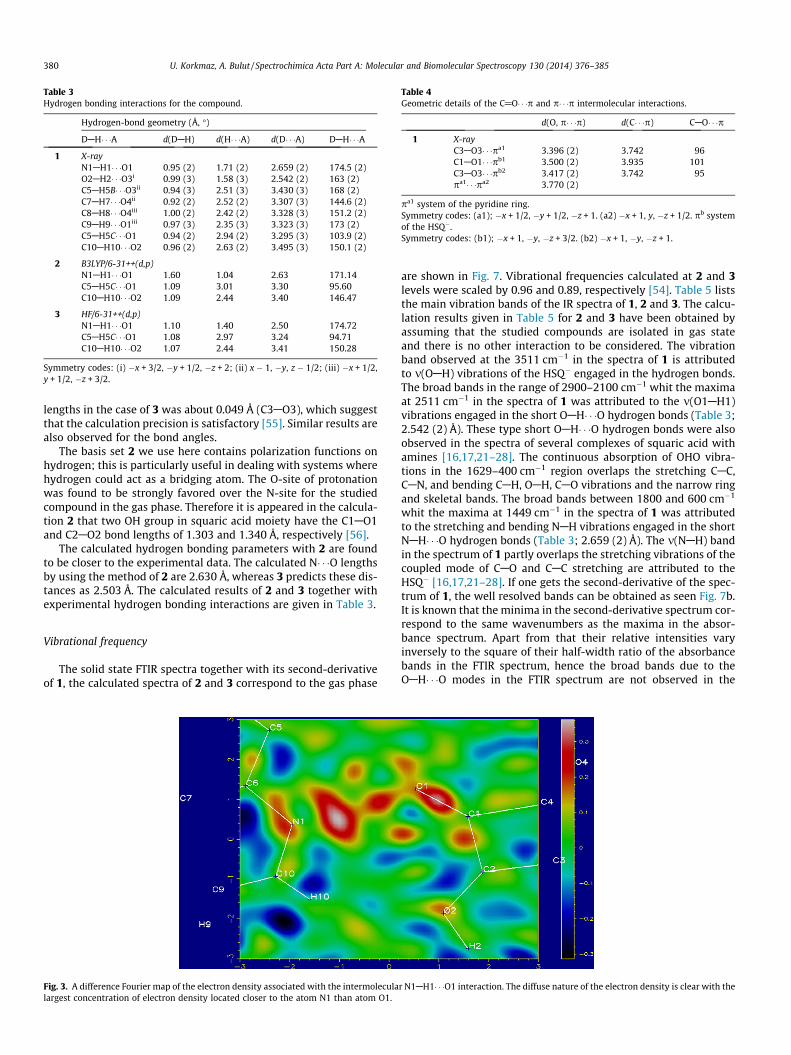
Table 3Hydrogen bonding interactions for the compound.
Hydrogen-bond geometry (Å, �)
DAH� � �A d(DAH) d(H� � �A) d(D� � �A) DAH� � �A
1 X-rayN1AH1� � �O1 0.95 (2) 1.71 (2) 2.659 (2) 174.5 (2)O2AH2� � �O3i 0.99 (3) 1.58 (3) 2.542 (2) 163 (2)C5AH5B� � �O3ii 0.94 (3) 2.51 (3) 3.430 (3) 168 (2)C7AH7� � �O4ii 0.92 (2) 2.52 (2) 3.307 (3) 144.6 (2)C8AH8� � �O4iii 1.00 (2) 2.42 (2) 3.328 (3) 151.2 (2)C9AH9� � �O1iii 0.97 (3) 2.35 (3) 3.323 (3) 173 (2)C5AH5C� � �O1 0.94 (2) 2.94 (2) 3.295 (3) 103.9 (2)C10AH10� � �O2 0.96 (2) 2.63 (2) 3.495 (3) 150.1 (2)
2 B3LYP/6-31++(d,p)N1AH1� � �O1 1.60 1.04 2.63 171.14C5AH5C� � �O1 1.09 3.01 3.30 95.60C10AH10� � �O2 1.09 2.44 3.40 146.47
3 HF/6-31++(d,p)N1AH1� � �O1 1.10 1.40 2.50 174.72C5AH5C� � �O1 1.08 2.97 3.24 94.71C10AH10� � �O2 1.07 2.44 3.41 150.28
Symmetry codes: (i) �x + 3/2, �y + 1/2, �z + 2; (ii) x � 1, �y, z � 1/2; (iii) �x + 1/2,y + 1/2, �z + 3/2.
