Steric Maps to Evaluate the Role of Steric Hindrance on the IPr NHC Ligand
10
Procedia Computer Science 18 (2013) 845 – 854 1877-0509 © 2013 The Authors. Published by Elsevier B.V. Selection and peer review under responsibility of the organizers of the 2013 International Conference on Computational Science doi:10.1016/j.procs.2013.05.249 2013 International Steric maps to evaluate Albert Poater a, *, Laura Falivene b , P. No a Institut de Química Computacional, Departament de Q Spain b Dipartimento di Chimica e Biologia, U c EaStCHEM School of Chemistry, University o d King Abdullah University of Science and Technolog Abstract Density functional theory (DFT) calculations of the structure of the prototype 1,3-bis carbene (NHC) ligand. The modification co substituent with phenyl groups, and here w environment and therefore the related catalyt significant structural difference between IPr olefin metathesis reactions, here by means o role and where it is a simple spectator, or a role/performance. Furthermore, this commun ligand. The optimization of these bulky new Keywords: NHC ligand; olefins metathesis; ruthenium c 1. Introduction Society is facing tremendous challenges in th With a growing population and dwindling resourc health care for everybody in an environmentally Seeking to contribute to solutions for the World characterization of more efficient catalysts for ole l Conference on Computational Science the role of steric hindrance on t h NHC ligand César A. Urbina-Blanco c , Simone Manzin olan c , Luigi Cavallo b,d Química, Universitat de Girona, Campus de Montilivi, E-17071 Gir n. E-mail: [email protected] Università di Salerno, Via Ponte don Melillo, 84084, Fisciano, Italy. of St Andrews, North Haugh, St Andrews, Fife, KY16 9ST, United K gy (KAUST), Physical Sciences and Engineering, Kaust Catalysis Ce 23955-6900, Saudi Arabia. s were used to predict and rationalize the effect of the s(2,6-diisopropylphenyl)imidazol-2-ylidene) (IPr) N onsists in the substitution of the methyl groups of or we plan to describe how such significant changes eff tic behaviour by simple steric maps. Bearing in mind r and IPr* ligands, that translated in different reactivi of DFT we characterize where the NHC ligand plays a at least its modification does not significantly change nication endeavours to modify further the skeleton of systems go to the limits of the DFT computational me catalysts; DFT calculations; IPr he 21 st century to maintain earth and its living beings in a su ces, the need to provide clean water, food and energy, as we y benign and sustainable way, will occupy the efforts of m d’s well-being from many different angles, we regard the efin metathesis. It is fundamental to reduce the waste for en he IPr ni c , Steven rona, Catalonia, . Kingdom. enter, Thuwal e modification N-heterocyclic rtho isopropyl fect the metal that there is a ity for several a more active e its catalytic the IPr NHC ethod. ustainable way. ell as affordable many scientists. e synthesis and nvironment, but Available online at www.sciencedirect.com
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Steric Maps to Evaluate the Role of Steric Hindrance on the IPr NHC Ligand

Procedia Computer Science 18 ( 2013 ) 845 – 854
1877-0509 © 2013 The Authors. Published by Elsevier B.V.Selection and peer review under responsibility of the organizers of the 2013 International Conference on Computational Sciencedoi: 10.1016/j.procs.2013.05.249
2013 International
Steric maps to evaluate
Albert Poatera,*, Laura Faliveneb, P. No
aInstitut de Química Computacional, Departament de QSpain
bDipartimento di Chimica e Biologia, UcEaStCHEM School of Chemistry, University o
dKing Abdullah University of Science and Technolog
Abstract
Density functional theory (DFT) calculationsof the structure of the prototype 1,3-biscarbene (NHC) ligand. The modification cosubstituent with phenyl groups, and here wenvironment and therefore the related catalytsignificant structural difference between IProlefin metathesis reactions, here by means orole and where it is a simple spectator, or arole/performance. Furthermore, this communligand. The optimization of these bulky new
Keywords: NHC ligand; olefins metathesis; ruthenium c
1. Introduction
Society is facing tremendous challenges in thWith a growing population and dwindling resourchealth care for everybody in an environmentallySeeking to contribute to solutions for the Worldcharacterization of more efficient catalysts for ole
l Conference on Computational Science
the role of steric hindrance on thNHC ligand
César A. Urbina-Blancoc, Simone Manzinolanc, Luigi Cavallob,d
Química, Universitat de Girona, Campus de Montilivi, E-17071 Girn. E-mail: [email protected] Università di Salerno, Via Ponte don Melillo, 84084, Fisciano, Italy.of St Andrews, North Haugh, St Andrews, Fife, KY16 9ST, United Kgy (KAUST), Physical Sciences and Engineering, Kaust Catalysis Ce
23955-6900, Saudi Arabia.
