
Protein crystal microspectrophotometry
8
Review Protein crystal microspectrophotometry ☆ Luca Ronda a , Stefano Bruno a , Stefano Bettati a,b , Andrea Mozzarelli a,b, ⁎ a Department of Biochemistry and Molecular Biology, University of Parma, Parma, Italy b National Institute for Biostructures and Biosystems, Rome, Italy abstract article info Article history: Received 8 November 2010 Received in revised form 10 December 2010 Accepted 11 December 2010 Available online xxxx Keywords: Protein function X-ray crystallography Microspectrophotometry Chromophoric protein Single crystal microspectrophotometry has emerged as a valuable technique for monitoring molecular events that take place within protein crystals, thus tightly coupling structure to function. Absorption and fluorescence spectra, ligand binding affinities and kinetic constants can be determined, allowing i) the definition of the experimental conditions for X-ray crystallography experiments and their interpretation, ii) the assessment of whether crystal lattice forces have altered conformational equilibria, iii) the comparison with data obtained in solution. Microspectrophotometric measurements using oriented crystals and linearly polarized light are carried out usually off-line with respect to X-ray data collection and are aimed at an in- depth characterization of protein function in the crystal, leading to robust structure–function relationships. The power of this approach is highlighted by reporting a few case studies, including hemoglobins, pyridoxal 5′-phosphate-dependent enzymes and acetylcholinesterases. This article is part of a Special Issue entitled: Protein Structure and Function in the Crystalline State. © 2010 Elsevier B.V. All rights reserved. 1. Introduction Many proteins contain endogenous chromophores, such as hemes, coenzymes, and amino acid-derived compounds. These proteins have attracted the interest of biochemists and biophysicists because their structural, dynamic and functional changes are signaled by modifica- tions of spectral properties. In fact, in most of the cases, the chromophore is located in the active site, thus reporting on chemical events that take place at the core of protein function. For example, the visible absorption spectrum of heme-containing proteins depends on their redox and ligation state (Fig. 1a), the catalytic intermediates of pyridoxal 5′-phosphate-dependent enzymes are associated with unique spectral properties of the coenzyme (Fig. 1b), and the protonation of the green fluorescent protein (GFP) chromophore affects both absorption (Fig. 1c) and emission spectra. When proteins do not contain chromophores, either cofactors and substrate chromophoric analogues [1,2] or exogenous chromophoric ligands can be exploited to monitor proteins in action [3,4]. Protein function is usually investigated in solution, whereas high resolution protein structure is usually determined in the crystalline state, with the exception of NMR-derived studies. In order to directly and unambiguously couple structural and functional information, investiga- tions aimed at characterizing protein mechanisms should be carried out also in the crystalline state. Given that 50% of the volume in a protein crystal is filled with solvent [5] and protein molecules in the crystalline lattice often retain a significant conformational flexibility [6], it is not surprising that they exhibit functional properties similar to those detected in solution. This implies that most of the conformational changes associated to binding, allosteric regulation and catalysis are not prevented by lattice forces. However, there are cases where a protein in the crystal is inactive because the substrate cannot penetrate the active site due to an adjacent protein molecule that occludes its access. Moreover, proteins can exhibit altered responses to substrates and ligands when a function-associated conformational transition is restrict- ed by lattice forces [7]. The latter phenomenon can turn into an advantage for the investigation when aimed at the characterization of the spectral and functional properties of individual tertiary and quaternary con- formations, either metastable or difficult to isolate in solution. Spectroscopic methods applied to investigate protein function in solution have been adapted to investigate proteins in the crystalline state. Given the reduced size of protein crystals, usually in the order of 10–100 μm, specific instrumentations have been developed, coupling a microscope for crystal mounting and positioning with a spectropho- tometer/spectrofluorimeter. Raman spectrometers have also been implemented in some microspectrophotometric apparatus [8]. These microspectrometers are designed to operate either on-line or off-line with respect to X-ray sources for diffraction data collection. The aims of Biochimica et Biophysica Acta xxx (2011) xxx–xxx Abbreviations: GAP, glyceraldehyde-3-phosphate; GAPDH, glyceraldehyde-3- phosphate dehydrogenase; MWC, Monod, Wyman and Changeux; OAS, O-acetylserine; OASS, O-acetylserine sulfhydrylase; PEG, polyethylene glycol; PLP, pyridoxal 5′- phosphate ☆ This article is part of a Special Issue entitled: Protein Structure and Function in the Crystalline State. ⁎ Corresponding author. Department of Biochemistry and Molecular Biology, University of Parma, Via GP Usberti 23/A, 43124 Parma, Italy. Tel.: + 39 0521905138; fax: + 39 0521905151. E-mail address: [email protected] (A. Mozzarelli). BBAPAP-38564; No. of pages: 8; 4C: 1570-9639/$ – see front matter © 2010 Elsevier B.V. All rights reserved. doi:10.1016/j.bbapap.2010.12.008 Contents lists available at ScienceDirect Biochimica et Biophysica Acta journal homepage: www.elsevier.com/locate/bbapap Please cite this article as: L. Ronda, et al., Protein crystal microspectrophotometry, Biochim. Biophys. Acta (2011), doi:10.1016/j. bbapap.2010.12.008
-
Upload
independent -
Category
Documents
-
view
7 -
download
0
Transcript of Protein crystal microspectrophotometry

Biochimica et Biophysica Acta xxx (2011) xxx–xxx
BBAPAP-38564; No. of pages: 8; 4C:
Contents lists available at ScienceDirect
Biochimica et Biophysica Acta
j ourna l homepage: www.e lsev ie r.com/ locate /bbapap
Review
Protein crystal microspectrophotometry☆
Luca Ronda a, Stefano Bruno a, Stefano Bettati a,b, Andrea Mozzarelli a,b,⁎a Department of Biochemistry and Molecular Biology, University of Parma, Parma, Italyb National Institute for Biostructures and Biosystems, Rome, Italy
Abbreviations: GAP, glyceraldehyde-3-phosphatephosphate dehydrogenase; MWC, Monod, Wyman andOASS, O-acetylserine sulfhydrylase; PEG, polyethylenphosphate☆ This article is part of a Special Issue entitled: ProteinCrystalline State.⁎ Corresponding author. Department of Biochemi
University of Parma, Via GP Usberti 23/A, 43124 Parmafax: +39 0521905151.
