
Posters - Karger Publishers
49
Posters P1-65 Fetal/Neonatal Endocrinology Sexual Dimorphism in Birth Size is Present from Early Gestation P.C. Hindmarsh 1 ; M.P.P. Geary* 1 ; J.C. P. Kingdom* 2 ; C.H. Rodeck* 1 ; T.J. Cole 1 ; Introduced by Peter Hindmarsh 1 University College London, London, United Kingdom; 2 Mount Sinai Hospital, Toronto, Canada Birth weight is sexually dimorphic but little is known of the differences in size at birth for other anthropometric measures and whether these differences develop in early or late gestation. Further little is known of the value of routine ultrasound assessment of fetal growth in predicting size at birth. We have stud- ied fetal growth in 1,650 low risk pregnancies by ultrasonography at 20 and 30 weeks gestation and related growth in utero to sex and size at term. In utero males had greater abdominal circumference and head size at 20 and 30 weeks gestation but not at 10 weeks (p ! 0.001). At term there were marked differ- ences in all body parameters measured (Table). Sex Weight (kg) Length (cm) Head circ. (cm) Quadriceps skinfold (mm) Arm circ. (cm) Male 3.54 50.6 35.0 7.26 10.6 Female 3.41 49.8 34.4 7.67 10.5 p ! 0.001 ! 0.001 ! 0.001 ! 0.001 ! 0.001 There were poor correlations between ultrasound measures at 20 and 30 weeks gestation and between these measures and those at birth (20 weeks with birth weight R2 12%; 30 weeks with birth weight R2 41%). Including growth rates between 20 and 30 weeks made no difference to these predictions. There is sexual dimorphism in anthropometric measures in the human fetus and these stem from about 20 weeks gestation. Males were leaner than females with larger head circumference. In a low risk population delivering at term routine ultra- sound examination is unhelpful in predicting intrauterine growth restriction. 20 Horm Res 2000;53(suppl 2):1–191 Posters
-
Upload
khangminh22 -
Category
Documents
-
view
3 -
download
0
Transcript of Posters - Karger Publishers

Posters
P1-65 Fetal/Neonatal Endocrinology
Sexual Dimorphism in Birth Size is Present from EarlyGestationP.C. Hindmarsh 1; M.P.P. Geary* 1; J.C. P. Kingdom* 2; C.H. Rodeck* 1;T.J. Cole 1; Introduced by Peter Hindmarsh1 University College London, London, United Kingdom; 2 MountSinai Hospital, Toronto, Canada
Birth weight is sexually dimorphic but little is known of the differences in sizeat birth for other anthropometric measures and whether these differencesdevelop in early or late gestation. Further little is known of the value of routineultrasound assessment of fetal growth in predicting size at birth. We have stud-ied fetal growth in 1,650 low risk pregnancies by ultrasonography at 20 and 30weeks gestation and related growth in utero to sex and size at term. In uteromales had greater abdominal circumference and head size at 20 and 30 weeksgestation but not at 10 weeks (p ! 0.001). At term there were marked differ-ences in all body parameters measured (Table).
Sex Weight(kg)
Length(cm)
Head circ.(cm)
Quadricepsskinfold (mm)
Arm circ.(cm)
Male 3.54 50.6 35.0 7.26 10.6Female 3.41 49.8 34.4 7.67 10.5p !0.001 !0.001 !0.001 !0.001 !0.001
There were poor correlations between ultrasound measures at 20 and 30 weeksgestation and between these measures and those at birth (20 weeks with birthweight R2 12%; 30 weeks with birth weight R2 41%). Including growth ratesbetween 20 and 30 weeks made no difference to these predictions. There issexual dimorphism in anthropometric measures in the human fetus and thesestem from about 20 weeks gestation. Males were leaner than females with largerhead circumference. In a low risk population delivering at term routine ultra-sound examination is unhelpful in predicting intrauterine growth restriction.
20 Horm Res 2000;53(suppl 2):1–191 Posters

39th Annual Meeting of the ESPE Horm Res 2000;53(suppl 2):1–191 21
P1-66 Fetal/Neonatal Endocrinology
Hypogonadotropic Hypogonadism (HH) DiagnosedPostnatally: Lack of Spontaneous Increase ofReproductive HormonesK.M. Main 1; N.E. Skakkebæk 1
1 Univ. Dept. of Growth and Reproduction, Rigshospitalet,Copenhagen, Denmark
Most cases of HH are not diagnosed until adolescence when clinical symptomsof the lack of pubertal increase in reproductive hormones becomes apparent.However, in healthy children, there is also a physiological rise of reproductivehormones early postnatally, the biological significance of which is yet poorlyunderstood. We here present two cases with HH which presented postnatallywith micropenis and cryptorchidism. They were diagnosed as HH due to acomplete lack of the early postnatal surge of gonadotropins and testosterone.There was no sign of any other pituitary insufficiencies. Case 1: Referreddirectly after birth for micropenis. Consanguinous Turkish parents, normalpregnancy and delivery (BW 3.6 kg). Family history of congenital heart diseaseand pes equinovarus. Penis occulta, length 20 mm, normal size scrotum,descended testicles, left hydrocele. Normal karyotype (46,XY). Case 2:Referred four months old for severe micropenis (short skin fold without palpa-ble corpus), hypoplastic scrotum, absent left and inguinal right gonade. Con-sanguinous Turkish parents, maternal history of hyposmia, irregular menstrua-tions and two miscarriages. External genitalia recorded as normal male at birthand one month old. Normal karyotype (46,XY). Hormonal results (standarddeviations (SD) are given in relation to values in 3 months old healthy boys):Serum LH and FSH were analysed by Oelfia (immunofluorometric assays),serum testosteron by RIA, inhibin B by specific ELISA. Case 1: Blood tests at2 weeks, 2 and 3 months of age showed unmeasurable serum testosterone, LH,FSH and low inhibin B (98–112 pg/ml , –2.9 to –3.1 SD). At 3 months sponta-neous FSH was 0.11 U/l (–3.2 SD). GnRH test at 4 months showed no increasein LH or FSH (0.09 and 0.08 U/l (–3.3 SD), respectively). Case 2: At 4 monthsserum testosterone and LH were unmeasurable, FSH was 0.18 U/l (–3.1 SD)and inhibin 37 pg/ml (–3.7 SD). HCG stimulation with 100 IU/kg twice weeklyfor 3 weeks induced a rise in testosterone to 8.01 nmol/ml (+1.8 SD). In conclu-sion, both patients showed a total absence of the normal postnatal surge inreproductive hormones, which strongly supports the diagnosis of HH. Thefamily history of hyposmia in case two indicates Kallmann’s syndrome. Wesuggest that repetitive basal serum measurements of gonadotropins and testos-terone during the early postnatal period may be clinically useful as a kind ofphysiological stimulation test, replacing provocation tests with GnRH andHCG.
P1-67 Fetal/Neonatal Endocrinology
Ovarian Hyporesponsiveness to Follicle-StimulatingHormone in Adolescent Girls Born Small for GestationalAgeM.V. Marcos* 1; L. Ibañez 2; N. Potau 3; F. de Zegher 4
1 Consorci Hospitalari de Terrassa, Barcelona, Spain; 2 University ofBarcelona, Barcelona, Spain; 3 Autonomous University of Barcelona,Barcelona, Spain; 4 University of Leuven, Leuven, Belgium
Girls with reduced prenatal growth are known to have, at birth, a small ovarianfraction of primordial follicles and, in adolescence, a uterus and ovaries ofsmall size. We have now examined whether reduced prenatal growth is alsofollowed by changes in the relationships among FSH, inhibin B and estradiol inadolescent girls. We studied 48 post-menarcheal girls (age 13.6 B 1.4 yr) whowere either born with an appropriate weight for gestational age (AGA; n = 33;mean weight 3.3 kg) or born small for gestational age (SGA; n = 15; meanweight 2.4 kg). Serum FSH, inhibin B and estradiol concentrations were mea-sured in the early follicular phase (range: day 5 B 3). SGA girls had, comparedto AGA girls, elevated serum FSH (7.2 B 0.7 vs 4.5 B 0.3 IU/ml; p = 0.0002),similar inhibin B (62.1 B 8.1 vs 60.7 B 6.5 pg/ml) and lower estradiol concen-trations (12.1 B 1.5 vs 21.2 B 2.4 pg/ml; p = 0.02). SGA girls thus displayed,early after menarche, a pattern that points to a hyporesponsiveness of the ovar-ian granulosa cell fraction and that is reminiscent of reproductive aging. Inconclusion, the gynecological correlates of prenatal growth restriction are here-with extended to include ovarian hyporesponsiveness to FSH in adolescence.
P1-68 Fetal/Neonatal Endocrinology
Endocrine Pancreas Development in Growth RetardedHuman FetusesF. Béringue* 1; M.C. Castellotti* 1; P. Czernichow 1, 2; M. Polak 1, 2
1 INSERM U457 and 2 Pediatric Endocrine Unit, Hôpital Robert Debré,Paris, France
The exact cause of glucose intolerance observed in adults born with intrauter-ine growth retardation (IUGR) is unknown but the possible mechanism is eith-er peripheral insulin resistance and/or an anomaly in the development of thepancreas during fetal life. To test the second hypothesis we have quantified theinsulin containing cells in deceased human fetuses with IUGR (!10th percen-tile; n = 18) (IUGRF) and in eutrophic fetuses (n = 15) (EF). The mean durationof gestation was identical in IUGRF and EF (36 gestational weeks). Paraffinembedded pancreatic tissues from fetuses of more than 32 weeks of gestationwere obtained from two foetopathology departments. Fetuses with malforma-tion, aneuploidy, presence of a genetic disease or samples with tissular lysiswere excluded. For each subject, six pancreatic sections regularly sampled with-in the organ were immunostained with an anti-insulin antibody. The total tis-sue and the insulin positive surfaces were measured by computer assisted quan-titative morphometry. The results were expressed in percentages. In order toestimate islet morphogenesis, the contribution of intra-islet and extra-islet insu-lin cells to the total insulin cell area was determined. The main finding was thatislets number per unit of surface was not different between the two groups offetuses (p = 0.52). In addition the percentage of pancreatic surface occupied byinsulin cells (%IS) was not correlated with gestational age (r = 0.07, p = 0.76 forIUGRF; r = 0.12, p = 0.67 for EF) nor with the weight (r = 0.07, p = 0.79 forIUGRF; r = 0.24, p = 0.39 for EF). Mean %IS was 2.6% in IUGRF group versus2.8% in the EF group (p = 0.66). The contribution of intra-islet insulin cells tothe total insulin area was identical in both groups (mean: 35% of the insulinsurface), which is against anomalies of morphogenesis in the IUGRF islets. Inconclusion, we showed that in EF no significant change in the pancreatic insu-lin surface was detected within the period examined. No difference wasdetected in the insulin surface nor in the islets organization between IUGRFand EF within the last two months of pregnancy, a period during which intra-uterine malnutrition becomes more apparent. These data are in accordancewith those showing an insulin secretion adapted to the degree of insulin resis-tance in the adults born with IUGR and are not in favor of a primary pancreaticorigin in the development of glucose intolerance later in life.

22 Horm Res 2000;53(suppl 2):1–191 Posters
P1-69 Fetal/Neonatal Endocrinology
Growth Hormone Profiles and Growth in Sick PretermInfants during the First Week of LifeR.J. Bolt* 1; M.M. van Weissenbruch 1; A. Cranendonk* 1;C. Popp-Snijders* 1; H.N. Lafeber* 1; H.A. Delemarre-van de Waal 1;1 University Hospital VU, Amsterdam, The Netherlands
Introduction: Little is known about the relation between the developingsomatotropic axis and growth in sick preterm infants. Objective: To studydifferences in the somatotropic axis between appropriate (AGA) and small-for-gestational age (SGA) ventilated preterm infants requiring supplementaloxygen. Methods: In 15 AGA and 7 SGA (birth weight !10th percentile cor-rected for gestational age (GA), gender and parity) preterm infants (GA 26–33weeks), blood samples were taken every 6 hours for 24 hours between day 3 to 7after birth (08.00, 14.00, 20.00, 02.00 and 08.00 hours). In all samples growthhormone (GH) and glucose were determined while IGF-1 and IGFBP-3 weredetermined in the first sample only. No differences between AGA (9 boys, 6girls) and SGA (4 boys, 3 girls) infants were found for gender, gestational age(29.0 B 1.9 vs. 29.2 B 1.5 weeks), mean fraction of inspired oxygen (0.34 B0.11 vs. 0.36 B 0.10) and nutritional intake (49 B 7 vs. 47 B 7 kcal/kg/day)during the first week of life. Values were analyzed with repeated measurementsANOVA and nonparametric tests. Results:
Table 1: GH (mU/l) and glucose values (mmol/l) (mean B SD)
08.00 14.00 20.00 02.00 08.00
GH AGA 75B49 69B46 77B45 72B41 65B34SGA* 117B58 104B58 112B73 133B60 109B43
Glucose AGA 6.7B2.0 6.1B2.8 5.7B1.6 5.4B1.3 5.9B0.9SGA 7.0B3.4 6.5B2.4 7.3B3.5 6.1B2.8 6.0B1.4
* p ! 0.05 (AGA vs. SGA)
SGA infants had significantly higher GH profiles compared to AGA infants(p = 0.027), glucose profiles were not different. Furthermore, no differenceswere found in SGA compared to AGA infants for IGF-1 (1.7 B 0.7 vs. 2.2 B0.7 nmol/l) and IGFBP-3 (0.66 B 0.19 vs. 0.75 B 0.22 mg/l). No significantcorrelations between GH, IGF-1 and IGFBP-3 were found. Mean weight gain(g/kg/day) and nutritional intake (kcal/kg/day) were not different in SGA com-pared to AGA infants during the first 2 weeks of life. Conclusion: Sick AGAand SGA preterms show comparable initial growth. However, sick SGA pre-term infants have higher GH profiles compared to sick AGA preterm infants.Either an altered GH sensitivity or an abnormal GH regulation as a conse-quence of intrauterine malnutrition might explain this finding.
P1-70 Fetal/Neonatal Endocrinology
Breast Tissue in Healthy 3-Months-Old Children:A Gender DifferenceI.M. Schmidt* 1; K.M. Main 1; A.-M. Haavisto* 1; M. Chellakooty* 1;U. Steendahl* 1; J. Toppari 2; N.E. Skakkebæk 1
1 Rigshospitalet, Copenhagen, Denmark; 2 University of Turku, Turku,Finland
Introduction: Lately, there has been considerable focus concerning theinfluence of hormones in early life on breast pathology in adulthood. It is well-known that many newborns have some degree of measurable breast tissue (BT),which is assumed to be related to maternal hormones. At 3 months of ageinfants have a peak in reproductive hormones. If BT is influenced by the endog-enous hormones at this age is unknown. Objectives: To determine the inci-dence of palpable BT in 3-months-old healthy children, and to examine genderdifference. Patients and Methods: A prospective cohort study of 301 girlsand 289 boys, aged 2.5–3.5 months was performed. BT was identified by palpa-tion and measured in mm using a small slide gauge. Diameters ! 3 mm wereconsidered unmeasurable. The methode of measuring was standardised at aworkshop attended by the four observers. Differences between groups wastested by Mann-Whitney or chi-square test. Results: The level of measure-ments varied considerably among the observers implicating methodologicaldifficulties. However, all observers found a gender difference with girls havinglarger BT than boys.
Observer A B C D
n 215 220 45 110
median (mm) girlsboys
5.54.7
5.95.4
3.73.4
9.07.6
95% confidence girlsinterval boys
1.0–8.61.0–8.0
1.0–10.31.0–9.0
1.0–7.11.0–6.8
1.0–14.91.0–14.5
girls 1 boys (p) 0.008 0.08 0.44 0.002
81% of all the children had mean BT 6 3 mm. Unmeasurable BT was morefrequent in boys than in girls (21.8% vs 15.3%) (p ! 0.05). Conclusion: Palpa-ble BT is a physiological condition in 3 months old healthy children, morepronounced in girls than in boys. We speculate that the gender difference in BTmay be related to the postnatal peak of reproductive hormones, including estra-diol, occuring at the this age.
P1-71 Fetal/Neonatal Endocrinology
Dynamic Postnatal Changes in Leptin Levels in PretermAGA and SGA Infants: A Marker of Nutrition?N. Murphy* 1; K.K. Ong* 2; A. Chong* 1; D.B. Dunger 2;T.G. Matthews* 1
1 Rotunda Hospital, Dublin, Ireland; 2 University of Cambridge,Cambridge, United Kingdom
Background: Leptin levels in cord blood correlate closely with adiposity atbirth and thus reflect level of intrauterine nutrition. Subsequently, in normalterm infants, during the period of physiological weight loss, leptin levels falldramatically, but then return to levels similar to those at birth by Day 30. Wemeasured postnatal changes in leptin levels in preterm neonates and examinedtheir relationship with postnatal nutrition. Methods: 34 AGA (mean B SDbirth weight: 1344 B 358 g) and 11 SGA (1074 B 358 g) preterm infants (gesta-tion 25–34 weeks) admitted to the Neonatal Unit, Rotunda Hospital, Dublin,had leptin levels measured by ELISA in cord blood samples and non-fastingvenous blood samples on days 1, 7, 14 and 28. All infants received intravenousB enteral feeding according to clinical guidelines, with the aim of providing120 kcal/kg in total. Results: Cord leptin levels in SGA infants (mean, SDrange: 131, 61–284 pcg/ml) were much lower than in AGA infants (759, 248–2321; p ! 0.0005). Postnatally, mean leptin levels rapidly declined to a nadirbetween Days 1 and 14 in both SGA (mean range: 41–58 pcg/ml) and AGAinfants (98–115 pcg/ml) despite the early introduction of intravenous B enteralfeeding which consistently provided on average 100 kcal/kg from Day 7 to Day28. SGA infants received more intravenous feeds (Day 14 mean B SD: 43.2 B28.8 kcal/kg) than AGA infants (22.2 B 29.5 kcal/kg, p = 0.05) but less enteralfeeds, which reflects the Unit’s cautious approach to enteral feeding in SGAinfants. However, there was no difference in total calories per kg (iv + enteral),nor in severity of illness (RDS, max FiO2, chronic lung disease, cerebral pathol-ogy or NEC) between AGA and SGA infants. By Day 28, compared to levels atbirth, SGA infants had increased in weight by 44% (mean B SD: 1544 B768 g) and leptin levels by 103% (mean, SD range: 266, 49–1437 pcg/ml). AGAinfants had increased in weight by 22.8% (1651 B 559 g); their leptin levels(273, 36–2092 pcg/ml) were higher than on Day 7 or Day 14, but were still 64%lower than levels at birth. Conclusions: Despite receiving near-target levels ofnutrition from Day 7, postnatal leptin remains at trough levels until at leastDay 14. AGA preterm infants still have much lower leptin levels on Day 28than at birth, suggesting that postnatal nutrition is reduced compared with nor-mal intrauterine conditions. In contrast, leptin levels and weights in SGAinfants were much higher at Day 28 than at birth, suggesting that their postna-tal nutrition had improved compared to in utero.

39th Annual Meeting of the ESPE Horm Res 2000;53(suppl 2):1–191 23
P1-72 Fetal/Neonatal Endocrinology
Reduced Placental Gene Expression of11ß-Hydroxysteroid Dehydrogenase Type 2 (11ß-HSD2)and 15-Hydroxyprostaglandin Dehydrogenase Type 1(PGDH) in PreeclampsiaE. Schoof* 1; M. Girstl* 1; M. Kirschbaum* 2; W. Frobenius* 3;H. Doerr 1; W. Rascher* 1; J. Doetsch* 1
1 University Children’s Hospital, Erlangen, Germany; 2 UniversityHospital for Gynecology and Obstetrics, Giessen, Germany;3 University Hospital for Gynecology and Obstetrics, Erlangen,Germany
Placental cortisol is converted to mineralocorticoid inactive cortisone by 11ß-HSD2. Cortisol, however, decreases PGDH gene expression in the placenta.PGDH is primarily responsible for the metabolism of prostaglandins in theplacenta and chorion. It was the objective of the study to examine changes of11ß-HSD2 gene expression and its possible impact on PGDH gene expressionin placental tissue of patients with preeclampsia. Patients and Methods:From 20 healthy women with normal pregnancy (36–42 weeks of gestation), 20patients with premature delivery following premature labor (18–34 weeks) and18 patients with preeclampsia (28–42 weeks) placental tissue was collected.11ß-HSD2 and PGDH mRNA expression was determined using quantitativereal-time PCR. The gene expression was analyzed in relation to the housekeep-ing genes ß-actin, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), andporphobilinogen deaminase (PBGD), and expressed in relative units (RU).Results: When comparing matched pairs, there were significantly lower pla-cental 11ß-HSD2/GAPDH mRNA levels in patients with preeclampsia than inthe controls (0.18 B 0.04 RU and 0.61 B 0.10 RU, resp., p = 0.0003). We alsofound a significant reduction in placental PGDH/GAPDH mRNA concentra-tions (0.28 B 0.15 RU and 0.50 B 0.18 RU, resp., p = 0.0003). PGDH/GAPDH and 11ß-HSD2/GAPDH mRNA levels correlated significantly (r =0.45, p = 0.004). Furthermore, 11ß-HSD2/GAPDH gene expression showed asignificant correlation to birth weight (r = 0.43, p = 0.01). All statistical resultscould be reproduced when analyzing the data in relation to ß-actin or PBGD.Conclusions: In preeclampsia, 11ß-HSD2 mRNA expression is reduced,leading to the known decrease in 11ß-HSD2 activity. In an autocrine or para-crine mechanism, the diminished conversion of cortisol may lead to reducedPGDH mRNA expression as found in the present study. Subsequent impairedmetabolism of cortisol and PGs may be involved in preterm delivery, reducedfetal growth, and local and systemic mineralocorticoid actions as hypertensionin the mother.
P1-73 Fetal/Neonatal Endocrinology
The Cortisol-Cortisone Shuttle in Preterm Infantsduring the First Week of Life: Its Relation withGestation and Body SizeR. J. Bolt* 1; M.M. van Weissenbruch 1; C.G.J. Sweep* 2;C. Popp-Snijders* 1; H.N. Lafeber* 1; H.A. Delemarre-van de Waal 1
1 University Hospital VU, Amsterdam, The Netherlands; 2 UniversityMedical Center St. Radboud, Nijmegen, The Netherlands
Introduction: It is well known that during the last trimester of pregnancycortisol is considered to be important to facilitate maturation of the fetus. It hasbeen hypothesized that a possible dysfunction of 11-beta-hydroxysteroid dehy-drogenase type 2 (11ß- HSD2) is involved in intrauterine growth retardation(IUGR) and in poor growth rates of children born with IUGR. Objective: Tostudy cortisol and cortisone blood levels, an indirect measure of the activity of11ß-HSD2, in preterm infants born before 33 weeks of gestation and the rela-tion with gestation and body size. Methods: In 22 appropriate-for-gestationalage (birth weight (BW) 1 –2 SD according to Dutch references; gestational age(GA) (mean B SD): 29.3 B 1.9 weeks; BW: 811–1985 grams) preterm infantsblood samples were obtained at 8am between day 3 to day 7 after birth for themeasurement of cortisol and cortisone by HPLC. In addition infants were sub-divided into 2 groups: group A: GA 26–30 weeks (n = 13); group B: 30–33weeks (n = 9). In 13 of 22 infants ACTH levels were obtained as well (group A: n= 8 and group B: n = 5). Results were analyzed with Pearson’s correlations andMann-Whitney-U tests. Results: Cortisol levels: 0.01–0.16 Ìmol/l, cortisone
levels: 14–140 nmol/l. No difference was found in cortisol levels between bothgroups (group A vs. group B: 0.05 B 0.03 vs. 0.08 B 0.05 Ìmol/l). Levels ofcortisone were significantly higher in group A: 99.7 B 35.2 nmol/l) comparedto group B: 64.6 B 40.1 nmol/l (p = 0.036). No significant correlation wasfound between cortisol and cortisone levels (r = 0.23; p = 0.30). In group A asignificant correlation (r = 0.65; p = 0.017) was found between levels of corti-sone and body mass index (BMI). This correlation was equal in group B butonly borderline significant (r = 0.64; p = 0.06). No significant difference in thelevel of ACTH was found between group A (6.7 B 1.8 pmol/l) and group B (7.3B 2.8 pmol/l). Conclusion: Cortisone levels in preterm infants born before 30weeks of gestation are significantly higher than in infants born between 30 and33 weeks of gestation. Levels of cortisol and ACTH, however, are comparablebetween both groups. These findings suggest an increased activity of 11ß-HSD2related to an immaturity of the adrenal cortex in extremely preterm infants.The correlation between cortisone and BMI in both groups may suggest a rela-tion between the activity of 11ß-HSD and the regulation of body size.
P1-74 Fetal/Neonatal Endocrinology
Serum Lipids in Short Children Born Small forGestational Age (SGA): Effect of GH Treatment?H.A. Wollmann* 1; G. Lang 2; M. Halder 3; M.B Ranke 3;O. Butenandt 3
1 University Children’s Hospital, Tübingen, Germany; 2 Pharmacia &Upjohn, Erlangen, Germany; 3 Haunersches Kinderspital, München,Germany
Long-term sequelae of children born SGA are associated with metabolic pro-gramming in perinatal life (Barker’s hypothesis). Besides insulin resistance,hyperlipidemia is one of the key pathophysiological features. Objectives: Tomeasure serum lipids before and during GH treatment in a cohort of SGAchildren. Patients and Methods: 99 (59m, 40f) prepubertal, short children(height SDS m: –3.2; f –3.7) born SGA (gestational age 38.0 B 3.3 weeks, birthweight 2031 B 416 g) with a mean age of 7.8 B 2.8 years with proven normalGH secretion were treated with rhGH [Genotropin®] in a supraphysiologicaldose (0.2 IU/kg/day s.c.) for 2 years within a controlled, multicenter, clinicaltrial. Cholesterol (CH) and triglycerides (TG) in serum were measured by rou-tine methods before and after 3, 6, 9, 12, 18 and 24 months of GH. In asubgroup of patients (n = 20) additionally Lp(a) was measured. Results: MeanTG conc (m 84.5; f 98.0 mg/dl) were in the normal range, not different for theboth sexes and there was no consistent effect of GH treatment. Basal CH(mean; SD; mg/dl) was normal for the group (182.7 B 36.7), however, 17% ofpatients had elevated values (237.8 B 22.7). GH treatment induced a signifi-cant decrase of CH for the whole group (169.1 B 30.1; p ! 0.05) as well as the‘elevated’ group (203.6 B 22.6; n = 17). Lp(a) was normal in 19 of the patients.Elevated concentrations in one patient increased further during GH treatment.Conclusion: A significant percentage of short children born SGA exhibit ele-vated basal cholesterol concentrations. GH treatment seems to have a benefi-cial effect on cholesterol, especially in those patients with primarily elevatedconcentrations. In the light of the proven influence of GH on adipose tissue andthe controversial data of GH treatment effects on plasma lipids, long-term,controlled trials are required for this high-risk population.

24 Horm Res 2000;53(suppl 2):1–191 Posters
P2-75 Fetal/Neonatal Endocrinology
Prospective Study of Placental Growth Hormone in 456Normal Pregnancies; Higher Levels in Women BearingFemale FoetusesM. Chellakooty* 1; L. Skibsted* 1; A. Juul 1; J.H. Petersen* 2;K.M. Main 1; A.M. Andersson* 1; S. Skouby* 1; N.E. Skakkebæk 1
1 Copenhagen University Hospital, Copenhagen, Denmark;2 University of Copenhagen, Copenhagen, Denmark
Introduction: During normal human pregnancy, a continuous placentalsecretion of a specific Growth Hormone (GH) variant suppresses the pulsatilepituitary GH secretion from approximately 20th week of gestation until term.This GH variant is only measurable in the maternal, not in the foetal, circula-tion and the physiological role and regulation of placental GH (pGH) is stillpoorly understood. Objective: To establish normal ranges for pGH in healthypregnant woman and to evaluate determinants of pGH. Subjects and Meth-ods: Serum concentrations of pGH at 18th and 28th week of gestation weremeasured using an immunoradiometric assay based on monoclonal antibodies(Biocode®, Liege, Belgium) in healthy pregnant women from a larger prospec-tive cohort study in which foetal ultrasonography, maternal factors and birthhistory records were registered. We selected a subcohort of 456 healthy preg-nant women with no pregnancy complications and normal blood pressure(! 140/90 mmHg) who delivered children with normal birth weight at 37–42weeks of gestation. Results: Placental GH levels were measurable in all preg-nant women at 18th week of gestation 4.60 (1.51) ng/ml (mean (SD)) andincreased significantly at 28th week of gestation to 9.74 (4.42) ng/ml (p !0.0001). The increase in pGH (¢pGH) was dependent on the gender of thefoetus; women carrying female foetuses having significantly higher changes inpGH 5.65 (3.84) ng/ml compared to male foetuses 4.63 (3.94) ng/ml, p !0.0001. In spite of these findings, estimated foetal size at 28 th week was signif-icantly higher in male foetuses 1271 (167) grams as compared to female foe-tuses 1221 (166) grams, p ! 0.002. Conclusion: In conclusion, we have foundthat 1) women carrying female foetuses had significantly higher levels of pla-cental GH in second trimester compared to women with male foetuses despitemale foetuses being significantly larger, and that 2) placental GH was measur-able in all pregnant woman already at 18th week of gestation. It remains to beelucidated if the gender related difference in hormone levels is of significancefor foetal growth and/or for the general well-being of the mother during preg-nancy.
P2-76 Fetal/Neonatal Endocrinology
Parathyroid Hormone and 1,25(OH)2 Vitamin D in Smallfor Gestational Age InfantsJ. Bel 1; A. Natal 1; W. Coroleu 1; M.L. Granada* 2; Introduced byJ.M. CuatrecasasDepartments of 1 Paediatrics and 2 Chemical Biochemistry, GermansTrias i Pujol Hospital, Autonomous University of Barcelona, Spain
Small for gestational age infants (SGA) have a deficit in bone mineral contentand lower serum phosphate (P) values from birth compared to appropriate forgestational age infants (AGA). A reduced placental phosphate transfer could bethe cause. Objective: To investigate the implication of parathyroid hormone(PTH) and 1,25(OH)2 vitamin D in the calcium-phosphate differences ob-served between SGA and AGA newborns. Patients and Methods: We stud-ied 20 AGA (gestational age: 34.0 B 1.8 weeks, birth weight: 2015 B 277 g) and28 SGA (37.0 B 1.5 weeks, 1903 B 348 g) admitted to the Neonatal Unit.Calcium (Ca) and P serum levels were measured in cord blood and at days 1and 3 of life. PTH and 1,25(OH)2 vitamin D were measured in cord blood andat day 3 of life. Results: The serum cord levels of Ca, P, PTH and 1,25(OH)2vitamin D were not different between both groups. At day 1, P values werelower (1.97 B 0.50 vs 2.45 B 0.12 mmol/l; p ! 0.01) and Ca values were higher(2.30 B 0.23 vs 2.15 B 0.22 mmol/l; p ! 0.05) in SGA group compared to AGAgroup. Ca and P levels did not correlate with gestational age. There were nodifferences in Ca, P and 1,25 (OH)2 vitamin D serum concentrations at day 3between both groups but PTH was lower in SGA infants (67.24 B 37.34 vs103.09 B 68.06 pg/ml; p ! 0.05). Conclusions: 1) PTH and 1,25(OH)2 vita-min D are not implicated in the placental mineral transfer deficit in SGA; 2)lower PTH values at day 3 may be a consequence of higher Ca values in SGAduring the first days of life.
P2-77 Fetal/Neonatal Endocrinology
Thyroid Ultrasound in Healthy Term InfantsR. Perry*; A. Hollman*; A. Wood*; M. Donaldson1 Royal Hospital for Sick Children, Glasgow, United Kingdom
Thyroid ultrasound assessment in babies with suspected congenital hypothy-roidism is non-invasive, can be performed on sick infants in the newborn nurs-ery and may be more informative than isotope scanning in babies who havereceived iodine for antiseptic procedures. Objective: To obtain normativedata in healthy newborns. Patients and Methods: 100 infants (51F:49M)gestation 37–42 weeks and birth weight 10–90th centile were scanned using alinear probe during the first week of life. With baby supine and neck extended,transverse and sagittal sections of each lobe were performed. Maximum length,transverse diameter (breadth) and anteroposterior diameter (depth) and vol-ume (length ! breadth ! /6) were calculated for each lobe. Results: Thethyroid gland was identified in all babies. The data were normally distributedwith no sex difference. Measurement of R and L lobes were virtually identicalfor the group as a whole, although individual babies showed considerable dis-crepancy (up to B 0.5 ml) between lobes.
Median SD Range
Single lobe length (cm) 1.93 0.24 0.9–2.5breadth (cm) 0.88 0.16 0.5–2.0depth (cm) 0.96 0.17 0.6–1.4volume (ml) 0.80 0.24 0.3–1.7
Volume of both lobes (ml) 1.62 0.40 0.7–3.3
Conclusion: Thyroid scanning is feasible and both lobes should be measured.These data provide a reference against which normal, hypoplastic and goitrousthyroid glands can be identified.
P2-78 Fetal/Neonatal Endocrinology
Socioeconomic Factors and Small for Gestational AgeInfants in LithuaniaR. Verkauskiene* 1; J. Kasparaviciene* 2; D. Mykolaityte* 2;D. Lashiene* 3; Introduced by K. Albertsson-Wikland1 Department of Pediatric Endocrinology, 2 Department of Pediatrics,and 3 Institute of Endocrinology, Kaunas University of Medicine,Kaunas, Lithuania
Children born small for gestational age form a very heterogeneous group. Inmore than one third of cases the intrauterine growth retardation cannot beexplained. Objective: To assess the association between some lifestile, socio-economic factors and infants small size at birth. Material and Methods: Thedata from the collaborative Swedish-Lithuanian project on postnatal growth ofSGA children constitute the study material. 71 small for gestational age (SGA)and 149 appropriate for gestational age (AGA) term infants, born during theperiod of two years (January 1998–February 2000) at Kaunas University Hos-pital were enrolled in a case-control study in Lithuania. SGA babies weredefined as having birth weight and/or birth length more than 2 standard devia-tions bellow the mean for the Swedish reference data. Maternal lifestyle factors,marital status, parental education and family income were compared betweenthe groups. Results: The proportion of mothers smoking 1–7 cigarettes perweek or more during pregnancy was significantly higher in the SGA group (OR4.7, 95 % CI 1.7–13.1). Maternal and paternal education of nine years or lesswere also significantly related to the risk of having an SGA baby (OR 5.9, 95%CI 2.2–15.8; OR 3.6, 95 % CI 1.8–7.2, respectively). Significant associationwas found between family income lower than 100 USD per household capitaper month and the SGA births (OR 2.2, 95% CI 1.2–3.9). However, no signifi-cant differences were found in maternal marital status and alcohol consump-tion habits during pregnancy between the two groups. Conclusions: Smokingand such socioeconomic factors, like parental education and family income, areimportant contributors to the intrauterine growth retardation.

39th Annual Meeting of the ESPE Horm Res 2000;53(suppl 2):1–191 25
P2-79 Fetal/Neonatal Endocrinology
Neonatal Ovarian Cyst in Preterm Infant: Surgery CanBe Avoided by LHRH-Agonist TreatmentP. Bretones* 1; M.G. Forest 2; Y. Morel 2; X. Cottin* 1
1 Centre Hospitalier Lyon Sud, Pierre-Bénite, France;2 INSERM U.329, Hôpital Debrousse, Lyon, France
Case report: We report the case of a female infant prematurely born at 27weeks of gestation. The baby was eutrophic: birth weight of 880 g, and birthheight of 33 cm. At 6 weeks of age, i.e. 33 weeks gestational age (GA), shepresented impressive symptoms of intestinal occlusion: a large abdominal masswas visible at first glance. The mass was palpated in the right iliac fossa. Ultra-sonography (US) revealed a 35 mm diameter cyst hiding the right ovary. On theleft ovary, another cyst (17 by 12 mm) was also found. After puncture of theright cyst (1.5 ml of fluid), the clinical status of the child improved. We believethat the situation resulted from an ovarian hyperstimulation syndrome, sinceboth serum estradiol (760 pmol/l) and serum LH levels (24 ÌU/ml) were veryhigh. Following the ovarian puncture, other cysts gradually developed again,one of them reaching 20 mm in diameter. At US, the uterus was stimulated.Curiously, there was no breast development, but an important oedema of thelabia majora. Growth was satisfactory being the usual catch-up growth seen inpreterm infants. Cerebral MRI eliminated a hypophyseal or hypothalamictumour. At 11 weeks of life (38 weeks of GA), an LH-RH agonist treatment wasstarted: 1.5 mg i.m. leuprorelin every 4 weeks. After already one month of treat-ment, US revealed that the cysts had vanished, and showed the presence ofsmall follicles. The pituitary-ovarian axis was inhibited (E2 = 22 to 36 pmol/l;LH = 0.6 to 1.4 ÌU/ml). Treatment was stopped after the 6th injection. Therewas no relapse. The child is now 3 years old with a height on the 50th centile,but advanced bone age (4.5 years). At US theovaries appeared normal. Con-clusion: It is often difficult for a paediatric surgeon to preserve the ovaries in apreterm infant in such a critical situation. To the best of our knowledge this isthe first attempt to treat successfully ovarian cysts. We believe that this treat-ment should be a first choice alternative in this condition.
P2-80 Fetal/Neonatal Endocrinology
Longitudinal Data for IGF-I and -II in Fetus between 21stand 35th Week of GestationB.C. Gohlke 1; H. Fahnenstich* 1; W.F. Blum 2; N. Albers 1
1 Department of Pediatrics, Bonn, Germany; 2 Department ofPediatrics, Giessen, Germany
Fetal IGF-I and -II was measured up to six times in the same individual duringthe second and third trimester of pregnancy providing longitudinal data for theIGFs. Patients and Methods: Patients were referred for repeated cordocen-tesis because of rhesus incompatibility. In 11 patients fetal blood was taken upto 6 times between 21st and 37th week of gestation. Growth was evaluated byultrasonography. All fetus were appropriate for gestational age at any time.Results: All patients showed a progressive increase of IGF-I and IGF-II levelsduring pregnancy with the main increase around the 30th week of gestation.
IGF-I (ng/ml) levels in all patients at the first (!30 wk) and last (130 wk) mea-surement:
First 25 27 19 36 23 28 35 47 29 30 28Last 56 76 30 44 33 30 70 52 59 38 61
IGF-II (ng/ml) levels in all patients at the first (!30 wk) and last (130 wk)measurement:
First 221 213 86 168 167 232 220 191 220 174 375Last 369 513 227 192 175 221 467 356 371 297 373
All data determined during the second trimester (!30 wk; n = 21 for IGF-I andn = 24 for IGF-II) were compared with data determined during the third tri-mester (130 wk; n = 28 for IGF-I and n = 27 for IGF-II) and a significantincrease was found for IGF-I (mean !30 wk): 33.8 ng/ml; mean (130 wk): 45.1
ng/ml; p ! 0.005) and IGF-II (mean !30 wk): 225.8 ng/ml; mean (130 wk):299.0 ng/ml; p ! 0.002). Conclusion: We present longitudinal data for IGF-Iand II for normal weight fetus (appropriate for gestational age) between 21stand 37th week of pregnancy. A significant gradual increase for IGF-I and -IIlevels was found. Mean data for IGF-I and -II determined during the thirdtrimester was significant higher than data determined during the second tri-mester.
P2-81 Fetal/Neonatal Endocrinology
A 120-kb Contiguous Gene Deletion on Chromosome11p Causing Severe Infantile Hyperinsulinism (HI),Sensorineural Hearing Loss (SHL), InflammatoryEnteropathy (IE) and a Renal Tubulopathy (RT) ProvidesNew Insights into the Control of the Development andStructure of These Organ SystemsK. Lindley* 1; M. Bitner-Glindzicz* 1; B. Glaser* 2; M. Dunne* 3;K. Hussain* 1; A. Aynsley-Green 1; Introduced by A. Aynsley-Green1 Institute of Child Health, London, United Kingdom; 2 HadassahMedical School, Jerusalem, Israel; 3 University of Sheffield,Sheffield, United Kingdom
Background: 40–50% of cases of infantile HI are caused by functional abnor-malities in the Katp channel in the ß-cell membrane. The channel’s proteincomplex of two subunits SUR1 and KIR6.2, is coded by genes located on chro-mosome 11P15.1. We report a new syndrome in 3 children with neonatal onsetHI, SHL, IE and RT in 2 consanguineous but unrelated families. All were unre-sponsive to diazoxide, chlorothiazide, glucagon, somatostatin and nifedipine;they were subjected to pancreatectomy. All had vomiting, diarrhoea, malab-sorption and bilateral deafness. Aim: To investigate the biological basis for thisnew syndrome. Results: In vitro electrophysiology of the ß-cell membranesshowed a severe abnormality of the KATP channel with no response to diazox-ide, chlorothiazide, glucagon, somatostatin, or verapamil. Audiology showedsevere bilateral sensorineural hearing loss; GI biopsy revealed severe inflam-matory enteropathy with distortion of epithelial architecture; there was also ageneralised aminoaciduria and increased urinary excretion of Retinol BindingProtein and NAG. Genetic analysis defined a small deletion on chromosome11p that includes the SUR1 gene, and overlaps with two loci implicated indeafness. Several transcripts map to the deleted region and are candidates forthe inflammatory enteropathy, deafness and renal tubulopathy. Analysis ofthese will be presented. Conclusions: The combination of HI with sensori-neural hearing loss, gastrointestinal enteropathy and renal tubulopathy repre-sents a new syndrome caused by a contiguous gene deletion on chromosome11p. The data provide new insights into the fundamental role played by thisregion for the normal development and function of several body systems.

26 Horm Res 2000;53(suppl 2):1–191 Posters
P2-82 Fetal/Neonatal Endocrinology
Steroid Use in Extremely Low Birth Weight PrematureInfantsS. R. Hintz* 1; R.L. Hintz 1
1 Stanford University, Palo Alto, CA, United States
Continued debate exists regarding the use of supraphysiologic glucocorticoidsin extremely low birth weight (ELBW) premature infants, especially when ini-tiated at less than one week of age. Prevalence of, indications for, and potentialcomplications from use of steroids in this population have not been well eluci-dated. Objectives: To evaluate prevalence and timing of glucocorticoid use inour neonatal intensive care unit (NICU) in ELBW preterm infants. Incidenceof HPA evaluation, as well as specific outcome variables, were reviewed.Patients and Methods: A retrospective chart review from 1/1/1997 to 12/31/1998 was preformed identifying infants 1) of gestational age !32 weeks andbirthweight !1000 grams, 2) inborn at Stanford University or transportedwithin 24 hours of life, and 3) who survived 172 hours. A total of 99 patientswere identified. Timing and type (decadron (DEC) or hydrocortisone (HC)) ofglucocorticoid treatment, number of steroid courses, incidence of necrotizingenterocolitis (NEC) and whether HPA evaluation was performed was evaluat-ed. Results: A total of 69 of the 99 patients identified (69.7%) received acourse of glucocorticoid sometime during their stay in the NICU. Thirty-six(36) of these infants (36.4%) received steroids prior to one week of age. Of theinfants treated early, most (83.3%) received HC. Almost half of all infantstreated with steroids (45 of 99, or 45.4%) received multiple courses. Interest-ingly, only 3 of all infants underwent evaluation with ACTH stimulation test-ing. NEC was diagnosed in 13 of the 99 infants; all of these infants had beentreated with steroids. Conclusions: Glucocorticoid treatment is overwhelm-ingly prevalent in the ELBW premature population, and common prior to oneweek of age, although adrenocortical function is rarely evaluated. Althoughthese infants are likely more ill, it is concerning that all infants with NECreceived steroids. Given these results, and recent information implicating earlyDEC use with poor neurodevelopmental outcome, we have now instituted aprospective study of the use of steroids in this ELBW population.
P2-83 Fetal/Neonatal Endocrinology
Effect of Recombinant Human Erythropoietin on SerumCortisol Levels in Very Preterm Infants with Anaemia ofPrematurityP. Ghirri* 1; M. Bernardini* 1; A. Cuttano* 1; C. Ciulli* 1; M. Vuerich* 1;U. Bottone* 1; A. Boldrini* 1; Introduced by G. Saggese1 Pisa, Italy
Recombinant human erythropoietin (R-HuEpo) seems to affect directly orindirectly CRH-ACTH-cortisol axis function in chronic haemodialysis patients(G Ramirez, J Clin End Metab, 1994). R-HuEpo seems to reduce the risk ofdeveloping chronic lung disease (CLD) in very low birth weight infants (G Grif-fiths, Arch Dis Child, 1997). In very preterm infants, low cortisol levels seem tobe associated with an increased risk of CLD. The aim of the study is to evaluateif R-HuEpo has any effect on serum cortisol levels in very preterm infants withanaemia of prematurity. Patients and Methods: Fifteen preterm infantswith anaemia of prematurity (gestational age: 25–29 weeks) were studied. 8were treated with subcutaneous R-HuEpo (3 times a week, 900 U/kg/wk for 6weeks), oral iron, folate and vit. B12; 7 received iron, folate and vit. B12 with-out R-HuEpo and were examined as control group. These infants were assignedrandomly to the two groups. R-HuEpo treatment was started at 4–5 weeks oflife. Cortisol levels were evaluated before, at the end of treatment and 3 weekslater, when blood samples were taken for laboratory monitoring. Cortisol wasmeasured by radioimmunoassay. Intra- and interassay coefficients of variationwere less than 10%. Results: R-HuEpo treatment induced a significantincrease in serum cortisol levels after 6 weeks (4.49 + 0.65 vs 11.59 + 2.95 Ìg/dl,mean + SD, p ! 0.001). The control group did not show any variation in serumcortisol levels (4.75 + 1.29 vs 4.90 + 0.69 Ìg/dl, p = n.s.). 3 weeks after the endof the treatment with R-HuEpo, cortisol levels decreased significantly (6.51 +0.81 Ìg/dl, p ! 0.001). The group treated with erythropoietin had an higherincrease of erythrocyte and haemoglobin concentrations and received fewerblood transfusions. Conclusions: R-HuEpo treatment seems to increaseserum cortisol levels in very preterm infants; this effect could be an importantfactor, in infants treated during the first weeks of life, to reduce the risk ofdeveloping CLD.
P2-84 Fetal/Neonatal Endocrinology
Neonatal Hyperinsulinism (HI) is Associated withAbnormal Cortisol Responses to Hypoglycaemia (HY)K. Hussain* 1; P. Hindmarsh 1; A. Aynsley-Green 1; Introduced byA. Aynsley-Green1 Institute of Child Health, London, United Kingdom
Background: HI is the commonest cause of severe persistent HY in the neo-nate. It is characterised by the inappropriate and excessive secretion of insulinin relation to the prevailing blood glucose concentration. The role played by thecounter-regulatory hormone cortisol at this time is not clear. Since neonates areable to generate a cortisol response to stressful stimuli it would be expected thatthey should also have a substantial cortisol responses to hypoglycaemia, partic-ularly when symptomatic. Aim: To assess the evolution of the cortisol counter-regulatory response during hypoglycaemia in infants with hyperinsulinism.Methods: Six neonates with HI were recruited into the study; mean birthweight was 3.1 kg (1.9–4.4 kg), gestational age range 38–41 weeks. Each of theseneonates underwent a diagnostic fast. Blood samples for cortisol assay weretaken before the hypoglycaemic episode, at the time of HY, and at 10 minuteintervals for 50 minutes after the episode was treated with intravenous dex-trose. All infants demonstrated symptoms at the time of HY. Results: Themean plasma cortisol level before HY was 150 nmol/l, at the point of HY it was162.5 B 66.1 nmol/l (Mean B SEM, 95% confidence interval –8.98 to 333.98);at 10, 20, 30, 40 and 50 minutes respectively the mean serum cortisol valueswere 362, 431, 393, and 405 nmol/l respectively. Plasma ACTH measured inone infant at the time of hypoglycaemia was !10 pmol/l. Conclusions: Neo-nates with HI have a delayed and possibly inadequate cortisol responses tosymptomatic hypoglycaemia. The cause of this is unclear, but it may well berelated to several factors. These include the rate of fall of glucose, and directeffects of insulin on the hypothalamus and pituitary. Further studies are neededto elucidate the mechanism and functional significance of this poor cortisolresponse, and to determine whether these infants should be treated with corti-sol.
P2-85 Fetal/Neonatal Endocrinology
Measurement of Cortisol Binding Capacity in thePerinatal PeriodP.C. Midgley* 1; J. Smith* 1; J. Armstrong 1; J. Yorke* 1; Introducedby E.Crowne1 University of Edinburgh, Edinburgh, United Kingdom
Background: Cortisol (F) levels are lower in the fetus than later in life. Levelsof corticosteroid binding globulin (CBG), as measured by RIA, are alsoreported to be lower in the fetus and neonate than in adults, but little data existson cortisol binding capacity (CBC). Methods: The method described by Ham-mond and Lahttenmaki 1 for measuring CBC in adults was adapted for use inumbilical cord and neonatal samples. This method involves removal of all ste-roids from plasma using dextran coated charcoal. The stripped plasma is thenincubated with tritium 3H cortisol, allowing the binding capacity to be saturat-ed. Results: 10 Ìl samples were used. The optimal incubation time was foundto be 8 mins, based on high affinity binding. Cortisol bound with 1000 timesmore affinity than cortisone (E) and 60 times more than 17 OH progesterone(17OHP). Our initial results suggest that there may be 2 distinct binding sites, alow affinity site and a high affinity site.
Results of perinatal samples
Source Cord blood First weekGestation Term 24 weeksn 137 7Median, nmol l –1
(range)47(29–119)
18(11–30)
Discussion/Conclusions: CBC can be measured in perinatal plasma, in lowsample volumes. CBC is low in relation to adults, and lower preterm than terminfants. This method gives a measure total plasma cortisol binding capacity(mostly due to CBG). There appeared to be 2 distinct lines on Scatchard analy-sis, suggesting 2 binding sites of differing affinity. Although CBC was not inhib-ited at physiological concentrations of E or 17OHP, it must be recognised thatthe numerous steroids that circulate in the perinatal period may influence Fbinding in vivo. 1 Clinica Chemica Acta 1983 132:101–110.