s were used to predict and rationalize the effect of thes(2,6-diisopropylphenyl)imidazol-2-ylidene) (IPr) Nonsists in the substitution of the methyl groups of or
we plan to describe how such significant changes efftic behaviour by simple steric maps. Bearing in mind r and IPr* ligands, that translated in different reactiviof DFT we characterize where the NHC ligand plays aat least its modification does not significantly changenication endeavours to modify further the skeleton of systems go to the limits of the DFT computational me
catalysts; DFT calculations; IPr
he 21st century to maintain earth and its living beings in a suces, the need to provide clean water, food and energy, as wey benign and sustainable way, will occupy the efforts of md’s well-being from many different angles, we regard theefin metathesis. It is fundamental to reduce the waste for en
he IPr
nic, Steven
rona, Catalonia,
. Kingdom.
enter, Thuwal
e modification N-heterocyclic rtho isopropyl fect the metal that there is a ity for several a more active e its catalytic the IPr NHC
ethod.
ustainable way. ell as affordable many scientists. e synthesis and nvironment, but
Available online at www.sciencedirect.com

846 Albert Poater et al. / Procedia Computer Science 18 ( 2013 ) 845 – 854
also for the reduction of the economic cost of transforming organic substrates. Finding new catalysts is a hard but exciting task, often driven by trial and error. In this respect, computational techniques can be used to screen novel catalyst architectures more rapidly to obtain insights that could help in the design and experimental synthesis of novel and improved catalysts. Bearing in mind the great work done in metathesis and mainly in Ru-based catalysts with N-heterocyclic carbenes (NHC, Figure 1),[1] here we wish to use Density Functional Theory (DFT) to characterize how adding steric hindrance to a particular NHC such as IPr ( IPr = 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene) affects its reactivity.[2,3] DFT-based prediction of the reactivity of new catalysts is not a new approach, and systematic methods are searched for a priori computational prediction to see whether a catalyst will be feasible experimentally, productive and fruitful in industry.[4]
Figure 1. Commonly used NHC in ruthenium olefin metathesis.
Taking into consideration that the latest big leap in the evolution of ruthenium based olefin metathesis catalysts was made by exchanging one of the phosphines in the first generation catalyst for a N-heterocyclic carbene ligand,[5,6] here we search how present computational tools might help improve the design of NHC ligands. The so-called second generation catalysts (those bearing a NHC ligand) are much more active in olefin metathesis than their first generation counterparts, and although many variations of the basic layout have been disclosed over the past 10 years,[7,8] a single universal catalyst superior to the rest of the field in all metathesis transformations has not been achieved, instead, a range of catalysts able to fulfil particular requests have been found. Here we want to examine if it is reasonable to continue exploring new NHC ligands with ever increasing complexity with respect to the common IPr, its saturated backbone relative SIPr and the 1,3-bis(2,4,6-trimethylphenyl)-4,5-dihydroimidazol-2-ylidene (SIMes) congener,[8,9] and rationalize the reactivity differences observed between IPr-, IPr*- and IPr*Tol-bearing olefin metathesis catalysts.[10]
1 2 3
Figure 2. Ruthenium based olefin metathesis catalysts with IPr (1), IPr* (2), and IPr*tol (3) NHC ligands.

847 Albert Poater et al. / Procedia Computer Science 18 ( 2013 ) 845 – 854
2. Computational Details
All DFT static calculations were performed at the GGA level with the Gaussian09 set of programs,[11] using the BP86 functional of Becke and Perdew.[12] The electronic configuration of the molecular systems was described with the standard split-valence basis set with a polarization function of Ahlrichs and co-workers for H, C, N, P, and Cl (SVP keyword in Gaussian).[ 13 ] For Ru we used the small-core, quasi-relativistic Stuttgart/Dresden effective core potential, with an associated valence basis set contracted (standard SDD keywords in gaussian09).[11] The geometry optimizations were performed without symmetry constraints, and the characterization of the located stationary points was performed by analytical frequency calculations. The reported energies have been optimized via single point calculations on the BP86 geometries with triple zeta valence plus polarization (TZVP keyword in Gaussian) using the M06 functional,[14] however estimating solvent effects with the polarizable continuous solvation model PCM using CH2Cl2 as solvent.[15] For all energies given throughout the text, zero point energies and thermal corrections calculated at the BP86 level were added to the M06 in solvent energies to approximate free energies in solvent.
Bearing in mind that Mayer Bond Order (MBO) theory gives insight into the strength of the bonds,[16] MBOs between two atoms A and B have been calculated through the eq 1,[17] where S is the atomic orbital overlap matrix and P is the density matrix. The sums run over the basis set functions belonging to a given atom A or B.
