
Structure factor calculations for a side-by-side model of B-DNA
11
Structure factor calculations for a side-by-side model of B-DNA S. R. Hubbard, R. J. Greenall* Department of Physics, University of York, York YO 1 5DD, UK A. K. Shrive, V. T. Forsyth and A. Mahendrasingam Department of Physics, University of Keele, Staffordshire ST5 5BG, UK Received 22 February 1994; revised 27 May 1994 In this paper, the side-by-side model of DNA proposed by Premilat and Albiser is investigated. The axial repeat of the model is equal to the c-axis repeat in the observed B-DNA unit cell in fibres. However, the model does not pack into the unit cell as efficiently as the B-DNA double helix does, nor is it as successful as the double helix in predicting the observed Bragg amplitudes. When the azimuthal orientations and the relative axial displacements of the two molecules in the unit cell are allowed to take general values, the best crystallographic R factor for the side-by-side model is 43.43% compared with 34.33% for the double helix. If constraints consistent with the accepted B-DNA space group, P21212~, are applied, the best R factors are 45.53% for the side-by-side model and 34.51% for the double helix. Therefore, the side-by-side model can be rejected as a possible conformation for B-DNA in crystalline fibres. Keywords: DNA double helical model; DNA side-by-side model; X-ray fibre diffraction Although the double helical (DH) Watson-Crick ~ model for DNA is well established, alternative models have been proposed from time to time. In particular, the side-by-side (SBS) models have been extensively investigated2-~°. In these models, the two polynucleotide chains are antiparallel and are linked by complementary Watson- Crick base pairs. The key characteristic of these models is that the molecule consists of alternating segments of right- and left-handed helix, each segment being about five nucleotide pairs in length. Until recently, the sole direct evidence for the structure of DNA came from fibre diffraction, and, indeed, this technique is still the only means of visualizing the conformation of polymeric DNA. In the case of B-DNA, fibre diffraction can be particularly powerful, since the fibres frequently contain regions of high crystallinity. The diffraction pattern contains discrete Bragg reflections which give three-dimensional information about the molecular structure ~ L12. The confidence we may have in such models is comparable to that which we may have in structures derived from crystallographic studies on DNA fragments ~3. Unfortunately, the majority of the SBS models proposed so far have a long-range twist associated with them, giving a helical structure whose pitch is approximately ten times greater than the pitch of DH B-DNA. Therefore, these models will not fit into *To whomcorrespondenceshould be addressed the observed unit cell of B-DNA, and can clearly be rejected as possible conformations for the crystalline form. However, B-DNA has also been observed in semi-crystalline fibres and in even less ordered forms, in which case the observed diffraction is related to the cylindrically averaged squared transform (CAST) of an individual molecule. All the proposers of SBS models have suggested that their models are superior to the double helix when calculated CASTs are compared with observed intensities from disordered specimens. This view has been challenged ~4 17, and the point has been made that any model for DNA should be tested against the high-quality crystalline diffraction data. The reasoning behind this has been discussed extensively elsewhere la. The proposal of an SBS model with no long-range twist by Premilat and Albiser 1° provides the first opportunity to calculate structure factors and to compare them with observed Bragg reflection amplitudes. In this paper, we report such calculations together with an investigation of the stereochemical feasibility of the model. Methods We have adopted the nucleotide naming convention that is customary for SBS models (Table 1). Premilat and Albiser ~ o have given the sugar and phosphate coordinates for residues A02 to A10. The coordinates for residue A01 were generated from those of A02 by application of the helix rise and rotation parameters of DH B-DNA 0141-8130/94/040195-11 :~ 1994Butterworth-Heinemann Limited Int. J. Biol. Macromol. Volume 16 Number 4 1994 195
Transcript of Structure factor calculations for a side-by-side model of B-DNA

Structure factor calculations for a side-by-side model of B - D N A
S. R. Hubbard, R. J. Greenal l * Department of Physics, University of York, York YO 1 5DD, UK
A. K. Shrive, V. T. Forsyth and A. Mahendras ingam Department of Physics, University of Keele, Staffordshire ST5 5BG, UK
Received 22 February 1994; revised 27 May 1994
In this paper, the side-by-side model of DNA proposed by Premilat and Albiser is investigated. The axial repeat of the model is equal to the c-axis repeat in the observed B-DNA unit cell in fibres. However, the model does not pack into the unit cell as efficiently as the B-DNA double helix does, nor is it as successful as the double helix in predicting the observed Bragg amplitudes. When the azimuthal orientations and the relative axial displacements of the two molecules in the unit cell are allowed to take general values, the best crystallographic R factor for the side-by-side model is 43.43% compared with 34.33% for the double helix. If constraints consistent with the accepted B-DNA space group, P21212~, are applied, the best R factors are 45.53% for the side-by-side model and 34.51% for the double helix. Therefore, the side-by-side model can be rejected as a possible conformation for B-DNA in crystalline fibres.
Keywords: DNA double helical model; DNA side-by-side model; X-ray fibre diffraction
Although the double helical (DH) Watson-Crick ~ model for DNA is well established, alternative models have been proposed from time to time. In particular, the side-by-side (SBS) models have been extensively investigated 2-~°. In these models, the two polynucleotide chains are antiparallel and are linked by complementary Watson- Crick base pairs. The key characteristic of these models is that the molecule consists of alternating segments of right- and left-handed helix, each segment being about five nucleotide pairs in length.
Until recently, the sole direct evidence for the structure of DNA came from fibre diffraction, and, indeed, this technique is still the only means of visualizing the conformation of polymeric DNA. In the case of B-DNA, fibre diffraction can be particularly powerful, since the fibres frequently contain regions of high crystallinity. The diffraction pattern contains discrete Bragg reflections which give three-dimensional information about the molecular structure ~ L12. The confidence we may have in such models is comparable to that which we may have in structures derived from crystallographic studies on DNA fragments ~3. Unfortunately, the majority of the SBS models proposed so far have a long-range twist associated with them, giving a helical structure whose pitch is approximately ten times greater than the pitch of DH B-DNA. Therefore, these models will not fit into
* To whom correspondence should be addressed
the observed unit cell of B-DNA, and can clearly be rejected as possible conformations for the crystalline form. However, B-DNA has also been observed in semi-crystalline fibres and in even less ordered forms, in which case the observed diffraction is related to the cylindrically averaged squared transform (CAST) of an individual molecule. All the proposers of SBS models have suggested that their models are superior to the double helix when calculated CASTs are compared with observed intensities from disordered specimens. This view has been challenged ~4 17, and the point has been made that any model for DNA should be tested against the high-quality crystalline diffraction data. The reasoning behind this has been discussed extensively elsewhere la.
The proposal of an SBS model with no long-range twist by Premilat and Albiser 1° provides the first opportunity to calculate structure factors and to compare them with observed Bragg reflection amplitudes. In this paper, we report such calculations together with an investigation of the stereochemical feasibility of the model.
Methods
We have adopted the nucleotide naming convention that is customary for SBS models (Table 1). Premilat and Albiser ~ o have given the sugar and phosphate coordinates for residues A02 to A10. The coordinates for residue A01 were generated from those of A02 by application of the helix rise and rotation parameters of DH B-DNA
0141-8130/94/040195-11 :~ 1994 Butterworth-Heinemann Limited Int. J. Biol. Macromol. Volume 16 Number 4 1994 195

determined from fibre diffraction ~9. Coordinates for atoms in the B-chain were generated using the diad axis which is perpendicular to the molecular axis in nucleotide pairs 1 and 6. Premilat and Albiser gave the coordinates of the nitrogen atom attached to CI' in each nucleotide but none of the other base atoms, so these must be derived. The coordinates of a standard untilted and untwisted Watson-Crick base pair 17 were rotated and translated until the vector from the purine N9 to the pyrimidine N1 coincided with the vector from the A-chain nitrogen to the B-chain nitrogen. The pseudodiad axis in the plane of the base pair was maintained perpendicular to the molecular axis. Since all the nucleotides are in the anti conformation, it was necessary to invert the base pairs in nucleotides 4-8, which lie in the left-handed region of the molecule. The base sequence (Table 1) and the choice of zero propeller twist are arbitrary and unlikely to have a significant effect on the calculations reported here. Coordinates for a DH B-DNA molecule with the same base sequence were derived from the fibre model described by Arnott and Hukins x9. Views of the two models are shown in Figure 1. To check the acceptability of the intramolecular stereochemistry, bond lengths and bond angles were calculated, and short van der Waals' contacts were located. A short contact is defined as one
Table 1 Sequence of bases used in the ten nucleotide repeat unit
A-strand B-strand
3 p 5 '
A01 A T B01 A02 G - - C B02 A03 C G B03 A04 T - - A B04 A05 C - - G B05 A06 T A B06 A07 G - - C B07 A08 A T B08 A09 T A B09 A10 C G B10 5' 3'
A = adenine; T = thymine; G = guanine; C = cytosine
(a)
in which the distance between two atoms is less than the sum of their van der Waals' radii. The van der Waals' radii were taken to be 1.4 ,~ for O, 1.5 ,~ for N, 1.6 A for C, 1.9 A for P and 2.0 A, for CH a.