39th Annual Meeting of the ESPE Horm Res 2000;53(suppl 2):1–191 27
P2-86 Fetal/Neonatal Endocrinology
Leptin Levels in Breast Milk in the First Two Months ofLifeV. Alloni* 1; A. Rapa* 1; A. Zavallone* 1; L. Strigini* 1; G. Bona 1
1 Dept of Medical Sciences, Pediatric Unit, Novara, Italy
Human milk contains several bioactive hormones, such as leptin, which wasmeasured in breast milk both in human and rat and was found passing frommaternal to suckling circulation in rats. Aim: To study the biological role andthe variation of leptin during lactation in early postnatal human growth. Mate-rials and Methods: Breast milk samples were collected from 18 mothers onday 3, 30, 60 after delivery at the beginning and at the end of feeding. Onthe same day their babies were weighted: median weight was 3055 g on day 3,4360 g on day 30 and 5270 g on day 60. Leptin levels were measured in wholeand skim breast milk by radioimmunoassay (Linco Research, Inc.). Whole milkwas sonicated (3 bursts, 5 sec duration/burst) to disrupt fat globules containingleptin. Skim milk was obtained after centrifugation (1500 g, 20 min, 4 °C).Results: Median leptin levels ng/ml were as follows.
Whole milk Skim milk p value
Day 3: beginning of feeding 1.82 1.09 0.03Day 3: end of feeding 2.67 0.98 0.01Day 30: beginning of feeding 5.26 0.96 0.002Day 30: end of feeding 8.96 1.07 0.01Day 60: beginning of feeding 12.40 1.13 0.09Day 60: end of feeding 9.72 1.22 0.007
An increasing trend in whole milk leptin values was showed from day 3 to day60 by Friedman ANOVA test (p ! 0.05). A positive correlation was foundbetween whole milk leptin concentrations and infant weight on day 30 (Spear-man r = 0.49, p = 0.04), but this correlation became negative on day 60 (Spear-man r = –0.71, p = 0.02). Conclusions: Our data showed that leptin concen-tration is always higher in whole than in skim milk for its partition in fat glob-ules. During lactation leptin levels progressively arise in whole milk, but not inskim milk. These data suggest a possible interference of breast milk lipid com-position in leptin availability. The relationship found between milk leptin andinfant weight was direct in the first month and inverse in the second month oflife and needs further investigations.
P2-87 Fetal/Neonatal Endocrinology
Analysis of the Methylation Status of The LIT1 Gene inLeukocyte DNA for the Diagnosis ofBeckwith-Wiedemann SyndromeV. Gaston* 1; Y. le Bouc 1; V. Soupre* 2; M.P. Vazquez* 2; C. Gicquel* 1
1 Laboratoire d’Explorations Fonctionnelles Endocriniennes,2Chirurgie maxillo-faciale, Hôpital Trousseau, Paris, France
Beckwith-Wiedemann syndrome (BWS) is an overgrowth disorder involvingdevelopmental abnormalities, tissue and organ hyperplasia and an increasedrisk of embryonic tumours (most commonly Wilms’ tumour). This multigenicdisorder is caused by dysregulation of the expression of imprinted genes in the11p15 chromosomal region. Other overgrowth syndromes as isolated hemihy-perplasia, are also caused by dysregulation of the 11p15 region. Diagnosis of11p15 overgrowth syndrome (OGS) is currently difficult for two major reasons:1) the availability of tissues affected by overgrowth; 2) the large spectrum ofgenetic (uniparental disomy, trisomy of 11p, mutation at p57KIP2) and epigen-etic (loss of imprinting of the IGF-II gene, hypermethylation of the H19 genepromoter) abnormalities. Recently, the loss of imprinting, with the loss ofmaternal aIlele-specific methylation, of the LIT1 gene (encoding an antisensetranscript within KvLQT1 and normally expressed from the paternal allele) hasbeen described in BWS patients. The aim of this study was to evaluate theusefulness of LIT1 methylation analysis in leukocyte DNA for the diagnosis of11p15 OGS. The allelic status of the 11p15 region and methylation status of theLIT1 and H19 genes were investigated in leukocyte DNA from 114 patientswith OGS. Patients were classified into three groups according to clinical data:complete BWS (CBWS) (n = 59), incomplete BWS (IBWS) (n = 37) and isolatedhemihyperplasia (IHH) (n = 18). Sixty-five (57%) patients (39/59 CWBS, 21/37
IWBS and 5/18 IHH) exhibited abnormal demethylation of the LIT1 gene. In19 of these 65 cases, demethylation of the LIT1 gene was related to 11p15uniparental disomy (12 CWBS,2 IWBS and 5 IHH). Among the 49 patientswith normal methylation of the LIT1 gene, 13 other patients were found tohave isolated hypermethylation of the H19 gene. Our results show that analysisof the methylation status of LIT1 and H19 genes in leukocyte DNA is useful inthe diagnosis of 11p15-related OGS, allowing diagnosis in about 70% ofpatients investigated for OGS.
P1-88 Growth Basic
Growth Patterns in Boys and Girls with AtopicDermatitisJ.A. Ellison* 1; L. Patel* 1; P.J. Foster* 1; T.J. David* 1; P.E. Clayton 1
1 University of Manchester, Manchester, United Kingdom
Findings consistent with the pattern seen in constitutional growth delay havebeen reported from prepubertal children with atopic dermatitis (AD). Objec-tive: To examine the growth pattern in AD from infancy to final adult height.Patients and Methods: We retrospectively studied hospital patients age 116years at the time of the study, who had been followed for at least 1 year duringchildhood and who had no other illness known to affect growth. From a data-base of 700 patients, 110 fulfilled these criteria (70 male, 40 female). Height(Ht) and weight (Wt) were converted to standard deviation scores (SDS) using1990 UK reference values and regression analysis was used to examine whetherthe mean trend was different from zero. Growth during late adolescence (peri-od between 14–18 years for females and 15–18 years for males) was assessed.Ht and Wt at age 118 years were compared with normal reference values.Results: Ht and Wt SDS trends for males and females showed a clear devia-tion below the zero line. For females, the fitted mean trend for the SDS wasquadratic for both Ht (p ! 0.0001) and Wt (p ! 0.0001) with the minimumvalue being at ages 10.6 years (mean Ht –1.4 SDS) and 9.4 years (mean Wt –0.6SDS) respectively. For males, the trend was linear, with a negative slope, for Ht(p ! 0.0001) and Wt (p ! 0.0001). Boys were short by 2 years of age, and lost afurther mean 0.8 SDS in both Ht and Wt by 18 years. The mean gain in Ht ofmale but not female patients during late adolescence was significantly greaterthan UK standards (males: 12.2 vs. 8.8 cm, p = 0.04; females: 6.6 vs. 4.5 cm, p =0.06), implying a late pubertal spurt. Patients’ mean Ht and Wt at a mean age of21 years compared with UK standards, revealed that male patients were short-er (mean 170.9 vs. 177.6 cm, p ! 0.001) and lighter (64.6 vs. 71.0 kg, p = 0.03)but female patients reached expected Ht (mean 160.4 vs. 163.8 cm) and Wt(56.4 vs. 57.9). However, 4 of 20 (20%) male and 3 of 11 (27%) female patientswere below the 3rd centile for Ht as adults. Conclusions: The pattern ofgrowth in patients with AD showed gender differences and also differed fromthe normal UK 1990 population. The gender difference may reflect the factsthat AD is more severe in boys and constitutional delay recovers more quicklyin girls. The significant gain in height in late adolescence, at a time when growthrate would be expected to be falling, suggests delayed growth. As a group,female patients reached expected height and weight but male patients wereshorter and lighter than the expected normal population.

28 Horm Res 2000;53(suppl 2):1–191 Posters
P1-89 Growth Basic
Catch-Up Growth in Liver-Transplanted ChildrenA. Wüsthof* 1; C. Schwarz* 1; M. Burdelski* 1; R.P. Willig 1
1 Children’s Hospital of the University of Hamburg, Hamburg,Germany
Children with end-stage liver disease have impaired growth rates. Objective:Evaluation of linear growth and weight gain after liver transplantation.Patients and Methods: A follow-up study was performed with 21 children,measuring their height, weight, skin folds and bone age before, one and twoyears after liver transplantation (mean age at transplantation: 14.4 months;range: 6–47 months). The dosage of corticosteroids was related to the growthrate. Results: Growth SDS corrected for target height was –4.6 before livertransplantation, improving after one year to –2.0 (n = 21) and after two years to–1.4 (n = 10). During the observation period a normalisation of liver functionoccured and the prednisolone dosage was reduced by the second year from amean of 2.6 mg/m2 to 1.0 mg/m2. The bone age of the children was retarded by4.5 months before transplantation and continued to be delayed after two years(7.6 months). The triceps skin folds improved from 5.0 cm (median) to 6.0 cmand 7.0 cm during the two years of observation. Before liver transplantation71% of all children had a weight below 2 SD; one year later only 19% and aftertwo years all assessed children were within normal ranges for weight. Conclu-sion: The failure to thrive in chronic liver disease can be reverted by livertransplantation. Normal liver function and decreasing dosages of corticoste-roids improve growth velocity and weight gain.
P1-90 Growth Basic
Growth Patterns in Early ChildhoodM. Hermanussen 1; S. Lange* 2
1 Aschauhof, Altenhof, Germany; 2 University of Kiel, Kiel, Germany
Particularly in infancy and early childhood, child growth tends to follow irregu-lar patterns with many centiles crossed before the later growth channels areachieved. Objective: To visualise the diversity of individual growth, and tosearch for characteristic patterns in early child growth. Patients and Meth-ods: Body length of 333 healthy girls and 329 healthy boys was measured longi-tudinally at least once every 3 months during the first year of life, at least twiceannually during the second year, and at least once annually, up to the age offive. Each individual growth curve was converted into a series of height SDS,and thereafter, into series of residual height SDS. Cluster analysis identifiedgroups (clusters, containing a minimum of at least 9 individuals) of childrenwith similar patterns of residual height SDS. Results: We identified 12 clus-ters that contained 230 out of the 333 female series of residual height SDS, eachcluster containing 10 to 47 girls. Growth of the other 103 girls was less homoge-neous and could not be assigned to clusters of at least 9 individuals. We identi-fied 10 clusters that contained 221 out of the 329 male series of residual heightSDS, each cluster containing 9 to 49 boys, 108 boys could not be assigned.Three male and 3 female clusters indicated towards a decline of residual heightSDS in early or mid-infancy followed by increasing residual height SDS afterthe age of 6 to 18 months. Other clusters showed steadily declining residualheight SDS up to the age of 4 with no evidence of intermittent growth spurts inearly childhood, and there were clusters with irregular patterns. Clusters arecomparably narrow; with SD that did not surpass 0.3 of the original height SDSof the background population. Conclusion: There is substantial diversity inearly childhood growth, but only a very limited number of characteristic growthpatterns appears possible, with unknown biological significance.
P1-91 Growth Basic
Patterns of Growth, Prevalence and Relative Risk ofShort Stature in Secondary School Graduates BornSmall for Gestational AgeV. Iotova 1; V. Tzaneva 1; K. Petrova* 1
1 Medical University, Varna, Bulgaria
Recent research has shown that being born small for gestational age (SGA)imposes considerable risk of adult short stature. Aim: To assess the prevalenceof SGA among secondary school graduates, to define the patterns of postnatal
growth and the relative risk of adult short stature. Patients and Methods:Overall 4,019 medical records of students from the last grade of all secondaryschools in Varna (population 375,000) were reviewed. Birth data were availablefor 2,255 (56.1%) full-term (38–42 weeks) healthy students from singletonpregnancies. After excluding those still growing, 72,920 measurements of 2,159students (969 boys) remained for analysis. Height and weight measurementswere expressed as SDS (SDSh and SDSw) according to national standard atbirth and to the study cohort thereafter (mean SDSh 0.2–0.9 above the availablestandards). Weight was logarithmically transformed to improve normal distri-bution. SGA was defined as length and/or weight at birth ^–2 SDS. Results:Overall 112 (5.19%) cases were identified, 35 (1.62%) only short (group I), 51(2.36%) only light (group II), and 26 (1.2%) both light and short at birth (groupIII). Group I showed best catch-up growth – only 8.6% of the children werebelow –2 SDSh at 3 months, while these were 23.1% in group III. Althoughfrom birth cases gained 1.62 SDSh, 10.7% of them were still short at finalheight compared to 1.4% of the reference group (p ! 0.0001). Mid-parentalSDSh was lower in the SGA children (–0.2 B 0.88 vs. 1.24 B 0.81, p ! 0.0001).Relative risk of short stature was:
RR 3 months 1 year 8 years Final height
Group I 9.3 13.9 6.0 9.5Group II 17.2 5.9 4.8 9.6Group III 14.3 13.6 15.6 13.9
The relative share of SGA children in the total short population at final heightwas 28.6%. Girls born SGA had earlier age at menarche (p = 0.05) and signifi-cantly lower weight at final height (p ! 0.0001). Conclusions: The prevalenceof SGA in our study is consistent with previously published. The SGA studentsrepresent a quarter of the short adolescent population at final height and theborn both short and light are at the greatest risk. The earlier start of menarcheand lower weight of SGA girls renders further investigation.
P1-92 Growth Basic
Leptin, IGF-I and IGFBP-3 Levels in ConstitutionalGrowth DelayA. Bideci* 1; P. Cinaz 1; A. Hasanoglu 2; L. Tümer 2; F.S Ezgü 2; E. Taç 3
Departments of 1 Pediatric Endocrinology and 2 Metabolism,3 Endocrinology Research Lab, Gazi University, Ankara, Turkey
Objective: This study was planned in order to investigate the role of IGF-I,IGFBP-3 and leptin, a product of ob gene and synthesized by fat tissue cells inconstitutional growth delay (CGD) which is the most frequent cause of shortstature in children. Patients and Methods: This study was conducted on 80CGD children (38 girls and 42 boys) aged 6 to 15 years (median age: 12.2 years)and 60 healthy children (30 girls and 30 boys) (median age: 11.8) served ascontrols. Serum IGF-I, IGFBP-3, insulin and plasma leptin levels were mea-sured by immunoradiometric assay. Results: Mean IGF-I and leptin levelswere significantly lower in CGD group compared with controls but meanIGFBP-3 level was not different in both groups. Mean leptin levels were 3.72 B2.29 in CGD and 4.86 B 3.08 in control group (p ! 0.05). There was a statisti-cally significant relationship between leptin levels and height, weight, and BMI(r: 0.53, 0.54, 0.45 respectively, p ! 0.05). Leptin levels was also correlated withboth chronological age, bone age and height age (r: 0.51, 0.44, 0.83 respectively;p ! 0.05). When evaluated according to pubertal status a significant differencewas found in IGF-I, leptin and IGFBP-3 levels between prepubertal and puber-tal delays. Leptin levels were significantly different in prepubertal CGD groupcompared with controls (2.41 B 1.03 and 3.18 B 1.18 respectively p B 0.05)but in pubertals only IGF-I levels was significantly different from controls. Lep-tin levels in the age groups of 9–11 and 13–15 were significantly different inCGD group than control group. Conclusions: As the weight of CGD patientswere also lower than control group, it is postulated that the reason of shortstature and delay in puberty may be this decrease in weight which is also thecause of low levels of leptin and IGF-I. It is emphasized also that leptin acted asa signal for the onset of puberty.

39th Annual Meeting of the ESPE Horm Res 2000;53(suppl 2):1–191 29
P1-93 Growth Basic
‘MATUROS 4.0’ – A CD Rom for Assessing SkeletalMaturityM. Bouchard* 1; M. Sempe* 1; Introduced by P. Chatelain1 Groupe Français d’Auxologie, Lyon, France
Bone age assessment is very useful in daily clinical practice but variationsamong observers and methods have introduced limitation and insufficient reli-ability. Objective: To provide physicians with a tool which gives the opportu-nity of an easier, prompter and more accurate evaluation of skeletal maturity inchildren. Method: Referring to the previous MATUROS software, this cur-rent approach is developing new functionalities.Using a computer that includesthe program, the physician has to examine only ten selected bones from hand-wrist radiograph in the simplified method, or twenty two in the complete classi-cal one. For each of them, the software displays typical pictures to compare andto assess the degree of maturity. So as to facilitate investigator’s performance,the software automatically proposes the three most appropriate stages. Theneach degree is shown on the screen in three forms: a reference radiograph, apicture and a descriptive text.After completion of evaluation, the software hasthe capacity of numerical calculation which enables the overall degree of matu-ration to be determined and expressed as a percentage of attainable maturitythat can then be converted into bone age. Conclusion: This new tool improvesboth feasibility and reliability of bone age determination in children and shouldhelp paediatricians and radiologists.
P1-94 Growth Basic
Differences of Short-Term Growth in Relation toVarying Doses of Dexamethasone Administration inPremature InfantsA. Keller* 1; C. Vogtmann* 1; E. Keller 1; M. Hermanussen 2; W. Kiess 1
1 University of Leipzig, Leipzig, Germany; 2 Aschauhof, Kiel,Germany
Objective: The negative influence of therapy with glucocorticoids on short-term growth is known since the 80’s. In neonatology, glucocorticoids are rou-tinely used for the prevention and treatment of bronchopulmonary dysplasia ofpremature infants. We intended to measure the putative direct effect of dexa-methasone on daily lower leg length and to elaborate whether the administra-tion of different doses of glucocorticoids is associated with alternations ofshort-term growth. Patients and Methods: We investigated 12 children (8boys, 4 girls), who received dexamethasone in different doses according to theindividual requirements. The mean administered dose of glucocorticoids was0.5 B 0.3 mg/kg body weight per day. The mean gestational age of the infantswas 26.8 B 2.2 weeks, the mean birth weight was 1066 B 371 g. Six infantsreceived a single dose, the other 6 patients received repeated doses of dexa-methasone (over a period of 1–4 days with intervals 15 days without treat-ment). All 12 premature infants underwent daily measurements of lower leglength performed by mini-knemometry and each complete investigation con-sisted of 4 single measurements. The measuring procedure is painless for theinfant and can be performed in the incubator. Results: Finally, 145 completeinvestigations were done by the same observers at 24-h-intervals over periodsof treatment with dexamethasone. The mean growth rate before therapy waslower (0.28 B 0.23 [=SEM] mm/d) than we usually observe in healthy prema-ture infants and is probably associated with impaired clinical condition. Wedivided the patients into tree subgroups dependent on doses of dexamethasoneand compared the changes of lower leg length (‘growth rate’) in these groups.
Group In = 10
Group IIn = 10
Group IIIn = 9
Dose of dexamethasone !0.75 mg/kg 0.75–1.0 mg/kg 11.0 mg/kgMean ‘growth rate’ under
therapy, mm/day(BSEM) –0.24 B0.18 * –0.22 B0.2 –0.40 B0.33
Mean ‘growth rate’ aftertherapy, mm/day(BSEM) 0.54 B0.22 * –0.09 B0.28 –0.06 B0.34
* p ! 0.05, Student’s t-test, SEM = standard error of the mean.
Conclusion: Our study demonstrates the effect of glucocorticoids on short-term growth dynamics of premature infants within 24 hours. Higher doses ofdexamethasone suppressed the short-term growth during and after therapy.However, low-dose-therapy also stopped the growth process, but 24 hours afteradministration of glucocorticoids we observed an increased growth velocitycorresponding to catch-up-growth. We hypothesize that the assessment ofshort-term growth by mini-knemometry can add to the better tailoring of drugtherapy in premature infants.
P1-95 Growth Basic
Triangulation of the Distal Radial Epiphysis PredictsSeverity of Growth Failure Due to Shox Deficiency inTurner SyndromeG. Binder 1; H. Fritsch* 1; R. Schweizer* 1; M.B. Ranke 1
1 University Childrens Hospital and Growth Research Center,Tübingen, Germany
The Madelung deformity is present in an patients with Leri-Weill syndrome, inless than 10 percent of Turner syndrome patients and in individuals with idio-pathic short stature caused by SHOX (short stature homeobox-containing gene)defects. A specific and indispensable radiographical sign of Madelung deformi-ty is the triangulation of the distal radial epiphysis. Objective: To evaluate theprevalence of the triangulation of the distal radial epiphysis in Turner syn-drome and to determine its diagnostic and predictive value for growth failure inTurner syndrome. Patients and Methods: We studied 184 radiographs ofthe left hand in 51 patients with Turner syndrome with a GP bone age olderthan 10.5 years. They were seen during the last 14 years in our clinic and weretreated with rhGH. The shape of the radial epiphysis was determined by defin-ing two medially and two laterally located distinct orientation points. The cal-culated ratio of their distances (= ratio of lateral length to medial length of thecalcified epiphysis) was the quantitative measure for the triangulation. Normalvalues were determined by analysis of 37 radiographs from patients with classi-cal GH deficiency, pathological values by the study of 7 radiographs frompatients with Leri-Weill syndrome and normal karyotype. Results: The meancalculated ratio of normal left hand radiographs was 2.54 B 0.44 with no boneage-dependent difference. The mean calculated ratio in patients with Leri-Weill syndrome was higher with 6.36 B 2.59 (p ! 0.008). In Turner syndrome,the mean ratio was higher than in controls, but lower than in Leri-Weill syn-drome with 3.19 B 0.70 (p ! 0.001 against controls). Five Turner patients metthe triangulation index found in Leri-Weill syndrome (!4.5). Three of these 5Turner individuals had reached final height and were significantly shorter(138.3, 145.7, 149.5 cm) than 30 fully-grown Turner patients with milder trian-gulation (151.6 B 4.8 cm) (p ! 0.01). One of them exhibited the full picture ofclassic Madelung deformity. Conclusion: We show for the first time thattriangulation of the distal radial epiphysis is a major sign of osteodysplasia inTurner syndrome. Our quantitative data demonstrate the close genetic rela-tionship between Turner syndrome and Leri-Weill syndrome (SHOX defect)and suggest that the severity of the epiphyseal triangulation predicts the degreeof growth failure.

30 Horm Res 2000;53(suppl 2):1–191 Posters
P1-96 Growth Basic
Growth Charts from Birth to 18 Years of Age forChildren with Down’s SyndromeÅ. Myrelid* 1; J. Gustafsson 1; B. Ollars 1; G. Annerén* 1
1 Uppsala University, Uppsala, Sweden
Growth in children with Down syndrome (DS) differs markedly from that ofnormal children. If standard growth charts are used for DS children the devel-opment of associated diseases influencing linear growth may be overlooked. Inthe present study Swedish DS specific growth charts are presented. The growthcharts are based on a combination of longitudinal and cross-sectional data from4832 examinations of 354 individuals with DS (203 M, 151 F), born between1970 and 1997. Mean birth lengths were 48 cm in both sexes. Final heights,161.5 cm for males and 147.5 cm for females, were reached at relatively youngages, 16 and 15 years, respectively. Mean birth weights were 3.0 kg for boys and2.9 kg for girls. A body mass index (BMI) 125 kg/m2 at 18 years of age wasobserved in 31% of the males and 36% of the females. Head growth wasimpaired resulting in a SDS for head circumference of –0.5 (Swedish standard)at birth decreasing to –2.0 at four years of age. Specific growth charts are impor-tant in evaluation of health of children having syndromes with impairedgrowth. Short stature is a cardinal sign of DS. Final height in DS-individuals isabout 18 cm below target height. The use of DS-specific growth charts is impor-tant for diagnosis of associated diseases, such as celiac disease and hypothy-roidism, which may further impair growth. Despite growth retardation the dif-ference in height between the sexes is the same as that of healthy individuals.Even though puberty appears somewhat early the charts show that DS individ-uals have a decreased pubertal growth rate. Our growth charts show that Euro-pean DS boys are taller than American boys with DS whereas European DSgirls, although being lighter, have a similar final height as the American girlswith DS.
P1-97 Growth Basic
Failure of Catch-Up Growth and Ongoing Bone Age inChildren after Orthotopic Liver TransplantationR. Odink 1; P.J.J. Sauer 2; H. van Soest 2; P. Peeters 1; C.M. Bijleveld 2
1 Groningen University Hospital and 2 Beatrix Children’s Hospital,Groningen, The Netherlands
Liver insufficiency as a chronic disease leads to impaired growth. Catch-upgrowth after a liver transplant is important in reaching a growth level whichis in accordance to nonaffected children. Objective: We evaluated heightadjusted by parental height and bone age in children at and during follow up(2 and 5 years) of orthotopic liver transplantation (OLT). The final height ifavailable and puberty when appropiate was recorded. Patients and Meth-ods: From 1982–2000 182 OLT operations were performed in 139 childrenunder 17 years. Fifty-four patients were excluded for follow up. Thirthy-onedied within two years of the transplant, twenty-three patients were transplantedafter 31-12-97 and the follow up time was considered to short. Retransplanta-tion was performed in 39 patients due to transplant related complications, 25 ofthem within 4 weeks. Height was expressed as height standard deviation scoreadjusted by parental height (HSDS-TH). Bone age was determined on yearlybasis. For bone age 20 Bones (Tanner & Whitehouse) was used. Final height(FH) had been reached when height velocity was less than 2 cm/year and/orbone age was completed. Results: HSDS-TH at OLT, 24 months and 60months post OLT was –1.91 (n = 139), –1.36 (n = 85) and –1.23 (n = 49). Heightexpressed for bone age was –1.57, –1.36 and –0.73 respectively. Final heighthad been reached in 21 patients and was –1.5. In 5 out of these 21 children whowere transplanted at an age !7.0 years FH was –2.2. Conclusions: Catch-upgrowth both 2 and 5 years after OLT is insufficient to reach a height within thetarget range (B1.3 SDS-TH) in the majority of children. Final height has beencompromised as well.
P2-98 Growth Basic
Secular Trend for Height and Weight in Italian Childrenfrom 1987 to 1999A. Luciano 1; M. Bolognani* 1; S. Cavalieri* 1; A. Bariani 1;P. Residori* 1; F. Bottazzi 1; G. Zoppi* 1
1 Major City Hospital Verona University, Verona, Italy
Secular changes in growth have occurred in most industrialised countries dur-ing the last century. The secular trend has slowed down after the second worldwar but is still continuing. Objective: To evaluate secular trend of height inannual groups of children and adolescents during the years 1987 to 1999.Patients and Methods: In 1999 7013 (3403 males and 3610 females) school-children, aged 3–18 years, from the Verona area, were recruited. Height andweight were evaluated, in annual groups (an average of 219 subjects for group),by a trained team, using standard measurement instruments and procedures.Data were compared to those obtained in 1987. Results: In males the heightincreased 4.6 B 1.98 cm (media B SD) with final height 176.8 (+0.98) cm; theweight increased 3.59 B 1.9 kg. In females the height increased 3.89 B 1.32 cm(media B SD) with final height 163.8 (+2.5) cm; the weight increased 2.59 (1.4)kg. Conclusions: These results suggest that the secular trend in height is stillcontinuing in our population. The final height has increased more in females(+2.5 cm) than in males (+0.98 cm), whereas weight has increased more inmales. In prepubertal age, secular trend of height, especially in males, showed ahigher increase toward final height.
P2-99 Growth Basic
The Effect of Metabolic Acidosis on the Local GrowthHormone (GH)-Insulin Like Growth Factor I (IGF-I) Axisin Skeletal Growth CentersG. Maor* 1; J. Green* 2
1 Department of Anatomy and Cell Biology, Faculty of Medicine,Technion; 2 Rambam Medical Center, Haifa, Israel
Chronic Metabolic Acidosis (CMA, e.g. uremia acidosis) causes growth retar-dation in children and plays a major role in the pathogenesis of renal osteodys-trophy. Given the importance of GH/IGF-I axis in maintaining skeletal lineargrowth as well as bone metabolism, we studied the effect of CMA on this endo-crine axis in skeletal growth centers. Due to the influence of the skeletal GH/IGF-I system on the function of PTH and vitamin D, we also evaluated theeffect of CMA on this interaction. We studied the murine mandibular condyle(6 days old ICR mice) as a model for endochondral ossification center. Organcultures were incubated in BGI medium at either neutral pH (pH F7.4) oracidic pH (pH F7.15 achieved by adding 2 mM HCI to the medium), and withor without 10 –8 or 10 –10 M of either PTH or vitamin D. After 24, 48, 72 and 96hours, condyles were thoroughly washed, fixed in formaldehyde and routinelyprocessed for paraffin embedding. Sections were evaluated for histology (byH&E staining), immunohistochemistry (IHC), and in situ hybridization (ISH).Following 3–4 days in acidic conditions, there was a marked reduction in thenumber of young chondrocytic population suggesting a defect in the processendochondral differentiation. IHC analysis revealed a marked reduction inIGF-1 receptor as well as in the GH receptor, which was evident after 48 hours.At 48 hours of acidosis (compared to non-acidosis conditions) there was also amarked reduction in the expression of IGF-1 both under basal (non-stimulated)conditions and following stimulation with GH. The expression of IGFBP-2 andIGFBP-4 was enhanced in CMA. The upregulation of the IGF-1R by IGF-1was blunted in the CMA. CMA also reduced the expression of both PTH recep-tors and vitamin D receptors. PTH (10 –10 M but not 10 –8 M) significantlystimulated the expression of IGF-I receptors. This effect was also blunted byCMA. In conclusion, CMA exerts an antianabolic effect on endochondral bonegrowth which is partly attributed to a state of resistance to GH, IGF-1 andPTH. These changes may contribute to the growth retardation caused byCMA.

0.0
39th Annual Meeting of the ESPE Horm Res 2000;53(suppl 2):1–191 31
P2-100 Growth Basic
Early Growth in Infants Born Small for Gestational Age(SGA) and Controls: No Correlation to Caloric IntakeM.-L. Grauer* 1; A. Sellmer* 2; H.A. Wollmann 2; M.B. Ranke 2
1 University Children’s Hospital, Tübingen, Germany; 2 Germany
While most children born SGA show catch-up growth during the first monthsof life and reach their genetically determined growth channel by the age of twoyears, some remain short. The aim of this study was to determine whether theextent of catch-up growth during the first year is correlated to nutrition. 98term-born healthy infants were included in the prospective study. They weresubdivided according to their birth weight (BW) into two groups: children bornSGA i.e. !10. percentile of Prader’s norms (n = 21, 8 m, 13 f, mean BW 2644 B343 g) and controls (n = 77, 42 m; 35 f; mean BW 3542 B 362 g). The mothersprotocolled their childrens meals for 3 consecutive days at the ages of 3, 6, 9 and12 months. At the same times, the childrens’ height and weight were recordedin a standardized fashion. The nutrition data were evaluated using EBIS soft-ware implemented on a PC with regard to caloric content, proteins, carbohy-drates and fat. Results: Although the SGA group was still significantly smallerand lighter than the controls at 12 months, catch-up growth was observed forlength and weight: Both groups gained in height SDS (Controls: + 0.4 SD, SGA+ 1 SD) over the observation period, the fastest growth occured during the first3 months. The controls gained 0.6 SD in weight over the first 3 months andrecurred to 0 SD over the rest of the observation period, while SGA childrengained 1.2 SD initially, then lost 0.4 SD. There were no significant differencesin energy uptake, protein, carbohydrate or fat intake in the two groups nor wasthere any sustained correlation between the energy or protein intake and lengthor weight gain. Conclusions: 1. The control group differed from Prader’sstandards. 2. Differences between the two groups show that incomplete catch-up growth occured in the SGA group. 3. The nutritional differences did notinfluence the outcome significantly in this generally well-nourished study popu-lation.
P2-101 Growth Basic
Longitudinal Standards from Birth to Maturity forHeight, Weight, Sitting Height and Height Velocity forGreek ChildrenS.K. Pantsiotou* 1; A.D. Koutras* 1; Introduced by K. Dacou1 Department of Clinical Therapeutics, Alexandra University Hospital,Athens, Greece
Objective: To produce longitudinal standard of growth suitable for monitor-ing greek children with growth problems. Patients and Methods: We beganmeasurements on 3/90 and finished 10/97. We measure 1222 children (boys583, girls 639) aged 0–17.5 years old. The measurements were every 4–12months. There are 8500 measurements (boys 3850, girls 4650) with meanrepeated measurements 7, range 2–22. We use the SPSS computer program forthe results. Results: Table with the mean values of somatometrics
Ageyears
Number
boys girls
Heightcm
boys girls
Weightkg
boys girls
Sit heightcm
boys girls
Heightvelocitycm/year
boys girls
199 192 51.7 51.2 3.5 3.3 38.3 37.51.0 149 167 77.8 75.7 10.5 9.6 49.9 48.5 15.9 15.92.0 139 175 89.9 88.5 13.5 12.8 55.6 54.4 12.2 10.43.0 95 98 98.1 96.9 15.7 15.2 58.6 58.5 8.9 8.84.0 83 82 106.3 105.7 18.1 18.0 62.0 61.7 7.6 7.85.0 90 95 112.7 112.2 20.6 20.8 64.7 64.5 6.8 7.06.0 129 116 118.9 119.2 22.8 22.9 67.0 66.9 6.5 6.47.0 134 153 125.3 124.7 25.3 25.6 69.2 69.2 6.2 6.08.0 200 208 131.4 130.6 28.9 29.2 72.0 70.5 6.1 5.49.0 260 539 137.6 136.0 33.1 33.0 73.5 72.5 5.4 5.6
10.0 477 710 142.0 141.9 36.7 36.6 75.7 75.4 5.3 6.211.0 234 306 146.7 148.3 41.2 42.5 77.7 78.3 5.5 6.811.5 237 277 149.3 151.9 43.2 44.2 78.7 80.0 6.0 6.912.0 396 500 152.8 154.6 45.3 46.3 80.2 91.6 6.4 6.313.0 170 169 159.8 159.5 51.7 50.7 83.4 84.5 7.3 4.214.0 239 271 167.1 162.4 58.4 54.0 87.3 86.6 7.3 2.415.0 119 163 173.2 164.1 67.0 57.0 90.9 87.8 5.5 1.416.0 100 93 175.9 165.4 71.9 58.0 92.9 88.4 3.0 0.917.0 60 62 176.5 165.5 77.0 60.0 94.0 89.0 2.0 0.3
The boys at the age of 17 years are 11 cm taller than girls. At the age of 17 yearsthe sitting height in the boys is 10.5 cm bigger than leg length and it is 53.9% ofstature. In the girls the sitting height is 12.5 cm bigger than leg length and it is53.8% of stature. The peak growth velocity in the boys is VPeak = 7.7 cm/yearand at the age of 13.5 years. In the girls the peak growth velocity is VPeak = 6.9cm/year. and two years earlier at the age of 11.5 years. Conclusion: Everynation must have his own longitudinal charts of growth because there are differ-ent genes and climate and different social background.
P2-102 Growth Basic
The Effect of Low Birth Weight on Menarcheal Status in14-Year-Old Polish GirlsS. Koziel* 1; E. Jankowska* 1; E. Barg* 2; Introduced by M. Maes1 Institute of Anthropology, Polish Academy of Sciences and2 Department of Endocrinology and Adolescence, Wroclaw MedicalUniversity, Wroclaw, Poland
The relationship between birth weight and the tempo of sexual maturation(controlled for such confounding factors as: the gestational age and the socio-economic background) has not been comprehensively revealed. Objective: Toevaluate the effect of low versus normal birth weight (with regard to the lengthof the gestation period and the social conditions) on the menarcheal status inPolish 14-year-old girls. Subjects and Methods: The study was carried outin a group of 790 14-year-old healthy girls, examined in 1997, randomly select-ed from primary schools in Wroclaw, Poland. The data were obtained througha questionnaire fulfilled by parents. A model of a binominal logit regression wasapplied where the pre- versus postmenarcheal status was a function of a) thebirth weight (below 2500 g versus 2500–4000 g); b) the socio-economic back-ground expressed by a level of mother’s education (a university or secondaryschool versus a trade or primary school); c) the gestational age (pre-term deliv-ered children i.e. below 38 Hbd versus full-term delivered ones). Results: Inour model there were neither effect of the mother’s education nor of the gesta-tional age on the menarcheal status (Wald’s statistics of those variables and allinteractions were not signjficant), whereas a significant effect of the low versusnormal birth weight was revealed (Wald’s statistic was 4.12; p ! 0.05). The oddsratio of the likelihood of the pre- versus postmenarcheal status at the age of 14as a result of low birth weight was 1.68 (95% CI – 1.03–2.77). Conclusions:The 14-year-old girls of low birth weight are more likely to be premenarchealthen postmenarcheal ones when compared with their age- matched colleaguesof normal birth weight (these data are controlled for confounding factors i.e. thegestational age and the socio-economic background).

32 Horm Res 2000;53(suppl 2):1–191 Posters
P2-103 Growth Basic
The Effects of Birth Weight, Gestational Age andSocio-Economic Status on Stature of 14-Year-OldPolish AdolescentsS. Koziel* 1; E. Jankowska* 1; E. Barg* 2; Introduced by M.O. Savage1 Institute of Anthropology, Polish Academy of Sciences and2 Department of Endocrinology and Adolescence, Wroclaw MedicalUniversity, Wroclaw, Poland
Objective: To assess the effect of birth weight (controlled for such confoundingfactors as: the gestational age and the socio-economic background) on the stature ofPolish 14-year-old healthy boys and girls. Subjects and Methods: Materialcomprised a group of 14-year-old healthy adolescents (797 boys and 819 girls),examined in 1997, randomly selected from primary schools in Wroclaw, Poland.Additional data were obtained through a questionnaire fulfilled by parents. A thee-way analysis of variance was applied, separately for both sexes, where the stature of14-year-old adolescents was a function of 3 factorial variables: a) the birth weight(below 2500 g versus 2500–4000 g); b) the socio-economic background expressedby a level of mother’s education; c) the gestational age (pre-term delivered childreni.e. below 38 Hbd versus full-term delivered ones). In order to estimate the relativerisk of being below the lower quartile of stature, the standardised odds ratios werecalculated, using a model of a binominal logit regression with 3 factors (listedabove). Results: Both the mother’s education and birth weight (but not gestation-al age) differentiated the stature of girls (F = 7.51; p ! 0.01 for mother’s educationand F = 8.51; p ! 0.01 for birth weight) and boys (F = 6.66; p ! 0.01 for mother’seducation and F = 12.85; p ! 0.001 for birth weight). All 3 factorial variablesexplained 2.95% and 5.00% of a total variance of male and female stature at theage of 14, respectively. In our model the standardised odds ratios of the likelihoodof being below the lower quartile of stature (controlled for both gestational age andmother’s education) at the age of 14 as a result of low versus normal birth weightwas 2.12 for boys (95% CI 1.68–2.56) and 2.08 for girls (95%CI 1.62–5.54). Con-clusions: Despite the lower socio-economic status has a disadvantageous effecton stature of l4-year-old adolescents, also children of lower birth weight (controlledfor the confounding factors i.e. the gestational age and the socio-economic back-ground) are at an increased risk of being shorter at the age of 4 as compared withtheir age-matched colleagues of normal birth weight.
P2-104 Growth Basic
Age-Dependent Regulation of Lipogenesis in RatAdipocytesA. Kamel* 1; S. Norgren* 1; H. Fakhari-Rad* 2; J. Galli* 2; C. Marcus 3
1 Huddinge University Hospital, Huddinge, Sweden; 2 KarolinskaHospital, Stockholm, Sweden; 3 Huddinge University Hospital,Stockholm, Sweden
The main function of adipose tissue is to form, store, and supply energy. Adiposetissue formation begins before birth and adipose tissue mass increases rapidly dur-ing infancy due both to an increase in adipocyte size and number. Objective: Thisstudy was designed to investigate the regulation of basal and insulin-induced lipo-genesis as well as glucose transporter protein expression in rat adipocytes fromdifferent age groups. Materials and Methods: The study included 30 maleweaned and 15 male adult Fischer rats. The lipogenesis experiments were per-formed under conditions at which glucose transport is rate limiting. Determinationof glucose transporters content (GLUT1 and GLUT4) in adipocytes was doneusing Western blotting method. Results: Basal lipogenesis and insulin-inducedlipogenesis were two times higher in weaned than in adult animals (p ! 0.05). Nodifference was detected either in GLUT1 or GLUT4 content between any of theage groups in rat adipocytes. Conclusion: Basal and insulin-stimulated lipogene-sis is increased in rat adipocytes early in life. This may be of importance for adiposetissue growth in early age.
P1-105 GH Secretion and Action
Relationship between the Morphology of PituitaryGland and the GH Response to GHRH plus Arginine inChildren and Adults with Congenital HypopituitarismM. Maghnie 1; B. Salati* 1; E. Stacul* 1; S. Bianchi* 1; C. Tinelli* 1;M. Autelli* 1; G. Aimaretti* 2; E. Ghigo* 2
1 University of Pavia, Pavia, Italy; 2 University of Turin, Turin, Italy
Objective: We evaluated the effect of GHRH+Arginine (GHRH+ARG) in 36patients with congenital GH deficiency (GHD) according to their pituitary
MRI findings consisting in anterior pituitary hypoplasia, pituitary stalk (PS)agenesis and posterior pituitary ectopia. Patients: Seventeen children (age 1–5.2 yrs) were evaluated at the time of diagnosis of GHD (mean age 3.6 B1.4 yrs) and 19 adults (age 15.9–28.6 yrs) with childhood-onset GHD were ree-valuated after completion of GH treatment at a mean age of 20.5 B 3.5 yrs.Eleven children had isolated GHD (IGHD) and 6 had MPHD while 7 adultshad IGHD and 12 had MPHD. Results: A residual vascular component of the(VPS) was visualized in 7 children and 7 adults with IGHD, whereas MRIshowed complete PS agenesis (PSA, both vascular and neural components) in10 children and 10 adults, of whom 16 with MPHD (6 children) and 4 childrenwith IGHD. In the children the median peak GH response to GHRH + ARG(7.6 Ìg/l, range 2.4–40.2) was significantly higher than in the adults (1.8 Ìg/l,range 0.8–37.4, p = 0.0039); it was also significantly higher in the IGHDpatients (18 Ìg/l, range 3.3–40.2) than in those with MPHD (1.9 Ìg/l, range0.8–7.6, p = 0.00004). In the patients with residual VPS the median peak GHresponses to GHRH+ARG (19.1 Ìg/l, range 1.6–40.2) was significantly higherthan in those with complete PSA (2.2 Ìg/l, range 0.8–8.8, p = 0.00005). Therewas a trend toward decrease in peak GH response to GHRH+ARG with age.Conclusions: We have confirmed that the partial integrity of the hypothalam-ic pituitary connections is essential for GHRH+ARG to express its GH releas-ing activity, and shown that this test is able to stimulate GH secretion to amajor extent in patients with GHD but with a residual VPS. This test is reliablein the diagnosis of congenital hypopituitarism in both children and adults whenassociated with complete PSA and MPHD. In young children (!6 years of age)pituitary responsiveness to GHRH+ARG does not exclude a congenital GHD;deterioraton of pituitary GH reserve with GH response of less than 10 Ìg/l after20 years of age makes this test very sensitive in the diagnosis of adult GHD.
P1-106 GH Secretion and Action
GH Regulates GH-Receptor Gene Transcription inPrimary Human Thyroid CellsC. Flück* 1; A. Eblé* 1; U. Marti* 2; U. Bürgi* 2; H.J. Peter* 3;P.E. Mullis 1
Divisions of 1 Paediatric Endocrinology and 2 Endocrinology andDiabetes, 3 Anna Seiler Haus, Inselspital, Bern, Switzerland
Background: The association between acromegaly and goitre is well knownand insulin-like growth factor-I (IGF-I) has been shown to act as thyroid growthfactor whereas, at least in vitro, its capacity to stimulate thyroid growth in theabsence cf TSH was limited. However, as GH causes IGF-I release and interac-tions between GH and thyroid hormones are well documented the direct effectof GH on growth and development of the thyroid gland is of interest. Aim andMethods: The aim of this study was to analyse the regulation of GH-receptorgene (OHR/GHBP) transcription by different concentrations of GH (0, 12.5,25, 50, 150, 500 ng/ml) with and without variable TSH concentrations (0.5, 2,20 mU/l) in primary human thyroid cells cultured in serum-free hormonally-defined medium. The incubation time was 6 hours and GWGHBP mRNAexpression was quantitatively assessed by using PCR amplification at an hourlyinterval. Results: Correlating with the GH-concentrations added a constantand significant increase of GH/GHBP gene transcription was found. After theaddition of 12.5 ng/ml GH, GHR/GHBP mRNA concentration remained con-stant over the incubation period of 6 hr but in comparison with the experimentswhere no GH was added there was a significant change of GHR/GHBP mRNAexpression. Following the addition of 25 ng/mL GH a slight increase of GHR/GHBP transcription products was seen which further increased in the experi-ments where higher GH concentrations were used. These data focusing onGHR/GHBP gene transcription derived from cDNA synthesis and quantita-tive PCR amplification were confinned by run-on experiments. Furthermore,cycloheximide did not affect these changes supporting the notion that GH stim-ulates GHR/GHBP gene transcription directly. In a second set of experiments,in combination with variable TSH levels, identical GH concentrations wereused and no difference in either GHR/GHBP mRNA levels or in transcriptionrate (run-on experiments) could be found. Conclusion: We report data show-ing that GH has a direct and dose dependent effect on OHR/GHBP gene tran-scription in primary thyroids cells. Furthermore, TSH does not a have a majorimpact on OHR/GHBP gene regulation. Therefore, these data support the ideathat OH and not only IGF-I may play an active and direct role in growth ofthyroid tissue leading to the occurrence of goitre in any clinical circumstanceswhere high levels of GH are present.

