
In vitro culture and expansion of human limbal epithelial cells

This article appeared in a journal published by Elsevier. The attachedcopy is furnished to the author for internal non-commercial researchand education use, including for instruction at the authors institution
and sharing with colleagues.
Other uses, including reproduction and distribution, or selling orlicensing copies, or posting to personal, institutional or third party
websites are prohibited.
In most cases authors are permitted to post their version of thearticle (e.g. in Word or Tex form) to their personal website orinstitutional repository. Authors requiring further information
regarding Elsevier’s archiving and manuscript policies areencouraged to visit:
http://www.elsevier.com/authorsrights

Author's personal copy
Simplifying corneal surface regeneration using a biodegradablesynthetic membrane and limbal tissue explants
Pallavi Deshpande a,1, Charanya Ramachandran b,1, Farshid Sefat a, Indumathi Mariappan b,Claire Johnson a, Robert McKean c, Melanie Hannah d, Virender S. Sangwan b,Frederik Claeyssens a, Anthony J. Ryan d, Sheila MacNeil a,*aDepartment of Materials Science and Engineering, Kroto Research Institute, North Campus, University of Sheffield, Broad Lane, Sheffield S3 7HQ, UnitedKingdomb Sudhakar and Sreekanth Ravi Stem Cell Laboratory, LV Prasad Eye Institute, Kallam Anji Reddy Campus, LV Prasad Marg, Hyderabad-500034, Indiac The Electrospinning Company Ltd., Rutherford Appleton Laboratory, Harwell Oxford, OX11 0QX, United KingdomdDepartment of Chemistry, Dainton Building, Brook Hill, University of Sheffield, S3 7HF, United Kingdom
a r t i c l e i n f o
Article history:Received 8 March 2013Accepted 25 March 2013Available online 13 April 2013
Keywords:MembraneCorneaStem cellTransplantationDegradation
a b s t r a c t
Currently, damage to the ocular surface can be repaired by transferring laboratory cultured limbalepithelial cells (LECs) to the cornea using donor human amniotic membrane as the cell carrier. Wedescribe the development of a synthetic biodegradable membrane of Poly D,L-lactide-co-glycolide (PLGA)with a 50:50 ratio of lactide and glycolide for the delivery of both isolated LECs and of cells grown outfrom limbal tissue explants. Both isolated LECs and limbal explants produced confluent limbal cultureswithin 2 weeks of culture on the membranes without the need for fibroblast feeder layers. Outgrowth ofcells from explants was promoted by the inclusion of fibrin. Membranes with cells on them broke downpredictably within 4e6 weeks in vitro and the breakdown was faster for a lower molecular weight (MW)(44 kg/mol) rather than a higher MW (153 kg/mol) PLGA. Membranes could be reproducibly produced,sterilised with gamma irradiation and stored dry at �20 �C for at least 12 months, and the ability tosupport cell outgrowth from explants was retained. We demonstrate transfer of cells (both isolated LECsand of cells grown out from limbal explants) from the membranes to an ex vivo rabbit cornea model.Characterisations of the cells by immunohistochemistry showed both differentiated and stem cellpopulations. A synthetic membrane combined with limbal explants in theatre would avoid the need fortissue banked human amniotic membrane and also avoid the need for specialist laboratory facilities forLEC expansion making this more accessible to many more surgeons and patients.
� 2013 Elsevier Ltd. All rights reserved.
1. Introduction
The overall aim of this work is to simplify and make safer andmore accessible the methodology for transplanting limbal epithe-lial cells (LECs) to resurface and regenerate the cornea for patientswho have lost vision due to limbal stem cell deficiency (LSCD).Currently there are only a small number of specialist ophthalmiccentres worldwide (we estimate less than 10 on the basis of pub-lished results) who have expertise in laboratory expansion of LECs
and of transplanting these cells to the cornea using either fibrin as asubstrate [1] or human amniotic membrane (hAM) [2,3].
An attractive alternative to using cultured cells, as was recentlydemonstrated [4], is outgrowth of cells from limbal explants in situ(on amniotic membrane) obviating the need for expansion of LECsin the laboratory at least for patients with unilateral LSCD.
Accordingly our specific aims are to develop a synthetic, steri-lised, rapidly degrading membrane that can be readily available touse as an alternative to the hAM and to explore regeneration of thecorneal epithelium both using laboratory cultured cells andoutgrowth of cells from small limbal explants. A combination of off-the-shelf synthetic membrane and culture of limbal epithelial cellsin situ could make this technique available to many more surgeonsand hence patients than can currently access treatment for LSCDonly through specialist centres.
* Corresponding author. Tel.: þ44 114 222 5943; fax: þ44 114 222 5945. .E-mail address: [email protected] (S. MacNeil).URL: http://www.shef.ac.uk/materials/staff/smacneil01
1 Drs Deshpande and Ramachandran contributed equally as first authors to thisstudy.
Contents lists available at SciVerse ScienceDirect
Biomaterials
journal homepage: www.elsevier .com/locate/biomateria ls
0142-9612/$ e see front matter � 2013 Elsevier Ltd. All rights reserved.http://dx.doi.org/10.1016/j.biomaterials.2013.03.064
Biomaterials 34 (2013) 5088e5106

Author's personal copy
It is necessary to review the current clinical situation to set thecontext for this study. Damage to or loss of the LECs of the eye canlead to extensive scar tissue formation often with vascularisation,an unstable epithelium and considerable pain [5,6].
Full thickness corneal transplantation (i.e. penetrating kerato-plasty) can temporarily restore vision in patients with LSCD buteventually fails due to insufficient or lack of recipient limbal stemcells necessary for sustained regeneration of the corneal epithelium[7,8]. To replace the loss of stem cells, the culture of LECs in thelaboratory and their subsequent transfer was established 16 yearsago [9] and this is now a treatment option available to patients insome specialised centres around the world [2,4,9]. Culture of cellsfrom the contralateral unaffected eye is undertaken whereverpossible (under regulated laboratory conditions approved by theappropriate regulatory bodies for each country) or from donor eyeswhen no autologous cells are available [10]. The latter patients thenrequire long term immunosuppression.
At its simplest if the loss of LECs is unilateral, then a biopsy canbe taken from the healthy contralateral eye, cells expanded in thelaboratory and then transplanted to the affected eye followingremoval of the scarred tissue. The condition of the eye post removalof the scarred tissue is such that cultured cells placed directly on itwould be unlikely to survive; hence cells are by preference placedonto a degradable substrate prior to transplantation. Of the varioussubstrates that have been explored, the hAM is the one mostcommonly used in clinical transplantation. A recent report by ourgroup (reviewing 200 patients) found a 76% success rate of LECtransplantation on the amniotic membrane after 1 year, droppingto 68% after 4 years [11].
There are likely to be many reasons why some patients continueto do well after several years and others do not including the initialaetiology of the condition which may dictate the extent of thedamage to the corneal stroma and the likelihood of recurrence ofthe condition. In addition, donor variability in the quality andprocessing of the fresh hAM may also contribute to the success ofthe treatment. Certainly the rate of breakdown of the amnioticmembrane is not consistent between different recipients.
For all of these reasons including the need to establish tissuebanks for processing the hAM, several groups are investigating al-ternatives to this material with both synthetic and natural mate-rials being explored. These include fibrin glue [1], collagenmembranes [12,13], thermo-sensitive substrates [14] and syntheticpolymer membranes [15,16]. Of these only hAM and fibrin arecurrently routinely used as carriers for cell transfer in the clinic forcorneal regeneration.
The desirable characteristics of a membrane for delivering LECsare that it provides secure attachment for the cells and supportstheir proliferation and migration. Additionally, it must be as lowrisk for clinical use as possible, i.e. ideally it must be sterilised usingan acceptable sterilisationmethodology. It must be capable of beingfixed to the eye post culture with cells and for it to be successful itneeds to degrade within a few weeks leaving LECs securelyattached to the underlying cornea without eliciting an immuneresponse. Thus we seek a membrane that will degrade predictablywithin a few weeks, support LEC attachment and subsequentlydeliver these cells securely attached to the cornea and which iscapable of being produced, sterilised and stored reproducibly.
We selected poly(D,L-lactide-co-glycolide) (PLGA) for this pur-pose because it is biodegradable and non-cytotoxic, FDA approvedand has been used for many years in products such as dissolvablesutures [17]. Such sutures are currently used in ocular surgeries andby varying the ratio of lactide to glycolide it is also possible topredict the degradation rates of these membranes in vivo [18].
In this study we examined the ability of these synthetic mem-branes to support the growth of isolated LECs, both in the presence
and absence of feeder cells, and also its ability to support outgrowthof LEC from intact limbal tissue explants placed directly on themembranes.We looked at the rate of breakdown of themembraneswith and without cells in vitro. We confirmed that both isolatedcells and cells outgrown from limbal explants would transfer fromthese membranes as they broke down onto an ex vivo rabbit corneamodel. Finally, we looked at the impact of g-irradiation on thephysical characteristics of the membrane and its breakdown in thepresence of cells and also the ability of these membranes to bestored in the presence of desiccant for long term storage.
2. Materials and methods
2.1. Polymers and electrospinning
The electrospun membranes were initially spun as previously described[16,18,19]. Briefly, poly (lactide-co-glycolide) 50:50 (Purac, The Netherlands) ofmolecular weight (MW) 44 kg/mol was dissolved in dichloromethane (Fisher Sci-entific, UK) at a concentration of 20% (wt/wt). The solutionwas then drawn into four5 ml syringes and blunt ended needles of internal diameter of 0.8 mm (Intertronics,UK) were fitted onto each syringe. The syringes were fitted onto a syringe pump(Genie Plus, Kent Scientific, Torrington, CT, USA) which was set to extrude thepolymer at 3000 ml/h and the voltage was set between 10 and 15 kV using a voltagegenerator (Genvolt, Bridgnorth, Shropshire UK). The jet of polymer released from thesyringe was deposited on a collector rotating at 300 rpm. The collector was wrappedwith a PTFE coated fabric (Bake-o-glide, Falcon Product Ltd, Haslingden, UK) so thatthe membranes could be easily removed after being spun. This resulted in a mat ofmembrane that was between 60 and 80 mm thick with fibre diameters of 2e5 mm. Inthese studies membranes were produced within the University of Sheffield underclean room conditions.
Once it was clear that these membranes were looking promising we establisheda collaboration with The Electrospinning Company to produce membranes ofreproducible quality for this study and we compared polymers of two MWs andincorporated sterilisation with g-irradiation. PLGA membranes were electrospun toour specifications by The Electrospinning Company (TEC) using Poly (D,L-lactide-co-glycolide) with a 50:50 ratio of lactide to glycolide at a low MW of 44 kg/mol and ahigh MW of 153 kg/mol. Henceforth, the membranes produced in Sheffield arereferred to as “in-house” membranes to distinguish them from the membranesproduced by the The Electrospinning Company which will be referred to as “TECmembranes”.
The TECmembranes producedwere 50 mm thickwith fibre diameters of 2e3 mm.Polymerswere dissolved in 1,1,1,3,3,3-hexafluoroisopropanol (HFIP) (SigmaeAldrich,Dorset, UK) by gentle stirring at room temperature to produce solutions of suitableviscosity for electrospinning (10 wt% and 20 wt% for the low MW and high MWpolymer respectively). Polymer solutions were loaded into 5 ml plastic syringes(BectonDickinson, Oxford, UK) and delivered at a constant feed rate of 800 ml/h, usinga programmable Harvard PHD4400 syringe pump (Harvard Apparatus, Kent, UK) viaPTFE tubing (1/1600 O.D.) to a blunt tipped stainless steel needle with an internaldiameter of 0.8 mm (Intertronics, Kidlington, Oxfordshire, UK). The tip of the needlewas in turn connected to a positive high voltage unit (Glassman High Voltage Inc.High Bridge, NJ, USA) and solutions were electrospun with an applied voltage of12.5 kV. Fibres were deposited onto a grounded, custom built rotating mandrel(120 mm in diameter, 250 mm in length) at a distance of 300 mm from the tip of theneedle, coated in 50 mm thick aluminium foil. Electrospinning was performed in anenvironmentally controlled, A1-Safetech, air recirculation cabinet (a1-envirosciencesGmbH,Düsseldorf, Germany) at an air temperature of 25 �C and a relative humidity of25%. Membranes were subsequently dried under vacuum at room temperature for48 h to ensure any residual solvent was completely removed, before being cut into22mmdiameter discs andmounted in 12-well, non-tissue culture treated plates (BDFalcon, Beckton Dickinson, Oxford, UK). The plates were then individually sealed inpolypropylene bags (Andrew James UK LTD, Bowburn, UK).
2.2. g-Irradiation of TEC membranes
Membranes (mounted in 12-well plates) fabricated by The ElectrospinningCompany were sterilised via g-irradiation at Synergy Health Plc. (Moray Road,Swindon, UK), with an external dose range of 25e40 kGy. The four types of mem-brane (44 and 153 kg/mol, sterilised and non-sterilised) were then comparedthroughout to assess the impact of sterilisation on their physical properties, theirability to support cells and also their breakdown when stored without cells at arange of temperatures.
2.3. Physical characterisation of membranes
2.3.1. Differential scanning calorimetry (DSC)DSC was used to obtain the glass transition temperature (Tg). Thermal transi-
tions were recorded on a PerkineElmer Pyris-1 connected to a controller model
P. Deshpande et al. / Biomaterials 34 (2013) 5088e5106 5089