The B-DNA unit cell contains two molecules 11.12, and it is orthogonal 2° with a=30.8_+0.1 A, b=22.5_+0.1 A, and c=33.7_+0.1A. The axes of both molecules are parallel to the crystalline c-axis. If one molecule is placed at the origin, then the fractional coordinates of the other arc (1/2, 1/2, Az/c), where Az is the displacement of the second molecule along the c-direction (Figure 2). It is also necessary to specify the azimuthal orientations (41 and 42) of the two molecules, where in each case the zero angle is dcfined by the crystalline a-axis. There is some evidence, which will be discussed below, that thc best DH B-DNA models satisfy 41 = 42 =90° and Az/c ,~ I/3, but we have also investigated general arrangements with arbitrary values of 41, 42 and Az.
In ordcr to assess the intermolecular stereochemistry within the unit cell, we defined a penalty function:
10{) P(4,, 42, A z ) = ~ - - (I)
where d~ is the distance between any two atoms, in different molecules, whose separation is less than the sum of their van der Waals' radii and where the summation is taken over all such close contacts. Although this penalty function is rather crude, it is sufficient to assess the acceptability of the molecular packing. We have calculated P with 0°~<41, 42~<360 ° and 0.0 A ~< Az < 34.0 A with an angular increment of 5 ° and a translational increment of 1.0 A.
The Fourier transform of an infinite helical molecule which has a polyatomic monomer and a diad axis perpendicular to the molecular axis is given by21:
where
(b) Figure 1 Views of the two models of B-DNA: (a) the DH model and (b) (d) the SBS model viewed perpendicular to the diad axis
(d) (c)
Side-by-side DNA: S. R. Hubbard et al.
the SBS model viewed parallel to the diad axis; (c) the DH model and
196 Int. J. Biol. Macromol. Volume 16 Number 4 1994

a
@ Figure 2 The B-DNA unit cell
In Equation (3), the summation is taken over all atoms in the asymmetric unit whose coordinates are (r j, 0j, z j), J , is the nth order Bessel function, and (R, ~, Z) is a point in reciprocal space. The transform is finite only on layer planes given by Z = l/c, where I is an integer.
We have used atomic scattering factors, fj(S), where S is the distance from the origin of reciprocal space, which were modified to account for the effects of scattering from solvent molecules using the method described by Langridge et al. 11'12. The scattering factors were also multiplied by a Deby~Waller factor, exp( - BS2/4), where B is the isotropic temperature factor. The number of Bragg reflections is insufficient to justify the assignment of individual B factors to each atom and so we have used the same B for all atoms. To determine whether the results presented below would be sensitive to the value of B, we calculated transforms with B=0, 4, 8, 12 and 16 A 2, but it was found that the effect was insignificant since the crystallographic R factors changed by no more than two percentage points 22. The results presented here were calculated with B = 4 A 2 in accordance with the value used by Langridge et al. 11'12.
The summation in Equation (2) was taken over those values of n which satisfy the helical selection rule n = ( l - N m ) / K where there are N asymmetric units in K turns of the helix and where m is any integer. In the case of DH B-DNA, where the asymmetric unit is a mononucleotide, we have N = 10 and K = 1. For the purpose of the Fourier transform calculation, we used an average base in which the atoms of all four bases were represented with the appropriate occupancy. The transform was calculated for l=0-10 with m=0,__l. In the case of the SBS model, where the asymmetric unit is 10 nucleotides, N = 1 and K = 1. The molecule is a one-fold helix, and hence every Bessel function contributes to each layer plane. The transform was calculated for /=0-10 with m=0,__1,__2, ...,__10. In both transform calculations, our choice of m values ensures that, when the selection rule permits J , to contribute to a layer plane, it will be included in the summation if In[ ~<20. Both transforms were calculated in the range R=0 .0A -1 to R=0 .4A -1 in steps of 0.0025 A-1. The CAST is then given by:
It(R) = ~ G~(R) (4) n
Side-by-side DNA: S. R. Hubbard et al.
The structure factors of a crystalline array of helical molecules with fractional unit cell coordinates rp and azimuthal orientations ~bp were calculated using the relationship:
( ;/ F(h) = ~ ~ G,~R) exp in • + exp i(2~h.rp- nc~o ) p n
(5) where h=(h, k, l) is the reciprocal lattice vector to the point with indices h, k and I. The observed structure factor intensities from a crystalline fibre are equivalent to those from a rotation pattern. In the case of the orthogonal B-DNA unit cell, the reflections with indices (hkl), (hkl), (hkl) and (/1/~1) will systematically overlap on such a pattern. In general, these reflections will not have the same intensities, and so, in the results given below, the amplitude of the (hkl) reflection is the square root of the sum of the squares of these structure factors. In addition, some reflections will overlap accidentally, and the intensity is then the sum of the intensities of these reflections. Sets of structure factors were calculated as a function of 41, ~b2 and Az using the same ranges and step sizes that were employed in the calculation of P(~I,~2,Az). The sets of structure factors were compared with the observed values given by Arnott and Hukins 2°, whose dataset contains 225 reflections extending to a resolution of approximately 3 A, and a crystallographic residual:
R - ~,k,llFol- klFcll (6)
~,ulfol was calculated, where IFol and IFcl are the observed and calculated structure factor amplitudes respectively and the summation was taken over all the observed reflections. The scale factor, k, was defined by:
k - ~a'IF°IIF°[ (7) ~hu[F=I 2
This choice of k minimizes the sum of the squares of the differences between the observed and calculated amplitudes.
Results
The mean bond lengths (Table 2) and angles (Table 3) in the SBS model have similar values to those of the DH B-DNA fibre diffraction model and to those of a synthetic B-DNA dodecamer 23, and the standard deviations of the bond lengths and angles in the SBS model are mostly quite small. The CI'-N bond length and the C2'-CI'-N bond angle have the largest standard deviations, arising from the difficulty of fitting flat base pairs into the SBS model, particularly at the bend regions in the structure. These minor anomalies could probably be alleviated by adopting a slightly more flexible method for fitting the base pairs into the structure than the one we have used.
The 21 close intramolecular contacts which were found in the SBS model are listed in Table 4. They occur between the C3', C4', C5' and O1' atoms in the sugar group of one nucleotide and the 03 ' and 05 ' atoms in the phosphate group of the adjacent nucleotide. There are close contacts between the 03 ' and 05 ' atoms in adjacent phosphate groups, and also in the bend regions, involving the methyl groups and the C5 atoms of the thymine bases
Int. J. Biol. Macromol. Volume 16 Number 4 1994 197

Side-by-side DNA: S. R. Hubbard et al.
