The Crystal Structure of Mercury(II) Chromate - Standard Input
Upload
independentCategory
view
0download
0
Stability of surface chromate ± A physicochemical investigationin relevance to environmental reservations about
calcined chromia catalysts
Mohamed I. Zakia,*, Muhammad A. Hasana, Nasr E. Fouadb
aChemistry Department, Faculty of Science, Kuwait University, PO Box 5969, Safat 13060, KuwaitbChemistry Department, Faculty of Science, Minia University, El-Minia 61519, Egypt
Abstract
The present investigation presents and correlates observed and reported characterization results of calcined, supported and
unsupported chromias obtained via various precursor compounds. Studies performed employed a range of surface and bulk
analytical techniques, in hopes of a proper assessment of the stability of surface chromate (Cr(VI)±O) species thus generated
to thermal decomposition, hydrolysis and reduction. Impacts on the redox catalytic activity were probed towards the
decomposition of H2O2 solutions and the CO oxidation in the gas phase. The results have revealed that chromate species
established on bulk and dispersed a-Cr2O3 particles enjoy high stability against thermal and chemical treatments, and yet
contribute to the formation of surface sites that are catalytically active in redox reactions. This should help lessening
environmental reservations about the industrial application of calcined chromia catalysts. # 1998 Elsevier Science B.V. All
rights reserved.
Keywords: Chromia; Calcined chromia catalysts; Surface chromate; Environmental reservations
1. Introduction
The title term `̀ surface chromate'' refers to Cr(VI)±
O species generated on oxidized surfaces of chromia
(Cr2O3) based catalysts, prepared by heating in air
(calcination) of adequate parent materials [1]. The
catalysts thus obtained are potential in redox reactions
[2] and promising in deep oxidation (combustion)
processes [3]. The activity and selectivity of the
catalysts depend critically on whether the calcined
chromia is supported or unsupported, and promoted or
unpromoted [4].
Calcination of chromia based catalysts leads not
only to Cr(IV) surface species, but also to Cr(IV) and
Cr(V) species [1,2,5±7]. However, the hexavalent
(chromate) species dominate at �6008C [8,9], assum-
ing monomeric �CrO2ÿ4 � [10,11] and/or polymeric
�Cr2�xO7�3x�2ÿ [12,13] surface structures. Under cer-
tain circumstances, they may form interaction species
with nearby Cr(III) species (e.g., chromium chromate
like [14]) and metal oxide support surfaces (e.g.,
aluminum chromate-like on alumina [10]). Conse-
quently formed Cr(VI)±Cr(III) pair-sites have been
considered to promote the redox catalytic activity of
chromia in several reaction incidents [14±16]. Accord-
ingly, a catalytic mechanism has been preferred [14],
whereby localized adsorption occurs on coordina-
Applied Catalysis A: General 171 (1998) 315±324
*Corresponding author. Tel.: (965) 4811188/5606; fax: (965)
4816482; e-mail: [email protected]
0926-860X/98/$19.00 # 1998 Elsevier Science B.V. All rights reserved.
P I I S 0 9 2 6 - 8 6 0 X ( 9 8 ) 0 0 0 8 8 - X

tively unsaturated Cr(III) sites [17], but electron avail-
ability occurs through interaction with nearby Cr(VI)
ions.
Despite numerous laboratory applications, the indus-
trial application of calcined chromia catalysts has been
hampered by the instability of surface chromate to
hydrolysis in aqueous media [15], reduction in reduc-
tive gas atmospheres [18], and high temperature treat-
ments in air [19].VolatileCr(VI)-compoundsare known
to be environmentally detrimental [19]. Some earlier
reports [5,20] claim, however, that the instability is the
concern of only a portion of surface chromate, and that
thermally and chemically stable chromate species
coexist on surfaces of calcined chromia. Based on
these reports, and without suf®cient experimental
control, irreducibility of calcined chromia in H2-TPR
experiments has been explained [21].
The present study explores observed and reported
characterization results of thermal and chemical sta-
bility of oxidized surfaces of supported and unsup-
ported chromias prepared by calcination in air.
According to previous studies [11,22,23], the chosen
parent materials yield calcination products exposing
variously structured surface chromate species estab-
lished on different substrates (a-Cr2O3, alumina and
silica particles). The thermal stability was probed by
thermogravimetry (TG), whereas the chemical stabi-
lity was tested versus hydrolysis and temperature-
programmed reduction in H2 atmosphere (H2-TPR).
The study correlates ®ndings with the catalytic per-
formance of test chromias in H2O2 decomposition
(liquid phase) and CO oxidation (gas phase) reactions.
Eventually, the study goals an objective assessment of
the environmental apprehensions of calcined chromia
catalysts.
2. Experimental
2.1. Calcined chromias
Supported and unsupported chromias examined in
the present study were obtained by calcination at 300±
6008C for 5 h (in a static atmosphere of air) of the
following parent materials:
� Unsupported chromias (denoted by Cr) were pro-
ducts of (I) chromia gel (Cr2O3�5H2O) prepared by
NH4OH alkalization of an aqueous solution of
AR-grade Merck Cr(NO3)3�9H2O [11], (II)
99.9% pure Riedel-de-Haen CrO3 [22], and (III)
AR-grade Merck (NH4)2Cr2O7 [23]. The resulting
chromias are discerned below by a designatory
combination of Cr, the indicative Roman numeral
(I)±(III) of the parent, and the calcination tem-
perature applied. Thus, CrI-600 means unsup-
ported chromia obtained by 6008C calcination
of the chromia gel.