Table 4Geometric details of the C@O� � �p and p� � �p intermolecular interactions.
d(O, p� � �p) d(C� � �p) CAO� � �p
1 X-rayC3AO3� � �pa1 3.396 (2) 3.742 96C1AO1� � �pb1 3.500 (2) 3.935 101C3AO3� � �pb2 3.417 (2) 3.742 95pa1� � �pa2 3.770 (2)
pa1 system of the pyridine ring.Symmetry codes: (a1); �x + 1/2, �y + 1/2, �z + 1. (a2) �x + 1, y, �z + 1/2. pb systemof the HSQ�.Symmetry codes: (b1); �x + 1, �y, �z + 3/2. (b2) �x + 1, �y, �z + 1.
380 U. Korkmaz, A. Bulut / Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 130 (2014) 376–385
lengths in the case of 3 was about 0.049 Å (C3AO3), which suggestthat the calculation precision is satisfactory [55]. Similar results arealso observed for the bond angles.
The basis set 2 we use here contains polarization functions onhydrogen; this is particularly useful in dealing with systems wherehydrogen could act as a bridging atom. The O-site of protonationwas found to be strongly favored over the N-site for the studiedcompound in the gas phase. Therefore it is appeared in the calcula-tion 2 that two OH group in squaric acid moiety have the C1AO1and C2AO2 bond lengths of 1.303 and 1.340 Å, respectively [56].
The calculated hydrogen bonding parameters with 2 are foundto be closer to the experimental data. The calculated N� � �O lengthsby using the method of 2 are 2.630 Å, whereas 3 predicts these dis-tances as 2.503 Å. The calculated results of 2 and 3 together withexperimental hydrogen bonding interactions are given in Table 3.
Vibrational frequency
The solid state FTIR spectra together with its second-derivativeof 1, the calculated spectra of 2 and 3 correspond to the gas phase
Fig. 3. A difference Fourier map of the electron density associated with the intermoleculalargest concentration of electron density located closer to the atom N1 than atom O1.
are shown in Fig. 7. Vibrational frequencies calculated at 2 and 3levels were scaled by 0.96 and 0.89, respectively [54]. Table 5 liststhe main vibration bands of the IR spectra of 1, 2 and 3. The calcu-lation results given in Table 5 for 2 and 3 have been obtained byassuming that the studied compounds are isolated in gas stateand there is no other interaction to be considered. The vibrationband observed at the 3511 cm�1 in the spectra of 1 is attributedto m(OAH) vibrations of the HSQ� engaged in the hydrogen bonds.The broad bands in the range of 2900–2100 cm�1 whit the maximaat 2511 cm�1 in the spectra of 1 was attributed to the m(O1AH1)vibrations engaged in the short OAH� � �O hydrogen bonds (Table 3;2.542 (2) Å). These type short OAH� � �O hydrogen bonds were alsoobserved in the spectra of several complexes of squaric acid withamines [16,17,21–28]. The continuous absorption of OHO vibra-tions in the 1629–400 cm�1 region overlaps the stretching CAC,CAN, and bending CAH, OAH, CAO vibrations and the narrow ringand skeletal bands. The broad bands between 1800 and 600 cm�1
whit the maxima at 1449 cm�1 in the spectra of 1 was attributedto the stretching and bending NAH vibrations engaged in the shortNAH� � �O hydrogen bonds (Table 3; 2.659 (2) Å). The m(NAH) bandin the spectrum of 1 partly overlaps the stretching vibrations of thecoupled mode of CAO and CAC stretching are attributed to theHSQ� [16,17,21–28]. If one gets the second-derivative of the spec-trum of 1, the well resolved bands can be obtained as seen Fig. 7b.It is known that the minima in the second-derivative spectrum cor-respond to the same wavenumbers as the maxima in the absor-bance spectrum. Apart from that their relative intensities varyinversely to the square of their half-width ratio of the absorbancebands in the FTIR spectrum, hence the broad bands due to theOAH� � �O modes in the FTIR spectrum are not observed in the
r N1AH1� � �O1 interaction. The diffuse nature of the electron density is clear with the

Fig. 4. The two-dimensional layered assembly in the crystals of 1, viewed down they-axes. The hydrogen bonds and weak interactions are shown as the dotted lines.
