
Stroke Genomics: Approaches to Identify, Validate, and Understand Ischemic Stroke Gene Expression
24
Review Article Stroke Genomics: Approaches to Identify, Validate, and Understand Ischemic Stroke Gene Expression *Simon J. Read, *Andrew A. Parsons, *David C. Harrison, *Karen Philpott, †Karen Kabnick, ‡Shawn O’ Brien, ‡Steven Clark, ‡Mary Brawner, ‡Stewart Bates, ‡Israel Gloger, §Jeffrey J. Legos, and §Frank C. Barone Neurology Center of Excellence for *Drug Discovery, †Bioinformatics, ‡Genetics Research, and §High Throughput Biology, GlaxoSmithKline, Harlow, U.K., and King of Prussia, Pennsylvania, U.S.A. Summary: Sequencing of the human genome is nearing completion and biologists, molecular biologists, and bioinfor- matics specialists have teamed up to develop global genomic technologies to help decipher the complex nature of patho- physiologic gene function. This review will focus on differen- tial gene expression in ischemic stroke. It will discuss inheri- tance in the broader stroke population, how experimental mod- els of spontaneous stroke might be applied to humans to identify chromosomal loci of increased risk and ischemic sen- sitivity, and also how the gene expression induced by stroke is related to the poststroke processes of brain injury, repair, and recovery. In addition, we discuss and summarise the literature of experimental stroke genomics and compare several ap- proaches of differential gene expression analyzes. These in- clude a comparison of representational difference analysis we have provided using an experimental stroke model that is rep- resentative of stroke evolution observed most often in man, and a summary of available data on stroke differential gene expres- sion. Issues regarding validation of potential genes as stroke targets, the verification of message translation to protein prod- ucts, the relevance of the expression of neuroprotective and neurodestructive genes and their specific timings, and the emerging problems of handling novel genes that may be dis- covered during differential gene expression analyses will also be addressed. Key Words: Differential display—Gene ex- pression—MCAO—Microarray gridding—Neuroprotective genes—Neurodestructive genes—Protein—Representative dif- ferential analysis—Stroke—Stroke in evolution—Suppressive subtractive hybridization—Target validation—Transcription— Translation. ISCHEMIC STROKE Genomics technology As efforts to complete sequencing of the human genome are nearing conclusion, there is an increased interest in the application of genomic approaches to aid in the discovery, development, and rationale of the use of drugs. As databases of differential gene ex- pression have expanded, so has the expectation of identifying novel drug targets for disease interven- tion. Indeed, significant work has already been per- formed to understand gene expression changes in the ischemic heart (Stanton et al., 2000) and the ischemic brain (Barone and Feuerstein, 1999; Soriano et al., 2000). Early epidemiologic studies of the 1970s provided ini- tial evidence for a genetic influence in stroke. The Framingham study was one of the first studies to suggest that a positive parental history of stroke contributed significantly to the risk of the offspring (Kannel et al., 1970). Thirty years later stroke remains an area of substantial unmet medical need. The complexity of stroke undoubtedly reflects the heterogeneity of the hu- man stroke population, the contribution of monogenic and polygenic disorders to this disease process, and the interactions of these with a multitude of environmen- tal factors. Received April 16, 2001; final revision received April 19, 2001; accepted April 19, 2001. Address correspondence and reprint requests to Dr. Simon J. Read, Neurology, HW2515, GlaxoSmithKline, New Frontiers Science Park North, Harlow CM19 5AW, U.K. or Dr. Frank C. Barone, High Throughput Biology, UW2521, GlaxoSmithKline, 709 Swedeland Road, King of Prussia, PA 19406, U.S.A. Abbreviations used: ANP, atrial natriuretic peptide; BNP, brain na- triuretic peptide; CADASIL, cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy; DWI, diffusion- weighted imaging; IEGS, immediate early genes; MCA, middle cere- bral artery; MCAO, middle cerebral artery occlusion; MELAS, mito- chondrial encephalopathy-lactic acidosis and strokelike syndrome; MRI, magnetic resonance imaging; pMCAO, permanent MCAO; PWI, perfusion-weighted imaging; RDA, representational difference analy- sis; SHR, spontaneously hypertensive rat; WKY, Wistar-Kyoto rats. Journal of Cerebral Blood Flow and Metabolism 21:755–778 © 2001 The International Society for Cerebral Blood Flow and Metabolism Published by Lippincott Williams & Wilkins, Inc., Philadelphia 755
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Stroke Genomics: Approaches to Identify, Validate, and Understand Ischemic Stroke Gene Expression

Review Article
Stroke Genomics: Approaches to Identify, Validate, andUnderstand Ischemic Stroke Gene Expression
*Simon J. Read, *Andrew A. Parsons, *David C. Harrison, *Karen Philpott, †Karen Kabnick,‡Shawn O’ Brien, ‡Steven Clark, ‡Mary Brawner, ‡Stewart Bates, ‡Israel Gloger,
§Jeffrey J. Legos, and §Frank C. Barone
Neurology Center of Excellence for *Drug Discovery, †Bioinformatics, ‡Genetics Research, and §High Throughput Biology,GlaxoSmithKline, Harlow, U.K., and King of Prussia, Pennsylvania, U.S.A.
Summary: Sequencing of the human genome is nearingcompletion and biologists, molecular biologists, and bioinfor-matics specialists have teamed up to develop global genomictechnologies to help decipher the complex nature of patho-physiologic gene function. This review will focus on differen-tial gene expression in ischemic stroke. It will discuss inheri-tance in the broader stroke population, how experimental mod-els of spontaneous stroke might be applied to humans toidentify chromosomal loci of increased risk and ischemic sen-sitivity, and also how the gene expression induced by stroke isrelated to the poststroke processes of brain injury, repair, andrecovery. In addition, we discuss and summarise the literatureof experimental stroke genomics and compare several ap-proaches of differential gene expression analyzes. These in-clude a comparison of representational difference analysis we
have provided using an experimental stroke model that is rep-resentative of stroke evolution observed most often in man, anda summary of available data on stroke differential gene expres-sion. Issues regarding validation of potential genes as stroketargets, the verification of message translation to protein prod-ucts, the relevance of the expression of neuroprotective andneurodestructive genes and their specific timings, and theemerging problems of handling novel genes that may be dis-covered during differential gene expression analyses willalso be addressed. Key Words: Differential display—Gene ex-pression—MCAO—Microarray gridding—Neuroprotectivegenes—Neurodestructive genes—Protein—Representative dif-ferential analysis—Stroke—Stroke in evolution—Suppressivesubtractive hybridization—Target validation—Transcription—Translation.
ISCHEMIC STROKE
Genomics technologyAs efforts to complete sequencing of the human
genome are nearing conclusion, there is an increasedinterest in the application of genomic approaches toaid in the discovery, development, and rationale of
the use of drugs. As databases of differential gene ex-pression have expanded, so has the expectation ofidentifying novel drug targets for disease interven-tion. Indeed, significant work has already been per-formed to understand gene expression changes in theischemic heart (Stanton et al., 2000) and the ischemicbrain (Barone and Feuerstein, 1999; Soriano et al.,2000).
Early epidemiologic studies of the 1970s provided ini-tial evidence for a genetic influence in stroke. TheFramingham study was one of the first studies to suggestthat a positive parental history of stroke contributedsignificantly to the risk of the offspring (Kannel et al.,1970). Thirty years later stroke remains an area ofsubstantial unmet medical need. The complexity ofstroke undoubtedly reflects the heterogeneity of the hu-man stroke population, the contribution of monogenicand polygenic disorders to this disease process, and theinteractions of these with a multitude of environmen-tal factors.
Received April 16, 2001; final revision received April 19, 2001;accepted April 19, 2001.
Address correspondence and reprint requests to Dr. Simon J. Read,Neurology, HW2515, GlaxoSmithKline, New Frontiers Science ParkNorth, Harlow CM19 5AW, U.K. or Dr. Frank C. Barone, HighThroughput Biology, UW2521, GlaxoSmithKline, 709 SwedelandRoad, King of Prussia, PA 19406, U.S.A.
Abbreviations used: ANP, atrial natriuretic peptide; BNP, brain na-triuretic peptide; CADASIL, cerebral autosomal dominant arteriopathywith subcortical infarcts and leukoencephalopathy; DWI, diffusion-weighted imaging; IEGS, immediate early genes; MCA, middle cere-bral artery; MCAO, middle cerebral artery occlusion; MELAS, mito-chondrial encephalopathy-lactic acidosis and strokelike syndrome;MRI, magnetic resonance imaging; pMCAO, permanent MCAO; PWI,perfusion-weighted imaging; RDA, representational difference analy-sis; SHR, spontaneously hypertensive rat; WKY, Wistar-Kyoto rats.
Journal of Cerebral Blood Flow and Metabolism21:755–778 © 2001 The International Society for Cerebral Blood Flow and MetabolismPublished by Lippincott Williams & Wilkins, Inc., Philadelphia
755

This review will briefly focus on the genetics of riskand sensitivity to ischemic stroke. It will discuss howgenetic history relates to the broader stroke populationand will provide a detailed discussion of the stroke ge-nomics literature. This review will describe how preclini-cal models of spontaneous stroke can be applied tohumans to identify the chromosomal loci of risk, andhow the changes in gene expression associated withstroke are associated with poststroke brain injury, reso-lution of brain injury, and brain recovery processes. Inaddition, it will provide detailed discussion of severaldifferential gene expression analysis techniques. Thiswill include a detailed comparison of the emerging tech-nology of representational difference analysis and its ap-plication to a stroke model that has been well-char-acterized and represents the type of stroke in evolutionmost often observed in humans. Issues will also be ad-dressed regarding validation of potential stroke targets,the relevance of the expression of neuroprotective andneurodestructive genes and their specific timings, and theemerging problems with handling novel and unknowngenes that may be discovered during analysis of differ-ential gene expression.
Preponderance and riskStroke is the third largest cause of death in the U.S.,
ranking only behind heart disease and all forms of can-cer. It is the leading cause of disability in the U.S. andhas the highest disease burden cost. Ischemic strokescomprise approximately 80% of all strokes. No medicaltreatment is approved for the treatment of ischemicstroke other than tPA, a thrombolytic factor, which has tobe administered within 3 hours after stroke. Only 1% to2% of stroke patients meet the criteria for treatment withthis thrombolytic agent. Aspirin and anticoagulants(where embolic phenomena are documented) are used aspreventative therapy. Estimates indicate that there areapproximately 775,000 stroke cases per year in the U.S.,with approximately 4 million surviving, but at an in-creased risk of a secondary cardiovascular event. In theU.S., stroke is costly, with an annual health care cost of$30 to $50 billion. Estimates indicate that stroke isresponsible for half of all patients hospitalized foracute neurologic disease (Stephenson, 1998; Fisher andBogousslavsky, 1998).
Stroke risk factors include both genetic and environ-mental factors. Stroke risk factors that can be treatedinclude high blood pressure, heart disease, cigarettesmoking, transient ischemic attacks, and high red bloodcell count. Risk factors for stroke that cannot be changedinclude increased age, gender (men have approximately a19% greater chance of stroke than women), race (blackshave a greater risk of death and disability from stroke),diabetes mellitus, previous stroke, and a family history ofstrokes. Other controllable risk factors are secondary risk
factors for stroke that contribute to heart disease, includ-ing high blood low-density lipoproteins (LDL)-cholesterol and lipids, physical inactivity, and obesity(Pancioli et al., 1998).
GENOTYPING IN STROKE
Genetics of increased stroke riskThe strongest evidence for a genetic risk to stroke is
derived from twin studies. Proband concordance rateshave long been used to identify the heritability of a traitor disorder. The concept of concordance is that for adisorder of genetic predisposition, the rate will be higherfor monozygotic twins than dizygotic twins. Aside fromgenetic influence, it is assumed that other factors, such asenvironmental exposure, will be approximately similarfor both types of twins (Hrubec and Robinette, 1984).Brass et al. (1992) confirmed an elevated probandwiseconcordance rate for stroke risk in monozygotic twinsover dizygotic twins (17.7% vs. 3.6%), confirming a ge-netic predisposition to stroke. Subsequent reassessmentof this cohort of patients 6 years later reported risk at-tributable to genetic influence, but with an increase in therole of environmental factors (Brass et al., 1992). A morerecent twin study by Carmelli et al. (1998) has refinedcohort analysis to stroke risk by assessing individualstroke phenotypes that may be influenced by genetic fac-tors. In this study, the phenotype of white matter hyper-intensity volumes using magnetic resonance imaging(MRI) was applied and genetic factors accounted for71% of the variation in this endpoint.
A large number of familial studies have verified that ahistory of paternal or maternal stroke is associated withoccurrence of stroke in offspring, and that a positivepaternal history of stroke was an independent prognosticpredictor of stroke (Kiely et al., 1993; Jousilahti et al.,1997; Liao et al., 1997). For example, Welin et al. (1987)reported that, in a cohort of men studied since 1913,maternal history of stroke increased relative risk bythreefold. Similarly, Khaw and Barrett-Connor (1986)reported that a positive family history of stroke in anyfirst degree relative is an independent predictor of strokemortality in women aged 50 to 79, but not in men. More-over, this study also identified that a family history ofstroke was an independent predictor of coronary heartdisease in men aged 50 to 64 years, indicating that geneticrisk factors for stroke may be shared with other cardio-vascular disorders that have a high genetic component.Indeed, studies of the relative risk of other cerebrovas-cular diseases with less heterogeneous phenotypes havedocumented strong patterns of inheritance. Bromberg etal. (1995) found that subarachnoid hemorrhage occurswith a relative risk of 6.6 in first degree relatives com-pared with second degree relatives. Indeed, defining spe-cific stroke subtypes may be key in elucidating the exact
S. J. READ ET AL.756
J Cereb Blood Flow Metab, Vol. 21, No. 7, 2001

degree of genetic contribution to any particular pheno-type. From twin studies, it appears that the extent towhich genetic factors may contribute to stroke risk varieswith age. These factors are caveats to the identificationof therapeutic targets from candidate gene strategies, andone must remember that a candidate gene approach forinheritance of risk factors may only be relevant to ahighly limited stroke subpopulation.
Simple strokelike diseases: single gene mutationsIdentification of possible genetic determinants of
stroke risk has been hampered by the lack of homologouspatient populations. Mendelian disorders with speci-fic strokelike phenotypes have been explored as gene-tic models of the more general population. These disor-ders include cerebral autosomal dominant arteriopathywith subcortical infarcts and leukoencephalopathy(CADASIL) (Tournier-Lasserve et al., 1993), mitochon-drial encephalopathy-lactic acidosis-and strokelike syn-drome (MELAS) (Hirano and Pavlakis, 1994), Sneddonsyndrome (Lossos et al., 1995), familial hemiplegicmigraine (Joutel et al., 1993), and hereditary coagulopa-thies (Hassan and Markus, 2000). Although these sub-groups contribute little to the overall prevalence ofstroke, genes identified from them are hoped to highlightpotential commonalties in the wider patient popula-tion. Studies on CADASIL and MELAS are examples ofsuch approaches.
CADASIL was originally described by Souranderand Walinder (1977) as an inherited, autosomally-domi-nant dementia with multiple infarcts. Epidemiologically,CADASIL is limited to sporadic identification in Europe(Chabriat et al., 1995; Dichgans et al., 1998) and NorthAmerica (Hedera and Friedland, 1997; Desmond et al.,1998). The principal symptoms of CADASIL are mi-graine with aura, ischemic stroke, and psychiatric symp-toms including dementia (Viitanen and Kalimo, 2000).In these patients, T2-weighted MRI discloses smallperiventricular white matter hyperintensities often in-volving the internal capsule (Chabriat et al., 1998). TheCADASIL gene, identified as Notch 3, is located at thechromosome loci 19p13.1 – 13.2 (Joutel et al., 1996;Dichgans et al., 1996). The Notch genes regulate thelin-12/sel-12 signalling pathway important in develop-ment, although the normal adult function of Notch genesremains unknown (Artavanis-Tsakonas et al., 1999). Aninteresting association of the Notch 3 gene with Alzhei-mer disease also has been discovered. Notch gene prod-ucts interact with the presenilin-1 pathway as substratesfor �-secretase. This enzyme is known to have a keypathologic role in the production of A� peptide, althoughthe modulatory role that Notch 3 may have in this diseaseprocess is undefined (Levitan and Greenwald, 1995;Viitanen and Kalimo, 2000). The Notch 3 gene encodesa transmembrane protein composed of 2,321 amino
acids, presumed to have a receptor function and locatedprimarily on smooth muscle cells (Viitanen and Kalimo,2000). In CADASIL, approximately 90% of patientshave missense mutations in extracellular domains ofthe protein product, whereas in approximately 70% ofpatients, the mutation is located within exons 3 and 4(Joutel et al., 1997). All known mutations associatedwith CADASIL result in removal or addition of cysteineresidues, and it is proposed that expression of thesemutated Notch 3 proteins results in cerebral vascularsmooth muscle dysfunction (Joutel and Tournier-Lasserve, 1998).
Whether abnormalities in Notch signaling impact onthe broader stroke population is currently unknown, al-though the pathogenesis of CADASIL, characterized byprogressive disruption of vascular endothelium, second-ary fibrosis, and thrombosis is typical of some strokesubpopulations (Ruchoux and Maurage, 1998). Interest-ingly, anticoagulant therapy has been tried in CADASILwithout positive results (Viitanen and Kalimo, 2000).More broadly, CADASIL also has close relations toAlzheimer disease, and signaling components of thepresenilin pathway are shared with the Notch pathway(De Strooper et al., 1999). The presenilin-1 regulated�-secretase cleaves both the Notch intracellular do-main and �-amyloid precursor protein for subsequenttranslocation to the nucleus and binding to DNA (DeStrooper et al., 1999). Therefore, although the pathologyof CADASIL may bear similarity to stroke, the cell biologyis also reminiscent of Alzheimer disease. Because vascularrisk factors, or disease, or both, can impact vascular de-mentia and Alzheimer disease, these relations are intriguing(Kudo et al., 2000; Schmidt et al., 2000; Skoog, 2000).
Mitochondrial encephalomyelopathy-lactic acidosis-and strokelike episodes is characterized by migrainelikeheadache, nausea, seizures and strokelike episodes. Le-sions are most commonly found in occipital and parietalregions, with high lactate levels found within lesionsunder proton nuclear magnetic resonance (Hassan andMarkus, 2000). Patients typically have mutations of mi-tochondrial DNA for the tRNA-leu gene at an A-G tran-sition mutation at nucleotide position 3243 (Ciafoaloni etal., 1992; Macmillan et al., 1993) and at a T-C transitionat 3271 (Sakuta et al., 1993). Kovalenko et al. (1996)speculated that as mutations accumulate, a gradual mi-tochondrial dysfunction develops. It is unclear howwidespread such mutations are in the broader strokepopulation. Indeed, cases of MELAS have been reportedwithout a family history, suggesting that these point mu-tations may be spontaneous (Rastenyte et al., 1998).Pharmacologic interventions have reflected a unique na-ture of MELAS within stroke and cardiovascular diseasesubpopulations. Antithrombotic therapy has been used inMELAS patients for cardiac complications associatedwith left ventricular dysfunction (Kosinski et al., 1995).
GENE EXPRESSION: ISSUES AND ANSWERS 757
J Cereb Blood Flow Metab, Vol. 21, No. 7, 2001