� Alumina-supported chromias (CrAl) were calci-
nation products of Degussa aluminum oxide C
(100 m2 gÿ1) coated with chromia gel (I) [11]
and wet-impregnated with CrO3 solution (II)
[22] at 10 mol% Cr2O3. They are indicated below
adopting the designatory system of the corre-
sponding unsupported chromias. Thus, CrAlII-
300 indicates the 3008C calcination product of
alumina impregnated with the CrO3 solution.
� Silica-supported chromias (CrSi) were calcination
products of Degussa aerosil-200 (200 m2 gÿ1)
coated with chromia gel (I) [11] and wet-impreg-
nated with CrO3 solution (II) [22] at 10 mol%
Cr2O3. For identification, the above designatory
system has been applied. Thus, CrSiI-600 signifies
the 6008C calcination product of silica coated with
the chromia gel.
2.2. Characterization methods and techniques
� X-ray powder diffractometry (XRD) was carried
out to elucidate the crystalline bulk structure of
test materials [24], employing a model D5000
Siemens diffractometer (35 kV and 30 mA)
equipped with Ni-filtered CuK� radiation.
� UV±visdiffuse reflectancespectroscopy(DRS)was
applied to identify surface chromate structures and
electronic interactions [11], using a Cary-5 Varian
spectrophotometer equipped with MgO-coated
integrating sphere and a data acquisition system.
� Thermogravimetry (TG) was performed to moni-
tor the thermal stability of surface chromate in a
stream of air (50 cm3 minÿ1) [22], by means of a
TGA-50 Shimadzu analyzer.
� Temperature-programmed reduction (TPR) was
the technique used to probe the chemical stability
of surface chromate in a stream of 5% H2/He at
30 cm3 minÿ1 [21], implementing a common flow
system equipped with a quartz microreactor and a
316 M.I. Zaki et al. / Applied Catalysis A: General 171 (1998) 315±324
Pd diffusion cell connected to a model 100 Carle
thermal conductivity detector.
� Colorimetry was the analytical method adopted to
estimate water-leachable surface chromate [6],
employing a Spekol±Carl±Seiz colorimeter.
� Iodometry was the analytical method applied to
titrate total surface chromate [7], using KI/0.12 N
HCl as a reagent and a solution of thiosulfate as a
titrant.
2.3. Catalytic activity tests
� In solution the catalytic activity was tested towards
H2O2(!H2O�12O2) decomposition at 20±408C
and zero-order kinetics; the reaction rate was
measured volumetrically [6].
� In the gas phase, CO(�12O2!CO2) oxidation in
oxygen-rich stream of He was the test catalytic
reaction, and the rate was followed using a com-
mon flow system equipped with plug-flow micro-
reactor, and a model 3400 Varian chromatograph
equipped with a TC-detector, propack-N column,
and a model 4290 Varian electric integrator [25].
3. Results and discussion
3.1. Crystalline bulk structure
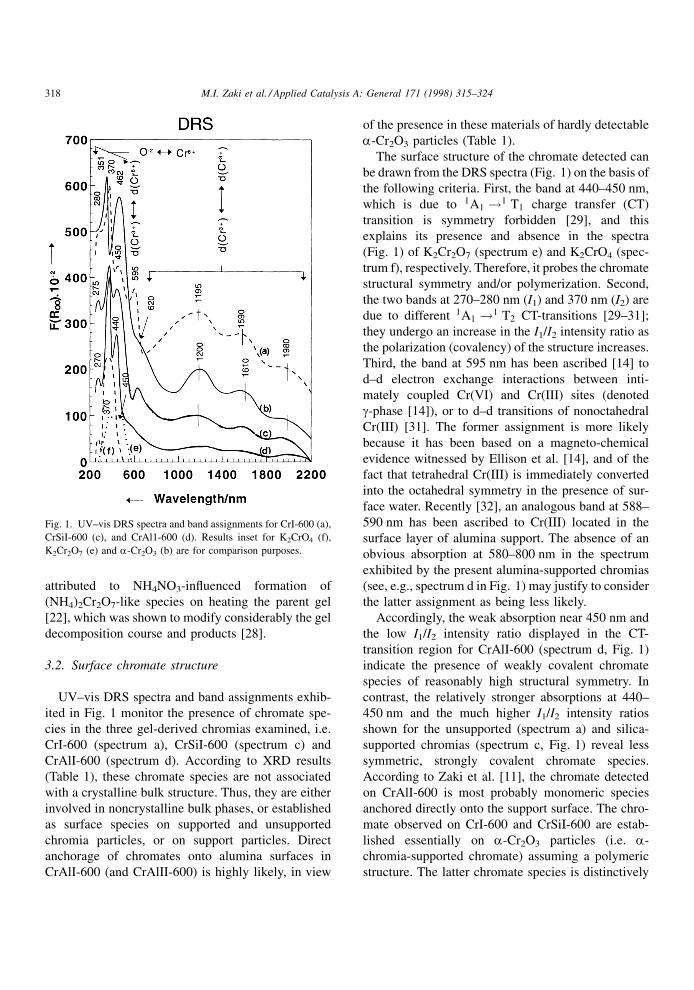
XRD-probed characteristics of crystalline bulk
structure for test chromias are compared in Table 1.