U. Korkmaz, A. Bulut / Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 130 (2014) 376–385 381
second-derivative spectrum [24–28]. There are several CAO bondsin the crystal structure of complex 1 belong to the hydrogen squa-rate units. The compound shows broad and very intense bands at1809 and 1439 cm�1, assignable to a coupled mode of CAO andCAC stretching are attributed to the HSQ�, which are calculated
Fig. 5. The ab initio calculation methods were also applie
at 1810, 1494 and 1843, 1469 cm�1 with 2 (Fig. 7c) and 3(Fig. 7d), respectively [22,23,25–27]. Comparing our theoreticaldata with experimental IR-spectra for HSQ� group in the com-pound, it can be concluded that the IR pattern can be bestdescribed as a superposition of the IR maxima of HSQ�. It shouldalso be noted here that slightly split in the stretching vibrationcompared to the corresponding vibration in K2C4O4 [57] indicatesthe symmetry is lower than D4h, confirmed by the X-ray analysis.
To make a comparison with experimental observations, westudied the correlation between the calculated and the experimen-tal data, and obtained correlation coefficients of 0.9984 and 0.9940for 2 and 3, respectively. As we can see from the correlation graphsin Fig. 8, the experimental fundamentals are found to have a goodcorrelation with the calculations.
Electronic absorption spectra
For the compound, the experimental electronic spectra in anaqueous solution exhibit for the carbonyl groups absorption band(335 nm) in the range of 200–800 nm (Fig. 9) [16–23]. The theoret-ical electronic excitation energies, oscillator strengths and natureof the first 10 spin-allowed singlet–singlet excitations were calcu-lated by the TD-DFT and TD-HF methods for the aqueous solution.TD-HF calculations are obtained from the absorption wavelengthsunder 265 nm. TD-HF calculations, the predicted absorption wave-lengths for compounds are not corresponding to the experimentalresults, which indicate that the HF-CIS model is not suitable to beused here to predict the electronic absorption spectra. TD-DFT cal-culations, the predicted absorption wavelengths in the range of231–332 nm, can predict the electronic absorption spectraapproximately.
The frontier molecular orbitals play an important role in theelectric and optical properties, as well as in UV–Vis spectra andchemical reactions [58]. According to the Molecular orbital coeffi-cients analyses based on DFT/B3LYP optimized structure indicatethat frontier molecular orbitals are mainly composed of p atomicorbitals, so electronic transitions from the HOMO � 2, HOMO � 1and HOMO to the LUMO are mainly derived from the n ? p* tran-sition. Fig. 10 shows the distributions and energy levels of theHOMO � 1, HOMO, LUMO and LUMO + 1 orbitals for the moleculein gas phase for compound. The positive phase is red and the neg-ative one is green. It is clear from Fig. 10 that while the HOMO andHOMO � 1 orbitals are delocalized on the HSQ�, in LUMO andLUMO + 1 electron cloud is completely delocalized on whole pyri-dine group in the compound. The value of the energy separationbetween the HOMO and LUMO is 2.21 eV. This large HOMO–LUMOgap clearly indicates the high excitation energies for many of theexcited states, good stability and a large chemical hardness forthe title compound.
d to the compound at (a) 2 and (b) 3 level of theory.