39th Annual Meeting of the ESPE Horm Res 2000;53(suppl 2):1–191 33
P1-107 GH Secretion and Action
Catch-Up and Catch-Down Growth Influence UrinaryGH and IGF-1 Excretion in 9-Year Old ChildrenP.C. Hindmarsh 1; C.H.D. Fall* 2; P.M. Clark* 2; A.W. Shiell* 2;C.M. Law* 2; P.E. Clayton 3
1 University College London, London, United Kingdom; 2 MRCEnvironmental Epidemiology Unit, Southampton, United Kingdom;3 Endocrine Sciences Research Group, Manchester, United Kingdom
Catch-up and catch-down growth is often observed in the first years of life andrepresents individual tracking towards genetic height. To what degree this pro-cess is influenced by or influences the GH-IGF-1 axis is unclear. We have stud-ied 24 h urinary GH and IGF-I excretion in 183 healthy children and relatedvalues to height at 9 years of age, birth length and parental stature. The tabledepicts 24 h uIGF-I excretion, current size and birth size. Midparental SDS areshown in parentheses.
BirthlengthSDS
Height SDS at 9 years
!–0.5 –0.5 to +0.5 1+0.5
!–0.5 651 (–0.6) 630 (0.1) 719 (0.5)–0.5 to +0.5 605 (–0.3) 662 (–0.1) 793 (0.2)
1+0.5 505 (–1.0) 609 (0.02) 837 (0.5)
uIGF, but not uGH, was related to current height (p ! 0.001) particularly whenparental size was considered (Top left cell to bottom right cell). uGH and uIGF-I were not related to birthlength. Catch-up growth (Top right cell) was associat-ed with high uIGF-I levels (p = 0.01) and occurred in children with tall parents(p ! 0.001). Catch-down growth, (Bottom left cell) was associated with the low-est uIGF-I values (p = 0.01) and occurred in children with short parents (p !0.001). uIGF-I at 9 years is related to current height and not birth size and isrelated to catch-up and catch-down in height between birth and 9 years, whichin turn is related to parental height. These observations suggest a key role forIGF-I in modulating post-natal growth.
P1-108 GH Secretion and Action
Effect of Age on Serum Concentrations of20 Kilodalton (20K) Growth Hormone (hGH)S. Murphy* 1; P.J. Pringle 2; M. Mellado* 3; I.C.A.F. Robinson 4;P.C. Hindmarsh 2; C.G.D. Brook 2
1 The Middlesex Hospital, London, United Kingdom; 2 UniversityCollege London, London, United Kingdom; 3 Universidad Autonoma,Madrid, Spain; 4 National Institute for Medical Research, London,United Kingdom
Background: Age-related changes in growth hormone secretion exist in man.Studies so far have examined the relationship between ageing and the secretionof 22KhGH. Several forms of hGH are secreted from the pituitary gland, themost abundant of which is 22K; 20K is the second. The fact that this isoform ofhGH has been evolutionarily conserved indicates that it is likely to have a phys-iological role, one, which may alter with age. We have used a newly developedimmunoassay, which is both sensitive and specific for 20KhGH to measure itsserum concentrations resulting from GHRH(1–29)NH2 stimulation. Methods:Healthy Caucasian male volunteers ranging from 20–30 years and 35–50 yearsof age were recruited and studied on 3 occasions. On 2 occasions, the volunteerswere given GHRH(1–29)NH2 (0.5 Ìg/kg IV) and, on the third, 0.9% saline wasgiven as a control. Blood samples were collected at 10-minute intervals for 30minutes prior to the administration of GHRH(1–29)NH2 or saline and at 5-min-ute intervals for 60 minutes thereafter. Results: Comparison of serum 20Kand 22K concentrations between the 2 age groups is shown in the table.
Age-group 20–30 35–50
Mean peak 20K (ng/ml) 1.5 (n = 14)range 0.6–3.65
0.83 (n = 5)range 0.3–1.9
Mean peak 22K (ng/ml) 21.8 (n = 14)range 6.1–55.4
2.9 (n = 7)range 0.1–5.6
% of peak as 20K 7.7range 2.6–22%
33range 5–91%
In the 20–30 year-old age-group there was a response of 20K and 22K toGHRH(1–29)NH2 in 12/14 tests and a response in 5/7 tests in the 35–50 year oldage-group. Conclusions: There is an age-related change in the response of20K and 22K to stimulation by GHRH(1–29)NH2. The proportion of 20K in thepeak response is greater in the older age group, but overall the degree of theresponse is reduced in this group.
P1-109 GH Secretion and Action
The Value of the Arginine Provocation Test for theDiagnosis of Growth Hormone Deficiency (GHD)M. Hoos* 1; W.J.M. Gerver 1; R. de Bruin* 1; K.R. Westerterp* 2;R. Donckerwolcke* 1
1 Departments of 1 Pediatrics and 2 Human Biology, UniversityMaastricht, Maastricht, The Netherlands
Objective: To explore, in idiopathic growth-retarded children, the relationbetween the arginine provocation test and their growth velocity in the yearbefore the test and the year after the test. In addition, to compare the test resultsof an arginine test with the test results of a clonidine and sleep test. Patientsand Methods: In the study 54 growth-retarded children were included withheight below –2 sds or with growth deviating more than 0.25 sds in one year.The maximum GH concentration during the arginine test was compared withyearly height increase before and after hGH therapy. One year height velocitieswere calculated before (¢SDS–1) and after (¢SDS+1 the test moment. Of thetotal group of 54 children, 27 started hGR therapy after the test. In 43 children¢SDS+1 could be calculated. All children were neither pubertal nor delayed forpuberty. In addition, in a subpopulation of 18 children clonidine and sleep testswere performed and the maximum values compared with the maximum valuesof the arginine tests. Results: See table for characteristics of the three patientgroups.
Mean values n No therapy n hGH therapy
Height sds 27 2.84 27 –3.19¢SDS–1 27 0.05 27 –0.02¢SDS+1 16 0.03 27 0.96
In both groups ¢SDS–1 was not significantly correlated to the maximum valueof the arginine test. ¢SDS+1 was only significantly correlated in the hGH thera-py group (Rsq = 0.32). No significant correlation was found between the maxi-mum value of the arginine test and the maximum value of the sleep or cloni-dine test. Conclusions: The variation in growth in the first year of treatmentcan only for 32% be explained by the maximum value of the arginine test.Using an arginine test, a clonidine test or a sleep test will lead to a differentconclusion in the diagnosis of GHD.

34 Horm Res 2000;53(suppl 2):1–191 Posters
P1-110 GH Secretion and Action
Can GH Values in Response to Provocative Test Definea Sub-Group of Children With Short Stature Due toPartial GH Insensitivity Syndrome?Y. Rakover 1, 2; A. Silbergeld 3; I. Lavi 4; M. Refat 2; I. Ben Shlomo 5
1 Pediatric Endocrine Unit, 2 Endocrine Laboratory, Ha’ Emek MedicalCenter, Afula, 3 Felsenstein Medical Research Center, 4 Tel AvivUniversity, Department of Community and Epidemiology, 5 CarmelMedical Center, Haifa, Department of Obstetrics and Gynecology,Ha’ Emek Medical Center, Afula, Israel
Background: The cause for short stature in children is unknown in the major-ity of the cases. GH-receptor (GHR) mutations have been reported in a fewchildren with apparently idiopathic short stature (ISS). These had normal orexaggerated GH response to provocative stimuli, low IGF-I and IGFBP-3 lev-els and low GHBP, suggestive of partial GH-insensitivity (GHI). Objective:We attempted to define a subgroup of children with partial GHI syndrome,based on their response to GH provocative stimuli. Patients and Methods:164 prepubertal children (97 boys, 67 girls) aged 7.2 (0.5–16.75) years werestudied. All bad short stature with height !3rd percentile. Basal blood sampleswere taken for IGF-I, IGFBP-3 and GHBP. All patients underwent a GH pro-vocative test with either clonidine, arginine or insulin. In a case of low GH peak(less than 9 ng/ml) repeated GH provocative test was preformed with a differ-ent stimulus. Subjects were divided into three groups: (A) Patients with peakGH concentration less than 9 ng/ml in two different provocative tests indicat-ing GH deficiency (GHD; n = 33). (B) Patients with peak GH between 9.1–19.9 ng/ml, indicating normal response (n = 78). (C) Patients with peak GH120 ng/ml, indicating exaggerated GH response (n = 53). Results: No signifi-cance differences were found between the groups in age, height (SD), ¢BA andparental height (SD). Patients with GHD were heavier (p = 0.039) and hadsignificantly higher BMI (SD) (p = 0.001). Maternal height (SD) and MPH (SD)were lower in the group of exaggerated response (p = 0.058, p = 0.04, respective-ly). No significant differences were found between the groups in all serum val-ues of basal GH, IGF-I, IGF-I (SD), IGFBP-3, GHBP (%) and GHBP (SD),apart from lower IGFBP-3 (SD) in the GHD group (p = 0.005). Height (SD)correlated negatively only with basal GH values in the whole group (r = –0.358,p ! 0.0001), in normal responders (r = –0.45, p ! 0.0001) and in the exaggeratedresponse group (r = –0.341, p ! 0.0001), but not in the GHD group. BMI (SD)correlated positively with IGF-I, IGFBP-3, GHBP, GHBP (SD) (r = 0.42, p !0.0001, r = 0.262, p ! 0.0001, r = 0.462, p ! 0.0001, r = 0.361, p ! 0.0001).Conclusion: Patients with exaggerated GH response had significantly lowerMPH (SDS) and shorter mothers. Height was correlated negatively with basalGH. GH values in response to provocative test could not be used to define asubgroup of children with GHI syndrome.
P1-111 GH Secretion and Action
Serum Immunofunctional hGH Levels Are QualitativelyComparable with Results of the Immunometric DelfiaKit in Healthy Adolescents and Young AdultsJ. Kratzsch* 1; J. Donaubauer* 2; T. Nowak* 3; H. Steinkamp 3;H. Willgerodt 2; W. Kiess* 2; E. Keller 2
1 Leipzig, Germany; 2 University of Leipzig, Leipzig; 3 Pharmacia &Upjohn GmbH, Erlangen, Germany
Objective: A recently developed immuno-functional assay (IFA) for the deter-mination of hGH measures only hGH forms that are capable of dimerizing GHreceptors. Therefore, IFA results seem to better represent biological GH effectsthan data of a ‘pure’ immunoassay. To evaluate the IFA as a diagnostic tool forGHD in adolescents we compared hGH levels measured by both methods.Patients and Methods: Three different GH stimulation tests were per-formed in 40 healthy volunteers (20 females, 20 males; aged 16–25 years): GH-releasing hormone (GHRH) combined with arginine (ARG), GHRH+pyrido-stigmine (PD), insulin tolerance test (ITT). Serum GH levels were measured bythe immunometric DELFIA kit (Wallac, Turku, Finland) and the IFA method(DSL, Sinsheim, Germany). Results: Maximum stimulated GH levels mea-sured by IFA (x) demonstrated a considerable inter-individual variation (me-dian/2.5th/97.5th percentile [ng/ml]) and were lower than DELFIA data (y):GHRH+ARG: 30.3/5.1/67.6, y = 1.48x + 6.7, r = 0.97; GHRH+PD: 23.8/4.0/
48.5, y = 1.50x + 4.8, r = 0.97; ITT: 12.3/2.8/37.7, y = 1.56 + 2.9, r = 0.98. IFA/DELFIA ratios of individual samples ranged between 0.15 and 0.85. Medianratio of unstimulated hGH levels at 0 min was significant lower than medianratio at the 60 min level after stimulation in all three tests (p ! 0.01). Conclu-sion: 1) IFA and DELFIA data are qualitatively comparable in healthy adoles-cents and young adults. 2) Differences in the standardization seem to be themain cause for the lower values observed in the IFA test. 3) High variations ofthe IFA/DELFIA ratio may reflect differences in the cross-reactivity of bothassays and/or alterations in the immunofunctionality of GH isoforms fromindividual subjects.
P1-112 GH Secretion and Action
The Effect of Different Sex Steroid Regimens on theStimulated Growth Hormone Levels in the Diagnosis ofGrowth Hormone DeficiencyE.N. Gönc* 1; N. Yordam 1; N. Kandemyr 1
1 Hacettepe University, Ankara, Turkey
The differential diagnosis of transient and permanent growth hormone defi-ciency (GHD) is a real problem of the children. Objective: To show the impor-tance of priming prior to GH stimulation tests in the diagnosis of GHD, theeffect of different doses and schedules of testosterone (T) on GH levels andfinal heights. Patients and Methods: Eighty-four prepubertal or early puber-tal boys whose heights were less than –2SDS and height velocities less than4 cm/year and who have failed in GH stimulation tests were included. Boneages were more than 9 years. The boys were grouped into two: the first groupconsisting of 41 boys and the second one 43 boys whose mean chronologicalages were 13.2 (1.7) years and 13.0 (1.6) years respectively. The first group wasprimed with 62.5 mg/m2 (low dose testosterone-LDT) and the second groupwith 125 mg/m2 depot T (high dose testosterone-HDT) im one week before thestimulation test. 21 boys out of 36 who have failed in GH stimulation tests afterone dose T (ODT) injection were given three doses of 62.5mg/m2 T (multipledose testosterone-MDT) injections monthly and retested. The patients werefollowed up 2.6 (1.4) years. Results: The mean GH levels increased after prim-ing with both LDT and HDT (p ! 0.001).The increment of mean GH levels byboth LDT and HDT were found to be similar (p = 0.443). The peak GH levelswere found to be elevated above 10 ng/ml in 22/41 (54%) and 26/43 (60%) whoreceived LDT and HDT respectively (p = 0.528). Normal GH responses wereobtained from 12/21 (57%) boys who received MDT. Thirteen of the ODT andsix of the MDT group have reached their final heights which were not signifi-cantly different from target heights (p = 0.421, p = 0.528). The predicted finalheights of the ones who have not reached their final heights yet were also in theexpected range of their target heights (p = 0.918, p = 0.715). Conclusion: LDTis as effective as HDT in priming. We suggest that sometimes MDT will benecessary to get normal responses in GH stimulation tests. Although this sub-ject is still in debate, lack of difference between final heights and target heightsof boys who have normal responses to GH stimulation tests after priming sug-gested that priming is a convenient method in diagnosis of GHD.

39th Annual Meeting of the ESPE Horm Res 2000;53(suppl 2):1–191 35
P2-113 GH Secretion and Action
Mammosomatotroph Adenoma of the PituitaryAssociated with Gigantism and HyperprolactinemiaA. Alikasifoglu* 1; N. Kandemir 1; N. Akalan* 1; N. Yordam 1
1 Hacettepe University, Ihsan Dogramacı Children’s Hospital, Ankara,Turkey
Pituitary tumours, especially invasive or macroadenomas, causing gigantismare rare in children. We describe an 11-year old girl with 6-year history ofgrowth acceleration, left visual loss and seizures due to an invasive mammoso-matotroph macroadenoma and also we present 4.5 years follow-up of thepatient. On physical examination at 11 years of age, her height was 177 cm (+5SD) and weight 89 kg. The enlargement of head, hands and feet was remark-able. Pubertal signs were present with stage II breast development and stage IIIpubic hair. There were complete visual loss and optic atrophy in the left eye.Magnetic resonance imaging (MRI) of the brain showed a tumor with suprasel-lar and parasellar extension. The tumor was fairly aggressive causing extensiveerosion of the sella. Prior to operation GH, PRL, IGF-I and IGFBP3 levels inthe serum were elevated (GH: 51 ng/ml. PRL: 1205 ng/ml (12–20.8), IGF-I:855 ng/ml (70–462), IGFBP3: 11.53 mg/l (2.71–6.34)). GH and PRL levelswere not suppressed during a glucose tolerance test. Serum T3, T4, TSH, ACTHand cortisol levels were normal. The patient was operated on frontotemporalcraniotomy with the subtotal removal of a large tumor adhering to optic nerve.Histologically the tumor was an acidophilic adenoma. After the operation GH,PRL, IGF-I did not drop significantly. Irradiation was applied to the skull post-operatively and then bromocriptine was administered. The initial bromocrip-tine dose was 10 mg/d and increased to 30 mg/d. During the follow-up ACTHdeficiency, secondary hypothyroidism and hypogonadism were developed, andreplacement therapy was given. At 15.5 years of age, her height was 188 cm (+4SD). GH level was still elevated (GH: 11.4 ng/ml), the PRL level had becomenearly normal (PRL: 23 ng/ml). The somatostatin analogue octreotide wasbegun l00 Ìg subcutaneously three times daily. At the last MRI the large size oftumor and adhesions to neighboring structures were prominent. The tumor wasextending the right cavernose sinus, and invading the left optic nerve. Leftinternal carotid artery was surrounded by the tumor which was also extendingto third ventricule floor.
P2-114 GH Secretion and Action
A Comparison of Growth Hormone Immunoactivity andBioactivityH. Mitchell* 1; J. Jones* 1; P.J. Pringle 1; P.C. Hindmarsh 1; C.G.D. Brook 1
1 University College London, London, United Kingdom
Immunoassays (IA) recognise specific epitopes on growth hormone moleculesbut can give no information about the biological activity. The aim of this studywas to compare GH immunoactivity with its in vitro somatogenic and lacto-genic bioactivities and the Immunofunctional Assay (IFA) system, which aimsto reflect GH bioactivity [1]. Immunoactive GH (NETRIA) was measured in45 patient samples obtained during Insulin Tolerance Tests. The MicrocultureTetrazolium Assay technique was used to measure in vitro lactogenic andsomatogenic bioactivities using Nb2 cells (expressing the rat prolactin receptor)and FDC-P1 cells (transfected with the human somatogenic receptor) respec-tively. The IFA is a two-site ELISA designed to assess simultaneously the integ-rity of both binding sites 1 and 2 on GH and hence the capability of GH tocause receptor dimerisation. IFA closely followed the lactogenic bioactivity andthe values from these assays were lower but parallel with the somatogenicbioactivity and NETRIA at lower concentrations. Differences between NE-TRIA and the other assays were exaggerated at peak GH concentrations. UsingBland-Altman plots to compare bioactivity to IFA, a consistent bias wasobserved from basal to peak GH concentrations. When bioactivity was com-pared to NETRIA, increasing bias was observed across a concentration range of0.4 to 85.4 mU/l GH. In this preliminary study it appears that best agreementwas observed between the GH determinations made by the lactogenic bioassayand the immunofunctional assay. NETRIA and somatogenic assays gave high-er readings.
1 Strasburger et al 1996; J. Clin. Endocrinol. Metab. 81;7:2613–2620.
P2-115 GH Secretion and Action
Spontaneous Hypoglycaemia (SHY) in Childhood isAccompanied by Paradoxical Cortisol and GrowthHormone ResponsesP. Hindmarsh 1; K. Hussain* 1; A. Aynsley-Green 1
1 London Centre for Paediatric Endocrinology and Metabolism, GreatOrmond St. Hospital and The Institute of Child Health, UniversityCollege London, UK
Background: Cortisol and growth hormone (GH) play essential roles in thecounter-regulatory responses to hypoglycaemia (HY). In adults, the glycaemicthreshold for the release of these hormones lies at the lower physiological levelof blood glucose. Provocation during the Insulin Tolerence Test (ITT) causesmaximum levels of both cortisol and GH at the time of HY in children. Little isknown of the response of these hormones to SHY. Aim: To study the responsesof cortisol and GH before, at and after fasting-induced SHY. Methods: 40children aged 6 months to 12 years without hyperinsulinism, GH or cortisolinsufficiency were referred for investigation of HY and subjected to a diagnos-tic fast. Ten children (age 6 months to 10 years) became hypoglycaemic: 7 had‘ketotic’ hypoglycaemia, 2 had glycogen storage disease type 1a and 1 had afatty acid oxidation disorder. Plasma cortisol and GH levels were measured atthe beginning of the fast and at hourly intervals. At the point of SHY (laborato-ry blood glucose concentration of !2.6 mmol/l), or if the child became symp-tomatic, blood samples were taken for assay of both hormones. The HY wascorrected and sampling continued at 10-min intervals for 50 minutes. Theresults of the GH were compared to 16 age matched children having an ITT fordiagnostic reasons. Results: Mean plasma cortisol value one hour before theSHY was 430.9 B 86.39 nmol/l (mean B SEM) and GH 5.4 B 1.07 mU/l,(95% confidence interval 3.0–7.87) in the 10 children. At the point of HY meancortisol value was 669 B 90.23 nmol/l (95% confidence interval 464–873) witha corresponding mean GH of 6.8 B 7.96 mU/l (95% confidence interval 1.6–11.45). In the age matched ITT children the GH responses were 29.29 B 1.977at the point of HY (95% confidence interval 25.05–33.25). The differences inthe mean GH responses at the point of HY were highly significant (p ! 0.01). Atten minutes the mean serum cortisol rose to 763 nmol/l; mean serum GHremained low at 3.06 mU/l. At 20, 30, 40 and 50 min the corresponding meanvalues for both hormones were 730, 759, 787, 835 nmol/l and GH 3.1, 3.7, 4.5and 6.2 mU/l respectively. Conclusions: In contrast to the magnitude andtime course of the GH response to HY seen during the ITT, there appears to bea paradoxical counter-regulatory response of GH and cortisol during SHY. Thecortisol response is appropriate and persists for at least 50 minutes, whereasthere is no GH response throughout the period of sampling. The cause of thispoor GH response is not clear, but it may be related to the slow speed of onset ofHY or to the presence of other substrates such as ketone bodies and non-esteri-fied fatty acids that inhibit GH secretion. Further work is needed to elucidatethe aetiology and functional consequence of this paradoxical response.

36 Horm Res 2000;53(suppl 2):1–191 Posters
P2-116 GH Secretion and Action
Results of Growth Hormone Stimulation Tests – ARG +GHRH, PD + GHRH and Insulin Tolerance Test – inAdolescent Healthy VolunteersJ. Donaubauer* 1; J. Kratzsch 1; H. Willgerodt 1; W. Kiess 1; T. Nowak* 2; E. Keller 1
1 University of Leipzig, Leipzig, Germany; 2 Pharmacia and UpjohnGmbH Erlangen, Erlangen, Germany
Objective: Re-evaluation of young adult patients with growth hormone defi-ciency (GHD) after cessation of linear growth is mandatory before committingthem to long-term adult growth hormone (GH) replacement therapy. But thereare only a few normative data published about growth hormone stimulation inadolescents. Patients and Methods: 40 healthy volunteers (20 females, 20males; aged 16–25 years) were tested with arginine (ARG) + growth hormone-releasing hormone (GHRH), pyridostigmine (PD) + GHRH and insulin toler-ance test (ITT), respectively, within eight weeks and a minimum of 48 hbetween each test. Growth hormone was measured using the Auto-DELFIAassay system. In the range of GH levels between 2.9 to 14.1 ng/ml we foundintraassay and interassay coefficients below 4.4%, respectively. Furthermorewe found a sensitivity of 0.03 ng/ml. For GH stimulation with ARG/GHRH weused 0.5 g/kg body weight arginine i.v. and 1 Ìg/kg body weight GHRH as i.v.bolus. For the PD/GHRH test we gave 120 mg Mestinon® orally plus 1 Ìg/kgbody weight GHRH i.v. For the ITT 0.1 IU/kg body weight Actrapid® i.v. wasadministered. Results: After stimulation with the different tests the followingmedian peak GH concentrations were recorded: 51.8 ng/ml (range 8.4–104.8)with ARG/GHRH; 40.4 ng/ml (range 9.1–77.2) with PD/GHRH and 20.3 ng/ml (range 8.8–58.8) with insulin. No side effects were observed with ARG/GHRH. The ITT performed with a relatively low dosage of 0.1 IU/kg bodyweight compared with previous studies led to a decrease of plasma glucose to2.2 mmol/l or even less. Additionally, typical signs of hypoglycaemia, but with-out severe side effects, were observed. After using PD/GHRH we observedclear symptoms due to increased cholinergic tonus in about 20% of the subjectstested. Conclusion: We provide normative data for peak stimulated GH con-centrations in healthy adolescents. The ARG/GHRH test seems to achieve amaximal stimulation of GH secretion with no side effects. The PD/GHRH testseems to be less suited for re-testing because of non-predictable side effects.
P2-117 GH Secretion and Action
Serum Concentrations of 20K Human Growth Hormone(hGH) – Implications for Measuring hGH?P.J. Pringle 1; S. Murphy* 1; M. Mellado* 2; I.C.A.F. Robinson* 3;C.G.D. Brook 1; P.C. Hindmarsh 1
1 University College London, London, UK; 2 Universidad Autonoma,Madrid, Spain; 3 National Institute for Medical Research, London, UK
Human growth hormone (hGH) is secreted from the pituitary in many forms, thepredominant of which is the monomeric 22 kilodalton form (22K) consisting of191 amino acids. The 2nd most abundant form is made up of the 20 kilodalton(20K) hGH, which is generated from an alternative splice site at exon 3 in thehGH gene. This results in a protein 178 amino acids long with internal deletion ofresidues 32–46. Other variants are derived from post-translational changesincluding dimerisation, glycosylation and deamidation. Some of the variabilityarising from the estimation of hGH by immunoassay may be due to the multipleforms of hGH found in the circulation. One solution might be to measure 22KhGH, the predominant form, exclusively. 20K hGH, the 2nd most abundantform, is equipotent for somatogenic bioactivity with 22K and is reported to cir-culate at 5–10% of total concentrations. 22K specific assays would therefore beexpected slightly to underestimate the amount of hGH in the circulation. Usingspecific immunoassays for 20K and 22K hGH, we have identified 4 individuals inwhom serum concentrations of 20K were raised (120%) and sometimes higherthan 22K hGH. Our subjects were healthy Caucasian male volunteers participat-ing in a study approved by the joint UCL/UCLH Committees on the Ethics ofHuman Research investigating the physiology of hGH variants. Blood sampleswere collected before and after administration of saline or GHRH(1–29)NH2(0.5 Ìg/kg). Unstimulated serum concentrations of 20K hGH were detectable inall samples and greater than 22K hGH in 73 of 83. After GHRH(1–29)NH2, thepeak response for both variants occurred concurrently in all subjects and 22K wasgreater than 20K hGH for the remainder of the test in three. In one subject,unstimulated 20K hGH concentrations ranged from 13.2–16.8 ng/ml. 22K hGHranged from 0.5–19.0 ng/ml and was greater in only 2 of 11 unstimulated samples.30 minutes after the peak response, the concentration of 20K was again greater
than 22K hGH due to a sharper decline in 22K hGH concentrations. Serial dilu-tion of samples in the 20K hGH assay showed parallelism. We have observed 20Kto be the predominant form of hGH in some individuals. Using assays calibratedto measure 22K hGH exclusively has the potential to give false negative results.Since 20K has similar somatogenic bioactivity as 22K hGH, this has implicationsfor the investigation of growth hormone secretion.
P2-118 GH Secretion and Action
Erythrocyte Polyamine Responses to Insulin in ShortChildren in Relation to Growth HormoneP.E. Garnier 1; S. Bottari 1; M. Bayle 1; J.L. Bosson 1; F. De Fraipont 1;M. Bost 1
1 CHU de Grenoble, Grenoble, France
Polyamines are related to tissue growth, protein synthesis and nucleic acid metab-olism. Ornithine decarboxylase (ODC) is the rate limiting enzyme of polyaminebiosynthesis. There are many evidences of stimulating effects of insulin andgrowth hormone (GH) on the activity of that enzyme. Objective: To see whetherthere is a response of erythrocytes polyamines, Putrescine (P), spermidine (Spd)and spermine (Sp), to insulin tolerance test (ITT) in GH deficient children (GHD)and in children with idiopathic short stature (ISS) and to compare between twogroups. Patients and Methods: ITT was performed in 13 ISS (8 boys, 5 girls)and 11 GHD (8 boys, 3 girls) aged 5–13.5 years, all prepubertal. P, Spd and Spwere measured in erythrocytes collected at the same time as blood drawn tomeasure GH, using HPLC. GHD were defined by GH peak responses ^20 UI/lduring two separate stimulation tests. Variables in both groups were compared byANOVA. Results: There were no significant variations of Spd or Sp during theITT and the Spd/Sp ratio was not modified. Interestingly, P increased significant-ly from time 0 to 30 min. in 7 ISS with a mean peak at 60 min (p ! 0.0003)(responders). By contrast, there was no response of P in GHD, except a low andlate rise at time 90 min in 3 of them. The difference between two groups wassignificant (p ! 0.025). No clinical or biological data were related to the rise of P inresponders and nothing allowed distinguishing between responders and non-responders ISS. Conclusions: 1. A significant response of P was shown in half ofthe ISS. Therefore, insulin seems able to stimulate P in patients having normalGH secretion. But it is not known why P does not rise in all of them. 2. The profileof the P response was significantly different in GHD and ISS. It might be assumedthat a normal GH secretion is necessary for a positive response of P to ITT,reflecting the influence of GH on ODC activity.
P2-119 GH Secretion and Action
Safety of the Insulin Tolerance Test: Glasgow’s 10 YearExperience with 550 TestsP. Galloway* 1; E. McNeill* 1; C. Hall* 1; M. Donaldson 1; Introducedby M.D.C Donaldson1 Royal Hospital for Sick Children, Glasgow, United Kingdom
In 1991 the Department of Health issued a circular, warning of the hazards ofthe insulin tolerance test (ITT) in the light of several fatalities in the UK. Sincethen many centres have abandoned its use in assessing growth hormone (GH)reserve. We have continued to use the ITT as our first line test, reserving thearginine test for children with epilepsy and those under 5 years. Since 1989, 550ITTs and 185 arginine tests have been performed. The annual numbers of ITTsperformed were 57–77 between 1989 and 1992, and 37–52 since 1993, as ourapproach to GH prescribing has become more conservative. No serious eventshave occurred in association with the ITTs and, in particular, no child has everfitted. Since our Endocrine Nurse specialist took up post in 1995, a strict proto-col has been followed giving Insulin 0.15 U/kg (0.1 U/kg if hypopituitarism issuspected) provided that capillary glucose is 13.5 mmol/l at –30 and 0 min,and venous access is secure. A doctor is present for the first 45 min and thenurse specialist throughout. Reviewing the 223 cases performed since 1995,demonstrated that the glucose nadir was documented at 15 min in 118 cases,0.6–2.2 (1.5, 1.7, 1.9) mmol/l [range and quartiles]. We, therefore, electivelygive 30 ml Lucozade to any hypoglycaemic child (repeated if required), whoseBM falls below 1.8 mmol/l, with intervention before 30 min being the norm.We believe the ITT, which offers a physiological stimulus to GH secretion, issafe if performed by experienced staff during dedicated time in a designatedarea. We have found it easier to administer than arginine, which readilyimpairs venous flow, often requiring recannulation. The problem with GHstimulation testing is not the procedure itself, but who should have it.

39th Annual Meeting of the ESPE Horm Res 2000;53(suppl 2):1–191 37
P2-120 GH Secretion and Action
Physical Exercise Induced GH Secretion in CAHPatients during Corticosteroid TherapyC. Bizzarri* 1; O. Porzio* 1; C. Martinez* 1; S. Benedetti* 1; S. Picone* 1;M. Cappa 1
1 Ospedale Bambino Gesù, Palidoro, Italy
It is well known that substitutive and suppressive corticosteroid treatment hasan inhibiting effect on GH secretion in humans and animals. Growth pattern isa main problem in the management of children with CAH. The aim of ourstudy was to evaluate growth hormone (GH) secretion after a physiologicalstimuli in patients (pts) with CAH. We analysed 47 pts with CAH, 17 males and30 females; mean age was 12 B 5 years. Thirty-nine pts were affected by classi-cal form of CAH, 8 by nonclassical form. All pts were treated with hydrocorti-son and ·-fluorocortison when needed. There were collected clinical and auxo-logical data and bone age for all. Classical Bruce’ test was performed in all pts.We evaluated basal and physical exercise induced GH, 17OHP, ACTH, PRLsecretion after 10’ from the end of the test. GH showed statistical significativecorrelation with bone age (p = 0.025), height (p = 0.01), and height-SDS (p =0.0009). There was any statistically significative correlation between GH peakand neither BMI, nor duration of physical exercise, nor maximum heart rate.PRL serum levels after the test were positively correlated to GH peak (p =0.004). GH peak is positively correlated to basal 17OHP levels (p = 0.02), stim-ulated 17OHP levels (p = 0.03) and stimulated ACTH levels (p = 0.03). Hydro-cortison dose (mg/m2/die) is not correlated with GH peak. These data suggestthat physical exercise induced GH secretion may be a good tool to explore thephysiological GH secretion in a patient that needs substitutive or suppressivecorticosteroid therapy.
P2-121 GH Secretion and Action
Relationships between in vitro and in vivo Responses toGH Treatment in Turner SyndromeA. Whatmore* 1; A.J. Woodall* 1; T. Huynh* 1; J.S. Freeth* 1; V. Tillmann 1; L. Patel* 1; D.A. Price 1; P.E. Clayton 1
1 University of Manchester, Manchester, United Kingdom
The initial and long-term growth response to GH in Turner syndrome (TS) isvariable. Only 25% of this variability can be explained by auxological and treat-ment factors (Ranke et al, 1993). The aim of this study was to assess whether invitro responses to GH in skin fibroblasts might predict clinical response to GH.Subjects: Primary cultures of skin fibroblasts were established from 10 TSgirls treated with GH for a mean 6.4 y (range 2.9 to 10.1) having started GH at amean age of 6.9 y (4.4 to 10.9). Cumulative annual changes in height, weightand BMI SD scores were used as clinical response variables. In vitro Assays:Thymidine incorporation (TI; a measure of cell proliferation) was measured inresponse to hypopituitary serum alone (HPS; 0.1%), HPS + GH (0–5000 ng/ml) and HPS + IGF-1 (10 ng/ml). Activation of mitogen activated proteinkinase (MAPK) was measured in response to both GH (0–2000 ng/ml) andEGF (10 ng/ml). Results: In vivo, the mean change in height SDS per year ontreatment (Tx) was +0.32 (+0.10 to +0.46) and the mean change in BMI SDSper year on Tx was +0.04 (–0.06 to +0.20). In vitro, the TI response to GH(expressed as area under the curve) correlated strongly with the response toIGF-1 (r2 = 0.80, p ! 0.001), but activation of MAPK in response to GH (orEGF) did not correlate with the TI response to either GH or IGF-1. No signifi-cant correlations were found between in vitro and in vivo responses (eithershort or long-term) to GH. However, TI response to HPS (as a measure of basalcell proliferation) was negatively correlated to both mid parental height SDS(p = 0.002) and change in height SDS per year on Tx (p = 0.038). MAPKresponse to EGF was positively correlated to basal BMI SDS (p = 0.016) andMAPK to GH was negatively correlated to change in BMI SDS per year on Tx(p = 0.043). Summary: In this small cohort of TS girls: (1) No significant cor-relations were found between in vitro and in vivo responses to GH, (2) the lackof correlation between MAPK and TI responses to GH may indicate that thesetwo end points of GH action may be regulated differently and (3) MAPK acti-vation was more closely related to changes in body composition than toheight.
P1-122 GH Deficiency or Insensitivity
Early Re-evaluation of GH Secretion Does Not Confirmthe Diagnosis in Most Children with Isolated GHDeficiency (GHD)O. Porzio* 1; M. Cappa 1; M. Maghnie 2; C. Tzialla* 2; C. Bizzarri* 1; Mr. Casini* 3; A. Catte* 3; S. Loche 3
1 Div. di Pediatria, Osp. Bambino Gesù, IRCCS, Palidoro, Italy;2 Clinica Pediatrica, Policlinico S Matteo, IRCCS, Pavia, Italy;3 Servizio di Endocrinologia Pediatrica, Osp. per le Microcitemie,Cagliari, Italy
We and others have shown that a high proportion of patients with childhood onsetGHD are no longer GHD when retested at the attainment of final height. Inaddition, we have shown that MRI findings of the hypothalamic-pituitary area inpatients with GHD rather than the response to pharmacological stimulation maybe the most important criteria upon which the decision to reevaluate the patientshoud be based (JCEM 1999;84:1324–1328). Objective: We hypothesized thatnormalization of the GB response to stimulation in patients with GHD and nor-mal MRI findings might occur much earlier. Patients and Methods: Thirtyprepubertal children (18 boys and 12 girls, age 5.2–10 y) with short stature (heightSDS –2.3 B 0.1; height velocity 4.8 B 0.2 cm/y), a GB response !10 Ìg/l after twostimuli (insulin, clonidine, or arginine), and normal MRI findings of the hypotha-lamic-pituitary area were retested after 1–6 months. Results: A GB response610 Ìg/l after retesting (range 10–21.3) was found in 26 patients, and a GBresponse !10 Ìg/l in only 4 patients (range 7.5–9). In 9 patients the GB response atdiagnosis was !7 Ìg/l to both tests employed. In 8 of them the GB response atretesting ranged from 10–21.3, and was 9.0 Ìg/l in the remainder. We could notfind any biochemical or auxological index at diagnosis which was predictive of theGB response at retesting. Conclusions: Our data indicate that patients withsubnormal GB responses to stimulation and normal MRI findings should be earlyretested to confirm the diagnosis of GHD.
P1-123 GH Deficiency or Insensitivity
Untreated Patients with Primary GH Insensitivity (LaronSyndrome) Have Reduced Maximal Aerobic CapacityZ. Laron 1; M. Gaides* 1; M. Scheinowitz* 1; R. Wagner* 1; I. Ben-Dov* 1
1 Tel Aviv University, Tel Aviv, Israel
The impact of GH deficiency and retreatment on physical performance in GHdeficient patients is not well understood (Woodhouse et al. JCEM1999;84:4570). Patients with Laron syndrome (LS) present a unique model totest the above, as they suffer from primary IGF-I deficiency resulting in dwarf-ism, acro- and organomicria, underdevelopment of the muscle mass and obesi-ty. We assessed maximal aerobic capacity in 8 untreated adults aged 21–48 yr,weight 41–61 kg, height 121–140 cm, BMI of 24–34 kg/m2; and one 10-year-oldgirl treated by IGF-I 180 Ìg/kg/d since age two. All underwent lung functionand incremental cycle ergometry (CPX, MedGraphics, USA) to the limit oftolerance. Lung function in all adults were low normal. Peak aerobic capacity inthe untreated patients revealed:
Absolute valuesm B SD
% Predicted values, m B SD
for age andweight
for age andheight
O2-uptake (ml/min) 909B473 53B20 59B20O2-uptake (ml/min/kg) 18B8 53B18 57B20AT‡ (ml/min) 516B183 31B7 34B7AT‡ (ml/min/kg) 11B3 31B6 33B7Heart rate (b/min) 157B14 – –Work rate (watts) 60B33 – –
‡ AT = Anaerobic threshold.
The IGF-I treated girl achieved near normal aerobic capacity. In conclusion:Maximal oxygen consumption and anaerobic threshold (based on weight orheight) were significantly reduced in all untreated LS patients. There was noventilatory constraint, therefore exercise limitation is assumed to be the resultof longstanding IGF-I deficiency affecting the cardiovascular and/or peripheralmuscular systems. The normal values in the IGF-I treated patient support thisassumption.
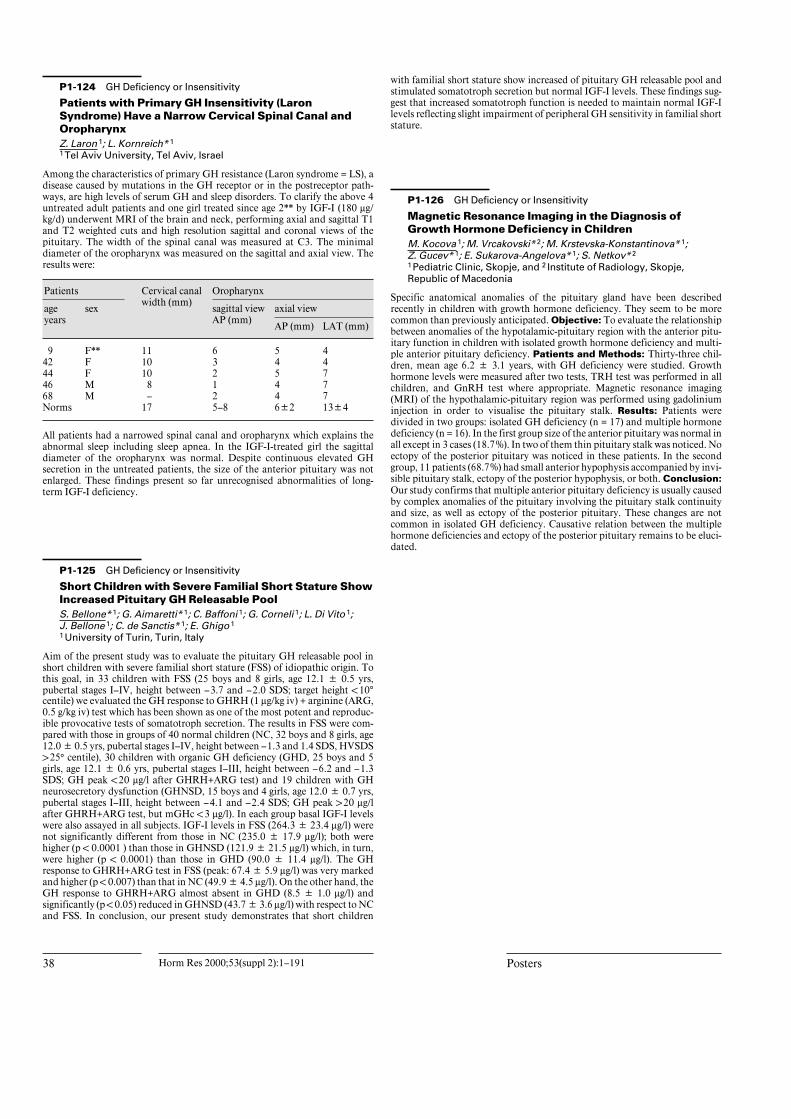
38 Horm Res 2000;53(suppl 2):1–191 Posters
P1-124 GH Deficiency or Insensitivity
Patients with Primary GH Insensitivity (LaronSyndrome) Have a Narrow Cervical Spinal Canal andOropharynxZ. Laron 1; L. Kornreich* 1
1 Tel Aviv University, Tel Aviv, Israel
Among the characteristics of primary GH resistance (Laron syndrome = LS), adisease caused by mutations in the GH receptor or in the postreceptor path-ways, are high levels of serum GH and sleep disorders. To clarify the above 4untreated adult patients and one girl treated since age 2** by IGF-I (180 Ìg/kg/d) underwent MRI of the brain and neck, performing axial and sagittal T1and T2 weighted cuts and high resolution sagittal and coronal views of thepituitary. The width of the spinal canal was measured at C3. The minimaldiameter of the oropharynx was measured on the sagittal and axial view. Theresults were:
Patients
ageyears
sex
Cervical canalwidth (mm)
Oropharynx
sagittal viewAP (mm)
axial view
AP (mm) LAT (mm)
9 F** 11 6 5 442 F 10 3 4 444 F 10 2 5 746 M 8 1 4 768 M – 2 4 7Norms 17 5–8 6B2 13B4
All patients had a narrowed spinal canal and oropharynx which explains theabnormal sleep including sleep apnea. In the IGF-I-treated girl the sagittaldiameter of the oropharynx was normal. Despite continuous elevated GHsecretion in the untreated patients, the size of the anterior pituitary was notenlarged. These findings present so far unrecognised abnormalities of long-term IGF-I deficiency.
P1-125 GH Deficiency or Insensitivity
Short Children with Severe Familial Short Stature ShowIncreased Pituitary GH Releasable PoolS. Bellone* 1; G. Aimaretti* 1; C. Baffoni 1; G. Corneli 1; L. Di Vito 1; J. Bellone 1; C. de Sanctis* 1; E. Ghigo 1
1 University of Turin, Turin, Italy
Aim of the present study was to evaluate the pituitary GH releasable pool inshort children with severe familial short stature (FSS) of idiopathic origin. Tothis goal, in 33 children with FSS (25 boys and 8 girls, age 12.1 B 0.5 yrs,pubertal stages I–IV, height between –3.7 and –2.0 SDS; target height !10°centile) we evaluated the GH response to GHRH (1 Ìg/kg iv) + arginine (ARG,0.5 g/kg iv) test which has been shown as one of the most potent and reproduc-ible provocative tests of somatotroph secretion. The results in FSS were com-pared with those in groups of 40 normal children (NC, 32 boys and 8 girls, age12.0 B 0.5 yrs, pubertal stages I–IV, height between –1.3 and 1.4 SDS, HVSDS125° centile), 30 children with organic GH deficiency (GHD, 25 boys and 5girls, age 12.1 B 0.6 yrs, pubertal stages I–III, height between –6.2 and –1.3SDS; GH peak !20 Ìg/l after GHRH+ARG test) and 19 children with GHneurosecretory dysfunction (GHNSD, 15 boys and 4 girls, age 12.0 B 0.7 yrs,pubertal stages I–III, height between –4.1 and –2.4 SDS; GH peak 120 Ìg/lafter GHRH+ARG test, but mGHc !3 Ìg/l). In each group basal IGF-I levelswere also assayed in all subjects. IGF-I levels in FSS (264.3 B 23.4 Ìg/l) werenot significantly different from those in NC (235.0 B 17.9 Ìg/l); both werehigher (p ! 0.0001 ) than those in GHNSD (121.9 B 21.5 Ìg/l) which, in turn,were higher (p ! 0.0001) than those in GHD (90.0 B 11.4 Ìg/l). The GHresponse to GHRH+ARG test in FSS (peak: 67.4 B 5.9 Ìg/l) was very markedand higher (p ! 0.007) than that in NC (49.9 B 4.5 Ìg/l). On the other hand, theGH response to GHRH+ARG almost absent in GHD (8.5 B 1.0 Ìg/l) andsignificantly (p ! 0.05) reduced in GHNSD (43.7 B 3.6 Ìg/l) with respect to NCand FSS. In conclusion, our present study demonstrates that short children
with familial short stature show increased of pituitary GH releasable pool andstimulated somatotroph secretion but normal IGF-I levels. These findings sug-gest that increased somatotroph function is needed to maintain normal IGF-Ilevels reflecting slight impairment of peripheral GH sensitivity in familial shortstature.
P1-126 GH Deficiency or Insensitivity
Magnetic Resonance Imaging in the Diagnosis ofGrowth Hormone Deficiency in ChildrenM. Kocova 1; M. Vrcakovski* 2; M. Krstevska-Konstantinova* 1; Z. Gucev* 1; E. Sukarova-Angelova* 1; S. Netkov* 2
1 Pediatric Clinic, Skopje, and 2 Institute of Radiology, Skopje,Republic of Macedonia
Specific anatomical anomalies of the pituitary gland have been describedrecently in children with growth hormone deficiency. They seem to be morecommon than previously anticipated. Objective: To evaluate the relationshipbetween anomalies of the hypotalamic-pituitary region with the anterior pitu-itary function in children with isolated growth hormone deficiency and multi-ple anterior pituitary deficiency. Patients and Methods: Thirty-three chil-dren, mean age 6.2 B 3.1 years, with GH deficiency were studied. Growthhormone levels were measured after two tests, TRH test was performed in allchildren, and GnRH test where appropriate. Magnetic resonance imaging(MRI) of the hypothalamic-pituitary region was performed using gadoliniuminjection in order to visualise the pituitary stalk. Results: Patients weredivided in two groups: isolated GH deficiency (n = 17) and multiple hormonedeficiency (n = 16). In the first group size of the anterior pituitary was normal inall except in 3 cases (18.7%). In two of them thin pituitary stalk was noticed. Noectopy of the posterior pituitary was noticed in these patients. In the secondgroup, 11 patients (68.7%) had small anterior hypophysis accompanied by invi-sible pituitary stalk, ectopy of the posterior hypophysis, or both. Conclusion:Our study confirms that multiple anterior pituitary deficiency is usually causedby complex anomalies of the pituitary involving the pituitary stalk continuityand size, as well as ectopy of the posterior pituitary. These changes are notcommon in isolated GH deficiency. Causative relation between the multiplehormone deficiencies and ectopy of the posterior pituitary remains to be eluci-dated.

