
Self-decontaminating layer-by-layer functionalized textiles based on WO 3-modified titanate...
13
Applied Catalysis A: General 391 (2011) 455–467 Contents lists available at ScienceDirect Applied Catalysis A: General journal homepage: www.elsevier.com/locate/apcata Self-decontaminating layer-by-layer functionalized textiles based on WO 3 -modified titanate nanotubes. Application to the solar photocatalytic removal of chemical warfare agents Mathieu Grandcolas a , Laura Sinault b , Franc ¸ ois Mosset b , Alain Louvet b , Nicolas Keller a , Valérie Keller a,∗ a Laboratoire des Matériaux, Surfaces et Procédés pour la Catalyse (LMSPC), CNRS, University of Strasbourg, 25 rue Becquerel, 67087 Strasbourg, France b Direction Générale de L’Armement (DGA), DGA CBRN Defense, BP 3, 91710 Vert-le-Petit, France article info Article history: Received 16 March 2010 Received in revised form 11 May 2010 Accepted 12 May 2010 Available online 27 May 2010 Keywords: Photocatalysis Titanate nanotubes WO3 Solar light Self-decontaminating textiles Layer-by-layer deposition Dimethylmethylphosphonate Yperite abstract Self-decontaminating photocatalytic textiles functionalized by the layer-by-layer approach with high aspect ratio WO 3 -modified titanate nanotubes have been developed and successfully used under solar light illumination for the photocatalytic degradation of organophosphorous and organosulfide chem- ical weapon agents. This multilayer building method, unlike both dipping and spray techniques, has been efficiently applied to textile substrates using polyethyleneimine as counter polyelectrolyte for homogeneously functionalizing textiles by a thin layer of a dense network of entangled WO 3 -modified titanate nanotubes, and preparing self-decontaminating photocatalytic textiles highly efficient toward the removal under solar light of the yperite blister live agent and of dimethylmethylphosphonate as neurotoxic agent simulant. Solar light responsive high surface WO 3 -modified titanate nanotubes were synthesized by hydro-thermally treating TiO 2 powder, with tungstate salt impregnation prior to the final calcination step. © 2010 Elsevier B.V. All rights reserved. 1. Introduction Attacks occurring both on the battlefield and in civilian areas have demonstrated the high occurrence of casualties, and have materialized the increase of the threat from chemical warfare agents (CWA) over the past century. Exposure to lethal doses can lead to collapse within seconds and even death within 10 min after a single deep inhalation or contact. Hence a need for efficient pro- tection technologies and for post-use decontamination procedures on exposed and thus contaminated materials is required for human beings as well as for equipments. Passive protection technologies, e.g. barriers, permselective membranes, filtration or adsorbents, although they could be efficient, led to maintain an important sur- face toxicity, and such technologies required thus to further set post-use decontamination, in addition to well-known restrictive drawbacks specific to each technology. Therefore, active protec- tion, i.e. with simultaneous degradation of the agent, should be preferred. ∗ Corresponding author. Tel.: +33 36885 736; fax: +33 36885 761. E-mail address: [email protected] (V. Keller). Of the current methods investigated or used, all present sig- nificant drawbacks: either they cannot be directly applied on the battlefield or they do not lead to destruction of the toxic agent but rather only to an adsorption or a skin barrier protection effect. By contrast, photocatalytic materials could be efficiently applied over textiles or paints for designing self-decontaminating materials with active protection resulting from their chemical (as well as biological) agent degradation properties under UV-A or solar illumination. It could thus be possible to envisage pro- tecting clothes that would be not only based on impermeable or semi-permeable barrier protection layers but also on a per- meable adsorptive protective over-garment, acting actively as a direct destructive layer toward toxic chemicals. Indeed, photo- catalysis has been successfully used for degrading organosulfur compounds such as diethylsulfide (DES), acting as a simulant for the live yperite CWA (blister agent, also known as mustard gas), as well as for degrading organophosphorous compounds such as dimethylmethylphosphonate (DMMP), acting as a simulant for live neurotoxic organophosphorous CWA (such as Soman, sarin, tabun and VX) [1–6]. It should be mentioned that photocatalysis has also already been applied, even if scarcely, on live CWA such as mustard gas [6–8]. In parallel to that, investigation on the photocatalytic oxi- 0926-860X/$ – see front matter © 2010 Elsevier B.V. All rights reserved. doi:10.1016/j.apcata.2010.05.028
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Self-decontaminating layer-by-layer functionalized textiles based on WO 3-modified titanate...

SWr
MNa
b
a
ARRAA
KPTWSSLDY
1
hmalatobeafpdtp
0d
Applied Catalysis A: General 391 (2011) 455–467
Contents lists available at ScienceDirect
Applied Catalysis A: General
journa l homepage: www.e lsev ier .com/ locate /apcata
elf-decontaminating layer-by-layer functionalized textiles based onO3-modified titanate nanotubes. Application to the solar photocatalytic
emoval of chemical warfare agents
athieu Grandcolasa, Laura Sinaultb, Francois Mossetb, Alain Louvetb,icolas Kellera, Valérie Kellera,∗
Laboratoire des Matériaux, Surfaces et Procédés pour la Catalyse (LMSPC), CNRS, University of Strasbourg, 25 rue Becquerel, 67087 Strasbourg, FranceDirection Générale de L’Armement (DGA), DGA CBRN Defense, BP 3, 91710 Vert-le-Petit, France
r t i c l e i n f o
rticle history:eceived 16 March 2010eceived in revised form 11 May 2010ccepted 12 May 2010vailable online 27 May 2010
eywords:
a b s t r a c t
Self-decontaminating photocatalytic textiles functionalized by the layer-by-layer approach with highaspect ratio WO3-modified titanate nanotubes have been developed and successfully used under solarlight illumination for the photocatalytic degradation of organophosphorous and organosulfide chem-ical weapon agents. This multilayer building method, unlike both dipping and spray techniques, hasbeen efficiently applied to textile substrates using polyethyleneimine as counter polyelectrolyte forhomogeneously functionalizing textiles by a thin layer of a dense network of entangled WO3-modifiedtitanate nanotubes, and preparing self-decontaminating photocatalytic textiles highly efficient toward
hotocatalysisitanate nanotubesO3
olar lightelf-decontaminating textilesayer-by-layer depositionimethylmethylphosphonate
the removal under solar light of the yperite blister live agent and of dimethylmethylphosphonate asneurotoxic agent simulant. Solar light responsive high surface WO3-modified titanate nanotubes weresynthesized by hydro-thermally treating TiO2 powder, with tungstate salt impregnation prior to the finalcalcination step.
© 2010 Elsevier B.V. All rights reserved.
perite
. Introduction
Attacks occurring both on the battlefield and in civilian areasave demonstrated the high occurrence of casualties, and haveaterialized the increase of the threat from chemical warfare
gents (CWA) over the past century. Exposure to lethal doses canead to collapse within seconds and even death within 10 min aftersingle deep inhalation or contact. Hence a need for efficient pro-
ection technologies and for post-use decontamination proceduresn exposed and thus contaminated materials is required for humaneings as well as for equipments. Passive protection technologies,.g. barriers, permselective membranes, filtration or adsorbents,lthough they could be efficient, led to maintain an important sur-ace toxicity, and such technologies required thus to further set
ost-use decontamination, in addition to well-known restrictiverawbacks specific to each technology. Therefore, active protec-ion, i.e. with simultaneous degradation of the agent, should bereferred.∗ Corresponding author. Tel.: +33 36885 736; fax: +33 36885 761.E-mail address: [email protected] (V. Keller).
