
Substituent effect on supramolecular motifs in series of succinimide polycyclic keto derivatives –...
8
Substituent effect on supramolecular motifs in series of succinimide polycyclic keto derivatives – Spectroscopic, theoretical and crystallographic studies Barbara Miroslaw a,⇑ , Anna E. Koziol a , Anna Bielenica b , Kamil Dziuba a , Marta Struga c a Faculty of Chemistry, Maria Curie-Sklodowska University, 20-031 Lublin, Poland b Department of Biochemistry, Medical University of Warsaw, Warszawa 02-097, Poland c Department of Pharmacogenomics, Medical University of Warsaw, 02-007 Warszawa, Poland highlights The substituent effect on supramolecular motifs in cyclic imides. The sequence of carbonyl stretching vibration bands in IR spectra was determined. The correlation between geometry and energetics of hydrogen bonds was analysed. Solvation free energies were computed using IEF PCM model. Binding energies for supramolecular clusters were calculated. graphical abstract article info Article history: Received 2 December 2013 Received in revised form 12 May 2014 Accepted 14 May 2014 Available online 28 May 2014 Keywords: Substituent effect Supramolecular synthon Hirshfeld surface analysis Solvation free energy PCM Binding energy abstract The substituent effect on the supramolecular arrangement in a series of polycyclic monoimide keto derivatives crystals was studied. Single crystal X-ray diffraction and IR spectroscopic experiments were performed for seven related compounds, as well as the Hirshfeld surface analysis and quantum chemical calculations at HF and DFT levels in vacuo, in solution and for small clusters. The presence of C@O group at the bridge of the main hydrocarbon skeleton implied the catemer motif of the N imide AHO imide hydrogen bond in case of smaller substituents (HA, MeOA, EtOA). For more voluminous groups ( i BuOA) or additional hydrogen bond acceptors (AcOA,O@) the steric hindrance increased and the imideimide interactions were no longer present in the solid state. The N imide AHO keto or N imide AHO ester hydrogen bonds were formed instead. The binding energy per one NAHO interaction calculated for supramolec- ular clusters at HF/6-31G(d,p) level was ca. 20 kJ mol 1 , indicating moderate strength of this hydrogen bond. The solvation free energies and induced dipole moments were computed at B3LYP/6-311+G(d,p) level using the integral equation formalism model (IEF PCM) considering three solvents of various polar- ity: non-polar chloroform, polar aprotic dimethyl sulfoxide (DMSO) and polar protic water. The relations between the vibrational spectra and the crystal structure have been discussed. The following sequence of carbonyl stretching modes in IR spectra has been derived from quantum chemical calculations: (1) at the highest frequencies – the symmetric vibration of two imide C@O bonds, (2) the vibrations of keto C@O bonds attached directly to the polycyclic hydrocarbon skeleton, (3) the asymmetric vibration of two imide C@O bonds, and (4) at the lowest frequencies – the vibration of ester C@O group. The characteristic http://dx.doi.org/10.1016/j.molstruc.2014.05.029 0022-2860/Ó 2014 Elsevier B.V. All rights reserved. ⇑ Corresponding author. Address: Faculty of Chemistry, Maria Curie-Sklodowska University, Maria Curie-Sklodowska sq. 3, PL. 20-031 Lublin, Poland. Tel.: +48 81 5375582. E-mail address: [email protected] (B. Miroslaw). Journal of Molecular Structure 1074 (2014) 695–702 Contents lists available at ScienceDirect Journal of Molecular Structure journal homepage: www.elsevier.com/locate/molstruc
Transcript of Substituent effect on supramolecular motifs in series of succinimide polycyclic keto derivatives –...

Journal of Molecular Structure 1074 (2014) 695–702
Contents lists available at ScienceDirect
Journal of Molecular Structure
journal homepage: www.elsevier .com/ locate /molst ruc
Substituent effect on supramolecular motifs in series of succinimidepolycyclic keto derivatives – Spectroscopic, theoretical andcrystallographic studies
http://dx.doi.org/10.1016/j.molstruc.2014.05.0290022-2860/� 2014 Elsevier B.V. All rights reserved.
⇑ Corresponding author. Address: Faculty of Chemistry, Maria Curie-Sklodowska University, Maria Curie-Sklodowska sq. 3, PL. 20-031 Lublin, Poland. Tel.: +48 81 5E-mail address: [email protected] (B. Miroslaw).
Barbara Miroslaw a,⇑, Anna E. Koziol a, Anna Bielenica b, Kamil Dziuba a, Marta Struga c
a Faculty of Chemistry, Maria Curie-Sklodowska University, 20-031 Lublin, Polandb Department of Biochemistry, Medical University of Warsaw, Warszawa 02-097, Polandc Department of Pharmacogenomics, Medical University of Warsaw, 02-007 Warszawa, Poland
h i g h l i g h t s
� The substituent effect onsupramolecular motifs in cyclicimides.� The sequence of carbonyl stretching
vibration bands in IR spectra wasdetermined.� The correlation between geometry
and energetics of hydrogen bondswas analysed.� Solvation free energies were
computed using IEF PCM model.� Binding energies for supramolecular
clusters were calculated.
g r a p h i c a l a b s t r a c t
a r t i c l e i n f o
Article history:Received 2 December 2013Received in revised form 12 May 2014Accepted 14 May 2014Available online 28 May 2014
Keywords:Substituent effectSupramolecular synthonHirshfeld surface analysisSolvation free energyPCMBinding energy
a b s t r a c t
The substituent effect on the supramolecular arrangement in a series of polycyclic monoimide ketoderivatives crystals was studied. Single crystal X-ray diffraction and IR spectroscopic experiments wereperformed for seven related compounds, as well as the Hirshfeld surface analysis and quantum chemicalcalculations at HF and DFT levels in vacuo, in solution and for small clusters. The presence of C@O group atthe bridge of the main hydrocarbon skeleton implied the catemer motif of the NimideAH� � �Oimide hydrogenbond in case of smaller substituents (HA, MeOA, EtOA). For more voluminous groups (iBuOA) oradditional hydrogen bond acceptors (AcOA, O@) the steric hindrance increased and the imide� � �imideinteractions were no longer present in the solid state. The NimideAH� � �Oketo or NimideAH� � �Oester hydrogenbonds were formed instead. The binding energy per one NAH� � �O interaction calculated for supramolec-ular clusters at HF/6-31G(d,p) level was ca. 20 kJ mol�1, indicating moderate strength of this hydrogenbond. The solvation free energies and induced dipole moments were computed at B3LYP/6-311+G(d,p)level using the integral equation formalism model (IEF PCM) considering three solvents of various polar-ity: non-polar chloroform, polar aprotic dimethyl sulfoxide (DMSO) and polar protic water. The relationsbetween the vibrational spectra and the crystal structure have been discussed. The following sequence ofcarbonyl stretching modes in IR spectra has been derived from quantum chemical calculations: (1) at thehighest frequencies – the symmetric vibration of two imide C@O bonds, (2) the vibrations of keto C@Obonds attached directly to the polycyclic hydrocarbon skeleton, (3) the asymmetric vibration of twoimide C@O bonds, and (4) at the lowest frequencies – the vibration of ester C@O group. The characteristic
375582.