E-mail address: [email protected] (A. Mozz
1570-9639/$ – see front matter © 2010 Elsevier B.V. Aldoi:10.1016/j.bbapap.2010.12.008
Please cite this article as: L. Ronda, etbbapap.2010.12.008
a b s t r a c t
a r t i c l e i n f oArticle history:Received 8 November 2010Received in revised form 10 December 2010Accepted 11 December 2010Available online xxxx
Keywords:Protein functionX-ray crystallographyMicrospectrophotometryChromophoric protein
Single crystal microspectrophotometry has emerged as a valuable technique for monitoring molecular eventsthat take place within protein crystals, thus tightly coupling structure to function. Absorption andfluorescence spectra, ligand binding affinities and kinetic constants can be determined, allowing i) thedefinition of the experimental conditions for X-ray crystallography experiments and their interpretation, ii)the assessment of whether crystal lattice forces have altered conformational equilibria, iii) the comparisonwith data obtained in solution. Microspectrophotometric measurements using oriented crystals and linearlypolarized light are carried out usually off-line with respect to X-ray data collection and are aimed at an in-depth characterization of protein function in the crystal, leading to robust structure–function relationships.The power of this approach is highlighted by reporting a few case studies, including hemoglobins, pyridoxal5′-phosphate-dependent enzymes and acetylcholinesterases. This article is part of a Special Issue entitled:Protein Structure and Function in the Crystalline State.
; GAPDH, glyceraldehyde-3-Changeux; OAS, O-acetylserine;e glycol; PLP, pyridoxal 5′-
Structure and Function in the
stry and Molecular Biology,, Italy. Tel.: +39 0521905138;
arelli).
l rights reserved.
al., Protein crystal microspectrophotometr
© 2010 Elsevier B.V. All rights reserved.
1. Introduction
Many proteins contain endogenous chromophores, such as hemes,coenzymes, and amino acid-derived compounds. These proteins haveattracted the interest of biochemists and biophysicists because theirstructural, dynamic and functional changes are signaled by modifica-tions of spectral properties. In fact, in most of the cases, thechromophore is located in the active site, thus reporting on chemicalevents that take place at the core of protein function. For example, thevisible absorption spectrum of heme-containing proteins depends ontheir redox and ligation state (Fig. 1a), the catalytic intermediates ofpyridoxal 5′-phosphate-dependent enzymes are associated withunique spectral properties of the coenzyme (Fig. 1b), and theprotonation of the green fluorescent protein (GFP) chromophoreaffects both absorption (Fig. 1c) and emission spectra. When proteinsdo not contain chromophores, either cofactors and substratechromophoric analogues [1,2] or exogenous chromophoric ligandscan be exploited to monitor proteins in action [3,4].
Protein function is usually investigated in solution, whereas highresolution protein structure is usually determined in the crystalline state,with the exception of NMR-derived studies. In order to directly andunambiguously couple structural and functional information, investiga-tions aimed at characterizing protein mechanisms should be carried outalso in the crystalline state. Given that 50% of the volume in a proteincrystal is filled with solvent [5] and protein molecules in the crystallinelattice often retain a significant conformational flexibility [6], it is notsurprising that they exhibit functional properties similar to thosedetected in solution. This implies that most of the conformationalchanges associated to binding, allosteric regulation and catalysis are notprevented by lattice forces. However, there are cases where a protein inthe crystal is inactive because the substrate cannot penetrate the activesite due to an adjacent protein molecule that occludes its access.Moreover, proteins can exhibit altered responses to substrates andligands when a function-associated conformational transition is restrict-edby lattice forces [7]. The latter phenomenoncan turn intoanadvantagefor the investigation when aimed at the characterization of the spectraland functional properties of individual tertiary and quaternary con-formations, either metastable or difficult to isolate in solution.
Spectroscopic methods applied to investigate protein function insolution have been adapted to investigate proteins in the crystallinestate. Given the reduced size of protein crystals, usually in the order of10–100 μm, specific instrumentations have been developed, coupling amicroscope for crystal mounting and positioning with a spectropho-tometer/spectrofluorimeter. Raman spectrometers have also beenimplemented in some microspectrophotometric apparatus [8]. Thesemicrospectrometers are designed to operate either on-line or off-linewith respect to X-ray sources for diffraction data collection. The aims of
y, Biochim. Biophys. Acta (2011), doi:10.1016/j.

2 L. Ronda et al. / Biochimica et Biophysica Acta xxx (2011) xxx–xxx
Please cite this article as: L. Ronda, et al., Protein crystal microspbbapap.2010.12.008
the two set-ups only partially overlap. The on-line instrumentmonitorsthe time evolution of chromophoric species and provides valuableinformation on the effects of radiation on metastable species duringkinetic crystallography [9,10]. The off-line instrument is aimed at adetailed characterization of the functional properties of a protein in thecrystal by determining spectra, ligand binding affinities and rate ofreactions to be compared with solution data and to be correlated withstructural data [11–15]. In the present review, we will focus on off-linemicrospectroscopy.
2. Instrumentation
Microspectrophotometerswere originally home-built by assemblinga microscope, a light source, a monochromator and a photomultiplier.Later, microspectrophotometers with excellent optics were manufac-tured by Leitz and Zeiss. Both companies have now discontinued theproduction of microspectrophotometers, presently produced by a fewsmaller companies. The main current commercial interest of thisinstrument resides in its application in forensic sciences for the analysisof fibers and varnish dyes. Indeed, most scientific police departmentscommonly use microspectrophotometers.
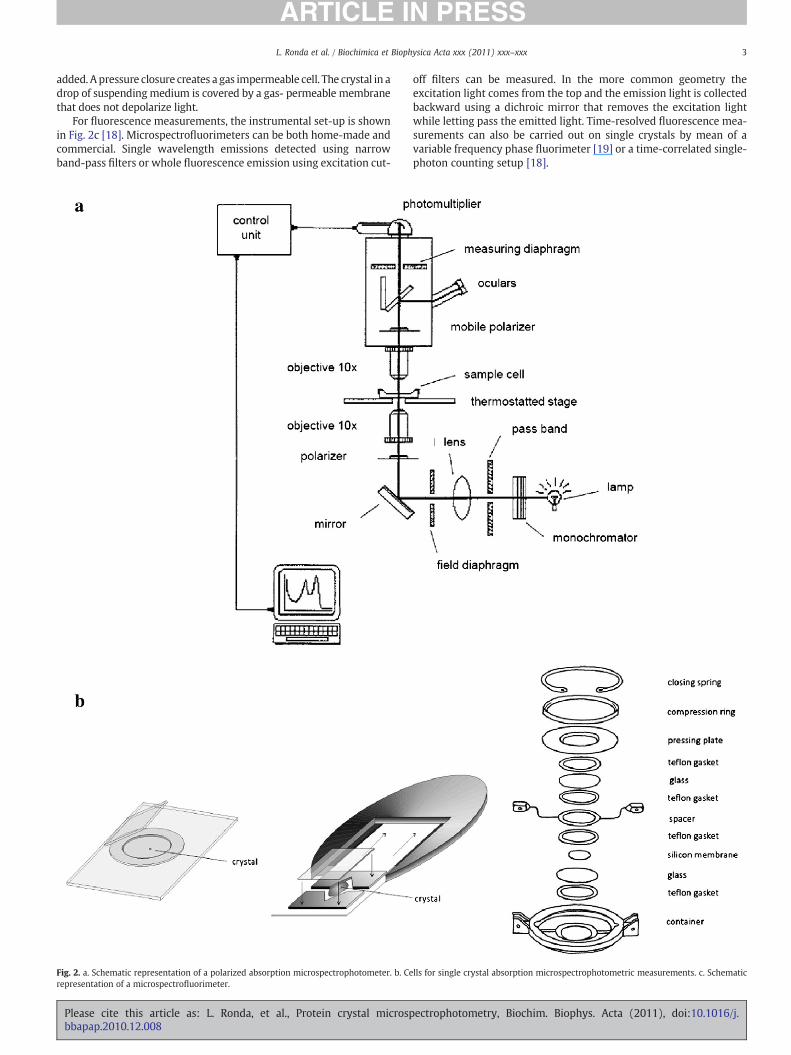