926-860X/$ – see front matter © 2010 Elsevier B.V. All rights reserved.oi:10.1016/j.apcata.2010.05.028
Of the current methods investigated or used, all present sig-nificant drawbacks: either they cannot be directly applied on thebattlefield or they do not lead to destruction of the toxic agentbut rather only to an adsorption or a skin barrier protectioneffect. By contrast, photocatalytic materials could be efficientlyapplied over textiles or paints for designing self-decontaminatingmaterials with active protection resulting from their chemical(as well as biological) agent degradation properties under UV-Aor solar illumination. It could thus be possible to envisage pro-tecting clothes that would be not only based on impermeableor semi-permeable barrier protection layers but also on a per-meable adsorptive protective over-garment, acting actively as adirect destructive layer toward toxic chemicals. Indeed, photo-catalysis has been successfully used for degrading organosulfurcompounds such as diethylsulfide (DES), acting as a simulant forthe live yperite CWA (blister agent, also known as mustard gas),as well as for degrading organophosphorous compounds such as
dimethylmethylphosphonate (DMMP), acting as a simulant for liveneurotoxic organophosphorous CWA (such as Soman, sarin, tabunand VX) [1–6]. It should be mentioned that photocatalysis has alsoalready been applied, even if scarcely, on live CWA such as mustardgas [6–8]. In parallel to that, investigation on the photocatalytic oxi-
4 talysis
db
obati[nttaamtriproscniltne[
rablimeieagmttcm
sotod
2
2
msDchw
56 M. Grandcolas et al. / Applied Ca
ation of nauseous organosulfides such as dimethylsulfides has alsoeen performed [9,10].
Since the discovery of carbon nanotubes, extensive researchn the synthesis of nanotubes made of materials other than car-on, and especially of oxides like MoO3, WO3, VOx, . . . as wells TiO2, has been reported [11–13]. Nanoscale 1D TiO2 struc-ures, including materials derived from TiO2, have been studiedn recent years for applications in high effect photovoltaic cells14,15], gas sensors or UV blockers. Such high aspect ratio TiO2anostructures have started recently to attract much attention forargeting promising applications in photocatalytic environmentalechnology [16–22], owning to a unique tubular structure withlarge surface-to-volume ratio resulting in a larger surface area
vailable in comparison to nanoparticles, to possible size confine-ent in radial direction, to enhanced electron transfer through the
ubular structure and globally to reduced photogenerated chargeecombination rates [23–25]. Thus, the high adsorption capac-ty as well as efficient charge separation yielding to reduce thehoto-electron and -hole recombination should have determinantoles in photocatalytic reactions [26]. Within this trend, the devel-pment of high aspect ratio TiO2 with high activity focused onolar applications has become one of the main challenges con-erning this material within this area. For instance, the search forew TiO2-based photocatalysts has already pointed out the pos-
tive influence of adding WO3 to TiO2, with regard to the oxideoading, for liquid and gas phase photocatalytic oxidation reac-ions [27–31]. Recently, Xiao et al. have prepared WO3/titanateanotube composites with enhanced photocatalytic properties,videnced in the case of the liquid phase Rhodamine B degradation20].
The electrostatic layer-by-layer (LbL) self-assembly method haseceived a growing interest and has been proved a well-establishedpproach to build composite or function-specific multilayer assem-lies such as multilayer thin films on planar substrates. The
ayer-by-layer self-assembly of polyelectrolytes was rigorouslynvestigated and rationalized by the group of Decher [32,33]. This
ethod consisted in the building of multilayer thin films throughlectrostatic interactions, by the alternate deposition of polyan-ons and polycations on the surface of a substrate, previouslyither negatively or positively charged. This by layer-by-layer self-ssembly has been extended to other charged systems, such aslobular proteins [34], organic molecules [35], mineral as well asetallic nanoparticles [36,37] and semiconductors [38], including
itanate nanotubes [16,17,39–41]. Amongst numerous anchorageechniques [42–47], the LbL deposition method was already suc-essfully applied for functionalizing textile fibers by photocatalyticaterials [6,48].The aim of this paper is thus to report on the preparation of
elf-decontaminating layer-by-layer functionalized textiles basedn WO3-modified titanate nanotubes with application to the pho-ocatalytic removal under solar light illumination of the DMMPrganophosphonate as live organophosphorous CWA simulant, andirectly of the live yperite organosulfide CWA.
. Experimental
.1. Photocatalyst preparation
Titanate nanotubes (TiNTs) were synthesized by the hydrother-al treatment of a TiO2 powder in a 10 M concentrated NaOH
olution at 130 ◦C [23]. In a typical synthesis, 1 g of TiO2 P25 fromegussa-Evonik) was added to 50 mL of solution in a Teflon auto-lave, stirred for 1 h, and digested at 130 ◦C for 24 h. After theydrothermal treatment, the white powder obtained was rinsedith distilled water, vacuum filtered and further washed with 1 M
A: General 391 (2011) 455–467
HCl. The material was rinsed with distilled water until neutral pHand finally overnight dried at 110 ◦C (H-TiNT).
The dried H-TiNT sample was impregnated withan ethanol/deionized water (25/75, vol.%) solution of(NH4)10W12O41·5H2O ammonium paratungstate pentahydrate,further dried at 110 ◦C for 2 h, before final calcination wasperformed in air at 380 ◦C for 2 h (WO3-TiNT).
2.2. Preparation of the functionalized photocatalytic textile
Coton/polyamide (50/50) military textile samples were pro-vided by the DGA. Prior to any photocatalytic material deposition,they were first cleaned with a commercially available detergent.The surface of the textile was then hydroxylated by submittingthe textile to a 0.5 M KOH solution treatment [49], rinsed withdistilled water, dried at ambient temperature for 15 h and furtherat 100 ◦C for 1 h. The as-prepared textiles were finally maintainedinside Ziploc© bags, especially for protecting them against exces-sive humidity capture and storage.
The deposition method by dipping consisted in immersing thetextile for 30 min under orbital stirring in an ethanolic/aqueous(50/50, vol.%) suspension of the nanomaterials at 10 g/L, previouslymechanically stirred for 24 h and ultrasonicated for 20 min. Thetextile was subsequently pressed under a rubber roll for mechani-cally forcing the suspension to impregnate the textile and then thenanoparticles to be in contact with the fibers, before being rinsedwith distilled water for removing the excess nanoparticles. Thisstep can be repeated several times if necessary. Finally, the textilewas dried at ambient temperature for 15 h and further at 100 ◦C for1 h.
The spray deposition method was based on the pulverizationof the suspension containing the photocatalytic material onto thetextile target, using a commercial spray gun, known as High Vol-ume Low Pressure (HVLP) device. The nanomaterial suspension wasthe ethanolic/aqueous (50/50, vol.%) suspension at 1 g/L, previouslymechanically stirred for 15 h and ultrasonicated for 20 min. Thetarget–spray distance was set at 10–20 cm (with a pulverizationnozzle diameter of 0.5 mm and pressurized air at about four bar)and the crossed-layers spray technique was used. After spraying,the textile was rinsed with distilled water for removing the excessnanoparticles. Each vaporization/rinsing cycle was followed by adrying at ambient temperature for 15 min and a further drying at100 ◦C for 1 h.
Layer-by-layer deposition was performed by the alternate sprayonto the textile substrate target of, either a cationic polyethylen-imine (PEI+) solution (8 g/L) or the ethanol/water (50/50, vol.%)suspension of WO3-TiNTs at 1 g/L, rectified at pH = 9. This sequencewas repeated until the number of layers was attained. After depo-sition of each layer (PEI+ or WO3-TiNTs), the substrate was rinsedthree times with pure water. One should note that after the depo-sition of the first polyelectrolyte layer, a compression step using arubber roll was performed for mechanically increasing the contactwith the textile. The final textile loaded with the photocatalyst wasfinally dried at 90 ◦C.