Fig. 6. Atom-by-atom superimposition of the structures calculated (red) (a = 2; b = 3) over the X-ray structure (black) for the compound. (For interpretation of the referencesto colour in this figure legend, the reader is referred to the web version of this article.)
Fig. 7. Infrared spectra of 2-Picolinium hydrogensquarate; (a) the solid-state spectrum of 1; (b) the second-derivative spectrum of 1; (c) frequencies calculated by the DFT/B3LYP/6-31++G(d,p) approach for 2; (d) frequencies calculated by the HF/6-31++G(d,p) approach for 3.
382 U. Korkmaz, A. Bulut / Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 130 (2014) 376–385

Table 5Experimental (mexp), calculated (mcalc), scaled (mscaled) vibrational frequencies (cm�1) and intensities (km/mol) of the calculated frequencies for the compound.
1 2 3
Assignments mexp mcalc mscaled Intensity mcalc mscaled Intensity
m(OAH)sq 3511w 3747 3587 96.56 4138 3682 150.99msym(CAH)pyr 3114 3222 3093 7.54 3407 3032 9.68masym(CAH)pyr 3053 3212 3083 7.23 3393 3019 11.40masmy(CH3) 3028sh 3142 3016 14.94 3303 2940 14.87msmy(CH3) 2959 3053 2931 6.40 3212 2859 4.37m(O1AH1) 2511m 2571 2468 4249.22 – – –(msym(CAC) + masym(CAO))sq + m(NAH) 1809m 1885 1810 207.47 2071 1843 377.02(masym(CAC) + masym(CAO))sq + m(NAH) – 1826 1753 367.26 2019 1797 1040.89(masym(CAC) + masym(CAO))sq + m(NAH) 1671s 1683 1616 1076.38 1969 1752 1056.87m(ring)pyr + r(NAH) 1625s 1654 1588 49.36 1926 1714 89.83(masym(CAC) + masym(CAO))sq + m(ring)pyr + m(NAH) + c(CH3) 1554s 1624 1559 114.83 1714 1525 3120.66(masym(CAC) + masym(CAO))sq + r(NAH) + m(ring)pyr 1439s 1556 1494 272.41 1650 1469 205.54r(NAH) + m(ring)pyr + c(CH3) 1410s 1496 1436 36.51 1608 1431 35.14c(CH3) – 1484 1424 9.12 1599 1423 7.25(masym(CAC) + masym(CAO))sq + m(ring)pyr + m(NAH) + c(CH3) 1385 1413 1356 556.71 1527 1359 2385.26r(OAH)sq 1293 1353 1298 117.57 1490 1326 118.07d(ring)pyr + d(CH3) 1058m 1133 1087 23.94 1226 1091 67.15m(CAN) + m(ring) 1041m 1077 1033 13.35 1199 1067 55.89hsq + d(ring)F 624s 654 628 27.08 749 666 180.46
Asym: asymmetric, sym: symmetric, m: medium, s: strong, w: weak, vs: very strong, m: stretching, h: ring breathing, d: bending, c: out-of-plane bending, r: rocking, a:scissoring, pyr: pyridine, sh:shoulder.
Fig. 8. Correlation graphic of calculated and experimental frequencies of title compound.
Fig. 9. UV–vis spectra of 1.