Compound R1 R2 R3 R4I H- H- H- H-II MeO- H- H- H-III EtO- H- H- H-IV iBuO- H- H- H-V AcO- H- H- H-VI AcO- H- H3C- H3C-
VII H- O= H- H-
Scheme 1. Molecular structure of com
696 B. Miroslaw et al. / Journal of Molecular Structure 1074 (2014) 695–702
peaks observed in imide experimental IR spectra at about 3080 cm�1 have been explained as overtoneand combination bands of mC@O stretching and cNAH out-of-plane bending vibrations.
� 2014 Elsevier B.V. All rights reserved.
Introduction competitive role of the keto substituent (attached directly to the
Bulky substituted cyclic imides are of great importance due totheir biological properties. For example Migrastatin (a naturallyoccurring compound) and its analogues are acetylcholine receptorantagonists and have potential anticancer activity [1,2]. A series ofisoindoledione based compounds were identified as potent andro-gen receptor antagonists [3]. Some derivatives with cyclic imideskeleton are aminopeptidase N inhibitors [4]. Norbormide and itsanalogs were studied due to their rat selective toxicant activity[5]. The II generation anxiolytics used in the treatment of psychoticand neurotic disorders, such as buspirone, gepirone or tandospi-rone, are polycyclic imide derivatives with a high affinity to the5-HT1A receptor [6–9]. Selected alkylarylpiperazinyl derivativesof 4-azatricyclo[5.2.2.02,6]undecane-3,5,8-trione show antidepres-sant activity and have no neurotoxic effect [10]. Other cyclic imidecompounds were evaluated on broad spectrum of pharmacologicalproperties, such as antibiotic, fungicidal, analgesic, anxiolytic, andcytostatic activities [11].
Polycyclic imides I–VII (Scheme 1) have been used as startingmaterials for synthesis of potential 5-HT1A receptor ligands [11–18]. The arylpiperazine derivatives have anxiolytic activity oncentral nervous system [11,16,17]. The O-methoxy-phenylpiperaz-inyl derivatives of II and VI show a similar 5-HT1A/5-HT2A bindingprofile as buspirone [11,16]. Imides I [12], IV and V [15] have beenevaluated in vitro for their cytotoxicity and anti-HIV-1 (HumanImmunodeficiency Virus type 1) activity. In addition to the biolog-ical assays, tetrahydro-1H-isoindole-1,3,5(2H,4H)-trione deriva-tives are used in polymer synthesis [18].
The knowledge about the most common supramolecularassemblies in solids is helpful in crystal engineering, solid-stateand computational chemistry etc. However, even the mostfrequently occurring supramolecular motifs (synthons), whichare observed for a given molecular fragment, should be only con-sidered as an indicator of possible intermolecular contacts [19].The supramolecular association preferences of amides [19,20], dii-mides [21] and simple derivatives of succinimide [22] were studiedpreviously. The secondary cyclic monoimides form both dimersand chains, through the intermolecular NimideAH� � �Oimide hydrogenbonds nearly equally often. The association motifs depend on theshape and hydrophobic character of the interacting molecules.They can be modified by the presence of substituents bonded tothe main hydrocarbon skeleton [22].
As a continuation of our studies on the intermolecular associa-tion preferences of the primary imide group [22], the analysis ofthe crystal and molecular structures of seven C10-substituted ketoderivatives of 4-aza-tricyclo [5.2.2.02,6]undecane-3,5-dione I–VII(Scheme 1) is presented. This study was aimed to determine the
R3 NH
R1
O
O
O
R2
R4
pounds I–VII.
polycyclic skeleton) on the intermolecular interaction modesbetween the imide groups in the solid state. To gain deeper insightinto relations between the geometry and energetics of hydrogenbonds in the analysed structures, the combination of Hirshfeld sur-face analysis, solid state IR spectroscopy and quantum-chemicalcalculations was used. The theoretical vibrational spectra were cal-culated in vacuo and in solution, using the Hartree–Fock (HF) anddensity functional theory (DFT) methods with the 6-311+G(d,p)basis set. The sequence of appearance of mC@O vibrations, comingfrom various functional groups, have been determined. The solva-tion free energy values and induced dipole moments in solutionshave been determined at B3LYP/6-311+G(d,p) level using the inte-gral equation formalism model (IEF PCM) [23]. Three solvents ofvarious polarity were included in these calculations: non-polarchloroform, polar aprotic dimethyl sulfoxide (DMSO) and polarprotic water. Additionally, the binding energy DE for a singleNAH� � �O hydrogen bond in each supramolecular cluster has beencalculated at HF/6-31G(d,p) level.
Results and discussion
Molecular structure
The atom-numbering scheme for the molecules I–VII is shownin Fig. 1. The molecular structure of I has already been publishedby our group [12], and its crystal structure is discussed here withmore details. The rigid hydrocarbon skeleton of the analysed mol-ecules had the cis,exo configuration. The C10-bonded keto oxygenatom was in the plane of the alkane fragment (C4AC9AC10AC7)with the largest deviation for I being of ca. 0.2 Å (Table S1). TheO-alkyl substituents (R1) at the bridgehead C4 atom had the ap,apconformation with respect to the C9AC10AC4 fragment. Torsionangles were close to 172� for substituted esters, and higher valueswere observed for the ether groups (Table S1).
Crystal structure
It is worth to notice that four of the seven mentionedcompounds crystallized in the non-centrosymmetric space groups(viz. P21 and P212121) as homo-enantiomeric crystals (Table S2).The synthesis reaction leading to the studied compounds is non-enantioselective [11–14,16,17,15]. The acetone solutions of ana-lysed compounds did not rotate the plane-polarized light. Thisindicated that both enantiomers were present in solution in equalquantities. The analysed compounds occurred in solids asconglomerates of enantiomeric crystals (III–V and VII) or as trueracemic compounds (I, II, VI) with both enantiomers in one crystalstructure.