Accordingly, the sole detectable CrOx crystalline
phase is a-Cr2O3. It assumes relatively strongest
crystallinity and largest particle size in CrII-600.
These properties are shown to have been suppressed
in low calcination temperature chromias, as well as
upon dispersion on support surfaces. Despite the
higher surface area of silica (200 m2 gÿ1), alumina
surfaces (100 m2 gÿ1) appear to be more capable of
dispersing a-Cr2O3 particles (Table 1). Consequently,
a-Cr2O3 is hardly detectable in CrAl-600 samples,
irrespective of the chromia precursor. Similar results,
obtained previously [22], have been interpreted on the
basis of stronger CrOx/support interactions in CrAl,
both during the precursor loading onto the support and
the subsequent calcination. It is to be noted that in all
supported chromias examined no CrOx/support inter-
action products were XRD-detectable. However, this
cannot exclude the likelihood of formation of surface
and/or noncrystalline bulk compounds, both being
XRD-undetectable. Compatibly, supported Cr(III)±O
species have been previously found [14,27] to exist in
various noncrystalline phases, namely d- (isolated
Cr(III) species), b- (Cr(III)-clusters), g- (Cr(III)
coupled with Cr(VI)) and a-phase (CrO1.5 species),
depending on the chromia content and calcination
temperature.
With respect to chromias derived from chromia gel
(NOÿ3 -contaminated), the a-Cr2O3 particle crystalli-
nity and size (Table 1) are signi®cantly higher than
those reported previously for corresponding chromias
obtained using a NOÿ3 -free gel [22]. This might be
Table 1
XRD-probed characteristics of crystalline bulk structure for unsupported and supported chromias
Chromia Chromia precursor Phase composition Cry.b (%) Dc (�2 nm)
CrI-400 Gela a-Cr2O3 55 28
CrI-600 a-Cr2O3 70 35
CrII-600 CrO3 a-Cr2O3 88 53
CrIII-400 (NH4)2Cr2O7 a-Cr2O3 63 34
CrIII-600 a-Cr2O3 75 42
CrAlI-600 Gela a-Cr2O3 12 10
g-Al2O3
CrAlII-600 CrO3 a-Cr2O3 12 15
g-Al2O3
CrSiI-600 Gela a-Cr2O3 58 30
CrSiII-600 CrO3 a-Cr2O3 73 44
aNH4NO3-contaminated.bCry.�crystallinity approximated relative to a well-crystalline Merck a-Cr2O3.cD�average particle size determined adopting XRD line broadening technique [26].
M.I. Zaki et al. / Applied Catalysis A: General 171 (1998) 315±324 317
attributed to NH4NO3-in¯uenced formation of
(NH4)2Cr2O7-like species on heating the parent gel
[22], which was shown to modify considerably the gel
decomposition course and products [28].
3.2. Surface chromate structure
UV±vis DRS spectra and band assignments exhib-
ited in Fig. 1 monitor the presence of chromate spe-
cies in the three gel-derived chromias examined, i.e.
CrI-600 (spectrum a), CrSiI-600 (spectrum c) and
CrAlI-600 (spectrum d). According to XRD results
(Table 1), these chromate species are not associated
with a crystalline bulk structure. Thus, they are either
involved in noncrystalline bulk phases, or established
as surface species on supported and unsupported
chromia particles, or on support particles. Direct
anchorage of chromates onto alumina surfaces in
CrAlI-600 (and CrAlII-600) is highly likely, in view
of the presence in these materials of hardly detectable
a-Cr2O3 particles (Table 1).
The surface structure of the chromate detected can
be drawn from the DRS spectra (Fig. 1) on the basis of
the following criteria. First, the band at 440±450 nm,
which is due to 1A1 !1 T1 charge transfer (CT)
transition is symmetry forbidden [29], and this
explains its presence and absence in the spectra
(Fig. 1) of K2Cr2O7 (spectrum e) and K2CrO4 (spec-
trum f), respectively. Therefore, it probes the chromate
structural symmetry and/or polymerization. Second,
the two bands at 270±280 nm (I1) and 370 nm (I2) are
due to different 1A1 !1 T2 CT-transitions [29±31];
they undergo an increase in the I1/I2 intensity ratio as
the polarization (covalency) of the structure increases.
Third, the band at 595 nm has been ascribed [14] to
d±d electron exchange interactions between inti-
mately coupled Cr(VI) and Cr(III) sites (denoted
g-phase [14]), or to d±d transitions of nonoctahedral
Cr(III) [31]. The former assignment is more likely
because it has been based on a magneto-chemical
evidence witnessed by Ellison et al. [14], and of the
fact that tetrahedral Cr(III) is immediately converted
into the octahedral symmetry in the presence of sur-
face water. Recently [32], an analogous band at 588±
590 nm has been ascribed to Cr(III) located in the
surface layer of alumina support. The absence of an
obvious absorption at 580±800 nm in the spectrum
exhibited by the present alumina-supported chromias
(see, e.g., spectrum d in Fig. 1) may justify to consider
the latter assignment as being less likely.
Accordingly, the weak absorption near 450 nm and
the low I1/I2 intensity ratio displayed in the CT-
transition region for CrAlI-600 (spectrum d, Fig. 1)
indicate the presence of weakly covalent chromate
species of reasonably high structural symmetry. In
contrast, the relatively stronger absorptions at 440±
450 nm and the much higher I1/I2 intensity ratios
shown for the unsupported (spectrum a) and silica-
supported chromias (spectrum c, Fig. 1) reveal less
symmetric, strongly covalent chromate species.
