
Recombinant C1 inhibitor in brain ischemic injury
11
Recombinant C1 Inhibitor in Brain Ischemic Injury Raffaella Gesuete, BD, 1 Claudio Storini, PhD, 1 Alessandro Fantin, BD, 1 Matteo Stravalaci, BD, 1 Elisa R. Zanier, MD, 1 Franca Orsini, BD, 1 Helene Vietsch, PhD, 2 M. L. Maurice Mannesse, PhD, 2 Bertjan Ziere, PhD, 2 Marco Gobbi, PhD, 1 and Maria-Grazia De Simoni, PhD 1 Objective: C1 inhibitor (C1-INH) is an endogenous inhibitor of complement and kinin systems. We have explored the efficacy and the therapeutic window of the recently available human recombinant (rh) C1-INH on ischemic brain injury and investi- gated its mechanism of action in comparison with that of plasma-derived (pd) C1-INH. Methods: rhC1-INH was administered intravenously to C57Bl/6 mice undergoing transient or permanent ischemia, and its protective effects were evaluated by measuring infarct volume and neurodegeneration. The binding profiles of rhC1-INH and pdC1-INH were assessed in vitro using surface plasmon resonance. Their localization in the ischemic brain tissue was determined by immunohistochemistry and confocal analysis. The functional consequences of rhC1-INH and pdC1-INH administration on complement activation were analyzed by enzyme-linked immunosorbent assay on plasma samples. Results: rhC1-INH markedly reduced cerebral damage when administered up to 18 hours after transient ischemia and up to 6 hours after permanent ischemia, thus showing a surprisingly wide therapeutic window. In vitro rhC1-INH bound mannose- binding lectin (MBL), a key protein in the lectin complement pathway, with high affinity, whereas pdC1-INH, which has a different glycosylation pattern, did not. In the ischemic brain, rhC1-INH was confined to cerebral vessels, where it colocalized with MBL, whereas pdC1-INH diffused into the brain parenchyma. In addition, rhC1-INH was more active than pdC1-INH in inhibiting MBL-induced complement activation. Interpretation: rhC1-INH showed a surprisingly wider time window of efficacy compared with the corresponding plasmatic protein. We propose that the superiority of rhC1-INH is due to its selective binding to MBL, which emerged as a novel target for stroke treatment. Ann Neurol 2009;66:332–342 C1 inhibitor (C1-INH) is a 478 amino acid glycopro- tein well known as an inhibitor of the C1 complex of the classical pathway of complement. 1,2 C1-INH also interacts with mannose-binding lectin (MBL)-asso- ciated serine proteases (MASPs). 3 Recent data indicate functional assembly of MBL-MASPs complex 4 and specifically MASP-2 5 as a major physiological target of C1-INH, thereby efficiently inhibiting the lectin path- way of complement activation. Although less effi- ciently, C1-INH also inhibits the alternative comple- ment pathway. 4,6 Other biological activities of C1- INH, independent of complement inhibition, 7 include inhibition of the kinin system and direct interference with the recruitment and migration of circulating leu- kocytes during inflammatory response 8,9 achieved by binding to selectins, molecules mediating the cellular adhesion to activated endothelium. A wide inflammatory response is known to be acti- vated in brain ischemia/reperfusion and to participate to the pathogenesis of the ischemic damage, including complement activation and inflammatory cell recruit- ment. 10 –12 We have previously shown that plasma- derived (pd) C1-INH administered at the beginning of the ischemic period decreased the ischemic volume (up to 90%) and the consequent neurological deficits in 2 strains of mice (1 outbred, CD1 and 1 inbred, C57Bl/6) after transient ischemia. 13,14 However, pdC1-INH has a narrow therapeutic window, because no protective ef- fects can be observed when administered 1 hour after the onset of ischemia. 13 We describe here the markedly improved protective effects of the human recombinant (rh) C1-INH in transient and permanent ischemia and provide data on a novel target of C1-INH action. From the 1 Mario Negri Institute for Pharmacological Research, Mi- lan, Italy; and 2 Pharming Technologies B.V., Leiden, The Nether- lands. Address correspondence to Dr De Simoni, Head of Laboratory of Inflammation and Nervous System Diseases, Mario Negri In- stitute, via G. La Masa, 19; 20156 Milan, Italy. E-mail: [email protected] Potential conflict of interest: Helene Vietsch, M. L. Maurice Man- nesse, and Bertjan Ziere are employees of Pharming Technologies B.V. Received Nov 12, 2008, and in revised form April 9, 2009. Accepted for publication April 17, 2009. Published online in Wiley Inter- Science (www.interscience.wiley.com). DOI: 10.1002/ana.21740 332 © 2009 American Neurological Association
Transcript of Recombinant C1 inhibitor in brain ischemic injury

Recombinant C1 Inhibitor in Brain IschemicInjury
Raffaella Gesuete, BD,1 Claudio Storini, PhD,1 Alessandro Fantin, BD,1 Matteo Stravalaci, BD,1
Elisa R. Zanier, MD,1 Franca Orsini, BD,1 Helene Vietsch, PhD,2 M. L. Maurice Mannesse, PhD,2
Bertjan Ziere, PhD,2 Marco Gobbi, PhD,1 and Maria-Grazia De Simoni, PhD1
Objective: C1 inhibitor (C1-INH) is an endogenous inhibitor of complement and kinin systems. We have explored the efficacyand the therapeutic window of the recently available human recombinant (rh) C1-INH on ischemic brain injury and investi-gated its mechanism of action in comparison with that of plasma-derived (pd) C1-INH.Methods: rhC1-INH was administered intravenously to C57Bl/6 mice undergoing transient or permanent ischemia, and itsprotective effects were evaluated by measuring infarct volume and neurodegeneration. The binding profiles of rhC1-INH andpdC1-INH were assessed in vitro using surface plasmon resonance. Their localization in the ischemic brain tissue wasdetermined by immunohistochemistry and confocal analysis. The functional consequences of rhC1-INH and pdC1-INHadministration on complement activation were analyzed by enzyme-linked immunosorbent assay on plasma samples.Results: rhC1-INH markedly reduced cerebral damage when administered up to 18 hours after transient ischemia and up to6 hours after permanent ischemia, thus showing a surprisingly wide therapeutic window. In vitro rhC1-INH bound mannose-binding lectin (MBL), a key protein in the lectin complement pathway, with high affinity, whereas pdC1-INH, which has adifferent glycosylation pattern, did not. In the ischemic brain, rhC1-INH was confined to cerebral vessels, where it colocalizedwith MBL, whereas pdC1-INH diffused into the brain parenchyma. In addition, rhC1-INH was more active than pdC1-INHin inhibiting MBL-induced complement activation.Interpretation: rhC1-INH showed a surprisingly wider time window of efficacy compared with the corresponding plasmaticprotein. We propose that the superiority of rhC1-INH is due to its selective binding to MBL, which emerged as a novel targetfor stroke treatment.
