
review article - Indian Drug Manufacturers' Association
72
INDIAN DRUGS 50(04) APRIL 2013 5 REVIEW ARTICLE SOLID LIPID NANOPARTICLES: EMERGING COLLOIDAL CARRIERS AS OCULAR DRUG DELIVERY SYSTEMS Swamy N.G.N.* and Abbas Z. (Received 30 August 2012) (Accepted 01 April 2013) ABSTRACT Numerous attempts have been made to improve the bioavailability from ocular drug delivery systems and to prolong the residence time of drugs applied topically onto the eye. Conventional ocular drug delivery systems such as eye drops and ointments are inefficient, whereas, systemic administration requires high doses which may result in significant toxicity. Therefore, a need arises to develop novel drug delivery carrier systems capable of increasing ocular bioavailability and decreasing both local and systemic cytotoxicity. Nanotechnology is expected to revolutionize ocular drug delivery. Solid lipid nanoparticles (SLNs) introduced in 1991 represent an alternative carrier system to traditional colloidal carriers, such as emulsions, liposomes and polymeric micro- and nanoparticles. SLNs do not show biotoxicity as they are prepared from physiological lipids and are ideal ocular drug delivery systems as they can enhance the corneal absorption of drugs and improve the ocular bioavailability of both hydrophilic and lipophilic drugs. SLNs have another advantage of allowing autoclave sterilization, an indispensible step in the formulation of ocular preparations. In this review a special attention has been given to the nature of lipids and surfactants commonly used for SLNs production. This article also reviews in detail the various fabrication methods, characterization, sterilization, and stabilization techniques for SLNs. In-vitro and in-vivo methods to study the drug release profile from SLNs have also been mentioned. A summary of previous studies involving the use of SLNs in ocular drug delivery is provided, along with a critical evaluation of SLNs as a potential colloidal ocular drug delivery system. *For correspondence Department of Pharmaceutics Government College of Pharmacy, No.2, P. Kalinga Rao Road, Subbaiah Circle, Bangalore – 560 027 E-mail: [email protected] Keywords: Ocular drug delivery, Solid lipid nanoparticles, nanostructured lipid carriers, Colloidal drug delivery, ocular controlled release. INTRODUCTION Ocular drug delivery is a challenge for pharmaceutical fraternity because of the complex nature and structure of the eye. Barriers such as the epithelial, aqueous–vitreous, blood–aqueous barrier, and blood–retinal barrier limits the entry of drugs via different routes to the eye. Usually deep drug penetration into the posterior chamber is necessary to treat glaucoma or uveitis and fight viral infections proliferated within the eye. Eye drops account for more than 90% of ocular preparations 1 . Although eye drops are cost effective, patient compatible and simple in formulation, a major fraction of the drug applied topically is washed away with tears or removed by other mechanisms. Ocular defense mechanisms limit the drug residence time over the cornea and reduce its absorption. From a conventional ophthalmic dosage form, only ∼5% of the drug enters the eye intact 2 . In many cases, posterior eye conditions are treated by intravenous or intravitreal administration of high doses of a drug, promising a high therapeutic index as in the case of antibiotics 3 . Drugs administered
-
Upload
khangminh22 -
Category
Documents
-
view
4 -
download
0
Transcript of review article - Indian Drug Manufacturers' Association

INDIAN DRUGS 50(04) ApRIl 2013 5
review article
solid lipid nanoparticles: emerging colloidal carriers as ocular drug delivery systems
swamy n.g.n.* and abbas Z.
(Received 30 August 2012) (Accepted 01 April 2013)
aBstract
Numerous attempts have been made to improve the bioavailability from ocular drug delivery systems and to prolong the residence time of drugs applied topically onto the eye. Conventional ocular drug delivery systems such as eye drops and ointments are inefficient, whereas, systemic administration requires high doses which may result in significant toxicity. Therefore, a need arises to develop novel drug delivery carrier systems capable of increasing ocular bioavailability and decreasing both local and systemic cytotoxicity. Nanotechnology is expected to revolutionize ocular drug delivery. Solid lipid nanoparticles (SlNs) introduced in 1991 represent an alternative carrier system to traditional colloidal carriers, such as emulsions, liposomes and polymeric micro- and nanoparticles. SlNs do not show biotoxicity as they are prepared from physiological lipids and are ideal ocular drug delivery systems as they can enhance the corneal absorption of drugs and improve the ocular bioavailability of both hydrophilic and lipophilic drugs. SlNs have another advantage of allowing autoclave sterilization, an indispensible step in the formulation of ocular preparations. In this review a special attention has been given to the nature of lipids and surfactants commonly used for SlNs production. This article also reviews in detail the various fabrication methods, characterization, sterilization, and stabilization techniques for SlNs. in-vitro and in-vivo methods to study the drug release profile from SlNs have also been mentioned. A summary of previous studies involving the use of SlNs in ocular drug delivery is provided, along with a critical evaluation of SlNs as a potential colloidal ocular drug delivery system.
*For correspondence department of pharmaceutics government college of pharmacy, no.2, p. Kalinga rao road, subbaiah circle, Bangalore – 560 027 e-mail: [email protected]
Keywords: Ocular drug delivery, Solid lipid nanoparticles, nanostructured lipid carriers, Colloidal drug delivery, ocular controlled release.
introduction
Ocular drug delivery is a challenge for pharmaceutical fraternity because of the complex nature and structure of the eye. Barriers such as the epithelial, aqueous–vitreous, blood–aqueous barrier, and blood–retinal barrier limits the entry of drugs via different routes to the eye. Usually deep drug
penetration into the posterior chamber is necessary to treat glaucoma or uveitis and fight viral infections proliferated within the eye.
Eye drops account for more than 90% of ocular preparations1. Although eye drops are cost effective, patient compatible and simple in formulation, a major fraction of the drug applied topically is washed away with tears or removed by other mechanisms. Ocular defense mechanisms limit the drug residence time over the cornea and reduce its absorption. From a conventional ophthalmic dosage form, only ∼5% of the drug enters the eye intact2.
In many cases, posterior eye conditions are treated by intravenous or intravitreal administration of high doses of a drug, promising a high therapeutic index as in the case of antibiotics3. Drugs administered

6 INDIAN DRUGS 50(04) ApRIl 2013
orally, intravenously, or through extravascular junctions barely reach the retina and the posterior chamber, thus delivering very low concentrations to the sites of action. periocular and intravitreal ocular administration routes offer advantages over conventional eye drops and ointments but there are still many associated drawbacks. A drug delivered via these routes is cleared rapidly from the site of action and repeated administration of high doses is often necessary4. Moreover, the intravitreal route of administration is considered invasive, which may cause endophthalmitis, cataracts, vitreous hemorrhages, and retinal detachment, especially if repeated exposure is necessary. Due to these reasons, this technique is less frequently used unless the therapeutic outcomes are extraordinary3,5. Sustained drug delivery devices including Ocusert®, Vitraser® and Retisert® offer several therapeutic improvements, but their use is somewhat limited due to a surgical procedure required to implant them. Therefore, it is increasingly desirable to develop novel delivery systems to achieve optimized treatment.
Drugs have to encounter several obstacles before reaching their target site to exert their pharmacological effect. The main obstacle is the ability to cross the tissue epithelium while maintaining stability. Novel drug delivery carriers such as solid lipid nanoparticles (SlNs) can help to alleviate these problems. SlNs are important because it was found that one of the criteria for a particle to enter the ocular mucosa, apart from its lipophilicity, is that it should be of submicron size6,7. For an ocular drug delivery system to be successful, it should have small particle size (less than 10 mcm) with a narrow size range8, should be non-irritant, adequately bioavailable, be compatible with ocular tissue, and cause no blurred vision1,9.
limitations for ocular absorption of drugs
surface removal of the dosage form
lacrimal secretions wash away topically applied drugs continuously and the excess of the lachrymal
fluid flows down the nasolacrimal duct swiftly3,10. Due to the presence of an extensive network of capillaries in the conjunctival sac and the nasal cavity, most of the drugs applied topically are absorbed into the systemic circulation, thereby reducing the ocular bioavailability to 5–10% only2,10. Systemic absorption from the ocular surface can cause side-effects, especially if the patient has various medication needs. For instance, timolol and other intra-ocular pressure reducing agents can cause cardiac and vascular complications, especially if the patient is susceptible11. Other pre-corneal factors limiting ocular drug absorption are drainage of the instilled solution; tear production (induced lacrimation), drug metabolism, and normal tear turn-over12. The human cul-de-sac can usually accommodate ∼30 μl of fluids, whereas, the instilled volume from eye drops is ∼50 μl. Nasolacrimal drainage further limits the ocular absorption. These limiting factors undermine therapeutic efficiency and reduce the pre-corneal half-life of drugs to ∼1–3 min8. The structure of the eye and the barriers for ocular drug absorption are presented in Fig. 1.
epithelial barrier
The epithelial layer is lipophilic and consists of tight junctions that limit the entry of hydrophilic drugs and macromolecules into the cornea and the aqueous humor12. In some eye conditions such as glaucoma and conjunctivitis, the corneal absorption increases significantly due to the morphological changes. This, along with the use of permeation enhancers and mucoadhesives, can be explored further to achieve better corneal penetrations.
Blood–aqueous barrier
This barrier is situated in the anterior segment of the eye and is composed of endothelial cells in the uvea. It limits the entry of hydrophilic drugs from the systemic circulation into the aqueous humor. This barrier gets disrupted sometimes due to inflammation and results in an enhanced temporary drug permeation3. Together with the blood–retinal barrier they make up the blood–ocular barrier.

INDIAN DRUGS 50(04) ApRIl 2013 7
Blood–retinal barrier
Blood-retinal barrier is situated in the posterior chamber; this barrier limits the entry of drugs from the systemic circulation to the retina. It is composed of retinal pigment epithelium (RpE) and the tight walls of the retinal capillaries13. Although drugs can reach the choroidal extravascular space easily through the leaky and extensive vasculature of the choroid, their retinal access is denied by RpE and retinal endothelia. The blood–retinal barrier, along with the blood–aqueous barrier, protects the eyes from the entry of xenobiotics and harmful substances3.
Systemic as well as topical administrations, intravitreal injections, local injections, lipophilic prodrugs, and ocular implants have all been used in retinal drug delivery14. These methods have their own disadvantages and limitations; there is as such a need to develop novel drug carriers which are safe, convenient, and efficient in crossing potential ocular barriers.
nanotechnology and its applications in ocular drug delivery
Nanotechnology is changing the perception of drug administration using conventional dosage forms
and has the potential to revolutionize the way we develop new therapies, as well as optimize existing ones. The term nanoparticle refers to a particulate drug delivery system where particle size is in the nanometer range (1–1000 nm). Nanoparticles are being investigated extensively in order to develop drug delivery systems capable of allowing penetration through physiological barriers. Nanoparticles are either in the form of matrix-dispersion (nanospheres) or a membrane-reservoir type (nanocapsules), where drugs can be dissolved, entrapped, encapsulated, and dispersed within the particles or adsorbed on the surface of these particles1.
In the course of preparation of SlNs a wide range of chemical and physiological materials have been used which include polymers, lipids, phospholipids, and metals. These multifunctional drug carriers are expected to accommodate high drug loads, help target them to the site of action, and promote sustained/controlled drug delivery while maintaining a minimum size of 30–300 nm15. physicochemical properties of SlNs such as particle size, surface net charge, shape, solubility, degree of ionization, and lipophilicity influence drug ocular absorption and determine the route of administration12.
Fig.1: the structure of the eye and barriers for drug absorption

8 INDIAN DRUGS 50(04) ApRIl 2013
In the recent years, scientists have envisaged keen interest in incorporating drugs and other therapeutics into nanoparticulate carriers, administered as modified eye drops which are cost effective and therapeutically efficient. The modified eye drops provide better penetration, extended ocular surface residence time, minimized drainage owing to mucoadhesive properties, simple administration, and patient compatibility16. Moreover, colloidal and particulate drug delivery systems can also be utilized for sub-conjunctival, periocular and intraocular injections17.
Nanoparticulate systems such as SlNs, niosomes, nanocapsules, nanospheres, dendrimers, nanosuspensions, liposomes and nanoemulsions have been employed in ocular drug delivery. These have alleviated problems associated with poorly soluble drugs, increasing their bioavailability while decreasing their administered dose and toxicity18. Nanotechnology-based delivery systems including SlNs can ensure sustained and controlled release of drugs and medicaments avoiding frequent administrations associated with conventional delivery systems. Further, nanomedicines have several advantages, such as the possibility of formulation as modified eye drops which are easily self-administered by the patient, elimination of the need for repeated administration, and offering protection against metabolic enzymes present on the ocular surfaces by constructing a protective barrier19,20,21.
liposomes are membrane-like lipid bilayer vesicles surrounding aqueous compartments. The common problems associated with liposomes are that they are in liquid form, which limits their pharmaceutical formulation feasibility. Moreover, most methods of sterilization are considered unsuitable for liposomes. The heating involved in autoclaving can irreversibly damage their vesicular structure and filtration reduces the vehicle to an average of 200 nm which limits their application22.
Hironaka et al. designed a sub-micron sized liposomal drug delivery system capable of delivering hydrophilic drugs to the posterior segment of the
eye21. Others have reported that acyclovir containing liposomes are more effective than simple eye drop preparations, due to better penetration and absorption through the cornea23.
Niosomes have been used to deliver drugs like gentamicin to the eye. Niosomes are formed from the self-assembling of nonionic amphiphiles in aqueous medium which are able to accommodate hydrophilic and hydrophobic drugs24,25,26. However, it is believed that SlNs have better stability, acceptable formulation feasibility, no toxicity, and superior release profile.
solid lipid nanoparticles
lipids have been used as an excipient or drug delivery vehicle for accommodating lipophilic drugs and elevating their poor physiological water solubility27. In the case of hydrophilic drugs, water in-oil emulsions and microemulsions were extensively investigated to solubilize them for potential ocular drug delivery. Although microemulsions have been known to scientists since 1928, the research for potential use in ocular delivery has only begun in the past decade. Despite their simple production, easy sterilization, and modest stability the use of microemulsions in ocular drug delivery is limited by the choice of ingredients possessing good ocular tolerability28. Oil-in-water emulsions and microemulsions require a high surfactant concentration to ensure formulation stability. This limits their ocular drug delivery applicability as the surfactants are usually not well tolerated.
SlNs are structured as a solid lipid core in nanometer ranges accommodating the drug stabilized by a layer of surfactants29. SlNs have several advantages over other colloidal carriers, such as the possibility of controlling drug release, drug targeting, long-term stability, good drug loading (whether hydrophilic or lipophilic), absence of biotoxicity due to the use of physiological lipids, possibility of sterilization by autoclaving, and easy large scale production30. In addition, due to their nano size range, SlNs can be an effective ocular drug delivery system by enhancing

INDIAN DRUGS 50(04) ApRIl 2013 9
corneal absorption, improving ocular bioavailability, prolonging the ocular retention time, and providing a sustained drug release profile31. SlNs also offer other advantages such as biodegradability, safety, low cost, simple production, and, most importantly, free dispersibility in aqueous media, enabling them to be formulated as modified eye drops.
SlNs are mainly fabricated from triglycerides in a specific orientation consisting of a polar core with polar heads facing towards the aqueous phase32. Many different lipids (triglycerides, hard fat types, partial glycerides, steroids, and waxes) and all classes of emulsifiers (ionic and non-ionic) have been used to prepare SlNs. In comparison to an aqueous eye drop, an ocular SlNs system can have extended residence time on the ocular surface and conjunctival sac leading to sustained release of the drug revealed by in vivo studies33. However, their efficiency for ocular drug delivery has not been studied extensively and necessitates further investigation34. Drug particles engulfed in SlNs do effectively cross the epithelium due to the SlNs lipophilic properties. Moreover, the epithelium is slightly negatively charged, hence cationic SlNs can increase the corneal residence time of the drug and increase its absorption levels.
Modification of SLNs and evolution of nanostructured lipid carriers (nlcs) and lipid drug conjugates (ldcs)
limited drug loading capacity, possibility of drug expulsion during phase modifications, and high water content of SlNs aqueous dispersions (70–90%) have led to the introduction of NlCs29,30. NlCs are produced by controlled addition of a spatially incompatible liquid lipid to the solid lipid component in order to accommodate a larger quantity of the drug and achieve a better release profile29,35. This can be achieved by increasing the space between the fatty acid chains of the glycerides, allowing more drugs to be accommodated, and formation of imperfect lipid crystals avoiding drug expulsion during storage35. Although NlCs contain up to 30% liquid lipids, the final product is in the solid state with no crystalline structure. Fig. 2 represents the difference in drug
particles encapsulated in a nanocapsule and drug particles dispersed in a nanosphere.
imperfect nlcs
Those special systems in which a liquid lipid (e.g. glycerides) is added to the solid lipid can be regarded as imperfect NlCs. They induce a larger distance between the fatty acid chains of the main lipid core, which causes imperfections in the crystalline structure of the lipid where a very large amount of the drug can be incorporated29. For example, Jenning et al. incorporated a medium length chain triglyceride oil in a matrix of a solid long chain glyceride, achieving higher payloads and controlled release properties36.
multiple type nlcs
In this case, liquid lipids are mixed in excess with solid lipids, resulting in nanocompartments of liquid lipid formed within the solid core which is already dispersed in the aqueous medium. The nanocompartments of liquid lipids are dispersed uniformly in the matrix of solid lipids, which are protected from degradation and potentiate prolonged release behaviour of the system35. This multiple system is beneficial for drugs that have higher solubility in liquid oils29. Excess of liquid lipids used in such cases inhibits drug expulsion, especially if a high concentration of drug is used in the preparation step. For example, NlCs of Compritol® 888 ATO can be prepared with incorporation of incompatible liquid lipids such as α-tocopherol or Miglyol® 812. These NlCs present a better drug protection and entrapment efficiency when compared to normal SlNs36,37.
structureless nlcs
This approach is to mix and melt together carefully selected liquid and solid lipids which upon cooling solidify but do not crystallize. These are regarded as structureless NlCs and can avoid drug expulsion caused by crystallization29.
lipid drug conjugates (ldcs)
Alternatively, lipid drug conjugates are developed by converting a hydrophilic drug to a lipophilic drug

10 INDIAN DRUGS 50(04) ApRIl 2013
conjugate or a prodrug by addition of an ester or amide group. These are either used alone or combined with other solid lipids and are expected to enhance biological transport and targeting of hydrophilic drugs29.
materials used in the preparation of slns
In general, the types of lipids, surfactants, and drugs used in the preparation of SlNs have their own influence on the final quality of the product. For instance, release of a lipophilic compound from a lipid carrier depends upon partition between the aqueous medium surrounding the carrier, surfactant, and the lipid exploited37.
lipids
In the formation of SlNs, the properties of lipids, their crystalline structure, concentration, and hydrophobicity characteristics are important factors37. Supercooling of SlNs is also an important feature to be ensured after melt homogenization, otherwise the final product would appear as a nanoemulsions rather than colloidally dispersed SlNs. Moreover, the solid matrix is expected to remain in the solid state at body temperatures so that controlled and sustained drug release properties could be achieved38. A comprehensive list of lipids used in the preparation of SlNs for various applications is available in the literature30.
triglycerides
Chemical structure of a triglyceride is presented in Fig. 3. Triglycerides are either short (< 5 carbons),
Fig.2: (a) drug particles encapsulated in a nanocapsules (b) drug particles dispersed in a
nanosphere
Fig. 3: chemical structure of triglycerides
medium (6–12 carbons), or long chain (> 12 carbons) and they might be synthetically hydrogenated to reduce oxidative degeneration39. The longer chain triglycerides have higher melting points, are believed to be more stable, and expected to produce better SlNs. Many triglycerides such as tricarpin40, trilaurin, trimyristin, tripalmitin, tristearin and hydrated coco-glucerides have been used in the preparation of SlNs41-45.
gelucire 44/14
Gelucires are semi-solid lipids often used in the preparation of NlCs for ocular drug delivery. Gelucires are saturated polyglycolized glycerides which consist of a mixture of mono-, di-, and tri-glycerides and di-fatty acid esters of polyethylene glycol. It is reported in the literature that they can enhance transdermal absorption of certain drugs46. This finding has been applied to the corneal permeation issue and some promising results were obtained.
compritol®888 ato
The solid lipid glycerol behenate (Compritol®888 ATO) is used very often to prepare SlNs. It is composed of a mixture of mono-, di-, and triacylglycerols with a very small amount of α-form which disappears under thermal stress owing to its thermodynamic instability47. The chemistry of compritol® 888 ATO is presented in Fig. 4.

INDIAN DRUGS 50(04) ApRIl 2013 11
particles are produced which increase the surface area of the system which requires excessive amounts of surfactants to coat the newly formed surfaces in order to stop the uncovered surfaces from colliding and agglomerating. In ocular drug delivery the use of cationic surfactants is favoured in order to induce a positive charge to the formulated SlNs52.This allows a longer residence time in the epithelium and better penetration to the posterior chamber of the eye. Stearylamine is used in the preparation of cationic SlNs for ocular and non-ocular drug delivery53.
the drug itself
One of the advantages of SlNs over other particular carrier systems is that both hydrophilic and lipophilic drugs can be incorporated. SlNs allow controlled/sustained release of drugs, thereby reducing the required administered dose to achieve therapeutic objectives. Drugs can be incorporated into SlNs through deposition between fatty acid chains, between lipid layers, or in imperfections37.
slns fabrication techniques
SlNs can be prepared by several methods which are enlisted below:
1. High shear homogenization
a) Hot homogenization technique
b) Cold homogenization technique
2. Ultrasonication
3. W/O/W multiple emulsion
4. Solvent emulsification-evaporation
5. Solvent emulsification-diffusion
6. Oil-in-water microemulsion
7. preparation of SlNs in co-flowing microchannels
8. preparation of SlNs by using supercritical fluid
Here are summarized a few important and widely used methods for the production of SlNs:
1. high pressure homogenization
High pressure homogenization technique has been used extensively to produce SlNs and
Fig.4: compritol® 888 ato consists of mixtures of monobehenate (a), dibehenate(b) and tribehenate
(c) of glycerol
Compritol usually crystallizes in its β´-modification which is very sensible at elevated temperatures. It can be useful to prepare SlNs with drugs which have solubility problems (e.g. Acyclovir) by using an optimized concentration of surfactants like poloxamer188. It has been shown that Compritol has higher loading capacities compared to stearic acid, monostearin, and tristearin37 and reportedly can be used to avoid re-crystallinity problems associated with other solid lipid used48.
cationic lipids
Cationic lipids are often used alone (e.g. stearic acid) or in combination (e.g. octadecylamine and N-[1-(2, 3-dioleoyloxy) propyl]-N, N, N-trimethylammonium chloride (DOTAp)) to induce a positive charge on SlNs. Cationic SlNs obtained using these cationic lipids have shown better corneal penetration and gene transfection properties49. The enhanced drug penetration through the cornea is probably due to the ionic interaction of cationic particles with negatively charged epithelial cells50.
Emulsifiers
Emulsifiers such as polysorbates (e.g. Tween®-80), polyoxyls (e.g. Cremophor® El), sodium lauryl sulfate, bile salts (e.g. cholic acid), poloxomer® 188, Brij®78, and saponins with HlB values ranging from 6–18 have been used quite often in preparation of SlNs33,39.Many of these emulsifiers are also used as corneal epithelium penetration enhancers. Studies have shown that higher concentrations of emulsifiers produce smaller sized particles and low concentrations of emulsifiers produce relatively larger particles51,52. During the emulsification process, smaller sized

12 INDIAN DRUGS 50(04) ApRIl 2013
nanoemulsions for parenteral use. High pressure homogenizers are widely available in pharmaceutical production plants and laboratories, and scaling up the method represents few problems. This technique is of two types (a) hot pressure homogenization (b) cold pressure homogenization. A preparatory step is involved in both hot and cold homogenization is the drug has to be dissolved or dispersed in the lipid melt. This step eliminates the need to use an organic solvent to dissolve the lipids which might cause toxicity concerns if residues remain.
(a) hot pressure homogenization
In hot homogenization the mixture of drug inside the lipid melt is first emulsified with a solution of proposed surfactant(s) using a high speed stirrer. The primary emulsion formed is subjected to high pressure homogenization in temperatures above the melting point of the lipid. The homogenization can be repeated a few times to achieve the desired sized particles, taking into account that extra cycles can actually yield bigger sized particles due to increased kinetic energy causing agglomeration. Finally, the hot oil-in-water emulsion is cooled to room temperature to produce a super cooled melt to yield SlNs30. One of the drawbacks of this method is the elevated temperatures required to melt the lipid which might further increase while applying high shear stress. This can affect the chemical stability of thermo-labile drugs and therapeutics such as antibiotics, peptides and proteins which are widely used in ophthalmic procedures.
(b) cold pressure homogenization
In cold homogenization excessive heating is avoided by solidifying the lipid-drug mixture in liquid nitrogen and grinding it to a powder with particle sizes below 100 nm before dispersing it in a cold aqueous solution containing the surfactant(s). Although cold homogenization can control the complex crystallization behavior of lipids during super-cooling and drug distribution through the aqueous phase during homogenization, it usually yields particles with higher average particle sizes30.
2. ultrasonication
In this method, solid lipid will be heated 5–100C above its melting point, and then added to a mixture of surfactants and water, previously heated at the same temperature. A pre-emulsion was obtained under stirring with an Ultra-Turrax T25 (Janke & Kunkel GmbH, Germany), at 8000 rpm for 5 min. A sonication probe (6mm diameter) was placed in this pre-emulsion, by means of an Ultrasonic processor VCX130 (Sonics, Switzerland). A power output with amplitude of 70% will be applied for 20 min, which leads to droplet breakage by acoustic cavitation, and subsequent formation of nanoparticles. For drug-loaded SlN, the drug will be added to the solid lipid before melting and sonication54.
Castelli F. et al. have prepared solid lipid nanoparticles (SlN) and nanostructured lipid carriers (NlC) of indomethacin by ultrasonication method. The mean particle size and percentage of drug encapsulation was determined55.
3. w/o/w multiple emulsion
W/O/W multiple emulsion is a relatively new method which has been utilized in recent years to prepare nanoemulsions and SlNs. Heating is not involved during the process, which makes it suitable for thermo-labile substances. The only disadvantages associated with this method are the use of organic solvent and the possibility of the presence of metal impurities from the sonicator30.
The method involves dissolving a drug in an aqueous solvent56. The aqueous solution is then emulsified in an oil phase containing the lipid(s) dissolved in an organic solvent to form a primary w/o emulsion. The primary w/o emulsion is then dispersed into an aqueous solution containing the surfactant. The w/o/w multiple emulsion system is mechanically agitated to allow complete evaporation of the organic solvent until SlNs are formed.
4. Solvent emulsification-evaporationSjostrom and Bergenstahl developed a method for
preparing lecithin stabilized nanoparticles containing