According to Zaki et al. [11], the chromate detected
on CrAlI-600 is most probably monomeric species
anchored directly onto the support surface. The chro-
mate observed on CrI-600 and CrSiI-600 are estab-
lished essentially on a-Cr2O3 particles (i.e. a-
chromia-supported chromate) assuming a polymeric
structure. The latter chromate species is distinctively
Fig. 1. UV±vis DRS spectra and band assignments for CrI-600 (a),
CrSiI-600 (c), and CrAl1-600 (d). Results inset for K2CrO4 (f),
K2Cr2O7 (e) and a-Cr2O3 (b) are for comparison purposes.
318 M.I. Zaki et al. / Applied Catalysis A: General 171 (1998) 315±324

pertained by the emergence of the 595 nm band
(spectra a and c, Fig. 1) assignable to the g-(Cr(VI)±
Cr(III)) phase. The absence of a well-resolved band
near 595 nm in the spectrum (d, Fig. 1) of CrAlI-600
sustains that the material contains predominantly
chromate anchored directly onto the alumina support.
Nevertheless, the presence in CrAlI-600 of chromate
species established on noncrystalline chromia cannot
be excluded with certainty.
3.3. Stability of surface chromate
3.3.1. Thermal stability
Fig. 2 compares TG curves for CrO3 and two
chromia gel samples, a NOÿ3 -free and a NOÿ3 -con-
taminated sample. The nitrate inclusion is inherited
from the preparation source conceded by the NOÿ3 -
containing gel [11]. The trioxide, CrO3, was chosen to
facilitate probing the thermal stability of initial chro-
mate, whereas the gel samples are to allow for obser-
vation of the thermal stability of topochemically
generated chromate via oxidation of initial Cr(III)
[24]. It is obvious from Fig. 2, that CrO3 is stable
to heating to 2508C, where it commences decomposi-
tion into meta-stable mixed-valency bulk phases at
250±4508C [23]. These meta-stable intermediates
contain Cr(VI) species, and are XRD-detectable
[22,23]. They decompose into a-Cr2O3 (Cr(III)) near
5008C [23].
TG curve of the NOÿ3 -free gel sample (Fig. 2)
indicates, according to Zaki and Fouad [28], that
heating in air (calcination) leads to overlapping dehy-
dration processes ending up with a monohydrate
(Cr2O3�H2O�2CrOOH) near 2508C. The monohy-
drate is topochemically oxidized into the g-phase
(Cr2O3�CrO3) via a complex series of disproportiona-
tion reactions [28] at 250±3808C. The g-phase thus
produced decomposes into a-Cr2O3 near 4008C(Fig. 2). None of the intermediate products was
XRD-detectable [23].
The inclusion of NH4NO3 is shown (Fig. 2) to
modify considerably the decomposition course of
the gel. The initial decomposition curve is notably
steepened (kinetically enhanced) and completed near
2008C (instead of 3008C for the NOÿ3 -free gel). On the
other hand, the product is suggested to consist essen-
tially of a-Cr2O3 with minority g-phase (Cr2O3�x).
Thus formation of the a-phase is markedly enhanced
to occur near 2008C from the contaminated gel,
instead of 4008C from the pure gel, and 5008C from
CrO3 (Fig. 2). A previous study [28] disclosed the
formation of a (NH4)2Cr2O7-like phase in the NOÿ3 -
contaminated gel, which was considered responsible
for the new decomposition course and products. Park
[33] examining gas and solid phase decomposition
products of pure (NH4)2Cr2O7 has found NH3 being
released to catalyze a reductive conversion of Cr(VI)
species into a-Cr2O3 near 2108C.
The above results indicate that Cr(VI)-containing
bulk phases are produced during the decomposition
course of the chromia precursors examined (Fig. 2).
These bulk phases are destabilized thermally at
�4508C, giving rise to a-Cr2O3 as the eventual
decomposition product. A thermo-analytic study of
supported chromias [22] has brought about similar
results. Accordingly, DRS-detected chromate species
Fig. 2. TG curves obtained for CrO3, and NH4NO4-free and ±
contaminated chromia gel samples under the conditions indicated.
M.I. Zaki et al. / Applied Catalysis A: General 171 (1998) 315±324 319
in the 6008C calcined chromias (Fig. 1) are domi-
nantly surface species. Characterization studies
employing surface-sensitive techniques, e.g. XPS
[8,9], indicated persistence of the chromate surface
species on calcined chromia to heating to temperatures
well above 6008C.