The most typical supramolecular motifs based on the Nimide
AH� � �Oimide hydrogen bonds observed in solid are the R22(8) ring
and the C(4) chain [19,20,22,24]. These types of interactions areenergetically favorable because of the phenomenon of intermolecu-lar Resonance Assisted Hydrogen Bond (RAHB) [25]. However, theR2
2(8) ring motif was not observed in the analysed group of crystalstructures. This is in accordance with our previous studies [22].The asymmetric substitution of the main hydrocarbon skeletonresulted in the catemeric self-assemblies. Moreover, the introduction
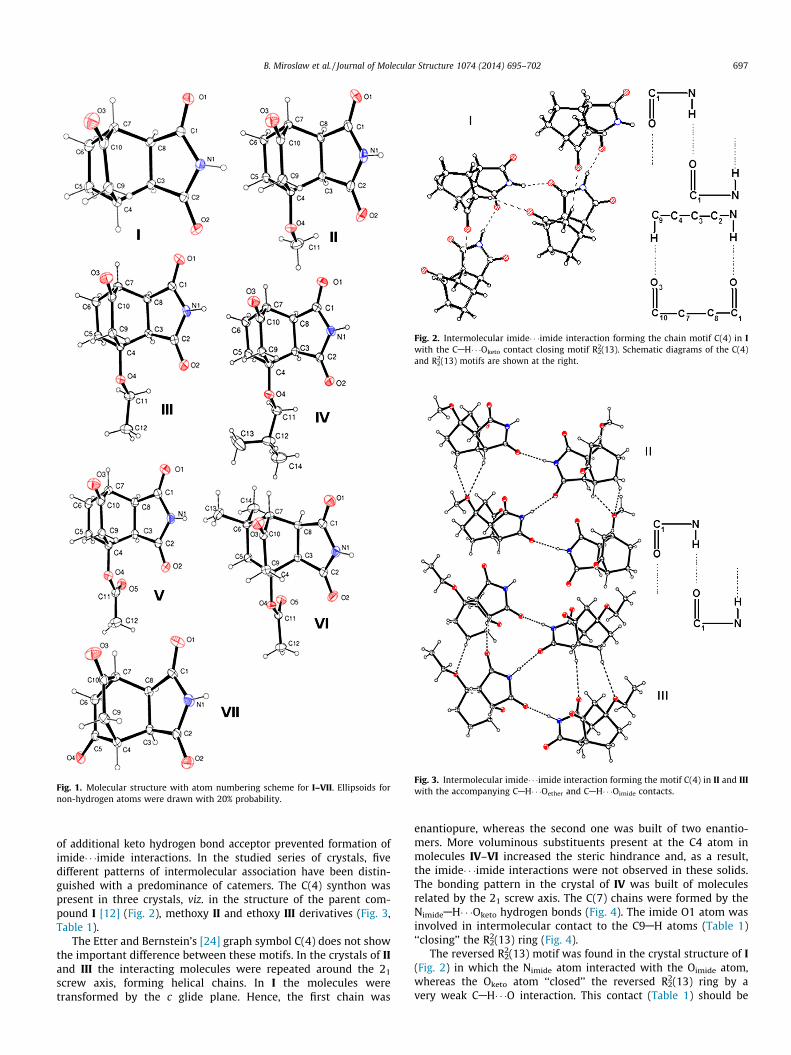
Fig. 1. Molecular structure with atom numbering scheme for I–VII. Ellipsoids fornon-hydrogen atoms were drawn with 20% probability.
Fig. 2. Intermolecular imide� � �imide interaction forming the chain motif C(4) in Iwith the CAH� � �Oketo contact closing motif R2
2(13). Schematic diagrams of the C(4)and R2
2(13) motifs are shown at the right.
Fig. 3. Intermolecular imide� � �imide interaction forming the motif C(4) in II and IIIwith the accompanying CAH� � �Oether and CAH� � �Oimide contacts.
B. Miroslaw et al. / Journal of Molecular Structure 1074 (2014) 695–702 697
of additional keto hydrogen bond acceptor prevented formation ofimide� � �imide interactions. In the studied series of crystals, fivedifferent patterns of intermolecular association have been distin-guished with a predominance of catemers. The C(4) synthon waspresent in three crystals, viz. in the structure of the parent com-pound I [12] (Fig. 2), methoxy II and ethoxy III derivatives (Fig. 3,Table 1).
The Etter and Bernstein’s [24] graph symbol C(4) does not showthe important difference between these motifs. In the crystals of IIand III the interacting molecules were repeated around the 21
screw axis, forming helical chains. In I the molecules weretransformed by the c glide plane. Hence, the first chain was
enantiopure, whereas the second one was built of two enantio-mers. More voluminous substituents present at the C4 atom inmolecules IV–VI increased the steric hindrance and, as a result,the imide� � �imide interactions were not observed in these solids.The bonding pattern in the crystal of IV was built of moleculesrelated by the 21 screw axis. The C(7) chains were formed by theNimideAH� � �Oketo hydrogen bonds (Fig. 4). The imide O1 atom wasinvolved in intermolecular contact to the C9AH atoms (Table 1)‘‘closing’’ the R2
2(13) ring (Fig. 4).The reversed R2
2(13) motif was found in the crystal structure of I(Fig. 2) in which the Nimide atom interacted with the Oimide atom,whereas the Oketo atom ‘‘closed’’ the reversed R2
2(13) ring by avery weak CAH� � �O interaction. This contact (Table 1) should be
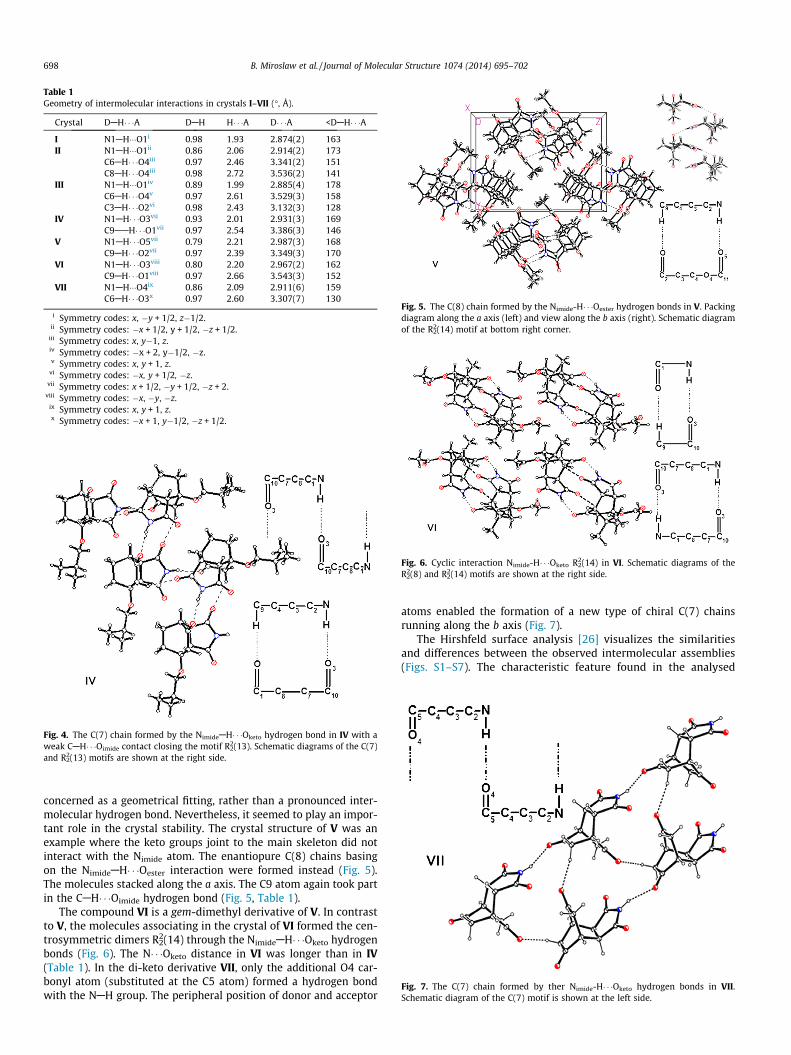
Table 1Geometry of intermolecular interactions in crystals I–VII (�, Å).