INDIAN DRUGS 50(04) ApRIl 2013 13
cholesteryl acetate. In this method the lipid component is dissolved in a water immiscible organic solvent and the drug is either dissolved or dispersed in the lipid solution. For preparation of cationic SlNs the main lipid core (e.g. precirol® ATO 5) is dissolved in an organic solvent (e.g. dichloromethane) and the cationic lipid (e.g. DODAB) can be dispersed in the aqueous phase containing the surfactant. This organic phase is then emulsified in an aqueous solution containing a biocompatible cosurfactant such as bile salts (e.g. sodium glycocholate and phosphatidylcholine). Emulsification can be achieved using a high speed stirrer Ultra-Turrax followed by high pressure homogenization. The system is mechanically stirred at room temperature until the solvent completely evaporates57.
The quality of the primary emulsion directly affects the final SlN average size. The smaller the droplets in the primary emulsions, the smaller will be the final SlN. Hence, it is important to optimize all parameters which influence the emulsification process such as
the lipid and emulsifier’s concentration and type. If optimized, this method can produce particles with an average size below 100 nm by precipitation into an o/w emulsion57. Fig. 5 depicts a schematic representation of multiple emulsion technique.
5. Solvent emulsification-diffusion
This method is based on the water miscibility property of certain organic solvents such as butyl lactate or benzylalcohol as the oil phase58,59. A primary oil-in-water emulsion is prepared containing the drug and the lipid phase solution in a water miscible solvent. This emulsion is then transferred into water. The hypothesis is that upon introduction to an aqueous phase, the water-miscible solvent will diffuse. Hence, the lipophilic material dissolved in the solvent will solidify due to the diffusion of solvent from droplets to the continuous phase59.
Trotta et al. used this method to encapsulate insulin into glyceride monostearate solid lipid micro- and nanoparticles using isobutyric acid as a
Fig.5: schematic representation of multiple emulsion technique

14 INDIAN DRUGS 50(04) ApRIl 2013
water-miscible solvent. The method is reliable for the entrapment of lipophilic and hydrophilic drugs if optimized59.
6. oil-in-water microemulsion
Oil-in-water microemulsion technique is an easy and suitable method for the preparation of SlNs. It does not require organic solvents; however, a pre-heating step is required which may be disadvantageous to thermolabile substances. Briefly, the oil components are melted at ∼10°C above their melting point and the drug to beloaded inside SlNs is mixed with the lipid melt. The lipid melt is then dispersed and homogenized in a hot aqueous phase containing the surfactant heated at the same temperature of the lipid melt simultaneously. The hot o/w microemulsion is then cooled rapidly while maintaining the mechanical stirring until SlNs are formed. Vighi et al. used this method to prepare SlNs suitable for gene delivery60.
The theory is that oil droplets are present in the hot o/w microemulsion. Upon sudden decrease of temperature, the nanoparticles are expected to crystallize rapidly; forming SlNs61. Marengo et al developed an apparatus for SlNs preparation which works by quenching of the warm o/w microemulsion into cold water. The warm o/w emulsion can be either cooled using an ice bath or it can be placed into cold water directly62,63.
7. Preparation of SLNs in co-flowing microchannels
preparation of SlNs using microchannels is under investigation so that critical procedures associated with earlier production methods such as high speeds, high pressures, high temperatures, and the use of toxic organic solvents might be avoided. Microchannels offer several advantages such as efficient mass transfer, stable flow field, and uniform concentration distribution, which results in continuous production of SlNs with a narrow size range64.
The basic principle behind this method is solvent displacement. As described by Zhang et al. the apparatus consists of a co-flowing microchannel system with inner and outer capillaries. The lipid is dissolved in a water-miscible solvent which is injected into the inner capillary. The aqueous phase containing the surfactant is injected into the outer capillary simultaneously. Displacement of the solvent from the lipid phase to the aqueous phase causes local super saturation and solidification of lipids resulting in the formation of SlNs65.
particles with different size and morphology can be prepared by altering parameters such as the velocities of liquids flow to adjust focused flow patterns. Due to the small diameter of microchannels it is often blocked by SlNs hindering continuous production. Gas–liquid slug flow was used by Yun et al. to overcome microchannel blockage66. Fig. 6 represents
Fig.6: Solid lipid production by liquid flow-focusing and gas displacement method

INDIAN DRUGS 50(04) ApRIl 2013 15
a schematic diagram of solid lipid production by liquid flow-focusing and gas displacement method.
8. preparation of slns by using super critical fluid
This method is relatively new technique for SlNs production and has the advantage of solvent-less processing. There are several variations in this platform technology for powder and nanoparticle preparation. SlNs can be prepared by the rapid expansion of supercritical carbon dioxide solutions (RESS) method. Carbon dioxide (99.99%) was the good choice as a solvent for this method63.
post-preparation procedures
Purification and separation of SLNs
purification is an important step in SlNs preparation to avoid toxicity associated with extra surfactants. Therefore, methods like ultrafiltration59, ultracentrifugation, and dialysis have been employed67.
sterilization
Sterilization is a required step for all ocular preparations. Ophthalmic products usually have a short shelf life and they must be used within 1 month after opening due to contamination risks. In many cases a sterilization technique has to be carefully chosen to ensure formulation sterility without degradation and aggregation of the solid lipids to avoid toxicity and instability30. Commonly used techniques for sterilization are autoclaving, filtration, and aseptic production γ-radiation.
One of the advantages of SlNs over other colloidal systems is that they can be sterilized by autoclaving, a commonly used, straightforward, and reliable technique. A study on trilaurine SlNs loaded with azidothymidinepalmitate showed that SlNs dispersions maintained good stability after autoclaving at 121°C for 20 min68. However, it should be noted that heating can induce physical instability and particle aggregation. An increase in the average particle size is usually observed after sterilization
by heating. During autoclaving SlNs melt and recrystallize in a controlled manner. Therefore, certain structural features assigned to SlNs by controlling the production parameters will be lost by autoclaving, the severity of which is defined by the composition of the SlNs69.
A filtration technique as well as aseptic preparation strategy can be also employed for SlNs sterilization, similar to the sterilization of parenteral emulsions and nutrition40.
Freeze drying of slns
Freeze-drying, also known as lyophilization, is a process that allows the stabilization of biomaterials so that they can be stored. lyophilization involves the removal of water by employing the use of a process known as sublimation, wherein a solid is converted to the vapor state without first passing through the liquid phase. lyophilization is utilized as a critical technique to convert the lipid dispersion to a solid state to extend the stability and to avoid particle aggregation30.
The freeze-drying process improves their long-term chemical and physical stability of SlNs. This means that degradation reactions such as hydrolysis are prevented and initial particle size is preserved. The freezing procedure also affects the crystal structure and properties of the lyophilizate60. Cooling may be done either rapidly or slowly. Rapid cooling can be performed by dipping the vial containing the preparation into liquid nitrogen or by adding the SlN dispersion dropwise to liquid nitrogen30. This results in the formation of small, heterogeneous crystals, whereas, slow cooling causes large crystals to form as is done by placing the vials in a freeze drier having a shelf temperature of −25°C for 24 h. Each type is associated with certain advantages and disadvantages. While rapid cooling decreases freezing out effects, it causes slower sublimation. Therefore, freeze-drying must be done in a sample-specific manner by optimizing the lyophilization process30.

16 INDIAN DRUGS 50(04) ApRIl 2013
The process of freeze-drying may have some unfavorable effects on the SlNs themselves; therefore it may be sometimes necessary to use certain protective materials known as cryoprotectors. These additives help to decrease particle aggregation and improve re-dispersion of the dry product60. Commonly used cryoprotective agents are sorbitol, mannose, trehalose, glucose, and polyvinylpyrrolidone (pVp).
spray-drying of slns
Spray-drying is used as an alternative method to increase SlNs stability. It involves the conversion of a solution or a suspension into a dry product. Although spray-drying is a widely used process in the pharmaceutical industry and is cheaper than lyophilization, it has rarely been used for SlNs formulations. Freitas and Muller described a process for carrying out spray-drying of SlNs wherein a product suitable for IV administration was obtained. They briefly summarized the steps into the following: (1) atomization of the feed into the spray, (2) spray–air contact, (3) drying of the spray, and (4) separation of the dried product from the drying gas. Several factors such as spray-drying parameters, the chemical nature of the lipid, and type of redispersion medium need to be taken into consideration in order to obtain a product which has optimum particle size and redispersion70.
drawbacks associated with slns
drug expulsion during storage
Crystalline behavior of lipids in the nanoscale is very complex. In SlNs, the solidification temperature is usually lower than that of bulk lipids71 and less ordered lipid modifications are often formed. However, drug expulsion during storage is one of the most common
Fig.7: drug expulsion during storage
issues associated with the lipid crystal transformation to a more stable β-modification with more a perfect crystalline lattice, leaving behind less space for drug accommodation. A typical diagram of drug expulsion during storage is presented drug in Fig. 7.
drug enriched shell and the burst release
Fig. 8 provides the explaination of burst release associated with solid lipid nanoparticles release profile. The burst effect is a common problem associated with SlNs release profiles. The burst effect is explained by the room temperature phase separation effect. It is proposed that when the hot o/w emulsion containing the drug is cooled to solidify, lipids crystallize to form SlNs with a drug-free core and a drug enriched shell. This soft deformable drug enriched surface shell was detected in a study by atomic force microscopy (AFM) and small angle X-ray scattering (SAXs) measured as a 15–18nm thick layer48. To achieve a prolonged release profile of SlNs, lower temperatures and surfactant concentrations during production are advised as optimal conditions30,35.
Fig.8: explanation of burst release associated with solid lipid nanoparticles release profile
Biotoxicity
The most important problem associated with most nanoparticulate drug delivery carriers is their lack of biodegradability, which makes them biotoxic72. Nanoparticles can induce cell toxicity by either one or a combination of the following mechanisms: adherence

INDIAN DRUGS 50(04) ApRIl 2013 17
to the cell membrane due to their nanometer scale or ionic charge, degradation, the release of cytotoxic degradation products, cellular internalization followed by degradation, and toxic effects inside the cells73. SlNs are usually made up of physiological lipids which are biodegradable with simple natural processes like enzyme digestion. Biodegradability of SlNs is one of the most important advantages in making them an outstanding drug delivery vehicle. These lipids and their degradation products are considered non-toxic to human body cells. However, due to their nano size range their biotoxicity is an important issue as the human body reacts very differently to nanoparticles as compared to larger particles of the same material. Certain tests (e.g. EpISKIN test and HET-CAM test) can be performed to evaluate topical and ocular toxicity and irritation caused by SlNs.
characterization and quality assessment of slns
Formulation stability of SlNs can be evaluated via particle size, zeta potential, and entrapment efficiency studies56. Characterization of structure and quality is a very important step in determining stability and release kinetics of SlNs.
A number of methods have been employed to measure SlN particle size and morphology, amongst them photon correlation spectroscopy (pCS)56, atomic force microscopy (AFM)60, transmission electron microscopy (TEM)56 and field flow- fractionation (FFF) being the most popular. Zeta potential measurement is equally important in order to predict stability of colloidal dispersions. It is known that particles of the same charges tend to repel each other and hence avoid aggregation. Measurement of crystallinity and lipid modification is important because these parameters influence drug incorporation, stability and release rates. Methods like differential scanning calorimetry (DSC) and X-ray scattering are often used to examine the status of lipids. In addition, nuclear magnetic resonance (NMR), and electron spin resonance (ESR) methods are used to assess the presence of other colloidal structures such as microparticles in the formulation30.
particle characterization
particle size
particle size measurement can be regarded as the easiest way to estimate SlNs stability in dispersion over a period of time. Methods widely used to measure particle size are photon correlation microscopy (pCS) and laser diffraction (lD)74.
Zeta potential
Electrokinetic potential of colloidal systems, referred to as zeta potential, determines the stability of colloidal dispersions. Zeta potential indicates the extent of particle–particle repulsion forces necessary to avoid agglomeration and aggregation. Higher zeta potential values, whether positive or negative, indicate higher dispersion stability. The zeta potential of an emulsion is determined by the chemical nature of the surfactant. Zeta potential of ±30 mV is considered to be sufficient to ensure physical stability of emulsion75. Apart from the stability of the dispersion, surface charges are important in molecular mechanisms of drug absorption. In ocular drug delivery, this is explained by the fact that the corneal epithelial cells are negatively charged. Therefore, to increase drug residence time and ultimately its penetration, cationic SlNs which are proposed to modify corneal morphology through ionic interactions can be used.
morphology of the particles
The following methods are used to determine particle size, size distribution and morphology of SlNs.
transmission electron microscopy (tem)
TEM evaluates particle morphology by examining the electrons that are transmitted through the specimen29. An image is produced by interpreting the interaction of the electrons passed through the specimen, which is visualized by an imaging device or detected by a special sensor.
scanning electron microscopy (sem)
This method offers excellent resolution and an easier sample preparation procedure for

18 INDIAN DRUGS 50(04) ApRIl 2013
morphological examination of SlNs. SEM measures electrons transmitted from the particle surfaces to evaluate their morphology29.
atomic force microscopy (aFm)
AFM produces a three-dimensional image of nanoparticles. It is a very sensitive device and spatial resolution of up to 0.01 nm can be achieved by measuring the force acting between the probing tip and the particle surface29.
crystalline properties of lipids
differential scanning calorimetry (dsc)
DSC can be described as a thermal analysis technique used in the investigation of melting, crystallization, solid-to-solid transition temperatures of lipids, and determination of the solid fat content of the excipient76. The thermal events that can be detected by this method may be endothermic phenomena such as melting or exothermic phenomena such as crystallization77.
DSC has various applications and is very successfully used in the pharmaceutical industry as a method of drug analysis and characterization of new delivery systems78. With specific reference to SlNs, DSC allows the study of their melting and recrystallization behavior. This is important as the crystallinity of a lipid matrix has an effect on the functional properties such as drug incorporation and release rates of the SlN derived from it.
The paper by Attama et al. describes the characterization of SlNs prepared with a mixture of theobroma oil and goat fat as the main lipids, phospholipon® 90G as the heterolipid, and polysorbate 80 as the mobile surfactant. Methods of characterization used were time-resolved particle size analysis, zeta potential and osmotic pressure measurements, differential scanning calorimetry (DSC), transmission electron microscopy (TEM), and isothermal heat conduction microcalorimetry (IMC)79.
wide angle X-ray diffraction (waXd)
This is a well-established method used to investigate the crystalline nature of the formulated SlNs79,80. It is used to qualitatively determine the crystalline ingredients of a dispersion according to their individual diffraction patterns. If a crystalline structure is identified, information about polymorphic modifications can be obtained subsequently. For example, Jenning et al., confirmed the presence of β´-modification of compritol using X-ray diffraction. This is based on the fact that the X-rays reflected due to the crystalline lipids appear well above the amorphous background of non-crystalline lipids81.
Drug encapsulation and loading efficiency
Determination of encapsulation efficiency is based on the separation of lipids from the aqueous phase of the dispersion achieved either by ultrafiltration, ultracentrifugation, gel filtration using a sephadex column, or dialysis29.
In ultrafiltration, the amount of drug loading can be calculated indirectly after centrifugation in a membrane concentrator82. Sometimes ultracentrifugation for a period of time is solely sufficient to separate SlNs from the aqueous phase. The amount of drug present in the aqueous continuous phase can be determined by a sensitive method like HplC or UV spectroscopy. The loading capacity can then be calculated considering the initial concentration of drug used. Attama et al.82 repeated these measurements thrice at 2-week intervals to allow the complete crystallization of the lipids in order to determine if the crystalline lattice modifications cause drug expulsion. The entrapment efficiency and drug loading can be calculated using equations (1) and (2)
%Entrapment efficiency = [(Initial drug weight– weight of free drug) / weight of initial drug] x 100% ........... (1)
%Drug loading = [(Initial drug weight – weight of free drug) / weight of lipid] x 100 % ............ (2)
Ultrafilitration and microdialysis are known to be the most accurate method to measure

INDIAN DRUGS 50(04) ApRIl 2013 19
entrapment efficiency of encapsulated systems83. Although ultracentrifugation is the fastest and easiest method to use, the results obtained are not reliable.
release studies
In vitro release studies
Drugs incorporated into SlNs are usually released by diffusion through the lipid matrix and/or biodegradation and surface erosion of the lipid matrix29. This should allow a sustained and controlled release of drug from the colloidal system. An immediate burst effect can release a major portion of the drug in a short period of time. This can be due to surface adsorption rather than encapsulation or dispersion. The aqueous solubility of most drugs is enhanced at higher temperatures. This causes a change in the partition coefficient of the drug and its position in the SlNs. Therefore, higher production temperatures promote drug localization on the surface of SlNs, causing a burst effect29.
In vitro release studies using a modified Franz diffusion cell
in vitro release studies can be performed in a modified Franz diffusion cell over a period of time82. A diffusion barrier like siliconized Spectrapore® MWCO 6000–8000 can be used to mimic physiological conditions. Alternatively, fresh cornea obtained from white male New Zealand rabbits can be used as the diffusion barrier mounted on a modified Franz-type cell84. A suitable buffer, such as phosphate buffer pH 7.4, is used as the acceptor medium which is magnetically stirred continuously. At specific time intervals, aliquots of samples containing the released drug are taken from the acceptor compartment and are quantified using a suitable method of determination such as HplC or spectroscopy.
In vivo release studies and toxicological tests
Animal studies are usually necessary to investigate the ocular bioavailability and release profile of drugs incorporated into different drug delivery vehicles. One animal used extensively for
ocular studies is the rabbit13, although larger animals are also employed in some studies85. The rabbit’s eye has obvious morphological and physiological differences with the human eye. The most important difference is the infrequent blinking rates, which can potentially affect preocular retention of the medicament13. Alternatively, release studies can be performed on a cell culture developed to mimic the eye physiology and its barriers. Ocular cell cultures avoid inter-species variability, and they can be used to study the release profile, mechanisms of cellular transport, metabolism, protein expression, and toxicity tests of SlNs and other ocular preparations13.
In vivo release
Attama et al.82 studied in vivo drug release of diclofenac sodium from SlNs by preparing a human cornea construct (HCC) using a method described in previous literature86,87.The use of HCC can resolve the problems associated with the use of rabbit’s cornea such as differences in physiological structure, enzymes presents, transporters, efflux proteins, surface proteins, and mucins.
The cornea of the eye is a multilayer barrier consisting of three layers, namely: the epithelium, the stroma, and the endothelium. This multilayered tissue can be engineered and created step-by-step in a Transwell cell culture insert using SV-40 immortalized human endothelial and epithelial cells and native fibroblasts (stromal cells). As a result, the human cornea construct prepared by this method will have a cellular structure resembling the real cornea with seven-to-nine layers of flattened epithelial cells, microvilli, and microplicae present. The method is promising since the permeability characteristics of HCC also closely resemble the permeation behaviour of excised porcine cornea86.
The HCC can be then mounted on a modified Franz diffusion cell and the amount of drug released to the acceptor medium can be obtained following a similar procedure as described before.

20 INDIAN DRUGS 50(04) ApRIl 2013
periocular retention of slns
In a method described in the literature two portions of 25 μl of fluorescent labeled SlNs were inserted into the lower conjunctival sac of a white male New Zealand rabbit at 90 s intervals38. A slit-lamp fitted with a blue filter was then used to examine the cornea. This test was performed on one eye of at least six rabbits, measuring the time course of fluorescence over the cornea88.
ocular tolerability and hydration levels
Irritation tendencies of SlNs preparations were tested on at least three rabbits by instilling 50 μl of SlNs in one eye while instilling a similar volume of physiological saline in the other eye as reference33. The eyes were examined using a slit-lamp at 0.5, 1, 3, and 12 h intervals and the irritancy was measured based on a scoring scale described in the literature89. Alternatively, the frequency of rabbit blinking in 5 min after SlNs administration to the lower cul-de-sac was measured against the frequency of blinking that occurred with the reference physiological solution or a phosphate buffer of pH 7.4 as a measure of ocular irritation84.
Hydration levels of the cornea can be used as a measure of SlNs/NlCs ocular toxicity. As described in the literature90, a healthy cornea maintains hydration levels between 76–80%; any value higher than 83% is associated with a certain degree of corneal injury. Hydration levels can be determined by calculating the difference in weight of the excised cornea before and after drying84.
case studies
Tobramycin (TOB) loaded SlNs were evaluated as carriers for topical ocular delivery33. The SlNs obtained were in the colloidal size range and contained 2.5% TOB as ion-pair complex with hexadecyl phosphate. Upon comparison with an equal dose of TOB administered by standard commercial eye drops, TOB-SlNs produced a significantly higher TOB bioavailability in the aqueous humour.
SlNs of diclofenac sodium (DNa) were prepared with a combination of homolipid from goat fat and phospholipid, and evaluated for delivery to the eye using bio-engineered human cornea, produced from immortalized human corneal endothelial cells (HENC), stromal fibroblasts and epithelial cells CEpI 17 Cl 4. Encapsulation efficiency was high and sustained release of DNa and high permeation through the bio-engineered cornea were achieved82.
Nanostructured lipid carriers for Ibuprofen84 were prepared by melted-ultrasonic method and investigated for its in vitro and in vivo characteristics. Gelucire 44/14 and Transcutol p enhanced the corneal permeability and stearylamine prolonged the pre-corneal retention time to some extent. Ibuprofen nanostructured lipid carriers displayed controlled-release property. The AUC of the optimized formulation of Ibuprofen nanostuctured lipid carriers was 3.99 times more than that of Ibuprofen eye drops.
Surface-modified SlNs containing timolol11 with and without phospholipid were formulated by melt emulsification with high-pressure homogenization. Results revealed that SlNs possessed small particles with low polydispersity indices, increased encapsulation efficiency and sustained in vitro release compared with unmodified lipid nanoparticles whose particles were greater than 160 nm. permeation of timolol from the surface-modified lipid nanoparticles across the cornea construct was sustained compared with timolol solution in distilled water.
Gatifloxacin loaded SlNs91 were prepared by o/w microemulsion technique with stearic acid (SlNs-A) and a mixture of stearic acid and Compritol (SlNs-B) as lipid matrix and poloxamer-188 as surfactant, using sodium taurocholate and ethanol as co-surfactant mixture, with a view to applying the SlNs in topical ocular drug delivery. SlNs composed of stearic acid and compritol have proved to be a good ocular drug delivery system considering the smaller particle size, particle size stability, and physiologically tolerable components.

INDIAN DRUGS 50(04) ApRIl 2013 21
Cyclosporine A (CsA)85 was successfully incorporated into cationic SlNs for ocular application. Due to the better characteristics like smaller particle size with narrow size distribution, high zeta potential and more stable lipid structure, Dynasan® 116 structured FD4 (0.1% CsA) formulation was chosen for in vivo studies. Sheep were used in in vivo studies and 200 ml of formulation was applied to sheep eyes (n=6) under veterinarian supervision. Release profiles were not decreased during 48 h indicating controlled and prolonged release of active agent from positively charged SlNs formulations due to increased residence time in eyes.
CsA loaded SlNs associated with chitosan (CS), were developed by high shear homogenization and ultrasound methods with CS in the aqueous phase and Compritol or precirol in the lipid phase to improve interaction and internalization in corneal cells. The penetration and permeation properties of the SlNs were assessed in vitro (cell culture) and ex vivo (excised pig cornea). CS-associated SlNs based on Compritol were biocompatible and enhanced the permeation/penetration of CsA along with a possible mechanism of internalization/uptake of the nanoparticles both in vitro and ex vivo92.
Methazolamide (MTA)93 SlNs were prepared by a modified emulsion-solvent evaporation method. The pharmacodynamics was investigated by determining the percentage decrease in intraocular pressure. The ocular irritation was studied by Draize test. Despite a burst release of SlNs, the pharmacodynamic experiment indicated that MTA–SlNs had higher therapeutic efficacy, later occurrence of maximum action, and more prolonged effect than drug solution and commercial product.
SlNs of baicalin (BA-SlNs)94 were prepared by emulsification / ultrasonication method for ocular drug delivery. The appearance of BA-SlNs was examined by the negative stain method. The results showed that the BA-SlNs had an average diameter of 91.42 ± 1.02 nm with a zeta potential of –33.5 ± –1.28 mV and the entrapment efficiency of 62.45 ±
1.67%. in vitro release studies indicated that the BA-SlNs retained the drug entity better than the baicalin ophthalmic solutions (BA-SOl). It was concluded that SlNs can be used as a carrier to enhance ocular bioavailability of baicalin.
SlNs of chloramphenicol95 were developed by a modified method of melt-emulsion ultrasonication and low temperature-solidification technique using glycerylmonostearate as the solid lipid, and poloxamer 188 as the surfactant employing response surface methodology design. in vitro release studies showed a burst release at the initial stage followed by a prolonged release of chloramphenicol from SlNs up to 48 hours. The results indicated that the SlNs could potentially be exploited as a delivery system with improved drug entrapment efficiency and controlled drug release.
With an aim to improve the ocular bioavailability of acyclovir96, SlNs and nanostructured lipid carriers (NlCs) were prepared by the modified hot O/W microemuslion method after optimizing a series of process parameters. The prepared nanoparticles were spherical and within the size range of 400– 777.56 nm. The drug release from SlNs and NlCs was a surface-based phenomenon. The results of the study suggest that SlNs can be successfully converted to physically superior NlCs, which have the potential to be developed further as ocular drug delivery systems for acyclovir.
conclusion
Solid lipid nanoparticles have the potential to offer a major contribution to the search for better ocular drug delivery systems. Recently, the US Food and drug administration (FDA) and European Medicines Agency (EMEA) have taken promising steps in order to lead academic research into more industrial and commercial aspects. What is required to end the long wait for a marketed ocular preparation based on colloidal carriers is a committed contribution from the scientists; interest and willingness of the pharmaceutical industry to combine and refine the academic findings into commercially viable ventures.