39th Annual Meeting of the ESPE Horm Res 2000;53(suppl 2):1–191 39
P1-127 GH Deficiency or Insensitivity
The Aetiology of Hypopituitarism Influences thePattern of Growth Hormone (GH) Responses toStimulation TestsS. Shalet 1; C. Lissett* 2; S. Saleem 2; A. Rahim* 2; B. Brennan 2
1 Christie Hospital, Manchester, United Kingdom; 2 United Kingdom
GH provocative tests remain the mainstay for the diagnosis of GH deficiency.At present the insulin tolerance test (ITT) is the ‘gold standard’, however, a vari-ety of other stimulation tests are used in clinical practice. Each necessitates aspecific ‘cut off’ derived from normative data, but there remains a widely heldview that the implications from a ‘failed’ test are independent of the nature ofthe stimulus. We sought to examine if this is the case in individuals with evi-dence of radiation damage to the GH axis. 162 non-acromegalic patients wereidentified who had undergone an AST and an ITT within a 3-month period, aspart of routine testing between 1975 and 1999. They were divided into thosetested before (n = 81, 48 M) and those tested after (n = 81, 36 M) completion ofgrowth and puberty. Patients were considered for inclusion in the study if theyhad a history of cranial irradiation and a GH response to one provocative test of!20 mU/l, taken as indicating that some damage to the GH axis may haveoccurred. The patients were compared with 2 control groups. The first com-prised 35 adults (18 male) for whom ethical committee approval and writtenconsent were obtained. The second control group was derived from our ownhistorical data. It included 16 children (10 male), all of whom were prepubertal.The median peak (range) GH response to the ITT was significantly greater (p !0.0001) than that to the AST in the adult controls: 64.8 (10.7–200) vs. 31.8(2.3–91) mU/l respectively. However, in the patients the GH responses weresimilar (p = 0.27): 5.3 (0.5–66.7) vs. 3.5 (0.5–33.4) mU/l to the ITT and ASTrespectively. In contrast to the pattern seen in the adult controls, the response toan ITT in childhood controls was of similar magnitude (p = 0.5) to that to theAST: 45.5 (21.0–104.0) vs. 50.5 (19.0–140.0) mU/l respectively. However in thepatients, the GH response to the AST was greater than that to the ITT (p !0.0001): 11.2 (1.8–44.8) vs. 7.8 (1.0–47.2) mU/l respectively. In our cohort of162 patients, 28 individuals, 14 children and 14 adults would be excluded fromthe potential benefits of GH therapy, if the AST in addition to the ITT was usedto define GH deficiency. In summary, we have demonstrated that the GHresponse to provocative testing is dependent on the nature of the insult to theGH axis. The GH response to an AST appears to be more resistant to the effectsof irradiation than that to the ITT. When investigating the impact of irradiationon GH secretory status the GH response to an AST may be a less sensitive guideto the functional ability of the GH axis.
P1-128 GH Deficiency or Insensitivity
Diagnostic Reliability of Reduced Number of GHMeasurements after Stimulation with GHRH plusArginineC. Baffoni* 1; S. Bellone* 1; C. Origlia* 1; L. Di Vito* 1; G. Corneli* 1; E. Arvat* 1; E. Ghigo* 1; G. Aimaretti* 1
1 University of Turin, Turin, Italy
Conventionally, the diagnosis of GH deficiency has to be demonstrated by failureof GH response to two provocative tests in childhood (arbitrary cut-off point!7–10 Ìg/l GH peak) and to single insulin-induced hypoglycemia test in adult-hood (arbitrary cut-off point !3 Ìg/l GH peak). Classical procedures of these testsinclude GH measurement every 15 min from baseline up to 90–180 min, i.e. atleast 7–10 assays for each testing session. We have previously shown that GHRHin combination with arginine (ARG) becomes one of the most potent and repro-ducible provocative test to evaluate the maximal secretory capacity of somato-troph cells. GHRH + ARG has been proposed as first step, single provocative testfor the evaluation of somatotroph function provided that appropriate cut-offlimit is assumed (with minimal variation across lifespan). Failure of GH responseto such a potent provocative test indicates that GH secretion is severely impairedallowing diagnosis of GHD. We show herein that the procedure of this test couldbe usefully reduced in clinical practice to three GH assays at 30, 45 and 60minutes after GHRH + arginine (1 Ìg/kg GHRH iv bolus at 0 min followed by0.5 g/kg arginine hydrochloride iv infusion from 0 to +30 min) even avoidingbasal GH measurement. In fact in a large population of normally growing chil-dren and adolescent (n = 92, 66 M and 26 F, age: 11.4 B 0.3 yrs), taking intoaccount data at +30, +45 and +60 min, GH values above the 3rd centile limitoccured in 99%. Similarly in young and middle-aged normal adults (n = 92, 54 Mand 38 F, age: 31.2 B 0.7 yrs), taking into account data at +30, +45 and +60 min,GH values above the 3rd centile limit of normality occurred in 99%. In bothgroups the mean peak of the GH response curve is at +45 min but some antici-pated or delayed responses may occur. Of course, this procedure (3 GH assays) ofperforming GHRH + arginine as single provocative test makes easier the clinicalpractice and further reduces costs. It has to be emphasized that, while the diagno-sis of severe GHD in adulthood relies on single provocative test only, in slowlygrowing short children a normal response to single GHRH + arginine as well astwo classical provocative tests does not rule out the existence of insufficient spon-taneous GH secretion over 24 h and the possibility that these subjects couldbenefit from rhGH treatment.
P1-129 GH Deficiency or Insensitivity
Growth Hormone and Possible GonadotropinDeficiency Associated with Ectodermal Dysplasia,Ectrodactyly, Clefting (EEC) SyndromeA. Bereket* 1; S. Turan* 1; Introduced by F. Darendeliler1 Marmara University, Istanbul, Turkey
EEC syndrome is characterized by ectodermal dysplasia, ectrodactyly and cleftlip/palate occurs sporadically or in autosomal dominant mode of transmission.GH deficiency has been described in two cases with EEC syndrome previously.We present another patient with EEC syndrome who has GH deficiency andmicropenis (possibly due to gonadotropin deficiency). Case Report: 8 3/12 year-old boy was referred for evaluation of micropenis. History was remarkable forpost-term delivery and parental consanguinity. Cleft lip and palate and polydac-tyly of both hands were noted at birth which were operated at 1 year and 5 years ofage respectively. He had frequent episodes of otitis media and aspiration pneu-monia. On physical examination: His height was 111.4 cm (!3%), his weight was22 kg (10%), Span: 97 cm, U/l segment ratio: 1.22. His bone age was 5 years. Inaddition to severe short stature, he has thin, sparse and dry hair, sparse eyebrows,congenital ptosis on the left eye, repaired cleft lip and palate, conical teeth withenamel hypoplasia, short forearm, short fingers, pectus excavatus and bilateraldiffuse rales on auscultation. His stretched penile length was 1.5 ! 0.5 cm, bothtestes were retractile and 2 ml in size. Radiological investigation showed polysyn-dactyly of metacarpals and radioulnar dysplasia. Endocrine evaluation: baselineFSH 0.25, LH !0.1, testosterone: !0.02 ng/ml. After HCG stimulation test tes-tosteron increased to 0.66 ng/ml. Thyroid function tests were within normal lim-its. GH stimulation tests revealed complete GH deficiency. Serum ACTH, corti-sol and urine specific gravity were normal. Penile size reached 3 ! 0.7 cm afterthree monthly injections of testosterone. The patient is presented due to rareassociation of hypopituitarism with EEC syndrome in addition to displayingfeatures novel to this syndrome such as ptosis and radioulnar dysplasia.

P1-130 GH Deficiency or Insensitivity
Growth Hormone Status in Children with SchwartzJampel Syndrome (Chondrodystrophic Myotonia)A. Özön* 1; N. Yordam 1; H. Topalo lu* 1
1 Hacettepe University, Ankara, Turkey
Schwartz-Jampel syndrome is a form of congenital myotonia characterized bychondrodystrophy, blepharophymosis, and severe joint limitation with con-tractures. Small stature of postnatal onset is another feature of the disease.Chondrodystrophy, orthopedic problems and other features are attributed tothe mechanical forces created by myotonia. Objective: To evaluate the growthhormone status in children with Schwartz-Jampel syndrome (SJS) and shortstature. Patients and Methods: Five children (3 males, 2 females) aged 2–16years were analysed. All had a height of less than –2 SDS below the mean (!–5in 2 children). Serum IGF-I and IGFBP-3 levels were determined in all chil-dren. Growth hormone status was evaluated by pharmacological stimulationtests using L-Dopa and clonidine, and a GHRH test was carried out in all chil-dren. Results: Peak growth hormone responses to pharmacological stimula-tion were subnormal in all children (!10 ng/ml). Two children whose heightSDS were below –5 had peak growth hormone levels less than 7 ng/ml (2 and5.75 ng/ml). GH response to GHRH was in the range of 20–50 ng/ml in 30minutes in all patients. Serum IGF-I and IGFBP-3 levels were below 5 percentfor age and sex in all but one of the children. Conclusion: Growth hormonedeficiency may be responsible from short stature at least in some cases withSJS. The deficiency in growth hormone secretion appears to be hypothalamic inorigin.
P1-131 GH Deficiency or Insensitivity
The Quality of Life of Hypopituitary Adult Male PatientsP. Bregani* 1; I. Achutegui* 1; M. Lukezic 1; R. De Angelis* 1; G. Viganò* 1; B. di Natale* 1; G. Chiumello 1
1 Milan, Italy
Historical data of hypopituitary patients report a low frequency of marital rela-tionships and of deep social ties. Aim: To evaluate the efficacy of hormonaltreatment as regards its indirect effects on the quality of life of patients affectedwith hypopituitarism who have received an optimal substitutive therapy.Patients and Methods: 8 male adult patients, mean age 25 years (range 17–36 years), with childhood-onset pituitary deficiencies, 6 of them showing multi-ple pituitary deficiencies and 2 isolated GH deficiency, who have been treatedwith GH and attained their final stature (mean 166 cm, range 154–180 cm) andreceived optimal substitutive therapy for their deficiencies except GH at thetime of the study, filled out the questionnaire by P. Herschbach and G. Hein-riich and had a psychological interview aimed at investigating each featurerelated to the quality of life to a deeper level. Results: The subjects’ perceptionof their physical characteristics results negative only as regards height with theexception of one subject (the tallest in the group); socialization with peersappears satisfying: all subjects state to be at ease sharing leisure time, sports,cultural or humanitarian interests with their friends. Their evaluation of sex-uality and male-female relationships, except in 3 cases, shows dissatisfaction todifferent degrees. Conclusions: This investigation elicits a positive datum:the capacity of coping with friends and peers implies that the subjects havedeveloped a sufficient degree of self-esteem and that body image, though theyresent their rather low stature, it does not negatively affect their global percep-tion of themselves in a significant way. It points also to a negative datum: theirsexual activity and male-female relationship are often associated with feelingsof insecurity and behavioural immaturity. To help these patients to attain moreconfidence and security in their sexual attitudes and behaviour we are planninga medical and psychological intervention.
P2-132 GH Deficiency or Insensitivity
Early Metabolic Changes after GH Withdrawal in Adultswith Childhood-Onset GH DeficiencyM. Lukezic* 1; S. Arrigoni* 1; A. Conversano* 1; F. Minicucci* 1; M. Sblendido* 1; C. Mora* 1; R. De Angelis* 1; B. di Natale* 1; G. Chiumello 1
1 Milan, Italy
At the end of the growth process, GH mainly shows its metabolic actions in theareas of lipid metabolism, cardiac structure and function, bone remodeling andskeletal muscle strength. Aim: To study the presence of metabolic and clinicalchanges in a group of young adults with childhood-onset GH deficiency (GHD)and off GH therapy. Patients and Methods: We studied a group of 11 malehypopituitary patients, mean age 26.6 + 5 years (range 16.9–35.9), with child-hood-onset GHD. 4/11 showed isolated GHD, 7/11 showed multiple pituitaryhormone deficiencies (7 GH, 6 Gn, 5 ACTH, 5 TSH, 2 ADH). All of them hadattained final stature, were reconfirmed GHD after the end of growth (GH peakafter ITT !3 ng/ml) and had stopped GH therapy from 7 months to 18.6 yearsbefore our study. We assessed: plasmatic lipid profile, body composition byDEXA, carotid intima-media thickness (IMT) by ultrasound, cardiac structureand function by means of 2-dimensional, M-mode and Doppler echo-cardiogra-phy. Control patients were found among the healthy population of the same agerange. Results: 1) Lipids: we found moderate hypercholesterolemia (tot choles-terol: 203 + 46 mg/dl); 2) Body composition: patients showed a significantincrease of fat mass % (mean GHD: 24 + 8 %, controls: 16 + 7 %, p = 0.037), morestriking for the limbs department (mean 29%). Lean mass was significantly lowerin the patient group than in the control (GHD: 256 + 38 g/cm, controls: 318 +16 g/cm, p = 0.0003). Bone density was not different from the controls’ both as agroup and in single cases (sBMAD of GHD: 0.170 + 0.02 g/cm3; controls: 0.178 +0.024 g/cm3); 3) IMT: patients showed a significantly higher value of IMT (GHD0.52 + 0.04 mm; controls: 0.46 + 0.02 mm ; p = 0.04); 4) Heart: Patients’ leftventricular mass index was not different from the controls’ as a group (GHD: 75 +25 g/m2, controls: 85 + 15 g/m2) but 2 patients had a significantly lower thannormal value (42 and 46 g/m2 respectively); a similar result was obtained forfractional shortening (patients = controls = 38 + 5%). A significant correlation wasfound between time elapsed from GH withdrawal and IMT, independent fromthe age of the patients and from IGF1 levels (r = 0.7, p = 0.03). For all the otherparameters (dyslipidemia, body composition, heart structure and function) nocorrelation was found both for time from GH withdrawal and for IGF1 levels.Conclusions: Our data show that the main signs of GH withdrawal are relatedto the lipolytic effects of the hormone. In fact, our patients show a mild incrementof body fat deposits, mainly in the limb area, mild hypercholesterolemia andinitial signs of atherosclerosis. We could not demonstrate a clear and homoge-neous impairment of cardiac structure and function in the group.
P2-133 GH Deficiency or Insensitivity
Patient of Isolated Growth Hormone Deficiency Showsa Gene Conversion between GH1 and GH2 GenesC. Quinteiro* 1; L. Castro* 2; L. Loidi* 3; M. de la Fuente* 3; J. Barreiro* 2; M. Pombo 2
1 Unidad de Medicina Molecular; 2 Paediatric Endocrinology andGrowth Unit, CHUS; 3 Molecular Medicine, Santiago de Compostela,Spain
The Growth Hormone (GH) is encoded by the GH1 gene (GHN) which islocated in tandem with the GH2 gene (GHV). Both genes share 95% sequencehomology and are located in chromosome 17q22–24. Unequal crossover andlarge gene deletions in this region have been described in cases of total GHdeficiency. Objective: Investigation of mutations in GH1 gene in a patient ofisolated GH deficiency. Patient and Method: A 13 years old boy with statureless than 2.2 SDS for his chronological age and sex with a decreased heightvelocity, delayed bone age (11 year), a peak of hGH level !10 ng/ml in twostimulation test, serum IGF-1 level was !2.2 SDS. DNA was prepared fromperipheral blood leucocytes using standard procedures. The GH gene wasamplified by PCR and the entire coding region and their intronic flanking areaswere sequenced by the dideoxi method. Results: We found a stretch ofsequence identical to the GH2 gene within exon 4 of the GH1 gene instead ofthe corresponding GH1 sequence. This mutation have probably arisen by asmall gene conversion between the two genes due to their great sequencehomology. Conclusion: This is the first report of a small gene conversionbetween GH1 and GH2 genes in a patient suffering isolated GH deficiency.
40 Horm Res 2000;53(suppl 2):1–191 Posters

39th Annual Meeting of the ESPE Horm Res 2000;53(suppl 2):1–191 41
P2-134 GH Deficiency or Insensitivity
Identification of Severely Growth Hormone Deficient(GHD) Subjects after the End of Pediatric hGHTreatmentA.F. Attanasio 1; S. Howell* 2; W.F. Blum 1; P. Frewer* 1; P.C. Bates* 1;C. Quigley* 1; S.M. Shalet 2
1 Lilly Research Center, Surrey, United Kingdom; 2 Christie Hospital,Manchester, United Kingdom
Therapy with recombinant human growth hormone (hGH) in GHD children isstopped at final height but some subjects may benefit from further treatment.For this purpose, confirmation of GHD diagnosis by stringent criteria is man-datory after termination of paediatric hGH treatment. In a multinational/mul-ticentre study, 167 hypopituitary GHD patients (age range 14–28 years) whohad been diagnosed and treated during childhood were screened by conven-tional GH stimulation tests (insulin, arginine, L-dopa, glucagon, clonidine orGHRH) and by serum IGF-I standard deviation score (SDS). Using a cut-offpoint of 3 Ìg/ml for all stimulation tests, 133 (79.6%) patients were GHD and34 (20.4%) non-GHD. Taking a cut-off point of !2 for IGF-I SDS, 134 (80.2%)patients were GHD. When stimulation test and IGF-I SDS criteria were bothapplied, 120 (71.8%) patients were GHD and 20 (12.0%) non-GHD, but thestimulation test and IGF-I results did not match in 27 (16.2%) patients. Whensimilar analyses were done in subgroups in relation to the most widely usedtests (insulin (n = 111) and arginine (n = 41) combination of the two diagnosticcriteria indicated 72.1% of insulin test patients and 70.7% of arginine testpatients as GHD. Statistically significant trends could be detected in the clini-cal presentation and demography of the patients. Diagnosis was idiopathic for63.3% and organic for 36.7% of the GHD (n = 120) group compared with80.0% idiopathic/20.0% organic for the non-GHD (n = 20) group (p = 0.151).Ratios for isolated GHD / multiple deficiency were 18.3%/81.7% for the GHDgroup compared with 80.0%/20.0% for the non-GHD group (p ! 0.001). In theGHD group 64% were male compared with 90% male in the non-GHD group(p = 0.023). No differences in height SDS were found between the groups. Theage at first GHD diagnosis was lower in the GHD group (8.2 B 3.9 years) thanthe non-GHD group (10.0 B 4.0 years, p = 0.054). The percentages who wereGHD or non-GHD were consistent across study centres. The proportion ofsubjects that will be severely GHD at retest depends primarily on selection, andhence may vary from one study to another. With the cut-offs used here, combi-nation of GH stimulation test (insulin or arginine) and IGF-I SDS give a 70%probability of identifying GHD. The severely GHD subjects were diagnosedearlier in childhood and were more likely to have multiple deficiencies.
P2-135 GH Deficiency or Insensitivity
Interruption of GH Treatment and Early Retesting HasLittle Value in Isolated GH DeficiencyM. Thomas* 1; M. Maes 2; J.P. Bourguignon 3
1 Belgian Study Group for Paediatric Endocrinology, Brussels,Belgium; 2 University of Louvain, Brussels, Belgium; 3 University ofLiège, Liège, Belgium
Different authors reported that GH deficiency in most patients with multiplepituitary hormone deficiency (MPHD) or organic hypopituitarism (OH) wasconfirmed upon GH retesting after cessation of growth while the majority ofpatients with isolated GH deficiency (IGHD) were no longer GH deficient. In aretrospective study of 10 MPHD and 33 IGHD diagnosed initially based onGH peak response to glucagon and to insulin tolerance test !10 ng/ml andheight velocity (HV) !10th centile, all MPHD were confirmed after cessationof growth (GH response !5 ng/ml) whereas only 24% of IGHD were confirmedusing the same criteria. We therefore designed a prospective study with GHretesting after the first year of GH therapy. Initial testing and retesting wereperformed after testosterone priming (50 mg proprionate) in boys and ethinyl-oestradiol (3 ! 50 Ìg) in girls. Glucagon retesting was performed 6 weeks afterinterruption of treatment which was resumed after 3 months except when GHpeak was 110 ng/ml or HV did not fall !5 cm/year. Among 13 patients withMPHD or OH initially investigated at a mean of 12.5 yrs, all were confirmed tobe GH deficient (!10 ng/ml). Among 16 patients with IGHD (pretreatmentHV 4.2 B 0.9 cm/yr, at 10.6 B 2.9 yrs, m B SD), 14 showed a gain in HV!2 cm/yr during the year of GH therapy (8.8 B 1.9 cm/yr) whereas 2 did not(5.1 and 5.3 cm) though their GH response at retesting was still !10 ng/ml.Only 3 had normalized their GH response (110 ng/ml) after a year. Two of
them however showed a marked fall in HV during the year off GH (2.9 and4.0 cm/yr). At a second retesting, GH response was !10 ng/ml and theyresumed GH therapy which increased HV (7.2 and 6.4 cm/yr). Among the 11other patients with GH response !10 ng/ml at retesting, only 8 resumed GHtherapy after 3 months and showed a HV of 7.7 B 1.4 cm/yr during the 2ndyear of therapy. Two patients did not want to resume therapy and their HV fellto 2.5 and 4.1 cm/yr. One patient did not resume therapy because HV appearedsufficient without treatment (6.5 cm/yr). In conclusion, interruption of GHtherapy and retesting after a year in patients with IGHD does not reliably indi-cate normalization of GH secretion. Some patients who still show a GH defi-cient response grow normally without therapy and the few who show a normal-ized response may still need GH therapy to achieve a satisfactory growth rate.
P2-136 GH Deficiency or Insensitivity
Growth Hormone (GH) Deficiency Due to Pituitary StalkInterruption Syndrome in Fanconi’s AnemiaS. Dupuis-Girod* 1; E. Gluckman* 1; J.C. Souberbielle* 1; R. Brauner 1
1 Hôpital Necker-Enfants Malades, Paris, France
Fanconi’s anemia (FA) is a rare autosomal recessive disease of variable pene-tration that may result in multiple congenital abnormalities, bone marrow fail-ure and increased susceptibility to cancer. Retarded growth has long been rec-ognized as one of the basic criteria for its diagnosis, together with a normal ordecreased GH response to pharmacological stimulation in the few casesreported. We analysed 5 patients with FA, growth retardation before chemo-therapy and bone marrow transplantation, biological GH deficiency and pitu-itary stalk interruption syndrome on magnetic resonance imaging. The meanage was 5.6 months and mean height –3.5 SD at diagnosis of FA, and 55months and –4 SD at diagnosis of GH deficiency. All 5 had facial dysmorphyand skeletal abnormalities, 3 boys had kidney abnormalities. Three boys hadmicrophallus and 2 of them had bilateral cryptorchidism, suggesting gonado-tropin deficiency. GH deficiency was isolated in 2 cases, associated with thyro-tropin deficiency in 3 cases. Pituitary adrenal function was normal in the 3cases evaluated. Conclusion: Pituitary stalk interruption syndrome has never before beendescribed in FA. These findings suggest a common genetic origin of the disease.GH treatment produced catch-up growth in 4 cases treated for more than oneyear.
P2-137 GH Deficiency or Insensitivity
Body Composition and Leptin Levels in PrepubertalChildren with Growth Hormone DeficiencyF. Karachaliou 1; E. Vlachopapadopoulou 1; E. Konstadellos* 2; A. Fotinou* 1; E. Paraskaki* 1; S. Michalacos* 1
1 Childrens Hospital P&A Kyriakou, Athens, Greece; 2 GeneralHospital of Nikaia, Athens, Greece
There are several indications that the GH and leptin systems are connected.Leptin is an adipose tissue secreted protein that plays an important role in bodyweight regulation and GH exerts a variety of metabolic actions on adipose tis-sue, including a lipolytic effect. Objective: To evaluate serum leptin concen-trations in association with body fat percentage in a group of GH-deficientchildren who usually present with adiposity. Patients and Methods: Ourstudy group consisted of 23 prepubertal GH deficient children (11 boys) withHSDS !–2, mean (sd) age 9.9 (3.2) years, mean BMI 19.2 (3.7) and meanpercent fat mass 28.8 (4.7)%. Twenty-two sex and age matched healthy childrenwith similar BMI served as controls. Serum leptin concentrations were deter-mined in duplicate by radioimmunoassay (Linco MA) and body compositionwas assessed by bioelectrical impedance (STA/BIA Akern). IGF-I and IGFBP-3serum levels were also determined using RIA (Nichol’s Institute kit). Statisticalanalysis was performed using SPSS 8.0 statistical package. Results: Leptinlevels were significantly increased in GH deficient children compared withcontrols (mean (SEM): 8.9 (1.4) vs 5.6 (1.3) ng/ml, M-W test: –2.1, p = 0.03).Leptin concentrations were positively correlated with BMI (r = 0.72, p ! 0.01)and percent fat mass (r = 0.66, p ! 0.01). No correlation was observed betweenleptin levels and HSDS, IGF-I and IGFBP-3 serum concentrations in GH defi-cient children. Conclusion: Increased serum leptin levels were observed inGH deficient children, which can be attributed to the alteration in body compo-sition associated with GH deficiency.

42 Horm Res 2000;53(suppl 2):1–191 Posters
P2-138 GH Deficiency or Insensitivity
Possible Explanations for the Deleterious Effect ofHeterozygous Mutations of the GH Receptor GeneV. Porra 1; B. Jouret 2; C. Molinas 3; F. Marin 2; E. Bieth 1; M. Tauber 2
1 Growth Research Center, Toulouse, France; 2 Unitéd’Endocrinologie, Génétique et Gynécologie Médicale, Hôpital desEnfants, Toulouse, France
Screening GH receptor (GHR) gene mutations in a population of childrentreated with GH with poor growth response led us to describe 2 patients among34 presenting with an heterozygous mutation of the GHR gene. Patient 1 car-ried a new described mutation in exon 7 Y222H, in the YGEFS motif which isequivalent of the WSXWS motif conserved in other member of the cytokinereceptor family. Patient 2 carried a mutation in exon 6 R161C, alreadydescribed in patients with Laron syndrome and Idiopathic Short Stature (ISS).This raises pathophysiological questions as to the involvement of the mutationin the partial GH resistance. Three hypotheses could be developped: i) theGHR mutation at the heterozygous state is sufficient to explain the phenotype,one of the possible mechanisms being a dominant negative effect. This explana-tion is possible in our families as the girls ( –2.2 SDS for height) inherited themutation from their respective fathers who were also short (–2.2 and –1.9SDS). ii) another defect in the GHR gene potentiates the deleterious effect ofthe mutation: in our 2 families, the patients inherited from their mothers aknown polymorphism in GHR: patient 1, in exon 6 (G168), patient 2, in exon10 (L5261). The exon 6 polymorphism had been shown to be more frequent inISS patients therefore in patient 1, the polymorphism could act as a potentiat-ing factor. iii) for patient 2, it could also be possible that the deleterious effect ofthe GHR mutation is potentiated by another gene involved in growth, as themother was also very short (–3.5 SDS).
P2-139 GH Deficiency or Insensitivity
The Changes of Serum ß-Endorphin Circadian Rhythmin Children with Short StatureL. Szewczyk 1; A. Szewczyk 1; A. Tarkowska 1; D. Witkowski 1
1 Medical Academy, Lublin, Poland
A few data of experiments indicate that opioids play an important role in adifferent neuroendocrine mechanisms and suggest also their neuromodulatoryrole in GH secretion (Bruhn 1989, Grossman 1991 , Wehrenberg 1985). Itseems to be interesting to make an attempt to monitor morning and nightchanges of ß-endorphin (B-E) levels – RIA Kit Incstar Corp. – in children withshort stature. The study concerned 55 children with height !3 percentile, poorheight velocity and delayed bone age and 20 peers with proper height. Growthhormone deficiency (GHD) was determined in 12 children with short stature,in the rest of the group GH secretion after stimulating tests was satisfied. Inchildren with normal height morning B-E level was M. = 12.85 pmol/l, at nightwas M. = 8.97 pmol/l. Morning/night B-E ratio was approximately 1.55. Inchildren with short stature decreased morning B-E levels (M. = 9.64 pmol/l)and nearly normal ranges of nocturnal B-E (M. = 8.61 pmol/l) were determined.Morning/night B-E ratio was decreased app. 1.13. In children with GHD morn-ing B-E levels (M. = 8.56 pmol/l) were distinctly decreased, nocturnal B-E levelswere increased (M. = 10.31 pmol/l) – what makes morning/night B-E ratioreversed = 0.88. It may indicate into inversion of circadian rhythm of B-Esecretion and different neuroendocrine situation in children with insufficientGH secretion. Unknown compensatory mechanisms are probably responsiblefor such a kind of neuroendocrine situation. Conclusion: In children withshort stature, especially with GHD, decreased morning B-E levels and reversedcircadian rhythm of B-E secretion were determined.
P2-140 GH Deficiency or Insensitivity
Neuropsychological Evaluation of Children withIsolated Growth Hormone DeficiencyM. Lukezic* 1; G. Pozzobon* 2; S. Forlani* 2; F. Somajni* 2; C. Lenti* 2;B. di Natale* 2; G. Chiumello 2; R. De Angelis* 2; Introduced byG. Chiumello1 Italy; 2 Milan, Italy
Growth hormone (GH) has both direct and indirect effects on the central ner-vous system, affecting some of its neurocognitive functions. GH receptors havebeen found also in the hippocampus, choroid plexus and putamen. Aim: Toevaluate the presence of neuropsychological dysfunctions in a group of childrenwith isolated GH deficiency. Patients and Methods: We studied a group of 7patients (5 M) with growth hormone deficiency with a mean age of 11.7 years(range 6.8–16.8), 3/7 prepubertal, on GH substitutive therapy at the time of thestudy (duration 2–6 years). Control group children were chosen among thehealthy population and were age and sex matched to the patients. All patientshad a GH peak after two standard stimulation test !5 ng/ml and their brainMRI was normal. The following areas were evaluated: IQ, visuo-spatial andverbal memory (both at short and long term), visuo-spatial and visuo-motorperformances, sorting ability and word fluency. Some of the tests were per-formed with a computer program (FePsy). Results: The patient’s IQ is signifi-cantly lower than the controls’, even if it remains in the normal range. Thedifference is more striking for the verbal IQ and for the comprehension andsimilarities subtests. Moreover, also the ‘recognition of words’, ‘card sortingtasks’ and ‘auditory reaction’ were significantly lower in the patients’ groupthan in the controls’. Conclusions: The presence of significant differences inthe verbal performances may be explained by the role of GH on the left cerebralhemisphere and in particular in the areas involved in the development and/ormainteinance of sorting strategies. GH deficiency in the early age could impairthe development of selective cognitive functions which are not restored by sub-stitutive GH therapy in the short term.
P1-141 GH Treatment
High Dose Growth Hormone Therapy Accelerates BoneAge and Induces Early Puberty in Children withIdiopathic Short StatureG.A. Kamp* 1; J.J.J. Waelkens* 2;S.M.P.F. de Muinck Keizer-Schrama 3; H.A. Delemarre-van de Waal 4; A.H. Zwinderman* 1; J.M. Wit 1
1 Leiden University Medical Centre, Leiden; 2 Catharina Hospital,Eindhoven, Netherlands; 3Sophia’s Children Hospital, Rotterdam;4 Free University, Amsterdam, The Netherlands
Long term GH treatment in children with Idiopathic Short Stature (ISS) resultsin a relative small gain in final height (on average 3–9 cm), which probably doesnot justify the expenses. Objective: We assessed the effect of short-term highdose GH treatment before puberty, in a study design seeking for an optimalcost-benefit ratio, on growth, bone maturation and pubertal onset. Patientsand Methods: Five-year results of a prospective controlled study are reported.26 boys and 9 girls were randomly assigned to a GH treatment group (n = 17) ora control group (n = 18). Inclusion criteria were: no signs of puberty, a height !2SDS, age 4–8 years for girls or 4–10 years for boys, a GH level 10 Ìg/ml afterprovocation and normal body proportions. During the first year of study, chil-dren assigned to the GH treatment group received low doses GH treatmentduring two periods of 3 months. Then, high dose GH treatment (6.0 IU/m2/day) was started and continued for at least two full years. When pubertyoccurred, GH treatment was discontinued at full years of study. Results:Height SDS for chronological age (CA) increased significantly (p ! 0.001) inGH treated children from –2.6 B 0.5 SDS to –1.3 B 0.5 after 2 years high doseGH treatment (n = 17), and to –1.4 B 0.5 SDS after 5 years of study (n = 8). Nochanges in height SDS were observed in controls. A rapid rate of bone matura-tion of 3.6 years/2 years in treated children compared to 2 years/2 years incontrols was observed in response to two years high dose GH treatment (p !0.001). Height SDS for bone age (BA) was not significantly different betweengroups during the study period. GH treated children entered puberty at a signif-icantly earlier age compared to controls (p ! 0.01). Conclusions: Our study ofhigh dose GH treatment before puberty accelerates bone age and induces anearly onset of puberty. These findings may limit the potential therapeutic bene-fit of GH treatment in ISS.

39th Annual Meeting of the ESPE Horm Res 2000;53(suppl 2):1–191 43
P1-142 GH Treatment
Growth and Body Composition during Seven Years ofGrowth Hormone Treatment in Prader-Willi SyndromeA.-C. Lindgren* 1; E.M. Ritzén 1
1 Pediatric Endocrinology Unit, Department of Woman and ChildHealth, Karolinska Hospital, Stockholm, Sweden
Eleven prepubertal children (3–12 years of age) with Prader-Willi Syndromehave been followed during 6–7 years of GH (Genotropin®, Pharmacia &Upjohn AB) treatment. Initially, they all participated in a randomised con-trolled GH trial with two different doses, 0.1 IU/kg/day for 8 subjects and 3subjects served as controls for the first year then treated with GH at a dose of0.2 IU/kg/day, conducted to assess the effects of GH treatment on growth, bodycomposition and behaviour. After 2 years of treatment, all children stoppedGH treatment for 6 months and were then restarted with GH at a dose of0.1 IU/kg/day. During the first year of GH treatment there was a dramaticincrease in height SDS and change in body composition in both groups. Theattained height percentile has persisted during the continued GH treatment–0.3 B 1.4 SDS and –0.8 B 1.5 SDS, respectively. Seven years after start ofGH treatment, the height is within average for age. Three children havereached final height, all within B2 SD of target height. During the first year ofGH treatment, ratio lean/fat improved remarkably, but deteriorated during the6 months period without treatment. Following the restart of GH treatment,ratio lean/fat reimproved and has persisted during treatment. In all 11 patients,fasting insulin, glucose and HbA1c remained within normal ranges duringthese years of GH treatment. In conclusion, GH treatment has proven to have afavourable longterm effect on growth and body composition in subjects withPWS. Treatment should be individualised, and close surveillance of glucosehomeostasis is needed, especially if the patient is severely obese.
P1-143 GH Treatment
Acceptability of the Easyject Autoinjector for theAdministration of Growth HormoneM. Colle 1; Easyject Study Group French Collaborative 1
1 Centre d’Endocrinologie Pédiatrique, Bordeaux, France
The treatment of children of short stature relies upon daily subcutaneous injec-tion of growth hormone (GH) at home. Injection using a classic syringe may bepainful, and can be stressful for both children and their parents. Objective: Toassess the acceptability and ease of use of an autoinjector compared with aclassic syringe for home administration of GH. Patients and Methods: Thestudy population included 77 children (37 boys, 40 girls) requiring GH for GHdeficiency (n = 69), Turner’s syndrome (n = 4) or chronic renal failure (n = 4).All were using a classic syringe prior to the study but used an autoinjector(Easyject™; Ares-Serono) for 30 days during the study. This autoinjector has novisible needle, and insertion of the needle and drug delivery take place with asingle click of a release button. Information concerning pain on injection (vi-sual analogue scale (VAS): 0 = no pain to 100 = maximal pain), ease of injectionand several related variables was obtained from a questionnaire completed bychildren and their families immediately before and after the 30-day study peri-od. Results: Pain on injection was significantly lower with the autoinjector(mean VAS 19.7) than with the syringe (mean VAS 32.7) (p = 0.0001). Theproportion of children reporting no pain during GH administration almostdoubled, from 23.7% with the syringe to 45.5% with the autoinjector (p =0.008). The majority (!50%) of respondents believed that the autoinjector hadimportant advantages over the syringe in terms of the child’s participation intreatment, the precision, speed and pain of injection, and the ability to adjustinjection depth. 73% of respondents felt that reduction in child anxiety was animportant advantage. Use of the autoinjector led to an increase in the propor-tions of children carrying out reconstitution (39% vs. 18.4%), dose adjustment(54.6% vs. 17.1%) and injection (53.3% vs. 25.0%) themselves. 54.7% ofrespondents found dose reconstitution easy, and over 90% found it simple toset the dose, push the button and release the needle from the skin. At the end ofthe study, 98.7% of the children wanted to keep the autoinjector. Conclu-sions: The autoinjector was well accepted by patients and their families, beingconvenient and easy to use, making treatment less stressful and significantlyreducing pain relative to the classic syringe. These benefits also encourage chil-dren to participate more in their own treatment.
P1-144 GH Treatment
rhGH Therapy and Muscular DiseaseG. Tonini 1; M. Lazzerini* 1; F. Bouquet* 2; M. Carrozzi* 2; G. Tamaro* 3; C. Malorgio* 1
1 Department of Paediatrics, 2 Paediatric Neuropsychiatry,3 Biochemical and Clinical Analysis Unit, IRCCS Burlo Garofolo,Trieste, Italy
Introduction: The rhGH therapy in GHD patients can increase muscularenzyme levels (CPK) this increasing could be only slight and transient (acciden-tally i.m. injection?) or more persistent in myositis, as a possible direct effect onmuscle by rhGH. The role of preservative substance, m-cresol, has beenexcluded. We observed two patients treated with rhGH for a GHD with anincrease of transaminases and then CPK, LDH and isoenzymes. Case 1: A8-year-old boy who showed an increase of transaminases (AST from 32 to 119U/l, ALT from 13 to 89 U/l), CPK 1145 U/l, LDH 855 U/l and muscular isoen-zymes (BB1 3.4%, BB2 8.4%, MB4 6.4%) 6 months after the beginning of ther-apy. Hepatitis and thyroid dysfunction were excluded. He was practising inten-sive physical activity without muscular pain or weakness. Both therapy andfitness were withdrawn and after 6 months CPK, LDH and isoenzymes werestill elevated. Occasional i.m. drug administration or a myositis were excluded.Biopsy revealed a mild fibrosis suggesting a primary myopathy. Distrophin wasnormal. Case 2: A 5-year-old boy with short stature and diagnosis of GHD wastreated with rhGH without improvement of height. He showed a significantincrease of transaminases (AST from 52 to 124 U/l, ALT from 26 to 179 U/l)after 1 year of treatment and increased CPK levels from 143 to 1260 U/l duringthe first 4 years of therapy, without symptoms of muscular disease. Before thetherapy were stopped (5th year of rhGH) CPK and LDH were still elevated(respectively 893 and 1214 U/l), also muscular isoenzymes were at pathologicallevels. Muscular biopsy was positive for myopathy (congenital disproportion ofthe different muscular fibres, Dubowitz type), distrophin was normal. Discus-sion and Conclusion: The clinical analysis in these patients suggests thatrhGH seems to be able to reveal a myopathy or to trigger fiber damage in non-symptomatic muscular structural disease (possibly mild), therefore we stronglysuggest to check in rhGH treated patients transaminases and muscularenzymes and, if they are increased, to complete diagnostic procedure also withmuscular biopsy. Moreover the variable increasing of the different isoenzymes,sometimes normal in percentage but all with elevated absolutely levels, second-ary to CPK increase, could also suggest a still not well known mitochondrialeffect of rhGH.
DL – onset GH
Age (years), start GH
44 Horm Res 2000;53(suppl 2):1–191 Posters
P1-145 GH Treatment
Final Height (FH) in Patients Who Received GrowthHormone (GH) Replacement for Radiation-Induced GHDeficiency following the Treatment of ChildhoodLeukaemiaT. Siebler* 1; B. Brennan 1; W.D.J. Ryder* 1; S.M. Shalet 1
1 Christie Hospital NHS Trust, Manchester, United Kingdom
Leukaemia is the most common childhood malignancy. A significant propor-tion of children who received cranial irradiation as part of their treatment isrendered GH deficient (GHD). If left untreated, these patients experience sig-nificant height loss of the order of 2 standard deviation scores (SDS). Aim: Todetermine if children with radiation-induced GHD following the treatment ofacute lymphoblastic and acute myeloid leukaemia benefit from GH replace-ment in terms of linear growth. Patients: 16 patients (12 male) were dividedinto two subgroups: [CI] n = 9, cranial irradiation only (18–24 Gy), [TBI] n = 7,total body irradiation (12–14.4 Gy). Median age at diagnosis was 3.6 (range1.6–9.9) years, at start of GH treatment 12.5 (6.7–14.4) years. Standard GHreplacement dose was 0.5 U/kg/week. The SDS for standing height was calcu-lated and SDS changes were examined for the indicated periods (DL = diagno-sis of leukaemia). The significance of median SDS changes was assessed usingthe Wilcoxon Signed Rank Test. Results: [median (range)]
Observation periodyears
Change in SDS
all patients, n = 16 [CI], n = 9 [TBI], n = 7
7.3 (2.1–10.7)–0.9 (–3.2–0.6) –1.2 (–3.2 – –0.3) –0.8 (–1.6–0.6)
onset GH – FH5.4 (3–9.1)
0.1 (–1.1–1.9) 0.7 (–1.1–1.5) –0.2 (–1.1–1.9)
DL – FH –1.1 (–2.6–2.5) –0.9 (–2.6–0.1) –1.5 (–1.8–2.5)
In all groups, SDS were significantly reduced before onset of GH treatment (DL– onset GH) (p ! 0.05) whereas no significant changes were observed for GHtreatment periods (onset GH – FH) (p 1 0.05), indicating a benefit from GHtreatment. In 11 patients GH status was reassessed after GH therapy. Sevenpatients were severely GH deficient (insulin tolerance test). Conclusions: Inour cohort total height loss was 1.1 SDS, about 1 SDS less than that seen inGH-untreated patients. In children receiving TBI, the adverse impact of GHDis compounded by radiation-induced skeletal dysplasia, however these patientsalso benefited from GH treatment. Patients who received only cranial irradia-tion showed an improvement in height SDS when treated with GH. Reassess-ment of GH status after GH therapy following CI or TBI is important and willhave an impact on the decision, whether or not to continue GH therapy in adultlife.
P1-146 GH Treatment
Changes in Basal Energy Expenditure (BEE) duringShort-Term Growth Hormone (GH) Therapy ReflectExtent of GH Deficiency and Response to GHR. Schweizer* 1; J. Ihle* 1; H.A. Wollmann 1; G. Binder 1; C.P. Schwarze* 1; M.B Ranke 1
1 Children’s Hospital University Tübingen, Tübingen, Germany
Objective: The metabolic effects of GH have not been sufficiently investi-gated in GH deficient (GHD) children. It is known that GH causes lean bodymass (LBM) to increase and fat mass to decrease. Since BEE is known to corre-late with LBM, we hypothesised that changes in BEE would indicate the extentof GHD as well as the response to GH treatment. Patients and Methods: Aspart of a complex anthropometrical evaluation BEE was measured by means ofindirect calorimetry using the device V max 229 (SensorMedics, medilabGmbH & Co, Würzburg). 11 children with GHD were investigated before andafter 4 (B1) months on GH replacement therapy. Characteristics of thepatients at start and on GH are shown in the table. Results: Basal BEE isnormal (98% of value for age and weight). The Changes in BEE are significant(p ! 0.001). Basal BEE does not correlate with basal IGF-I concentrations ormaximum GH levels to two GH stimulation tests. However, there is a signifi-cant correlation between ¢IGF-I and ¢BEE (R = 0.74, p = 0.008) and a positivecorrelation between ¢BEE and ¢height [SDS] (R = 0.58, p = 0.06) (see Fig.).
Median Range
12.1 3.7 16.8Peak GH (Ìg/l) 4.9 1.5 6.7Dose (IE/kg*week) 0.56 0.26 0.67Height (SDS), start –3.04 –4.24 –1.92BMI (SDS), start –0.79 –1.51 3.67IGF-I (SDS), start GH –3.28 –7.62 –0.97Delta IGF-I (Ìg/l) 97 30 228BEE (kcal/d), start GH 994 741 1415Delta BEE (kcal/d) 240 72 463
Conclusions: Changes to GH in lean body mass during short-term GH thera-py reflect the extent of GH deficiency (in terms of IGF-I deficiency). Theobserved changes in BEE suggest, that this parameter is a predictor of GHresponsiveness.
P1-147 GH Treatment
Six Month Results of High Dose hGH Treatment in Girlswith Turner Syndrome in a Multicentre Study in FranceL. di Nicola* 1; C. Gendrel* 2; M. David 3
1 Grandis Biotech France, Clichy, France; 2 Grandis Biotech FranceS.A., Paris, France; 3 Hopital Lyon Sud, Lyon, France
Turner Syndrome is ordinarily treated in France with dose 1.0 IU/kg/week;with this dose (in first year), growth velocity is about 8.0 cm/year and decreasessignificantly during the second year with this standard dose. 30 girls with Turn-er syndrome were enrolled in above study at 20 centres in France and treatedwith Granditropin™ 15 IU (5 mg) liquid formulation of hGH filled in car-tridges of 1.5 ml and daily administrated with Grandipen™ (B.D.). Objective:The aim of this study is to evaluate the beneficial effect of high dose hGHtreatment on growth velocity during the first two years of therapy. Patients willreceive during 24 months, 2.0 IU/kg/w Granditropin™ 15 IU (5 mg). Metabolicparameters (lipids, IGFI, IGFBP3, HbA1C) are evaluated every 6 months andoral glucose tolerance is carried out before start and after 24 months of treat-ment. Patients and Methods: The girls with Turner syndrome received dur-ing two years double dose (2.0 IU/kg/week) compared with the standard recom-mended dose. They had growth retardation (–2 DS) or delayed growth velocity(–1 DS/AC). Results: Data on 13 patients who had completed 6 months thera-py can be presented as follows:
Parameters Baseline 6 months Change 0 to 6
CA 8.9B4.3 9.4B4.3 –HA 5.8B3.0 6.6B3.0 0.8B0.2HSDS 3.4B9.8 2.8B1.0 0.6B0.4HV 4.4B3.3 11.1B3.3 –
Conclusion: After 6 months therapy, growth velocity was significantlyincreased to 11.1 B 3.3 cm/year compared with results obtained with standarddoses (8 cm/y). The local tolerance of new liquid hGH formulation was excel-lent.