CADASIL and MELAS demonstrate that several rela-tively rare “strokelike” mendelian syndromes can beused to explore potential genetic determinants of stroke.Parallel strategies have been adopted with similar suc-cess in other more complex multifactorial polygenictraits such as hypertension (Dominiczak et al., 2000).Genes such as 11�-hydroxylase in glucocorticoid reme-diable aldosteronism have been shown to mediate thehereditary hypertension in these patients (Lifton et al.,1992). However, as in stroke genetics, narrowing hetero-geneity and studying single gene and mendelian disor-ders has been found to limit the application to thebroader patient population.
Ischemic stroke: a disease having complexgenetic associations
In common with many diseases, there are individualswith complex genetic profiles and with complex profilesof poststroke gene expression that can contribute to therisk of ischemic stroke and increased cerebral ischemicstroke sensitivity, respectively. Candidate gene studies inheterogeneous stroke populations negate issues of lim-ited patient population by the a priori choice of a func-tionally relevant gene and its relation with a particularphenotype. This is often termed ‘association‘ and is astatistical measure of the dependence of a particular phe-notype (for example, ischemic stroke with the presenceof a particular candidate gene/allele). Therefore, associa-tion can be positive (that is, has a significant statisticalrelationship/association between the gene of choice andphenotype) (see Table 1), or negative (that is, has nosignificant relationship/association between gene/alleleand phenotype) (see Table 2). Candidate gene choice isfrequently driven by accepted stroke risk factors (forexample, hypertension, hemostasis, and abnormalities inlipid metabolism), and indeed, significant positive asso-
ciations of numerous markers with ischemic stroke havebeen identified, including ApoE �2 allele and D/D ge-notype of angiotensin converting enzyme-1 (Table 1).However, there are also numerous examples of negative(or lack of) associations of genes with ischemic stroke.For example, a negative association was identified forsome hemostasis factors (for example, Factor V, Q506polymorphism, and Factor VII R353 Q polymorphism)and some hypertension factors (for example, angioten-sinogen and M235T polymorphism) (Table 2). Cur-rently, it is difficult to identify clear patterns of candidategene associations with ischemic stroke. Furthermore, thereproducibility of these gene expression associations indifferent patient populations (for example, different raceor genetic backgrounds) is not known. The bewilderingcombination of possible outcomes for candidate geneassociation studies is emphasized by the genomic andphenotypic heterogeneity of the global stroke population.Studies are typically designed with case controls or bycohorts to enable close approximation of phenotype be-tween affected and nonaffected individuals. Superim-posed upon these levels of variation are issues in thetiming of stroke onset, in the variability of environmentalinfluences and penetrance (that is, not all individuals ofa given genotype will express the phenotype). Finally,although the human genome project continues apace(Genome International Sequencing Consortium, 2001),identifying functionality of gene products lags consider-ably. Current estimates propose only approximately 10%of the human genome has been ascribed function(Hassan and Markus, 2000). Certainly a lot of workneeds to be done in this area, and issues related to strokegenomics that include risk and the expression of genesunderlying brain vulnerability and ischemic sensitivitymust be considered.
TABLE 1. Candidate genes with a positive association with ischemic stroke
Gene marker Phenotype PolymorphismRiskfactor Result
Localize to1p36-35 Reference
Apolipoprotein E Ischemic stroke e2/e3/e4 e2 Positive No Couderc et al. (1993)Ischemic stroke e2/e3/e4 e4 Positive No Pedro-Botet et al. (1992)Ischemic stroke e2/e3/e4 e4 Positive No Margaglione et al. (1998)
ACE Ischemic stroke II/ID/DD DD Positive No Margaglione et al. (1996)Ischemic stroke II/ID/DD DD Positive No Nakata et al. (1997)Ischemic stroke II/ID/DD DD Positive No Sharma et al. (1994)Ischemic stroke CT2/3 2/2 Positive No Elbaz et al. (1999)
Fibrinogen Ischemic stroke G455A hz Positive No Kessler et al. (1997)Factor V Ischemic stroke Q506 Positive No Albucher et al. (1996)Prothrombin Ischemic stroke G20210 Positive No DeStefano et al. (1998)ANP Multiple subtypes
includingischemic stroke
G664A Positive Yes Rubattu et al. (1999a)
Candidate gene studies use the a priori choice of a potential pathophysiologically relevant gene and its relation with a particular stroke phenotype.This is often termed “association” and is a statistical measure of the dependence of a particular phenotype, for example, ischemic stroke with thepresence of a particular candidate gene/allele. This table represents positive associations (that is, gene polymorphisms demonstrated to have asignificant relationship) with the occurrence of ischemic stroke. ACE, angiotensin converting enzyme; hz, homozygotes; ANP, atrial natriureticpeptide.
S. J. READ ET AL.758
J Cereb Blood Flow Metab, Vol. 21, No. 7, 2001

PRECLINICAL MODELS OFSPONTANEOUS STROKE
It is with these caveats in mind that studies have fo-cused on animal models of spontaneous stroke, in whichenvironmental and genetic variability can be controlled.Bioinformatic approaches using synteny can facilitatethe matching of “stroke loci” found in stroke-pronerats to candidate genes on the human chromosome. Het-erogeneity of risk factors and life events in humanshas made it advantageous to study rodent models. Highlyhomogeneous populations of stroke-prone rats havebeen isolated from the incompletely inbred, spontane-ously hypertensive rat (SHR) and then inbred further forthis phenotype. Initial studies using the stroke-prone ratindicated that the degree of functional collateral bloodflow after occlusion of the middle cerebral artery(MCAO) was inherited in an autosomally recessivemanner (Coyle et al., 1984). The authors studied luminaldiameters in vascular anastomoses between middle andanterior cerebral arteries and hypothesized that thecollateral flow phenotype was determined by a sin-gle gene not directly linked to hypertension. Further ge-netic comparisons between strains were hampered byheterogeneity.
Narrowing the genotype by further crossing SHR ratswith stroke-prone animals allowed cosegregation ofgenes defining various stroke phenotypes and for homo-geneity of alleles for hypertension (Nagaoka et al.,1976). Two separate groups have used these inbredpopulations for identification of genes associated withmanifestation of specific stroke phenotypes. Rubattu etal. (1996) performed a genome-wide screen in an F2
cross, obtained by mating stroke-prone and SHR rats, inwhich latency to stroke was used as a phenotype. Theyidentified three major quantitative trait loci (QTLs)—STR1, STR2, and STR3. Of these, STR-2 and STR-3conferred a protective effect against stroke in the pres-ence of stroke-prone alleles, and STR-2 colocalized withthe candidate gene encoding atrial natriuretic peptide
(ANP) and brain natriuretic peptide (BNP). Furthermore,interactions between alleles from within STR1 and STR2suggested that this phenotype was a reasonable model ofthe polygenicity of stroke in man. Follow-up sequencingto characterize ANP and BNP as candidates for strokerevealed point mutations in ANP and no differences inBNP. In vitro functional studies indicated less ANP pro-moter activation in endothelial cells from stroke-pronerats versus SHR, with significantly less ANP expressionin the brain and no difference in BNP expression (Ru-battu et al., 1999c). To determine the in vivo significanceof the STR-2 lowered ANP promoter activation instroke-prone animals compared with stroke-resistant ani-mals, Rubattu et al. (1999b) performed a cosegrega-tion analysis of stroke occurrence in SHR stroke-prone rats/SHR stroke-resistant F2 descendents andANP expression. It was found that reduced expression ofANP did cosegregate with the appearance of “early”strokes in F2 animals; therefore, although lowered ANPexpression may be part of the phenotype of the “protec-tive” STR-2 QTL, it is unlikely that this is the primaryprotective mechanism in these animals. Parallel humanstudies of the role of ANP in cerebrovascular diseasehave confirmed that variation in the ANP gene may rep-resent an independent risk factor for “cerebrovascularaccidents” in humans (Rubattu et al., 1996) (Table 1) andmay emphasize the utility of this cohort of animals as amodel of ANP dysfunction in multiple subtypes ofstroke.
Two other groups have used a modified model of thestroke-prone animal, using F2 hybrids derived fromcrossing the stroke-prone SHR with Wistar-Kyoto(WKY) rats (Ikeda et al., 1996; Jeffs et al., 1997). Ikedaet al. (1996) used brain weight poststroke as the pheno-type for linkage analysis, after the discovery that F2 ani-mals had higher levels of brain edema formation post-stroke. Evidence of the linkage of phenotype to a gene onchromosome 4 was found that contributed to the severityof edema and was independent of blood pressure andSTR3 identified by Rubattu et al. (1996). Jeffs et al.
TABLE 2. Candidate gene studies with negative association to ischemic stroke
Gene marker Phenotype PolymorphismRiskfactor Result
Localize to1p36-35 Reference
eNOS Ischemic stroke Glu298-Asp Negative No MacLeod et al. (1999)Methylenetetra-hydrofolate Ischemic stroke C677T Negative Yes Nakata et al. (1998)
Ischemic stroke C677T Negative Yes DeStefano et al. (1998)Angiotensinogen Ischemic stroke M235T Negative No Nakata et al. (1997)Factor V Ischemic stroke Q506 Negative No Kontula et al. (1995)
Ischemic stroke Q506 Negative No Fisher et al. (1996)Factor VII Ischemic stroke R353Q Negative No Corral et al. (1998)Factor XIII Ischemic stroke Val34-Leu Negative No Catto et al. (1998)Prothrombin Ischemic stroke G20210A Negative No Poort et al. (1996)
Studies of candidate genes (that is, a priori chosen) for the study of a relation to ischemic stroke. In this table, however, negative associations (thatis, no significant relation was demonstrated) between gene/allele and the occurrence of ischemic stroke were identified. eNOS, endothelial nitric oxidesynthase.
GENE EXPRESSION: ISSUES AND ANSWERS 759
J Cereb Blood Flow Metab, Vol. 21, No. 7, 2001

(1997) designed studies to identify the genetic compo-nent responsible for large infarct volumes in the stroke-prone rat in response to a focal ischemic insult. To dothis, they performed a genome scan in an F2 cross, de-rived from the stroke-prone rat and the normotensiveWKY rat. In contrast with Rubattu and coworkers, theywere only able to identify one major QTL responsible forlarge infarct volumes. This QTL was located on rat chro-mosome 5, and like STR-2, it colocalized with ANP andBNP and was blood pressure independent. Unlike STR-2, this locus showed a much higher significance (lod16.6) and accounted for 67% of phenotypic variance.Subsequent studies identified that infarct volumes in theF1 rats were approximately identical to those of thestroke-prone animals, suggesting a dominant mode ofinheritance (Gratton et al., 1998).
Authors have argued over the significance of the over-lap of STR2 identified from Rubattu et al. (1996) withthe QTL identified by Jeffs et al. (1997) on chromosome5. It is unclear how the two phenotypes studied—latencyto stroke (that is, relative risk) (Rubattu et al., 1996) andsize of infarct after occlusion (that is, sensitivity to focalischemia) (Jeffs et al., 1997)—should physiologically re-late to each other. This may only become apparent whenindividual genes can be cosegregated with each pheno-type. Currently, altered ANP expression seems to play arole in the phenotype described by Rubattu et al. (1996),but has been excluded from a role in the colony used byJeffs et al. (1997; Brosnan et al., 1999).
What can be concluded from each of these stroke-prone rat models? Certainly, each represents a uniqueand valid “model” of stroke for the study of inheritanceand for a role of candidate genes in particular strokephenotypes. Neither colony represents a definitive modelof human stroke, although progress has been made withtitrating identified candidate genes in these stroke-pronecolonies to the human population (Rubattu et al., 1999a).One such research strategy we have used is the analysisof genomic synteny between the rat and human genome.This bioinformatic approach seeks to align regions ofhomology using evolutionary conserved markers and hasbeen applied with some success in relating animal mod-els to human genetics of other disease paradigms, forexample, noninsulin-dependent diabetes (Ktorza et al.,1997). Relating identified loci from stroke-prone animalsto the human genome offers a strategy for potential iden-tification of candidate genes. For example, the STR2region of rat chromosome 5 shows well-conserved geneorder and synteny with the human chromosome region1p35–36. The high level of synteny between these re-gions makes this region ideal for rat–human comparativeanalysis. Sequence tagged sites localized to this regionhave been identified and mapped to human transcriptclusters. As many as 132 transcripts have been identifiedin this region. The main candidates are listed in Table 3.
Interestingly, only a few candidate genes identified at1p35–36 have been examined in association studies.ANP recently has been assessed for association withmultiple subtypes of stroke (Rubattu et al., 1999a). Thepolymorphism G664A, responsible for a valine–methionine substitution in pro-ANP peptide was found tobe positively associated with the occurrence of stroke(Table 1). In contrast, methylenetetrahydrofolate, an-other marker located at 1p35–36, was negatively associ-ated with occurrence of stroke (Table 2). Further studiesmay elucidate the predictability of markers of 1p35–36and association with stroke (Table 3).
In contrast, rat–human synteny in the regions of the ratSTR-1 and STR-3 loci are not well conserved, as severaldisruptions of synteny appear to have been introducedduring evolution. It may be difficult to determine exactregions of synteny between these rat loci and humanchromosomal loci, and thus it may be difficult to ex-trapolate the candidate genes from rat to human. Humanchromosomal regions syntenic with STR1 span regionsof two human chromosomes, around 16p11 and 19q13.Human synteny with the STR3 region also appears to bedisrupted, with regions of synteny mapping telomericallyto opposite arms of chromosome 7 (7p21 & 7q35). Ofcourse, this is a problem of animal modeling of humandiseases in general and is not restricted only to ischemicstroke.
GENE EXPRESSION IN THE EVOLUTION OFPOSTSTROKE BRAIN INJURY
Cerebral ischemia is a powerful stimulus for the denovo expression and up-regulation of numerous genesystems (Barone and Feuerstein, 1999; Koistinaho andHokfelt, 1997). In terms of isolation of gene candidatesfor a neuroprotection strategy, interpretation of expres-sion changes has proven difficult. The multitude of ani-mal models of ischemia with varying genetic heteroge-neity and infarct pathophysiology is also complicated byspatial and temporal variations that has largely con-founded interpretation (Sharp et al., 2000; Iadecola andRoss, 1997). Furthermore, assays of differential expres-sion have varying sensitivity to the relative fold increaseor decrease in mRNA expression. As a result, “fishing”exercises will often result in “catches” of differentialgene expression that vary depending on the assay used.
With this bewildering array of complexity as a caveat,the following section addresses that assessment of ani-mal model(s) that might be used, the appropriate assaysavailable for differential gene expression analysis, thetarget confirmation methodology that is necessary afterthe identification and confirmation of a differentially ex-pressed gene (that is, a “hit”), and ultimately, the func-tional assessment of these genes in the disease process. Ahierarchical critical path that depicts the path from target
S. J. READ ET AL.760
J Cereb Blood Flow Metab, Vol. 21, No. 7, 2001