Table 2 Bond lengths of the SBS and DH models
Bond lengths (A) SBS residue O5'-C5' C5'-C4' C4'-O1' C4'-C3' O1'-C1' C3'-C2' C3'-O3' C2'-C1' CI'-N O5'-P O3'-P P-O1P P-O2P
A01 1.44 1.51 1.45 1.52 1.42 1.52 1.42 1.56 1.45 1.54 1.60 1.48 1.48
A02 1.44 1.51 1.45 1.52 1.42 1.53 1.42 1.56 1.48 1.60 1.60 1.48 1.48
A03 1.44 1.51 1.46 1.52 1.42 1.53 1.42 1.60 1.42 1.60 1.60 1.49 1.48
A04 1.43 1.51 1.46 1.53 1.41 1.54 1.42 1.62 1.56 1.60 1.60 1.49 1.48
A05 1.44 1.51 1.45 1.50 1.42 1.62 1.38 1.55 1.45 1.59 1.60 1.49 1.48
A06 1.45 1.51 1.45 1.52 1.42 1.53 1.43 1.49 1.47 1.59 1.60 1.49 1.48
A07 1.45 1.51 1.44 1.53 1.43 1.53 1.42 1.62 1.44 1.59 1.61 1.48 1.48
A08 1.45 1.50 1.45 1.54 1.42 1.53 1.42 1.56 1.57 1.60 1.60 1.49 1.47
A09 1.45 1.51 1.45 1.52 1.42 1.52 1.43 1.62 1.43 1.60 1.60 1.48 1.48
A10 1.44 1.51 1.45 1.53 1.42 1.53 1.42 1.56 1.48 1.60 1.48 !.47
Mean 1.44 1.51 1.45 1.52 1.42 1.54 1.42 1.57 1.48 1.58 1.60 1.48 1.48
s.d. 0.01 0.01 0.01 0.01 0.00 0.03 0.01 0.04 0.05 0.04 0.01 0.01 0.00
DHfib, e 1.45 1.51 1.46 1.53 1.41 1.52 1.42 1.53 1.49 1.60 1.59 1.48 1.48
DHollg o 1.44 1.50 1.43 1.52 1.42 1.53 1.43 1.52 1.48 1.59 1.60 1.48 1.48
s.d. 0.01 0.01 0.01 0.01 0.01 0.01 0.01 0.01 0.01 0.01 0.01 0.01 0.00
s.d. =standard deviation
Table 3 Bond angles of the SBS and DH models
Bond angles (degrees)
i i i i 0 i i i i i i i SBS ~ ~ ~ :- ~- ~- ~- - ;- N ~ residue © c~ ~ © ~ r) ~ © © ~ ~
~ ~. ~. ~, ~ ~ ~-- . . . . =. ~ ~. o
A01 110 109 116 106 110 101 1t2 105 108 113 103 119 111 110 109 103 110 112 114 117
A02 110 109 116 106 110 100 112 105 108 113 103 120 112 110 109 101 109 114 113 119
A03 110 109 116 107 110 100 112 105 107 113 100 119 124 109 110 102 109 113 114 120
A04 110 109 116 106 110 100 112 105 111 113 100 119 101 110 109 101 110 114 113 119
A05 110 109 117 109 110 97 116 106 106 110 103 121 112 109 109 101 110 113 114 119
A06 110 109 116 107 110 100 112 107 107 112 106 119 116 110 109 102 110 113 114 119
A07 110 109 116 107 110 100 112 104 106 113 100 119 111 109 109 102 109 113 114 119
A08 110 109 116 107 109 100 112 98 111 113 101 119 150 110 110 101 109 113 114 119
A09 110 109 116 106 110 100 112 105 107 113 101 119 106 109 109 102 109 113 113 119
AI0 110 109 116 107 110 100 112 106 108 113 104 119 112 110 110 - 110
Mean 110 109 116 107 110 100 112 105 108 113 102 119 116 110 109 102 110 113 114 119
s.d. 0 0 0 1 0 1 1 2 2 1 2 1 13 0 0 1 1 1 0 1
DHfib~ e 110 109 116 104 110 103 112 108 108 110 102 119 114 109 110 102 116 110 110 119
DHoliso 106 108 116 105 107 102 112 102 105 114 101 121 112 110 109 102 117 110 108 120
s.d. 2 2 1 1 2 2 1 2 2 2 2 1 2 2 1 1 1 2 1 1
s.d. =standard deviation
a t n u c l e o t i d e p o s i t i o n s A 0 4 a n d B08, a n d t h e C2 ' a t o m s in n u c l e o t i d e s A03 a n d B09, respec t ive ly . T h e s h o r t e s t c lose c o n t a c t in t h e s t r u c t u r e is t h e s e p a r a t i o n of 2.48 A b e t w e e n t h e a t o m s A 08 O 1' a n d A 0 9 O5 ' , w i t h t he s u m of t he v a n d e r W a a l s ' r ad i i o f t h e s e a t o m s b e i n g 2.80 A. T h e r e a r e n o ve ry s h o r t c o n t a c t s , a n d t he r e a s o n a b l e n a t u r e o f t he S BS m o d e l ' s s t e r e o c h e m i s t r y , in t e r m s o f i n t e r n a l n o n - b o n d e d c o n t a c t s , is c o n f i r m e d b y e x a m i n a t i o n o f t he c lose c o n t a c t s in t he D H m o d e l . I n th i s s t r u c t u r e , t h e r e is a c lose c o n t a c t b e t w e e n O 1 P in r e s idue n a n d C 2 ' in r e s i d u e n + 1. T h e i n t e r a t o m i c d i s t a n c e is 2.68 A w h e r e a s t h e s u m of t h e v a n d e r W a a l s ' r ad i i is 3.00 A. D u e to t h e he l i ca l s y m m e t r y , t h e r e a r e 18 s u c h c o n t a c t s . I n a d d i t i o n , t h e r e a r e f o u r i d e n t i c a l
c o n t a c t s b e t w e e n t h e t h y m i n e m e t h y l g r o u p s a n d t h e C 2 ' in a d j a c e n t res idues . I n th i s case, t he i n t e r a t o m i c d i s t a n c e is 3.24 A w h e r e a s t h e s u m of t h e v a n d e r W a a l s ' r ad i i is
3.60 A. T h e C A S T s o f t he S B S a n d D H m o d e l s o f B - D N A
are s h o w n in Figure 3. T h e l aye r l ine i n t e n s i t i e s h a v e b e e n s e p a r a t e d o n t he ve r t i ca l ax is to i n d i c a t e t he s p a c i n g of t he l aye r l ines l o n t h e m e r i d i o n a l axis b y 1/P, w h e r e P is t h e p i t c h l eng th . H o w e v e r , t h e i n t e n s i t i e s o n al l t h e l aye r l ines a r e o n t h e s a m e r e l a t i ve scale. C o m p a r i s o n of t h e C A S T s w i t h t h e o b s e r v e d i n t e n s i t i e s c o r r e c t e d for t he effects o f p a c k i n g (see F i g u r e 6 of L a n g r i d g e et al. 12) s h o w s t h a t t he D H t r a n s f o r m p r o v i d e s g o o d q u a l i t a t i v e a g r e e m e n t w i t h t he o b s e r v e d in tens i t i e s , w h e r e a s t he SBS
1 9 8 Int. J. Biol . M a c r o m o l . V o l u m e 16 N u m b e r 4 1 9 9 4
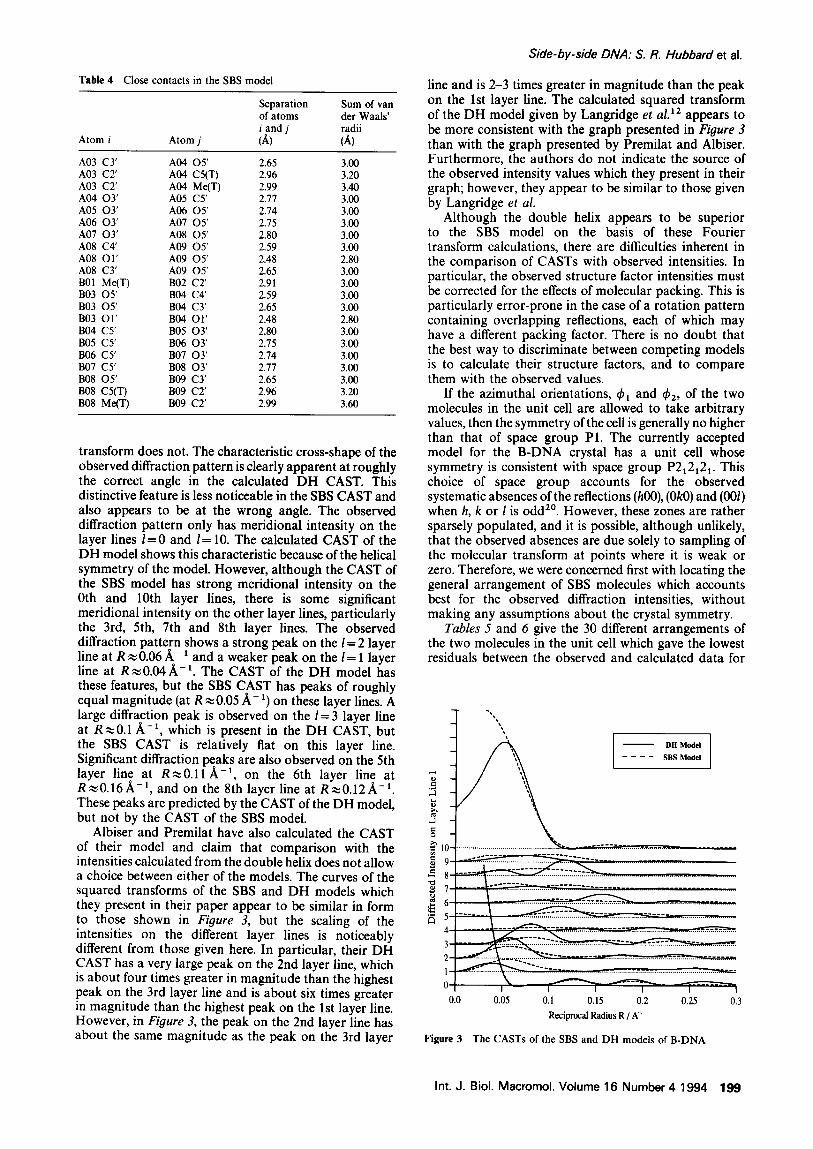
Table 4 Close contacts in the SBS model
Atom i Atom j
Separation of atoms i and j (A)
Sum of van der Waals' radii (A)
A03 C3' A04 05 ' 2.65 3.00 A03 C2' A04 C5(T) 2.96 3.20 A03 C2' A04 Me(T) 2.99 3.40 A04 O3' A05 C5' 2.77 3.00 A05 03 ' A06 05 ' 2.74 3.00 A06 03 ' A07 05 ' 2.75 3.00 A07 O3' A08 O5' 2.80 3.00 A08 C4' A09 05 ' 2.59 3.00 A08 O1' A09 O5' 2.48 2.80 A08 C3' A09 05 ' 2.65 3.00 B01 Me(T) B02 C2' 2.91 3.00 B03 05 ' B04 C4' 2.59 3.00 B03 05 ' B04 C3' 2.65 3.00 B03 O1' B04 O1' 2.48 2.80 B04 C5' B05 03 ' 2.80 3.00 B05 C5' B06 03 ' 2.75 3.00 B06 C5' B07 03 ' 2.74 3.00 B07 C5' B08 03 ' 2.77 3.00 B08 05 ' B09 C3' 2.65 3.00 B08 C5(T) B09 C2' 2.96 3.20 B08 Me(T) B09 C2' 2.99 3.60
transform does not. The characteristic cross-shape of the observed diffraction pattern is clearly apparent at roughly the correct angle in the calculated DH CAST. This distinctive feature is less noticeable in the SBS CAST and also appears to be at the wrong angle. The observed diffraction pattern only has meridional intensity on the layer l ines /=0 and l= 10. The calculated CAST of the DH model shows this characteristic because of the helical symmetry of the model. However, although the CAST of the SBS model has strong meridional intensity on the 0th and 10th layer lines, there is some significant meridional intensity on the other layer lines, particularly the 3rd, 5th, 7th and 8th layer lines. The observed diffraction pattern shows a strong peak on the l = 2 layer line at R ~ 0.06 A- 1 and a weaker peak on the l = 1 layer line at R ~0.04 A,-1. The CAST of the DH model has these features, but the SBS CAST has peaks of roughly equal magnitude (at R ~0.05 A-1) on these layer lines. A large diffraction peak is observed on the l= 3 layer line at R,,~0.1 A -1, which is present in the DH CAST, but the SBS CAST is relatively fiat on this layer line. Significant diffraction peaks are also observed on the 5th layer line at R~0.11/~ -1, on the 6th layer line at R~0.16A -1, and on the 8th layer line at R~0.12/~ -1. These peaks are predicted by the CAST of the DH model, but not by the CAST of the SBS model.