39th Annual Meeting of the ESPE Horm Res 2000;53(suppl 2):1–191 45
P1-148 GH Treatment
The KIGS Database-A Tool for Expansion of Knowledgeon Rare DiseasesO. Neyzi* 1; F. Darendeliler 1; I. Sipila 2; P. Larsson* 3; A.C. Lindgren* 3; Introduced by I. Sipila1 Istanbul Faculty of Medicine, Capa, Istanbul, Turkey; 2 UniversityHospital of Helsinki, Finland; 3 Pharmacia & Upjohn, Stockholm,Sweden
The Pharmacia & Upjohn (KIGS) database comprises a large number of chil-dren treated with growth hormone (GH). Objective: This paper presentsgrowth data and the impact of GH treatment in two rare diseases registered inKIGS. Patients and Methods: There are 17 (1 F, 16 M) cases of Aarskogsyndrome and 8 (3 F, 5 M) cases of Williams syndrome registered in KIGS.Growth hormone (Genotropin®) was started at a mean age of 8.7 B 3.2 years ina dose of 0.7 IU/kg/wk in Aarskog syndrome and at a median age of 6.0 B 3years in a dose of 0.6 IU/kg/wk in Williams syndrome. The growth data of thesepatients on Genotropin were evaluated. Results: In Aarskog syndrome meanheight SDS showed an improvement of –2.8 to –2.2 over a median period of3.4 years. Initial height – mean parental height SDS changed by 0.7 over thera-py. Mean height SDS in Williams syndrome improved from –4.0 to –2.1 over amedian period of 5.4 years. Initial height – mean parental height SDSimproved by 1.8 in these patients. No adverse effects related to treatment werenoted in either group. Conclusions: The data on these two relatively largeclusters of patients are examples of documentation provided by the KIGS data-base of long outcome in rare diseases. These cases also represent the firstreported groups on GH treatment for these diseases.
P1-149 GH Treatment
Responses of Bone Turnover Markers and Bone MineralDensity to Growth Hormone (GH) Therapy in Childrenwith Isolated GH Deficiency and Multiple PituitaryHormone DeficiencyN. Kandemir 1; E.N. Gönc* 1; N. Yordam 1
1 Hacettepe University, Ankara, Turkey
A reduction in bone mass has been reported in children with growth hormonedeficiency (GHD). However the effect of the severity of GHD and associationof the other hormone deficiencies on bone status has not been investigatedbefore in children. Objective: To examine the effects of GHD and rhGHreplacement therapy on bone turnover markers and bone mineral density inchildren with isolated GHD (IGHD) and multiple pituitary hormone deficien-cies (MPHD). Patients and Methods: This study was performed in 41 GHDchildren (28 males and 13 females) with a mean age of 10.52 B 3.95 years.Twenty-eight children had IGHD and 13 MPHD. Serum levels of calcium,phosphorus, alkaline phosphatase (ALP), PTH, bone ALP (BALP), carboxy-terminal propeptide of type I procollagen (PICP), osteocalcin (OSC) wereassessed before and every 4 months of therapy for 1 year. Bone mineral density(BMD) was measured before and at 6 and 12 months of therapy. Results: Themean age of the patients with IGHD and MPHD was similar at the time of thediagnosis but peak GH response to stimulation test and height SDS were signif-icantly lower in the MPHD group. There weren’t significant difference at base-line levels of bone turnover markers and BMD between two groups of patients.GH therapy increased significantly serum ALP, BALP, OSC, PIPC levels at 4,8 and 12 months of therapy. BMD was also increased significantly both at 6and 12 months. No difference was found between groups regarding response ofbone turnover markers and BMD to GH therapy. Conclusion: GH therapyhas significant effect on bone formation and mineralization of patients bothwith IGHD and MPHD. However the lack of the difference between groupscan be due to short duration of treatment.
P1-150 GH Treatment
Follow-Up Height of IUGR Children afterDiscontinuation of Growth Hormone TreatmentF. Darendeliler 1; S. Kabatas* 1; R Bundak 1; F. Bas 1; N. Saka 1; H. Günöz 1
1 Department of Pediatrics, Istanbul Faculty of Medicine, Istanbul,Turkey
Growth hormone (GH) treatment has been used in intrauterine growthretarded (IUGR) children to promote growth with success in several short-termclinical trials. Objective: To evaluate the growth data of IUGR children fol-lowing two years of GH therapy. Patients and Methods: 16 children (12 F,4 M) who had received GH (Genotropin®) at age 5.3 (1.3) yrs in a dose of0.2 IU/kg/day for two years (Group I) and 10 (6 F, 4 M) controls of age 4.3(1.7) yrs without treatment (Group 2) were followed after completion of thetrial over a median period of 4 years. Results: Height SDS of the GH treatedgroup showed an increase from –3 (0.5) to –2.0 (0.7) (p ! 0.001) over 2 yrs oftherapy. Off therapy it decreased to –2.9 (0.8) (p: 0.001) at a mean age of 11.1(1.9) yrs. The difference between the initial and recent height SDS in this groupwas not different. Height SDS of the control group, –3.1 (0.9) initially did notchange over the two year observation period. At follow-up 7 control childrenreceived GH in a similar fashion for one year. Height SDS of these cases afterGH therapy increased to –2.6 (1.7) (p: 0.02) and at age 10.7 (1.8) yrs itdecreased to –2.8 (1.3). The overall change in height SDS in this group wasinsignificant. There was no significant difference between the recent heights ofthe two groups at final examination. Conclusions: Height SDS showed a sig-nificant increase on GH therapy in IUGR however decelerated after discontin-uation of therapy. This finding shows that to improve final height GH shouldbe given continuously.
P1-151 GH Treatment
Del (18p) Syndrome with GH-Deficiency: Report of SixCasesO. Jaramillo* 1; C. Sevin* 1; L. Telvi 1; J.-L. Chaussain 1
1 Hôpital Saint-Vincent de Paul, Paris, France
Del 18p was found in 6 patients investigated for short stature from –1,8 to –6SD. All presented with various degrees of facial dysmorphism characteristic ofthe syndrome and an IQ ranging from normal low to severe mental retardation.Endocrine studies demonstrated a partial and isolated GH deficiency (GHD)in 4 patients. MRI scan showed brain abnormalities in 3 of the four patientswith GHD. 2 patients, one with and another without GHD, received rGHreplacement therapy at a dose of 0.4 and 0.9 IU/kg/week, respectively. Heightof both patients showed a significant increase, from –3.8 to –2.25 SD for theformer and –2.25 to the mean for the later. We concluded that GH deficiencymight be a contributing factor to short stature in these patients. Pituitary func-tion has to be evaluated in all patients with the 18p– Syndrome, in order todetect those that may benefit from rGH therapy. Introduction: More than100 cases of 18p– have been reported until recently. Clinical features of thesepatients are variable. Patients presented characteristic dysmorphia with mildto sever mental retardation. A short stature was found constantly. The etiologyof the growth retardation is unknown. We describe 6 patients with short stature(–1.8 to –6 SD) and 18p monosomy (5 cases with del (18p) and 1 case with der18,t(13;18)(q11;p11.2)). Four patients had a partial and isolated GH deficien-cy, and 2 of them showed an abnormal pituitary gland on the MRI examina-tion. Two patients, out of 6, (one with GHD and one with normal GH secre-tion) received GH treatment (0.7–0.9 IU/kg/wk) for 5 years. Height went upfrom –4 SD to –2 SD for the GH deficient patient and from –2 SD to averagefor the non GH deficient patient. To date 8 cases of 18p monosomy associatedwith short stature and GH deficiency have been described. Three of themshowed abnormalities of the development of the pituitary gland. Four patientshad GH treatment with satisfactory results. The two cases reported, in additionto the 4 of the literature, showed that patients with 18p monosomy and shortstature should be considered for GH treatment.

Leptin SD score
Baseline
46 Horm Res 2000;53(suppl 2):1–191 Posters
P1-152 GH Treatment
Quality of Life in Children Treated for Idiopathic ShortStature: A Two Year Controlled Randomized Follow-UpStudyN.C.M. Theunissen* 1; G.A. Kamp* 2; H.M. Koopman* 3; K.A.H. Zwinderman* 4; T.G.C. Vogels* 5; J.M. Wit 2
1 Utrecht University, Utrecht, 2 Leiden University Medical Center,Leiden, 4 Leiden University, Leiden, 5 TNO Prevention and Health,Leiden, The Netherlands
Objective: Changes in Health Related Quality of Life (HRQoL) and self-esteem were studied in children with idiopathic short stature (ISS) participatingin a prospective randomized controlled study on the effect of Growth Hormone(GH) treatment. Sample and Method: The sample consisted of 33 prepuber-tal children (age 4 to 10 years old at start) with ISS (height !–2 SD). The chil-dren were randomly assigned to a treatment or control group. The children ofthe GH treatment group underwent an extensive biochemical assessment toverify their GH responsiveness during the first year. Thereafter, high dose GHtherapy (6 IU/m2
W d) (Pharmacia & Upjohn, Stockholm, Sweden) was startedand given for at least two years. HRQoL and self-esteem were assessed threetimes: shorthly after randomization (T1), after one (T2) and two years (T3).Children with ISS, their parents and the paediatrician completed question-naires. These results were compared to those of a large and representative pop-ulation sample. Results: At T1, children with ISS did not have a lowerHRQoL and self-esteem than the norm population, except for social function-ing as reported by child and parents. Children, parents and paediatricianassessed changes differently; although the paediatrician reported an improve-ment of HRQoL and self-esteem in the children treated for short stature, theparents reported no change, and the children in the treatmente group reportedthe same or sometimes even worse HRQoL or self-esteem than the controlgroup. According to linear regression analyses, changes in HRQoL and selfesteem between T2 and T3 hardly related to growth (objectively measured or asperceived by the child). Instead, changes in several HRQoL and self-esteemscales related to the height appreciation by the child her/himself. Conclu-sions: The assumption that GH treatment improves HRQoL in children withISS could not be supported in this study.
P1-153 GH Treatment
A Randomised, Controlled Study in Short Children BornSmall for Gestational Age (SGA): Effect of GrowthHormone Treatment (GHRx) versus no Treatment onSerum Leptin Levels, Dietary Intake and BodyCompositionN.J.T Arends* 1; W.F. Blum 2; A.C.S. Hokken-Koelega 1
1 Sophia Children’s Hospital, Rotterdam, The Netherlands;2 University of Giessen, Giessen, Germany
Aim: To study baseline values and the effect of 1 year GHRx vs no treatmenton serum leptin levels, dietary intake and body composition in short childrenborn SGA. Patients and Methods: 82 (40 M/42 F) prepubertal children withshort stature (–1.88 SD) born SGA, mean age 5.5 year, were randomised into aGH-group (n = 59) or a control group (n = 23). GH group started with GHRx ina dose of 3 IU/m2/day. Control group remained untreated for 3 years. Leptin,BMI, sum of 4 skinfolds and dietary intake were determined. In a subgroup of39 children body composition was determined by Dual Energy X-ray Absorp-tiometry (DEXA) type Lunar DPX-L PED using the paediatric medium scanmode. Lean body mass (LBM), amount of body fat (Fat) and % body fat (% Fat)were measured. Results:
GH group
at start after 1 year
Control group
at start after 1 year
–0.60 (1.56) –1.16 (1.18) 0.04 (1.08) –0.19 (1.29)BMI SD score –1.36 (1.05) –1.06 (1.04)* –1.08 (1.13) –0.91 (1.08)Skinfolds SD score –1.24 (0.64) –1.89 (0.57)*# –1.03 (1.08) –1.05 (1.01)% fat SD score –1.04 (0.80) –1.69 (0.71)*† –0.92 (1.03) –0.77 (1.04)Fat SD score –1.33 (0.42) –1.56 (0.45) –1.22 (0.52) –1.12 (0.59)LBM SD score –2.64 (0.51) –1.76 (0.46)*# –2.62 (0.48) –2.63 (0.42)
Results are means with SD in parentheses.* Diff. from start (p ! 0.001); # diff. in 1 year change between groups (p ! 0.001); † diff.
in 1 year change between groups (p ! 0.05). Dietary intake increased sign. in GH group butremained unchanged in control group. Leptin, fat mass and LBM at start were sign. lowercompared to normal children.
Conclusion: Leptin levels in short SGA children are lower compared to nor-mal children. GHRx results in a decrease in serum leptin levels, a decrease inthe amount of body fat and an increase in LBM and dietary intake. A furtherdecrease in leptin levels during GHRx may be a trigger to stimulate dietaryintake or GHRx may normalise leptin sensitivity in short SGA children.
P1-154 GH Treatment
Adult Height after Long-Term Recombinant GrowthHormone Treatment for Isolated Idiopathic GrowthHormone Deficiency in Prepubertal Children: Results ofthe French RegistryJ.-C. Carel 1; E. Ecosse* 1; M. Nicolino 2; M. Tauber 3; J. Leger 4; S. Cabrol 5; I. Bastie-Sigeac* 1; J.-L. Chaussain 1; J. Coste* 1
1 Groupe Hospitalier Cochin, Paris, France; 2 Hôpital Debrousse,Lyon, France; 3 Hôpital des Enfants, Toulouse, France; 4 HôpitalRobert Debré, Paris, France; 5 Hôpital Trousseau, Paris, France
We analyzed adult height (AH) results in all 1,855 French prepubertal childrentreated for idiopathic isolated growth hormone (GH) deficiency (GHD): treat-ments were initiated from July 87 to December 92 and interrupted prior toDecember 96. Only 11% of the patients had complete GHD, defined as GHpeaks ! 5 ng/ml at 2 pharmacological tests. Patients were separated accordingto completion of treatment until scheduled end (48%) or early interruption(52%). Recombinant GH was used at a mean (BSD) dose of 0.42 B 0.11 IU/kg/w. AH, defined by a growth velocity !1 cm/yr or a bone age x 16 yr (F) orx 18 yr (M), could be recorded in 75% of the patients. The main results arepresented on the table (mean B SD) and indicate a significant catch-up growthof early interrupters, after cessation of the treatment.
Treated untilscheduled end
Interruptedprematurely
n 881 974Age (yr) 12.1B2.0 11.6B2.1Height SDS –2.7B0.8 –2.7B0.8Growth velocity (cm/yr) 4.4B1.6 4.1B1.4Peak GH 9.3B6.5 10.4B7.8
End of tx Duration of treatment 4.3B1.4 2.9B1.4Height SDS –1.7B0.8 –2.0B0.9
AH % with AH recorded 83% 68%Age (yr) 20.1B2.7 20.3B2.4Height SDS –1.6B0.9 –1.6B1.0
A multivariate analysis of height gain in SDS from initiation of treatment toAH identified the following predictive factors (r2 = 0.54): target height, birthweight, age at baseline (stronger influence in girls), bone age delay at baseline,weight at baseline (negative influence), duration of treatment, use of an initialdose x0.7 IU/kg/w. Treatment associated variables (duration and high initialdose) accounted for 4.6% of outcome variance. Our results indicate thatpatients treated for GHD have a significant spontaneous growth potential thatshould be taken into account when measuring of the effect of GH on AH. Usageof G doses closer to current practices was associated with improved outcome.

N
At start
39th Annual Meeting of the ESPE Horm Res 2000;53(suppl 2):1–191 47
P2-155 GH Treatment
The Transatlantic Gap: Small Differences in Diagnoses –Large Differences in GH TherapyM.B. Ranke 1; M.E. Geffner 1; A.-C. Lindgren 1; A. Lindberg 1; D.M. Brown 1; M. Davenport 1; P. Chatelain 1; W. Cutfield 1; D.A. Price 1; K. Albertsson-Wikland 1; P. Wilton 2
1 University Children’s Hospital, Tuebingen, Germany; 2 For theInternational and the United States Board of KIGS, Germany
By Feb. 2000 a total of 32,229 children were enrolled in KIGS (Pharmacia &Upjohn International Growth Data Base), the world-wide pharmaco-epidemio-logical survey of rhGH. From 1996 to 1999, 2,422 children were enrolled fromthe USA and 5,714 from the ‘rest of the world’ (ROW). It is of interest whethersimilarities or differences prevail. The following diagnostic groups were record-ed; (*: p ! 0.01; ** p ! 0.001) [diagnosis, (KIGS code): N, (%)] US-ROW:1) idiopGHD (1.1.): 1,378 (56.8) – 2,459 (43.1)*; 2) NSD (1.2.): 209 (8.6) – 164(2.9)*; 3) acquGHD (2.2.): 196 (8.1) – 650 (11.4); 4) ISS (3.1.): 144 (5.9) – 271(4.7) ; 5) TS (3.2.1.): 229 (9.3) – 867 (15.1)*. Some characteristics (medians) ofthe patients are listed.
Idiopathic GHD
US ROW
Turner Syndrome
US ROW
1,378 2,459 229 867Sex = male % 72.4 65.4 – –Birth WT SDS –0.5 –0.6** –0.9 –1.1*MPH SDS –0.2 –0.7** 0.3 0.1*Max GH ng/ml 6.3 6.5 11.0 11.5CA Yrs 10.9 10.6 9.7 10.1Height SDS –2.4 –2.4 –2.6 –2.5Height SDS (TS Ra) – – 0.2 0.3GH dose U/kg/wk 0.89 0.62** 1.05 0.94**HV 1st yr cm/yr 9.3 8.7** 7.7 7.5
The most frequently used GH stimulation tests are: USA: Clonidine (22.2%),L-Dopa (20.8%) – ROW: Insulin (26.4%), Arginine (22.3%). Conclusions:Diagnostic paths, indications for treatment and treatment modalities may beinfluenced by medical traditions and directions given by medical administra-tive authorities in various countries. The comparison between GH treatedpatients in the USA and ROW shows a remarkable similarity with regard to thepatient characteristics. Diagnostic procedures and treatment modalities, how-ever, differ. Although standardization in medical practice is desired, outomeresearch in children treated with GH may best be served by heterogeneity ofdata.
P2-156 GH Treatment
Head Circumference in Growth Hormone DeficientChildren – The Effect of Growth Hormone TherapyN. Wright* 1; R. Lockwood* 1; M. Pickering 1; J.K.H. Wales 1;Introduced by JKH Wales1 Sheffield Children’s Hospital, Sheffield, United Kingdom
Objective: It is presumed that children with Growth Hormone (GH) deficien-cy exhibit ‘head sparing’ as in other forms of growth failure. The aim of thisstudy was to examine the effect of GH deficiency and replacement therapy onhead circumference (HC). Methods: Data were obtained for 111 individualstreated for GH deficiency between 1973–1999. Individuals GH deficient fol-lowing surgery/irradiation were excluded. All were measured prospectively bythe same auxologist using identical techniques. Height (Ht), weight and HCwere converted to SDS scores (1990 UK standards). Individuals were dividedinto 3 age bands (!8 years, 8–12 years, 112 years) according to their age whenstarting GH replacement. The mean SDS score for each age group at the start oftreatment was compared with the mean SDS score after 6 months, 1 year, 2years and 4 years using paired t-tests. Results: Children with GH deficiencyhad small heads with sparing of the head compared to the reduction in height.Mean HC SDS –1.67 (95% CI –1.38 to –1.96) compared to mean height SDS–3.37. There was catch up growth in height on GH in all groups but only theyoungest children showed catch up growth of the head.
Age !8 years(n = 33)
Ht SDS HC SDS
Age 8–12 years(n = 22)
Ht SDS HC SDS
Age 112 years(n = 58)
Ht SDS HC SDS
treatment–3.80
(B1.13)–2.17
(B1.45)–3.24
(B1.38)–1.50
(B2.13)–3.17
(B1.22)–1.44
(B1.29)6 months –3.21*
(B1.11)–1.93*
(B1.60)–2.86*
(B1.28)–1.71
(B1.80)–3.07*
(B1.23)–1.45
(B1.31)1 year –2.83*
(B1.17)–1.83*
(B1.59)–2.81*
(B1.35)–1.69
(B1.81)–2.83*
(B1.23)–1.41
(B1.33)2 years –2.31*
(B1.34)–1.55*
(B1.38)–2.68*
(B1.39)–1.57
(B1.87)–2.57*
(B1.37)–1.59
(B1.28)4 years –1.93*
(B1.44)–1.30*
(B1.49)–2.59*
(B1.47)–1.61
(B1.64)–2.51*
(B1.35)–1.41
(B1.89)
* p value for difference between mean SDS score and mean at start treat-ment !0.001.
Conclusions: All age groups showed catch up growth in height but only thegroup who started GH replacement before the age of 8 years showed catch upgrowth of the head. In children who started GH after the age of 8 years therewas no catch up growth of the head, despite catch up growth in height.
P2-157 GH Treatment
Bone Mineral Metabolism in Patients with Turner’sSyndrome at Final Height: GH Therapy and/orEstrogens. Longitudinal DataR. Bergamaschi 1; L. Mazzanti 1; R. Caudarella* 1; E. Scarano* 1; A. Buffa* 1; A. Masi* 1; S. Valgimigli* 2; E. Cacciari 2
1 Department of Pediatrics and 2 3rd Internal Medical Clinic,University of Bologna, Italy
Osteopenia and impaired skeletal growth are characteristic features of patientswith Turner syndrome. To assess the effects of hormone therapies on bonemineral content, we evaluated patients with Turner syndrome at final height.Subjects and Methods: 33 patients, 14–32 yrs of age (BA 15–18 yr), werestudied. All the patients reached final height (FH). Karyotypes were: 45,X in 10patients (30.3%), X-mosaicism in 5 (15.2%) and X-structural abnormalities in18 (54.5%). 22 patients (group A) received GH (1 UI/kg/week) and estrogen(EE) (0.01–0.05 mg/day) therapy for 5.4 B 2.2 yrs (range 2.33–9 yrs) and 11(group B) only estrogens for 9.7 B 1.4 yrs (range 8–12.5 yrs). 27 of these ptswere followed-up for 4 yrs after final height. All the patients underwent pelvicsonography. Bone mass was measured at distal (BMC and BMD) and ultradis-tal (UBMC and UBMD) radius levels by single X-ray absorptiometry. Bloodsamples were obtained to assay: electrolytes, alkaline phosphatase (AP) and itsisoenzymes, PTH, CT, osteocalcin, estradiol, LH and FSH. Electrolytes, creati-nine and citrates were evaluated on a 24-hour urine sample and collagen cross-links in the first morning urine sample. Results: Bone mineral content (BMCand UBMC) was significantly higher in patients treated with GH vs thosetreated with EE alone. The multiple regression analysis showed that BMC wasinfluenced by GH (p ! 0.0001) and EE-therapy (p = 0.006) according to theirduration. The evaluation in follow-up (64 yrs) permitted us to show that BMC,UBMC and UBMD were influenced by EE-therapy according to its duration(p ! 0.001). In our study detectable ovaries with a real function did not seem toexert any influence on bone mineralization. Evaluating hormonal and bio-chemical parameters: alkaline phosphatase, osteocalcin and urine collagencrosslinks correlated negatively with EE-therapy. Conclusions: In conclusionthe main effect on bone mineral metabolism seemed to be exerted by GH,according to its duration. In fact, BMC and UBMC were significantly higher inpatients who reached their final height with GH, than those treated with EE-therapy alone. No influence seemed to be exerted by endogenous estrogens.EE-therapy seemed to influence positively bone mineralization above all atultradistal level (trabecular bone level), confirming their importance in main-taining bone mass. There is probably a threshold for estrogens to effect bonemineral status.

48 Horm Res 2000;53(suppl 2):1–191 Posters
P2-158 GH Treatment
The Predictiveness of Quantitative Tests of GrowthHormone (GH) Secretion for Response to GH Therapyand Final Attained HeightZ. Zadik 1
1 Kaplan Medical Center, Jerusalem, Israel
Growth hormone (GH) secretion is an essential factor mediating normal lineargrowth during childhood. Growth hormone release from the pituitary, is pulsa-tile throughout most of the day. Between the secretory pulses, GH concentra-tions are very low, even in normal individuals. In order to improve the diagnos-tic effectiveness of plasma GH measurements for identification of GH defi-ciency two different approaches have been developed: 1) pharmacological stim-ulation of GH release (PT) and 2) measurement of spontaneous GH secretion(SGS). While failure to respond to 2 different diagnostic GH PT is required forthe diagnosis of GH deficiency (GHD). SGS is still considered a research tool.It is also now theoretically appreciated that body mass, nutritional status, ageand gonadal steroids affect GH secretion. Few recent published consensusguidelines on the diagnosis of GHD consider a uniform and strict cutoff pointfor all PT at all ages. In order to address this question we studied 1515 subjects0.2–60 years of age, 40% females, 411 normal growing children with a largespectrum of pubertal stages, growth velocity and stature we evaluated G statusby 24-hour GH secretion, PT, IGF-I and GH binding protein. Nutritional sta-tus was evaluated in 70% of the children, 73% of the children reached finalheight. We compared the normal response range to arginine, clonidine, gluca-gon and ITT. The effect of Pubertal stage on GH response was also evaluated.GH response to PT was high in the first months of life similar to that found atthe pubertal age. Similar to SGS, PT changes with age. IGF-I follows a similarpattern in children 12 years. In children !2 years both IGF-I and GHBP arelow while PT is high denoting GH resistance. GH response clonidine arginineand ITT. Nutritional status affects the normative range of PT and SGS, nocorrection factor was is useful in obese patients, but IGF-I was helpful. follows asimilar. GH secretion by PT and SGS is not an all or non phenomenon butrather a continuum from severe deficiency to very high levels, as is growthvelocity and any parameter studied. In 5% of normally growing children (at anyheight or growth rate), GH secretion is in the GHD range. In some of thesepatients, GH secretion normalizes once they enter puberty. There producibilityof the SGS was greater than that of the PS (0.759 vs. 0.525 respectively). Insome patients GH response was consistently lower to arginine but not to cloni-dine implementing the possibility of a specific mechanism behind the differ-ential effect. Performing 4 PT in a patient reduced an erroneous diagnosis to!4%. Markers of GH status correlate with growth velocity (PS r = 0.145, SGSr = 0.453), but not with final height. It is concluded that in order to avoid depri-vation of therapy from patients in need, normative data should be specific toage, pubertal status and PS. In spite of the relatively low reproducibility of thePT, they are the easiest to perform. A combination of clinical evaluation, PT,IGF-I and nutritional status will help us to choose the best population for GHtherapy.
P2-159 GH Treatment
Final Height of Growth Hormone (GH)-Treated ChildrenAccording to Pituitary Magnetic Resonance Imagingand GH SecretionS. Rouleau* 1; R. Coutant* 1; J.M. Limal 1
1 University Hospital, Angers, France
Final height of 64 GH-treated children was analyzed according to the pituitaryMRI findings in addition to the GH response to provocative tests. Forty-threetreated children had idiopathic (non-acquired) GH deficiency (GHD), eithertotal (GH peak below 5 Ìg/l, n = 15) or partial (GH peak between 5 and 10 Ìg/l,n = 28). Congenital hypothalamic-pituitary abnormalities (stalk agenesis) werefound in 9 children with total GHD, the others had normal MRI. To study therole of GHD in the short stature of patients diagnosed with partial GHD, wecompared growth of these children to that of 21 treated and 42 untreated chil-dren with idiopathic short stature (ISS, GH peak over 10 Ìg/l). Mean GH dosewas 0.47 IU/kg/week given for a mean of 4.5 years. Children with total GHDwere shorter and younger at the time of evaluation than those with partialGHD. Although final heights were similar (–1.4 B 0.9 vs. –1.5 B 0.9 SDS,
total vs. partial GHD), height gain was better in children with total GHD (p !0.05). However, only patients with total GHD and abnormal pituitary MRIhad a better height gain than those with partial GHD (+2.9 B 1.0 vs. +1.3 B0.7 SDS, p ! 0.01). These results underline the heterogeneity of the idiopathicGHD group. By contrast, children with ISS and those with partial GHD hadcomparable auxological characteristics, age at evaluation, and target height.Treated children with ISS or partial GHD had similar final height (–1.8 B 0.6SDS vs. –1.5 B 0.9 SDS, p 1 0.05) and height gain (+1.0 B 0.7 SDS vs. +1.3 B0.7 SDS, p 1 0.05), whereas untreated children with ISS had a final height of–1.9 B 0.9 SDS (p 1 0.05, untreated vs. treated ISS) and a height gain of +0.6B 0.8 SDS (p = 0.05, untreated vs. treated ISS). In a multivariate analysis offinal height in children with either partial GHD or ISS adjusting for height SDSat evaluation, GH treatment was significantly associated (p = 0.008) with a gainof 0.36 SDS (for ISS) to 0.50 SDS (for partial GHD). Our observations suggestthat the short stature of many of the children diagnosed with partial GHD ispartly idiopathic. They should be treated with higher GH dose than that cur-rently used in GHD in order to reach a clinically significant height gain.
P2-160 GH Treatment
Growth Hormone (GH) Administration and Quality ofLife in Children with GH DeficiencyM. Manrique* 1; Introduced by C. Azcona1 Madrid, Spain
Introduction: Administration of GH subcutaneously on a daily basis can beannoying and painful and therefore interfere, in some cases, with an adequatetherapy compliance and quality of life. Objective: To contrast the administra-tion of GH with and without needle, and quality of life of patients. Materialand Methods: 20 children, (6 females and 14 males) with GH deficiency(GHD). GHD was diagnosed as GH peak level !10 ng/ml after two pharmaco-logical studies. A 21-item questionnaire was performed to assess psychologicalresponses to daily administration of GH focus on therapy compliance, opinionon ease of preparation, affective responses to GH administration, side effectsand overall preference. The items were as follows: Are you concerned aboutbeing shorter than your friends? Do you have less physical endurance thanyours friends? Do your friends keep you apart from their games? Would youlike to be taller? Would you prefer to inject the GH dose by yourself? Wouldyou prefer a GH device without needles? Do you find troublesome the dailyGH administration? Do you prepare the GH by yourself? What do you think ofhaving to prepare the GH dose? Who is the person in charge of administeringthe GH dose to you? How many days did you forget the GH administration inthe last month? Did you postpone the GH administration? Why did you post-pone the GH administration? Did you find GH administration to be painful?How do you feel whenever you think of having to receive the GH dose or whenyou receive the dose? How often did the GH administration cause bruising inthe last month? Is the bruising painful? How often did the GH administrationcause bleeding in the last month? Do you think that the method you use isaccurate? Do you think that the device could be improved? Has you social lifebeen altered by the method of GH administration? Results: I: 20 yes, 0 no. II:2 yes, 18 no. III: 10 yes, 10 no. IV: 20 yes, 0 no. V: 1 yes, 18 no. It doesn’tmatter. VI: 2 no, 18 yes. VII: 7 yes, 13 no. VIII: 6 yes, 14 no. IX: 11 easy, 9complex. X: 8 the patient, 9 the parents. XI: 1–2 days 6, 3–4 days 5, none 9.XII: 9 yes, 11 not. XIII: holidays 7, to leave out 4. XIV: 9 yes, 11 no. XV: 11wrong, 9 not wrong. XVI: a) without needle: Rarely 4, frequent 14, not 2; b)with needle less frequent. XVII: 8 yes, 10 no. XVIII: a) without needle: 2 adrop; 8 sometimes; rest not; b) with needle less frequent. XIX: a) without nee-dle: Yes 10, doubtful 10; b) with needle is more accurate. XX: 13 yes, 7 don’tknow. XXI: 4 yes, 16 no. Conclusions: Children usually prefer a system with-out needle. If the method without needle is adequately learned does not causemany problems, but bruising. The system with needle causes more rejectionand pain, bruising is less frequent, it also produces bleeding, the dose is alwayswell injected and there are less problems to measure the doses.

39th Annual Meeting of the ESPE Horm Res 2000;53(suppl 2):1–191 49
P2-161 GH Treatment
Bone Mass and Growth Hormone Treatment of YoungAdult Survivors of Childhood LymphoblasticLeukaemiaS.C.C.M. van Coeverden* 1; C.M. de Ridder* 1; J.J.G. Hoorweg-Nijman 1; G. Kardos* 2;H.A. Delemarre-van de Waal 1
1 Institute of Endocrinology, Reproduction and Metabolism and2 Department of Pediatrics, University Hospital Vrije Universiteit,Amsterdam, The Netherlands
Objectives: The aim of our study was to evaluate bone mass developmentduring growth hormone (GH) treatment in young adults with a history of acutelymphoblastic leukaemia (ALL). Patients and Methods: Bone mineral den-sity (BMD g/cm2) of 4 males and 2 females, aged 24.1 B 3.5, who had a symp-tom-free interval of at least 5 years were evaluated. Bone mineral density (g/cm2) of the lumbar spine, the femoral neck and trochanter, and the total bodywere measured at t = –30, t = 0, t = +6, t = +12 and t = +21 months, using dualenergy X-ray absorptiometry with the Hologic 2000 QDR. BMD was expressedas SOS adjusted for sex and age. A BMO value below –1 SD at least at 2 skeletallocations was one of the inclusion criteria for GH therapy. Five patients havehad cranial radiotherapy of the skull during ALL treatment. Growth hormonedeficiency (GHD) was diagnosed in 3 subjects, defined as the maximum levelof GH was below 9 mU/l after stimulation with growth hormone releasing hor-mone or after an insuline tolerance test. During treatment with GH for 21months (t = 0–t = 21), the level of insulin like growth factor (IGF-I) was heldconstant at + 1 SD according to age and sex. Results: One GHD subject hadcomparable BMD values of the lumbar spine at t = –30, 0 and +12 months, butthe BMD decreased during the last 9 months of GH treatment. BMD of thelumbar spine of the additional 5 subjects decreased in the period before GHtreatment. BMD increased in all 5 subjects in the first year of GH treatment.Between 12 and 21 months of GH treatment, BMD decreased again in 4 out ofthese 5 subjects. Similar results were found for the other skeletal sites. In sum-mary, bone mass remained at the same level after the 21 months of GH treat-ment in the young adults with a history of acute lymphoblastic leukaemia, butbone mass decreased in the 2.5 years before the GH therapy. Conclusion: Theincrease in bone mass during the first year of GH therapy did not persist duringthe 9 months thereafter in 5 out of 6 young adults with a history of acute lym-phoblastic leukaemia.
P2-162 GH Treatment
Acute Pancreatitis after Growth Hormone Treatment:Disease or Treatment LinkedC. de Beaufort* 1; P. Beck* 1; R. Seligman* 1; Introduced byC. Heinrichs1 Clinique Pediatrique, Luxembourg, Luxembourg
Acute pancreatitis is rarely observed in association with growth hormone treat-ment (about 10 cases in literature: Turner, GHD, idiopathic short stature).Objective: Case description of acute pancreatitis after the start of growth hor-mone treatment. Case History: Change of the growth chart over a 2 year peri-od and progressive tiredness in a 7 year old girl lead to the diagnosis of partialgrowth hormone deficiency (two insufficient stimulation tests max GH 5.8 ng/ml, a reduced IGF1 level). Further hormonal testing revealed no abnormalitiesand growth hormone treatment was started. Shortly afterwards she developedan acute pancreatitis with amylase of 788 U/I and lipase of 375 U/I. Growthhormone treatment was stopped. Further analysis revealed major metabolicdisturbances and the diagnosis of a mitochondrial cytopathy (deficiency ofcomplex I), confirmed by genetic analysis (mutation T323l). Pancreatic inflam-mation regressed and specific cofactor treatment was started. Conclusion:Increased anabolism, induced by growth hormone treatment in a child withgrowth hormone deficiency and a mitochondrial cytopathy, probably causedthe pancreatitis in this case.
P3-163 GH Treatment
Growth Efficacy and Metabolic Changes after TwoYears of Growth Hormone Treatment (GH) in PatientsBorn Small for Gestational Age (SGA) Are NotDependent of the GH Secretory StatusC. Luzuriaga* 1; M. Oyarzbal 2; J. Bel* 3; M. Granada* 3; A. Tejerina* 1;M. Chueca* 2; R.M. Espadero* 4; A. Ulied* 4
1 University Hospital Valdecilla, Santander, Spain; 2 Hospital Virgendel Camino, Pamplona, Spain; 3 Hospital Germans Tras i Pujol,Badalona, Spain; 4 Pharmacia and Upjohn, Sant Cugat del Vallès,Spain
Objective: To assess growth and metabolic response (IGF-I, IGFBP-3,IGFBP-1, GHBP) over two years of GH treatment (Genotropin®) in patientsborn SGA compared with a non-treated control group. Subjects and Meth-ods: Inclusion criteria: Boys and girls with birth weight and/or length !P10.Gestational age 135 weeks. Height ^3rd percentile (Tanner). Height velocity(HV) ^50 percentile in relation to bone age. Bone age (BA) 3–9 years for girlsand 3–10 years for boys. Height control at least 6 months prior to inclusion.Patients were selected by GH secretion. Patients with GH peak 110 ng/ml (GHpharmacological test: exercise + propranolol) underwent a night GH secretionprofile. Eight patients with neurosecretory dysfunction diagnosis (NSD), (GHpeak 110 ng/ml and integrated secretion (IS) !3 ng/ml) were treated withrhGH at dosage of 1.4 IU/kg/week. Patients with GH normosecretion (GHpeak 110 ng/ml and IS 13 ng/ml) were randomised to GH treatment at thesame dosage, 17 patients (NS-treat), or to no GH treatment, 17 patients (con-trol group). Results: Gestational age, birth weight and length were similar andnot statistically different in all three groups. At baseline age (mean and SD) was8.0 (2.1); 9.2 (2.4) and 9.4 (2.0) years and height SDS was: –2.6 (0.4), –2.5 (0.3)and –2.3 (0.4) in NSD, NS-treat and control group respectively. BA wasdelayed in all groups. HV (cm/year): 4.8 (0.8), 4.4 (1.6), and 4.8 (0.9); was notstatistically different. IGF-I in SDS was: –2.3 (0.7), –1.7 (1.1) and –1.4 (1.0). At1 year height SDS changed to –1.7 (0.5), –1.7 (0.4) in the treated groups andremained unchanged in controls: –2.3 (0.4), p ! 0.0001. At 2 years height SDSwas –1.2 (0.5), –1.3 (0.6) in the treated groups and –2.2 (0.6) in controls, p !0.0001. IGF-I SDS values at 1 year were –0.4 (0.8), 0.8 (1.1) in treated groupsand –1.2 (1.5) in controls. At 2 years IGF-I SDS was 1.3 (0.5), 1.5 (0.7) and –0.3(1.8) respectively; differences were found between baseline and 24-month val-ues in the treated groups: p ! 0.000 and not significant in the control group.Mean GHBP values did not change throughout the study period. Conclusion:Two-year growth hormone treatment improved height SDS and IGF-I SDS inSGA children compared to the non-GH-treated control group, regardless ofprevious GH secretory status.
P3-164 GH Treatment
Final Height after Two Regimens of rhGH TreatmentG. Radetti* 1; F. Buzi 2; B. Felappi* 2; C. Paganini* 1
1 Regional Hospital of Bolzano, Bolzano, Italy; 2 University of Brescia,Brescia, Italy
Objective: To evaluate the effects of two regimens of rhGH treatment on thefinal height of GH deficient children. Patients and Methods: Out of twocohorts of GH deficient patients (cohort A: 85 patients treated in Bolzano, witha weekly rhGH dose of 1 IU/kg body weight; cohort B: 73 patients treated inBrescia, with a weekly rhGH dose of 0.5 IU/kg body weight), we selected 2groups (group A and group B respectively), each including 12 patients who atthe beginning of treatment were matched for age (mean B SD, group A: 12.2 B1.8 years; group B: 12.1 B 2.1 years; NS), sex (2 girls and 10 boys in each group)and height SDS (group A: –2.15 B 0.61; group B –2.33 B 0.44; NS). Finalheight was assessed when bone age was more than 17 years in boys and 15 yearsin girls and when no further height gain was observed in the last 6 months.Results: Final height was 170.12 B 5.17 cm (height SDS –0.48 B 0.33) ingroup A and 164.54 B 8.92 cm (height SDS 1.37 B 0.95) in group B (p ! 0.01).The height gain (the difference in final height SDS minus height SDS at thebeginning of treatment) was 1.69 B 0.78 in group A and 0.96 B 0.93 in group B(p ! 0.05). The difference between the final height and the target height was1.34 B 2.39 cm (0.19 B 0.35 SD) in group A and –5.06 B 9.39 cm (–1.03 B1.46 SD) in group B (p ! 0.05 for both). Conclusions: Children treated withhigher doses of rhGH show higher final height and height gain, while reachingthe target height. The economical impact of this kind of treatment, however,has to be taken into account when evaluating the results.

Height for CA at diagnosis
Height for CA
50 Horm Res 2000;53(suppl 2):1–191 Posters
P3-165 GH Treatment
Different Influence of Genetic Background on FinalHeight in Male and Female Patients Treated for GHDeficiencyS. Gualandi* 1; S. Zucchini 1; M. Bal* 1; M. Gennari* 1; E. Scarano* 1;S. Nanni* 1; E. Cacciari 1
1 Department of Pediatrics, University of Bologna, Bologna, Italy
To evaluate the influence of father and mother’s height on final results afterhGH treatment in GH deficient subjects, we retrospectively studied 109 sub-jects (73 M, 36 F; 69 prepurbertal, 40 pubertal) affected by idiopathic and iso-lated GH deficiency after final height (FH) was reached. All subjects, born atterm with appropriate birth weight, were treated with hGH at a constant doseof 20 U/m2/week for a median period of 56 months (range 36–132). At diagno-sis mean age was 11.0 B 2.09 years. We separately evaluated in males andfemales the influence on FH of parental height and of various auxologicalparameters. Results: Males (SDS): height for CA at diagnosis –2.00 B 0.42;FH –1.12 B 0.76. Females (SDS): height for CA at diagnosis –2.22 B 0.48; FH–1.37 B 0.79. Correlations between FH and various auxological parameters:
Males (n = 73) Females (n = 36)
r = 0.39 p ! 0.0001 r = 0.47 p ! 0.005Height for BA at diagnosis r = 0.20 p ! 0.05 NSFather’s height r = 0.40 p ! 0.0001 NSMother’s height r = 0.45 p ! 0.0001 NS
mother’s height height for CAr2 = 0.22 p ! 0.0001 r2 = 0.20 p ! 0.01father’s height
Multiple regression analysis r2 change = 0.06 p ! 0.05height for CAr2 change = 0.07 p ! 0.01
By considering response to treatment (FH-height at diagnosis) multiple regres-sion analysis identified in males the variable father’s height (r2 = 0.13; p ! 0.005),while in females no influencing variable was found. In both males and females nocorrelation was found between FH and therapy duration, GH peaks after phar-macological tests or mean nocturnal GH concentration and growth velocity after1 year of treatment. Conclusions: In male and female subjects final heightafter GH treatment was differently influenced by the genetic background. Inmales the final result was partly explained by mother and father’s height, whilein females the variable considered seemed to have little influence.
P3-166 GH Treatment
Growth Hormone (GH) Treatment in Children withHuman Immunodeficiency Virus (HIV)-AssociatedFailure to ThriveG. Pinto 1; S. Blanche* 1; I. Thiriet* 1; J.C. Souberbielle* 1; O. Goulet* 1; R. Brauner 1
1 Hôpital Necker-Enfants Malades and Laboratoires Serono, Paris,France
The frequency of wasting syndrome has decreased dramatically since the intro-duction of triple drug therapy. However, some patients still have a decreasedgrowth rate despite efficient therapy and nutritional support. This preliminarystudy was done to check the acceptability of a daily subcutaneous injection ofGH and to obtain initial data on its tolerance and efficacy in children withHIV-associated growth failure. GH (Saizen®; 0.2 IU/kg/day) was given for 28days to 5 prepubertal children with HIV-associated growth failure and normalOH peak after stimulation, before using triple drug therapy. Three of themwere on enteral tube feeding. There were significant changes (p ! 0.05), com-pared to before treatment, in weight (0.86 B 0.25 kg), plasma insulin-likegrowth factor I (IGF I, 117 B 25 ng/ml) and its GH-dependent binding protein(IGFBP-3) (0.54 B 0.06 mg/l) concentrations. The changes in fat free mass(1.22 B 0.44 kg) and plasma leptin (–1.8 B 1.7 ng/ml) concentrations were notsignificant. The laboratory parameters surveyed remained in the normal range.Mean CD4 lymphocytes count increased from 114 to 132/mm3 (NS) and meanplasma virus load (log RNA HIV/ml) from 4.95 to 5.1 (NS). Conclusion: GHtreatment was well accepted, with no adverse effects. GH can produce signifi-cant short term increases in weight and in plasma IGF I and IGFBP-3 concen-trations. Other studies using more prolonged treatment are needed to evaluatethe effects of GH therapy on growth rate and immune function.
P3-167 GH Treatment
Birth Weight Affects Final Height in Patients Treated forGH Deficiency: Differences between Males and FemalesS. Zucchini 1; A. Cicognani 1; P. Pirazzoli 1; S. Gualandi* 1; M. Gennari* 1; D. Tassinari 1; M. Bal* 1; E. Cacciari 1
1 Department of Pediatrics, University of Bologna, Italy
To assess possible relationships between birth weight (BW) and final height(FH) in patients treated for GH deficiency, we examined final results in 148GH-treated patients (age at diagnosis 11.1 B 2.1 years; 94 males, 54 females)affected by idiopathic and isolated GH deficiency. They were born at termsmall (BW !3rd centile, 2,369 B 278 g, n = 30), appropriate (BW 3rd–90thcentile, 3,207 B 299 g, n = 110) or large for gestational age (BW 190th centile,4,225 B 160 g, n = 8). All patients reached FH and were treated with hGH at adose of 20 U/m2/week for a median period of 59 months (36–132). The 3groups of patients started therapy at a similar height for chronological and boneage and had comparable target height. Results:
Small (n = 30) Normal (n = 110) Large (n = 8)
–2.14 (0.65) –2.02 (0.59) –2.16 (0.55)Height for BA –0.97 (1.35) –0.56 (1.27) –0.71 (1.02)Target height –1.31 (1.05) –1.01 (1.0) –1.19 (1.13)Final height* –1.77B0.87 –1.20B0.78 –0.63B0.83Height increment** 0.38 (1.34) 0.84 (0.99) 1.72 (1.55)
Data are expressed in SDS as meanBSD or median (IQR).* p ! 0.01. ** p ! 0.001.
In the whole group of patients there was a positive correlation between BW andFH (r = 0.33; p ! 0.0001), height increment during therapy (r = 0.30; p !0.0001) and difference FH-TH (r = 0.25; p ! 0.001). The correlations with BWwere confirmed in male patients examined separately: with FH (r = 0.36; p !0.0001) and height increment (r = 0.31; p ! 0.001). On the contrary, in femalesthere was only a weak correlation between BW and height increment (r = 0.26;p ! 0.05). Conclusions: In our patients treated for idiopathic and isolated GHdeficiency birth weight significantly affected the final results, both in terms ofFH and height increment during treatment. The influence of BW on the finalresults was mainly due to the group of male patients.
P3-168 GH Treatment
Isolated Idiopathic Growth Hormone Deficiency(IIGHD): Re-evaluation during Growth Hormone (GH)TherapyI. Rica* 1; A. Vela* 1; I. Carpintero* 1; P. Martul 1
1 Hospital de Cruces, Baracaldo (Bizkaia), Spain
Diagnosis of IIGHD is difficult in many cases. A great proportion of patientsclassified as deficients, do not remain in this condition when GH is measuredafter growth has ended and therapy has been discontinued. Aims: To test GHsecretion after clonidine administration in a group of patients affected byIIGHD during treatment with GH. Patients and Methods: In 21 isolatedidiopathic GH deficient patients diagnosed at 9.2 B 3.2 years of age which hadbeen on GH treatment for 4.8 B 3.2 years, therapy was stopped for 1 month, tostudy GH secretion after clonidine administration. Short mid-parental heightwas associated in 76% of patients. When diagnosis was confirmed (GHpeak !10 ng/ml) GH administration was restarted immediately. Results:1) IIGHD was confirmed in 61,9% of patients. In this subgroup, therapysupression during one month did not cause any problem, since height (ex-pressed as SDS) remained unchanged one year after. 2) Patients in which defi-ciency was not confirmed were older, both in terms of chronological and boneage, at the time of GH re-evaluation. We could not find difference in any otherauxological variable between this subgroup, and patients remained deficient.Height (expressed as SDS) in this subgroup, one year after GH therapy hadbeen stopped, was similar as at the time of supression. Conclusions: In 38.1%of patients previously diagnosed as isolated idiopathic growth hormone defi-cient, diagnosis was not confirmed before growth had ended. GH therapysupression during one month was enough to revaluate GH secretion after phar-macological stimuli. The height (SDS) of patients who remained deficient didnot get worse because of GH supression for one month.