identification to target confirmation and validation is de-picted schematically in Fig. 1.
ISCHEMIC STROKE MODELS: THE SEARCHFOR CLINICAL RELEVANCE
The failure of several putative neuroprotective agentsin recent large multicentered clinical trials (Clark et al.,2000; Lees et al., 2000) has led to critical reexamina-tion of the predictability of many preclinical modelsof ischemia (Feinklestein et al., 1999). Heterogeneityin the human stroke population and the multitude ofwell-defined animal models of ischemia have led to at-tempts to refine model choice as related to patient sub-groups (DeGraba and Pettigrew, 2000; Parsons et al.,2000). In an effort to stratify patient groups that can bepredicted using specific animal models, authors have fo-cused on the use of MRI signatures, in particular, perfu-
sion- (PWI) and diffusion-weighted imaging (DWI) mis-matches. Two main groups of acute stroke patients areidentifiable, those with evolving infarcts in which lesionPWI > DWI, or those with a stabilized infarct in whichPWI � DWI (Baird and Warach, 1998; Albers, 1999).Such PWI and DWI assessments have been proposed tocorrelate to the extent of salvageable tissue, with ap-proximately 70% of patients exhibiting PWI lesions >DWI at 6 hours poststroke, and approximately 50% ofpatients exhibiting this mismatch at 24 hours poststroke(Albers, 1999).
Applying such imaging paradigms to animal modelsof focal ischemia should enable translation of preclinicalpathophysiology into predictive outcomes in the appro-priate patient population. Detailed comparisons of thedevelopment of PWI/DWI signatures between animalmodels of ischemia are difficult to establish because of
TABLE 3. 1p36-p35 postional candidates with a biologic rationale in stroke
Homology Rationale in stroke
CD30L receptor precursor Nerve growth factor receptor superfamilyTumor necrosis factor receptor 2 Mouse KO increases neuronal damage in response
to insultsHuman MASP-2 serine protease
protein—complement processing proteaseDepletion of complement system improves
outcome after cerebral ischemiagp40 mucin—putative influenza virus receptor Role in cell adhesionHuman Tropomyosin-related protein—exclusively
neuronal/brain expressionMay be related to sustained contraction during
cerebral vasospasmHuman protease proMch6 (Mch6)/Caspase-9 Critical upstream activator of the caspase cascade
in vivoEphrin receptor EphA2/tyrosine-protein kinase
receptor ECKRole in neuronal development
Human PDGF-associated proteinEndothelin-converting enzyme E1 VasoconstrictorWNT4 protein precursor Possible role in synaptic plasticity. Linked to JNK
signalling and indirectly to Notch (CADASIL)Ephrin receptor EphB2/Tyrosine-protein kinase
receptor ERKRole in neuronal development
Stathmin—v high brain expression Phosphorylated by CAM kinase IICorticosteroid binding protein—yeastPutative bicistronic heat shock proteins Stress responsePlatelet-activating factor receptorDishevelled-1 Possible role in synaptic plasticity. Linked to JNK
signaling and indirectly to NotchAtrial Natriuretic Peptide A Localized with LOD peak hypertensionBrain Natriuretic Peptide B Localized with LOD peak hypertensionComplement component 1, q subcomponent, alpha
polypeptide (C1QA)Depletion of complement system improves
outcome after cerebral ischemia5,10-Methylene-tetrahydrofolate reductase Heterozygous mutations are significant cause of
stroke in general populationBrain-specific angiogenesis inhibitor (BAI2) Regulator of angiogenesisPlatelet phospholipase A2, group IIA Antiplatelet agents modify stroke riskSodium hydrogen exchanger-1 pH regulator, acidity assoc. with postischemic
damagePlatelet-activating factor receptor (PTAFR) PAF involved in arterial thrombosis. Antiplatelet
agents modify stroke risk
Identification of human candidate genes that are syntenic to the STR2 region of rat chromosome 5 identifiedby Rubattu et al. (1996) in a cohort of SHR-stroke prone animals. STR2 shows well-conserved gene order andsynteny with the human chromosome region 1p35-36 (between D1S503-D1S2667). The high level of syntenybetween these regions makes this region ideal for rat–human comparative analysis. Sequence tagged siteslocalized to this region have been identified and mapped to human transcript clusters. Many transcripts, spe-cifically 132 of them, have been identified in this region, with the main candidates listed above. KO, knockout;PDGF, platelet-derived growth factor; SHR, spontaneously hypertensive rats.
GENE EXPRESSION: ISSUES AND ANSWERS 761
J Cereb Blood Flow Metab, Vol. 21, No. 7, 2001

the use of various rat strains, anesthetics, and modes ofischemia induction. However, broad comparisons arepossible by exploring the development of DWI signal asa marker of lesion volume with respect to time. There-fore, data in certain animal models of focal stroke canshow a delay in the development of DWI hyperintensity(that is, brain lesion size) that lags behind a perfusiondeficit (that is, PWI changes associated with stroke andfocal ischemia). This delay is attributable, in part, to therelative contribution of insufficient collateral flow andthe periinfarct depolarizations that eventually signifi-cantly injure the poorly perfused, ischemic brain duringinfarct evolution (Hossman, 1996; Parsons et al., 2000).
Photothrombotic ischemia by the rose-bengal methodproduces a highly consistent focal infarct. Diffusion-weighted imaging lesion develops primarily during thefirst 24 hours, with an expanding volume of DWI deficitcontinuing over a subsequent 3 to 7 days (van Bruggen etal., 1992; De Ryck et al., 2000; Lee et al., 1996). Theextensive thrombosis produced in this model in associa-tion with profound blood–brain barrier breakdown may
limit the application of identified gene expression in thismodel to the clinic (that is, lesion development is rapidwith little penumbra area to impact upon). Direct MCAOby proximal electrocoagulation of the MCA produces anexpanding DWI lesion, with an initial marked expansionat 4 hours followed by a small increase from 4 to 24hours (Gill et al., 1995). Modification of this occlusion todistal MCAO in SHR produces a rapidly evolving infarctwith a near maximal lesion observed after 1 hour (Chan-dra et al., 1999).
Available embolic models of focal stroke using intra-arterial injection of thrombin (Zhang et al., 1997), aged(Jiang et al., 2000) or fibrin-rich (Busch et al., 1998)clots have reported approximately similar expansion ofDWI hyperintensity. For example, after thrombin injec-tion, DWI hyperintensity is apparent 80 minutes postad-ministration, with the volume gradually expanding up to24 hours (Zhang et al., 1997). Intraluminal suture occlu-sion produces a range of DWI lesion progression depen-dent on whether the filament is introduced through thecommon carotid artery (Koizumi et al., 1986) or the
FIG. 1. Hierarchical organization of target identification and confirmation. After selection of an animal model, appropriate to clinicalsubpopulation (1), broad mRNA fishing strategies are adopted using differential expression assays such as representational differenceanalysis (RDA), microarrays, subtractive hybridization (SSH), and differential display (DD). (2) Across these assays, commonalties inidentified hits (3) are explored as a technique of prioritising subsequent confirmation studies (see Fig. 2). Comprehensive expressionanalysis using reverse transcription-polymerase chain reaction (RT-PCR) (4) allows confirmation of identified hits and fully quantifiedtemporal profiling. Protein confirmation (5) by ELISA, Western blotting, and immunohistochemistry follows mRNA profiling to confirmtranslation. In an ischemic brain, pooling of mRNA and uncoupling of translation can occur (see Fig. 3). Finally, functional studiesencompassing target gene knockout (KO), adenoviral transfection, and in vivo pharmacology (6) complete validation of a potential tar-get gene.
S. J. READ ET AL.762
J Cereb Blood Flow Metab, Vol. 21, No. 7, 2001

external carotid artery (Longa et al., 1989). Permanentocclusion through Koizumi suture MCAO produces arapid evolution of DWI hyperintensity within minutes,followed by maximal expansion by 2 hours (Neumann-Haefelin et al., 2000; Li et al., 2000). In comparison,permanent MCAO (pMCAO) by the method of Longa etal. (1989) evokes an initial rapid expansion of DWI hy-perintensity during the first 30 minutes followed by finalinfarct volume reached at 7 hours (Gyngell et al., 1995;Kohno et al., 1995a,b). A close inspection of this modelsidentifies it as exhibiting a mismatch similar to that inhumans, that is, representing the type of infarct in evo-lution that should provide information relevant to humanstroke (Parsons et al., 2000). For this reason we havedecided to use this model for our recent differential geneexpression studies (Bates et al., 2001).
METHODOLOGIES FOR DIFFERENTIAL GENEEXPRESSION IN FOCAL STROKE
The detection of genes that are differentially expressedbecause of stroke can be identified using the simpler(that is, well established and straightforward) techniquessuch as Northern blotting, reverse transcription-polymerase chain reaction (RT-PCR), in situ hybridiza-tion, and so on. These techniques involve the selectionand study of a specific gene of interest (that is, based onprevious data that provides a biological rationale forstudy in stroke or another specific disease). However,several more complex screening techniques are nowavailable that can identify groups of differentially ex-pressed genes, both known and unknown. These com-plex screening techniques include subtractive librar-ies/subtractive hybridisation, differential hybridisation,serial analysis of gene expression, representational dif-ferential analysis (RDA), and differential display (Feu-erstein et al., 1996). For example, the identification ofdifferentially expressed genes in stroke has used the sim-pler and complex techniques of differential displayanalysis, subtractive hybridization, DNA microarrays,and representational difference analysis. A completesummary of stroke-associated gene expression by alltechniques is listed in Table 4. Variations in assay andthreshold of detection often result in the isolation of genesets that differ according to assay selection. Therefore, toensure maximum confidence in the detection of adaptiveup-regulation of gene expression, a pragmatic approachshould be adopted. For example, commonalties in iden-tified genes and pathways should be identified acrossseveral differential expression assays, or independentcross-validation of a gene’s up-regulation should be em-phasized, or both, rather than relying on the results of asingle differential expression technique. Table 4 providesa list of only those gene and message expressions thathave been confirmed within or between laboratories us-
ing more than one expression detection technique. Abrief summary of the more complex techniques is pro-vided below.
Differential displayDifferential display is a means of comparing all poly
(A)+ mRNA between experimental and control popula-tions. mRNA is converted into first strand cDNA throughreverse transcription plus oligo(dT)-anchored primersfollowed by PCR with multiple sets of primers. The PCRproducts then are displayed with control and experimen-tal samples side-by-side on high resolution denaturinggel, this way differential gene expression is immediatelyapparent. This technique has been applied with successin isolating differentially expressed products after ex-perimental MCAO. Wang et al. (1995b), using mRNAdifferential display after rat focal MCAO, isolated a genethat encodes adrenomedullin, a member of the calcitoningene-related peptide (CGRP) family. This was followedup by independent temporal studies using Northernanalysis, which confirmed that expression of mRNA lev-els increased in the ischemic cortex at 3 hours and 6hours post-MCAO. Levels remained elevated for up to15 days post-MCAO. Immunohistochemical studies toconfirm protein expression localized adrenomedullin toischemic neuronal processes. In functional studies, syn-thetic adrenomedullin microinjected onto preconstrictedrat pial arteries produced dose-dependent relaxation ofthese vessels. In addition, intracerebroventricular admin-istration of adrenomedullin, before and after MCAO, in-creased the degree of focal ischemic injury.
Other groups also have used this technique in ischemiato identify differentially expressed mRNA such as serineprotease inhibitors (SPI-3), zinc transporter gene (ZnT-1), and ADP-ribosylation factor like gene (ARF4L)in models of gerbil forebrain ischemia (Tsuda et al.,1996, 1997; Katayama et al., 1998). Proteosome expres-sion was identified after rat photochemical ischemia(Keyvani et al., 2000). Transcription factor (SEF-2) andproteosome expression (p112) after rat global ischemia(Wigle et al., 1999) and chemokine identification (ST-38) after rat MCAO (Utans-Schneitz et al., 1998) alsowere demonstrated.
Differential display, although a labor-intensive tech-nique, is very useful. For example, differential display isideal for examining several RNA samples simulta-neously and has been used extensively for temporal,dose-response, and multiple treatment studies. Also, al-though differential display is “semiquantitative,” onlyrelatively small amounts of total RNA (approximately 15�g) are required. However, as can be observed from theliterature, problems with interpretation of data have beenidentified. High rates of false positives that can not beconfirmed by RT-PCR or Northern blotting. Modifica-tions to this approach have been used, such as subtracted
GENE EXPRESSION: ISSUES AND ANSWERS 763
J Cereb Blood Flow Metab, Vol. 21, No. 7, 2001