Crystal DAH� � �A DAH H� � �A D� � �A <DAH� � �A
I N1AH���O1i 0.98 1.93 2.874(2) 163II N1AH���O1ii 0.86 2.06 2.914(2) 173
C6AH� � �O4iii 0.97 2.46 3.341(2) 151C8AH� � �O4iii 0.98 2.72 3.536(2) 141
III N1AH���O1iv 0.89 1.99 2.885(4) 178C6AH� � �O4v 0.97 2.61 3.529(3) 158C3AH� � �O2vi 0.98 2.43 3.132(3) 128
IV N1AH� � �O3vii 0.93 2.01 2.931(3) 169C9AAH� � �O1vii 0.97 2.54 3.386(3) 146
V N1AH� � �O5vii 0.79 2.21 2.987(3) 168C9AH� � �O2vii 0.97 2.39 3.349(3) 170
VI N1AH� � �O3viii 0.80 2.20 2.967(2) 162C9AH� � �O1viii 0.97 2.66 3.543(3) 152
VII N1AH���O4ix 0.86 2.09 2.911(6) 159C6AH� � �O3x 0.97 2.60 3.307(7) 130
i Symmetry codes: x, �y + 1/2, z�1/2.ii Symmetry codes: �x + 1/2, y + 1/2, �z + 1/2.
iii Symmetry codes: x, y�1, z.iv Symmetry codes: �x + 2, y�1/2, �z.v Symmetry codes: x, y + 1, z.
vi Symmetry codes: �x, y + 1/2, �z.vii Symmetry codes: x + 1/2, �y + 1/2, �z + 2.
viii Symmetry codes: �x, �y, �z.ix Symmetry codes: x, y + 1, z.x Symmetry codes: �x + 1, y�1/2, �z + 1/2.
Fig. 4. The C(7) chain formed by the NimideAH� � �Oketo hydrogen bond in IV with aweak CAH� � �Oimide contact closing the motif R2
2(13). Schematic diagrams of the C(7)and R2
2(13) motifs are shown at the right side.
Fig. 5. The C(8) chain formed by the Nimide-H� � �Oester hydrogen bonds in V. Packingdiagram along the a axis (left) and view along the b axis (right). Schematic diagramof the R2
2(14) motif at bottom right corner.
Fig. 6. Cyclic interaction Nimide-H� � �Oketo R22(14) in VI. Schematic diagrams of the
R22(8) and R2
2(14) motifs are shown at the right side.
Fig. 7. The C(7) chain formed by ther Nimide-H� � �Oketo hydrogen bonds in VII.Schematic diagram of the C(7) motif is shown at the left side.
698 B. Miroslaw et al. / Journal of Molecular Structure 1074 (2014) 695–702
concerned as a geometrical fitting, rather than a pronounced inter-molecular hydrogen bond. Nevertheless, it seemed to play an impor-tant role in the crystal stability. The crystal structure of V was anexample where the keto groups joint to the main skeleton did notinteract with the Nimide atom. The enantiopure C(8) chains basingon the NimideAH� � �Oester interaction were formed instead (Fig. 5).The molecules stacked along the a axis. The C9 atom again took partin the CAH� � �Oimide hydrogen bond (Fig. 5, Table 1).
The compound VI is a gem-dimethyl derivative of V. In contrastto V, the molecules associating in the crystal of VI formed the cen-trosymmetric dimers R2
2(14) through the NimideAH� � �Oketo hydrogenbonds (Fig. 6). The N� � �Oketo distance in VI was longer than in IV(Table 1). In the di-keto derivative VII, only the additional O4 car-bonyl atom (substituted at the C5 atom) formed a hydrogen bondwith the NAH group. The peripheral position of donor and acceptor
atoms enabled the formation of a new type of chiral C(7) chainsrunning along the b axis (Fig. 7).
The Hirshfeld surface analysis [26] visualizes the similaritiesand differences between the observed intermolecular assemblies(Figs. S1–S7). The characteristic feature found in the analysed
B. Miroslaw et al. / Journal of Molecular Structure 1074 (2014) 695–702 699
crystal structures was that the Z-oriented C@O bonds, viz. O1imide
and O3keto, were preferred in the intermolecular NAH� � �O contacts,whereas the opposite side of the molecule was involved in hydro-phobic interactions.
IR spectroscopy
Spectroscopic studies on imides in solid, solution and low-temperature Ar and N2 matrices have been carried out by manygroups of scientists focusing on the mNAH and mC@O vibrationregions [27–37]. The experiments performed in solution (in water,pentane, chloroform and methanol) by Hulme et al. [27] have notconfirmed the existence of hydrogen bonded dimers, irrespectivelyof whether the solvent was polar or non-polar. The analysis ofmC@O vibration region indicated only a partial solvation. Theaggregation process of imide derivatives in solutions and low-tem-perature Ar and N2 matrices has been studied by Glice, Bajdor, Leset al. [32–34]. The conclusion was, that the additional broad bandsat about 3200 and 3100 cm�1 originate from NAH moiety involvedin hydrogen bonding and may be used as indicators of supramolec-ular dimerization. The intensity of these peaks increase with soluteconcentration. Simultaneously, the intensity of a band around3400 cm�1 which is characteristic of the non-interacting monomer,decreases.
Correlations have been found between the solid state IR data ofI–VII and the geometry and symmetry of the observed intermolec-ular interactions types (Fig. S8, Table 2).
In crystals of I, II and III, the difference between the symmetryof the association motifs (C(4) NAH� � �Oimide chains) can be
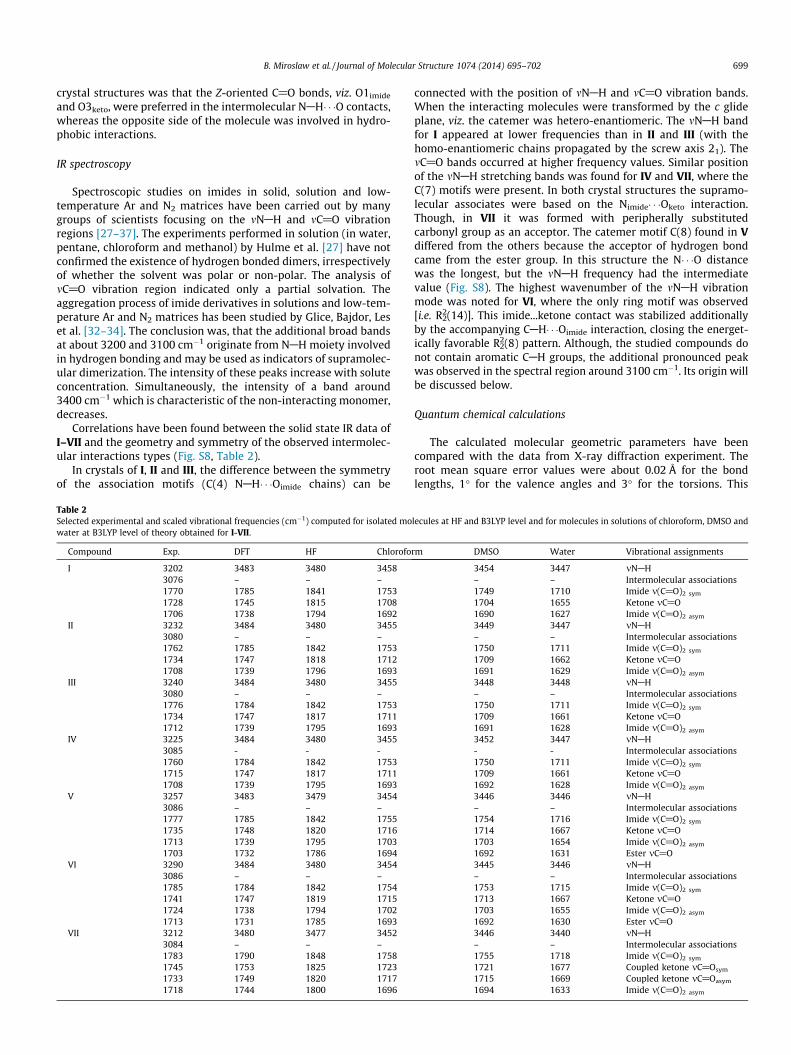
Table 2Selected experimental and scaled vibrational frequencies (cm�1) computed for isolated mowater at B3LYP level of theory obtained for I-VII.