U. Korkmaz, A. Bulut / Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 130 (2014) 376–385 383
Molecular electrostatic potential
The molecular electrostatic potential (MEP) is related to theelectronic density and is a very useful descriptor in understandingsites for electrophilic attack and nucleophilic reactions as well ashydrogen-bonding interactions [59,60]. Being a real physicalproperty, V(r) can be determined experimentally by diffraction orby computational methods [61]. To predict reactive sites for elec-trophilic and nucleophilic attack for the title molecule, MEP was
calculated by applying the DFT(B3LYP) method with using 6-31++G(d,p) basis set for the optimized geometry. The negative(orange) regions of MEP were related to electrophilic reactivityand the positive (blue) regions to nucleophilic reactivity sitesshown in Fig. 11. As can be seen in Fig. 11, there are two possiblesites on the title compound for electrophilic attack. The negativeregions are mainly around O3 and O4 atoms. The positive regionis mainly around H7 atom. The maximum values of negative andpositive regions are �0.0817 and 0.0604 a.u., respectively. Theseresults provide information concerning the region where the com-pound can have intra- or intermolecular interaction and metallicbonding. So, the MEP map confirms the existence of intra- andintermolecular interactions observed in the solid state. Theseresults obtained in MEP analysis clearly consistent with the X-ray experimental results given in Table 3.
Nonlinear optical properties
The values of dipole moment (ltot), linear polarizability (atot)and first-order hyperpolarizability (btot) of the molecule were cal-culated at the level of 2 by using Gaussian 03W program. Theobtained values for ltot, atot and btot are 10.9319 D, 20.9656 Å3
and 2.3985 � 10�30 cm5/esu, respectively.

Fig. 10. Molecular orbital surfaces and energy levels using the DFT/B3LYP methodfor the HOMO � 1, HOMO, LUMO and LUMO + 1 of compound.
Fig. 12. TG, DTG and DTA curves of 1.
384 U. Korkmaz, A. Bulut / Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 130 (2014) 376–385
The urea is accepted as a prototype molecule for nonlinear opti-cal materials and results were compared with its values [62]. Thecalculation results of ltot, atot and btot for urea at the level of 2are 3.8583 D, 4.9991 Å3 and 3.2637 � 10�31 cm5/esu, respectively.Applying the same method to the molecule, it has been found thatthe value of btot is 7.3490 times greater than that of urea. Thisresult clearly indicates that the title compound is a strong candi-date to develop a nonlinear optical material.
Thermal analysis
The thermal analysis (TG/DTA) of C6H8N+�C4HO�4 molecule wascarried out with the temperature gradient of 10 �C �min�1 andin the range of 50 �C and 600 �C under the nitrogen atmosphere.Fig. 12 gives the TGA and its differential thermogravimetric spec-trum of the compound. It can be followed from Fig. 12 that thereis intensive mass loss in the temperature range of 200 �C and300 �C indicating that the structure does not have any crystalwater and stable up to 200 �C. The TGA curve shows an endother-mic transition in the region of 200 �C and 300 �C. The sharp endo-thermic peak around 210 �C can be attributed to the melting pointof 1. The decomposition and evaporation of the compound occur
Fig. 11. Molecular electrostatic pote
over 300 �C. These result clearly express that the compound of 1can be used as nonlinear optical material below 200 �C [20].
Conclusion
The experimental and theoretical investigation of newly syn-thesized squarate salt of 2-Picolinium hydrogensquarate (1),C6H8Nþ � C4HO�4 , were reported in this study. The single X-raydiffraction study clearly indicated that the packing is obtained bythe OAH� � �O homonuclear hydrogen bonds that forms D2
2ð10Þ (a-dimer) type ring give contribution as both donor and acceptor tothe crystal packing (Fig. 2) via HSQ� anions as an acceptor bridge.Glancing at acceptor–donor distances in hydrogen bonding in thestructure (Table 3), one can conclude that CAHB interactions aredominant in the crystal packing. The hydrogen bonds make themolecular assembly to produce a unidimensional construction inthe supramolecular view, while the p-stacking interactions extendthis to bidimensionality.
The calculation methods were compared with the experimentalresults. The experimental NAH and OAH vibration frequencies
ntial map calculated at 2 level.