TABLE 4. Summary of differential gene expression changes identified to date by techniques that measure transcriptiondifferences after focal ischemia stroke
Gene Model
Early RNAexpression<24 hours
Late RNAexpression>24 hours
RNA/proteincross-validation
techniques
Proteindemonstrated/
verified Reference
IEGNGFI-A pMCAO Yes No In situ No Honkaniemi et al., 1997NGFI-B pMCAO Yes No In situ No Honkaniemi et al., 1997NGFI-C pMCAO Yes No In situ No Honkaniemi et al., 1997NGFI-C pMCAO Yes No Oligonucleotide array
In situNo Soriano et al., 2000
Nurr-1 pMCAO Yes No In situ No Honkaniemi et al., 1997erg-2 pMCAO Yes No In situ No Honkaniemi et al., 1997erg-3 pMCAO Yes No In situ No Honkaniemi et al., 1997Zif268, c-fos pMCAO Yes No Northern blotting No Wang et al., 1995NK-kB tMCAO Yes No Immunocytochemistry
gel shift analysisYes Stephenson et al., 2000
NF-kB tMCAO Yes (subunitspecific)
Yes (subunitspecific)
Western blottingimmunohistochemistrygel shift analysis
Yes Gabriel et al., 1999
Activating transcriptionfactor
tMCAO Yes(decrease)
No ImmunocytochemistryWestern blotting
Yes Martin-Villalba et al.,1999
c-fos, c-jun, zif268 pMCAO Yes No Northern blotting No Collaco-Moraes et al.,1994
CytokinesIL-1 receptor pMCAO Yes (subunit
specific)No RT-PCR No Wang et al., 1997
IL-1RA pMCAO Yes Yes RT-PCR No Wang et al., 1997IL-1RA pMCAO Yes RT-PCR No Medhurst et al., 2000IL-1B pMCAO Yes Yes Northern blotting No Liu et al., 1993bIL-1B pMCAO Yes No In situ No Buttini et al., 1994IL-1� pMCAO Yes No RT-PCR No Zhai et al., 1997IL-1� tMCAO Yes Yes Northern blotting No Wang et al., 1994IL-2 pMCAO No change No change RT-PCR No Zhai et al., 1997IL6, Zif 268, c-fos pMCAO Yes No Northern blotting No Wang et al., 1995dCINC MCAO Yes Northern blotting No Liu et al., 1993aIL-10 pMCAO Yes (6
hours)No RT-PCR No Zhai et al., 1997
TNF-� pMCAO Yes Yes Immunohistochemistryin situ
RT-PCR
Yes Buttini et al., 1996
TNF-� tMCAO Yes Yes Northern blotting No Wang et al., 1994TNF-� pMCAO Yes No RT-PCR No Zhai et al., 1997TNF-� pMCAO Yes Yes Northern blotting
immunohistochemistryYes Liu et al., 1994
TGF-�1 pMCAO Yes SYBR� green RT-PCRRDA
No Bates et al., 2001
TGF-� pMCAO No Yes Northern blotting No Wang et al., 1995cLIF tMCAO Yes Yes RT-PCR
Western blotimmunohistochemistry
Yes Suzuki et al., 2000
LIF pMCAO Yes Yes TaqMan� RT-PCRRDA
No Bates et al., 2001
SOCS-3 pMCAO Yes Yes TaqMan� RT-PCRRDA
No Bates et al., 2001
Inflammation GenesCOX-1 tMCAO No change No change RT-PCR No Nogawa et al., 1997COX-2 tMCAO Yes (6–24
hours peak)Yes RT-PCR
immunocytochemistryYes Nogawa et al., 1997
COX-2 pMCAO Yes SYBR� green RT-PCRRDA
No Bates et al., 2001
MCP-1 tMCAO Yes Yes ELISA Yes Yamagami et al., 1999MCP-1 pMCAO Yes Yes Northern blotting
RT-PCRimmunocytochemistry
Yes Kim et al., 1995
MCP-1 pMCAO No Yes Northern blotting No Wang et al., 1995aMCP-1 pMCAO Yes Yes TaqMan� RT-PCR
RDANo Bates et al., 2001
iNOS tMCAO Yes (6–12hours peak)
No RT-PCR No Nogawa et al., 1996
S. J. READ ET AL.764
J Cereb Blood Flow Metab, Vol. 21, No. 7, 2001
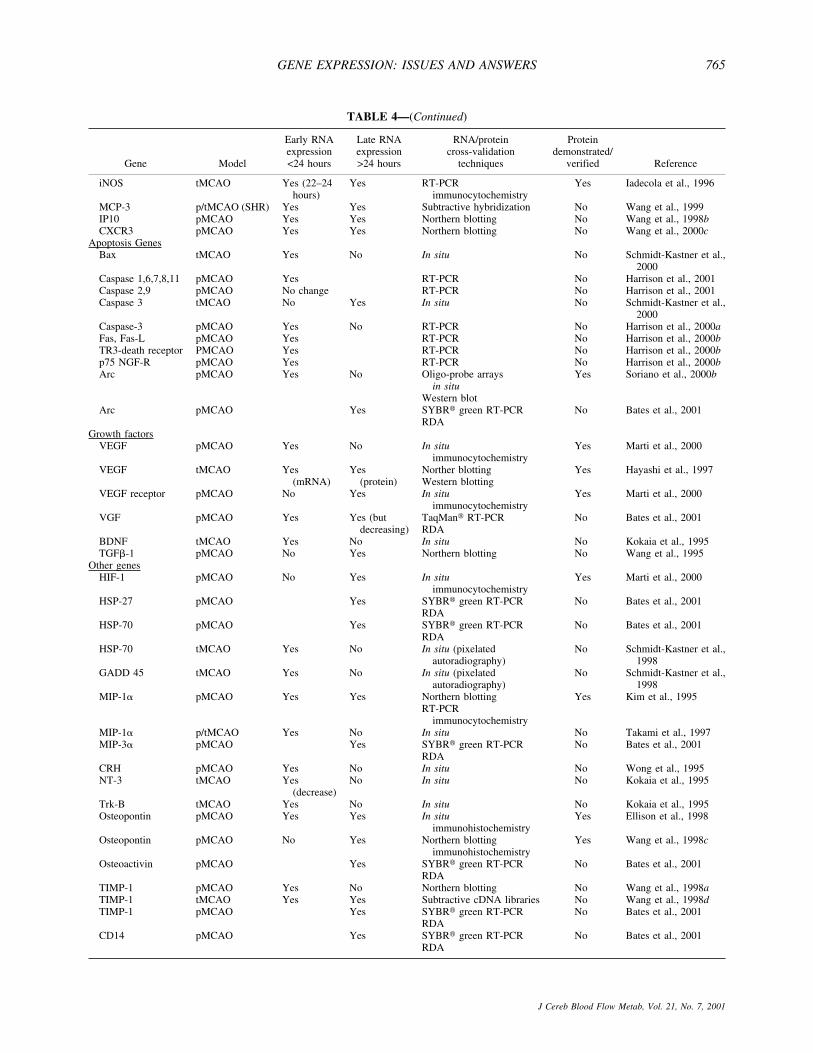
TABLE 4—(Continued)
Gene Model
Early RNAexpression<24 hours
Late RNAexpression>24 hours
RNA/proteincross-validation
techniques
Proteindemonstrated/
verified Reference
iNOS tMCAO Yes (22–24hours)
Yes RT-PCRimmunocytochemistry
Yes Iadecola et al., 1996
MCP-3 p/tMCAO (SHR) Yes Yes Subtractive hybridization No Wang et al., 1999IP10 pMCAO Yes Yes Northern blotting No Wang et al., 1998bCXCR3 pMCAO Yes Yes Northern blotting No Wang et al., 2000c
Apoptosis GenesBax tMCAO Yes No In situ No Schmidt-Kastner et al.,
2000Caspase 1,6,7,8,11 pMCAO Yes RT-PCR No Harrison et al., 2001Caspase 2,9 pMCAO No change RT-PCR No Harrison et al., 2001Caspase 3 tMCAO No Yes In situ No Schmidt-Kastner et al.,
2000Caspase-3 pMCAO Yes No RT-PCR No Harrison et al., 2000aFas, Fas-L pMCAO Yes RT-PCR No Harrison et al., 2000bTR3-death receptor PMCAO Yes RT-PCR No Harrison et al., 2000bp75 NGF-R pMCAO Yes RT-PCR No Harrison et al., 2000bArc pMCAO Yes No Oligo-probe arrays
in situWestern blot
Yes Soriano et al., 2000b
Arc pMCAO Yes SYBR� green RT-PCRRDA
No Bates et al., 2001
Growth factorsVEGF pMCAO Yes No In situ
immunocytochemistryYes Marti et al., 2000
VEGF tMCAO Yes(mRNA)
Yes(protein)
Norther blottingWestern blotting
Yes Hayashi et al., 1997
VEGF receptor pMCAO No Yes In situimmunocytochemistry
Yes Marti et al., 2000
VGF pMCAO Yes Yes (butdecreasing)
TaqMan� RT-PCRRDA
No Bates et al., 2001
BDNF tMCAO Yes No In situ No Kokaia et al., 1995TGF�-1 pMCAO No Yes Northern blotting No Wang et al., 1995
Other genesHIF-1 pMCAO No Yes In situ
immunocytochemistryYes Marti et al., 2000
HSP-27 pMCAO Yes SYBR� green RT-PCRRDA
No Bates et al., 2001
HSP-70 pMCAO Yes SYBR� green RT-PCRRDA
No Bates et al., 2001
HSP-70 tMCAO Yes No In situ (pixelatedautoradiography)
No Schmidt-Kastner et al.,1998
GADD 45 tMCAO Yes No In situ (pixelatedautoradiography)
No Schmidt-Kastner et al.,1998
MIP-1� pMCAO Yes Yes Northern blottingRT-PCR
immunocytochemistry
Yes Kim et al., 1995
MIP-1� p/tMCAO Yes No In situ No Takami et al., 1997MIP-3� pMCAO Yes SYBR� green RT-PCR
RDANo Bates et al., 2001
CRH pMCAO Yes No In situ No Wong et al., 1995NT-3 tMCAO Yes
(decrease)No In situ No Kokaia et al., 1995
Trk-B tMCAO Yes No In situ No Kokaia et al., 1995Osteopontin pMCAO Yes Yes In situ
immunohistochemistryYes Ellison et al., 1998
Osteopontin pMCAO No Yes Northern blottingimmunohistochemistry
Yes Wang et al., 1998c
Osteoactivin pMCAO Yes SYBR� green RT-PCRRDA
No Bates et al., 2001
TIMP-1 pMCAO Yes No Northern blotting No Wang et al., 1998aTIMP-1 tMCAO Yes Yes Subtractive cDNA libraries No Wang et al., 1998dTIMP-1 pMCAO Yes SYBR� green RT-PCR
RDANo Bates et al., 2001
CD14 pMCAO Yes SYBR� green RT-PCRRDA
No Bates et al., 2001
GENE EXPRESSION: ISSUES AND ANSWERS 765
J Cereb Blood Flow Metab, Vol. 21, No. 7, 2001

differential display, which removes unregulated cDNAby mRNA subtraction before differential display (Wangand Uhl, 1998). Nevertheless, confidence in an isolatedcandidate gene can be improved by using other indepen-dent follow-up assays of gene expression, or other dif-ferential expression techniques in parallel, or both. Con-firmation across several assays will aid the identificationof false-positives and improve the confidence that a spe-cific gene putatively up-regulated in ischemic stroke issignificant.
Subtractive hybridization and suppressionsubtractive hybridization
Subtractive hybridization compares qualitative differ-ences in gene expression between two experimentalparadigms. This is usually achieved by hybridization ofbiotinylated “driver” cDNA to the mRNA pool from the“target” tissue. Duplexes of driver cDNA and targetmRNA are then removed, resulting in a pool of targetmRNA expressed only by the target tissue (Barr andEmanuel, 1990). Down-regulated mRNA are determinedby performing the reaction in reverse. Modifications tothe assay include suppression subtractive hybridizationand RDA (see next subsection), where the PCR replacesphysical subtraction methods to enrich for differentiallyexpressed transcripts. Such modifications emphasize dif-ferential mRNA of both low and high abundance, ratherthan biasing selection of only highly expressed genes, asis the case with the more basic subtractive hybridizationmethodology.
Suppression subtractive hybridization has been suc-cessfully used to identify candidate genes with putativeroles in experimental cerebral ischemia. Wang et al.(1999) identified the induced expression of a rat homo-logue to human monocyte chemotactic protein-3 (MCP-3) in ischemic brain. Independent Northern analysisidentified increases in MCP-3 mRNA observed 12 hourspostischemia, with 49-fold and 17-fold increases overcontrol in permanent and temporary MCAO, respec-tively. Moreover, significant induction of MCP-3 in the
ischemic cortex was sustained up to 5 days after isch-emic injury. In other models, subtractive hybridizationhas been less widely used to identify candidate genes.This may be because of the difficulty of the subtractionapproach, although false positives are less frequent. Nev-ertheless, Abe et al. (1996) have used subtractive hybrid-ization to successfully identify a novel cDNA clone(pGSH3) expressed only after ischemia in the gerbil cor-tex. Basal cortical levels were found to be low, but 8hours after a 10-minute transient forebrain ischemia thegene expression became prominent in the cerebral cor-tex. Analysis of DNA sequence revealed that the pGSH3insert had a 91.3% homology with a 72-kd human heatshock protein (hsp70) gene.
Representational difference analysis (a form ofsubtractive hybridization)
Representational difference analysis is a relativelynovel PCR-coupled, genome subtractive process (Hu-bank and Schatz, 1994; Lisitsyn et al., 1995) that untilonly very recently (Bates et al., 2001) had not been usedto assay differential expression in models of cerebralischemia. Representational difference analysis is concep-tually similar to subtractive hybridization, but the un-availability of a commercially produced kit for RDA hasmeant that it has been less broadly exploited. Represen-tational difference analysis was originally established tomonitor differences in genomic DNA content betweenindividuals, it was later modified to identify differencesin gene expression (Hubank and Schatz, 1994; Lisitsyn etal., 1993).
The robust gene expression changes that characterizethe MCAO model has also proved amenable to RDA, aswe have recently been able to show (Bates et al., 2001).Subtracting ischemic cortex from rats 24 hours afterpMCAO from similarly treated tissue from sham-operated animals, we were able to identify a number ofcandidate ischemia-regulated transcripts. Primary confir-mation of the accumulation of these gene products in theischemic cortex was confirmed using SYBR Green RT-
TABLE 4—(Continued)
Gene Model
Early RNAexpression<24 hours
Late RNAexpression>24 hours
RNA/proteincross-validation
techniques
Proteindemonstrated/
verified Reference
CD44 pMCAO Yes SYBR� green RT-PCRRDA
No Bates et al., 2001
GADD45� pMCAO Yes SYBR� green RT-PCRRDA
No Bates et al., 2001
Xin pMCAO Yes SYBR� green RT-PCRRDA
No Bates et al., 2001
This table only lists increased message expression that has been validated within or between laboratories independently. The importance of thisvalidation in addition to the verification of translated protein for candidate genes is emphasized in the text. IEG, immediate early gene; pMCAO,permanent middle cerebral artery occlusion; tMCAO, temporary MCAO; IL-1, interleukin-1; RT-PCR, reverse transcription-polymerase chainreaction; TNF-�, tumor necrosis factor-alpha; TGF, transforming growth factor; RDA, representational difference analysis; COX, cyclooxygenase;SHR, spontaneously hypertensive rats; VEGF, vascular endothelial growth factor; BDNF, brain-derived nerve growth factor; HSP, heat shock protein;TIMP-1, tissue inhibitor of matrix metalloproteinase-1.
S. J. READ ET AL.766
J Cereb Blood Flow Metab, Vol. 21, No. 7, 2001

PCR, followed by the more comprehensive time-courseanalysis using TaqMan RT-PCR in selected cases. Sev-eral genes identified through this approach previouslyhad been reported to increase after MCAO, such as heatshock proteins (hsp27 and hsp70) and others (MCP-1,MIP3�, cyclooxygenase-2 (COX-2), TGF-�1, tissue in-hibitor of matrix metalloproteinase-1 (TIMP-1), andArc), and several were first identified to be MCAO-induced in this study (LIF, SOCS-3, VGF, CD44, CD14,CD81, osteoactivin, GADD45�, and Xin) (Bates et al.,2001). These gene expressions and follow-up verifica-tions of these and other differentially expressed genes arealso listed, with appropriate references, in Table 4, andare discussed in more detail below.
Array technologiesAll of the above strategies identify small numbers of
differentially expressed genes. However, large numbersof DNA fragments (110 to 450bp) are produced in theprocess that need to be confirmed and frequently ex-tended to full lengths to obtain gene identity. Althoughall of these technologies are useful for isolating candi-date genes, they are of limited use in providing a broadcharacterization of the expression of large numbers ofgenes within a particular model.
Array-based technology allows such analysis. It pro-vides for a full analysis of gene expression within a studyincluding time-response profiling, drug treatment analy-sis, and so on. Whether using arrays of oligonucleotides(Lockhart et al., 1996; Lipshutz et al., 1999) or genefragments (Schena et al., 1995), the technology allowsparallel expression monitoring of several hundred genesat a time. The limitations and biases of the technique areobviously in the selection of genes to study on the array.Technology has been advancing rapidly in the area ofcDNA array analysis and now short oligomers can betransferred to glass slides, allowing rapid development ofimportant tools for genotyping and mRNA expressionanalysis (Young, 2000).
Soriano et al. (2000) pioneered this technique in theapplication of studying gene expression in the Tamura(1981) model of rat MCAO. The authors used an oligo-nucleotide probe array with 750 predetermined genesoptimized for gene expression in rat bone and cartilage.The chip (ROEZ002; Hoffman-LaRoche Limited, Basel,Switerland) was used to monitor gene expression after 3hours of permanent focal ischemia. To determine genesdifferentially expressed as a consequence of ischemia,the authors took tissue from the ipsilateral frontal andparietal cortices and compared the tissue with corre-sponding regions on the contralateral side. A significantchange in transcription was defined as a twofold orgreater increase or decrease in expression compared withthe contralateral hemisphere. The authors describe a sig-nificant up-regulation of 24 genes in the parietal, frontal
cortices, and striatum with particularly robust changes inc-fos, NGFI-A, NGFI-C, Krox20, NGFI-B, Nor-1, COX-2, and Arc.
The current study clearly demonstrates the utility ofarray technology for broad characterization of the regu-lation of genes during experimental cerebral ischemia.The use of arrays optimized for bone and cartilage genesunfortunately limited the usefulness to 15% of the totalgene representation on the array. Nevertheless, key genefamilies such as the phosphatases (MKP-1 and MKP-3)and the chemokines (MCP-1 and MIP-1�) were repre-sented and expression profiles agreed with previous find-ings (Wiessner et al., 1995; Gass et al., 1996; Kim et al.,1995). No change in housekeeping genes GAPDH and�-actin were found (ipsilateral vs. contralateral) at this3-hour time point.
In our laboratories, we have further assessed the utilityof microarrays. After MCAO, again using the Longa etal. (1989) technique and commercially available micro-array grids from Affymax, we conducted differentialgene expression analysis. Ipsilateral cortex samples werepooled from MCAO animals and sham-operated animalsat 24 hours post-pMCAO. In common with Soriano et al.(2000), we identified several immediate early genes(IEGs) that were up-regulated, including c-fos (2.9-fold)and NGFI-A (2.9-fold). As reflected in later time pointsstudied, fold induction of the IEGs was less than at 3hours poststroke as studied by Soriano et al. (2000). Sev-eral heat shock proteins were found to be up-regulated,specifically HSP-27 (20.9-fold) and HSP-70 (9.8-fold),which were shown to be up-regulated after stroke byother methods that assess gene expression (Table 4). Inaddition, our microarray experiment revealed 74genes/sequence candidates that are up-regulated in theMCAO model 24 hours postocclusion. Several candidategenes previously shown to be up-regulated or down-regulated in stroke were confirmed to be regulated at thelevel of transcription in this experiment. TIMP-1, Arc,osteopontin, glial fibrillary acidic protein (GFAP), VGF(NGF-induced), interferon-induced protein, calmodulin(and calmodulin binding proteins), and COX-2 havebeen shown to be up-regulated by us using microarrays,and in some cases by others using various techniques(see Table 4). Furthermore, many structural genes ortheir regulators (forms of actin and tubulin, vimentin,ARP2/3) and genes involved in basic cell metabolism(ribosomal proteins, polyA-binding protein, elongationfactors, cysteine oxygen oxidoreductase, ribosomalRNA, mitochondrial cytochrome oxidase) were shown tobe affected after stroke.
Interestingly, as documented by Soriano et al. (2000),differential expression of Arc also was found (2.6-fold).However, it should be noted that this was at 24 hourspostocclusion. Using the Tamura et al. (1981) method ofocclusion, Soriano and coworkers documented that Arc
GENE EXPRESSION: ISSUES AND ANSWERS 767
J Cereb Blood Flow Metab, Vol. 21, No. 7, 2001