(1) The electrophilicity of the complexes was evaluated thanks to the Parr electrophilicity index shown in eq 2.[18]
(2)
where and are the chemical potential and the molecular hardness, respectively. In the framework of DFT,[19] and for a N-electron system with total electronic energy E are defined as the first and second derivatives of the energy with respect to N at a fixed external potential.[20] In numerical applications, and are calculated with the finite difference formulas of eq 3, which are based on Koopmans’ approximation,[21]
and (3)
where ε and εL are the energies of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO), respectively. Over the last years, conceptual DFT has been used to explain the reactivity pattern, and in particular the regioselectivity in chemical reactions.[22]
3. Results and Discussion
DFT calculations have initially been performed on the geometry of complexes 1-3 in Figure 3 and the optimized structure of complex 1 is overall in excellent agreement with the X-ray structure (rmsd=0.033 Å on distances and 0.8° on angles).[23,24] The effect of the coordination of IPr and IPr* in the Ru catalysts was first evaluated by analysing the strength of the Ru-NHC bond. As expected, the Ru-IPr bond is somewhat shorter than the Ru-IPr* and the Ru- IPr*Tol bonds for the complexes containing PPh3, while the difference is minimal in the later complexes. To have a better understanding of the different stability of the Ru-NHC and Ru-P bonds in 1-3, we performed a Mayer Bond Order (MBO) analysis.[16,17] The analysis showed that the Ru-P bond trans to the NHC ligand, displays MBO values of 0.550, 0.587, and 0.581 for 1-3 respectively; while the Ru-NHC MBOs are 0.817, 0.777, and 0.776. These results are in line with the evidence that the modifications on the IPr ligand weaken the Ru-NHC bond significantly (comparing systems 2 and 3 with 1), thus enhancing interaction of the other labile ligand with the metal. Focusing on the MBOs of the Ru-indenylidene bond, 1.472, 1.456, and 1.457 for 1-3 respectively indicates that replacing the IPr ligand by the sterically demanding IPr* and IPr*Tol ligands impacts the strength of the Ru-NHC and Ru-P bonds, but has minor influence on the Ru-alkylidene bond. The weakened Ru-IPr* and Ru-IPr*Tol bonds in 2 and 3 can be ascribed to steric repulsion between the bulky ortho-CHPh2 groups of these NHC ligands and PPh3.
,2
2
ημω =
)(2
1HL εεμ +≅ ,)(
2
1HL εεη −≅

848 Albert Poater et al. / Procedia Computer Science 18 ( 2013 ) 845 – 854
1 2 3
Figure 3. 1-3 catalysts.
The energy cost for the dissociation of PPh3 was evaluated next (Figure 4), spanning from 14.9 for 1 to 12.0 for 2 and 19.4 kcal/mol for 3. Although the differences are not big, they are significant. Surprisingly, the energies follow the trend 3 (IPr*Tol) > 1 (IPr) > 2 (IPr*). The value for 2 is uncertain because it was expected that the substitution of IPr by IPr* could improve the activation significantly, however, the dissociation is disfavored by 2.9 kcal/mol. In contrast, and as expected, dissociation of PPh3 from 3 requires 4.5 kcal/mol more than for 1. Although the barrier for dissociation could be helpful for the representation of the thermodynamic of the labile ligand dissociation, the large size of the modified IPr ligands, specially the IPr*Tol, excludes the search for these barriers. However, here conceptual DFT supposes a great tool to overcome such problems and it is a cheap tool to compare the reactivity between similar catalysts from an electronic point of view, and could avoid the study of all the species involved in the olefin metathesis pathway for future comparisons with other NHC ligands. Thus, although this modification of the IPr ligand deals principally with the steric influence of the IPr* ligand, we also examined simple electronic properties such as chemical hardness and electrophilicity for complexes 1-3.[18-21] The chemical hardness values are 0.0123, 0.0126, and 0.0148 a.u., which means that complexes containing the modified IPr ligands are less hard, and thus tend to be less reactive, specially complex 3 with the IPr*Tol ligand. On the other hand, the electrophilicity values are 0.719, 0.701, and 0.670 a.u., and these values confirm that the affinity to react with a nucleophile is similar for all complexes, following the trend of the bulkier the NHC ligand the less reactive the system is, mainly due to the reduction of the free space around the Ru center. Thus, these low differences confirm that the diverse catalytic performance of these complexes should be mainly related to different steric properties. Moving to the olefin metathesis reaction itself, for the sake of clarity we used ethylene as the olefin substrate in the calculations.[25] Coordination of ethylene to the 14e- substrate, see Figure 4, is an endothermic step for all complexes except for 3, where there is a release of 1.8 kcal/mol.
Figure 4. Energy profile of olefin metathesis of complexes 1-3 with ethylene (energies are in kcal/mol).

849 Albert Poater et al. / Procedia Computer Science 18 ( 2013 ) 845 – 854
The following step is the metallacycle formaalthough small differences can be found. For all CI1, but for system 1, with IPr, the metallacycle of the bulky IPr*Tol in 3, the metallacycle is 3.1IPr* in system 2 this difference reduces to just 1detected. The main goal of the geometrical modificationIPr* effected the reactivity. To analyze the sterispace defining the steric map were located with thcoordination sphere around the metal, which is thvolume of a given ligand, which is a number toccupied by this ligand.[27] A modified versioevaluating the %VBur in the single quadrants arouSplitting the total %VBur into quadrant contributioand allows to understand how changing the ligandalready introduced topographic steric maps to chmaps were calculated for the free ligands first, wwell as in the next 14e species, as well as for the m
Figure 5. St
ation, and again, the complexes behave similarly with an especies the metallacycle is more stable than the coordinatiois more stable than the CI species, by 2.8 kcal/mol. Then, kcal/mol less stable than the CI intermediate, whereas for
1.0 kcal/mol. However, these energy differences are scarce
n of the IPr ligand was to understand how the modified steriic influence of the ligand we used topographic steric mapshe SambVca package developed by us.[26] This program anhe place where catalysis occurs. It is normally used to calcuthat quantifies the amount of the first coordination spher
on of SambVca allows the user to perform a more detailnd the Ru center, as well as the steric map of the NHC in th
ons quantifies any asymmetry in the way the ligand wraps ard from IPr to IPr* modifies the shape of the reactive pockeharacterize Ru-complexes relevant to olefin metathesis.[26
without symmetry constraints, and then for the ligands in commetallacycle intermediates.