Albiser and Premilat have also calculated the CAST of their model and claim that comparison with the intensities calculated from the double helix does not allow a choice between either of the models. The curves of the squared transforms of the SBS and DH models which they present in their paper appear to be similar in form to those shown in Figure 3, but the scaling of the intensities on the different layer lines is noticeably different from those given here. In particular, their DH CAST has a very large peak on the 2nd layer line, which is about four times greater in magnitude than the highest peak on the 3rd layer line and is about six times greater in magnitude than the highest peak on the 1st layer line. However, in Figure 3, the peak on the 2nd layer line has about the same magnitude as the peak on the 3rd layer
Side-by-side DNA." S. R. Hubbard et al.
line and is 2-3 times greater in magnitude than the peak on the 1st layer line. The calculated squared transform of the DH model given by Langridge et al. lz appears to be more consistent with the graph presented in Figure 3 than with the graph presented by Premilat and Albiser. Furthermore, the authors do not indicate the source of the observed intensity values which they present in their graph; however, they appear to be similar to those given by Langridge et al.
Although the double helix appears to be superior to the SBS model on the basis of these Fourier transform calculations, there are difficulties inherent in the comparison of CASTs with observed intensities. In particular, the observed structure factor intensities must be corrected for the effects of molecular packing. This is particularly error-prone in the case of a rotation pattern containing overlapping reflections, each of which may have a different packing factor. There is no doubt that the best way to discriminate between competing models is to calculate their structure factors, and to compare them with the observed values.
If the azimuthal orientations, 41 and 42, of the two molecules in the unit cell are allowed to take arbitrary values, then the symmetry of the cell is generally no higher than that of space group P1. The currently accepted model for the B-DNA crystal has a unit cell whose symmetry is consistent with space group P212121. This choice of space group accounts for the observed systematic absences of the reflections (h00), (0k0) and (001) when h, k or I is odd 2°. However, these zones are rather sparsely populated, and it is possible, although unlikely, that the observed absences are due solely to sampling of the molecular transform at points where it is weak or zero. Therefore, we were concerned first with locating the general arrangement of SBS molecules which accounts best for the observed diffraction intensities, without making any assumptions about the crystal symmetry.
Tables 5 and 6 give the 30 different arrangements of the two molecules in the unit cell which gave the lowest residuals between the observed and calculated data for
, d
• "~ lO - - "" -" - -----. ::.'...-.... =.~.~ - .... '
2 ~ ~ - = w . : . ' ~ " 2 ' ' ' ~ : - = ~ : ~ . . . . . . . . . . ?" . . . . . . . . . . . . • . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . - . . . .
0[ -- I - - l I I I I
0.0 0.05 0.1 0. ! 5 0.2 0.25 0.3
Reciprocal Radius R / A t
Figure 3 The CASTs of the SBS and DH models of B-DNA
Int. J. Biol. Macromol. Volume 16 Number 4 1994 199

Side-by-side DNA: S. R. Hubbard et al.
Table $ The 30 arrangements of the molecules in a crystalline SBS model which had the lowest values of the crystallographic residual R. The corresponding values of the penalty function P are given for each of these arrangements
q~l ~b2 Az R(~bl,~b2,Az ) P(~bl,tb2,Az ) (degrees) (degrees) (A) (%) (A- 2)
60.00 50.00 23.00 43.42 1222 30.00 30.00 23.00 43.68 1102 50.00 50.00 23.00 44.06 1594 60.00 60.00 23.00 44.12 1332 50.00 60.00 23.00 44.15 1328 50.00 80.00 6.00 44.24 2080 60.00 90.00 13.00 44.32 885
140.00 70.00 4.00 44.37 311 50.00 80.00 7.00 44.43 1943 50.00 60.00 11.00 44.43 1264 50.00 50.00 11.00 44.49 1546 60.00 270.00 23.00 44.49 800 60.00 50.00 23.00 44.51 1222 80.00 50.00 7.00 44.54 1086 30.00 30.00 23.00 44.57 1102 50.00 80.00 26.00 44.61 1329 60.00 60.00 11.00 44.62 1281 30.00 130.00 33.00 44.62 1025 50.00 300.00 33.00 44.63 556
130.00 60.00 4.00 44.69 548 90.00 240.00 23.00 44.71 1992
300.00 30.00 4.00 44.72 1329 60.00 90.00 14.00 44.72 650 60.00 270.00 23.00 44.72 800 20.00 230.00 23.00 44.73 1517 60.00 60.00 23.00 44.73 1332 90.00 80.00 23.00 44.74 875 60.00 70.00 23.00 44.78 1316 30.00 140.00 33.00 44.78 1091 80.00 50.00 6.00 44.80 944
Table 6 The 30 arrangements of the molecules in a crystalline DH model which had the lowest values of the crystallographic residual R. The corresponding values of the penalty function P are given for each of these arrangements
~bl q~2 Az R(~bl,q~2,Az) P(~bl,~2,Az) (degrees) (degrees) (,~) (%) (A -2)
20.00 20.00 23.00 34.33 109 90.00 90.00 23.00 34.52 113 50.00 90.00 14.00 34.71 66 20.00 270.00 13.00 34.85 59
160.00 200.00 14.00 34.89 70 50.00 50.00 23.00 34.90 198 90.00 230.00 24.00 34.91 295 90.00 20.00 4.00 34.92 58
130.00 20.00 13.00 34.94 108 20.00 310.00 4.00 34.95 127 90.00 200.00 33.00 35.02 128 20.00 130.00 33.00 35.07 303 20.00 200.00 6.00 35.08 82 90.00 230.00 3.00 35.16 66 50.00 270.00 31.00 35.22 65 60.00 200.00 24.00 35.23 335 90.00 270.00 6.00 35.26 110 20.00 160.00 24.00 35.30 198 20.00 60.00 14.00 35.31 152 90.00 50.00 7.00 35.32 296 20.00 90.00 30.00 35.36 110
160.00 20.00 31.00 35.37 69 20.00 160.00 3.00 35.38 71 60.00 90.00 14.00 35.39 368
130.00 200.00 30.00 35.51 107 60.00 20.00 7.00 35.58 331
130.00 200.00 17.00 35.60 265 90.00 50.00 20.00 35.61 66 50.00 230.00 6.00 35.65 149 20.00 50.00 14.00 35.69 276
the SBS and D H B - D N A models, respectively. The corresponding value of the penalty function P(~t,~2,Az) is also given for each of these arrangements. It is clear that, considering all the possible molecular arrangements, the R factor for the D H model assumes significantly lower values than the R factor for the SBS model. The lowest residuals for the D H model are approximately 10 percentage points less than the lowest residuals for the SBS model. We have found several hundred arrangements of D H D N A within the unit cell whose R factors are lower than 43,42%, which is the best value achieved by any arrangement of SBS molecules. Also, the arrangements giving the lowest R values for the D H model tend to have small values of the penalty function P, which indicates that there are few close intermolecular contacts in these arrangements. However, for the SBS model, almost all the arrangements giving the lowest R values have large values of P, suggesting that there is considerable steric hindrance between the molecules. These close contacts usually occur between O 1 P and O 2 P a toms in the phosphate groups of adjacent strands, but the P, 0 3 ' and CY atoms are also involved in some cases. Clearly, given a general arrangement of the molecules in the crystalline form of B-DNA, the SBS model is significantly worse then the D H model in account ing for the observed X-ray diffraction pat tern from these fibres, and its packing into the unit cell is also much less satisfactory.