Ann Neurol 2009;66:332–342
C1 inhibitor (C1-INH) is a 478 amino acid glycopro-tein well known as an inhibitor of the C1 complex ofthe classical pathway of complement.1,2 C1-INH alsointeracts with mannose-binding lectin (MBL)-asso-ciated serine proteases (MASPs).3 Recent data indicatefunctional assembly of MBL-MASPs complex4 andspecifically MASP-25 as a major physiological target ofC1-INH, thereby efficiently inhibiting the lectin path-way of complement activation. Although less effi-ciently, C1-INH also inhibits the alternative comple-ment pathway.4,6 Other biological activities of C1-INH, independent of complement inhibition,7 includeinhibition of the kinin system and direct interferencewith the recruitment and migration of circulating leu-kocytes during inflammatory response8,9 achieved bybinding to selectins, molecules mediating the cellularadhesion to activated endothelium.
A wide inflammatory response is known to be acti-vated in brain ischemia/reperfusion and to participate tothe pathogenesis of the ischemic damage, includingcomplement activation and inflammatory cell recruit-ment.10–12 We have previously shown that plasma-derived (pd) C1-INH administered at the beginning ofthe ischemic period decreased the ischemic volume (upto 90%) and the consequent neurological deficits in 2strains of mice (1 outbred, CD1 and 1 inbred, C57Bl/6)after transient ischemia.13,14 However, pdC1-INH has anarrow therapeutic window, because no protective ef-fects can be observed when administered 1 hour afterthe onset of ischemia.13
We describe here the markedly improved protectiveeffects of the human recombinant (rh) C1-INH intransient and permanent ischemia and provide data ona novel target of C1-INH action.
From the 1Mario Negri Institute for Pharmacological Research, Mi-lan, Italy; and 2Pharming Technologies B.V., Leiden, The Nether-lands.
Address correspondence to Dr De Simoni, Head of Laboratoryof Inflammation and Nervous System Diseases, Mario Negri In-stitute, via G. La Masa, 19; 20156 Milan, Italy. E-mail:[email protected]
Potential conflict of interest: Helene Vietsch, M. L. Maurice Man-nesse, and Bertjan Ziere are employees of Pharming TechnologiesB.V.
Received Nov 12, 2008, and in revised form April 9, 2009. Acceptedfor publication April 17, 2009. Published online in Wiley Inter-Science (www.interscience.wiley.com). DOI: 10.1002/ana.21740
332 © 2009 American Neurological Association

Materials and MethodsDrugspdC1-INH (molecular weight [MW] � 74kDa) was sup-plied by Baxter-Immuno, Pisa, Italy. The activity was 1U/mg, 1U being the activity of 1ml of normal plasma. rhC1-INH (MW � 67 kDa; Pharming Technologies B.V.,Leiden, The Netherlands) was produced in the milk of trans-genic rabbits (Oryctolagus cuniculus). The specific promoterdriving the expression of the hC1-INH transgene is the bo-vine �S1-casein promoter, which is specific for the secretionof caseins in the milk. rhC1-INH was then isolated by stan-dard centrifugation, filtration, and chromatography steps.The activity of rhC1-INH was 6 U/mg. Estimated purity,assessed by sodium dodecyl sulfate polyacrylamide gel elec-trophoresis and Coomassie Blue staining, was �98% forrhC1-INH and about 70% for pdC1-INH.
AnimalsMale C57Bl/6 mice (26–28g, Harlan, Calco, Italy) wereused. Procedures involving animals and their care were con-ducted in conformity with institutional guidelines that are incompliance with national and international laws and poli-cies.11,15
Physiological ParametersThirty minutes after intravenous injection of 15U of rhC1-INH or saline, mice were anesthetized by 3% isoflurane inN2O/O2 (70/30%), and blood was withdrawn from thecommon carotid artery and loaded on G3� cartridges (i-STAT Corporation, East Windsor, NJ). The hemogas anal-ysis was then performed by the i-STAT 1 analyzer (i-STATCorporation).
Transient and Permanent Focal IschemiaTransient ischemia was achieved by middle cerebral artery(MCA) occlusion (tMCAO) as previously described13–15 bymeans of a siliconized filament (7-0, Doccol Corporation,Redlands, CA) introduced into the MCA and advanced toblock it for 30 minutes. Adequacy of the vascular occlusion/reperfusion was measured by laser Doppler flowmetry (Tran-sonic BLF-21, Perimed, Jarfalla, Sweden) in the MCA terri-tory. In this model, surgery is associated with a mortality rateof 31%. For permanent MCAO (pMCAO), the left MCAwas permanently occluded by electrocoagulation.11 The mor-tality rate for this model is 13.5%. In both ischemia models,sham-operated (sham) mice received identical anesthesia andsurgery without artery occlusion. Mice were maintained at37°C during surgery.
Quantification of Infarct SizeTwenty-micrometer coronal brain sections obtained fromperfused brains were cut and stained with neutral red (Neu-tral Red Gurr Certistain, BDH, Poole, UK).13,15 Infarct vol-umes were calculated by the integration of infarct areas oneach brain slice as previously described.11,13
Fluoro-Jade LabelingFluoro-Jade (FJ) labeling was carried out on 20�m sec-tions.13,14,16 Semiquantitative scores were used to identify
different degrees of positivity: ��� � high, �� � inter-mediate, � � low, � � no positivity.