Author's personal copy
CCA7 (PerkineElmer, Wellesley, MA, USA). The samples were heated in an inertatmosphere of Nitrogen (30ml/min flow rate) from 0 to 100 �C at a rate of 10 �C/min;each sample was re-heated. Samples of 5e10 mg of each TEC PLGA membrane wereplaced in a crimped aluminium pan and subjected to the heat gradient. Thermo-grams were obtained and subsequently analysed.
2.3.2. Gel permeation chromatographyThe polymer molecular weight distribution for the PLGA (50:50) samples either
as polymer discs (TEC) or crystals (before spinning) were determined by gelpermeation chromatography (PerkineElmer, Wellesley, MA, USA) equipped with arefractive index detector (PerkineElmer, Wellesley, MA, USA). The dried sampleswere dissolved in tetrahydrofuran (THF, Fisher Scientific, Loughborough, UK) with aflow rate marker of toluene at a concentration of 8 mg/ml and eluted through thecolumn at a flow rate of 1 ml/min at 37 �C. Polystyrene standards (Polysciences,Warrington, PA, USA) were used to obtain a primary calibration curve. All samples ofthe same polymer type were analysed in a single run.
2.3.3. Wettability of TEC membranesFor determination of the wettability of membranes, the contact angle of a 3 ml
drop of deionised water (dH2O), phosphate buffer saline (PBS) or media (DMEM)was measured using a contact angle goniometer (ramé-hart, USA). Five sampleswere used for each test and the average value was reported with a standard devi-ation (�SD).
2.3.4. Biomechanical properties of membranesTECmembranes were cut into rectangles with dimensions of 30� 10mm2 and a
Bose Electroforce 3100 tensile test machine (Bose, MN, USA) was used to carry outmechanical tests on PLGA membranes. The membranes were clamped in place viastainless steel clamps and pulled apart at a rate of 1 mm/10 s with grip distance of7 mm. The force exerted was measured via a 22 N small-load cell and the samplethicknesses measured using a micrometer. A total number of five samples were usedfor each experimental condition.
2.4. Culture of limbal epithelial cells
In the evaluation of these membranes experiments were undertaken both in theUK with rabbit epithelial cells (where access to human cells for experimentationproved challenging) and with human cells in India (where corneas were obtainedfrom the Ramayamma International Eye Bank, LV Prasad Eye Institute, Hyderabad).
2.4.1. Rabbit limbal epithelial cells (rLEC)Eyes from New Zealand white rabbits were obtained from Hook Farm, Hook,
Berkshire UK. The rabbits weighed between 2.4 and 2.6 kg. The eyes were cleaned toremove excess fat and tissue and then disinfected using 3% videne antiseptic solu-tion (Ecolab, Swindon, UK). Rabbit limbal epithelial cells (rLEC) were isolated aspreviously described [16,20]. Briefly, the limbal rim of the cornea was excised intofour segments and placed into 2.5 mg/ml of Dispase II (Roche Diagnostics Ltd.,Burgess Hill, UK) in Dulbecco’s Modified Eagle Medium (Biosera, Ringmer, UK) at37 �C for 45 min. The limbal cells were then scraped off the tissues into phosphatebuffered saline using a pair of blunt forceps. The solution was centrifuged at 200 gfor 5 min. The supernatant was discarded and the cells were resuspended in freshepithelial culture medium. The cells were seeded into flasks containing growtharrested NIH3T3 murine fibroblasts as a feeder layer. The culture media for the rLECconsisted of Dulbecco’s Modified Eagle Medium (DMEM GlutaMAX�, Gibco LifeTechnologies Ltd., Paisley UK) and Ham’s F12medium (Biosera, Ringmer, UK) in a 1:1ratio supplemented with 10% foetal calf serum (Biosera, Ringer, UK), 10 ng/ml EGF,5 mg/ml insulin, 2.5 mg/ml Amphoterin B and 100 IU/ml penicillin and 100 mg/mlstreptomycin (SigmaeAldrich, Poole, UK) Cells were used between P0 and P2. Forexplant culture, the limbal tissue was chopped into small pieces of approximately0.5 mm and placed on electrospun membranes and cultured for 14 days.
2.4.2. Human limbal epithelial cells (hLEC)Limbal cells were isolated from both cadaver corneas and normal individuals
undergoing cataract surgery after obtaining IRB approval and informed consent. TheLECs were cultured as described previously [21]. For suspension cultures, limbaltissues were incubated in 2.5 mg/ml dispase for 1 h at 37 �C and cells were scrapedusing blunt forceps. The isolated cells were cultured in the presence of a feeder layerof irradiated NIH3T3 mouse fibroblasts (ATCC, Manassas, VA) and P0 cells were usedfor experiments. For explant culture, the limbal tissue was chopped into small piecesof approximately 0.5 mm and placed on either the hAM or electrospun membranesand cultured for 14 days.
2.5. Cell culture on membranes
In-house membranes were cut into discs of 22 mm diameter and placed in a 12well tissue culture plate along with pieces of Bake-o-Glide (cut to fit these wells)underneath to prevent sticking of the membrane to the tissue culture plastic overtime. In house membranes used were uncoated, coated with fibrin glue or pre-seeded with growth arrested NIH3T3 mouse feeder cells. Medical grade stainless
steel rings of 22 mm outer diameter were placed on the membranes to prevent anycontraction during culture and to provide wells of 10 mm diameter into which toseed the cultured LEC. For the fibrin coating, a 1:1 mixture of fibrinogen (18.75 mg/ml) and thrombin (2.5 U/ml) was pipetted onto the membranes. The drops werespread thinly on the membranes using a cell scraper. For establishing feeder cul-tures, growth arrested 3T3s were seeded at a density of 2.4 � 104 cells per cm2 areaof PLGAmembrane or hAM (in some experiments) the day before seeding 1.25�105
of isolated LEC into the centre of the stainless steel rings and cultured for twoweeks.For explant culture, eight to ten explants of approximately 0.5 mm were placed inthe centre of the membrane at a distance of about 2 mm from each other andcultured for 2 weeks. At the end of the culture period, the cells were fixed with 3.7%formaldehyde and processed for immunocytochemistry.
2.6. Assessment of cell outgrowth from explants using rose bengal staining
Cell outgrowth from explants on membranes was established by staining themembranes using 1% Rose Bengal (SigmaeAldrich, UK and USA) for 5 min. Thesamples were then washed several times with PBS to remove any excess RoseBengal. These stained membranes were then photographed to obtain a rapidphotographicmethod which was assessed quantitatively using Image J (NIH, USA) toestimate the extent of outgrowth from explants onto themembranes. The differencein the areas was averaged and the data expressed as mean � SEM.
2.7. Identification of label retaining cells using BrdU pulse chase
After 1 week, cells cultured on hAM and fibrin coated in-house membranes werepulsed with BrdU (5 mM) for 24 h to identify the number of proliferating cells. Afterthe 24 h pulse, the cultures were chased for 7 days by switching to BrdU free me-dium for quantifying the label retaining cells. One set of PLGA membranes and hAMwere fixed and stained immediately after pulse and the other set was fixed andstained for BrdUpositive cells at the end of the chase period. All samples in duplicatewere fixed in 4% paraformaldehyde before being processed for BrdU staining. Inbrief, samples were incubated in 2NHCl at 37 �C for 1 h to denature DNA followed byneutralization in borate buffer (pH 8.5) for 20min. BrdUwas detected using a mouseanti-BrdU antibody, followed by secondary antibody incubation for 1 h. Propidiumiodide was used as the nuclear counterstain. At least 10 representative images weretaken for each sample using a Zeiss LSM 510 META confocal microscope and a 10�objective. The BrdU positive and propidium iodide positive cells were quantified bycounting the number of cells using the Image J software (NIH, USA). The percentBrdU positive cells was calculated by dividing the number of BrdU positive cells bythe total number of cells. The experiment was repeated twice with tissues takenfrom different donors (N ¼ 3).
2.8. In vitro breakdown of membranes
TEC Membranes were cut into 18 mm diameter disks using a stainless steel corkborer and placed in media in a 12 well plate. rLEC were then seeded into stainlesssteel rings of 10 mm internal diameter at a density of 104 cells/well on membranesand cultured for a period of up to 6 weeks (at 37 �C). Breakdown was assessed bylight microscopy, by physically picking up the membranes, by weight loss and bySEM imaging by measuring the fibre thickness.
2.8.1. Light microscopy and membrane handlingrLEC were cultured on membranes for 6 weeks at 37 �C in a humidified atmo-
sphere. Physical changes in the membranes were recorded daily by taking opticalmicrographs and membrane handling was carried out simply by using a pair offorceps after they were removed from culture (inwet conditions) and scored as easyto handle (þþþ), fragile (þþ) or very brittle (þ).
2.8.2. SEM of membranesThe morphology, microstructure, fibre diameter, fibre integrity and degradation
rate of non-sterile and g-irradiated PLGA membranes were examined by SEM.Following periods of culture, specimens were fixed using 3.7% formaldehyde solu-tion and prepared for SEM imaging. Specimens were washed with 2 ml of 0.1 Msodium cacodylate buffer for 5 min at room temperature then fixed once more in2.5% glutaraldehyde (in 0.1 M phosphate buffer) for 30 min at 4 �C. One wash with2 ml of sodium cacodylate was then carried out to rinse away any remainingglutaraldehyde. Secondary fixation was carried out in 1% osmium tetraoxide for 2 h.A final wash was performed with sodium cacodylate and samples were dehydratedin ascending grades of alcohol, freeze dried overnight, bisected and mounted on12.5 mm stubs. The samples were sputter coated with approximately 25 nm of goldand then examined using a scanning electronmicroscope (Philips/FEI XL-20 SEM) atan accelerating voltage of 10e15 kV.
2.8.3. Membrane weight loss as an indicator of membrane breakdownThe extent of breakdown was expressed as a percentage of weight loss of the
dried membranes after incubation with cells in media for varying periods of time.Membranes were weighed at the beginning of the experiment and then eachmembrane was weighed on a weekly basis after removing it from culture and was
P. Deshpande et al. / Biomaterials 34 (2013) 5088e51065090