transcript levels decrease to basal levels by 24 hours.Given the putative role of Arc in mediation of cytoskel-etal changes underlying neuronal plasticity (Fosnaugh etal., 1995), these findings emphasize temporal differencesbetween models that may reflect different pathophysi-ologic processes (that is, evolving vs. terminal strokes asdiscussed earlier) (Parsons et al., 2000). Below we willdiscuss in more detail model to model differences, theimportance of the poststroke timings of RNA sampling,different experimental paradigms in stroke that mighthelp in the discovery of genes that have roles in brainprotection or tolerance, and the specific studies that canbe used to look for genes that might contribute to recov-ery and plasticity of the brain postinjury.
Between assay variation and increasing confidencein identified gene targets
In the preceding sections we have discussed many ofthe techniques available for detection of differential geneexpression and some of the “within assay” issues asso-ciated with each technique. However, in generating andcomparing gene expression data, there are several issuesthat warrant discussion, including the significant vari-ability between techniques and the identification of false-positive and false-negative results. Assays of differentialexpression have an inherent variability dependent on as-say methodology, sensitivity, and reaction efficiency.When exploring disease paradigms that are powerfulstimulators of gene expression, such as cerebral isch-emia, the tendency is to highlight vast gene sets that areup-regulated and differentially expressed. Given thelarge number of genes identified, it is difficult to confirmall differentially expressed genes and false-positive andfalse-negative differential gene expression becomes anissue. False-positives can be broadly defined as geneswhose differential expression is not subsequently con-firmed by an independent study (that is, by RT-PCR,Northern analysis, in situ hybridization, and so on). Incontrast, false negatives are genes that are, in fact, dif-ferentially expressed, but are not detected as such by thecomplex assay used (for example, subtractive hybridisa-tion, RDA, DNA microarray, and so on).
To manage these issues, we have used the strategy ofusing multiple assays of differential expression on thesame RNA pool and then cross-validating all of thenumerous differentially expressed products between as-says. Confidence in particular products can then be in-creased by identifying commonalties in expressionacross assays. This approach is represented in Fig. 2,where targets are listed according to their detectionacross several assays of differential expression. Genesidentified across all three assays (SSH, RDA, andgridding) are prioritized first, as they are associated withhigh levels of confidence. A table of descending confi-dence in hits can be constructed, which could include
products such as TIMP-1, MCP-1, and hsp-27, priori-tized first for subsequent levels of confirmation (for ex-ample, Taqman RT-PCR, protein expression). This tech-nique for handling large numbers of “hits” circumvents(that is, avoids) issues of biasing the identification ofdifferential expression to a single assay and also inter-assay variability. Subsequent analysis by Taqman RT-PCR confirmed that the robust “hits” (that is, differentialgene expression identified across all assays) had particu-larly high fold increases in expression versus naïve rats(for example, MCP-1 were high at 603-fold), whereasother lower fold increases were also still robust “hits,”including HSP-70 at 20.9-fold, GFAP at 18.1-fold, andVGF at 7.3-fold versus naïve. Figure 2 also highlightsthat this strategy has been particularly useful for identi-fying false negatives (that is, products differentially ex-pressed but not detected as such in assays). For example,in our studies, GFAP was identified to be up-regulated instroke by gridding, whereas SOCS-3 was identified bysubtractive hybridization, but by harnessing both tech-niques in a unified approach we were able to confirm(that is, using TaqMan RT-PCR) that both of these genesindeed did accumulate in the ischemic cortex afterpMCAO. This “complimentary techniques” approach attarget validation maximizes the coverage of differentialgene expression by minimizing the losses caused by thetechnical vagaries of any single technique.
Confirmation: an integral part of differential geneexpression analysis
The techniques cited above for the identification ofdifferentially expressed mRNA represent one of severalstarting points for an integrated approach to the study ofgene expression in stroke models. All data derived bythese methods require confirmation in an independentstudy to remove false positives, and this usually formspart of a broader analysis of expression of the gene thathas been identified (Soriano et al., 2000; Wang et al.,1995b, 1998a,d).
Traditional methods for analyzing gene expression in-clude techniques such as Northern blotting, RNAse pro-tection assay, in situ hybridization, and semiquantitativeRT-PCR. All of these methodologies have been used onnumerous occasions to study the expression of individualgenes or small groups of genes in stroke models. How-ever, the differential screening methodologies ideallygenerate large numbers of hits that require rapid confir-mation in a higher throughput system.
Recently, real-time quantitative RT-PCR techniqueshave become available, such as the use of Taqmanprobes or SYBR green (Gibson et al., 1996), to monitoran accumulating PCR product in real time, hence allow-ing an accurate comparison of initial PCR template num-bers. These assays can be performed in 96- or 384-wellformat and are highly amenable to the use of robots,
S. J. READ ET AL.768
J Cereb Blood Flow Metab, Vol. 21, No. 7, 2001

reducing operator time and error. With these new tech-niques, it is possible to perform rapid confirmation ofmany differentially expressed genes simultaneously or toundertake a detailed expression analysis of a single hit(Medhurst et al., 2000). Taqman RT-PCR analysis hasbeen extensively applied to the temporal profiling ofcaspase expression after MCAO in rats (Harrison et al.,2000c, 2001), and SYBR green has been used to confirmdifferentially expressed genes identified by RDA (Bateset al., 2001). Glial fibrillary acidic protein up-regulationafter MCAO, identified as differential expression iden-tified by microarray gridding, was confirmed by TaqManRT-PCR (Fig. 2). Figure 2 shows the temporal profile ofGFAP expression after MCAO (ipsilateral and contralat-eral cortices) and sham-operated animals (ipsilateral andcontralateral cortices). In the ipsilateral cortex, GFAPexpression increases over time to 24 hours at which timeexpression is 18.1-fold greater than in naïve animals.
Taqman methodology is a PCR-based technique that ismore sensitive than other confirmatory technologies suchas Northern blotting. Typically, Taqman RT-PCR uses
approximately 50 ng total RNA per gene, whereas North-ern blotting uses 10 to 20 �g. In addition, Taqman RT-PCR is as sensitive as in situ hybridization, with theadded advantage of higher throughput. Perhaps most im-portantly for disease paradigms such as MCAO, wheregene expression can exceed 600-fold over that observedin naïve animals (for example, MCP-1), Taqman PCRcan quantitate gene expression over 5 to 6 orders ofmagnitude without multiple dilution series as necessi-tated by other assays (Medhurst et al., 2000). Clearly,PCR-based technologies such as Taqman RT-PCR andSYBR Green RT-PCR, although in their infancy in ap-plication to the study of cerebral ischemia (Bates et al.,2001; Harrison et al., 2000a,b, 2001), offer many advan-tages versus other assays for confirmation and expansionof data on differential gene expression.
These techniques are also of value in testing hypoth-eses about genes already known to be regulated in strokemodels or where differential expression is suggested byother biologic evidence. The sensitivity of PCR-basedmethodologies suggests that sufficient RNA can be
FIG. 2. Example of prioritization of mRNA “fishing” hits before subsequent confirmation. Commonalities are identified across each assayof differential expression with greater levels of confidence assigned to “hits” highlighted with all techniques (subtractive hybridization(SSH), representational difference analysis (RDA), and gridding). This strategy is particularly useful for achieving the maximum breadthof fishing and highlighting false-negatives. In this example, glial fibrillary acidic protein (GFAP) was not identified by RDA or SSH, butmicroarray grids did highlight a differential expression. Subsequent analysis by reverse transcription-polymerase chain reaction (RT-PCR)demonstrated elevated expression in ipsilateral ischemic cortex (MCAO L) as compared with the contralateral control cortex (MCAO R)and with sham-operated cortices (SHAM L and R). HSP, heat shock protein; COX, cyclooxygenase; MCAO, middle cerebral arteryocclusion.
GENE EXPRESSION: ISSUES AND ANSWERS 769
J Cereb Blood Flow Metab, Vol. 21, No. 7, 2001

isolated from a single animal to allow the simultaneousassessment of several hundred genes. Thus, a large bodyof data can be gathered and the expression of many dif-ferent genes can be compared in a single study withoutdrawbacks such as variation between studies, operators,and cohorts of animals. The major drawback of highthroughput quantitative RT-PCR is that although it al-lows for the rapid assessment of changes in gene expres-sion at the level of mRNA, it is not able to provideinformation on the precise cellular localization of suchchanges. In the case of the Longa et al. (1989), strokemodel that has been extensively studied in our laborato-ries, the cell-type and intracellular location(s) of changesin gene expression are likely to be invaluable. Neuronesand oligodendrocytes die within the ischemic infarct(Bartus et al., 1995; Mandai et al., 1997), particularlyafter 12 hours of ischemia. Astrocytes and microglia aredecreased in number in the core region of the lesion and
proliferation of both of these cell types occurs in themarginal areas (Davies et al., 1998).
Polymorphonuclear leukocytes, and later macro-phages, invade the lesion after approximately 12 hoursand for days after (Clark et al., 1993; Davies et al., 1998;Kochanek and Hallenbeck, 1992). Changes in gene ex-pression have to be understood in the context of theevolving cell types present at the time. Ultimately, thishas to involve techniques, such as in situ hybridizationand immunohistochemistry, which allow the localizationof expression to be viewed in relation to the structure ofthe evolving lesion and the identification of the types ofcells in which expression is occurring.
Confirmation and localization of protein expression:transcription to translation verification
Confirmation of protein expression and time course oftranslation is of primary importance. This is especially
FIG. 3. Uncoupling of transcription and translation can occur in stroke and disease, therefore, message expression information alone isnot adequate. Confirmation of protein expression and time course of translation is of primary importance. During ischemic compromise,transcription and translation can become uncoupled because of the energetic demand of assembly of functional protein. After permanentocclusion of the middle cerebral artery (pMCAO) in normotensive Sprague-Dawley rats (by the “thread” method of Longa et al., 1989),TaqMan reverse transcription-polymerase chain reaction analysis of IL-1� and IL-6 mRNA shows an increase in mRNA levels in ischemicrelative to sham-operated animals at 3 and 6 hours post-pMCAO. However, protein determination by ELISA at 6 hours post-pMCAO failsto confirm a concomitant increase in IL-1� protein (that is, not translated to protein within a similar time frame), although IL-6 translationwas verified with measured protein levels being significantly elevated at 6 hours. Solid bars represent ischemic cortex mean ± SE. Openbars represent the contralateral control cortex mean ± SE. Bars represent the data from 5 to 6 rats. *P < 0.05, statistically significantdifference as compared with the contralateral control cortex (analysis of variance, then Dunnett’s test).
S. J. READ ET AL.770
J Cereb Blood Flow Metab, Vol. 21, No. 7, 2001

pertinent given the severe energy compromise in theischemic brain. During this state, transcription and trans-lation can become uncoupled because of the energeticdemand of assembling functional protein. This will betemporally and spatially dependent, and has been exten-sively reviewed by Koistinaho and Hokfelt (1997) andSharp et al. (2000).
Uncoupling of mRNA transcription and protein trans-lation after pMCAO using the technique of Longa et al.(1989) is shown in Fig. 3. From previous and currentdata, it is apparent that both IL-1� (Liu et al., 1993b;Butinni et al., 1994; Wang et al., 1994) and IL-6 (Wanget al., 1995d) show an early increase in mRNA levels inischemic versus sham-operated animals, 3 and 6 hourspost-MCAO. However, protein determination by ELISAat 6 hours post-pMCAO fails to confirm a concomitantincrease in IL-1� protein, although IL-6 protein levelsare significantly elevated in ischemic animals at this timepoint, as expected. Issues associated with the sensitivityof protein detection assays must be considered for someproteins. However, from a family of related proteins,sampled from an identical cortical region, with an appar-ently similar mRNA expression profile over the first 6hours post-pMCAO, completely opposing protein pro-files have been obtained. An example of both messageand protein up-regulation like that seen for IL-6 (that is,coupling of transcription and translation) can also beprovided for tumor necrosis factor-� (TNF-�). TNF-�message and protein expression (that is, protein detectedby the very sensitive immunohistochemical technique)can be detected at a similar and early time points post-stroke (Butinni et al., 1996; Liu et al., 1994) (see Table4). With the advent of the sequencing of the full humangenome (Genome International Sequencing Consortium,2001), we now find that only approximately 30,000genes produce the much more copious and diverse num-ber of proteins. Ultimately, it is proteins that are pivotalin cellular function; thus, proteomic analysis in additionto mRNA analysis points the way ahead.
Functional studies, transgenic studies, and invivo pharmacology
There are already many examples from the literaturewhere transgenic animal studies, or pharmacologic stud-ies, or both, have followed up or coincided with geneexpression studies to demonstrate involvement of spe-cific gene expression in focal stroke injury or protection.For example, COX-2–deficient mice exhibit reduced sus-ceptibility to brain injury (Iadecola et al., 2001). Notably,IL-1ra was shown to be neuroprotective in brain injury(Relton and Rothwell, 1992) long before the altered ex-pression of the IL-1 system in stroke was demonstrated(Wang et al., 1997). Also, IL-6 has been shown to beneuroprotective in stroke (Ali et al., 2000; Loddick et al.,1998). In addition, blocking thyrotropin-releasing hor-
mone provides significant protection against ischemicbrain damage and associated neurologic deficits (Borlon-gan et al., 1999; Yonemori et al., 2000). Finally, post-stroke treatment with brain-derived nerve growth factor(BDNF) reduces brain injury to pMCAO (Yamashita etal., 1997). Genes for all of these proteins have beenshown to be up-regulated in stroke models. These studiesdemonstrate the full circle and cross-validation of differ-ential gene expression technology that is necessary inthis research, and are directly relevant to the discussionson new directions and models provided below.
ADDITIONAL MODELS FOR PROBINGDIFFERENTIAL GENE EXPRESSION
Neuroprotective and neurodestructivegene expression
As pointed out previously (Barone and Feuerstein,1999), focal ischemia is a powerful stimulus to elicitresponses in the brain in the form of multiple gene ex-pression changes. Focal ischemia is a powerful reformat-ting and reprogramming stimulus for the brain. There arebroad and robust gene expression responses that occurafter focal stroke that are exhibited as temporal episodesor “waves” of expression of different groups of genes(Barone and Feuerstein, 1999). These “waves” arelargely composed of increased expression of inflamma-tory cytokines, including IL-6 and IL-1ra. In addition,growth factors (for example, BDNF) that might be ex-pected to play a neuroprotective role after stroke alsoincrease in this time frame. The increased cytokine geneexpression appears to drive leukocyte infiltration, a post-stroke brain response to injury, and is associated withsecondary brain injury and repair processes after stroke.Later waves of new gene expression include mediatorsthat appear to be important in tissue remodeling (that is,resolution of ischemic tissue injury) and perhaps recov-ery of function. These issues are important in relation tothe models suggested below for future differential geneexpression analysis.
Preconditioning stress in brain tolerance strategiesA short ischemic event can result in subsequent isch-
emic tolerance, a resistance to severe ischemic tissueinjury. This phenomenon, known as ischemic precondi-tioning, has been described in several organs, especiallybrain and heart, and may represent a fundamental pro-tective response to injury after previous stress (Kitagawaet al., 1990, 1991; Yellon et al., 1998; Lawson andDowney, 1993). Ischemic preconditioning is a powerfulinducer of ischemic brain tolerance to severe stroke asreflected by preservation of brain tissue and motor func-tion for up to 7 days after the initial preconditioning
GENE EXPRESSION: ISSUES AND ANSWERS 771
J Cereb Blood Flow Metab, Vol. 21, No. 7, 2001

stimulus (Barone et al., 1998). Ischemic tolerance is de-pendent on de novo protein synthesis at the precondi-tioned brain site, which contributes to the neuroprotec-tion. These models of brain protection provide an oppor-tunity to identify novel protective gene expressionassociated with the development of brain tolerance. It hasbeen suggested that neurotrophic factors, stress proteins,and cytokines contribute to the tolerance response toischemia and other forms of stress to the brain (Barone etal., 1998; Currie et al., 2000; Yanamoto et al., 2000;Chen et al., 1996; Chen and Simon, 1997; Wang et al.,2000a,b). For example, ischemic tolerance is associatedwith increased expression of the neuroprotective protein,IL-1ra, and a reduced postischemic expression of theearly response genes, c-fos and zif268. These brain tol-erance models are amenable to the differential gene ex-pression approaches described above. For example,Wang et al. (1998d) applied the suppressive subtractivehybridization methodology to discover genes responsiblefor ischemic tolerance after preconditioning. Using sup-pressive subtractive hybridization, TIMP-1 was identi-fied. Northern analysis confirmed that TIMP-1 mRNAwas significantly elevated at 24 hours and 2 days afterpreconditioning, which corresponded well to the onset ofischemic tolerance.
Later gene changes: strategies in brain recovery,plasticity, and recovery of function
Although initially neurologic functional deficits occurafter stroke, there is a significant recovery of brain func-tion that both occurs spontaneously and improves withtraining after stroke (Barone and Feuerstein, 1999;Rossini et al., 1999a,b; Dobkin, 2000; Hunter et al.,2000). Sampling tissue during functional brain recovery(that is, at later time periods poststroke) might be ex-pected to provide an opportunity to identify novel genesimportant for brain regeneration or plasticity. Thesewould be amenable to the differential gene expressionanalyses, but would be profiled at later poststroke timepoints or under treatment conditions shown to facilitatesuch brain regeneration and recovery.
Gene therapyIn recent years, the emerging technology of gene
therapy has provided some insights into the use of in vivogene transfer. Therapeutic neovascularization for isch-emic diseases has been particularly encouraging. In ani-mal models, angiogenesis and improved outcome in isch-emic tissues has been produced by intramuscular injec-tions of plasmid or adenoviral vectors encoding vascularendothelial growth factor (VEGF) (Byun et al., 2001;Gowdak et al., 2000). Similarly, in clinical trials, VEGFgene transfer augments the population of circulating en-dothelial progenitor cells and transiently increasesplasma levels of VEGF (Kalka et al., 2000). Further-more, myocardial transfer of naked plasmid DNA
phVEGF (165) has been found to augment perfusion ofischemic myocardium and reduce the size of defectsdocumented at rest by single-photon emission computedtomography imaging (Vale et al., 2000). Ex vivo genetransfer, using the modification of cultured cells and sub-sequent implantation into a host organism, is a provenstrategy for recovery from central nervous system injury.Recovery from long-term rodent hemiparkinsonism byimplantation of tyrosine transfected myoblasts after6-OHDA lesioning was found to improve behavioraldeficit for up to 13 months (Cao et al., 2000).
In vivo gene transfer, the delivery of a gene directly torecipient somatic cells, has also been explored for neu-roprotection and recovery from central nervous systeminjury. The delivery of the protooncogene bcl-2 has beenexamined in gerbil models using adeno-associated virusvectors. Transduction both pre- and postforebrain isch-emia was found to prevent DNA fragmentation in hip-pocampal CA1 neurones, commonly associated with celldeath induced by ischemia (Shimizaki et al., 2000). Theadenoviral transfection of the endogenous cytokine an-tagonist, IL-1ra, has also demonstrated neuroprotectionin transient focal cerebral ischemia and reperfusion in themouse (Yang et al., 1997, 1999). In addition, the neuro-protective potential of HSP70 on brain injury, includingthat produced by brain ischemia, has been demonstrated.Transgenic animals and gene transfer technology used tooverexpress HSP70 clearly protects the brain (Yenari etal., 1998, 1999).
THE ELUSIVENESS OF NOVEL GENETARGETS AND THE FUTURE
Initially, the movement to discover differential geneexpression in brain destruction or protection was be-lieved to be driven by the novel or unknown genes thatwould provide pathways to completely novel and propri-etary therapeutics. Although this may be true, the com-plexity of understanding and applying resources to genefragments in the hopes of ultimately reaching this thera-peutic nirvana has not yet occurred. We have identifiedseveral potentially novel genes that have been differen-tially expressed in ischemic or tolerant brain tissue buthave not made further progress with these to date. Basi-cally, the odds for success are much better for the pursuitof known genes as therapeutic targets, as resourcing canbe based on a solid biologic rationale. Unknown genesoften may not provide a significant rationale to moveforward without the full-length gene in the current re-source restricted research environment. Many other fac-tors can cause investing resources into work on unknowngenes costly, risky, and difficult to pursue. Some of theseinclude the absence of any understanding of an identifi-able function for the unknown protein, and if it is, in fact,associated with a novel gene, and lack of any information
S. J. READ ET AL.772
J Cereb Blood Flow Metab, Vol. 21, No. 7, 2001