22 INDIAN DRUGS 50(04) ApRIl 2013
reFerences1. Bourlais C.l., Acar l., Zia H., Sado p.A., Needham T.,
leverge R.: Ophthalmic drug delivery systems—recent advances. prog. retin. eye res. 1998: 17(1): 33–58.
2. Gaudana R., Jwala J., Boddu S.H., Mitra A.K., Gaudana R., Boddu S.H.S.: Recent perspectives in ocular drug delivery. pharm. res. 2009; 26(5):1197–1216.
3. Urtti A.: Challenges and obstacles of ocular pharmacokinetics and drug delivery. adv. drug deliv. rev. 2006; 58(11):1131–1135.
4. Moshfeghi A.A., peyman G.A.: Micro- and nanoparticulates. adv. drug deliv. rev. 2005; 57(14): 2047–2052.
5. Yasukawa T., Kimura H., Tabata Y., Ogura Y.: Biodegradable scleral plugs for vitreoretinal drug delivery. adv. drug deliv. rev. 2001; 52(1):25–36.
6. Calvo p., Thomas C., Alonso M.J., Vila Jato J.l., Robinson J.: Study of the mechanism of interaction of poly-E-caprolactonenanocapsules with the cornea by confocal laser scanning microscopy. int. J. pharm. 1994; 103(3):283–291.
7. Alonso M.J.: Nanomedicines for overcoming biological barriers. Biomed pharmacother. 2004; 58(3): 168–172.
8. Zimmer A., Kreuter J.: Microspheres and nanoparticles used in ocular delivery systems. adv. drug deliv. rev. 1995; 16: 61–73.
9. Sahoo S.K., Dilnawaz F., Krishnakumar S.: Nanotechnology in ocular drug delivery. drug discov. today. 2008; 13(3-4):144–151.
10. Urtti A., Salminen l.: Minimizing systemic absorption of topically administered ophthalmic drugs. surv. ophthalmol. 1993; 37(6): 435–456.
11. Attama A.A., Reichl S., Muller-Goymann C.C.: Sustained release and permeation of timolol from surface-modified solid lipid nanoparticles through bioengineered human cornea. curr. eye res. 2009; 34(8):698–705.
12. Mainardes R.M., Urban M.C.C., Cinto p.O., Khalil N.M., Chaud M.V., Evangelista R.C., DaflonGremiao M.p.: Colloidal carriers for ophthalmic drug delivery. curr. drug target. 2005; 6(3): 363–371.
13. HornofM.,Toropainen E., Urtti A.: Cell culture models of the ocular barriers. eur. J pharm. Biopharm. 2005; 60(2): 207–225.
14. Duvvuri S., Majumdar S., Mitra A.K.: Drug delivery to the retina: challenges and opportunities. expert opinBiolther. 2003; 3(1): 45–56.
15. Debbage p. Targeted drugs and nanomedicine: present and future. curr. pharm. des. 2009; 15(2):153–172.
16. Nagarwal R.C., Kant S., Singh p.N., Maiti p., pandit J.K.: polymeric nanoparticulate system: a potential approach for ocular drug delivery. J. controlled rel. 2009; 136(1):2–13.
17. Janoria K.G., Hariharan S., Dasari C.R., Mitra A.K.: Recent patents and advances in ophthalmic drug
delivery. recent patents drug deliv. Formul. 2007; 1(2): 161–170.
18. Wissing S.A., Kayser O., Muller R.H.: Solid lipid nanoparticles for parenteral drug delivery. adv. drug deliv. rev. 2004; 56(9):1257–1272.
19. Kaur I.p., Garg A., Singla A.K., Aggarwal D.: Vesicular systems in ocular drug delivery: an overview. int. J. pharm. 2004; 269(1):1–14.
20. Araujo, J., Gonzalez E., Egea M.A., Garcia M.l., Souto E.B.: Nanomedicines for ocular NSAIDs: safety on drug delivery. nanomedicine. 2009; 5(4): 394–401.
21. Hironaka K., Inokuchi Y., Tozuka Y., Shimazawa M., Hara H., Takeuchi H.:Design and evaluation of a liposomal delivery system targeting the posterior segment of the eye. J. controlled rel. 2009; 136(3): 247–253.
22. Meisner D., Mezei M.: liposome ocular delivery systems. adv. drug deliv. rev. 1995; 16(1): 75–93.
23. law S.l., Huang K.J., Chiang C.H.: Acyclovir-containing liposomes for potential ocular delivery: corneal penetration and absorption. J. controlled rel. 2000; 63(1-2):135–140.
24. Abdelbary G., El-Gendy N.: Niosome-encapsulated gentamicin for ophthalmic controlled delivery. aaps pharmscitech. 2008; 9(3): 740–747.
25. Carafa M., Santucci E., Alhaique F., Coviello T., Murtas E., Riccieri F.M., lucania G., Torrisi M.R.: preparation and properties of new unilamellar non-ionic/ionic surfactant vesicles. int. J. pharm. 1998; 160(1): 51–59.
26. Uchegbu I.F., Vyas S.p.: Non-ionic surfactant based vesicles (niosomes) in drug delivery. int. J. pharm. 1998; 172(1-2): 33–70.
27. Chen M.l.: lipid excipients and delivery systems for pharmaceutical development: a regulatory perspective. adv. drug deliv. rev. 2008; 60(6): 768–777.
28. Vandamme T.F.: Microemulsions as ocular drug delivery systems: recent developments and future challenges. progr. retin. eye res. 2002; 21(1):15–34.
29. Sawant K.K., Dodiya S.S.: Recent advances and patents on solid lipid nanoparticles. recent patents drug deliv Formulation. 2008; 2(2): 120–135.
30. Mehnert W., Mader K.: Solid lipid nanoparticles: production, characterization and applications. adv. drug deliv. rev. 2001: 47(2-3):165–196.
31. Kaur I.p., Kanwar M., Kaur I.p., Kanwar M.: Ocular preparations: the formulation approach. drug dev.ind. pharm. 2002; 28(5):473–493.
32. Wadhwa S., paliwal R., paliwal S.R., Vyas S.p.: Nanocarriers in ocular drug delivery: an update review. curr. pharm. des. 2009; 15(23):2724–2750.
33. Cavalli R., Gasco M.R., Chetoni p., Burgalassi S., Saettone M.F.: Solid lipid nanoparticles (SlN) as ocular delivery system for tobramycin. int. J. pharm. 2002; 238(1-2):241–245.

INDIAN DRUGS 50(04) ApRIl 2013 23
34. Velpandian T., Intraocular penetration of antimicrobial agents in ophthalmic infections and drug delivery strategies. expert opin. drug deliv. 2009; 6(3): 255–270.
35. Muller R.H., Radtke M., Wissing S.A.: Nanostructured lipid matrices for improved microencapsulation of drugs. int. J. pharm. 2002; 242(1-2):121–128.
36. Jenning V., Mader K., Gohla S.H.: Solid lipid nanoparticles (SlNTM) based on binary mixtures of liquid and solid lipids: a 1H-NMR study. int. J. pharm. 2000; 205:15–21.
37. Vyas S.p., Rai S., paliwal R., Gupta p.N., Khatri K., Goyal A.K., Vaidya B.: Solid lipid nanoparticles (SlNs) as a rising tool in drug delivery science: one step up in nanotechnology. curr. nanosci. 2008; 4(1): 30–44.
38. Bunjes H., Westesen K., Koch M.H.J.: Crystallization tendency and polymorphic transitions in triglyceride nanoparticles. int. J. pharm. 1996; 129(1-2):159–173.
39. Hauss D.J. Oral lipid-based formulations. adv. drug deliv. rev. 2007; 59(7): 667–676.
40. Domb A.J.: long acting injectable oxytetracycline-liposphere formulations. int. J. pharm. 1995; 124(2):271–278.
41. Westesen K., Bunjes H.: Do nanoparticles prepared from lipids solid at room temperature always possess a solid lipid matrix? int. J. pharm. 1995; 115(1):129–131.
42. Heiati H., Tawashi R., Shivers R.R., phillipsN.C.: Solid lipid nanoparticles as drug carriers. I. Incorporation and retention of the lipophilic prodrug 3’-azido-3’ deoxythymidinepalmitate. int. J. pharm. 1997; 146(1):123–131.
43. Westesen K., Bunjes H., Koch M.H.J.: physicochemical characterizationof lipid nanoparticles and evaluation of their drug loading capacity and sustained release potential. J. controlled rel. 1997; 48(2-3):223–236.
44. Westesen K., Siekmann B.: Investigation of the gel formation of phospholipid-stabilized solid lipid nanoparticles. int. J. pharm. 1997; 151(1):35–45.
45. Almeida A.J., Runge S., Muller R.H.: peptide-loaded solid lipid nanoparticles (SlN): influence of production parameters. int. J. pharm. 1997; 149(2): 255–265.
46. Nilufer Y., Aysegul K., Yalcin O., Ayhan S., Sibel A.O., Tamer B.: Enhanced bioavailability of piroxicam using Gelucire 44/14 and labrasol in vitro and in vivo evaluation. eur. J. pharm. Biopharm. 2003; 56(3): 453–459.
47. Souto E.B., Mehnert W., Muller R.H.: polymorphic behavior of Compritol 888® ATO as bulk lipid and as SlN and NlC. J. microencapsul. 2006; 23(4):417–433.
48. ZurMuhlen A., Schwarz C., Mehnert W.: Solid lipid nanoparticles (SlN) for controlled drug delivery—drug release and release mechanism. eur. J. pharm. Biopharm. 1998; 45(2): 149–155.
49. Del pozo-Rodríguez A., Delgado D., Solinis M.A., Gascon A.R., pedraz J.l.: Solid lipid nanoparticles for retinal gene
therapy: transfection and intracellular trafficking in RpE cells. int J pharm. 2008; 360(1-2): 177–183.
50. Cortesi R., Argnani R., Esposito E., Dalpiaz A., Scatturin A., Bortolotti,F., lufino M., Guerrini R., Cavicchioni G., Incorvaia C., Menegatti E., Manservigi R.: Cationic liposomes as potential carriers for ocular administration of peptides with anti-herpetic activity. int. J. pharm. 2006; 317(1): 90–100.
51. Helgason T., Awad T.S., Kristbergsson K., McClements D.J., Weiss J.: Effect of surfactant surface coverage on formation of solid lipid nanoparticles (SlN). J. colloid interface sci. 2009; 334(1):75–81.
52. Radomska-Soukharev A.: Stability of lipid excipients in solid lipid nanoparticles. adv. drug deliv. rev. 2007; 59(6): 411–418.
53. li X., Nie S.F., Kong J., li N., Ju C.Y., pan W.S.: A controlled- release ocular delivery system for ibuprofen based on nanostructured lipid carriers. int. J. pharm. 2008; 363(1-2): 177–182.
54. Mulla J.S., Khazi I.M., Sharma N.K., Hiremath S.p., Jamakandi V.G.: Solid lipid Nanoparticles: Methods of preparation. iJndd. 2011; 3(3): 170-175.
55. Castelli F., puglia C., Sarpietro M.G.: Characterization of indomethacin-loaded lipid nanoparticles by differential scanning calorimetry. int. J. pharm. 2005; 304:231-238.
56. lv Q., Yu A., Xi Y., li H., Song Z., Cui J., Cao F., Zhai G.: Development and evaluation of penciclovir-loaded solid lipid nanoparticles for topical delivery. int. J. pharm. 2009; 372(1-2):191–198.
57. Sjostrom B., Bergenstahl B.: preparation of submicron drug particles in lecithin-stabilized o/w emulsionsI. Model studies of the precipitation of cholesteryl acetate. int. J. pharm. 1992; 88(1-3): 53–62.
58. Hu F.Q., Hong Y., Yuan H.: preparation and characterization of solid lipid nanoparticles containing peptide. int. J. pharm. 2004; 273(1-2): 29–35.
59. Trotta M., Cavalli R., CarlottiM.E., Battaglia l., Debernardi F.: Solid lipid micro-particles carrying insulin formed by solvent-in water emulsion-diffusion technique. int. J. pharm. 2005; 288(2):281–288.
60. Vighi E., Ruozi B., Montanari M., Battini R., leo E.: Re-dispersible cationic solid lipid nanoparticles (SlNs) freeze dried without cryoprotectors: characterization and ability to bind the pEGFp-plasmid. eur. J. pharm. Biopharm. 2007; 67(2):320–328.
61. Kuo Y.C., Chen H.H.: Entrapment and release of saquinavir using novel cationic solid lipid nanoparticles. int. J. pharm. 2009; 365(1-2): 206–213.
62. Marengo E., Cavalli R., Caputo O., Rodriguez l., Gasco M.R.: Scale-up of the preparation process of solid lipid nanospheres. partI.int. J. pharm. 2000; 205(1-2):3–13.
63. Cavalli R., Marengo E., Rodriguez l., Gascom R.: Effects of some experimental factors on the production process

24 INDIAN DRUGS 50(04) ApRIl 2013
of solid lipid nanoparticles. eur. J. pharm. Biopharm. 1996; 42(2): 110–115.
64. Yun J., Zhang S., Shen S., Chen Z., Yao K., Chen J.: Continuous production of solid lipid nanoparticles by liquid flow-focusing and gas displacing method in microchannels.chem. eng. sci. 2009; 64(19): 4115–4122.
65. Zhang S.H., Shen S.C., Chen Z., Yun J.X., Yao K.J., Chen B.B., Chen J.Z.: preparation of solid lipid nanoparticles in coflowingmicrochannels. chem. eng.J. 2008; 144(2): 324–328.
66. Schwarz C., Mehnert W.: Freeze-drying of drug-free and drug loaded solid lipid nanoparticles (SlN). int. J. pharm. 1997; 157(2):171–179.
67. Heydenreich A.V., Westmeier R., pedersen N., poulsen H.S., Kristensen H.G.: preparation and purification of cationic solid lipid nanospheres—effects on particle size, physical stability and cell toxicity. int. J. pharm. 2003; 254(1): 83–87.
68. Heiati H., Tawashi R., phillips N.C.: Drug retention and stability of solid lipid nanoparticles containing azidothymidinepalmitate after autoclaving, storage and lyophilization. J. microencapsul. 1998; 15(2):173–184.
69. Muller R.H., Mader K., Gohla S.: Solid lipid nanoparticles (SlN) for controlled drug delivery—a review of the state of the art. eur. J. pharm. Biopharm. 2000; 50(1): 161–177.
70. Freitas C., Muller R.H.: Spray-drying of solid lipid nanoparticles (SlN™). eur. J. pharm. Biopharm. 1998; 46(2):145–151.
71. Bunjes H., Koch M.H.J.: Saturated phospholipids promote crystallization but slow down polymorphic transitions in triglyceride nanoparticles. J. controlled rel. 2005; 107(2):229–243.
72. pardeike J., Hommoss A., Müller R. H.: lipid nanopar t ic les (SlN, NlC) in cosmet ic and pharmaceutical dermal products. int. J. pharm. 2009; 366(1-2): 170–184.
73. lherm C., Muller R.H., puisieux F., Couvreur p.: Alkylcyanoacrylate drug carriers: II. Cytotoxicity of cyanoacrylate nanoparticles with different alkyl chain length. int. J. pharm. 1992; 84(1):13–22.
74. pouton C.W., porter C.J.H.: Formulation of lipid-based delivery systems for oral administration: materials, methods and strategies. adv. drug deliv. rev. 2008; 60(6):625–637.
75. li X., lin X., Zheng l., Yu l., lv F., Zhang Q., liu W.: Effect of poly(ethylene glycol) stearate on the phase behavior of monocaprate/Tween80/water system and characterization of poly(ethylene glycol) stearate-modified solid lipid nanoparticles. colloid surf. a. physicochem. eng. aspect. 2008; 317(1-3): 352–359.
76. Jannin V., Musakhanian J., Marchaud D.: Approaches for the development of solid and semi-solid lipid-based
formulations. adv. drug deliv. rev. 2008; 60(6): 734–746.
77. ColemanN.J., Craig D.Q.M.: Modulated temperature differential scanning calorimetry: a novel approach to pharmaceutical thermal analysis. int. J. pharm. 1996; 135(1-2):13–29.
78. Gill p.S., Sauerbrunn S.R., Reading M.: Modulated differential scanning calorimetry. J. therm. anal. calorim. 1993; 40(3): 931–939.
79. Attama A.A., Schicke B.C., paepenmuller T., Muller-Goymann C.C.: Solid lipid nanodispersions containing mixed lipid core and a polar heterolipid: characterization. eur. J. pharm. Biopharm. 2007; 67(1):48–57.
80. Bunjes H., Unruh T.: Characterization of lipid nanoparticles by differential scanning calorimetry, X-ray and neutron scattering. adv. drug deliv. rev. 2007; 59(6): 379–402.
81. Attama A.A., Muller-Goymann C.C.: Investigation of surface modified solid lipid nanocontainers formulated with a heterolipid-templatedhomolipid. int. J. pharm. 2007; 334(1-2):179–189.
82. Attama A.A., Reichl S., Muller-Goymann C.C.: Diclofenac sodium delivery to the eye: in vitro evaluation of novel solid lipid nanoparticle formulation using human cornea construct. int. J. pharm. 2008; 355(1-2):307–313.
83. liu X., Zhang Y., Tang X., Zhang H.: Determination of entrapment efficiency and drug phase distribution of submicron emulsions loaded silybin. J. microencapsul. 2009; 26(2): 180–186.
84. li X., Nie S.F., Kong J., li N., Ju C.Y., pan W.S.: A controlled- release ocular delivery system for ibuprofen based on nanostructured lipid carriers. int. J. pharm. 2008; 363(1-2): 177–182.
85. Basaran E., Demirel M., Sirmagul B., Yazan Y.: Cyclosporine-A incorporated cationic solid lipid nanopart icles for ocular del ivery.Journal of microencapsulation. 2010; 27(1): 37–47
86. Reichl S., Bednarz J., Muller-Goymann C.C.: Human corneal equivalent as cell culture model for in vitro drug permeation studies. B. J. ophthalmol. 2004; 88: 560–565.
87. Reichl S., Dohring S., Bednarz J., Muller-Goymann C.C.: Human cornea construct HCC—an alternative for in vitro permeation studies? A comparison with human donor corneas. eur. J. pharm. Biopharm. 2005; 60(2): 305–308.
88. Giunchedi p., ConteU., Chetoni p., Saettone M.F.: pectin microspheres as ophthalmic carriers for piroxicam: evaluation in vitro and in vivo in albino rabbits. eur. J. pharm. sci. 1999; 9(1): 1–7.
89. Bottari F., Giannaccini B., Cristofori B., Saettone M.F., Tellini N.: Semisolid ophthalmic vehicles. I—A study of eye irritation in albino rabbits of a series of gel-type

INDIAN DRUGS 50(04) ApRIl 2013 25
aqueous bases. Farmaco— edizionepratica. 1978; 33(10):434–446.
90. Ronald D.S., Hong-Shian H.: Corneal penetration behavior of beta-blocking agents I: physicochemical factors. J. pharm. sci. 1983; 72(11):1266–1272.
91. Kalam M.A., Sultana Y., Ali A., Aqil M., Mishra A.K., Chuttani K.: preparation, characterization, and evaluation of gatifloxacin loaded solid lipid nanoparticles as colloidal ocular drug delivery system. J drug target.2010;18(3):191-204.
92. Sandri G., Bonferoni M.C., Gokce E.H., Ferrari F., Rossi S., patrini M., Caramella C.: Chitosan-associated SlN: in vitro and ex vivo characterization of cyclosporine A loaded ophthalmic systems.J. microencapsulation. 2010; 27(8): 735–746
93. li R., Jiang S., liu D., Bi X., Wang F., Zhang Q., Xu Q.: A potential new therapeutic system for glaucoma:
solid lipid nanoparticles containing methazolamide. J. microencapsulation. 2011; 28(2): 134–141.
94. liu Z., Zhang X., Wu H., li J., Shu l., liu R., li l., li N.: preparation and evaluation of solid lipid nanoparticles of baicalin for ocular drug delivery system in vitro and in vivo. drug dev.ind. pharm. 2011; 37(4): 475–481.
95. Hao J., Fang X., Zhou Y., Wang J., Guo F., li F., peng X.: Development and optimization of solid lipid nanoparticle formulation for ophthalmic delivery of chloramphenicol using a Box-Behnken design. Int J nanomedicine. 2011; 6: 683-692.
96. Seyfoddin A., Al-Kassas R.: Development of solid lipid nanoparticles and nanostructured lipid carriers for improving ocular delivery of acyclovir. drug dev. ind. pharm. 2012; Early online: 1-12.
idma puBlicationsSr. No. Name of Publications Cost in `
1. IDMA BULLETIN(Published on 7, 14, 21 and 30th of every month)Each Copy 25/-ANNUAL SUBSCRIPTION:Members 1000/- p.a.Non Members 3000/- p.a.
2. INDIAN DRUGS (Published on 28th of every month)Each Copy 75/-ANNUAL SUBSCRIPTION:Members & Educational institutions 1000/- p.a.Non Members 3000/- p.a.
3. IDMA APA ForumAnnual Membership 500/-Life Membership 5000/-
4. TECHNICAL MONOGRAPHS:NO. 1: STABILITY TESTING OF EXISTING DRUG SUBSTANCES AND PRODUCTS 400/-
NO. 2: PRIMARY & SECONDARY CHEMICAL REFE RENCE SUBSTANCES 400/-NO. 3: INVESTIGATION OF OUT OF SPECIFICATION (OOS) TEST RESULTS 400/-NO. 4: PHARMACEUTICAL PREFORMULATION ANALYTICAL STUDIES 400/-NO. 5: ENVIRONMENTAL MONITORING IN CLEANROOMS 400/-NO. 6: CORRECTIVE/PREVENTIVE ACTIONS (CAPA) GUIDELINE 400/-
5. IDMA MEMBERS DIRECTORY 1,000/-6. 51ST ANNUAL PUBLICATION 2013 1,000/-
Kindly note: Courier charges are applicable. We do not mail any publication against VPP Payment. All payments to be made in advance as DD only in favour of “INDIAN DRUG MANUFACTURERS’ ASSOCIATION” payable at Mumbai
For More Details Please Contact: PUBLICATIONS DIVISION. Ms. Prachi INDIAN DRUG MANUFACTURERS’ ASSOCIATION
102-B, “A”- Wing, Poonam Chambers, Dr A B Road, Worli, Mumbai 400018. Tel: 24944624 / 24974308 – Extn. 103, Fax: 022- 24950723 / E-mail: [email protected] / Website: www.idma-assn.org

26 INDIAN DRUGS 50(04) ApRIl 2013
original research articles
evaluation oF antioXidant potential oF Caralluma attenuatasama v.*, rajesh B., Krishnaiah a., ravikiran a., reddy B. m. and mullangi r.
(Received 12 November 2012) (Accepted 12 February 2013)
aBstract
Antioxidant activity of ethanolic extract of Caralluma attenuata was evaluated for its free radical scavenging property in different in vitro models. The extract showed good dose dependent free radical scavenging property in all models except in inhibition of nitric oxide radical and lipid peroxidation. IC50 values are found to be 10, 14, 110 μg/ml in DppH, super oxide and hydroxyl radical scavenging assays, respectively. Measurement of total phenolic compounds by Folin Ciocalteau phenol reagent indicated that 1 g of extract contain 450 mg of GAE/g. The results indicate that the antioxidant property of extract may be due to high content of phenolic compounds.
Keywords: Caralluma attenuata, Asclepiadaceae, free radicals, antioxidant, phenolic compounds.
introduction
The exogenous chemicals and endogenous metabolic process in the human body might produce highly reactive oxygen species, which include free radicals such as superoxide anion radical (O2
.-), hydroxyl radical (OH.), non free radical species such as hydrogen peroxide (H2O2), are various forms of activated oxygen1,2,3. The importance of free radicals and reactive oxygen species (ROS) has attracted increasing attention over the past decade. These molecules are exaterbating factors in cellular injury and aging process4. The free radicals are known to implicated in causations of several diseases such as liver cirrhosis, inflammation, atherosclerosis, diabetes, cancer, neurodegenerative deceases and so forth5. Almost all organisms are well protected
*For correspondence g. pulla reddy college of pharmacy mehdipatnam, hyderabad- 500 028, andhra pradesh. e-mail: [email protected]
against free radical damage by enzymes such as superoxide dismutase and catalase (or) compounds such as ascorbic acid, tocoferol and glutathione6. When the mechanism of antioxidant potential becomes unbalanced by factors such as ageing, deterioration of physiological functions may occur resulting in diseases and accelerates ageing. Several plant extracts and plant products and antioxidant supplements have been shown to possess significant antioxidant potential7,8.
Synthetic antioxidants are widely used because they are effective and cheaper than natural types. However the safety and toxicity of synthetic antioxidants have been important concerns9. Unfortunately, it has been shown that they are able to enhance carcinogenesis, cause pulmonary damage, liver necrosis, hemorrhagic death and neophasia in rats10. Much attention has been focused on the use of antioxidants especially natural anti oxidants which inhibit lipid peroxidation (or) to protect the human body from the oxidative damage by free radicals. Now research is focused on the extraction of natural antioxidants that would be less toxic and more effective than the commonly applied synthetic