teric maps vs geographic physical maps.
energy release, on intermediate in the presence
r the less bulky and no trend is
ic properties of . The points in
nalyzes the first ulate the buried re of the metal led analysis by he Ru-systems. round the metal et.[28] We have 6,28] The steric mplexes 1-3 as

850 Albert Poater et al. / Procedia Computer Science 18 ( 2013 ) 845 – 854
For an easy comprehension, these steric maps allow comparative analyses between different complexes. Indeed, steric maps could be considered as the classical geographic physical maps (see Figure 5), that depict the physical features like various landforms and water bodies present on the Earth’s surface. Different colors, lines, tints, shading and spot elevations are used to show the elevation and to differentiate lowlands from the mountains in physical maps. We used the same philosophy to build the steric maps of the complexes. The Ru center is at level zero, and the ligands are placed at the same plane or below the metal. In this framework, brown areas indicate zones where the ligand protrudes like a mountain towards the reacting groups, thus limiting the space at their disposal, whereas blue areas indicate empty zones where the ligand retracts like a lake from the reacting groups. The map of the free NHC ligand for IPr* reported in Figure 6 shows clearly that is much more sterically demanding than IPr, whereas for IPr*Tol this comparison is not so clear. The buried %VBur is 50.0% for IPr* and and 51.0% for IPr*Tol, whereas a value of only 42.2% is found for IPr. Bearing in mind that the topographic maps characterize the ligand surface offered to the substrate, it is clear that the modified IPr ligands have an intrinsic strong steric hindrance that after binding to the metal precludes coordination of big substrates.
(a) (b) (c)
Figure 6. Topographic steric maps of the free (a) IPr, (b) IPr* and (c) IPr*Tol NHC ligands. The isocontour curves of the steric maps are in Å. The xz plane is the mean plane of the NHC ring, whereas the yz plane is the plane orthogonal to the mean plane of the NHC ring, and passing through the carbene C atom of the NHC ring. The carbene C atom of the NHC ring is at the origin.
Focusing on the steric maps in Figure 6 in more detail,(1994) the IPr map shows that the two quadrants on the right are slightly more hindered, but this asymmetry is negligible compared to the asymmetry in the IPr* map, where the distribution of the steric bulk around the metal is remarkably different and the hindrance is much more localized into two quadrants (top right and bottom left quadrants %VBur~70%, top left and bottom right quadrants %VBur~29%). Thus, although the %VBur values are similar for IPr* and IPr*Tol the steric maps are key to further understand that the simple para substitution of the phenyl groups in IPr*Tol with respect to IPr* allows more reactive surface for a substrate around the metal with IPr*Tol. Figure 7 reports the steric maps for the three ligands in catalysts 1-3. The maps are very different from those of the free NHCs. In particular the IPr* ligand is able to rearrange in less sterically demanding conformations with the aromatic groups on the NHC somewhat bent to adopt a final position further away from the metal. The %VBur 29.1, 28.6 and 28.7 for complexes 1-3, respectively, thus with no significant differences. To further evaluate differences between IPr and IPr* we calculated the steric maps for the 14e- species, see again Figure 8. In these systems the modified IPr* and IPr*Tol ligands create higher steric constraints than IPr. In particular, they again adopt a quite asymmetric folding, creating a groove into which the olefin has to coordinate. Differently, the IPr ligand shapes a flat surface to host the incoming olefin. Overall, this analysis indicates that modified IPr ligands can be seen as rather flexible ligands that are able to exert steric pressure to push away the incoming substrate. At the same time, they are not too rigid, so that they can retract away to make space for other ligands. This analysis highlights the intrinsic dynamic behavior of the modified IPr ligands, indicating that the interaction between the complex and the substrate requires a conformation rearrangement of the complex, more consistent with an induce fit model rather than with a key and lock model.