ImmunohistochemistryImmunohistochemistry was performed on 20�m sections.Primary antibodies used were: monoclonal anti–glial fibril-lary acidic protein (anti-GFAP, 1:2000, Chemicon Interna-tional, Temecula, CA), biotinylated monoclonal anti-CD45(1:800, BD Biosciences Pharmigen, San Jose, CA), antihu-man C1-INH (1:800, Dako, Glostrup, Denmark), andmonoclonal anti-MBL-A and anti-MBL-C (both 1:100, Hy-cult biotechnology, the Nederlands). CD45 and C1-INHpositivity was revealed by DAB (Vector Laboratories, Burlin-game, CA).14 Immunohistochemistry for CD45 was per-formed with nickel ammonium sulfate enhancement.13 Dou-ble immunofluorescence for GFAP and C1-INH wasperformed using the secondary antibody Alexa 488 fluoro-conjugated goat antimouse immunoglobulin (Ig)G (Molecu-lar Probes, Eugene, OR) for GFAP, whereas immunoreac-tions for C1-INH were revealed by Tyramide SignalAmplification (TSA) kit (NEL705A, Perkin Elmer). Doubleimmunofluorescence for C1-INH and MBL-A/MBL-C wasperformed using the secondary antibody Alexa 594 fluoro-conjugated goat antirabbit IgG (Invitrogen, Karlsruhe, Ger-many) for C1-INH, whereas immunoreactions for MBL-Aand MBL-C were revealed by TSA kit. Alexa 488 fluorocon-jugated Griffonia simplicifolia isolectin IB4 (Invitrogen) wasused to identify cerebral vessels.
Quantification of CD45-Positive CellsCD45-immunopositive cells were counted in cortical sections(anteroposterior (AP) coordinates: �0.70mm and �0.20mmfrom bregma). Nine fields at 20� were analyzed for eachmouse.17
Confocal AnalysisImmunofluorescent staining was acquired by an IX81 micro-scope equipped with a confocal scan unit FV500 with 3lines: Ar-Kr (488nm), He-Ne red (646nm), and He-Negreen (532nm) (Olympus, Tokyo, Japan). Colocalization wasrevealed using a scanning sequential mode to eliminate pos-sible bleed-through effect (Fluoview, Olympus).
Functional MBL/MASP-2 AssayBlood samples were collected from the vena cava in 10mMethylenediamine tetraacetic acid and 0.125% Polybrene(Sigma, St. Louis, MO). Plasma was separated by centrifu-gation for 15 min at 2,000 g at 4°C and immediately storedat �80°C. Human functional MBL/MASP-2 complexeswere assayed in 1:900 diluted samples by enzyme-linked im-munosorbent assay (ELISA) (HK 327, Hbt).
Study Design and Blinding of In Vivo StudiesMice were assigned to the following experimental groups: 1)mice receiving saline or 15U of rhC1-INH and killed 30minutes after injection, for physiological parameter measure-ment, n � 5; 2) ischemic mice receiving saline at the begin-ning of tMCAO, killed at 48 hours, serving as controls fortime window of efficacy, FJ staining, anti-CD45 immuno-histochemistry, dose-response, and comparison, n � 6; 3)
Gesuete et al: Recombinant C1-INH in Cerebral Ischemia 333

ischemic mice receiving 15U of rhC1-INH at different timepoints from tMCAO (0, 3, 6, 18, and 24 hours) and killedat 48 hours, for time window of efficacy, FJ staining, andanti-CD45 immunohistochemistry (3 hours after ischemiagroup), n � 6; 4) ischemic mice receiving 5 or 10U ofrhC1-INH 3 hours after tMCAO and killed at 48 hours, fordose-response, n � 6; 5) ischemic mice receiving 15U ofpdC1-INH 3 hours after tMCAO and killed at 48 hours,n � 6; 6) ischemic mice receiving saline at the beginning ofpMCAO and killed at 7 days, as controls for time window ofefficacy and FJ staining, n � 6; 7) ischemic mice receiving15U of rhC1-INH at different time points after pMCAO (0,3, 6, and 18 hours) and killed at 7 days, for time window ofefficacy and FJ staining, n � 6; 8) ischemic mice receiving15U of rhC1-INH or pdC1-INH at the beginning of tM-CAO and killed at 30 minutes, for anti-human C1-INH im-munohistochemistry and double immunofluorescence (C1-INH�GFAP), n � 3; 9) ischemic mice receiving 15U of
Fig 1. Effect of human recombinant (rh) C1 inhibitor (C1-INH) on infarct volume 48 hours after transient middle cerebral arteryocclusion (tMCAO). (A) Time window of efficacy: infarct volume in mice receiving saline or 15U/mouse of rhC1-INH at differenttime points from the beginning of ischemia. Data are reported as scatter dot plots and mean (bars), n � 6. (B) Dose-response andcomparison between rhC1-INH and plasma-derived (pd) C1-INH: infarct volume in mice receiving saline or 5, 10, or 15U/mouseof rhC1-INH or 75U/mouse of pdC1-INH 3 hours after ischemia. Saline and 15U rhC1-INH groups are the same as in (A).Data are reported as in (A), n � 6. (C) A representative ischemic lesion is outlined in typical brain slices of mice treated withsaline and rhC1-INH 3 hours after ischemia. (D) Ischemic area was assessed in 7 serial coronal sections in mice receiving saline or15U/mouse of rhC1-INH 3 hours after ischemia. Data are reported as mean � standard deviation, n � 6. *p � 0.05, **p �0.01, ***p � 0.001 versus saline.
Table. Physiological Parameters Measured in ArterialBlood of Mice Receiving Intravenous Injection 30Minutes Previously of 15U rhC1-INH or VehicleAlone (200 �L Saline)
Saline rhC1-INH
pH 7.42 � 0.05 7.42 � 0.04
pCO2 (mm Hg) 25.06 � 2.83 29.82 � 5.24
pO2 (mm Hg) 171 � 4.88 164.3 � 19.52
sO2 % 100 99.67 � 0.52
rhC1-INH � human recombinant C1 inhibitor; pCO2 �partial pressure of carbon dioxide; pO2 � partial pressure ofoxygen; sO2 � oxygen saturation.
