
POSTER SESSION 1 - CiteSeerX
441
36th International Symposium on High Performance Liquid Phase Separations and Related Techniques Budapest Congress & World Trade Centre 19–23, June, 2011 • Budapest, Hungary POSTER SESSION 1 P1-G-001-MO A Critical Evaluation of Sub-2 μm Particles for Packed Column Supercritical Fluid Chromatography Michael Jones 1,2 , Norman Smith 1 , Cristina Legido-Quigley 1 Andrew Aubin 2 1 Pharmaceutical Science Division, School of Biomedical and Health Sciences, King’s College London 2 Waters Corporation, London, UK 34 Maple Street, Milford, MA 01757, USA The use and benefits of sub-2 μm chromatographic particles in reversed phase chroma- tography (RPLC) is well established. Often, packed column supercritical fluid chromatog- raphy (pSFC) is performed using 5 μm particles typically with columns of 150–250 mm in length. Although many users have had success using this approach, limitations in analyti- cal SFC instrument design and column availability have prevented the adoption of small particle pSFC. Earlier attempts at sub-2 μm pSFC have resulted in many false starts due to the lack of commercially available instrumentation optimized for sub-2 μm particle sta- tionary phase analysis. However, the use of SFC has seen a resurgence of use within the pharmaceutical industry due to the complementarily to HPLC as well as “Green” chemis- try aspects [1]. pSFC has been readily adopted for chiral and more recently, achiral sepa- rations for purification analysis. The driving factors have been decreases in run time; increase in chromatographic efficiencies and for purification, shorter evaporation times and less solvent usage [2–5]. As for analytical pSFC, many systems identified in publica- tion utilize instrument configurations limited by large system volume and extra column volumes affecting the ability to achieve optimal chromatographic performance. The scope of this work explores the impact of a low dwell volume and low extra column volume instrument configuration with intent to determine any benefits in chromatographic effi- ciency, resolution, sensitivity for sub 2 μm particles stationary phases. The advantages of overcoming these instrumental performance limitations will allow a true assessment of pSFC for compatible sub-2 μm stationary phases. Presented here, an evaluation of column efficiencies using columns packed with different particle size packing materials run under SFC conditions. A series of van Deemter plots were constructed for each of the different particle size columns showing the increased efficiency at smaller particles sizes. Various column internal diameters are explored to determine a best practical approach for pharmaceutical analysis. Measured pressure drops and temperature changes across the various columns are also reported. Techniques are also discussed regarding the difficulties involved during the experimen- tal process.
-
Upload
khangminh22 -
Category
Documents
-
view
0 -
download
0
Transcript of POSTER SESSION 1 - CiteSeerX

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-001-MO
A Critical Evaluation of Sub-2 μμm Particles for Packed ColumnSupercritical Fluid Chromatography
Michael Jones1,2, Norman Smith1, Cristina Legido-Quigley1
Andrew Aubin2
1Pharmaceutical Science Division, School of Biomedical and Health Sciences,King’s College London
2Waters Corporation, London, UK34 Maple Street, Milford, MA 01757, USA
The use and benefits of sub-2 μm chromatographic particles in reversed phase chroma-tography (RPLC) is well established. Often, packed column supercritical fluid chromatog-raphy (pSFC) is performed using 5 μm particles typically with columns of 150–250 mm inlength. Although many users have had success using this approach, limitations in analyti-cal SFC instrument design and column availability have prevented the adoption of smallparticle pSFC. Earlier attempts at sub-2 μm pSFC have resulted in many false starts due tothe lack of commercially available instrumentation optimized for sub-2 μm particle sta-tionary phase analysis. However, the use of SFC has seen a resurgence of use within thepharmaceutical industry due to the complementarily to HPLC as well as “Green” chemis-try aspects [1]. pSFC has been readily adopted for chiral and more recently, achiral sepa-rations for purification analysis. The driving factors have been decreases in run time;increase in chromatographic efficiencies and for purification, shorter evaporation timesand less solvent usage [2–5]. As for analytical pSFC, many systems identified in publica-tion utilize instrument configurations limited by large system volume and extra columnvolumes affecting the ability to achieve optimal chromatographic performance. The scopeof this work explores the impact of a low dwell volume and low extra column volumeinstrument configuration with intent to determine any benefits in chromatographic effi-ciency, resolution, sensitivity for sub 2 μm particles stationary phases. The advantages ofovercoming these instrumental performance limitations will allow a true assessment ofpSFC for compatible sub-2 μm stationary phases.
Presented here, an evaluation of column efficiencies using columns packed withdifferent particle size packing materials run under SFC conditions. A series of vanDeemter plots were constructed for each of the different particle size columns showingthe increased efficiency at smaller particles sizes. Various column internal diameters areexplored to determine a best practical approach for pharmaceutical analysis. Measuredpressure drops and temperature changes across the various columns are also reported.Techniques are also discussed regarding the difficulties involved during the experimen-tal process.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
1. Harris CM. The SFC comeback. Pharmaceuticals give supercritical fluid chromatography a fighting chance.Anal Chem. 2002 Feb 1;74(3):87A–91A
2. B. Bolanos, M. Greig, M. Ventura, W. Farrell, C.M. Aurigemma,H. Li, T.L. Quenzer, K. Tivel, J.M.R. Bylund,P. Tran, C. Pham, D.Phillipson, Int. J. Mass Spectrom. 238 (2004) 85.
3. M. Ventura, W. Farrell, C. Aurigemma, K. Tivel, M. Greig, J. Wheatley,A. Yanovsky, K.E. Milgram, D.Dalesandro, R. DeGuzman, P.Tran, L. Nguyen, L. Chung, O. Gron, C.A. Koch, J. Chromatogr. A 1036(2004) 7.
4. T. Wang, M. Barber, I. Hardt, B. Kassel, Rapid Commun. Mass Spectrom. 15 (2001) 2067.5. C. White, J. Burnett, J. Chromatogr A. 1074 (2005) 175–185

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-002-TU
A Highly Automated Multi-Pump, Multi-Detector, Super-Critical FluidChromatography (SFC) System for Purification in Drug Discovery
Xu Zhang, Qing Ping Han, Mark J. HaywardLundbeck Research
USA – 215 College Rd. – Paramus, NJ 07652, USA
When building our gradient SFC/MS based chiral purification platform for drug discovery,we designed specifications based solely on the existing practices of the medicinal chem-ists and business need. Specifically, we concluded that we must routinely inject ≥ 100 mgand yield ≥ 10 g per day of high purity (95–99% ee) for any number of compounds(1–100). These specifications tend to require gradient operation and significantly exceedthose of the off the shelf instrumentation more commonly used in this setting, particularlywith regard to mass per injection for many compounds. So, we set out to find better waysto use the available instruments and ultimately meet our business need. While achievingthis requires attention to detail in all the parameters, one crucial part of the implementa-tion is optimizing the injection process using a separate pumping sub-system capable ofusing mixtures CO2 and modifier flowing through the injection valve to enhance solubili-ty. Benefits of this “at-column dilution” approach to sample injection are flexible solventchemistry, automation, and ability to load high masses to the head of the preparativecolumn. Success at optimizing the injection process allows one to then focus attention onthe separation to achieve high speed (high velocity, 4 mm/s) while maintaining highresolution. The key principle involved is addressing the capacity issues while simulta-neously applying all known techniques to achieve both high speed and separation effi-ciency. The primary approaches used to achieve simultaneous high speed and resolutionare the same ones used at analytical scale, namely volume reduction and small stationaryphase particles used with shorter columns. Maintaining sufficient buffer capacity on-column is most readily addressed by creating a separate pumping sub-system for on-linebuffer mixing. Control of the rate of adsorption / desorption of compounds to / from thestationary phase is achieved by preheating the eluent system. Application of the aboveconcepts allows routine, sustained purification rates of 1 g/hr per SFC/MS. Enantiomericexcesses of greater than 95% are achieved with > 99% success rate. Further productivityenhancement is the result of other automation features including collected mass estima-tion by ELSD, UV waste detection, and immediate site-wide access to data. The impact ofthe accumulation of these benefits being delivered by a purification expert is at least a 10fold improvement in time based human efficiency relative to medicinal chemists doingthe purifications on their own and the differences appear to be even greater when com-pared to chiral synthesis. Data demonstrating the principles, set up, analyses, and applica-tions will be provided.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-003-MO
Applications of a New HILIC Stationary Phase
Ken Butchart, Mark WoodruffFortis Technologies Ltd,
45 Coalbrookdale Road, Clayhill Industrial Park, Neston, Cheshire, CH64 3UG, UK
Hydrophilic Interaction Chromatography (HILIC) is a popular and growing area of interestfor the retention and separation of polar analytes than are not achievable on traditionalreversed phase stationary phase systems. Moving towards the use of normal phase condi-tions, HILIC offers the ability to retain very polar molecules without the need for complexmobile phase systems, ion-pair reagents or other buffers that weaken the ability to utilisethe advantages of MS as a detection technology.
We discuss the use of a new HILIC stationary phase for the separation and retention ofpolar analytes, we compare this to other LC techniques for the retention of polar analytes,such as high pH and ion-pair chromatography, and we highlight the relative strengthsand weaknesses.
Applications highlighting the unique selectivity that can be achieved with simple mo-bile phases and a HILIC stationary phase are shown. Examples of the molecular structuresthat can be successfully retained are shown in comparison with their reversed phasecomparisons. The advantages of HILIC chromatography as a technique are discussed.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-004-TU
Basic Study of Macroporous Spongy Monolith on HPLC Separationand its Application for Effective Concentration of PAHs
Takuya Kubo1, Tetsuya Tanigawa1,2, Keita Kato1, Yoshiyuki Watabe3,Yoshitomo Tanaka4, Ken Hosoya1
1Graduate School of Environmental Studies, Tohoku University,Aoba 6-6-20, Aramaki, Aobaku, Sendai 9088579, Japan
2Chemco Scientific Co., Ltd.,Osaka, Japan
3Analytical Applications Department, Shimadzu Corporation,Kyoto, Japan
4Glory Industry Co., Ltd.,Osaka Japan
As well-known, the high throughput analyses are pretty important in several fields suchenvironmental, pharmaceutical, chemical industry, and medicals. Especially, the efficiencyby high throughput on the sample preparation process is strongly required. In our previ-ous studies, the novel separation media based on the sponge-like macroporous polymermaterials (spongy monoliths) have been reported as the effective adsorption media forsample preparations. In these studies, we have clarified the advantages of spongy mono-liths in point of higher permeability compared with any other commercially availableparticles and/or silica monolithic materials, and we also demonstrated the effective sam-ple pretreatment of an endocrine disrupter, bisphenol A under the high throughputconcentration.
On the other hand, we also found that one of problems of spongy monoliths wasmuch lower adsorption capacity against any other media. According to the morphologicalanalyses of spongy monoliths, it was easily understood that the problem of adsorptioncapacity was depended on the lower specific surface area. Also, we had not examined thefundamental retention properties of spongy monolith.
In this present study, we report the fundamental retention properties of spongy mono-lith by HPLC evaluations to improve the problems. According to the detailed examinationon HPLC evaluations, the spongy monolith consisted of poly(ethylene-co-vinyl acetate)and having macroporous co-continuous structures showed the selective retention abilityfor poly aromatic hydrocarbons (PAHs), especially coplanar compounds, whereas lowerretentions were observed for typical hydrophobic and hydrophilic compounds comparedto commonly used C18 medium. Moreover, we demonstrate the effective pretreatment ofbenzo[a]pyrene using a spongy monolith as a pretreatment column of on-line columnswitching HPLC system.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-005-MO
Design of Linear Peptidic Affinity Ligands Based onSingle-Chain Antibody Fragment Sequence Information
Katrin Kurz, Frank Hilbrig, Ruth FreitagProcess Biotechnology, University of Bayreuth,
Germany
In recent years, the downstream processing of biotechnological products and in particularrecombinant therapeutics has become a major bottleneck in bioprocess design. The firststep in many of the established isolation procedures is based on specific affinity interac-tion, which combines high enrichment with superior selectivity for the target molecule.Amongst the most popular examples is Protein A affinity chromatography for antibodycapture. However, not for all putative biotech products are suitable affinity ligands avail-able. In such cases, affinity ligands can be identified via screening of ligands libraries.Currently most popular for this purpose is the screening of peptide or antibody librariesdisplayed on filamentous phages.
Peptidic ligands thereby identified possess the advantages of easy and automatedsynthesis and well-known strategies for purification. The coupling of the ligands to chro-matographic stationary phases e.g. Sepharose is also straightforward. By comparison,ligands based on antibodies or single chain fragments variables (scFv) are expensive andmore difficult to prepare. In consequence such ligands are less often used, even thoughtheir specific affinity to the target molecule is often superior to that of linear peptides.
In an effort to combine the benefits of both classes of affinity ligand we started withscreening an scFv-phage-library for candidates that capture the bovine pancreatic trypsininhibitor (BPTI). As platform we chose the Biacore system to immobilize BPTI and performthe enrichment of specifically interactive phages. For those scFv that generated the bestsignals in a subsequent ELISA the sequences were determined. Subsequently the comple-mentary determining regions (CDR) of the light and heavy chain were prepared via solidphase peptide synthesis and covalently connected by an adequate linker. To define di-mension and sequence of this linker, calculations based on already published scFv X-raystructures were performed. In addition candidates were evaluated using sequence tostructure alignments. Subsequently Biacore studies were performed to determine bindingkinetics and affinities. These results showed considerably higher KD-values for the scFv-derived ligands than for ligands identified in an analogous manner using phage-displayedpeptide-libraries.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-006-TU
Development of a Comprehensive Two-DimensionalLiquid Chromatography / Mass Spectrometry System
and its Applications in Analyzing Triglycerides
Qin Yang, Xiaoli Hou, Xianzhe Shi, Yuanhong Shan, Guowang XuCAS Key Laboratory of Separation Science for Analytical Chemistry,Dalian Institute of Chemical Physics, Chinese Academy of Sciences,
457 Zhongshan Road, Dalian 116023, China
In the present work, an on-line comprehensive silver-ion liquid chromatography × re-versed-phase liquid chromatography system was constructed to analyze triglycerides(TAGs). The column parameters and mobile phases were optimized. The system coupledwith atmospheric pressure chemical ionization mass spectrometry (APCI MS) was appliedto analyze an edible peanut oil and a mouse liver extract. TAG structures with differentcarbon chains were identified based on TAGs’ retention behaviors and their MS frag-ments. The results showed developed comprehensive two-dimensional LC system demon-strated unique selectivity for TAG compounds and satisfactory peak capacity.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-007-MO
Electrospun Nanofibers as Sorbent Material for MiniaturizedSolid Phase Extraction Devices
Nelson Torto, Samuel ChigomeDepartment of Chemistry, Rhodes University,
P.O. Box 94, Grahamstown 6140, South Africa
One of the main focuses of current sample preparation trends is the development ofminiaturized sorbent based techniques with greater selectivity and sorptive capacity. Solidphase extraction (SPE) is very popular for sample preparation of liquid samples due to itssimplicity of operation, high selectivity and good reproducibility [1]. A possible way ofmaking SPE meet the current sample preparation requirements would be to use a smallersorbent bed mass that has a large specific surface area. An alternative sorbent fabricationtechnique that produces a sorbent material that possesses a large specific surface area aswell as exhibiting chemical and morphological properties that can be easily modified isseen as the key to miniaturization of SPE. Electrospinning is seen as a suitable sorbentfabrication technique due to the fact that it can fabricate any material that forms a meltor solution, into nanofibrous form. Furthermore, it has the ability to incorporate a varietyof functionalities and morphological modifications thus presenting a platform to tune thesorbent for specific applications.
Our research focuses on the development of sorbent based sample preparation techniquesthat rely on electrospinning as the sorbent fabrication technique. The poster will focus onsome of our efforts towards development of miniaturized SPE devices. Electrospun polysty-rene copolymer fibers were fabricated by a combination of emulsion polymerisation andelectrospinning. The resultant fibers (5 – 10 mg) were packed into a 200 μl micropipette tip(micro column SPE device) or into a 500 μl SPE barrel (micro disk SPE device). Besides poly-styrene copolymer fibers, a second disk SPE device that used electrospun nylon 6 fibers (1.5mg) as sorbent bed was developed. The developed SPE devices were used to investigate theextraction of analytes from microliter volumes of biological and aqueous matrices. For exam-ple hydrocortisone, cortisone acetate, prednisone, prednisolone and 19-nortestosterone wereused as model analytes to evaluate the performance of electrospun polystyrene fiber basedSPE. Extraction recoveries up to 80.13% in plasma and 93.43% in water were obtained andthe limits of detection ranged from 0.75 to 1.29 ng/ml [2]. The results demonstrated theviability of the use of electrospun nanofibers for miniaturization of SPE devices.
1. I. Liska, J. Chromatogr., A, 2000, 885, 3–16.2. Chigome S, Darko G, Buttner U, Torto N, Anal Methods, 2010, 2: 623–626.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-008-TU
Evaluation of Diol and Polyethylene Glycol Columnsfor the Analysis of Ionisable Solutes
by Different Chromatographic Modes
Alberto dos Santos Pereira1, Nobukazu Higashi2, Kazuhisa Mitsui2,Hirooki Kanda3, Frank David1, Pat Sandra1
1Research Institute for Chromatography,Kennedypark 26, 8500 Kortrijk, Belgium
2Japan Tobacco Inc.Tobacco Science Research Center 6-2, Umegaoka, Aoba-ku, 227-8512 Yokohama, Japan
3Gerstel K.K.,2-13-18 Nakane, Meguro-ku, 152-0031 Tokyo, Japan
In our search for optimal separation of ionisable solutes, commercially available diol- andpolyethyleneglycol-silica columns were evaluated for a large number of model solutesusing different separation modes namely reversed-phase LC (RP-LC), hydrophilic interac-tion chromatography (HILIC), per-aqueous LC (PALC), normal phase LC (NP-LC), super-critical fluid chromatography (SFC) and enhanced fluidity chromatography (EFC).
Main selectivity differences between both phases are related to the more hydrophobicnature of PEG compared to diol. Notwithstanding this, both phases can be applied for theanalysis of ionisable solutes but the optimal operation mode depends on this hydropho-bicity.
As an example, the diol column with a pure aqueous mobile phase (RP-LC or PALCconditions) can constitute an alternative to hydrophilic interaction chromatography(HILIC) with a mobile phase rich in acetonitrile. On the other hand, this approach is notsuccessful for compounds with logP > 3. For phenolic compounds, a reduced plate heightof 1.9 at uopt of 0.2 mm/s was calculated on a 25 cm x 7.8 mm ID, 5 μm diol column.
Changing the pH of the mobile phase can induce significant differences in selectivity,as well as in efficiency, while the retention is shortened by adding an organic modifier. Inthe framework of green chromatography, the biodegradable ethanol can be used withoutloss in efficiency when compared to acetonitrile. In true reversed phase conditions the useof ethanol or methanol give better retention and similar efficiency as by using acetonitrilewhich is opposite to what is normally observed under HILIC conditions.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-009-MO
Evaluation of New UHPLC Mixed-Mode Hybrid Stationary Phasesin Pharmaceutical Analysis
Lucie Nováková, Hana VlckováDepartment of Analytical Chemistry, Faculty of Pharmacy, Charles University,
Heyrovského 1203, 500 05, Hradec Králové, Czech Republic
In early beginning of UHPLC (ultra-high performance liquid chromatography) in 2004, whenthe technique was first commercially introduced, there were only few stationary phases avail-able. Initially they covered mostly reverse-phase separations. Recently, the range of applicabili-ty of UHPLC stationary phases has been widely extended including normal phase, ion-ex-change and HILIC applications as well [1]. The attention has been attracted to mixed modestationary phases, which allow for multiple retention process to occur simultaneously due tosurface modification. Such modification enables to obtain further selectivity and to add dimen-sionality in 2D separations [2]. Typical mixed-mode stationary phases contain C18 reversechain and simultaneously SAX and/or WAX group. Such sorbents are also widely used for SPEisolation technique. New UHPLC mixed-mode stationary phase based on hybrid support wasintroduced in 2010 by Waters including a new family of BEH CSH analytical columns [3].
In this study the selectivity, retention properties, peak shape and loading capacity forbases were evaluated using two UHPLC mixed-mode BEH CSH stationary phases modifiedby C18 and Phenyl groups. The data were compared with the data obtained on otherUHPLC hybrid stationary phases (BEH C18, BEH C8, BEH Phenyl and BEH Shield RP18) atboth basic and acidic conditions using conventional HPLC buffers (10 mM ammoniumacetate) as well as low concentration additives such as e. g. 0.1 – 0.01% formic or aceticacid and 1 mM solution of ammonium acetate, which are widely used in LC-MS applica-tions. Pharmaceutically important compounds encompassing all acids, bases and neutralswere included into the study. As expected due to properties of BEH CSH sorbent (whichpossess positively charged surface besides RP group), much improved peak shapes andweaker retention was obtained for bases even at very low concentration of acidic addi-tives. Such conditions are ideally suited for LC-MS applications of bases, where typicallychromatographic conditions (retention and good selectivity at basic pH) and LS-MSconditions (ionization at acidic pH) are not in agreement if conventional RP separation isused. On the other hand, acids were more strongly retained and for some compounds thepeak shape was influenced negatively. Further, the behavior of acidic, basic and neutralsolutes is discussed using various additives at both basic and acidic pH for all above statedcolumns. Method robustness after pH change from basic to acidic was also evaluated.
1. L. Nováková, H. Vlcková, Anal. Chim. Acta 656 (2009) 8–35.2. P. G. Stevenson, J. N. Fairchild, G. Guiochon, J. Chromatogr. A (2011) in press3. P. C. Iraneta, K. D. Wyndham, D. R. McCabe, T. H. Walter, A review of Waters hybrid particle technology,
Part 3, Waters corporation, 2010, Milford, USA
Acknowledgement: The authors gratefully acknowledge research project MSM002162082 and technicalsupport of Waters, Czech Republic.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-010-TU
Evaluation of UHPLC and Core-Shell Particles
Ken Butchart, Mark WoodruffFortis Technologies Ltd,
45 Coalbrookdale Road, Clayhill Industrial Park, Neston, Cheshire, CH64 3UG, UK
In recent years much has been made of speed in ultra high pressure liquid chromatogra-phy (UHPLC) which has become ever more popular. Analysts can now utilise smallerparticles to increase efficiency of the separation, leading to increased sensitivity, speedand/or resolution. Another option that has appeared is the use of “fused-core” or “core-shell” particles, which have the potential benefit of efficiency without the pressure ofUHPLC particles.
In this poster we discuss the differences, options and limitations of fully porous particlesvs core-shell particles in terms of their ability to aid us in method development, since thespeed from both of these options is not in doubt. We highlight how UHPLC particles candiffer significantly from each other and how we can use these differences to aid us. Pos-sessing a high surface area, fully porous particle offers certain advantages, peak capacity,loadability and maybe most importantly scalability from UHPLC through analytical scaleto prep scale. This means less method development for analysts on the various stages ofthe project. We show how there is a compromise in choosing between retention, speed,resolution and pressure
We highlight the performance criteria that can lead to robust, reproducible methoddesign especially when transferring a method worldwide.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-011-MO
Fast Method Development of Rooibos Tea Phenolicsusing a Variable Column Length Strategy
Deirdre Cabooter1,2, André de Villiers2, Martha Kallili2, Gert Desmet1
1Vrije Universiteit Brussel, Department of Chemical Engineering,Pleinlaan 2, B-1050 Brussels, Belgium
2Stellenbosch University, Department of Chemistry & Polymer Science,Private Bag XI, 7602 Matieland, South Africa
The development of a method for the separation of the 15 main phenolic compoundsfound in rooibos tea is presented. The method development process is significantly en-hanced by using the recently introduced automated column coupler in combination withthe variable column length strategy. This strategy consists of performing the initial scout-ing runs, wherein the best separation conditions are determined, on a short column andsubsequently fine-tuning the separation on longer columns to benefit from their higherseparation performance. The method development process can further be expedited byoperating each column length at the maximum pressure, in this case 1000 bar. It is dem-onstrated that the temperature gradients that develop in the different column lengthsunder these ultra-high pressures do not affect the selectivity of the separation, eventhough the sample under consideration is highly temperature sensitive. This observationsuggests that the effect of the temperature increase due to viscous heating on separationselectivity is compensated by the effect of pure pressure.
The final obtained method is to the best of our knowledge the first method describedin literature that leads to the baseline separation of all 15 phenolic compounds. Applyingthis method to unfermented and fermented aqueous rooibos tea extracts, in combinationwith a Q-TOF mass spectrometry detector, some 30 phenolic compounds were tentativelyidentified in the rooibos tea samples.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-012-TU
Injection Sequence for Optimizing Performancein UHPLC Separations
Jason A. Anspach, A. Carl Sanchez, Tivadar FarkasPhenomenex,
411 Madrid Ave, Torrance, CA 90501, USA
Over the last 7 to 10 years there has been a lot of work done in bringing forth ultra highperformance separations. In the beginning these separations were performed using totallyporous particles that were below 2 μm in particle size. To use these particles, specialinstrumentation was developed that delivers the high pressure and lowered band disper-sion in comparison to previous generation instrumentation. Recently, the use of core-shellmaterials has become a popular alternative to the use of small particles for UHPLC separa-tions. By using core-shell materials, performances equal to that of sub 2 μm materials areobtained at pressures that are compatible with 400 bar instrumentation. On both the 400bar generation, and 1000 bar generation instruments, there is a significant amount ofband dispersion generated in the injection system. In this paper we will demonstrate aperformance optimizing injection sequence (POISe) which effectively eliminates thedispersion due to the injector, allowing for higher performance UHPLC separations onboth 400 and 1000 bar generation instruments.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-013-MO
Ion Chromatography – Analysis of Aqueous Solutions of Amines
Maciej Chrubasik, Ewa Micek, Justyna TarnowskaInstitute for Chemical Processing of Coal,
Zamkowa 1a Street, 41-803 Zabrze, Poland
Ion Chromatography offers many interesting solutions for industry. One of them is theanalysis of basic compounds like amines, which is of particular interest when nitrogendistribution has to be monitored. High selectivity of IC towards ions enables trace analysisof amines in their mixtures with aprotic organic compounds. Moreover due to high sensi-tivity of modern detectors samples can be diluted up to 100 000 times which eliminatesmatrix effect.
In recent years there is a great growth of interest in clean coal technologies, especiallyin removal of carbon dioxide from exhaust gas. Research conducted in our Institute fo-cused on CO2 absorption in amino alcohols like: ethanoloamine (MEA) and methyldi-ethanoloamine (MDEA). In present report we would like to show our early attempts inseparation of complex mixtures of amines and their derivatives as well as our attempts intheir quantitative analysis. The first problem we have approached was a non-linear corre-lation between concentration of amine and peak area. This is a well known phenomenawhen eluent suppresion is used. Another problem was peak tailing which make it difficultto establish peak end and decreased precision of results. In that case the utility of eluentcontaining organic solvent was examined.
The results presented in this paper were obtained from research work co-financed by the National Centre ofResearch and Development in the framework of Contract SP/E/1/67484/10 – Strategic Research Programme –Advanced technologies for obtaining energy: Development of a technology for highly efficient zero-emissioncoal-fired power units integrated with CO2 capture.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-014-TU
Maximizing Peak Capacity with UHPLC
Siji Joseph1, Edgar Naegele2, Syed Lateef1, Christian Gotenfels2
1Agilent Technologies India Pvt Ltd,RMZ Centennial, Block C, 3rd floor, ITPL Road, Mahadevapura Post, Bangalore-68
2Agilent Technologies,Waldbronn, Germany
The most commonly used measure for the performance of a chromatographic separationis plate counts. This value mainly depends on the particle size, column length and thepacking quality of the column. However, plate counts are not an effective indicator ofgradient separations. Peak capacity is a performance measure that describes the numberof peaks that can be separated during a gradient run with a certain resolution and is aparameter used to evaluate the performance of a chromatographic separation. Higherpeak capacity values are important for the separation of complex samples with an un-known number and variety of analytes.
In this study, the effect of changing gradient parameters, such as flow rate, gradienttime, column length and slope of the gradient, on peak capacity are measured and dis-cussed in the context of current literature. Agilent ZORBAX Eclipse Plus columns with aninner diameter of 2.1 mm, 1.8 μm particle size and various column lengths (50 mm, 100mm and 150 mm) were used for this evaluation. An Agilent 1290 Infinity LC was used forthe study because of its broad flow rate and pressure-range.
The study was performed using a typical standard spike mix of small molecules. Theindividual components of the sample were uracil, phenol, methyl paraben, ethyl paraben,propyl paraben, butyl paraben, heptyl paraben, N, N-diethyl-m-toluamide and toluene. Inthis study, the peak width is uniform throughout the gradient chromatogram and as amid-eluting peak, the peak width of N, N-diethyl-m-toluamide was very close to theobserved average peak width value. Therefore, the peak width measured at 4σ of N,N-diethyl-m-toluamide was selected for the peak capacity calculation.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-015-MO
Multidimensional HPLC+GC Coupling System for Automated On-LineClean-Up, Derivatization and Analysis of Complex Samples
Josep Mª Gibert1, Nieves Sarrión1, Ariadna Galve1, Roger Gibert1, David Alonso1,Jesús Villén2, José Manuel Cortés2, Lorena Colmenero Fernández1
1KONIK-Tech S.A., R&D and Applications Lab.,Av. Cerdanyola, 73 08172 St. Cugat, Barcelona, Spain
2Escuela Técnica Superior de Ingenieros Agrónomos, Universidad de Castilla-La Mancha.Campus Universitario s/n. 02076 Albacete, Spain
The innovative multidimensional on-line KONIK K2 HPLC+GC system, based on the pat-ented TOTAD interface (Patent Nº: US 6,402,947 B1 and others), marries in synergy theseparation and fractionation potential of HPLC to the separation and selective detection andquantification capabilities of HRGC. The TOTAD interface has facilitated the full automatiza-tion of the LC-GC system, minimizing thus sample handling and shortening analysis time.
The HPLC stage, prior to the introduction into the GC column, contribute to simplifythe sample preparation and its conditioning. The HPLC column and mobile phases cancontribute totally or partially to the sample clean up depending on the complexity of thematrix. Liquid samples can be directly introduced into the HPLC without any manipula-tion thus guaranteeing the sample integrity while improving quantification of analytes.The trap interface do the rest, selectively or universally, trapping the compounds of inter-est which are, after the complete solvent elimination, desorbed and transferred to the GCsystem for their analysis using MS or selective detectors.
The K2 HPLC+GC system has been successfully applied to the analysis of compoundseasily chromatographied by GC, but its use can be extended to the determination ofpolar/non-volatile substances (through on-line derivatization). Polar/Non-volatile substanc-es can be trapped on suitable absorbents and derivatised under stopped flow conditionsand further eluted into the GC column (patent applied for) allowing the automatic analy-sis of a much larger range of middle size molecules in this system. This opens an interest-ing range of applications of the LC-TOTAD-GC system in pharmaceutical and forensicanalysis by selective derivatization of compound classes via esterification and acetylationor a more universal derivatization through a sylilation step.
In this work, a summary of main applications performed with this system with on-linederivatization if necessary is presented: analysis of pesticides in water, olive oil, vegetablesand urine samples, PAHs in mineral oil, separation and characterization of petroleum frac-tions, analysis of phenolic compounds in cocoa samples, screening of impurities on solid-doseillicit drugs and determination of several drugs in biological samples among others. Qualityparameters of the technique and analysis of real samples are presented for these applications.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-016-TU
Multidimensional Orthogonal High PerformanceLiquid Chromatography with Backward Elution (Eluent Back-Flush)
as a New Solution for General Elution Problems
Marian Kaminski, Grzegorz Boczkaj, Maciej Trznadel,Mariusz Jaszczołt, Sebastian Zalewski
Department of Chemical and Process Engineering, Chemical Faculty,Gdansk University of Technology,
80-233 Gdansk, 11/12 Narutowicza Str., Poland
For many years a backward elution is appreciated in the separation science, especially ingas chromatography for light gases multi-column separation systems. In HPLC a so-calledeluent backflush (BF), is almost not applied for concern of sorbent bed degradation in thechromatographic HPLC column. The most popular exceptions are the IP 391 [1], PN-EN12916 [2] or in the preparative scale the ASTM D6369 [3] standard test methods. In ourresearch team we modified the PN-EN 12916, and the every next separation is proceededin the opposite direction in relation to previous separation. This resulted in many effectiveapplications from both – preparative as well as analytic high-performance liquid chroma-tography [4–7].
The main advantages of this solution are the assurance of total elution of the componentsintroduced in the mixture to the column on the supposition that the chemisorptions orprecipitation of the components doesn’t occur. In the effect a collection of the target frac-tion in the eluent back-flush with high elution strength eluent is possible. This solutionprovides also an equal level of the sorption activity of stationary phase in the every separa-tion, which gives a constancy and the repeatability of the chromatographic conditions.
Actually produced monolithic columns are as a rule stable. This eliminated the risk of theinstability of the packing and lets to the utilization of all advantages of the eluent back-flush.
The paper presents the results of research over the optimum conditions of the eluentback-flush implementation. The specific conditions of separation, in which applying BF issuitable and has a measurable time and economic advantages are characterized andsummarized as well. A developed preparative and analytical applications, mainly forpetroleum products and industrial sewages, are also presented.
1. IP 391:95, Petroleum Product – Determination of aromatic hydrocarbon types in middle distillations – Highperformance liquid chromatography method with refractive index detection;
2. PN-EN 12916:2000, Petroleum Product – Determination of aromatic hydrocarbon types in middle distilla-tions – High performance liquid chromatography method with refractive index detection;
3. ASTM D 6379, Standard Test Method for Determination of Aromatic Hydrocarbon Types in Aviation Fuelsand Petroleum Distillates-High Performance Liquid Chromatography Method with Refractive Index Detec-tion;