Compound Exp. DFT HF Chlorofo
I 3202 3483 3480 34583076 – – –1770 1785 1841 17531728 1745 1815 17081706 1738 1794 1692
II 3232 3484 3480 34553080 – – –1762 1785 1842 17531734 1747 1818 17121708 1739 1796 1693
III 3240 3484 3480 34553080 – – –1776 1784 1842 17531734 1747 1817 17111712 1739 1795 1693
IV 3225 3484 3480 34553085 - - -1760 1784 1842 17531715 1747 1817 17111708 1739 1795 1693
V 3257 3483 3479 34543086 – – –1777 1785 1842 17551735 1748 1820 17161713 1739 1795 17031703 1732 1786 1694
VI 3290 3484 3480 34543086 – – –1785 1784 1842 17541741 1747 1819 17151724 1738 1794 17021713 1731 1785 1693
VII 3212 3480 3477 34523084 – – –1783 1790 1848 17581745 1753 1825 17231733 1749 1820 17171718 1744 1800 1696
connected with the position of mNAH and mC@O vibration bands.When the interacting molecules were transformed by the c glideplane, viz. the catemer was hetero-enantiomeric. The mNAH bandfor I appeared at lower frequencies than in II and III (with thehomo-enantiomeric chains propagated by the screw axis 21). ThemC@O bands occurred at higher frequency values. Similar positionof the mNAH stretching bands was found for IV and VII, where theC(7) motifs were present. In both crystal structures the supramo-lecular associates were based on the Nimide� � �Oketo interaction.Though, in VII it was formed with peripherally substitutedcarbonyl group as an acceptor. The catemer motif C(8) found in Vdiffered from the others because the acceptor of hydrogen bondcame from the ester group. In this structure the N� � �O distancewas the longest, but the mNAH frequency had the intermediatevalue (Fig. S8). The highest wavenumber of the mNAH vibrationmode was noted for VI, where the only ring motif was observed[i.e. R2
2(14)]. This imide...ketone contact was stabilized additionallyby the accompanying CAH� � �Oimide interaction, closing the energet-ically favorable R2
2(8) pattern. Although, the studied compounds donot contain aromatic CAH groups, the additional pronounced peakwas observed in the spectral region around 3100 cm�1. Its origin willbe discussed below.
Quantum chemical calculations
The calculated molecular geometric parameters have beencompared with the data from X-ray diffraction experiment. Theroot mean square error values were about 0.02 Å for the bondlengths, 1� for the valence angles and 3� for the torsions. This
lecules at HF and B3LYP level and for molecules in solutions of chloroform, DMSO and
rm DMSO Water Vibrational assignments
3454 3447 mNAH– – Intermolecular associations1749 1710 Imide m(C@O)2 sym
1704 1655 Ketone mC@O1690 1627 Imide m(C@O)2 asym
3449 3447 mNAH– – Intermolecular associations1750 1711 Imide m(C@O)2 sym
1709 1662 Ketone mC@O1691 1629 Imide m(C@O)2 asym
3448 3448 mNAH– – Intermolecular associations1750 1711 Imide m(C@O)2 sym
1709 1661 Ketone mC@O1691 1628 Imide m(C@O)2 asym
3452 3447 mNAH- - Intermolecular associations1750 1711 Imide m(C@O)2 sym
1709 1661 Ketone mC@O1692 1628 Imide m(C@O)2 asym
3446 3446 mNAH– – Intermolecular associations1754 1716 Imide m(C@O)2 sym
1714 1667 Ketone mC@O1703 1654 Imide m(C@O)2 asym
1692 1631 Ester mC@O3445 3446 mNAH– – Intermolecular associations1753 1715 Imide m(C@O)2 sym
1713 1667 Ketone mC@O1703 1655 Imide m(C@O)2 asym
1692 1630 Ester mC@O3446 3440 mNAH– – Intermolecular associations1755 1718 Imide m(C@O)2 sym
1721 1677 Coupled ketone mC@Osym
1715 1669 Coupled ketone mC@Oasym
1694 1633 Imide m(C@O)2 asym
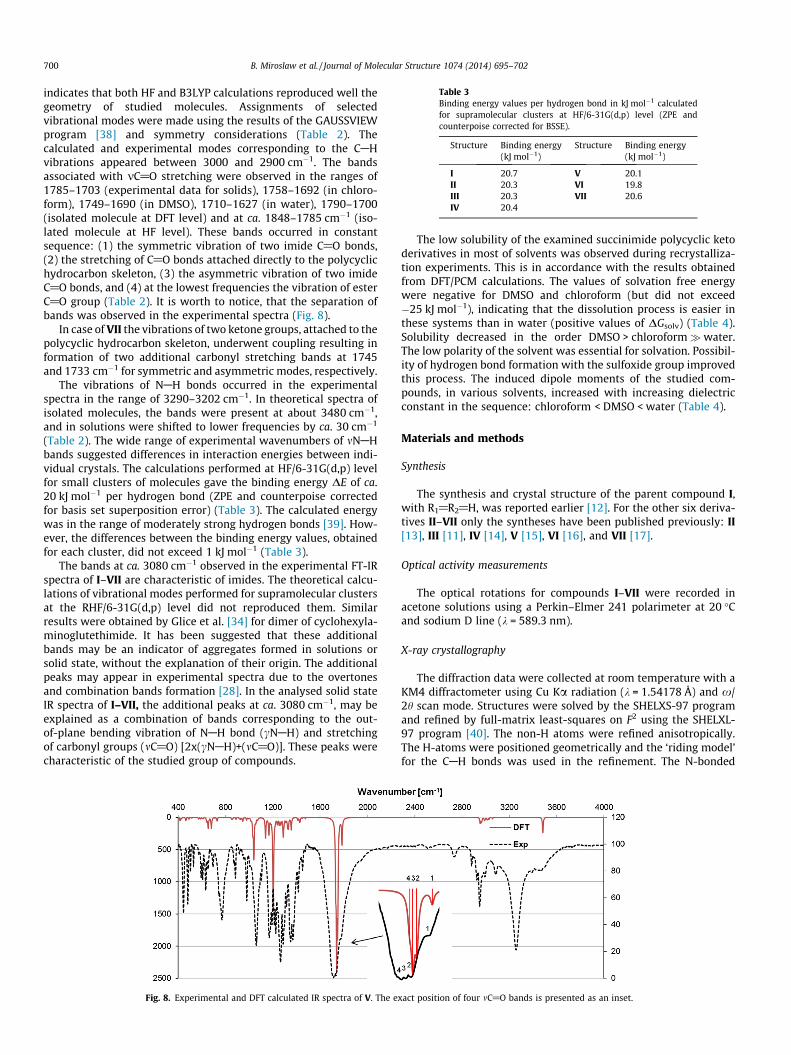
Table 3Binding energy values per hydrogen bond in kJ mol�1 calculatedfor supramolecular clusters at HF/6-31G(d,p) level (ZPE andcounterpoise corrected for BSSE).