334 Annals of Neurology Vol 66 No 3 September 2009

rhC1-INH at the beginning of tMCAO and killed at 30minutes for double immunofluorescence (C1-INH�MBL-Aor MBL-C), n � 3; 10) sham mice receiving saline and isch-emic mice receiving saline or 15U of rhC1-INH or pdC1-INH at the beginning of tMCAO and killed at 30 minutes,for blood collection for ELISA, n � 7.
Mice were allocated to surgery and treatment groups, withsurgery and treatment distributed equally across cages anddays. The surgery was performed by an investigator blindedto the treatment allocation. The injection solutions (pdC1-INH, rhC1-INH, or saline) were masked to the researcherwho performed the injections. Investigators who measuredinfarct size, FJ evaluation, and CD45 counts were blinded tothe treatment.
Surface Plasmon Resonance (SPR)Binding studies were carried out using the ProteOn XPR36Protein Interaction Array system (Bio-Rad Laboratories, Her-cules, CA), based on surface plasmon resonance (SPR) tech-nology. rhMBL (R&D Systems, Minneapolis, MN), rhP-selectin (R&D), and bovine serum albumin (BSA) (Sigma)were covalently immobilized onto 3 different flow cell sur-faces of the same gas liquid chromatography sensor chip(Bio-Rad), using amine-coupling chemistry, with immobili-
zation levels of about 4,000, 3,500 and 5,000 resonanceunits (RU, 1RU � 1pg protein/mm2), respectively. rhC1-INH and pdC1-INH were injected simultaneously over thedifferent proteins immobilized on the chip. The injectedanalytes flowed at a rate of 30�l/min at 25°C. The runningbuffer was 10mM Tris buffer containing 1.2mM CaCl2,150–300mM NaCl, and 0.005% Tween 20, pH 7.4. Thesensorgrams (time course of the SPR signal in RU) were nor-malized to a baseline value of 0. The signal observed on thesurfaces immobilizing MBL and rhP-selectin were correctedby subtracting the nonspecific response observed on the ref-erence surface immobilizing BSA. Parallel injections of vehi-cle alone allowed correction for binding-independent re-sponses (ie, drift effects). The resulting sensorgrams werefitted by the simplest 1:1 interaction model (Langmuirmodel, analysis software by Proteon, Columbus, OH) to ob-tain the corresponding association and dissociation rate con-stants (Kon and Koff).
StatisticsData are expressed as mean � standard deviation (SD). One-way analysis of variance (ANOVA) followed by Dunnett orTukey as post hoc tests were used for ischemic volume dataand ELISA, respectively. Two-tailed unpaired t test was used
Fig 2. Fluoro-Jade labeling 48 hours after transient middle cerebral artery occlusion (tMCAO). (A) Representative images of degen-erating neurons by Fluoro-Jade staining in the dentate gyrus of hippocampus of ischemic mice receiving saline (Sal) or 15U/mouseof human recombinant C1 inhibitor at different time points from the beginning of tMCAO. Scale bar: 50�m. (B) Semiquantita-tive evaluation of Fluoro-Jade staining in 3 different brain areas performed 48 hours after transient ischemia, n � 6. � � nopositivity, � � low positivity, �� � intermediate positivity, ��� � high positivity.
Gesuete et al: Recombinant C1-INH in Cerebral Ischemia 335

for quantification of CD45-positive cells and 2-way ANOVAfollowed by Bonferroni post hoc test for ischemic area data.
ResultsPhysiological ParametersArterial blood pH, partial pressure of carbon dioxide,partial pressure of oxygen, and oxygen saturation valueswere not affected by intravenous administration of15U of rhC1-INH (Table).
Transient Ischemia (tMCAO)The administration of rhC1-INH (15U) at the begin-ning of the ischemic period significantly decreased theischemic volume by 70% (Fig 1A). The inhibitormaintained its activity when given 3, 6, or 18 hoursafter ischemia, with ischemic volumes reduced by 75%,56%, and 41%, respectively. No protective effect wasdetected when rhC1-INH was administered 24 hoursafter ischemia. Dose-response studies indicated a signif-icant protective effect with 10U (52% decrease of isch-emic volume), but not with 5U (Fig 1B) when given 8hours after ischemia. At this time point, pdC1-INH(15U) was not effective, thus confirming previous re-sults obtained with a different protocol of ischemia.13
The protective action of rhC1-INH was evidentthroughout the lesion, at each anteroposterior levelconsidered (Fig 1D). FJ staining showed that degener-
ating cells were massively present in the striatum, cor-tex, and hippocampus in all saline-treated mice (n � 6,Fig 2). No FJ-positive cells were observed in hip-pocampus or cortex of mice injected with rhC1-INH3–6 hours after ischemia. A reduction of FJ-positiveneurons in cortex was still detected when the inhibitorwas administered 18 hours, but not 24 hours, afterischemia. At all time points considered, striatumshowed a large presence of positive cells, in both saline-and rhC1-INH–treated mice.
CD45-positive cells were present in the cortex ofmice 48 hours after ischemia. They displayed 2 mor-phologies: a leukocyte-like shape (arrow, Fig 3A) cor-responding to cells with a rounded cell body withoutbranches, and a microglia-like shape (arrowhead, Fig3A) having a small cell body and several branches. Inaddition, the leukocyte-like cells showed a higher ex-pression of CD45 compared with microglia-likecells.18,19 rhC1-INH administered 3 hours after isch-emia markedly decreased the number of leukocyte-likecells, but not microglia-like cells. No CD45-positivecells were observed in sham-operated mice and in thecontralateral hemisphere of ischemic mice (Fig 3).18,19
Permanent Ischemia (pMCAO)rhC1-INH significantly decreased the ischemic volumealso in a pMCAO model (Fig 4). Mice receiving rhC1-
Fig 3. Evaluation of inflammatory cells by anti-CD45 immunostaining. Immunohistochemistry was performed in cortex of ischemicmice receiving saline (A) or human recombinant C1 inhibitor (rhC1-INH) (B) 3 hours after transient middle cerebral artery occlu-sion (tMCAO). Forty-eight hours after ischemia, CD45-positive cells are clearly visible in saline-treated mice (A), whereas a markedreduction in these cells is apparent in rhC1-INH–treated ischemic mice (B). Note that CD45-immunopositive cells show eitherleukocyte-like (arrow in A) or microglia-like (arrowhead in A) morphology. Scale bar (A and B): 100�m. (C) Quantification ofCD45-positive cells based on their morphology. Data are expressed as mean � standard deviation, n � 6, 18 frames/mouse.**p � 0.01 versus saline.