The procedure adopted here is the s tandard method for testing proposed models against fibre diffraction data. However, it might be argued that it is biased, since the summat ion in the residual calculation is taken only over those Bragg reflections whose observed intensities are greater than the background. Any reflection with a lower intensity is deemed to have a zero amplitude, and it is omitted from the summation. If a proposed model predicted that such a reflection should have a significant intensity, this would go undetected. To overcome this deficiency, we have also tested a slightly modified procedure. The list of observed Bragg reflections was expanded to include all reflections which fell within the 3 A sphere. The amplitudes of the reflections which appeared in the Arnot t and Hukins set were unchanged, and any new reflections were assigned a zero amplitude. When residuals were calculated with this new dataset (results not shown), we found that 10-15 percentage points were added to the values obtained with the unmodified Arnot t and Hukins data for both the D H and SBS models. Similar results (not shown) were obtained when the new reflections were assigned an amplitude of 200, which is approximately half the threshold. Thus, a l though this modified procedure significantly worsens the residuals of the D H and SBS models, it does not affect our earlier conclusion that the D H model is superior to the SBS model.
We then considered molecular orientations which are consistent with the symmetry of the space group P21212 r This space group requires that the D N A molecule should contain at least one diad axis perpendicular to the molecular axis in each axial repeat unit (as both the D H and SBS models do), and that the diad axes of the two molecules in the unit cell should be parallel to each other and to either the a-axis (corresponding to ~b~ = (~2 = 0 ° ) or the b-axis (corresponding to q~x = ~b2 =90°). There is no limitation on the relative axial displacement, Az, of the two molecules, but extra absences, not required by
200 Int. J. Biol. Macromol. Volume 16 Number 4 1994

Side-by-side DNA." S. R. Hubbard et al.
Table 7 Values of the crystallographic residual R calculated for the crystalline DH and SBS models, in the molecular arrangements with q~l = ~2 = 90°, at different values of Az. The corresponding values of the penalty function P are also given for each of these arrangements. If the space group of the crystalline form is assumed to be P212t2~, then the allowed arrangements are those such that ~ =~b 2 =00,90 °
DH DNA SBS DNA
Az R(90°,90°,Az) P(90°,90°,Az) Az R(90°,90°,Az) P(90°,90°,Az) (A) (%) (A-2) (A) (%) (A-2)
0.0 58.14 0 0.0 67.29 792 1.0 49.27 270 1.0 57.28 1017 2.0 47.82 1023 2.0 54.52 3615 3.0 44.66 2465 3.0 51.74 2147 4.0 42.85 4737 4.0 51.93 2098 5.0 48.63 3161 5.0 57.64 1522 6.0 46.73 2317 6.0 55.31 1449 7.0 46.01 1233 7.0 56.19 1413 8.0 46.77 706 8.0 57.42 1792 9.0 41.96 145 9.0 51.87 1576
10.0 37.07 119 10.0 47.74 1453 11.0 36.06 107 11.0 46.82 2602 12.0 40.61 0 12.0 53.07 657 13.0 42.10 376 13.0 52.96 513 14.0 43.93 1239 14.0 53.69 599 15.0 46.93 2686 15.0 54.99 862 16.0 49.56 4192 16.0 54.47 1037 17.0 57.59 5489 17.0 62.75 1200 18.0 48.05 3571 18.0 54.19 981 19.0 45.34 2093 19.0 54.61 821 20.0 44.33 876 20.0 53.93 601 21.0 41.72 244 21.0 53.02 546 22.0 39.80 0 22.0 51.79 752 23.0 34.51 113 23.0 45.53 3439
24.0 37.44 116 24.0 47.87 1468 25.0 45.69 341 25.0 56.07 1743 26.0 44.80 907 26.0 55.67 1550 27.0 47.65 1676 27.0 56.89 1396 28.0 48.55 2779 28.0 57.17 1533 29.0 46.79 3404 29.0 56.49 1522 30.0 43.65 4116 30.0 52.27 2267 31.0 44.35 1818 31.0 51.17 2542 32.0 49.52 820 32.0 56.06 1959 33.0 50.37 122 33.0 58.75 781
this space group, are observed to occur in the diffract ion pa t t e rn f rom crysta l l ine B - D N A fibres when h + k is odd and I = 3m, where m is any integer. If these absences were systematic , they would imply that , for each molecule at (x, y, z), an ident ical one would exist a t ( x + 1/2, y + 1/2, z + 1/3). Hence, they require tha t Az = c/3. If one molecule were at the or igin with az imutha l o r ien ta t ion q~l, then the second molecule would be at (1/2, 1/2, 1/3), with az imutha l o r ien ta t ion ~b2=~bl=~ . In this case, the s t ructure fac tor of the Bragg reflection, F(hkl), is of the form:
F(hkl)=~.,, G,,t(R)exp{in(~+;-q~)}
x[l+expf i2 (h+k-+Li I( \2 2 3 / ; I (8)
This express ion clearly predic ts the absence of reflections
for which h+k is odd and l=3m. However , these reflections are only general ly no t sys temat ica l ly absent , and the list given by Arno t t and Huk ins conta ins eight reflections, namely (4,1,3), (3,2,3), (0,5,3), (4,1,6), (8,1,6), (7,2,6), (1,0,9) and (4,1,9), whose indices satisfy the above rule, and yet which have very significant intensities. The impl ica t ion of this is tha t the a r r angemen t of the molecules in the unit cell is subject only to the weaker cons t ra in t tha t Az ~ c/3.
Mos t of the a r r angemen t s of bo th D H and SBS D N A which have the lowest res iduals a p p e a r to occur when Az = 23.0 A (Tables 5 and 6). However , the a r r angemen t (q~l,~b2,Az) is equivalent to tha t with (~bl,~b2,c-Az). Hence, the a r r angemen t (~bl,~b2,23.0) is equivalent to (~bl,~b2,10.7). The value of 10.7 ~ is r easonab ly close to the accepted value of Az ~ c/3.
Tables 7 and 8 show the values of the R factor ca lcula ted for the crysta l l ine forms of bo th the D H and SBS mode l s of B - D N A at different values of Az, where
Int. J. Biol. Macromol. Volume 16 Number 4 1994 201

Side-by-side DNA: S. R. Hubbard et al.
Table 8 Values of the crystallographic residual R calculated for the crystalline DH and SBS models, in the molecular arrangements with ~1 =~b2 =0°, at different values of Az. The corresponding values of the penalty function P are also given for each of these arrangements, which form the other set of possible arrangements consistent with P212121 being the space group of the crystalline form
DH DNA SBS DNA
Az R(O °,0 °,Az) P(0 °,0°,Az) Az R(0 °,0°,Az) P(0°,0°,Az) (A) (%) (A-2) (A) (%) (A-5)
0.0 70.44 6186 0.0 69.60 4013 1.0 61.96 3882 1.0 60.03 4254 2.0 57.64 2405 2.0 57.68 3787 3.0 50.05 1622 3.0 53.40 3983 4.0 50.60 933 4.0 52.93 3846 5.0 56.70 737 5.0 58.14 3575 6.0 53.74 922 6.0 57.24 3615 7.0 53.12 8537 7.0 56.91 4599 8.0 54.96 1556 8.0 56.50 3899 9.0 51.04 831 9.0 51.69 1457
10.0 45.32 602 10.0 49.11 1225 11.0 44.05 1094 i1.0 49.09 1142 12.0 50.85 1894 12.0 53.05 1049 13.0 52.73 3990 13.0 54.07 1101 14.0 52.64 3798 14.0 56.46 717 15.0 55.97 1996 15.0 59.24 370 16.0 56.30 1143 16.0 60.01 50 17.0 63.84 755 17.0 67.52 19 18.0 55.48 1325 18.0 59.34 130 19.0 55.04 2262 19.0 58.46 468 20.0 53.39 5190 20.0 55.80 808 21.0 52.13 3130 21.0 53.57 1057 22.0 49.43 1485 22.0 52.54 1054 23.0 42.50 583 23.0 48.01 1137 24.0 45.50 749 24.0 48.95 1216 25.0 54.22 967 25.0 55.05 1664 26.0 53.56 2110 26.0 55.25 3126 27.0 54.18 2902 27.0 57.80 51179 28.0 55.84 831 28.0 58.76 3636 29.0 54.59 662 29.0 56.55 3586 30.0 50.56 1006 30.0 52.66 3853 31.0 50.81 1658 31.0 53.98 3684 32.0 59.90 2641 32.0 59.30 3943 33.0 64.05 4715 33.0 61.73 4088
$ 1 = $ 2 = 9 0 ° or 0 °, respectively. The corresponding values of the steric hindrance penalty function are also given for these arrangements. The DH molecular arrangements in which t~l =~b2=90 ° have their lowest R value of 34.51% at Az=23 .0A or 10.7A. This arrangement also corresponds roughly to a minimum in the penalty function P, indicating that there is no steric hindrance between the two molecules. In the case of the SBS model, the values of the residual are approximately 10 percentage points higher than for the DH model. The lowest value of the residual in this arrangement, R=45.53%, also occurs at Az=23.0A. However, the penalty function P has a very large value, showing that there are many close intermolecular contacts. In the second possible arrangement, with t# t= t#2=0 °, the residuals for the DH model are significantly higher than in the first arrangement, with the lowest value being R = 42.50%, which again occurs at Az = 23.0 A. The value of the penalty function in this arrangement, P = 583 A- 2, indicates that there are a number of close intermolecular contacts. The SBS model has markedly higher residuals than the DH model in this second arrangement, with the lowest value of the residual being R--48.01%, at Az = 23.0 A; the value of the penalty function is 1137 A - 2 in this case.