2.3. Characterization techniques
X-ray diffraction (XRD) measurements were carried out on a D8Advance Bruker diffractometer, in a �/2� mode and using the K�1radiation of Cu at 1.5406 Å.
Scanning electron microscopy (SEM) was carried out on a Jeol
JSM-6700F working at 1–10 kV, equipped with a CCD camera. Thesample was previously coated with carbon and then deposited ona standard holder for observation.The surface area and porosimetry measurements were carriedout on an ASAP2010 Micromeritics using N2 as adsorbant at liq-

M. Grandcolas et al. / Applied Catalysis
F(Tc
uomttad
teec
Tr
with a HP Plot Q and HP-5MS columns, and coupled to a Ther-
ig. 1. XRD patterns of the crude titanates synthesized by the hydrothermal methodNa-TiNTs), and further after washing with HCl until neutralization and drying (H-iNTs) and WO3 (4 wt.%)-TiNTs calcined at 380 ◦C. The TiO2 P25 is reported foromparison, exhibiting the usual anatase and rutile diffraction peaks.
id N2 temperature. Before the N2 adsorption, the material wasutgassed overnight at 130 ◦C in order to desorb the impurities oroisture from its surface. The surface area was calculated from
he nitrogen adsorption isotherms using the B.E.T. method (SBET),he micropore surface area was derived using the t-plot method,nd the pore size distribution was obtained using the B.J.H. methoduring the desorption isotherm branch.
X-ray photoelectron spectroscopy (XPS) surface characteriza-ion was performed on a MULTILAB 2000 (TERMO) spectrometerquipped with Al K� anode (h� = 1486.6 eV). The subtraction of thenergy shift due to electrostatic charging was determined using the
ontamination carbon C1s band at 284.6 eV as reference.Transmission electron microscopy (TEM) was performed on aopcon 002B microscope working with 200 kV and a point-to-pointesolution of 0.17 nm. The sample was sonically dispersed in an
Fig. 2. TEM images of the raw Na-titanate nanosheets (
A: General 391 (2011) 455–467 457
ethanol solution before a drop of the solution was deposited onto acopper grid covered by a holey carbon membrane for observation.
The UV–Vis absorption spectra of the materials were recordedon a Cary 100 Scan UV/Vis spectrophotometer from Varianequipped with a DRA-CA-301 Labsphere diffuse reflectance cell.Liquid phase measurements were also performed using a usualdouble beam and double Quartz cell system, for comparing thesedimentation rate of different one-dimension nanostructures inan ethanol/water solution.
Isoelectrical point (IEP) measurements were performed on aMalvern© ZetaSizer with automatic titration.
Contact angle measurements were performed with a CAM100 contact angle goniometer (Sodexim) working according tothe sessile drop technique, using a 2 �L microdrop and a 1 mLmicronic screwed syringe (Hamilton, 13.23 �L/revolution). Thevalues reported corresponded to contact angles at steady-state con-ditions, i.e. 60 s after the drop deposition.
2.4. Experimental device and procedure
The photocatalytic test procedure was adapted from a proto-col developed for surface decontamination tests. First, 10 g/m2 ofsimulant or live warfare agents was deposited as droplets on thephotocatalytic textile using a microsyringe. Taking into accountthe 2 × 2 cm geometrical surface of the textile, this correspondedto 5.1 �L of agent per textile sample. The substrate was thensubmitted to the illumination for different durations. After illumi-nation, one proceeded to the extraction of the adsorbed chemicalmolecules from the photocatalytic textile. The optimized extractionprotocol consisted in immersing twice the photocatalytic substrateinto 10 mL of pure isopropanol under orbital stirring for 30 min. Theproducts extracted from the photocatalytic substrate were furtheranalyzed by gas chromatography (Agilent GC-6890N) equipped
mal Conductivity Detector and a MS-5973N Mass Spectrometer,respectively. Performing several injections of the same extractionsolution evidenced the good reproducibility of both extraction andinjection procedures. Quantifying the remaining extracted pol-
A, B) and of HCl washed and dried H-TiNTs (C, D).

458 M. Grandcolas et al. / Applied Catalysis A: General 391 (2011) 455–467
F he TiNd cted fr
lpytw2Pw
t
3
3
r
ig. 3. TEM images of (A–E) the WO3-TiNTs (4 wt.%) calcined at 380 ◦C, and (F) tegradation of the tubular morphology with the appearance of nanoparticles extra
utant led to express the self-decontamination efficiency of thehotocatalytic textile as the evolution of the DMMP and residualperite concentrations in the extracted solution as a function ofhe time under illumination. Similar procedures were also followedith bare non-functionalized textile, with or without illumination.
4 W simulating solar light illumination (Model TL5 HO 24W/965,hilips) and 24 W UV-A (Model PL-L 24W/10/4P Philips) lampsere used as illumination sources.
The photocatalytic tests toward the yperite were performed athe DGA CBRN Defense Center (MoD/DGA).
. Results and discussion
.1. Material characterization
Fig. 1 shows the XRD pattern of the one-dimensional mate-ial at different steps of the synthesis procedure, leading to the
Ts calcined at 380 ◦C with no prior impregnation with tungsten, evidencing theom nanotubes.
final WO3-modified nanotube photocatalyst, whereas its mor-phology was shown with TEM images in Figs. 2A–D and 3A–E.The TEM images evidenced that the hydrothermal synthesis wastotally selective to produce one-dimensional nanomaterials, withno nanopowder being observed in the sample. Combining XRDand TEM characterization showed that the raw material obtaineddirectly after synthesis consisted of one-dimensional sodiumtitanate nanosheets of Na2Ti3O7 type (with characteristic diffrac-tion peaks at 30.2◦, 34.4◦, 35.5◦, 38.1◦ and 41.6◦), with 30 nmthickness and several hundreds of nm length for mean character-istics, while no nanotubes were observed beside the nanosheets[50–52]. After further washing with HCl until neutralization and
drying at 110 ◦C, this Na-containing structure was transformed intoanother structure which could be indexed to NaxH2−xTi3O7 nan-otubes [53] or rather to H2Ti3O7 nanotubes according to otherreports [54,55] and to the absence of any residual sodium whichwas not detected by electron dispersive spectroscopy (EDS) during
M. Grandcolas et al. / Applied Catalysis A: General 391 (2011) 455–467 459
F�H
Mottpatnanl
aa
FT
ig. 4. (A) N2 adsorption–desorption isotherms of (�, ©) the dried H-TiNTs and (�,) the WO3 (4 wt.%)-TiNT calcined at 380 ◦C. (B) Pore size distribution of the dried-TiNTs (�) and the WO3 (4 wt.%)-TiNTs calcined at 380 ◦C (�).
ET analysis, elementary analysis and XPS in the present study. Thene-dimensional tubular nature of the material was confirmed byhe broad peak observed at low diffraction angles around 10◦. Theransformation into nanotubes resulted from interlayered total (orartial) cation exchange between Na+ and H+ leading to bendingnd rolling up of the nanosheets during HCl washing. This step wasotally selective toward the formation of nanotubes (Fig. 2C). Theanotubes obtained displayed lengths of several hundreds of nmnd mean diameters of 10–20 nm, with a multilayered wall thick-
ess of <2 nm corresponding to 2–6 titanate sheets in average, inine with the results reported in the literature [56,57].Impregnation with the tungstate salt followed by calcination
t 380 ◦C does not seem to affect the XRD pattern of the washednd dried TiNTs, due to the good dispersion of tungsten and/or its
ig. 6. Evolution with time of the UV–Vis absorption maximum of an ethanolic/aqueousiO2 P25, (�) H-TiNTs and (©) WO3-TiNTs.