HV (cm/year)
Cystinosis
39th Annual Meeting of the ESPE Horm Res 2000;53(suppl 2):1–191 51
P3-169 GH Treatment
Psychological Evaluation after Long-Term GrowthHormone Treatment in Women with TSY.K. Van Pareren* 1; Th. Sas* 1; K.J. Simis* 1; M.B. Hofstra* 1;S.M.P.F. De Muinck Keizer-Schrama 1; K. Schrama 1; F.M.E. Slijper* 1;Introduced by S.M.P.F. De Muinck Keizer-Schrama1 Sophia Children’s Hospital, Rotterdam, The Netherlands
Introduction: It is known that girls and women with Turner Syndrome (TS)are at risk for the development of psychological problems. Aim: In a pilotstudy, we evaluated perceived physical attractiveness, emotional and behavior-al problems, social perception in TS women after long-term growth hormonetreatment (GHT). Patients: 14 girls with TS, mean (SD) age 20.3 (1.4) years,final height 158 (4.6) cm, BMI-SDS 1.7 (1.6). All women were using adult doseHRT at time of study. Mean (SD) duration of GHT 3.6 (0.7) years (dose: 6IU/m2/day; F0.0675 mg/kg/day). Methods: 2–3 years after final height (FH)was reached several self-report questionnaires were filled in, measuring emo-tional and behavioral problems (YASR), personality characteristics (Big Five),social perception (HSPP-A), depression (CDI), coping (UCL) and perceivedbodily appearance (BCS). Fifteen of the original 19 TS participants of the GH-study agreed to participate. The four non participants did not significantly dif-fer from the participants in age or FH. One woman was excluded due to mentalretardation. The results of the questionnaires were compared with a normalsubsample of the general population as described by Simis et al. (n = 128) andfor the YASR as described by Hofstra et al. (n = 257). Both sub-samples wereselected on same age-group and sex. Except for the YASR, results are indicatedas high when above 90% of the score found in the female population subsampleor as low when below 10%. Results: (1) Education: 7/14 finished high school,the rest have an associate’s degree. (2) YASR: Five of the 14 women (36%) hada YASR total problem score in the clinical range, compared to 24/257 of thepopulation sub-sample (9.3%; p = 0.01). Furthermore the TS women scoredsignificantly higher on the Internalizing scale, Anxious/Depressed scale andWithdrawn scale compared to the normal female population (p ! 0.05). (3) BigFive: 4/14 (29%) scored high on the Agreeableness scale (p = 0.09). (4) HSPP-A:6/14 (43%; p = 0.002) scored low on the Close Friendship scale and 4/14 on theOverall Self-Esteem scale (p = 0.09). On the total HSPP score 4/14 scored low(p = 0.09). (5) CDI: 4/14 (29%) scored high on the total CDI score (p = 0.09).(6) UCL: 4/14 have a high score on the Avoidance scale and 4/14 score low onthe Passive Reaction Pattern scale (p = 0.09). (7) BCS: 13/14 are neutral to verysatisfied with their height and 12/14 with their overall appearance. Conclu-sions: (1) TS women, after long-term GHT, report more emotional and behav-ioral problems than the normal female population. (2) TS women tend to havean internalizing behavioral pattern and tend to be more agreeable, anxious,depressed, withdrawn and to have less close friendships and lower self-esteemthan the general female population of their age. (3) Their coping strategy seemsto be characterized by a high degree of avoidance and passiveness. (4) Afterlong-term GHT most TS women are satisfied with their overall appearance.
P3-170 GH Treatment
Growth Hormone (Saizen®) Is Effective in TreatingGrowth Failure across the Spectrum of Chronic RenalDiseaseD. Geary 1; F. Murray 2; P. Saenger 3;Saizen® Chronic Renal Failure Study Group 1
1 University of Toronto, Toronto, Canada; 2 Serono Laboratories Inc.,Norwell, United States; 3 Albert Einsteins College of Medicine
Several studies have shown that treatment with recombinant human growthhormone (rhGH) accelerates height velocity (HV) in children with growth fail-ure due to chronic renal disease (CRD). To date, however, only one study hasinvestigated the use of rhGH across the spectrum of CRD. Furthermore, theresponses to rhGH of children with different underlying disorders have notpreviously been compared. Objective: To evaluate the efficacy and safety ofrhGH (Saizen®) in the treatment of growth failure due to different forms ofCRD. Also, to compare the response to rhGH in children with cystinosis withthat in children in other diagnostic categories. Study Design: Eighty-one chil-dren with growth retardation due to chronic renal insufficiency (CRI), end-stage renal disease (ESRD) or renal transplantation (RT) were entered in a mul-
ticentre, prospective trial of rhGH (Saizen®; Ares-Serono). Efficacy was evalu-ated over a 2-year period and safety was evaluated over 5 years. Results: Keyefficacy results are summarised in Table 1.
Table 1
Baseline At 1 year At 2 years
4.6B3.1 9.0B3.6 7.5B2.9Patients with HV SDS 10 14% 80% 72%Patients with HV SDS 1baseline – 96% 94%Height SDS –3.7B1.7 –3.0B1.7 –2.5B1.5
Increases in HV and HV SDS were significant for all patient groups in year 1and for the CRI and RT groups in year 2. The improvement in mean heightSDS was significant in all groups in both years. The growth response in childrenwith cystinosis was similar to that in children with other underlying disorders(Table 2).
Table 2. Year 2 vs. baseline
¢ height SDS ¢ HV (cm/year) ¢ HV SDS
1.6B2.5 3.6B7.7 2.7B7.6Others 1.1B1.0 2.9B3.1 3.5B2.9
The incidence of GH-related adverse events was similar to that reported inprevious trials of rhGH in childhood CRD. Conclusion: The results of thisstudy confirm that growth hormone (Saizen®) is effective in treating growthfailure in children who have CRI or ESRD or who have undergone RT, includ-ing those with cystinosis. Therefore, they support the use of this agent across thespectrum of CRD.
P3-171 GH Treatment
Non Conventional Use of Growth Hormone in ShortGirls May End with a Shorter than Expected OutcomeZ. Zadik 1; R. Dovev 2; M. Chen* 3
1 Kaplan Medical Center, Rehovot, Israel; 2 Pediatric Endocrinology,Rehovot, Israel; 3 Endocrine Lab, Rehovot, Israel
Background: None of the published work so far could offer a way to predictthe final outcome of non GH deficient children treated with GH. In most of thepublished papers there were no real time controls, treatment period was vari-able or age range of patients was wide. Objective: To evaluate final heightoutcome of short pubertal girls without classical GH deficiency. Methods: Wetreated 20 pubertal girls, Tanner stage 3 13–14.5 years with GH until finalheight (FH) was reached (2–3 years). 40 age matched untreated girls served ascontrols. 6 patients (group A) were treated with 0.1 IU GH/day, 6 patients(group B) were treated with 0.2 IU GH/day and 6 patients (group C) weretreated with 0.1 IU GH/day and long acting LRH analogue. The following vari-ables were studied for analysis: Chronologic age (CA), bone age (BA), heightSD, IGF-I, GH-binding protein, 24 hour integrated concentration of GH(ICGH). The difference between the predicted adult height (PAH) before treat-ment and achieved final height was compared in the 3 treatment groups andtheir controls. Final height was also compared to siblings’ final height.Results: Calculating the gain in FH in all the patients as a group resulted in anet gain of –1.1 B 0.5 cm over PAH and target height (TH) but –1.1 B 0.5 cmwhen compared to controls. As a group none of the variables had any predictingvalue. Separating the patients into 3 groups revealed that in group A FHreached was +2 cm to –1 cm when compared to TH but –3 to –4 cm as com-pared to sibling heights and controls. Height gain in group C was +4 to +1(significant p ! 0.05, but small), group B had a reduced final height –3 to –6 cm(p ! 0.05) as compared to TH and PH but –4 to –8 cm as compared to siblings’heights and controls. Pubertal length was significantly shorter in the GHtreated 0.2 IU/day group as compared to the control (1.6 B 0.3 vs. 2.2 B 0.5 p! 0.03). GH treatment in the pubertal age may have a deleterious effect on finalheight in GH sufficient children.

Boys
CA, years
A (n = 22)
A (n = 8)
52 Horm Res 2000;53(suppl 2):1–191 Posters
P3-172 GH Treatment
Final Height in Children with Chronic Renal FailureTreated with Growth HormoneA. Papathanasiou 1; C. Theodoridis 1; C. Hadjiathanasiou 1; E. Kousta* 1; C. Stefanidis* 1
1 Departments of Endocrinology and Nephrology, Aglaia KyriakouChildren’s Hospital, Athens, Greece
Chronic renal failure causes severe growth retardation, which persists in mostchildren even after successful renal transplantation, resulting in final heightbelow the normal range (3rd percentile) in approximately 70% of the patients.While GH is extensively used over the past years in CRF patients, few dataexist on the effect of rhGH on final height of these patients. Aim of theStudy: To evaluate the effect of rhGH on final height of growth retarded chil-dren with CRF. Patients and Methods: From a total of 20 growth retardedpatients with CRF who were treated with rhGH for 1–8 years, 6 patients (4boys, 2 girls) reached final or near final height after 3.5 B 1.4 years of treat-ment. 4 patients were on conservative treatment and 2 were transplanted. Atthe beginning of therapy 4 were prepubertal and 2 had entered puberty (Tannerstage 2). Mean chronological age (CA) at therapy initiation was 11.8 B 1.9(boys) and 11.2 B 3 (girls), mean bone age 9.9 B 1.6 and 9.7 B 3.1, height SDS(CA) –2.28 B 1.1 and –2 B 0.4 and target height SDS (THSDS) –0.56 B 0.75and –1.2 B 1.05 respectively. Results: Height SDS increased in all patientsduring therapy compared to pretreatment SDS (p ! 0.01). After the end oftherapy height SDS decreased slightly but remained normal (1–2 SD) till FH.Boys achieved a mean FH of 163.58 B 6.39 and girls 152.11 B 1.2 cm.Although mean FH SDS was less than TH SDS, 5 out of 6 patients reachednormal FH (13rd percentile) and 3 of them exceeded TH.
HtSDS (CA)at start
HtSDS (CA)end of therapy
FH (cm) FH SDS TH SDS
–2.28B1.11 –1.08B1.47 163.58B6.39 –1.48B1.17 –0.56B0.75Girls –2.00B0.14 –1.25B0.35 152.15B1.20 –1.60B0.14 –1.21B1.05
Conclusions: rhGH treatment in growth retarded children with CRF inducespersistent catch-up growth, resulting in normal adult height in the majority ofpatients. Further studies are necessary to confirm these results on a larger num-ber of patients.
P3-173 GH Treatment
Dose-Dependent Growth Response of Two-Year GHTreatment in Children with IUGRA. Papatheodorou* 1; C.G. Hadjiathanasiou 1; C. Theodoridis 1;A. Papathanasiou 1; M. Tsiarta* 1; D. Dellios* 1; C. Kostalos* 1
1 Department of Endocrinology, P and A Kyriakou Children’sHospital, Athens, Greece
A significant proportion of children with IUGR show persistent growth failureand eventually short stature. In recent years, administration of rhGH hasshown potential in improving height prognosis of these children. Aim: Toassess the response of IUGR to 2 different GH therapy dose regimens over aperiod of 2 years. Subjects and Methods: Fifteen prepubertal children (9boys, 6 girls) of a mean chronological age (CA) of 6.9 B 3.1 years with IUGR(length and/or weight at birth !–2 SD for gestational age) were included in thestudy. All children had height !3rd percentile, normal GH secretion in at leastone provocative stimulus and normal IgFI levels. None had history of congeni-tal infection, chromosomal abnormality or chronic illness. Five childrenreceived a daily subcutaneous rhGH dose of 0.1 IU/kg (group A) and 10 chil-dren received a daily dose of 0.2 IU/kg (group B), after randomization. Clinicaland laboratory evaluation was carried out every 3 months, while bone age (BA)was assessed at baseline and at the end of the 1st and 2nd year of treatment.
The results are presented in the table below:
Group A (n = 5)
0 m 12 m 24 m
Group B (n = 10)
0 m 12 m 24 m
8.8B3.1 9.8B3.1 10.8B3.1 6.0B2.8 7.0B2.8 8.0B2.8BA, years 7.8B2.6 8.7B2.7 9.9B3.1 5.1B2.9 6.3B2.9 7.4B2.8HV, cm/year 2.8B1.3 9.3B1.9* 6.5B1.7* 5.0B1.5 11.2B2.3* 7.8B1.5*Ht SDS/CA –2.6B0.9 –2.1B0.9* –1.9B0.9* –2.4B0.4 –1.3B0.5* –0.9B0.5*Ht SDS/BA –1.7B0.8 –1.0B0.9* –1.0B1.0* –1.2B1.1 –0.3B1.5* –0.1B1.3*
* p ! 0.005.
No serious side effects were observed during the treatment period. Conclu-sion: rhGH administration at doses of 0.1 and 0.2 IU/kg/day for 2 years inshort children with IUGR resulted in significant improvement of HV and HtSDS/CA, without causing disproportional BA maturation. After the 2nd year oftreatment HV had declined, but was still above the baseline level. Using HtSDS/BA as a prognostic factor for final height, these results suggest significantand dose-dependent improvement of the predicted adult height. The responseto therapy for a longer period, as well as HV and BA maturation after discontin-uation, will determine more accurately its effect on the final height.
P3-174 GH Treatment
First Year Results of a Multicentre Dose-ResponseStudy with the Liquid GH Formulation Granditropin(TM) 15 IU (5 mg) in Prepubertal GHD Patients in FranceL. di Nicola* 1; C. Gendrel* 1; P. Ryziger* 1; J.L. Chaussain 2
1 Grandis Biotech France SA, Clichy, France; 2 Division of PaediatricEndocrinology, Saint-Vincent de Paul Hospital, Paris, France
80 patients were enrolled in the above study at 35 centres in France and treatedwith Granditropin™ 15 IU (5 mg), a liquid formulation of hGH filled in car-tridges of 1.5 ml, administrated with Grandipen™ (BD). Objective: The aimof this study is to evaluate the beneficial effects of two different doses of hGHduring the first year of treatment and during 5 years follow-up period.Patients and Therapy: Prepubertal GHD patients with growth retardation^–2 SDS (proved by 2 pharmacological stimulation tests !10 mg/ml hGHserum concentration) were included in this French collaborative multicentrestudy. Both groups were similar in terms of population (boys and girls) as wellas in terms of bone age and chronological age:
Group A: C.A.: 8.9B2.7; B.A.: 6.8B2.7Group B: C.A.: 7.1B3.5; B.A.: 5.6B2.8
The patients were randomly allocated to Granditropin™ 0.7 IU/kg/week(group A) or Granditropin™ 1.4 IU/kg/week (group B). Results: After 6months Granditropin™ therapy we are able to present the results of 46 patientsand after 1 year for 13 patients:
Groupn = 46
Baseline
HV HSDS
6 months
HV HSDS
3.8B0.7 –3.0B0.7 11.5B3.4 –2.3B0.6B (n = 24) 4.2B1.3 –3.0B0.8 13B2.3 –2.2B0.7
Groupn = 13
Baseline
HV HSDS
12 months
HV HSDS ¢SDS
4B1.0 –3.0B0.4 9.3B1.4 –2.2B0.3 0.8B0.4B (n = 5) 3.4B0.4 –3.5B1.0 11.1B1.8 –2.2B1.0 1.3B0.2
Conclusion: From the results, we see that there is a significant differencebetween mean gain in HSDS at month 12 between Groups A and B (p = 0.04)and between mean HV values at month 12 (p = 0.01). However, a long-termfollow-up period will be requested to demonstrate the beneficial effect of highdose treatment on growth velocity and increase in height. The local tolerance ofnew liquid formulation was very good.

1
ES
39th Annual Meeting of the ESPE Horm Res 2000;53(suppl 2):1–191 53
P3-175 GH Treatment
Dwarfism with Gloomy Face: Phenotypic Aspects andResponse to Growth Hormone TreatmentW. Elchemeitelli* 1; V. Cormier-Daire* 1; J.C. Souberbielle* 1; G. Pinto 1; M. Le Merrer* 1; R. Brauner 1
1 Hôpital Necker-Enfants Malades, Paris, France
Dwarfism with gloomy face was described as a novel entity within the 3-Msyndrome in patients with familial consanguinity, history of intra-uterinegrowth retardation (IUGR), extreme short stature and macrocephaly withoutmental retardation, hypothyroidism nor GH deficiency. Objective: To de-scribe the initial clinical and laboratory features of patients with dwarfism withgloomy face and their response to GB treatment. Patients and Methods:The 8 patients descended from 5 families. In all cases the patients’ parents werefirst cousins. Among them, 7 received GH treatment at ages ranging from 3 to7.3 years at doses of 1.2 to 1.4 IU/kg/week. Results: Out of 8 patients, 7 had ahistory of IUGR. 6 had a head circumference (HC) over the 90th percentile atbirth. All patients had normal target heights (–1 to +0.5 SD). The patients’features at the first evaluation were:
Patient Gender Age,years
Height,SD
BMI,ZS
HC,SD
GHp,ng/ml
IGF-I,ng/ml
F 1.5 –6 –1.2 +2 16.5 872 F 2.8 –5 –0.2 +2 10.03 F 10.2 –4 –0.5 +1 17.2 3884 F 1.6 –4 0.3 10.0 1085 M 3.8 –4 –1.6 0 186 M 1.5 –5 –0.5 –1.5 29.2 1067 F 5.9 –3 0.4 +1 18 1608 F 1.6 –2.5 0.1 +1 14.2 146
GHp = GH peak value after stimulation test.
Under GH treatment, the growth rate increased from –2.6 B 1.2 SD to 2.0 B2.0 SD (p ! 0.05) during the 1st year, and to –0.8 B 0.5 SD (NS) during the 2ndyear. In patients for whom data was available, plasma IGF-I levels alsoincreased (IGF-I at 269, 244 and 196 ng/ml in patients 1, 2 and 4 respectively).Conclusion: In patients with dwarfism with gloomy face, the growth rateincreased significantly during the first year of GH treatment. The increase ingrowth rate and in IGF-I levels suggests that dwarfism with gloomy face is notsecondary to resistance to GH. The efficiency of GH treatment beyond a oneyear period needs further evaluation.
P3-176 GH Treatment
Incidence of Antithyroid Antibodies in TurnerSyndrome Girls: Influence of Growth HormoneTreatmentA. Wisniewski* 1; A. Lecka* 1; W. Skawinski* 1; T.E. Romer 1
1 The Children’s Memorial Health Institute, Warsaw, Poland
In Turner syndrome (TS) patients a high incidence of circulating antithyroidantibodies has been reported [1]. The influence of long-term treatment withgrowth hormone (GH) on thyroid function in TS is unknown. Aim: We investi-gated thyroid antibodies in 47 TS during the long-term growth hormone (GH)therapy and compared with age-matched never treated patients. Patients andMethods: At the start of GH therapy the mean TS age (CA) was 7.32 (2.03)years, range from 3.11 to 10.00 years, all patients were clinically and biochemi-cally euthyroid [mean TSH 2.56 (1.12) mU/ml, range from 1.10 to 5.10], andno goitre was detected. Two patients had elevated titers of A-TPO and ATGantibodies (4%). The presence of thyroid antimicrosomal (A-TPO) and antithy-reoglobulin (ATG) antibodies (thyroid AA) was assessed by Boyden’s passivehaemagglutination system (Thymune-T and M, Murex, UK). Results: Aftermean 7.29 (0.69) years of GH therapy the positive titers of thyroid AA werepresent in 15 out of the 47 patients (31.9%). In 32 patients with normal anti-body titers, 1 patient with compensated hypothyroidism with TSH of 7.5 mU/ml was found. In 15 thyroid AA positive patients also 1 had elevated TSH level(13.5 mU/ml) with normal thyroid hormone levels. The results were comparedwith the incidence of thyroid AA in 35 TS never treated with GH, anabolic
steroids or estrogens (control group). Age of the subject in control group was13.46 (2.35) years (mean), range from 10.21 to 18.17 years and it was not differ-ent from the age of GH treated TS at the time of thyroid AA measurement. Inthe control group 6 patients were positive for thyroid AA (17.1%). The differ-ence with GH treated group is statistically significant. Conclusion: Antithy-roid antibodies are frequently present in GH treated Turner syndromepatients.
1 Papendieck LG, Iorcansky S, Coco R, Rivarola MA, Bergada C: High inci-dence of thyroid disturbances in 49 children with Turner syndrome. JPediatr 1987;111:258–261.
P3-177 GH Treatment
Cardiac Effects of Growth Hormone Treatment inChildrenP. Neou 1; C. Trapali 2; I. Zevlekari 1; I. Polychroni 1; L. Stamoyiannou 1;A. Margetakis 2
Departments of 1 Pediatrics and 2 Cardiology, P and A KyriakouChildren’s Hospital, Athens, Greece
Growth hormone is known to stimulate myocardial hypertrophy and increasesmyocyte contractility. Many adults with growth hormone deficiency (GHD)have reduced ventricular mass and lower ejection fraction, while GH replace-ment appears to correct many of these abnorma1ities. There are few studies onthe cardiovascular effects of GH therapy in children with GHD. On purpose toevaluate the effects of GH treatment in children with GH deficiency in the leftventricular function or morphology, during 1 year of therapy, we investigated18 children (12 boys) with mean age 7.4, who received rGH 0.5 IU/kg/week.Evaluation of cardiac parameters for myocardial mass and cardiac contractilityat the initiation of therapy, six months and 1 year later, by M-Mode echocar-diographic study showed the following results:
0–6 months
t-value p
6–12 months
t-value p
0–12 months
t-value p
2.08 0.045CO 2.29 0.029CI 3.25 0.003Mass –2.48 0.019Mass/BSA 2.28 0.031IVSS –3.14 0.004 –3.37 0.002IVSD –2.89 0.007Height –2.2 0.036BSA –2.27 0.031LVPWS –2.15 0.041 –2.33 0.027
FS = Shortening fraction (%), CO = cardiac output, CI = cardiac index,Mass = left ventricular mass, Mass/M2 = mass/mg, IVSS = intraventricularseptum systolic, IVSD = intraventricular septum diastolic, LVPWS = left ven-tricular posterior wall systolic.
These results revealed a significant time effect for CO, CI and FS with valuesdecreasing at midterm (at 6 months) and normalized at the end of the studyperiod. A significant increasing of IVSS, IVSD and LVPWS provided at theend of the study. Also at the end of the study there was a significant increase incardiac mass but when this parameter was determined as Mass/BSA there wasan inversing to decrease. That is the result of significant increasing of BSAwhile hight remains stable.
CA, years
54 Horm Res 2000;53(suppl 2):1–191 Posters
P3-178 GH Treatment
Two-Year GH Therapy in Short Normal Children: Effecton Final HeightA. Papatheodorou 1; C. Evagelopoulou 1; C. Theodoridis 1; C. Hadjiathanasiou 1; A. Papathanasiou 1; D. Dellios 1
1 Department of Endocrinology, A. Kyriakou Children’s Hospital,Athens, Greece
Growth hormone (GH) in short normal children improves HN during therapy.However, results of the effect on final height is still limited. We report the resultof two years therapy with rhGH on final height in 9 (4 M/5 F) prepubertal shortchildren without GH deficiency (Group A). Mean CA at the start of treatmentwas 11.3 B 1.7 years. GH was given for 2 years in a dose of 0.1 IU/kg/day s.c.Twenty-two (15 M/7 F) age and sex matched short children without GH defi-ciency have served as controls (Group B). Auxological data were obtained fromall children every three months until they reached final height (FH). Results:
Group A
M (n = 4) F (n = 5)
Group B
M ( n = 15) F (n = 7)
11.1B1.7 11.4B0.9 12.4B1.4 11.7B1BA, years 10.0B1.5 8.9B1.0 10.8B1.5 10.0B1.3Start Ht SDS/CA –2.4B0.3 –2.9B0.6 –2.2B0.6 –2.5B0.5Start Ht SDS/BA –1.6B0.7 –0.5B0.9 –0.8B0.7 –1.1B1Final Ht SDS –1.6B0.6 –1.0B0.4 –1.0B0.7 –1.1B0.4Target Ht SDS –1.0B0.7 –1.8B0.8 –1.1B0.8 –1.1B1.2
In treated boys FH was 0.6SD less than their TH while in treated girls FH was0.8 SD above their TH. In untreated boys and girls (Group B) final height wasnot different from their TH. Conclusion: It seems that 2 years GH therapyhad no real beneficial effect on FH in short normal non GH deficient childrendespite previously reported accelerated growth velocity during treatment. Ingirls there is some beneficial effect, which could be explained by the presence ofdelayed HA at the start of GH treatment.
P3-179 GH Treatment
Prader-Labhart-Willi Syndrome: Response to GrowthHormone Treatment in Patients with Classical GrowthHormone DeficiencyH. Schmidt* 1; S. Bechtold* 1; S. Arleth* 1; H.P. Schwarz 1
1 Division of Pediatric Endocrinology, Dr. von HaunerschesKinderspital, Ludwig-Maximilians University, Munich, Germany
In patients with Prader-Labhart-Willi syndrome (PLWS), divergent results ofgrowth hormone (GH) treatment have been reported. Objective: To evaluatethe effect of a conventional GH dose in patients with PLWS and classical GHdeficiency. Patients and Methods: Between 1992 and 1998, 20 patients withproven PLWS presented in our clinic. In 10 (8 females, 2 males, aged 7–15years), GH deficiency was suspected on clinical grounds, 9 had maximum GHlevels !10 ng/ml in two provocative tests (clonidine and arginine), 7 had ele-vated adrenal androgen levels, 6 had a hypoplastic or empty sella in cranialMRI. All 10 patients received GH at a dose of 2 IU/m2/day, 9 for 2 years and 3for up to 4 years. Results: Height velocity SDS increased from baseline –1.74B 1.93 to 2.65 B 1.74 and 1.78 B 2.08 after 1 and 2 years, respectively. HeightSDS rose from baseline –3.47 B1.34 to –2.78 B 1.21 and –2.27 B 1.02. Con-comitant marked increments in IGF-1 levels from 64 B 24 ng/ml to 350 B 143and 274 B 129 were measured. Bone age progressed normally with 2.13 yearsafter 2 years. Predicted adult height increased by 5.1 cm (1.9–9.1). BMI did notchange during 2 years of therapy. It was 22.60 B 5.44 kg/m2 before and 22.93B 5.15 and 22.71 B 5.01 kg/m2 during therapy. However, in 3 patients treatedfor 4 years, the beneficial effect of GH on height velocity and height SDS per-sisted, but BMI increased dramatically. Conclusions: In PLWS patients andclassical GH deficiency based on short stature, abnormal growth velocity,decreased IGF-1 levels and abnormal OH provocative tests, a GH dose of 2IU/m2/day has long- term positive effects on growth velocity and height gained,but will most likely not affect obesity.
P3-180 GH Treatment
Changes of GH Molecular Forms after RecombinantHuman GH (rhGH) InjectionM. Bozzola 1; F. Buzi 2; G. Radetti 3; L. Tatò 4; S. Pagani* 1; M. Stabile* 1; G. Rondini* 1
Peadiatric Departments of 1 Pavia, 2 Brescia, 3 Bolzano, and 4 Verona,Italy
We evaluated 13 prepubertal children, 6.47 B 1.70 years old, with total (n = 5)and partial (n = 8) GH deficiency prior to and 2, 4, 6 hours after the first subcu-taneous injection (0.1 IU/kg) of rhGH, i.e. 22 kD (22K) molecule. Serum GHlevels were measured using an immunofluorometric assay (IFMA), an enzyme-linked immunosorbent assay (ELISA) and a Nb2 cell bioassay which measured22K-GH, 20K-GH and GH biological activity (bioGH), respectively. The per-centage of 20K on total GH (20K%) was calculated as the ratio 20K/(22K+20K) ! 100. We observed a significant increase (p ! 0.001) in 22K val-ues (ng/ml B SD; baseline: 1,737 B 1,859; 2 h: 13,796 B 7,997; 4 h: l7,792 B10,109; 6 h: l4,059 B 9,121) as well as in bioGH levels (baseline: 4,284 B1,704; 2 h: 10,994 B 8.009; 4 h: 14,228 B 10,271; 6 h: 10,383 B 4,550), and astrong positive correlation between 22K and bioGH concentrations at baseline,2 h, 4 h and 6 h (p ! 0.014, r = 0.99), suggesting that the administered hormonepreserves its biological activity. No changes in 20K values were found suggest-ing that the injected hormone is not degraded to 20K in the circulation. As aconsequence of the absence of variations of 20K, 20K% decreased in an oppo-site fashion to the increment of 22K and bioGH levels (regression analysis: p !0.025, r = 0.98). These data suggest that, in spite of serum 22K and bioGHvariations, rhGH injection does not influence spontaneous secretion of the 20 kmolecule.
P3-181 GH Treatment
The Influence of Hormonal Therapy on Height inPatients with Turner Syndrome with Respect to BoneAge ProgressionA. Gawlik* 1; B. Koehler 1
1 Silesian School of Medicine, Katowice, Poland
Introduction: It is known that growth hormone (GH) combined with oxandro-lone (Ox) therapy improves final height in patients with Turner syndrome (TS)more efficiently than either of the two hormones alone. Estrogens (E) are absent inmost cases of TS. They are responsible for pubertal growth acceleration in healthygirls, that is not observed in TS. Both the dose and the age of therapy beginning arestill controversial. Aim: Comparison of each mode of therapy with respect toefficacy in height velocity (HV) improvement considering bone age (BA) acceler-ation. Patients and Methods: We observed 41 patients with TS during 60.1 B10.2 months divided into 5 groups: not treated –22; GH –5; GH + Ox – 6; GH +Ox + E – 2; Ox + E – 6. We administered GH (1 IU/kg/week) to patients below 3 pc(!–2 hSDS), Ox to patients from the age of 9 (0.05–0.0625 mg/kg/day), E (trans-dermal therapy) from the BA of 11 (BA assessed according to Greulich-Pyle).Results: Upon the height measures and the age of patients we calculate theincrease of hSDS value (Ranke growth chart for TS) during the course of therapy(¢hSDS/t [1/month]). One way ANOVA revealed stastistically significant differ-ences between ¢hSDS/t for the specified groups (p ! 0.000001). The ¢hSDS/tvalues for each group were: non treated: –0.002 B 0.03; GH: 0.077 B 0.021;GH + Ox: 0.110 B 0.011; GH + Ox + E: 0.096 B 0.012; Ox + E: 0.035 B 0.020.To evaluate the influence on BA we compared the increase of BA against chrono-logical age and ‘height age’ (HA) increase (HA – the age at which patient will reachthe given height without therapy – constant hSDS value). The regression analysisrevealed that the best results are obtained in GH + Ox + E patients (DBA/DHA =0.105). In other groups DBA/DHA values were: Ox + E: 0.152; GH + Ox: 0.249;GH: 0.54; non treated: 0.63. Conclusion: Considering the influence on bone ageand height velocity it is clearly visible that each mode of therapy is better than notherapy in Turner syndrome patients.

39th Annual Meeting of the ESPE Horm Res 2000;53(suppl 2):1–191 55
P1-182 Growth and Genetics
Shox Gene Deletions in Leri-Weill DyschondrosteosisC. Falcinelli* 1; S. Mariani* 1; S. Madeo* 1; C. De Sanctis* 2; A. Forabosco* 1; S. Bernasconi 1
1 Modena, Italy; 2 Torino, Italy
Leri-Weill Dyschondrosteosis (LWD, OMIM 127300) is an autosomal domi-nant skeletal dysplasia characterised by disproportionate short stature, me-somelic limb shortening and Madelung deformity of the forearms. It has beenshowed that LWD is associated to the haploinsufficiency of SHOX gene (Shortstature HOmeoboX-containing gene) which maps in the pseudoautosomal regionof the X and Y-chromosomes. We studied four families with LWD phenotype.Three polymorphic sequences were tested to detect deletions in the pseudoautos-omal region containing SHOX gene. Two of them, DXYS233 and DXYS234, areextragenic and one, CASHOX, is intragenic. In three families a deletion contain-ing SHOX was demonstrated. It was terminal in two cases and interstitial in theremaining one. In the fourth family the deletion was hypothesised since the noninformativity of the polymorphisms. In two of the three families, where the pres-ence is sure, the deletion was de novo, while in the third family it was familial. Ourfindings confirm the high frequency of deletions involving SHOX gene in LWDexcluding the possibility of genetic heterogeneity.
P1-183 Growth and Genetics
Mutations in the Short Stature Homebox Gene inDyschondrosteosis but Not in HypochondroplasiaG. Grigelioniene* 1; O. Eklöf* 3; S.A. Ivarsson* 4; O. Westphal* 5; D. Kedra* 2; J.P. Dumanski* 6; L. Hagenäs* 1; Introduced by O. Söder1 Pediatric Endocrinology Unit, 2 Centrum for Molecular Medicine,3 Pediatric Radiology Unit, Karolinska Hospital, Stockholm; PediatricEndocrinology Units, 4 University Hospital Malmö, Lund University,and 5 Gothenburg University Hospital, Gothenburg; 6 Department ofGenetics and Pathology, Rudbeck Laboratory, Uppsala University,Uppsala, Sweden
Hypochondroplasia (HCH) and dyschondrosteosis (DCS) are two common skele-tal dysplasias with disproportionate short stature in which diagnosis might be diffi-cult to establish especially in early childhood. Mutations in the FGFR3 gene arefound in 40–70% of HCH cases, whereas point mutations/deletions of the SHOXgene are detected in DCS. The genetic defects for a large proportion of the individ-uals with HCH are not known yet. Objective: To perform mutational analysis ofthe SHOX gene in individuals with DCS and individuals with HCH, negative forthe known HCH mutations in the FGFR3 gene. Patients and Methods: TheSHOX gene of 4 probands with DCS and of 18 probands with HCH was examinedusing hemizygosity/heterozygosity assay, direct sequencing and Southern blotting.The auxological and radiological phenotype of these patients was carefully deter-mined. Results: Two novel mutations in individuals with DCS were found. Onewas a frameshift mutation due to deletion of G at the position 272 of the cDNAsequence, resulting in a premature stop codon at the position 75 of the amino acidsequence. The second mutation was a C to G transversion at the position 485,coding for a leucine to valine substitution at the position 132 of the amino acidsequence. Two other individuals with DCS and the individuals with HCH dit notshow any mutations/deletions of the SHOX gene. The individuals with HCH andDCS were similar in gross body proportions (height, sitting height percentage of thetotal height and arm span), but the individuals with DCS hat shortening of theforearm bones with or without Madelung deformity. The phenotype of the patientswith both skeletal dysplasis was varying from severe to moderate short stature andbody disproportion. Conclusions: Two novel mutations in the SHOX gene werefound in the patients with DCS. Our results suggest that DCS as well as HCH areclinically and genetically heterogenous disorders.
P1-184 Growth and Genetics
Growth Hormone Treatment in Dyschondrosteosis TypeLeri-Weill with SHOX Gene DeletionK. Klemp* 1; K.P. Ullrich* 1; K. Gerlach* 2; E. von Büren* 3; Introducedby W. von Petrykowski1 Children’s Hospital, Helios Clinic, Gotha, Germany; 2 Childrens’Hospital, Helios Clinic, Erfurt, Germany; 3 Schwarz Pharma AG,Monheim, Germany
The Leri-Weill syndrome is a rare autosomal dominant disease with dispropor-tional growth deficiency and Turner syndrome like symptoms. We report on a
16.6 years old boy. At the age of 12 the disproportional growth deficiency wasfirstly documented, his height was 129 cm (–3.4 SD). The growth hormone(GH) secretion was pathological in 2 standard provocative tests and in the over-night profile. Genetic analysis revealed the loss of the terminal p-arm of chro-mosome Y (deletion), in 4 of 30 metaphases the karyotype 45XO and in 1metaphase a ring-chromosome Y. The chromosomal formula: 46, Xidic(Y)(p 11.2, 11.3), ish idic(Y) (DYZ3++, AMELY++ [25]/45,X [4]/46,X,r(Y) [1].Typical pathological signs in the radius, tibia, ulna and fibula reinforced thesuspected diagnosis. After start of GH-treatment (0,4–0,5 IU/kg/week, CA 13.3y, BA 11.5 y) catch-up growth began resulting in an improvement of HtSDS to–2.6 at 14.6 years and –2.9 at 16.6 years without bone age acceleration.
GH treatment can improve growth in children suffering from dyschondrosteo-sis type Leri-Weill with SHOX gene deletion and GH-deficiency without dete-riorating the bone length disproportion.
P1-185 Growth and Genetics
Increased Growth in Children with Leri-Weill Syndromeafter Treatment with Recombinant Human GrowthHormoneA. Linglart* 1; J.-E. Toublanc 1
1 Hôpital Saint-Vincent-de-Paul, Paris, France
Léri-Weill syndrome (LWS) is an autosomal dominant disorder characterizedby mesomelic dysplasia and forearm deformity (Madelung Deformity). Alter-ations of SHOX, Short Stature Homeobox-containing gene on the pseudo-auto-somal region of the chromosome X, were recently described in families withidiopathic short stature or LWS. Haploinsufficiency of SHOX is thought to bethe mechanism of short stature and shortened extremities in LWS as in Turnersyndrome. We report here three non-related subjects (two girls and one boy)with LWS, treated with recombinant growth hormone (rGH). Two wereincluded for intra-uterine growth retardation and one for growth hormone defi-ciency. Karyotype were performed to eliminate Turner syndrome. In two cases,mothers present the same forearm deformity. Skeletal radiologic features wereassessed at different periods of the growth. rGH therapy was started beforepuberty, at 2 years 10/12, 4 years 9/12, and 8 years 5/12. The hindsight rangedfrom 2 to 6.7 years. Two subjects were treated using 1.2 UI/kg/week. For one,the growth rate increased in the first year from –3 SDS to –1.5 SDS. After thatinitial period, growth velocity stabilized, and she kept growing on the –1.5 SDScurve. The second patient is still accelerating from –2.5 SDS to –0.8 SDS. Thelast subject has been treated using a lower dosage of 1 UI/kg/week during eight-een months, increasing the height from –2.5 SDS to the mean curve. Growthvelocity was, then, maintained upper than the mean using 0,7 UI/kg/week. Thebone age remained correlated to the chronological age. Skeletal features of LWSdevelopped during growth and were apparently not modified by rGH therapy.These observations suggest an efficiency of rGH therapy in LWS, even with lowdosage. If SHOX haploinsufficiency lead to short stature in LWS as in Turnersyndrome, an height improvement is expected after rGH therapy in both dis-eases.

Controls
56 Horm Res 2000;53(suppl 2):1–191 Posters
P1-186 Growth and Genetics
Analysis of the Intragenic SHOX Microsatellite inPatients with Short StatureB. Ezquieta* 1; A. Oliver* 2; E. Cueva* 1; D. de Sotto* 1; J.M. Varela* 1;J. Fernandez* 1; R. Gracia* 1; Introduced by M. OyarzabalServicio de 1 Bioquımica y 2 Endocronologia Pediatrica,Hospital La Paz, Madrid, Spain
Haploinsufficiency for the SHOX-PHOG gene has been suggested to be animportant genetic factor giving rise to the commonest sign of Turner Syndrome(ST), the short stature. Deletions of this gene have been found in the Leri WellSyndrome (LWS), in which short stature is found together with mesomelic dys-plasia and Madelung deformity. The intragenic microsatellite in the 5) untrans-lated region of the SHOX gene has been analysed in three groups of short-stature patients: 1) 57 TS patients, 27 mosaicisms included, 2) isolated short-stature (73 patients), and 3) short-stature patients with skeletal disproportion(77 patients). An additional group of controls (30 individuals) was studied toestablish the heterozygosity of the marker. Two additional microsatellites,DXS 1055 and 1192, were also analysed. 93% of Turner girls (group 1) werehemizygous, including mosaicisms; the four patients left showed a dispropor-tion of alleles compatible with the existence of 45X lines, suggesting that SHOXmicrosatellite might be a good molecular indicator for the syndrome. Withrespect to the other groups: Two alleles were found in most patients from group2 (92%), being the heterozygosity in these patients similar (p = 0.997) to that inthe control group (7%). Contrarily, patients with short stature and skeletal dis-proportion (group 3) were hemizygous in a higher (p = 0.027) proportion thanproportionated short patients (27% vs. 8%). Two hemizygous patients fromgroup 3 were found to be LWS (with mesomelic dysplasia and Madelung defor-mity) and the segregation of alleles in parents documented the haploinsufficien-cy. Haploinsufficiency for the SHOX gene was also demonstrated in anothergirl from group 1 with partial Xp monosomy. A very good response to growthhormone therapy has been observed in this girl, as it has been documented forother patients with SHOX haploinsufficiency.
P1-187 Growth and Genetics
Molecular Heterogeneity of Familial Isolated GrowthHormone Deficiency, Type II: A Novel IVS3 +2T→CSplicing Mutation in the GH-1 GeneO.V. Fofanova 1; O.V Evgrafov* 2; A.V. Polyakov* 2; A.B Poltoraus* 3;V.A. Peterkova 1; I.I. Dedov* 1
1 Department of Pediatrics, Endocrinology Research Center;2 DNA-Dianostics Laboratory, Research Center for Medical Genetics,3 Engelgardt Institute of Molecular Biology, Moscow,Russian Federation
Studies on molecular basis of isolated GH-deficiency (IGHD) are of great inter-est because of apparent heterogeneity of GH-1 gene abnormalities both infamilial and sporadic cases. Objective: To carry out screening for mutationsin the GH-1 gene in Russian children with IGHD. Patients and Methods:28 children with severe IGHD and early onset of growth retardation were stud-ied. The cohort included 10 familial (8 families) and 18 sporadic cases. Rt SDScorresponded to (–) 3.22 B 1.2 at 1 yr and (–) 4.27 B 0.9 at 2 yr of life. DNAfragments, covering each of 4 exons of GH-1 (exons 2, 3, 4, 5) and boundaryregions were amplified using PCR. Direct DNA sequencing was performedusing ABI PRISM 373 DNA Sequencer (Perkin Elmer, USA). Results: Severalnovel splicing mutations in the GH-1 gene were identified in studied patients.We report here our finding of a heterozygous second base transition +2T→C ofthe intron 3 donor splice site of GH-1 in a boy with familial case of IGRD, typeII. This novel IVS3 +2T→C mutation is located in a highly conserved region ofTCCgtg of exon 3/IVS3 boundary. IVS3 +2T→C mutation is close to the dom-inant-negative IVS3 +1G→A mutation in IGRD II, causing skipping of exon 3(Cogan J.D. et al., 1995). We speculate that IVS3 +2T→C transition has thesame effect on GH mRNA splicing with dominant-negative effect on OR secre-tion. Conclusions: Our findings (1) extend the spectrum of GH-1 mutationsin IGRD described so far; (2) strongly indicate to the molecular-genetic hetero-geneity of IGHD, type II, in Russian patients.
P1-188 Growth and Genetics
Candidate Gene Studies in Small for Gestational Age(SGA) and Short Stature Appropriate for GestationalAge (AGA) Subjects – Analysis of IGF-I, IRS1, 11ßHSD2and Glucokinase MarkersL. Johnston* 1; J. Dahlgren* 2; L. Gelander* 2; M. Savage 1; K. Albertsson Wikland 2; A. Clark* 1
1 St Bartholomew’s Hospital, London, United Kingdom; 2 GrowthResearch Centre, Gothenburg, Sweden
Size at birth and postnatal growth are strongly influenced by genetic factorsaccording to epidemiological studies. This study aims to identify genesinvolved by studying three groups of children (short children born AGA, shortchildren born SGA and SGA children with catch-up growth). Individual candi-date genes were studied using an association study (case-control) approach,comparing each of the groups with a normal control group. These candidategenes were identified for the following reasons: IGF-I is a major fetal growthfactor. IRS1 is an important signalling molecule of the insulin and IGF type1receptors and has been associated with insulin resistance. 11ßHSD2 isexpressed at high levels in the normal placenta, converting maternal cortisol tocortisone thereby protecting the fetus from the growth inhibitory effects of cor-tisol. Furthermore 11bHSD2 RNA expression has been reported to be reducedin placentae of SGA deliveries. Glucokinase mutations have been reported toassociate with reduced birth size. Subjects: 468 children from the GrowthResearch Centre, Gothenburg were studied. Subjects with dysmorphic syn-dromes or abnormal karyotypes were excluded.
n Birth lengthBSD
Birth weightBSD
Height at 2–4 yrBSD
Adult heightBSD
115 –0.09B1.04 –0.01B1.03 –0.37B1.31Short AGA 178 –0.68B0.83 –0.48B0.85 –2.81B0.61Short SGA 116 –2.68B0.98 –2.42B0.89 –3.32B1.01SGA catch-up 59 –2.38B0.68 –2.31B0.87 –0.44B0.88
Methods: Dinucleotide repeat markers were selected for each candidate gene.Intronic markers were used for all four genes and additional microsatellitemarkers were idenitifed for 11ßHSD2 and IGF-I using Genemap99(www.ncbi.nlm.nih.gov). Markers were amplified by PCR using fluorescentoligonucleotide primers and analysed by pooling PCR reactions before electro-phoresis on the ABI 377 DNA Analyser. Results: The IGF-I intronic markerand microsatellite marker associate with the short SGA phenotype (p = 0.0009and p = 0.05 respectively) but not the SGA catch-up or short AGA group. Themarkers for IRS1, glucokinase and the two markers for 11ßHSD2 do not asso-ciate with any of the three phenotypic groups. Conclusions: These resultssuggest that the IGF-I gene may influence growth in SGA children who havepoor postnatal growth, but does not influence growth in those with postnatalcatch-up growth. The IRS1, 11ßHSD2 and glucokinase genes to not associatewith any of the three phenotypic groups suggesting that they do not influencethese abnormal growth patterns in this population.
Case 1
Case 1
39th Annual Meeting of the ESPE Horm Res 2000;53(suppl 2):1–191 57
P1-189 Growth and Genetics
GH Binding Activity and Positive hGH Growth Responsein Familial GH Resistance (Laron Syndrome) withHomozygous D152H MutationL. Baldazzi 1; M. Barbaro 1; A. Pasini 1; C. Fabiano 1; P. Pirazzoli 1; A. Balsamo 1; A. Cicognani 1; E. Cacciari 1
1 Department of Pediatrics, S. Orsola Hospital, University of Bologna,Italy
Laron syndrome is a rare autosomal recessive growth syndrome. This conditionis due to mutations in the growth hormone receptor (GHR) gene. The molecu-lar defects of GHR have been identified in extracellular, transmembrane andintracellular domains of patients showing extreme short height and displayingthe classic features of Laron phenotype. We report clinical and endocrinologi-cal features of two patients.
Table 1
Age (CA) Bone age (BA) Heigth for CAsds
Height for BAsds
15.25 11 –6.11 –3.27Case 2 18.5 13 –6.36 –3.02
Table 2
GH, ng/ml(basal)
GH, ng/ml(arginine peak)
GH, ng/ml(L-Dopa peak)
IGF-1, ng/ml
6.1 19.2 13.2 UndetectableCase 2 3.7 20.2 30.4 Undetectable
These patients performed an IGF-I generation test (4 UI/day for 7 days). TheIGF-I levels increased from undetectable to 32 ng/ml (case 1) and to 35 ng/ml(case 2). These results induced to perform a therapeutical trial for both boyswith rGH (1 UI/kg/ week) for 6 months. The growth response was 4.3 cm forcase 1 and 2.5 cm for case 2. After these results a new trial of 6 months oftherapy was repeated with a growth response of 3.1 and 1.6 cm (with a growthrate/yr respectively of 7.4 and 4.1 cm). These patients presented normal levelsof circulating GH-binding protein with high sensitivity (case 1: 26%, case 2:32.5%). GHBP was usually considered undetectable in the majority of theLaron patients. Genomic DNA from patients was amplified by PCR anddirectly sequenced. The same mutation was identified in both patients, result-ing in the substitution of a high conserved aspartate residue by histidine atposition 152 (D152H) in extra ellular domain. This mutation has beenreported to be responsible for preventing the process of dimerization GH-GHreceptors considered essential for the GH signaling pathway. Conclusions:These results seem to provide the evidence that high doses of rGH in humanscan obtain an acceptable growth response in patients with D152H mutation.
P1-190 Growth and Genetics
The Parental Origin of Monosomy X in TurnerSyndrome: Lack of Correlation with the Response toGrowth Hormone TherapyC.G. Hadjiathanasiou 1; C. Theodoridis 1; A. Tsezou* 2; C. Kyrou* 1; A. Papathanasiou 1; N. Moschonas* 3; P.C. Patsalis* 4; S. Kitsiou* 2
1 P. and A. Kyriakou Childrens Hospital, Athens, Greece; 2 Universityof Athens, Athens, Greece; 3 University of Crete, Heraklion, Crete,Greece; 4 The Cyprus Institute of Neurology and Genetics, Nicosia,Cyprus
To correlate the origin of the retained X chromosome in Turner syndrome (TS)with phenotype, pretreatment height and response to recombinant humangrowth hormone (rhGH) therapy, systematic clinical assessment and molecularstudies were carried out in 33 Greek TS children (18 with 45,X and 15 withX-mosaicisms) and their parents. PCR analysis, using microsatellite markerson X chromosomes (DXS 101 and DXS 337), revealed that the intact X wasmaternal (Xm) in 15/30 and paternal (Xp) in 15/30 children, while 3/33 fami-lies were non-informative. No significant relationship was found between par-
ental origin of the retained X and any of the clinical and ultrasonographicalphenotypic features assessed. In 17 of these children who completed at least 2years of rhGH therapy (0.7 IU/kg/wk), we found no differences between Xmand Xp groups in the improvement of height velocity, which was significant inboth. In conclusion, in our series of TS, we observed no preferential loss of thepaternal or maternal X chromosome. In addition, we found no relationshipbetween the origin of the retained X chromosome and the response to rhGHtherapy or their phenotype.
P1-191 Growth and Genetics
Endocrine Findings and Growth Response to CombinedGH and GnRH Agonist Treatment in 2 Siblings with aNovel Mutation of the GHRH ReceptorW.H. Stokvis-Brantsma* 1; O. Blankenstein* 3; W. Oostdijk 1; F. Roelfsema* 2; R. Pfäffle 3; J.M. Wit 1
Departments of 1 Pediatrics and 2 Endocrinology, Leiden UniversityMedical Center, Leiden, The Netherlands; 3 Department of Pediatrics,RWTH Aachen, Aachen, Germany
A mutation in the GHRH-Receptor is a rare cause of isolated GH deficiency.We present clinical and endocrine characteristics of 2 siblings with such muta-tion. Objective: To evaluate the endocrine findings at presentation, theresponse to treatment and endogenous GH secretion in adulthood. Patientsand Methods: Two children of non-consanguineous Moroccan parents, a boyof 16.0 yr and his sister of 14.8 yr, presented with extremely short stature (–5.7SDS and –7.7 SDS, resp. ). Target height was –1.4 SDS. Both children were inearly puberty (G2P4A2, testicular volume 15 ml; B2P1A1). After extraction ofgenomic DNA each exon of the coding sequence of the GHRH-R gene wasamplified by PCR and subjected to SSCP analysis, followed by completesequencing. For the 24-hour GH profile an ultrasensitive assay was used andthe profile was analysed with a multi-parameter deconvolution method.Results: Serum IGF-I was 1.9 (–6.3 SDS) and 3.5 nmol/l (–4.8 SDS). Exerciseand arginine tolerance tests showed low GH peaks of 2.8 and 5.0 mU/l in theboy and 0.5 and 1.3 mU/l in the girl. Thyroid function was normal, but theTRH test showed a low TSH peak (5.7 mU/l) with a normal rise of prolactin.Adrenal function was normal. Bone age was 14.0 and 11.5 ‘yrs’, respectively CTscans of the sellar region showed a partial empty sella, later confirmed by MRI.DNA analysis showed a recessive point mutation G–C in the first position ofthe donor-splicing site of intron 7, resulting in an extremely truncated protein.Both children were treated with GH for 5 years, 2 U/m2 for 2 years and 3 U/m2
thereafter. A GnRH agonist (Triptorelin, 3.75 mg/4 wks) was added for 2.5years. Final height SDS of both patients was –2.0. At retesting in adulthood, ani.v. injection of GHRH led to a very small rise of serum GH in the male (0.12 to0.18 mU/l), while in the female no response was noted (0.09 and 0.06 mU/l).The GH profile was characterised by an increased number of pulses. Basalsecretion was similar to age- and gender-matched controls, but the pulsatilesecretion was 7-fold lower in the male and 30-fold lower in the female. Conclu-sion: This novel mutation in the GHRH-receptor gene in two siblings led tosevere growth retardation. Combined treatment of GH and GnRHa resulted ina final height close to target height, despite a late start of treatment at anadvanced bone age. The presence ofGH peaks points to the role of somatostatinin GH pulsatility.