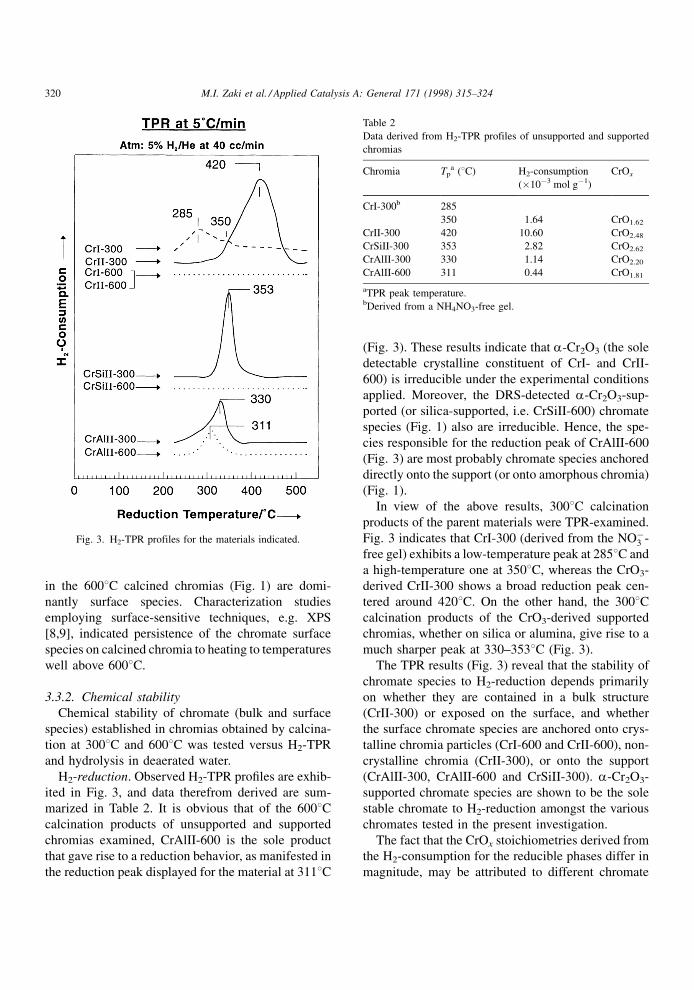
3.3.2. Chemical stability
Chemical stability of chromate (bulk and surface
species) established in chromias obtained by calcina-
tion at 3008C and 6008C was tested versus H2-TPR
and hydrolysis in deaerated water.
H2-reduction. Observed H2-TPR pro®les are exhib-
ited in Fig. 3, and data therefrom derived are sum-
marized in Table 2. It is obvious that of the 6008Ccalcination products of unsupported and supported
chromias examined, CrAlII-600 is the sole product
that gave rise to a reduction behavior, as manifested in
the reduction peak displayed for the material at 3118C
(Fig. 3). These results indicate that a-Cr2O3 (the sole
detectable crystalline constituent of CrI- and CrII-
600) is irreducible under the experimental conditions
applied. Moreover, the DRS-detected a-Cr2O3-sup-
ported (or silica-supported, i.e. CrSiII-600) chromate
species (Fig. 1) also are irreducible. Hence, the spe-
cies responsible for the reduction peak of CrAlII-600
(Fig. 3) are most probably chromate species anchored
directly onto the support (or onto amorphous chromia)
(Fig. 1).
In view of the above results, 3008C calcination
products of the parent materials were TPR-examined.
Fig. 3 indicates that CrI-300 (derived from the NOÿ3 -
free gel) exhibits a low-temperature peak at 2858C and
a high-temperature one at 3508C, whereas the CrO3-
derived CrII-300 shows a broad reduction peak cen-
tered around 4208C. On the other hand, the 3008Ccalcination products of the CrO3-derived supported
chromias, whether on silica or alumina, give rise to a
much sharper peak at 330±3538C (Fig. 3).
The TPR results (Fig. 3) reveal that the stability of
chromate species to H2-reduction depends primarily
on whether they are contained in a bulk structure
(CrII-300) or exposed on the surface, and whether
the surface chromate species are anchored onto crys-
talline chromia particles (CrI-600 and CrII-600), non-
crystalline chromia (CrII-300), or onto the support
(CrAlII-300, CrAlII-600 and CrSiII-300). a-Cr2O3-
supported chromate species are shown to be the sole
stable chromate to H2-reduction amongst the various
chromates tested in the present investigation.
The fact that the CrOx stoichiometries derived from
the H2-consumption for the reducible phases differ in
magnitude, may be attributed to different chromate
Fig. 3. H2-TPR profiles for the materials indicated.
Table 2
Data derived from H2-TPR profiles of unsupported and supported
chromias
Chromia Tpa (8C) H2-consumption
(�10ÿ3 mol gÿ1)
CrOx
CrI-300b 285
350 1.64 CrO1.62
CrII-300 420 10.60 CrO2.48
CrSiII-300 353 2.82 CrO2.62
CrAlII-300 330 1.14 CrO2.20
CrAlII-600 311 0.44 CrO1.81
aTPR peak temperature.bDerived from a NH4NO3-free gel.
320 M.I. Zaki et al. / Applied Catalysis A: General 171 (1998) 315±324

compositions and structures. It is likely that O/Cr
ratios �2.2 (i.e., CrO2.2±CrO2.6) originate from sur-
face and bulk chromates, whereas O/Cr ratios �1.8
(i.e., CrO1.62±CrO1.8) are due mostly to surface chro-
mate species. This likelihood ®nds a strong support by
the fact that highly stable chromate species are exhib-
ited largely by CrO3-derived chromias, a precursor
that produces bulk chromates on heating at 250±4508C(Fig. 2).