INDIAN DRUGS 50(04) ApRIl 2013 27
antioxidants to fight the so called ‘oxidative stress’ which is a highly pathological. Flavonoids and other plyphenols are paid the greatest attention due to their non toxicity and potential for implementation in human diet11.
Caralluma attenuate Wight. (Synonym: Caralluma fimbriata Husk.) belongs to family Asclepiadaceae, is a thick, succulent, perennial fleshy herb growing wild in dry hilly regions of Hyderabad and in several districts of Andhra pradesh, India. locally it is known as Kundeti Kommu. Ramesh et al., has reported the significant anti-inflammatory and antinociceptive activity of the flavonoid glycoside isolated from this plant12,13. Earlier we have reported the significant hypoglycemic effects of C. attenuata in normal and diabetic rats and we also reported the analgesic effects produced by C. attenuata and act both at peripheral and central levels14,15. Jaykar et al., have also reported the significant antihyperglycemic effects of C. attenuata in normal and alloxan induced diabetic rats16. These reported pharmacological activities may be related to the antioxidant activity of C. attenuata extract, although this was not fully substantiated. In fact there is no report on antioxidant activity of C. attenuata and in this report we have evaluated a systematic investigation on the antioxidant potential of ethanolic extract of C. attenuata in various in vitro models.
materials and methods
Fresh whole plants of C. attenuata were collected from Osmania University campus, Hyderabad, A.p, India. Dr. prabhakar Reddy Taxonomist Department of Botany, Osmania University, Hyderabad, performed the botanical identification. The voucher specimen (GpR-CA-99) is being maintained in the Department of pharmacognosy and phytochemistry of G. pulla Reddy College of pharmacy, Mehdipatnam, Hyderabad for future reference.
preparation of extract
The fresh whole plants of C. attenuata (1 kg) were chopped and crushed in a blender to a pasty
mass and extracted with 3 l of ethyl alcohol by cold maceration for 7 days. The resulting extract was filtered and concentrated under reduced pressure. The yield of C. attenuata ethanolic extract (CAE) was 4%. The dried CAE was dissolved in methanol and used for present studies. The ethanolic extract was qualitatively tested for the presence of different chemical groups of compounds as per standard methods17.
chemicals
All the chemicals used were analytical grade. 2, 2-Diphenyl-1-picryl hydrazyl (DppH) was obtained from Sigma Chemical, USA. Nitroblue tetrazolium, phenazine metho sulphate, nicotinamide adenine dinucleotide were obtained from loba chemicals ltd., Mumbai, India. potassium dihydrogen phosphate, ethylenediaminetetraacetic acid, hydrogen peroxide, ferric chloride, ascorbic acid, deoxyribose, trichloroacetic acid, thiobarbituric acid, catechin, curcurmin, sodium nitroprusside, potassium ferricyanide and butylated hydroxyl toluene were pocured from SD Fine Chem, Mumbai, India.
determination of dpph radical scavenging activity
The free radical scavenging activity of CAE was measured by 2, 2-diphenyl-1-picryl hydrazyl (DppH) according to the method of Aquino et al18. DppH in its radical form has an absorption peak at 515nm, which disappears on reduction by an antioxidant compound. A 90μM solution of DppH in methanol was prepared and 2 ml of this solution was added to 1 ml of various concentrations of CAE (10-100 μg/ml) and reference compounds. After 10 min, absorbance was measured at 515nm. Ascorbic acid (1-10 μg/ml) used as a reference material. The percentage of inhibition of DppH+ in the reaction medium is calculated by comparing with control.
% Inhibition = Absorbance of blank - Absorbance of test /absorbance of blank X 100 ---- (1)

28 INDIAN DRUGS 50(04) ApRIl 2013
determination of super oxide anion radical scavenging activity
Measurement of super oxide anion scavenging activity of CAE was done according to the method described by Nshimiki et al19 with modifications. About 1 ml of 156 mM nitro blue tetrazolium (NBT) solution in phosphate buffer (100 mM, pH 7.4), 1 ml 468 μM nicotinamide adenine dinucleotide (NADH) in phosphate buffer (100 mM, pH 7.4) and 3 ml of various concentrations of CAE (10 to 100 μg/ml) and reference compound (1 to 10 μg/ml) were mixed and reaction was started by adding 100 μl of 60 mM phenazine metho sulphate (pMS) in phosphate buffer (100 mM, pH 7.4). The reaction mixtures were incubated at 250C for 5 min and absorbance at 560nm was measured against control. Ascorbic acid is used as reference compound. The percentage of inhibition was determined using formula (1).
determination of hydroxyl radical scavenging activity
The hydroxyl radical scavenging activity was measured by studying the competition between deoxyribose and test compounds (CAE) for hydroxyl radicals generated by Fe+3 - ascorbate - EDTA-H2O2 (Fentone reaction) according to the method of Kunchandy and Rao20. The assay was performed by adding 0.1 ml of 1 mM of EDTA, 0.01 ml of 10 mM ferric chloride, 0.1 ml of 10 mM of hydrogen peroxide, 0.36 ml of 10 mM of 2-deoxy-2-ribose, 1 ml of various concentrations of CAE (100 to 1000 μg) and reference compound (10 to 100 μg), 0.33 ml of phosphate buffer (50 mM, pH 7.4) and 0.1 mM of ascorbic acid in sequence. The mixture incubated at 370C for 60 min. To 1 ml incubated mixture added with 1 ml of thiobarbituric acid (0.5%) and 1 ml of trichloro acetic acid (10%). The reaction mixture was heated at 1000C for 20 min. After cooling the absorbance measured at 532nm against control containing deoxyribose and buffer. Catechin was used as a positive control. Reactions were carried out in triplicate. The percentage inhibition was determined by formula (1).
determination of inhibition of lipid peroxidation
Reaction mixture (4 ml) containing 0.5 ml of liver homogenate (25% w/V), in Tris-HCl buffer (40 mM, pH 7), 3.2 ml of various concentrations of CAE and reference compound (10-100 μg) and 100 μl of each of 0.15 M KCl, 15 mM FeSO4 and 6 mM ascorbic acid were incubated for 1 h at 37oC. A 1 ml of TCA (10%) was added to the reaction mixture and the samples were centrifuged at 3000 rpm for 20 min at 4oC to remove insoluble proteins. To the supernatant solution 1 ml of TBA (0.5%) was added and the volume was made to 4 ml with distilled water and kept in water bath at 90oC for 20min. After cooling the colored complex was extracted with 2 ml of butanol. The absorbance of alcoholic layer was measured at 532nm21. Butylated hydroxyl toluene used as a reference compound. The percentage inhibition of lipid peroxidation was determined by comparing the results of test compound with control using formula (1).
determination of reducing power
The reducing power of CAE was determined according to the method Oyaizu22. A 1 ml of different concentrations of CAE (to produce final concentration 100-1000 μg) in methanol were mixed with 2.5 ml of potassium ferricyanide (1%) and 2.5 ml of phosphate buffer (0.2 M, pH 6.6). The mixture was incubated at 500C for 20 min. To this 2.5 ml of trichloro acetic acid (10%) was added and centrifuged at 3000 rpm for 20 min. An aliquot (2.5 ml) of supernatant was taken and mixed with 2.5 ml of distilled water and 0.5 ml of ferric chloride (0.1%). Absorbance was measured at 700nm. Curcumin was used as reference compound. Higher absorbance of reaction mixture indicates greater reducing power.
determination of nitric oxide radical scavenging activity
Nitric oxide generated from sodium nitroprusside in aqueous solution at physiological pH interacts with oxygen to produce nitrite ion, which can be measured by Griess reactions23. A 2 ml of sodium nitroprusside

INDIAN DRUGS 50(04) ApRIl 2013 29
Fig. 1: dpph radical scavenging effects of Caralluma attenuata
Fig. 2: dpph radical scavenging effects of ascorbic acid
Fig. 3: super oxide radical scavenging effects of Caralluma attenuata
Fig. 4: super oxide radical scavenging effects of ascorbic acid
Fig. 5: hydroxyl radical scavenging effects of Caralluma attenuata
Fig. 6: hydroxyl radical scavenging effects of catechin
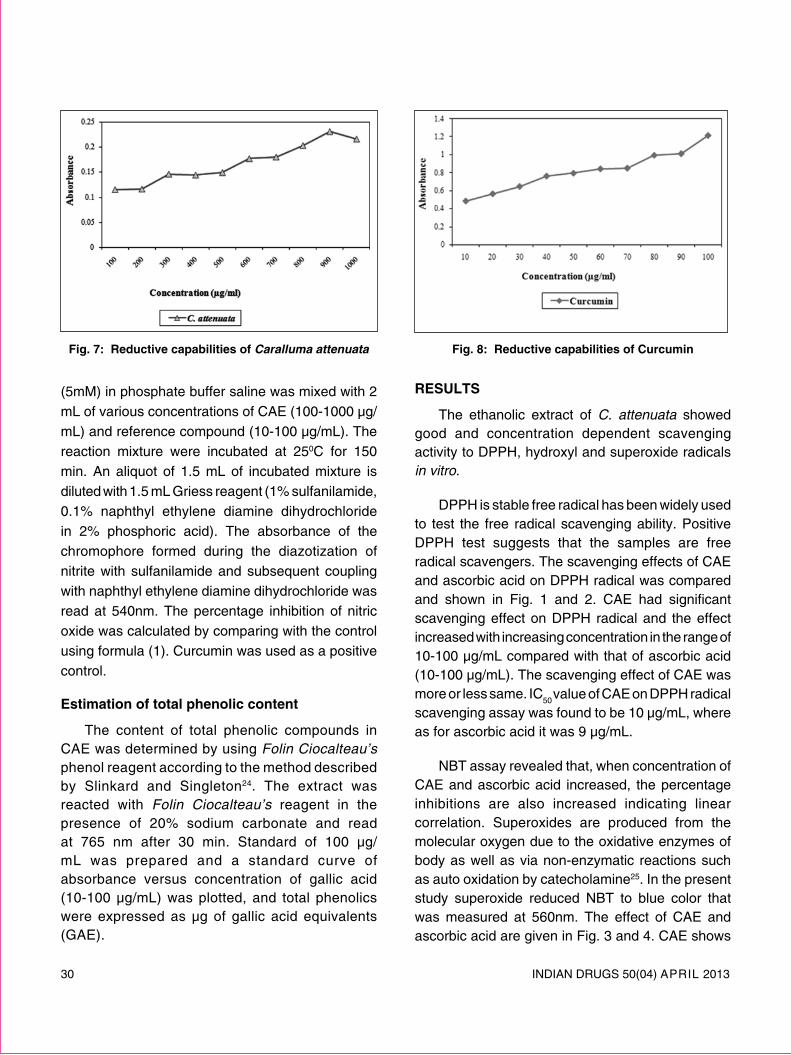
30 INDIAN DRUGS 50(04) ApRIl 2013
(5mM) in phosphate buffer saline was mixed with 2 ml of various concentrations of CAE (100-1000 μg/ml) and reference compound (10-100 μg/ml). The reaction mixture were incubated at 250C for 150 min. An aliquot of 1.5 ml of incubated mixture is diluted with 1.5 ml Griess reagent (1% sulfanilamide, 0.1% naphthyl ethylene diamine dihydrochloride in 2% phosphoric acid). The absorbance of the chromophore formed during the diazotization of nitrite with sulfanilamide and subsequent coupling with naphthyl ethylene diamine dihydrochloride was read at 540nm. The percentage inhibition of nitric oxide was calculated by comparing with the control using formula (1). Curcumin was used as a positive control.
estimation of total phenolic content
The content of total phenolic compounds in CAE was determined by using Folin Ciocalteau’s phenol reagent according to the method described by Slinkard and Singleton24. The extract was reacted with Folin Ciocalteau’s reagent in the presence of 20% sodium carbonate and read at 765 nm after 30 min. Standard of 100 μg/ml was prepared and a standard curve of absorbance versus concentration of gallic acid (10-100 μg/ml) was plotted, and total phenolics were expressed as μg of gallic acid equivalents (GAE).
results
The ethanolic extract of C. attenuata showed good and concentration dependent scavenging activity to DppH, hydroxyl and superoxide radicals in vitro.
DppH is stable free radical has been widely used to test the free radical scavenging ability. positive DppH test suggests that the samples are free radical scavengers. The scavenging effects of CAE and ascorbic acid on DppH radical was compared and shown in Fig. 1 and 2. CAE had significant scavenging effect on DppH radical and the effect increased with increasing concentration in the range of 10-100 μg/ml compared with that of ascorbic acid (10-100 μg/ml). The scavenging effect of CAE was more or less same. IC
50 value of CAE on DppH radical
scavenging assay was found to be 10 μg/ml, where as for ascorbic acid it was 9 μg/ml.
NBT assay revealed that, when concentration of CAE and ascorbic acid increased, the percentage inhibitions are also increased indicating linear correlation. Superoxides are produced from the molecular oxygen due to the oxidative enzymes of body as well as via non-enzymatic reactions such as auto oxidation by catecholamine25. In the present study superoxide reduced NBT to blue color that was measured at 560nm. The effect of CAE and ascorbic acid are given in Fig. 3 and 4. CAE shows
Fig. 8: reductive capabilities of curcuminFig. 7: reductive capabilities of Caralluma attenuata

INDIAN DRUGS 50(04) ApRIl 2013 31
strong superoxide radical scavenging activity. CAE was found to possess good scavenging activity on superoxide anion at all the test concentrations. CAE at concentrations ranging from 10-100 μg/ml inhibited the production of superoxide anion radicals by 46.72 to 82.32. The IC50
value of CAE and ascorbic acid on superoxide radical was found to be 14 and 0.6 μg/ml, respectively.
Hydroxyl radicals are the major reactive species causing lipid oxidation and abnormal biological damage26. Hydroxyl radicals are formed in free solution and were detected by their ability to degrade 2-deoxy-2-ribose into fragments that on heating with TBA at low pH form pink chromogen27. The CAE and catechin removed the hydroxyl radicals from sugar and prevented the degradation. The results are shown in Fig. 5 and 6. The CAE was capable of scavenging hydroxyl radicals at all concentrations. IC50
value of CAE was found to be 110 μg/ml, where as the catechin IC50
value was 70 μg/ml.
The measurement of reductive ability is a significant indicator for determination of potential antioxidant activity. The reaction involves Fe+3 to Fe+2 conversions in the presence of antioxidant. Fig. 7 and 8 show the reductive capability of CAE and curcumin. The reducing power of CAE was increased with increasing concentration in a range of 100-1000 μg/ml. However the reducing power of CAE is lower compared to curcumin (10-100 μg/ml).
The results of present study do not show any promising nitric oxide radical scavenging property and inhibition of lipid peroxidation. The qualitative phytochemical analysis indicates the presence of steroids and/or terpinoids, flavonoids and their glycosides, tannin, carbohydrates in CAE. polyphenols particularly flavonoids and tannins are well known natural antioxidants. The total phenolic content of CAE was found to be 450 mg of GAE/g.
discussion
Free radicals are chemical entities that can exist separately with one or more unpaired electrons. The propagation of free radicals can brings about thousands of reactions and thus make us excessive tissue damage, lipid, protein, DNA are all susceptible to attack free radicals28,29. Antioxidants may offers resistant against oxidative stress by scavenging the free radicals, inhibiting lipid peroxidation etc.
The results obtained from present studies are clearly indicated that ethanolic extract of C. attenuata had powerful antioxidant activity against various antioxidant systems in vitro. These studies indicate that CAE effectively scavenged superoxide and hydroxyl radicals in vitro. These radicals are generated inside the body during the normal metabolism (or) in presence of xenobiotics. The stable free radical DppH was also scavenged by CAE extract. However, the result of the present study does not show any promising nitric oxide radical scavenging property and inhibition of lipid peroxidation. These differences can be explained by understanding the nature and generation of radicals as well as studying the differences in physical and chemical properties of the naturally occurring antioxidants. The stable radicals like DppH reacts stoichometrically with antioxidants which are good hydrogen donors30,31.
The reducing property of CAE implies that, it is capable of donating hydrogen atom in a dose dependent manner. The content of phenolic compounds in the extract may be a contributing factor towards antioxidant activity because the phenolic compounds are known to have direct antioxidant property due to the presence of hydroxyl groups, which can function as hydrogen donor32,33.
preliminary phytochemical analysis indicates the presence of tannins, flavonoids, steroids and/ (or) their glycosides. A flavone glycoside known as luteolin-4’-O-neohesperdoside was reported13. However the

32 INDIAN DRUGS 50(04) ApRIl 2013
phenolic antioxidants such as flavonoids and tannins are well known to scavenge the free radicals dose dependently. Thus these ingredients may contribute to the antioxidant potential of C. attenuata ethanolic extract and presence of these phenolic compounds in the ethanolic extract is confirmed by Folin Ciocalteau’s reagent test.
acKnowledgement
Management of G. pulla Reddy College of pharmacy for providing the necessary research facilities.
reFerences1. Gulcin I, Buyukokurogiu M.E., Oktay M. and Kufrevioglu
O.I.: On the in vitro antioxidant properties of melatonin, J pineal res. 2002, 33,167-171.
2. Halliwell B. and Gutteridge J.M.: Free Radicals in Biology and Medicine. 2nd ed., Claredon press, Oxford, 1989.
3. Yildirim A., Mavi A., Oktay M., Kara A.A., Algur O.F. and Bialoglu V.: Comparison of antioxidant and antimicrobial activities of tilia (Tilia argentia Desf Ex DC), Sage (Salvia triloba l.) and black tea (Camellia Sinensis) extracts, J agri Food chem. 2000, 48, 5030-5034.
4. lai l.S., Chou S.T. and Chao W.W.: Studies on the antioxidative activities of Hsian-tsao (Mesona procumbens Hemsl) leaf gum, J agri Food chem. 2001, 49, 963-968.
5. preethi K.C., Kuttan G. and Kuttan R.: Antioxidant potential of an extract of Calendula officinalis flowers in vitro and in vivo, pharm Biol. 2006, 44, 691-697.
6. Schmidt E., leopold J., Buchbauer G., Eller G.A., Stoilova I, and Krastanov A.:.Composition and antioxidant activities of the essential oil of Cinnamon (Cinnamomum zeylanicum Blume) leaves from Sri lanka, J essential oil-Bearing plants. 2006, 9, 170-182.
7. Sabu M.C. and Kuttan R.: Antioxidant activity of Indian herbal drugs in rats with alloxan induced diabetis, pharm Biol. 2003, 41, 500-505.
8. Ramanathan., Sambath Kumar., Thangavel., Sivakumar. and RajaGopal Shanmaga.: Antimicrobial and antioxidant activities of Careya arborea Roxb. stem bark, ind J pharmacol and therp. 2006, 5, 35-41.
9. Imaida K., Fukushima S., Shivai T., Ohtam M., Nakanishi K. and Ita N.: promoting activities of butylated hydroxyl anisole and butylated hydroxyl toluene on 2-stage urinary bladder carcinogenesis and inhibition of Y-glutamyl transpeptidase-positive Foci development in the liver of rats, carcinogenesis, 1983, 4, 895-899.
10. Ito N., Hirose M., Fukushima S., Tsuda H., Shirai T. and Tatemastu M.: Studies of antioxidants: Their carcinogenic and modifying effect on chemical carcinogenesis, Food chem toxicol. 1985, 24, 1071-1077.
11. pier-Giorgio p. Flavonoids as antioxidants, J nat prod. 2000, 63, 1035-1042.
12. Ramesh M., Rao Y.N., Kumar M.R., Mohan G.K., Kumar B.R., Rao A.V.N.: Flavone glycoside from three Caralluma species, Biochem syst ecol. 1999, 27, 85-86.
13. Ramesh M, Rao Y.N, Kumar M.R, Rao A.V.N, prabhakar M.C. and Rao C.S.: Antinociceptive and anti-inflammatory activity of flavonoid isolated from Caralluma attenuata, J ethnopharmacol. 1998, 2, 63-66.
14. Venkatesh S., Reddy G.D., Reddy B.M., Ramesh M. and Rao A.V.: Antihyperglycemic activity of Caralluma attenuate, Fitoterapia. 2004, 74, 274-279.
15. Sama V., Rajakumari p., Reddy B.M. and Ramesh M.: Antinociceptive effect of Caralluma attenuata in mice - A possible mechanism, ind drugs. 2010, 47, 23-27.
16. Jaykar B., Rajkapoor B. and Suresh B.: Effect of Caralluma attenuata in normal & alloxan induced diabetic rats, J herb pharmacother. 2004, 4, 35-40.
17. Kokate C.K.: practical pharmacognosy. 5th ed. Vallabh prakashan, New Delhi, India, 2004.
18. Aquino R., Morelli S., lauro M.R., Abdo S., Saija A. and Tomaino A.: phenolic constituents and antioxidant activity of an extract of Anthurium versicular leaves, J nat prod. 2001, 64, 1019-1023.
19. Nshimiki M., Rao N.A., Appaji N. and Yagi K. The occurance of superoxide anion in the reaction of reduced phenazine methosulphate & molecular oxygen, Biochem Biophys res comm. 1972, 46, 849-854.
20. Kunchandy E. and Rao M.N.A.: Oxygen radical scavenging activity of Curcumin, int J pharmacog. 1990, 58, 237-240.
21. prashanth Kumar V., Sashidhara S., Kumar M.M. and Shidhara B.Y.: Effect if luffa echinata on lipid peroxidation and free radical scavenging activity, J pharm pharmcol. 2000, 52, 891.
22. Oyaizu M.: Studies on product of browning reaction prepared from glucose amine, Japanese J nut. 1986, 44, 307-315.
23. Green l.C., Wagner D.A., Glogow S.J., Skipper p.l., Wishnok J.K. and Tannenbaum.: Analysis of nitrate, nitrite and 15 N nitrate in biological fluids, anal Biochem. 1982, 126, 131-138.
24. Slinkard K. and Singleton V.l.: Total phenol analyses; automation and comparison with manual methods, american J enol viticult. 1977; 28: 49-55.
25. Hemmani T. and parihar M.S.: Reactive oxygen species and oxidative DNA damage, ind J physiol pharmacol. 1998, 42, 440-444.

INDIAN DRUGS 50(04) ApRIl 2013 33
26. Aurand l.W., Boonme N.H. and Gidding G.G.: Superoxide and singlet oxygen in milk lipid peroxidation, J dairy sci, 1977, 60, 363-369.
27. Halliwel B., Gutteridge J.M.C. and Aruoma O.I.: The deoxyribose method: A simple test tube assay for determination of rate constants for reaction of hydroxyl radicals, anal Biochem. 1987, 165, 215-219.
28. Cotran R.S., Kumar V. and Collins T.: Robbin’s pathological Basis of Diseases. 6th ed Thomson press (I) ltd, Noida, India, 1999.
29. Yu B.p., Suescun E.A. and Yang S.Y.: Effect of age-related lipid peroxidation on membrane fluidity and phospholipase A2: modulation by dietary restriction, mech ageing dev. 1992, 65, 17-21.
30. Blois M.S.: Antioxidant determination by the use of stable free radical, nature. 1958, 26, 1199-1204.
31. Gardner p.T., Mcphail D.B. and Duthie G.G.: Electron spin resonance assessment of the antioxidant potential of teas in aqueous and organic media. J sci Food agri. 1998, 76, 257-261.
32. Duh p.D., Tu Y.Y. and Yen G.C. : Antioxidant activity of aqueous extract of Harng Jyur (Chrysanthemum morifolium Ramat), lebensmittel wissenschaft und technologie. 1999, 32, 269-277.
33. Dreosi I.E. Antioxidant polyphenols in tea, coca and wine, nutrition, 2000, 16, 692-695.
l l l
disso india 2013
Society of pharmaceutical Dissolution Science (SpDS) is organizing a Two day International Annual Symposium titled as “DISSO INDIA 2013” on 3rd & 4th May 2013 at Hotel lalit, Mumbai.
For registering as a delegate, please log on to: www.spds.in and avail this Golden opportunity of learning advancements in Dissolution Science and to interact with the Global & National Experts in this area.
pharmasynth wins rajiv gandhi national Quality award
pharmasynth Formulations ltd., a patriotic & progressive, Delhi based pharmaceutical Company having their registered office at A-10/15, Jhilmil Indl Area, Delhi 110 095, has been selected as winner of the prestigious Rajiv Gandhi National Quality Award for the year 2011-12. This was announced by prof K V Thomas, Hon’ble Union Minister for Consumer Affairs, Food and public Distribution, in New Delhi.
This Award is instituted by the Bureau of Indian Standards in 1991 to give Special recognition to those who are considered to be the leaders of Quality movement in India. This is a National Award of International repute, assessed in line with Malcolm Baldrige National Award of USA, Deming prize of Japan and European Quality Award. According to Mr. A. K. Gupta, Managing Director of the Company, “Fulfillment of environment & social responsibilities are also the integral part of Quality”.