A scheme of a microspectrophotometer for UV–vis spectroscopy isreported in Fig. 2a. The light source is usually a stabilized Xenon lamp.The light beam passes through a monochromator and a slit and isfocused by a lens on amirror that deflects the light path fromhorizontalto vertical. The beam travels through an objective that acts as a lightcondenser, focusing it on the measuring field at the level where thesample is placed. The stage can rotate, move the sample in the x,y planeand be thermostatted. A polarizer can be inserted before the objective toobtain linearly polarized light, an absolute requirement for thecollection of reliable spectra on protein crystals (see below). Afterpassing through either the sample or a reference solution, usually thecrystal suspending medium, the radiation is collected by an upperobjective and masked before hitting the photomultiplier. The secondmask allows the selection of a measuring field that is fully containedwithin the crystal. The double masking avoids scattered light, increasesthe spectral quality and significantly reduces the signal background. Aremovable second polarizer (analyzer), filtering linearly polarized lightat 90° with respect to the first polarizer, can be used for properorientation of the crystal (electric vector of the polarized light parallel toone of the crystal optical axes). Crystals can be mounted in cells,designed differently according to the desired measurements. Threedifferent cells are shown in Fig. 2b. The cell shown on the left consists ofa ring of inert material attached to a quartz surface. By removing thecover slip, a crystal can be easily inserted in the small chamber and thesuspending medium can be quickly replaced. The cell shown at thecenter (Fig. 2b) is composed of a quartz window glued to a chamberwith inlet and outlet channels that allow the fast exchange of thesuspendingmedium.The crystal is properly located in ordernot tomoveduring the solution exchange. The cell on the right (Fig. 2b) is theDvorak–Stotler flow cell [16] and is used whenever spectra are to becollected on protein crystals under anaerobic conditions or at definedgas mixtures generated by a gas mixer [17]. In this cell, the crystal isplaced on a quartz window, then a spacer, a metal ring containing aninlet and outlet line, another spacer, and a second quartz window are
Fig. 1. a. Absorbance spectra of a solution containing human hemoglobin A, 100 mMHepes, 1 mM EDTA, pH 7, at 20 °C. The absorption spectra of oxyhemoglobin (dash–dot–dot line), deoxyhemoglobin (solid line) and aquomet hemoglobin (dashed line)are reported. b. Absorbance spectra of a solution containing the PLP-dependent enzymeOASS-A from Haemophilus influenzae, 100 mMHepes, pH 6.5, in the absence (solid line)and presence of 100 mM cysteine (dash–dot–dot line) or 2 mM O-acetylserine (dashedline). c. Absorbance spectra of a solution containing GFPmut2 (Aequorea victoria GFPmutants S65A, V68L, and S72A), 50 mM Tris, 50 mM MES, and 20% w/v MPD at pH 4.8(dashed line) and 8.6 (solid line).
ectrophotometry, Biochim. Biophys. Acta (2011), doi:10.1016/j.
3L. Ronda et al. / Biochimica et Biophysica Acta xxx (2011) xxx–xxx
added. A pressure closure creates a gas impermeable cell. The crystal in adrop of suspendingmedium is covered by a gas- permeable membranethat does not depolarize light.
For fluorescence measurements, the instrumental set-up is shownin Fig. 2c [18]. Microspectrofluorimeters can be both home-made andcommercial. Single wavelength emissions detected using narrowband-pass filters or whole fluorescence emission using excitation cut-
Fig. 2. a. Schematic representation of a polarized absorption microspectrophotometer. b. Cerepresentation of a microspectrofluorimeter.
Please cite this article as: L. Ronda, et al., Protein crystal microspbbapap.2010.12.008
off filters can be measured. In the more common geometry theexcitation light comes from the top and the emission light is collectedbackward using a dichroic mirror that removes the excitation lightwhile letting pass the emitted light. Time-resolved fluorescence mea-surements can also be carried out on single crystals by mean of avariable frequency phase fluorimeter [19] or a time-correlated single-photon counting setup [18].
lls for single crystal absorption microspectrophotometric measurements. c. Schematic
ectrophotometry, Biochim. Biophys. Acta (2011), doi:10.1016/j.

Fig. 2 (continued).
4 L. Ronda et al. / Biochimica et Biophysica Acta xxx (2011) xxx–xxx
For quantitative absorption measurements on protein crystals it ismandatory to use polarized light and to properly orient the crystal sothat the electric vector of the incident light is parallel to any of thecrystal optical directions, i.e. the three directions along which linearlypolarized light travels through the anisotropic crystal withoutdepolarization [20]. These directions are visually identified by rotatinga crystal on the stage using white light and cross-polarizers. Onlyabsorption intensities measured with linearly polarized light onoriented crystals are directly proportional to the chromophoreconcentration, following the Beer–Lambert law as in solution. In thecrystal, the isotropic extinction coefficient (i.e. the solution extinctioncoefficient) is equal to 1/3(εa+εb+εc)=1/3(εx+εy+εz) where a, band c are crystal optical axes and x, y and z are three perpendiculardirections. In the case of orthorhombic crystals, these directionscoincide with the crystallographic axes. In principle, spectra should berecorded along the three optical axes in order to reconstruct theisotropic spectrum, which is superimposable to the solution spectrumin the absence of lattice-induced equilibrium changes affectingspectral properties. However, due to crystal geometry, generallyonly two perpendicular directions are accessible. The use of eitherunpolarized light or partially polarized light and unoriented crystalsleads to spectra that depend on crystal size and orientation andprovide only qualitative meaningful information on intermediatespecies accumulation and reaction time courses.
Single crystal polarized absorption microspectophotometers usesingle beams, with, in some cases, a reference line that corrects forlight fluctuations. Therefore, first, two reference spectra are collectedon the crystal suspending solution near the crystal edges usinglinearly polarized light with perpendicular electric vectors. Then,spectra are collected on the oriented protein crystal and absorptionspectra are calculated. Depending on chromophore orientation withincrystals and on the direction of the transition dipole vectors of thechromophore, absorption intensity along the two directions can varysignificantly, with the limiting case where absorption along one of thedirection is negligible, indicating that the transition dipole moment ispredominantly projecting along one of the perpendicular directions.