Hydrolysis. Hydrolysis of surface chromate was
tested against 3 h shaking of known amounts of var-
ious chromias in distilled water. Leached chromate
species were determined in solution by colorimetry
[6]. To check on the presence of unleachable chromate
species, surface chromates remaining on the solid
residue were determined by shaking with KI/0.12 N
HCl solution, and a subsequent titration of liberated
iodine by a standard thiosulfate solution [7]. Quanti-
tative estimates based on the results are summarized in
Table 3. It is worth noting that of the 6008C calcined
supported and unsupported chromias, CrAlII-600 is
the sole product that showed positive test results. Thus
chromate species established on the other 6008Ccalcined chromias were stable to hydrolysis. Accord-
ingly, one may conclude that chromate species stabi-
lized against H2-reduction (Fig. 3 and Table 2), are
equally stable to hydrolysis and KI-reduction.
The hydrolysis results (Table 3) are in line with the
TPR results, and, therefore, support their conclusions.
Of the 3008C calcined chromias, the supported chro-
mias assume higher total contents of surface chromate
than the unsupported chromias. The highest content of
hydrolyzable chromate is shown for CrSiII-300,
whereas the lowest for CrII-300 (Table 3). The pro-
portion of hydrolyzable to total surface chromate
contents (Table 3) implies that alumina surfaces are
more capable of stabilizing surface chromate than
silica surfaces, and the stability is improved at the
higher calcination temperature of 6008C. Similar
results have very recently been reported by JoÂzÂwrak
and Dalla Lana [34].
3.4. Redox catalytic activity
3.4.1. H2O2 decomposition activity
Chromias tested as H2O2 decomposition catalysts in
the present study were chosen from the unsupported
set of chromias to avoid likely complications due to
hydration and amphoteric interactions of support
materials. The elected materials, which include stable
and unstable chromate species, were catalytically
tested before and after leaching by water. The leached
chromate species also were tested as a homogeneous
catalyst for H2O2 decomposition. Plots of volume of
oxygen liberated (VO2) versus time (t) determined at
408C for solid catalysts only, are displayed in Fig. 4.
The data in Fig. 4 help to arrange the water-
unleached catalysts in the following descending order
of the H2O2 decomposition activity:
CrI-500 > CrI-400 > CrIII-400 > CrIII-500:
These catalysts can be arranged in the following
descending order of total content of surface chromate
[7]:
CrI-400 > CrI-500 > CrIII-400 > CrIII-500:
Except for CrI-400, the two orders meet, thus
indicating that the higher the surface total content
of chromate species the higher the catalyst activity
towards H2O2 decomposition.
Water leaching of surface chromate species from
CrI-400 and CrI-500 resulted in a marked improve-
ment of the activity of the former catalyst and in a
slight suppression of the activity of the latter catalyst.
This implies that the relationship between the catalytic
activity and the surface content of chromate species
passes through a maximum, which seems to be located
at CrI-500. Thus, CrI-500 represents a catalyst case for
which the loss on surface chromate content is com-
pensated for by improved energetics of active sites
(Cr(VI)±Cr(III)) [35].
A similar upward swing to that occurring from the
initial linearity of the VO2ÿt plot of CrI-400 (Fig. 4)
Table 3
Quantitative estimates for surface chromate generated on unsup-
ported and supported chromias
Chromia CrO4(w)a
(�10ÿ4 g gÿ1)
CrO4(t)b
(�10ÿ4 g gÿ1)
CrO4(w)/CrO4(t)
(%)
CrI-300 27 84 32
CrII-300 17 70 24
CrSiII-300 147 190 77
CrAlII-300 65 149 44
CrAlII-600 23 119 19
aWater-leached surface chromate detected colorimetrically [6].bTotal surface chromate detected iodometrically [7].
M.I. Zaki et al. / Applied Catalysis A: General 171 (1998) 315±324 321
has been considered [15] to imply an in situ generation
of a homogeneous catalyst (soluble chromate) for
H2O2 decomposition. Indeed, chromate species lea-
ched out of CrI-400 and CrI-500 were used in a
separate, but similar, set of experiments to catalyze
H2O2 decomposition. They were found to catalyze the
decomposition, following an induction period of time
(a few minutes) in which a wine red colored inter-
mediate was developed [36]. The VO2ÿt plots (not
shown) were linear, and the slope (rate constant) was
directly proportional to the amount of chromate in
solution.
The above results underline the importance of sur-
face chromate to the H2O2 decomposition activity of
calcined chromia. They also indicate that unstable
surface chromate species are not important to the
formation of the catalyst active sites (cf. CrI-400),
i.e. electron-mobile Cr(VI)±Cr(III) ion-pairs moni-
tored by the DRS-band at 595 nm (Fig. 1).