34 INDIAN DRUGS 50(04) ApRIl 2013
potential diuretic eFFect oF CurCuma amaDa roXB. rhiZome eXtracts
Bommannavar p.* and patil K.
(Received 09 July 2012) (Accepted 23 February 2013)
aBstract
The present study was undertaken to establish the diuretic activity of alcoholic and aqueous extract of dried rhizomes of Curcuma amada Roxb in rats. Alcoholic and aqueous extracts of rhizomes were administered to experimental male Wistar rats orally at doses of 250 and 500 mg/kg and compared with furosemide (10 mg/kg) as the reference standard. The parameters measured for diuretic activity were total urine volume, urine electrolyte concentration such as sodium, potassium and chloride have been evaluated. The rats treated with alcoholic and aqueous extract of Curcuma amada in a dose of 250 and 500 mg/kg showed higher urine output when compared to the respective control. Both alcoholic and aqueous extracts have showed a significant dose-dependent increase in the excretion of electrolytes when compared to the control group. The result indicates that alcoholic and aqueous extract is an effective natriuretic and kaliuretic diuretic, which supports the traditional claim about the Curcuma amada Roxb being used as diuretics.
Keywords: Aqueous extract, diuretic activity, alcoholic extract, Curcuma amada, electrolyte analysis, furosemide, nephron reabsorption.
introduction
Drugs inducing a state of increased urine flow are called as diuretics1. These agents are ion transport inhibitors that decrease the reabsorption of sodium at different sites in the nephron. As a result, sodium and other ions such as chloride enter the urine in greater amounts than normal along with the water, which is carried passively to maintain osmotic equilibrium. Diuretics thus increase the volume of urine and often change its pH as well as the ionic composition of the urine and blood 2.
Diuretics are one of the most commonly used drugs in modern therapeutics. Their major clinical uses are in managing disorders involving abnormal fluid retention (edema) or in treating hypertension in
*For correspondence Kle university’s college of pharmacy Belgaum - 590010, Karnataka e-mail: [email protected]
which their diuretic action causes a decreased blood volume, leading to a reduction in blood pressure. Thus, they are widely used in the treatment of congestion heart failure, acute and chronic renal failure, ascites, nephritic syndromes, glaucoma, and toxaemia of pregnancy, premenstrual tension and hypertension3.
Curcuma amada Roxb (Family: Zingerberaceae), commonly known as Mango ginger, Ama-haldi in Hindi, and Ambarasini in Kannada, is a rhizomatous aromatic herb with a leafy tuft, 60-90 cm in height, with leaves long, petiolate, oblong- lanceolate, tapering at both ends, globrous and green on both sides. The flowers are white or pale yellow in spikes in the centre of the tuft of leaves4. This herb is cultivated throughout India predominantly in Karnataka, Gujarat and also found wild in parts of West Bengal, Uttar pradesh and Tamil Nadu4-6. The Curcuma amada rhizome has been extensively used as carminative, stomachic, appetizer, demulcent, aphrodisiac, laxative, expectorant, anti-inflammatory and antipyretic. They are useful in vitiated conditions of pitta, anorexia, dyspepsia, flatulence, colic, bruises, wounds, chronic ulcers, skin diseases, pruritus, fever,

INDIAN DRUGS 50(04) ApRIl 2013 35
constipation, strangury, hiccough, cough, bronchitis, sprains, gout, halitosis, otalgia and inflammation due to injuries. They are used externally in the form of paste in contusions and sprains. Rhizomes contain essential oils contain resin, sugar, gum, starch, albuminoids and organic acids. Essential oil contains d-α-pinene (18 %), cis and trans- Ocimene (47.2 %), linalool (11.2 %), linalyl acetate (9.1 %), Safrol (9.3 %), d-Camphor, 1-β-Curcumene, d-Curcumene, Myrcene isolated from the rhizomes7. The ether extract of Curcuma amada in a dose of 1g given orally in the form of an aqueous suspension could lower the blood cholesterol level in rabbits. The extracts of rhizomes of C. amada had shown CNS-depressant activity in mice8. Recently, difurocumenonol and amadannulen compounds, were been isolated and characterised which were reported to possess highly antioxidant and antimicrobial activity 9.
Since, the diuretic effect of Curcuma amada rhizome has never been experimentally confirmed. The main aim of the present investigation was to evaluate the claimed diuretic activity of Curcuma amada rhizome in rats and thus validate its use as diuretic in traditional and folk system.
Furosemide tablets (lasix® 40 mg), a loop diuretic, was selected as the reference drug, since it is used clinically in many pathologies. The alcoholic and aqueous extract of Curcuma amada rhizome was administered by oral route at the dose of 250 and 500 mg/kg body weight to determine its acute effect in urine output and excretion of Na+, K+, Cl- ions in rats.
material and methods
plant material
The fresh rhizomes of Curcuma amada Roxb were collected from local areas of pune, Maharashtra, India during July 2010 and authenticated by Dr. Harsha. Hegde, Scientist ‘B’ Research Officer, Regional Medical Research Centre, Nehru Nagar, Belgaum, Karnataka, India with a voucher specimen No. RMRC-549 was deposited in the herbarium.
extraction procedure
The fresh rhizomes were first washed thoroughly with water, chopped into thin slices and shade dried at room temperature. The dried rhizomes were reduced to a coarse powder, separately in a mechanical grinder. About 500 g of dried coarse powder was extracted with 90% V/V alcohol by using soxhlet extractor. The extract was concentrated to 3/4th of its original volume by distillation. The concentrated extract were taken in a china dish and evaporated on a thermostat controlled water bath. The marc obtained after alcoholic extract was dried at room temperature and subjected to maceration process using distilled water in a conical flask, for about 7 days at room temperature. The material was filtered through a muslin cloth and filtrate was concentrated in a china dish and evaporated on a thermostat controlled water bath till it forms a thick paste. The yield of both the extracts was found to be 1/5th of the crude drug and extracts were subjected to preliminary phytochemical studies10,11,12.
storage of extract
The alcoholic and aqueous extract of Curcuma amada Roxb CAAl and CAAQ respectively was stored in tightly closed amber coloured glass bottles and was kept in a refrigerator.
pharmacological study
preparation of suspension of curcuma amada roxb for oral administration: Suspension of Curcuma amada Roxb extracts was prepared by suspending the alcoholic and aqueous extract with 2% gum acacia in glass mortar with gradual addition of distilled water to make up the volume. The same procedure was used for the preparation of vehicle without addition of extracts. Similarly, accurately weighed quantity of furosemide was suspended in distilled water to make up the volume. The volume of the drug solution was calculated based upon the body weight of animals.
route of administration: The suspension of the extracts, drug and vehicle were administered per orally13.
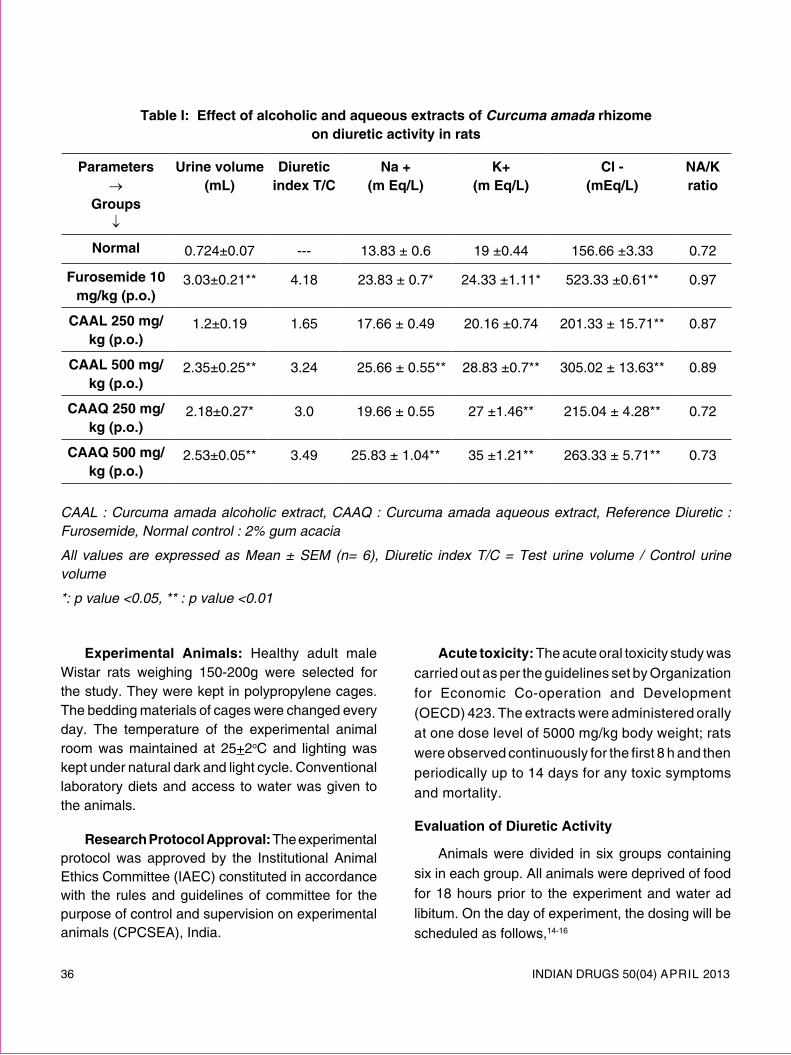
36 INDIAN DRUGS 50(04) ApRIl 2013
table i: effect of alcoholic and aqueous extracts of Curcuma amada rhizome on diuretic activity in rats
parameters →
groups ↓
urine volume (ml)
diuretic index t/c
na + (m eq/l)
K+ (m eq/l)
cl - (meq/l)
na/K ratio
normal 0.724±0.07 --- 13.83 ± 0.6 19 ±0.44 156.66 ±3.33 0.72
Furosemide 10 mg/kg (p.o.)
3.03±0.21** 4.18 23.83 ± 0.7* 24.33 ±1.11* 523.33 ±0.61** 0.97
caal 250 mg/kg (p.o.)
1.2±0.19 1.65 17.66 ± 0.49 20.16 ±0.74 201.33 ± 15.71** 0.87
caal 500 mg/kg (p.o.)
2.35±0.25** 3.24 25.66 ± 0.55** 28.83 ±0.7** 305.02 ± 13.63** 0.89
caaQ 250 mg/kg (p.o.)
2.18±0.27* 3.0 19.66 ± 0.55 27 ±1.46** 215.04 ± 4.28** 0.72
caaQ 500 mg/kg (p.o.)
2.53±0.05** 3.49 25.83 ± 1.04** 35 ±1.21** 263.33 ± 5.71** 0.73
CAAL : Curcuma amada alcoholic extract, CAAQ : Curcuma amada aqueous extract, Reference Diuretic : Furosemide, Normal control : 2% gum acacia
All values are expressed as Mean ± SEM (n= 6), Diuretic index T/C = Test urine volume / Control urine volume
*: p value <0.05, ** : p value <0.01
experimental animals: Healthy adult male Wistar rats weighing 150-200g were selected for the study. They were kept in polypropylene cages. The bedding materials of cages were changed every day. The temperature of the experimental animal room was maintained at 25+2oC and lighting was kept under natural dark and light cycle. Conventional laboratory diets and access to water was given to the animals.
research protocol approval: The experimental protocol was approved by the Institutional Animal Ethics Committee (IAEC) constituted in accordance with the rules and guidelines of committee for the purpose of control and supervision on experimental animals (CpCSEA), India.
acute toxicity: The acute oral toxicity study was carried out as per the guidelines set by Organization for Economic Co-operation and Development (OECD) 423. The extracts were administered orally at one dose level of 5000 mg/kg body weight; rats were observed continuously for the first 8 h and then periodically up to 14 days for any toxic symptoms and mortality.
evaluation of diuretic activity
Animals were divided in six groups containing six in each group. All animals were deprived of food for 18 hours prior to the experiment and water ad libitum. On the day of experiment, the dosing will be scheduled as follows,14-16

INDIAN DRUGS 50(04) ApRIl 2013 37
Group I served as normal control was administered with 2% gum acacia, Group II was administered with reference diuretic Furosemide 10 mg/kg, Group III was treated with Curcuma amada alcoholic extract (CAAl) 250 mg/kg, Group IV was treated with Curcuma amada alcoholic extract (CAAl) 500 mg/kg, Group V was treated with Curcuma amada aqueous extract (CAAQ) 250 mg/kg and Group VI was treated with Curcuma amada aqueous extract (CAAQ) 500 mg/kg.
Immediately after the dosing, animals were placed in metabolic cages and urine was collected after 5 hours from dosing. During this period no water or food was made available to the animal.
determination of parameters
The urine samples from the experimental rats were collected and the total urine volume collected was measured and recorded. The urine concentration of Na+ and K+ were measured by flame photometry and Cl- concentration was estimated by titration with silver nitrate using potassium chromate solution as indicator17.
statistical analysis
Data obtained by the various parameters was analyzed by one way analysis of variance test (ANOVA) followed by Dunnett’ test. P≤0.05 was considered as significant.
results
The acute toxicity study showed no mortality at 5000mg/kg in Curcuma amada alcoholic and aqueous extracts, therefore lD50 has been considered to be more than 5000mg/kg.
The present study showed that furosemide-treated rats showed a significant increase in urine volume and urinary excretion of sodium potassium and chloride as compared to the normal group. The alcoholic and aqueous extracts of Curcuma amada rhizome at the dose of 500 mg/kg showed significant (P<0.01) increase in the urine volume when compared with normal group and was near to
standard drug. The aqueous extract at the dose of 250mg/kg showed significant (P<0.05) increase in urine volume, wherein the alcoholic extract at 250 mg/kg dose does not show significant increase in the urine volume in the comparison of normal group that was shown in Table I .
In terms of the effect on urinary electrolyte excretion the alcoholic and aqueous extract of Curcuma amada rhizome 500 mg/kg showed significant (P<0.01) increase in sodium output in urine when compared with normal group and was highly significant to standard group. The alcoholic and aqueous extracts at the dose of 250 mg/kg did not show significant increase in the sodium output in urine when compared with standard group. However both extracts dose of Curcuma amada rhizome showed significant (P<0.01) increase in potassium output in urine that were significant to standard group when compared to normal group. All the dose of alcoholic and aqueous extract of Curcuma amada rhizome showed significant (P<0.01) output of chloride ion in urine when compared to the normal group as cited in Table I.
discussion
The aim of this study was to test the diuretic activity of Curcuma amada rhizome in rats. The present study data indicate that Curcuma amada rhizome extracts showed dose depended diuretic effect. By definition, diuretics are the drugs that increase the rate of urine flow; however, clinically useful diuretics also increase the rate of excretion of sodium (natriuresis) and an accompanying anion, usually chloride15. The result obtained from present study indicates that the alcoholic and aqueous extract of Curcuma amada rhizome at the dose of 500 mg/kg have shown to increase urine output over a period of 5 hours as shown in Table I. It also increased the sodium, potassium and chloride excretion significantly. The control of sodium is important in regulating of blood pressure, the control of potassium is required to maintain proper function of cardiac and skeletal muscles. The regulation of electrolyte concentration

38 INDIAN DRUGS 50(04) ApRIl 2013
is also intimately related to renal control of acid-base balance. The potassium lose that occur with many diuretics may lead to hypokalemia. For this reason, generally potassium sparing diuretics are recommended. In the present study, treatment with the alcoholic and aqueous extract of Curcuma amada rhizome resulted in elevated levels of potassium in urine, which may increase the risk of hypokalemia and hence its potassium sparing capacity has to be investigated. The preliminary phytochemical study and the previous studies have demonstrated that there are several phytoconstituent, which could be responsible for the diuretic effect in plants such as flavonoids, terpenoids, saponins, tannins or organic acids 18. The effect may be produced by stimulation of regional blood flow or initial vasodilatation, or by producing inhibition of tubular reabsorption of water and anions19, that results in being diuresis.
Based on our study we can conclude that, the above mentioned alcoholic and aqueous extract of Curcuma amada rhizome have showed dose dependent diuretics activity with increase in urine output and electrolyte excretion that can be the basis of its therapeutic application in various renal ailments such as nephritis, burning micturation, different oedematous diseases and hypertension.
acKnowledgements
The authors are thankful to the Dean and principal of KlE University College of pharmacy, Belgaum for providing necessary facilities to carry out the present research work.
reFerences 1. Harvey A.R, pamela C. C, Howland D.R., Mycek J.M.
lippincott’s Illustrated Reviews pharmacology, 2nd ed, lippincott’s William’s and Wilkin’s, UK 1997.
2. Rang p.H., Dale M. M., Ritter M. J., Flower J.R, Henderson G.: Text Book of pharmacology, 7th ed, Churchill living stone, UK 2003.
3. Brunton l.l., lazo J.S., parker K.l.: The pharmacological Basis of Therapeutics.11th ed, the McGraw- Hill companies, USA 2006.
4. Arya Vaidya Sala. Indian Medicinal plants a Compendium of 500 species, Orient longman ltd, Chennai 1997.
5. Chatterjee A., parkashi S.C.: The Treatise on Indian Medicinal plants. Vol-6, CSIR New Delhi 2003.
6. Agarwal V.S.: Drugs plants of India., Kalyani publisers, New Delhi 1997.
7. Kapoor l.D.: Handbook of Ayurvedic Medicinal plants. CRC press llC, US 2005.
8. Kiritikar K.R., Basu B.D., Singh B., Singh M.p.: Indian Medicinal plants. 2nd ed, Dehradun 1980.
9. policegoudra R.S., Divakar S., Aradhya S.M, Identification of diflurocumenonol, a new antimicrobial compound from mango ginger (Curuma amada Roxb) rhizome, J applied microbio. 2007, 102:1595-1602.
10. Quality Standards of Indian Medicinal plants. Evaluation of crude Drugs. Indian council of medicinal research, New Delhi Appendix I 2003, 1 : 236-37.
11. Evans C.W. Trease and Evans pharmacognosy, 15th ed, Elsevier, New Delhi 2005.
12. Kokata C.K. practical pharmacognosy, 4st ed ,Vallabh prakashan; Mumbai 1994.
13. Vogel H.G., Vogel W.H.: “Drug Discovery and Evaluation pharmacological Assays” 2nd ed, Springer-Verlag Berlin publication 2008.
14. Afzal M, Khan N. A., Ghufran A. et al. Diuretic and Nephroprotective effect of Jawarish Zarooni Sada- A poly herbal Unani formulation. J. ethanopharmacol. 2004, 91: 219-223.
15. Ruckmani K.B., Jaykar, Anandan R.: Diuretic Activity of Moringa Oleifera lam. indian drugs. 1997, 34 (5):289-290.
16. Swapnadeep p., Jain D.C and Joshi S.B.: RASAYANA J. chem. 2009, 2 (1):53-56.
17. Skoog D. A.; West D. M.; Holler F. J.: Fundamentals of Analytical Chemistry, 7th ed, Thomson learning, Inc, USA 1996.
18. Maghrani M., Zuggwugh N., Hslbui M., Edduls M.: Acute diuretic effect of aqueous extract of Retama raetam in normal rats. J. ethanopharmacol., 2005, 99:31-35.
19. Stanic G., Samarzija I.: Diuretic activity of Satureja montana subsp. Montana extracts and oil in rats. phytother res., 1993, 7: 363-366.

INDIAN DRUGS 50(04) ApRIl 2013 39
synthesis and evaluation oF some novel 1, 3, 4-oXadiaZole derivatives For antimicroBial and antidiaBetic activitiespattan s.r*, Kasar p.n, randale B.d, shinde p.m, pattan J.s, atre m.a. and Balsane a.s.
(Received 21 December 2012) (Accepted 04 February 2013)
aBstract
A series of 1, 3, 4-Oxadiazole derivatives have been synthesized and evaluated for antimicrobial and antidiabetic activities. The newly synthesized compounds have been characterized by IR, 1H NMR and elemental analysis. All compounds have shown promising antimicrobial and antidiabetic activities when compared with standard drug ciprofloxacin and acarbose respectively. Compounds A2,B2,C1,D1,D2,E1 and E2 have shown promising antimicrobial activity while compounds A1,A2,B1,C1,D1,E1 and E2 have shown promising antidiabetic activity.
Keywords: 1, 3, 4-Oxadiazole, antimicrobial, antidiabetic, 1H NMR.
introduction
Diabetes mellitus is a condition in which the pancreas no longer produces enough insulin or cells stop responding to the insulin that is produced, so that glucose in the blood cannot be absorbed into the cells of the body. Symptoms include frequent urination, lethargy, excessive thirst, and hunger. The treatment includes changes in diet, oral medications, and in some cases, daily injections of insulin.
Oxadiazoles and thiadiazole derivatives attracted organic chemists very much due to their biological and chemotherapeutic importance. Thiadiazole and related fused heterocycles are of interest as potential bioactive molecules. In many cases, heterocyclic fusion of ring resulted in compounds with wide spectrum of biological activities. Some of the activities possessed by heterofused thiadiazole include antimicrobial, antidiabetic, antifungal, anticancer, anti-inflammatory, antitubercular activity etc1.
*For correspondence department of pharmaceutical chemistry pravara rural college of pharmacy, pravaranagar a/p-loni, tal- rahata, dist. - ahmednagar pin- 413736, maharashtra. e-mail: [email protected]
1,3,4-oxadiazole have been reported to possess diverse biological and pharmacological activities like antimicrobial2, antifungal3 and antitubercular4
activities. Here we attempted to substitute different aldehyde to synergize their biological and pharmacological activities. Beyond this we have clubbed the active pharmaceutical ingredients with oxadiazole derivatives for getting the best results. This observation promoted the synthesis of a few compound with the presumption that incorporation of active pharmaceutical ingredients with oxadiazole nuclei would produce new compounds with significant antimicrobial and antidiabetic activities. The synthesized compounds were evaluated for various biological activities like antimicrobial and antidiabetic activities.
eXperimental section
The melting points were determined by open capillary method and are recorded. Homogeneity of compounds was checked on silica gel TlC plates. IR spectra were reported on Thermo Nicolet IR 200 spectrophotometer using KBr disc method. 1H NMR spectra were recorded on Varian-NMR-Mercury 300 with CDCl3 as solvent and DMSO as internal standard.Ten different derivatives were synthesized- Table I, elemental analysis & other physicochemical properties were checked (Table II).

40 INDIAN DRUGS 50(04) ApRIl 2013
preparation of 2-amino-5-aryl-1, 3, 4-oxadiazole5, 6 (i)
A mixture of semicarbazide (0.1mol), aryl carboxylic acid (0.1mol), & conc. sulphuric acid (10 drops) was refluxed for 1 hr & poured onto crushed ice. The solid separated out was filtered, washed with water & recrystallized from ethanol to give I.
preparation of (4-aryl-oxadiazole-1-amino) acetone (ii)
A mixture of 2-amino-4-aryl-1,3,4-oxadiazole (10mol) & chloro acetone (10mol) in presence of anhydrous potassium carbonate in dry acetone was refluxed for 12 hrs, the reaction mixture was cooled and the separated solid was filtered and recrystallized from ethanol to give product II.
preparation of (iii) compound
A mixture of compound (II) (10mol), malononitrile (10mol), benzaldehyde (10mol) & ammonium acetate in ethanol (50ml) was refluxed for 10 hour the reaction mixture was cooled & poured onto crushed ice; the product separated was filtered & recrystallized from ethanol to give product (III).
preparation of (iv) compound
The mixture of product (III) (0.01mol) and aldehyde (0.01mol) was refluxed in ethanol (10ml) for 2 hr, the resultant solution was cooled, the solid separated was filtered and recrystallized from petroleum ether to give product (IV).
preparation of (v) compound7
Sodium borohydride (0.02mol) was added to product (IV) (0.01mol) and methanol (10ml); the mixture was stirred for 30 min at ambient temperature. The solid was separated on pouring the reaction mixture into ice cold water which was filtered and recrystallized from ethanol to give product (V).
spectral data
a1:IR(KBr)cm-1:3393.14(NH-str),3090.17(Ar-CHstr), 2602.26(CH2 str), 2197.49(Ar-C=N str),
1666.21(ring C-N str), 1575.33 (Ar-C-C str), 1362.32 (C=O str), 1099.23 (Ar-C-O-C str).
a2:IR (KBr) cm-1: 3511.04(OH-str), 3468.35(NH-str), 3068.35(Ar-CH str), 2664.18 (CH2 str), 2210.99(Ar-C=N str), 1688.37(C=O str),1511.92 (Ar-C-C str), 1279.54(Ring C-N str),1032(Ar-C-O-C str).
B1: IR (KBr) cm-1: 3367.01 (NH str), 3063.37(Ar-CH str), 2998.72(CH2 str), 1778.05(C=O str),1551.42(Ar-C-C str), 1324.80( Ring C-C str), 1080.91 ( Ar-C-O-C str).
B2:IR(KBr) cm-1: 3511.74(OH str), 3463.39 (NH str), 3063.37 (Ar CH str), 2834.35 (CH2 str), 1771.03(C=O str),1516.74 (Ar-C-C str), 1384.64(Ring C-N str), 1080.91(Ar-C-O str).
c1:IR(KBr) cm-1: 3560.91(OH str), 3339.28(NH-str), 3028.55(Ar-CH str),2896.91(CH2 str), 1674.87(Ar C-C str), 1535.31(C=O str), 1372.21(Ring C-N str), 1084.18(Ar-C-O-Cstr), 1H NMR: δ 10.0-10.1(s 1H OH Alcohol),δ 7.7-7.8(s 1H CH pyridine), δ 7.2-7.6(s 4H CH2 methylene), δ 6.8-6.91(s 4H CH aryl), δ 4.2-4.3(s 1H NH Sec. amine), δ 4.3-4.4(s 1H NH pyridine).
c2:IR(KBr) cm-1: 3516.56(OH str), 3437.37(NH-str), 3032.51(Ar-CH str), 2967.91(CH2 str), 1697.05 (C=O str),1599.61(Ar C-C str),1384.34(Ring C-N str), 1028.17(Ar-C-O-Cstr).
d1:IR(KBr) cm-1: 3459.60(NH-str), 3023.84(Ar-CHstr), 2930.31(CH2 str), 1778.05(C=O str), 1511.92(Ar C-C str), 1307.50(Ring C-N str), 1089.58(Ar-C-O-C str).
d2:IR(KBr) cm-1: 3507.88(OH str), 3463.53(NH-str), 3050.78(Ar-CHstr), 2956.74(CH2 str), 1760.86(C=O str), 1508.06(Ar C-C str), 1347.68(Ring C-N str), 1052.67(Ar-C-O-Cstr), 1H NMR: δ 8.9-9.0(s 1H OH Alcohol), δ 7.7-7.8(s 1H CH pyridine), δ 7.5-7.6(s 12H CH benzene), δ 6.8-6.9(s 1H NH pyridine), δ 3.9-4.0(s 1H NH Sec. amine), δ 4.3-4.5(s 4H CH2 methylene), δ 5.3-5.4(s 2H NH pri. amine), δ 3.8-3.9(s 3H CH3 Methoxy).