Fig. 5. Zeta potential as a function of the pH for determining the isoelectrical point.(�) H-TiNT, (©) WO3-TiNTs and (�) TiO2 P25.
low amount deposited, with the exception of both line broaden-ing and diffraction signal lowering at low diffraction angles. Thisslight change at low diffraction angles resulted from the intrinsicdehydration of the nanotubes during the thermal treatment, lead-ing to a slight contraction of the nanotubes, and as a result, of thecrystallographic cell. This led thus to a slight increase in the thick-ness of the walls from 2 to almost 5 nm (Fig. 3B–D). According to themechanism reported by Peng and co-workers for the TiNT synthesis[58], this inter-wall dehydration and nanotube contraction was alsoaccompanied by a slight decrease in the external diameter of theTiNTs. This was confirmed by TEM analysis, which evidenced thatthe temperature treatment led to a decrease in the inter-wall dis-tance of the nanotubes from 9.5 to 8 Å, respectively, in agreementwith the literature [59].
Fig. 3A evidenced that the morphology of the nanotubes wastotally maintained during the calcination at 380 ◦C of the tungsten-impregnated titanate nanotubes, without any agglomeration ofparticles located on the outer wall of the TiNTs and it was worth not-ing that the calcined nanotubes were open at both ends (Fig. 3B,C).The high magnification image (Fig. 3E) reveals decoration of theTiNT with nano-islands of WO3 particles, whereas the presence oftungsten in the whole sample was confirmed by EDS analysis (notshown). One should note that calcining the dried TiNTs with noprior tungstate salt impregnation led to the progressive destructionof the tubular morphology, which turned continuously into TiO2nanoparticles (Fig. 3F). This indicated that the presence of tung-sten dispersed at the TiNTs surface stabilized the morphology ofthe TiNTs during the temperature treatment, and increased theirthermal stability [60,61].
Fig. 4A shows the nitrogen adsorption–desorption isotherms ofboth WO3-TiNTs and H-TiNTs materials, corresponding to a mainlymesoporous solid according to the well-accepted isotherm classi-fication. The dried H-TiNTs and the WO3-TiNTs had high specificsurface areas of 412 and 274 m2/g, respectively, with no microp-
(50/50) suspension (neutral pH) containing different nanostructures at 1 g/L. (�)

4 talysis A: General 391 (2011) 455–467
osatnmastetcotcb
mTfcwrcosbito
smtsi
60 M. Grandcolas et al. / Applied Ca
rosity, only resulting from the external and internal geometricalurfaces developed by the nanostructures. They had a monomodalnd mesoporous pore size distribution (Fig. 4B), corresponding tohe internal diameter of the nanotubes—the internal volume of theanotubes being seen as a very high aspect ratio nanopore, in agree-ent with the microscopy measurements. As the specific surface
rea of the nanotubes only resulted from both external and internalurfaces, this decrease in specific surface area could be attributedo the slight decrease in size of both external and internal diam-ters of the nanotubes during the calcination at 380 ◦C. Althoughhe quantitative mean pore sizes derived from Fig. 4B should beonsidered cautiously, the decrease in size of the internal diameterf the nanotubes was also evidenced by the qualitative change inhe monomodal pore size distribution of the nanotubes during thealcination treatment, with a slight shift toward lower pore sizeseing observed.
Fig. 5 shows the zeta potential as a function of the pH for deter-ining the IEP of the WO3-TiNT sample (and of both H-TiNT and
iO2 P25 samples as reference), as important parameter to knowor further applying WO3-TiNTs onto the textile by the LbL pro-ess. The H-TiNTs have an isoelectrical point of 5.3, in agreementith the literature [16,17], indicating the presence of a slight acidity
esulting from the chemical composition of the TiNT surface, whenompared to the quasi-neutral surface of TiO2 P25, for which an IEPf 6.8 was measured. By contrast, adding 4 wt.% of WO3 decreasedtrongly the isoelectrical point of the material down to 3.4. It shoulde noted that WO3 is often used in heterogeneous catalysis for acid-
fying the surface of oxide catalysts [62], the isoelectrical point ofhe bare oxide being evaluated between 0.2 and 0.5 leading thexide to be considered as an acid oxide [63].
The isoelectrical point could also be seen as the pH at which the
tability of nanoparticles in suspension were the weakest, due to aore pronounced aggregation phenomena resulting mainly fromhe Van der Waals forces existing between nanoparticles in suspen-ion. Indeed, according to the theory of electrostatic stabilizationn solution [64], a high zeta potential value resulted in increased
Fig. 8. Photography and SEM images of 2 × 2 cm coton
Fig. 7. Light absorption properties of WO3-TiNTs, H-TiNTs and TiO2 P25 materials.
electrostatic repulsions between the particles, and thus led to abetter stability of the suspension [65]. Therefore, it will be neces-sary to use an impregnation solution with a pH far away as possiblefrom 5.3 to 3.4, for looking for a homogeneous coverage of the tex-tile with TiNTs and WO3-TiNTs, respectively. Such a high pH willallow the electrostatic interactions and therefore the agglomera-tion of nanoparticles in suspension to be limited. The increase inthe electrostatic repulsion between the particles when adding WO3to the TiNTs was also evidenced by investigating by UV–Vis thesedimentation rate in a neutral ethanol/water solution, and thus
the stability of the suspension. The sedimentation of the nanos-tructures with time caused a progressive decrease in the UV–Visabsorption maximum of the suspension. Fig. 6 shows the betterstability with time of the WO3-TiNTs ethanolic/aqueous (50/50)/polyamide (50/50) raw military textile pieces.

M. Grandcolas et al. / Applied Catalysis A: General 391 (2011) 455–467 461
4 wt.%
ssst
Wrsttatuadapn
3t
3
ct
Fig. 9. SEM images of the photocatalytic textiles with WO3 (
uspension when compared to both TiO2 P25 and H-TiNT suspen-ions, due to the large difference between the IEP and the pH of theolution, the most pronounced decantation rate being observed forhe TiO2 P25 suspension, with close IEP and pH.
Fig. 7 shows the UV–Vis absorption spectra recorded for theO3 (4 wt.%)-TiNT, the H-TiNT and the TiO2 P25 reference mate-
ials. It was noteworthy that the WO3-TiNTs showed an importanthift toward the visible light part of the spectrum compared tohe reference TiO2 P25 or to the tungsten-free H-TiNTs, withhe enlargement of the absorption tail up to about 500 nm. Thebsorption of the composite occurred in the UV-A as well as inhe visible light region of the spectrum, suggesting the potentialse of these nanocomposites under solar light irradiation (UV-And visible light wavelengths) conditions. It will be proposed andetailed elsewhere in a forthcoming article that this visible lightbsorption could result from the existence of a WxTiyOx superficialhase located at the interface between the nanotubes and the WO3anoparticles.
.2. Characterization of WO3-TiNT functionalized photocatalyticextiles
.2.1. Morphological characterizationFig. 8 shows the morphological characteristics of the
oton/polyamide (50/50) military textiles. The textile struc-ure consisted of 10 �m mean diameter fibers entangled to form
)-TiNTs obtained by (A) dipping and (B–E) spray procedures.
the two-dimension network of the woven textile. One could notethe smooth surface of the fibers, even if some surface asperities orirregularities could be observed.
The SEM images of the photocatalytic textiles obtained by bothdipping and spray methods are reported in Fig. 9. The dippingmethod did not lead to a homogeneous coverage of the fibers,even after three successive dippings of the textile into the sus-pension, with the simultaneous presence on the fiber surface oflarge particle aggregates and of naked zones with no photocat-alytic material. Even if large amounts of photocatalyst could bedeposited by this easy-of-use technique, the strong in homogeneityof the coverage as well as a bad anchorage and a weak mechani-cal resistance of the deposit, were fully redhibitory. By contrast,the use of the spray deposition method resulted in a more homo-geneous coverage of the fibers when compared to the dippingtechnique (Fig. 9B–D). Despite this first improvement in termsof homogeneity – although only partly obtained and observed atthe fiber surface – higher resolution images shows the formationof balls made of agglomerated nanotubes rather than a homoge-neous coverage of the fibers by a network of entangled nanotubes(Fig. 9E).