Author's personal copy
dried using vacuum oven. The loss of weight was calculated and expressed as apercentage of the initial weight. The weight loss percentages of the specimens werecalculated from the weights obtained before and after degradation using thefollowing formula (1) in which W1 and W2 are the sample weights before and afterdegradation, respectively.
Weight lossð%Þ ¼ ðW1 �W2Þ=W1 � 100 (1)
2.9. Storage of membranes
PLGA membranes (g-irradiated and non-sterile) were stored at a wide range oftemperatures from �80 �C to þ50 �C in order to assess the storage shelf life of themembranes over 12 months. Additional membranes were stored at 37 �C (both in adry oven and a humidified incubator). In the case of the latter, membranes wereplaced in an empty petri dish within an incubator containing 5% CO2 with highhumidity. Water absorption by membranes was detected using silica orange desic-cant. Fibre integrity was assessed by using SEM.
2.10. Use of ex vivo cornea organ culture to assess cell transfer from membranes tocornea
2.10.1. Organ culture modelRabbit eyes were prepared as described above. The eyes were then immersed in
0.14% ammonium hydroxide (SigmaeAldrich, Poole, UK) for 5 min after which theywere rinsed with PBS. Using a sclerotome knife, the epithelium on the cornea as wellas the limbal region was scraped away. The corneo-scleral button with approxi-mately 4 mm of the sclera was excised and placed epithelial side down as describedpreviously [20] onto a sterile cup. Approximately 500 ml of the epithelial culturemedium containing 0.5% agar (SigmaeAldrich, Poole, UK) and collagen I extractedfrom rat tail (Fluka, Poole, United Kingdom) (1 mg/ml final concentration) wasadded onto the cornea for support and when set was inverted upright into a 35-mmdish (Nunc; Loughborough, UK). The corneas were cultured in the epithelial culturemedium. Every experiment included a positive control which was an unwoundedcornea and a negative control, a wounded cornea.
2.10.2. Transfer of laboratory expanded cells from membranes onto ex vivo organcultures
For the transfer of cultured cells, membranes of 22 mm diameter were placed in12 well petri dishes and a stainless steel ring of 22 mm outer diameter and 10 mminner diameter was placed on top. 1.25 � 105 rabbit limbal epithelial cells were thenseeded into the centre of these rings and cultured for 5 days. At the end of 5 days, themembranes with cells were placed on the denuded rabbit corneas either cell side upor cell side down and cultured either at an aireliquid interface or submerged. After 4weeks in culture the corneas were fixed using 3.7% buffered formaldehyde and thenparaffin embedded and processed for conventional histology. Sections were cut ofbetween 4 and 6 microns and then stained with haematoxylin and eosin (H&E) orprocessed for immunohistochemistry.
2.10.3. Transfer of cells from limbal explants from membranes onto ex vivo organcultures
Limbal explants were placed on the membranes as described above. The mem-branes were placed on the denuded corneas and cultured for 4 weeks after whichthey were fixed using 3.7% buffered formaldehyde, imaged using optical coherencetomography and then paraffin embedded and processed for conventional histology,sectioning the samples between 4 and 6 microns. The samples were then eitherstainedwith haematoxylin and eosin (H&E) or processed for immunohistochemistry.
2.11. Characterisation of corneal cells by immunochemistry
To characterise the cells cultured on hAM and membranes, cells were stainedwith species compatible antibodies for actin filaments, cytokeratin 3 (rabbit) orcytokeratin 12 (human) (marker for differentiated epithelial cells), as well as p63and p63a (stem cell markers). Briefly, cells were treated with 0.5% triton X for 10minafter which they were blocked with 10% goat’s serum in PBS (SigmaeAldrich, UK) or5% BSA in PBS (SigmaeAldrich, USA) for 1 h. The cells were incubated in theappropriate primary antibody diluted in 1% blocking medium overnight at 4 �C andthen incubated in a biotin conjugated secondary antibody for 1 h followed by astreptavidin conjugated tertiary antibody for 30e45 min. To stain the actin fila-ments, cells were incubated in phalloidin FITC for 30 min. Finally, the cells werecounterstained with propidium iodide and imaged using a Zeiss LSM 510 METAconfocal microscope.
To characterise the cells transferred onto the corneas, sections were dewaxed inxylene after which they were rehydrated in 100% ethanol, 70% ethanol and thenwater. Sections were delineated with a Dako pen and the samples were treated with0.05% trypsin (SigmaeAldrich, UK) for 20 min at 37 �C. The samples were rinsed inPBS and blocked using 10% goat’s serum (SigmaeAldrich, UK) for 1 h. The sectionswere then incubatedwith either mousemonoclonal antibody for cytokeratin 3 (1:50dilution) (Merck Millipore, UK), p63 (1:150 dilution) (Merck Millipore, UK) or mousemonoclonal antibody for vinculin (1:150) (Sigma, Aldrich, UK) diluted in 1% goat’s
serum for 1 h at room temperature and then rinsed with PBS. Next the sections weretreated with a biotinylated anti mouse secondary antibody (1:1000) (Vector labs,Peterborough, UK) for 1 h and then a streptavidin FITC conjugated tertiary antibody(1:100) (Vector labs, Peterborough, UK) for half an hour. The cells were counter-stained with propidium iodide at a concentration of 1 mg/ml for 10 min and imagedusing a Zeiss LSM 510 META confocal microscope at laser wavelength 488 nm and543 nm.
2.12. Statistical analysis
For quantifying the extent of cell outgrowth from explants onto the membraneStudents t-test was performed to compare between the uncoated and fibrin coatedmembranes. For the BrdU dataset, a 2-factor ANOVAwith Bonferroni correctionwasperformed. Fibre diameter results were tested for normality using a KolmogorovSmirnov test. Results that showed normal distribution (p > 0.05) were analysedusing SPSS via a Oneway Analysis of Variance (ANOVA) followed by a post HocBonferroni test. KruskaleWallis test and serial Mann Whitney tests were used fornon-normally distributed results (p< 0.05). All statistical tests were performed suchthat a p value of <0.05 was considered as indicating a significant difference.
3. Results
3.1. Breakdown of TEC PLGA membranes with and without cellspresent
Fig. 1A shows that PLGA membranes with 50:50 of lactic andglycolic acid rapidly broke down as expected when placed in mediaat 37 �C in a CO2 gassed incubator. Similar results were seen withmembranes both with and without cells and the data shownthroughout Fig. 1 are for those with cells present.
Membrane fibre diameters were initially 2e3 mm and themembranes had a depth of 50 mm and these measurements werevery consistent from batch to batch. Results for membrane break-down (Fig. 1A and B) and loss of mass (Fig. 1C) show that g-irra-diation significantly accelerated breakdown and is summarised inTable 1.
There was no apparent change in the fibre diameter by day 7(Fig. 1A) but as early as day 14 it was evident that polymer fibreshad absorbed moisture and some swelling was evident. Sterilisedfibre diameters increased to 13.10 mm� 0.86 SD and 7.84 mm� 0.52SD for lower MWand higher MW polymers respectively, comparedto 10.10 mm � 1.67 SD and 5.28 mm � 0.84 SD for these non-sterilised fibres (Fig. 1A and Table 1). Over 6 weeks fibres clearlylost integrity and coalesced. Breakdown of the 44 kg/mol mem-branes was faster than that of the 153 kg/mol and the impact ofsterilisationwas highly significant (p < 0.001) as depicted in Fig. 1Cand summarised in Table 1.
Fig. 1B shows the gross appearance of the membranes with cellson them over 6 weeks. This figure depicts a g-irradiated PLGAmembrane (molecular weight 44 kg/mol) over 6 weeks whencultured with rLEC. Membranes are shown placed on glass cover-slips for ease of handling and it is evident that these maintain somestructural integrity for the first 3 weeks, but by 4 weeks they areclearly breaking down and would be very hard to handle.
We assessed the ease of handling the membranes by simplypicking them up with forceps. Membranes incubated with cellscould be handled for a few weeks (from 2 to 5 weeks depending ontheir molecular weight and whether they were irradiated or not)before they became brittle and hard to handle. The low molecularweightmembrane could be handled for up to 2weeks with care butonly 1week if g-irradiated. The highermolecularweightmembranecould be handled for up to 5 weeks with care, but only 3 weeks if g-irradiated. From this it appears that g-irradiation increased thebrittleness and fragility of the membranes and reduced the periodfor which they could be handled by approximately a week.
Additionally, each week a sample was sacrificed to assessmembrane mass by drying membranes and weighing them tocalculate the percentage weight loss. Fig. 1C which shows a steady
P. Deshpande et al. / Biomaterials 34 (2013) 5088e5106 5091

Author's personal copy
increase in weight loss for non-sterile lowMWmembranes (44 kg/mol) throughout the 6 weeks studied with a mean % loss of48.32% � 1.2%SD by week 6, whereas, g-irradiated samples had amean % weight loss of 78.43% � 2.73% SD by week 6 (p < 0.01).Similarly there was steady increase for the non-sterile higher MWmembranes (153 kg/mol) throughout the 6 weeks studied with amean % loss of 37.29% � 1.81%SD by week 6, whereas, g-irradiatedsamples had a mean % weight loss of 75% � 2.81% SD by week 6(p < 0.01).
3.2. Impact of sterilisation and spinning on membrane physicalproperties
Table 2 summarises the impact of sterilisation and electro-spinning on the physical properties of the membranes. As shown inTable 2, the initial glass transition temperature Tg for PLGA withlow or high molecular weight before spinning was 44.5 �C and47.7 �C respectively. After spinning these temperatures droppedsignificantly to 31.1 �C and 40.4 �C. The reduction in Tg post spin-ning was significant for both low and high molecular weightpolymers (p < 0.01). g-irradiation of these electrospun membranesdid not cause any further changes in transition temperature.
The molecular weights of the 153 kg/mol and 44 kg/mol PLGAwere determined by gel permeation chromatography (GPC) andassessed at 102 kg/mol and 39 kg/mol respectively (see Table 2).Electrospinning did not significantly affect the molecular weight ofeither of these polymers but g-irradiation post spinning
significantly reduced the molecular weight estimate for the 153 kg/mol PLGA to 27 kg/mol. For the 44 kg/mol PLGA the reduction wasfrom 38 kg/mol to 26 kg/mol.
The polydispersity index for the 153 kg/mol PLGA was 1.38 andthis significantly increased post spinning to 1.86, but was notsignificantly increased by subsequent g-irradiation (2.05). For the44 kg/mol PLGA, the polydispersity index was 1.64. Post spinningtherewas only a slight increase to 1.72, but it significantly increasedto 1.97 by subsequent g-irradiation (p < 0.05) (Table 2).
Table 2 shows the ultimate tensile strength and Young’smodulus for the membranes before and after g-irradiation. For thehigh molecular weight membrane g-irradiation significantlyincreased tensile strength (from 3.68 � 0.16 to 4.59 � 0.17 N/mm2)(p < 0.05). For the low molecular weight membrane ultimate ten-sile strength slightly increased from 4.13 � 0.79 to 4.96 � 1.76 N/mm2 by g-irradiation but this was not significant. With respect toYoung’s modulus, g-irradiation did not affect Young’s modulus forthe high molecular weight membrane, but did significantly in-crease the Young’s modulus from 62.98� 4.4 to 95.5� 0.98 N/mm2
(p < 0.001) for the low molecular weight membrane.
3.3. Wettability of membranes
The use of contact angles to indicate the wettability of themembranes was assessed using deionised water (dH2O), PBS andmedia (DMEM) and results are summarised in Table 3. For the153 kg/mol PLGA it was clear that g-irradiated membranes showed
Fig. 1. Breakdown of PLGA membranes over 6 weeks. In these experiments membranes were placed in culture media and their morphology and mass examined over 6 weeks. Ashows SEM of the fibres of non-sterilised and g-irradiated for 44 kg/mol and 153 kg/mol of PLGA over 6 weeks. B shows the visual appearance of g-irradiated PLGA (molecularweight 44 kg/mol) over 6 weeks when cultured with rLEC cells. Membranes plus cells are shown on glass coverslips. C shows percentage weight loss of the membranes both non-sterile and g-irradiated comparing low (44 kg/mol) and high (153 kg/mol) molecular weight membranes over 6 weeks of culture with rLEC cells.
P. Deshpande et al. / Biomaterials 34 (2013) 5088e51065092