that the novel gene/protein has any involvement in thepathophysiology of stroke. In spite of all this, it is clearthat among these potential unknowns clearly are the po-tential novel therapeutic targets of the future. As such,novel and unknown genes might be a potential site ofcollaboration between the academic and industrial labo-ratories, thus providing potential novel gene productsthat can be evaluated for tissue distribution, function, andrelevance in tissue injury and protection.
Clearly the methodology is currently available to iden-tify differential gene expression in stroke in addition toother conditions of pathophysiology and disease. How-ever, this methodology needs to be used with the caveatsdiscussed above. If one does operate cautiously as wehave suggested, there is the potential for many signifi-cant opportunities for biologic discovery, not only re-lated to stroke but in many other conditions such as endorgan failure in various cardiovascular diseases, in areassuch as oncology, and perhaps extending further to othercomplex problems such as substance abuse, tolerance,addiction, and drug dependency.
Acknowledgments: The authors thank Sue Tirri forassisting in the preparation of this review, and Dr. AndyMedhurst for his helpful discussions on differential expressiontechnology.
REFERENCES
Abe K, Kawagoe J, Itoyama Y, Kogure K (1996) Isolation of an isch-emia-induced gene and early disturbance of mitochondrial DNAexpression after transient forebrain ischemia. Adv Neurol 71:485–503
Albers GW (1999) Expanding the window for thrombolytic therapy inacute stroke. The potential role of acute MRI for patient selection.Stroke 30:2230–2237
Albucher JF, Guirard-Chaumeil B, Chollet F, Cadroy Y, Sie P (1996)Frequency of resistance to activated protein C due to factor Vmutation in young patients with ischemic stroke. Stroke27:766–767
Ali C, Nicole O, Docagne F, Lesne S, MacKenzie ET, Nouvelot A,Buisson A, Vivien D (2000) Ischemia-induced interleukin-6 as apotential endogenous neuroprotective cytokine against NMDA re-ceptor-mediated excitotoxicity in the brain. J Cereb Blood FlowMetab 20:956–966
Artavanis-Tsakonas S, Rand MD, Lake RJ (1999) Notch signaling: cellfate control and signal integration in development. Science284:770–776
Baird AE, Warach S (1998) Magnetic resonance imaging of acutestroke. J Cereb Blood Flow Metab 18:583–609
Barone FC, White RF, Spera PA, Ellison J, Currie RW, Wang X,Feuerstein GZ (1998) Ischemic preconditioning and brain toler-ance: temporal histological and functional outcomes, protein syn-thesis requirement, and interleukin-1 receptor antagonist and earlygene expression. Stroke 29:1937–1950
Barone FC, Feuerstein GZ (1999) Inflammatory mediators and stroke:new opportunities for novel therapeutics (review). J Cereb BloodFlow Metab 15:819–834
Barr FG, Emanuel BS (1990) Application of a subtraction hybridizationtechnique involving photoactivatable biotin and organic extractionto solution hybridization analysis of genomic DNA. Anal Biochem186:369–373
Bartus RT, Dean RL, Cavanaugh K, Eveleth D, Carriero DL, Lynch G(1995) Time-related neuronal changes following middle cerebral
artery occlusion: implications for therapeutic intervention and therole of calpain. J Cereb Blood Flow Metab 15:969–979
Bates S, Read SJ, Harrison DC, Topp S, Morrow R, Gale D, MurdockP, Barone FC, Parsons AA, Gloger IS (2001) Molecular charac-terisation by subtractive hybridisation of the Zea Longa model ofpermanent middle cerebral artery occlusion in the rat. Identifica-tion of novel gene expression following experimental ischemia.Mol Brain Res (in press)
Borlongan CV, Stahl CE, Redei E, Wang Y (1999) Prepro-thyrotropin-releasing hormone 178–199 exerts partial protection against cere-bral ischemia in adult rats. Neuroreport 10:3501–3505
Brass LM, Isaacsohn JL, Merikangas KR, Robinette CD (1992) Astudy of twins and stroke. Stroke 23:221–223
Bromberg JE, Rinkel GJ, Algra A, Greebe P, van Duyn CM, Hasan D,Limburg M, ter Berg HW, Wijdicks EF, van Gijn J (1995) Sub-arachnoid haemorrhage in first and second degree relatives of pa-tients with subarachnoid. BMJ 311:288–289
Brosnan MJ, Clark JS, Jeffs B, Negrin CD, Van Vooren P, Arribas SM,Carswell H, Aitman TJ, Szpirer C, Macrae IM, Dominiczak AF(1999) Genes encoding atrial and brain natriuretic peptides as can-didates for sensitivity to brain ischemia in stroke-prone hyperten-sive rats. Hypertension 33:290–297
Busch E, Kruger K, Allegrini PR, Kerskens CM, Gyngell ML, Hoehn-Berlage M, Hossmann KA (1998) Reperfusion after thrombolytictherapy of embolic stroke in the rat: magnetic resonance and bio-chemical imaging. J Cereb Blood Flow Metab 18:407–418
Buttini M, Sauter A, Boddeke HW (1994) Induction of interleukin-1beta mRNA after focal cerebral ischaemia in the rat. Brain Res MolBrain Res 23:126–134
Buttini M, Appel K, Sauter A, Gebicke-Haerter PJ, Boddeke HW(1996) Expression of tumor necrosis factor alpha after focal cere-bral ischaemia in the rat. Neuroscience 71:1–16
Byun J, Heard JM, Huh JE, Park SJ, Jung EA, Jeong O, Gwon HC, KimDK (2001) Efficient expression of the vascular endothelial growthfactor gene in vitro and in vivo, using an adeno-associated virusvector. J Mol Cell Cardiol 33:295–305
Cao L, Zhao YC, Jiang ZH, Xu DH, Liu ZG, Chen SD, Liu XY, ZhengZC (2000). Long-term phenotypic correction of rodent hemipar-kinsonism by gene therapy using genetically modified myoblasts.Gene Ther 7:445–449
Carmelli D, DeCarli C, Swan GE, Jack LM, Reed T, Wolf PA, MillerBL (1998) Evidence for genetic variance in white matter hyperin-tensity volume in normal elderly male twins. Stroke 29:1177–1181
Catto AJ, Kohler HP, Bannan S, Stickland M, Carter A, Grant PJ(1998) Factor XIII Val 34 Leu: a novel association with primaryintracerebral hemorrhage. Stroke 29:813–816
Chabriat H, Tournier-Lasserve E, Vahedi K, Leys D, Joutel A, NibbioA, Escaillas JP, Iba-Zizen MT, Bracard S, Tehindrazanarivelo A(1995) Autosomal dominant migraine with MRI white-matter ab-normalities mapping to the CADASIL locus. Neurology 45:1086–1091
Chabriat H, Levy C, Taillia H, Iba-Zizen MT, Vahedi K, Joutel A,Tournier-Lasserve E, Bousser MG (1998) Patterns of MRI lesionsin CADASIL. Neurology 51:452–457
Chandra S, White RF, Everding D, Feuerstein GZ, Coatney RW, SarkarSK, Barone FC (1999) Use of diffusion-weighted MRI and neu-rological deficit scores to demonstrate beneficial effects of isra-dipine in a rat model of focal ischemia. Pharmacology 58:292–299
Chen J, Graham SH, Zhu RL, Simon RP (1996) Stress proteins andtolerance to focal cerebral ischemia. J Cereb Blood Flow Metab16:566–577
Chen J, Simon RP (1997) Ischemic tolerance in the brain. Neurology48:306–311
Ciafaloni E, Ricci E, Shanske S, Moraes CT, Silvestri G, Hirano M,Simonetti S, Angelini C, Donati MA, Garcia C (1992) MELAS:clinical features, biochemistry, and molecular genetics. Ann Neurol31:391–398
Clark RK, Lee EV, Fish CJ, White RF, Price WJ, Jonak ZL, FeuersteinGZ, Barone FC (1993) Development of tissue damage, inflamma-tion and resolution following stroke: an immunohistochemical andquantitative planimetric study. Brain Res Bull 31:565–572
Clark WM, Raps EC, Tong DC, Kelly RE (2000) Cervene (Nalmefene)
GENE EXPRESSION: ISSUES AND ANSWERS 773
J Cereb Blood Flow Metab, Vol. 21, No. 7, 2001

in acute ischemic stroke: final results of a phase III efficacy study.The Cervene Stroke Study Investigators. Stroke 31:1234–1239
Collaco-Moraes Y, Aspey BS, de Belleroche JS, Harrison MJ (1994)Focal ischemia causes an extensive induction of immediate earlygenes that are sensitive to MK-801. Stroke 25:1855–1860
Corral J, Gonzalez-Conejero R, Lozano ML, Rivera J, Vicente V(1998) Genetic polymorphisms of factor VII are not associatedwith arterial thrombosis. Blood Coagul Fibrinolysis 9:267–272
Couderc R, Mahieux F, Bailleul S, Fenelon G, Mary R, Fermanian J(1993) Prevalence of apolipoprotein E phenotypes in ischemic ce-rebrovascular disease. A case-control study. Stroke 24:661–664
Coyle P, Odenheimer DJ, Sing CF (1984) Cerebral infarction aftermiddle cerebral artery occlusion in progenies of spontaneouslystroke-prone and normal rats. Stroke 15:711–716
Currie RW, Ellison JA, White RF, Feuerstein GZ, Wang X, Barone FC(2000) Benign focal ischemic preconditioning induces neuronalHsp70 and prolonged astrogliosis with expression of Hsp27. BrainRes 863:169–181
Davies CA, Loddick SA, Stroemer RP, Hunt J, Rothwell NJ (1998) Anintegrated analysis of the progression of cell responses induced bypermanent focal middle cerebral artery occlusion in the rat. ExpNeurol 154:199–212
DeGraba TJ, Pettigrew LC (2000) Why do neuroprotective drugs workin animals but not humans? Neurol Clin 18:475–493
De Ryck M, Verhoye M, Van der Linden AM (2000) Diffusion-weighted MRI of infarct growth in a rat photochemical strokemodel: effect of lubeluzole. Neuropharmacology 39:691–702
De Strooper B, Annaert W, Cupers P, Saftig P, Craessaerts K, MummJS, Schroeter EH, Schrijvers V, Wolfe MS, Ray WJ, Goate A,Kopan R (1999) A presenilin-1-dependent gamma-secretase-likeprotease mediates release of Notch intracellular domain. Nature398:518–522
DeStefano V, Chiusolo P, Paciaroni K, Casorelli I, Rossi E, MolinariM, Servidei S, Tonali PA, Leone G (1998) Prothrombin G20210Amutant genotype is a risk factor for cerebrovascular ischemic dis-ease in young patients. Blood 91:3562–3565
Desmond DW, Moroney JT, Lynch T, Chan S, Chin SS, Shungu DC,Naini AB, Mohr JP (1998) CADASIL in a North American family:clinical, pathologic, and radiologic findings. Neurology 51:844–849
Dichgans M, Mayer M, Muller-Myhsok B, Straube A, Gasser T (1996)Identification of a key recombinant narrows the CADASIL generegion to 8 cM and argues against allelism of CADASIL andfamilial hemiplegic migraine. Genomics 32:151–154
Dichgans M, Mayer M, Uttner I, Bruning R, Muller-Hocker J, RunggerG, Ebke M, Klockgether T, Gasser T (1998) The phenotypic spec-trum of CADASIL: clinical findings in 102 cases. Ann Neurol44:731–739
Dobkin BH (2000) Functional rewiring of brain and spinal cord afterinjury: the three Rs of neural repair and neurological rehabilitation.Curr Opin Neurol 13:655–659
Dominiczak AF, Negrin DC, Clark JS, Brosnan MJ, McBride MW,Alexander MY (2000) Genes and hypertension: from gene map-ping in experimental models to vascular gene transfer strategies.Hypertension 35:164–172
Elbaz A, Amarenco P (1999) Genetic susceptibility and ischemicstroke. Curr Opin Neurol 12:47–55
Ellison JA, Velier JJ, Spera P, Jonak ZL, Wang X, Barone FC, Feuer-stein GZ (1998) Osteopontin and its integrin receptor alpha(v)beta3 are upregulated during formation of the glial scar afterfocal stroke. Stroke 29:1698–1706
Feinklestein SP, Fisher M, Furland AJ, Goldstein LB, Gorelick PB,Kaste M, Lees KR, et al (1999) Recommendations for standardsregarding preclinical neuroprotective and restorative drug devel-opment. Stroke 30:2752–2758
Feuerstein GZ, Barone FC, Wang X (1996) Role of molecular biologyin strategies to discover novel targets for drug discovery. In: Newfrontiers in stress research; modulation of brain function (A Levy,E Grauer, D Ben-Nathan, R deKloet, eds), New York: HardwoodAcademic Publishers, pp 285–293
Fisher M, Fernandez JA, Ameriso SF, Xie D, Gruber A, Paganini-HillA, Griffin JH (1996) Activated protein C resistance in ischemic
stroke not due to factor V arginine506–>glutamine mutation.Stroke 27:1163–1166
Fisher M, Bogousslavsky J (1998) Further evolution toward effectivetherapy for acute ischemic stroke. JAMA 279:1298–1303
Fosnaugh JS, Bhat RV, Yamagata K, Worley PF, Baraban JM (1995)Activation of arc, a putative “effector” immediate early gene, bycocaine in rat brain. J Neurochem 64:2377–2380
Gabriel C, Justicia C, Camins A, Planas AM (1999) Activation ofnuclear factor-kappaB in the rat brain after transient focal isch-emia. Molec Brain Res 65:61–69
Gass P, Eckhardt A, Schroder H, Bravo R, Herdegen T (1996) Tran-sient expression of the mitogen-activated protein kinase phospha-tase MKP-1 (3CH134/ERP1) in the rat brain after limbic epilepsy.Mol Brain Res 41:74–80
Genome International Sequencing Consortium (2001) Initial sequenc-ing and analysis of the human genome. Nature 409:860–921
Gibson UE, Heid CA, Williams PM (1996) A novel method for realtime quantitative RT-PCR. Genome Res 6:995–1001
Gill R, Sibson NR, Hatfield RH, Burdett NG, Carpenter TA, Hall LD,Pickard JD (1995) A comparison of the early development ofischemic damage following permanent middle cerebral artery oc-clusion in rats as assessed using magnetic resonance imaging andhistology. J Cereb Blood Flow Metab 15:1–11
Gowdak LH, Poliakova L, Wang X, Kovesdi I, Fishbein KW, ZacheoA, Palumbo R, Straino S, Emanueli C, Marrocco-Trischitta M,Lakatta EG, Anversa P, Spencer RG, Talan M, Capogrossi MC(2000) Adenovirus-mediated VEGF(121) gene transfer stimulatesangiogenesis in normoperfused skeletal muscle and preserves tis-sue perfusion after induction of ischemia. Circulation 102:565–571
Gratton JA, Sauter A, Rudin M, Lees KR, McColl J, Reid JL, Domin-iczak AF, Macrae IM (1998) Susceptibility to cerebral infarction inthe stroke-prone spontaneously hypertensive rat is inherited as adominant trait. Stroke 29:690–694
Gyngell ML, Busch E, Schmitz B, Kohno K, Back T, Hoehn-BerlageM, Hossmann KA (1995) Evolution of acute focal cerebral isch-aemia in rats observed by localized 1H MRS, diffusion-weightedMRI, and electrophysiological monitoring. NMR Biomed8:206–214
Harrison DC, Medhurst AD, Bond BC, Campbell CA, Davis RP, Phil-pott KL (2000a) The use of quantitative RT-PCR to measuremRNA expression in a rat model of focal ischemia–caspase-3 as acase study. Mol Brain Res 75:143–149
Harrison DC, Roberts J, Campbell CA, Crook B, Davis R, Deen K,Meakin J, Michalovich D, Price J, Stammers M, Maycox PR(2000b) TR3 death receptor expression in the normal and ischemicbrain. Neuroscience 96:147–160
Harrison DC, Davis RP, Bond BC, Campbell CA, James MF, ParsonsAA, Philpott KL (2001) Caspase mRNA expression in a rat modelof focal cerebral ischemia. Mol Brain Res 89:133–146
Hassan A, Markus HS (2000) Genetics and ischemic stroke. Brain123:1784–1812
Hayashi T, Abe K, Suzuki H, Itoyama Y (1997) Rapid induction ofvascular endothelial growth factor gene expression after transientmiddle cerebral artery occlusion in rats. Stroke 28:2039–2044
Hedera P, Friedland RP (1997) Cerebral autosomal dominant arteri-opathy with subcortical infarcts and leukoencephalopathy: study oftwo American families with predominant dementia. J Neurol Sci146:27–33
Hirano M, Pavlakis SG (1994) Mitochondrial myopathy, encephalop-athy, lactic acidosis, and stroke-like episodes (MELAS): currentconcepts. J Child Neurol 9:4–13
Honkaniemi J, States BA, Weinstein PR, Espinoza J, Sharp FR (1997)Expression of zinc finger immediate early genes in rat brain afterpermanent middle cerebral artery occlusion. J Cereb Blood FlowMetab 17:636–646
Hossmann KA (1996) Periinfarct depolarizations. Cerebrovas BrainMetab Rev 8:195–208
Hrubec Z, Robinette CD (1984) The study of human twins in medicalresearch. New Engl J Med 310:435–441
Hubank M, Schatz DG (1994) Identifying differences in mRNA ex-pression by representational difference analysis of cDNA. NuclAcids Res 22:5640–5648
S. J. READ ET AL.774
J Cereb Blood Flow Metab, Vol. 21, No. 7, 2001