Fig. 10 shows the SEM images of the WO3 (4 wt.%)-TiNTs func-tionalized photocatalytic textile obtained by the LbL multilayerdeposition method (10 double-layers with PEI as polyelectrolyte).It was worth noting the full and homogeneous coverage of thefibers by the network of entangled nanotubes, with neither remain-
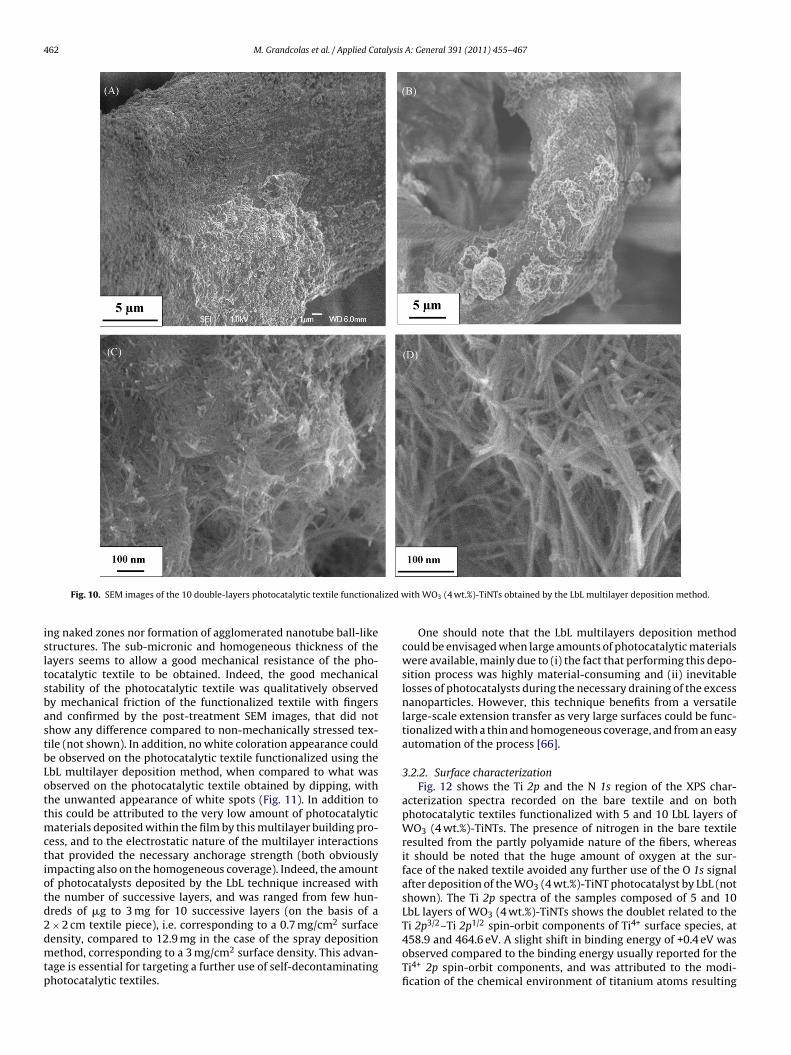
462 M. Grandcolas et al. / Applied Catalysis A: General 391 (2011) 455–467
lized w
isltsbastbLottmctiotd2dmtp
Fig. 10. SEM images of the 10 double-layers photocatalytic textile functiona
ng naked zones nor formation of agglomerated nanotube ball-liketructures. The sub-micronic and homogeneous thickness of theayers seems to allow a good mechanical resistance of the pho-ocatalytic textile to be obtained. Indeed, the good mechanicaltability of the photocatalytic textile was qualitatively observedy mechanical friction of the functionalized textile with fingersnd confirmed by the post-treatment SEM images, that did nothow any difference compared to non-mechanically stressed tex-ile (not shown). In addition, no white coloration appearance coulde observed on the photocatalytic textile functionalized using thebL multilayer deposition method, when compared to what wasbserved on the photocatalytic textile obtained by dipping, withhe unwanted appearance of white spots (Fig. 11). In addition tohis could be attributed to the very low amount of photocatalytic
aterials deposited within the film by this multilayer building pro-ess, and to the electrostatic nature of the multilayer interactionshat provided the necessary anchorage strength (both obviouslympacting also on the homogeneous coverage). Indeed, the amountf photocatalysts deposited by the LbL technique increased withhe number of successive layers, and was ranged from few hun-reds of �g to 3 mg for 10 successive layers (on the basis of a
× 2 cm textile piece), i.e. corresponding to a 0.7 mg/cm2 surfaceensity, compared to 12.9 mg in the case of the spray depositionethod, corresponding to a 3 mg/cm2 surface density. This advan-age is essential for targeting a further use of self-decontaminatinghotocatalytic textiles.
ith WO3 (4 wt.%)-TiNTs obtained by the LbL multilayer deposition method.
One should note that the LbL multilayers deposition methodcould be envisaged when large amounts of photocatalytic materialswere available, mainly due to (i) the fact that performing this depo-sition process was highly material-consuming and (ii) inevitablelosses of photocatalysts during the necessary draining of the excessnanoparticles. However, this technique benefits from a versatilelarge-scale extension transfer as very large surfaces could be func-tionalized with a thin and homogeneous coverage, and from an easyautomation of the process [66].
3.2.2. Surface characterizationFig. 12 shows the Ti 2p and the N 1s region of the XPS char-
acterization spectra recorded on the bare textile and on bothphotocatalytic textiles functionalized with 5 and 10 LbL layers ofWO3 (4 wt.%)-TiNTs. The presence of nitrogen in the bare textileresulted from the partly polyamide nature of the fibers, whereasit should be noted that the huge amount of oxygen at the sur-face of the naked textile avoided any further use of the O 1s signalafter deposition of the WO3 (4 wt.%)-TiNT photocatalyst by LbL (notshown). The Ti 2p spectra of the samples composed of 5 and 10LbL layers of WO3 (4 wt.%)-TiNTs shows the doublet related to the
Ti 2p3/2–Ti 2p1/2 spin-orbit components of Ti4+ surface species, at458.9 and 464.6 eV. A slight shift in binding energy of +0.4 eV wasobserved compared to the binding energy usually reported for theTi4+ 2p spin-orbit components, and was attributed to the modi-fication of the chemical environment of titanium atoms resulting
M. Grandcolas et al. / Applied Catalysis A: General 391 (2011) 455–467 463
F meth
fiadtetbmeMPlreio
3
3
ttu(wectwrLot
ig. 11. Comparison between a classical spray deposition method (left) and the LbL
rom the presence of PEI. In addition, one could note the increase inntensity of the Ti 2p signal with increasing the number of layers, asresult from the increase of the amount of photocatalytic materialeposited with successive layers. In parallel to that, an increase inhe N 1s signal at a binding energy of 404.2 eV was observed andvidenced the increase of the amount of PEI deposited, in addi-ion to the polyamide from the naked fibers. Thus, XPS appeared toe a suitable technique to evidence the increase of photocatalyticaterial deposited with successive layers, as observed by differ-
nt authors during the building of polyelectrolyte films. Chen andcCarthy have studied the deposition of poly(styrene sulfonate)
SS and poly(allyl hydrochloric) PAH on poly(ethylene terephta-ate) (PET) and used the N 1s and S 1s peaks for determining theelative concentrations in PSS and PAH, as well as the number of lay-rs within the multilayer film built [67]. They showed the increasen the amount of nitrogen and sulfur within the film as a functionf the number of layers deposited.