Author's personal copy
a more rapid decrease in contact angle with time when PBS orDMEM were used. These were completely wet after 10 min for PBSand 20 min for DMEM. For the 44 kg/mol PLGA all membranesshowed a similar reduction in contact angle over 20e25min. Again,results were slightly faster if DMEM was used rather than PBS ordH2O. For these sterilised membranes they were largely wet within15 min. Accordingly, all membranes were soaked in media for30 min before use with cells or explants.
3.4. Culture of isolated limbal epithelial cells on PLGA membranes
To determine the ability of the PLGA membranes to support cellgrowth, LEC (both human and rabbit) isolated using enzymedigestionwere cultured at a density of 1.25�105 cells/cm2 either inthe presence or absence of feeder cells, as it is well known that thepresence of feeder cells is critical for maintaining the isolatedepithelial stem cells in culture [22,23]. Fig. 2 shows that in thepresence of feeder cells, the hLEC grew in colonies and expanded toform a monolayer on the hAM at the end of 14 days. Immuno-staining of these cells showed typical cobblestone morphology ofepithelial cells (Panel 2A) and a heterogeneous population of cellscomprising of both stem cells and differentiated cells (Panels 2B to
2D). The marker expression in cells cultured in the absence offeeder cells was comparable to the cells cultured on feeders (Panels2E to 2H). However, it was observed that the number of p63apositive cells (Panel 2H) was noticeably reduced (by approximately40%) when compared to the LEC cultured on feeders.
Human LEC cultured on the PLGA membranes showed a similarmorphology (Panels 2I and 2M) both in the presence and absence offeeder cells. The membranes also supported a heterogeneouspopulation of cells with the presence of both differentiated andstem cells similar to that of cells cultured on hAM (Panels 2J to 2P).Similar cell surface expression patterns were also obtained withrLEC cultured on the membranes (Panels 2Q to 2X)
3.5. Explant culture of limbal epithelial cells on PLGA membranes
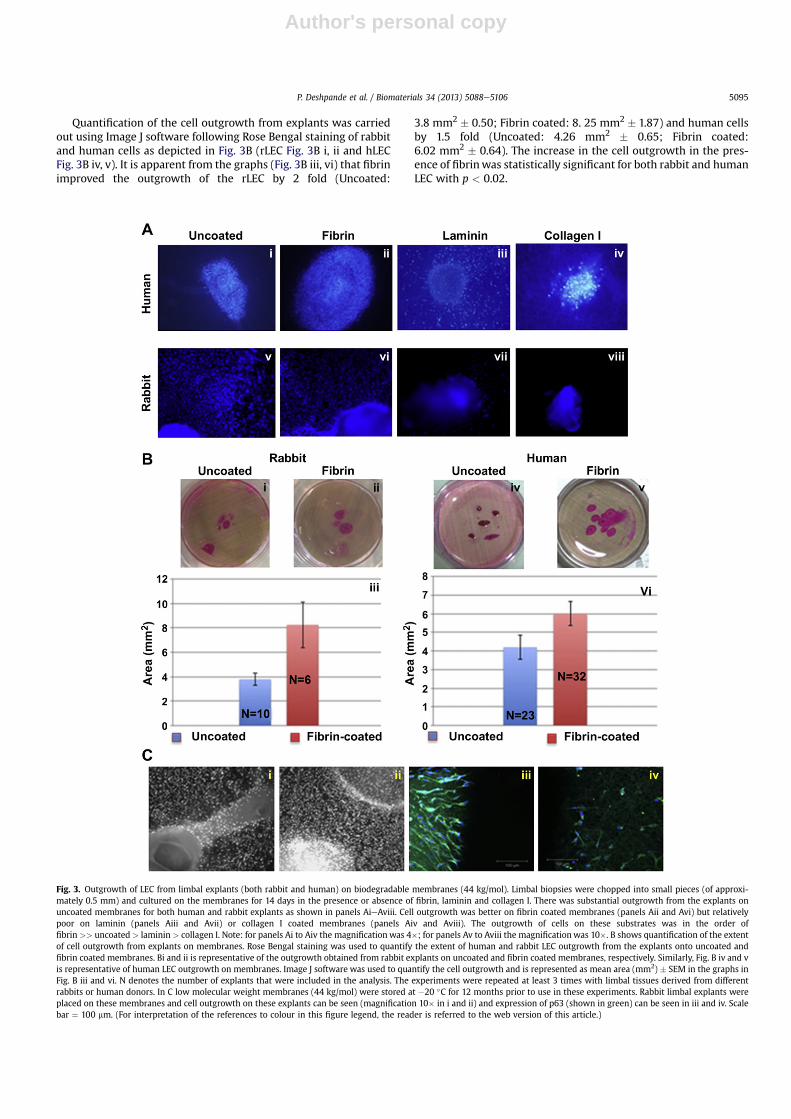
One of the current techniques used for expanding limbal stemcells for transplantation is to culture the epithelial cells fromwholelimbal tissue [3,4,24]. It is thought that since the limbal niche isintact, that the niche cells will provide the necessary cues formaintaining the stem cell population in vitro without the need forfeeder cells. As seen in Fig. 3A, there was abundant cell outgrowthfrom explants on the membranes. Further to determine if the
Table 2Characterisation of the impact of g-irradiation on PLGA membranes prepared from low (44 kg/mol) and high (153 kg/mol) molecular weight polymer including transitiontemperature (Tg), molecular weight (MW), polydispersity (PD), ultimate tensile strength (UTS) and Young’s modulus (E).
153 kg/mol 44 kg/mol
Before spinning Post spinningnon-sterile
Post spinning gammairradiated
Before spinning Post spinningnon-sterile
Post spinning gammairradiated
Tg (�C) 47.67 � 0.13 40.44 � 1.64 38.33 � 1.11 44.50 � 1.77 31.09 � 2.01 30.19 � 1.68MW (�103) 102.45 104.05 27.25 38.76 37.73 26.46PD 1.38 1.86 2.05 1.64 1.72 1.97UTS (N/mm2) e 3.68 � 0.16 4.59 � 0.17 e 4.13 � 0.79 4.96 � 1.76E (N/mm2) e 75.48 � 3.53 72.29 � 2.65 e 62.98 � 4.4 95.58 � 0.98
Table 1.Membrane mass, mean % of weight loss (�SD), fibre diameter (mm � SD) and degradation of non-sterile and g-irradiated electrospun PLGA (50/50) membranes with low(44 kg/mol) and high (153 kg/mol) molecular weight polymer over periods of 6 weeks. The approximate mass of membranes was assessed as follows: membranes of 75e100%of the original mass are indicated as ‘þþþþ’; 50e75% of the original mass as ‘þþþ’; 25e50% of the original mass as ‘þþ’; and 0e25% of the original mass as ‘þ’; no membranedetected is indicated as ‘0’. In the case of breakdown, no degradation also scored as none ¼ 0, some þ or extensive þþ.
PLGA (50/50) scaffolds Time point Scaffold mass Mean % of weightloss (�SD)
Fibre diameter (mm � SD) Degradation
Non-Sterile (MW: 153 kg/mol) Day 0 þþþþ 0.00 2.39 � 1.55 0Week 1 þþþþ 0.00 4.16 � 0.95 0Week 2 þþþþ 4.01 � 0.21 5.28 � 0.84 0Week 3 þþþþ 12.13 � 0.36 6.57 � 1.24 0Week 4 þþþþ 17.41 � 0.39 6.94 � 2.01 0Week 5 þþþ 27.03 � 1.09 7.34 � 1.77 þWeek 6 þþþ 37.50 � 1.21 8.67 � 1.41 þ
Gamma irradiated (MW: 153 kg/mol) Day 0 þþþþ 0.00 2.55 � 0.93 0Week 1 þþþþ 2.51 � 0.29 6.11 � 1.12 0Week 2 þþþþ 4.85 � 0.91 7.84 � 0.52 0Week 3 þþþþ 20.19 � 1.35 12.1 � 1.46 0Week 4 þþþ 39.44 � 1.46 13.72 � 0.82 þWeek 5 þþþ 44.18 � 1.71 11.53 � 1.66 þWeek 6 þþ 73.41 � 2.94 9.21 � 1.02 þþ
Non-Sterile (MW: 44 kg/mol) Day 0 þþþþ 0.00 2.74 � 0.89 0Week 1 þþþþ 2.43 � 0.36 11.51 � 1.11 0Week 2 þþþþ 11.53 � 0.31 10.10 � 1.67 0Week 3 þþþþ 15.26 � 0.54 8.94 � 0.57 0Week 4 þþþþ 22.41 � 0.79 Fibres swollen & merged þWeek 5 þþþ 38.49 � 1.20 Fibres swollen & merged þWeek 6 þþþ 48.32 � 1.20 Fibres swollen & merged þ
Gamma-irradiated (MW: 44 kg/mol) Day 0 þþþþ 0.00 2.76 � 1.25 0Week 1 þþþþ 9.1 � 0.43 10.85 � 1.64 0Week 2 þþþþ 13.81 � 0.47 13.10 � 0.86 0Week 3 þþþþ 21.93 � 1.51 Fibres swollen & merged 0Week 4 þþþ 31.45 � 0.93 Fibres swollen & merged þWeek 5 þþ 70.18 � 2.39 Fibres swollen & merged þþWeek 6 þ 78.43 � 2.73 Fibres swollen & merged þþ
P. Deshpande et al. / Biomaterials 34 (2013) 5088e5106 5093