U. Korkmaz, A. Bulut / Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 130 (2014) 376–385 385
involving the hydrogen bond slightly different from those of calcu-lated ones. The result also put forth that TD-HF method is not aconvenient method for calculation for the electronic absorptions,whereas TD-DFT method is a much better method to obtain elec-tronic absorption parameters giving very close values to the exper-imental results. The UV data indicated that the electronictransition in the compound is n ? p*. The values of dipole moment(ltot), linear polarizability (atot) and first-order hyperpolarizability(btot) of the molecule were calculated at the level of 2. Applying thesame method to the molecule, it has been found that the value offirst-order hyperpolarizability is 7.3490 times greater than thatof urea. The thermal analysis study showed that the melting pointof the compound 1 is 210 �C and is indicating that the 1 is usefulfor making NLO devices below 200 �C.
Supplementary material
CCDC 947689 contains supplementary crystallographic data(excluding structure factors) for the structure reported in this arti-cle. These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/data_request/cif, by e-mailing [email protected] or by contacting The Cambridge CrystallographicData Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +441223 336033.
Acknowledgements
We would like thank to Ondokuz Mayıs University ResearchFund (PYO.FEN.1904.12.020) for financial support. Special thanksalso goes to Dr. Necmi Dege for collecting XRD data and to Dr.Yıldıray Topçu for supplying UV–vis and thermal analysis data.
References
[1] G.R. Desiraju, Crystal Engineering: The Design of Organic Solids, Elsevier,Amsterdam, 1989.
[2] M.C. Etter, Acc. Chem. Res. 23 (1990) 120.[3] V. Bertolasi, P. Gilli, V. Ferrety, G. Gilli, Act. Cryst. B57 (1995) 1004.[4] G.A. Jeffrey, An Introduction to Hydrogen Bonding, Oxford University Press,
New York, 1997.[5] G.R. Desiraju, C.V.K. Sharma, The Crystal as a Supramolecular Entity, 1995.[6] G.R. Desiraju, Nature 412 (2001) 397.[7] C.B. Aakeröy, Acta. Cryst. B53 (1997) 569.[8] J.M. Lehn, Angew. Chem. Int. Ed. Engl. 29 (1990) 1304.[9] G.R. Desiraju, Angew. Chem. Int. Ed. Engl. 34 (1995) 2311.
[10] J.C. MacDonald, G.M. Whitesides, Chem. Rev. 94 (1994) 2383.[11] M.T. Reetz, S. Hooger, K. Harms, Angew. Chem. Int. Ed. Engl. 33 (1994) 181.[12] A. Bulut, O.Z. Yesilel, N. Dege, H. Icbudak, H. Olmez, O. Buyukgungor, Acta.
Cryst. C59 (2003) o727.[13] S. Mathew, G. Paul, K. Shivasankar, A. Choudhury, C.N.R. Rao, J. Mol. Struct. 641
(2002) 263.[14] V. Bertolasi, P. Gilli, V. Ferretti, G. Gilli, Acta. Cryst. B57 (2001) 591.[15] D.M.S. Martins, D.S. Middlemiss, C.R. Pulham, C.C. Wilson, M.T. Weller, P.F.
Henry, N. Shankland, K. Shankland, W.G. Marshall, R.M. Ibberson, K. Knight, S.Moggach, M. Brunelli, C.A. Morrison, J. Am. Chem. Soc. 131 (2009) 3884.
[16] T. Kolev, R.W. Seidel, B.B. Koleva, H. Mayer-Figge, M. Spiteller, W.S. Sheldrick, J.Mol. Struct. 931 (2009) 45.
[17] B.B. Koleva, T. Kolev, R.W. Seidel, T. Tsanev, H. Mayer-Figge, M. Spiteller, W.S.Sheldrick, Spectrochim. Acta A71 (2008) 695.
[18] D.S. Chemla, J. Zyss, Nonlinear Optical Properties of Organic Molecules andCrystals, Academic Press, New York, 1987.