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
4. R. Kartanowicz, A. Przyjazny, M. Kaminski, Application of high-performance liquid chromatography withultraviolet diode array detection and refractive index detection to the determination of class compositionand the analysis of gasoline, J. Chromatogr. A 1029 (2004) 77;
5. M. Kaminski, E. Gilgenast, A. Przyjazny, G. Romanik, Procedure for and results of simultaneous determina-tion of aromatic hydrocarbons and fatty acids methyl esters in diesel fuels by high performance liquidchromatography, J. Chromatogr. A 1122 (2006) 153;
6. R. Kartanowicz, E. Gilgenast, M. Kaminski, Group-type analysis of middle distillates by test method IP-391/EN-12916/ASTM D 6379 in terms of resolution and selectivity of chromatographic columns, ChemAnal. (Warsaw) 52 (2007) 265;
7. M. Kaminski, Simple test for determination of the degree of distortion of the liquid-phase flow profile incolumns for preparative liquid chromatography, J. Chromatogr. A 589 (1992) 61.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-017-MO
Optimized Supercritical Fluid Chromatographic (SFC) Separationof Organic Light Emitting Diode (OLED) Materials
Dennis Lorenzen, Axel Thomasberger, Bernhard SchubachMerck KGaA, HPC A022/001,
Frankfurter Str. 250, 64293 Darmstadt, Germany
Temperature and pressure conditions were systematically investigated for SFC separa-tion of several OLED materials. Different organic solvents were used as organic modifierto the CO2.
The separation power of the system was improved by comparing different stationaryphases, like reversed phase, middle polar phase, HILIC and normal phase. Because of lowbackpressure of SFC we are able to couple up to three columns.
With this optimized conditions we achieved a half width of the peaks around six sec-onds. The theoretical plates of this separation system are over 200000 per meter.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-018-TU
Robustness Criterion vs. Experimental Design in Robustness Testingof RP-HPLC Method for Ramipril and its Impurities Determination
Biljana Jancic-Stojanovic, Tijana Rakic, Sava Vemic, Nad– a Kostic,Ana Vemic, And– elija Malenovic
University of Belgrade, Faculty of Pharmacy, Department of Drug Analysis,Vojvode Stepe 450, Belgrade, Serbia
In this paper, the innovative concept for robustness evaluation is presented. The newapproach was applied on RP-HPLC method developed for separation of ramipril and itsfive impurities. The method capability to provide adequate separation when the experi-mental conditions are slightly changed was tested in two phases. Firstly, the data fromthe optimization procedure were used to determine the functional dependence of theselected response (minimum effective resolution) upon different factors. The rate ofchange of the response was tracked by appropriate derivatives of the functions. Thesignificance of the factors was estimated graphically and numerically, calculating therobustness criterion. Afterwards, Plackett-Burman experimental plan was applied for thethorough estimation of the method robustness. Experimental design included seven realfactors (acetonitrile content in the mobile phase, pH of the mobile phase, triethylamineand potassium dihydrogen phosphate content in the water phase, the flow of the mobilephase, column temperature and wavelength of the detection) and four dummy factors.The influential factor effects were identified utilizing the Dong algorithm and Paretocharts. Simultaneous comparison of the results obtained from two different approacheswas done. The robustness criterion was found to be reliable parameter for screening themethod robustness, especially during the method optimization where it can support theselection of the adequate optimum. On the other hand, the experimental design in test-ing the robustness allows the detailed assessment that includes a great number of thefactors influences and should be done to confirm the results obtained by robustnesscriterion.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-019-MO
Search of Suitable Two-Dimensional Chromatographic Systemsfor the Separation of Biomass Compounds
David Angot1, Agnes Fonverne1, Amélie Dechenaux2, Sabine Heinisch2
1IFP Energies Nouvelles – Lyon, Direction Physique et Analyse,Rond-Point de l’échangeur de Solaize – BP3 – 69360 Solaize, France
2Laboratoire des Sciences Analytiques, CNRS UMR 5180-UCBL, Université de Lyon,43 boulevard du 11 novembre 1918, 69622 Villeurbanne Cedex, France
The development of modern chemistry during the half of the twentieth century wasfounded on the use of crude oil and gas as raw materials for the synthesis of mass marketchemical products. Now, the dwindling of petroleum resources makes it essential to findother substitutes for these fossil products and consequently, renewable resources enablingthe production of chemical synthons. Biomass, as the only source of renewable carbon,and in particular lignocellulosic biomass, shows great promise for large-scale productionunits. Various processes have already been investigated as biomass pyrolysis, hydrother-mal conversion or enzymatic treatment, without forgetting additional catalytic hydro-treatments which aim at reducing the large number of produced molecules and signifi-cantly moving the production towards valuable products. As far as we know, these valu-able products are often present in a variable amount in a very complex mixture from achemical point of view with a large scale of polarities and families (acids, aldehydes,ketones, lactones, alcools, furans, phenols, cresols, catechols, esters…) and the identifica-tion of these interesting molecules from this mixture will be a tough challenge. The use ofseparative techniques is straightforward, but the development of bidimensional approach-es must be taken on because classical one-dimensional separation has not sufficient peakcapacity and selectivity.
Because of the presence of compounds with elevated molecular masses or with athermo sensibility in this kind of complex mixture comprehensive LC x LC has beenchoosen compared to comprehensive GC x GC technique.
LC x LC involves the separation of a sample by using two liquid chromatographicmethods. The two methods should differ in their selectivity which essentially depends onthe chosen system defined by three parameters: the stationary phase, the mobile phaseand the temperature. Ideally, the retention times in the two separation dimensions arecompletely independent and the dimensions are qualified as “orthogonal”.
In this study, a screening of the potential conditions in terms of chromatographicmode (HILIC and RPLC), stationary phases (silica- and non silica-based columns), mobilephases (buffered or not) and temperature (within a large range) will be described withthe aim of assessing the degree of orthogonality and maximizing the practical peak ca-pacity. A comparison of a great number of coupled chromatographic systems obtained

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
with a sample contained 38 compounds, representative of biomass samples will be pre-sented. 9 stationary phases (Hypercarb, Zr PBD, Zr Carb, PLRP-S, Xbridge BEH C18,Kinetex PFP, Acquity BEH Shield C18, Zorbax SDB CN, Acquity CSH phenyhexyl), 2 tem-peratures (30 °C and 65 °C or 30 °C and 80 °C) and 2 organic phases (acetonitrile andmethanol) have been studied in gradient elution mode; that means 630 possible combi-naisons. The data will be interpreted in term of peak capacity, which is an excellent indi-cator of the separated power for one bidimensional system and one sample.
First results of comprehensive LC x LC obtained in the optimized conditions will bepresented.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-020-TU
Sensitive Analysis of Trace Metal Ions in a Saline Sample CombiningOnline Dye Complexation and Transient Isotachophoresis
Kihwan Choi, Asif Riaz, Sunyoung Cho, Jihye Kim, Doo Soo ChungDepartment of Chemistry, Seoul National University,
Seoul 151-747, Korea
For the determination of trace metal ions in capillary electrophoresis, chromophoriccomplexation has been used since most metal ions are neither naturally absorbing UV/Visnor fluorescent. By mixing with a dye, 4-(2-pyridylazo) resorcinol (PAR), the metal ionssuch as Ni2+, Fe2+ and Zn2+ in a saline sample were converted into anionic complexeshaving strong absorbance near 500 nm. A large volume of the metal-PAR complex samplesolution injected into a coated capillary was stacked isotachophoretically and separatedunder a reverse potential. The salt anion (chloride) and PAR in the sample matrix acted asthe leading and terminating electrolytes, respectively. However, when another complex-ing agent 4-(2-thiazolylazo) resorcinol (TAR) was used for metal ions including Cd2+, themobilities of metal-TAR complexes were lower than that of TAR. Thus, a long plug of thesaline sample containing the trace metal ions, but devoid of TAR, was injected instead ofpretreating the sample with TAR. Since the mobility of TAR fell between the mobilities ofthe anionic leading electrolyte (chloride in the sample) and the anionic terminating elec-trolyte (CHES in the background electrolyte), a highly concentrated zone of TAR wasformed at the rear of the sample matrix. This isotachophoretically concentrated TAR plugswept the metal cations migrating to the cathode by forming anionic metal-TAR complex-es, thus yielding highly efficient metal-TAR peaks. Combination of the dye complexationand transient isotachophoresis provided excellent detection limits for trace metal ions inthe low ppb range with absorbance detection. Our method was successfully applied tothe analysis of urine samples without pretreatment or desalting steps.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-021-MO
Separation Modes in Ice Chromatography
Tetsuo Okada, Yuiko Tasaki, Taiki Shamoto, Yuji Miyazaki, Satsuki TakahashiDepartment of Chemistry, Tokyo Institute of Technology,
Meguro-ku, Tokyo 152-8551, Japan
Water-ice is involved not only in physical processes in the global environments such as thedetermination of snow crystal morphology, thunderstorm, and forest heave, but also inchemical processes such as nitrogen circulation, ozone layer depletion, and the functionof antifreezing biopolymers. These suggest that water-ice can be an adsorbent for variouscompounds and that a liquid phase coexistent with ice provides a new class of reactionmedium. From such perspectives, we have devised ice chromatography. The surface of icehas the dangling bonds potentially acting as hydrogen bond donors or acceptors, and,thus, ice can be used for adsorption chromatography. With a dopant, a liquid phase isevolved and thermodynamically coexistent with solid ice. Such ice can be used for parti-tion-type chromatography. Some functionality can be hereby added to ice chromato-graphic stationary phase.
As already reported, some compounds are adsorbed on the ice surface by hydrogenbonds and are separated with ice chromatography. The compounds having two or morepolar groups can be separated with a hexane-based mobile phase. When an appropriatedopant is incorporated in the ice stationary phase, a solution phase is developed de-pending on the type and concentration of a dopant as well as on temperature. Solutesare partitioned into the liquid phase, the sizes of which range from 0.1 μm to 4 μm. Thevolume of the liquid phase is controllable with varying the temperature and the concen-tration of a dopant, and thus the balance between adsorption and partition mechanismis also varied with these experimental parameters. The liquid phase coexistent with theice phase enhances the functionality of the ice stationary phase. Of particular interest isthat the incorporation of an appropriate chiral selector into ice enables chiral ice chro-matographic separation. Our studies have indicated that chiral recognition occurs in theliquid phase evolved in the ice stationary phase. An appropriate salt (KCl, NaCl, etc) anda chiral selector (e.g. β-cyclodextrin) are simultaneously incorporated in the ice station-ary phase to facilitate the chiral recognition in the liquid phase evolved in the ice sta-tionary phase. This allows successful ice-chromatographic separation of enantiomers ofsome chiral compounds. The ice stationary phase supported by a porous material prom-ises the further versatility of ice chromatography. The results obtained by this approachare also presented.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-022-TU
Separation of Monosaccharides by Ionic Liquid-ImmobilizedMicroporous Polymers
Minglei Tian, Kyung Ho RowDepartment of Chemical Engineering, Inha University,
253 Yonghyun-Dong, Nam-Ku, Incheon 402-751, Korea
Monosaccharides (fructose, glucose, galactose, ribose and xylose) are the most basic unitsof biologically important carbohydrates. Fructose, glucose and galactose are the threeimportant dietary monosaccharides. Especially, bioethanol, a renewable energy source,can be produced by glucose. Xylose with its free carbonyl group is already used in lots offood additives. So separation of these monosaccharides is necessary.
In previous HPLC researches, the most widely used method is anion-exchange chro-matogram. However, the low separation efficiency, low stability and high cost of tradi-tional stationary phases need to be solved. In this case, development of a functionalmodified, eco-friendly and low energy cost stationary phase is the most convenientmethod.
Functional ionic liquid-modified microporous polymers will be introduced in this sepa-ration. Microporous polymer has a large surface area which can contain large amount offunctional groups. Ionic liquids (ILs), as excellent functional chemicals, received lots ofresearch attentions in the fields of analytical chemistry, sample preparation, organicsynthesis, liquid-phase extraction and chromatographic separation. Their characteristics,such as hydrophobicity, miscibility with several inorganic/organic solvents, π-π interac-tions between analyte and functional groups of the ILs, are widely applied.
Ionic liquid with different functional groups (-CH3, -COOH, -NH2, et al.) will be immo-bilized on the surface of microporous polymers. According to the effect of the ILcations/anions, the interaction of functional groups and the polarities, the five mono-saccharides will be separated. The adsorption behavior of all monosaccharides on thestationary phases will be investigated. Then the mobile phase component and tempera-ture will be optimized to improve the performance for the separation.
Acknowledgements: This research was supported by Basic Science Research Program through the NationalResearch Foundation (NRF) of Korea funded by the Ministry of Education, Science and Technology (2011-0002642).

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-023-MO
Simple Separation and Detection Techniquesfor the Analysis of Carbohydrates
Ian N. Acworth, Bruce Bailey, Marc Plante, Christopher CraftsESA – A Dionex Company,
22 Alpha Rd., Chelmsford, MA 01824, USA
Carbohydrates are difficult to analyze because they are very polar compounds, exhibitsimilar structural characteristics, and do not have a suitable chromophore. Their analysisis often performed directly using high-performance anion-exchange chromatography withpulsed amperometric detection (HPAE-PAD) with a high-pH mobile phase. This techniquehas been approved for use in a variety of official methods for the analysis of carbohydratesin foods. Another common technique is to derivatize the sample with a suitable fluores-cent tag and use HPLC with fluorescence detection for the analysis. This approach canassist with the separation of sugar isomers and provide sensitive detection, althoughderivatization can contribute to increased assay variability. HPLC column technology forthe separation of carbohydrates has advanced during the past several years. New hydro-philic interaction liquid chromatography (HILIC) mode separations are providing a uniqueway to separate polar carbohydrates using simple organic/water mobile phases. Thischromatographic approach not only provides suitable separation, but also enables the useof sensitive nebulizer based detectors (i.e., mass spectrometry [MS] or charged aerosoldetection [CAD]). The work presented here describes the use of the Corona® CAD® detec-tor for the measurement of simple carbohydrates. The method has a limit of detection of< 10 ng on column and a wide dynamic range that covers ng to μg levels with highreproducibility. The determination of more complex carbohydrates – for example, glycansliberated from glycoproteins – can be achieved using a combination of MS and CADdetection. Because the CAD is a universal detector, there is no need to form fluorescenttagged derivatives thus simplifying their analysis. The CAD is used for quantitative analy-ses while the MS is used for structural verification. Data showing the utility of the LC-MS-CAD platform for analysis of glycoproteins will be presented.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-024-TU
Simultaneous Analysis of Metal-Chelate Complexes and their Ligandsusing High=Performance Ion Chromatography
Renáta Tófalvi, Annamária Sepsey, Krisztián Horváth, Péter HajósDepartment of Analytical Chemistry, University of Pannonia,
POB 158, 8201 Veszprém, Hungary
The trace analysis of metal-complexes has long been an area of interest for analyticalchemists and environmental researchers. When basic solution contains an excess of astrong complexing anion of high charge such as ethylenediaminetetraacetate (EDTA) ion,most metal ions will occur as anionic complexes and can be separated by anion exchange[1]. Hence this method provides simultaneous metal and anion separation using sup-pressed conductivity detection. This study incorporates an ion-exchange system with acarbonate eluent at various concentrations and pH levels. An advantageous condition ofthe method is that the same basic pH-range is favourable to the stability of the metalcomplexes and also to elution of the components. A theoretical framework is developedwhen complexation effects are present in the sample [2]. The EDTA and the trans-1,2-diamine-cyclohexane-tetraacetic acid (DCTA) are excellent chelating agents, they can alsoform stable chelate with different metal ions. Because aminopolycarboxylic acids are apotential risk to the environment, it is important also to develop an effective analyticaltechnique for determination of these complexes and for determination of their ligands inexcess. Several factors effect the retention by the separation of the complex anions. Thereare complex formation reactions, ion-exchange equilibria and protolysis depending on pHin the same time. The aim of this work is the optimization of a simultaneous chromato-graphic separation and identification of metal ions precomplexed by the ligand EDTA orDCTA, free ligands and non-metallic species by suppressed anion-exchange chromatogra-phy. The method was utilized to separate CuEDTA2−, CuDCTA2−, ZnEDTA2−, ZnDCTA2−,AlEDTA−, AlDCTA−, Cl−, piruvate and maleate anions. The capacity factors of complex ionswere determined for wide ranges of pH values and eluent concentrations. The collectedfractions of peaks were also identified for free multivalent ligands with ATR-FTIR methodand metal ions by atomspectroscopy.
1. P. Hajos, G. Revesz, O. Horvath, Peear J., Sarzanini C. J. Chrom. Sci., 34,( 1996),291,2. P. Hajos, G. Revesz, Sarzanini C., Sacchero G., Mentasti E J. Chrom., 640 (1993) 15–25
Acknowledgements: The authors acknowledge the financial support of the Hungarian Scientific ResearchFund (OTKA, No.81843).