Author's personal copy
addition of extracellular matrix components would enhance theoutgrowth, the membranes were coated with laminin, collagen I orwith fibrin glue. As shown in Fig. 3A i, the best outgrowth was seenin membranes coated with fibrin when compared to the laminincoated (Panel 3A iii) or uncoated membrane (Panel 3A i).Outgrowth was least on membranes coated with collagen I (Panel3A iv). Similar results were obtained with explant culture of rLEC(Panels 3A v to 3A viii).
The outgrowth from the explantswas graded based on the numberof explants giving out cells and the extent of cell migration on themembranes. As summarised in Table 4 the extent of outgrowthwas inthe following order fibrin >> uncoated > laminin > collagen I. Sinceonlyfibrin improved theoutgrowth fromtheexplants compared to theuncoated membranes, all subsequent experiments were conductedwith fibrin coated membranes using uncoated membranes as acomparison.
Table 3Contact angles of non-sterile and g-irradiated electrospun PLGA (50/50) membraneswith low (44 kg/mol) and high (153 kg/mol) molecular weight polymer using a 3 ml drop ofdeionised water (dH2O), media (DMEM) and PBS. The average value was reported with a standard deviation (�SD).
PLGA (50/50) Contact angle
0 min 5 min 10 min 15 min 20 min 25 min
Non-Sterile (MW: 153 kg/mol) dH2O 99��4.11 92��3.84 84��2.11 69��2.25 38��1.95 33��2.05DMEM 54��1.25 53��3.24 51��2.65 47��1.98 45��2.57 40��3.21PBS 100��2.45 77��3.95 52��2.36 50��1.58 46��3.27 40��2.48
Gamma Irradiated (MW: 153 kg/mol) dH2O 86��2.01 63��1.45 44��2.84 34��1.95 23��1.29 7��2.25DMEM 47��2.54 45��3.21 38��1.54 23��2.67 0� 0�
PBS 95��2.68 32��3.48 0� 0� 0� 0�
Non-Sterile (MW: 44 kg/mol) dH2O 107��3.94 96��1.21 90��4.58 71��2.25 53��2.38 17��1.50�
DMEM 91��3.66 40��2.29 33��3.32 20��1.58 0� 0�
PBS 100��3.24 93��1.25 70��3.68 36��4.01 27��3.98 16��2.54Gamma Irradiated (MW: 44 kg/mol) dH2O 99��1.59 93��1.50 86��1.19 70��2.54 40��1.88 0�
DMEM 103��4.58 96��1.66 77��3.67 63��2.69 30��2.61 0�
PBS 101��1.35 94��2.32 85��2.65 60��2.44 30��3.12 15��4.2
Fig. 2. Growth of isolated limbal epithelial cells (both human and rabbit) on biodegradable membranes (44 kg/mol) compared to growth of cells on hAM for 14 days in the presenceand absence of NIH3T3 feeder cells. The presence of feeder cells promoted the growth of a monolayer of epithelial cells on hAM. Actin staining with phalloidin (Panel A) shows thetypical morphology of the epithelial cells. The cells were positive for CK12 (Panel B), p63 (Panel C) and p63a (Panel D) showing the presence of a mixed population of cells in culture.A similar staining pattern was seen in cells cultured on hAM in the absence of feeder cells (Panels EeH). The marker expression in cells cultured on membranes in the presence orabsence of feeders was comparable to the cells cultured on hAM (Panels IeP). Further, the characteristics of rabbit LEC cultured on membranes were comparable to that of humans(Panels QeX) with the exception that the p63a staining was cytoplasmic rather than nuclear in rabbit cells. Notes: Red: Propidium iodide (Panels A, E, I, M, Q and U); Row 1:magnification 10�; Rows 2,3 and 4: magnification 40�. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
P. Deshpande et al. / Biomaterials 34 (2013) 5088e51065094
Author's personal copy
Quantification of the cell outgrowth from explants was carriedout using Image J software following Rose Bengal staining of rabbitand human cells as depicted in Fig. 3B (rLEC Fig. 3B i, ii and hLECFig. 3B iv, v). It is apparent from the graphs (Fig. 3B iii, vi) that fibrinimproved the outgrowth of the rLEC by 2 fold (Uncoated:
3.8 mm2 � 0.50; Fibrin coated: 8. 25 mm2 � 1.87) and human cellsby 1.5 fold (Uncoated: 4.26 mm2 � 0.65; Fibrin coated:6.02 mm2 � 0.64). The increase in the cell outgrowth in the pres-ence of fibrinwas statistically significant for both rabbit and humanLEC with p < 0.02.
Fig. 3. Outgrowth of LEC from limbal explants (both rabbit and human) on biodegradable membranes (44 kg/mol). Limbal biopsies were chopped into small pieces (of approxi-mately 0.5 mm) and cultured on the membranes for 14 days in the presence or absence of fibrin, laminin and collagen I. There was substantial outgrowth from the explants onuncoated membranes for both human and rabbit explants as shown in panels AieAviii. Cell outgrowth was better on fibrin coated membranes (panels Aii and Avi) but relativelypoor on laminin (panels Aiii and Avii) or collagen I coated membranes (panels Aiv and Aviii). The outgrowth of cells on these substrates was in the order offibrin >> uncoated > laminin > collagen I. Note: for panels Ai to Aiv the magnification was 4�; for panels Av to Aviii the magnificationwas 10�. B shows quantification of the extentof cell outgrowth from explants on membranes. Rose Bengal staining was used to quantify the extent of human and rabbit LEC outgrowth from the explants onto uncoated andfibrin coated membranes. Bi and ii is representative of the outgrowth obtained from rabbit explants on uncoated and fibrin coated membranes, respectively. Similarly, Fig. B iv and vis representative of human LEC outgrowth on membranes. Image J software was used to quantify the cell outgrowth and is represented as mean area (mm2) � SEM in the graphs inFig. B iii and vi. N denotes the number of explants that were included in the analysis. The experiments were repeated at least 3 times with limbal tissues derived from differentrabbits or human donors. In C low molecular weight membranes (44 kg/mol) were stored at �20 �C for 12 months prior to use in these experiments. Rabbit limbal explants wereplaced on these membranes and cell outgrowth on these explants can be seen (magnification 10� in i and ii) and expression of p63 (shown in green) can be seen in iii and iv. Scalebar ¼ 100 mm. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
P. Deshpande et al. / Biomaterials 34 (2013) 5088e5106 5095

Author's personal copy
Finally, to confirm the suitability of membranes stored at�20 �Cto support cell culture, limbal explants were grown on membranesof 44 kg/mol (non-sterilised) which had been stored at �20 �C for12 months. The results clearly show cell outgrowth from explantsonto these membranes after 2 weeks (Fig. 3C i, ii) and that cellsexpressed the proliferation marker p63 (Fig. 3C iii, iv) suggestingthat the membrane is able to support the growth of limbalepithelial cells even after 12 months in storage.
3.6. Characterisation of cells growing out from explants onmembranes
Cells cultured from explants on membranes and hAM for 14days were stained using specific markers to identify the cell types.Rabbit cells on membranes had typical epithelial morphology(Panels 4A and 4E) when cultured on uncoated and fibrin coatedmembranes. There was positive staining for epithelial specific CK3(Panels 4B and 4F) and stem cell markers p63 (Panels 4C and 4G)and p63a (Panels 4D and 4H). There was no perceptible differencein the nature of the cell populations cultured on uncoated or fibrincoated membranes. The hLEC cultured on the PLGA membranes(Panels 4M to 4T; uncoated and fibrin coated) showed similarmarker expression as the rabbit cells. The expression patterns andcell populations identified in hAM cultured (Panels 4I to 4L) andmembrane cultured cells were comparable. It was noted that inrLEC the p63a staining was consistently cytosolic rather than nu-clear, as seen in hLEC, evenwhen an identical staining protocol wasfollowed (Panels 4D and 4H vs Panels 4L, 4P and 4T).
3.7. Label retaining cells on hAM and PLGA membranes
BrdU has been used previously to identify stem cells [25,26]. Asa thymidine analogue, BrdU is taken up specifically by the dividingcells and retained for longer by the slow dividing or quiescent stem
Table 4Extent of cell outgrowth from explants on PLGA membranes.
Matrix coating Human (n � 3) Rabbit (n � 3)
Uncoated þþ þþFibrin þþþ þþþLaminin þ/� þ/�Collagen I e e
The extent of cell outgrowth from explants examined in 3 or more experiments wasinitially scored qualitatively as.� ¼ No outgrowth.�/þ ¼ Some outgrowth.þþ ¼ Outgrowth from many explants.þþþ ¼ Outgrowth from most explants.(Quantitative assessment of the influence of fibrin on outgrowth was then assessedin subsequent experiments.)
Fig. 4. Characterization of cells grown out from rabbit and human limbal explants cultured on membranes: Explant culture of LEC was carried out for 14 days on hAM and onuncoated and fibrin coated membranes. The rabbit cells cultured on uncoated and fibrin coated membranes showed typical epithelial morphology (Panels A and E). Markerexpression showed the presence of both differentiated (Panels B and F) and stem cell populations (Panels C, G, D and H). Similar expression patterns were also noted in the humanLEC cultured on hAM (Panels IeL) and membranes (Panels MeT). Magnification �20.
P. Deshpande et al. / Biomaterials 34 (2013) 5088e51065096

Author's personal copy
cells while it is lost from the rapidly dividing cells. This allows forthe identification and quantification of the stem cell population incultured cells or in the whole tissue.
The number of label retaining cells at the end of the pulse period(24 h) and at the end of the chase period (7 days) was quantified bycounting the number of BrdU positive cells on hAM and on mem-branes. The percent positive cells were calculated by dividing theBrdU positive cells by the total number of cells (identified usingpropidium iodide) in the same field of view as shown in Fig. 5. Thepercent BrdU positive cells on hAM was 52.1 � 3% at the end of thepulse, which reduced to 34.2 � 1.9% by day 14. On the membrane,the percent BrdU positive cells was 47.5 � 2.7% which reduced to29.3 � 2.1% by day 14. This decrease in label retaining cells at theend of chase period was statistically significant (p < 0.0001) whilethere was no significant influence of the substrate (hAM vs mem-brane) on the number of label retaining cells (p ¼ 0.125).
3.8. Transfer of cell suspensions and cells grown out from explantson TEC membranes to cornea
Experiments then focussed on whether cells would leave themembranes to attach to the rabbit cornea model. Fig. 6A shows therabbit organ culture model and the experimental design (Fig. 6B)for culturing cells or explants on the membranes prior to placingthem on the corneas for 4 weeks of culture.
In examining cell transfer to the cornea both low and high MWmembranes were used and the cultured cells were placed cell sideup or cell side down and also examined in submerged culture andat an aireliquid interface.
Fig. 6CeL shows that after 4 weeks of culture of rabbit cells onmembranes on the rabbit cornea models there was still a consid-erable amount of membrane remaining as can be seen from theloosely connected layers seen post sectioning and histology butthere was clear attachment of some of these rabbit cells initiallycultured on the membranes onto the stroma of the corneas in allcases. There was no difference in cell transfer whether the mem-branes were placed cell side up (Fig. 6CeF) or cell side down(Fig. 6GeJ) however there was a difference in the distribution ofcells within the membranes whether the corneas were placed ataireliquid interface (Fig. 6C, D, G, H) or submerged (Fig. 6 E, F, I, J).Submerged, the cells were seen spread throughout the layers of themembrane. With the aireliquid culture most of the cells were seenin the lower layers of the membrane attached to the cornea.
In the case of using the high MW membranes there were a lownumber of cells transferred compared to using the lower MW in allcases whether the membranes were placed cell side up at aireliquid interface (Fig. 6K, L) and submerged or down at aireliquidinterface and submerged (not shown).
Fig. 6 MeV shows results for outgrowth of cells from explantsand their transfer to the cornea. After 4 weeks, cells had grown out
Fig. 5. Quantification of label retaining cells cultured from human limbal explants: Explant culture of LEC was carried out for 14 days on hAM and on fibrin coated membranes. Onthe 7th day of culture, BrdU (5 mM) was added to the cells and incubated for 24 h (pulse). The cells were further cultured in BrdU-free medium for 7 days (chase). Cells were fixedeither at the end of the pulse or chase period and stained for BrdU positive cells. The number of positive cells was counted from images as shown in the figure. Percent labelretaining cells at the two time points was calculated by dividing the number of BrdU positive cells by the total number of cells identified using propidium iodide and plotted in thegraph. Magnification �10.
P. Deshpande et al. / Biomaterials 34 (2013) 5088e5106 5097

Author's personal copy
Fig. 6. Transfer of cells from membranes onto ex vivo rabbit cornea model. A shows an ex vivo rabbit cornea model (i), a histology section of an unwounded cornea (ii) and a wounded (epithelium removed) cornea (iii). B shows theexperimental design of delivering the cells to the cornea via the membrane. C shows H&E of transfer of rabbit limbal cells cultured on membranes for 5 days then transferred onto the cornea organ culture model for 4 weeks. Cells wereplaced on the low molecular weight (44 kg/mol) membranes cell-side up, at either an air liquid interface (C,D) or submerged in medium (E,F) and also placed cell-side down, at either an aireliquid interface (G,H) or submerged inmedium (I,J) and high molecular weight (153 kg/mol) membranes cell side up at aireliquid interface (K,L). Explants were placed on the low molecular weight (44 kg/mol) membranes cell-side up, at either an aireliquid interface (M,N)or submerged in medium (O,P) and also placed cell-side down, at either an aireliquid interface (Q,R) or submerged in medium (S,T) and high molecular weight (153 kg/mol) membranes cell side up at aireliquid interface (U,V). Imagesshown are a section of the cornea taken at 40� magnification (C,E,G,I,K,M,O,Q,S,U), and a panoramic view of the whole corneas (D,F,H,J,L,N,P,R,T,V) on the membranes. Insets (M,O,Q,S,U) show images taken at 10� magnification.
P.Deshpande
etal./
Biomaterials
34(2013)
5088e5106
5098