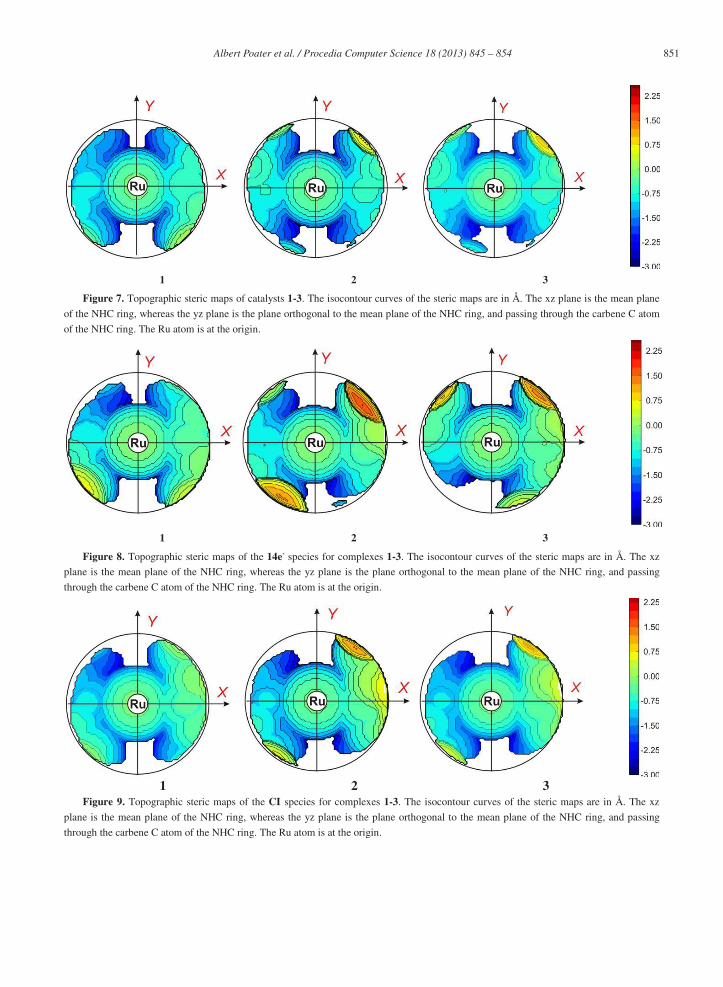
851 Albert Poater et al. / Procedia Computer Science 18 ( 2013 ) 845 – 854
1 2 3
Figure 7. Topographic steric maps of catalysts 1-3. The isocontour curves of the steric maps are in Å. The xz plane is the mean plane
of the NHC ring, whereas the yz plane is the plane orthogonal to the mean plane of the NHC ring, and passing through the carbene C atom
of the NHC ring. The Ru atom is at the origin.
1 2 3
Figure 8. Topographic steric maps of the 14e- species for complexes 1-3. The isocontour curves of the steric maps are in Å. The xz
plane is the mean plane of the NHC ring, whereas the yz plane is the plane orthogonal to the mean plane of the NHC ring, and passing
through the carbene C atom of the NHC ring. The Ru atom is at the origin.
1 2 3
Figure 9. Topographic steric maps of the CI species for complexes 1-3. The isocontour curves of the steric maps are in Å. The xz
plane is the mean plane of the NHC ring, whereas the yz plane is the plane orthogonal to the mean plane of the NHC ring, and passing
through the carbene C atom of the NHC ring. The Ru atom is at the origin.

852 Albert Poater et al. / Procedia Computer Science 18 ( 2013 ) 845 – 854
1 2 3
Figure 10. Topographic steric maps of the Metallacycle species for complexes 1-3. The isocontour curves of the steric maps are in Å.
The xz plane is the mean plane of the NHC ring, whereas the yz plane is the plane orthogonal to the mean plane of the NHC ring, and
passing through the carbene C atom of the NHC ring. The Ru atom is at the origin.
Experimentally, it was observed that complexes bearing the IPr* ligand are only more active than complexes bearing the IPr ligand when low hindered substrates are used, and that complex 1 was the most active of all.10 However, when bulkier substrates were used, the IPr bearing complexes showed always superior activity. This can be easily explained by analysis of the steric maps and of the energy profiles we calculated. Even though the energy barrier of the phosphine dissociation is similar and even higher for IPr*, from the steric maps the bulkier IPr* ligand favors dissociation of bulky labile ligands such as PPh3, and still can coordinate well small substrates, thus resulting in good catalytic performances. Consistently, coordination of bulky substrates is more difficult for the IPr* bearing complexes, thus making the reaction proceed significantly slower than with their IPr counterparts. In Figures 8-10 the steric maps of the 14e-, CI and Metallacycle species show that surprisingly the %VBur values for IPr*Tol are lower than for IPr*. For the 14e- species are 33.0, 35.3 and 34.4 for complexes 1-3, respectively; for the CI 30.9, 32.4, and 31.3, whereas for the Metallacycle intermediate 32.5, 33.5, and 32.9. Thus, the results of IPr*Tol look promising because they seem to collect the advantages of the higher steric pressure of IPr* with respect to IPr. The para substitution of the phenyl groups allows to increase the free surface around the metal because the modification renders the IPr*Tol less flexible than IPr* as can be observed in the steric maps of the free ligand species in Figure 7. However, bearing in mind previous studies,29 we must point out that the differences between IPr* with respect to IPr*Tol are subtle, and the a priori expected higher sterical hindrance of IPr*Tol is not translated into new relevant insights, suggesting that the modification of the NHC must be focused in the part nearer to the metal core.