Ligand binding titrations can be easily carried out on proteincrystals and straightforwardly compared with solution data. The only
Please cite this article as: L. Ronda, et al., Protein crystal microspbbapap.2010.12.008
precaution is to make sure that the equilibration of ligand concen-tration between the suspending solution and the inner crystalchannels is completed. It should be pointed out that crystal proteinconcentration is very high with respect to solution experiments,reaching values of the order of 5–20 mM. As a rule of thumb, a cubicprotein crystal of 100 μm contains the same number of molecules as100 μl of a 10−6 M solution. This should be taken into account whenthe saturation of the crystallized protein with a ligand in solution isrequired. Kinetic experiments in the crystalline state might be biasedby the slow diffusion of the ligand through the crystal channels.Reactions that take place on a time scale of the order of minutes arelikely not to be diffusion-limited, whereas the faster ones should becarefully controlled by evaluating the dependence of the reactionrates on the crystal thickness. A useful equation for the determinationof the maximum crystal thickness required to avoid substratediffusion limitations was previously reported [21,22].
Very fast reactions in the crystalline state can be triggered by laserlight flashes and monitored by absorption/fluorescence/Ramanspectroscopy and, concomitantly, by time-resolved crystallography.Examples are molecular events initiated by photolysis of cagedcompounds (e.g. caged-protons, caged-substrates), bond photolysis(CO and O2 in heme proteins), photocycles in bacteriorhodopsin [23]and photoreceptor proteins [24–26].
3. Case studies
Home-built microspectrophotometers were originally developed todetect the localization and concentrationofDNA inside a cell treatedwiththe Feulgendye [27]. Later, the techniquewas exploited to determine theabsorption dipole transition moment directions of cytochrome c [28],myoglobin [29] and hemoglobin crystals [30]. The first reportedcharacterization of protein function by microspectrophotometry wasthe monitoring of the acylation of chymotrypsin in the crystalline stateusing chromophoric substrate analogues [31]. Since then, manychromophore-containing protein crystals have been investigated. Thegeneral goal is to characterize protein function in the crystal, either for adirect structure-function relationship or for establishing the
ectrophotometry, Biochim. Biophys. Acta (2011), doi:10.1016/j.
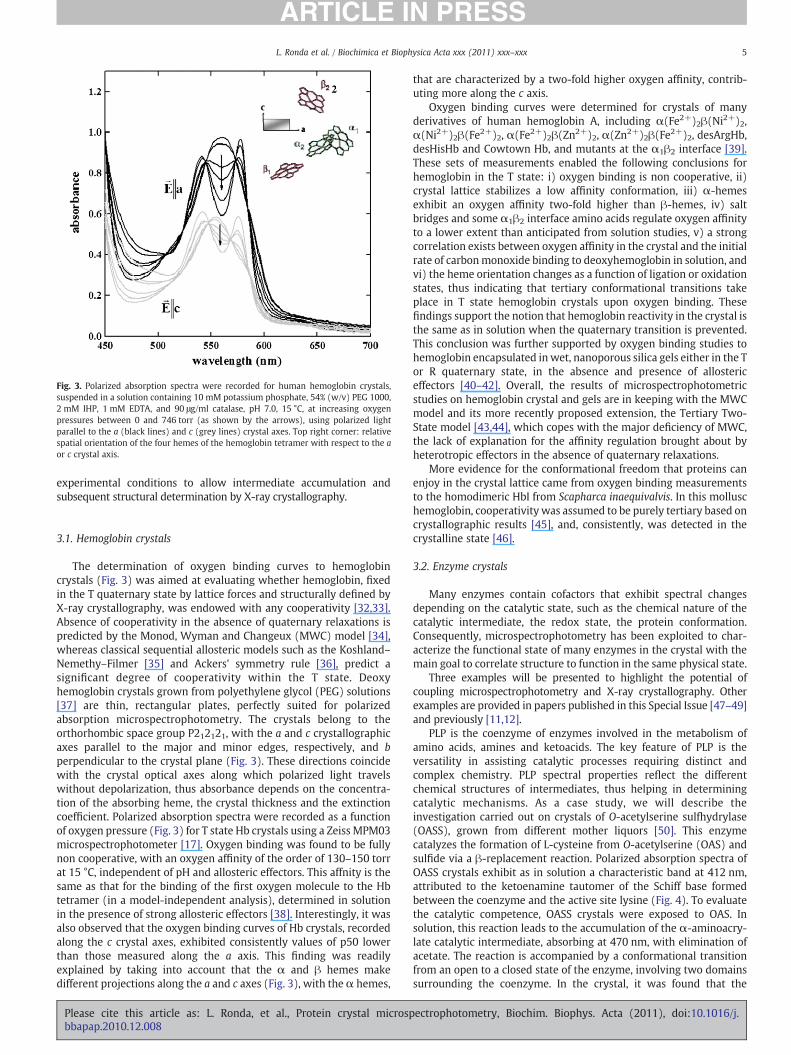
Fig. 3. Polarized absorption spectra were recorded for human hemoglobin crystals,suspended in a solution containing 10 mM potassium phosphate, 54% (w/v) PEG 1000,2 mM IHP, 1 mM EDTA, and 90 μg/ml catalase, pH 7.0, 15 °C, at increasing oxygenpressures between 0 and 746 torr (as shown by the arrows), using polarized lightparallel to the a (black lines) and c (grey lines) crystal axes. Top right corner: relativespatial orientation of the four hemes of the hemoglobin tetramer with respect to the aor c crystal axis.
5L. Ronda et al. / Biochimica et Biophysica Acta xxx (2011) xxx–xxx
experimental conditions to allow intermediate accumulation andsubsequent structural determination by X-ray crystallography.
3.1. Hemoglobin crystals
The determination of oxygen binding curves to hemoglobincrystals (Fig. 3) was aimed at evaluating whether hemoglobin, fixedin the T quaternary state by lattice forces and structurally defined byX-ray crystallography, was endowed with any cooperativity [32,33].Absence of cooperativity in the absence of quaternary relaxations ispredicted by the Monod, Wyman and Changeux (MWC) model [34],whereas classical sequential allosteric models such as the Koshland–Nemethy–Filmer [35] and Ackers' symmetry rule [36], predict asignificant degree of cooperativity within the T state. Deoxyhemoglobin crystals grown from polyethylene glycol (PEG) solutions[37] are thin, rectangular plates, perfectly suited for polarizedabsorption microspectrophotometry. The crystals belong to theorthorhombic space group P212121, with the a and c crystallographicaxes parallel to the major and minor edges, respectively, and bperpendicular to the crystal plane (Fig. 3). These directions coincidewith the crystal optical axes along which polarized light travelswithout depolarization, thus absorbance depends on the concentra-tion of the absorbing heme, the crystal thickness and the extinctioncoefficient. Polarized absorption spectra were recorded as a functionof oxygen pressure (Fig. 3) for T state Hb crystals using a Zeiss MPM03microspectrophotometer [17]. Oxygen binding was found to be fullynon cooperative, with an oxygen affinity of the order of 130–150 torrat 15 °C, independent of pH and allosteric effectors. This affnity is thesame as that for the binding of the first oxygen molecule to the Hbtetramer (in a model-independent analysis), determined in solutionin the presence of strong allosteric effectors [38]. Interestingly, it wasalso observed that the oxygen binding curves of Hb crystals, recordedalong the c crystal axes, exhibited consistently values of p50 lowerthan those measured along the a axis. This finding was readilyexplained by taking into account that the α and β hemes makedifferent projections along the a and c axes (Fig. 3), with the α hemes,
Please cite this article as: L. Ronda, et al., Protein crystal microspbbapap.2010.12.008
that are characterized by a two-fold higher oxygen affinity, contrib-uting more along the c axis.