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-025-MO
Size-Exclusion Chromatography of Silica-Based Nanoparticles
Marcell Pálmai1, Tibor Kremmer1, Lívia Nagyné Naszályi1, Judith Mihály1,Imola Szigyártó1, Péter Németh2
1Department of Biological Nanochemistry,2Electron Microscope Laboratory
Chemical Research Center of Hungarian Academy of Sciences (CRC-HAS),Institute of Nanochemistry and Catalysis,
H-1025 Budapest, Pusztaszeri út 59–67., Hungary
In the last decades size-exlusion liquid chromatography (SEC, GPC, GFC) proved to be anefficient method in the separation and characterization of natural and synthetic macro-molecules and biopolymers. It has also been observed that various macroporous station-ary phases were suitable for the separation of nanoparticles of different origin and compo-sition (silica, gold etc.). In the present work unmodified and modified (aminopropyl,squaric acid) Stöber silica nanoparticles were prepared and separated using size-exlusionchromatography. In order to find the optimal stationary and mobile phases for nano-particles the chromatographic properties of oligosaccharide (Sepharose CL-2B, -6B), silicagel (VYDAC 218WP54) and polystyrene-divinylbenzene copolymer based (PLgel 500 Å)columns eluted with solvent systems of different polarity were compared. Nanoparticleswere also analyzed and characterized by infrared spectroscopy (IR), ultraviolet-visiblespectroscopy (UV-Vis), transmission electron microscopy (TEM), dynamic light scattering(DLS) and density gradient centrifugation. SEC of silica nanoparticles prepared with arelatively narrow size distribution (nearly 20 nm) resulted in characteristic separationpatterns depending on the type, particle and pore size of stationary phase used. A bulkseparation of silica nanoparticles from a smaller fraction of aggregates was achieved onSepharose CL-6B having significantly bigger (40–165 μm) particle as well as pore size(exclusion limit 1000 KDa for dextrans). PLgel 500 and VYDAC 218WP54 columns withwide pores (50 and 30 nm, respectively) and smaller (5 μm) particle size resulted in moredetailed subfractionation of nanoparticles. Our results demonstrate that like macro-molecules and biopolymers SEC is an appropriate and useful method either for purifyingor fractionation of silica-based nanoparticles according to their particle size.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-026-TU
Study of Axial Dispersion with Peak Parking in Different Columns
Nándor Lambert, Attila FelingerUniversity of Pécs, Faculty of Sciences, Department of Analytical and Environmental Chemistry,
Ifjúság útja 6., 7624 Pécs, Hungary
By means of the so called arrested flow or peak parking method the axial dispersion canbe measured in a simple way. By this method a sample pulse is injected onto the col-umn, and continuous flow of the mobile phase at constant velocity under isocraticconditions is used to transport the concentration zone of the sample to the middle ofthe column. Then the flow is interrupted, and the band is let free to diffuse during agiven peak parking time. Then the flow is resumed and the sample zone is detected.The kinetic parameters can be derived from the band broadening of the injected sampleunder static conditions.
The band broadening during the parking time is represented by the increase in vari-ance, which is equal to the second central moment, proportional to the parking time, andoriginates from axial diffusion of the solute molecules during the parking period. Whenthe flow channels in the column are tortuous and constricted, the influence of their struc-tural characteristics on molecular diffusion must be taken into account. This effect isexpressed by the obstructive factor.
In the present study, the axial molecular diffusivity in THF of several polystyrene sam-ples with different molecular weights has been determined. A porous and a core-shellcolumn were used to observe the differences in the dispersion.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-027-MO
Switching from Constant Flow Rate to ConstantPressure Elution Mode
Matthias Verstraeten1, Ken Broeckhoven1, Frederic Lynen2, Konstantin Choikhet3,Monika Dittmann3, Klaus Witt3, Klaus Landt3, Pat Sandra2,4, Gert Desmet1
1Vrije Universiteit Brussel, Department of Chemical Engineering,Elsene, Belgium
2Pfizer Analytical Research Centre (PARC),Gent, Belgium
3Agilent Technologies,Waldbronn, Germany
4Research Institute for Chromatography (RIC),Kortrijk, Belgium
Constant pressure gradient elution was recently introduced to the field of liquid chroma-tography [1, 2]. In this operating mode, the pressure drop is maintained at a set pressureduring gradient separations. This results in an increase in flow rate for a decreasing mo-bile phase viscosity, while still the same selectivity can be obtained as in the customaryconstant-flow gradient elution. The latter is achieved by running the same mobile phasegradient vs. eluent volume.
In the presented series of measurements the main focus was on the method transferfrom constant flow gradient elution to constant pressure gradient elution. The contribu-tion will focus on the separation of complex and real-life samples (such as anti-oxidants,steroids, wine and cocoa butter) and investigates the benefits and drawbacks of theconstant pressure mode for these samples. Accuracy and reproducibility of retention timesand peak area are investigated for concentration sensitive detectors (UV) as well as formass-sensitive detectors (CAD and MS). Calibration curves and limits of detection aremeasured and compared for both elution modes. Effects of the differences in pressure andtemperature (viscous heating) on the selectivity and resolution are noticed and thorough-ly discussed.
1. K. Broeckhoven, M. Verstraeten, K. Choikhet, M. Dittmann, K. Witt, G. Desmet, Kinetic performance limitsof constant pressure versus constant flow rate gradient elution separations. Part I: Theoretical, J. Chrom. A,1218 (2011) 1153–1169.
2. M. Verstraeten, K. Broeckhoven, M. Dittmann, K. Choikhet, K. Witt, G. Desmet, Kinetic performance limitsof constant pressure versus constant flow rate gradient elution separations. Part II: Experimental, J. Chrom.A, 1218 (2011) 1170–1184.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-028-TU
The HPLC Study on Spontaneous Peptidization of Phenylglycine,Phenylalanine, and Phenylglycine – Phenylalanine Systems
Mieczysław Sajewicz, Monika Gontarska, Teresa KowalskaInstitute of Chemistry, Silesian University,
9 Szkolna Street, 40-006 Katowice, Poland
Among chiral compounds investigated within the framework of our research project(profens, amino acids, hydroxy acids), phenylalanine plays a particular role due to itsrecognized biological importance, whereas phenylglycine is an important substrate toproduce drugs and cosmetics.
In our earlier studies [1–4], we described the results of investigating the remarkablephenomenon of spontaneous in vitro oscillatory chiral conversion of the selected opticallypure amino acids (e.g., L-phenylalanine, L-alanine, L-tyrosine, L- and D-phenylglicine),when dissolved in the abiotic aqueous and non-aqueous media, and then aged for certainperiods of time at ambient temperature, in the stoppered glass vials. These investigationswere carried out with aid of the chiral thin-layer chromatography and polarimetry. Later,we managed to experimentally prove that the spontaneous in vitro oscillatory chiral con-version of L- and D-phenylglycine is accompanied by the spontaneous oscillatory poly-condensation of these compounds and our polycondensation results originated from thenon-chiral high-performance liquid chromatography [5].
High-performance liquid chromatography with the diode array detection (HPLC-DAD)is a reliable and accurate enough tool to monitor and quantify oscillatory reactions inpurely organic and colorless solutions, basically with the UV-absorbing analytes. In thisstudy, we employed HPLC with the two different detectors (diode array (DAD) and evapo-rative light scattering (ELSD)) to present the chromatographic results of chemical transfor-mation with L-phenylalanine and L-phenylglycine, when dissolved in 70% aqueous etha-nol and in pure dichlomethane, and then stored for certain periods of time in thestoppered glass vials at ambient temperature. Additionally, the LC-MS analytical systemwas employed to confirm the presence of the condensation products in the investigatedsamples by means of the respective mass spectra.
1. M. Sajewicz, D. Kronenbach, M. Gontarska, T. Kowalska. J. Planar Chromatogr. 21: 43–47 (2008)2. M. Sajewicz, E. John, D. Kronenbach, M. Gontarska, T. Kowalska. J. Liq. Chromatogr. Relat. Technol. 31:
1986–2005 (2008)3. M. Sajewicz, D. Kronenbach, D. Staszek, M. Wróbel, G. Grygierczyk, T. Kowalska. J. Liq. Chromatogr.
Relat. Technol. 31: 2006–2018 (2008)4. M. Sajewicz, M. Gontarska, D. Kronenbach, T. Kowalska, Acta Chromatogr., 21, 151–160 (2009)5. M. Sajewicz, M. Gontarska, D. Kronenbach, M. Leda, T. Kowalska, I.R. Epstein, J. Syst. Chem., 1:7 (2010);
DOI:10.1186/1759-2208-1-7
The work of one author (M.G.) was partially supported by the PhD scholarship granted to her in 2010 withinthe framework of the ‘University as a Partner of the Economy Based on Science’ (UPGOW) project, subsidizedby the European Social Fund (EFS) of the European Union.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-029-MO
The Retention Behaviour of Acidic Polar Compoundson Zirconia-Based Stationary Phases
Radim Kucera, Petra Kovaríková, Michal Klivický, Jirí KlimešCharles University in Prague, Faculty of Pharmacy in Hradec Králové,
Department of Pharmaceutical Chemistry and Drug Analysis,Heyrovského 1203, CZ-50005 Hradec Králové, Czech Republic
The most separations in HILIC mode are performed on silica-based supports. Nevertheless,recently published results have indicated that the metal oxides stationary phases alsopossess the ability to interact with hydrophilic compounds under HILIC conditions. Thispaper primarily describes the retention behaviour of model hydrophilic analytes (4-amino-sulfonic acid, 4-aminobenzoic acid, 4-hydroxybenzoic acid, 3,4-diaminobenzoic acid,3-aminophenol and 3-nitrophenol) on the polybutadine modified zirconia in HILIC. Theresults were simultaneously compared with a bare zirconia and a silica-based HILIC phase.The mobile phase strength, pH and the column temperature were systematically modifiedto assess their impact on the retention of model compounds. It was found that the reten-tion of our model hydrophilic analytes on both zirconia phases was mainly governed byadsorption while on the silica-based HILIC phase partitioning was primarily involved. Theability of ligand-exchange interactions of zirconia surface with a carboxylic moiety influ-enced substantially the response of carboxylic acids on the elevated temperature as wellas to the change of the mobile phase pH in contrast to the silica phase. However, no ornegligible ligand-exchange interactions were observed for sulfanilic acid. The results ofthis study clearly demonstrated the ability of modified zirconia phase to retain polar acidiccompounds under HILIC conditions, which might substantially enlarge the applicationarea of the zirconia-based stationary phases.
The work was implemented by the financial support of the Czech Ministry of Education, project MSM0021620822.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-030-TU
TLC / HPTLC / MS / LT-ELSD Coupling:New Development and Optimization
François Bretin1, Francis Maquin1, Pierre Bernard-Savary2,Eric Verette3, Henry Gangloff3, Véronique de Nailly4
1SANOFI-AVENTIS, Research Center,13 quai Jules Guesde, 94400 Vitry sur Seine, France
2CHROMACIM-CAMAG,L’Ancienne Eglise, 38340 Pommiers la Placette, France
3SEDERE SAS,B.P.27, parc Volta, 9 rue Parmentier – 94141 Alfortville Cedex, France
4BCP INSTRUMENTS,24 rue de la Mouche, 69540 Irigny, France
Mass spectrometer coupled with chromatography is supporting the chemist decision, foridentification or follow-up in a synthesis mixture. Most of the time, HPLC/UHPLC is uti-lized and coupled with MS/DAD/ELSD. On the other hand TLC/HPTLC is still widely usedby the chemists, as a rapid, reliable, and easy handling method to follow the reactionprocess steps. For this reason, a transposition is then necessary for lead candidates identi-fication and impurities follow-up from TLC/HPTLC to HPLC/UHPLC-MS/DAD/ELSD. Formany years, off-line coupling TLC/HPTLC with MS was used by scraping the silica contain-ing the spot.
More recently an interface has been developed to semi-automatically extract zones ofinterest from a TLC/HPTLC plate and direct them online into a mass spectrometer forsubstance identification or elucidation. For further investigation of “not visible” com-pounds (i.e. non chromophoric or hardly ionisable analytes), the system has been com-pleted by a Low-Temperature Evaporative Light-Scattering Detector (LT-ELSDTM). Indeed,LT-ELSD is a universal (not selective) detector and may be considered as the “eye” of theanalyst. In such a way, the analyst may have absolute confidence in the extraction/elutionprocess for a given spot when the peak appears on the detector signal. LT-ELSD is there-fore a quite relevant additional equipment enabling the analyst to distinguish between acompound which is not eluted from the plate and one which is not ionised enough to bedetected by MS.
The present work fully describes the TLC/HPTLC/MS/LT-ELSD coupling system which isrunning in open access on a pharmaceutical research site and particularly emphasizes theadvantages regarding reliability of the data, automation and overall throughput.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-031-MO
Towards the Comprehensive Two-Dimensional LCxMEKC Separationsusing On-Line Preconcentration andProgrammed Separation Conditions
Petr Cesla, Jan Fischer, Pavel JanderaUniversity of Pardubice, Faculty of Chemical Technology, Department of Analytical Chemistry,
Studentská 573, CZ-53210 Pardubice, Czech Republic
Complex samples subjected to two-dimensional liquid chromatography separations oftencontain compounds widely differing in retention. Programmed elution conditions in bothdimensions can greatly improve the overall sample resolution and peak capacity over theisocratic mode, but the gradient conditions should be carefully optimized to meet thecompatibility of transferred fractions with the separation conditions in the second dimen-sion. When gradient elution is applied in the first dimension of two-dimensional LCxMEKCsystem, increasing concentration of organic solvent in the mobile phase and in fractionstransferred from LC influences the electroosmotic flow, partitioning equilibria of samplesin micelles and properties of the micelles, which results in shifts of migration times duringthe consecutive runs in the second MEKC separation dimension. The effect becomes moresignificant with increasing length of the injected sample zone, i.e. during the on-linepreconcentration step of transferred LC fraction in the second dimension MEKC separa-tion. In present work, the on-line sweeping preconcentration step with injection of signifi-cant part of the volume of transferred fractions from LC was applied in the recently devel-oped LCxMEKC method. The changing composition of micellar pseudostationary phase inbackground electrolyte was employed to compensate the influence of increasing concen-tration of acetonitrile on MEKC separation in second dimension of two-dimensionalLCxMEKC system. The optimized LCxMEKC method with programmed separation condi-tions in both dimensions was demonstrated on the separation of natural antioxidants.
Authors acknowledge the financial support by the Ministry of Education, Youth and Sports of Czech Republic,project No. MSM0021627502 and by the Czech Science Foundation, project No. 203/09/P199.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-032-TU
A Novel Economic Approach towards Free-Flow-Electrophoresis Chips
Stefan Jezierski, Leonid Gitlin, Detlev BelderUniversity of Leipzig, Institute of Analytical Chemistry,
Weissenfelser Strasse 67, 04229 Leipzig, Germany
Microfluidics and miniaturised separation methods are new and attractive fields of scien-tific research. Unfortunately, the fabrication of microfluidic structures is demanding withregard to technical effort and production costs. Usually, meticulous and time consumingprocesses like structuring and bonding require a cleanroom facility. Therefore manyworkgroups may not consider tackling this interesting area of research.
With our new approach it is possible to produce microfluidic systems in a fast andeconomical way. A cleanroom is no longer necessary, and the technique can be replicatedin every common laboratory, with only a printer and a UV light source being needed. Thepresented procedure has the potential for rapid prototyping and creating various proto-types with different designs in a short time period. Furthermore bonding is not crucialanymore. After one photolithographic production step the created chips are ready to use.Thanks to multiple cycles, it is possible to integrate functional components into the micro-fluidic systems or to modify surface characteristics.
To demonstrate the possibilities of this approach we present the production of a micro-fluidic freeflow electrophoresis (μFFE) chip. The challenge in producing structures for μFFEis to create a separation bed which is spatially detached from the electric contacts. Due tothe chosen setup, resulting gases from electrolysis will not enter the separation bed.Nevertheless, the electric contact to couple the electric field is granted, which enables theelectrophoresis. The presented chip was created in three easy cycles using different poly-ethylene-glycol (PEG) diacrylates and one PEG methylether methacrylate in a photolitho-graphic process. Thereby, it was possible to create a conductive wall between the elec-trodes and the separation bed. The height of the systems is below 40 μm. With theachieved chip it was possible to separate a mixture of fluorescent dyes and fluorescentlylabelled amino acids, achieve results analogous to commonly produced μFFE chips.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-033-MO
A Novel Microfluidic-Based Chip Device for Rapidand Automated Characterization of N-Linked Glycans
from Monoclonal Antibodies
Stephan Buckenmaier, Michael Frank, Tom van de Goor, Lukas TrojerAgilent Technologies,
Hewlett-Packard Strasse 8, D-76337 Waldbronn, Germany
Monoclonal antibodies (mAbs) are glycoproteins bearing complex oligosaccharide moi-eties within their structure, whose presence and profile can have significant impact ontherapeutic efficacy. Glycosylation is influenced by many factors such as the cell line inwhich the mAb is produced as well as specific production parameters including pH ortemperature. Thus, characterization of glycan profiles is of vital importance throughoutthe various phases of development of these therapeutic mAbs. Here we present a fast andautomated generic method for N-glycan characterization utilizing a novel microfluidicchip device coupled to a time-of-flight (TOF) mass spectrometer.
Two different microfluidic chip designs were investigated. Both incorporated a 310nLenzyme reactor packed with immobilized PNGase F beads, a 160nL enrichment column,and a 43 x 0.075mm separation column that directly connects to the ESI sprayer. Bothcolumns were packed with porous graphitized carbon material.
Residence time of the mAb on the enzyme reactor determines the deglycosylationefficiency. In design-1, the enzyme reactor was placed directly in-line with the glycanenrichment column and was therefore operated in a flow through mode. Thus residencetime is a function of the flow used to load the mAb onto the chip. Data have shown thata period of 6 seconds obtained at a loading flow of 1 μL/min could efficiently deglycosy-late the majority of many mAbs studied. However, some did not react quantitatively andrequired further lowering the flow rate. This was detrimental for analysis speed.
Design-2 uses a novel concentric rotor-in-rotor design, which allows for switching enrich-ment column and enzyme reactor independently into and out of the flow path. In this way,residence time and loading flow are decoupled. Sample injection and loading of the enzymereactor required 1 minute at 3 μL/min flow rate. The residence time was varied by switchingthe enzyme reactor to bypass. Data showed that a 4 minute period resulted in the completedeglycosylation of all mAbs studied. Transfer of cleaved N-glycans from the enzyme reactorto the enrichment column and subsequent separation of the glycans took 7 minutes. Dataprocessing, which was facilitated by matching accurate masses with a glycan database,typically needed less than 5 minutes per sample. This makes a total of about 20 minutes foran assay that traditionally could take one half to several days.
Furthermore, data will be presented comparing results obtained using this microfluidicbased LC/MS method to those generated with traditional profiling assays.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-034-TU
A Novel NMR Chip for Chemical Reaction Analysisin Extended-Nano Space
Shota Yoshioka, Kazuma Mawatari, Takehiko KitamoriDepartment of Applied Chemistry, School of Engineering,
The University of Tokyo, Japan
Chemistry in nano-size spaces has been getting much attention by its scientific interestand many applications such as microchip based reactor, chromatographic separationdevices, biological devices and others. Recently, specific and unique chemical propertiesof molecules confined in 10 – 1000 nm scale spaces (called extended-nano spaces) havebeen revealed by our group. Surface properties are dominant factor of the molecularbehavior or reactions in the extended-nano spaces. These confined molecules play adifferent role from that in bulk space. Therefore, extended-nano spaces are expected tobe a promising new reaction field. Although attractive unique properties of molecules inthe extended-nano spaces have been researched extensively, there were only a few re-ports of chemical reactions utilizing extended-nano spaces. The problem arises from adifficulty of reaction investigation in the extended-nano spaces using conventional meth-ods due to its extremely low amount of reaction sample (~ nL) and difficulty to handlesamples accurately in nano spaces.
Herein, we report a development of a novel on-chip analytical system for chemicalspecies confined in the extended-nano spaces by combining with nuclear magnetic reso-nance (NMR) spectroscopy. The NMR chip is constructed from thermally-bonded threequartz glass plates and has several thousands of extended-nano channels for chemicalreactions between the glass plates. In order to test the availability of the NMR chip forreactions in nano spaces, ethanol was injected into the extended-nano channels by apressure driven method and evaluated by 1H-NMR spectroscopy equipped with a general-ly-used tunable 5 Φ probe. The NMR spectrum of ethanol in extended-nano spaces wasrecorded using the NMR chip placed in a sample tube filled with D2O as an externalsolvent for NMR lock and gradient shimming. The broad peaks of ethanol were observeddue to unadjusted shimming for extremely small volume of sample inside of the NMRchip, however, the obtained results is enough to investigate molecular behavior in nanochannels. Hence, we apply this NMR chip for nano-scale chemical reaction which includesmixing, reacting and analyzing processes in the extended-nano spaces and demonstrateits availability for the development of chemistry in the extended-nano space.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-035-MO
Application of the HPLC-chip LC-MS Technology for Globaland Targeted Investigations of Proteomes
Hartmut Schlüter1, Steffen Ehlert2, Ullrich Tallarek2, Verena Richter1,Diana Hildebrand1, Maria Trusch1
1University Medicine Hamburg-Eppendorf, Inst. of Clin. Chemistry,Hamburg-Eppendorf, Germany
2Philipps University, Inst. of Analyt. Chemistry,Marburg, Germany
The enormous complexity of tryptic peptide mixtures of protein extracts requires a liquidchromatography system separating the peptides with high resolution. The HPLC-chiptechnology was developed to meet these demands. It consists of a microfluidic polyimide-based microchip integrating a reversed-phase-trapping column connected via a valve witha reversed-phase separation channel, and an integrated ESI-tip. The latter is formed bydirect laser ablation of the polyimide films and thus guarantees a very stable and robustelectrospray. For studying the performance of the HPLC-chip integrated in a LC-MS sys-tem a low-complexity sample of tryptic albumin peptides was used at different concentra-tions to test the detection limits, peak capacities, and reproducibility. Furthermore trypticpeptides from a protein fraction with high-complexity (a human plasma protein fraction)were used for investigating the performance of the HPLC-chip in the LC-MS systemequipped with ESI-ion-trap mass spectrometer. Additionally the HPLC-chip LC-MS systemwas used for the analysis of the proteome of lipid rafts from renal tissue and for thequantification of peptides via an HPLC-chip-ESI-triple-quadrupole-MS operated in theselected reaction monitoring mode (SRM). The systematic studies of the performance ofthe HPLC-chip LC-MS system yielded highly reproducible separations with satisfying peakcapacities and sequence coverage, particularly for low sample amounts. These beneficialproperties are also obvious when looking at the results of the global and targeted analysisof proteomes: The investigation of a lipid rafts proteome by the HPLC-chip LC-MS systemyielded several hundred peptides stemming from membrane proteins. In a plasma proteinfraction several hundred peptides derived from plasma proteins were identified. SRMexperiments with the HPLC-chip-ESI-triple-quadrupole-MS showed a low detection limit inthe atomol range, a large dynamic range and a high reproducibility combined with ro-bustness making the system suited for targeted proteomic analysis.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-036-TU
Bead-Based Liquid Chromatography in Microfluidic Devices
Sebastian Thürmann, Detlev BelderUniversity of Leipzig, Institute of Analytical Chemistry,
Johannisallee 29, 04103 Leipzig, Germany
Miniaturization of well established separation techniques has been a major focus of re-search efforts. After electrophoresis was successfully transferred to lab-on-chip systems inthe nineties, chromatographic techniques shifted in the centre of interest. For incorporat-ing stationary phases in microfluidic channels porous polymer monoliths (PPMs) as well assilica beads are mainly used. Integrating the chromatographic bed and using high pres-sures of up to 400 bar is a challenging field of work. Especially the integration of particlebased columns imposes high demands on the technical equipment and the design of thechip. Numerous approaches have been published so far [1–5]. Our effort was it to achievea simple manufacturing process and implementing chromatographic material in any chiplayout. An additional aim was to use glass as chip material because of its high rigidity,chemical robustness, electrical properties, transparency, and the ability to support electro-osmotic flow.
In this context we present high pressure resistant bead-based beds for chip chromatog-raphy. The frits were manufactured by creating PPMs via photomask or laser-assistedphotopolymerisation. The PPMs are composed of butyl acrylate as monomer and 1,3-butanediol diacrylate as crosslinker. The experiments were carried out in simple crosslayout glass chips with channel dimensions of 20 x 50 μm. The high pressure resistantPPMs with a length of 50 μm could be introduced with a resolution of ~ 5 μm in order tocreate spatially-resolved retraining segments within the microchannel. The reversed-phasechromatographic bed was evaluated with regard to its capability for high-speed [6] andhigh-pressure chromatographic separations on chip via mass spectrometric or fluores-cence detection.
1. Ceriotti L, Rooij NF, Verpoorte E Anal Chem 2002, 74, 639–647.2. Jemere AB, Oleschuk RD, Harrison DJ Electrophoresis 2003, 24, 3018–3025.3. Ro KW, Chang WJ, Kim H, Koo YM, Hahn JH Electrophoresis 2003, 24, 3253–3259.4. Jindal R, Cramer SM Journal of Chromatography A 2004, 1044, 277–285.5. Gaspar A, Piyasena ME, Gomez FA Anal Chem 2007, 79, 7906–7909.6. Fritzsche S, Hoffmann P, Belder D Lab Chip 2010, 10, 1227–1230.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-037-MO
Comparison of Glycan Distribution in Therapeutic MonoclonalAntibodies by LC-MS using mAb-Glyco-Chip and CE-LIF
Shiaw-Lin Wu1, Yi Wang1, Sam Tep2, Zoran Sosic2, Yelena Lyubarskaya2,Ning Tang3, William S. Hancock1, and Barry Karger1
1Barnett Institute and Department of Chemistry and Chemical Biology,Boston, MA 02115, USA
2Analytical Development, Biogen Idec Inc.Cambridge, MA 02142, USA
3Agilent Technologies,Santa Clara, CA 95051, USA
A robust, high throughput method is needed to measure the consistency of the N-glycandistribution in therapeutic monoclonal antibodies (mAbs). A new chip-based LC-MSapproach has been developed for direct analysis of intact mAbs, in which immobilizedPNGase F and graphitized carbon are integrated together in the chip to on-line releasethe glycans for mass spectrometric analysis. In addition to using a QTOF MS for analysis(precursor ion measurement), we also applied the same separation platform with a QQQmass spectrometer for MRM measurement (to further improve the high throughputcapability). The pros and cons of the glycan analysis using the new LC-MS approach arecompared to routine CE-LIF method (off-line release of glycans).
Intact monoclonal antibodies (2 μg) were directly loaded on an Agilent mAb-Glyco-Chip consisting of immobilized PNGase F and a porous graphitized carbon column, whichwas coupled to an Agilent QTOF 6520 mass spectrometer. For MRM analysis, the chipwas coupled to an Agilent QQQ 6460 mass spectrometer. The precursor and product ionswith their corresponding charge states and glycan structures have been determined byQTOF, such as G0 glycan using m/z 731.8 (2+) as precursor, and m/z 204.1 (N-acetylglucosamine) and m/z 366.1 (N-acetyl glucosamine plus hexose) and m/z 528.2 (N-acetylglucosamine plus 2 hexose) as product ions for MRM transitions. A Beckman PA8000 CEwith laser-induced fluorescence detector (LIF) was used to measure the released glycans(off-line).
Overall, this new glyco-chip based assay offers a rapid method for glycan characteriza-tion and analysis, as the intact mAbs are analyzed directly; thereby, eliminating samplepreparation and allowing for complete analysis in less than 30 minutes.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-038-TU
Determination of Low-Molecular-Mass Amines using MicrochipElectrophoresis and Contactless Conductivity Detector
Ahmed O. AlnajjarDepartment of Chemistry, College of Science, King Faisal University,
P.O. Box 400, Hofuf 31982, Saudi Arabia
Microchip electrophoresis with contactless conductivity detection was successfully dem-onstrated for the determination of dimethylamine and other low-molecular-mass amines.Derivatization of the compounds or the addition of a visualization agent as for indirectoptical detection schemes were not needed. Non-charged chiral selectors were employedsuch as hydroxypropylated cyclodextrin (CD) and 18-crown-6-tetracarboxylic acid. Acidicbuffer solutions based on lactic or citric acid were used. The detection limit was in therange of 0.01 to 0.05 mg L−1. The analytical characteristics including recovery, intermedi-ate precision, linear dynamic ranges, linearity and selectivity were demonstrated. Theapplicability of the described method to biological samples was also studied.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-039-MO
Development and Prevalidation of a Nano-LC-Chip-MS/MS Methodfor High Sensitivity Hepcidin Quantitation
Virginie Houbart1, Frédéric Lecomte2, Gaël Cobraiville3, Anne-Catherine Servais1,Philippe Hubert2, Marianne Fillet1,3
1Laboratory of Analytical Pharmaceutical Chemistry, Department of Pharmacy,University of Liège, Belgium
2Laboratory of Analytical Chemistry, Department of Pharmacy,University of Liège, Belgium
3Clinical Chemistry, GIGA Research,University of Liège, Belgium
Recently, an increasing interest has been observed for microfluidic LC systems in the fieldof bioanalysis. Miniaturisation presents undeniable advantages as increased efficacy dueto lower peak dispersion, reduced solvent and sample volume consumption and highsensitivity. The major trend to overcome the lack of robustness of such systems is theintegration of all fittings and chromatographic components in one single piece. LC-chip isthe first integrated microfluidic technology for high throughput nanoflow liquid chroma-tography. Briefly, the centrepiece of the system is the chip itself that integrates the sampleenrichment column, the separation column, the commutation valve and the nanoelectro-spray tip that allows the coupling with a mass spectrometer. This integrated design reduc-es dramatically the dead volumes of the system and consequently increases sensitivity andpeak efficiency that are crucial features for the quantitation of peptides present in verysmall amounts in biological matrices. HPLC-chip coupled to an ion trap mass spectrome-ter was therefore chosen to develop a robust and sensitive quantitation method ofhepcidin, a peptide marker of clinical disorders linked to iron metabolism.
Method development was performed in several steps. First, stable isotope labelled hepcidinwas chosen as internal standard. Then, the detection was optimised by infusion of hepcidinand internal standard. The linear gradient giving rise to the best results in terms of peak inten-sity, peak shape and carry-over was then determined by experimental design methodologyusing a face-centered composite design. Three factors were considered: the amount of formicacid in the mobile phase, the initial composition of the mobile phase and the gradient time.Fifteen conditions were tested and led to the optimal parameters. A second design of experi-ments was then applied in order to determine the best sample composition. A full-factorialdesign was created to investigate three factors (proportion of acetonitrile, proportion andnature of the acidic additive) in order to maximise peak intensity and minimise carry-over. Thisapproach resulted in sharp and intense peaks and low carry-over.
Finally, the method was prevalidated using accuracy profiles in order to demonstratethe ability of the technique to quantify low concentration hepcidin. Good results withrespect to accuracy, trueness and precision were obtained, as well as a very low limit ofquantitation (25 attomoles of loaded material).

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-040-TU
Development of a Micro-Pillar Array Separation Devicefor Commercially Available Micro-HPLC Systems
Hiroshi Kobayashi, Hiroo WadaShinwa Chemical Industries Ltd., Kyoto, Japan
A micro-pillar array separation device was developed for the purpose of conventionalusage in high performance liquid chromatography (HPLC). After the creation of micro-pillar arrays in nano-fluidic channels on silicon wafers utilizing so-called “MEMS” (micro-electromechanical systems) technology, the surfaces of the micro-pillar arrays were modi-fied with dimethyloctadecylchlorosilane (ODS) for reversed-phase liquid chromatography.The investigation of retentive properties and separation performances of ODS modifiedseparation device were carried out using commercially available nano-flow HPLC instru-ments consisting of a split-less flow nano pump (DiNa S, KYA), a 20 nL volume injector(VICI) and a direct on-capillary cell detector (CE-2070, JASCO) in methanol/water mobilephases condition.
For the investigation of retentive properties, solutions of uracil, amylbenzene/butyl-benzene, triphenylene/o-terphenyl, and caffeine/phenol mixtures were used as the indica-tors of unretained solute (t0), surface coverage (αA/B), steric selectivity (αT/O), and residualsilanol group activity (αC/P), respectively. As a result, relatively higher surface coverage(αA/B ≈ 1.6) and higher steric selectivity (αT/O ≈ 1.7) were observed in comparison with aconventional silica based particulate packed column in methanol/water = 80/20 mobilephase. In the investigation of dynamics of chromatographic performance, theoreticalplate numbers greater than 25,000 were observed for butylbenzene with a retentionfactor of 1.4 at a linear velocity of 1.0 mm/s, a column length of 310 mm, and a mobilephase consisting of methanol/water = 80/20. Plate heights close to 12 μm were observedin 0.5–1.5 mm/s linear velocities, and these were similar to those for 5 μm particulatepacked columns.
Other detection systems, i.e., an optical fiber fluorescence detector (FLE1000, NSG) anda contactless conductivity detector (C4D system, eDAQ), were utilized for investigation of amicro-pillar array separation device. Applications will be introduced in the session.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-041-MO
Development of Laplace Pressure Valve for Chromatography Systemin Extended-Nano Space
Shogo Kubota, Kazuma Mawatari, Xu Yan, Takehiko KitamoriDepartment of Applied Chemistry, School of Engineering,
The University of Tokyo, Japan
The study on genomics, proteomics, and metabolomics of a single cell has been startedwith great interests of various biological fields. Extended-nano space that is 102 – 103 nm-sized enclosed space fabricated on a glass plate is thought to be suitable for these analy-ses. We have clarified the effectiveness of extended-nano space as a separation column ofa liquid chromatography system so far [1]. Then, another key technique is a sampleinjection. Sample volume is assumed to become smaller to be femtoliter to several hun-dreds attoliter. To achieve this, a valve in extended-nano space is indispensably necessary.However, mechanical valves or conventional chemical valves made by partial photo-chemical surface modification cannot be applied due to the insufficient spatial resolution.In this report, we conceived a new hydrophobic patterning method by combining chemi-cal modification with nanostructure fabricated on the bottom of the extended-nanochannel, and developed a valve that we call a Laplace valve in extended-nano space. TheLaplace pressure valve uses the Laplace pressure difference arising at the interface ofhydrophobic patterning.
Firstly, we verified the enhancement of hydrophobicity on a nanostructured surface.Nanopillars (width 60 nm, pitch 90 nm, height 60 nm) were fabricated on a fused silicaplate by EB lithography and dry etching techniques. The whole surface was chemicallymodified by trimethylsililimidazole, and static contact angles (C.A.) of water sessile dropson the surface with/without pillars were measured. The C.A. on the flat surface was 88°,while the C.A. on the surface with the nanopillars was 98°. These results showed that thenanopillars could enhance the hydrophobicity even though the intrinsic C.A. was smallerthan 90°. Secondly, the microchip consisting of microchannels as reservoirs, nanopillarsthat would induce strong hydrophobicity, and extended-nano channel (width 2 μm,depth 200 nm) overlaid on the pillar area as a separation column were fabricated. Aftermodification by octadecyltrichlrosilane, water was pushed into the nanochannel. Weobserved by an optical microscope that water was stopped at the interface of the pillararea. We think that it was due to a large Laplace pressure induced there. The value of theLaplace pressure is now under measurement.
1. M. Kato et al., Anal. Chem., 82, 543–547 (2010)