Conclusions
Screening of new catalysts in silico is an area that could assist experimental tests in targeting preferred structural types. In this contribution, the catalytic performance of ruthenium IPr, IPr*, and IPr*Tol complexes were tested in such a manner for an olefin metathesis reaction. The calculations allowed the rationalization of the experimental findings and thus validate the computational mode, as well as to predict a new NHC ligand such as IPr*Tol. These results also highlight the potential of computational methods to assist in the development of novel catalyst types in silico thereby providing a significant resource-saving tool to rational catalyst design.

853 Albert Poater et al. / Procedia Computer Science 18 ( 2013 ) 845 – 854
Acknowledgements
The research leading to these results has received funding from the European Community's Seventh Framework Programme (FP7/2007-2013) under grant agreement n° CP-FP 211468-2 EUMET. A.P. and L.C. thank BSC (QCM-2010-2-0020), and the HPC team of Enea for using the ENEA-GRID and the HPC facilities CRESCO in Portici (Italy) for access to remarkable computational resources. A.P. thanks the Spanish MICINN for a Ramón y Cajal contract (RYC-2009-05226), European Commission for a Career Integration Grant (CIG09-GA-2011-293900), and Generalitat de Catalunya (2011BE100793). SPN is a Royal Society Wolfson Research Merit Award holder.
References
[1] a) J.-L. Herisson and Y. Chauvin, Makromol. Chem., 1970, 141, 161: b) A. Fürstner, Angew. Chem., Int. Ed., 2000, 39, 3012; c) R. R.
Schrock and A. H. Hoveyda, Angew. Chem., Int. Ed., 2003, 42, 4592; d) A. H. Hoveyda and R. R. Schrock, Chem.–Eur. J., 2001, 7, 945; e) T. M. Trnka and R. H. Grubbs, Acc. Chem. Res., 2000, 34, 18; f) R. H. Grubbs, Handbook of Olefin Metathesis, Wiley-VCH, Weinheim, Germany, 2003.
[2] R. Credendino, A. Poater, F. Ragone and L. Cavallo, Catal. Sci. Technol., 2011, 1, 1287. [3] a) H. Jacobsen, A. Correa, A. Poater, C. Costabile and L. Cavallo, Coord. Chem. Rev., 2009, 253, 687; b) H. Jacobsen, A. Correa, A.
Poater, C. Costabile and L. Cavallo, Coord. Chem. Rev., 2009, 25, 2584; c) A. S. K. Hashmi, A. M. Schuster, S. Gaillard, L. Cavallo, A. Poater and S. P. Nolan, Organometallics, 2011, 30, 6328; d) G. C. Fortman, A. Poater, J. W. Levell, S. Gaillard, A. M. Z. Slawin, I. D. W. Samuel, L. Cavallo and S. P. Nolan, Dalton Trans., 2010, 39, 10382; e) R. S. Ramón, Sylvain Gaillard, A. Poater, L. Cavallo, A. M. Z. Slawin and S. P. Nolan, Chem. Eur. J., 2011, 17, 1238; f) P. Nun, S. Dupuy, S. Gaillard, A. Poater, L. Cavallo and S. P. Nolan, Catal. Sci. Technol., 2011, 1, 58; g) P. Nun, S. Gaillard, A. Poater, L. Cavallo and S. P. Nolan, Org. Biomol. Chem., 2011, 9, 101; h) A. Poater, L. Falivene, C. A. Urbina-Blanco, S. Manzini, S. P. Nolan and L. Cavallo, Dalton Trans., 2013, DOI: 10.1039/c3dt32980a.
[4] Y. Chu, W. Heyndrickx, G. Occhipinti, B. K. Alsberg and V. R. Jensen, J. Am. Chem. Soc., 2012, 134, 8885. [5] a) T. Weskamp, W. C. Schattenmann, M. Spiegler and W. A. Herrmann, Angew. Chem. Int. Ed., 1998, 37, 2490; b) J. Huang, E. D.
Stevens, J. L. Petersen and S. P. Nolan, J. Am. Chem. Soc., 1999, 121, 2674; c) M. Scholl, S. Ding, C. W. Lee and R. H. Grubbs, Org. Lett., 1999, 1, 953.