Structure Binding energy(kJ mol�1)
Structure Binding energy(kJ mol�1)
I 20.7 V 20.1II 20.3 VI 19.8III 20.3 VII 20.6IV 20.4
700 B. Miroslaw et al. / Journal of Molecular Structure 1074 (2014) 695–702
indicates that both HF and B3LYP calculations reproduced well thegeometry of studied molecules. Assignments of selectedvibrational modes were made using the results of the GAUSSVIEWprogram [38] and symmetry considerations (Table 2). Thecalculated and experimental modes corresponding to the CAHvibrations appeared between 3000 and 2900 cm�1. The bandsassociated with mC@O stretching were observed in the ranges of1785–1703 (experimental data for solids), 1758–1692 (in chloro-form), 1749–1690 (in DMSO), 1710–1627 (in water), 1790–1700(isolated molecule at DFT level) and at ca. 1848–1785 cm�1 (iso-lated molecule at HF level). These bands occurred in constantsequence: (1) the symmetric vibration of two imide C@O bonds,(2) the stretching of C@O bonds attached directly to the polycyclichydrocarbon skeleton, (3) the asymmetric vibration of two imideC@O bonds, and (4) at the lowest frequencies the vibration of esterC@O group (Table 2). It is worth to notice, that the separation ofbands was observed in the experimental spectra (Fig. 8).
In case of VII the vibrations of two ketone groups, attached to thepolycyclic hydrocarbon skeleton, underwent coupling resulting information of two additional carbonyl stretching bands at 1745and 1733 cm�1 for symmetric and asymmetric modes, respectively.
The vibrations of NAH bonds occurred in the experimentalspectra in the range of 3290–3202 cm�1. In theoretical spectra ofisolated molecules, the bands were present at about 3480 cm�1,and in solutions were shifted to lower frequencies by ca. 30 cm�1
(Table 2). The wide range of experimental wavenumbers of mNAHbands suggested differences in interaction energies between indi-vidual crystals. The calculations performed at HF/6-31G(d,p) levelfor small clusters of molecules gave the binding energy DE of ca.20 kJ mol�1 per hydrogen bond (ZPE and counterpoise correctedfor basis set superposition error) (Table 3). The calculated energywas in the range of moderately strong hydrogen bonds [39]. How-ever, the differences between the binding energy values, obtainedfor each cluster, did not exceed 1 kJ mol�1 (Table 3).
The bands at ca. 3080 cm�1 observed in the experimental FT-IRspectra of I–VII are characteristic of imides. The theoretical calcu-lations of vibrational modes performed for supramolecular clustersat the RHF/6-31G(d,p) level did not reproduced them. Similarresults were obtained by Glice et al. [34] for dimer of cyclohexyla-minoglutethimide. It has been suggested that these additionalbands may be an indicator of aggregates formed in solutions orsolid state, without the explanation of their origin. The additionalpeaks may appear in experimental spectra due to the overtonesand combination bands formation [28]. In the analysed solid stateIR spectra of I–VII, the additional peaks at ca. 3080 cm�1, may beexplained as a combination of bands corresponding to the out-of-plane bending vibration of NAH bond (cNAH) and stretchingof carbonyl groups (mC@O) [2x(cNAH)+(mC@O)]. These peaks werecharacteristic of the studied group of compounds.
Fig. 8. Experimental and DFT calculated IR spectra of V. The e
The low solubility of the examined succinimide polycyclic ketoderivatives in most of solvents was observed during recrystalliza-tion experiments. This is in accordance with the results obtainedfrom DFT/PCM calculations. The values of solvation free energywere negative for DMSO and chloroform (but did not exceed�25 kJ mol�1), indicating that the dissolution process is easier inthese systems than in water (positive values of DGsolv) (Table 4).Solubility decreased in the order DMSO > chloroform�water.The low polarity of the solvent was essential for solvation. Possibil-ity of hydrogen bond formation with the sulfoxide group improvedthis process. The induced dipole moments of the studied com-pounds, in various solvents, increased with increasing dielectricconstant in the sequence: chloroform < DMSO < water (Table 4).
Materials and methods
Synthesis
The synthesis and crystal structure of the parent compound I,with R1@R2@H, was reported earlier [12]. For the other six deriva-tives II–VII only the syntheses have been published previously: II[13], III [11], IV [14], V [15], VI [16], and VII [17].
Optical activity measurements
The optical rotations for compounds I–VII were recorded inacetone solutions using a Perkin–Elmer 241 polarimeter at 20 �Cand sodium D line (k = 589.3 nm).
X-ray crystallography
The diffraction data were collected at room temperature with aKM4 diffractometer using Cu Ka radiation (k = 1.54178 Å) and x/2h scan mode. Structures were solved by the SHELXS-97 programand refined by full-matrix least-squares on F2 using the SHELXL-97 program [40]. The non-H atoms were refined anisotropically.The H-atoms were positioned geometrically and the ‘riding model’for the CAH bonds was used in the refinement. The N-bonded
xact position of four mC@O bands is presented as an inset.

Table 4Dipole moments and free energies of solvation of I–VII calculated in various media atB3LYP/6-311+G(d,p) level of theory.
Compound Medium Dipole moment [Debye] DGsolv [kJ mol�1]
I in vacuo 4.50 –Chloroform 5.81 �10.50DMSO 6.07 �19.25Water 6.91 23.51
II in vacuo 5.07 –Chloroform 6.87 �6.99DMSO 7.24 �15.94Water 8.27 27.87
III in vacuo 5.09 –Chloroform 6.90 �11.09DMSO 7.30 �17.87Water 8.39 28.20
IV in vacuo 4.92 –Chloroform 6.70 �15.02DMSO 7.09 �18.54Water 8.28 30.79
V in vacuo 3.23 –Chloroform 3.96 �8.16DMSO 4.05 �20.50Water 4.64 32.43
VI in vacuo 3.42 –Chloroform 4.20 �8.58DMSO 4.35 �18.66Water 4.96 36.44
VII in vacuo 1.82 –Chloroform 2.60 �7.41DMSO 2.87 �24.23Water 3.16 27.53
B. Miroslaw et al. / Journal of Molecular Structure 1074 (2014) 695–702 701
H-atoms were located in the difference maps. A summary of crystaldata and relevant refinement parameters is given in Table S2.Selected parameters describing molecular geometry and intermo-lecular interactions in crystals I–VII are presented in Table S1and Table 1, respectively.
The experimental details and final atomic parameters forII–VII have been deposited with the Cambridge CrystallographicData Centre, No. CCDC 960530–960535. Copies of the data canbe obtained free of charge on application to The Director, CCDC,12 Union Road, Cambridge CB2 1EZ, UK, (Fax: +44 1223 336 033;E-mail: [email protected] or www: http://www.ccdc.cam.ac.uk).