[19] J.F. Nicoud, Mol. Cryst. Liq. Cryst. 156 (1988) 257.[20] C.R. Raja, P. Paramasivam, N. Vijayan, Spectrochim. Acta Part A 69 (2008) 1146.[21] G. Gilli, V. Bertolasi, P. Gilli, V. Ferretti, Acta. Cryst. B57 (2001) 859.[22] U. Korkmaz, _I. Uçar, A. Bulut, O. Büyükgüngör, Struct. Chem. 22 (2011) 1249.
[23] U. Korkmaz, A. Bulut, J. Mol. Struct. 1050 (2013) 61.[24] P. Barczynski, Z. Dega-Szafran, A. Katrusiak, M. Szafran, J. Mol. Struct. 1013
(2012) 95.[25] Z. Dega-Szafran, G. Dutkiewicz, Z. Kosturkiewicz, J. Mol. Struct. 1029 (2012)
28.[26] Z. Dega-Szafran, G. Dutkiewicz, Z. Kosturkiewicz, M. Szafran, J. Mol. Struct.
1007 (2012) 113.[27] P. Barczynski, Z. Dega-Szafran, A. Katrusiak, M. Szafran, J. Mol. Struct. 998
(2011) 240.[28] B.B. Koleva, T. Kolev, R.W. Seidel, M. Spiteller, H. Mayer-Figge, W.S. Sheldrick, J.
Phys. Chem. A 113 (2009) 3088.[29] Stoe & Cie X-AREA (Version 118) and X-RED (Version 104). Stoe & Cie,
Darmstadt, Germany, 2002.[30] G.M. Sheldrick SHELXL97, Program for Crystal Structure Refinement.
University of Göttingen, Germany, 1997.[31] Brandenburg K, DIAMOND, Demonstrated Version, Crystal Impact GbR, Bonn,
Germany, 2005.[32] C.F. Macrae, I.J. Bruno, J.A. Chisholm, P.R. Edgington, P. McCabe, E. Pidcock, L.
Rodriguez-Monge, R. Taylor, J. van de Streek, P.A. Wood, J. Appl. Cryst. 41(2008) 466.
[33] M.J. Frisch, G.W. Trucks, H.B. Schlegel, G.E. Scuseria, M.A. Robb, J.R. Cheeseman,J.A. Montgomery Jr., T. Vreven, K.N. Kudin, J.C. Burant, J.M. Millam, S.S. Iyengar,J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G.A.Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa,M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J.E. Knox,H.P. Hratchian, J.B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R.E.Stratmann, O. Yazyev, A.J. Austin, R. Cammi, C. Pomelli, J.W. Ochterski, P.Y.Ayala, K. Morokuma, G.A. Voth, P. Salvador, J.J. Dannenberg, V.G. Zakrzewski, S.Dapprich, A.D. Daniels, M.C. Strain, O. Farkas, D.K. Malick, A.D. Rabuck, K.Raghavachari, J.B. Foresman, J.V. Ortiz, Q. Cui, A.G. Baboul, S. Clifford, J.Cioslowski, B.B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R.L.Martin, D.J. Fox, T. Keith, M.A. Al-Laham, C.Y. Peng, A. Nanayakkara, M.Challacombe, P.M.W. Gill, B. Johnson, W. Chen, M.W. Wong, C. Gonzalez, J.A.Pople, Gaussian 03, Revision E.01, Gaussian Inc., Wallingford, C.T., 2004.
[34] R. Dennington II, T. Keith, J. Millam, Gauss View, Version 4.1.2, Semichem Inc.,Shawnee Mission, KS, 2007.