Hunter AJ, Hatcher J, Virley D, Nelson P, Irving E, Hadingham SJ,Parsons AA (2000) Functional assessments in mice and rats afterfocal stroke. Neuropharmacology 39:806–816
Iadecola C, Zhang F, Casey R, Clark HB, Ross ME (1996) Induciblenitric oxide synthase gene expression in vascular cells after tran-sient focal cerebral ischemia. Stroke 27:1373–1380
Iadecola C, Ross ME (1997) Molecular pathology of cerebral ischemia:delayed gene expression and strategies for neuroprotection. AnnNew York Acad Sci 835:203–217
Iadecola C, Niwa K, Nogawa S, Zhao X, Nagayama M, Araki E,Morham S, Ross ME (2001) Reduced susceptibility to ischemicbrain injury and N-methyl-D-aspartate-mediated neurotoxicity incyclooxygenase-2-deficient mice. Proc Natl Acad Sci U S A98:1294–1299
Ikeda K, Nara Y, Matumoto C, Mashimo T, Tamada T, Sawamura M,Nabika T, Yamori Y (1996) The region responsible for stroke onchromosome 4 in the stroke-prone spontaneously hypertensive rat.Biochem Biophys Res Comm 229:658–662
Jeffs B, Clark JS, Anderson NH, Gratton J, Brosnan MJ, Gauguier D,Reid JL, Macrae IM, Dominiczak AF (1997) Sensitivity to cerebralischemic insult in a rat model of stroke is determined by a singlegenetic locus. Nature Genet 16:364–367
Jiang Q, Zhang RL, Zhang ZG, Ewing JR, Jiang P, Divine GW, KnightRA, Chopp M (2000) Magnetic resonance imaging indexes oftherapeutic efficacy of recombinant tissue plasminogen activatortreatment of rat at 1 and 4 hours after embolic stroke. J CerebBlood Flow Metab 20:21–27
Jousilahti P, Rastenyte D, Tuomilehto J, Sarti C, Vartiainen E (1997)Parental history of cardiovascular disease and risk of stroke. Aprospective follow-up of 14371 middle-aged men and women inFinland. Stroke 28:1361–1366
Joutel A, Bousser MG, Biousse V, Labauge P, Chabriat H, Nibbio A,Maciazek J, Meyer B, Bach MA, Weissenbach J (1993) A gene forfamilial hemiplegic migraine maps to chromosome 19. Nat Genet5:40–45
Joutel A, Corpechot C, Ducros A, Vahedi K, Chabriat H, Mouton P,Alamowitch S, Domenga V, Cecillion M, Marechal E, Maciazek J,Vayssiere C, Cruaud C, Cabanis EA, Ruchoux MM, WeissenbachJ, Bach JF, Bousser MG, Tournier-Lasserve E (1996) Notch3 mu-tations in CADASIL, a hereditary adult-onset condition causingstroke and dementia. Nature 383:707–710
Joutel A, Vahedi K, Corpechot C, Troesch A, Chabriat H, Vayssiere C,Cruaud C, Maciazek J, Weissenbach J, Bousser MG, Bach JF,Tournier-Lasserve E (1997) Strong clustering and stereotyped na-ture of Notch3 mutations in CADASIL patients. Lancet350:1511–1515
Joutel A, Tournier-Lasserve E (1998) Notch signalling pathway andhuman diseases. Sem Cell Develop Biol 9:619–625
Kalka C, Masuda H, Takahashi T, Gordon R, Tepper O, Gravereaux E,Pieczek A, Iwaguro H, Hayashi SI, Isner JM, Asahara T (2000)Vascular endothelial growth factor(165) gene transfer augmentscirculating endothelial progenitor cells in human subjects. Circ Res86:1198–1202
Kannel WB, Wolf PA, Verter J, McNamara PM (1970) Epidemiologicassessment of the role of blood pressure in stroke. The Framing-ham study. JAMA 214:301–310
Katayama T, Imaizumi K, Tsuda M, Mori Y, Takagi T, Tohyama M(1998) Expression of an ADP-ribosylation factor like gene,ARF4L, is induced after transient forebrain ischemia in the gerbil.Mol Brain Res 56:66–75
Kessler C, Spitzer C, Stauske D, Mende S, Stadlmuller J, Walther R,Rettig R (1997) The apolipoprotein E and beta-fibrinogen G/A-455gene polymorphisms are associated with ischemic stroke involvinglarge-vessel disease. Arterioscl Thromb Vasc Biol 17:2880–2884
Keyvani K, Reinecke S, Abts HF, Paulus W, Witte OW (2000) Sup-pression of proteasome C2 contralateral to ischemic lesions in ratbrain. Brain Res 858:386–392
Khaw KT, Barrett-Connor E (1986) Family history of stroke as anindependent predictor of ischemic heart disease in men and strokein women. Am J Epidem 123:59–66
Kiely DK, Wolf PA, Cupples LA, Beiser AS, Myers RH (1993) Fa-milial aggregation of stroke. The Framingham Study. Stroke24:1366–1371
Kim JS, Gautam SC, Chopp M, Zaloga C, Jones ML, Ward PA, WelchKM (1995) Expression of monocyte chemoattractant protein-1 andmacrophage inflammatory protein-1 after focal cerebral ischemiain the rat. J Neuroimmun 56:127–134
Kitagawa K, Matsumoto M, Tagaya M, Hata R, Ueda H, Niinobe M,Handa N, Fukunaga R, Kimura K, Mikoshiba K (1990) “Ischemictolerance” phenomenon found in the brain. Brain Res 528:21–24
Kitagawa K, Matsumoto M, Kuwabara K, Tagaya M, Ohtsuki T, HataR, Ueda H, Handa N, Kimura K, Kamada T (1991) “Ischemictolerance” phenomenon detected in various brain regions. BrainRes 561:203–211
Kochanek PM, Hallenbeck JM (1992) Polymorphonuclear leukocytesand monocytes/macrophages in the pathogenesis of cerebral isch-emia and stroke. Stroke 23:1367–1379
Kohno K, Hoehn-Berlage M, Mies G, Back T, Hossmann KA (1995a)Relationship between diffusion-weighted MR images, cerebralblood flow, and energy state in experimental brain infarction.Magn Reson Imaging 13:73–80
Kohno K, Back T, Hoehn-Berlage M, Hossmann KA (1995b) A modi-fied rat model of middle cerebral artery thread occlusion underelectrophysiological control for magnetic resonance investigations.Magn Reson Imaging 13:65–71
Koistinaho J, Hokfelt T (1997) Altered gene expression in brain isch-emia. Neuroreport 8:i–viii
Koizumi J, Yoshida Y, Nakaazawa T Ooneda G (1986) Experimentalstudies of ischemic brain edema. 1. A new experimental model ofcerebral embolism in rats which recirculation can be introduced inthe ischemic area. Jpn J Stroke 8:1–8
Kokaia Z, Zhao Q, Kokaia M, Elmer E, Metsis M, Smith ML, SiesjoBK, Lindvall O (1995) Regulation of brain-derived neurotrophicfactor gene expression after transient middle cerebral artery occlu-sion with and without brain damage. Exp Neurol 136:73–88
Kontula K, Ylikorkala A, Miettinen H, Vuorio A, Kauppinen-MakelinR, Hamalainen L, Palomaki H, Kaste M (1995) Arg506Gln factorV mutation (factor V Leiden) in patients with ischemic cerebro-vascular disease and survivors of myocardial infarction. ThrombHaemost 73:558–560
Kosinski C, Mull M, Lethen H, Topper R (1995) Evidence for cardio-embolic stroke in a case of Kearns-Sayre syndrome. Stroke26:1950–1952
Kovalenko SA, Tanaka M, Yoneda M, Iakovlev AF, Ozawa T (1996)Accumulation of somatic nucleotide substitutions in mitochondrialDNA associated with the 3243 A-to-G tRNA(leu)(UUR) mutationin encephalomyopathy and cardiomyopathy. Biochem Biophys ResComm 222:201–220
Ktorza A, Bernard C, Parent V, Penicaud L, Froguel P, Lathrop M,Gauguier D (1997) Are animal models of diabetes relevant to thestudy of the genetics of non-insulin-dependent diabetes in humans?Diabetes Metab 23(Suppl 2):38–46
Kudo T, Imaizumi K, Tanimukai H, Katayama T, Sato N, Nakamura Y,Tanaka T, Kashiwagi Y, Jinno Y, Tohyama M, Takeda M (2000)Are cerebrovascular factors involved in Alzheimer’s disease? Neu-robiol Aging 21:215–224
Lawson CS, Downey JM (1993) Reconditioning: state of the art myo-cardial protection [review]. Cardiovasc Res 27:542–550
Lee LY, Patel SR, Hackett NR, Mack CA, Polce DR, El-Sawy T,Hachamovitch R, Zanazonico P, Sanborn TA, Parikh M, IsomOW, Crystal RG, Rosengart TK (2000) Focal angiogen therapyusing intramyocardial delivery of an adenovirus vector coding forvascular endothelial growth factor 121. Ann Thorac Surg 69:14–24
Lee VM, Burdett NG, Carpenter A, Hall LD, Pambakian PS, Patel S,Wood NI, James MF (1996) Evolution of photochemically inducedfocal cerebral ischemia in the rat. Magnetic resonance imaging andhistology. Stroke 27:2110–2118
Levitan D, Greenwald I (1995) Facilitation of lin-12-mediated signal-ling by sel-12, a Caenorhabditis elegans S182 Alzheimer’s diseasegene. Nature 377:351–354
Li FH, Liu KF, Silva MD, Omae T, Sotak CH, Fenstermacher JD,Fisher M (2000) Transient and permanent resolution of ischemiclesions on diffusion-weighted imaging after brief periods of focalischemia in rats - correlation with histopathology. Stroke31:946–953
Liao D, Myers R, Hunt S, Shahar E, Paton C, Burke G, Province M,
GENE EXPRESSION: ISSUES AND ANSWERS 775
J Cereb Blood Flow Metab, Vol. 21, No. 7, 2001

Heiss G (1997) Familial history of stroke and stroke risk. TheFamily Heart Study. Stroke 28:1908–1912
Lifton RP, Dluhy RG, Powers M, Ulick S, Lalouel JM (1992) Themolecular basis of glucocorticoid-remediable aldosteronism, aMendelian cause of human hypertension. Trans Assoc Am Physi-cians 105:64–71
Lipshutz RJ, Fodor SP, Gingeras TR, Lockhart DJ (1999) High densitysynthetic oligonucleotide arrays. Nature Genet 21:20–24
Lisitsyn NA (1995) Representational difference analysis: finding thedifferences between genomes. Trends Genet 11:303–307
Liu T, Young PR, McDonnell PC, White RF, Barone FC, FeuersteinGZ (1993a) Cytokine-induced neutrophil chemoattractant mRNAexpressed in cerebral ischemia. Neurosci Lett 164:125–128
Liu T, McDonnell PC, Young PR, White RF, Siren AL, Barone FC,Feuerstein GZ (1993b) Interleukin-1� expression in ischemic ratcortex. Stroke 24:1746–1751
Liu T, Clark RK, McDonnell PC, Young PR, White RF, Barone FC,Feuerstein GZ (1994) Tumor necrosis factor-alpha expression inischemic neurons. Stroke 25:1481–1488
Lockhart DJ, Dong H, Byrne MC, Follettie MT, Gallo MV, Chee MS,Mittmann M, Wang C, Kobayashi M, Horton H, Brown EL (1996)Expression monitoring by hybridization to high-density oligo-nucleotide arrays. Nature Biotechnol 14:1675–1680
Loddick SA, Turnbull AV, Rothwell NJ (1998) Cerebral interleukin-6is neuroprotective during permanent focal cerebral ischemia in therat. J Cereb Blood Flow Metab 18:176–179
Longa EZ, Weinstein PR, Carlson S, Cummins R (1989) Reversiblemiddle cerebral artery occlusion without craniectomy in rats.Stroke 20:84–91
Lossos A, Ben Hur T, Ben Nariah Z, Enk C, Gomori M, Soffer D(1995) Familial Sneddon’s syndrome. J Neurol 242:164–168
MacLeod MJ, Dahiyat MT, Cumming A, Meiklejohn D, Shaw D, StClair D (1999) No association between Glu/Asp polymorphism ofNOS3 gene and ischemic stroke. Neurology 53:418–420
Macmillan C, Lach B, Shoubridge EA (1993) Variable distribution ofmutant mitochondrial DNAs (tRNA(Leu[3243])) in tissues ofsymptomatic relatives with MELAS: the role of mitotic segrega-tion. Neurology 43:1586–1590
Mandai K, Matsumoto M, Kitagawa K, Matsushita K, Ohtsuki T, Ma-buchi T, Colman DR, Kamada T, Yanagihara T (1997) Ischemicdamage and subsequent proliferation of oligodendrocytes in focalcerebral ischemia. Neuroscience 77:849–861
Margaglione M, Celentano E, Grandone E, Vecchione G, Cappucci G,Giuliani N, Colaizzo D, Panico S, Mancini FP, Di Minno G (1996)Deletion polymorphism in the angiotensin-converting enzymegene in patients with a history of ischemic stroke. ArteriosclThromb Vasc Biol 16:304–309
Margaglione M, Seripa D, Gravina C, Grandone E, Vecchione G, Cap-pucci G, Merla G, Papa S, Postiglione A, Di Minno G, Fazio VM(1998) Prevalence of apolipoprotein E alleles in healthy subjectsand survivors of ischemic stroke: an Italian Case-Control Study.Stroke 29:399–403
Marti HJ, Bernaudin M, Bellail A, Schoch H, Euler M, Petit E, RisauW (2000) Hypoxia-induced vascular endothelial growth factor ex-pression precedes neovascularization after cerebral ischemia. Am JPathol 156:965–976
Martin-Villalba A, Herr I, Jeremias I, Hahne M, Brandt R, Vogel J,Schenkel J, Herdegen T, Debatin KM (1999) CD95 ligand (Fas-L/APO-1L) and tumor necrosis factor-related apoptosis-inducingligand mediate ischemia-induced apoptosis in neurons. J Neurosci19:3809–3817
Medhurst AD, Harrison DC, Read SJ, Campbell CA, Robbins MJ,Pangalos MN (2000) The use of TaqMan RT-PCR assays for semi-quantitative analysis of gene expression in CNS tissues and diseasemodels. J Neurosci Methods 98:9–20
Nagaoka A, Iwatsuka H, Suzuoki Z, Okamoto K (1976) Genetic pre-disposition to stroke in spontaneously hypertensive rats. Am JPhysiol 230:1354–1359
Nakata Y, Katsuya T, Rakugi H, Takami S, Sato N, Kamide K, OhishiM, Miki T, Higaki J, Ogihara T (1997) Polymorphism of angio-tensin converting enzyme, angiotensinogen, and apolipoprotein Egenes in a Japanese population with cerebrovascular disease. Am JHypertens 10:1391–1395
Nakata Y, Katsuya T, Takami S, Sato N, Fu Y, Ishikawa K, TakiuchiS, Rakugi H, Miki T, Higaki J, Ogihara T (1998) Methylenetetra-hydrofolate reductase gene polymorphism: relation to blood pres-sure and cerebrovascular disease. Am J Hypertens 11:1019–1023
Neumann-Haefelin T, Kastrup A, de Crespigny A, Yenari MA, RingerT, Sun GH, Moseley ME (2000) Serial MRI after transient focalcerebral ischemia in rats: dynamics of tissue injury, blood-brainbarrier damage, and edema formation. Stroke 31:1965–1972
Nogawa S, Zhang F, Ross ME, Iadecola C (1997) Cyclo-oxygenase-2gene expression in neurons contributes to ischemic brain damage.J Neurosci 15:2746–2755
Nogawa S, Forster C, Zhang F, Nagayama M, Ross ME, Iadecola C(1998) Interaction between inducible nitric oxide synthase andcyclooxygenase-2 after cerebral ischemia. Proc Natl Acad Sci USA95:10966–10971
Pancioli AM, Broderick J, Kothari R, Brott T, Tuchfarber A, Miller R,Khoury J, Jauch E (1998) Public perception of stroke warningsigns and knowledge of potential risk factors. JAMA 279:1288–1292
Parsons AA, Irving EA, Legos JJ, Lenhard SC, Chandra S, SchaefferTR, Haimback RE, White RF, Hunter AJ, Barone FC (2000) Acutestroke therapy: Iissues for translating pre-clinical neuroprotectionto therapeutic reality. Curr Opin Invest Drugs 1:452–463
Pedro-Botet J, Senti M, Nogues X, Rubies-Prat J, Roquer J,D’Olhaberriague L, Olive J (1992) Lipoprotein and apolipoproteinprofile in men with ischemic stroke. Role of lipoprotein(a), triglyc-eride-rich lipoproteins, and apolipoprotein E polymorphism.Stroke 23:1556–1562
Poort SR, Rosendaal FR, Reitsma PH, Bertina RM (1996) A commongenetic variation in the 3�-untranslated region of the prothrombingene is associated with elevated plasma prothrombin levels and anincrease in venous thrombosis. Blood 88:3698–3703
Rastenyte D, Tuomilehto J, Sarti C (1998) Genetics of stroke–a review.J Neurol Sci 153:132–145
Relton JK, Rothwell NJ (1992) Interleukin-1 receptor antagonist inhib-its ischemic and excitotoxic neuronal damage in the rat. Brain ResBull 29:243–246
Rossini PM, Pauri F, Cicinelli P, Pasqualetti P, Traversa R, Tecchio F(1999a) Neuromagnetic recordings and magnetic brain stimulationin the evaluation of sensorimotor hand area interhemispheric dif-ferences: normative, experimental and patients’ data. Electroen-cephal Clin Neurophysiol 50(Suppl):210–220
Rossini PM, Pauri F, Pizzella V, Pasqualetti P, Cicinelli P, Traversa R(1999b) An integrative neuroimaging approach to restorative neu-rology. Electroenceph Clin Neurophysiol 49(suppl):19–24
Rubattu S, Volpe M, Kreutz R, Ganten U, Ganten D, Lindpaintner K(1996) Chromosomal mapping of quantitative trait loci contribut-ing to stroke in a rat model of complex human disease. NatureGenet 13:429–434
Rubattu S, Ridker P, Stampfer MJ, Volpe M, Hennekens CH, Lind-paintner K (1999a) The gene encoding atrial natriuretic peptideand the risk of human stroke. Circulation 100:1722–1726
Rubattu S, Giliberti R, Ganten U, Volpe M (1999b) Differential brainatrial natriuretic peptide expression co-segregates with occurrenceof early stroke in the stroke-prone phenotype of the spontaneouslyhypertensive rat. J Hypertens 17(12 Pt 2):1849–1852
Rubattu S, Lee-Kirsch MA, DePaolis P, Giliberti R, Gigante B, Lom-bardi A, Volpe M, Lindpaintner K (1999c) Altered structure, regu-lation, and function of the gene encoding the atrial natriureticpeptide in the stroke-prone spontaneously hypertensive rat. CircRes 85:900–905
Ruchoux MM, Maurage CA (1998) Endothelial changes in muscle andskin biopsies in patients with CADASIL. Neuropathol Appl Neu-robiol 24:60–65
Sakuta R, Goto Y, Horai S, Nonaka I (1993) Mitochondrial DNAmutations at nucleotide positions 3243 and 3271 in mitochondrialmyopathy, encephalopathy, lactic acidosis, and stroke-like epi-sodes: a comparative study. J Neurol Sci 115:158–160
Schena M, Shalon D, Davis RW, Brown PO (1995) Quantitative moni-toring of gene expression patterns with a complementary DNAmicroarray. Science 270:467–470
Schmidt R, Schmidt H, Fazekas F (2000) Vascular risk factors indementia. J Neurol 247:81–87
S. J. READ ET AL.776
J Cereb Blood Flow Metab, Vol. 21, No. 7, 2001