IR spectroscopy
The solid state FTIR spectra were recorded over the rangeof 4000–400 cm�1 using the FT-IR 1725X spectrometer(Perkin–Elmer). Samples for FTIR spectra measurements wereprepared as KBr discs. The selected IR vibrational modes withsuggested assignment are presented in Table 2.
Computational methods
All the calculations were performed without imposing anymolecular symmetry by using the Gaussian 09 Program Package[41] at supercomputing facility in the Academic Computer CentreCYFRONET, AGH, Krakow. The ground state molecular geometry(in vacuo), as well as the theoretical IR spectra were calculatedfor the analysed compounds by using the Hartree–Fock (HF) andBecke 3-Lee–Yang–Parr (B3LYP) levels of theory with the standard6-311+G(d,p) basis set [42]. Initial geometry was taken from theX-ray crystallography data and minimized without any constraintsin the potential energy surface at Hartree–Fock level, with the6-311+G(d,p) basis set. This geometry was then re-optimized atB3LYP level, using the same basis set 6-311+G(d,p) for betterresults. The optimized structural parameters were used in the
harmonic vibrational frequency calculations at the HF and DFT lev-els to characterize stationary points, giving the IR frequencies dataand respective intensities. The calculated vibrational frequencieswere scaled by 0.9059 and 0.9688 for HF/6-311+G(d,p) andB3LYP/6-311+G(d,p), respectively (Table 3) [43]. Analogouscalculations at HF/6-31G(d,p) level were repeated for small supra-molecular clusters of I–VII (as shown in Figs. 2–7) to obtain thebinding energy DE accounting for each intermolecular interactionin which the ZPE and counterpoise (CP) correction for basis setsuperposition error [44] were included. To calculate the energyper one hydrogen bond the simplification was made, that the dom-inant intermolecular interaction was the NAH� � �O (Table 3). Toperform solvation analysis the integral equation formalism model(IEF PCM) implemented in Gaussian 09 was applied [23]. Three sol-vents with different dielectric constants (e) were considered: non-polar chloroform (e = 4.71), polar aprotic dimethyl sulfoxide(e = 46.83) and polar protic water (e = 78.36). The computationswere performed in the solvent media at B3LYP/6-311+G(d,p) level.The free energies of solvation and induced dipole moments(Table 4) were calculated for all compounds in the selectedsolvents, as well as the harmonic IR frequencies (Table 2).
Conclusions
The substituent effect on the crystal packing and the intermo-lecular interaction mode in a series of seven crystals of polycyclicmonoimide keto derivatives was studied. The additional ketone,ether or ester substituents, introduced in a systematic way intothe hydrocarbon skeleton, played the essential role on the resultingintermolecular interaction motifs. In the studied structures, thesubstitution at the bridge position by an additional carbonyl groupforced the catemeric motif of the NimideAH� � �Oimide hydrogen bond.However, when the substituents were more voluminous (iBuOA)or additional hydrogen bond acceptors (AcOA, O@) were present(i.e. the molecule became more asymmetric in shape), the sterichindrance increased. As the result the imide� � �imide interactionswere no longer present in the solid state. The NimideAH� � �Oketo orNimideAH� � �Oester hydrogen bonds were formed instead. In general,the contacts involved the less blocked O1 and O3 atoms. Thecrystal structure was additionally stabilized by weak CAH� � �Ointermolecular interactions that completed the hydrogen bondpattern. The different types of supramolecular motifs were variedin the solid state FT-IR spectra with the different positions of bandscorresponding to the mNAH (3290–3202 cm�1) and mC@O (1790–1700 cm�1) vibrations. The computed at HF/6-31G(d,p) level inter-action energy per one NAH� � �O hydrogen bond was about20 kJ mol�1. The theoretical calculations of IR spectra facilitatedthe spectral assignment of the vibrational modes. The followingsequence of the mC@O stretching modes has been determined:
1. imide m(C@O)2 sym – at the highest frequencies the symmetricvibration of two imide C@O bonds,
2. ketone mC@O – the vibrations of C@O bond attached to thepolycyclic hydrocarbon skeleton,
3. imide m(C@O)2 asym – the asymmetric vibration of two imideC@O bonds,
4. ester mC@O – at the lowest wavenumbers the stretching of esterC@O group.
The characteristic peak at ca. 3080 cm�1 observed inexperimental spectra of cyclic imides in solid state and in solutionshas been suggested previously by Bajdor, Glice, Les et al. as anindicator of formation of intermolecular associations [32–34].However, the theoretical calculations of harmonic frequenciesperformed for small molecular clusters did not explain its source.

702 B. Miroslaw et al. / Journal of Molecular Structure 1074 (2014) 695–702
It originates most probably from overtone and combination of theout-of-plane bending vibration of NAH group (cNAH) andstretching of C@O bond (mC@O) [2x(cNAH) + (mC@O)], and ischaracteristic for cyclic imides.
The solvation free energies were computed by DFT/PCMcalculations to determine the solubility of I–VII, which decreasedin the order DMSO > chloroform�water. The induced dipolemoments in solutions increased with increasing dielectricconstant: chloroform < DMSO < water. The presented results maybe useful in understanding the interaction of similar compoundswith active centres in biological systems.
Acknowledgements
The access to the supercomputing facility at the AcademicComputer Centre CYFRONET, AGH, Krakow, (Grant No. MNiSW/SGI3700/UMCSLublin/071/2012) is gratefully acknowledged.
Appendix A. Supplementary material
Supplementary data associated with this article can be found, inthe online version, at http://dx.doi.org/10.1016/j.molstruc.2014.05.029. These data include Tables S1–S2, Figures S1–S8, MOL filesand InChiKeys of the most important compounds described in thisarticle.
References
[1] K. Nakae, Y. Nishimura, S. Ohba, Y. Akamatsu, J. Antibiot. 59 (2006) 685.[2] D. Shan, L. Chen, J.T. Njardarson, C. Gaul, X. Ma, S.J. Danishefsky, X.Y. Huang,
Proc. Natl. Acad. Sci. USA 102 (2005) 3772.[3] M.E. Salvati, A. Balog, W. Shan, D.D. Wei, D. Pickering, R.M. Attar, J. Geng, C.A.
Rizzo, M.M. Gottardis, R. Weinmann, S.R. Krystek, J. Sack, Y. An, K. Kish, Bioorg.Med. Chem. Lett. 15 (2005) 271.
[4] H. Miyachi, M. Kato, F. Kato, Y. Hashimoto, J. Med. Chem. 41 (1998) 263.[5] D. Rennison, S. Bova, M. Cavalli, F. Ricchelli, A. Zulian, B. Hopkins, M.A. Brimble,
Bioorg. Med. Chem. 15 (2007) 2963.[6] L.A. Riblet, D.P. Taylor, M.S. Eison, H.C. Stanton, J. Clin. Psychiatry 43 (1982) 11.[7] J. Turlo, T. Zawadowski, Farmaco 51 (1996) 815.[8] K. Ishizumi, A. Kojma, F. Antoku, Chem. Pharm. Bull. 39 (1991) 2288.[9] G. P. Stack, M. A. Abou-Gharbia, E. J. Podlesny, U.S. Patent 4,748,240, 1988.
[10] A.J. Bojarski, B. Kuran, J. Kossakowski, A. Kozioł, E. Jagiełło-Wójtowicz, A.Chodkowska, Eur. J. Med. Chem. 44 (2009) 152.