[6] a) H. Clavier, C. A. Urbina-Blanco and S. P. Nolan, Organometallics, 2009, 28, 2848; b) J. Broggi, C. A. Urbina-Blanco, H. Clavier, A. Leitgeb, C. Slugovc, A. M. Z. Slawin and S. P. Nolan, Chem. Eur. J., 2010, 16, 9215; c) C. A. Urbina-Blanco, X. Bantreil, H. Clavier, A. M. Z. Slawin and S. P. Nolan, Beilstein J. Org. Chem., 2010, 6, 1120; d) C. A. Urbina-Blanco, A. Leitgeb, C. Slugovc, X. Bantreil, H. Clavier, A. M. Z. Slawin and S. P. Nolan, Chem. Eur. J., 2011, 17, 5045; e) C. A. Urbina-Blanco, S. Manzini, J. P. Gomes, A. Doppiu and S. P. Nolan, Chem. Commun., 2011, 47, 5022; f) J. Wappel, C. A. Urbina-Blanco, C. A.; M. Abbas, J. H. Albering, R. Saf, S. P. Nolan and C. Slugovc, Beilstein J. Org. Chem., 2010, 6, 1091; (g) X. Bantreil, A. Poater, C. A. Urbina-Blanco, Y. D. Bidal, L. Falivene, R. A. M. Randall, L. Cavallo, A. M. Z. Slawin and C. S. J. Cazin, Organometallics, 2012, 31, 7415; h) A. Leitgeb, M. Abbas, R. C. Fischer, A. Poater, L. Cavallo and C. Slugovc, Catal. Sci. Technol., 2012, 2, 1640; i) C. Samojłowicz, M. Bieniek, A. Pazio, A. Makal, K. Wozniak, A. Poater, L. Cavallo, J. Wójcik, K. Zdanowski and K. Grela, Chem. Eur. J., 2011, 17, 12981; j) R. S. Ramón, S. Gaillard, A. Poater, L. Cavallo, A. M. Z. Slawin and S. P. Nolan, Chem. Eur. J., 2010, 16, 14354; k) A. Poater, N. Bahri-Laleh and L. Cavallo, Chem. Comm., 2011, 47, 6674; l) A. Poater and L. Cavallo (2010) In: V. Dragutan, A. Demonceau, I. Dragutan, E. S. Finkelshtein (eds) NATO ASI series A. Springer, Dordrecht, p 275; m) A. Poater, R. Credendino, C. Slugovc, L. Cavallo, Dalton Trans., 2013, DOI: 10.1039/c3dt32884h.
[7] a) C. A. Urbina-Blanco, A. Leitgeb, C. Slugovc, X. Bantreil, H. Clavier, A. M. Z. Slawin and S. P. Nolan, Chem. Eur. J., 2011, 17, 5044; b) S. Manzini, C. A. Urbina-Blanco, A. Poater, A. M. Z. Slawin, L. Cavallo and S. P. Nolan, Angew. Chem., Int. Ed., 2012, 51, 1042; c) A. Poater and L. Cavallo, Theor. Chem. Acc., 2012, 131, 1155; d) A. Poater, F. Ragone, M. Garrido, S. Pérez, M. Poch, A. Correa, L. Cavallo, Proc. Comp. Sci., 2011, 4, 1222; e) L. Falivene, A. Poater, C. S. J. Cazin, C. Slugovc and L. Cavallo, Dalton Trans., 2013, DOI: 10.1039/c2dt32277c.
[8] a) C. Samojowicz, M. Bieniek and K. Grela, Chem. Rev. 2009, 109, 370; b) G. C. Vougioukalakis and R. H. Grubbs, Chem. Rev. 2010, 110, 1746.
[9] A. Poater, F. Ragone, A. Correa and L. Cavallo, Dalton Trans., 2011, 40, 11066. [10] S. Manzini, C. A. Urbina-Blanco, A. M. Z. Slawin and S. P. Nolan, Organometallics, 2012, 31, 6514. [11] Gaussian 09, Revision A.1, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V.
Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L.

854 Albert Poater et al. / Procedia Computer Science 18 ( 2013 ) 845 – 854
Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox, Gaussian, Inc., Wallingford CT, 2009.
[12] a) A. D. Becke, Phys. Rev. A, 1988, 38, 3098; b) J. P. Perdew, Phys. Rev. B, 1986, 33, 8822; c) J. P. Perdew, Phys. Rev. B, 1986, 34, 7406.
[13] A. Schafer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571. [14] Y. Zhao and D. Truhlar, Theor. Chem. Acc., 2008, 120, 215. [15] a) V. Barone and M. Cossi, J. Chem. Phys. A, 1998, 102, 1995; b) J. Tomasi and M. Persico, Chem. Rev., 1994, 94, 2027. [16] a) A. Poater and L. Cavallo, J. Mol. Catal. A, 2010, 324, 75; b) A. Poater, J. Phys. Chem. A, 2009, 113, 9030; c) A. Poater, F. Ragone,
A. Correa and L. Cavallo, J. Am. Chem. Soc., 2009, 131, 9000; d) A. Poater, A. G. Saliner, L. Cavallo, M. Poch, M. Solà and A.P. Worth, Curr. Med. Chem., 2012, 19, 5219; e) A. Casitas, A. Poater, M. Solà, S. S. Stahl, M. Costas and X. Ribas, Dalton Trans., 2010, 39, 10458; f) A. Poater, S. Moradell, E. Pinilla, J. Poater, M. Solà, M. A. Martínez and A. Llobet, Dalton Trans., 2006, 1188.
[17] I. Mayer, I. Chem. Phys. Lett., 1983, 97, 270; b) I. Mayer, Int. J. Quantum Chem., 1984, 26, 151. [18] R. G. Parr, L. von Szentpaly and S. Liu, J. Am. Chem. Soc., 1999, 121, 1922. [19] P. Geerlings, F. De Proft and W. Langenaeker, Chem. Rev., 2003, 103, 1793. [20] a) R. G. Parr and W. Yang, Density Functional Theory of Atoms and Molecules; Oxford University Press: New York, 1989; b) R. G.