Once we had confirmed that the best axial displacement of both models was approximately c/3, we attempted to define it as accurately as possible. Residuals were
calculated for values of Az between 10.5 A and 11.5 A in steps of 0.1A with ~bl=~b2=90 °. In each case, three different sets of observed structure factors were used in calculation of the residual. Dataset 1 consisted of the original observed structure factors of Arnott and Hukins 2°. Dataset 2 included the extra absences discussed above, assigning a zero amplitude to each. Dataset 3 was the same except that the extra absences were assigned a value of 200. The results of this analysis are shown in Table 9. When dataset 1 was used, the lowest values of the residual occurred at Az = 10.7 A for both the DH and SBS models. Inclusion of the extra absences did not substantially modify this conclusion: the optimum value of Az changed to 11.2 A when the absences were included with zero amplitude and to 11.0 A when they were included with an amplitude of 200. Therefore, we conclude that the optimum arrangements of the DH and SBS models consistent with space group P212~21 occur when ~b t =~b2 =90 ° and Az= 10.7 A.
The observed and calculated structure factors for the optimum arrangements of both the DH and SBS models are given in Table 10. The residuals between the calculated and observed structure factor amplitudes with these arrangements of the two models are R=45.53% for the SBS model and R=34 .51% for the DH model. The corresponding values of the penalty function P are 3439A -2 for the SBS model and l13A -2 for the DH model.
202 Int. J. Biol. Macromol. Volume 16 Number 4 1994

Table 9 Residuals and penalty functions of the DH and SBS models when Az ~ c/3 and q~l = ~b2 = 90 °. See text for explanation of the datasets
Dataset 1 Dataset 2 Dataset 3 R(90°,90°,Az) R(90°,90°,Az) R(90°,90°,Az) P
(%) (%) (%) (/~-2) Az (./~) DH SBS DH SBS DH SBS DH SBS
10.5 34.96 45.90 44.87 56.74 41.35 52.32 117 2165 10.6 34.52 45.59 43.40 55.44 39.99 51.14 115 2740 10.7 34.51 45.51 42.31 54.17 39.12 50.03 113 3439 10.8 34.99 45.58 41.59 52.98 38.68 49.08 111 3847 10.9 35.52 46.14 40.93 52.14 38.33 48.62 109 3355 11.0 36.06 46.80 40.22 51.44 38.07 48.46 107 2602 11.1 36.63 47.46 39.50 50.70 38.30 48.85 104 1985 11.2 37.19 48.19 38.73 49.99 39.63 50.45 0 1500 11.3 37.75 48.91 39.72 51.16 39.77 50.70 0 1188 11.4 38.23 49.59 41.53 53.18 39.83 50.80 0 1057 11.5 38.81 50.26 43.28 55.12 40.91 51,86 0 942
Discussion
The testing of a proposed model for fibrous DNA falls into two stages. In the first stage, the internal stereochemistry must be checked against standard bonded and non-bonded distances, and then the Fourier transform of the molecule must be calculated to see that it conforms, at least in general, with the observed distribution of intensities in the diffraction pattern. Both these procedures essentially treat the molecule as an isolated object. Tests of this type have been used exclusively in assessing the acceptability of previous SBS models. Most SBS models have had reasonable stereochemistry; none of them could have been rejected on the basis of stereochemistry alone, since small adjustments to the coordinates of a few atoms may alleviate any incorrect covalent bond lengths or close non-bonded contacts. The more controversial part of the first stage is the comparison of observed and calculated diffraction intensity distributions. Those who have proposed SBS models have made the point that the fibre diffraction data on which the double helix is based are of such poor quality that it is impossible to use them to discriminate between DH and SBS models. However, they have all used diffraction data from disordered or semi-crystalline fibres. The degree to which a calculated CAST agrees with such data is to some extent subjective, although it must be emphasized that, in our view, no SBS model has satisfactorily accounted for even this low- quality data. The latest model 1° does not appear to be significantly more successful than its predecessors in this regard.
The second stage of testing can only be undertaken when crystalline diffraction data are available. Once the dimensions of the unit cell and the number of molecules contained within it are known, the packing can be examined. It is clear that the Premilat and Albiser model cannot be packed into the observed B-DNA cell, since significant intermolecular short contacts are present for nearly all relative displacements and orientations of the two molecules, although it is possible that a least-squares refinement of the model would reduce the number and severity of these contacts. The failure of the SBS model to predict the observed structure factor intensities is a more serious deficiency. Its R factor is worse than that of the DH model by 9 percentage points (43.42% for SBS and 34.33% for DH) when general arrangements of the molecules are allowed, and by 11 percentage points
Side-by-side DNA: S. R. Hubbard et al.
(45.53% for SBS and 34.52% for DH) when the symmetry of the cell is constrained to that of space group P212121. On the basis of these figures, the model with the higher R factors could be decisively rejected even if, for example, both models were double helices having a mononucleotide asymmetric unit. However, the SBS molecule has 10 nucleotides in the asymmetric unit and therefore has 10 times more degrees of freedom than the DH model. We have not used standard statistical tests to evaluate the confidence that we may have in these models, but there is no doubt that the SBS model could be rejected with greater than 99% confidence.
Several points can be made about the agreement between the observed and calculated structure factors which may be of significance in the design of any future models for DNA. For the purposes of this discussion, we have arbitrarily divided the Bragg reflections into weak (Fo < 2000), medium (2000 ~< Fo ~< 5000) and strong (Fo>5000) spots. There are 73, 61 and 17 reflections, respectively, in these categories.
The SBS model does not account adequately for the general distribution of intensities in the B-DNA diffraction pattern. In the case of the weak reflections, the SBS model over-estimates (+ ) the amplitudes of 54 spots and under-estimates ( - ) the amplitudes of 19 spots. For the medium reflections, the figures are + 18 and - 4 3 , and for the strong reflections they are + 0 and - 17. The results for the DH model are + 39 and - 3 4 in the case of weak spots, +30 and - 3 1 for medium spots, and + 4 and - 1 3 for strong spots. It is clear that the amplitudes calculated from the SBS model are significantly skewed, with a tendency to over-estimate the weak reflections and to under-estimate the medium and strong reflections. It is of particular note that all the strong reflections are under-estimated. In contrast to this, the amplitudes calculated from the DH model are more evenly distributed about the observed values.
Before discussing the level of agreement between the observed and calculated amplitudes of specific reflections, it is useful to consider the possible sources of error in the measurement of intensities and the calculation of transforms. The major contribution to errors in the observed intensities arises from the difficulty in determining the background scattering in a fibre diffraction pattern. The result of this baseline error is that weak intensities are measured to an accuracy of about +5 0 % of F o, and the error in strong reflections is probably about 10-15%. Errors in the calculated amplitudes may arise from the approximation made in accounting for the scattering from water and from neglecting any contribution from ions in the fibre. Water molecules and ions in ordered positions in the unit cell may make significant contributions to individual reflections. However, this effect should be small, especially since lithium was the counter-ion in the fibres from which the Arnott and Hukins data were recorded. A more significant error may arise, in the case of the DH model, from the assumption that the molecule is a perfectly regular helix. This is unlikely to be true for a DNA molecule with a random base sequence, since oligonucleotide crystals show that small-scale polymorphism is present which is dependent on the local base sequence (e.g. ref. 23). Therefore, in a DNA polymer, there are likely to be sequence-dependent perturbations of the helical structure which will not be taken into account when calculating the structure factors.
Int. J. Biol. Macromol. Volume 16 Number 4 1994 203

PO
2 T
ab
le
10
Ob
serv
ed
an
d c
alcu
late
d s
tru
ctu
re f
acto
r a
mp
litu
de
s fo
r th
e S
BS
an
d D
H
B-D
NA
mo
de
ls
d(hk
l) lE
e[
IF01
d(
hkl)
IF
¢l
IFcl
d(
hkl)
IF
cl
IFJ
d(hk
l)
IF¢l
IF
¢I
d(hk
l)
IF01
IF
J d(
hkl)
IF
0[
IFJ
hkl
(A)
[Fo[
SB
S D
H
hkl
(A)
IFol
SB
S D
H
hkl
(A)
IFol
SB
S D
H
hkl
(A)
lEo[
SB
S D
H
hkl
(A)
IF.I
SB
S D
H
hkl
(A)
IF.l
SBS
DH
2 0
0 15
.40
1930
29
6 67
3
0 1
9.82
1
06
1
1902
82
7 3
2 2
6.92
53
1 1
69
8
1210
5
3 3
4.38
32
17
1917
26
19
3 3
5 4.