INDIAN DRUGS 50(04) ApRIl 2013 41
scheme

42 INDIAN DRUGS 50(04) ApRIl 2013
table i: list of synthesized compounds
compound ar r ar’
A1 - -
A2
B1 - -
B2
C1 - -
C2
D1
D2 - -
E1
E2 - -
Table I: List of synthesized compounds
Compound Ar R Ar’
A1
-
-
A2
OH
H3CO
B1
-
-
B2
N OH
H3CO
C1
-
-
C2
OH
OH
H3CO
D1 NH2
D2
-
- OH
H3CO
Table I: List of synthesized compounds
Compound Ar R Ar’
A1
-
-
A2
OH
H3CO
B1
-
-
B2
N OH
H3CO
C1
-
-
C2
OH
OH
H3CO
D1 NH2
D2
-
- OH
H3CO
Table I: List of synthesized compounds
Compound Ar R Ar’
A1
-
-
A2
OH
H3CO
B1
-
-
B2
N OH
H3CO
C1
-
-
C2
OH
OH
H3CO
D1 NH2
D2
-
- OH
H3CO
Table I: List of synthesized compounds
Compound Ar R Ar’
A1
-
-
A2
OH
H3CO
B1
-
-
B2
N OH
H3CO
C1
-
-
C2
OH
OH
H3CO
D1 NH2
D2
-
- OH
H3CO
Table I: List of synthesized compounds
Compound Ar R Ar’
A1
-
-
A2
OH
H3CO
B1
-
-
B2
N OH
H3CO
C1
-
-
C2
OH
OH
H3CO
D1 NH2
D2
-
- OH
H3CO
Table I: List of synthesized compounds
Compound Ar R Ar’
A1
-
-
A2
OH
H3CO
B1
-
-
B2
N OH
H3CO
C1
-
-
C2
OH
OH
H3CO
D1 NH2
D2
-
- OH
H3CO
Table I: List of synthesized compounds
Compound Ar R Ar’
A1
-
-
A2
OH
H3CO
B1
-
-
B2
N OH
H3CO
C1
-
-
C2
OH
OH
H3CO
D1 NH2
D2
-
- OH
H3CO
Table I: List of synthesized compounds
Compound Ar R Ar’
A1
-
-
A2
OH
H3CO
B1
-
-
B2
N OH
H3CO
C1
-
-
C2
OH
OH
H3CO
D1 NH2
D2
-
- OH
H3CO
Table I: List of synthesized compounds
Compound Ar R Ar’
A1
-
-
A2
OH
H3CO
B1
-
-
B2
N OH
H3CO
C1
-
-
C2
OH
OH
H3CO
D1 NH2
D2
-
- OH
H3CO
Table I: List of synthesized compounds
Compound Ar R Ar’
A1
-
-
A2
OH
H3CO
B1
-
-
B2
N OH
H3CO
C1
-
-
C2
OH
OH
H3CO
D1 NH2
D2
-
- OH
H3CO
Table I: List of synthesized compounds
Compound Ar R Ar’
A1
-
-
A2
OH
H3CO
B1
-
-
B2
N OH
H3CO
C1
-
-
C2
OH
OH
H3CO
D1 NH2
D2
-
- OH
H3CO
Table I: List of synthesized compounds
Compound Ar R Ar’
A1
-
-
A2
OH
H3CO
B1
-
-
B2
N OH
H3CO
C1
-
-
C2
OH
OH
H3CO
D1 NH2
D2
-
- OH
H3CO
Table I: List of synthesized compounds
Compound Ar R Ar’
A1
-
-
A2
OH
H3CO
B1
-
-
B2
N OH
H3CO
C1
-
-
C2
OH
OH
H3CO
D1 NH2
D2
-
- OH
H3CO
Table I: List of synthesized compounds
Compound Ar R Ar’
A1
-
-
A2
OH
H3CO
B1
-
-
B2
N OH
H3CO
C1
-
-
C2
OH
OH
H3CO
D1 NH2
D2
-
- OH
H3CO
Table I: List of synthesized compounds
Compound Ar R Ar’
A1
-
-
A2
OH
H3CO
B1
-
-
B2
N OH
H3CO
C1
-
-
C2
OH
OH
H3CO
D1 NH2
D2
-
- OH
H3CO
Table I: List of synthesized compounds
Compound Ar R Ar’
A1
-
-
A2
OH
H3CO
B1
-
-
B2
N OH
H3CO
C1
-
-
C2
OH
OH
H3CO
D1 NH2
D2
-
- OH
H3CO
Table II: Physical and analytical characterization data of synthesized
compounds
E1
N
F
N
NH
O
E2
-
- OH
H3CO
Compound Mol.Formula Mol.
Wt.
M.P 0C
Rf
Value
Yield
%
Elemental analysis
%
C H N
A1 C28H22N6O
458.51
269-
272
0.06 80.12 73.35
(73.05)
4.84
(4.50)
18.33
(18.13)
A2 C29H24N6O3
504.53
139-
142
0.55 82.65 69.04 4.79 16.66
B1 C27H23N7O
461.51 269-
272
0.55 86.45 70.27
(70.20)
5.02
(5.05)
21.24
(21.20)
B2 C22H24N6O4
436.46 149-
150
0.67 67.23 60.54 5.54 19.25
C1 C28H22N6O2
474.51 149-
150
0.51 68.82 70.87 4.67 17.71
C2 C29H24N6O4
520.53 159-
161
0.62 75.68 66.91 4.65 16.14
Table II: Physical and analytical characterization data of synthesized
compounds
E1
N
F
N
NH
O
E2
-
- OH
H3CO
Compound Mol.Formula Mol.
Wt.
M.P 0C
Rf
Value
Yield
%
Elemental analysis
%
C H N
A1 C28H22N6O
458.51
269-
272
0.06 80.12 73.35
(73.05)
4.84
(4.50)
18.33
(18.13)
A2 C29H24N6O3
504.53
139-
142
0.55 82.65 69.04 4.79 16.66
B1 C27H23N7O
461.51 269-
272
0.55 86.45 70.27
(70.20)
5.02
(5.05)
21.24
(21.20)
B2 C22H24N6O4
436.46 149-
150
0.67 67.23 60.54 5.54 19.25
C1 C28H22N6O2
474.51 149-
150
0.51 68.82 70.87 4.67 17.71
C2 C29H24N6O4
520.53 159-
161
0.62 75.68 66.91 4.65 16.14
Table II: Physical and analytical characterization data of synthesized
compounds
E1
N
F
N
NH
O
E2
-
- OH
H3CO
Compound Mol.Formula Mol.
Wt.
M.P 0C
Rf
Value
Yield
%
Elemental analysis
%
C H N
A1 C28H22N6O
458.51
269-
272
0.06 80.12 73.35
(73.05)
4.84
(4.50)
18.33
(18.13)
A2 C29H24N6O3
504.53
139-
142
0.55 82.65 69.04 4.79 16.66
B1 C27H23N7O
461.51 269-
272
0.55 86.45 70.27
(70.20)
5.02
(5.05)
21.24
(21.20)
B2 C22H24N6O4
436.46 149-
150
0.67 67.23 60.54 5.54 19.25
C1 C28H22N6O2
474.51 149-
150
0.51 68.82 70.87 4.67 17.71
C2 C29H24N6O4
520.53 159-
161
0.62 75.68 66.91 4.65 16.14
Table II: Physical and analytical characterization data of synthesized
compounds
E1
N
F
N
NH
O
E2
-
- OH
H3CO
Compound Mol.Formula Mol.
Wt.
M.P 0C
Rf
Value
Yield
%
Elemental analysis
%
C H N
A1 C28H22N6O
458.51
269-
272
0.06 80.12 73.35
(73.05)
4.84
(4.50)
18.33
(18.13)
A2 C29H24N6O3
504.53
139-
142
0.55 82.65 69.04 4.79 16.66
B1 C27H23N7O
461.51 269-
272
0.55 86.45 70.27
(70.20)
5.02
(5.05)
21.24
(21.20)
B2 C22H24N6O4
436.46 149-
150
0.67 67.23 60.54 5.54 19.25
C1 C28H22N6O2
474.51 149-
150
0.51 68.82 70.87 4.67 17.71
C2 C29H24N6O4
520.53 159-
161
0.62 75.68 66.91 4.65 16.14

INDIAN DRUGS 50(04) ApRIl 2013 43
table ii: physical and analytical characterization data of synthesized compounds
compound mol.Formula mol. wt. m.p 0c rf value yield % elemental analysis %c h n
A1 C28H22N6O 458.51 269-272 0.06 80.12 73.35 (73.05)
4.84 (4.50)
18.33 (18.13)
a2 C29H24N6O3 504.53 139-142 0.55 82.65 69.04 4.79 16.66
B1 C27H23N7O 461.51 269-272 0.55 86.45 70.27 (70.20)
5.02 (5.05)
21.24 (21.20)
B2 C22H24N6O4 436.46 149-150 0.67 67.23 60.54 5.54 19.25
c1 C28H22N6O2 474.51 149-150 0.51 68.82 70.87 4.67 17.71
c2 C29H24N6O4 520.53 159-161 0.62 75.68 66.91 4.65 16.14
d1 C28H23N7O 473.52 189-191 0.53 80.12 71.02 4.90 20.71
d2 C29H25N7O3 519.55 239-241 0.62 80.34 67.04 4.85 18.87
e1 C36H35N9O2 652.72 199-201 0.66 71.28 66.76 (66.71)
5.12 (5.15)
20.02 (20.06)
e2 C36H34FN9O4 675.71 239-241 0.72 75.22 63.99 5.07 18.66
e1:IR(KBr) cm-1:3305.40(NH-str),3019.68(Ar-CHstr),2848.56(CH2-str)1802.15(C=O -str), 1573.87(Ar C-C str),1358.42(Ring C-N str),1087.55(Ar-C-O-Cstr), 1H NMR: δ 8.9-9.0(s 1H NH Sec. amine), δ 7.6-7.7(s 1H CH pyridine), δ 7.5-7.6(s 10H CH aryl), δ 7.0-7.1 (s CH dihydroquinone), δ3.6-3.7(s 4H CH2 methylene),δ 2.7-2.8(s 4H CH piperazine), δ 1.9-2.0(S 1H NH piperazine) δ 0.5-0.8(S 5H CH Cyclopropane).
e2:IR(KBr)cm-1:3516.56(OHstr),3459.53(NH-str),3028.66(Ar-CHstr),2870.52(CH2str) 1700.94 (C=O str), 1525.42(Ar C-C str),1367.28(Ring C-N str),1050.11(Ar-C-O-Cstr).
antimicrobial activity3,4
Antimicrobial activity study was carried out by cup plate agar diffusion method using nutrient agar. The compounds were tested in vitro for their antimicrobial activity against two microorganisms viz Escherichia coli (NCTC10418) and Staphylococcus aureus (NCTC 5671) which are pathogenic to human beings. Standard drug Ciprofloxacin was used for comparision (Table III).
table iii: anti-bacterial activity of synthesized compounds
compound code
Zone of inhibition at 100µg/ml.
( mm)
Zone of inhibition at 100µg/ml.
( mm)
e.coli S.aureus a.niger C.albicansa1 16 14 17 16
a2 21 24 16 18
B1 19 12 18 22
B2 24 25 16 20
c1 22 22 12 18
c2 19 18 16 18
d1 24 25 19 18
d2 23 22 16 18
e1 24 24 24 26
e2 23 26 24 23
Ciprofloxacin 28 26 - -
griseofulvin - - 26 27
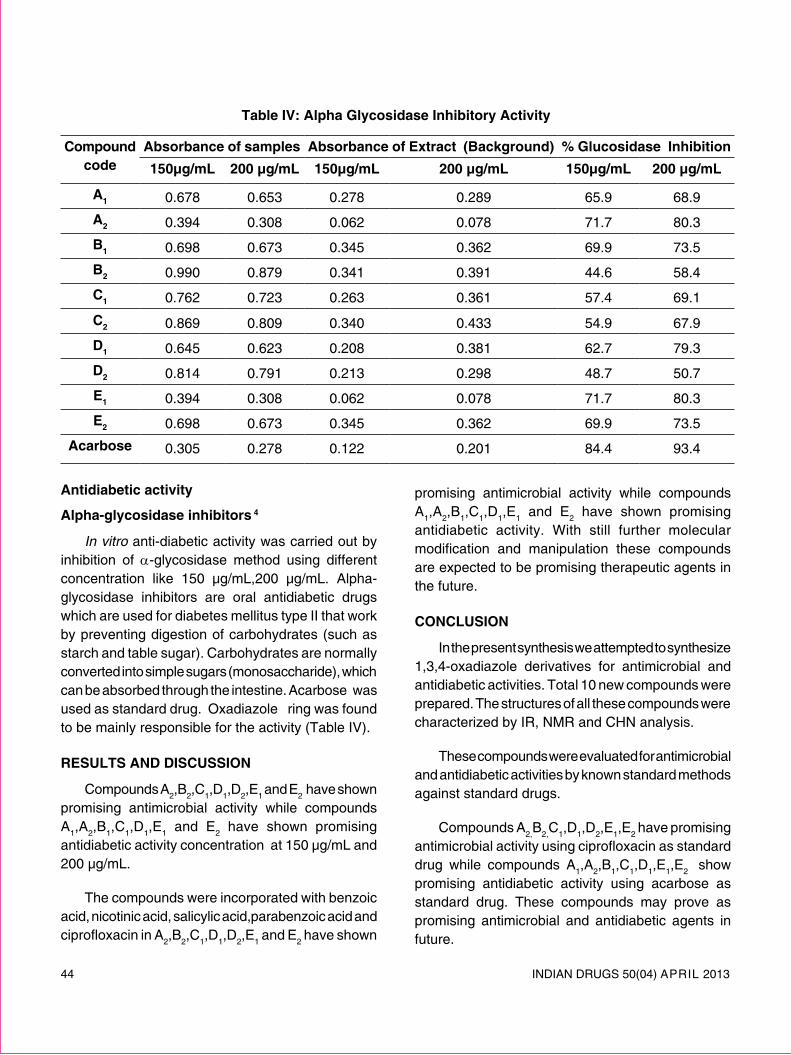
44 INDIAN DRUGS 50(04) ApRIl 2013
antidiabetic activity
alpha-glycosidase inhibitors 4
in vitro anti-diabetic activity was carried out by inhibition of α-glycosidase method using different concentration like 150 μg/ml,200 μg/ml. Alpha-glycosidase inhibitors are oral antidiabetic drugs which are used for diabetes mellitus type II that work by preventing digestion of carbohydrates (such as starch and table sugar). Carbohydrates are normally converted into simple sugars (monosaccharide), which can be absorbed through the intestine. Acarbose was used as standard drug. Oxadiazole ring was found to be mainly responsible for the activity (Table IV).
results and discussion
Compounds A2,B2,C1,D1,D2,E1 and E2 have shown promising antimicrobial activity while compounds A1,A2,B1,C1,D1,E1 and E2 have shown promising antidiabetic activity concentration at 150 μg/ml and 200 μg/ml.
The compounds were incorporated with benzoic acid, nicotinic acid, salicylic acid,parabenzoic acid and ciprofloxacin in A2,B2,C1,D1,D2,E1 and E2 have shown
promising antimicrobial activity while compounds A1,A2,B1,C1,D1,E1 and E2 have shown promising antidiabetic activity. With still further molecular modification and manipulation these compounds are expected to be promising therapeutic agents in the future.
conclusion
In the present synthesis we attempted to synthesize 1,3,4-oxadiazole derivatives for antimicrobial and antidiabetic activities. Total 10 new compounds were prepared. The structures of all these compounds were characterized by IR, NMR and CHN analysis.
These compounds were evaluated for antimicrobial and antidiabetic activities by known standard methods against standard drugs.
Compounds A2,B2,C1,D1,D2,E1,E2 have promising antimicrobial activity using ciprofloxacin as standard drug while compounds A1,A2,B1,C1,D1,E1,E2 show promising antidiabetic activity using acarbose as standard drug. These compounds may prove as promising antimicrobial and antidiabetic agents in future.
table iv: alpha glycosidase inhibitory activity
compound code
absorbance of samples absorbance of extract (Background) % glucosidase inhibition150µg/ml 200 µg/ml 150µg/ml 200 µg/ml 150µg/ml 200 µg/ml
a1 0.678 0.653 0.278 0.289 65.9 68.9
a2 0.394 0.308 0.062 0.078 71.7 80.3
B1 0.698 0.673 0.345 0.362 69.9 73.5
B2 0.990 0.879 0.341 0.391 44.6 58.4
c1 0.762 0.723 0.263 0.361 57.4 69.1
c2 0.869 0.809 0.340 0.433 54.9 67.9
d1 0.645 0.623 0.208 0.381 62.7 79.3
d2 0.814 0.791 0.213 0.298 48.7 50.7
e1 0.394 0.308 0.062 0.078 71.7 80.3
e2 0.698 0.673 0.345 0.362 69.9 73.5
acarbose 0.305 0.278 0.122 0.201 84.4 93.4
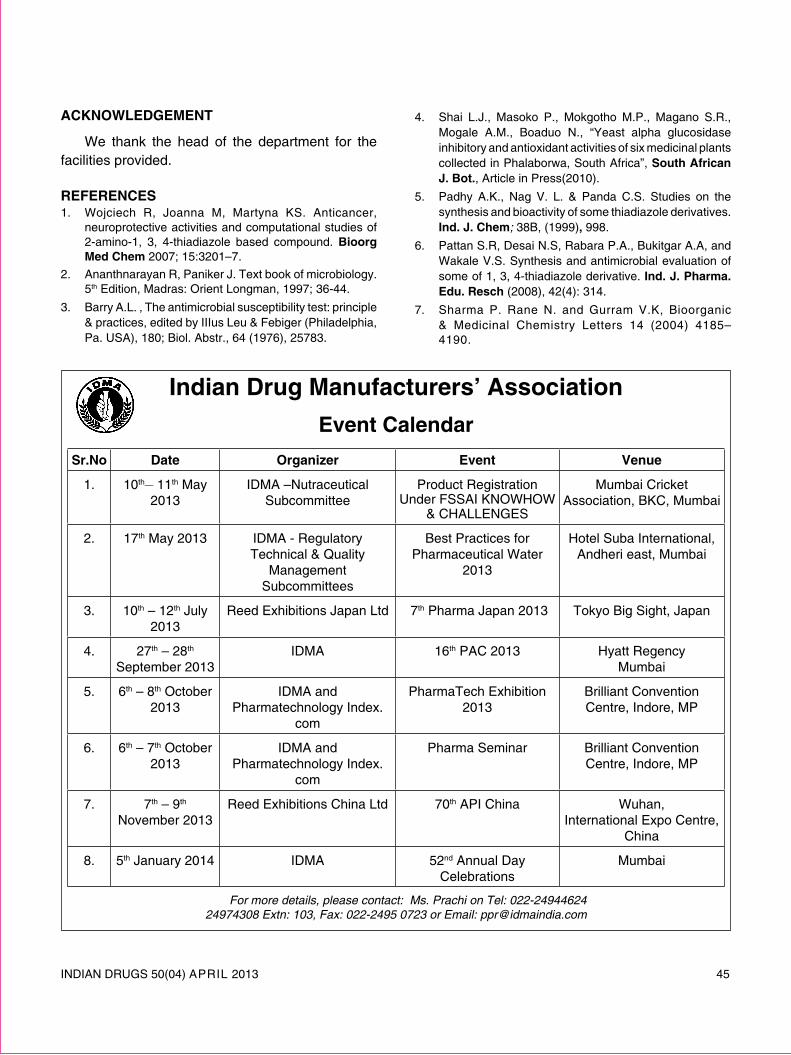
INDIAN DRUGS 50(04) ApRIl 2013 45
acKnowledgement
We thank the head of the department for the facilities provided.
reFerences1. Wojciech R, Joanna M, Martyna KS. Anticancer,
neuroprotective activities and computational studies of 2-amino-1, 3, 4-thiadiazole based compound. Bioorg med chem 2007; 15:3201–7.
2. Ananthnarayan R, paniker J. Text book of microbiology. 5th Edition, Madras: Orient longman, 1997; 36-44.
3. Barry A.l. , The antimicrobial susceptibility test: principle & practices, edited by IIIus leu & Febiger (philadelphia, pa. USA), 180; Biol. Abstr., 64 (1976), 25783.
4. Shai l.J., Masoko p., Mokgotho M.p., Magano S.R., Mogale A.M., Boaduo N., “Yeast alpha glucosidase inhibitory and antioxidant activities of six medicinal plants collected in phalaborwa, South Africa”, south african J. Bot., Article in press(2010).
5. padhy A.K., Nag V. l. & panda C.S. Studies on the synthesis and bioactivity of some thiadiazole derivatives. ind. J. chem; 38B, (1999), 998.
6. pattan S.R, Desai N.S, Rabara p.A., Bukitgar A.A, and Wakale V.S. Synthesis and antimicrobial evaluation of some of 1, 3, 4-thiadiazole derivative. ind. J. pharma. edu. resch (2008), 42(4): 314.
7. Sharma p. Rane N. and Gurram V.K, Bioorganic & Medicinal Chemistry letters 14 (2004) 4185–4190.
indian drug manufacturers’ associationevent calendar
sr.no date organizer event venue
1. 10th__ 11th May 2013
IDMA –Nutraceutical Subcommittee
product Registration Under FSSAI KNOWHOW
& CHAllENGES
Mumbai Cricket Association, BKC, Mumbai
2. 17th May 2013 IDMA - Regulatory Technical & Quality
Management Subcommittees
Best practices for pharmaceutical Water
2013
Hotel Suba International, Andheri east, Mumbai
3. 10th – 12th July 2013
Reed Exhibitions Japan ltd 7th pharma Japan 2013 Tokyo Big Sight, Japan
4. 27th – 28th September 2013
IDMA 16th pAC 2013 Hyatt RegencyMumbai
5. 6th – 8th October 2013
IDMA and pharmatechnology Index.
com
pharmaTech Exhibition 2013
Brilliant Convention Centre, Indore, Mp
6. 6th – 7th October 2013
IDMA and pharmatechnology Index.
com
pharma Seminar Brilliant Convention Centre, Indore, Mp
7. 7th – 9th November 2013
Reed Exhibitions China ltd 70th ApI China Wuhan,International Expo Centre,
China
8. 5th January 2014 IDMA 52nd Annual Day Celebrations
Mumbai
For more details, please contact: Ms. Prachi on Tel: 022-24944624 24974308 Extn: 103, Fax: 022-2495 0723 or Email: [email protected]

46 INDIAN DRUGS 50(04) ApRIl 2013
novel uv spectrophotometric method For estimation oF sitagliptin phosphate and simvastatin By area under curve method
chopde v. v.*, patil p. m., rathod s. d. and chaudhari p. d.
(Received 16 August 2012) (Accepted 23 February 2013)
aBstract
A simple, versatile, accurate, precise and economic method for simultaneous determination of sitagliptin phosphate and simvastatin in fixed dose combination products was developed. The absorbance values at 267 nm and 239 nm for sitagliptin phosphate and simvastatin. The combination is also estimated by AUC method it involved measurement of area under curve in the wavelength range is 264-270 nm (λ1-λ2) and 236-242 nm (λ3-λ4) sitagliptin phosphate and simvastatin respectively. This method obeyed Beer’s law in the concentration range of 10-60 μg /ml for sitagliptin phosphate and 2-12 μg /ml for simvastatin. The results of analyses have been validated statistically for linearity, accuracy, precision, lOD and lOQ of the proposed method.
Keywords: Simvastatin (SIMVA), Sitagliptin phosphate (SITA), methanol, distilled water, Area under curve method.
introduction
Sitagliptin chemically is (3R) -3-amino-1-[3- (trifluoromethyl)-6,8-dihydro-5h- [1,2,4] triazolo [3,4-c] pyrazin-7-yl]-4-(2,4,5-trifluorophenyl) butan- 1-one (Fig. 1) , an oral Anti-diabetic agent that blocks Dipeptidyl peptidase-4 (Dpp-4) activity. Sitagliptin increased incretin levels (Glp-1 and GIp) which inhibit glucagon release, in turn decreases blood glucose, but more significantly increases insulin secretion 1-3.
Simvastatin (SIMVA) is chemically is 2,2-Dimethyl butanoic acid (1S,3R,7S,8S,8aR)-1,2,3,7,8,8ahexahydro-3,7-dimethyl-8-[2-[(2R,4R)- tetrahydro-4- hydroxy-6 oxo2H pyran-2yl]ethyl]1-napthalenyl ester (Fig. 2); Simvastatin used as a HMG-CoA reductase inhibitors Simvastatin is used in the treatment of primary hypercholesterolemia and is effective in reducing total and lDl-Cholesterol as well as plasma triglycerides and apolipoprotein B 4-6.
*For correspondence p. e. society’s modern collage of pharmacy, yamunanagar sector no 21, nigdi, pune. e-mail: [email protected]
Fig. 1: structure of sitagliptin phosphate
Fig. 2: structure of simvastatin

INDIAN DRUGS 50(04) ApRIl 2013 47
Fig. 3: area under curve spectra of simva
Fig. 4: area under curve spectra of sita
A literature research revealed different analytical methods such as simultaneous and HplC7-11 for quantitative determination of sitagliptin phosphate and simvastatin. The present work deals with simple, rapid, accurate, precise, less expensive and economical multicomponent mode of analysis for sitagliptin phosphate and simvastatin in tablet by using area under curve method.
material and methods
instrumentation
A Jasco UV/Visible double beam spectrophotom-eter (UV model – 1700) and 1cm UV matched quartz cells were used. Gift samples of SITA and SIMVA were obtained from Merck pharmaceutical, Sinner Maharashtra, India.
reagents and chemicals
All chemicals and reagents used were of analytical grade and purchased from SD Fine Chemicals, Mumbai, India. Marketed formulation containing sitagliptin phosphate 100mg and simvastatin 40 mg was used as sample.
preparation of standard stock solution
Accurately weighed 10 mg of SITA and SIMVA was transferred to 100 ml volumetric flask separately, dissolved in 40 ml methanol and 60 ml distilled water by sonicater, sonicate up to 10 minute. The volume was adjusted with the same up to the mark to give final strength i.e. 100 μg/ml.
selection of wavelength for analysis
By appropriate dilutions with distilled water were prepared for each drug from the standard stock solution and scanned in the spectrum mode from 400 nm to 200 nm. The sampling wavelengths selected for analysis of SITA and SIMVA were 264 nm- 270 nm (λ
1- λ2) and 236-242 nm (λ3- λ4) respectively. Fig. 3, 4 and 5 represents the spectra SIMVA, SITA and spectra of mixture along with their selected AUC range.