336 Annals of Neurology Vol 66 No 3 September 2009

INH up to 6 hours after ischemia showed a significantdecrease (38% on average) of the infarct area, whereasno protective effect was detected when rhC1-INH was
administered 18 hours after ischemia. Administrationof rhC1-INH, up to 6 hours after ischemia, signifi-cantly reduced the area containing FJ-positive cells (Fig
Fig 4. Effect of human recombinant C1 inhibitor (rhC1-INH) on infarct volume 7 days after permanent middle cerebral arteryocclusion (pMCAO). (A) Time window of efficacy: infarct volume in mice receiving saline or 15U/mouse of rhC1-INH at differenttime points after ischemia. Data are reported as scatter dot plots and mean (bars), n � 6. (B) A representative ischemic lesion isoutlined in typical brain slices of mice treated with saline and rhC1-INH 3 hours after ischemia. (C) Ischemic area was assessed in7 serial coronal sections in mice receiving saline or 15U/mouse of rhC1-INH 3 hours after ischemia. Data are reported as mean �standard deviation, n � 6. *p � 0.05, **p � 0.01 versus saline
Fig 5. Fluoro-Jade labeling 7 days after permanent middle cerebral artery occlusion (pMCAO). (A) Representative images ofFluoro-Jade staining in the cortex of ischemic mice receiving saline or 15U/mouse of human recombinant C1 inhibitor at differenttime points after ischemia. The ischemic area is outlined. Scale bar: 1mm. Inserts show Fluoro-Jade–positive cells at higher magnifi-cation. (B) Semiquantitative evaluation of Fluoro-Jade staining performed in cortex of ischemic mice 7 days after permanent isch-emia, n � 6. � � no positivity, � � low positivity, �� � intermediate positivity, ��� � high positivity.
Gesuete et al: Recombinant C1-INH in Cerebral Ischemia 337
5), but not the density of degenerating neurons in thearea close to the occlusion site (Fig 5A, inserts), indi-cating that the protective action was localized in theperiphery of the damaged area.
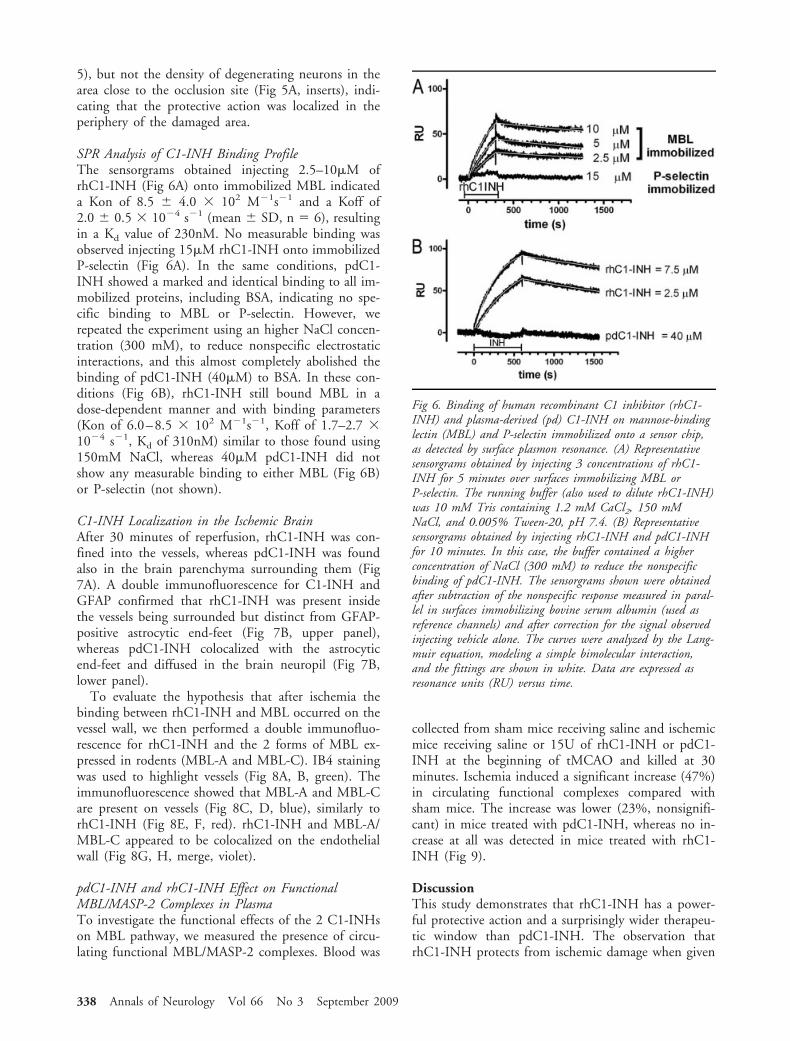
SPR Analysis of C1-INH Binding ProfileThe sensorgrams obtained injecting 2.5–10�M ofrhC1-INH (Fig 6A) onto immobilized MBL indicateda Kon of 8.5 � 4.0 � 102 M�1s�1 and a Koff of2.0 � 0.5 � 10�4 s�1 (mean � SD, n � 6), resultingin a Kd value of 230nM. No measurable binding wasobserved injecting 15�M rhC1-INH onto immobilizedP-selectin (Fig 6A). In the same conditions, pdC1-INH showed a marked and identical binding to all im-mobilized proteins, including BSA, indicating no spe-cific binding to MBL or P-selectin. However, werepeated the experiment using an higher NaCl concen-tration (300 mM), to reduce nonspecific electrostaticinteractions, and this almost completely abolished thebinding of pdC1-INH (40�M) to BSA. In these con-ditions (Fig 6B), rhC1-INH still bound MBL in adose-dependent manner and with binding parameters(Kon of 6.0–8.5 � 102 M�1s�1, Koff of 1.7–2.7 �10�4 s�1, Kd of 310nM) similar to those found using150mM NaCl, whereas 40�M pdC1-INH did notshow any measurable binding to either MBL (Fig 6B)or P-selectin (not shown).