Parr, R. A. Donnelly, M. Levy and W. E. Palke, J. Chem. Phys., 1978, 68, 3801; c) R. G. Parr and R. G. Pearson, J. Am. Chem. Soc., 1983, 105, 7512.
[21] T. Koopmans, Physica, 1934, 1, 104. [22] a) A. Poater, M. Duran, P. Jaque, A. Toro-Labbé and M. Solà, J. Phys. Chem. B, 2006, 110, 6526-6536; b) P. W. Ayers and R. G. Parr.
J. Am. Chem. Soc. 2000, 122, 2010; (c) A. Poater, F. Ragone, A. Correa and L. Cavallo, J. Am. Chem. Soc., 2009, 131, 9000; d) M. Costas, X. Ribas, A. Poater, J. M. L. Valbuena, R. Xifra, A. Company, M. Duran, M. Solà, A. Llobet, M. Corbella, M. A. Uson, MJ. Mahia, X. Solans, X. P. Shan, J. Benet-Buchholz, Inorg. Chem., 2006, 45, 3569; e) A. Poater, A. Gallegos Saliner, R. Carbó, J. Poater, M. Solà, L. Cavallo and A. P. Worth, J. Comput Chem., 2009, 30, 275; f) A. Poater, A. Gallegos Saliner, M. Solà, L. Cavallo, A. P. Worth, Expert Opin. Drug Deliv., 2010, 7, 295; A. Poater and L. Cavallo, Theor. Chem. Acc. 2013, DOI 10.1007/s00214-013-1336-x.
[23] Standard deviations for distances and angles: sn-1 = [ i=1 N (CV - EV)2/(N - 1)]1/2, where CV means calculated value, EV means experimental value (X-ray data), and N is the number of distances or angles taken into account.
[24] a) X. Sala, E. Plantalech, I. Romero, M. Rodríguez, A. Llobet, A. Poater, M. Duran, M. Solà, S. Jansat, M. Gómez, T. Parella, H. Stoeckli-Evans and J. Benet-Buchholz, Chem. Eur. J., 2006, 12, 2798; b) J. Mola, M. Rodríguez, I. Romero, A. Llobet, T. Parella, A. Poater, M. Duran, M. Solà and J. Benet-Buchholz, Inorg. Chem., 2006, 45, 10520; c) A. Poater and L. Cavallo, Inorg. Chem., 2009, 48, 4062; d) A. Poater, X. Ribas, A. Llobet, L. Cavallo and M. Solà, J. Am. Chem. Soc., 2008, 130, 17710; e) I. Serrano, M. I. López, I. Ferrer, A. Poater, T. Parella, X. Fontrodona, M. Solà, A. Llobet, M. Rodríguez and I. Romero, Inorg. Chem., 2011, 50, 6044; f) J. Duran, A. Polo, J. Real, J. Benet-Buchholz, J. Poater and M. Solà, Eur. J. Inorg. Chem. 2003, 414, 4147; g) J. Rich, M. Rodríguez, I. Romero, X. Fontrodona, P. W. N. M. van Leeuwen, Z. Freixa, X. Sala, A. Poater and M. Solà, Eur. J. Inorg. Chem., 2013, DOI:10.1002/ejic.201201154; h) J. Mola, I. Romero, M. Rodríguez, F. Bozoglian, A. Poater, M. Solà, T. Parella, J. Benet-Buchholz, X. Fontrodona and A. Llobet, Inorg. Chem., 2007, 46, 10707.
[25] O. M. Aagaard, R. J. Meier and F. Buda, J. Am. Chem. Soc., 1998, 120, 7174. [26] A. Poater, B. Cosenza, A. Correa, S. Giudice, F. Ragone, V. Scarano and L. Cavallo, Eur. J. Inorg. Chem., 2009, 1759. [27] a) L. Cavallo, A. Correa, C. Costabile and H. Jacobsen, J. Organomet. Chem., 2005, 690, 5407; b) A. Poater, F. Ragone, S. Giudice,
C. Costabile, R. Dorta, S. P. Nolan and L. Cavallo, Organometallics, 2008, 27, 2679; c) A. Poater and L. Cavallo, Dalton Trans., 2009, 41, 8878; d) X. Luan, R. Mariz, M. Gatti, C. Costabile, A. Poater, L. Cavallo, A. Linden and R. Dorta, J. Am. Chem. Soc., 2008, 130, 6848; e) J. Bosson, A. Poater, L. Cavallo and S. P. Nolan, J. Am. Chem. Soc., 2010, 132, 13146.
[28] a) F. Ragone, A. Poater and L. Cavallo, J. Am. Chem. Soc., 2010, 132, 4249; b) A. Poater, F. Ragone, R. Mariz, R. Dorta and L. Cavallo, Chem. Eur. J., 2010, 16, 14348.
[29] P. Liu, J. Montgomery and K. N. Houk, J. Am. Chem. Soc., 2011, 133, 6956.