50
1287
48
3 36
0 1
2 7
4.38
i
O 3 o_.
< o_.
e-" 3 ¢1)
Z
e- 3 O" o 4~
tO
¢,D
3 1
0 9.
34
2467
21
81
2506
3
1 1
9.00
96
5 18
91
804
1 3
2 6.
69
6 2
3 4,
31
3325
13
99
4545
5
1 5
4.46
12
87
1047
36
7 3
0 7
4.36
2 2
0 9.
08
3754
31
47
3192
2
2 1
8.77
42
9 11
27
936
4 1
2 6.
69
1394
22
50
663
0 5
3 4.
18
3641
28
2 58
9 1
4 5
4.28
55
77
2158
33
92
3 1
7 4.
28
4 0
0 7.
70
2896
2
18
1
2684
0
3 1
7.32
2
3 2
6.26
15
01
2758
71
1 1
5 3
4.14
31
10
4101
40
96
5 2
5 4.
22
2 2
7 4.
25
1 3
0 7.
29
1394
1
97
8
3090
3
2 1
7.40
91
0 16
10
1157
5
0 2
5.79
2
5 3
4.03
4
3 5
4.20
56
84
3724
43
55
1 3
7 4.
02
4 2
0 6.
35
2145
1
71
1
14
85
1
3 1
7,12
3
3 2
5.70
7
1 3
4.03
39
68
4371
40
37
6 0
5 4.
08
4 1
7 4.
02
3 3
0 6.
06
858
2032
58
6 4
1 1
7.12
75
1 93
1 14
05
5 1
2 5.
60
2124
27
73
2135
3
5 3
3.87
17
16
4463
27
05
6 1
5 4.
02
1214
19
50
1353
2
3 7
3.92
5 1
0 5.
94
536
1233
12
1 2
3 l
6.61
96
5 1
27
1
1414
1
4 2
5.26
28
96
2453
32
65
0 6
3 3.
56
3217
99
1 18
14
0 5
5 3.
74
5 1
7 3.
74
0 4
0 5.
63
858
1226
67
1 4
2 1
6.24
64
3 13
20
1685
5
2 2
5.15
2
6 3
3.47
1
5 5
3.72
2
0 8
4.06
2 4
0 5.
28
5791
46
17
6026
5
0 1
6.06
85
8 1
10
8
2110
4
3 2
5.12
33
25
2500
44
78
8 2
3 3.
46
7 0
5 3.
68
1896
32
62
498
2 1
8 4.
00
6 0
0 5.
13
6649
29
13
4073
3
3 1
5.96
64
3 99
5 12
69
6 1
2 4.
80
5 5
3 3,
46
3646
30
48
5645
6
3 5
3.59
1
2 8
3.91
5 3
0 4.
76
2896
26
25
3224
1
4 1
5.46
1
82
3
2634
21
05
3 4
2 4.
73
3217
2
10
1
3771
0
7 3
3,09
5
4 5
3.54
24
67
2472
27
57
3 0
8 3.
90
4 4
0 4.
54
2038
1
00
8
1288
5
2 1
5.33
0
5 2
4.35
5
6 3
3.08
1
1 6
5.37
75
1 12
29
1304
3
1 8
3.84
7 1
0 4.
32
751
2176
51
3 4
3 1
5.3
1
2788
30
13
234
1 5
2 4.
30
1 7
3 3.
07
2 0
6 5.
28
1501
23
7 11
61
2 2
8 3.
82
3 5
0 4.
12
2467
17
44
2558
2
4 1
5.22
16
09
2115
21
81
7 0
2 4.
26
1896
17
29
2542
8
4 3
3.06
20
48
3741
34
37
0 2
6 5,
03
1930
15
43
2412
3
2 8
3.68
8 0
0 3.
85
7614
19
14
3985
6
1 1
4.95
2
5 2
4.18
1
1 4
7.64
7 3
0 3.
80
3 4
1 4.
88
2038
18
16
3118
7
1 2
4.18
16
09
4067
34
51
2 1
4 7.
02
6 4
0 3.
79
4075
34
04
4094
5
4 1
4.12
1
50
1
2571
32
00
6 3
2 4.
11
1609
18
06
3280
1
2 4
6.59
0 6
0 3.
75
5899
1
64
9
2904
7
2 1
4,07
1
50
1
2894
24
56
5 4
2 4.
03
3 0
4 6.
51
2 6
0 3.
64
4 5
1 3.
86
3 5
2 4.
00
3 1
4 6.
26
8 2
0 3.
64
8 0
1 3.
83
2427
1
63
8
3269
7
2 2
3.98
45
04
2807
39
71
2 2
4 6.
18
5 5
0 3.
63
1093
9 26
92
6990
8
1 1
3.77
4
5 2
3.79
1
3 4
5.51
9 1
0 3.
38
6 4
1 3,
77
8 0
2 3.
75
2199
29
32
2035
4
1 4
5,51
4
6 0
3.37
31
10
11
67
19
50
7 3
1 3.
77
3754
36
70
4915
1
1 3
9.55
38
61
2467
48
19
5 2
4 4.
55
322
2146
19
1 2
2 6
4.78
16
09
3435
29
28
0 3
8 3.
67
858
1444
75
4 4
0 6
4.54
51
48
875
2769
1
3 8
3.65
751
3246
87
3 1
3 6
4.45
4
1 8
3,65
643
1459
62
4 4
1 6
4.45
36
46
2433
47
25
2 3
8 3.
57
4 2
6 4.
21
5362
28
37
6537
5
0 8
3,48
536
2707
15
44
5 1
6 4.
08
643
16
08
20
50
4 3
8 3.
32
5 3
6 3.
63
1501
15
09
1127
2
4 8
3.29
965
2175
26
73
1 5
6 3.
49
3217
19
88
2681
1
0 9
3.72
2
5 6
3,42
0
2 9
3.55
1517
27
07
2303
1394
33
00
1342
2359
25
22
1358
1609
20
96
749
858
1214
60
858
267
222
4612
26
22
329
7888
60
21
5343
4502
24
47
6201
4550
27
71
2719
5470
43
35
3243
18
23
23
48
3463
2896
18
49
1900
1930
16
99
1529
2359
89
4 77
6
2574
1
12
1
2515
1 7
0 3.
20
1 6
1 3.
70
2 0
3 9.
08
4826
73
6 45
08
4 3
4 4,
53
1930
23
74
1564
7
1 6
3.42
18
23
1857
29
74
3 1
9 3.
48
8 4
0 3.
18
1180
14
49
2218
2
6 1
3.62
0
2 3
7.95
43
97
3578
45
25
3 4
4 4.
26
2896
30
63
2371
3
5 6
3.32
2
2 9
3.46
28
96
6155
37
31
7 5
0 3.
15
5 5
1 3.
61
3 1
3 7.
18
2788
29
42
4140
7
4 4
3.21
28
96
2269
25
09
7 2
6 3.
31
1716
1
93
5
2679
1
3 9
3.33
9 3
0 3.
11
2359
1
71
8
2507
8
2 1
3.62
2
2 3
7.06
20
38
19
81
22
92
0 1
5 6,
46
643
389
56
7 3
6 3.
14
4 1
9 3,
33
2145
22
53
773
10 0
0
3.08
3
6 1
3.50
39
68
4955
39
31
4 0
3 6.
35
2 1
5 5.
95
643
2193
39
16
6 4
6 3.
14
1 1
10
3.3
1
3 7
0 3.
07
1 0
2 1
4.7
8
3432
27
24
2832
3
2 3
6.29
18
20
1424
19
79
1 2
5 5,
68
8 1
6 3.
14
2252
36
47
2485
2
0 10
3.
29
1756
9 16
512
1716
7
6 6
0 3.
03
2882
43
08
710
0 1
2 1
3.4
9
4933
39
68
4127
1
3 3
6.11
3
0 5
5,63
64
47
4686
75
09
2 6
6 3.
06
2 1
10
3.26
1 0
1 22
.74
26
81
10
56
2757
1
1 2
12
.35
28
96
2078
30
02
4 1
3 6.
11
965
3693
23
35
3 1
5 5.
47
965
779
1152
8
2 6
3.06
0
2 10
3.
23
8192
76
00
8454
0 1
1 18
.71
3861
2
10
1
2739
2
0 2
11
.37
34
32
1959
20
59
4 2
3 5
.53
10
72
4040
70
0 2
2 5
5.41
96
5 92
0 54
8 5
5 6
3.05
16
09
2426
20
26
1 2
10
3.21
1 1
1 15
.99
1072
36
24
2064
2
1 2
10
.15
46
12
2217
47
68
3 3
3 5.