[11] J. Kossakowski, M. Jarocka, Farmaco 56 (2001) 785.[12] J. Kossakowski, A. Bielenica, B. Miroslaw, A.E. Koziol, I. Dybala, M. Struga,
Molecules 13 (2008) 1570.[13] J. Kossakowski, M. Jarocka-Wierzba, Acta Pol. Pharm. Drug Res. 60 (2003) 367.[14] J. Kossakowski, A. Wojciechowska, Annales UMCS Sectio AA LIX (2004) 98.[15] A. Bielenica, M. Struga, B. Mirosław, A.E. Kozioł, J. Kossakowski, G. Sanna, P. La
Colla, G. Giliberti, Acta Pol. Pharm. Drug Res. 70 (2013) 809.[16] J. Kossakowski, E. Hejchman, Acta Pol. Pharm. Drug Res. 60 (2003) 61.[17] T. Zawadowski, J. Kossakowski, S. Rump, I. Jakowicz, A. Plaznik, Acta Pol.
Pharm. Drug Res. 52 (1995) 43.[18] E.M. Smith, Tetrahedron Lett. 40 (1999) 3285.
[19] F.H. Allen, W.D.S. Motherwell, P.R. Raithby, G.P. Shields, R. Taylor, New J. Chem.23 (1999) 25.
[20] F.H. Allen, P.R. Raithby, G.P. Shields, R. Taylor, Chem. Commun. (1998) 1043.[21] J.C. MacDonald, G.M. Whitesides, Chem. Rev. 94 (1994) 2383.[22] M. Struga, B. Miroslaw, M. Pakosinska-Parys, A. Drzewiecka, P. Borowski, J.
Kossakowski, A.E. Koziol, J. Mol. Struct. 965 (2010) 23.[23] (a) E. Cancès, B. Mennucci, J. Math. Chem. 23 (1998) 309;
(b) E. Cancès, B. Mennucci, J. Tomasi, J. Chem. Phys. 107 (1997) 3032;(c) B. Mennucci, E. Cancès, J. Tomasi J. Phys. Chem. B 101 (1997) 10506;(d) S. Miertus, E. Scrocco, J. Tomasi, Chem. Phys. 55 (1981) 117;(e) R. Cammi, J. Tomasi, J. Comput. Chem. 16 (1995) 1449.
[24] M.C. Etter, J.C. MacDonald, J. Bernstein, Acta Cryst. B46 (1990) 256.[25] V. Bertolasi, P. Gilli, V. Ferretti, G. Gilli, Acta Cryst. B51 (1995) 1004.[26] M.A. Spackman, J.J. McKinnon, Cryst. Eng. Comm. 4 (2002) 378.[27] A.T. Hulme, A. Johnston, A.J. Florence, P. Fernandes, K. Shankland, C.T. Bedford,
G.W.A. Welch, G. Sadiq, D.A. Haynes, W.D.S. Motherwell, D.A. Tocher, S.L. Price,J. Am. Chem. Soc. 129 (2007) 3649.
[28] I. Petrov, O. Grupce, T. Stafilov, J. Mol. Struct. 142 (1986) 275.[29] P.T. McKittrick, J.E. Katon, Appl. Spectrosc. 44 (1990) 812.[30] R.A. Nyquist, S.L. Fiedler, Vib. Spectrosc. 8 (1995) 365.[31] P. Naumov, G. Jovanovski, Curr. Org. Chem. 5 (2001) 1059.[32] M.M. Glice, A. Les, K. Bajdor, J. Mol. Struct. 450 (1998) 141.[33] K. Bajdor, M.M. Glice, A. Les, L. Adamowicz, R. Kołos, J. Mol. Struct. 511–512
(1999) 107.[34] M.M. Glice, K. Bajdor, A. Les, T. Stepanenko, M.J. Nowak, H.Y. Aboul-Enein, Z.
Chilmonczyk, J. Mol. Struct. 560 (2001) 137.[35] I.G. Binev, B.A. Stamboliyska, Y.I. Binev, E.A. Velcheva, J.A. Tsenov, J. Mol. Struct.
513 (1999) 231.[36] B.A. Stamboliyska, Y.I. Binev, V.B. Radomirska, J.A. Tsenov, I.N. Juchnovski, J.
Mol. Struct. 516 (2000) 237.[37] W. Wysocka, A. Przybył, G. Wojciechowski, B. Brzezinski, J. Mol. Struct. 516
(2000) 157.[38] A. Frisch, A. B. Nielson, A. J. Holder, GAUSSVIEW User Manual, Gaussian Inc.,
Pittsburgh, PA, 2000.[39] (a) G.A. Jeffery, An Introduction to Hydrogen Bonding, Oxford University Press,
New York, 1997;(b) G.R. Desiraju, T. Steiner, The Weak Hydrogen Bond, Oxford University Press,Oxford, U.K., 1999;(c) G.R. Desiraju, Acc. Chem. Res. 35 (2002) 565;(d) M.M. Deshmukh, S.R. Gadre, J. Phys. Chem. A 113 (2009) 7927.
[40] G.M. Sheldrick, Acta Cryst. A64 (2008) 112.[41] M.J. Frisch, G.W. Trucks, H.B. Schlegel, G.E. Scuseria, M.A. Robb, J.R. Cheeseman,
G. Scalmani, V. Barone, B. Mennucci, G.A. Petersson, H. Nakatsuji, M. Caricato,X. Li, H.P. Hratchian, A.F. Izmaylov, J. Bloino, G. Zheng, J.L. Sonnenberg, M.Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y.Honda, O. Kitao, H. Nakai, T. Vreven, J.A. Montgomery Jr., J.E. Peralta, F. Ogliaro,M. Bearpark, J.J. Heyd, E. Brothers, K.N. Kudin, V.N. Staroverov, R. Kobayashi, J.Normand, K. Raghavachari, A. Rendell, J.C. Burant, S.S. Iyengar, J. Tomasi, M.Cossi, N. Rega, J.M. Millam, M. Klene, J.E. Knox, J.B. Cross, V. Bakken, C. Adamo,J. Jaramillo, R. Gomperts, R.E. Stratmann, O. Yazyev, A.J. Austin, R. Cammi, C.Pomelli, J.W. Ochterski, R.L. Martin, K. Morokuma, V.G. Zakrzewski, G.A. Voth,P. Salvador, J.J. Dannenberg, S. Dapprich, A.D. Daniels, Ö. Farkas, J.B. Foresman,J.V. Ortiz, J. Cioslowski, D.J. Fox, Gaussian 09, Revision A.02, Gaussian Inc,Wallingford, CT, 2009.
[42] A.D. Becke, J. Chem. Phys. 98 (1993) 5648;C. Lee, W. Yang, R.G. Parr, Phys. Rev. B 37 (1988) 785;S.H. Vosko, L. Wilk, M. Nusair, Can. J. Phys. 58 (1980) 1200;P.J. Stephens, F.J. Devlin, C.F. Chabalowski, M.J. Frisch, J. Phys. Chem. 98 (1994)11623.
[43] J.P. Merrick, D. Moran, L. Radom, J. Phys. Chem. A 111 (2007) 11683.[44] S.F. Boys, F. Bernardi, Mol. Phys. 19 (1970) 553.




