48 INDIAN DRUGS 50(04) ApRIl 2013
Fig. 5: area under curve spectra of mixture
Fig. 6: standard calibration curve of sita
Fig. 7: standard calibration curve of simva
linearity
For each drug, appropriate aliquots of standard stock solutions were transferred to a series of 10 ml volumetric flasks. The volume was made up to the mark with distilled water to obtain working standard solutions for each drug of concentrations of 1-100 μg / ml for both SITA and SIMVA. Six sets of each concentration of the drugs were prepared separately. The absorbances of the working standard solutions of each concentration were measured at the selected analytical wavelengths. The standard calibration curves of absorbance vs concentration were plotted using the mean of these six independent observations. The concentration range over which the drugs obeyed Beer- lambert’s law was found to be between 10 to 60 μg/ml for SITA and 2 to 12 μg/ml for SIMVA. Standard calibration curve shown in (Fig. 6 and 7)
methods
area under curve method12
For the s imu l taneous determination using the area under curve (AUC) method, suitable dilutions of the standard stock solutions (100 μg/ml) of SITA and SIMVA were prepared separately in methanol: water (40:60V/V). The solutions of drugs were scanned in the range of 200-400 nm. For Area Under Curve method, calibration curve was plotted and the sampling wavelength ranges selected

INDIAN DRUGS 50(04) ApRIl 2013 49
for estimation of SITA and SIMVA are 264-270nm (λ1-λ2) and 236-242nm (λ3-λ4) and area were integrated between these selected wavelength ranges for both drugs and absorptivity value shown in Table I, which showed linear response with increasing concentration hence the same wavelength range were used for estimation of tablet formulations.
CSITa = ( X SM236-242 x aUC M
264-270 ) - ( X SM264-270 x aUC M
236-242)
( X SM236-242 x XST
264-270 ) - ( X SM264-270 x XST 236-242)
CSIMVa = ( X ST264-270 x aUC M
236-242) - ( X ST236-242 x aUC M
264-270 )
( X SM236-242 x XST
264-270 ) - ( X SM264-270 x XST 236-242)
Where,
C SITa = concentration of Sitagliptin phosphate
C SIMVa = concentration of Simvastatin
aUC M264-270 = aUC of mixture at wavelength range 264-270 nm
aUC M236-242 = aUC of mixture at wavelength range 236-242 nm
X ST264-270 = aUC of SITa at wavelength range 264-270 nm
Concentration of SITa in gm/100ml
X ST236-242= aUC of SITa at wavelength range 236-242 nm
Concentration of SITa in gm/100ml
X SM264-270 = aUC of SIMVa at wavelength range 264-270 nm
Concentration of SIMVa in gm/100ml
X SM236-242= aUC of SIMVa at wavelength range 236-242 nm
Concentration of SIMVa in gm/100ml
Substituting the standard absorptivity values,
CSITa = (442.50 x aUC M264-270 ) - (452.43 x aUC M
236-242)
830
CSIMVa = (154.25 X aUC M236-242) - (119.27 X aUC M
264-270)
830
Using the equations 3 and 4, the concentration of SITA and SIMVA in the solutions can be calculated respectively.
table i: standard absorptivity values of sita and simva
drugs *absorptivity value at
(264 nm-270 nm)
* absorptivity value at
(236 nm-242 nm)
SITA 154.25 119.27
SIMVA 452.43 442.10
*Average of six determinations
validation of the method
Marketed tablets containing 40 mg of SIMVA and 100 mg of SITA were used. Twenty tablets were weighed and average weight was calculated. The tablets were triturated to a fine powder. An accurately weighed quantity of powder equivalent to 100 mg of SITA and 40 mg of SIMVA were transferred to 100 ml volumetric flask and dissolved in 40 ml of methanol solution by sonicating for 10 min and volume was then adjusted up to 100 ml with distilled water. The solution was filtered through Whatman filter paper No. 41. Summary of validation parameter shown in Table II.
accuracy
For accuracy of method, recovery study was carried out by applying the method of drug samples to which known amount of SITA and SIMVA were added at 80 %, 100 % and 120 % levels. At each level of the amount, six determinations were carried out by applying each sample 6 times and standard deviation (SD) was calculated.
precision
precision of the method was studied as intra-day and inter-day variations. Intra-day precision was determined by analyzing the 20 μg/ml of SITA and 8 μg/ml of SIMVA solutions for three times in the same day. Inter-day precision was determined
.... (1)
.... (2)
.... (3)
.... (4)
50 INDIAN DRUGS 50(04) ApRIl 2013
by analyzing the 20 μg/ml of SITA and 8 μg/ml of SIMVA solutions daily for three days over the period of week.
sensitivity
The sensitivity of measurements of SITA and SIMVA by the use of the proposed method was estimated in terms of the limit of Quanfication (lOQ) and limit of Detection (lOD). The lOQ and lOD were calculated using equation lOD = 3.3 x N/B and lOQ = 10 x N/B, where, ‘N’ is standard deviation of the peak areas of the drugs (n = 3), taken as a measure of noise, and ‘B’ is the slope of the corresponding calibration curve.
repeatability
Repeatability was determined by analyzing 20 μg/ml and 8 μg/ml concentration of SITA and SIMVA solution for six times.
results and discussion
method validation
The proposed method was validated as per ICH guidelines. The solutions of the drugs were prepared
as per the earlier adopted procedure given in the experiment.
linearity studies
The linear regression data for the calibration curves showed good linear relationship over the concentration range 10-60 μg/ml for SITA and 2-12 μg/ml for SIMVA. linear regression equation was found to be Y = 0.004 X-0.005 (r2 = 0.990) of SITA and for SIMVA Y = 0.050X +0.069 (r2 = 0.992). The results are tabulated under Table II.
accuracy
The solutions were reanalyzed by proposed method; results of recovery studies are reported in Table III which showed that the % amount found was 99.50% and 99.35 with % R.S.D. < 2 for SITA and SIMVA.
precision
The precision of the developed method was expressed in terms of % relative standard deviation (% RSD). These result shows reproducibility of the assay. The % R.S.D. values found to be less
table ii: summary of validation parameter
parameter sita simva
Absorption (nm) 264-270 nm 236-242 nm
linearity range (μg/ml) 10-60 μg/ml 2-12 μg/ml
Standard regression equation y= 0.004x- 0.005 y= 0.050x+ 0.069
Correlation coefficient (r2) r2= 0.990 r2= 0.992
Accuracy (% recovery± *SD) 99.50±0.088 99.35±0.0141
precision (% CV± *SD) 99.94±0.014 99.89±0.014
Repeatability (% mean± *SD) 102.3 ± 1.2 101.1 ± 1.07
*lOD 0.00125 0.0694
*lOQ 0.00379 0.2104
*Average of six determinations

INDIAN DRUGS 50(04) ApRIl 2013 51
table iii: determination of accuracy by percentage recovery method for sita and simva
ingredients tablet amount (µg/ml)
amount added
(µg/ml)
level of addition
amount recovered
(µg/ml)
percentage recovery
average % recovery
*s.d
% rsd
sita 20 16 80 % 36 83.94 99.50 ±0.088 0.081
20 20 100 % 40 99.79
20 14 120 % 44 111.79simva 8 6.4 80 % 14.4 85.56 99.35±0.0141 0.015
8 8 100 % 16 99.87
8 9.6 120 % 17.6 112.63
*Average of six determinations
than 2, so that indicate this method precise for the determination of both the drugs in formulation. The results are tabulated under Table II and IV.
sensitivity
The lOD and lOQ for SITA and SIMVA were found to be 0.00125 and 0.00379, 0.0694 and 0.2104. The results are tabulated under Table II.
repeatability
Repeatability was determined by analyzing 20 μg/ml concentration of SITA solution and 8 μg/ml concentration of SIMVA for six times and the % amount with % R.S.D. less than 2. The results are tabulated under Table II.
conclusion
This area under curve method is quite simple, accurate, precise and reproducible. The area under curve method has been developed for quantification
table iv: inter-day and intra-day precision
drugs inter-day intra-day
mean *s.d. *%r.s.d. mean *s.d. *%r.s.d.
sita 99.94 0.014 0.014 99.97 0.10 0.10
simva 99.89 0.014 0.014 99.94 0.007 0.006
*Average of six determinations
of sitagliptin phosphate and simvastatin in tablet formulation. The validation procedure confirms that this is an appropriate method for their quantification in the bulk and marketed formulation. The relative std. deviation (RSD) for all parameters was found to be less than two, which indicates the validity of method and assay results are also within the limit so the proposed method can be used for routine quantitative simultaneous estimation of both the drugs in multi-component pharmaceutical.
acKnowledgement
The authors are thankful to Merck pharmaceuticals, Sinner, Maharashtra, India for providing free gift sample of sitagliptin phosphate and simvastatin.
reFerences1. Herman G, Stevens C, Van Dyck K, Bergman A, Yi B,
De Smet M, pharmacokinetics and pharmacodynamics of sitagliptin, an inhibitor of dipeptidyl peptidase IV, in healthy subjects: results from two randomized, double-

52 INDIAN DRUGS 50(04) ApRIl 2013
blind, placebo-controlled studies with single oral doses. clin. pharmacol.ther. 2005, 78, 675-688.
2. Herman G, Bergman A, liu F, Stevens C, Wang A, Zeng W, pharmacokinetics and pharmacodynamic effects of the oral Dpp-4 inhibitor sitagliptin in middleaged obese subjects. J. clin. pharmacol. 2006, 46, 876-886.
3. Stella Vincent H, James Reed R, Bergman AJ, Elmore CS, Metabolism and excretion of Dipeptidase 4 inhibitor (14C) sitagliptin in humans. drug metab. dispo. : 2007, 35, 533-538.
4. Ochiai H.: Determination of Simvastatin and Its Active Metabolites in Human plasma by Column- Switching High performance liquid Chromatography with Fluorescence Detection after Derivatization with 1-Bromoacetylpyrene. J. chromatogram Biomed. sci.: 1997, 694, 211-217.
5. Tang, Y., Xiang, l., Wen, Sun, Min Y.U., Zuo-gang lI.; Rp-HplC determination of sitagliptin phosphate; chinese J. pharma. analysis: 2009; 08, 121-125.
6. Sekaran B.C., Rani p; Development and Validation of Spectrophotometric Method for determination of Dpp-4 Inhibitor, Sitagliptin, in its pharmaceutical Dosage Form; int. J. pharma. sci. : 2010, 2(4), 138-142.
7. Basavaiah K., Tharpa K.: The Development and Validation of visible Spectrophotometric methods for
Simvastatin determination in pure and the tablet dosage forms; chemical industry and chemical engineering Quarterly: 2008, 14(3), 205-210.
8. Srinivas C., Mohd A.B.; Development and Validation of Simvastatin in Microemulsion formulation using Rp-HplC; int. J. pharma. sci. : 2010, 4(1), 398-403.
9. Madhukar A., Swapna V.: Sensitive Analytical Method Development and Validation of Simvastatin Bulk Drug by Rp-HplC: J. pharm. resch, 2012, 5(2), 906-907.
10. Sharma S., Kothari A., Harsoliya. M.S.; Simultaneous UV Spectrophotometric Method for Estimation of Sitagliptin phosphate and Simvastatin in Tablet dosage form; J. pharm. resch: 2012, 5(1), 444-446.
11. Mane V.B., Babar S. and Kulkarni N. Development of UV Spectrophotometric method for the simultaneous estimation of Simvastatine and Ezetimibe in tablet dosage form by simultaneous Equation and Absorbance ratio method; int. J. pharm tech resch, 2011, 3(3),s 1459-1466.
12. Tambe V., Vichare V., Kandekar U. and Dhole S.; novel UV Spectrophotometric method for estimation of ramipril and hydrochlorothiazide by simultaneous equation and area under curve method : int.J. appl.pharma., 2010, 2(4), 20-22.
now availaBle ! idma-apa guidelines / technical monographs
TECHNICAl MONOGRApH NO. 1 TECHNICAl MONOGRApH NO. 2 staBility testing oF eXisting primary & secondary drugs suBstances and products chemical reFerence suBstances
TECHNICAl MONOGRApH NO. 3 TECHNICAl MONOGRApH NO. 4 investigation oF out oF pharmaceutical preFormulation speciFication (oos) test results analytical studies
TECHNICAl MONOGRApH NO. 5 TECHNICAl MONOGRApH NO. 6 environmental monitoring in corrective/preventive actions cleanrooms (capa) guideline
Copies are available at IDMA Office, Mumbai. We do not mail any publications against Vpp payment. All payments to be made in advance as DD only in favour of “indian drug manuFacturers’ association” at Mumbai.
For More Details please Contact: puBlications division. ms. prachi Tel.: 022 - 2494 4624 / 2497 4308 Ext.: 103, Fax: 022 - 2495 0723 E-mail: [email protected]

INDIAN DRUGS 50(04) ApRIl 2013 53
short notes
comparative In vItro antiBacterial activity oF ethanolic eXtract oF Plumbago zeylenICa
aBstract
Ethanolic extract of leaf, flower and root of Plumbago zeylenica l. (white variety) traditionally used in the treatment of gastrointestinal disorders were screened for antibacterial activity against bacterial pathogens Staphyllococcus aureus, Escerchia coli, Klebsiella pneumoniae, Enterobacter aurugens, Proteus vulgaris, Shigella sonnei, Salmonella typhi which are known to cause different type of gastrointestinal disorders. The results showed that leaf and root extracts exhibited strong antibacterial action at concentration of 2-8 mg/ ml of the bacterial pathogen tested. However, powdered flower extract showed moderate activity. Thus the present data reveals that leaves and flowers are equally potential for the treatment of bacterial pathogens, which causes intestinal catarrh, indigestion, and colic intestinal abscesses.
Keywords: Plumbago zeylenica, antibacterial activity, ethanolic extract.
introduction
In many developing countries where living conditions are crowded and hygiene is poor, various gastrointestinal disorders including diarrhea, dysentery, poisoning, internal abscesses caused by bacterial pathogen are among the main cause of morbidity and mortality1. In India diarrhea and dysentery is a serious health problem in rural area and is the second cause of disease among all age groups. Much more herbs used traditionally in Indian system of medicine for the treatment of gastrointestinal disorders.
Plumbago zeylenica. l. (white variety) Family: plumbaginaceae is a tropical shrub and more wide spread and cultivated in gardens through out India. Plumbago zeylenica used to treat fever, loss of appetite, strong digestive stimulant, edema, piles, parasitic infections and obstinate skin diseases including leprosy. pharmacological studies indicated that Plumbago zeylenica roots extract has antispasmodial2, antimicrobial3, antioxidant4, antiheliobacter5, antiviral6, antifungal7, anti-inflammatory8, antihyperglycemic9, hypolipidaemic and antiatherosclerotic activity10 and
phytochemical investigations showed to contain plumbagin11, cytotoxic napthoquinones and plumbagic acid glucosides12. leaves and flowers are widely used as medicinal herbs in Indian traditional medicines is believed to have function for intestinal catarrh, indigestion, colic, intestinal abscesses, jaundice, intestinal parasites, piles, urinary calculi, polyuria, spermaturia, vaginal discharges, deficient lactation, virulent skin diseases, poisoning13. In this study we investigated the comparative in vitro antibacterial activity of ethanolic extracts of leaves and flowers along with roots.
materials and methods
plant material
The leaves, flowers and roots of Plumbago zeylenica were collected from Satpura hills near Chopda (MS) India. The fresh leaves, flowers and roots are authenticated by Dr. R.M. Bagul, Botanist, Department of Botany, ASC College, Chopda .The specimen voucher was deposited at the Department of pharmacognosy, Smt. S.S. patil College of pharmacy, Chopda.
preparation of ethanolic extract
Shade dried and coarsely powdered leaves (200 g), flowers (200 g), roots (200 g) were extracted

54 INDIAN DRUGS 50(04) ApRIl 2013
with 90% ethanol by process of continuous hot extraction using Soxhelt. Excess of organic solvents removed by concentration of extracts under reduced pressure at temperature not more than 400C. The residue resulting was packed in to glass vial and stored in a desiccator over silica gel until use.
test strains
Different bacterial strains as Staphyllococcus aureus (ATCC 29737), Escerchia coli (ATCC 8739), Klebsiella pneumoniae (ATCC 10031), Enterobacter aurugens (ATCC 13048), Proteus vulgaris (ATCC 6380), Shigella sonnei (MTCC 2957), Salmonella typhi (MTCC 733) were obtained from the polymed lab, Department of Microbiology, Jalgaon-425 002 (MS), India.
antibacterial testing
Antibacterial activity was measured using a dilution in agar technique. The extract for testing were dissolved in dimethyl sulfoxide (DMSO) and serially diluted in melted Muller Hilton agar culture medium in petri dishes to obtain the final concentration of 2, 4, 6, 8 mg/ ml. All bacterial strain was grown in Muller Hilton Broth (MHB) for 8 h at 370C. Bacterial suspension were prepared equivalent to 104 CFU were prepared by dilution with MHB and diluted inoculum was added to petri dishes and incubated for 24 h at temperature 370C. After incubation all petri dishes were observed for zones of growth inhibition, and the diameter of this zone was measured in millimeters with ruler. Results were expressed as the percentage of inhibition of bacterial growth, determined by comparing it with normal bacterial growth in the control plate (growth in this control plate was considered as 100%). The minimum inhibitory concentration (MIC) was determined as the lowest concentration that completely inhibited macroscopic growth of bacteria.
results and discussion
The percentage yields of ethanolic extract of the leaves, flower and root of Plumbago zeylenica
table i: percentage yield of ethanolic extract of various plant parts of Plumbago
zeylenica
plant species part extracted
percentage yield (w/w)
Plumbago zeylenica leaves 17.9
Plumbago zeylenica Flowers 15.2
Plumbago zeylenica Roots 20.2
table ii: percentage inhibition of bacterial growth by ethanolic extract of various plant
parts of Plumbago zeylenica
sr. no.
Bacterial strains Plumbago zeylenica (% inhibition at 8 mg/ml)
leaves Flowers roots
1. Staphyllococcus aureus ATCC 29737
57.1 55.2 57.2
2. Escerchia coli ATCC 8739
66.8 49.3 67.8
3. Klebsiella pneumoniae ATCC 10031
25.0 46.1 58.2
4. Enterobacter aurugens ATCC 13048
100.0 55.3 100.0
5. Proteus vulgaris ATCC 6380
67.4 58.5 78.0
6. Shigella sonnei MTCC 2957
100.0 41.2 100.0
7. Salmonella typhi MTCC 733
27.2 23.6 36.4
Results are reported as mean (n=4) & expressed as % inhibition.
Staphyllococcus aureus ATCC 29737, Escerchia coli ATCC 8739, Klebsiella pneumoniae ATCC 10031, Enterobacter aurugens ATCC 1304, Proteus vulgaris ATCC 6380, Shigella sonnei MTCC 2957, Salmonella typhi MTCC 733

INDIAN DRUGS 50(04) ApRIl 2013 55
l. (white variety) were given in Table I. The crude ethanolic extracts of Plumbago zeylenica l. were used for screening of antibacterial activity against eight bacterial species. percentage inhibition of bacterial growth by ethanolic extract of the leaves, flower and root of Plumbago zeylenica l. (white variety) were shown in Table II. The plant differ significantly in their activity against the tested strain and the maximum antibacterial activity was observed at concentration 8 mg / ml. All the extracts have shown potential activity against all bacterial strains with percentage inhibition ranging from 23.6 to 100.0%. Among these leaf extract shown 100.0% inhibition of growth against Shigella sonnei and Enterobacter with MIC value 2-8 mg/ml same as of root extract. Also leaf extract exhibited moderate activity against Escerchia coli, Proteus vulgaris, Staphyllococcus aureus, which was very comparative to the activity of root extract. Crude extract of flowers shown moderate potential against all strains of bacteria. Thus our findings confirm the traditional therapeutic potential of leaves and flowers along with root of Plumbago zeylenica l. Fractionation of the active crude extract of leaves and flowers to isolate and identify the component responsible for the antibacterial activity, are in process.
acknowledgement
The authors wish to thank Dr. R.M. Bagul, Department of Botany ASC College, Chopda for
identification of plant species. We also thanks to Mr. Sumit Kabra, Department of Microbiology, polymed laboratories, Jalgaon- 425 002 (MS), India for giving the bacterial strains.
reFerences1. World Health Organization, 2002 WHO strategy on
tradition medicine. World health organization Geneva. 2002. p. 57.
2. Simonsen H.T., Nordskjold J.B., Smitt U.W., Nyman U., palpu p., Joshi p., Varughese G. J ethnopharmacol 2001; 74: 195.
3. Ahmad I, Mehmood Z, Mohammad F. J ethnophamacol 1998; 62(2):183.
4. Tilak J.C., Adhikari S, Devasagavam T.p. redox rep 2004; 9(4): 219.
5. Wang Y.C., Huang T.l. Immunological Med Mocrobiol 2005; 43(3): 407.
6. Gebre-Mariam T, Neubert R, Schmidt p.C., Wutler p, Schmidtke M.J ethnopharmacol 2005; 72:130.
7. Mehmood Z, Ahmed I, Mohammad F, Ahmad S. pharmaceut Biol 1999; 37: 237.
8. Oyedapo O.O. int J pharmacognosy 1996; 34365.
9. Olagunju J. A., Jobi A.A., Oyedapo O.O. phytother res 1999; 13:346.
10. Sharma I, Gusain D, Dixit V.p. indian J physiol pharmcol 1991; 35:10.
11. Hsieh Y.J., lin l.C., Tsai T.H., J chromatogr a 2005; 12: 141.
12. lin l.C., Yaug l.l., Chou C. J phytochemistry 2003; 62 (3): 619.
13. Encyclopedia of Indian Medicinal plants, Khare Cp (editor) Springer, Germany 2004. p. 376.
smt. s.s. patil college of pharmacy, vadnere g.p.*, pathan a.r., Kulkarni B.u. and singhai a.K. chopda, dist-jalgaon (ms)-425107 e-mail: [email protected]
*For correspondence
(Received 16 January 2013) (Accepted 23 February 2013)

56 INDIAN DRUGS 50(04) ApRIl 2013
isolation and characteriZation oF Betaine From dried seeds oF aChyrantheS aSPera
aBstract
Marker compounds play an important role in standardization of herbal extracts and herbal formulations. The isolation of marker/s from crude extract or fractions of an extract is often a lengthy and expensive process and it many times requires numerous separation steps involving use of different separation techniques. The present work describes new method of isolation of betaine, a chemical marker from the plant Achyranthes aspera by preparative TlC and its characterization by chemical and spectroscopic techniques like IR, NMR and mass spectroscopy.
Keywords: Marker, Apamarga, Achyranthes aspera, Betaine, Amaranthacae, Chirchita
introduction
Achyranthes aspera linn is a variable, erect, annual to perennial herb which can grow up to 30-120 cm tall1,2. It is distributed throughout the old world tropics, including Malaysia and Australlia and introduced into tropical America. In India it is distributed throughout along roadsides and waste places3. The plant is used as diuretic, carminative, digestive, expectorant, bitter tonic, anti-inflammatory and a killer of intestinal worms.
The plant contains alkaloids betaine and achyranthine. It also contains hentriacontane, ecdysterone and two glycosides of oleanolic acid and amino acids4. Achyranthine is yellow coloured semisolid which is highly hygroscopic and unstable. Betaine is present in considerable amount and can be used as marker for standardization of extracts and formulations containing Achyranthes aspera. According to literature survey, betaine supplements decrease homocysteine levels in homocystinuria and in chronic renal failure patients. They have multiple effects on homocysteine metabolism in lewis rats5,6. Betaine along with ethanol is found to decrease hepatic triglyceride, lipid peroxide levels and serum transaminase activities and to increase GSH levels in guinea pigs7.
The present paper discusses new method for isolation of betaine, a marker compound from Achyranthes aspera and its characterization.
eXperimental
procurement of plant material
The seeds of Achyranthes aspera were obtained from three different geographical sources as given below:
S1: Seeds were procured from Valsad district of Gujarat in the month of November 2009.
S2: Seeds were procured from Virudunagar district of Tamil Nadu in the month of November 2009.
S3: Seeds of were procured from local market of Mumbai, Maharashtra in the month of November 2009.
The different herbaria of S1, S2 and S3 were deposited at Botanical Survey of India, pune under names AAGSOD4, AAGSOD5 and AAGSOD6 respectively and samples of seeds were identified by prof. Shirodkar.
extraction of alkaloids from achyranthes aspera
20 gm of powdered seeds were extracted with 200 ml of acidified ethanol (pH 4 was made by HCl) by heating at 700C for 5hr. It was then filtered. To this ammonia solution was added (25% Exrapure, specific gravity 0.91) till pH 10 to get the precipitate of alkaloids. The precipitate of the crude alkaloid was filtered and dried8.