C1-INH Localization in the Ischemic BrainAfter 30 minutes of reperfusion, rhC1-INH was con-fined into the vessels, whereas pdC1-INH was foundalso in the brain parenchyma surrounding them (Fig7A). A double immunofluorescence for C1-INH andGFAP confirmed that rhC1-INH was present insidethe vessels being surrounded but distinct from GFAP-positive astrocytic end-feet (Fig 7B, upper panel),whereas pdC1-INH colocalized with the astrocyticend-feet and diffused in the brain neuropil (Fig 7B,lower panel).
To evaluate the hypothesis that after ischemia thebinding between rhC1-INH and MBL occurred on thevessel wall, we then performed a double immunofluo-rescence for rhC1-INH and the 2 forms of MBL ex-pressed in rodents (MBL-A and MBL-C). IB4 stainingwas used to highlight vessels (Fig 8A, B, green). Theimmunofluorescence showed that MBL-A and MBL-Care present on vessels (Fig 8C, D, blue), similarly torhC1-INH (Fig 8E, F, red). rhC1-INH and MBL-A/MBL-C appeared to be colocalized on the endothelialwall (Fig 8G, H, merge, violet).
pdC1-INH and rhC1-INH Effect on FunctionalMBL/MASP-2 Complexes in PlasmaTo investigate the functional effects of the 2 C1-INHson MBL pathway, we measured the presence of circu-lating functional MBL/MASP-2 complexes. Blood was
collected from sham mice receiving saline and ischemicmice receiving saline or 15U of rhC1-INH or pdC1-INH at the beginning of tMCAO and killed at 30minutes. Ischemia induced a significant increase (47%)in circulating functional complexes compared withsham mice. The increase was lower (23%, nonsignifi-cant) in mice treated with pdC1-INH, whereas no in-crease at all was detected in mice treated with rhC1-INH (Fig 9).
DiscussionThis study demonstrates that rhC1-INH has a power-ful protective action and a surprisingly wider therapeu-tic window than pdC1-INH. The observation thatrhC1-INH protects from ischemic damage when given
Fig 6. Binding of human recombinant C1 inhibitor (rhC1-INH) and plasma-derived (pd) C1-INH on mannose-bindinglectin (MBL) and P-selectin immobilized onto a sensor chip,as detected by surface plasmon resonance. (A) Representativesensorgrams obtained by injecting 3 concentrations of rhC1-INH for 5 minutes over surfaces immobilizing MBL orP-selectin. The running buffer (also used to dilute rhC1-INH)was 10 mM Tris containing 1.2 mM CaCl2, 150 mMNaCl, and 0.005% Tween-20, pH 7.4. (B) Representativesensorgrams obtained by injecting rhC1-INH and pdC1-INHfor 10 minutes. In this case, the buffer contained a higherconcentration of NaCl (300 mM) to reduce the nonspecificbinding of pdC1-INH. The sensorgrams shown were obtainedafter subtraction of the nonspecific response measured in paral-lel in surfaces immobilizing bovine serum albumin (used asreference channels) and after correction for the signal observedinjecting vehicle alone. The curves were analyzed by the Lang-muir equation, modeling a simple bimolecular interaction,and the fittings are shown in white. Data are expressed asresonance units (RU) versus time.
338 Annals of Neurology Vol 66 No 3 September 2009

up to 18 hours after tMCAO and up to 6 hours afterpMCAO is particularly relevant because the therapeu-tic window is a critical factor in the development ofdrugs for stroke. It is noteworthy that C1-INH is al-ready used in humans for the therapy of C1-INH de-ficiency (hereditary or acquired angioedema).1,20,21 FJstaining showed that 3 cerebral areas are mainly af-fected by tMCAO: striatum; the core of the lesion; andcortex and hippocampus, including the penumbra.22
The protective effects of rhC1-INH on ischemic vol-ume were associated with a rescue of degenerating neu-rons in the ischemic penumbra. In permanent isch-emia, FJ-positive cell density was the same throughoutthe lesioned area, but a concentric reduction of the in-jured area was detected after rhC1-INH treatment, in-dicating that the action of the inhibitor was exerted inthe periphery of the lesion, similarly to what happenedin transient ischemia.
rhC1-INH decreased the infiltration of CD45-
positive peripheral inflammatory cells in the brain pa-renchyma, a phenomenon critically involved in tissuedamage, because leucocytes produce cytotoxic mole-cules that promote cell death.23,24 rhC1-INH de-creased the number of leukocyte-like but notmicroglia-like cells. Our observation that microglia hasa fainter CD45 immunostaining compared with re-cruited leukocytes is in agreement with previous studiesdemonstrating that microglial cells express lower levelsof CD45 antigen than circulating leukocytes and that,within resident microglia, CD45 expression is en-hanced only in the activated subpopulation.18,19 Thus,as expected, rhC1-INH reduces the inflammatory re-sponse in the ischemic brain. Actually, C1-INH hasbeen shown to directly affect multiple aspects of theinflammatory response, including complement activity,kinin system, phagocytosis, leukocyte rolling, andtransmigration across the endothelium.7–9 WhetherC1-INH may act also on noninflammatory targets, in-
Fig 7. Plasma-derived C1 inhibitor (pdC1-INH) and human recombinant (rh) C1-INH localization by immunohistochemistry andconfocal analysis. (A) Immunohistochemistry for rhC1-INH and pdC1-INH performed after 30 minutes of reperfusion. In cortex,the immunoreactivity indicates that the recombinant inhibitor is present inside brain capillaries, whereas the plasmatic inhibitordiffuses in the brain parenchyma. Scale bar: 50�m. (B) Double immunofluorescence for rhC1-INH or pdC1-INH with glial fibril-lary acidic protein (GFAP) performed after 30 minutes of reperfusion. The lack of colocalization between GFAP and rhC1-INHconfirms that the inhibitor is localized in the brain vessels, whereas the presence of colocalization between pdC1-INH and the astro-cytic end-feet surrounding the vessel suggests an extravasation of the inhibitor. Scale bar: 10�m.