[35] H.B. Schlegel, J. Comput. Chem. 3 (1982) 214.[36] R. Ditchfield, W.J. Hehre, J.A. Pople, J. Chem. Phys. 54 (1971) 724.[37] A.D. Becke, J. Chem. Phys. 98 (1993) 5648.[38] C. Lee, W. Yang, R.G. Parr, Phys. Rev. B37 (1988) 785.[39] Y.-X. Sun, Q.-L. Hao, Z.-X. Yu, W.-X. Wei, L.-D. Lu, X. Wang, Mol. Phys. 107
(2009) 223.[40] J.P. Abraham, D. Sajan, V. Shettigar, S.D. Dharmaprakash, I. Nemec, I.H. Joe, V.S.
Jayakumar, J. Mol. Struct. 917 (2009) 27.[41] A.B. Ahmed, H. Feki, Y. Abid, H. Boughzala, C. Minot, Spectrochim. Acta Part A
75 (2010) 293.[42] A.B. Ahmed, H. Feki, Y. Abid, H. Boughzala, C. Minot, A. Mlayah, J. Mol. Struct.
920 (2009) 1.[43] S.G. Sagdinc, A. Esme, Spectrochim. Acta Part A 75 (2010) 1370.[44] K.S. Thanthiriwatte, K.M. Nalin de Silva, J. Mol. Struct. Theochem. 617 (2002)
169.[45] P. Barczynski, Z. Dega-Szafran, A. Katrusiak, M. Szafran, J. Mol. Struct. 1018
(2012) 28.[46] _I. Uçar, A. Bulut, O. Büyükgüngör, Acta. Cryst. C61 (2005) m266.[47] P. Gilli, V. Bertolasi, V. Ferretti, G. Gilli, J. Am. Chem. Soc. 116 (1994) 909.[48] G. Gilli, P. Gilli, J. Mol. Struct. 552 (2000) 1.[49] J.A. Silva, A.C. Santos, A.T. Marques, M.R. Silva, A.M. Beja, A.J.F.N. Sobral, J.
Chem. Cryst. 38 (2008) 301.[50] C.E. Silva, H.F. Dos Santos, N.L. Speziali, R. Diniz, L.F.C. de Oliveira, J. Phys.
Chem. A114 (2010) 10097.[51] A.L. Spek, Acta Cryst. D65 (2009) 148.[52] S.Y. Lee, Bull. Kor. Chem. Soc. 19 (1998) 93.[53] (a) S.Y. Lee, B.H. Boo, J. Phys. Chem. 100 (1996) 15073;
(b) S.Y. Lee, B.H. Boo, J. Phys. Chem. 100 (1996) 8782.[54] M. Kurt, S. Yurdakul, J. Mol. Struct. 654 (2003) 1.[55] F.F. Jian, P.S. Zhaou, Q. Yu, Q.X. Wang, K. Jiao, J. Phys. Chem. A108 (2004) 5258.[56] S.A.K. Elroby, H.A. Ewais, S.G. Aziz, Chin. Sci. Bull. 57 (2012) 1665.[57] M. Ito, B. West, J. Am. Chem. Soc. 85 (1963) 2580.[58] I. Fleming, Frontier Orbitals and Organic Chemical Reactions, Wiley, London,
1976.[59] E. Scrocco, J. Tomasi, Adv. Quantum. Chem. 11 (1978) 115.[60] F.J. Luque, J.M. Lopez, M. Orozco, Theor. Chem. Acc. 103 (2000) 343.[61] P. Politzer, P.R. Laurence, K. Jayasuriya, J. McKinney, Environ. Health Perspect.
61 (1985) 191.[62] L.S. Pu, Acs Sypos. Series 455 (1991) 331.
















![Bis[(1 S *,2 S *)- trans -1,2-bis(diphenylphosphinoxy)cyclohexane]chloridoruthenium(II) trifluoromethanesulfonate dichloromethane disolvate](https://static.fdokumen.com/doc/165x107/63360a7bcd4bf2402c0b5520/bis1-s-2-s-trans-12-bisdiphenylphosphinoxycyclohexanechloridorutheniumii.jpg)