.3. Self-decontaminating photocatalytic properties
.3.1. Influence of the deposition methodFig. 13A shows the influence of the deposition method on
he kinetic of the photocatalytic removal of DMMP from func-ionalized textiles. It shows the evolution as a function of timender UV-A illumination, of the extracted DMMP concentrationi.e. the remaining DMMP toxicity) over the textile functionalizedith TiO2 P25 by spray deposition and by LbL deposition (10 lay-
rs). After a 90 min illumination, only 20 ppm of DMMP, and byontrast 90 ppm of DMMP, remained at the surface of the pho-ocatalytic textile functionalized by LbL and spray, respectively,
hereas a duration of 120 min of illumination was necessary foremoving totally the toxicity from the textile functionalized by thebL deposition method and in contrary, a remaining concentrationf 60 ppm of DMMP was extracted from the spray-functionalizedextile after a similar illumination duration. In addition, the high
od (right) for functionalizing the photocatalytic textile with WO3 (4 wt.%)-TiNTs.
performances shown by the multilayer film were obtained witha very low amount of photocatalyst compared to that depositedin the spray coating, i.e. 3 vs. 12.9 mg (0.7 vs. 3 mg/cm2 of surfacedensity), probably as a result of the high and homogeneous photo-catalytic material dispersion obtained through the LbL depositionmethod. This evidenced the interest of using this technique forproviding self-decontamination properties to the textile by surfacefunctionalization.
3.3.2. Influence of the number of layersFig. 13B shows the influence of the number of LbL multilayers on
the photocatalytic removal of DMMP from functionalized textiles.It shows the evolution as a function of time under solar light illu-mination, of the extracted DMMP concentration (i.e. the remainingDMMP toxicity) over the naked textile without photocatalyst andboth photocatalytic textiles functionalized by LbL with 5 and 10layers of WO3-TiNT materials. The evolution of the DMMP concen-tration with time over the naked textile corresponded to a blanktest, for which the progressive disappearance of DMPP resultedfrom the natural evaporation of DMMP due to the heating of thetextile through the illumination. 30 min was necessary to achievethe full evaporation of the DMMP.
The DMMP evaporation was confirmed by performing contactangle measurements with DMMP (and also water for comparison)microdrops over the naked textile. No change in contact angle wasobserved during the elapsed duration in the dark whatever themicrodrop nature. By contrast, the decrease with time under solarlight illumination, of the contact angle between the DMMP micro-drop and the surface of the naked fibers was more rapid than withthe water microdrop (Fig. 14), indicating that the DMMP microdrop
was very sensitive toward evaporation from the textile.By contrast, the duration under solar light illumination neces-sary for reaching the complete removal of the DMMP from thetextile was decreased down to 12 and 7 min, over the photo-catalytic textile functionalized by LbL with 5 and 10 layers of

464 M. Grandcolas et al. / Applied Catalysis A: General 391 (2011) 455–467
Fa5
Wlpspatitt
tfdtaDmu
3
sotltu
Fig. 13. Photocatalytic removal of DMMP: evolution as a function of time under illu-mination of the extracted DMMP concentration (i.e. the remaining DMMP toxicity).(A) under UV-A illumination over the textile functionalized with TiO2 P25 by spraydeposition (�) and by LbL deposition (10 layers) (�). (B) under solar illuminationover the naked textile without photocatalyst (�), and both textiles functionalizedby LbL with WO3-TiNTs (five layers) (©) and (10 layers) (�).
Fig. 14. Evolution as a function of the duration under solar light illumination, of
ig. 12. XPS spectrum of (A) the Ti 2p1/2–Ti 2p3/2 region, and (B) the N 1s region, ofcoton/polyamide (50/50) naked textile (�), and both photocatalytic textiles with(�) and 10 layers (©) of WO3 (4%)-TiNTs.
O3-TiNTs, respectively. The increase in the number of LbL multi-ayers was thus beneficial for improving the self-decontaminationroperties of the functionalized textile. This was attributed to theub-micronic thickness of the LbL multilayer film, which allowedrobably the total of the WO3-TiNT photocatalytic materials to bectivated by the solar light irradiation. However, increasing fur-her the number of LbL multilayers was not performed, for takingnto account the possible deterioration of the mechanical proper-ies of the functionalized textile with increasing the thickness ofhe grafted photocatalytic film.
One should note that DMMP evaporation also occurred simul-aneously to the photocatalytic degradation over the textile fibersunctionalized with WO3-TiNTs. Comparison with water micro-rop evaporation was also reported. In terms of contact angle athe solar light illuminated DMMP/WO3-TiNT interface, the contactngle decreased very rapidly down to 0◦, i.e. disappearance of theMMP microdrop, within a period of 8 min. This was in close agree-ent with the absence of DMMP extracted from textile after 7 min
nder solar light illumination reported in Fig. 13B.
.3.3. Tests toward the yperite live chemical warfare agentFig. 15 shows the evolution as a function of time under UV-A and
olar light illumination, of the extracted residual yperite in �g/cm2
ver the naked textile without photocatalyst and the photocatalytic
extile functionalized with WO3-TiNTs by LbL deposition (LbL-10ayers). One should note that the self-decontamination efficiency ofhe WO3-TiNTs functionalized textiles toward yperite was highernder solar light than under pure UV-A illumination, with 20 andthe contact angle of water and DMMP drops over the naked textile and the photo-catalytic textile functionalized with WO3 (4%)-TiNTs (LbL-10 layers by spray): (�)H2O/naked textile, (�) H2O/photocatalytic textile, (�) DMMP/naked textile and (©)DMMP/photocatalytic textile. One should note that the interpolation had no physicalmeaning and remained a way of clarifying the graph.

M. Grandcolas et al. / Applied Catalysis A: General 391 (2011) 455–467 465
Fig. 15. Photocatalytic removal of yperite: evolution as a function of time under illu-mination of the residual yperite concentration in �g/cm2 (i.e. the remaining yperitettt
3rdatptwttahfTl
3
pHotpppn
This study was performed with the photocatalytic textile func-
oxicity) over the naked textile without photocatalyst (�, �), and the textile func-ionalized with WO3-TiNTs (LbL-10 layers) (�, ©). Squares and circles correspondo UV-A and solar illumination, respectively.
7 min of exposure being necessary for achieving a total yperiteemoval, respectively. Exposure durations required were longer foregrading yperite than in the case of DMMP. This was preferentiallyttributed to the more important persistence of yperite at the tex-ile surface when compared to that of DMMP, rather than to a lowerhotocatalytic reactivity. Indeed, total evaporation of yperite overhe naked textile occurred within 90 min whatever the light range,hile only 30 min was necessary in the case of DMMP. In addition
o that, as the heat-assisted evaporation of yperite and DMMP fromhe naked textile were only slightly differing between both UV-And solar light exposure, we could put forward that the stronglyigher efficiency obtained under solar light illumination resulted
rom the enhanced solar light activation shown by the WO3-iNTs photocatalytic material compared to that through pure UV-Aight.
.3.4. Reaction pathwayThe literature reports different reactional pathways for the
hotocatalytic degradation of organophosphorous compounds.arada et al. have proposed in the case of the degradationf dimethyl-2,2-dichlorovinyl phosphonate and dimethyl-2,2,2-richloro-1-hydroxyethyl phosphonate, two organophosphorous
esticides close from DMMP, that the electron transfer from thehosphonate group to the TiO2 could lead to the formation ofhosphonate radicalar cations [68]. This direct reaction mecha-ism through photogenerated holes resulted in the formation ofFig. 16. Mechanistic pathway involving addit
Fig. 17. XPS spectrum of (A) the P 1s region of the photocatalytic textiles coatedwith WO3 (4%)-TiNTs (LbL-10 layers): (�) fresh, (�) after 2 h of DMMP removal pho-tocatalytic test under solar light illumination and (©) after basic aqueous washingat pH = 12.
acetal species. This was not taken into account by previous studies,performed in diluted aqueous media, for which the predominantdegradation pathway was through the hydroxyl radicals.