INDIAN DRUGS 50(04) ApRIl 2013 57
isolation of alkaloid from achyranthes aspera
The crude alkaloid thus obtained was sonicated with 5X 50 ml of distilled water for 20 min. The alkaloid being water soluble gets extracted in water. Aqueous layer showed some coloured impurities which were removed using a charcoal column. The charcoal column was washed with 5X 25 ml of distilled water. The clear water extracts were pooled and evaporated to dryness under reduced pressure to yield crude alkaloid.
tlc studies
To check the purity of isolated alkaloid, TlC studies were carried out. Methanol: chloroform (7:3) was selected as mobile phase which gave good resolution with optimum Rf.
effect of time on % yield
The effect of time of extraction on % yield of the alkaloid was checked. The reflux extraction was carried out for different time intervals such as 3 hr, 4 hr and 5 hr in order to see at which time interval maximum yield is obtained.
effect of defatting on % yield
The powdered seeds were defatted with petroleum ether (60- 800C) to remove organic impurities and then the extraction of powder was carried out using acidified alcohol. The effect of defatting on % yield was also checked.
variation in % yield of marker compound with respect to geographical source
The seeds of Achyranthes aspera were obtained from three different geographical regions. percentage yield of marker compound was calculated for all the three samples of seeds and compared.
Purification of alkaloid
The crude alkaloid was purified by preparative TlC using methanol: chloroform (7:3) as mobile phase followed by repeated recrystallization with methanol.
characterization of alkaloid
The isolated pure alkaloid was characterized by chemical (melting point), chromatographic and spectral studies (IR, HNMR and mass spectroscopy).
results and discussion
Alkaloids of seeds of A. aspera were extracted by conventional reflux extraction technique. Extraction was carried out by acidified alcohol. preliminary phytochemical screening of alcoholic extract was carried out. It showed the presence of alkaloid, carbohydrates and glycosides.
The % extractive yield obtained was 0.056 %. TlC studies of isolated compound reveled presence of orange coloured band at Rf value 0.82 (Fig. 1).
The extraction time was optimized by checking the yield of compound at 3 hr, 4 hr and 5 hr time interval. (Table I).The yield of the alkaloid was 0.055 % after 3 hr which was found to decrease at 4 hr and very slight increase in the % yield was observed at 5 hr. Although there was no significant increase in the % yield of the compound was observed, it was decided to set extraction time for 5 hr.
Defatting of drug prior to extraction could not increase the yield significantly. Variation in yield of isolated compound with respect to three different geographical sources was checked. The yield of isolated compound obtained from sample S1 (Gujarat variety-0.076 %) was maximum followed by sample S2 (Tamil Nadu variety-0.068) and S3 (Maharashtra variety-0.056 %). From this it can be concluded that the amount of chemical constituents in plant varies with the geographical source.
The isolated compound was further purified by preparative TlC followed by recrystallization using methanol. The solvent system used for preparative TlC was same as that used for TlC.
After purification, IR, NMR and mass spectra of the isolated compound were obtained. (Fig. 2 and 3 respectively). IR spectroscopy confirmed the
58 INDIAN DRUGS 50(04) ApRIl 2013
presence of COO- group (1624.21 cm-1) and C-N stretching (1238.41 cm-1). HNMR showed presence of nine equivalent methyl protons and two methylene protons. (Table II). Mass spectrum confirmed presence of M+H peak at m/z value of 118 confirming the molecular weight of the compound (Table III). Based on spectroscopic studies the structure of the isolated alkaloid was confirmed as betaine (I).
(I) Chemical Name: N,N,N-
trimethylammonioacetate ( Trimethylglycine OR Betaine)
Molecular weight: 117 g/mol
table i: effect of time on the yield of isolated compound
extraction time (hr)
% yield of isolated compound a
3 0.055±0.005
4 0.047±0.000
5 0.056±0.003
a Each value is the mean± S.E.M. of three readings
table ii: interpretation of hnmr spectrum
sr. no. δ values inference
1. 3.27 (-3CH3, s)
2. 3.8 (-CH2, s)
table iii: interpretation of mass spectrum
sr. no. m/z values inference
1. 140 [M+Na]+ peak
2. 118 [M+H]+ peak
3. 74 [M+H-COO]+ peak
Fig. 1: TLC profile of isolated compound
Fig. 2: nmr spectrum of isolated compound
Fig. 3: mass spectrum of isolated compound
conclusion
An alkaloid, betaine was successfully isolated from dried seeds of Achyranthes aspera. The yield of

INDIAN DRUGS 50(04) ApRIl 2013 59
the compound was compared for three samples from different geographical sources. Betaine can be used as a marker for standardization of extracts or formulations containing Achyranthes aspera.
acKnowledgements
Authors are thankful to National Medicinal plants Board (NMpB), Department of AYUSH, New Delhi for financial assistance.
Authors are thankful to prof. Shirodkar, Botanical Survey of India, pune for authenticating the plant material.
reFerences1. pullaiah T., Ramakrishnaiah V., Sandhyarani S. and Rao
p. N., Flora of Guntur District, Andhra pradesh, India, Regency publications, New Delhi, 2000
2. Kakkar, K.K.: Apamarga-Medicinal Uses and Therapeutic Efficacy: A Review, ind. drugs. 2001, 38(2), 56-62
3. Warrier p.K., Nambiar V.p. K. and Ramankutty C., Indian Medicinal plants, Orient longman pvt. ltd., Chennai, Vol 1, 1993
4. Chatterjee A. and pakrashi S., The Treatise on Indian Medicinal plants, publications & Information Directorate, New Delhi , Vol 1, 1995.
5. Slow S., lever M., lee M. B., George p. M., Chambers S. T.: Betaine analogues alter homocysteine metabolism in rats, int. J. Biochem. cell. Biol. 2004, 36 (5), 870–880
6. lawson-Yuen A, levy H. l.: The use of betaine in the treatment of elevated homocysteine, mol. genet. metab. 88 (3), 2006, 201–207
7. Balkan J., Oztezcan S., Kucuk M., Kbas U. C., Toker N. K., and Uysal, M.: The effect of betaine treatment on triglyceride levels and oxidative stress in the liver of ethanol-treated guinea pigs, exp. toxic. pathol. 2004, 55(6), 505–509
8. Harbone J. B., phytochemical Methods A Guide to Modern Techniques of plant Analysis, 3rd Edition, Springer (India) pvt. ltd., New Delhi, 2005
c. u. shah college of pharmacy desai s. s., tatke p.a.* and gabhe s.y. s. n. d. t. women’s university sir vithaldas vidhyavihar, Juhu road santacruz (w), mumbai-400 049 e-mail:[email protected]
*For correspondence
(Received 31 January 2013) (Accepted 08 March 2013)
available dvd of ip – 2010 DVD of Ip – 2010 is available from the office of the Secretary-cum-Scientific Director, I.p.Commission, Sector-23, Raj Nagar, Ghaziabad – 201002, (U.p.), @ Rs.25,000/-per DVD.For more details please visit the website: www.ipc.gov.in; E.mail Id: [email protected] or contact to The publication Division, I.p.Commission, Tel.Nos.: 0120-2783392, 2783400, Extn.309, 308 & 112.

60 INDIAN DRUGS 50(04) ApRIl 2013
IDMA Bulletin XLIV (15) 15 to 21 April 2013 29
INDIAN DRUG MANUFACTURERS’ ASSOCIATION 102B, Poonam Chambers, Dr Annie Besant Road, Worli, Mumbai – 400018 INDIA Tel: +91-22-4944624/4974308 Fax: +91-22-4950723 Email: [email protected] / [email protected], Web: www.idma-assn.org
A One Day seminar on “BEST PRACTICES FOR PHARMACEUTICAL WATER”
On Friday, 17th May 2013 at Hotel Suba International, Andheri east, Mumbai.We are pleased to inform you that Indian Drug Manufacturers’ Association (IDMA); Regulatory & Technical and Quality Management Subcommittees are organizing a One Day Seminar on “BEST PRACTICES FOR PHARMACEUTICAL WATER”on Friday, 17th May 2013 at Hotel Suba International , Andheri east, Mumbai .
Water is a basic necessity for pharma operations. Sources are varied and the quality may differ seasonally.Water gets easily contaminated and in turn contaminates the product in which it is used.
Even by simply storing water, it gets contaminated. Equipments also need to be cleaned with purified water.It makes good science and business sense to Validate Water Systems.
Topics for the Seminar to be presented by eminent speakers“Standards for Drinking Water” Mr. Kapil Bhargava, Former Deputy, Drugs Controller, India “Inspection of Pharmaceutical water system” Dr Rakesh Tirpude, Asst.Commissioner, FDA, Maharashtra“Modern Microbial Evaluation of Pharmaceutical Waters” Dr G M Warke , Managing Director, Himedia Labs“Using Six Sigma approach to Water Purification” Mr Pravin Manker, Pharmaceutical Consultant.“High purity water generation systems” Mr Satish Bhadri, Managing Director, Envirochem Tech Solutions (I) Pvt. Ltd.“ WPU Distribution, from Design to Delivery” Mr.Shoeb Kurawadwala, Managing Director, Christ Nishotech water systems Pvt. LtdQ&A session
Panel Discussion
This one day seminar will demystify all aspects of Pharmaceuticals Water systems validation. Be it DM, RO, UV, Distillation, storage & distribution. With their first hand experience the expert faculty will simplify this complicated activity.Excellent opportunity for the R & D Scientists, Engineers, QA, Production, QC & Regulatory Affairs.
Registration FeesIDMA Members ` 2000/-
Non Members ` 2500/- Please join in large numbers and support us.
We look forward to your active participation, support and co-operation as in the past and request you to kindly send the registration forms with the payment at the earliest. The cheque/DD to be drawn on “Indian Drug Manufacturers’ Association”
For more details please contact:Ms. Prachi Rane
(Cell : +91 9867634383) E-mail: [email protected]
Mr. Anup Shetty(Cell +91 9892207797)
E-mail: [email protected]

INDIAN DRUGS 50(04) ApRIl 2013 61
IDMA Bulletin XLIV (15) 15 to 21 April 2013 30
REGISTRATION FORM
To, The Secretary General Indian Drug Manufacturers’ Association 102/B, A Wing, Poonam Chambers, Worli, Mumbai 400 018. Tel. # 022 - 24974308 / 24944624 Fax # 022 - 24950723 E-mail: [email protected] / [email protected] Date: _____________
Dear Sir,
One day seminar on “BEST PRACTICES FOR PHARMACEUTICAL WATER” on Friday, 17th May 2013 from 9.30 a.m. to 5.30 p.m. at Hotel Suba International, Mumbai.
~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
Kindly register the name/s of the following person/s from our company to participate in the above programme:
SR. NO. NAME DESIGNATION
Our Cheque/DD* no._____________________ dated _____________________ for Rs. _________________ is enclosed.
Thanking you, Yours faithfully,
(Name & Designation)
Name of the Company :Address :
Tel No. : Fax No. : E-Mail:
The subsidized Registration fee for the same would be as follows: -
` 2000/- per person (For IDMA Members)
` 2500/- per person (For Non IDMA Members)
Note: Participation fee is neither refundable nor adjustable against future programmes. However, changes in nominations are accepted. Kindly use photocopies of this form for additional registrations. The cheque/DD should be drawn in favour of “Indian Drug Manufacturers’ Association”. *Outstation parties to remit by DD please.

62 INDIAN DRUGS 50(04) ApRIl 2013
IDMA Bulletin XLIV (14) 08 to 14 April 2013 38
INDIAN DRUG MANUFACTURERS’ ASSOCIATION
IDMA-APA-PAC 2013“16th PHARMACEUTICAL ANALYSTS’ CONVENTION”
On
27th & 28th September 2013At
Hotel Hyatt Regency, Sahar Airport Road, Mumbai
We are pleased to inform you that Indian Drug Manufacturers’Association (IDMAand Association of Pharmaceutical Analysts (APA) have pleasure in announcing the 16th Pharmaceutical Analysts’ Convention (PAC) 2013 on Friday & Saturday, 27th & 28th September 2013, at Hotel Hyatt Regency, Sahar Airport Road, Andheri, Mumbai.
The main theme for this year is “GENERICS THE GAME CHANGER”Excellent opportunity for the Professionals working in the Pharmaceutical industry, R&D Institutions, Government Laboratories and Approved Testing Laboratories, Academicians, Machine Manufacturers, API, Excipients & Intermediates Manufacturers.
Registration fees, Exhibits, sponsorship details etc. will be informed shortly:
For more details, please contact: Ms. Prachi Rane
Sr. Manager- Publications & PR, Cell: +91 9867634383,
E-mail: [email protected]
INDIAN DRUG MANUFACTURERS’ ASSOCIATION (IDMA) 102/B, A Wing; Poonam Chambers; Worli; Mumbai 400 018, India.
Tel: +91 22-24974308 / 24944624; Fax: +91 22-24950723 email: [email protected], web: www.idma-assn.org
IDMA takes pleasure in announcing the much awaited “16th PAC-2013”
Kindly block your dates

INDIAN DRUGS 50(04) ApRIl 2013 63
IDMA Bulletin XLIV (14) 08 to 14 April 2013 39
INDIAN DRUG MANUFACTURERS’ ASSOCIATION
QUALITY EXCELLENCE AWARDS SCHEME 2013TO REINFORCE OUR STATUS OF “PHARMACY TO THE WORLD”
WE NEED TO RAISE THE BAR of
PERFORMANCE & PRACTICES.
“FROM MEETING STANDARDS to
SETTING STANDARDS”
WE INVITE OUR MEMBERS TO PARTICIPATE IN THIS UNIQUE SCHEME
MOTIVATE YOUR TECHNICAL PERSONNEL
INNOVATIVE IDEAS FOR UPGRADATIONS DESPITE CONSTRAINTS
IMBIBE LATEST INTERNATIONAL PRACTICES
ACCELERATE APPROVALS BY USFDA, MHRA,WHO…
ADD A USP TO YOUR COMPANY IN ATTRACTING PERSONNEL
QUALIFY for PLATINUM, GOLD or SILVER AWARDS.
CHALO KUCH KHAS KARAIN YES, WE NEED TO DO SOMETHING SPECIAL!
WE CAN DO IT
A confidential report on some salient improvement suggestions will be made available to MD/CEO after the visit by the experts to the participating units
DETAILS WILL BE ANNOUNCED SHORTLY
Have you renewed your membership for the Current Year 2013-2014?If not, please do so :
Kindly Contact: Tel.: 022 - 2494 4624 / 2497 4308 /Ext.: 103 / Fax: 022 - 2495 0723 E-mail: [email protected]

64 INDIAN DRUGS 50(04) ApRIl 2013
ideal For:lFood Business Operators (FBOs) / Nutra products
Manufacturers
CEOs / Directors
Medical Directors / Advisors
Regulatory personnel
Marketing & Sales personnel
l practicing Clinicians / Dieticians
l Medical Students / Nutritionists
delegate Fees:l IDMA Member: ` 10,000/-
l Non-IDMA Member: ` 12,000/-
l Academic Institute Delegate / Student: ` 4,000/-
l early Bird discounts (idma / non-idma members):
mBefore 30th April 2013: 10% discount
l Group registration benefits (cannot be combined with discounts):
Does Food Safety and Standards Act, 2006 Affect You or Your Business?
indian drug manuFacturers’ association nutraceutical suBcommittee
organizes two day seminar
on
PRODUCT REGISTRATION UNDER FSSAI: KNOWHOW & CHALLENGES
on
Friday & saturday - 10th & 11th may 2013At Mumbai Cricket Association (MCA) Club Bandra-Kurla Complex, Mumbai (India)
a golden opportunity for you to understand the Fssai act in-depth, its impact & how to successfully adapt the same!
mFor every 3 delegates registered from same organization, 1 delegate registration is complimentary.
mFor every 5 Medical Dr / Dieticians registered from same organization / institution / group private practice, 2 delegate registrations will be complimentary.
sponsorship details:l enclosures / table space: ` 30,000/- for 2 days; 2 delegates’ registration complimentary.
l tea (3 sponsors): ` 20,000/- per tea; 1 delegate’s registration complimentary.
l lunch (2 sponsors): ` 50,000/-; 3 delegates’ registration complimentary.
l sessions (1 sponsor): ` 70,000/-; 2 delegates’ registration complimentary & 5-minute
corporate CD can be played during 2 breaks of choice.
l seminar (1 sponsor): ` 1,50,000/-; 4 delegates’ registration complimentary, 5-minute
corporate CD can be played during 2 breaks of choice and standee on stage.
For more details about the seminar & registration etc., please contact:Ms Prachi Rane Cell: +91-98676 34383 e-mail: [email protected]
Mr Anup Shetty Cell: +91-98922 07797 e-mail: [email protected]
Early Bird Discounts

INDIAN DRUGS 50(04) ApRIl 2013 65
programme day 1 : pre- lunch
schedule agenda 08.30 - 09.00 Registration
09.00 - 09.05 welcome - Dr Sumedh Gaikwad, Member - Nutraceutical Subcommittee (IDMA)
09.05 - 09.15 opening remarks - mr manish doshi, President - IDMA
09.15 - 09.30inauguration Mr Y Venkateswara Rao,Head, R & D Wipro Consumer Care (Bangalore) Mr Vinay Kumar,vice President, Technical & Regulatory Affairs - Amway India Ent pvt ltd (Gurgaon)
09.30 - 10.00Keynote address: nutraceuticals - a distinct enviable category Mr Anuj Reddy,vice President, Business Dev & Strategy–Wockhardt ltd (Mumbai)
10.00 - 10.30 TEA BREAK
10.30 - 13.00
session i : chairpersons Mr Mahesh Zagade,Commissioner, FDA Maharashtra (Mumbai) Dr. Rajendra pratap Gupta,Chairman - Government Industry Dialogue & President - Disease Management Association of India (Navi Mumbai)
10.30 - 11.00advantage : pharma to nutra Mr R S Gadgil, Director - Energya Nutrition (Thane)
11.00 - 11.30consumer perception to nutraceuticals - an insight Ms priti Mohile, MD - MediaMedic Communications (Mumbai)
11.30 - 12.00worldwide glimpse of nutra regulation Dr Baidyanath Mishra, GM, Medical & Regulatory Affairs - Olive lifesciences pvt ltd (Bangalore)
12.00 - 12.45tumultuous transition From pFa to Fssai regime Mr Ganesh Kamath, Director - Vital Neutraceuticals pvt ltd (Thane)
Product registration under Fssai: KnoWHoW & cHaLLenges
Organized by nutraceutical suBcommittee oF idma
on Friday & Saturday - 10th & 11th May 2013 at Mumbai Cricket Association
Bandra -Kurla Complex; Bandra (East); Mumbai 400051
1. dr r K sanghavi, Consultant, NeuroMarketing (Healthcare) - Chairman 2. mr sandeep gupta, Group Vp, Business Develop & Strategic Initiatives (Geltec / Universal Medicare) -
vice Chairman3. ms priti mohile, Managing Director (MediaMedic Communications) - Member
4. dr sumedh gaikwad, Director, Medical Services (Themis Medicare) - Member
5. dr aziz Keshvani, Medical Director (Raptakos Brett) - Member
6. mr subramanian r vaidya, Director (Bliss GVS) / partner (Sunayan) - Member
7. mr ramesh s gadgil, Director ((Energya Nutrition) - Member
8. mr ganesh v Kamath, Director (Vital Neutraceuticals) - Member
9. mr milapsingh heero, Advisor, Marketing - (McW Healthcare) - Member
nutraceutical subcommittee :

66 INDIAN DRUGS 50(04) ApRIl 2013
12.45 - 13.00open session Clarifications
13.00 - 14.00 lUNCH BREAK
day 1 : post - lunch
14.00 - 16.15session ii : chairpersons Mr Ais Kumar, Dy Director, FSSAI - Regional Office West Zone (Mumbai) Dr S W Deshpande,Complete Legal Solution, pharmalex (Mumbai)
14.00 - 14.35product approvals: existing & new Dr priti Baijal, Head Business Development, Medical & Regulatory Affairs CHC - Boehringer Ingelheim Ind pvt ltd (Mumbai)
14.35 - 15.05product labeling & label claims Mr Sanjay Singh, Head F&D, Nutraceuticals & Herbals - plethico pharm ltd (Indore)
15.05 - 15.35Fssai : rda By utl Dr K Madhavan Nair, Scientist - National Institute of Nutrition (Hyderabad)
15.35 - 16.15product composition: nutrition & nutraceuticals Dr R K Sanghavi, Consultant, Neuro Marketing - Healthcare (Mumbai)
16.15 - 16.45 TEA BREAK
16.45 - 17.30
Clarifications - FSSAI: Vox Populi Dr R K Sanghavi,Consultant, NeuroMarketing - Healthcare (Mumbai) Mr Sandeep Gupta,Group vP, Business Develop & Strategic initiatives - Geltec pvt ltd / Universal Medicare pvt ltd (Bangalore / Mumbai) Mr Ganesh Kamath, Director - Vital Neutraceuticals (Thane) Dr priti Baijal, Head Business Development, Medical & Regulatory Affairs CHC - Boehringer Ingelheim Ind pvt ltd (Mumbai) Mr Sanjay Singh, Head F&D, Nutraceuticals & Herbals - plethico pharm ltd (Indore) Dr K Madhavan Nair, Scientist - National Institute of Nutrition (Hyderabad)
day 2 : pre - lunch
09.00 - 09.15inauguration Dr pradip Chakraborty, Director, product Approval - FSSAI (Delhi)
09.15 - 10.00Keynote address: nutraceuticals – Functioning of Fssai Dr pradip Chakraborty, Director, product Approval - FSSAI (Delhi)
10.00 - 10.45 TEA BREAK
session iii : chairpersons Dr R K Sanghavi, Chairman, Nutraceutical Subcommittee - IDMA Mr Sandeep Gupta, vice Chairman, Nutraceutical Subcommittee - IDMA (Mumbai)
10.45 - 12.45
FSSAI Act Clarifications: panelists Ms Vinod Kotwal, Director, Codex - FSSAi (Delhi) Dr pradip Chakraborty, Director, product Approval - FSSAI (Delhi) Mr S Dave, Advisor - FSSAI (Delhi) Dr Manish paliwal, Director, Regulatory Affairs - pfizer ltd (Mumbai) Dr Neeraj pant, Head, Regulatory Affairs & Medical (South Asia) - Reckitt Benckiser India ltd (Gurgaon) Dr priti Baijal, Head Business Development, Medical & Regulatory Affairs CHC - Boehringer Ingelheim Ind pvt ltd (Mumbai) Dr Anil pareekh, President, Medical, Regulatory & Clinical Research - IpCA lab ltd (Mumbai)
12.45 - 12.55summing up Dr R K Sanghavi, Chairman, Nutraceutical Subcommittee - IDMA
12.55 - 13.00vote oF thanKs Mr Subramanian R Vaidya, Member, Nutraceutical Subcommittee - IDMA
13.00 - 14.00 lUNCH BREAK

INDIAN DRUGS 50(04) ApRIl 2013 67
registration Form
To, Date: __________The Secretary Generalindian drug manufacturers’ association102/B, A Wing; poonam ChambersWorli; Mumbai 400 018 Tel. # 022 - 24974308 / 24944624Fax # 022 - 24950723E-mail: [email protected] / [email protected]
Dear Sir,
Subject: Workshop on: “product registration under Fssai: Knowhow & challenges” on 10th & 11th May 2013 at: MCA Club, BKC, Mumbai
Kindly register the name/s of the following person/s from our Company to participate in the abovementioned conference.
sr. no. name designation1.
2.
3.
4.
5.
Our cheque / DD No: _____________, dated: ___/___/_____ drawn on: _____________________ is
enclosed.
Thanking You,
Yours faithfully
Name of the Company:
Address:
Tel No: Fax No: email:
note: participation fee is neither refundable nor adjustable against future programmes. However, changes in nominations are accepted. Kindly use photocopies of this form for additional registrations. The cheque/DD to be drawn on “indian drug manufacturers’ association”. *Outstation parties to remit by DD please.

68 INDIAN DRUGS 50(04) ApRIl 2013
(Rates in Rupees per insertion)Position (Print Area Dimensions) B/W Colour
Full Page (18 cm x 23.5 cm) 5,000 7,500Half Page(19 cm x 11.5 cm) (Horizontal) 2,000 5,000
Half Page (9 cm x 23.5 cm) (Vertical) 2,000 5,000
Quarter Page (9 cm x 11.5 cm) 1,000 4,500Inside Front Cover - 10,000Inside Back Cover - 7,500Back Cover 10,000 15,000
terms and conditions
a) All payments by DD in advance only to be made in favour of indian drug manufacturers’ association, payable at Mumbai
b) 25% discount for IDMA members on display advertisements
c) Series discounts of 10% available for 12 or more insertions for Non Members (valid for 12 months from date of release)
d) please provide positives/Artwork/Typeset matter for advertisements.
e) advertisement material must reach us 7 days before the date of publication
f) positioning of the Advt. other than Cover positions will be at our discretion.
g) Only Colour Advts will be entertained on Cover positions.
CONTACT: PUBLICATIONS DIVISION. Ms. Prachi INDIAN DRUG MANUFACTURERS' ASSOCIATION
102-B, Poonam Chambers, Dr A B Road Worli, Mumbai 400 018Tel: 24944624/24974308 Extn: 103 Fax: 24950723
Email: [email protected]: www.idma-assn.org
INDIAN DRUGSPUBLISHED ON 28TH OF EVERY MONTH
ADVERTISEMENT TARIFF(With effect from 7th August, 2012)
Magazine size :21.5 cm x 27.5 cm

INDIAN DRUGS 50(04) ApRIl 2013 69

70 INDIAN DRUGS 50(04) ApRIl 2013

INDIAN DRUGS 50(04) ApRIl 2013 71
For Advertising in the Classified Columns and Series Advertisement
Discount Please Contact : Advertising Dept.
INDIAN DRUGS Tel.: 022 - 2494 4624 / 2497 4308
Ext.: 103 Fax: 022 - 2495 0723 E-mail: [email protected]

72 INDIAN DRUGS 50(04) ApRIl 2013

INDIAN DRUGS 50(04) ApRIl 2013 73

74 INDIAN DRUGS 50(04) ApRIl 2013
Boost Performance Complementary analytical methods find more and different compounds, faster
Save Time Shared MS and integrated software enable the fastest possible technology switching
Reduce Costs Single software package and aqueous solvents greatly reduce startup and operating costs
AMPLIFY YOUR
Agilent CE/MS unites electrophoretic separation power and versatile MS performance into a fully integrated single-vendor solution. Using CE/MS as orthogonal technique provides greater insight into complex chromatographic challenges.
FINDINGS
INFINITELY BETTER TECHNOLOGIESIntegrated CE/MS for orthogonal analysis is one of a wave of innovative Agilent technologies for ensuring your analytical lab stays at the forefront of separation potential.
Watch video www.agilent.com/chem/infinity-cems
© Agilent Technologies, Inc. 2013

INDIAN DRUGS 50(04) ApRIl 2013 75
IDMA Bulletin XLIII (38) 08 to 14 October 2012 38

76 INDIAN DRUGS 50(04) ApRIl 2013
RNI REGN.NO.11547/63 REGD.NO.MH/MR/WEST/98/2012-2014
published and posted on 28th of Every Month This issue posted at Mumbai patrika Channel Sorting Office on 28.04.2013




