58 Horm Res 2000;53(suppl 2):1–191 Posters
P2-192 Growth and Genetics
Clinical and MRI Imaging Follow-Up in a Girl with Pit-1Gene MutationA. Salvatoni* 1; A. Facchinetti* 2; M. Maghnie 3; R. Rovelli* 1; L. Nespoli* 1
1 Insubria University, Varese, Italy; 2 Ospedale di Circolo, Varese,Italy; 3 IRCCS Policlinico S.Matteo, Pavia, Italy
Case Report: We report a case of a seven years old girl with multiple pituitaryhormone deficiency due to point mutation in the POU homeodomain of thePit-1 gene (R271W). The first MRI imaging, made at 2.5 months of age,showed a normal pituitary gland from the point of view of morphology; how-ever the height of the gland was 2 mm corresponding to the lower limit of thenormal range. The girl was treated with L-thyroxine from the age of 3 monthsand rhGH from the age of 9 months. 3 years later the growth curve from –3SDachieved the 50th percentile and her height is now between the 50th and the75th percentile. Neurological development is normal. The dosage of L-thyrox-ine ranged from 5 to 3 Ìg/kg/day and that of rhGH from 0.5 to 0.3 U/kg/week.We repeated the MRI at the age of 7 years in order to re-evaluate the state of thepituitary gland. Its height was 2.6 mm, still corresponding to the lower limit forthe normal range for the age (2–6 mm). Discussion and Conclusions:Although Pit-1 mutation is associated with severe pituitary hypoplasia in adult-hood while a gland of normal size has been reported in infants. This observa-tion supported the hypothesis that Pit-1 mutation affects the activation of hor-mone expression, but not the development of somatotrophs and lactotrophs.Our case shows that this mutation does not cause progressive pituitary hypopla-sia at least up to the age of our patient.
P2-193 Growth and Genetics
Hypopituitarism in a Brazilian Girl Caused by a NewMutation in Prop-1 (F88S)M.G.F. Osorio* 1; P. Kopp 2; S. Marui* 1; A.C. Latronico* 1; B. Mendonca* 1; I. Arnhold* 1
1 Faculdade de Medicina da Universidade de Sao Paolo, Sao Paulo,Brazil; 2 Northwestern University, Chicago, IL, United States
Hypopituitarism results from mutations in the homeodomain transcriptionfactors that direct embryological development of the anterior pituitary gland.Mutations in Prop-1 cause deficiencies of GH, PRL, TSH and gonadotropins.Only 8 different Prop-1 mutations have been reported in humans. We studied aBrazilian girl with growth failure since 1.5 yr who presented at 4.9 yr with aheight of 92.5 cm (–3.1 SDS) and bone age 2.5 yr. She was the third child bornat term with a length of 51 cm and weight of 3.65 kg. At 4.9 yr, GH peak afterclonidine and insulin-induced hypoglycemia tests was low (1.6 ng/ml). AfterTRH+GnRH+insulin administration peak values of PRL 11 ng/ml (normal20–35) and TSH 4.4 U/l (normal 5.6–33) were also low and of cortisol 41 Ìg/dl(normal 20–35) was normal. Basal T4 was 6.5 mg/dl, T3 125 ng/dl and IGF-I 49ng/ml. Combined T4 and GH therapy was followed by catch-up growth. At14.1 yr her height was 150.0 cm (–1.5 SDS), bone age 12 yr, breasts Tanner I,pubic hair Tanner II. After a GnRH+insulin test peak LH !0.6 U/l and FSH1U/l were low and cortisol 22 Ìg/dl remained normal. At 9 yr lateral skull X-rayhad shown a wide, open sella with a volume above average for age and a MRIshowed normal pituitary stalk and posterior lobe and a hypoplastic asymmetricanterior pituitary lobe (height 3 mm, normal for age 4.5 B 0.6 mm). GenomicDNA was isolated from peripheral blood of the patient, her parents, 2 sistersand 20 normal subjects. Each of the 3 exons of the Prop-1 gene were amplifiedby PCR and sequenced in a ABI Prism 310 automatic DNA sequencer.Sequencing of exon 2 of the patient revealed a homozygous transition(263T →C) which led to the substitution of the phenylalanine 88 by a serine.Consistent with autosomal recessive inheritance, the parents, who were firstcousins, and 2 sisters, all of normal height, were heterozygous for the mutationwhich was absent in 40 alleles of normal subjects. Phenylalanine 88 of Prop-1constitutes the hydrophobic core of the first alpha-helix of the homeodomainand is highly conserved among homeodomain of transcription factors. Replace-ment by the polar serine amino acid is predicted to alter the structure of the firstalpha helix and impair binding of Prop-1 to target DNA sequences. The F88Smutation is the human PROP1 mutation located closest to that of the Amesmouse (mProp1 S83P). We conclude that deficiencies of GH, PRL, TSH andgonadotropins in this girl were caused by the novel F88S mutation in Prop-1.
P2-194 Growth and Genetics
Concordance in Genotype but Discordance inPhenotype in Two Siblings with Prop-1 MutationF.G. Riepe* 1; O. Blankenstein* 2; C.J. Partsch 1; R.W. Pfäffle 2; W.G. Sippell 1
1 University of Kiel, Kiel, Germany; 2 RWTH Aachen, Aachen,Germany
Mutations of the Prop-1 gene cause combined pituitary hormone deficiency(CPHD) of GH, TSH, PRL and gonadotropins. Congenital hypoplasia of theanterior pituitary gland is the most common MR imaging (MRI) finding inpatients with Prop-1 mutations. Patients, Methods and Results: We stud-ied two brothers by PCR, SSCP and automated sequencing and found com-pound heterozygosity for the mutations ¢A150/¢AG301/302 in exon 2 result-ing in a complete loss of function of Prop-1. Presenting at age 3.5 and 1 years,both showed marked growth failure and moderate developmental delay.CPHD was diagnosed by subnormal GH, TSH, PRL, LH and FSH levels afterstimulation. Both patients had marked catch-up growth and development onGH and T4 replacement. MRI (2 mm thick slices) was done repeatedly in bothboys. In the older brother, MRI at 12 and 13 years showed a constant hypopla-sia of the anterior pituitary lobe (pituitary height 2 mm, normal range for age5.3 B 0.8 mm), normal position and size of posterior lobe, stalk and hypothala-mus. MRI of the younger brother at 9 and 10 years showed a constant enlarge-ment of the anterior pituitary gland (pituitary height 16 mm, normal range forage 4.5 B 0.6 mm), but otherwise normal morphology. Discussion: Bothpatients have the same Prop-1 mutations and an identical pattern of CPHD butshow constant differences in their pituitary morphology. Although sellarenlargement rarely occurs in familiar hypopituitarism due to Prop-1 mutations,the present discordance in brothers bearing the same mutation is puzzling. Itmay be speculated that abnormal expression of other early transcription factorslike P-Lim/Lhx-3 could be involved in the pituitary morphogenesis of Prop-1deficient individuals.
P2-195 Growth and Genetics
Prop-1 Mutations in Multiple Pituitary HormoneDeficiency (MPHD). Diverse Phenotype and PituitaryAnatomyA. Voutetakis* 1; A. Sertedaki 2; M. Maniati-Christidi 1; C. Dacou-Voutetakis 2
1 Athens University, Athens, Greece
Prop-1 gene mutations cause defective ontogenesis of the pituitary gland withresultant deficient synthesis of GH, TSH, prolactin, gonadotrophins and possi-bly ACTH. We have initiated a molecular genetics study of subjects withMPHD, without evidence of perinatal insult. Eleven children, with MPHD(low GH, TSH, prolactin, gonadotrophins and progressive ACTH insufficien-cy, now aged 19.8 years (SD 6.1) have been evaluated. PCR amplification ofexon 2 of the Prop-1 gene and direct DNA sequencing revealed that 4 of thepatients were homozygotes for the common 296GA deletion previouslydescribed. The parents of the patients were heterozygotes for the deletion.Another patient with the full phenotype and partial ACTH deficiency wasfound to be heterozygote for the same deletion. No other mutation in exons 2and 3 were recognised on this patient and exon 1 is under investigation. Corti-sol values during glucagon were normal at a mean age 16.3 years (SD 4.5) in 3patients with available values. In one patient peak value decreased from 18.2 to8.2. MRI findings: small pituitary in one patient and small cysts in the pituitaryin another patient, which normalised after 2 years of follow up. In two siblingsan adenoma was present in early life (0.8 cm in diameter), which disappeared atage 9 and 7 years, respectively. The data showed that diversity in phenotypicexpression and pituitary anatomy characterises Prop-1 296GA deletion.

HSDS1
39th Annual Meeting of the ESPE Horm Res 2000;53(suppl 2):1–191 59
P2-196 Growth and Genetics
Pseudotumor of the Pituitary Associated with Prop-1DeletionC. Teinturier* 1; S. Vallette* 2; C. Adamsbaum* 1; T. Brue* 2; P.F. Bougnères 1
1 Hopital Saint-Vincent-de-Paul, Paris, France; 2 Hospital de laConception, Marseille, France
Hypopituitarism associated with pituitary mass in childhood is frequently theconsequence of craniopharyngioma or Rathke’s cleft cyst. A 6 year old boy,born from non consanguineous parents had hypopituitarism associated with anintra-sellar pseudo-tumor. Height was 100 cm (–4 SDS). He had small penilelength (3.5 cm) and inguinal testes. GH peak after ornithine test was 1.1 ng/ml.TSH peak after TRH test was 4.7 UI/ml. Gonadotropins peaks after Gn-RHtest were 0.5 UI/l). PRL peak after TRH test was 20.6 ng/ml. Cortisol wasnormal (13.5 g/dl). MRI showed a round intrasellar mass (14 mm), with anhyperintense T1, hypointense T2 signal, respecting the chiasma. The posteriorlobe was still seen posteriorly to the intrasellar mass. The child received hor-monal replacement therapy. No surgery was attempted. During the following 4years, the MRI disclosed a marked reduction of the mass (5 mm) and ACTHdeficiency appeared. Direct sequencing of the Prop-1 gene showed an homozy-gous AG 99 deletion (2-BP del 301 AG). Prop-1 gene is involved in the develop-ment of the pituitary gland and is responsible for CPHD. Most of the reportedcases are associated with hypophyseal hypoplasia but it can also be associatedwith pituitary mass, which can be misleading with a true tumor.
P1-197 Growth and Steroid Hormones
Growth in Prepubertal Asthmatic Children with InhaledGlucocorticosteroid Treatment. A Comparison betweenBeclomethasone Dipropionate and BudesonideM.-T. Saha* 1; H. Lenko 2; Introduced by Hanna Liisa Lenko1 Tampere University Hospital, Tampere, Finland; 2 Tampere, Finland
We have recently reported retarded growth in children treated with inhaledglucocorticosteroids for asthma. The aim of the present study was to evaluatewhether differences in growth existed between children receiving different glu-cocorticosteroids. From our original material of 201 children, 180 (124 M, 56F) received beclomethasone dipropionate (Bdp) and 19 (11 M, 8 F) budesonide(Bud). All children were prepubertal. The mean height and height velocity stan-dard deviation score when starting the medication (HSDS1, HVSDS1) andyearly thereafter (HSDS2–5, HVSDS2–5) during follow-up of five years for theseparate groups were:
Bdp Bud Bdp Bud
–0.02 +0.25 HVSDS1 +0.04 +0.16HSDS2 –0.20 +0.20 HVSDS2 –0.82 –0.12HSDS3 –0.33 +0.21 HVSDS3 –0.36 +0.01HSDS4 –0.41 +0.52 HVSDS4 –0.53 +0.05HSDS5 –0.40 +0.44 HVSDS5 –0.17 +0.84
The growth retardation seen in both groups seemed to be more profound inchildren receiving beclomethasone dipropionate. However, the result is merelysuggestive, since the groups were not equal in size. Additionally, children in thebud group were slightly younger and their asthma was more severe comparedwith children in the bdp group. However, since individual sensitivity to boththe effects and side-effects of inhaled glucocorticosteroids may play an impor-tant role, careful follow-up of all children receiving glucocorticosteroids isessential. If deterioration in growth velocity is observed, the possibility ofchanging medication, even between glucocorticosteroids, should also be kept inmind.
P1-198 Growth and Steroid Hormones
Adrogen Receptor Expression in the Epiphyseal Plate ofGrowing Male and Female Rats and Its Regulation byGonadectomyB.C.J. van der Eerden 1; N.P. van Til* 1; A.O. Brinkmann* 2; C.W.G.M. Lowik* 1; M. Karperien* 1; J.M. Wit 1
1 Leiden University Medical Center, Leiden, The Netherlands;2 Erasmus University, Rotterdam, The Netherlands
Androgens have a pronounced stimulatory effect on longitudinal bone growth.This effect is believed to be mediated by the androgen receptor (AR). It ispresently unclear whether the AR is expressed in growth plate chondrocytes.We have therefore evaluated the expression of the AR by immunohistochemis-try in female and male rat tibial growth plates isolated at various timepointsduring postnatal development and are studying its regulation by ovariectomy(OV-X) and orchidectomy (ORCH-X). Tibial growth plates of 1, 4, 7, 12 and 16weeks old female and male rats were isolated and immunohistochemistry wasperformed. In the tibial growth plates of 1, 4, and 7 weeks old rats abundantstaining for the AR was observed in late proliferating and hypertrophic chon-drocytes as well as in the growth plate stem cells. After puberty, in 12 and 16weeks old rats staining was reduced and confined to a few early hypertrophicchondrocytes. The staining was predominantly found in the cytoplasm. Thestaining patterns were similar between the two sexes, with male rats having ahigher staining intensity of AR, especially in the stem cell zone. The specificityof the staining was controlled by performing immunohistochemistry on testisin which abundant staining was observed in spermatogonia in sequential stagesof differentiation in concordance with literature while omission of the first anti-body did not reveal any staining. We subsequently studied whether AR expres-sion is regulated by sex-steroids. For this, rats were SHAM operated or gonad-ectomised at 3 weeks of age and 3 weeks later tibial growth plates were isolated.Compared to the SHAM-operated rats, the OV-X rats (n = 5) had a significantlyhigher body weight increase (173 vs. 137%; p ! 0.001) and growth plate width(0.52 vs. 0.43 mm; p = 0.007). In contrast, the ORCH-X rats had a lower bodyweight increase (169 vs. 186%; p = 0.011) and a smaller growth plate width(0.52 vs. 0.43 mm; p = 0.002) compared to controls. Staining for AR in tibialgrowth plates of the OV-X and ORCH-X rats was confined to late proliferatingand hypertrophic chondrocytes as well as to growth plate stem cells, which issimilar to the untreated rats. Results concerning the relative AR expression inthe OV-X and ORCH-X rats compared to their SHAM operated controls arecurrently underway. We conclude that AR is present in tibial growth plates atvarious timepoints during postnatal development of male and female rats, sug-gesting that part of the growth promoting effects of androgens are mediated bydirect actions on growth plate chondrocytes. The expression of the AR stronglydeclines after puberty. In males more AR expression seems present, suggestingthat male rats tend to rely more on direct actions of androgens on growth platechondrocytes than do females.

60 Horm Res 2000;53(suppl 2):1–191 Posters
P1-199 Growth and Steroid Hormones
Growth Patterns and Final Height in Congenital AdrenalHyperplasia (CAH). Results of a Multicenter StudyG. Hargitai* 1; J. Slyom 1; T. Battelino 2; J. Lebl 3; Z. Pribilincova* 4; R. Hauspie* 5; J. Kovacs* 6; F. Waldhauser 7; H. Frisch 7; MEWPE-CAHStudy Group1 Semmelweis University, Budapest, Hungary; 2 University ofLjubljana, Ljubljana, Slovenia; 3 Charles University, Prague, CzechRepublic; 4 University Children’s Hospital, Bratislava, Slovakia; 5 FreeUniversity of Brussels, Brussels, Belgium; 6 University of Szeged,Szeged, Hungary; 7 University of Vienna, Vienna, Austria
Longitudinal growth and bone age development are the most important clinicalparameters to monitor adequate glucocorticoid replacement in children with CAH.Objective: To analyze the growth pattern and final height in a large group of treatedCAH patients. Patients and Methods: A data base of 600 patients with CAH wascreated in 5 Middle European countries and growth data of 341 treated patients with21-OH deficiency (salt wasting (SW) and simple virilizing (SV)) were analyzed retro-spectively. Centiles were constructed in a longitudinal/cross sectional way and anadditional longitudinal analysis was done to evaluate the pubertal growth spurt bythe use of particular statistical methods (Preece-Baines method). Results: Growthof SW patients was impaired in infancy and early childhood (0–3 years of age), butfollowed normal patterns in childhood until puberty. In contrast, children with theSV-CAH had normal patterns of growth in infancy and early childhood and wereconsiderably taller than healthy references during childhood. In the longitudinalstudy, peak height velocity in both, boys and girls had a normal magnitude, but itoccurred at an earlier age than in the standard population. Final height of patientswith CAH was decreased in comparison to the reference and to the individual targetheight. No correlation was found between final height and age at start of therapy inSV patients or between final height and the patients’ year of birth. Bone age wasadvanced, especially in SV patients. Conclusion: Characteristic growth patternsfor treated SV and SW CAH children were identified, the pubertal growth spurt wasdecreased with respect to the early occurrence and final height was reduced.
P1-200 Growth and Steroid Hormones
IGF-I and IGF-Binding Proteins during High DoseEstrogen Treatment in Constitutionally Tall GirlsR. Rooman* 1; L. Op De Beeck* 1; M. Martin* 1; M. Du Caju 1
1 Antwerp University Hospital, Edegem, Belgium
High doses of estrogens have been used to reduce the final height of constitutionallytall girls. The mechanism of this growth inhibition is essentially unknown. Objec-tive: To investigate the effect of estrogens on IGF-I and IGF binding proteins(IGFBP’s). Patients and Methods: Twenty-four constitutionally tall girls wereincluded in this longitudinal study. Ethinyloestradiol 0.1 mg was given every day. In16 patients blood samples were collected at the start of therapy and after 3 months oftreatment. In 8 other patients samples were collected at the end of therapy and 3months afterwards. Sera were analyzed by Western ligand blot and by Westernimmunoblot for IGFBP-3. Total IGF-I , ‘free’ IGF-I , IGFBP-3 and IGFBP-2 weremeasured by immunological methods. The results were analyzed by the Student t-testfor paired data. Results:
Start 3 months on End 3 months off
IGF-I (ng/ml) 528B31 370B27 243B29 354B27IGFBP-3 (ng/ml) 3,919B106 3,963B112 3,205B218 3,664B123IGFBP-2 (ng/ml) 556B46 305B21 349B47 504B52
Free IGF-I fell to 35 B 4% after 3 months of estrogen therapy and rose again whenthe treatment was stopped. The changes in IGFBP-2 and IGFBP-3 were confirmedby ligand blot analysis. In addition a strong induction of a 24 kDa band corre-sponding to IGFBP-4 was found. In the 30 kDa area 4 other binding species weredetected. The 2 bands with the lowest molecular weight were increased after 3months of estrogen therapy . One of these bands is probably an IGFBP-3 fragmentthat was identified by Western immunoblot. Immunoblots for IGFBP-1, IGFBP-4and IGFBP-5 failed to detect any of the 3 remaining bands. Conclusion: Estro-gens modulate several components of the IGF system such as IGF-I, IGFBP-2,IGFBP-4 and possibly IGFBP-3 proteolysis. The important decrease in total IGF-Iand free IGF-I are most likely the result of an inhibitory effect of estrogens onIGF-I synthesis. Besides the decrease in IGF-I, the induction of IGFBP-4 may alsocontribute to the growth inhibitory effects of estrogens.
P1-201 Growth and Steroid Hormones
24-Hour Urine Cortisol Metabolites in ChildrenO.D. Wolthers 1; C. Heuck* 2; J.W. Honour 3
1 Randers Central Sygehus, Randers, Denmark; 2 Aarhus, Denmark;3 UCL, London, United Kingdom
Systemic glucocorticoids may suppress urine cortisol metabolites. Objective:To assess whether administration regimen may influence the suppressiveeffects of prednisolone on 24-hour urine cortisol metabolites in children. Sub-jects and Methods: Four girls and four boys aged 10–15 years with pubertalstages I–IV were studied in an open 2 period cross-over trial with a 1-day runin, a 4-day period of 5 mg prednisolone in the morning and a 4-day period of5 mg prednisolone in the evening with a 3-week washout. During run in and onthe last day of treatment periods urine was collected during 24 h. Cortisolmetabolites were determined by gas chromatographic analysis. Results: Thefollowing cortisol metabolites were detected: 11ß-hydroxyandrosterone, 11ß-hydroaetiocholanolone, tetrahydrocortisone, tetrahydrocortisol, allo-tetrahy-drocortisol, ·-cortolone, ß-cortolone + ß-cortol, ·-cortol. Total cortisol metabo-lites (SEM) were 7,927 (800) (run in), 5,320 (649) (prednisolone in the morn-ing) and 4,970 (787) Ìg (prednisolone in the evening). The differences betweenrun in and prednisolone treatments were: run in – morning: 2,606 (720) Ìg,95% confidence interval 903–4,309 Ìg, t = 3.6, p = 0.009; run in – evening:2,956 (624) Ìg, 95% confidence interval 1,480–4,432 Ìg; t = 4.7; p = 0.002);between treatments: 350 (696) Ìg, 95% confidence interval 903–4,309 Ìg, t =–0.5, p = 0.63. Conclusion: Prednisolone in the morning or in the eveningcauses a similar suppression of 24-hour urine cortisol metabolites.
P1-202 Growth and Steroid Hormones
Glucocorticoids Modulate the Cytotoxic Effects of aVariety of DNA Damaging Agents in Both Normal andMalignant Cells in VitroH. Robson* 1; O.B. Eden 1; E. Anderson 1; S.M. Shalet 1
1 Christie Hospital NHS Trust, Manchester, United Kingdom
We have previously reported that in vitro pre-treatment of normal rat growthplate chondrocytes with glucocorticoids (GC) reduces the adverse effects of anumber of cytotoxic drugs commonly used in the treatment of childhood cancers.Extrapolation of these experimental results suggests that GCs could be used toprotect children undergoing intensive chemotherapy against the long termadverse effects on growth. However, little is known about the modulatory effectsof GCs on the chemosensitivity of malignant cells. In the present study we exam-ined the sensitivity of human osteosarcoma cell lines (HOS, MG63, U2OS,G292) to Dexamethasone (dexa) and whether DNA damage induced by Cispla-tin (cDDP), Etoposide (VP16) and Doxorubicin (doxo) is reduced by this agentin these cells. The cytotoxic drugs reduced cell number in all four osteosarcomacell lines although the degree to which this occurred was highly variable. Dexaalso exhibited significant differences in terms of its effect on cell number, havingno effect in two of the cell lines, an inhibitory effect in the MG63 cell line and astimulatory effect in the G292 cell line. In the latter, cell numbers were increasedby 72.7 B 8.2% (mean B SD; p ! 0.01) above the untreated control at a dexaconcentration of 0.05 mg/ml. To determine whether GC responsiveness wasinfluenced by the ligand and non-ligand binding glucocorticoid receptor (GR) ·and ß isoforms, specific antibodies and dual immunofluorescence techniqueswere used. GR· was found in the nucleus and cytoplasm, whilst GRß wasexpressed only in the nucleus, of non-mitotic cells. Using confocal microscopyand dual immunofluorescence GR· and ß did not co-localise within the nucleuswith GRß being uniquely located within the nucleoli. This suggests not only thatGC responsiveness is not influenced merely by the presence or absence of theGRs in these cells but also that there is no direct interaction between these tworeceptor isoforms. By pretreating the osteosarcoma cells with dexa for 48 h priorto adminstration of the cytotoxics, a significant reduction (p ! 0.005) in the toxiceffects of VP16, cDDP and doxo, by between 5 and 45% in 3 of 4 osteosarcomacell lines was observed. Preliminary data indicate that this modulatory effect ofdexa is not due to a reduction in the amount of DNA damage incurred, asassessed using the Alkaline Comet Assay (Tail Moment pre- vs. non pre-treatedcells (mean B SD): VP16, 128.3 B 30.0 vs. 114.3 B 31.1; doxo, 127.3 B 27.0 vs.123.2 B 22.9; cDDP, 114.9 B 35.0 vs. 124.9 B 36.8). In conclusion these find-ings clearly have important implications for treatment schedules involving bothcytotoxic drugs and GCs in childhood cancers and highlights the need to under-stand the mechanisms by which GCs differentially influence cell growth andcellular survival mechanisms.

Group A
39th Annual Meeting of the ESPE Horm Res 2000;53(suppl 2):1–191 61
P1-203 Growth and Steroid Hormones
Estrogen Induced Hyperprolactinemia Leading toProlactinoma in Tall Girls Treated with EstrogensR. Odink 1; W.M. van Waarde 1; C Rouwé 1
1 Beatrix Children’s Hospital, Groningen, Netherlands
Estrogen treatment for tall stature is the treatment of choice for familial tallstature in girls. Estrogens are reported to play a role in hyperprolactinemia andprolactinomas in women. Objective: We evaluated basal prolactin levels bothbefore, during and after estrogen treatment in familial tall girls. MRI imagingwas performed when hyperprolactinemia had been found. Patients andMethods: Twenty-four girls were included in this study. Mean age was 12.8yrs (10.3–14.6) Estrogens in a dose of 100 Ìg per day were given to the tall girls.During 10 days each month a progestogen was added to induce regular vaginalbleeding. Prolactin was measured before, during and after treatment whenavailable (normal value adult women: !600 Mu/l). Results: Mean age at thestart was 12.8 years (10.3–16.8). Prolactin concentrations at start, during andafter treatment were: –280 (range: 65–1,220), 640 (range: 210–2,300) and 430(range 65–1,680) respectively. One girl experienced galactorrhea after treat-ment, (prolactin: 1,680) and with MRI imaging she appeared to have a micoad-enoma. Dopamine agonist treatment (Quinagolide) was instituted. This re-sulted in prolactin concentrations of !100 Mu/l and the clinical symptoms dis-apppeared. Conclusions: Estrogens in a dose of 100 microgram per dayinduce prolactin secretion in girls with familial tall stature. As a consequence inthese girls prolactinoma’s may be induced by estrogen.
P1-204 Growth and Steroid Hormones
Bone Mineral Density and Age in Congenital AdrenalHyperplasiaB. Pawlaczyk 1; M. Orkiszewski 1; M. Orkizewska 1; E. Pawlaczyk-Wroblewska 1; Z. Drozdz* 1
1 Institute of Pediatrics, University of Medical Science in Poznan,Poland
Among disturbances connected with treatment of congenital adrenal hypertro-phy one of the most interesting are bone structure disturbances. These distur-bances create differences between bone and calendar age. Data about teeth ageare very small. We examined 22 girls, and 5 boys, age 2–17 y, suffering fromcongenital adrenal hypertrophy. 19 of them presented salt loosing syndrome.They were treated by hydrocortison in dosage 15–25 mg/m2 of body surface,and 9·-fluorocortison in dosage 0.05–0.1 mg/24 h. All our patients we con-trolled somatic development using skeletal, teeth and height age. In a group of12 (10 girls and 2 boys) we made ultrasound densitometry. Skeletal age wassignificantly lower (p ! 0.014) in a group of 15 patients (81.5%). In a group of7 persons skeletal age was higher than calendar age (difference of l–2 years), and5 patients did not present any differences. Teeth age in 19 persons was normal,1 patient had lower, and 7 had higher teeth age than calendar age. Skeletal agein 20 persons was decreased, in 3 it was increased and in 4 it was adequate tocalendar age. Analysis of skeletal and height age shows that this parameters arenot connected with kind of enzymatic disturbance. Teeth age in most of exam-ined persons was adequate with calendar age, but it was not adequate withskeletal age, and is not connected with kind of enzymatic disturbance. Densi-tometry shows in all patients osteopenia, based on WHO norm T-score 1.12–2.48, one boy 16 years old had 2.51 standard deviations lower density thanmax. for his age. Lower bone density and disturbances in bone structure areeffected by chronic steroid therapy, and they caused secondary osteoporosis.From our observations we find that Polish Osteoporosis Society propositionabout wit D dosage 400–800 units/24 h, and calcium 1.2 g/24 h and alendron-ian sodium (Fosamax) – 10 mg/day, are necessary.
P1-205 IGF and Growth Factors
IGF-I and Its Binding Proteins 1 and 3 in Adolescentswith IDDM and Diabetic ComplicationsA. Niedzwiedzka* 1; H. Dziatkowiak* 1; Introduced by M. Maes1 Department of Pediatric Endocrinology, Polish-American Children’sHospital, Krakow, Poland
Numerous studies suggest that growth factors could take part in developmentof diabetic complications. The aim of the study was evaluation of Insulin-Growth-Factor I (IGF-I) and its binding proteins I and 3 (IGFBP-1, IGFBP-3)
in adolescents with IDDM and diabetic complications. 32 patients and 13 con-trols were examined. Diabetic patients were divided into two groups: group A(17 adolescents) with diabetic complications and group B (15 adolescents) withIDDM without complications. Mean age and glycosylated hemoglobin(HbA1c) (SD) were respectively: 18 (1.5), 9.8 (1.8) in group A and 17.97 (1.8),9.46 (1.3) in group B. Mean age in control group C was 17.35 (2.7). All patientsin group A had microalbuminuria, two of them had also retinopathy. Bloodsamples were taken at 8.00 a.m., after 8 h fasting. The blood concentrations ofIGF-I, BP-1 and BP-3 and urine albumin concentration were measured byradioimmunoassay; HbA1c by HPLC method. Diabetic patients were exam-ined by an experienced ophthalmologist. There were no differences regard toage and body mass index (BMI) between the three groups. Mean values of BMI(SO) were in group A: 21.11 (4.7), in group B: 22.57 (3.4) and in group C 21.65(2.5). Main results are in table (with SD in parentheses).
IGF-I [ng/ml] IGFBP-I [ng/ml] IGFBP-3 [Ìg/ml]
312.0 (150.6) 90.3 (61.4) 8.3 (3.4)Group B 375.4 (92.7) 39.1 (46.3) 7.4 (1.4)Group C 454.5 (139.8) 51.5 (21.7) 4.2 (0.9)
Data were statistically analysed by t-Student test. There were found differencesin IGF-I values between groups A and C (p = 0.027) and in IGFBP-1 valuesbetween groups A and B (p = 0.034). IGFBP-3 values differ in group B and C(p = 0.003). There were found no differences between other parameters in thethree groups. Our data indicate that IGF serum level depends mainly on insulinaction. Examined patients belong to the group with poor metabolic control ofdiabetes (HbA1c 18%). IGFBP-1 concentrations were different in IpDMgroups, in spite of the similar values of HbA1c and were significantly higher inpatients with diabetic complications. This study cannot exclude the role oflocally produced IGF-I (in eye and kidney) in development of microangiopa-thy. Presented data could indicate the participation of circulated IGFBP-1 inorigin of diabetic complications.
P1-206 IGF and Growth Factors
Responsiveness to IGF-I and IGF-II and IGFBP-3Secretion of Skin Fibroblasts of Children with SotosSyndromeL. de Boer* 1; E. Willemstein* 1; M. Karperien* 1; T. Maassen* 1; J.M. Wit 1
1 University Medical Center Leiden, Leiden, The Netherlands
Sotos syndrome is an overgrowth syndrome of which the aetiology is still unknown.An important characteristic is excessive growth of length and head circumferencein utero, followed by rapid growth in the first year and tall stature in childhood,adolescence and adulthood. IGF-I and -II are known to be important foetal growthfactors. In Sotos syndrome serum somatomedin activity and IGF-I has beenreported normal and no data are available on serum IGF-II. Objective: To inves-tigate whether overgrowth in Sotos syndrome is associated with changes in differ-ent characteristics of skin fibroblasts in terms of responsiveness to IGF-I and IGF-II and IGFBP-3 secretion in comparison to control fibroblasts. Patients andMethods: Skin fibroblasts of 5 children with Sotos syndrome and 6 healthy con-trols with normal stature were cultured on 9-cm tissue culture plates in DMEMcontaining 10% FCS. Fibroblasts were cultured for 48 h in 24-well plates inDMEM 10% FCS and then starved for 72 h in DMEM containing 0.1% BSA. Afterstimulation with IGF-I and IGF-II for 24 h, [3H]thymidine was added, the cellswere lysed and the samples were counted. Mitogenic responses were expressed infold stimulation. For IGFBP-3 measurements in media, cells were cultured asabove and seeded on 6-well plates. After culture in DMEM 10% FCS for 24 h, cellswere starved for 120 h in DMEM 0.1% BSA. Media were collected and stored at–20 °C. After ultrafiltration, the medium was concentrated 10 times and IGFBP-3levels were measured by RIA. For mitogenic responses the Student t-test was per-formed after log transformation and for IGFBP-3 secretion the Mann-Whitneytest. Results: [3H]thymidine incorporation in response to IGF-I (10 ng/ml) andIGF-II (10 ng/ml) in Sotos fibroblasts was not significantly different from controls:median 4.5 (range 2.29–6.7) vs. 4.1 (2.8–12.7) for IGF-I and 4.7 (1.3–7.3) vs. 4.0(2.5–14.3) fold stimulation for IGF-II, p = 0.56 and p = 0.74. Fibroblasts ofpatients with Sotos syndrome tended to secrete less IGFBP-3 than controls, but thedifference did not reach significance: median 2.7 ng/ml (range 1.1–2.8) vs. 3.6 ng/ml (0.7–9.1), p = 0.50. Studies in an additional number of 20 patients with Sotossyndrome are in progress. Conclusions: These preliminary data indicate thatovergrowth in patients with Sotos syndrome is not associated with increasedresponses to IGF-I and IGF-II in skin fibroblasts, but IGFBP-3 secretion tends tobe lower. Data on 25 patients will be presented.

62 Horm Res 2000;53(suppl 2):1–191 Posters
P1-207 IGF and Growth Factors
Sotos Syndrome Is Associated with Elevated SerumIGFBP-3 and Normal Serum IGF-IL. de Boer* 1; M. Frohlich* 1; J.M. Wit 1
1 Leiden University Medical Center, Leiden, The Netherlands
Sotos syndrome is characterized by rapid intrauterine growth in terms of lengthand head circumference, tall stature in childhood and adulthood, advancedbone age, mental retardation and dysmorphic features. In view of the impor-tance of IGF’s for intrauterine and postnatal growth, a disturbance in IGF-I orIGF-II secretion in Sotos syndrome is plausible. Previous studies have shownthat the biological activity in plasma of these growth factors (‘somatomedinactivity’) is normal. This was later confirmed by sporadic reports on serumIGF-I. No data have been reported on serum IGF-II and IGF binding proteins(IGFBP’s). Objective: To investigate whether overgrowth in Sotos syndromeis associated with abnormal serum levels of IGF-I and IGFBP-3. Patients andMethods: Nineteen patients (5 F, 14 M) with a mean age of 12 years (range2.1–36.4) were enrolled in this study. The diagnosis was based on at least threeof the following criteria: typical facial characteristics, rapid growth in the firstyear, advanced bone age and mental retardation. The serum IGF-I was mea-sured by RIA (Incstar) and IGFBP-3 by RIA (Nichols). Results are expressed asmean (SEM) SDS. Results: Mean IGF-I SDS was –0.3 (0.34) (n = 19), notsignificantly different from the reference. Mean IGFBP-3 SDS was 1.6 (0.32)(n = 16), significantly greater than the reference population (p ! 0.001). Fiveboys (age 4.1–15.2 years) showed a value 12SDS. Determinations of serumIGF-II and other IGFBP’s are in progress. Conclusions: These data confirmearlier reports on normal IGF-I serum levels in patients with Sotos syndrome.A novel finding is that Sotos syndrome is associated with elevated IGFBP-3levels. Further studies are needed to unravel the pathophysiological signifi-cance of this finding.
P1-208 IGF and Growth Factors
Serum IGF-I in Turner Syndrome: The Age DependentPattern during Growth Hormone TherapyS. Pruhov* 1; J. Lebl 1; M. Pechov* 1
1 Charles University, Praha, Czech Republic
Patients with Turner syndrome (TS) are growth retarded in spite of normalcirculating IGF-I levels. Their growth failure might be attributed to a decreasedsensitivity to IGF-I in distinct cell lines. GH therapy in TS may help to over-come the IGF-I resistance. Objective: To evaluate age-dependent pattern ofserum IGF-I levels during GH or GH+estradiol (E2) treatment in GH-treatedgirls with Turner syndrome. Patients and Methods: 56 patients with TS (age4.4–18.3; median 11.9) were studied on 138 occasions at least 3 month apartduring their GH treatment (1 IU/kg/week). Patients with clinical signs of spon-taneous puberty (Tanner stage B 11 without estrogen substitution) wereexcluded from the study. The patients were treated either with GH only or withGH+E2. Circulating IGF-I levels were expressed as SDS compared to back-ground population. Results: The individual IGF-I levels are shown in Fig. 1(full circles – GH only, opened squares – GH+E2, area between the curved lines– mean B 1 SD). Mean SDS of IGF-I levels is shown in Fig. 2.
Conclusions: In Turner girls on long-term GH therapy, circulating IGF-I lev-els are consistently markedly elevated compared to age-specific standards. Theage-dependent pattern of IGF-I levels is conserved in TS during GH therapy inspite of their lack of estrogens but the onset of pubertal increase tends to appearearlier. It might be contributed to adrenarche. With respect to clinical experi-ence, there is no correlate in an increasing growth velocity. Later on, the estro-gen substitution does not lead to a further increase of IGF-I levels in spite of itsknown modest growth promoting effect. The long term risks of the markedlyelevated IGF-I levels remain unknown.
P1-209 IGF and Growth Factors
Final Stature in a Patient with Growth HormoneDeficiency (GHD) Type 1A Treated with RecombinantHuman Insulin-Growth Factor I (IGF)M.F. Messina* 1; M. Wasniewska* 1; F.A. De Luca* 1; M. Valenzise* 1;F. Lombardo* 1; F. De Luca 1
1 Institute of Pediatrics, University of Messina, Messina, Italy
IGF is a novel drug which has been used in the treatment of only two diseases:GH insensitivity syndrome and GH-N gene deletion. Although IGF is knownto represent the elective therapy and to accelerate growth velocity in these con-ditions, its long-term results in terms of final height (FH) are not well estab-lished to now. A 7-mth girl with GHD type 1A was referred to our Centre forsevere height failure (–5.0 SDS) and treated with biosynthetic GH, showing aninitial excellent growth response which lasted for a long time (Acta Paediatr 91,80:1235). After 46 months of therapy she developed GH antibodies and hergrowth rate decreased to 0.2 cm/month (Acta Paediatr 92, 81:730). At the age of7.6 yrs GH treatment was definitively discontinued and one year later (height–4.0 SDS, growth velocity 1.2 cm/year) she started on IGF at a dose of 40 Ìg/kgmice daily, subcutaneously. Therapy was rigorously administered after break-fast and dinner in order to avoid or minimize the hypoglycaemic effects anddoses were gradually augmented up to 120 Ìg/kg twice daily during the follow-ing years. Although a satisfactory height acceleration (5.8 cm/yr) was recordedduring the first 2 yrs of therapy, no significant catch-up was observed andheight deficiency at the time of puberty onset (age 10.8 yrs. bone age 10.5 yrs )was superimposable to the one measured at the time of IGF therapy start (–4.0SDS). During puberty (duration 3 yrs) no growth spurt occurred and totalpubertal height gain (9.2 cm) was very disappointing, with a FH (128.6 cm)widely lower with respect to target height (153.6 cm). During the entire IGFtherapy period (5 yrs) no significant side effects were recorded and only mildand sporadic headaches were reported. In conclusion, we may assert that:1) IGF treatment even though prolonged for a long period is relatively safe;2) Patients with GHD type 1A under IGF show a satisfactory statural growthduring the first yrs, as also reported by others (Horm Res 99;51:128); 3) Due tothe absence of a significant pubertal growth spurt, the long term results in termsof FH are very disappointing.