No acetal having been observed in the by-products in thepresent study, and as mass spectrometry (MS) coupled to Gas Chro-matography performed after isopropanol extraction evidenced thepresence of methylmethylphosphonates (MMP) and methylphos-phonic acid (MPA), we preferred to propose that the DMMPdegradation mechanism involved mainly hydroxyl radicals. Indeed,the formation of such by-products could be explained by the suc-cessive addition reactions of hydroxyl radicals on the phosphorouscenter, followed by the removal of the methoxy group (Fig. 16).This reaction mechanism involved the formation of MMP, MPAand methanol, this latter having been also observed by MS in theextracted products. This confirmed the role of the hydroxyl radi-cals in the present reaction conditions. The subsequent formationof phosphates from MPA could be possible by splitting/oxidation ofthe C–P binding, although this seems not to occur through hydroxylradicals, but through photogenerated holes or superoxyd radicals[4]. The MMP intermediate by-product could also probably lead tothe formation of phosphates.
3.3.5. Regeneration of the photocatalytic textile
tionalized by LbL with WO3 (4%)-TiNTs (10 layers). Fig. 17 shows theXPS P 1s region of the fresh, after 2 h of DMMP removal photocat-alytic test under solar light illumination and after the regenerativebasic aqueous washing at pH = 12. The P 1s signal of the photo-
ive reactions through hydroxyl radical.

4 talysis
caumttTmpswtstwa
4
bdoutW
dst3
paahios
itailrpstWeoc
A
(at(aySh
[
[[
[[
[[[[
[[
[
[
[
[
[
[
[[
[[
[
[[
[
[
[
[
[
[[
[[[[[
66 M. Grandcolas et al. / Applied Ca
atalytic textile after 2 h of test revealed a broad peak at 138 eV,ttributed to the presence at the textile surface, of phosphates asltime oxidation state of phosphor atoms resulting from the deepineralization of DMMP molecules, and that would result with
ime in the photocatalyst deactivation by active site blocking, andhus in the loss of the self-decontaminating properties of the textile.he XPS analysis allowed the efficiency of the regenerative treat-ent to be evidenced, with the disappearance of the P 1s broad
eak at 138 eV binding energy assigned to the phosphate surfacepecies, evidencing the refreshment of the photoactive surface. Thisas confirmed by a subsequent post-washing photocatalytic test
hat led to the total removal of DMMP for 10 min of illumination,imilarly than over the fresh photocatalytic textile. This showedhat a simple regeneration method such as a washing with basicater at pH = 12 could be processed for recovering the efficiency
nd the fresh surface of the photocatalytic textile.
. Conclusion
The layer-by-layer or multilayer building method haseen applied to develop photocatalytic textiles with self-econtaminating properties toward the photocatalytic degradationf organophosphorous and organosulfide chemical weapon agentsnder solar light illumination, obtained by functionalizing theextile fibers by a thin layer of a dense network of entangled
O3-modified titanate nanotubes.Solar light activated WO3-TiNTs with 10–20 nm mean outer
iameter and a high specific surface area of 274 m2/g have beenynthesized by hydrothermal treatment of TiO2 powder, with poly-ungstate salt impregnation prior to the final calcination step at80 ◦C.
In this layer-by-layer approach, polyethyleneimine was used asositively charged counter polyelectrolyte, alternatively depositeds multi double-layers with the WO3-modified titanate nanotubequeous suspension rectified at pH = 9. The layer-by-layer approachas been preferred to both dipping and spray deposition methods
n terms of homogenous functionalization of the textile as well asf photocatalytic self-decontaminating efficiency under artificialolar-like illumination.
The layer-by-layer functionalized photocatalytic textiles exhib-ted under solar light high self-decontaminating performancesoward the removal of the neurotoxic agent-simulating DMMP withcomplete removal of the toxicity within 7 min under standard-
zed protocol. That decontamination process can be used for yperiteive agent destruction under the same conditions, with a completeemoval of the toxicity within 20 min. The very low amount ofhotocatalytic materials deposited within the film and the electro-tatic nature of the multilayer interactions are of high interest forargeting the future development of self-decontaminating textiles.
orks are ongoing to further optimize the multilayer building andvaluate the self-decontaminating efficiency toward the removalf chemical and biological agents of interest for military as well asivilian purposes.
cknowledgements
The authors deeply thank the Direction Générale de l’ArmementDGA) for financial support and its continuous interest in the study,s well as for providing military textiles and performing the testsoward the yperite live CWA at the DGA CBRN Defense Center
MoD/DGA). P. Bernhardt and T. Romero (LMSPC) are gratefullycknowledged for performing XPS characterization and SEM anal-sis, respectively. Dr Corinne Ulhaq and Driss Ihiawakrim (IPCMS,trasbourg) are also thanked for performing TEM images andelping in IEP measurements, respectively. Dr. N. Chaoui (LMSCL,[
[
[
A: General 391 (2011) 455–467
Forbach, UPV) is acknowledged for its involvement in the contactangle measurement.
References
[1] A.V. Vorontsov, E.V. Savinov, L. Davydov, P.G. Smirniotis, Appl. Catal. B: Environ.32 (2001) 11–24.
[2] A.V. Vorontsov, C. Lion, E.N. Savinov, P.G. Smirniotis, J. Catal. 220 (2003)414–423.
[3] J. Zhou, K. Varazo, J.E. Reddic, M.L. Myrick, D.A. Chen, Anal. Chim. Acta 496(2003) 289–300.
[4] K.E. O’Shea, S. Beightol, I. Garcia, M. Aguilar, D.V. Kalen, W.J. Cooper, J. Pho-tochem. Photobiol. A 107 (1997) 221–226.
[5] T.N. Obee, S. Satyapal, J. Photochem. Photobiol. A 118 (1998) 45–51.[6] M. Grandcolas, A. Louvet, N. Keller, V. Keller, Angew. Chem. 48 (2009) 161–164.[7] B. Cojocaru, V.I. Parvulescu, E. Preda, G. Epure, V. Somoghi, E. Carbonell, M.
Alvaro, H. Garcia, Environ. Sci. Technol. 42 (2008) 4908–4913.[8] S. Neatu, V.I. Parvulescu, G. Epure, N. Petrea, V. Somoghi, G. Ricchiardi, S. Bor-
diga, A. Zecchina, Appl. Catal. B: Environ. 91 (2009) 546–553.[9] C. Cantau, S. Larribau, T. Pigot, M. Simon, M.T. Maurette, S. Lacombe, Catal. Today
122 (1–2) (2007) 27–38.10] C. Cantau, T. Pigot, R. Brown, P. Mocho, M.T. Maurette, F. Benoit-Marque, S.
Lacombe, Appl. Catal. B: Environ. 65 (1–2) (2006) 77–85.11] Y. Feldman, E. Wasserman, D.A. Srolovitch, Science 267 (1995) 222–225.12] M. Niederberger, H.J. Huhr, F. Krumeich, F. Bieri, D. Gunther, R. Nesper, Chem.
Mater. 12 (2000) 1995–2001.13] T. Kasuga, M. Hiramatsu, A. Hoson, Langmuir 14 (1998) 3160–3163.14] M.R. Hoffmann, S.T. Martin, W. Choi, D.W. Bahnemann, Chem. Rev. 95 (1995)
69–96.15] B. O’Regan, M. Graetzel, Nature 353 (1991) 737–740.16] M. Miyauchi, H. Tokudome, Thin Solid Films 515 (2006) 2091–2096.17] H. Tokudome, M. Miyauchi, Chem. Lett. 33 (2004) 1108–1109.18] M. Zhang, Z.S. Jin, J.W. Zhang, X.Y. Guo, J.J. Yang, W. Li, X.D. Wang, Z.J. Zhang, J.