Oxygen binding curves were determined for crystals of manyderivatives of human hemoglobin A, including α(Fe2+)2β(Ni2+)2,α(Ni2+)2β(Fe2+)2, α(Fe2+)2β(Zn2+)2, α(Zn2+)2β(Fe2+)2, desArgHb,desHisHb and Cowtown Hb, and mutants at the α1β2 interface [39].These sets of measurements enabled the following conclusions forhemoglobin in the T state: i) oxygen binding is non cooperative, ii)crystal lattice stabilizes a low affinity conformation, iii) α-hemesexhibit an oxygen affinity two-fold higher than β-hemes, iv) saltbridges and some α1β2 interface amino acids regulate oxygen affinityto a lower extent than anticipated from solution studies, v) a strongcorrelation exists between oxygen affinity in the crystal and the initialrate of carbonmonoxide binding to deoxyhemoglobin in solution, andvi) the heme orientation changes as a function of ligation or oxidationstates, thus indicating that tertiary conformational transitions takeplace in T state hemoglobin crystals upon oxygen binding. Thesefindings support the notion that hemoglobin reactivity in the crystal isthe same as in solution when the quaternary transition is prevented.This conclusion was further supported by oxygen binding studies tohemoglobin encapsulated in wet, nanoporous silica gels either in the Tor R quaternary state, in the absence and presence of allostericeffectors [40–42]. Overall, the results of microspectrophotometricstudies on hemoglobin crystal and gels are in keeping with the MWCmodel and its more recently proposed extension, the Tertiary Two-State model [43,44], which copes with the major deficiency of MWC,the lack of explanation for the affinity regulation brought about byheterotropic effectors in the absence of quaternary relaxations.
More evidence for the conformational freedom that proteins canenjoy in the crystal lattice came from oxygen binding measurementsto the homodimeric HbI from Scapharca inaequivalvis. In this molluschemoglobin, cooperativity was assumed to be purely tertiary based oncrystallographic results [45], and, consistently, was detected in thecrystalline state [46].
3.2. Enzyme crystals
Many enzymes contain cofactors that exhibit spectral changesdepending on the catalytic state, such as the chemical nature of thecatalytic intermediate, the redox state, the protein conformation.Consequently, microspectrophotometry has been exploited to char-acterize the functional state of many enzymes in the crystal with themain goal to correlate structure to function in the same physical state.
Three examples will be presented to highlight the potential ofcoupling microspectrophotometry and X-ray crystallography. Otherexamples are provided in papers published in this Special Issue [47–49]and previously [11,12].
PLP is the coenzyme of enzymes involved in the metabolism ofamino acids, amines and ketoacids. The key feature of PLP is theversatility in assisting catalytic processes requiring distinct andcomplex chemistry. PLP spectral properties reflect the differentchemical structures of intermediates, thus helping in determiningcatalytic mechanisms. As a case study, we will describe theinvestigation carried out on crystals of O-acetylserine sulfhydrylase(OASS), grown from different mother liquors [50]. This enzymecatalyzes the formation of L-cysteine from O-acetylserine (OAS) andsulfide via a β-replacement reaction. Polarized absorption spectra ofOASS crystals exhibit as in solution a characteristic band at 412 nm,attributed to the ketoenamine tautomer of the Schiff base formedbetween the coenzyme and the active site lysine (Fig. 4). To evaluatethe catalytic competence, OASS crystals were exposed to OAS. Insolution, this reaction leads to the accumulation of the α-aminoacry-late catalytic intermediate, absorbing at 470 nm, with elimination ofacetate. The reaction is accompanied by a conformational transitionfrom an open to a closed state of the enzyme, involving two domainssurrounding the coenzyme. In the crystal, it was found that the
ectrophotometry, Biochim. Biophys. Acta (2011), doi:10.1016/j.

Fig. 5. Polarized absorbance spectra of crystals of Bacillus stearothermoplilus holo-GAPDHin the absence and presence of substrates and inhibitors. The spectrumof a GAPDH crystalsuspended in a solution containing 2.7 M ammonium sulfate and 0.1 M Tris, pH 8.2 (solidline) was recorded with the polarized light perpendicular to the b axis of the crystal. Thecrystal was then soaked in a solution containing either 10 mM iodoacetic acid (dashedline) or 60 mM GAP and 1 mM sodium arsenate (dash–dot–dot line).
Fig. 4. Polarized absorbance spectra of OASS crystals grown in 30% w/v PEG (Mr 4000),100 mM Tris–HCl, 130 mM Li2SO4, and 5% DMSO, pH 7, using polarized light parallel tothe a (black lines) and b (grey lines) crystal axes, in the absence (spectra centered at412 nm) and presence (spectra centered at about 470 nm) of 18 mM OAS.
6 L. Ronda et al. / Biochimica et Biophysica Acta xxx (2011) xxx–xxx
reactivity and the associated spectral changes depend on the type ofcrystals. In fact, OASS crystals grown in 30% w/v PEG (Mr 4000),100 mM Tris–HCl, 130 mM Li2SO4, and 5% DMSO, pH 7, exhibited awell defined band at 470 nm (Fig. 4), indicating that the conforma-tional transition is fully allowed in the crystalline state. In contrast,OASS crystals grown in 20% w/v PEG (Mr 8000), 100 mM sodiumcacodylate, 200 mM calcium acetate, and 7.8% glycerol, pH 7, and usedin the crystallographic study [51], did not show any reaction. OASScrystals grown in 30% w/v PEG (Mr 4000), 100 mM Tris–HCl, and130 mM Li2SO4, pH 7 [52], cracked upon OAS addition. This exampleclearly indicates how small differences in crystallization conditionscan dramatically affect lattice geometry and forces, thus modulatingenzyme crystal reactivity and the capability to accumulate catalyticintermediates. These findings also indicate the relevance of carryingout microspectrophotometric studies prior to X-ray data collection, asthese measurements direct the selection of protein crystals thatexhibit unaltered or less altered catalytic competence with respect tosolution.
Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) is a glyco-litic, NAD+-dependent enzyme catalyzing the oxidative phosphory-lation of glyceraldehyde-3-phosphate (GAP) to 1,3-phosphoglycerate.The reaction pathway involves the formation of a metastable acyl-enzyme. The three-dimensional structure of the enzyme was one ofthe first to be determined for a protein [53], leading to the definitionof the so-called Rossman fold, i.e. the nucleotide binding domain [54].Several attempts were made exploiting substrate analogues todetermine the structure of the acyl-enzyme, a key intermediate inthe catalytic mechanism [55]. The natural acyl-enzyme structure hasbeen recently determined as a result of a microspectrophotometricstudy that helped tune the experimental conditions for the accumu-lation of the acyl-enzyme and the evaluation of its stability [56]. Theholo-enzyme GAPDH crystals exhibit a very low and broad absorptionband originated by the charge transfer interaction between thenicotinamide ring and the active site cysteine (Fig. 5), as in solution,indicative of a catalytically active enzyme. This band is abolishedwhen the enzyme reacts with iodoacetic acid, which alkylates thereactive cysteine (Fig. 5). The reaction of GAPDH crystals with GAPleads to the formation of NADH, absorbing at 340 nm, indicating thatthe hydride transfer occurs in the crystal (Fig. 5). The amount of acyl-enzyme, estimated from NADH absorption intensity, was found to be
Please cite this article as: L. Ronda, et al., Protein crystal microspbbapap.2010.12.008
dependent on GAP concentration [56]. The time-course is slowbecause the reaction is rate-limited by GAP diffusion through thecrystal water channels. Exploiting the results of the microspectro-photometric study, GAPDH crystals were suspended in a highlyconcentrated GAP solution for a defined time, then flash-cooled inliquid nitrogen before X-ray data acquisition at a synchrotron source.The crystallographic analysis revealed an occupancy for the acyl-enzyme that was in good agreement with that determined spectro-scopically. Structural data allowed the postulation of a flip-flopcatalytic mechanism involving distinct conformations of an active siteloop and the 3-phosphate of the substrate [56].
Acetylcholinesterase is the enzyme degrading acetylcholine, thussilencing the nerve-impulse. The enzyme is a target of drugs againstAlzheimer disease, pesticides and warfare gases. The enzyme structurerevealed a very narrow active site access route [57,58], suggesting theoccurrence of a transient conformational change in order to allowsubstratepenetration.With the aimofdetecting this structural transition,a kinetic crystallographic study was carried out [59], exploiting cagedarsenocholine, a precursor of an analogue of the product choline. Therelease of arsenocholine took place upon photolysis by laser light at266 nm. Crystals of acetylcholine esterasewere incubatedwith the cagedcompound and flash-cooled at 100 K in a gaseous stream of N2. Then,crystals were warmed up to room temperature and concomitantlyilluminated with laser light. Crystals were flash-cooled at different timesof illumination and maintenance at room temperature. To monitor theextent of arsenocholine release from the caged compound, absorptionspectra were recorded at 100 K on single crystals monitoring theincreasing absorption of a chromophoric moiety of the caged compoundabsorbing at 310 nm. The evolving structures allowed the detection of atransient widening of the active site entrance and the formation of apossible back-door for product release [59].
Other proteins containing chromophoric molecules have beenrecently investigated by either single crystal microspectrophotometryor microspectrofluorimetry [18]. These include green fluorescentproteins [48,60], plant light harvesting complex [24], bacteriorhodopsin[23], myoglobin [61–63], methylamine dehydrogenase [64], and copperamine oxidase [65]. The aims of these investigations range from the
ectrophotometry, Biochim. Biophys. Acta (2011), doi:10.1016/j.

7L. Ronda et al. / Biochimica et Biophysica Acta xxx (2011) xxx–xxx
assessment of the effect of synchrotron radiations on the redox state of aprotein to the comparison of spectroscopic and crystallographicinformation towards the identification of transient species.
4. Conclusions
Off-line single crystal polarized absorption microspectophotometry,and, less frequently, microspectrofluorimetry and Raman spectroscopy,have emergedas essential techniques tounveil the functional propertiesof proteinswithin the unique conditions of the crystalline lattice, whereconformational changesmight be prevented. As the structural informa-tion is usually exploited to gain insight into biological mechanisms, acareful and detailed microspectrophotometric analysis is highlyrecommendable to obtain robust and quantitative protein structure–function relationships.
Acknowledgements
This work was supported by grants from Fondazione Cariparma toA.M. andMinistero dell'Università e della Ricerca (COFIN 2007 to A.M.and COFIN 2008 to S.B.).
References
[1] S.A. Bernhard, H. Gutfreund, The optical detection of transients in trypsin- andchymotrypsin-catalyzed reactions, Proc. Natl Acad. Sci. USA 53 (1965)1238–1243.
[2] S.A. Bernhard, S.J. Lau, H. Noller, Spectrophotometric identification of acyl enzymeintermediates, Biochemistry 4 (1965) 1108–1118.
[3] M.E. Kirtley, D.E. Koshland Jr., The introduction of a “reporter” group at the activesite of glyceraldehyde-3-phosphate dehydrogenase, Biochem. Biophys. Res.Commun. 23 (1966) 810–815.
[4] R.S. Zukin, P.R. Hartig, D.E. Koshland Jr., Effect of an induced conformationalchange on the physical properties of two chemotactic receptor molecules,Biochemistry 18 (1979) 5599–5605.
[5] B.W. Matthews, Solvent content of protein crystals, J. Mol. Biol. 33 (1968)491–497.
[6] A. Schmidt, V.S. Lamzin, Internal motion in protein crystal structures, Protein Sci.19 (2010) 944–953.
[7] M. Paoli, R. Liddington, J. Tame, A. Wilkinson, G. Dodson, Crystal structure of Tstate haemoglobin with oxygen bound at all four haems, J. Mol. Biol. 256 (1996)775–792.
[8] J.E. McGeehan, D. Bourgeois, A. Royant, P. Carpentier, Raman-assisted crystallog-raphy of biomolecules at the synchrotron: instrumentation, methods andapplications. Biochim. Biophys. Acta. this issue.
[9] M.J. Ellis, S.G. Buffey, M.A. Hough, S.S. Hasnain, On-line optical and X-rayspectroscopy with crystallography: an integrated approach for determiningmetalloprotein structures in functionally well defined states, J. SynchrotronRadiat. 15 (2008) 433–439.
[10] R.L. Owen, A.R. Pearson, A. Meents, P. Boehler, V. Thominet, C. Schulze-Briese, Anew on-axis multimode spectrometer for macromolecular crystallographybeamlines of the Swiss Light Source, J. Synchrotron Radiat. 16 (2009) 173–182.
[11] A. Mozzarelli, G.L. Rossi, Protein function in the crystal, Annu. Rev. Biophys.Biomol. Struct. 25 (1996) 343–365.
[12] A.R. Pearson, A. Mozzarelli, G.L. Rossi, Microspectrophotometry for structuralenzymology, Curr. Opin. Struct. Biol. 14 (2004) 656–662.
[13] D. Bourgeois, A. Royant, Advances in kinetic protein crystallography, Curr. Opin.Struct. Biol. 15 (2005) 538–547.