Author's personal copy
from the explants in all cases for both the low (Fig. 6MeT) and high(Fig. 6U,V) MWmembranes. In the case of the lowMWmembrane,there appeared to be more cells growing out from the membranesin the cell side up (Fig. 6MeP) position rather than in the cell sidedown position (Fig. 6QeT). However there did not appear to be anydifference in cell transfer to the corneas whether the corneas wereplaced at an aireliquid interface (Fig. 6M, N, Q, R) or submerged(Fig. 6O, P, S, T). In the case of transfer using the high MW mem-branes the cells present did not appear to be as many as seen usingthe lower MW. This was true for all conditions whether the ex-plants were placed at an aireliquid interface or submerged andwith explant side up or down. As a representative example, cellstransferred with explants facing up at an aireliquid interface and isshown in Fig. 6U, V. For this reason immunohistochemistry wascarried out only on the corneas treated with the lower MWmembrane.
3.9. Characterisation of cells transferred to organ culture cornea
The cells cultured on the lower molecular weight membranesand then transferred onto the organ culture showed expression ofcytokeratin 3 which is a differentiation marker (Fig. 7AeD) as wellas p63 which is a stem cell and transient amplifying cell marker(Fig. 7 EeH) suggesting that these cells retain the stem/epithelialcell phenotype after culture on these membranes and transfer tothe cornea models.
To establish whether the transferred cells were well adhered tothe cornea, the cells were stained with vinculin, a focal adhesionmarker. Expression of vinculin was observed in the cytoplasm ofthe cells and appeared in nearly all the cells within the membraneand on the cornea (Fig. 7IeL).
Similarly for the cells that had grown out of the explants ontothe membranes and then onto the corneas these also expressedcytokeratin 3 confirming that these cells are limbal epithelial cells(Fig. 7MeP) and they also expressed the p63 marker (Fig. 7QeT)and vinculin (Fig. 7UeX) exactly as for the suspension cultured cellsshown in Fig. 7AeL.
Scanning electron microscopy showed a multilayer of cells onthe surface of the unwounded cornea (Fig. 8A) and there did notappear to be many cells on the wounded cornea (Fig. 8B). Mem-branes could be clearly seen on the corneas in the case of cellscultured on the corneas placed cell side up (Fig. 8C, D) and down(8E, F). The same was seen for the explants on the membranes(Fig. 8GeJ). In some cases, the explants on the membranes could beseen (Fig. 8G). There did not appear to be any obvious difference inseeding the cells/explants on the membrane cell side or cell sidedown. The cells in all cases appeared to be well adhered to thecornea.
3.10. Effect of temperature, humidity and sterilisation on ability ofTEC membranes to survive long term storage
The storage of membranes under a range of temperatures(�80 �C, �20 �C, þ4 �C, room temperature, þ37 �C moistincubator,þ37 �C dry oven andþ50 �C) was investigated looking atmembranes stored in the presence of desiccant for periods of up to12months. On the basis of SEM images, fibre integrity was assessedas fully intact (þþþ), some indication of fibre swelling (þþ), fibresmerging (þ) or no evidence of intact fibres remaining (�). Anexample of these is shown in Fig. 9 for membranes stored for a yearat �20 �C in the presence of desiccant. There was no visible ormeasurable change in fibre diameter irrespective of MW orwhether membranes were g-irradiated or not. Similar results wereseen at �80 �C (not shown).
Fig. 10 summarises all storage experiments for these biode-gradable membranes. The inclusion of silica orange desiccantproved useful in seeing changes in water content in the environ-ment. At 50 �C the 153 kg/mol membranes were clearly breakingdown by 2 weeks whether sterilised or not and the 44 kg/mol hadcompletely collapsed. At 37 �C (under dry conditions) membranesshowed some evidence of breakdown by 2 weeks and under moistconditions fibres had completely merged by 2 weeks. Placingmembranes within a moist environment at 37 �C resulted in a rapidcollapse of the fibres, whereas the same membranes maintained inculture media (with a bicarbonate pH buffering system) survivedfor several weeks both with and without cells. In the absence of anybuffering system it is likely that these membranes became highlyacidic due to the presence of CO2 and H2O. At room temperature(21 �C in our laboratory) membranes started to take up water andbecome brittle after 2 months. At 4 �C g-irradiated membraneswere stable for 3 months (both 44 kg/mol and 153 kg/mol). Thenon-sterile membranes were also stable for 3 months (for the153 kg/mol) and less stable for the 44 kg/mol (only 1 month).
For membranes stored for a year at �20 �C in the presence ofdesiccant there was no visible or measurable change in fibrediameter irrespective of MW or whether membranes were g-irra-diated or not. Similar results were seen at �80 �C.
4. Discussion
This study describes the development, characterisation andin vitro evaluation of a biodegradable synthetic polymer membraneas an alternative for the use of hAM for delivering cultured (andexplant derived) LECs for corneal regeneration. The study demon-strates that it is possible to achieve transfer of LECs expanded onthese simple synthetic PLGA membranes using the suspensionculture technique or from cells grown out from explants placed onthese membranes, onto an ex vivo wounded rabbit cornea model.Membranes could be produced and sterilised to breakdown in apredictable fashion approximately 6 weeks once cultured withcells. Additionally, they could be stored dry for at least a yearat �20 �C and still support cell outgrowth from explants.
Previous work from our laboratory has shown that the ratio ofPLA to PGA can be altered to achieve predictable breakdown ofmembranes both in vitro and in vivo [18]. For the current purposeswe aimed to have the membranes breakdown over 4e6 weeks,leaving epithelial cells attached to the corneal stroma. This is theapproximate time that the hAM takes to breakdown in vivo whentransplanted in the clinic (clinical observation). Accordingly amembrane of 50% PLA and 50% PGAwas produced with fibres of 2e3 mm in diameter and a thickness of 50 mm.
From the current study it was clear that membrane breakdownbegan when fibres began to take up moisture irrespective ofwhether cells were present or not. Degradation, marked by pre-dictable swelling of the PLGA fibres, was evident by twoweeks withcomplete breakdown occurring by six weeks in vitro (see Fig. 1).Initial experiments then looked at the ability of the membranes tosupport the growth of LECs using cells isolated by enzyme digestionand cultured in the presence or absence of feeder cells. We foundthat it took about two weeks for the cells to form a confluent layeron the membranes in the laboratory and membranes could behandled well for this period. The cells were cultured for 5 days onthe membranes prior to transfer on to the cornea. Membranescould be handled and transferred to cornea models quite easily atthis time.
As can be seen in Fig. 2, the cells cultured on hAM in the pres-ence of feeder cells gave rise to a monolayer of epithelial cellsexhibiting typical morphology. These cultured cells comprised of amixture of differentiated (CK3/12 positive), transient amplifying
P. Deshpande et al. / Biomaterials 34 (2013) 5088e5106 5099

Author's personal copy
Fig. 7. Characterisation of cells transferred from membranes to ex vivo rabbit cornea. A shows the characterisation of cells cultured on low molecular weight membranes (44 kg/mol) post transfer onto corneas. Figures show expression of cytokeratin 3 (AeD), p63 (EeH) and vinculin (I-,L) (show in green) post transfer onto corneas. Cells were cultured onmembranes for 5 days and then transferred onto corneas for 4 weeks. B shows characterisation of cells grown out from explants placed on lowmolecular weight membranes (44 kg/
P. Deshpande et al. / Biomaterials 34 (2013) 5088e51065100

Author's personal copy
and stem cells (p63, p63a positive cells). It is well known that whenthe epithelial stem cells are isolated by separation from their nichethey require feeder cells to provide support to maintain the stem-ness of these cells [22,23,27]. The feeder cells support the expan-sion and maintenance of the stem cell population by providingthem with the necessary growth factors and extracellular matrixproteins [28]. Therefore, the absence of feeder cells can lead todifferentiation of the stem cells resulting in their depletion. In ourexperiments, we found that when cells were seeded at a density of1.25�105 cells/cm2 that cells expressing p63awas retained in boththe presence and absence of feeder cells. At this cell density, theabsence of feeder cells did not seem to result in a complete loss ofthe stem cell population. It was observed, however, that there was asubstantial (roughly 40%) decrease in the p63a positive cells infeeder-free suspension cultures of LECs when compared to thecultures with feeder cells. Together these results show that themembranes provide a good substrate for the LECs to grow on. Moreimportantly, the finding that the membranes are able to maintainthe stem cell population in vitro is critical given that the aim oftransplantation is to correct the stem cell deficiency.
Growth inhibited mouse NIH3T3 cells are the most commonlyused feeder cells and a specific strain of these cells (NIH3T3-J2) hasbeen FDA approved for use in the clinic. These cells have beenshown to support the maintenance of stem cells in vitro and havebeen used in the expansion of the LECs for transplantation in pa-tients [29]. To reduce the use of animal derived cells and productsfor the expansion of the limbal stem cell, our group has been usingan explant culture technique for culturing the LEC for trans-plantation [3,11,30]. In this technique the limbal epithelial cells arenot separated from their niche therefore do not require the use offeeder cells for maintaining the stem cell population. This makes ita much simpler technique for establishing LEC cultures comparedto the feeder culture technique. A study looking at the long termoutcome of transplantation surgeries conducted with explantcultured LECs showed these results to be comparable to theoutcome achieved using feeder cells [11,29].
A key contribution of this study however is a further develop-ment and simplification of LEC transplantation. We have recentlydemonstrated that by using an explant culture technique it ispossible to completely remove the intermediate step of cellexpansion in the laboratory, allowing the surgeon to use limbalexplants on an amniotic membrane in a single stage operation intheatre for patients with unilateral LSCD [4]. The significance of thisis that it potentially opens up the technique to many moreophthalmology centres worldwide. The substitution of a syntheticmembrane for a human donor tissue will make this techniquefurther accessible to centres which do not have access to tissuebanks in addition to making the procedure safer for patients thanusing a donor hAM.
Accordingly we next determined the ability of the PLGA mem-branes to support the outgrowth of LECs from intact limbal ex-plants. We found that there was good outgrowth from the explants,which further improved in the presence of fibrin glue but not whenthe membranes were coated with collagen I or laminin (Fig. 3A).Quantification of the extent of cell outgrowth using Rose Bengalstain showed that the outgrowth in the presence of fibrin wassignificantly more when compared to the uncoated membrane(Fig. 3B). This improvement in outgrowth with fibrin glue is to beexpected since it has been shown that fibrin alone can be used as asubstrate for culturing these cells for transplantation [1]. More
importantly, these results show that the addition of fibrin glue tothe membranes is not detrimental to the growth of the limbalepithelial cells on the membranes. This is of particular importancebecause fibrin glue is commonly used in the clinic not only forsecuring the hAM to the corneal surface without sutures but alsofor adhering the limbal explants to the hAM (as was recentlydescribed in simple limbal epithelial transplantation) [4].
Marker expression studies were then conducted to characterisethe cells cultured from explants on hAM and membranes. The cellswere stained for differentiated and stem cells using antibodiesagainst CK3/CK12, p63 and p63a respectively. Fig. 4 shows a mixedpopulation of cells, similar to hAM, was present on the membranesboth in the presence and absence of fibrin glue. The positivestaining with specific stem cell marker confirms the ability of thePLGA membranes to provide sufficient support to maintain thestem cells in vitro similar to hAM. We noticed that p63a staining inthe rabbit LEC was exclusively cytoplasmic while in the human LECit was found to be exclusively nuclear. These discrepancies werenoted despite the use of similar protocols for staining the cells fromboth species. To get an estimate of the stem cell population incultures established on the fibrin coated membranes and hAM, weused the BrdU pulse chase technique. This is an accepted techniquefor identifying stem cells since these cells are relatively slowdividing and therefore retain the label longer allowing for theiridentification in culture [26]. Fig. 5 shows that approximately 50%of the cells took up the label during the pulse period, indicating thepercentage of cells in the S-phase of cell cycle, and this proportionwas reduced to about 30% at the end of the chase period of 7 days.Previous studies on explant derived limbal cells cultured on hAMestimated the percent of label retaining cells to be between 11 and50% at the end of 7 days of chase [25,31]. The main reason for thehuge difference in these estimates can be attributed to the varyingpulse periods of 1 daye3 days between studies. Increasing thechase to 21 days further reduced this number tow2e4% [25] whichis very close to what is estimated to be the actual stem cell popu-lation in the limbus [32]. This decrease in the percent of cells couldbe due to the fact that rapidly dividing cells such as the transientamplifying cells also take up BrdU during incubation but lose thenucleotide with time due to repeated cell proliferation. In thisexperiment, at the end of the pulse chase periods the percentage ofBrdU positive cells were comparable between cultures establishedon hAM and on syntheticmembranes. This allows us to suggest thatthe membrane is similar to the hAM in supporting the growth andmaintenance of the stem cell population.
Different groups using the amniotic membrane have reporteddifferent success rates [27,33,34]. While there may be many vari-ables involved, some of this variation may be due to variability inthe hAM used e this may relate to the initial location of the am-niotic membrane used, its processing and storage [35,36] as well asany donor variation [35,37].
Consequently, there have been a number of studies seeking toreplace the use of hAM to treat LSCD. These include the use ofcontact lenses [20,38], both coated and non-coated, the use ofnatural polymers such as recombinant cross-linked collagen [13],fish scale collagen [39], fibrin [1], myogel [40], silk [41] and syn-thetic polymers such as poly (N-isopropylacrylamide) [14], acopolymer of poly(N-isopropylacrylamide-co-n-butyl methacry-late) and polyethylene glycol (PEG) [42]. At the time of writinghowever, only two methodologies are in routine clinical use e
fibrin [1] and amniotic membrane [4,27,43e45].
mol) post transfer onto corneas. Explants were placed on the membranes and then subsequently placed on corneas and cultured for 4 weeks. Images show expression of cytokeratin3 (MeP) and P63 (QeT) and vinculin (UeX) (shown in green). Membranes were placed cell side uppermost or cell side down at aireliquid interface or submerged. Cells have beencounterstained with propidium iodide (red) and magnification is 40�. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version ofthis article.)
P. Deshpande et al. / Biomaterials 34 (2013) 5088e5106 5101