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-042-TU
Development of Polymer-Modification Methodfor Novel Separation Device on Extended-Nano Space
Junpei Katagiri, Tatsuhiro Yamamoto, Kazuma Mawatari, Takehiko KitamoriSchool of Engineering, The University of Tokyo,7-3-1, Hongo, Bunkyo, Tokyo, 113-8656, Japan
Recently, high efficient separation method for ultra-low volume of samples is required forvarious applications such as single cell analysis and proteomics. We have been focused on101–102 nm scale channels (so-called extended-nano space), and developed extended-nano chromatography that utilizes surface of extended-nano channel as stationary phase.It has large number of theoretical plates (106 plate/m) and achieves high separationcapability for ultra-low volume of sample. However, it is difficult to apply extended-nanochromatography to separation of various biomacromolecules, since it is not enough toseparate clearly these samples, which have different charges. The objective of our study isto develop polymer-modification method by surface-initiated atom transfer radical poly-merization (SI-ATRP) on extended-nano space for novel separation mode, which utilizesmultiple effects of polymer. Herein, we report the availability confirmation of this SI-ATRPmethod on quartz glass surface of microchannel.
The polymer modification using SI-ATRP was performed as described below. An initia-tor, (1-trichlorosilyl-2-(m,p-chloromethylphenyl)ethane), was firstly modified on the sur-face of microchannel of 400 μm-width and 2 μm-depth, which was fabricated on quartzglass plate. Reaction solution of monomer (N,N’-dimethylacrylamide), catalyst(CuCl/CuCl2 and tris[2-(dimethylamino)ethyl]amine) was introduced into microchanneland polymerization reaction was performed.
Polymer modification on microchannel surface was evaluated by the resistance toprotein adsorption. A solution of Texas Red-labeled bovine serum albumin was introducedinto unmodified and polymer-modified microchannels. Phosphate buffered saline wasthen introduced for washing. After washing, reduction of fluorescent intensity was notobserved on unmodified microchannel due to nonspecific adsorption of proteins onto thesurface. In contrast, fluorescent intensity was decreased on polymer-modified micro-channel. These results indicate that resistance to protein adsorption was caused by suc-cessful polymer modification on microchannel. Therefore, we have confirmed the avail-ability of SI-ATRP method onto quarts glass channel surface for novel separation mode inextended-nano chromatography.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-043-MO
Electrochromatographic Properties of Monoliths Preparedby Radiation-Induced Free Radical Polymerization
for Lab-on-a-Chip Applications
Claudia Ernst, Detlev BelderUniversity of Leipzig, Institute of Analytical Chemistry
Weissenfelser Strasse 67, 04229 Leipzig, Germany
Porous polymer monoliths as stationary phases have gained increasing interest in recentyears. There are several polymerization techniques regarding their preparation, the freeradical polymerization being the best established. The radical polymerization process canbe initiated either chemically, by heat or ultraviolet light (UV). Applying UV light, themonoliths can be prepared and functionalized within defined zones, which makes thistechnique in particular attractive for Lab-on-a-Chip purposes. However, this polymeriza-tion technique is limited to devices transparent to UV light and the polymerization pro-cess itself restricts the monolith preparation to tubings of small diameter. Furthermore,the added photo initiator remains within the polymeric structure, eventually leading tolight absorption phenomena.
Recently, an initiator-free approach to prepare polymer monoliths for high perfor-mance liquid chromatography using electron beam induced polymerization was devel-oped [1]. This technique affords the polymerization both within opaque microfluidicdevices and large scale tubings. Though, until today no efforts have been taken to com-pare the separation performance of monoliths prepared by electron beam or UV initiatedpolymerization.
For this purpose porous polymer monoliths were prepared within capillaries (I.D. 75 μm)using a 1.2 MeV electron accelerator. The total dose was optimized according to the flowresistance of the monoliths compared to monoliths formed by UV polymerization. After-wards, electrochromatographic separations of several test mixtures (substituted anilines,substituted benzenes, polycyclic aromatic hydrocarbons) were performed on a homebuiltcapillary electrochromatography (CE) unit. Scanning electron micrographs were taken toachieve further morphological information. Forthcoming objective is to transfer the gainedresults to microfluidic chips for chip electrochromatography (ChEC) coupled to an electro-spray ionization mass spectrometer (ESI-MS) or on-chip UV detection. Finally, preliminaryresults in chip electrochromatography are presented.
1. R. Bandari, C. Elsner, W. Knolle, C. Kühnel, U. Decker, M. R. Buchmeiser, J. Sep. Sci. 2007, 30, 2821–2827.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-044-TU
Evaluation of Microfluidic Chip Chromatography-Mass Spectrometryof N- and O-Glycans using Porous Graphitised Carbon, Hydrophilic
Interaction Media and Online Anion Exchange Fractionation
Oscar G Potter1, Rudolf Grimm2, Pauline M Rudd1
1National Institute for Bioprocessing Research and Training,Foster’s Avenue, Blackrock, Dublin 4, Ireland
2Agilent Technologies,Santa Clara, CA 95051, USA
The increased sensitivity achieved by nanoLC and nanospray ionisation as compared tohigher flow rate chromatography with electrospray ionisation (ESI) is highly valuable in theanalysis of N- and O-linked glycans due to their poor ionisation efficiency and the fact thatsample quantities are often limited. Microfluidic chip nanoLC-MS addresses the ease-of-useand reproducibility issues that are sometimes associated with nanospray ionisation and hasrecently been demonstrated as a convenient platform for integrating online sample prepara-tion steps including endoglycosidase digestion, sample cleanup and enrichment [1].
Amidst this rapid progress, there remains some uncertainty about the best approach toseparation conditions and the choice of stationary phases. Separation quality is veryimportant in LC-MS of glycans because it may be necessary to separate structural isomersand other isobaric structures, whilst coelution of different glycans can lead to ionisationcompetition and may also reduce the viability of LC-MS/MS or LC-MSn methods.
Porous graphitised carbon (PGC) [2] and hydrophilic interaction (HILIC) [3] stationaryphases are leading contenders for microfluidic chip-LC-MS of glycans. These two station-ary phases are compared with regards to their ability to separate N- and O-linked glycans.It is found that the different mobile phase requirements of these two types of stationaryphase have strong implications for achieving stable nanospray ignition, especially whennegative mode ionisation is required.
Online weak anion exchange fractionation is achieved by inserting a capillary columnbetween the autosampler and the microfluidic chip. This approach is shown to assist peakassignment and to dramatically reduce the number of co-eluting species by allowingneutral, mono-, di-, tri- and tetra-sialated glycans to be run separately in the secondseparation dimension. The resulting multidimensional separation is more convenient fornanoLC-MS/MS and it also allows individual optimisation of the high resolution separationand detection conditions for the different glycan fractions.
1. Bynum, M.A., et al., Characterization of IgG N-Glycans Employing a Microfluidic Chip that IntegratesGlycan Cleavage, Sample Purification, LC Separation, and MS Detection. Analytical Chemistry, 2009.81(21): p. 8818–8825.
2. Chu, C.S., et al., Profile of native N-linked glycan structures from human serum using high performanceliquid chromatography on a microfluidic chip and time-of-flight mass spectrometry. Proteomics, 2009.9(7): p. 1939–1951.
3. Staples, G.O., et al., Improved Hydrophilic Interaction Chromatography LC/MS of Heparinoids Using aChip with Postcolumn Makeup Flow. Analytical Chemistry, 2010. 82(2): p. 516–522.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-045-MO
Evolution of Nano-Scale Chemistries for Information-RichUPLC-Based MS Analyses
Martin Gilar, Jim Murphy, Geoff GerhardtWaters Corporation,
34 Maple St, Milford, MA 01757, USA
Proteomics researchers are concerned small changes in expression and post-translationalmodifications (PTM) that may be in low concentrations in acomplex mixture of proteinsor proteome. For drug metabolism pharmacokinetics (DMPK) or metabolic identification(met i.d.), increasingly researchers are working with small samples of body fluids or tis-sues during development and in clinical trials.
Researchers have turned to nano- and capillary-scale chromatography with sub-2-micron packing materials (nanoUPLC) and high resolution MS to detect and quantifysmall changes in metabolism or protein expression. Typical nanoUPLC uses reversedphase (RP) columns with internal diameters of 300 micron to 75 micron to overcomedispersive losses incurred typical analytical-scaleLC. A standard approach is to use a shortpre-column to trap the analytes and flush unwanted solutes to waste. Then the analytesare displaced from the trapping column to an analytical column packed with sub-2-mi-cron particles to obtain better chromatographic resolution and higher peak capacityseparations that result in better data quality from the MS.
This can be accomplished with planar microfluidic devices that remove the user inter-action with fittings, tubings and emitter placement and deliver UPLC performance. Whenextremely sample-limited, this can be done at nanoflow on an 80 micron chromatograph-ic channel at 300–500 nL/min or at 300 micron or capillary-scale for high sensitivityLC/MS/MS analyses of bioanalytical samples at flow rates of 8–12 μL/min.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-046-TU
From Ordered Pillars to Random Packings: Effect of Confinementon Transcolumn Velocity Bias and Separation Efficiency
Anton Daneyko, Siarhei Khirevich, Alexandra Höltzel, Ulrich TallarekPhilipps-Universität Marburg, Department of Chemistry,
Hans-Meerwein-Strasse, D-35032 Marburg, Germany
The unconfined chromatographic support is a mathematical abstraction: the system doesnot have any bounds and thus is infinite. Bulk, unconfined systems allow to simplify themathematical and numerical analysis of the properties of mass transport in porous mediasuch as particulate packings or micro-pillar arrays. Yet they are useful to study bulk trans-port properties of these materials. In addition, the use of unconfined systems reduces thedemand for computer power one needs to perform computational fluid dynamics simula-tions, allowing to employ desktop computers.
We have set up a detailed, pore scale three-dimensional study to investigate the impactof the (three-dimensional) confinement as well as the resulting consequences due to thewall effects and the associated transcolumn velocity bias on the achievable separationefficiencies [1–3]. We were also interested in the limits of the applicability of unconfinedsystems as a proper model of the separation supports in real, i.e., confined chromato-graphic devices, because comparisons between different column designs and formatsoften mix up bulk and confined system performances, which is inadequate [4].
We record plate heights of both confined and unconfined hard sphere packings andmicro-pillar arrays obtained in our computer simulations using a very efficient, highlyparallel simulation methodology on a high-performance computing platform. Efficientparallelization allowed us to quantitate flow and transport in both types of systems as wellas the engendered effects on eddy dispersion due to the supports confinement by quanti-tative numerical analysis methods. These simulations employ the correct dimensionality(3D) in the velocity field calculations and modeling of advective-diffusive mass transportwith a random-walk particle-tracking approach.
The results of these three-dimensional simulations demonstrate a substantial increase ofthe plate height data as we incorporate the confinement of the supports, both for packedbeds and ordered micro-pillar arrays. Depending of the realized packing microstructure,the confined packed beds can show separation efficiencies comparable with the perfectlyordered, but confined micro-pillar arrays.
1. S. Khirevich, A. Höltzel, D. Hlushkou, U. Tallarek, Anal. Chem. 2007, 79, 9340–9349.2. S. Khirevich, A. Höltzel, A. Seidel-Morgenstern, U. Tallarek, Anal. Chem. 2009, 81, 7057–7066.3. S. Khirevich, A. Höltzel, U. Tallarek, Philos. Trans. R. Soc. Lond. A 2011, in press (doi:
10.1098/rsta.2011.0027).4. J. Eijkel, Lab Chip 2007, 7, 815–817.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-047-MO
High Sensitivity Characterization of Propanolol and AssociatedMetabolites Utilizing Integrated Microfluidic LC/MS/MS
Paul D. Rainville1,2, Michael Tomany1, Norman W. Smith2,David Cowan2, Robert S. Plumb1,2
1Waters Corporation34 Maple Street Milford, MA 10757, USA
2Kings College London150 Stamford Street, SE1 9NH, UK
Miniaturization of chromatographic separation systems provides a means of greatly in-creasing sensitivity in LC/MS. Here we demonstrate the use of an integrated microfluidicdevice for the LC/MS/MS investigation of the in vitro microsomal metabolism of themodel drug propranolol using a sample volume of 1 μL of a 1 μMol incubation. Thesystem was capable of obtaining high quality MS and MS/MS data from the injection oftest drug substance containing sufficient information to correctly derive the structure ofthe drug metabolites. The positive ESI LC/MS TIC full scan trace for the gradient LC analy-sis of 1 μL injection of the 1 μMol incubation of propranolol resulted in some thirteenpeaks detected with the average peak width was determined to be in the region of 6–7seconds at the base, thus producing a peak capacity in the region of 10 peaks/ minute or120 for a 10 minute separation. Although the peak capacity is less than that of standardanalytical scale sub 2 μm particle LC it is still sufficient to provide a high quality separa-tion. A comparative study whereby the same volume was injected onto the micro fluidicdevice and a standard analytical UPLC demonstrated a 30 fold increase in sensitivity forthe micro fluidic format. The identification of the drug metabolites was confirmed by areview of the MS/MS spectra obtained from the survey “scan mode” analysis. The frag-mentation of propranolol gave rise to three major peaks at m/z = 183, 157 and 116 andthese ions were used as diagnostic fragment ions to search for drug related metabolites inthe sample. Utilizing the m/z = 116 ion as a diagnostic ion we further detected a greaternumber of potentially drug related metabolites than the others (eight in total). These datastudy suggest that these types of micro LC/MS/MS device are robust enough for routineapplications, and well suited to the analysis of small samples.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-048-TU
HPLC on a Compact Disc: Fabrication and Feasibility
Phillip Morgan1, Dominic Banks2, Robert Flanagan1, Peter Myers3
1Toxicology Laboratory, Department of Clinical Biochemistry, King’s College Hospital,Denmark Hill, London SE5 9RS, UK.
2Institute for Global Health and Infection, University of Liverpool,Daulby Street, Liverpool, UK
3Department of Chemistry, University of Liverpool,Crown Street, Liverpool, UK
Miniaturisation of HPLC offers advantages in that volumes of sample and reagents are re-duced, and sensitivity and speed of analysis are increased. Moreover, such systems may behighly portable and thus suited to ‘point-of-care’ testing (POCT). Such a device for themeasurement of the anti-psychotic clozapine and its N-demethyl metabolite, norclozapine,in plasma would offer improved patient management without the need for additional vene-puncture. We have investigated a liquid chromatographic POCT device based on compactdisc (CD) technology. Microfluidic channels were engraved into the surface of standardsized, uncoated polycarbonate CDs. Two complimentary discs were joined using double-sided, pressure sensitive adhesive film (PMMA, 50 μm thick). The analytical columns weremanufactured from glass capillaries (100 x 0.5 mm i.d., 0.7 mm o.d.) narrowed at one endand fritted using a single large porous silica particle (100 μm diameter). Centrifugal forcewas used to dry-pack Waters Spherisorb S5W HPLC packing into the capillary, before cuttingto length (12 mm) and mounting onto the disc. The column was sealed into the disc usingpolydimethylsiloxane (PDMS). Whole blood (ca. 50 μL) was introduced through a septummounted on the disc. Centrifugal force was applied to separate the plasma and cells, and tomove a fixed volume of plasma onto the head of the analytical column. A combination of‘burst’ and sacrificial valves were utilised, the latter through the use of a focussed 200 mW(660 nm) laser to open a path through the PMMA film. Methanol (as the eluent) was intro-duced to the separation column, and the plasma eluted using centrifugal force. The systemdemonstrates the integration of all aspects of an HPLC analysis except detection onto asimple disc without the need for electrodes or pumping systems. However, the reducedvolumes and path lengths make detection of drugs in such systems at the required sensitivi-ty a challenge. Options for analyte detection include capacitively coupled contactless con-ductivity (C4D), and chemiluminescence. Of these, C4D is most suited to the system.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-050-TU
Method of Development for Toner-Based Microchips
Una Crowley1, Mark Naussbaum1,2, Jeremy D. Glennon1
1Innovative Chromatography Group, Irish Separation Science Cluster (ISSC),Department of Chemistry and the Analytical & Biological Chemistry Research Facility (ABCRF),
University College Cork, Ireland2 Department of Chemistry, Hillsdale College,
Hillsdale, MI 49242, USA
Since the introduction of micro-total analytical systems (μTAS) and lab-on-a-chip technol-ogy, microfluidic devices have been applied in almost every area of separation science.The initial development of these devices was based on glass or quartz devices that areoften expensive to design; however, the introduction of new types of materials, such asplastics, offered a new way for fast prototyping and disposable devices.
One such development of microfluidic devices is based on the lamination of laser-printed polyester films. The advantages of these microchips are numerous and include fastanalysis times, minute consumption of electrolyte, cheap and quick and ease of reproduc-tion. In this work, a laser printer is used to selectively deposit toner onto polyester film,which is then laminated against another printed polyester film. The toner melts and bindsthe two polyester films and allows the blank regions to become channels for microfluidics.The depth and width of these channels are approximately 12 μm and 200 μm respective-ly. Multiple formats of printing and ink colours were tested in order to establish a methodto create well defined channels, and several types of software, including Corel Draw andMicrosoft Paint. The electrophoretic mobilities of fluorescein and morin were investigatedthrough the channels using appropriate voltages. The resulting toner-based microchipsdemonstrated a potential viability and cheaper alternative for chemical assays, coupledwith several detection methods, particularly Chip-Electrophoresis-Chemiluminescence(CE-CL) detection.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-051-MO
Microfluidic Liquid Junction System for CE/MS
Jakub Grym, František ForetInstitute of Analytical Chemistry ASCR v.v.i.,
Brno, Czech Republic
Mass spectrometry (MS) represents one of the most important tools for bioanalysis. Atpresent electrospray is the dominant method for on-line coupling of microscale separa-tions with MS. Commercially available MS instrumentation is, in many respects, matureand provides robust MS coupling with chromatographic separations. On the other handcapillary electrophoresis/MS coupling is still an evolving technology. In this work we havedesigned experimental systems suitable for electrospray coupling of microcolumn separa-tions and especially capillary electrophoresis. This system is based on hybrid microfluidicsdesign where the microfabricated part serves as a manifold for attachment of the separa-tion capillary and connection of the electrode reservoirs. The device is designed as ahybrid capillary/microfluidics manifold where the microfabricated part incorporates self-aligning liquid junction for precise positioning of the CE separation capillary and fusedsilica electrospray needle and connections for the external electrode reservoirs. This inter-face is constructed as an external part attached to the sampling orifice of the mass spec-trometer via a miniature subatmospheric electrospray chamber. The performance ofsystem was characterized with respect to the signal intensity and practical use for analysesof peptides, proteins and oligosaccharides.
This work was supported by Ministry of Education, Youth and Sports (LC06023) and institute research planAV0Z40310501.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-052-TU
Micro-Scaled High-Throughput Screening of Digestion Conditionsfor Insulin Analog Precursor
Lidia Gurba, Michał Odrowaz-Sypniewski, Anna Bierczynska-Krzysik,Grazyna Płucienniczak, Bozena Tejchman-Małecka, Dorota Stadnik
Institute of Biotechnology and Antibiotics,Staroscinska 5, Warsaw, Poland
The traditional gel electrophoresis is widely used for purification and separation ofpeptides and proteins. However, despite advances in technology, this technique is stillvery time-consuming and requires many manual operations. Intensive development ofmicro-analytical systems, also known as Lab-on-a-chip, has contributed to the introduc-tion of new equipment for separation and analysis of biomolecules. In these devices, somestages of the whole analytical procedure were completely automated and sampling,separation and detection take place on a single chip with minimal sample and reagentsconsumption.
The aim of our research was to screen enzymatic cleavage conditions for an insulinanalog precursor with the use of a chip-based capillary electrophoresis system Bioanalyzer2100 (Agilent). Our research project was carried out on the recombinant Gly(A22)-Arg(B31)-Arg(B32)-human insulin precursor, which is an intermediate in production ofrecombinant insulin analogs. Depending on digestion conditions three different analogs:single chain GlyA22-ArgB31-ArgB32 human insulin, two chain GlyA22-ArgB31-ArgB32 humaninsulin and two chain GlyA22-ArgB31 human insulin could be obtained. The following diges-tion conditions were evaluated: enzyme to protein ratio, temperature and incubationtime. The products of hydrolysis were additionally identified using MALDI-TOF/TOF MS(Applied Biosystems).
The studies confirmed that the new chip-based platform for gel electrophoresis can beapplied successfully for screening of digestion conditions for recombinant proteins. More-over, the system enables distinction between a single and two chain protein form.
Acknowledgement: This study was supported by EU within the European Regional Development Fund (POIG.01.01.02-00-007/08-00).

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-053-MO
On-Line Analysis of Cocaine and its Metabolite from Biological Fluidsusing a Chip-SPE-LC-MS System
Valérie Thibert, Florence Chapuis-Hugon, Valérie PichonDept. of Analytical and Bioanalytical Sciences and Miniaturization ESPCI ParisTech,
UMR PECSA 7195 (CNRS – UPMC – ESPCI ParisTech),10 Rue Vauquelin 75005 Paris, France
Analysis of compounds at ultra-trace levels in complex matrices usually requires a samplepre-treatment to clean-up and/or to concentrate the target compounds before theirchromatographic analysis. The miniaturization of these steps became of increasing inter-est thanks to the sensitivity gain, the reduction of reagents consumption, and the possibil-ity to integrate different stages of the analysis in one chip system. Recently, a chip inte-grating an enrichment channel, a separation channel and a nanoelectrospray tip wasdeveloped. Our objective was to apply this on-chip LC-MS system to the analysis of smallmolecules in complex matrices. Therefore, cocaine and its main metabolite, benzo-ylecgonine, were chosen as model molecules and analysed on chip from hair extracts andurine.
The design of the chip is well adapted to trace analysis in complex matrices since largevolumes can be injected (1–2 μL). Indeed, the compounds are first trapped in an enrich-ment channel and then eluted by the mobile phase to the analytical channel for thechromatographic separation. The conditions of chromatographic separation were firstoptimized before studying the enrichment step. Then, the volume of the solution perco-lated through the enrichment channel and the amount of organic solvent in the samplewere studied to obtain an optimal enrichment to have a sufficient clean-up without loss oftarget compounds.
The potential of the developed on chip SPE-LC-MS method was demonstrated with theobtention of very low limits of quantification and a gain factor around 40 compared toconventional LC-MS. In the case of hair extracts, cocaine was analyzed with a LOQ of only12 pg/mg, and in the case of urine samples, benzoylecgonine was determined with aLOQ of 3.2 ng/mL. Great sensitivity was obtained and also a very easy and quick samplepre-treatment was used.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-054-TU
Poly(ethylene-Glycol) Coated Microfluidic Devicesfor Microchip Electrophoresis
Marcel Schulze, Detlev BelderUniversity of Leipzig, Institute of Analytical Chemistry
Weissenfelser Str. 67, 04229 Leipzig, Germany
Miniaturized separation techniques are very attractive due to the unexcelled potential forhigh speed separations on a small system. One example for successful miniaturization ofa classical separation technique is microchip electrophoresis (MCE). However, some chal-lenges remain with the use of glass microchips, because of the similar surface characteris-tic as in fused silica capillaries used in capillary electrophoresis (CE). Due to the negativelycharged silanol groups on the channel surface at a pH above 2–3, unwanted adsorptionof cationic or/and hydrophobic analytes to the surface occurs, which results in bandbroadening, poor reproducibility, peak tailing, and reduced efficiency [1].
These effects can be reduced by coating of the surface. One of the commonly usedcoating agents for CE/MCE is Poly(ethylene-glycol) (PEG). PEG is a neutral molecule whichsuppresses the unwanted interaction of analytes with the surface. For the modification ofthe microfluidic surface two different methods are known. One possibility is the dynamicmodification of the channels by adding the coating material to the separation buffer orsimply flush the channels with the coating agent prior to electrophoretic separation.Another strategy is to covalently bond the PEG by introducing linker group on the glasssurface [2, 3].
In this context we present a new, one step method for permanent coating of micro-fluidic channel in a chip. Therefore a glass chip is flushed with the coating solution andheated up to 200 °C in order to induced polymerization. The modified surface showeda reduced electroosmotic flow. This effect could be exploited to separate chiral FITC-labeled amino acids with a separation length of 5.8 cm. Furthermore a satisfactorysuppression of the interaction of proteins to the modified channel wall of the microchipsis shown by performing separation of three native basic proteins with fluorescenceexcitation at 266 nm.
1. V. Dolník, Electrophoresis 2004, 25, 3589–3601.2. T. T. Razunguzwa, M. Warrier, A. T. Timperman, Analytical Chemistry 2006, 78, 4326–4333.3. R. J. Zeng, Z. F. Luo, D. Zhou, F. H. Cao, Y. M. Wang, Electrophoresis 2010, 31, 3334–3341.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-056-TU
The Challenge of Hyphenating a Continuous Flow Systemwith Capillary Liquid Chromatography for Detection
of Enzymatic Regulation in House Dust
Katja Oeste1,2, Paul Ermisch1, Christoph Portner1,Thorsten Teutenberg1, Thomas Letzel2
1Institut für Energie- und Umwelttechnik e. V.(Institute of Energy and Environmental Technology, IUTA),
Bliersheimer Strasse 60, 47229 Duisburg, Germany2Technische Universität München,
Weihenstephaner Steig 23, 85354 Freising-Weihenstephan, Germany
The presented project is focused on the development of a fast and cost-efficient analyticalmethod to detect enzymatic regulation in house dust, e. g. substances interacting withenzymes. House dust, which acts like a diffusive sampler for semi or low volatile organiccompounds, will be used as a model matrix. Furthermore, the toxicological effects of e. g.mycotoxins, biocides, and plasticizer have to be considered.
To detect regulatory active substances, this method hyphenates the separation by high-temperature HPLC (HT-HPLC) with a continuous-flow enzymatic assay and mass spec-trometry [1, 2].
In order to maintain the enzymatic activity of the assay, a reduction of organic solventin the mobile phase up to 20% is necessary. Hence, high-temperature HPLC is used toensure the elution of non-polar compounds and to permit the biological function of theassay [3].
This presentation is focused on the hyphenation of a continuous-flow-system withcapillary-ToF-MS. The challenge is to minimize band-broadening of the separated sub-stances in the continuous-flow-system while simultaneously ensuring a sufficient mixingbetween the inhibitor, enzyme and substrate.
1. A.R. de Boer et al. 2005. Anal. Chem. 77 (24), 7894–79002. T. Letzel. 2008. Anal. Bioanal. Chem. 390, 257–2613. T. Teutenberg 2010. High Temperature Liquid Chromatography: A User’s Guide for Method Development.
RSC Chromatography Monographs

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-057-MO
UHPLC-MS with High Pressure Microfluidic Metal Chips
Faizy Ahmed1, Greg Staples2, Reid Brennen1, Karen Seaward2, Jon James2,Hongfeng Yin1, Lynette Martinez1, Liz Carr1, Sue Post1, Qing Bai1,
John Mannion1, Kevin Killeen1
1Agilent Technologies,5301 Stevens Creek Blvd. Santa Clara, CA, USA
2Agilent Technologies,1400 Fountain Grove Pkwy; Santa Rosa, CA, USA
We have fabricated the first metal microfluidic chips capable of UHPLC operation andhave characterized their LC-MS performance.
Microfluidic chips have enabled a new class of reproducible integrated workflow devic-es that combine sample preparation, enrichment and HPLC separation with ESI for highsensitivity Nano-LC-MS applications. These devices have most commonly been fabricatedusing polymer, ceramic, and glass materials but the next generation of higher capacityand high throughput microfluidic chips for LC-MS requires materials and structures capa-ble of UHPLC operation with higher pressure limits. In this work, we describe the fabrica-tion and performance of diffusion-bonded metal chips packed with sub-2 μm silica bond-ed phases for high performance Nano- and micro flow LC-MS operation. These novelmetal devices exhibit state of the art performance in peak capacity and throughput formicrofluidic LC/MS chips.
Metal microfluidic chips with integrated frits, transfer channels, ports, and 43 mm to150 mm long columns were fabricated by diffusion-bonding multiple layers of stainlesssteel resulting in 25 x 50 x 0.4 mm devices whose features were patterned on each layerusing lithographic electro-chemical machining to create sizes ranging from less than 5 μmto 150 μm channels with very smooth surfaces.
These metal chips were slurry-packed with 1.8 μm Zorbax 300-SB C18 at pressures ofup to 1,000 bars. Sixty nL (12 fmol, 200 fmol/μL) of tryptic digest of BSA sample wasloaded using the 60 nL loop. A 2%–42% acetonitrile gradient in 20 or 60 minute, fol-lowed by a 85% acetonitrile clean-up step, were used with an Agilent 6520 QTOF LC-MSequipped with an HPLC-Chip Cube source as MSD.
The metal chips were tested to pressure limits over 2,000 bars. Fabricated metal chipswere operated at pressures over 356 bar with 0.5 and 1.0 μL/min flow rates during LC-MSruns. Extracted ion currents for peptides were measured to determine chromatographicresolution. Peak widths, FWHM, for two representative BSA peptide peaks, 461.7 and395.235 m/z, were 2.5 ± 0.2 sec (20min gradient) and 5.5 ± 0.5 sec (60 min gradient)over multiple chips. Peak capacities of up to 256 peaks were obtained with the 60 minutegradient.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-058-TU
Using 200 μμm ID cHiPLC Columns for Increased Sample Throughputin Peptide Quantitation
Remco van Soest, Nicole Hebert, Erika LinEksigent, division of AB Sciex,
5875 Arnold Road, Dublin, CA 94568, USA
In this study we present the results of an investigation of the effect of larger ID chip basedcolumns in nanoLC systems on throughput and sensitivity for peptide quantitation. Cur-rently available chip columns using a 75 μm i.d. x 15 cm requires 300nL/min flow rateand longer run times. In this work we used 200 μm x 15 cm and 200 μm x 5 cm chipcolumns packed with 3 μm C18 particles or 2.7 μm HALOTM fused core particles. Samplethroughput and sensitivity were investigated at flow rates ranging from 300 nl/min to 3.5μl/min using current available chip columns and the larger ID columns. A reduction inanalysis time of 20% can be obtained through minimizing the system delay time when aflow rate of 1.5 μl/min is used with a 200 μm i.d. column compared with using a 75 μmi.d. column with 300 nl/min flow rate. Sensitivity at the higher flow rate is reduced pro-portional to the flow rate. Reducing the column length from 15 cm to 5 cm, in conjunc-tion with a faster gradient, decreases run times by a factor of four, at the cost of a three-fold loss in peak capacity. Sensitivity improved by almost a factor of two because ofnarrower peaks. For many applications the increased throughput could outweigh thereduced peak capacity and sensitivity. Additionally, the use of a larger diameter columnwill allow for higher sample loading, which may be used to combat the decreased sensi-tivity seen at higher flow rates.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-059-MO
Analysis of Chiral Amino Acids in Cerebrospinal Fluid Samples Linkedto Different Stages of Alzheimer Disease
Shorena Samakashvili1, Clara Ibáñez2, Carolina Simó2, Francisco J. Gil-Bea3,Bengt Winblad3, Angel Cedazo-Mínguez3, Alejandro Cifuentes2
1Department of Physical and Analytical Chemistry, School of Exact and Natural Sciences,Tbilisi State University, Tbilisi, Georgia
2Laboratory of Foodomics, Institute of Food Science Research (CSIC),Nicolas Cabrera 9, 28049 Madrid, Spain
3NVS Department, KI-Alzheimer’s Disease Research Center, Karolinska Institute,14186 Stockholm, Sweden
In this work, chiral micellar electrokinetic chromatography with laser induced fluorescencedetection (chiral-MEKC-LIF) is used to investigate D- and L-amino acids contents in cere-brospinal fluid (CSF) samples related to different Alzheimer disease (AD) stages. CSFsamples were taken from i) control subjects (S1 pool, n = 33 individuals), ii) subjectsshowing a mild cognitive impairment (MCI) who remained stable (S2 pool, n = 26 indi-viduals), iii) subjects showing a MCI that progressed to AD (S3 pool, n = 13 individuals)and iv) subjects diagnosed with AD (S4 pool, n = 27 individuals). The optimized proce-dure only needs 10 μL of CSF and it includes sample cleaning, derivatization with FITCand chiral-MEKC-LIF separation in a background electrolyte composed of 100 mM sodiumtetraborate, 80 mM SDS and 20 mM β-CD at pH 10.0. The majority amino acids usuallyfound in CSF samples (namely, D- and L-forms of Arg, Lys, Leu, Gln, Ser, Ala, Glu and Asptogether with the non-chiral amino acids gamma-aminobutyric acid (GABA) and Gly)were separated with efficiencies up to 703 000 plates/m, high sensitivity (LODs in the nMrange) and good resolution (values ranging from 2.6 to 9.5). Using this method, L-Arg,L-Leu, L-Gln, GABA, L-Ser, D-Ser, L-Ala, Gly, L-Lys, L-Glu and L-Asp were detected in allthe CSF samples. S3 and S4 samples (i.e., AD subjects) showed significant lower amountsof L-Arg L-Lys, L-Glu and L-Asp compared to the non-Alzheimer S1 and S2 samples, show-ing the S4 group the lowest amounts of L-Arg L-Lys, L-Glu and L-Asp. Moreover, GABAwas significantly higher in AD subjects with the highest amount also found for S4. Nosignificant differences were observed for the rest of amino acids including D-Ser. Based onthe obtained chiral-MEKC-LIF data, it was possible to correctly classify all the samples intothe four groups. These results demonstrate that the use of enantioselective procedures asthe one developed in this work can provide some new light on the investigations of AD,including the discovery of new biomarkers related to different stages of AD.
Acknowledgements: Sh.S. thanks Shota Rustaveli Georgia National Science Foundation for her Young Scien-tists Grant (Project #04/03).

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-060-TU
Capillary Electrophoretic Enantioseparationof Aminonaphthol Analogs
István Ilisz1, Gábor Fodor1, Zoltán Pataj1, István Szatmári2,Ferenc Fülöp2, Lajos Szente3, Antal Péter3
1Department of Inorganic and Analytical Chemistry, University of Szeged,Dóm tér 7, H-6720 Szeged, Hungary
2Institute of Pharmaceutical Chemistry, University of Szeged,Eötvös u. 6, H-6720 Szeged, Hungary
3CycloLab R&D Ltd,Illatos út 7, H-1097 Budapest, Hungary
The synthesis and application of new chiral ligands in asymmetric transformations iscurrently a field of great interest in organic chemistry. In the past decade, modified three-component Mannich reactions, based on the aminoalkylation of 2- or 1-naphthol, havebecome of considerable importance for the formation of a C-C bond under mild experi-mental conditions. The areas of application of these aminonaphthols differ with the na-ture of the amino group; for example the enantiomers of secondary and tertiary amino-naphthols have been widely applied as chiral catalysts in the enantioselective alkylation orarylation of benzaldehyde. Since this asymmetric catalytic activity of aminonaphtholanalogs depends strongly on their stereochemistry, there is a clear need for the elabora-tion of precise separation and identification methods by which their configurations can beassigned.
Among the numerous possible resolving techniques, capillary electrophoresis (CE) is arelatively new technique, which complements the most frequently applied high perfor-mance liquid chromatographic methods. CE is a method of analysis that offers severaladvantages e.g. small sample size, low reagent consumption, fast and efficient separa-tions, outstanding plate numbers and relatively inexpensive analysis for wide variety ofsample types.
The present paper describes direct capillary zone electrophoretic method for theenantioseparation of 1-(phenylethylamino)methyl-2-naphthol analogs or 1-(naphthyl-ethylamino)methyl-2-naphthol analogs containing two chiral centers, with the applicationof different cyclodextrins, as chiral additives. To achieve baseline separation of the stereo-isomers the electrophoretic conditions were optimized. The effects of different parameterson the selectivity, such as the nature of the buffer, the mobile phase composition, pH,concentration of chiral additives were examined and are discussed.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-061-MO
Chiral HPLC Determination of Nabumetone and its Phase IMetabolites In Vitro. Application to Study of Stereospecificity
of Carbonyl Reductases Involved in Nabumetone Biotransformation
Milan Nobilis1,2, Veronika Holmanová2, Barbora Szotáková2,Chamseddin Chamseddin3, Thomas Jira3, Eliška Matoušová2, Jirí Kuneš2, Milan Pour2
1Institute of Experimental Biopharmaceutics, Joint Research Center of PRO. MED. CS Praha a.s.and Academy of Sciences of the Czech Republic,
Heyrovského 1207, CZ-500 03 Hradec Králové, Czech Republic2Charles University, Faculty of Pharmacy,
Heyrovského 1203, CZ-500 03 Hradec Králové, Czech Republic3Ernst-Moritz-Arndt-University, Institute of Pharmacy,
F.-L.-Jahn-Str. 17, D-17489 Greifswald, Germany
For the evaluation of stereospecificity of rat and human cytosolic carbonyl reductases andactivity of microsomal oxidases involved in nabumetone biotransformation, a new chiralhigh-performance liquid chromatographic method was developed and validated. The pre-pared LLE-HPLC-PDA-(FL) method enabled extraction, separation and ultraviolet or fluores-cence detection of prochiral nabumetone and its five phase I metabolites including enantio-mers of two chiral biotransformation products of carbonyl reduction. Methyl ester of napro-xen served as an internal standard. Diethyl ether was used for liquid-liquid extraction (LLE)of biomatrices. Chiralcel OD-R 250 mm × 4.6 mm column with a mobile phase metha-nol-1M NaClO4/HClO4 aqueous solution pH = 3 (75:25, v/v) were employed in isocraticsufficient separation of nine analytes. The whole analysis lasted 55 minutes at the flow rateof 0.5 ml/min. The column effluent was monitored using a photodiode-array detector (scanor single wavelength at λ = 265 nm) and fluorescence detector (for lower concentrations ofanalytes) in tandem. The results of in vitro nabumetone biotransformation in cytosole andmicrosomal fraction of liver cells of two species (rat vs. human) were compared and enantio-meric excess of chiral nabumetone metabolites was evaluated. Stereospecificity in the ratcytosolic carbonyl reductases to nabumetone was observed: enantiomeric excess of (+)-reduced nabumetone vs. (−)-reduced nabumetone was found to be 10% vs. 90%). ChiralHPLC was successfully employed in drug-metabolism-tracking. The proposed chiral HPLC-PDA-FL method complements our previous chromatographic methods developed for thestudy of nabumetone phase I [1] and phase II biotransformation [2].
1. Nobilis M., Kopecký J., Kvetina J., Svoboda Z., Pour M., Kuneš J., Holcapek M., Kolárová L. J. Pharm.Biomed. Anal. Vol. 32/4–5 (2003) 641–656
2. Nobilis M., Holcapek M., Kolárová L., Kopecký J., Kuneš M., Svoboda Z., Kvetina J, J. Chromatogr. A1031/1–2 (2004) 229–236
This work was supported by MSM 0021620822 (MŠMT, Czech Republic).