Gesuete et al: Recombinant C1-INH in Cerebral Ischemia 339

ducing a consequent reduction of the inflammatory re-sponse, is not known presently. It is noteworthy thatthis inhibitor is effective in ischemia/reperfusion injuryof different organs and tissues (eg, skeletal muscle,liver, heart, and intestine), and its protective action hasalways been associated with its pleiotropic inhibitoryactivity against inflammation.25–27
The most important difference in the structure ofrhC1-INH and pdC1-INH is the degree of glycosyla-tion, which is lower in the former, being produced in aheterologous system.28 Importantly, these modifica-tions are located in the amino-terminal domain of theprotein, whereas the carboxyl-terminal domain, con-taining the serpin activity,1 is not affected. The re-duced level of sialylation and the presence of oligom-
annose structures results in exposure of terminalgalactose and mannose residues,28 and could thereforeaffect the binding affinity for lectins. We report thatrhC1-INH binds MBL with a relatively high affinity(230nM), whereas pdC1-INH does not show anybinding up to 40�m. Importantly, rhC1-INH andMBL colocalize in the ischemic brain, thus supportingthe existence of in vivo binding. In addition, the ad-ministration of rhC1-INH completely antagonizesischemia-induced activation of circulating functionalMBL/MASP-2 complexes, whereas pdC1-INH hasonly a small effect. It is possible that the binding ofrhC1-INH to MBL suppresses complement activationeither by preventing interaction with MASP-2 or bykeeping C1-INH at a site where it can more efficientlyinhibit MASP-2. Nevertheless, the possibility that thefunctional advantages of rhC1-INH involve its specificinteraction with MBL is supported by the evidencethat MBL-deficient mice show a reduced susceptibilityto ischemic-reperfusion injury in different organs.25–27
Restoration of ischemia-induced muscle injury inMBL-deficient mice was obtained after reconstitutionwith rhMBL,29 suggesting that activation of MBLpathway is both necessary and sufficient to develop lo-cal damage. On the other hand, the observations thatC1q-deficient mice are not protected after ischemic
Fig 9. Effect of plasma-derived C1 inhibitor (pdC1-INH) andhuman recombinant (rh) C1-INH on plasmatic functional(MBL)/(MBL)-associated serine protease(MASP)-2 levels.MBL/MASP-2 levels were determined after 30 minutes ofreperfusion by enzyme-linked immunosorbent assay performedon plasma samples obtained from saline-treated sham miceand saline-, pdC1-INH–, or rh-C1-INH–treated ischemicmice. Data are expressed as mean � standard deviation, n �7. *p � 0.05.
Fig 8. Mannose-binding lectin (MBL)-A/MBL-C and humanrecombinant C1 inhibitor (rhC1-INH) localization by confo-cal analysis. Double immunofluorescence for MBL-A/MBL-Cand rhC1-INH performed after 30 minutes of reperfusion. Incortex, the immunofluorescence indicates that MBL-A (blue,C), MBL-C (blue, D) and rhC1-INH (red, E, F) are presenton brain capillaries (IB-4, green, A, B) and colocalize (violet,G, H) suggesting an in vivo binding between rhC1-INH andMBL-A/C on the endothelial wall. Left panels: scale bar �20 �m. Right panels: scale bar � 100 �m.
340 Annals of Neurology Vol 66 No 3 September 2009

damage,13,29 and that in these mice C1-INH is stillprotective,13 indicate that the complement classicalpathway is not significantly involved.
It has been previously shown that pdC1-INH inter-acts with P-selectin with low affinity.8 We did not de-tect any binding of eith rh or pdC1-INH to P-selectin,likely because the affinity is too low to be measured bySPR in our conditions. These data suggest that inter-action with P-selectin is probably not involved in theneuroprotective properties of rhC1-INH, and that thesuperiority of rhC1-INH over plasmatic protein is dueto the high affinity of the former—but not of the lat-ter—for MBL.
The observation that pdC1-INH diffuses into thebrain parenchyma whereas rhC1-INH remains con-fined—likely because of its binding with MBL—on thebrain vessels suggests that the superior protective actionof the latter is accomplished in the vessel compartment.These data are particularly relevant because increasingevidence shows that MBL binds to endothelial cells30
and that this binding is crucial for cytotoxic dam-age.31,32 Thus, increased MBL deposition has beendemonstrated on endothelial cells immediately afterischemia.31 Moreover, MBL and C3 colocalize on vas-cular endothelium both in vitro on hypoxic/reoxygen-ated human endothelial cells and in vivo followingmyocardial ischemia/reperfusion.31,32 These data sug-gest that MBL deposition on the vascular endotheliumleads to lectin pathway activation and amplifies isch-emic damage. Our data indicate indeed that rhC1-INH may bind MBL deposed on endothelial cells earlyafter ischemia, and that this binding may directly pre-vent the lectin pathway activation, thus providing long-lasting protection against ischemic damage.
In conclusion, rhC1-INH, which possess a distinctglycosylation pattern and binding profile, is superior tothe corresponding plasmatic protein in the protectionagainst cerebral injury. This can be due to a directbinding to MBL and, consequently, a more efficientinhibition of complement lectin pathway. The surpris-ingly wide time window of efficacy of rhC1-INH rep-resents a crucial aspect for the possible therapeutic useof this drug in the treatment of stroke.
This study was partially supported by Pharming Technologies B.V.
H.V., M.L.M.M., and B.Z. of Pharming TechnologiesB.V. provided the drug and participated in discussionof the data.
References1. Caliezi C, Wuillenin WA, Zeerleder S et al. C1 esterase
inhibitor: an anti-inflammatory agent and its potential use inthe treatment of diseases other than hereditary angioedema.Pharmacol Rev 2000;52:91–112.
2. Wagenaar-Bos IG, Hack CE. Structure and function of C1-inhibitor. Immunol Allergy Clin North Am 2006;26:615–632.
3. Matsushita M, Thiel S, Jensenius JC, et al. Proteolytic activitiesof two types of mannose-binding lectin-associated serine pro-tease. J Immunol 2000;165:2637–2642.