33
2574
29
58
3434
3
2 5
5.04
1
0 7
4,76
85
8 10
80
289
3 0
10
3.20
78
12
4128
43
55
2 0
1 14
.01
536
401
1004
0
2 2
9.36
1
28
7
790
915
5 1
3 5.
25
2252
40
55
2852
0
3 5
5.01
58
40
5802
46
41
1 1
7 4.
65
1716
26
22
1487
1
3 10
3.
06
2 1
1 11
.89
322
5650
63
9 1
2 2
8.95
0
4 3
5.03
13
94
1117
60
4 1
3 5
4.95
2
1 7
4.50
16
09
1536
19
41
4 1
10
3.06
15
01
2332
10
95
0 2
1 10
.67
643
301
190
3 0
2 8.
77
2882
49
21
1859
2
4 3
4.78
18
23
1102
11
82
4 1
5 4.
95
5362
37
16
2433
1 2
1 10
.08
4 0
2 7.
00
6 0
3 4.
67
3003
25
20
1094
2
3 5
4.77
19
30
1389
15
21
• h,
k a
nd I
den
ote
the
Mil
ler
indi
ces
of e
ach
refl
ectio
n •
d(hk
l) is
the
spa
cing
of
the
crys
tal
plan
es h
kl a
nd
is
give
n by
:
[-h
2 k 2
12
] -~
,2
d(hk
l)=L-
a~ +
~+
~J
• tF
ol is
the
obs
erve
d st
ruct
ure
fact
or a
mpl
itud
e of
eac
h re
flec
tion
(ref
. 20
). T
he
refl
ectio
ns
in t
he l
ist
for
whi
ch n
o va
lue
of I
Fol i
s gi
ven
are
over
lapp
ing
refl
ectio
ns,
whi
ch o
ver
lap
wit
h th
e ne
xt r
efle
ctio
n in
the
lis
t fo
r w
hich
a v
alue
of
IFol
is
cite
d. T
his
valu
e of
IFo
l cor
resp
onds
to
the
squa
re r
oot
of t
he t
otal
int
ensi
ty o
f th
e o
ver
lap
pin
g r
efle
ctio
ns
• IF
cl S
BS
is t
he s
truc
ture
fac
tor
ampl
itud
e of
eac
h re
flec
tion,
ca
lcul
ated
for
the
SB
S m
odel
hav
ing
the
arr
ange
men
t: ~
b~ =
~b 2
=9
0.0
0 °
and
Az=
10.
7 A
. T
he
resi
dual
R b
etw
een
the
calc
ulat
ed a
nd
obs
erve
d st
ruct
ure
fact
or a
mpl
itud
es f
or t
his
arra
ng
emen
t of
the
SB
S m
odel
= 4
5.51
%
• N
Fcl
DH
is
the
stru
ctur
e fa
ctor
am
plit
ude
of e
ach
refl
ectio
n,
calc
ulat
ed f
or t
he D
H m
od
el h
avin
g th
e ar
rang
emen
t: ~
b~ =
~b 2
= 9
0.00
° an
d A
z =
10.
7 A
. T
he
resi
dual
R b
etw
een
the
calc
ulat
ed a
nd o
bser
ved
stru
ctur
e fa
ctor
am
plit
udes
for
thi
s ar
rang
emen
t of
the
DH
mod
el =
34.
51%

We have arbitrarily defined reflections for which F c is outside the range Fo++_½Fo as at variance with the observed amplitude given the possible sources of error discussed above. In the category of weak reflections, the SBS model predicts that 41 reflections satisfy this criterion, whereas 38 do so for the DH model. In view of the large errors of measurement associated with weak reflections, we do not believe that these apparently large numbers of deficiencies in the predicted amplitudes cast serious doubt on the acceptability of either of the models. However, in the case of the SBS model, three reflections, namely (2,1,1), (1,1,4) and (2,2,4), have predicted amplitudes which are very large, although only small amplitudes are observed. The DH model has one reflection, (2,1,5), which suffers from this problem.
The SBS and DH models also predict that 20 and 12 medium reflections, respectively, and 5 and 2 strong reflections, respectively, have calculated amplitudes which lie outside the range defined above. The presence of strong reflections in the diffraction pattern indicates that there is a high concentration of atoms in the corresponding crystalline planes, so the inability of any model to account for their observed amplitudes must be regarded as a particularly serious deficiency. It is noteworthy that the SBS model does not account adequately for nearly one-third of the strong reflections. It may be pertinent to observe that four of the five reflections are equatorial, namely the (6,0,0), (8,0,0), (0,6,0) and (5,5,0) spots. Indeed, there is only one strong equatorial reflection, (2,4,0), for which the SBS model accounts satisfactorily. In contrast, the DH model only fails to account for one equatorial reflection, (0,6,0). Since these reflections arise from the projection of the structure along the c-axis, they are sensitive to the rotational symmetry of the molecule. It is tempting to conclude that the failure of the SBS model and the success of the D H model in predicting equatorial amplitudes arise simply because the DNA molecule has the high rotational symmetry possessed by the DH model but not by the SBS model.
The other area of the diffraction pattern in which there is a concentration of strong reflections is the 10th layer line. Diffraction on this layer line is dominated by the stacking of the bases, although the positions of the phosphate groups can have a significant effect 17. Both models account reasonably well for these strong reflections. This is not unexpected since the base stacking is similar in both cases, with Watson-Crick base pairs approximately perpendicular to the helical axis.
C o n c l u s i o n
The work presented here underlines the point that
Side-by-side DNA: S. R. Hubbard et al.
crystalline diffraction data should be used when testing proposed models since they provide an objective and quantitative measure of the acceptability of the models. It also illustrates the degree of confidence that we may have in the large number of double helical conformations that have been determined from crystalline fibrous diffraction data. The model described by Premilat and Albiser, although ultimately unsuccessful in displacing the double helix as the best model for B-DNA in fibres, has allowed us to set the standard against which any future model should be judged.
Acknowledgements We are grateful to the Science and Engineering Research Council for the provision of a Research Studentship to S.R.H.
References 1 Watson, J.D. and Crick, F.H.C. Nature 1953, 171, 737 2 Rodley, G.A., Scobie, R.S., Bates, R.H.T. and Lewitt, R.M. Proc.
Natl Acad. Sci. USA 1976, 73, 2959 3 Sasisekharan, V. and Pattabiraman, N. Curr. Sci. 1976, 45, 779 4 Sasisekharan, V. and Pattabiraman, N. Nature 1978, 275, 179 5 Bates, R.H.T., Lewitt, R.M, Rowe, C.H., Day, J.P. and Rodley, G.A.
J. Proc. R. Soc. N Z 1977, 7, 273 6 Bates, R.H.T., McKinnon, G.C., Millane, R.P. and Rodley, G.A.
Pramana 1980, 14, 233 7 Sasisekharan, V., Pattabiraman, N. and Gupta, G. Proc. Natl
Acad. Sci. USA 1978, 75, 4092 8 Albiser, G. and Premilat, S. Biochem. Biophys. Res. Commun.
1980, 95, 1231 9 Miilane, R.P. and Rodley, G.A. NucL Acids Res. 1981, 9, 1765
10 Premilat, S. and Albiser, G. Biochem. Biophys. Res. Commun. 1982, 104, 22
11 Langridge, R., Marvin, D.A., Seeds, W.E., Wilson, H.R., Hooper, C.W., Wilkins, M.H.F. and Hamilton, L.D.J. Mol. Biol. 1960, 2, 38
12 Langridge, R., Wilson, H.R., Hooper, C.W., Wilkins, M.H.F. and Hamilton, L.D.J. Mol. Biol. 1960, 2, 19
13 Fuller, W. and Mahendrasingam, A. in 'Topics in Nucleic Acid Structure', Part 3 (Ed S. Neidle), Macmillan Press, Basingstoke, 1987, p 101
14 Greenall, R.J., Pigram, W.J. and Fuller, W. Nature 1979, 282, 880 15 Arnott, S. Nature 1979, 278, 780 16 Arnott, S. Trends Biochem. Sci. 1980, 231 17 Greenall, R.J. PhD Thesis, University of Keele, 1982 18 GreenaU, R.J. in 'Topics in Nucleic Acid Structure', Part 3
(Ed S. Neidle), Macmillan Press, Basingstoke, 1987, p 133 19 Arnott, S. and Hukins, D.W.L Biochem. Biophys. Res. Commun.
1972, 47, 1504 20 Arnott, S. and Hukins, D.W.L J. Mol. Biol. 1973, 81, 93 21 Cochran, W., Crick, F.H.C. and Vand, V. Acta Crystallogr. 1952,
5, 581 22 Hubbard, S.R. DPhil Thesis, University of York, submitted 1994 23 Drew, H.R., Wing, R.M., Takano, T., Broka, C., Tanaka, S.,
Itakura, K. and Dickerson, R.E. Proc. Natl Acad. Sci. USA 1981, 78, 2179
Int. J. Biol. Macromol. Volume 16 Number 4 1994 205




