Author's personal copy
With amniotic membrane, the most abundantly used substratefor LEC transplantation, the initial success rate of deliveringcultured LEC is very high, but a recent study by Sangwan et al. [4]shows that long term results are less encouraging. As acknowl-edged earlier, there may be many factors which explain how
patients vary in their long term response to the treatment e theaetiology of the condition that led to stem cell loss for example,may be a major factor and the state of the eye pre-transplantationand the inflammatory response to the cultured LEC cells. Also, thesuccess rate is thought to be dependent on the type of cells used to
Fig. 8. Scanning electron microscopy of unwounded cornea (A), wounded cornea (B), cells cultured on membranes and then placed on corneas in the up position and cultured ataireliquid interface (C) and submerged (D) and in the down position and then cultured at aireliquid interface (E) and submerged (F). Explants were also placed on the membranesand then placed on corneas in the up position and cultured at aireliquid interface (G) and submerged (H) and in the down position cultured at aireliquid interface (I) and sub-merged (J). Scale bar is 100 mm. In all cases cells or explants were placed on membranes and subsequently placed on corneas and cultured for 4 weeks.
P. Deshpande et al. / Biomaterials 34 (2013) 5088e51065102

Author's personal copy
treat LSCD. Patient’s own oral mucosal [14] in the case of bilateralinjuries have been used but the use of the oral cells has not beenvery successful leading to neovascularisation or stromal haze[46,47].
As stated previously we confirmed that cells would transferfrom the membranes to a cornea model (see Fig. 6). After 4 weeksin culture the membranes were still visible as several layers ofloosely connected fibres containing cells throughout them but inall cases there were several layers of limbal cells attached to thecornea. Immunohistochemistry confirmed that these had differ-entiation markers indicative of limbal cells and proliferationmarkers (Fig. 7). This mixed population of cells would contain
transiently amplifying cells and stem cells. Adhesion of the cells tothe basement membrane is also an important factor. Fig. 7 showsthe expression of vinculin, a structural protein that has beenshown to increase during the healing process [48] and is found incadherin mediated cellecell and integrin mediated cell-matrixadhesion complexes [49,50]. Vinculin is usually expressed at thecytoplasmic side of the focal contacts but in newly adhering,motile cells which lack focal adhesions it is visible in the cyto-plasm and as the cells spread, they develop focal adhesions [51].As this can be seen in the transferred cells in this study it suggeststhat the cells are still motile and have not anchored completely tothe basement membrane.
Fig. 9. Comparison of fibre integrity of non-sterile and g-irradiated electrospun PLGA (50/50) membranes with low (44 kg/mol) and high (153 kg/mol) molecular weight polymerat �20 �C during storage over periods of 12 months, scale bar ¼ 20 mm. Changes in water absorption by membrane were detected using silica orange desiccant.
P. Deshpande et al. / Biomaterials 34 (2013) 5088e5106 5103

Author's personal copy
As the methodology of LEC culture and transplantation hasevolved over the years, one assumption has been that it is necessaryto transfer an intact, well organised, multi-layered epithelium forclinical success. We suggest that this is not essential. Other studiesbased on skin for example show that stratification can happenin vivo on patient, if sufficient stem cells with proliferative capacityare transplanted [52] This is true also for the corneal epitheliumwhere we have previously shown that limbal cells transferred as amonolayer were able to stratify in vivo [53]. In this study wediscovered that irrespective of the orientation of the cells in themembrane in all cases cells migrated through the layers of themembrane and formed a multicellular layer on the underlyingcornea. This suggests that the membrane provides a good envi-ronment for cell proliferation and migration without the cellsdifferentiating so far that they are unable to attach to the surface ofthe cornea.
Of particular relevance to this point, recent work in LV Prasadused freshly excised explants placed directly on the amnioticmembrane with fibrin glue in theatre. Cells grew out from the ex-plants, achieving a confluent epithelium by 6 weeks which wasshown to be stable even after 9 months of surgery. Using the ex-plants directly on ready-to-use PLGA membrane would completelyeliminate the need to expand cells in the laboratory and would alsoeliminate the requirement of the amniotic membrane. In the cur-rent study we show that we have obtained a multi-layeredepithelium within 4 weeks of culture in vitro. We anticipate that
explants placed on themembrane and held in place with fibrin glue(possibly protected by a bandage contact lens) will regenerate anepithelium as was the case for explants placed on the amnioticmembrane.
With respect to the production of a synthetic polymer there areseveral key issues which need to be addressed before membranescan be used clinicallyethey either need to be produced understerile conditions or, more economically viable, sterilised using anaccepted sterilisation methodology. As we are aware that anysterilisation methodology is likely to impact negatively on theseand other biomaterials we compared polymers of two molecularweights (153 kg/mol and 44 kg/mol).
g-irradiation was chosen as the preferred method of steri-lisation. The choice of sterilisation methodology for polymers isoften chosen depending on their applications. The common steri-lisation methods are ethylene oxide (EO) gas [54], dry heat [55],steam [56], organic solvents (ethanol) [57], plasma treatment [58]and gamma irradiation [59]. Typical sterilisation methods areoften not suitable for polymer membranes [60]. For example,autoclaving and ethylene oxide treatment have been shown toadversely affect polymer structures with deformation at elevatedtemperatures, and deterioration due to decreased molecularweight [54,60]. Similarly dry heat and steam sterilisation can causeextensive degradation [55,56]. Plasma treatment sterilisation usedrecently may change the structure of the polymer due to the heatgenerated during the process [58]. Ethanol is a convenient method
Fig. 10. Effect of temperature and length of storage and g-irradiation on integrity of PLGA (50/50) membranes (both 153 and 44 kg/mol) examined over 12 months of storage.Membrane integrity was scored as fully intact fibres (þþþ), some fibre swelling (þþ), fibre merging (þ) or no intact fibres (�).
P. Deshpande et al. / Biomaterials 34 (2013) 5088e51065104

Author's personal copy
to sterilise membranes for experimental use but this is not a rec-ognised sterilisation methodology that can be taken to the clinic.Peracetic acid has also been tested. This is clinically approved and isa wet-sterilisation methodology, but this also reduces membranestrength [60].
In this study we have shown that g-irradiation does not affectthe interaction of the cells with the membranes. There is a reduc-tion in terms of molecular weight, transition temperature andbiomechanical properties but fortunately in producing carriermembranes for cornea tissue engineering these effects of g-irra-diation are an advantage as they slightly increase the rate ofbreakdown of the PLGA membranes without affecting culture ofcells on these membranes.
Finally, for a membrane to have a wide distribution it is neces-sary for it to be stored for a considerable amount of time to beeconomically viable. Accordingly, we examined the storage of thedry membranes with desiccant at a range of temperatures, delib-erately chosen to span very cold (�80 �C and �20 �C) through tovery warm (50 �C). The results were very clear as summarised inFigs. 9 and 10. Membranes were stable at �20 �C and �80 �C for atleast 12months (the longest period examined in this investigation).At progressively higher temperatures the membranes were stablefor shorter periods of time. It was interesting to note that mem-branes stored at 37 �C under dry conditions were stable for 3e4weeks whereas those at 37 �C (moist incubator) had completely losttheir structure by twoweeks. In contrast membranes cultured withcells in media (buffered to pH 7.4) at 37 �C (moist incubator) for upto 6 weeks were more stable. We suggest this is explained by thepH bicarbonate buffered environment of the cell cultures (pH 6.8e7.2), whereas the membranes kept at 37 �C under moist conditionshad no pH buffering and broke down very quickly. We suggest thatthis is due to a build up of carbonic acid on the membrane, thusleading to acid catalysed hydrolysis of the polymer membrane.
5. Conclusion
Our study shows that a simple synthetic, electrospunmembraneof PLGA sterilised with g-irradiation can support the growth oflimbal epithelial cells and their transfer to the cornea delivering apopulation of cells of mixed differentiation and proliferativephenotype. Under such circumstances membranes began to visiblybreakdown after 2e4 weeks, losing 75% of their weight by 6 weeksin vitro. Further, membranes could be stored dry at �20 �C(or �80 �C) for at least a year and retained the ability to supportoutgrowth of limbal explants. This synthetic sterilised carrier offersa lower disease risk alternative for clinical use and wider accessi-bility, particularly when combined with tissue explant outgrowthto simplify LEC transplantation so that it may become accessible tomore surgeons and help benefit more patients.
Acknowledgements
We would like to thank the Wellcome Trust for funding thiswork through an Affordable Healthcare in India award, ProfessorDorairajan Balasubramanian and Dr Ilida Ortega Asencio for theirsupport, Dr Nicola H Green for her help with confocal microscopyand Alison Weather from Hook Farm for supplying the rabbit eyes.
References
[1] Rama P, Bonini S, Lambiase A, Golisano O, Paterna P, De Luca M, et al.Autologous fibrin-cultured limbal stem cells permanently restore the cornealsurface of patients with total limbal stem cell deficiency. Transplantation2001;72(9):1478e85.
[2] Koizumi N, Inatomi T, Suzuki T, Sotozono C, Kinoshita S. Cultivated cornealepithelial stem cell transplantation in ocular surface disorders. Ophthal-mology 2001;108(9):1569e74.
[3] Fatima A, Sangwan V, Iftekhar G, Reddy P, Matalia H, Balasubramanian D,et al. Technique of cultivating limbal derived corneal epithelium on humanamniotic membrane for clinical transplantation. J Postgrad Med 2006;52(4):257e61.
[4] Sangwan VS, Basu S, MacNeil S, Balasubramanian D. Simple limbal epithelialtransplantation (SLET): a novel surgical technique for the treatment of uni-lateral limbal stem cell deficiency. Br J Ophthalmol 2012;96(7):931e4.
[5] Dua HS, Azuara-Blanco A. Autologous limbal transplantation in patients withunilateral corneal stem cell deficiency. Br J Ophthalmol 2000;84(3):273e8.
[6] Dua H, Saini J, Azuara-Blanco A, Gupta P. Limbal stem cell deficiency: concept,aetiology, clinical presentation, diagnosis and management. Curr Ophthalmol2000;48(2):83e92.
[7] Brown S, Tragakis M, Pearce D. Corneal transplantation for severe alkali burns.Trans Am Acad Ophthalmol Otolaryngol 1972;76(5):1266e74.
[8] Brown S, Bloomfield S, Pearce D. A follow-up report on transplantation of thealkali-burned cornea. Am J Ophthalmol 1974;77(4):538e42.
[9] Pellegrini G, Traverso CE, Franzi AT, Zingirian M, Cancedda R, De Luca M. Long-term restoration of damaged corneal surfaces with autologous cultivatedcorneal epithelium. Lancet 1997;349(9057):990e3.
[10] Basu S, Fernandez MM, Das S, Gaddipati S, Vemuganti GK, Sangwan VS.Clinical outcomes of xeno-free allogeneic cultivated limbal epithelial trans-plantation for bilateral limbal stem cell deficiency. Br J Ophthalmol2012;96(12):1504e9.
[11] SangwanVS, BasuS,Vemuganti GK, SejpalK, SubramaniamSV, BandyopadhyayS,et al. Clinical outcomes of xeno-free autologous cultivated limbal epithelialtransplantation: a 10-year study. Br J Ophthalmol 2011;95(11):1525e9.
[12] Dravida S, Gaddipati S, Griffith M, Merrett K, Madhira S, Sangwan V, et al.A biomimetic scaffold for culturing limbal stem cells: a promising alternativefor clinical transplantation. J Tissue Eng Regen Med 2008;2(5):263e71.
[13] Levis HJ, Brown RA, Daniels JT. Plastic compressed collagen as a biomimetic sub-strate forhuman limbal epithelial cell culture. Biomaterials2010;31(30):7726e37.
[14] Nishida K, Yamato M, Hayashida Y, Watanabe K, Yamamoto K, Adachi E, et al.Corneal reconstruction with tissue-engineered cell sheets composed ofautologous oral mucosal epithelium. NEJM 2004;351(12):1187e96.
[15] Zajicova A, Pokorna K, Lencova A, Krulova M, Svobodova E, Kubinova S, et al.Treatment of ocular surface injuries by limbal and mesenchymal stem cellsgrowing on nanofiber scaffolds. Cell Transplant 2010;19(10):1281e90.
[16] Deshpande P, McKean R, Blackwood KA, Senior RA, Ogunbanjo A, Ryan AJ,et al. Using poly(lactide-co-glycolide) electrospun scaffolds to deliver culturedepithelial cells to the cornea. Regen Med 2010;5(3):395e401.
[17] Mohan M, Angra S. Vicryl suture in ophthalmic surgery. Indian J Ophthalmol1979;27(3):24e8.
[18] Blackwood KA, McKean R, Canton I, Freeman CO, Franklin KL, Cole D, et al.Development of biodegradable electrospun scaffolds for dermal replacement.Biomaterials 2008;29(21):3091e104.
[19] Selim M, Bullock AJ, Blackwood KA, Chapple CR, MacNeil S. Developingbiodegradable scaffolds for tissue engineering of the urethra. BJU Int2011;107(2):296e302.
[20] Deshpande P, Notara M, Bullett N, Daniels JT, Haddow DB, MacNeil S. Devel-opment of a surface-modified contact lens for the transfer of cultured limbalepithelial cells to the cornea for ocular surface diseases. Tissue Eng Part A2009;15(10):2889e902.
[21] Mariappan I, Maddileti S, Savy S, Tiwari S, Gaddipati S, Fatima A, et al. In vitroculture and expansion of human limbal epithelial cells. Nat Protoc 2010;5(8):1470e9.
[22] Balasubramanian S, Jasty S, Sitalakshmi G, Madhavan HN, Krishnakumar S.Influence of feeder layer on the expression of stem cell markers in culturedlimbal corneal epithelial cells. Indian J Med Res 2008;128(5):616e22.
[23] Sharma SM, Fuchsluger T, Ahmad S, Katikireddy KR, Armant M, Dana R, et al.Comparative analysis of human-derived feeder layers with 3T3 fibroblasts forthe ex vivo expansion of human limbal and oral epithelium. Stem Cell Rev2012;8(3):696e705.
[24] Nakamura T, Inatomi T, Sotozono C, Koizumi N, Kinoshita S. Successful pri-mary culture and autologous transplantation of corneal limbal epithelial cellsfrom minimal biopsy for unilateral severe ocular surface disease. Acta Oph-thalmol Scand 2004;82(4):468e71.
[25] Kim HS, Jun Song X, de Paiva CS, Chen Z, Pflugfelder SC, Li DQ. Phenotypiccharacterization of human corneal epithelial cells expanded ex vivo fromlimbal explant and single cell cultures. Exp Eye Res 2004;79(1):41e9.
[26] Meller D, Pires RT, Tseng SC. Ex vivo preservation and expansion of humanlimbal epithelial stem cells on amniotic membrane cultures. Br J Ophthalmol2002;86(4):463e71.
[27] Sangwan V, Matalia H, Vemuganti G, Fatima A, Ifthekar G, Singh S, et al.Clinical outcome of autologous cultivated limbal epithelium transplantation.Indian J Ophthalmol 2006;54(1):29e34.
[28] Takagi R, Yamato M, Kushida A, Nishida K, Okano T. Profiling of extracellularmatrix and cadherin family gene expression in mouse feeder layer cells: typeVI collagen Is a candidate molecule inducing the colony formation of epithelialcells. Tissue Eng Part A 2012;18(23e24):2539e48.
[29] Rama P, Matuska S, Paganoni G, Spinelli A, De Luca M, Pellegrini G. Limbalstem-cell therapy and long-term corneal regeneration. NEJM 2010;363(2):147e55.
P. Deshpande et al. / Biomaterials 34 (2013) 5088e5106 5105