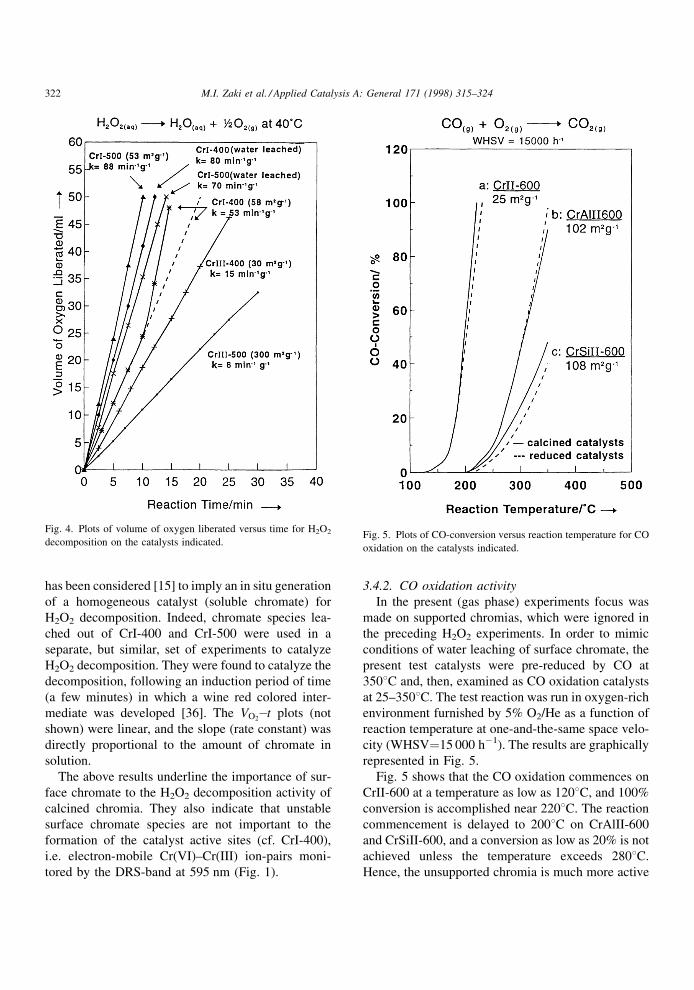
3.4.2. CO oxidation activity
In the present (gas phase) experiments focus was
made on supported chromias, which were ignored in
the preceding H2O2 experiments. In order to mimic
conditions of water leaching of surface chromate, the
present test catalysts were pre-reduced by CO at
3508C and, then, examined as CO oxidation catalysts
at 25±3508C. The test reaction was run in oxygen-rich
environment furnished by 5% O2/He as a function of
reaction temperature at one-and-the-same space velo-
city (WHSV�15 000 hÿ1). The results are graphically
represented in Fig. 5.
Fig. 5 shows that the CO oxidation commences on
CrII-600 at a temperature as low as 1208C, and 100%
conversion is accomplished near 2208C. The reaction
commencement is delayed to 2008C on CrAlII-600
and CrSiII-600, and a conversion as low as 20% is not
achieved unless the temperature exceeds 2808C.
Hence, the unsupported chromia is much more active
Fig. 4. Plots of volume of oxygen liberated versus time for H2O2
decomposition on the catalysts indicated.Fig. 5. Plots of CO-conversion versus reaction temperature for CO
oxidation on the catalysts indicated.
322 M.I. Zaki et al. / Applied Catalysis A: General 171 (1998) 315±324

as a CO-oxidation catalyst than the supported chro-
mias. When the results shown in Fig. 5 are compared
at a given temperature (2808C), the activity of the
supported chromias can be ranked in the following
descending order:
CrAlII-600�r� � CrAlII-600�c�> CrSiII-600�c� > CrSiII-600�r�;
where r�reduced and c�calcined.
These results indicate that the CrII-600 catalyst,
which contains chromate species established on large
a-Cr2O3 particles (Tables 1 and 3) and involved in
electronic interactions with nearby Cr(III) sites
(Fig. 1), exhibits higher CO oxidation activity than
the catalysts containing similar sites however exposed
on noncrystalline chromias (CrAlII-600) or on small
a-Cr2O3 particles (CrSiII-600). A pre-reduction of the
catalysts with CO alters, but slightly, their CO oxida-
tion activity. Since the CO-treatment of calcined
chromia catalysts under similar conditions to those
applied here has been found [13,37] to eliminate
surface chromate species, particularly those estab-
lished on surfaces other than of crystalline chromia,
the catalysis results may attribute the CO oxidation
activity largely to thermally and chemically stable
chromates. Thus the unstable chromate species seem
not to contribute importantly to the generation of
redox catalytic sites (presumably Cr(VI)±Cr(III)).
4. Conclusion
Calcination of unsupported or supported chromias
(on silica and alumina surfaces) at 6008C generates
high valency surface CrOx species mostly of Cr(VI).
These species are anchored onto surfaces of the
chromia phase (crystalline or noncrystalline) and/or
surfaces of the support material. Chromates produced
on surfaces of noncrystalline chromia and silica sur-
faces are the least stable to thermal decomposition,
hydrolysis and chemical reduction. In contrast, chro-
mates established on crystalline a-Cr2O3 particles
interact electronically with nearby Cr(III)±O species
to form thermally and chemically persistive interac-
tion species (Cr(VI)±Cr(III)) thus facilitating the elec-
tron-mobile environment demanded by surface redox
reactions. Elimination of unstable chromates by a
brief CO-reduction at 3508C, or by hydrolysis at room
temperature, does not signi®cantly alter the surface
redox activity. Thus, chromia catalysts synthesized by
calcination and a subsequent elimination of unstable
chromates would still possess suf®cient catalytic
potential in redox processes, and pose no acute threat
to the environment.
Acknowledgements
MIZ and MAH thank Kuwait University for a grant
(SC. 076) and the SAF (SLC 063) of the Faculty of
Science for technical assistance. NEF thanks Prof. H.
KnoÈzinger of Munich University for the permission
granted to carry out the CO-oxidation and H2-TPR
experiments.
References
[1] M.I. Zaki, N.E. Fouad, G.C. Bond, S.F. Tahir, Thermochim.
Acta 285 (1996) 167.
[2] K. KoÈhler, M. Maciejewski, H. Schneider, A. Baiker, J. Catal.