4. Nielsen EW, Waage C, Fure H, et al. Effect of supraphysiologiclevels of C1-inhibitor on the classical, lectin and alternativepathways of complement. Mol Immunol 2007;44:1819–1826.
5. Kerr FK, Thomas AR, Wijeyewickrema LC, et al. Elucidationof the substrate specificity of the MASP-2 protease of the lectincomplement pathway and identification of the enzyme as a ma-jor physiological target of the serpin, C1-inhibitor. Mol Immu-nol 2008;45:670–677.
6. Jiang H, Wagner E, Zhang H, Frank MM. Complement 1 in-hibitor is a regulator of the alternative complement pathway. JExp Med 2001;194:1609–1616.
7. Davis AE III, Cai S, Liu D. C1 inhibitor: biologic activitiesthat are independent of protease inhibition. Immunobiology2007;212:313–323.
8. Cai S, Davis AE III. Complement regulatory protein C1 inhib-itor binds to selectins and interferes with endothelial-leukocyteadhesion. J Immunol 2003;171:4786–4791.
9. Cai S, Dole VS, Bergmeier W, et al. A direct role for C1 in-hibitor in regulation of leukocyte adhesion. J Immunol 2005;174:6462–6466.
10. D’Ambrosio AL, Pinsky DJ, Connolly ES. The role of the com-plement cascade in ischemia/reperfusion injury: implications forneuroprotection. Mol Med 2001;7:367–382.
11. Storini C, Bergamaschini L, Gesuete R, et al. Selective inhibi-tion of plasma kallikrein protects brain from reperfusion injury.J Pharmacol Exp Ther 2006;318:849–854.
12. Lo EH, Dalkara T, Moskowitz MA. Mechanisms, challengesand opportunities in stroke. Nat Rev Neurosci 2003;4:399–415.
13. De Simoni MG, Rossi E, Storini C, et al. The powerful neu-roprotective action of C1-inhibitor on brain ischemia-reperfusion injury does not require C1q. Am J Pathol 2004;164:1857–1863.
14. De Simoni MG, Storini C, Barba M, et al. Neuroprotection bycomplement (C1) inhibitor in mouse transient brain ischemia.J Cereb Blood Flow Metab 2003;23:232–239.
15. Storini C, Rossi E, Marrella V, et al. C1 inhibitor protectsagainst brain ischemia-reperfusion injury via inhibition of cellrecruitment and inflammation. Neurobiol Dis 2005;19:10–17.
16. Schmued LC, Albertson C, Sikker W. Fluoro-Jade: a novel flu-orochrome for the sensitive and reliable histochemical localiza-tion of neuronal degeneration. Brain Res 1997;751:37–46.
17. Capone C, Frigerio S, Fumagalli S, et al. Neurosphere-derivedcells exert a neuroprotective action by changing the ischemicmicroenvironment. PLoS One 2007;2:e373.
18. Sedgwick JD, Schwender S, Imrich H, et al. Isolation and di-rect characterization of resident microglial cells from the normaland inflamed central nervous system. Proc Natl Acad Sci U S A1991;88:7438–7442.
19. Stein VM, Baumgartner W, Schroder S, et al. Differential ex-pression of CD45 on canine microglial cells. J Vet Med APhysiol Pathol Clin Med 2007;54:314–320.
20. Agostoni A, Aygoren-Pursun E, Binkley KE, et al. Hereditaryand acquired angioedema: problems and progress: proceedingsof the third C1 esterase inhibitor deficiency workshop and be-yond. J Allergy Clin Immunol 2004;114:S51–S131.
21. Zuraw BL. Current and future therapy for hereditary angio-edema. Clin Immunol 2005;114:10–16.
22. Hata R, Maeda K, Hermann D, et al. Evolution of brain in-farction after transient focal cerebral ischemia in mice. J CerebBlood Flow Metab 2000;20:937–946.
Gesuete et al: Recombinant C1-INH in Cerebral Ischemia 341

23. Huang J, Upadhyay UM, Tamargo RJ. Inflammation in strokeand focal cerebral ischemia. Surg Neurol 2006;66:232–245.
24. Wen YD, Zhang HL, Qin ZH. Inflammatory mechanism inischemic neuronal injury. Neurosci Bull 2006;22:171–182.
25. Hart ML, Ceonzo KA, Shaffer LA, et al. Gastrointestinalischemia-reperfusion injury is lectin complement pathway de-pendent without involving C1q. J Immunol 2005;174:6373–6380.
26. Moller-Kristensen M, Wang W, Ruseva M, et al. Mannan-binding lectin recognizes structures on ischaemic reperfusedmouse kidneys and is implicated in tissue injury. Scand J Im-munol 2005;61:426–434.
27. Walsh MC, Bourcier T, Takahashi K, et al. Mannose-bindinglectin is a regulator of inflammation that accompanies myocar-dial ischemia and reperfusion injury. J Immunol 2005;175:541–546.
28. Koles K, van Berkel PH, Pieper FR, et al. N- and O-glycans ofrecombinant human C1 inhibitor expressed in the milk oftransgenic rabbits. Glycobiology 2004;14:51–64.
29. Chan RK, Ibrahim SI, Takahashi K, et al. The differing roles ofthe classical and mannose-binding lectin complement pathwaysin the events following skeletal muscle ischemia-reperfusion.J Immunol 2006;177:8080–8085.
30. Oroszlan M, Daha MR, Cervenak L, et al. MBL and C1q com-pete for interaction with human endothelial cells. Mol Immu-nol 2007;44:1150–1158.
31. Collard CD, Montalto MC, Reenstra WR, et al. Endothelialoxidative stress activates the lectin complement pathway: role ofcytokeratin 1. Am J Pathol 2001;159:1045–1054.
32. Collard CD, Vakeva A, Morrissey MA, et al. Complement ac-tivation after oxidative stress: role of the lectin complementpathway. Am J Pathol 2000;156:1549–1556.
342 Annals of Neurology Vol 66 No 3 September 2009




