[14] T. De la Mora-Rey, C.M.Wilmot, Synergy within structural biology of single crystaloptical spectroscopy and X-ray crystallography, Curr. Opin. Struct. Biol. 17 (2007)580–586.
[15] A.R. Pearson, R.L. Owen, Combining X-ray crystallography and single-crystalspectroscopy to probe enzyme mechanisms, Biochem. Soc. Transactions 37(2009) 378–381.
[16] J.A. Dvorak, W.F. Stotler, A controlled-environment culture system for highresolution light microscopy, Exp. Cell Res. 68 (1971) 144–148.
[17] L. Ronda, S. Bruno, S. Faggiano, S. Bettati, A. Mozzarelli, Oxygen binding to hemeproteins in solution, encapsulated in silica gels, and in the crystalline state,Methods Enzymol. 437 (2008) 311–328.
[18] A. Royant, P. Carpentier, J. Ohana, J. McGeehan, B. Paetzold, M. Noirclerc-Savoye, X.Vernede, V. Adam, D. Bourgeois, Advances in spectroscopic methods for biologicalcrystals. 1. Fluorescence lifetime measurements, Appl. Cryst. 40 (2007)1105–1112.
[19] M.A. Perozzo, K.B. Ward, R.B. Thompson, W.W. Ward, X-ray diffraction and time-resolved fluorescence analyses of Aequorea green fluorescent protein crystals,J. Biol. Chem. 263 (1988) 7713–7716.
[20] J. Hofrichter, W.A. Eaton, Linear dichrosim of biological chromophores, Annu. Rev.Biophys. Bioeng. 5 (1976) 511–560.
Please cite this article as: L. Ronda, et al., Protein crystal microspbbapap.2010.12.008
[21] W. Hoogenstraaten, A.E. Sluyterman, The activity of papain in the crystalline state,Biochim. Biophys. Acta 171 (1969) 284–287.
[22] B. Pioselli, S. Bettati, T.V. Demidkina, L.N. Zakomirdina, R.S. Phillips, A. Mozzarelli,Tyrosine phenol-lyase and tryptophan indole-lyase encapsulated in wet nano-porous silica gels: selective stabilization of tertiary conformations, Protein Sci. 13(2004) 913–924.
[23] R. Efremov, V.I. Gordeliy, J. Heberle, G. Bueldt, Time-resolved microspectroscopyon a single crystal of bacteriorhodopsin reveals lattice induced differences in thephotocycle kinetics, Biophys. J. 91 (2006) 1441–1451.
[24] T. Barros, A. Royant, J. Standfuss, A. Dreuw, W. Kuhlbrandt, Crystal structure ofplant light-harvesting complex shows the active, energy-transmitting state,EMBO J. 28 (2009) 298–306.
[25] A. Moglich, X. Yang, R.A. Ayers, K. Moffat, Structure and function of plantphotoreceptors. Annu. Rev. Plant Biol. (2010) 61 21–47.
[26] S. Yeremenko, I.H. van Stokkum,K.Moffat, K.J.Hellingwerf, Influence of the crystallinestate onphotoinduceddynamicsof photoactive yellowprotein studiedbyultraviolet-visible transient absorption spectroscopy, Biophys. J. 90 (2006) 4224–4235.
[27] A.W. Pollister, H. Ris, Cold Spring Harb. Symp. Quant. Biol. 12 (1947) 147.[28] W.A. Eaton, R.M. Hochstrasser, Electronic spectrum of single crystals of
ferricytochrome-c, J. Phys. Chem. 46 (1967) 2533–2539.[29] W.A. Eaton, R.M. Hochstrasser, Single-crystal spectra of ferrimyoglobin complexes
in polarized light, J. Phys. Chem. 49 (1968) 985–995.[30] M.W. Makinen, W.A. Eaton, Optically detected conformational changes in
haemoglobin single crystals, Nature 247 (1974) 62–64.[31] G.L. Rossi, S.A. Bernhard, Are the structure and function of an enzyme the same in
aqueous solution and in the wet crystal? J. Mol. Biol. 49 (1970) 85–91.[32] A. Mozzarelli, C. Rivetti, G.L. Rossi, E.R. Henry, W.A. Eaton, Crystals of haemoglobin
with the T quaternary structure bind oxygen noncooperatively with no Bohreffect, Nature 351 (1991) 416–419.
[33] C. Rivetti, A. Mozzarelli, G.L. Rossi, E.R. Henry, W.A. Eaton, Oxygen binding bysingle crystals of hemoglobin, Biochemistry 32 (1993) 2888–2906.
[34] J. Monod, J. Wyman, J.P. Changeux, On the nature of allosteric transitions: aplausible model, J. Mol. Biol. 12 (1965) 88–118.
[35] D.E. Koshland Jr., G. Nemethy, D. Filmer, Comparison of experimental binding dataand theoretical models in proteins containing subunits, Biochemistry 5 (1966)365–385.
[36] G.K. Ackers, F.R. Smith, The hemoglobin tetramer: a three-state molecular switch forcontrol of ligand affinity, Annu. Rev. Biophys. Biophys. Chem. 16 (1987) 583–609.
[37] A. Brzozowski, Z. Derewenda, E. Dodson, G. Dodson, M. Grabowski, R. Liddington,T. Skarzynski, D. Vallely, Bonding of molecular oxygen to T state humanhaemoglobin, Nature 307 (1984) 74–76.
[38] M.C. Marden, B. Bohn, J. Kister, C. Poyart, Effectors of hemoglobin — separation ofallosteric and affinity factors, Biophys. J. 57 (1990) 397–403.
[39] S. Bettati, C. Viappiani, A. Mozzarelli, Hemoglobin, an “evergreen” red protein,Biochim. Biophys. Acta 1794 (2009) 1317–1324.
[40] C. Viappiani, S. Bettati, S. Bruno, L. Ronda, S. Abbruzzetti, A. Mozzarelli, W.A. Eaton,New insights into allosteric mechanisms from trapping unstable proteinconformations in silica gels, Proc. Natl Acad. Sci. USA 101 (2004) 14414–14419.
[41] S. Bettati, A. Mozzarelli, T state hemoglobin binds oxygen noncooperatively withallosteric effects of protons, inositol hexaphosphate, and chloride, J. Biol. Chem.272 (1997) 32050–32055.
[42] S. Bruno, M. Bonaccio, S. Bettati, C. Rivetti, C. Viappiani, S. Abbruzzetti, A.Mozzarelli, High and low oxygen affinity conformations of T state hemoglobin,Protein Sci. 10 (2001) 2401–2407.
[43] W.A. Eaton, E.R. Henry, J. Hofrichter, S. Bettati, C. Viappiani, A. Mozzarelli,Evolution of allosteric models for hemoglobin, IUBMB Life 59 (2007) 586–599.
[44] E.R. Henry, S. Bettati, J. Hofrichter, W.A. Eaton, A tertiary two-state allostericmodel for hemoglobin, Biophys. Chem. 98 (2002) 149–164.