39th Annual Meeting of the ESPE Horm Res 2000;53(suppl 2):1–191 63
P1-210 IGF and Growth Factors
Relationship of Changes in IGF-I, IGFBP-3, ALS andLeptin to Inflammatory and Nutritional Markers duringEnteral Feeding in Children with Crohn’s DiseaseK. Bannerjee* 1; N.M. Croft* 2; K. Babinska* 1; R. Edwards* 3; C. Camancho-Hübner 1; I.R. Sanderson* 4; M.O. Savage 3
Departments of 1 Paediatric Endocrinology and 2 Gastroenterology,and 3 NETRIA UNIT St. Bartholomew’s Hospital and the RoyalLondon School of Medicine and Dentistry, London, United Kingdom
Aims: Growth failure is common in Crohn’s disease, but the relative contribu-tions of nutritional status and inflammatory activity are difficult to differen-tiate. Exclusive enteral feeding with casein based feeds is known to suppressdisease activity and improve growth and nutrition. This study aimed to showthat changes in growth factors and associated proteins are temporally related todecrease in inflammation (CRP , ESR) rather than improvement of nutrition.Seven children with active small bowel Crohn’s disease being treated with a sixweek course of an enteral feed (AL110, Nestle UK) were studied at day 0, 3, 7,14, 21, 28 and 2, 4 months. The weight, paediatric Crohn’s disease activityindex (PCDAI), triceps skin folds (TSFT), mid upper arm circumference(MUAC) and IGF-I, IGFBP-3, ALS, Leptin and CRP, ESR were measured ateach visit. Results: At day 7, there were significant decreases in PCDAI andESR and an increase in IGF-I (all p 0.05). CRP was decreased on day 14 andfat stores (MUAC, TSFT) did not increase until day 21 and leptin at 4 months(p = 0.01). Conclusions: Changes in clinical status, IGF-I and inflammation(ESR) occurred before any significant changes in nutritional, parameters.These data suggest that the beneficial effect of enteral nutrition in childhoodCrohn’s disease may be primarily due to suppression of inflammation ratherthan to improvement of nutritional status.
P1-211 IGF and Growth Factors
Cord Blood Insulin-Like Growth Factors (IGF) and Sizeat Birth: Opposing Effects of IGF-II and Soluble IGF-IIReceptor LevelsK. Ong* 1; J. Kratzsch* 2; W. Kiess 2; C. Scott* 3; Study TeamALSPAC 4; D. Dunger 1
1 University of Cambridge, Cambridge, United Kingdom; 2 Universityof Leipzig, Leipzig, Germany; 3 University of Sydney, Sydney,Australia; 4 University of Bristol, Bristol, United Kingdom
Experimental knock-out studies in rodents demonstrate the importance ofimprinted genes encoding IGF-II and its specific inhibitor, the non-signallingIGF-II/mannose-6-phosphate receptor (IGF2R) in fetal growth; igf2 is exclu-sively paternally expressed and promotes fetal growth, while the maternallyexpressed igf2r inhibits fetal growth. However the roles of these factors inhuman fetal growth is yet unclear. Methods: In 199 normal term (37–42weeks gestation) singletons from the large Avon Longitudinal Study of Preg-nancy and Childhood (ALSPAC) birth cohort, we measured cord blood levelsof insulin, IGF-I, IGF-II, IGFBP-1, IGFBP-3 and soluble IGF2R and exam-ined their relationships to weight, length, head circumference, ponderal index(PI) and placental weight at birth. The molar ratio of IGF-II to IGF2R wascalculated as an estimate of their expected interaction. Results: No sex differ-ences in cord levels were seen. Insulin levels decreased and IGFBP-1 increasedwith gestational age. IGF-I levels were significantly related to insulin (r = 0.32,p ! 0.0005), IGFBP-1 (r = –0.36, p ! 0.0005), IGFBP-3 (r = 0.36, p ! 0.0005)and IGF-II (r = 0.29, p ! 0.0005). IGF-II levels were also related to IGFBP-3(r = 0.45, p ! 0.0005) and soluble IGF2R (r = 0.20, p ! 0.005). Insulin was mostclosely related to PI (r = 0.31, p ! 0.0005), while IGF-I was positively related toall parameters of size at birth. IGF-II was weakly related to PI (r = 0.18, p !0.05) and placental weight (r = 0.18, p ! 0.05), and the molar ratio of IGF-II tosoluble IGF2R was also related to birth weight (r = 0.15, p ! 0.05). Correlationsbetween IGF’s and size at birth were much stronger in non-primiparous preg-nancies and in these, IGF-I (r = 0.52, p ! 0.0005), IGFBP-3 (r = 0.41, p !0.0005) and the IGF-II to IGF2R ratio (r = 0.40, p ! 0.0005) were most closelyrelated to placental weight, together accounting for 39% of its variance. Con-clusions: Our data demonstrate for the first time the opposing influences ofIGF-II and IGF2R on normal human fetal growth. It is known that familial
birth weight correlations improve with increasing parity. As associationsbetween IGF’s and size at birth were stronger in multiparous pregnancies, wesuggest that the familial influence on size at birth IGF’s may be mediated viathe IGF system.
P1-212 IGF and Growth Factors
Relative Effects of Early and Later Weight Gain onCirculating IGF-I and Leptin Levels at 5 YearsK. Ong* 1; J. Kratzsch* 2; W. Kiess 2; Study Team ALSPAC 3; D.B. Dunger 1
1 University of Cambridge, Cambridge, United Kingdom; 2 University of Leipzig, Leipzig, Germany; 3 University of Bristol,Bristol, United Kingdom
Rate of weight gain and growth during infancy is predictive of rate of growthand maturation in later childhood. We proposed that early growth rates mayhave a long-term influence on levels of circulating growth factors. Methods:Children in the large Avon Longitudinal Study of Pregnancy and Childhood(ALSPAC) birth cohort have been regularly measured for weight and height(length up to 2 years) since birth. At 5 yrs, a non-fasting venous blood samplewas collected for measurement of IGF-I and leptin levels, and fat mass andfat-free mass were estimated from 4 skinfold thickness measurements. Com-plete hormone and growth data were available for 524 children (294 boys, 230girls). Multiple regression models were used to compare the effects of fat massvs. fat-free mass, and early vs. late growth on levels of each hormone; partialcorrelation coefficients (ß) are displayed. Sex and gestation adjusted standarddeviation scores (SDS) were calculated for weight at 0 and 2 yrs. Postnatalcatch-up growth was defined by a gain in weight SDS 0–2 yrs of 10.67 SDS, andcatch-down growth by a change !–0.67 SDS. Results: Girls had higher meanlevels of IGF-I (160 ng/ml) and leptin (1.47 ng/ml) than boys (IGF-I: 138 ng/ml,p ! 0.0005) (leptin: 0.88 ng/ml, p ! 0.0005). All subsequent analyses areadjusted for sex. IGF-I levels were more closely related to fat-free mass (ß =0.22, p ! 0.0005) than to fat mass (ß = 0.12, p ! 0.05), while leptin levels wereclosely related to fat mass (ß = 0.57, p ! 0.0005) but not to fat-free mass (ß =0.01, NS). IGF-I levels at 5 yrs were as closely related to early growth between 0and 2 yrs (weight gain: ß = 0.14, p ! 0.005; length gain: ß = 0.26, p ! 0.0005) asto growth 3–5 yrs (weight gain ß = 0.23, p ! 0.0005; length gain: ß = 0.19, p !0.0005). In contrast, leptin levels were more closely related to 3–5 yrs weightgain (ß = 0.40, p ! 0.0005) or length gain (ß = 0.20, p ! 0.0005) than to 0–2 yrsweight gain (ß = 0.17, p ! 0.0005) or length gain (ß = 0.09, p ! 0.05). Thus, evenon allowing for current size (weight, height or fat-free mass at 5 yrs), childrenwho showed postnatal catch-up growth 0–2 yrs had higher mean IGF-I levels at5 yrs (157 ng/ml) than those who showed little/no change in weight SDS (147ng/ml) or catch-down growth (140 ng/ml) (p trend adjusted for fat-free mass =0.008). Conclusions: Irrespective of current body size, circulating levels ofIGF-I, but not leptin, in 5-yr old children are influenced by rate of growth 0–2yrs. These data may help to explain the positive influence of high infancyweight gain and nutrition on rates of growth and maturation in later child-hood.

P1-213 IGF and Growth Factors
Interleukin-6 (IL-6) Levels and the IGF-IGFBP System inChronic Inflammatory Bowel Disease (CIBD) inChildhoodM.E. Street 1; G.L. de’Angelis* 1; C. Camacho-Hübner 3; M.O. Savage 3; F. Fornaroli* 1; S. Bernasconi 2; G. Giovannelli 1
Departments of Paediatrics, Universities of 1 Parma, and 2 Modena,Italy; 3 Department of Endocrinology, St Bartholomew’s Hospital,London, United Kingdom
Ulcerative colitis (UC) and Crohn’s disease (CD) are characterised by alteredcytokine levels in the serum and in the intestinal mucosa. In some patients thisclinical condition has been associated with impaired linear growth. The aim ofthis study was to assess the serum levels of IL-6, IGF-I, IGFBP-2 and IGFBP-3in patients with CIBD in the acute phase and in remission versus a controlgroup. The subjects with CIBD were divided into 2 groups. Patients were con-sidered ‘acute’ if the ESR was 120 mm/h and the C reactive protein (CRP) wasabove 1.5 mg/dl (N = 19, 7 M, 12 F, 11 with UC and 8 with CD, chronologicalage, CA, mean B SEM: 14.71 B 0.80 yr, pubertal stage (S)1, n = 3, S2 n = 1, S3n = 3, S4 n = 4, S5 N = 8). The subjects in ‘remission’ were 18 (12 M, 7 F, 9 UC,7 CD, CA: 15.35 B 1.07 yr, S1 n = 4, S3 n = 1, S4 n = 4, S5 n = 9). A controlgroup of 7 healthy subjects was studied (5 M, 2 F, CA: 12.81 B 1.40 yr; S1 n =2, S4 n = 4, S5 n = 1). Males and females were analysed together. IGF-I levelsdid not differ between the 3 groups. However, if the subjects with acute CDwere evaluated separately the levels were significantly lower than in those withCD in remission and controls (148.2 B 29.8 ng/ml versus 342.9 B 45.7 ng/mland 445.2 B 51.6 ng/ml, respectively, p ! 0.05). IGFBP-2 levels were signifi-cantly higher in the acute subjects versus those in remission and the controls(731.2 B 100.8 ng/ml versus 421.5 B 37.6 ng/ml and 445.2 B 51.6 ng/ml,respectively, p ! 0.05). Also patients with acute UC had higher levels than thosein remission and the controls. IGFBP-3 levels were similar in the 3 groupsalthough subjects with acute CD had significantly lower levels compared topatients with acute UC (3060.0 B 232.6 ng/ml versus 3977.3 B 152.8 ng/ml,p ! 0.05). IL-6 levels were significantly higher in the acute subjects versus thosein remission and the controls (5.8 B 0.7 pg/ml versus 3.0 B 0.4 pg/ml and2.2 B 0.4 g/ml respectively, p ! 0.05), in subjects with acute UC versus patientswith CD and UC in remission and controls (7.1 B 1.2 pg/ml versus 2.5 B0.5 pg/ml and 2.2 B 0.4 pg/ml, p ! 0.05), and in acute CD (5.8 B 0.7 pg/ml)versus controls. IGFBP-2 was positively correlated with the ESR (r2 = 0.55, p !0.05) and the IL-6 levels in the entire group (r2 = 0.58, p ! 0.05). The ESR andthe IL-6 levels were also significantly correlated in the entire group (r = 0.62, p! 0.05). In conclusion, the IGF-IGFBP system undergoes changes in CIBD inthe acute phase and these changes seem to be correlated with markers of inflam-mation and the proinflammatory cytokine IL-6.
P1-214 IGF and Growth Factors
Interleukin (IL)-1ß and IL-6 Modulate Insulin-LikeGrowth Factor Binding Protein (IGFBP) Secretion inCaco-2 CellsM.E. Street 1; F. Miraki-Moud* 2; I.R. Sanderson* 3; M.O. Savage 4; G. Giovannelli 1; S. Bernasconi 5; C. Camacho-Hübner 2
Departments of 1 Endocrinology and 2 Paediatric Gastoenterology,St Bartholomew’s Hospital, London, United Kingdom;Departments of Paediatrics, Universities of 3 Parma and 4 Modena,Italy
Chronic inflammation is characterised by modifications in cytokine concentra-tions, whereas growth is mainly dependent on the GH-IGF axis. It is wellknown that IGFBPs modulate IGF-I biological actions, however, the regulationof the IGF system by cytokines is less well established. The aim of the presentstudy was to evaluate the effect of cytokines (IL-1ß and IL-6) on IGFBPs pro-duced by a colonic epithelial intestinal cell line (Caco-2) at different states ofcell differentiation. Experiments were carried out on day 4 (undifferentiatedstate) and day 14 (differentiated state). IGF-I at 50 ng/ml was used as a control.IGFBP secretion was determined by Western Ligand Blotting (125I-IGF-I) andimmunoblotting. Changes in IGFBP intensity were determined by scanningdensitometry. IGFBP gene expression was determined by RT-PCR. Caco-2cells expressed and secreted IGFBP-2, and IGFBP-4. The carcinoembryonic
antigen was used as a marker of differentiation and increased significantly inthe conditioned media (CM) from day 4 to day 14 (0.2 B 0.04 ng/ml/104 cellsversus 0.6 B 0.07 ng/ml/104 cells, p = 0.001). This change was associated withan increase in IGFBP-2 and IGFBP-4 secretion. On day 4 IL-1ß (1 ng/ml)reduced IGFBP-2 secretion (–29.0 B 8.3% versus serum free media, SFM, p !0.05). Similar changes were observed with IL-6 but at higher concentrations(50 ng/ml). IGFBP-4 was significantly reduced after stimulation with IL-1ß at 1and 50 ng/ml (–14.1 B 2.3% and –55.0 B 17% versus SFM, respectively,p ! 0.05), and a similar effect was found with IL-6 at 50 ng/ml. When IL-1ß andIL-6 were used in combination IGFBP-2 was significantly reduced at 1 and 10ng/ml. IGFBP-4 was significantly reduced after stimulation with IL-1ß and IL-6 at 1 ng/ml. On day 14, IGFBP-2 was reduced by both cytokines at the higherconcentration (50 ng/ml). IGFBP-4 was significantly reduced after stimulationwith IL-1ß and IL-6 at 10 and 50 ng/ml. When IL-1ß and IL-6 were used incombination both IGFBP-2 and IGFBP-4 were significantly reduced at 10 ng/ml. IGF-I at 50 ng/ml reduced significantly IGFBP-2 and IGFBP-4 in all theexperiments as previously reported. In summary, IL-1ß and IL-6 regulateIGFBP-2 and IGFBP-4 secretion in Caco-2 cells. The level at which cytokinesact to regulate IGFBP levels is under investigation. The effect of decreasedIGFBP secretion on IGF-I action in these colonic epithelial cells remains to bedetermined.
P2-215 IGF and Growth Factors
The Insulin-Like Growth Factor Receptor Colocalizeswith Estrogen Receptor (ER) · and ER ß in Cells of theAnterior PituitaryJ.A. Chowen 1; A.I. Arroba* 1; L.M. Garcıa-Segura* 2; J. Argente 1
1 Hospital Niño Jesus, and 2 Instituto Cajal, CSIC, Madrid, Spain
It is becoming increasingly evident that insulin-like growth factor I (IGF-I) andsex steroids interact within many types of tissues. Not only can sex steroidsmodulate the production of IGF-I and it’s receptors in some tissues, and vice-versa, but the interaction of these two factors can involve activation of the samereceptors and/or utilization of the same intracellular signaling pathways. Wehave previously reported that IGF-I modulates the effect of estrogen on prolac-tin production. Objectives: The purpose of this study was to determine if theIGF receptor is colocalized with one or both types of estrogen receptor in theanterior pituitary. Methods and Materials: Postpubertal male rats were per-fused and the pituitaries remove and cut on a cryostat (35 Ìm). Double immu-nocytochemistry was performed on floating sections. Sections were incubatedfor 3 days at 4 ° C with a monoclonal antibody towards the IGF receptor (di-luted 1:100) and polyclonal antibodies towards the estrogen receptor (ER) ·(diluted 1:1000) or ER ß (1:250). Sections were then incubated with secondaryantibodies with fluorescent labels for 1.5 h at room temperature. Sections wereobserved in a scanning confocal microscope. Results: The IGF receptor colo-calizes with both ER · and ER ß in the anterior pituitary in specific populationsof cells. Although ER · is expressed in a much larger number of cells than ER ß,the percentage of cells which colocalize ER · and the IGF receptor and ER ßand the IGF receptor is more similar. Furthermore, not all IGF receptor posi-tive cells express ERs. Conclusions: Specific populations of anterior pituitarycells express both the IGF-I and estrogen receptors. Therefore, these two hor-mones may interact within the same cells to modulate hormone production.Further investigation is needed to determine what type of cell is expressingboth receptor types.
64 Horm Res 2000;53(suppl 2):1–191 Posters

S
39th Annual Meeting of the ESPE Horm Res 2000;53(suppl 2):1–191 65
P2-216 IGF and Growth Factors
The Effect of Steroid Treatment on the GH-IGF Axis ofChildren with a Reduced GFRP. Kapila* 1; L. Rees* 1; Introduced by Camacho-Hubner1 Institute of Child Health, London, United Kingdom
Impaired linear growth in children with uremia and those receiving chronicsteroid treatment post-transplantation has been well established. The mecha-nisms for the growth failure in such patients is less clear; it has been proposedthat steroids reduce growth hormone (GH) secretion, whilst uremia may causeresistance to its actions. What is uncertain is whether a reduced glomerularfiltration rate (GFR) and steroid treatment have a cumulative effect on growth.Aim: To compare the GH-IGF axis between children on dialysis (D), chronicrenal failure (CRF) or post renal transplantation (T) with children with normalGFR receiving steroid treatment (S). Patients and Methods: 78 childrenattending the Nephrology Out-patients Clinics at Great Ormond Street Hospi-tal were recruited into the study. Patient details (n, age: mean B SD, steroiddose, GFR ml/min/1.73 m2) were as follows: 10 S (11.5 B 2, 0.42 B 0.2 mg/kg/alt. day, NA); 31 CRF (11.7 B 4, NA, 24 B 12); 27 T (12.2 B 3, 0.25 B 0.2mg/kg/alt. day, 42 B 22) and 10 D (10.8 B 4, NA, NA). Pubertal status rangedfrom pre- to mid-puberty. Blood samples were obtained at 9 a.m. and analysedfor IGF-I, IGFBP-1, IGFBP-3, acid-labile subunit (ALS), and insulin. Kruskal-Wallis statistical test was used for the comparison. Results: The data areexpressed as median (interquartile).
Group Ht SDS IGF-Ing/ml
IGFBP-1ng/ml
IGFBP-3mg/l
ALSmg/l
InsulinmU/l
–1.0 (1.3)* 479 (295) 29.6 (21.8)* 4.3 (1.3)* 47 (10.7)* 12.2 (16.9)T –2.56 (3.3) 417 (343) 68.5 (56.8) 5.0 (0.9) 28.9 (9.1) 14.1 (8.0)C –2.26 (2.2) 350 (257) 79.7 (51.2) 5.3 (0.9) 42 (44.6) 10.7 (6.4)D –1.03 (2.8) 417 (203) 131 (62.3) 5.8 (1.7) 28.6 (7.1) 11.8 (7.1)
* Significant difference between groups (p ! 0.05).
Conclusion: Steroids may add to the effect of GFR on Ht SDS by a mecha-nism other than increasing levels of IGFBP-1 and -3.
P2-217 IGF and Growth Factors
The Effect of Body Composition and Serum Leptin onIGF-1 Response to Growth Hormone Administration inPrepubertal ChildrenR. Coutant* 1; F. Boux de Casson* 1; O. Douay* 1; S. Rouleau* 1;J.L. Ginies* 1; M. Audran* 1; J.M. Limal 1
1 University Hospital, Angers, France
The growth hormone-IGF-1 axis is involved in the regulation of growth andbody composition through its anabolic and lipolytic effects. Whereas metabolicor hormonal factors affecting growth hormone secretion have been extensivelystudied, data on factors influencing the IGF-1 response to GH administrationare limited. We report the IGF-1 response 24 hours after a single administra-tion of growth hormone (6 IU/m2 subcutaneously) in 51 healthy short normalchildren (32 boys, 19 girls) aged 9.7–16.5 years with decreasing growth velocitybefore the pubertal growth spurt. The aim of this study was to analyze the effectof body composition using dual energy X-ray absorptiometry, as well as ofserum leptin concentration on this response. Mean height was –2.3 B 1.0 SDscore. Children were all at Tanner stage I. Mean serum baseline IGF-1 concen-tration was 288 B 103 ng/ml and mean serum IGF-1 increase in response toGH administration was 131 B 75 ng/ml, corresponding to a 47.6 B 30.7 %increase over the baseline value. A significant positive correlation was foundbetween IGF-1 response to GH, and body mass index, total body fat (r = 0.53,p ! 0.01) or percent of fat mass (r = 0.56, p ! 0.001), indicating that metabolicor hormonal factors related to fat mass may control GH sensitivity. In thisrespect, the IGF-1 response to GH was also positively correlated to baselineserum C peptide (r = –0.39, p ! 0.05) and serum leptin (r = 0.43, p ! 0.05), andnegatively to plasma free fatty acids (r = –0.39, p ! 0.05). Conversely, the GHpeak to insulin tolerance test negatively correlated to baseline serum C peptide(r = –0.40, p ! 0.01) and serum leptin (r = –0.41, p ! 0.05). We demonstrated a
significant positive correlation between GH sensitivity and body fat. Leptinand insulin may be part of the mechanisms relating storage of energy and IGF-1secretion. Our observations suggest that the level of nutritional status has oppo-site effects on GH production and GH sensitivity.
P2-218 IGF and Growth Factors
Values of Serum IGF-I, IGFBP-1, IGFBP-3 and Insulin inChildren with Normal Weight and Overweight between4 and 10 YearsG. Cesaretti* 1; G. Saggese 1
1 University of Pisa, Pisa, Italy
The children with an overweight may show an alteration of metabolic charac-teristics since their first years of life. Objective: The evaluation of the presenceof metabolic change in a group of overweight children in comparison to a groupof subjects with normal weight. Patients and Methods: We measured IGF-I,IGFBP-1, IGFBP-3 and insulin levels in a cross-sectional study of 126 healthyprepubertal subjects (69 M, 57 F) aged 4–10 years: 77 with a normal weight(No) and 49 with a simple overweight (Ob). The children were divided intothree groups: (A) aged between 4 and 6 years; (B) aged between 6 and 8 years;(C) aged between 8 and 10 years. Results: Both in No and in Ob subjects,IGF-I, IGFBP-3 and insulin levels were significant higher in groups B and Cthan in group A (p ! 0.01 and p ! 0.02, respectively), in group B than in group Aand in group C than in group B (p ! 0.01). On the contrary, IGFBP-1 concentra-tions were lower in groups B and C than in group A (p ! 0.01), in group B thanin group A and in group C than in group B (p ! 0.01). Besides IGF-I, IGFBP-3and insulin levels increased with age (p ! 0.01), whereas IGFBP-1 decreasedwith age (p ! 0.01) both in No and in Ob groups. The values of IGF-I, IGFBP-3and insulin were higher in Ob subjects than in No subjects (p ! 0.01), whereasIGFBP-1 levels were lower in Ob subjects (p ! 0.01). In Ob subjects the valuesof IGF-I and IGFBP-3 were directly correlated with BMI (p ! 0.01), whileIGFBP-1 values were inversely correlated (p ! 0.01) with BMI. A positive cor-relation between BMI and insulin levels and a negative correlation betweeninsulin and IGFBP-1 values were showed considering only the subjects with anoverweight higher than +30%. Conclusion: In overweight subjects, our datashowed the presence of some metabolic modifications: particularly IGFBP-3,insulin and IGF-I, even correlated positively with age as in subjects with nor-mal weight, resulted higher than in normal children, whereas the IGFBP-1 val-ues, even correlated negatively with age as in subjects with normal weight,resulted lower than in normal subjects. Besides, significant relations betweenBMI and insulin and between insulin and IGFBP-1 were shown only in thosesubjects with a relevant overweight, suggesting that the serum levels of IGFBP-1 may be a precocious indicator of insulin resistance.

66 Horm Res 2000;53(suppl 2):1–191 Posters
P2-219 IGF and Growth Factors
A New Syndrome of Hemihypertrophy and SevereNon-Ketotic Hypoglycaemia (NKH) withoutHyperinsulinism in Childhood – Possible Role ofUp-Regulation of IGF-I Receptor FunctionA. Aynsley-Green 1; F. Cameron* 1; O. Bodamer* 2;C. Camacho-Hubner 3; M. Soos* 4; S. O’Rahilly* 4
1 Institute of Child Health, London, United Kingdom; 2 Baylor Collegeof Medicine, Houston, United States; 3 St. Bartholomew’s Hospital,London, United Kingdom; 4 Addenbrooke’s Hospital, Cambridge,United Kingdom
Background: Persistent NKH in childhood is rare and is usually due to either anenzymatic defect in fatty acid oxidation, or a failure of glucose-insulin secretioncoupling mechanisms that switch off insulin secretion as blood glucose (BG) levelfalls. The former is associated with low blood levels of ketones (K) yet high levels offatty acids (FFA). The latter has the characteristic anabolic ‘footprint’ of hypoke-tonaemia, hypofattyacidaemia and hyperinsulinism with low levels of branchedchain amino acids (BCAs) and IGFBP-1 when hypoglycaemic. Insulin alsodecreases hepatic production of glucose whilst increasing peripheral consumption.Exceptionally rare children present with NKH due to a tumour causing eitherexcessive insulin secretion or an overproduction of IGF-II. We report a 3-year oldboy with a new syndrome of hemi-hypertrophy with severe, persistent NKH with-out hyperinsulinism or overproduction of IGF-II. Results: When hypoglycaemic(2.6 mmol/l) low levels of K, FFA, BCA were found; insulin species were belowassay limits of sensitivity; IGFBP-1 levels were elevated 3-fold (mean 154 ng/ml);IGF-I and IGFBP-3 levels were low, whereas IGF-II was normal. Serum pro-IGF-II profile assessed by size-exclusion chromatography was also normal. Radiologicinvestigations including CT head, chest, abdomen and an abdominal ultrasoundwere all normal. Karyotypic studies demonstrated a normal 46, XY karyotype withno evidence of 11p15 duplication. Hepatic glucose production during primed con-stant infusion of (6,6)-D2-glucose whilst fasting was completely suppressed; BGincreased by 4.6 mmol/l after glucagon when hypoglycaemic. The boy was unre-sponsive to diazoxide and somatostatin with ongoing instability despite contin-uous glucagon infusions. Normoglycaemia could only be maintained by 2-hourlyfeeds and nocturnal gastrostomy infusions. Conclusions: The profile of dynamicand static data points to a new syndrome probably caused by an activating muta-tion of IGF-I receptor. Work is ongoing to dissect the locus of abnormality. How-ever, we suggest that greater attention should be paid to studies of insulin andIGF-I receptor in children with severe NKH in whom insulin levels may be sup-pressed or only marginally elevated.
P2-220 IGF and Growth Factors
Evaluation of Insulin-Like Growth Factors and TheirBinding Proteins in Cerebrospinal Fluid of Children withHydrocephalusF. Hochhaus* 1; W.F. Blum 2; M. Elmlinger 3; O. Butenandt 4;E. Ring-Mrozik* 5; Introduced by O.Butenandt1 Augustenburger Platz 1, Germany; 2 Eli Lilly Germany, BadHomburg, Germany; 3 Children’s Hospital, University of Tübingen,Tübingen, Germany; 4 Children’s Hospital, University of Munich,Munich, Germany; 5 Department of Pediatric Surgery, Ingolstadt,Germany
Introduction: Insulin-like growth factors IGF-I and IGF-II, as well as the insulin-like growth factor binding proteins (IGFBPs) are present in different parts of thenerve tissue and in cerebrospinal fluid (CSF). Increased intracranial pressure (ICP)causes hypoxy and ischemia of the brain. This is the first evaluation of IGFs andBPs in CSF of children with hydrocephalus. Patients and Methods: CSFs ofchildren with hydrocephalus (14 m, 23 f, median age 0.4 years) were collectedintra-operatively in case ICP occurred and surgical intervention was required.IGF-I, IGF-II, IGFBP-2 and IGFBP-3 were measured either by RIA or ELISAmethod, protein by BCA assay. Results: Median concentration (ng/ml) of IGF-Iwas 125, IGF-II was 58 B 8, IGFBP-3 was 960 B 237, IGFBP-2 was 459 B 102and protein was 253 B 117 mg/dl. Significant correlations (p ! 0.01) were IGF-Iversus IGF-II (r = 0.548), IGF-I versus IGFBP-2 (r = 0.81), IGF-II versus IGFBP-3(r = 0.427). Only protein versus IGFBP-2 correlated significantly (r = 0.934, p !0.01). No differences were found in age groups (! 1 year n = 25, 1 1 year n = 12).Comparison of IGFs/BPs with established data shows 10-fold increased concentra-tions for IGF-I, 2-fold for IGFBP-2 and 30-fold for IGFBP-3. (B1SDS). Conclu-sions: Our results confirm that there is a strong positive relationship betweenIGF-I and -II and IGFBP-2 and -3, but an inverse one as in human sera. Whetherelevated levels of IGFs and their binding proteins in CSF are signs of local produc-tion by brain cells or by the circulatory system remains unclear.
P2-221 IGF and Growth Factors
IGF-I Gene Expression in Adipose Tissue of Obese andLean Zucker RatsS. Tenoutasse* 1; A.L. Hubert* 2; J.P. Thissen* 2; Introduced byC. Heinrichs1 HUDERF, Brussels, Belgium; 2 UCL, Brussels, Belgium
The mechanisms underlying the maintenance of normal to high rates of lineargrowth in spite of low GH secretion in obese children are unknown. Among theanimal models of early-onset obesity, obese Zucker (fa/fa) rats are particularlyappropriate because their linear growth shows this relative GH-independency.Because IGF-I is highly expressed in adipose tissue, we wondered whether overex-pression of IGF-I in adipose tissue of these animals might contribute to normallinear growth in spite of low GH. To answer this question, we determined serumIGF-I and IGF-I expression (mRNA) in different adipose depots and in liver of 6-and 12-week-old male Zucker obese (fa/fa) and lean (Fa/?) rats (n = 4/group). Inlean controls, IGF-I expression in white adipose tissue (WAT) increased with age(epididymal: +81%, p ! 0.05; inguinal: +204%, p ! 0.05; perirenal: +115%, p !0.001, vs. lean at 6 weeks), but not in brown adipose tissue (BAT). In all adiposedepots, IGF-I expression was lower in obese rats than in lean controls at both ages(epididymal: –52% at 6 weeks and –55% at 12 weeks, NS, and p ! 0.05; inguinal:–59% at 6 weeks and –55% at 12 weeks, p ! 0.05 for both; perirenal: –75% at 6weeks and –73% at 12 weeks, p ! 0.05 for both, vs. lean at each age). As in adiposetissue, IGF-I expression in the liver of lean animals increased also with age (+47 %,p ! 0.001, vs. lean at 6 weeks). In contrast to adipose tissue, liver IGF-I mRNA wasnot reduced in 6-week-old obese rats (–6%, NS, vs. lean), but only at 12 weeks(–50%, p ! 0.001 vs. lean). Serum IGF-I paralleled liver IGF-I mRNA levels (6weeks: obese = 613 B 24 vs. lean = 569 B 29 ng/ml, NS; 12 weeks: obese = 701 B53 vs. lean = 1055 B 33 ng/ml, p ! 0.001). The discrepancy between normal IGF-ImRNA in liver and decreased IGF-I mRNA in adipose tissue at 6 weeks indicatesthat other factors than decreased GH secretion are responsible for the low adiposeIGF-I expression in young obese rats. In conclusion, adipose tissue of obese Zuckerrats does not overexpress IGF-I gene, indicating that adipose IGF-I productiondoes not contribute to explain the normal growth of these animals despite lowGH.
P2-222 IGF and Growth Factors
IGF-I/IGFBP-3 Molar Ratios in Children with GrowthHormone Deficiency (GHD) on GH TherapyT. Rohrer* 1; J. Dötsch* 1; M. Marx* 1; K. Zepf* 1; H.G. Dörr 1
1 University Erlangen, Erlangen, Germany
Objective: Epidemiological studies have shown that elevated IGF-I levels inadults are associated with an increased risk of developing cancers. The presentstudy was designed to assess the short-term effect of GH therapy on IGF-I andIGFBP-3 levels and on IGF-I/IGFBP-3 molar ratios in children with GHD.Patients and Methods: Analyses of auxological parameters, serum IGF-I andIGFBP-3 levels in 24 children with idiopathic GHD (IGHD, 9 f, 15 m, Tannerstage I) and 26 children with organic GHD due to different brain tumours (OGHD,9 f, 17 m, Tanner stage I). All patients had max. GH levels ! 5 ng/ml in 2 provoca-tive tests. Mean hGH dose (IU/kg/week) was 0.5 in OGHD and 0.6 in IGHD.IGF-I and IGFBP-3 levels were measured by RIA at start and after 1 year of hGH.Standard deviation scores (SDS) were calculated for all IGF-I and IGFBP-3 valuesin relation to healthy children. IGF-I/IGFBP-3 molar ratios (molecular weights:IGF-I: 7.5 kD, IGFBP-3: 31.3 kD) were assessed according to A. Juul (Ph.D. thesis1995). Results: (SDS; mean B SD) IGF-I levels increased from –3.0 B 1.5(OGHD), and –4.0 B 2.4 (IGHD) to 1.3 B 1.8 (OGHD; min/max: –1.8/5.9) and0.1 B 1.7 (IGHD; min/max: –4.2/2.6), respectively, after 1 year of hGH (p ! 0.01).The increase in IGF-I SDS was approximately +4 SDS in both groups. IGFBP-3levels increased from –2.5 B 2.3 to 1.0 B 1.4 (OGHD) and –2.8 B 2.7 to 0.7 B 1.2(IGHD). Height velocity SDS increased significantly in both groups (¢ HV-SDS:+5.1 OGHD; +6.0 IGHD) after 1 year of hGH. Baseline molar IGF-I/IGFBP-3ratios increased significantly from 0.16 B 0.06 to 0.27 B 0.1 (OGHD) and from0.13 B 0.1 to 0.3 B 0.2 (IGHD). Baseline ratios were either below –2 SD or in thelower normal range. After 1 year on hGH, IGF-I/IGFBP-3 molar ratios increasedbut remained in the normal range in almost all children. Only one child had a ratioof 0.62 (12 SD). Conclusions: After 1 year of hGH, there is a supraphysiologicalincrease of both IGF-I and IGFBP-3 levels. This increase is necessary to provideadequate growth. The IGF-I/IGFBP-3 molar ratios remained in the normal rangein almost all children suggesting that the IGF-I increase is attenuated by the simul-taneous IGFBP-3 increase.

39th Annual Meeting of the ESPE Horm Res 2000;53(suppl 2):1–191 67
P2-223 IGF and Growth Factors
Growth Factors Increase Androgen ReceptorTransactivation in Human Androgen Target Cell LinesF. Orio* 1; B. Térouanne* 1; Ch. Belon* 1; J.C. Nicolas* 1; Ch. Sultan 2
1 INSERM U439 et Service d’Hormonologie, Hôpital Lapeyronie,Montpellier, France; 2 Unité d’Endocrinologie Pédiatrique, HôpitalA. de Villeneuve, Montpellier, France
Micropenis is an acknowledged feature of congenital isolated growth hormonedeficiency: low GH and IGF-I levels are reported to decrease androgen biosyn-thesis, but reduced androgen responsiveness of external genitalia has not yetbeen ruled out. Objective: This work was designed to find out whether IGF-Iand other growth factors could increase transactivation of androgen receptor intarget cells as human prostate cell lines. Materials and Methods: We firstperformed the characterization of IGF-I receptors by ligand binding assayswith 125I-IGF-I. Then we evaluated the effects of IGF-I, IGF-II, EGF on stimu-lation of the AR-mediated gene transcription in human prostatic tumor (PC3,DU-145) and normal human epithelial (PNT1A) cell lines. These cell lines weretransiently cotransfected with the androgen-inducible luciferase reporter gene(MMTV-LUC), the pSG5hAR expression vector and the pCMV-B-Galactosi-dase control plasmid. After transfection, cells were submitted to hormonalinduction by R 1881, a synthetic androgen, at different concentrations andIGF-I, IGF-II or EGF at a concentration of 50 ng/ml. The same experimentswere performed on PALM cell line, stably expressing AR and MMTV-Lucifer-ase. Results: PC-3 and PNTlA cell lines expressed specific binding sites forIGF-I with similar dissociation constants (Kd = 0.1 nM) while DUl45 cell lineshowed two kinds of affinity (Kd = 0.02 nM and Kd = 0.7 nM) which mayindicate two different populations of IGF-I receptors. In all experiments, noneof the tested growth factors (IGF-I, IGF-II, EGF) alone stimulated reportergene activity . Conversely, in PNT1A and in DUl45 cell lines, IGF-1 or EGFacted synergistically in combination with low concentration of R 1881(10–11 M). This cooperation between AR and growth factors was not observedin the PC3 and PALM cells. Conclusions: There is an interaction betweenIGF-I and EGF and the AR and its ligand. This cooperation between growthfactors and AR is cell-dependent. Nevertheless, our data suggest a cross-linkbetween the androgen nuclear transcription pathway and the growth factorpathway, which may be partly involved in external genitalia development inthe neonatal as well as pubertal period.
P2-224 IGF and Growth Factors
Interleukin-6 Overproduction Reduces IGF-I andIGFBP-3 Serum Levels in Children with Perinatal HIV-1Infection and Growth FailureR. Salti* 1; G. Bindi* 1; F. Galluzzi* 1; S. Stagi* 1; F. Masoni* 1;C. La Cauza 1; M. de Martino* 1; L. Galli* 1; A. Verrotti* 2; F. Chiarelli 2
1 Department of Paediatrics, University of Florence, Florence, Italy;2 Department of Medicine, Division of Paediatrics, University ofChieti, Chieti, Italy
Some animal models show that the proinflammatory interleukin (IL)-6 nega-tively interferes with insulin-like growth factor-I (IGF-I) activity or secretionhence leading to growth impairment. In this study spontaneous and phyto-hemagglutinin (PHA)-stimulated IL-6 release by cultured peripheral bloodmononuclear cells was related to height velocity, bone age, IGF-I and IGFBP-3serum level standard deviation scores (SDS) of 32 children [aged 91 (median;range 13–151) months] with human immunodeficiency virus-type 1 (HIV-1)perinatal infection and severe disease. Spontaneous and PHA-stimulated IL-6release inversely correlated with height velocity (r = –0.759; p ! 0.011 and r =–0.463; p = 0.008, respectively), bone age (r = –0.487; p ! 0.005 and r = –0.443;p ! 0.011, respectively), IGF-I (r = –0.773; p ! 0.0001 and r = –0.650; p !0.0001, respectively) and IGFBP-3 (r = –0.506; p ! 0.003 and r = –0.725; p !0.0001, respectively) SDS. Ten children with height velocity SDS ^–2, com-pared to 22 children with height velocity SDS 1–2, showed higher spontaneous(1,959 B 198 vs. 1,023 B 327 pg/ml; p ! 0.0001) and PHA- stimulated (2,276B 363 vs. 1,467 B 628 pg/m; p ! 0.0001) IL-6 release and lower IGF-I (–2.1 B0.3 vs. –1.07 B 0.7; p ! 0.0001) and IGFBP-3 (–2.6 B 0.8 vs. –0.9 B 1.3; p !0.0001) SDS, irrespective of CD4-positive T-lymphocyte counts, viral load, liv-er disease, or nutrition status. In conclusion, our data suggest that IL-6 overpro-
duction may be a mechanism of IGF-I and IGFBP-3 down-regulation andimpaired linear growth in children with perinatal HIV-1 infection. Growth-promoting strategies, including targeted anticytokine treatments, could bedevised for such children.
P1-225 Adrenals
Comparison of 2 Different Ways of HydrocortisoneAdministration in Patients with Congenital AdrenalHyperplasia (CAH)M. Razzaghy-Azar 1; J. Baghai 1; Introduced by D. Knorr1 H. Aliasghar Hospital, Iran University of Medical Science, Tehran,Iran
ACTH is secreted with most activity during night (02.00–09.00) and steroidmedication at night seems to be more effective in suppressing ACTH than atother times. On the other hand the patient compliance is not very well withmore divided doses of a medicine. Objective: To find an easy and better wayto treat and control CAH. Patients and Methods: We compared 2 differentways of hydrocortisone administration in 67 patients (43 girls, 24 boys) withCAH. Height SDS, growth velocity index (GVI = patient growth velocity/nor-mal growth velocity for age ! 100), body mass index (BMI), weight-for-heightindex (WHI = weight/median weight for the height age ! 100) and serum levelof 17-QH progesterone at 08.00 h were measured after treatment in 2 differentways in the same patients. The first way ‘old treatment (OT)’ was 3 divideddoses with the largest portion in the morning, the ‘new treatment’ (NT as usedby M. New) was twice daily, with the largest portion at night. The morning dosewas at 07.00 h and the night dose at 21.00 h in NT and OT and the afternoondose was at 15.00 h in OT. The data were analyzed by paired t test. Serum17OH-progesterone level was grouped to !10 ng/ml and 110 ng/ml and ana-lyzed with 2 test. The duration of study for OT was 20.8 B 5.6 months and forNT was 20.6 B 5.7 months. The dosage of hydrocortisone for OT was 16 B 5mg/m2/24 h and for NT was 15 B 3 mg/m2/24 h. Results: The level of 17OH-progesterone !10 ng/ml was; observed in 82.7% of cases with NT but 65.4% ofcases with OT, the difference was significant (p = 0.002). Height SDS at the endof OT and NT was –0.46 B 1.6 and –0.53 B 1.5, respectively, the differencewas not significant. GVI was 83 B 35.7 in OT and 92 B 40.5 in NT, the differ-ence was not significant. BMI at the end of OT was 19.7 B 5 and at the end ofNT was 20.6 B 5.7, the difference was significant (p ! 0.000). WHI was 118.87B 25.5 in OT and 122 B 36 in NT, the difference was significant (p = 0.023).The patients did not complain of fatigue in both ways of treatment. Conclu-sion: 17OH-P was in better control with hydrocortisone administration twicedaily with the largest portion at night while growth velocity did not decrease.Weight gain was more with twice daily regimen although BMI was not at therange of obesity.

68 Horm Res 2000;53(suppl 2):1–191 Posters
P1-226 Adrenals
Familial Glucocorticoid Deficiency (FGD): NovelMutations in the Adrenocorticotropin Receptor Gene(MC2R)A. Huebner 1; R. Dunckelmann* 2; S. Nolte-Buchholtz* 1; H. Petzold* 1;H.G. Doerr 3
1 University Children’s Hospital, Dresden, Germany; 2 Department ofPaediatrics, Hospital Speyer, Speyer, Germany; 3 UniversityChildren’s Hospital, Erlangen, Germany
Familial glucocorticoid deficiency (FGD; MIM*202200) is an autosomal reces-sive syndrome of adrenal unresponsiveness to adrenocorticotropin (ACTH)characterized by glucocorticoid deficiency, high plasma ACTH concentrations,but preservation of mineralocorticoid function. Several homozygous and com-pound heterozygous mutations within the ACTH receptor gene (MC2R) havebeen described in some, but not all FGD patients, suggesting that FGD is agenetically heterogeneous disorder. Patients: We report on two novel casesfrom independent families with typical features of FGD. The male patient de-riving from non-consanguineous Caucasian parents (family 1) presented at theage of 13 months with a severe hypoglycaemia. Hyperpigmentation was notapparent despite undetectable cortisol levels and an ACTH level !1000 pg/ml.The female patient deriving from consanguineous Turkish parents (family 2)was admitted at the age of 10 months with a hypoglycaemic shock and hyper-pigmentation. Basal and stimulated cortisol were undetectable in the presenceof ACTH levels between 2000 and 3000 pg/ml. In both patients renin and elec-trolytes remained normal. The boy is of normal height whereas the girl appearstall for the family. Both children remain well on hydrocortisone replacement.Mutation analysis: In patient 1, we identified a homozygous cytosine to thy-mine transition in the first position of codon 137. This results in an arginine bytryptophan substitution in the second intracellular loop (R137W). In patient 2,a homozygous deletion of thymine at position 1255 was detected resulting in aframeshift with a premature stop codon (1255delT) of the fifth transmembranedomain of the MC2-R. Segregation studies within the families revealed hetero-zygosity in the parents supporting the hypothesis that the mutations are thecause of FGD in these patients. Discussion: Mutations of the second intracel-lular loop have been shown to result in a loss of signal transduction, a similareffect is supposed for the R137W mutation. The one-base-pair deletion atnucleic acid position 1255 results in a truncated receptor and consequently incomplete loss of receptor function which is reflected by the clinical course of thepatient. The tall stature of the girl (patient 2) is in accordance with other FGDpatients bearing MC2R mutations.
P1-227 Adrenals
Aldosterone Synthase Deficiency Type I with NoMutations in the CYP11B2 GeneM. Wasniewska* 1; T. Arrigo 1; M.F. Messina* 1; G. Crisafulli* 1;F. De Luca 1
1 Institute of Pediatrics, University of Messina, Messina, Italy
Isolated hypoaldosteronism is an autosomal recessively inherited disorder ofterminal aldosterone (A) synthesis leading to a severe salt-wasting syndrome.There are two biochemically different forms which are termed A synthase defi-ciency (ASD) I and II and differ in that 18-hydroxycorticosterone (18-OH-B) isdeficient in ASD I but overproduced in ASD II. Genetic studies of the patientswith ASD I have always documented some mutations in the CYP11B2 genewhich encodes A synthase. On the contrary the patients with ASD II have beenoccasionally reported to have no mutations in coding regions or to have muta-tions that do not modify A synthase activity. In a girl with severe salt-wastingsyndrome during the first weeks of life and a biochemical pattern pathogno-monic of ASD I (very low 18-OH-B serum levels, i.e. 20 pg/ml; abnormally highcorticosterone/18-OH-B serum ratio, i.e. 306.5; abnormally low 18-OH-B/Aserum ratio, i.e. 2.1) we were not able to find any mutation in the CYP11B2gene, which is surprising and seems to be inconsistent with the previouslyreported correlation of the phenotype and genotype in ASD I. These data sug-gest that phenotype-genotype relationships in isolated hypoaldosteronism arenot yet well understood, as already postulated by other authors at the light oftheir findings in ASD II patients. A plausible question which may be raised onthe basis of our data is whether other genes apart from CYP11B2 are involvedin the regulation of terminal A synthase.
P1-228 Adrenals
POMC Deficiency, a Striking Phenotype: AdrenalInsufficiency, Infantile Obesity and Red Hair. A CaseReportP.M.V.M. Theunissen* 1; J. Kooijman* 2; S.B. van der Meer* 1;Introduced by W.J. Gerver1 Department of Pediatrics, Atrium Medical Centre, Heerlen,The Netherlands; 2 University Hospital Maastricht, Maastricht,The Netherlands
A one-year-old boy is described with symptoms of adrenal insufficiency, infan-tile obesity and red hair. Pro-opio-melanocortin (POMC) deficiency was diag-nosed and confirmed by mutation analysis showing a homozygous C3804Amutation in the POMC gene on chromosome 2. This mutation affects the trans-lation of the POMC gene and interferes with the synthesis of the POMC-derived peptides such as adrenocorticotrophin and melanocortin. Adrenocorti-cotrophin deficiency causes adrenal insufficiency via the melanocortin MC2-receptor, while lack of alpha-melanocyte stimulating hormone causes alterationin pigmentation (MC-1 receptor) and probably excessive food intake (MC-4receptor). Furthermore, our patient showed an increased growth velocity, forwhich we have no apparent explanation. This is the third patient reported witha genetic defect in the POMC gene causing adrenal insufficiency and a strikingphenotype.
P1-229 Adrenals
Treatment of Hirsutism, Hyperandrogenism,Oligomenorrhea, Dyslipidemia and Hyperinsulinism inNon-Obese, Adolescent Girls: Effect of FlutamideN. Potau 1; M.V. Marcos* 2; F. de Zegher 3; L Ibañez 4
1 Autonomous University of Barcelona, Barcelona, Spain; 2 ConsorciHospitalari de Terrassa, Barcelona, Spain; 3 University of Leuven,Leuven, Belgium; 4 University of Barcelona, Barcelona, Spain
Precocious pubarche (PP) in girls is a risk factor for subsequent functional ovar-ian hyperandrogenism, which is usually accompanied by hyperinsulinism anddyslipidemia. We assessed the effects of low-dose flutamide treatment (250 mgdaily for 18 months) on hormonal and metabolic variables in 18 non-obese,adolescent girls [age, 16.8 B 0.3 years; body mass index (BMI), 21.6 B 0.6kg/m2], with functional ovarian hyperandrogenism (diagnosis by GnRH-ago-nist test) after PP. Flutamide treatment was accompanied by a marked decreasein the hirsutism score, serum free androgen index, testosterone, androstene-dione and dehydroepiandrosterone (DHEA) levels (p ! 0.001), and by anincrease in sex hormone-binding globulin (SHBG) concentrations (p = 0.0001).There were no substantial changes in the pattern of menstrual cycles, serumgonadotropin, estradiol or DHEA-sulfate concentrations, and there was nodetectable effect on the 17-hydroxyprogesterone response to GnRH-agonist.Serum triglycerides, total cholesterol and LDL-cholesterol levels decreased dur-ing flutamide treatment (p = 0.01), but HDL-cholesterol, fasting glycemia/insu-linemia, and the insulin response to a glucose load remained unaltered. Flutam-ide was well tolerated. In conclusion, low-dose flutamide treatment was foundto be an effective and safe approach to reduce hirsutism and circulating andro-gens, LDL-cholesterol and triglycerides in girls with functional ovarian hyper-androgenism after PP. However, flutamide failed to increase HDL-cholesterollevels or to decrease hyperinsulinemia, i.e. to affect two major risk factors forsubsequent cardiovascular disease.




