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-062-TU
Chiral HPLC-FD Method Validation for Determinationof Several Beta-Blockers and Fluoxetine in Biodegradation Assays
Ana Rita Lado Ribeiro1,2, Paula Maria Lima Castro2, Carlos Magalhães Afonso3,Maria Elizabeth Tiritan1,3
1Centro de Investigação em Ciências da Saúde (CICS), Instituto Superior de Ciências da Saúde-Norte;Rua Central de Gandra 1317, 4585-116 Gandra, Portugal
2Centro de Biotecnologia e Química Fina (CBQF), Escola Superior de Biotecnologia,Universidade Católica Portuguesa; Rua Dr. António Bernardino de Almeida, 4200-072 Porto, Portugal
3Centro de Química Medicinal da Universidade do Porto (CEQUIMED-UP),Laboratório de Química Orgânica e Farmacêutica, Departamento de Química,
Faculdade de Farmácia, Universidade do Porto, Rua Aníbal Cunha 164, 4050-047 Porto, Portugal
Chiral pharmaceuticals and the fate and effects of their enantiomers in the environment arestill largely unknown [1, 2]. Enantiomers have different interactions with enzymes, receptorsand any chiral molecules leading to different biological activities and affecting organisms ina different manner. Thus, biodegradation tends to be enantioselective in contrast to abioticdegradation. The methods developed to quantify the enantiomeric fraction in the environ-ment and to follow biodegradation are scarce [3]. Thus in this work we describe the devel-opment and validation of HPLC methods that allow the enantiomeric separation of widelyused drugs namely four beta-blockers: alprenolol (ALP), propranolol (PHO), metoprolol(MET) and atenolol (ATE) and the antidepressant fluoxetine (FX). The macrocyclic antibioticvancomycin CSP (ASTEC Chirobiotic V 5 μm) was used under polar organic mode (meth-anol:ethanol:triethylamine:acetic acid.50:50 v/v) and fluorescence detection for enantio-meric fraction quantification. The developed methods were established using a minimalmedium inoculated with activated sludge as a matrix which is the condition used in thebiodegradation studies. The vancomycin CSP was able to resolve ALP and PHO as well asMET, ATE and FX in two chromatographic runs. The chromatographic parameters obtainedhave shown the separation factor (α) between 1.12 and 1.34 and resolution (Rs) between1.30 and 4.35. The methods demonstrated to be selective and linearity with r2 higher than0.999 for the range selected. The method detection limits were between 2.5 to 10 ng/mL.These methods were applied to follow the biodegradation of the target chiral compoundsduring 15 days. The biodegradation assays were performed using activated sludge from aWWTP and the results indicate the higher degradation extents for the S-enantiomer forms.
1. Kümmerer, K., J Environ Manage 90, 2354–2366 (2009).2. Stanley, J. K and Brooks, B. W., Integr Environ Assess Manag 5: 3, 364–373 (2009).3. Hashim, N.H., Shafie, S. and Khan, S.J., EnvironTechnol 31:12, 1349–1370 (2010).
Ackowledgments: A.R. Ribeiro acknowledges FCT, Portugal for the grant (SFRH/BD/64999/2009), QREN-POPH, European Social Fund and MCTES.This work was also financially supported by CESPU (09-GCQF-CICS-09) and PTDC/EBBEBI/111699/2009

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-063-MO
Chiral Separation of Binaphthyl Catalysts using New HPLC ChiralStationary Phases Based on Derivatized Cyclofructans
Lucie Janecková1, Kveta Kalíková2, Jirí Vozka2, Zuzana Bosáková1,Daniel W. Armstrong3, Eva Tesarová2
1Department of Analytical Chemistry, Faculty of Science, Charles University in Prague,128 43 Prague 2, Albertov 2030, Czech Republic
2Department of Physical and Macromolecular Chemistry, Faculty of Science, Charles University in Prague,128 43 Prague 2, Albertov 2030, Czech Republic
3Department of Chemistry and Biochemistry, University of Texas at Arlington,Box 19065, Arlington, TX 76019-0065, USA
A new class of chiral selectors, cyclofructans (CFs), have been recently introduced forapplication in separation techniques such as liquid chromatography and capillary electro-phoresis. Native cyclofructans have rather limited capabilities as chiral selectors, butaliphatic- and aromatic-functionalized CFs possess interesting enantioseparation possibili-ties for a variety of compounds.
In this work, three different CF-based chiral stationary phases were used for chiralseparation of binaphthyl derivatives. The employed columns differ in the derivatizationgroup and/or the size of CF cavity, i.e. isopropyl-CF6 (IP-CF6), R-naphthylethyl-CF6 (RN-CF6) and dimethylphenyl-CF7 (DMP-CF7) were used. Binaphthyl derivatives as an inter-esting group of analytes perform axial chirality due to restricted rotation around thesingle bond in the binaphthyl skeleton. They have been widely used to control asymmet-ric processes. The substituents and their position in the binaphthyl skeleton significantlyaffect their properties. The chromatographic behavior of binaphthyl derivatives was stud-ied in the normal phase mode, the mobile phases were composed of n-hexane and pro-pane-2-ol in various volume ratios and the addition of TFA was also tested. The CF-basedCSPs performed good separation capabilities and some excellent enantioseparations ofbinaphthyl derivatives were achieved.
Acknowledgements: The Grant Agency of the Charles University in Prague, projects No. 356411 and SVV261204, the Ministry of Education, Youth and Sports of the Czech Republic, projects Kontakt LH11018,Centrum 1M06011, RP 14/63 and the long-term project MSM0021620857 are gratefully acknowledged forthe financial support.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-064-TU
Chiral Separation of Citalopram by Capillary Electrophoresis
Xiaolan Deng, Erwin Adams, Ann Van SchepdaelLaboratory for Pharmaceutical Analysis, Faculty of Pharmaceutical Sciences,
Katholieke Universiteit Leuven,Leuven, Belgium
Enantiomeric purity control is critical issue in the pharmaceutical industry especially sincemore and more drugs are used as single-isomer drugs instead of the racemic drugs, whichcould improve the efficacy, biological activity, and potency of the drug. The S-enantiomerof citalopram, is used for the treatment of major depressive disorder. In contrast to theracemic drug citalopram, escitalopram shows greater efficacy and faster onset of action.
A rapid capillary zone electrophoresis method has been developed capable of quantify-ing 0.05% of R-enantiomer and assaying the main component in escitalopram tablets.Many chemical and instrumental parameters influencing enantioseparation were investi-gated, which include chiral selectors, buffer composition and pH, applied voltage, capil-lary length and temperature. Optimal separation conditions were obtained by using a 20mM phosphate buffer adjusted to pH 7.0, containing 1.6% (w/v) sulfated-β-cyclodextrinin combination with short end injection. Separation was achieved in less than 2.5 min.After a wide range validation (stability of the solutions, selectivity, linearity, precision,accuracy), the proposed method was then applied to escitalopram commercially availabletablets, to quantify the main component to determine and enantiomeric impurity in thetablets.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-065-MO
Chiral Separation of Clinically Important Compounds using CE– Determination of the Most Effective Chiral Selector
Christoforos A. Hadjistasi, Constantina P. Kapnissi-ChristodoulouUniversity of Cyprus,
P.O. Box 20537, 1678, Nicosia, Cyprus
The goal of this research is the development of electrophoretic methods for improvedchiral separation of analytes that are extremely important in clinical analysis. Chiral com-pounds are common in several synthetic and biological systems. It has been shown thatusually only one enantiomer is active, while the other might be less active, inactive or hasdamaging side effects. Fucose is a rare carbohydrate essential for proper cell-to-cell com-munication. Fucose can be found in the skin, brain cells, kidneys, retina of the eye, and inthe breast milk. In addition, it has a dynamic role in the immune system and in the redblood cell function. Pipecolic acid is a human metabolite that is found in biofluids. Inhuman blood stream, the L-enantiomer is mostly present, while the D-enantiomer isprimarily excreted. It has been shown that the D-enantiomer originates from metabolismof intestinal bacteria and from dietary sources. Even though high levels od D-pipecolicacid are not found in plasma of a normal adult, they are significantly increased in urine ofpeople with chronic liver disease. Therefore, the separation of the (−) form from the (+)for the compounds mentioned above is of greatest importance. During recent years thepopularity of CE has increased markedly due to its short analysis time, excellent separa-tion efficiency, small sample and solvent volumes, simple instrumentation and variousseparation modes. In this study, micellar electrokinetic chromatography (MEKC) andcyclodextrin electrokinetic chromatography (CD-EKC) were used for the chiral separationof fucose and pipecolic acid. The optimum conditions were obtained by investigatingdifferent parameters and particularly the type of the chiral selector. Different chiral selec-tors were used as additives in the mobile phase, such as polymeric surfactants and cyclo-dextrins. The most effective chiral selector was determined in regards to resolution, analy-sis time and reproducibility.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-066-TU
Chiral Separation of Huperzine A using CE– Method Validation and Application in Pharmaceutical Formulations
Despina Tsioupi, Constantina P. Kapnissi-ChristodoulouUniversity of Cyprus,
P.O.Box 20537, 1678, Nicosia, Cyprus
The aim of this work is the development, validation and application of a MEKC methodfor the chiral separation of Huperzine A. Huperzine A is an important compound that isused to treat Alzheimer’s disease. However, only the (−) form of this compound is biologi-cally active and behaves as a potential acetylcholinesterase inhibitor. Therefore, the sepa-ration of the (−) form from the (+) form is of greatest importance. Optimal conditions, inregards to resolution and analysis time, were established by altering several experimentalparameters such as temperature, field strength, pH and type and concentration of back-ground electrolyte (BGE) and chiral selector. A major problem that had to be solved inthis study was the low intensity and efficiency of the peaks. More parameters were exam-ined, such as the addition of modifiers, in order to optimize further the separation inregards of efficiency. Baseline enantioseparation of the racemic compound was achievedby applying a 20 kV voltage across the fused-silica capillary that was filled with a BGEconsisting of a 50 mM acetate buffer at pH 5.0, supplemented with 0.2% (w/v)poly(sodium N-undecanoyl LL-alanyl-valinate) and 10% (v/v) tert-butanol at 25 °C. Themethod was then validated in terms of reproducibility, linearity, LOD and LOQ. Finally,the optimum conditions mentioned above were applied to a pharmaceutical formulation,in order to demonstrate the ability of the method to control the purity of the (−)-Huper-zine A in pharmaceutical formulations.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-067-MO
Chiral Separations of Un-Derivatized Peptideson Chiral Zwitterionic Stationary Phases
Stefanie Wernisch, Reinhard Pell, Wolfgang LindnerInstitute for Analytical Chemistry, University of Vienna,
Währinger Straße 38, A-1090 Vienna, Austria
Recently developed zwitterionic chiral stationary phases (ZWIX CSPs) obtained through thefusion of Cinchona alkaloids (weak anion exchangers, WAX) and aminosulfonic acids (strongcation exchangers, SCX) and their subsequent immobilization onto porous silica support canbe used in the HPLC enantiomer separation of free (un-derivatised) peptides. Their mostfavourable characteristics are straightforward application and comparatively short retentiontimes. An intramolecular counterion effect (IMCI) of the selectors’ acidic and basic ionexchanging sites on the respective carboxylic and amino groups of the peptides allows us tominimize the amount of buffer salts required for the elution of the charged analytes [1].
We chose homologous series of small all-L and all-D peptides (< 10 amino acid resi-dues) based on alanine, valine and phenylalanine to investigate structure-enantioselect-ivity relationships of a total of seven related zwitterionic chiral stationary phases. The CSPset included Quinine- and pseudo-enantiomeric Quindine-based selectors which offer thepossibility of reversed elution orders [2] and, in addition, increasing intramolecular dis-tance between the SCX and WAX sites introduced by homologous aminosulfonic acid sidechains (aminoethane, aminopropane- and aminobutanesulfonic acid). To complete theCSP set, we included a ZWIX CSP with a chiral center close to the sulfonic acid group(aminohexanesulfonic acid) which exhibits increased enantiomer selectivity for free aminoacids compared to its achiral sulfonic acid analogue.
We investigated the chromatographic performance of these CSPs for peptide enantiomerseparations in the polar-organic mobile phase mode and were able to show that selectivityand resolution increase with the bulkiness of the peptide side chain (Ala-n < Val-n < Phe-n)and with the length of the spacer group between WAX and SCX site of the selector.
In addition, we investigated the peculiarity of Pro-n (n = 1–4), X-Pro and Pro-X peptides,since the secondary amino acid proline (Pro) is of special interest in biological systems due toits rigid structure and its ability to form almost equally stable cis and trans isomers of thepeptide bond [3]. Thus, poly-L-proline motifs of a peptide can form both left- (3 amino acidsper turn) and right-handed helices (3.3 residues per turn) [4]. Therefore, we were especiallyinterested in the chiral separation of proline-containing peptides and report the outcome of aseries of experiments performed with various stereoisomer mixtures on ZWIX CSPs.
1. Christian V. Hoffmann et al., Analytical Chemistry 80, 22 (2008): 8780–8789.2. Michael Lämmerhofer et al., Journal of Chromatography A 741, 1 (1996): 33–48.3. John F. Brandts, et al., Biochemistry 14, 22 (1975): 4953–4963.4. Alexei A. Adzhubei et al., Journal of Molecular Biology 229, 2 (1993): 472–493.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-068-TU
Chiral Stationary Phases Based on Derivatized Cyclofructanfor Chiral HPLC Separation
Kveta Kalíková1, Lucie Janecková2, Radim Geryk2,Daniel W. Armstrong3, Eva Tesarová1
1Department of Physical and Macromolecular Chemistry, Faculty of Science, Charles University in Prague,128 43 Prague 2, Albertov 2030, Czech Republic
2Department of Analytical Chemistry, Faculty of Science, Charles University in Prague,128 43 Prague 2, Albertov 2030, Czech Republic
3Department of Chemistry and Biochemistry, University of Texas at Arlington,Box 19065, Arlington, TX 76019-0065, USA
Derivatized cyclofructans (CFs) have been recently introduced as a new class of chiralselectors with great application potential [1, 2]. In principle CF-based chiral stationaryphases (CSPs) can be used in all three modes (normal, reversed phase and polar organic),which brings many advantages.
In this work we used three CF-based CSPs that differ in the type of the derivatizationgroups and also the number of fructofuranose units in the macrocyclic ring. RN-CF6(contains R-naphthylethyl carbamate group), IP-CF6 (isopropyl carbamate modified CF6)and DMP-CF7 CSPs (dimethylphenyl carbamate functionalized CF7), where CF6 and CF7means six and seven fructofuranose units, respectively, were evaluated in the term ofinteraction possibilities in the normal phase mode. The applied mobile phases were com-posed of n-hexane/propane-2-ol 80/20 (v/v) and n-hexane/propane-2-ol/trifluoroaceticacid 80/20/0.5 (v/v/v). Dominant interaction types that play a role in the separationmechanism were revealed by a linear free energy relationship method (LFER). Further-more, different enantioselectivities of these CSPs were evaluated by injection of differentchiral pharmaceuticals to the chromatographic systems.
1. P. Sun, C. Wang, Z.S. Breitbach, Y. Zhang, D.W. Armstrong, Anal. Chem. 81 (2009) 10215.2. P. Sun, D.W. Armstrong, J. Chromatogr. A 1217 (2010) 4904.
We gratefully acknowledge financial support of the KONTAKTAM2010, project no. LH11018, Grant Agency ofthe Czech Republic, grant no. 203/08/1428, Grant Agency of the Charles University, grant no. 356411 andlong-term research plan of the Ministry of Education, Youth and Sports of the Czech Republic, MSM0021620857.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-069-MO
Combination of Capillary Electrophoresis, Molecular Modellingand Nuclear Magnetic Resonance to Study the Interaction
Mechanisms between Single-Isomer Anionic CD Derivativesand Basic Drug Enantiomers in a Methanolic Background Electrolyte
Anne-Catherine Servais1, Anne Rousseau1, Georges Dive2, Michel Frederich3,Jacques Crommen1, Marianne Fillet1
1Laboratory of Analytical Pharmaceutical Chemistry, Dept. of Pharmaceutical Sciences, CIRM,University of Liege,
CHU, B36, B-4000 Liege, Belgium2Center for Protein Engineering, University of Liege,
B6a, B-4000 Liege, Belgium3Laboratory of Pharmacognosy, Dept. of Pharmaceutical Sciences, CIRM, University of Liege,
CHU, B36, B-4000 Liege, Belgium
It is now well-established that nonaqueous capillary electrophoresis (NACE) is a very powerfultool for enantioseparations [1]. NA electrolytes enable to use chiral selectors and analytes witha low solubility in water, to reduce adsorption onto the capillary wall and to generate reducedelectric current. Moreover, organic solvents with lower dielectric constants than water consti-tute, in principle, a more favourable environment for chiral discrimination due to their abilityto promote intermolecular interactions [2]. If the analytical interest of CDs is obvious, theirenantiomer recognition pattern in NA systems is still not fully understood. In order to improveour knowledge of the mechanisms of enantiomer recognition pattern in nonaqueous systems,an approach combining NACE, molecular modelling and nuclear magnetic resonance (NMR)was undertaken. Bupivacaine and propranolol were selected as model compounds and theirinteractions with two single-isomer highly charged β-CD derivatives, namely heptakis(2,3-di-O-methyl-6-O-sulfo)-β-CD (HDMS-β-CD) and heptakis(2,3-di-O-acetyl-6-O-sulfo)-β-CD (HDAS-β-CD), were studied. In the case of propranolol, NMR experiments suggested that externalcomplexes were formed in the methanolic BGE containing HDMS-β-CD while in the presenceof HDAS-β-CD, the alkyl chain of R-propranolol was assumed to enter into the CD macrocyclethrough the wider opening [3]. The structures of the CD-bupivacaine complexes were evaluat-ed by 2-D Rotating-frame Overhauser Effect SpectroscopY (ROESY) experiments. From theseexperiments, it can be assumed that inclusion complexes are not formed, whatever the CDderivative used. Molecular modelling was performed at the RHF/MINI-1 or B3LYP/6-31G(d)level. External as well as inclusion type complexes with the alkyl chain of propranolol into theCD cavity were located. Interaction energies calculated for bupivacaine and propranololcorrelated with the enantiomer migration order observed in the NACE experiments using bothanionic CD derivatives.
1. M. Lämmerhofer, J. Chromatogr. A 1068 (2005) 3.2. A. Rizzi, Electrophoresis 22 (2001) 3079.3. A.-C. Servais, A. Rousseau, M. Fillet, K. Lomsadze, A. Salgado, J. Crommen, B. Chankvetadze, Electrophore-
sis 31 (2010) 1467.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-070-TU
Comparison of the Charge State Distributionin Commercially Available Sulfated Cyclodextrins
used as Chiral Resolving Agents in CE
Roy Estrada1,2, Gyula Vigh1
1Chemistry Department, Texas A&M University,College Station, TX 77842, USA
2TX Molecular LLP,Deer Park, TX 77536, USA
Sulfated cyclodextrins are one of the most commonly used chiral resolving agents incapillary electrophoresis. Most of the commercially available sulfated cyclodextrins arerandomly sulfated and are complex mixtures of homologues of varying charge states witha distribution of isomers within each charge state. Each isomer has its own electrophoreticand complexation behavior and when present in a mixture, the different isomers mayhave an antagonistic effect on the separation of an enantiomeric pair. Previous attemptshave failed to unequivocally characterize the charge state distribution of sulfated cyclo-dextrins, especially those with higher degrees of sulfation.
The charge state distributions of randomly sulfated cyclodextrins purchased fromAldrich and Beckman Coulter as well as single isomer sulfated cyclodextrins purchasedfrom TM Chemicals LP were investigated using Hydrophilic Liquid Interaction Chromatog-raphy (HILIC). Groups of different charge states were resolved for the highly sulfatedspecies and isomers in each charge group were partially separated. The charge statedistribution is wide for the randomly sulfated cyclodextrins while the single isomer sulfat-ed cyclodextrins yielded single and well defined charge state groups.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-071-MO
Cyclodextrin Based Cation-Exchanger Chiral Columns
Julianna Szemán1, Júlia Visy2, Éva Jámbor3, Gábor Fodor4,Róbert Ohmacht3, Gábor Varga5
1CycloLab R&D Ltd,Illatos út 7, Budapest, H-1097, Hungary
2Institute of Biomolecular Chemistry, Chemical Research Center, Hungarian Academy of Sciences,Budapest, H-1525 P.O.B. 17, Hungary
3Institute of Biochemistry, Department of Biochemistry and Medical Chemistry, University of Pécs,Szigeti u. 12, Pécs, H-7624, Hungary
4Department of Inorganic and Analytical Chemistry, University of Szeged,Dóm tér 7, Szeged, H-6720, Hungary
5ChiroQuest Ltd,Rumbach S. 7, 1/23, Budapest, H-1075, Hungary
Novel cation-exchanger cyclodextrin (CD) bonded stationary phases have been preparedby a one-step chemical process. The silylating agents containing the selectors have beenprepared and reacted with bare silica. As these special silylating agents do not containisomers in the spacer part, the resulted coverage is well defined, energetically homoge-neous, and free of non-specific interactions.
Our aim was to develop a robust and reproducible production procedure. The qualityof the applied carboxymethylated β-cyclodextrin reagents was also thoroughly studied byusing different techniques (capillary electrophoresis, HPLC), because a slight difference inthe degree and pattern of substitution of the CD reagent can cause significant differencein the efficiency of chiral separation. The influence of the derivatization procedure on theproperties of the phases has been studied. The changes of the pore structure duringderivatization, variation of surface coverage and ion-exchange capacity of the phases bydifferent reaction conditions have been investigated.
The prepared carboxymethylated β-cyclodextrin bonded stationary phase proved to be veryeffective for enantioseparation of basic drug substances. Using this new stationary phase thecombined effect of inclusion complex formation and cation exchange results in more selectiveinteractions between the analyte and the bound ionic CD selector compared to the CD bond-ed stationary phases having no ionic groups. Although, the inclusion interaction becomesweaker at high organic solvent content of the mobile phase, the retentive ability and theselectivity of this stationary phase are preserved due to the ionic forces, which is a significantadvantage over the common CD phases. For example, high resolution value, Rs = 7.0 couldbe achieved between the peaks of 2R,3S and 2S,3R Voriconazole; and Rs = 4.5 was found incase of Chloropheniramine enantiomers on a 250x4 mm analytical column. Moreover, thesemi-preparative separation of these compounds was also efficiently fulfilled.
Acknowledgement: This work was financially supported by Jedlik Ányos grant 00180/2007,NKFP_07_A3_NATURSEP.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-072-TU
Direct Chiral Determination of Salbutamol, Salmeterol and Atenololby Two-Dimensional (LC-LC) High Performance
Liquid Chromatography
Yun Yang1,2, Noelia Rosales-Conrado1, Vanesa Guillén-Casla1,María Eugenia de León-González1, Luis Vicente Pérez-Arribas1,
Luis María Polo-Díez1
1Department of Analytical Chemistry, Faculty of Chemistry, Complutense University of Madrid,E-28040 Madrid, Spain
2Institute of Modern Separation Science, Northwest University,Shaanxi Key Laboratory of Modern Separation Science,
Key Laboratory of Synthetic and Natural Functional Molecule Chemistry of Ministry of Education,Xi’an 710069, China
β2-Adrenoceptor agonists such as salbutamol and salmeterol are used in the treatment ofasthma. These drugs are marketed as racemic mixtures although only the R-(−)-enantiomers are pharmacologically active. The β2-agonist activity of the R-(−)-enantiomersis ca. 40 times greater than that of its S-(+)-enantiomer. (R)-salbutamol and(S)-salbutamol cause smooth muscle to relax and contract, respectively; the two isomersact on different receptors and thus on different pathways resulting in the opposing ef-fects. Atenolol belongs to commonly known group of β-blockers and it is used to treathypertension, sinus tachycardia, arrhythmias, coronary heart disease and myocardialinfarction where it acts preferentially upon the β-adrenergic receptors in the heart.(S)-enantiomer possesses much greater affinity for binding to the β-adrenergic receptorsthan the (R)-antipode. In spite of the fact that nearly complete therapeutic activity residesin (S)-enantiomers, most of the β-blockers are also marketed as racemic mixtures andapplied in therapy as such. The two enantiomers should be considered as different drugsas there are differences in their stereoselective mechanism.
Macrocyclic antibiotics, in particular vancomycin, have been extensively used as chiralstationary phases (CSPs) for adrenoreceptor agonist and antagonist. These CSPs havedemonstrated a broad selectivity in reversed phase, normal phase, polar ionic and polarorganic modes. A common problem of CSPs is their intrinsically limited chemical selectivi-ty and efficiency. This means that complex mixtures containing diverse enantiomer pairsare difficult to analyze only in one run due to peak overlapping. This problem could beovercome by heart cut two dimensional HPLC.
Reversed phase LC (RPLC) is not often the method of choice for the analysis of com-pounds having high polarity. Hydrophilic interaction chromatography (HILIC) has beengaining interest as an alternative to RPLC for analysis of polar and hydrophilic analytes asit could be the case of salbutamol, salmeterol and atenolol.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
In this work, racemic of target compounds were previously separated in a primarycolumn (KinetexTM HILIC 2.6 μm, 150 x 2.1 mm) using a mixture of MeOH:ACN:ammo-nium acetate buffer (5 mM, pH 6) 90:5:5 (v/v/v) as mobile phase at a flow rate of 0.40mL min−1. Enantiomeric separations were carried out by transferring part of the respectiveracemic peaks through a switching valve to a vancomycin chiral column (ChirobioticTM V2.6 μm, 150 x 2.1 mm) using MeOH:ammonium acetate buffer (2 mM, pH 4) 97:3 (v/v)as mobile phase at a flow of 0.5 mL min−1. UV detection was done at 227 nm in both LCdimensions.
Detection limits were between 0.12 and 0.24 μg mL−1 for salbutamol and salmeterolenantiomers, respectively. Linearity ranges were in the concentration range 0.5–10mg L−1. Intra-day and inter-day reproducibility for the whole LC-LC system, expressed asrelative standard deviation (RSD), were between 4.8 and 16%.
Y. Yang thanks the Connec (“Connecting Europe and China through Interuniversity Exchange”) programmefor financial support.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-074-TU
Enantioselective 2D-HPLC Determination of N-Methylaspartic Acidand N-Methylglutamic Acid in Mammals and Bivalves
Reiko Koga1, Yurika Miyoshi1, Kyoko Ueno1, Masashi Mita2,Wolfgang Lindner3, Kenji Hamase1
1Graduate School of Pharmaceutical Sciences, Kyushu University,Fukuoka 812-8582, Japan
2Shiseido Co., Ltd.,Tokyo 105-0021, Japan
3Institute of Analytical Chemistry, University of Vienna,A-1090 Vienna, Austria
By the progress of analytical technologies, several free D-amino acids, the enantiomersof L-amino acids, have been found in mammals including human beings. Their distribu-tions and functions have also been elucidated and these D-amino acids are now expect-ed as candidates of novel biomarkers and physiologically active substances. N-Methyl-D-aspartic acid (NMDA) is one of the synthetic D-amino acid analogues and it is wellknown as a potent agonist of NMDA subtype of the glutamate receptors. In addition toits role as a neurotransmitter, NMDA has also been reported to regulate hormonalsecretion, such as prolactin and luteinizing hormone. However, the presence of natural-ly occurring NMDA in various living organisms, tissue distribution and metabolism arestill unclear, and their clarification is highly expected. In the present study, we haveestablished a 2D-HPLC system for the analysis of N-methylaspartic acid (NMA) andN-methylglutamic acid (NMG) enantiomers, and intrinsic amounts of NMDA analogueswere determined in the tissues and physiological fluids of mammals and bivalves. Theamino acids in the biological samples were derivatized with 4-fluoro-7-nitro-2,1,3-benzoxadiazole (NBD-F) and their NBD-derivatives were detected at 530 nm with exci-tation at 470 nm. In the present 2D-HPLC system, a microbore-monolithic ODS column(0.53 mm i.d. x 1000 mm) was used to separate NBD-derivatives of NMA and NMGfrom impurities in the first dimension, and these fractions were automatically introducedto an enantioselective column as the second dimension. For the enantiomer separationsof NMA and NMG, we tested various Pirkle type and anion exchange type enantio-selective columns (Sumichiral OA-2500S, OA-2500R, OA-3100S, OA-3200S and OA-3300S, 1.5 mm i.d. x 250 mm, having amino acid derivatives, and Chiralpak QN-1-AX,QN-2-AX, QD-1-AX and QD-2-AX, 1.5 mm i.d. x 150 mm, having quinine and quinidinederivatives as the chiral selectors). As a result, separations of both NMA and NMG wereobtained by Sumichiral OA-2500S, OA-2500R and Chiralpak QN-2-AX columns. There-fore, a 2D-HPLC system has been constructed by combining the microbore-monolithicODS column and these enantioselective columns, and was applied to the plasma andcerebral cortex of rat, and to the mantle and foot of S. broughtonii. In the rat plasma