157 (1995) 301.
[3] E. Zhecheva, M.D. Shibanova, D. Mekhandzhiev, B.M.
Kadenatsi, Kinet. Catal. 33 (1992) 388.
[4] R.L. Burwell Jr., G.L. Haller, K.C. Taylor, J.F. Read, Adv.
Catal. 20 (1969) 1.
[5] J.D. Carruthers, K.S.W. Sing, J. Fenerty, Nature (London) 213
(1967) 16.
[6] J. Deren, J. Haber, H. Podgorecka, J. Burzyk, J. Catal. 2
(1963) 161.
[7] R.B. Fahim, R.M. Gabr, M.I. Zaki, S.A.A. Mansour, J.
Colloid Interface Sci. 81 (1981) 468.
[8] K. Jagannathan, A. Srinivasan, C.N.R. Rao, J. Catal. 69
(1981) 418.
[9] R. Rahman, M.H. Mohamed, M. Ahmed, A.M. Aitani, Appl.
Catal. 12 (1995) 203.
[10] A. Iannibello, S. Marengo, P. Tirtarelli, G. Morelli, A.
Zecchina, J. Chem. Soc., Faraday Trans. I 80 (1984) 2209.
[11] M.I. Zaki, N.E. Fouad, J. Leyrer, H. KnoÈzinger, Appl. Catal.
21 (1986) 359.
[12] H.L. Kraus, H. Stach, Inorg. Nucl. Chem. Lett. 4 (1968) 393;
Z. Anorg. Chem. 34 (1969) 280.
[13] A. Zecchina, E. Garrone, E. Ghiotti, C. Morterra, E. Borello,
J. Phys. Chem. 79 (1975) 466.
[14] A. Ellison, J.O.V. Oubridge, K.S.W. Sing, Trans. Faraday Soc.
66 (1970) 1004; A. Ellison, K.S.W. Sing, J. Chem. Soc.,
Faraday Trans. I 74 (1978) 2807.
[15] R.B. Fahim, M.I. Zaki, R.M. Gabr, Surf. Technol. 12 (1981)
317.
[16] R.B. Fahim, M.I. Zaki, R.M. Gabr, Appl. Catal. 4 (1982) 189.
M.I. Zaki et al. / Applied Catalysis A: General 171 (1998) 315±324 323

[17] A.A. Davydov, Yu.M. Schekochikhin, N.P. Keier, Kinet.
Katal. 13 (1972) 1088.
[18] A.S. Megahed, M.Sc. Thesis, Minia University, Egypt, 1994.
[19] Fact Sheet No. 210, A Publication of California Department
of Toxic Substance Control, Office of Pollution Prevention
and Technology, Sacramento/CA, USA, May 1993.
[20] D. Mihajlova, A. Andreev, Bull. Bulg. Acad. Sci. 2 (1976)
265.
[21] N.E. Fouad, H. KnoÈzinger, M.I. Zaki, Z. Phys. Chem. 186
(1994) 231.
[22] N.E. Fouad, H. KnoÈzinger, M.I. Zaki, S.A.A. Mansour, Z.
Phys. Chem. 171 (1991) 75.
[23] M.I. Zaki, R.B. Fahim, J. Thermal Anal. 31 (1986) 825.
[24] M.I. Zaki, A.A.M. Ali, Colloids and Surfaces 119 (1996) 39.
[25] N.E. Fouad, Ph.D. Thesis, Minia University, Egypt, 1989.
[26] H.P. Klug, L.E. Alexander, X-ray Diffraction Procedures, 2nd
ed., Wiley, New York, 1974, pp. 618±692.
[27] C.P. Poole Jr., D.S. MacIver, Adv. Catal. 17 (1967) 223.
[28] M.I. Zaki, N.E. Fouad, Thermochim. Acta 95 (1985) 73.
[29] Z.G. Szabo, K.K. Kamaras, S.Z. Szebeni, I. Ruff, Spectro-
chim. Acta 34 (1978) 4756.
[30] R.A. Schoonheydt, in: F. Delannay (Ed.), Characterization of
Heterogeneous Catalysts, Marcel Dekker, New York, 1981,
pp. 125±167.
[31] B.M. Weckhuysen, L.M. De Ridder, R.A. Schoonheydt, J.
Phys. Chem. 97 (1993) 4756.
[32] V.V. Mikholaichuk, A.V. Isarov, Yu.V. Plyuto, A.A. Chuiko,
Teoreticheskaya i Eksperimental'naya Khim. (Engl. Transl.)
27 (1991) 729.
[33] H. Park, Bull. Chem. Soc. Jpn. 45 (1972) 2749.
[34] W.K. JoÂzÂwrak, I.G. Dalla Lana, J. Chem. Soc., Faraday Trans.
93 (1997) 2583.
[35] G.C. Bond, Heterogeneous Catalysis: Principles and Applica-
tions, 2nd ed., Oxford Science Publishers, Oxford, 1990,
pp. 55±57.
[36] R.B. Fahim, M.I. Zaki, R.M. Gabr, Surf. Technol. 14 (1981) 289.
[37] H.E. Curry-Hyde, H. Musch, A. Baiker, Appl. Catal. 65
(1990) 211.
324 M.I. Zaki et al. / Applied Catalysis A: General 171 (1998) 315±324
Copyright © 2022 FDOKUMEN