Mol. Catal. A: Chem. 217 (2004) 203–210.19] J.G. Yu, H.G. Yu, B. Cheng, C. Trapalis, J. Mol. Catal. A: Chem. 249 (2006) 135–142.20] M.W. Xiao, L.S. Wang, X.J. Huang, Y.D. Wu, Z. Dang, J. Alloys Compd. 470 (1–2)
(2009) 486–491.21] C.-T. Hsieh, W.-S. Fan, W.-Y. Chen, J.Y. Lin, Sep. Purif. Technol. 67 (3) (2009)
312–318.22] M. Qamar, C.R. Yoon, H.J. Oh, N.H. Lee, K. Park, D.H. Kim, K.S. Lee, W.J. Lee, S.J.
Kim, Catal. Today 131 (1–4) (2008) 3–14.23] Z.R. Tian, J.A. Voigt, J. Liu, B. Mckenzie, H. Xu, J. Am. Chem. Soc. 125 (2003)
12384–12385.24] B.D. Yao, Y.F. Chan, X.Y. Zhang, W.F. Zhang, Z.Y. Yang, N. Wang, Appl. Phys. Lett.
82 (2003) 281–283.25] T. Kasuga, M. Hiramatsu, A. Hoson, T. Sekino, K. Nihara, Adv. Mater. 11 (1999)
1307–1311.26] T. Tachikawa, S. Tojo, M. Fujitsuka, T. Sekino, T. Majima, J. Phys. Chem. B: Lett.
110 (2006) 14055–14059.27] Y.R. Do, W. Lee, K. Dwight, A. Wold, J. Solid State Chem. 108 (1994) 198–201.28] C. Martin, G. Solana, V. Rives, G. Marci, L. Palmisano, A. Sclafani, Catal. Lett. 49
(1997) 235–243.29] Y.T. Kwon, K.Y. Song, W.I. Lee, G.J. Choi, Y.R. Do, J. Catal. 191 (2000) 192–199.30] F. Bosc, D. Edwards, N. Keller, V. Keller, A. Ayral, Thin Solid Films 495 (2006)
272–279.31] N. Keller, E. Barraud, F. Bosc, D. Edwards, V. Keller, Appl. Catal. B: Environ. 70
(2007) 423–430.32] G. Decher, Science 277 (1997) 1232–1237.33] G. Decher, J.B. Schlenoff, Multilayer Thin Films Sequential Assembly of
Nanocomposite Materials, Wiley-VCH, Weinheim, Germany, 2003, 524p.34] F. Caruso, K. Niikura, D. Neil Furlong, Y. Okahata, Langmuir 13 (1997)
3427–3433.35] K.U. Fulda, A. Kampes, L. Krasemann, B. Tieke, Thin Solid Films 327–329 (1998)
752–757.36] K.M. Chen, X. Jiang, L.C. Kimerling, P.T. Hammond, Langmuir 16 (2000)
7825–7834.37] M.D. Musick, D.J. Pena, S.L. Botsko, T.M. McEvoy, J.N. Richardson, M.J. Natan,
Langmuir 15 (1999) 844–850.38] T. Cassagneau, T.E. Mallouk, J.H. Fendler, J. Am. Chem. Soc. 120 (1998)
7848–7859.39] R. Ma, T. Sasaki, Y. Bando, J. Am. Chem. Soc. 126 (2004) 10382–10388.40] S. Hou, C.C. Harrell, L. Trofin, P. Kohli, C.R. Martin, J. Am. Chem. Soc. 126 (2004)
5674–5675.41] A. Yu, G.Q.M. Lu, J. Drennan, I.R. Gentle, Adv. Funct. Mater. 17 (2007) 2600–2605.42] K.T. Meilert, D. Laub, J. Kiwi, J. Mol. Catal. A: Chem. 237 (2005) 101–108.43] T. Yuranova, D. Laub, J. Kiwi, Catal. Today 122 (2007) 109–117.44] W. Daoud, J. Xin, J. Am. Ceram. Soc. 87 (2004) 953–955.45] H. Imai, H. Hirashima, J. Am. Ceram. Soc. 82 (1999) 2301–2306.
46] D. Mihailovic, Z. Saponjic, M. Radoicic, T. Radetic, P. Jovancic, J. Nedeljkovic, M.Radetic, Carbohydr. Polym. 79 (3) (2010) 526–532.47] M.J. Uddin, F. Cesano, F. Bonino, S. Bordiga, G. Spoto, D. Scarano, A. Zecchina, J.
Photochem. Photobiol. A: Chem. 189 (2007) 286–294.48] K.C. Krogman, N.S. Zacharia, D.M. Grillo, P.T. Hammond, Chem. Mater. 20 (2008)
1924–1930.

talysis
[
[[[
[[[
[[[[
[[
[
[[
Industry Press, Beijing, 2005, pp. 74–85.
M. Grandcolas et al. / Applied Ca
49] Y. Tsuge, J. Kim, Y. Sone, O. Kuwaki, S. Shiratori, Thin Solid Films 516 (2008)2463–2468.
50] S. Anderson, Acta Crystallogr. 15 (1962) 194–201.51] H. Izawa, S. Kikkawa, M. Koizumi, J. Phys. Chem. 86 (1982) 5023–5026.52] A. Sauvet, S. Baliteau, C. Lopez, P. Fabry, J. Solid State Chem. 177 (2004)
4508–4515.53] X. Sun, Y. Li, Chem. Eur. J. 9 (2003) 2229–2238.54] Q. Chen, G.H. Du, S. Zhang, L.M. Peng, Acta Crystallogr. B 58 (2002) 587–593.
55] Q. Chen, G.H. Du, R.C. Che, Z.Y. Yan, M. Peng, Appl. Phys. Lett. 79 (2001)3702–3704.56] D. Bavykin, J.M. Friedrich, Adv. Mater. 18 (2006) 2807–2824.57] Q. Chen, W. Zhou, L.M. Peng, Adv. Mater. 14 (2002) 1208–1211.58] S. Zhang, Q. Chen, L.-M. Peng, Phys. Rev. B 71 (2005) 014104–014115.59] Y. Suzuki, S. Yoshikawa, J. Mater. Res. 19 (2004) 982–985.
[[
[[
A: General 391 (2011) 455–467 467
60] M. Grandcolas, PhD Dissertation, Strasbourg University, 2009.61] Y. Suzuki, B.P. Pichon, M. Grandcolas, N. Keller, V. Keller, S. Yoshikawa, Trans.
Mater. Res. Soc. Jpn. 34 (3) (2009) 545–549.62] L.H. Gielgens, M.G. van Kampen, M.M. Broek, R. van Hardeveld, V.J. Ponec, J.
Catal. 154 (1995) 201–207.63] M. Kosmulski, Chemical Properties of Material Surfaces, Marcel Dekker, 2001.64] J. Ren, J. Shen, S.C. Lu, Science and Technology of Particles Dispersion, Chemical
65] H. Zhu, C. Zhang, Y. Tang, J. Wang, B. Ren, Y. Yin, Carbon 45 (2007) 226–228.66] K.C. Krogman, N.S. Zacharia, S. Schroeder, P.T. Hammond, Langmuir 23 (2007)
3137–3141.67] W. Chen, T. McCarthy, Macromolecules 30 (1997) 78–86.68] H. Harada, T. Hisanaga, K. Tanaka, Water Res. 24 (1990) 375–387.




