[45] E. Chiancone, D. Verzili, A. Boffi, W.E. Royer Jr., W.A. Hendrickson, A cooperativehemoglobin with directly communicating hemes. The Scapharca inaequivalvishomodimer, Biophys. Chem. 37 (1990) 287–292.
[46] A. Mozzarelli, S. Bettati, C. Rivetti, G.L. Rossi, G. Colotti, E. Chiancone, Cooperativeoxygen binding to Scapharca inaequivalvis hemoglobin in the crystal, J. Biol. Chem.271 (1996) 3627–3632.
[47] L. Ronda, N.P. Bazhulina, E.A. Morozova, S.V. Revtovich, V.O. Chekhov, A.D. Nikulin,T.V. Demidkina, A. Mozzarelli, Exploring methionine gamma-lyase structure–function relationship via microspectrophotometry and X-ray crystallography.Biochim. Biophys. Acta. this issue.
[48] S. Bettati, E. Pasqualetto, G. Lolli, B. Campanini, R. Battistutta, Structure and singlecrystal spectroscopy of greenfluorescent proteins. Biochem. Biophys. Acta. this issue.
[49] C. Micalella, S. Martignon, S. Bruno, B. Pioselli, R. Caglio, F. Valetti, E. Pessione, C.Giunta, M. Rizzi, X-ray crystallography, mass spectrometry and single crystalmicrospectrophotometry: A multidisciplinary characterization of catechol 1,2dioxygenase. Biochim. Biophys. Acta. this issue.
[50] A. Mozzarelli, S. Bettati, A.M. Pucci, P. Burkhard, P.F. Cook, Catalytic competence ofO-acetylserine sulfhydrylase in the crystal probed by polarized absorptionmicrospectrophotometry, J. Mol. Biol. 283 (1998) 135–146.
[51] P. Burkhard, G.S. Rao, E. Hohenester, K.D. Schnackerz, P.F. Cook, J.N. Jansonius,Three-dimensional structure of O-acetylserine sulfhydrylase from Salmonellatyphimurium, J. Mol. Biol. 283 (1998) 121–133.
[52] G.S. Rao, J. Mottonen, E.J. Goldsmith, P.F. Cook, Crystallization and preliminary X-raydata for the A-isozyme ofO-acetylserine sulfhydrylase from Salmonella typhimurium,J. Mol. Biol. 231 (1993) 1130–1132.
[53] M. Buehner, G.C. Ford, D. Moras, K.W. Olsen, M.G. Rossmann, Structuredetermination of crystalline lobster D-glyceraldehyde-3-phosphate dehydroge-nase, J. Mol. Biol. 82 (1974) 563–585.
ectrophotometry, Biochim. Biophys. Acta (2011), doi:10.1016/j.

8 L. Ronda et al. / Biochimica et Biophysica Acta xxx (2011) xxx–xxx
[54] M.G. Rossmann, D. Moras, K.W. Olsen, Chemical and biological evolution ofnucleotide-binding protein, Nature 250 (1974) 194–199.
[55] T. Skarzynski, P.C. Moody, A.J. Wonacott, Structure of holo-glyceraldehyde-3-phosphate dehydrogenase from Bacillus stearothermophilus at 1.8 A resolution,J. Mol. Biol. 193 (1987) 171–187.
[56] S. Moniot, S. Bruno, C. Vonrhein, C. Didierjean, S. Boschi-Muller, M. Vas, G. Bricogne,G. Branlant, A. Mozzarelli, C. Corbier, Trapping of the thioacylglyceraldehyde-3-phosphate dehydrogenase intermediate from Bacillus stearothermophilus. Directevidence for a flip-flop mechanism, J. Biol. Chem. 283 (2008) 21693–21702.
[57] J.L. Sussman, M. Harel, F. Frolow, C. Oefner, A. Goldman, L. Toker, I. Silman, Atomicstructure of acetylcholinesterase from Torpedo californica: a prototypic acetyl-choline-binding protein, Science 253 (1991) 872–879.
[58] J.P. Colletier, D. Fournier, H.M. Greenblatt, J. Stojan, J.L. Sussman, G. Zaccai, I.Silman, M. Weik, Structural insights into substrate traffic and inhibition inacetylcholinesterase, EMBO J. 25 (2006) 2746–2756.
[59] J.P. Colletier, A. Royant, A. Specht, B. Sanson, F. Nachon, P. Masson, G. Zaccai, J.L.Sussman, M. Goeldner, I. Silman, D. Bourgeois, M. Weik, Use of a ‘caged’ analogueto study the traffic of choline within acetylcholinesterase by kinetic crystallog-raphy, Acta Crystallogr. 63 (2007) 1115–1128.
[60] V. Adam, P. Carpentier, S. Violot, M. Lelimousin, C. Darnault, G.U. Nienhaus, D.Bourgeois, Structural basis of X-ray-induced transient photobleaching in a
Please cite this article as: L. Ronda, et al., Protein crystal microspbbapap.2010.12.008
photoactivatable green fluorescent protein, J. Am. Chem. Soc. 131 (2009)18063–18065.
[61] H.P. Hersleth, Y.W. Hsiao, U. Ryde, C.H. Gorbitz, K.K. Andersson, The influence of X-rays on the structural studies of peroxide-derived myoglobin intermediates,Chem. Biodivers. 5 (2008) 2067–2089.
[62] H.P. Hersleth, Y.W. Hsiao, U. Ryde, C.H. Gorbitz, K.K. Andersson, The crystal structureof peroxymyoglobin generated through cryoradiolytic reduction of myoglobincompound III during data collection, Biochem. J. 412 (2008) 257–264.
[63] H.P. Hersleth, T. Uchida, A.K. Rohr, T. Teschner, V. Schunemann, T. Kitagawa, A.X.Trautwein, C.H. Gorbitz, K.K. Andersson, Crystallographic and spectroscopicstudies of peroxide-derived myoglobin compound II and occurrence of proton-ated FeIV O, J. Biol. Chem. 282 (2007) 23372–23386.
[64] A.R. Pearson, R. Pahl, E.G. Kovaleva, V.L. Davidson, C.M. Wilmot, Tracking X-ray-derived redox changes in crystals of a methylamine dehydrogenase/amicyanincomplex using single crystal UV–Vis microspectrophotometry, J. SynchrotronRadiat. 14 (2007) 92–98.
[65] Y.C. Chiu, T. Okajima, T. Murakawa, M. Uchida, M. Taki, S. Hirota, M. Kim, H.Yamaguchi, Y. Kawano, N. Kamiya, S. Kuroda, H. Hayashi, Y. Yamamoto, K.Tanizawa, Kinetic and structural studies on the catalytic role of the asparticacid residue conserved in copper amine oxidase, Biochemistry 45 (2006)4105–4120.
ectrophotometry, Biochim. Biophys. Acta (2011), doi:10.1016/j.




