Schmidt-Kastner R, Zhao W, Truettner J, Belayev L, Busto R, Gins-berg MD (1998) Pixel-based image analysis of HSP70, GADD45and MAP2 mRNA expression after focal cerebral ischemia: hemo-dynamic and histological correlates. Molec Brain Res 63:79–97
Schmidt-Kastner R, Truettner J, Zhao W, Belayev L, Krieger C, BustoR, Ginsberg MD (2000) Differential changes of bax, caspase-3 andp21 mRNA expression after transient focal brain ischemia in therat. Molec Brain Res 79:88–101
Sharma P, Carter ND, Barley J, Brown MM (1994) Molecular approachto assessing the genetic risk of cerebral infarction: deletion poly-morphism in the gene encoding angiotensin 1-converting enzyme.J Hum Hypertens 8:645–648
Sharp FR, Lu A, Tang Y, Millhorn DE (2000) Multiple molecularpenumbras after focal cerebral ischemia. J Cereb Blood FlowMetab 20:1011–1032
Shimazaki K, Urabe M, Monahan J, Ozawa K, Kawai N (2000) Adeno-associated virus vector-mediated bcl-2 gene transfer into post-ischemic gerbil brain in vivo: prospects for gene therapy of isch-emia-induced neuronal death. Gene Ther 7:1244–1249
Skoog I (2000) Vascular aspects in Alzheimer’s disease. J NeuralTransm Suppl 59:37–43
Soriano MA, Tessier M, Certa U, Gill R (2000) Parallel gene expres-sion monitoring using oligonucleotide probe arrays of multipletranscripts with an animal model of focal ischemia. J Cereb BloodFlow Metab 20:1045–1055
Sourander P, Walinder J (1977) Hereditary multi-infarct dementia.Lancet 1:1015
Stanton LW, Garrard LJ, Damm D, Garrick BL, Lam A, Kapoun AM,Zheng Q, Protter AA, Schreiner GF, White RT (2000) Alteredpatterns of gene expression in response to myocardial infarction.Circ Res 86:939–945
Stephenson D, Yin T, Smalstig EB, Hsu MA, Panetta J, Little S, Cle-mens J (2000) Transcription factor nuclear factor-kappa B is acti-vated in neurons after focal cerebral ischemia. J Cereb Blood FlowMetab 20:592–603
Stephenson J (1998) Rising stroke rates spur efforts to identify risks,prevent disease. JAMA 279:1239–1240
Suzuki S, Tanaka K, Nogawa S, Ito D, Dembo T, Kosakai A, FukuuchiY (2000) Immunohistochemical detection of leukemia inhibitoryfactor after focal cerebral ischemia in rats. J Cereb Blood FlowMetab 20:661–668
Takami S, Nishikawa H, Minami M, Nishiyori A, Sato M, Akaike A,Satoh M (1997) Induction of macrophage inflammatory proteinMIP-1alpha mRNA on glial cells after focal cerebral ischemia inthe rat. Neurosci Lett 227:173–176
Tamura A, Graham DI, McCulloch J, Teasdale GM (1981) Focal ce-rebral ischaemia in the rat: 1. Description of technique and earlyneuropathological consequences following middle cerebral arteryocclusion. J Cereb Blood Flow Metab 1:53–60
Tournier-Lasserve E, Joutel A, Melki J, Weissenbach J, Lathrop GM,Chabriat H, Mas JL, Cabanis EA, Baudrimont M, Maciazek J(1993) Cerebral autosomal dominant arteriopathy with subcorticalinfarcts and leukoencephalopathy maps to chromosome 19q12.Nature Genet 3:256–259
Tsuda M, Kitagawa K, Imaizumi K, Wanaka A, Tohyama M, Takagi T(1996) Induction of SPI-3 mRNA, encoding a serine protease in-hibitor, in gerbil hippocampus after transient forebrain ischemia.Mol Brain Res 35:314–318
Tsuda M, Imaizumi K, Katayama T, Kitagawa K, Wanaka A, TohyamaM, Takagi T (1997) Expression of zinc transporter gene, ZnT-1, isinduced after transient forebrain ischemia in the gerbil. J Neurosci17:6678–6684
Utans-Schneitz U, Lorez H, Klinkert WE, da Silva J, Lesslauer W(1998) A novel rat CC chemokine, identified by targeted differen-tial display, is upregulated in brain inflammation. J Neuroimmunol92:179–190
Vale PR, Losordo DW, Milliken CE, Maysky M, Esakof DD, SymesJF, Isner JM (2000) Left ventricular electromechanical mapping toassess efficacy of phVEGF(165) gene transfer for therapeutic an-giogenesis in chronic myocardial ischemia. Circulation 102:965–974
van Bruggen N, Cullen BM, King MD, Doran M, Williams SR, GadianDG, Cremer JE (1992) T2- and diffusion-weighted magnetic reso-
nance imaging of a focal ischemic lesion in rat brain. Stroke23:576–582
Viitanen M, Kalimo H (2000) CADASIL: hereditary arteriopathy lead-ing to multiple brain infarcts and dementia. Ann N Y Acad Sci903:273–284
Wang XB, Uhl GR (1998) Subtracted differential display: genes withamphetamine-altered expression patterns include calcineurin. MolBrain Res 53:344–347
Wang XK, Yue TL, Barone FC, White RF, Gagnon RC, Feuerstein GZ(1994) Concomitant cortical expression of TNF-alpha and IL-1beta mRNAs follows early response gene expression in transientfocal ischemia. Mol Chem Neuropathol 23:103–114
Wang XK, Yue TL, Barone FC, Feuerstein GZ (1995a) Monocytechemoattractant protein-1 (MCP-1) mRNA expression in rat isch-emic cortex. Stroke 26:661–665
Wang XK, Yue TL, Barone FC, White RF, Clark RK, Willette RN,Sulpizio AC, Aiyar NV, Ruffolo RR Jr, Feuerstein GZ (1995b)Discovery of adrenomedullin in rat ischemic cortex and evidencefor its role in exacerbating focal brain ischemic damage. Proc NatlAcad Sci U S A 92:11480–11484
Wang XK, Yue TL, White RF, Barone FC, Feuerstein GZ (1995c)Transforming growth factor ß1, exhibits delayed gene expressionfollowing focal cerebral ischemia. Brain Res Bull 36:607–609
Wang, XK, Yue TL Young PR, Barone FC, Feuerstein GZ (1995d)Expression of interleukin 6, c-fos and zif268 mRNA in rat isch-emic cortex. J Cereb Blood Flow Metab 15:166–171
Wang XK, Barone FC, Aiyar NV, Feuerstein GZ (1997) Interleukin-1receptor and receptor antagonist gene expression after focal strokein rats. Stroke 28:155–161
Wang XK, Barone FC, White RF, Feuerstein GZ (1998a) Subtractivecloning identifies tissue inhibitor of matrix metalloproteinase-1(TIMP-1) increased gene expression in focal stroke. Stroke29:516–520
Wang XK, Ellison J, Siren AL, Lysko PG, Yue TL, Barone FC, Shatz-man A, Feuerstein GZ (1998b) Prolonged expression of interferoninducible protein-10 in ischemic cortex after permanent occlusionof the middle cerebral artery in the rat. J Neurochem 71:1194–1204
Wang XK, Louden C, Yue TL, Ellison JA, Barone FC, Solleveld H,Feuerstein GZ (1998c) Delayed expression of osteopontin afterfocal stroke in the rat. J Neurosci 18:2075–2083
Wang XK, Yaish-Ohad S, Li X, Barone FC, Feuerstein GZ (1998d)Use of suppression subtractive hybridization strategy for discoveryof increased tissue inhibitor of matrix metalloproteinase-1 geneexpression in brain ischemic tolerance. J Cereb Blood Flow Metab18:1173–1177
Wang XK, Li X, Yaish-Ohad S, Sarau HM, Barone FC, Feuerstein GZ(1999) Molecular cloning and expression of the rat monocyte che-motactic protein-3 gene: a possible role in stroke. Mol Brain Res71:304–312
Wang XK, Li X, Currie RW, Willette RN, Barone FC, Feuerstein GZ(2000a) Application of real-time polymerase chain reaction toquantitate induced expression of interleukin-1beta mRNA in isch-emic brain tolerance. J Neurosci Res 59:238–246
Wang XK, Li X, Erhardt JA, Barone FC, Feuerstein GZ (2000b) De-tection of tumor necrosis factor-alpha mRNA induction in isch-emic brain tolerance by means of real-time polymerase chain re-action. J Cereb Blood Flow Metab 20:15–20
Wang XK, Li X, Schmidt D, Foley JJ, Barone FC, Ames RS, Sarau HM(2000c) Identification and molecular characterization of ratCXCR3: discovery of increased expression and interferon-inducible protein-10 binding are increased in focal stroke. MolPharmacol 57:1190–1198
Welin L, Svardsudd K, Wilhelmsen L, Larsson B, Tibblin G (1987)Analysis of risk factors for stroke in a cohort of men born in 1913.N Engl J Med 317:521–526
Wiessner C, Neumann-Haefelin T, Vogel P, Back T, Hossmann KA(1995) Transient forebrain ischemia induces an immediate-earlygene encoding the mitogen-activated protein kinase phosphatase3CH134 in the adult rat brain. Neuroscience 64:959–966
Wigle D, Ho W, Lo D, Francis J, Eubanks JH, Wallace MC (1999)Altered expression levels of SEF-2 and p112 in the rat hippocam-pus after transient cerebral ischemia: identification by mRNA dif-ferential display. J Cereb Blood Flow Metab 19:435–442
GENE EXPRESSION: ISSUES AND ANSWERS 777
J Cereb Blood Flow Metab, Vol. 21, No. 7, 2001

Wong ML, Loddick SA, Bongiorno PB, Gold PW, Rothwell NJ,Licinio J (1995) Focal cerebral ischemia induces CRH mRNA inrat cerebral cortex and amygdala. Neuroreport 6:1785–1788
Yamagami S, Tamura M, Hayashi M, Endo N, Tanabe H, Katsuura Y,Komoriya K (1999) Differential production of MCP-1 and cyto-kine-induced neutrophil chemoattractant in the ischemic brain aftertransient focal ischemia in rats. J Leukocyte Biol 65:744–749
Yamashita K, Wiessner C, Lindholm D, Thoenen H, Hossmann KA(1997) Post-occlusion treatment with BDNF reduces infarct size ina model of permanent occlusion of the middle cerebral artery in rat.Metab Brain Dis 12:271–280
Yanamoto H, Nagata I, Sakata M, Zhang Z, Tohnai N, Sakai H, Kiku-chi H (2000) Infarct tolerance induced by intra-cerebral infusion ofrecombinant brain-derived neurotrophic factor. Brain Res859:240–248
Yang GY, Zhao YJ, Davidson BL, Betz AL (1997) Overexpression ofinterleukin-1 receptor antagonist in the mouse brain reduces isch-emic brain injury. Brain Res 751:181–188
Yang GY, Mao Y, Zhou LF, Ye W, Liu XH, Gong C, Lorris BA (1999)Attenuation of temporary focal cerebral ischemic injury in the
mouse following transfection with interleukin-1 receptor antago-nist. Mol Brain Res 72:129–137
Yellon DM, Baxter GF, Garcia-Dorado D, Heusch G, Sumeray MS(1998) Ischemic preconditioning: present position and future di-rections. Cardiovasc Res 37:21–33
Yenari MA, Fink SL, Sun GH, Chang LK, Patel MK, Kunis DM, OnleyD, Ho DY, Sapolsky RM, Steinberg GK (1998) Gene therapy withHSP 72 is neuroprotective in rat models of stroke and epilepsy.Ann Neurol 44:584–591
Yenari MA, Giffard RG, Sapolsky RM, Steinberg GK (1999) Theneuroprotective potential of heat shock protein 70 (HSP 70). MolMed Today 5:525–531
Yonemori F, Yamaguchi T, Nakayama H, Narita K, Hojo S, Tamura A(2000) Effect of JTP-2942, a novel thyrotropin-releasing hormoneanalog, on motor deficits after chronic focal cerebral ischemia inrats. J Cereb Blood Flow Metab 20:74–81
Young RA (2000) Biomedical discovery with DNA arrays. Cell102:9–15
Zhang Z, Zhang RL, Jiang Q, Raman SB, Cantwell L, Chopp M (1997)A new rat model of thrombotic focal cerebral ischemia. J CerebBlood Flow Metab 17:123–135
S. J. READ ET AL.778
J Cereb Blood Flow Metab, Vol. 21, No. 7, 2001




