Author's personal copy
[30] Sangwan VS, Vemuganti GK, Iftekhar G, Bansal AK, Rao GN. Use of autologouscultured limbal and conjunctival epithelium in a patient with severe bilateralocular surface disease induced by acid injury: a case report of unique appli-cation. Cornea 2003;22(5):478e81.
[31] Galindo EE, Theiss C, Pauklin M, Wettey FR, Steuhl KP, Meller D. Correlation ofcell cycle kinetics with p63 expression in human limbal epithelial cellsexpanded on intact human amniotic membrane. Ophthalmic Res 2009;41(2):83e90.
[32] Pellegrini G, Golisano O, Paterna P, Lambiase A, Bonini S, Rama P, et al.Location and clonal analysis of stem cells and their differentiated progeny inthe human ocular surface. J Cell Biol 1999;145(4):769e82.
[33] Shimazaki J, Aiba M, Goto E, Kato N, Shimmura S, Tsubota K. Transplantationof human limbal epithelium cultivated on amniotic membrane for the treat-ment of severe ocular surface disorders. Ophthalmol 2002;109(7):1285e90.
[34] Lee SH, Tseng SC. Amniotic membrane transplantation for persistent epithelialdefects with ulceration. Am J Ophthalmol 1997;123(3):303e12.
[35] Connon CJ, Doutch J, Chen B, Hopkinson A, Mehta JS, Nakamura T, et al. Thevariation in transparency of amniotic membrane used in ocular surfaceregeneration. Br J Ophthalmol 2010;94(8):1057e61.
[36] Hopkinson A, McIntosh RS, Tighe PJ, James DK, Dua HS. Amniotic membranefor ocular surface reconstruction: donor variations and the effect of handlingon TGF-b content. Invest Ophthalmol Vis Sci 2006;47(10):4316e22.
[37] Gicquel J, Dua H, Brodie A, Mohammed I, Suleman H, Lazutina E, et al.Epidermal growth factor variations in amniotic membrane used for ex vivotissue constructs. Tissue Eng Part A 2009;15(8):1919e27.
[38] Di Girolamo N, Bosch M, Zamora K, Coroneo M, Wakefield D, Watson S.A contact lens-based technique for expansion and transplantation of autolo-gous epithelial progenitors for ocular surface reconstruction. Transplantation2009;87(10):1571e8.
[39] Krishnan S, Sekar S, Katheem MF, Krishnakumar S, Sastry TP. Fish scalecollagen-a novel material for corneal tissue engineering. Artif Organs2012;36(9):829e35.
[40] Francis D, Abberton K, Thompson E, Daniell M. Myogel supports the ex-vivoamplification of corneal epithelial cells. Exp Eye Res 2009;88(3):339e46.
[41] Liu J, Lawrence BD, Liu A, Schwab IR, Oliveira LA, Rosenblatt MI. Silk fibroin asa biomaterial substrate for corneal epithelial cell sheet generation. InvestOphthalmol Vis Sci 2012;53(7):4130e8.
[42] Sudha B, Madhavan H, Sitalakshmi G, Malathi J, Krishnakumar S, Mori Y, et al.Cultivation of human corneal limbal stem cells in Mebiol gel�ea thermo-reversible gelation polymer. Indian J Med Res 2006;124(6):655e64.
[43] Sangwan V, Vemuganti G, Singh S, Balasubramanian D. Successful recon-struction of damaged ocular outer surface in humans using limbal and con-juctival stem cell culture methods. Biosci Rep 2003;23(4):169e74.
[44] Tseng S, Prabhasawat P, Barton K, Gray T, Meller D. Amniotic membranetransplantation with or without limbal allografts for corneal surface
reconstruction in patients with limbal stem cell deficiency. Arch Ophthalmol1998;116(4):431e41.
[45] Sridhar MS, Bansal AK, Sangwan VS, Rao GN. Amniotic membrane trans-plantation in acute chemical and thermal injury. Am J Ophthalmol2000;130(1):134e7.
[46] Dobrowolski D, Wylegala E, Wowra B, Orzechowska-Wylegala B. Cultivatedoral mucosa epithelium transplantation (COMET) in bilateral limbal stem celldeficiency. Acta Ophthalmol 2011;89(s248):0.
[47] Nakamura T, Inatomi T, Cooper LJ, Rigby H, Fullwood NJ, Kinoshita S.Phenotypic investigation of human eyes with transplanted autologous culti-vated oral mucosal epithelial sheets for severe ocular surface diseases. Oph-thalmol 2007;114(6):1080e8.
[48] Zieske JD, Bukusoglu G, Gipson IK. Enhancement of vinculin synthesis bymigrating stratified squamous epithelium. J Cell Biol 1989;109(2):571e6.
[49] Ziegler WH, Liddington RC, Critchley DR. The structure and regulation ofvinculin. Trends Cell Biol 2006;16(9):453e60.
[50] Weiss EE, Kroemker M, Rüdiger A-H, Jockusch BM, Rüdiger M. Vinculin is partof the cadherin-catenin junctional complex: complex formation betweenalpha-catenin and vinculin. J Cell Biol 1998;141(3):755e64.
[51] Babic I, Sharma S, Black DL. A role for polypyrimidine tract binding protein inthe establishment of focal adhesions. Mol Cell Biol 2009;29(20):5564e77.
[52] MacNeil S. Progress and opportunities for tissue-engineered skin. Nature2007;445(7130):874e80.
[53] Fatima A, Vemuganti GK, Iftekhar G, Rao GN, Sangwan VS. In vivo survival andstratification of cultured limbal epithelium. Clin Exp Ophthalmol 2007;35(1):96e8.
[54] Athanasiou KA, Niederauer GG, Agrawal CM. Sterilization, toxicity, biocom-patibility and clinical applications of polylactic acid/polyglycolic acid co-polymers. Biomaterials 1996;17(2):93e102.
[55] Mann A, Kiefer M, Leuenberger H. Thermal sterilization of heat-sensitiveproducts using high-temperature short-time sterilization. J Pharm Sci2001;90(3):275e87.
[56] Tardi C, Drechsler M, Bauer KH, Brandl M. Steam sterilisation of vesicularphospholipid gels. Int J Pharm 2001;217(1e2):161e72.
[57] Lee J, Chae G, Khang G, Kim M, Cho S, Lee H. The effect of gamma irradiationon PLGA and release behavior of BCNU from PLGA wafer. Macromol Res2003;11(5):352e6.
[58] Holy CE, Cheng C, Davies JE, Shoichet MS. Optimizing the sterilization of PLGAscaffolds for use in tissue engineering. Biomaterials 2000;22(1):25e31.
[59] Chen L, Apte RN, Cohen S. Characterization of PLGA microspheres for thecontrolled delivery of IL-1a for tumor immunotherapy. J Contr Release1997;43(2e3):261e72.
[60] Shearer H, Ellis M, Perera S, Chaudhuri J. Effects of common sterilizationmethods on the structure and properties of poly(D, L lactic-co-glycolic acid)scaffolds. Tissue Eng 2006;12(10):2717e27.
P. Deshpande et al. / Biomaterials 34 (2013) 5088e51065106
Copyright © 2022 FDOKUMEN




