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
and cerebral cortex, the amounts of 4 enantiomers were trace, on the other hand,relatively high amounts of both NMA and NMG enantiomers were found in the S.broughtonii. The present 2D-HPLC system is suitable for the studies to elucidate thedetailed distributions and metabolism of NMA and NMG enantiomers, and further studyusing various living organisms is in progress.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-075-MO
Enantioseparation of Amino Acids by Molecularly ImprintedElectrochromatography with Electrochemical Detection
Fengjun Shang1 John, H. T. Loung,2 Mila Pravda,1 Jeremy D. Glennon1
1Innovative Chromatography Group, Irish Separation Science Cluster (ISSC),Department of Chemistry and the Analytical and Biological Chemistry Research Facility (ABCRF),
University College Cork,Cork, Ireland
2Biotechnology Research Institute, National Research Council Canada, Montreal,Quebec, H4P2R2, Canada
Molecular imprinting technology offers the unique opportunity to tailor chiral stationaryphases with predefined chiral recognition properties by employing the enantiomers ofinterest as binding-site-forming templates. Compared with conventional chiral selectors,molecularly imprinted polymer (MIPs) have several advantages such as ease of prepara-tion, scalability, chemical robustness, low-cost production, and flexibility to design variousself-supporting formats [1]. In this study, MIP-coated capillaries were prepared by in situmolecular imprinting using acrylamide (AM) as a functional monomer and ethylene glycoldimethacrylate (EDMA) as a cross-linker. A boron-doped diamond (BDD) electrode, fea-turing a high current density, wide potential window, low background current, extremeelectrochemical stability and high resistance to fouling, was employed as a sensitiveamperometric detector [2]. The effects of the buffer pH, buffer concentration, bufferadditives, separation voltage and detection potential on the enantioseparation of trypto-phan were investigated.
1. Maier, N. M.; Lindner, W. Anal. Bioanal. Chem. 2007, 389, 377-397.2. Shang, F., Zhou, L.; Mahmoud, K. A.; Hrapovic, S.; Liu, Y.; Moynihan, H. A.; Glennon, J. D.; Luong, J. H. T.
Anal. Chem. 2009, 81, 4089-4098.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-076-TU
High Performance Chiral Columns for Efficient Enantioseparation
Yong Wang, Feng Ai, Siu-Choon Ng, Timothy T.Y. TanSchool of Chemical and Biomedical Engineering, Nanyang Technological University,
62 Nanyang Drive, 637459, Singapore
The current talk focuses on the design and fabrication of high performance chiral col-umns, and the demonstration of their enhanced enantioseparation efficiency. Two ap-proaches were adopted by our group in designing these columns: 1. Fabrication of“small” CSP materials for improved separation efficiency, and 2. Formulation of newstrategy for immobilization of cyclodextrin moeities on silica particles for enhanced stabili-ty of CSP. Sub-1-micron mesoporous silica particles were prepared as CSP and their col-umns could be operated at a relative low back-pressure (< 8000 psi) at a high flow rate of2.0 ml/min when used in ultra-high pressure liquid chromatography. The CSP also exhib-ited rapid chiral separation (within 10 min) and good resolution of 6 different neutral andbasic drugs. In another aspect, our group has devised a simple and effective protocol forthe preparation of 5- and 3-micron native β-cyclodextrin (β-CD) chiral stationary phasesusing “click” chemistry. Compared to existing methods, the current approach can achieveregioselective immobilization of specific sites on the CD rims and therefore ensure batch-to-batch reproducibility. The column was subsequently used for the enantioseparation ofchiral drugs in high performance liquid chromatography and the 3-micron columnshowed better separation efficiency compared to that of 5-micron column.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-077-MO
High Throughput Chiral Screening:A Comparison between LC and SFC
Melissa Dunkle1, Gerd Vanhoenacker1, Alberto dos Santos Pereira1,William Farrell2, Frank David1, Pat Sandra1
1Research Institute for Chromatography,Kennedypark 26, 8500 Kortrijk, Belgium
2Pfizer Global R&D,La Jolla, California, USA
Chiral separations are one of the most prevalent applications of packed column Super-critical Fluid Chromatography (SFC). It has replaced chiral HPLC analysis in many industri-al applications due to the fact that SFC is considered a green technique, it is generallyfaster than HPLC, and there is less solvent usage and waste generation. However, it isknown that in some cases, chiral HPLC analysis can give a better separation than chiralSFC. In this work, generic chiral high throughput screening methods were developed forboth HPLC and SFC, and the results obtained from the two techniques were compared.Different chiral stationary phases were used for the screening method, and automatedcolumn selection was performed. 50 different randomly selected chiral samples wereanalyzed using both the SFC and HPLC screening methods. The features of both tech-niques in terms of speed and selectivity will be presented.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-078-TU
Highly Sensitive Chiral Analysis in Microscale Electrophoresisusing Large-Volume Sample Stacking with Electroosmotic Flow Pump
Takayuki Kawai, Jun Ito, Kenji Sueyoshi, Fumihiko Kitagawa, Koji OtsukaDepartment of Material Chemistry, Graduate School of Engineering, Kyoto University
A3-222 Katsura, Nishikyo-ku, Kyoto 615-8510, Japan
In conventional chiral analysis by capillary/microchip electrophoresis, the poor sensitivityis one of major problems. To overcome its disadvantage, several online preconcentrationtechniques have been developed. However, these techniques usually reduce the effectiveseparation length due to the large-volume sample injection, resulting in an insufficientenantioseparation. On the other hand, large-volume sample stacking with electroosmoticflow (EOF) pump (LVSEP) [1] provides an efficient preconcentration without loss of resolu-tion, which is suitable for highly sensitive chiral analysis. In this study, we applied LVSEPto cyclodextrin (CD)-modified capillary zone electrophoresis (CD-CZE) and CD-modifiedmicellar electrokinetic chromatography (CD-MEKC) to achieve high enantio-resolutionand high detectability.
In the LVSEP analysis, an EOF-suppressed capillary is filled with a low ionic strength (I)solution containing anionic analytes. After the inlet and outlet vials are filled with a highI background solution (BGS) containing a chiral selector, a constant voltage is applied.The analytes are concentrated around the anodic-side sample/BGS boundary according tothe difference in the electric field strength between the two zones. The focused analytesmove toward the cathode and BGS is introduced into the capillary by a temporarily en-hanced EOF in the low I sample solution. After the low I solution without the analytes isremoved out of the capillary, the EOF is suppressed again due to the high I BGS. Theanalytes start moving toward the anode and are separated by CD-CZE or CD-MEKC.
The LVSEP-CD-CZE analysis of racemic warfarin was performed with 2,6-di-O-methyl-β-CD as a chiral selector. The racemates were completely separated with a 1000-fold sensi-tivity increase without loss of resolution compared with the conventional CD-CZE analysis.The LVSEP-CD-MEKC analysis of racemic amino acids was also carried out by using γ-CDand sodium dodecyl sulfate as a chiral selector and a surfactant, respectively. As a typicalresult, Arg, Asn, Met, and Leu enantiomers were well optically resolved with up to a1200-fold sensitivity improvement in contrast with conventional CD-MEKC. These resultsindicate the good applicability of LVSEP to several separation modes in microscale electro-phoresis.
1. Kawai, T. et al. Anal. Chem. 2010, 82, 6504–6511.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-079-MO
High-Performance Liquid Chromatographic Enantioseparationof Isoxazoline-Fused Cispentacin Derivatives
on Chiral Stationary Phases
László Sipos2, Melinda Nonn2, Ferenc Fülöp2, Daniel W. Armstrong3, Antal Péter1
1Department of Inorganic and Analytical Chemistry, University of Szeged,Dóm tér 7, H-6720 Szeged, Hungary
2Institute of Pharmaceutical Chemistry, University of Szeged,Eötvös utca 6, H-6720 Szeged, Hungary
3Department of Chemistry and Biochemistry, University of Texas at Arlington,Arlington, TX 76019-0065, USA
β-Amino acids and their foldameric oligomers are currently at the focus of research inter-est, because they are important constituents of natural products such as alkaloids,peptides and β-lactam antibiotics. β-amino acids can be used as building blocks for thepreparation of modified (unnatural) analogs of biologically active peptides. More recently,a novel class of β-peptide analogues adopting predictible and reproducible folding pat-terns is being evaluated as a potential source of new drugs and catalysts. As a result of thewide-ranging utility of these compounds, much attention has been paid to their enantio-selective synthesis, which requires analytical methods for control of the enantiopurity ofthe final products.
High-performance liquid chromatography (HPLC) is one of the most useful techniquesfor separation of stereoisomers, including enantiomers. A number of review articles andbooks deal with the methods and results of the direct enantioseparation of various com-pounds on chiral stationary phases (CSPs) and many attempts have been made to inter-pret how these CSPs operate with respect to molecular recognition.
The enantiomers of eight isoxazoline-fused 2-aminocyclopentanecarboxylic acids weredirectly separated on macrocyclic glycopeptide antibiotic-based CSPs such as teicoplanin(Astec Chirobiotic T and T2), teicoplanin aglycone (Chirobiotic TAG), vancomycin (Chiro-biotic V) and vancomycin aglycone (Chirobiotic VAG) as chiral selectors. The mobilephase composition – reversed-phase mode (0.1% TEAA (pH = 4.1)/MeOH), polar-organicmode (100% MeOH) and polar-ionic mode (MeOH/AcOH/TEA) – effect of mobile phaseadditieves and structure of the analyte on the separation were investigated. The enantio-and diastereoselectivity of macrocyclic glycopeptide antibiotic-based CSPs were discussed.The sequence of elution of the enantiomers was determined in all cases.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-080-TU
HPLC Enantioseparation and a Temperature-Induced Inversionof the Elution Sequence of 1-(Phenylethylamino)-
or 1-(Naphthylethylamino)methyl-2-Naphthol Analogs
Anita Aranyi1, István Ilisz1, Zoltán Pataj1, István Szatmári1,Ferenc Fülöp1, Antal Péter1
1Department of Inorganic and Analytical Chemistry, University of Szeged,Dóm tér 7, H-6720 Szeged, Hungary
2Institute of Pharmaceutical Chemistry, University of Szeged,Eötvös u. 6, H-6720 Szeged, Hungary
The search for new chiral ligands which can be efficiently applied in biology and asym-metric catalysis is currently a field of great interest in organic chemistry. The asymmetricaddition of diethylzinc to aldehydes in the presence of catalytic amounts of chiral catalystshas attracted considerable attention.
The three-component modified Mannich reaction is a convenient method to prepareaminoalkyl-naphthols. Through the use of chiral amines, nonracemic aminonaphtholderivatives have been prepared which catalyze the asymmetric addition of diethylzinc toaryl aldehydes. Numerous efforts have been made to use chiral ligands such as biarylalcohols, β-amino alcohols, amino thiols, amines and o-hydroxybenzylamines.
Since the asymmetric catalytic and biological activities of aminonaphthol analogsdepend strongly on their stereochemistry, there is a clear need for analytical methods bywhich their configurations can be identified. High-performance liquid chromatography(HPLC) on chiral stationary phases (CSPs) is an effective analytical tool for the resolutionof chiral compounds on both analytical and preparative scales.
The enantiomers of five 1-(phenylethylamino)methyl-2-naphthol analogs or1-(naphthylethylamino)methyl-2-naphthol analogs containing two chiral centers weredirectly separated on CSPs containing the chiral selectors cellulose tris-(3,5-dimethyl-phenyl) carbamate (Lux Cellulose-1), cellulose tris-(3-chloro-4-methylphenyl) carbamate(Lux Cellulose-2) and amylose tris-(5-chloro-2-methylphenyl) carbamate (Lux Amylose-2).
Experiments were performed in normal-phase mode in a wide temperature range inorder to study the effects of temperature on the separations. Apparent thermodynamicparameters and Tiso values were calculated from plots of lnk or lnα versus 1/T. Somemechanistic aspects of the chiral recognition process are discussed with respect to thestructures of the analytes. The sequence of elution of the enantiomers was determined inall cases and a temperature-induced inversion of the elution sequence was observed. Thethree newly commercialized CSPs display complementary character leading to highlysuccessful resolution.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-081-MO
HPLC Enantioseparation and Chiral Recognition Mechanismof Novel Xanthone Derivatives on Macrocyclic Antibiotic
Chiral Stationary Phases
Carla Fernandes1,2, Maria Elizabeth Tiritan1,3, Quezia Cass4,Miguel Fernandes5, Madalena Pinto1,2
1Centro de Química Medicinal – Universidade do Porto (CEQUIMED-UP), Portugal2Departamento de Ciências Químicas, Laboratório de Química Orgânica e Farmacêutica,
Faculdade de Farmácia, Universidade do Porto, Portugal3Centro de Investigação em Ciências da Saúde, Instituto Superior de Ciências da Saúde-Norte, (CICS-ISCS-N),
Rua Central de Gandra 1317, 4585-116-Gandra PRD, Portugal4Departamento de Química, Universidade Federal de São Carlos, SP, Brasil
5Centro de Química da Madeira, Universidade da Madeira, Portugal
The need to generate both enantiomers in high enantiomeric excess (ee) for biologicalassays is crucial in the early phases of the drug discovery process [1]. Therefore the devel-opment of efficient methodologies for synthesis with high yields and high ee, as well asenantioselective resolution of chiral compounds by HPLC, is a very important task. Inrecent years, chiral stationary phases (CSPs) with covalently bonded macrocyclic antibiot-ics have turn into versatile and selective tools for the resolution and evaluation of theenantiomeric purity of different classes of chiral compounds [2–3]. Computational studiesbased on the chromatographic parameters have demonstrated to be very useful to under-stand the chiral recognition phenomenon [4].
This work described the synthesis of a small library of chiral xanthone derivatives(CXDs), in enantiomerically pure form, by coupling two carboxyxanthones with bothenantiomers of commercially available chiral reagents. These reactions were carried outwith the coupling reagent O-(benzotriazol-1-yl)-N-N-N’-N’-tetramethyluronium tetra-fluoroborate (TBTU) [5] at room temperature. Fourteen new CXDs were synthesized invery high yield (above 90%) and short reaction time. The structure elucidation of theCXDs was established by spectroscopic methods (IR, 1H NMR, 13C RMN, HMBC, HSQC,and mass spectrometry).
A chiral HPLC method using four macrocyclic antibiotics CSPs (Chirobiotic T, Chiro-biotic TAG, Chirobiotic V and Chirobiotic R), under multimodal elution, has been investi-gated for determination of the enantiomeric purity of the synthesized compounds.
The analyses were performed at room temperature in isocratic mode and UV and CDdetection at a wavelength of 254 nm. The best enantioselectivity and resolution wereachieved on Chirobiotic R, under normal phase, with α = 1.67 and Rs = 2.50.
The optimized chromatographic conditions allowed the determination of the ee ofeight CXDs, always higher than 99%.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
In order to better understand the chromatographic parameters at a molecular level,and the structural features associated with the chiral recognition mechanism, computa-tional modeling studies by molecular docking technique were carried out using VDock.These studies shed light on the mechanisms involved in the enantioseparation. Theseresults will be discussed in this work.
1. I Agranat, SR Wainschtein, DDT, 2010, 15(5–6), 163.
2. DW Armstrong, et al., Chirality, 1995, 7, 474.
3. TJ Ward, AB Farris III, J. Chromatogr. A, 2001, 906, 73.
4. M Lammerhofer, J. Chromatogr. A, 2010, 1217, 814.
5. F Albericio, et al., Tetrahedron, 2001, 57, 9607.
Ackwoledgments: FCT (I&D, nº4040/2007), FEDER, POCI, FCT-Grices/Capes 00770 29/05/08, for financial
support.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-082-TU
Hydroxyethylcellulose as a Dynamic Coating Agentin Chiral Capillary Electrophoresis
Tímea Czipó-Takács, Mónika Babják, Mária GazdagGedeon Richter Plc.,
H-1475 Budapest, P. O. Box 27, Hungary
Capillary electrophoresis has become a powerful analytical technique in the field of chiralseparation in the last decades. Apart from the well-known advantages of the technique(great efficiency, rapidity, low sample and solvent consumption) the poor reproducibilityof the migration time and resolution could cause serious problems in the method devel-opment and in the routine work too.
Cyclodextrins as chiral selectors are widely used for the separation of enantiomers. Onthe basis of the literature and our practice mainly in case of charged cyclodextrins thereproducibility of the method is a problematic issue.
Using hydroxyethylcellulose (HEC) as dynamic coating agent the electroosmotic flowcan be eliminated in the capillary. Therefore, the migration time does not depend on thecharge of the capillary’s inner wall and the reproducibility increases.
The separations were performed in bare fused silica capillaries with background electro-lyte containing HEC, carboxymethyl-beta- or carboxymethyl-gamma-cyclodextrins and, insome cases, using of buffer was also advantageous. For separation reversed polarity wasused because of the negative charged cyclodextrins and the lack of electroosmotic flow.
After optimization of the temperature, the composition of buffer and pH the methodswere suitable for separation of enantiomeric pairs of several drug molecules.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-083-MO
LC Method for the Enantiomeric Purity Determinationof S-Amlodipine with Special Emphasis on Reversal
of Enantiomer Elution Order using Chlorinated Cellulose-BasedChiral Stationary Phases and Polar Non-Aqueous Mobile Phases
Katina S.S. Dossou1, Patrick A. Edorh2, Patrice Chiap3, Bezhan Chankvetadze4,Anne-Catherine Servais1, Marianne Fillet1, Jacques Crommen1
1Laboratory of Analytical Pharmaceutical Chemistry, Department of Pharmaceutical Sciences,University of Liege,
CHU, B36, B-4000 Liege, Belgium2Laboratory of Environmental Toxicology, Departement of Biochimistry and Cellular Biology,
University of Abomey-Calavi,Cotonou, Benin
3Advanced Technology Corporation (A.T.C.), University Hospital of Liege,Liege, Belgium
4Institute of Physical and Analytical Chemistry, School of Exact and Natural Sciences,Tbilisi State University, Tbilisi, Georgia
Amlodipine, a long-acting L-type calcium-channel antagonist, is commercially available asa racemic mixture of (R)- and (S)-isomers. The comparison of the R(+)- and S(−)-isomersactivities has shown that the S(−)-isomer is 1000 times more potent than the R(+)-isomer[1, 2]. Thus, the chiral switch from racemic amlodipine to its S-enantiomer becomesevident since better tolerability and antihypertensive effects are expected.
A LC method was developed and prevalidated for the enantiomeric purity determina-tion of S-amlodipine in polar organic solvent chromatography (POSC) using chlorinecontaining cellulose-based chiral stationary phases (CSPs). The concentration of formicacid (FA) (0.01–0.2%) in the mobile phase containing acetonitrile as the main solvent,was found to influence the elution order of amlodipine enantiomers as well as theenantioresolution. A reversal of the enantiomer elution order of amlodipine was onlyobserved with CSPs with both electron-withdrawing groups (chloro) and electron-donat-ing group (methyl) on the phenyl moieties of the chiral selector, namely cellulose tris(3-chloro-4-methylphenylcarbamate) and cellulose tris(4-chloro-3-methylphenylcarbamate).The highest enantioresolution (Rs: 4.1) value was obtained at the lowest FA concentration(0.01%) using CSP with cellulose tris(4-chloro-3-methylphenylcarbamate) as chiral selec-tor and the enantiomeric impurity, R-amlodipine, eluted first under these conditions.Therefore, the mobile phase selected for the prevalidation of the method consisted ofACN/0.1% DEA/0.01% FA and the temperature was set at 25 °C.
The prevalidation of the method by means of the strategy based on total measurementerror and the accuracy profile led to the selection of the weighted (1/X2) linear regressionas response function of the calibration curve within the matrix. The method was found to

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
be selective and the limit of quantification was found to be about 0.5 μg/mL (0.05%) forR-amlodipine while the limit of detection was close to 0.2 μg/mL (0.02%). Moreover, nosignificant matrix effect was observed.
1. X.P. Zhang, K.E. Loke, S. Mital et al., J Cardiovasc Pharmacol. 39 (2002) 208–2142. L. Adik-Pathak, J Assoc Physicians India. 52 (2004) 187–188.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-084-TU
Research on the Metabolism of4-(Methylnitrosamino)-1-(3-Pyridyl)-1-Butanone to the
Enantiomers of 4-(Methylnitrosamino)-1-(3-Pyridyl)-1-ButanolIn Vitro in Human Bronchial Epithelial Cells using
Chiral Capillary Electrophoresis
Youyou Yang1, Cong Yu1, Meng Zhou2, Ning Li2, Jie Liao2, Yu Bai1, Huwei Liu1
1Beijing National Laboratory for Molecular Sciences,Key Laboratory of Bioorganic Chemistry and Molecular Engineering of Ministry of Education,
Institute of Analytical Chemistry, College of Chemistry and Molecular Engineering,Peking University, Beijing 100871, China
2Medical Experiment & Analysis Center, General Hospital of Chinese PLA,Beijing 100853, China
4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol (NNAL), a major metabolite of 4-(methyl-nitrosamino)-1-(3-pyridyl)-1-butanone (NNK), has a chiral center in its molecule that willlead to the formation of NNAL enantiomers. As tobacco specific N-nitrosamines (TSNAs),NNK and NNAL are the most pulmonary carcinogen in tobacco products and smoke. Inthis paper, a chiral capillary electrophoresis (CE) method modified with highly sulfatedβ-cyclodextrin (S-β-CD) was developed to investigate the stereoselective formation ofNNAL from NNK in vitro in normal human bronchial epithelial (NHBE) cells. Combinedwith solid phase extraction (SPE) of the cell samples, NNK and NNAL enantiomers werebaseline separated under the optimum CE conditions, with satisfactory recovery (72.5–112.9% for NNK and (±)-NNAL) and low detection limit (2.5–3 μg/mL for NNK and (±)-NNAL). The cytotoxicity of NNK in NHBE cells was investigated through the CCK-8 assayand proved to be highly dependent on the NNK’s concentration. The metabolic resultsobtained from CE analysis demonstrated that NNK was preferentially metabolized to (+)-NNAL through carbonyl reduction. Meanwhile, the ratio of (+)-NNAL/(−)-NNAL wasconstant and independent of NHBE cells’ incubation time with NNK but could variedaccording to the concentration of NNK. This chiral CE method could be useful for thestudy on toxicology and metabolic transformation of related TSNAs.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-085-MO
Separation of Phenotropil on Polysaccharide BasedChiral Stationary Phases
Helena Kažoka1, Oksana Rotkaja1,2, Grigorij Veinberg1,Maksim Vorona1, Anton Lebedev1
1Latvian Institute of Organic Synthesis,21 Aizkraukles Street, LV-1006 Riga, Latvia
2Latvian University,48 Kr.Valdemara Street, LV-1013 Rıga, Latvia
It is known that optical antipodes of biologically active enantiomers are less active ortotally inactive and their presence in pharmaceutical substance should be avoided. That iswhy the stereochemical resolution of racemic drugs is a method of choice in the discoveryof enantiomers responsible for pharmacological activity. Appropriate resolution of thepsycho-stimulating drug – Phenotropil [2-(4-phenyl-2-oxopyrrolidin-1-yl)acetamide] andcomparative investigation of pharmacological properties for separate antipodes wasperformed in Latvian Institute of Organic Synthesis and it led to the discovery ofR-Phenotropil as the biologically active component of racemic drug [1].
The absence of published data about chromatographic separation of S- andR-Phenotropil stimulated our efforts aimed at the elaboration of chiral HPLC methodologyuseful for qualitative and quantitative analysis of its racemic mixture and separateenatiomers. Among the available HPLC methods for the separation of enantiomers the useof chiral stationary phases (CSPs) has become the most important tool for determinationof the optical purity of organic compounds and polysaccharide based CSPs are the mostdominant and widely used.
The aim of this work was to explore the suitability of polysaccharide based CSPs for theenantiomeric separation of Phenotropil. Coated stationary phases Lux Cellulose-1 and LuxCellulose-2, or a similar phase, but with chemically bound selector, Chiralpak IC, was usedunder polar organic (PO) and normal phase (NP) mode. The investigations showed thatthe different nature and position of the substituent introduced in the phenyl ring of thecarbamate derivatives has a major influence on the chiral recognition ability of the CSPs.On CSPs Lux Cellulose-2 and Chiralpak IC under PO mode the best separation wasachieved with 100% 2-propanol as a mobile phase. CSP Lux Cellulose-1 had the bestcapability to resolve enantiomers with 100% acetonitrile, but under NP mode was unableto separate S- and R-Phenotropil. In conclusion, even though CSP Lux Cellulose-2 wasapplicable for the separation of enantiomers, Chiralpak IC column was found to be themost convenient for the assessment of optical purity of R-Phenotropil under NP mode.
1. G. Veinberg, M. Vorona, M. Dambrova, L. Karina, L. Zvejniece, A. Chernobrovijs, I. Kalvinsh, LR Pat.13630 (2006).
Acknowledgement: This work was supported by the ERDF grant Nr. 2DP/2.1.1.1.0/10/APIA/VIAA/059.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-086-TU
Simultaneous Discrimination of Jasmonic Acid Stereoisomersin Wounded Tobacco Leaves by LC-QTOF-MS
Yehua Han, Yu Bai, Yuan Liang, Meiping Zhao, Huwei LiuBeijing National Laboratory for Molecular Sciences,
Key Laboratory of Bioorganic Chemistry and Molecular Engineering of Ministry of Education,Institute of Analytical Chemistry, College of Chemistry and Molecular Engineering,
Peking University, Beijing 100871, P. R. China
Jasmonic acid (JA), an essential plant hormone controlling the plant defense signalingsystem and developmental processes, has stereospecific bioactivities that have not beenwell understood mainly due to the limitation in separation and detection methodologies.In this work, a liquid chromatography – quadrupole time-of-flight mass spectrometry (LC-QTOF-MS) detection based on a chiral column was successfully developed for theenantioseparation of racemic JA. Although no pure JA stereoisomers were commerciallyavailable, all the separated JA stereoisomers were identified directly by collecting thepeaks and detecting then by CD, as well as MS spectra. Satisfactory results were obtainedin terms of sensitivity and repeatability. This established LC-QTOF-MS method was latersuccessfully applied to the study of the naturally occurring JA stereoisomers in woundedtobacco leaves, which is anticipated to be applied to a wide range of plant samples tostudy the stereochemistry related bioactivities of JA.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-087-MO
Simultaneous Enantioselective 2D-HPLC Analysis of Neuro-activeAmino Acids in Mouse Central Nervous System
using Various Pirkle-Type Chiral Stationary Phases
Yurika Miyoshi1, Kyoko Ueno1, Jumpei Sasabe2, Masashi Mita3,Sadakazu Aiso2, Kenji Hamase1
1Graduate School of Pharmaceutical Sciences,Kyushu University, Fukuoka, Japan
2Keio University School of Medicine,Tokyo, Japan
3Shiseido Co., Ltd.,Tokyo, Japan
N-Methyl-D-aspartate (NMDA) receptors are related to neuronal diseases such as schizo-phrenia, amyotrophic lateral sclerosis (ALS) and Alzheimer’s disease. For a long time, onlyL-glutamate (L-Glu) and glycine (Gly) were noticed as neuro-active amino acids of NMDAreceptors. However, with the advancement of analytical technologies, D-serine (D-Ser)and D-alanine (D-Ala) have been found in mammalian brain, and these D-amino acidswere reported to regulate the neurotransmission of NMDA receptors. Therefore, for thedevelopment of novel drugs and diagnostic markers focusing on neuro-active amino acidenantiomers, a sensitive and selective analytical method for the simultaneous determina-tion of D-Ser and D-Ala in addition to L-Glu and Gly is expected. For the purpose, a 2D-HPLC system with various enantioselective columns is useful, because the intrinsicamounts of D-amino acids are trace in most of the tissue samples. In the present work,enantiomer separations of neuro-active amino acids were studied using various Pirkle-typeenantioselective columns, and the established 2D-HPLC system was applied to the deter-mination of the neuro-active amino acid enantiomers in mouse brain. Mouse tissues werehomogenized in MeOH and amino acids were fluorometrically derivatized with 4-fluoro-7-nitro-2,1,3-benzoxadiazole (NBD-F) in the presence of 400 mM sodium borate buffer (pH8.0). NBD-amino acids were determined using a 2D-HPLC system combining a monolithicODS column (0.53 mm i.d. x 750 mm) and a Pirkle-type enantioselective column (1.5mm i.d. x 250 mm). NBD-amino acids were detected by the fluorescence (ex. 470 nm,em. 530 nm). Among 9 Pirkle-type enantioselective columns tested (Sumichiral OA-2000S, 2500S, 3100S, 3200S, 3300S, 4100SR, 4500SR, 4700SR, 4900SR), all neuro-activeamino acid enantiomers could be separated using OA-2500S, 3100S, 3200S, 4100SR,4700SR columns. Especially, good separation factors were obtained by the OA-3200Scolumn (Ser; 1.28, Glu; 1.26, Ala; 1.28). According to these results, a 2D-HPLC systemhas been established using OA-3200S or OA-2500S column, and intrinsic neuro-activeamino acids in the spinal cord of ALS model mouse were determined. As a result, the

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
amount of D-Ser in the model mouse was higher than that of control mouse (ALS model;10.1 nmol/g, control; 6.2 nmol/g). Moreover, intrinsic D-Ser in the spinal cord of themodel mouse was increased in the terminal stage compared with that in the mouse ofearly stage of ALS. These results suggest that D-Ser is the candidate of diagnostic markerof ALS, and further investigations using the human clinical samples are in progress.
1. J. Sasabe, T. Chiba, M. Yamada, K. Okamoto, I. Nishimoto, M. Matsuoka, S. Aiso, EMBO J. 26 (2007) 4149.2. K. Hamase, Y. Miyoshi, K. Ueno, H. Han, J. Hirano, A. Morikawa, M. Mita, T. Kaneko, W. Lindner, K. Zaitsu,
J. Chromatogr. A 1217 (2010) 1056.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-088-TU
Stereochemical Characterization of Fluorinated 2-Arylpropionic Acidsby Enantioselective HPLC Analysis and ECD Detection
Carlo Bertucci1, Marco Pistolozzi1, Daniele Tedesco1, Riccardo Zanasi2,Renzo Ruzziconi3, Anna Maria Di Pietra1
1Department of Pharmaceutical Sciences, University of Bologna, Italy2Department of Chemistry and Biology, University of Salerno, Italy
3Department of Chemistry, University of Perugia, Italy
Enantioselective HPLC coupled with a detection system based on the simultaneous mea-surement of UV absorption and electronic circular dichroism (ECD) allows a completestereochemical characterization of chiral compounds [1]. In the present communicationwe report the development of enantioselective HPLC methods for the resolution of aseries of 2-(fluoroaryl)propionic acids [2]. Different chiral stationary phases were tested:Chiralcel® OJ, Chiralcel® OD, (S,S)-Whelk-O® 1, and α1-acid glycoprotein (AGP).
The highest enantioselectivity values were obtained with the (S,S)-Whelk-O® 1 columnfor some of the examined compounds. However, the results allow the application of thedeveloped methods for a reliable determination of the enantiomeric excess for all theexamined compounds. In the case of rac-2-(6-fluorophenanthren-1-yl)propionic acid (1),the absolute configuration of the enantiomeric fractions was determined by ECD analysisand time-dependent density functional theory (TD-DFT) calculations on the(S)-enantiomer of 1 [3].
The experimental ECD spectrum of the second-eluted fraction on the (S,S)-Whelk-O® 1column was found to be in excellent agreement with the theoretical ECD spectrum of(S)-1; therefore, the absolute configuration of the first- and second-eluted enantiomers onthe (S,S)-Whelk-O® 1 column was assessed as (R)-1 and (S)-1, respectively, and the elutionorders of the enantiomeric forms of 1 were determined on all the different chiral station-ary phases.
1. Salvadori, P., Bertucci,C., Rosini, C., Circular dichroism coupled with HPLC: a powerful tool for analytical,stereochemical and spectroscopic determinations, in: Circular dichroism: principles and applications, eds.Nakanishi, K., Berova N., and Woody. R.W., VCH Publishers, New York, 1994, 541–560.
2. Ricci, G., and Ruzziconi, R., J. Org. Chem. 2005, 70, 611–623.3. Autschbach, J., Chirality 2009, 21, E116–E152.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-089-MO
Study of Tert-Butyl Calix[n]arene Derivativesand Molecularly Imprinted Polymers in the Chiral Separation
of Oral Anticoagulants and ββ-Blockers
Ioana Daria Tiuca, Bogdan Cezar Iacob, Ede Bodoki, Radu Oprean“Iuliu Hatieganu” University of Medicine and Pharmacy,Faculty of Pharmacy, Analytical Chemistry Department,
Cluj-Napoca, Romania
Orally administered anticoagulants (acenocoumarol, warfarin) show narrow therapeuticindex and their enantiomers exhibit significantly different pharmacokinetic and pharma-codynamic behavior, which also applies for β-blockers.
Calixarenes, in comparison with other chiral selectors (cyclodextrins, crown ethers), showthe advantage of higher molecular flexibility needed in the formation of transient inclusioncomplexes with the selectand, as well as the ease of hydrogen bond formation contributingto the stability of the formed complexes. On the other hand, molecularly imprinted poly-mers (MIP) represent a versatile, inexpensive and reproducible chiral selector alternative.
In each case, the most suitable chiral selector was choosen according to their specificinteraction with the studied enantiomers investigated by ATR-FT-IR and Raman spectros-copy, assisted with data obtained by molecular modeling.
The efficiency of the chiral selectors has been evaluated by chromatographic and electro-phoretic techniques (TLC, CEC). Calixarene derivatives were immobilized on the pre-coatedsilica thin layer or on the inner surface of the fused silica capillary. Trimethoxysilane derivativeof calixarenes have been synthesized at 80 °C in HClO4 catalysis under pure nitrogen, fol-lowed by their immobilization at 150 °C (open tubular CEC capillaries and TLC plates).
Furthermore, two chromatographic stationary phases based on MIP for the separationof β-blockers were evaluated. The monolithic stationary phase was prepared by the in situcopolymerization using methacrylamide as a functional monomer, piperazinediacrylamide as a cross-linking agent, phosphate buffer 50 mM pH = 7.0, ammoniumsulfate as a porogen and vinylsulfonic acid as charge providing agent. Polymerization wasinitiated by addition of ammonium peroxodisulfate and N,N,N’,N’-tetramethylethylenedi-amine. Results were compared with the ones obtained with a new open tubular MIPcapillary containing the same stationary phase in the form of a thin film of polymer. Theuse of a dilute monomer mixture ensured the formation of an open tubular type MIPlayer, without the need of an external source of pressure for CEC experiments.
In both cases, a chemometric approach was employed to optimise the CEC parameterssuch as pH of the buffer, organic solvent and salt concentration on the electroosmoticflow (EOF) and chiral recognition selectivity. Under the optimal conditions, baselineresolutions of the studied chiral compounds were achieved.
This work was supported by CNCSIS-UEFISCSU, project number PN II-RU 469/2010.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-090-TU
The Application of a Newly Developed IsopropylCarbamate-Cyclofructane6-Based (IP-CF6) Chiral Stationary Phase
for HPLC Enantioseparation of Betti Base Analogs
Zoltán Pataj1, István Ilisz1, István Szatmári2, Ferenc Fülöp2,Daniel W. Armstrong3, Antal Péter1
1Department of Inorganic and Analytical Chemistry, University of Szeged,Dóm tér 7, H-6720 Szeged, Hungary
2Institute of Pharmaceutical Chemistry, University of Szeged,Eötvös u. 6, H-6720 Szeged, Hungary
3Department of Chemistry and Biochemistry, University of Texas at Arlington,Arlington, TX 76019-0065, USA
Aminonaphthol analogs (Betti bases) and their derivatives display good enantioselectivityin asymmetric catalytic reactions, and especially in catalytic asymmetric alkylations,alkenylations, Michael additions, aldol reactions and cyclopropanations. Preparation ofthe enantiomers of the Betti bases and their N-substituted derivatives is therefore ofappreciable significance. Moreover, Betti base derivatives provide convenient access tomany useful synthetic building blocks because the amino and phenolic hydroxy groupscan be converted into a wide variety of compounds.
The cyclofructane (CF) selectors consist of six or more (usually seven or eight) β-(2-1)-linked D-fructofuranose units. Common abbreviations for these compounds are CF6,CF7, CF8, etc. These selectors are members of the macrocyclic oligosaccharides like thecyclodextrins, which are perhaps the best known members of this class. CF6, with sixD-fructofuranose units, contains a 18-crown-6 ether core. The 18-crown-6 ring serves asthe skeleton core of CF6, with six fructofuranose units attached on its rim. The fructo-furanose units are alternatively pointing towards and away from the molecular center,which are described as “inward-inclined” and “outward-inclined”. It was recently re-ported that derivatized-CF-bonded CSPs permitted effective enantiomeric separationsfor a variety of compounds, and especially for chiral molecules containing a primaryamine functional group. Such columns can be used in normal-phase and polar-organicmodes.
The direct separation of the enantiomers of Betti base were performed on a newlydeveloped chiral stationary phase containing IP-CF6 as chiral selector, with n-heptane/al-cohol/TFA as mobile phase.
The effects of the mobile phase composition, the nature and concentration of thealcoholic and acidic modifiers, and the structures of the analytes on the retention andresolution were investigated. In some cases separations were carried out at constantmobile phase compositions in the temperature range 5–40 °C. Thermodynamic parame-

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
ters and Tiso values were calculated from plots of lnk or lnα versus 1/T. −Δ(ΔH°) rangedfrom 2.8 to 3.2 kJ mol−1, −Δ(ΔS°) from 7.7 to 10.1 J mol−1 K−1 and −Δ(ΔG°) from 0.2 to 0.5kJ mol−1. It was found that the enantioseparations were enthalpically driven. The sequenceof elution of the stereoisomers determined in some cases was (R) < (S).

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-091-MO
Use of Chiral Ionic Liquids as Additives in Capillary Electrophoresis
Ioannis Stavrou, Constantina P. Kapnissi-ChristodoulouUniversity of Cyprus,
P.O. Box 20537, 1678, Nicosia, Cyprus
In this study, different chiral ionic liquids are synthesized, characterized and used asadditives in mobile phases for improved chiral separations in Capillary Electrophoresis(CE). A number of chiral ionic liquids, such as L- and D-alanine tert butyl ester NTF2
(NTF2: bis(trifluoromethane)sulfonimide), L-phenylalanine ethyl ester NTF2, etc, are initial-ly synthesized and characterized using NMR. The ionic liquids mentioned above are thenadded in the mobile phase in order to improve the chiral separation of different analytesin regards to efficiency, resolution and analysis time. Several chromatographic parame-ters, such as type and concentration of the ionic liquid, the chiral selector and the back-ground electrolyte, are evaluated in order to further optimize the separation. In addition,efficiencies and resolutions are estimated with and without the use of ionic liquids asadditives. These values are then compared in order to demonstrate the importance ofionic liquids as additives for more efficient separations.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-092-TU
A Novel Non-Card Based Format for Dried Blood Spots Analysis
Michel Wagner, Emmanuel Varesio, Gérard HopfgartnerLife Sciences Mass Spectrometry, School of Pharmaceutical Sciences,
University of Geneva, University of Lausanne,20 Boulevard d’Yvoy, CH-1211 Geneva 4, Switzerland
Dried blood spots (DBS) have been introduced sixty years ago as sample collection fornewborn screening. DBS consist in the deposition of a droplet of capillary blood, usuallyobtained by finger pricking, on a paper card. After solid-liquid extraction (SLE) of analyteswith an adequate solvent, the samples are analysed by chromatography or mass spectro-metric techniques. Contrarily to classical blood sampling techniques DBS samples can bestored at room temperature and easily shipped. DBS have shown their usefulness for alarge number of applications besides newborn screening, including quantitation of drugs,for instance therapeutic drug monitoring or, more recently, pharmaco and toxicokinetics.
Liquid chromatography requires for analysis liquid extracts. Therefore a portion or theDBS spot is usually punched out of the card, and analytes are extracted as describedabove. Compared to liquid-liquid extraction (LLE), SLE is less selective and endogenouscompounds can be co-extracted with the analytes, leading to possible matrix effect withmass spectrometric detection. The removal of phospholipids during the sample prepara-tion is of particular interest in method development, because those compounds present athigh concentration levels in blood and can be one of the major causes of matrix effects inatmospheric pressure ionisation.
We present herein a novel, tube based format, which allows either sample collection onpaper and analyte extraction, in a single device. The integration of sample collection andsample preparation in a single device eliminates the need of a punching step and pre-vents any related cross contamination or loss of sample. Moreover, our format, in additionto SLE, allows applying paper supported micro liquid-liquid extraction (μLLE) on filterpaper in a simple way. The concept can also be extended to the 96 well format.
Extraction efficiencies were evaluated for both SLE and μLLE, using a mixture of 18analytes, representative of 5 distinct chemical classes (amphetamines, cocaine and metab-olites, tricyclic antidepressants, benzodiazepines, antiretroviral drugs). The selectivity ofthe extraction procedure was studied by assessing phospholipid removal and monitoringinterfering compounds. Finally, the potential of our format for quantitative purpose isdemonstrated for analysis of saquinavir in blood.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-093-MO
A Rapid Method for the Detection of Metabolite of Sulfur Mustard1,1’-Sulfonylbis[2-S-(N-Acetylcysteinyl)Ethane] in Biofluids
via LC-MS/MS
Igor Rodin1, Arkady Braun1, Oleg Shpigun1, Irina Ananieva1, Igor Rybalchenko1Department of Chemistry, Lomonosov Moscow State University,
Moscow, Russia2“Lumex Ltd.”,Moscow, Russia
Sulfur mustard is a widely known chemical warfare agent. In Russia and former USSRrepublics heavy stocks of ammunition containing this agent are stored. Industrial destruc-tion of these dangerous objects has been taking place recently. Control of environmentaland human safety in regions located near factories is an important analytical task. Anotherreason of creating analytical technique for the determination of chemical warfare agentsis a threat of application of sulfur mustard in acts of terror.
After human intoxication by sulfur mustard it metabolizes producing various metabo-lites. Some of them are not stable, but one – 1,1’-sulfonyl-bys-[2-S-(N-acetylcysteinyl)etanis stable enough to be found in plasma for several days. This compound is the mostsuitable for being used as a sulphur mustard intoxication marker.
In present work LC-MS/MS technique was used for the determination of 1,1’-sulfonyl-bys-[2-S-(N-acetylcysteinyl)etan in plasma samples. The procedure of sample pretreat-ment included steps of plasma deproteinization with 10% water solution of trichloraceticacid, and solid phase extraction to clean the sample. HPLC separation was carried out ona reversed-phase column (Synergi RP Hydro (150x2.1 mm) using water-acetonitrile as amobile phase (pH 5.4). The detection in a negative electrospray ionization mode providedgood analytical signal. Deprotonised molecular ion (m/z = 443, as a parent ion) and itsfragments (m/z = 163.5 and 127) were used for the detection in MRM. Limit of detectionwas about 0,1 ng/ml. Validation of technique was carried out using plasma of rats, intoxi-cated by sulfur mustard (about ½ LD 50).
1,1’-sulfonyl-bys-[2-S-(N-acetylcysteinyl)etan was found in rat plasma and urea up to144 hours after intoxication.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-094-TU
Analysis of 1αα,25-Dihydroxyvitamin D3 (Calcitriol)in a Biological Fluid using LC-MS/MS
Joanna Denbigh, Tony Edge, Joanne Gartland, Tim Liddicoat, Kim PhippsThermoFisher Scientific,
Runcorn, Cheshire, WA7 1TA, UK
Vitamin D plays important roles in bone health and a variety of other pathophysiologicalconditions such as diabetes and cardiovascular disease. There are two common forms ofvitamin D, cholecalciferol (D3) and ergocalciferol (D2). Vitamin D3 is produced in the skinof vertebrates after exposure to ultraviolet B light from the sun or artificial sources, andcan be obtained from diet, as it occurs naturally in a small range of foods. In some coun-tries staples such as milk, flour and margarine are artificially fortified with vitamin D, andit is also available as a supplement in tablet form. In the body, vitamin D3 undergoesmetabolism to 25-hydroxyvitamin D3 in the liver, and then is further metabolised to1α,25-dihydroxyvitamin D3 in the kidney. 1α,25-dihydroxyvitamin D3 is the biologicalactive form of vitamin D3 and is useful for evaluation of several diseases including chronicrenal failure, sarcoidosis, hypoparathyroidism, and rickets. Measurement of 1α,25-di-hydroxyvitamin D3 is very challenging due the presence of interfering substances inserum. It has been historically measured by radio receptor assay and radio immunoassaywhich require extensive time consuming sample preparation to remove isobaric interfer-ing substances and matrix effects. With the increased interest today to monitor vitamin Din the clinical environment, the production of a robust assay is essential to ensure thatappropriate quantitation is obtained. Liquid chromatography-tandem mass spectrometry(LC-MS/MS) is widely recognised to be the technique of choice for the analysis of ste-roids. A simple and sensitive method has been developed which uses solid phase extrac-tion coupled to LC-MS/MS which is an accurate and precise measurement of 1α,25-di-hydroxyvitamin D3 (calcitriol) in serum. Extraction efficiencies and matrix effects will alsobe highlighted.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-095-MO
Analysis of 25-Hydroxy Vitamin D2 and D3 in Human Serumusing UHPLC/SQ MS with Field-Free APCI Ion Source
Avinash Dalmia1, Sean Daugherty2, Daniel Pentek1
1PerkinElmer,710 Bridgeport av. Shelton, CT 06484, USA
2PerkinElmer,Chalfont Road, Seer Green, Beaconsfield, HP9 2FX, UK
Vitamin D (where D refers to D2 or D3) is a fat soluble vitamin. The major physiologicalfunction of vitamin D metabolites is to maintain calcium and phosphate homeostasis andits status has been associated with variety of diseases including cancer, diabetes, cardio-vascular disease, osteoporosis and multiple sclerosis. 25-Hydroxy vitamin D is the metabo-lite measured in blood to determine the vitamin D status of patients. Conventional tech-niques such as immunoassay or LC/UV lack adequate sensitivity, specificity and speed foranalysis of vitamin D. This paper describes a fast 5 min UHPLC method with single quad-rupole MS equipped with a field free APCI source for selective, sensitive measurement of25(OH)D2 and 25(OH)D3 with detection limits of 1 ng/ml or lower.
The separation and measurement of both 25-hydroxyvitamin D2 and D3 was carriedusing PerkinElmer FlexarTM FX-10 LC system and Flexar SQ 300 MS equipped with aPerkinElmer field-free APCI source. The field-free APCI source parameters such as tempera-ture and capillary exit voltage were optimized for measurements.The separation wascarried out in 5 minutes using PerkinElmer Brownlee HRES Analytical DB column (2.1 mmx 50 mm, 1.9 μm) at flow rate of 0.5 mL/min. The extraction of both hydroxy vitaminsfrom human serum was carried out using a simple protein precipitation /LLE method. Afixed amount of 25-Hydroxy vitamin D3–d6 was used as an internal standard. Recovery ofanalytes from the extraction procedure was 90% or better with no ion suppression ofanalyte response from matrix after extraction. The minimum detection limit of bothanalytes in human serum was lower than 1 ng/mL. The response was linear over therequired concentration range of 3 ng/mL to 75 ng/mL.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-097-MO
Analysis of Triazophos Residue (Pesticide) in Tomatousing High-Performance Liquid Chromatography
Chandra Mohineesh, Anupuma Raina, Jaya Raj, T.D. DograDepartment of Forensic Medicine & Toxicology
A.I.I.M.S., New Delhi-29, India
Qualitative and quantitative analysis of pesticides in fruits and vegetable is important forpublic health and safety reasons. Besides being beneficial for increased crop yield as wellas in vector control programme, it has resulted in several healths’ related problems. In thisstudy, the blended tomato (25 g) was spiked with Trizophos (pesticide) and mixed withanhydrous sodium sulphate (15 g) and extracted with ethyl acetate (67 ml). A Dionexreverse phase High performance liquid chromatography having Photodiode Array Detec-tor(PDA) was introduced for identification and quantification of Triazophos (pesticide).Separation was performed on reverse phase C-18 column, using acetonitrile and water(70:30) as mobile phase at a flow rate 1 ml−min. 245 nm UV_VIS wavelength is used.Column oven temperature was maintained at 35 °C. 20 μl injection volume was used forthe analysis. Result shows 87 percent recovery of Triazophos. Calibration curve that con-structed for the analyte spiked into sample followed linear relationship with good correla-tion coefficients.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-098-TU
CE-LIF Assays Based on Boronic Acid Functionalized Dyesfor Microbe Surface Glycoprotein Targets
Christa L. Colyer1, Shingo Saito1,2, Tara L. Massie1, Xiuli Lin1,Takeshi Maeda3, Hiroyuki Nakazumi3
1Department of Chemistry, Wake Forest University,Winston-Salem, NC, USA 27109
2Graduate School of Science and Engineering, Saitama University,255 Shimo-Okubo, Sakura-ku, Saitama 338-8570, Japan
3Graduate School of Engineering, Osaka Prefecture University,1-1 Gakuen-cho, Naka-ku, Sakai, Osaka 599-8531, Japan
Analytical tools that can be adapted to a wide range of analyte types and sizes are invaluablein an increasingly interdisciplinary scientific landscape. Such tools must be able to deliverhigh efficiency and high sensitivity measurements, especially for bioanalytical targets. Tofacilitate such measurements, it is possible to exploit specific interactions between sugar‘handles’ on bioanalytes and new fluorescent probes for analysis by capillary electrophoresiswith laser-induced fluorescence detection (CE-LIF). For example, novel squarylium and tri-azolylcoumarin probes functionalized with a boronic acid moiety can selectively respond tocarbohydrate and glycoprotein targets through the formation of reversible cyclic boronateester complexes, demonstrating an increased fluorescence response for detection. To begin,spectrofluorometric experiments were conducted by mixing a new functionalized squaryliumdye with simple monosaccharides, resulting in the largest (10-fold) enhancement in emissionobserved for fructose and the smallest for glucose, with binding constants (obtained bytitration of the dye with monosaccharide) of 102.8 and 100.9 M−1 for fructose and glucose,respectively. Enhanced fluorescence emission was also observed for the glycoprotein mucinand for intact Bacillus globigii (BG) spores in aqueous solution. Surface glycoproteins are aconserved feature of Bacillus spores, regardless of the presence of an exosporium layer. Sporespecific carbohydrates rhamnose, 3-O-methyl rhamnose, and galactosamine, which areabsent from vegetative cells, are hypothesized to be constituents of the Bacillus glycoproteinBclA74, and so these carbohydrates, along with Bg spores (also known as Bacillus atrophaeus,a commonly used simulant of biothreat agent Bacillus anthracis (Ba)) are identified as suitabletargets for labeling with our new boronic acid-based dyes in the CE-LIF assays described inthis paper. In preliminary studies with nonselective probes, migration times were under 10minutes for all dye-spore complexes, with net mobilities ranging from (−) 3.5 x 10−4 to 6.9 x10−4 cm2V−1s−1. In all cases, the effects of solution pH, concentration, and buffer additivesmust be considered to determine optimal conditions not only for CE separation efficiency,but also for LIF detection sensitivity, and selectivity of the probe-microbe target association.This work has important implications for analyses in the areas of clinical chemistry, environ-mental science, homeland security, and forensics.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-099-MO
Comparison of Different Commercial Sorbents for the In-LineSolid Phase Extraction in Capillary Electrophoresis Determination
of Barbiturate Drugs in Biological Samples
Igor Botello, Francesc Borrull, Marta Calull, Carme AguilarRovira i Virgili University, Department of Analytical Chemistry and Organic Chemistry,
Faculty of Chemistry,Marcel.lí Domingo s/n 43007 Tarragona, Spain
The source of contamination by residues of drugs of abuse in the environment is mainlyfrom the consumers, because these drugs are excreted by the urine. The study of the levelsof these drugs in different biological fluids provides information about the consumption ofillicit drugs. This information allows the investigation of intoxication or the determination ofthe causes and circumstances of death. In the analysis of these illicit drugs in different matri-ces, capillary electrophoresis (CE) has been found to be a useful approach.
The main problem in the analysis of biological samples lies in the fact that in this kindof complex matrices, interfering endogenous compounds are normally present in a higherconcentration than the target analytes. Therefore, it is necessary to develop sensitive andselective methods for the extraction, separation and determination of these drugs inhuman body fluids when these compounds are present at low concentration levels. Toimprove the concentration sensitivity of CE an interesting approach is by means of in-linesolid phase extraction (SPE-CE).
In this work we present a comparison study of different commercial sorbents such asOasis HLB, mixed mode Oasis sorbents and C18 for in-line SPE in the determination of thethree barbiturates in biological samples. For each sorbent studied, different parametersaffecting the SPE performance such as sample pH, sample injection time or the elutionconditions were optimized in order to obtain high sensitivity enhancement factors. In allcases, the SPE-CE extractor consisted on a short length (2 mm) of a capillary (packed withthe sorbent) near to the inlet within the separation capillary. The efficiencies of the differ-ent sorbents are discussed and the validation with the more adequate extractor devicedemonstrated good linearity, low detection limits as well as satisfactory precision in termsof repeatability and reproducibility. The methodology developed resulted simple andeffective for the determination of the studied barbiturates.
1. M.C. Breadmore, M. Dawod, J.P. Quirino, Electrophoresis 32 (2011) 127.2. A. Macià, F. Borrull, M. Calull, F. Benavente, E. Hernández, V. Sanz-Nebot, J. Barbosa, C. Aguilar, J. Sep.
Sci. 31 (2008) 872.3. R. Ramautar, G.W. Somsen, G.J. Jong, Electrophoresis 31 (2010) 44.4. C. Postigo, M.J. López, D. Barceló, Env. Inter. 36 (2010) 74.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-100-TU
Confirmatory Quantitation of Benzodiazepinesin Post-Mortem Matrices using HPLC/MS with Online Cleanup
Guifeng Jiang, Terry Zhang, Kathryn PrestonThermo Fisher Scientific,
355 River Oaks Parkway, San Jose, CA 95134, USA
In this presentation we demonstrate the use of the Transcend TLX system with TurboFlowtechnology and single quadrupole mass spectrometry (TLX//MS) for the cleanup, separa-tion, detection, and quanitation of 23 benzodiazepines within 20 minutes, achievingresults typically associated only with triple quadrupole mass spectrometry. The TLX/MSmethod employed achieves consistent ppb level quantitative results against internalstandards from molecular and confirming ions in blood and urine matrices with area RSDbetter than 5%. For the analysis discussed in this presentation, confirming ions are creat-ed through in-source collision induced dissociation (CID). Quantitative results of confirm-ing ions are within 5% of the quantitative results from molecular ions. For the analysis ofblood samples, the unfiltered supernatant from a simple protein precipitation is injecteddirectly onto the TurboFlow column employed in the system. For the evaluation of urinesamples, the undiluted and unfiltered supernatant from hydrolyzed urine is injecteddirectly onto the TurboFlow column. Using the confirmatory quantitative method de-scribed in this presentation, benzodiazepines were identified in three post-mortem bloodsamples and two post-mortem urine samples. The utilization of TurboFlow technologydrastically reduces the cleanup required for the analysis of benzodiazepines, eliminatescontamination of the MS source from strongly hydrolyzed samples, and significantlyimproves limits of quantitation thus allowing confirmatory quantitation with singlequadrupole mass spectrometry.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-101-MO
Determination of Antiphycotic Drugs in Brain Tissueby LC-ESI-MS-MS: Screening and Quantitation of Samples
of Forensic Interest
M. Carmen Sampedro, M. Aranzazu Goicolea, Ramón J. BarrioDepartment of Analytical Chemistry, Faculty of Pharmacy, University of the Basque Country,
01006 Vitoria-Gasteiz, Spain
A quantitative LC-MS-MS method has been developed for the simultaneous determinationof 14 antipsychotic in human post-mortem brain tissue. The drugs have been chosen asthe commonly used antipsychotic according to data from the European Medicines Agency(EMEA). As protein precipitant agent was used formic acid in acetonitrile (1%). The pre-cipitant agent was added to samples and were homogenised with a sonifier cell disruptor.Then, the samples were mechanical shaken during 5 minutes and centrifuged. The super-natants were transferred to HybridSPE cartridges. The eluates were dry until dryness andredissolved in mobile phase. The extracts were centrifuged at 1050 s−1 at 4 °C and ana-lyzed by LC-MS-MS. The chromatographic separation was performed in 21 min on a C8column, applying gradient elution with formate ammonium and acetonitrile and flow rategradient. Triple Quadrupole mass spectrometry was employed to generate tandem massspectrometric (MS/MS) data of the target analytes and to selected the ion signals of m/z.Quantitation of the analytes was performed operating in dynamic multiple reaction moni-toring (dynamic MRM) mode using electrospray ionization interface. Calibration curves inspiked brain tissue were linear in the range 8–8000 ng/g (r > 0.991) for all drugs, within-and between-day coefficients of variation were lower than 25% for all drugs at the LLOQ.The LLOQ in matrix ranged 2 ng/g to 80 ng/g. The method was successfully applied tohuman post-mortem brain tissues, for forensic investigations.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-102-TU
Determination of Isomeric Synthetic Cannabinoid Metabolitesin Hydrolyzed Urine by LC/MS/MS
Ty Kahler1, Amanda Rigdon1, Paul Kennedy2, Rick Lake1,Steve Kozel1, Chris Denicola1
1Restek.Bellefonte, PA, USA2Cayman Chemical,Ann Arbor, MI, USA
In the past few years, a new product called ‘K2’, ‘spice’, or ‘legal marijuana’ has gainedpopularity. These herbal mixtures are not marketed for human consumption, but are inreality meant to be smoked. Even though these blends are marketed as containing onlynatural products, many have been found to contain synthetic cannabinoids such as JWH-018, JWH-073, and CP-47, 497, which have been sprayed onto the herbal mixture. It isthese unlisted ingredients that are responsible for the cannabis-like high experienced bythe user.
Toxicological testing for these compounds and their metabolites is of particular interestnot only to drug enforcement laboratories, but also to laboratories servicing centers foraddiction management and pre-employment drug testing. Synthetic cannabinoids arerapidly and almost completely metabolized prior to excretion, which means that testingmust be performed for specific metabolites rather than the parent drug itself.
Since synthetic cannabinoids are relatively new, limited research has been performed todetermine the exact urine metabolite profile for any given parent compound. Adding tothe complexity of the analysis, many of the metabolites for a given parent are mono-hydroxylated isomers of that parent compound. These metabolites are isobaric com-pounds that share a very similar fragmentation pattern. Some laboratories simply analyzethese isomers as one group, with little to no chromatographic resolution between theisomers, however, when required, specific isomers may have to be separately identifiedand quantified.
The method presented in this poster chromatographically separates all isomeric metab-olites of JWH-018 and JWH-073 in hydrolyzed urine matrix. A dilute-and-shoot methodol-ogy was used for sample analysis. After hydrolysis, spiked samples were diluted 20x. Theinstrument used for analysis was a Shimadzu UFLCXR coupled with an AB Sciex API 4000MS/MS. The column used for analysis was a Pinnacle DB Biphenyl 50 mm x 2.1 mm RP-HPLC column. The total run time for the method was 8.5 minutes. The MS was operatedin positive ionization mode, monitoring three transitions for each metabolite. Preliminaryvalidation experiments established LODs of 1 – 5 ng/mL. Linearity (R2) was > 0.997 for allanalytes, and accuracy was 85% – 120% for all analytes at all calibration levels.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-103-MO
Determination of Selenoaminoacids by HPLC Fluorescencein Plasma of Sheeps Supplemented with Selenium
Claudia Valdez Flores, Erika F. Pérez Becerril, Alma L. Revilla VázquezUniversidad Nacional Autónoma de Mexico,Universidad 3000, Mexico, 04510, Mexico
The chemical analogy between sulfur and selenium is important from a biological stand-point, because selenium can replace sulfur in amino acids such as selenomethionine,selenocysteine and selenocystine. Therefore is a priority to know the amount of selenium.
In this study a method by high performance liquid chromatography (HPLC) and fluo-rescence detection was developed, using a pre-column derivatization with the reagent6-aminoquinolyl-N-hydroxysuccinimidyl carbamate to selenoaminoacids and amino acidswhich are present in plasma of sheeps.
To evaluate the applicability, 12 pelibuey sheeps divided into three groups were used:1) control group, 2) group supplemented with sodium selenite, and 3) group supple-mented with selenoyeast. The administration was conducted over seven days and samplesof blood for 11 days were taken. Total selenium in plasma was measured by atomic ab-sorption spectrophotometry with hydride generator; the results showed a strong increasein the concentration of selenium in the groups supplemented with both kind of seleniumcomparing with the control group. This analysis allowed selecting the samples for HPLC,those that presented higher concentration of selenium.
In conclusion this method is capable of identification amino acids and quantificationselenoaminoacids in concentration range of 61 to 142 μg/L for selenomethylseleno-cysteine, 50 to 252 μg/L for selenocystine and 20 to 103 μg/L for selenomethionine. Insamples, the presence of selenomethionine and selenomethylselenocysteine was ob-served.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-104-TU
Determination of t,t-muconic Acid in Urine Samples
Silvia Marten, Mareike NaguschewskiKnauer GmbH,
Hegauer Weg 38, 14163 Berlin, Germany
The ubiquity of Benzene in the environment due to its occurrence in mineral oil andcombustion processes results in health issues [1]. Not only the environmental benzeneconcentration is of interest, but also the control of human benzene exposure, especially atwork places. It is important to establish a method that is sensitive enough to detect hu-man exposure to very low benzene concentrations as it is already toxic in low doses [2].Metabolites like tt-MA can be easily extracted from urine matrices using liquid-liquidextraction techniques with ethyl acetate for example [3]. The determination of t,t-muconic acid in urine samples could be successfully accomplished on different columntypes. A column tandem of spherical sub 2 μm columns with different modifications canbe used as well as a fused core high speed column with C18 modification. The separationof the target compound from the complex urine matrix in which it exists could be carriedout in less than in 6 minutes on the fused core 1.7 μm C18 column.
1. Tharnpoophasiam, P. et al.; Southeast Asian J Trop Med Public Health, Vol. 35, No. 3, 717–723 (Septem-ber 2004)
2. Hu, X. et al.; Biomed Environ Sci, Vol. 19, 292–296 (2006)3. Boogard, P. J. et al.; Environ Health Perspectives; Vol. 104, Supplement 6, 1151–1157 (December 1996)

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-106-TU
Development of an Automated Ion Trap MSn Based ScreeningMethod for Clinical and Forensic Toxicology
Jürgen Kempf1, Susanne Vogt1, Anna Sandhaas1, Wolfgang Weinmann2,Birgit Schneider3, Petra Decker3, Sebastian Götz3,
Arnd Ingendoh3, Carsten Baessmann3
1Institute of Legal Medicine, University Medical Center Freiburg, Germany2Institut für Rechtsmedizin, Universität Bern, Switzerland
3Bruker Daltonik GmbH, Bremen, Germany
Screening applications in clinical and forensic environments require rapid and unambiguousresults that can be generated even by unskilled users. Liquid chromatography-tandem massspectrometry (LC-MS/MS) combined with library search is an emerging screening technolo-gy in these fields. It provides more valuable information than LC-UV detection while cover-ing a broader and in some respect complementary range of analytes when compared toGC-MS. This study focuses on applying and challenging a robust and easy-to-use Ion-Trap-based solution for the detection and identification of common drugs, drugs of abuse andtheir metabolites in shortest time as possible in a fully automated environment.
Serum and urine extracts are separated using a UHPLC-system (Dionex RSLC) connect-ed to an amaZon X ion trap MS instrument generating data dependent MS2 and MS3
spectra in both positive and negative mode in a single run. Identification of the generatedspectra was performed using SmileMS library search software (GeneBio). For methoddevelopment several chromatographic columns were tested for resolution and peak shapeusing a mixture of 10 substances covering the desired mass range (100 – 800 amu) andboth polarities. Additionally, the effects of eluent composition and eluent buffer concen-tration in positive and negative mode were examined to find the most effective method.
A fast 8 minutes gradient using formic acid, ammonium acetate and acetonitrile aseluent and a Dionex Acclaim RLSC C18 100 x 2.1 mm column were used for chromato-graphic separation to generate a mass spectral library of about 800 compounds.
A subset of 200 compounds were defined and spiked into human serum matrix at 3different concentration levels (low therapeutic, therapeutic and elevated). All compoundswere combined into mixtures containing 5–10 substances each and then analyzed.
It has been shown, that low therapeutic doses (low ng/ml range) of commonly usedand miss-used benzodiazepines like diazepam could be successfully detected and identi-fied in all three MS-stages. Also low-dose benzodiazepines like flunitrazepam could bedetected and identified in spiked serum samples. The final results will show statisticalevaluation of the data listing identifications at different concentration levels, false positiveand false negative rates. The presented library screening method offers a fast, reliable andsensitive procedure for clinical and forensic analysis. The combination of MS2/MS3 spectraand retention time allows identification of drugs and metabolites at therapeutic levels.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-107-MO
Enantiomeric Resolution, Identification, and Quantitationof Chiral Illicit Drugs using SFC APCI MS/MS
Lakshmi Subbarao1, John McCauley1, Harbaksh Sidhu2,Rui Chen1, Jacquelyn Runco1
1Waters Corporation,New Castle, DE, USA2Waters Corporation,Pittsburgh, PA, USA
Illicit drugs can include active ingredients from bona fide registered pharmaceuticalshaving therapeutic uses or active ingredients that are banned from all use under variousinternational conventions or national laws. Many of the illicit drugs occur as optical iso-mers with different psychotropic activities. The enantiomeric purity and impurity profile inthe drugs can provide valuable intelligence to the law enforcement to determine thesynthetic route as well as the manufacturing practice for the illicit drugs. It is thereforeimportant to develop a rapid, reliable, and sensitive analytical method for the separation,identification, and quantitation of chiral illicit drugs.
Widely accepted as the technique of choice for chiral separation, supercritical fluidchromatography (SFC) has found its use in many stages of pharmaceutical industry, fromdiscovery to development. On average, SFC is 3–10 times faster than normal phase HPLCfor chiral separations due to the low viscosity and high diffusivity of supercritical CO2, themain mobile phase used in SFC. The co-solvent used in SFC, most commonly alcohols,also enables a facile coupling between SFC and MS detectors. However, the SFC chiralapplications in forensic arena are limited in scope. This is, at least in part, due to thelimitations in instrument design that prevents the low level detection often required inforensic applications.
Presented here are examples of chiral separation, identification, and quantitation ofillicit drugs using SFC APCI MS and SFC APCI MS/MS. Methadone, 3, 4-methylenedioxyamphetamine, hexobarbital, and tetramisole were chosen as the representative illicitdrugs in this study. Chiral columns of different size particles (5 μm and 3 μm) and lengthswill be evaluated with respect to the chromatographic efficiency and speed of analysis.The interface between SFC and MS and the choice of ionization will be evaluated. Quanti-tative analyses including limit-of-detection (LOD), limit-of-quantitation (LOQ), linearity,calibration curve, precision, accuracy, reproducibility, stability and recovery will be pre-sented and compared with those obtained from HPLC and CE. Key system attributes thatneed to be addressed to enable fast analysis with high detection sensitivity will also bediscussed.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-108-TU
Fabrication of Hierarchically Porous TiO2 Monolithsfor HPLC Columns
George Hasegawa1, Kei Morisato2, Kazuyoshi Kanamori1, Kazuki Nakanishi11Kyoto University,
Kitashirakawa, Sakyo-ku, Kyoto, 606-8502, Japan2GL Sciences Inc., Japan
Considerable attention has been focused on porous titania (TiO2) owing to its uniquecharacteristics and wide applications to electronics, catalysis, sensing, lasing, and separa-tion science. Especially, in the field of separation, titania is of great importance as thestationary phase for high-performance liquid chromatography (HPLC) columns because ofthe following reasons. First, titania possesses excellent stability both to heat and extremepH and also shows high mechanical strength. In addition, specific adsorption of organo-phosphate compounds provides a particular advantage to titania columns.
As another current trend in the separation field, monolithic columns with flow-throughpores which consist of a single piece of silica (SiO2) or polymer have become increasinglypopular over the last decade because they are superior to the traditional microparticulatepacked columns from the viewpoint of permeability. While the independent control ofsizes of particles and interstitial voids is not possible with particle-packed columns, mono-lithic columns can exhibit high performance at substantially low pressure owing to theindependent controllability of pore size and volume. However, there are very few reportson the porous monoliths of non-SiO2 metal oxides as monolithic separation media be-cause of their difficulty in synthesis due to high reactivity of non-SiO2 metal alkoxides.
Recently, we have also developed the pathway of fabricating hierarchically poroustitania monoliths by the sol-gel reaction utilizing a chelating agent and mineral saltsunder a mild condition accompanied by phase separation. By utilizing our novel synthesismethod, hierarchically porous titania monoliths consisting of anatase crystallites can beobtained without any thermal treatment after a hot-water treatment and drying. In thisstudy, we evaluated the separation efficiency of the hierarchically porous titania monolithsprepared using our method in HPLC. Separation of polar benzene derivatives in the nor-mal phase mode and that of organophosphates were investigated with the monolithictitania columns calcined at different temperatures.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-109-MO
Forensic Screening of Isobaric Compounds in Crude Samplesby Ultrahigh-Resolution UHPLC-QTOF Technology
Zoltan Czentnar1, Marcus Macht1, Anna Pelander2,Petra Decker1, Carsten Baessmann1
1Bruker Daltonik GmbH,Bremen, Germany
2University of Helsinki,Helsinki, Finland
Comprehensive forensic screening by LC/TOFMS based on a large target database of ~700standard compounds has been established as a routine method. Compound identification isdone on accurate mass, isotope pattern and retention time information. Although themethod was proven to be very reliable (~7000 samples p.a.), the target database contains afew isobaric compound pairs with similar retention time, which require a mass resolutionmuch higher than R = 10,000 achievable with standard ESI-TOF systems. We investigated onreal-life samples which resolution is needed in those cases to reduce the number or evencompletely avoid any false negatives in screening of complex samples.
17 target compounds in 8 groups are found in the target database which require ahigh mass resolution to be distinguished. Their molecular weights ranged from 211 to414 Da. A mass resolution up to ~50.000 was required to separate all compound groupsclearly. Since the MS was coupled to a fast UHPLC separation, this mass resolution mustbe achieved at simultaneously high data acquisition rate of 10–20 Hz. Automated dataprocessing was performed using dedicated screening software.
The mass accuracy of the systems allows the use of highly selective EIC trace widthsdown to 0.5 mDa. Observed average mass accuracies were mostly better than 0.5 ppmand independent of the acquisition rate. 12 of the 17 target compounds were detectedeven at 0.1 pg/μl, whilst 14 compounds gave linear calibration curves for the full concen-tration range (~ 4 orders of magnitude), demonstrating the suitability of the system forquantitative analysis. Additionally, the QTOF was successfully applied to an example ofinterference between a database compound and a supposed metabolite: according toexperience for urine samples with positive findings of doxepin the identification of theinternal standard dibenzepin is affected by an obviously coeluting compound of similarmass on standard TOF systems. The QTOF method resolves the two mass peaks andallows for confirmation of the assumed metabolite sum formula.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-110-TU
HPLC Determination of Benzodiazepines in Vitreous Humorusing Microwave Assisted Extraction
Purificación Fernández1, Estrella Núñez1, Ana M. Bermejo1,Rosa A. Lorenzo2, Antonia M. Carro2
1Institute of Legal Medicine, Forensic Toxicology Service, Faculty of Medicine,C/ San Francisco, s/n, 15782-Santiago de Compostela, Spain
2Department of Analytical Chemistry, Faculty of Chemistry,Avda, de las Ciencias s/n, 15782-Santiago de Compostela, Spain
In the last years there has been an increase of patients with benzodiazepines intoxication,causing a considerable interest in developing methods for their determination. The vitre-ous humor is a clean fluid of easy collection and high stability, making it very useful forthe detection of substances when the urine and blood are not available (hemorrhagicshocks, burns, stuffed) or they are altered by cadaveric phenomena (decomposition). Asimple preparation procedure using microwave energy (MAE) is proposed for the investi-gation of alprazolam, bromazepam, diazepam, lorazepam, lormetazepam and tetrazepamin vitreous humor; the working conditions as regards solvent, volume of solvent, tempera-ture and extraction time were optimized using a factorial design, selecting: ethyl acetate,11 mL, 98 °C and 14 minutes, respectively. The benzodiazepines were quantified by highperformance liquid chromatography with diode array detection (HPLC-DAD), using anXBridge® RP18 column (250x4.6 mm i.d. 5 μm particle size) and a mobile phase com-posed of acetonitrile and phosphate buffer pH 6.0 in gradient mode. The analytical meth-od was validated by determining the linearity, precision, accuracy and limits of detection(LOD) and quantitation (LOQ). The detector response was linear over the concentrationrange of 0.1–20 μg/mL in vitreous humor; the coefficients of variation and the relativeerrors were lower than 10.2% and 12%, respectively; LOD ranged from 3 to 19 ng/mL,and LOQ was the lowest concentration in the calibration curve. The analytes recoveries,calculated at two different concentrations (1 and 10 μg/mL), ranged from 81% to 101%.Finally, the method here proposed was applied to eight vitreous humor samples obtainedfrom benzodiazepines users.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-111-MO
Lipidomic Profiling of Patients with Cardiovascular Diseases
Katerina Netušilová, Miroslav Lísa, Michal HolcapekUniversity of Pardubice, Faculty of Chemical Technology, Department of Analytical Chemistry,
Studentská 573, 532 10 Pardubice, Czech Republic
Lipids are important compounds in the organism due to their wide range of biologicalfunctions, i.e., the main source of energy, structural components of cell membranes,important signaling molecules, etc. Disruption of lipids metabolism can lead to seriousdiseases, i.e., diabetes mellitus, cardiovascular diseases, cancer, Alzheimer’s disease, obesi-ty, etc. The goal of this work is the lipidomic profiling of erythrocytes, plasma and lipo-protein fractions (very-low-density, low-density and high-density lipoproteins) samples todistinguish healthy patients and patients with different types of cardiovascular diseases.Differences in lipid composition are determined based on changes in fatty acids andclasses of lipids profiles. Gas chromatography with flame ionization detection (GC/FID) isthe standard method used for profiling of fatty acids in a biological material as fatty acidmethyl esters (FAME) after their transesterification with sodium methoxide. The length ofthe column, the type and flow rate of carrier gas and the temperature program are im-portant parameters for the separation of FAME using GC/FID. These conditions wereoptimized using a model mixture of FAME standards and plasma samples of healthyvolunteers. The analysis of lipid classes was performed with ultra high performance liquidchromatography – mass spectrometry (UHPLC/MS) using hydrophilic interaction liquidchromatography based on their polarity. In total, 25 fatty acids in the GC/FID analysis and6 classes of lipids in the HILIC analysis were detected. The data from individual patientswere processed using Principal Component Analysis (PCA).
This work was supported by the project MSM0021627502 sponsored by the Ministry of Education, Youth andSports of the Czech Republic and by the project P206/11/0022 sponsored by the Czech Science Foundation.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-112-TU
Quantitative Analysis of Underivatized 1,25-DihydroxyvitaminD3 and D2 in Blood by UHPLC and Triple QuadrupoleMass Spectrometer Utilizing Ion Funnel Technology
Andre Szczesniewski, Monika DittmannAgilent Technologies, Waldbronn, Germany
Vitamin D exists in two major forms – vitamin D3 and D2. Vitamin D3 is obtained from thediet and produced by the skin after exposure to ultraviolet (UV) light from the sun. Vita-min D2 is obtained from a diet containing plants and fungi. Vitamin D is first metabolizedin the liver to form 25-Hydroxyvitamin D (25-OHD) and subsequently in kidneys to 1,25-Dihydroxyvitamin D (1,25-(OH)2D). The analysis of vitamin D levels in blood is done bythe measurement of these two major metabolites.
There are several complications to deal with when quantifying underivatized 1,25-(OH)2D – a complex biological matrix, isobaric isomers and low picogram per milliliterlevels for the analyte of interest. To help deal with biological matrix complexity, 2-dimen-sional liquid chromatography was used to reduce the amount of interference sentthrough to the mass spectrometer. 1,25-(OH)2D was first enriched on a loading columnfor a short time before being eluted onto an analytical column. In order to achieve sepa-ration of 1,25-(OH)2D from its isobaric isomers, it was necessary to run a 12 minutegradient of methanol and water containing ammonium formate on an analytical columncontaining sub two micron particle packing. This was achieved through the use of anUltra High Performance Liquid Chromatography (UHPLC) system. Detection was per-formed using a mass spectrometer utilizing ion funnel technology to provide the neces-sary sensitivity.
The 1,25-(OH)2D ammonium adduct was chosen as the precursor ion (m/z 434). Initial-ly, the transition 434 > 399 was selected for quantitation based on response, but severalinterfering peaks with the same retention time as 1,25-(OH)2D were noted. The transition434 > 381 had half the response but no interfering peaks and an equivalent signal-to-noise ratio. Ultimately, transitions to m/z 381 and m/z 363 were used for quantitationand conformation respectively. This decision allowed for greatly improved reproducibilitywhile maintaining excellent sensitivity. The limit of detection in a neat solution was 1pg/ml using a 100 μl injection (100 fg on column). In spiked serum samples, the limit ofdetection was found to be 5 pg/ml (500 fg on column). Calibration curves for neat andextracted samples showed excellent linearity and reproducibility.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-113-MO
Quantitative Profiling of Eicosanoids in Plasmaby Liquid Chromatography – Quadrupole/Time-of-Flight-Mass
Spectrometry
Su Hyeon Lee1,2, Young Wan Ha1, Won-Yong Lee2,Bong Chul Chung1, Man Ho Choi1
1Biomolecule Function Research Center,KIST, 39-1 Hawolkok-dong, Seoul 136-791, Korea;
2Department of Chemistry, Yonsei University,50 Yonsei-ro, Seoul 120-749, Korea
Eicosanoids as the metabolites of arachidonic acid regulates vascular function, inflamma-tion, hypertension and other pathologies. A validated quantitative analysis of 30eicosanoids including 18 prostaglandins (PGs), 4 leukotrienes (LTs), 2 thromboxanes (TXs)and 6 hydroxyeicosatetraenoic acids (HETEs), were developed with liquid chromatogra-phy-quadrupole/time-of-flight-mass spectrometry (LC-QTOF-MS). The sensitive andselective method was achieved in negative ionization of the high-resolution selected ionmonitoring (HR/SIM) mode. All eicosanoids were separated through an 1.9 μm particleC18 column (50 × 2.1 mm) at 30 °C and gradient elution with mobile phase consisted of0.1% formic acid in 95% water (solvent A) and 0.1% formic acid in 95% acetonitrile(solvent B) at a flow rate of 0.25 mL/min within 55 min run. The LC-QTOF-MS basedeicosanoids analysis was found to be linear ranged from 0.9929 to 0.9994, while theinstrument detection limits (IDL) were below 1 ng/mL in all eicosanoids analyzed. Thepresent method could be applicable in the integration with the evaluation of inflamma-tion and cardiovascular diseases.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-114-TU
Rapid Screening of Derivatised Anabolic Steroids in Equine Urineusing Ultra Performance Liquid Chromatography
– Tandem Mass Spectrometry
Colton H. F. Wong, David K. K. Leung, Francis P. W. Tang,Jenny K. Y. Wong, Nola H. Yu, Terence S. M. Wan
Racing Laboratory, The Hong Kong Jockey Club, Sha Tin Racecourse,Sha Tin, N.T., Hong Kong, China
Liquid chromatography-mass spectrometry (LC-MS) has been successfully applied to thedetection of anabolic steroids in biological samples [1–5]. However, the detection ofsaturated hydroxysteroids, such as androstanediols, by Electrospray Ionisation (ESI) isdifficult because of their poor ability to ionise. In view of this, chemical derivatisation hasbeen used to enhance the detection sensitivity of hydroxysteroids by LC-MS [6–8]. Thispaper describes the development of a highly sensitive ultra performance liquid chroma-tography – tandem mass spectrometry (UPLC-MS-MS) method for the screening of ana-bolic steroids in equine urine by incorporating a chemical derivatisation step, usingpicolinic acid as the derivatisation reagent.
The method involved solid-phase extraction (SPE) of both free and conjugated anabolicsteroids in equine urine using an Abs Elut Nexus cartridge. The conjugated steroids in theeluate were hydrolysed by methanolysis [9] and the resulting extract was further cleanedup by liquid-liquid extraction. The steroids in the extract were derivatised with picolinicacid to form the corresponding picolinoyl esters and analysed by UPLC-MS-MS in thepositive ESI mode with selected-reaction-monitoring (SRM). Separation of the targetedsteroids was performed on a BEH C18 UPLC column (10 cm L x 2.1 mm ID with 1.7 μmparticles). Over 30 anabolic steroids (including 17β-estradiol, 5(10)-estrene-3β,17α-diol,5α-estrane-3β,17α-diol, 5α-estran-17α-ethyl-3α,17β-diol, 5α-androstan-17α-methyl-3α/β,17β-diols, androstanediols, nandrolone and testosterone) spiked in negative equineurine at low to sub ppb levels could be consistently detected. The instrument turnaroundtime was 10.5 minutes inclusive of post-run equilibration. With exceptional separationachieved by the UPLC system, matrix interferences were minimal for the selected transi-tions at the expected retention times. As detection was performed with an UPLC systemcoupled to a fast-scanning triple quadrupole mass spectrometer, the method could easilybe expanded to accommodate additional steroid targets. Method validation data, includ-ing sensitivity, reproducibility and recovery, will be presented.
1. N.H. Yu, E.N.M. Ho, D.K.K. Leung, T.S.M. Wan, J Pharm Biomed Anal, 2005, 37, 1031–8.2. E.N.M. Ho, D.K.K. Leung, T.S.M. Wan, N.H. Yu, J Chromatogr A, 2006, 1120, 38–53.3. N.H. Yu, E.N.M. Ho, F.P.W. Tang, T.S.M. Wan, A.S.Y. Wong, J Chromatogr A, 2008, 1189, 426–434.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
4. F. Guan, C.E. Uboh, L.R. Soma, Y. You, Y. Liu, X. Li, J Mass Spectrom, 2010, 45, 1270–9.5. N. Deshmukh, I. Hussain, J. Barker, A. Petroczi, D.P. Naughton, Steroids, 2010, 75, 710–4.6. A. Honda, T. Miyazaki, T. Ikegami, J. Iwamoto, K. Yamashita, M. Numazawa, Y. Matsuzaki, J Steroid Bio-
chem Mol Biol, 2010, 121, 556–64.7. K. Yamashita, K. Yamazaki, S. Komatsu, M. Numazawa, J Am Soc Mass Spectrom, 2010, 21, 249–53.8. C. Zu, H.N. Praay, B.M. Bell, O.D. Redwine, Rapid Commun Mass Spectrom, 2010, 24, 120–8.9. P.W. Tang, D.L. Crone, Anal Biochem, 1989, 182, 289–294.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-115-MO
Rapid Selective Diagnosis of Bacillus Anthracis Infectionby LC-MS Detection of Endoproteinase Activity of Antibody Captured
Anthrax Lethal Factor in Serum
Zsuzsanna Kuklenyik1, Anne E. Boyer1, Renato Lins2, Conrad P. Quinn1,Maribel Gallegos-Candela1, John R. Barr1
1Centers for Disease Control and Prevention,4770 Buford Hghwy, Atlanta, GA 30341, USA
2Battelle Atlanta Analytical Services,2987 Clairmont Rd NE, Atlanta, GA 30329, USA
Diagnosing and treating anthrax at the earliest stage of disease is critical. We developed amethod for early diagnosis of anthrax infection by detecting anthrax lethal factor (LF) atthe attomole/mL concentrations in plasma or serum. The method uses monoclonal anti-body-magnetic bead capture and quantification of LF endoproteinase activity by liquidchromatography (LC) separation and tandem mass spectrometry (MS/MS) detection ofpeptide cleavage products. The limit of detection was 0.0025 ng/mL using 200 μL serumor plasma. With this highly magnified approach LF detection and anthrax diagnosis maybe possible as early as 12 h after spore exposure, as shown in both rabbits and rhesusmacaques with inhalation anthrax. The detection limit and response rate are affected byreaction time and bead surface concentration of LF. By controlling the magnetic beadamount and particle size, and controlled substrate cleavage reaction times a stable linearcalibration range from 0.05–2.5 ng/mL can be achieved when measured after 2 h, andfrom 0.005–1.0 ng/mL after 18 h incubation time. The coefficient of variation for qualitycontrol samples was 6–12%. The level of accuracy and precision of the LF measurementallows tracking the course of clinical treatment in patients with advanced inhalationanthrax and evaluation of the efficacy of anthrax therapeutics.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-117-MO
The Development of the Method for the Analysis of Neopterinand its Derivatives by Means of UHPLC-MS/MS and UHPLC-FD
Helena Tomšíková, Hana Vlcková, Lucie NovákováDepartment of Analytical Chemistry, Faculty of Pharmacy in Hradec Králové, Charles University in Prague,
Heyrovského 1203, 500 05 Hradec Králové, Czech Republic
Neopterin [2-amino-6-(1, 2, 3 – trihydroxypropyl)-1H-pteridin-4-one] belongs to thefamily of pteridines, which have become important in cancer screening research in thelast three decades. Concentrations of different pteridine derivatives can indicate thepresence of cancer [1]. Neopterin is released by monocytes/macrophages after stimula-tion with interferon-γ and plays a role in the physiological functions [2]. It reflects theimmune activation status. Measurement of neopterin concentrations thus enables thestudy of the immune system and its interaction within the pathogenesis of tumour devel-opment [1]. The determination of neopterin is the most important in clinical applicationssuch as the monitoring of malignant diseases and chronic infections, differential diagnosisof acute viral and bacterial infections, monitoring of immune-stimulatory therapy, prog-nostic indication of HIV infections and early indications of complications in allograft recipi-ents [2–4]. Neopterin can be easily detected in serum and urine [2, 4].
The aim of the study was determination of neopterin and its derivatives (dihydro-biopterin, biopterin and dihydroneopterin) by means of UHPLC-MS (Ultra High PerformanceLiquid Chromatography with Mass Spectrometry) and UHPLC-FD (with fluorescence detec-tion). The development of the chromatographic separation method was focused on optimi-zation of HILIC (Hydrophilic Interaction Chromatography) conditions (mobile phase, pH,flow rate etc.) on different stationary phases (BEH HILIC and BEH Amide). The best separa-tion of pteridine derivates was obtained in a pH range of 4.8 – 7.8. Optimized conditions forUHPLC-FD were: acetonitrile and 50mM ammonium acetate buffer pH 6.8 (85:15) as themobile phase for isocratic elution at flow rate 0.4 ml/min. Optimal conditions for UHPLC-MSwere achieved by using 23% 1mM ammonium acetate (pH 4.8) and acetonitrile. It wasbetter to use higher concentrations of ammonium acetate buffer for FD, while MS detectionneeded lower concentrations in order to prevent signal suppression. As regard to columnsorbents, BEH HILIC stationary phases demonstrated lower retention properties and theinsufficient selectivity compared to BEH Amide. The sensitivity and linearity of both LC-FDand LC-MS were compared.
1. Gamagedara S, Gibbons S, Ma YClin Chim Acta. 412 (2011) 120–8.2. Hamerlinck FF. Exp Dermatol. 8 (1999) 167–76.3. Sucher R. et al., Cancer Lett. 287 (2010) 13–22.4. Tomandl J, Tallova J, Tomandlova M, Palyza V, J. Sep. Sci., 26 (2003) 674–678.5. Nováková L, Kaufmannová I, Jánská R, J Sep Sci. 33(2010) 765–72.
The authors gratefully acknowledge the financial support of the project GA UK 384111/2011.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-118-TU
Unambiguous Determination of Milk Phospholipidsby HILIC Coupled to an Hybrid Mass Analyzer
Filomena Cichello1, Francesco Cacciola1, Paola Donato2,1,Paola Dugo1,2, Luigi Mondello1,2
1Dipartimento Farmaco-chimico, University of Messina,viale Annunziata, 98168 Messina, Italy
2University Campus Bio-Medico,Rome, Italy
In HILIC chromatography, retention mechanisms consist in a combination of hydrophilicinteraction, ion-exchange, and reversed-phase retention, which results in unique selectivi-ty for polar analytes such as phospholipids (PLs). The challenge in their analysis arise fromtheir low abundance with respect to the nonpolar triglycerides, and the simultaneousoccurence of a number of positional and structural isomers. Taking into account the highvariety of fatty acids that could be found bound to the particular structure of a single PLsgroup, the number of different compounds that should be identified is extremely great.As a result, PLs analysis typically involves different steps, consisting in fat extraction frommilk, isolation of PL fraction from the other lipid classes, separation and detection of thedifferent phospholipid classes and or species.
In this contribution, we achieved fully characterization of PLs classes, by high-resolutionchromatography on a shell packed HILIC column (2.7 μm). Fast-scanning and sensitivedetection was attained on-line by ESI ionization followed by a hybrid mass spectrometer(IT-TOF). The ESI-generated molecular ion species and fragments provided useful informa-tion in determining the distinct PLs profile of cow and donkey milk samples.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-119-MO
Stability of Linear Polyacrylamide Coated Capillaries in Acidic Mediain the Presence of Organic Modifiers and Surfactants
Philippe Anres, Nathalie Delaunay, Pierre GareilLaboratory of Physicochemistry of Electrolytes, Colloids and Analytical Sciences (PECSA),
Chimie ParisTech, CNRS,11 rue Pierre et Marie Curie, 75231 Paris Cedex 05, France
Since its first introduction in 1985 by Hjerten [1], linear polyacrylamide (LPA) coatedcapillaries have been widely used because of the unique properties of LPA (i) to suppresselectroosmotic flow (EOF) almost entirely and (ii) to reduce considerably adsorption ofhydrophobic compounds such as proteins or surfactants (SDS, ionic liquids). Both of theseproperties are mainly linked to its neutral and very hydrophilic character. These propertiesand the simplicity of the coating procedure made LPA-coated capillaries a prime choicefor in-situ preconcentration techniques in capillary electrophoresis involving stacking andsweeping, where EOF suppression is required to enhance performances.
Usually, with this kind of capillary, pH of background electrolytes (BGEs) are restrictedto the 4–8 range because it appears the stability of the coating is an issue for long lifeutilisation at pH below 4 and higher that 8. Indeed, in high acidic or basic conditions,acrylamide moieties can be hydrolysed into carboxylic groups, which alter capillary prop-erties regarding EOF and wall adsorption. However, to the best of our knowledge nostudies on the stability of capillaries in this pH range have been reported. Moreover, inMEKC and sweeping techniques organic modifiers are often used to tailor selectivity andno report is available on the stability of LPA-coated capillaries under such conditions.
The aim of this work was to study the stability of LPA coated capillary with a view totheir use with acidic background electrolytes (pH 2–4) in the presence of organic modifi-ers (acetonitrile and methanol), anionic surfactants (SDS) and long chain ionic liquid(dodecyldimethylimidazolium bromide). To this end, capillaries were coated followingHjerten’s procedure and allowed to be submitted for 70 hours to the different conditionsstudied, while Vigh’s tests were performed every hour to follow the evolution of EOF.
The results obtained allow concluding the coating procedure gives capillaries whereEOF is efficiently reduced and stable over 70 hours of utilisation. Consequently, this kindof capillaries can be used for preconcentration techniques in acidic media with organicmodifiers and surfactants. This opens up new possibilities to implement field enhancedsample injection (FESI)-Sweep-MEKC to preconcentrate compounds and next to separatethem in acidic media [2] and the utilisation of such capillaries will, for sure, gives betterresults in term of reproducibility and sensitivity enhancement, because the EOF is morestable and reduced comparing to non coated capillaries at pH in the range 2–4.
1. S. Hjerten, M-D. Zhu, J. Chromatogr. 1985, 346, 265.2. J.P. Quirino, S. Terabe, Anal. Chem. 2000, 72, 1023.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-120-TU
A Sensitive Method for the Determination of Ochratoxin Aby Capillary Zone Electrophoresis using Fluorescence Detection
and its Application to Certain Foods
Muzaffer Tuncel1, Dilek Dogrukol-Ak1, Rasime Demirel3, Elif Mine Oncu2,Ulku Dilek Uysal2, Merih Kivanc3
1Anadolu University, Faculty of Pharmacy, Department of Analytical Chemistry,26470, Eskisehir, Turkey
2Anadolu University, Faculty of Science, Department of Analytical Chemistry,26470, Eskisehir, Turkey
3Anadolu University, Faculty of Pharmacy, Department of Microbiology,26470, Eskisehir, Turkey
A capillary zone electrophoretic method with fluorescence detection for the determinationof ochratoxin A and its application to food samples is described in this study. Separationwas performed in an electrolyte consisting of borate buffer (40 mM, pH 9.24) and aceto-nitrile (10%, v/v) using a fused silica capillary column (59 cm of total and 43 cm of effec-tive length with internal diameter 75 μm). Fluorescence intensity of ochratoxin A wasdetected with excitation filter of 200–400 nm and emission light filtered through a 495nm bandpass filter. The best peak morphologies and short retention time were observedby using optimum conditions such as 25 kV of applied potential, 8 s of injection time and25 °C of temperature. The detection step requires only about 8 min without washingprocesses. Validation of the method was investigated as precision, accuracy and linearityin the mentioned conditions and quality control in cherry juice matrix. Sample clean-upwas performed by using an immunoaffinity column providing 50 times pre-concentrationof analyte in samples. Linearity of the method was shown in the range of 4.0 – 40.0ng/mL for ochratoxin A with a good correlation coefficient (0.9993). Recovery of methodwas found to be in the range of 105 and 93% with good precision. The limit of quanti-fication of CZE-FL method was 4 ng/mL in matrix. The method was applied to differentfood samples such as wine, various fruit juices, cheese, bread and instant coffee for thedetermination of ochratoxin A and their results were compared to those of the HPLCmethod. It was found that there was statistically insignificant difference between themethods.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-121-MO
Application of Micellar Electrokinetic Chromatography Methodfor Determination of Oxidized and Reduced Glutathione
in Human Blood
Paulina Chyla, Dariusz Wideł, Ewelina Blonska, Zygfryd WitkiewiczInstitute of Chemistry, Jan Kochanowski University,
Poland, 15 Swietokrzyska Str., 25-406 Kielce, Poland
Micellar electrokinetic chromatography (MEKC), was applied for glutathione analysis inhuman blood. MEKC is a modification of capillary electrophoresis, where the samples areseparated by differential partitioning between micelles and a surrounding aqueous buffersolution (mobile phase). Glutathione (GSH) is in a constant state of metabolic turnover.Because it is actively synthesized, it also must be degraded. In the first step of GSH syn-thesis, an amide linkage is formed between cysteine and glutamate catalyzed by g-gluta-mylcysteine synthetase.
GSH synthetase catalyzes the reaction between amine residue of glycine and the cyste-ine carboxyl from g-glutamylcysteine dipeptide to form GSH. GSH is transported out ofthe cell and degraded by the membrane-bound enzyme g-glutamyltransferase, whichremoves the g-glutamyl moiety, and by dipeptidases, which remove the glycine moiety.
Glutathione is present in most of the plants and animals tissues that constitute humandiet. Plasma aromatic and sulfur containing amino acids are good indicators of proteinanabolism/catabolism, while blood reduced and oxidized glutathione reflect oxidativestatus in an organism. The reduced glutathione/oxidized glutathione (GSH/GSSG) ratio isused to evaluate the oxidative stress status in biological systems.
A review of literature revealed that there were some studies on specific determinationof glutathione using micellar electrokinetic chromatography and on-column reaction with2,29-dipyridyldisulfide [1].
Blood samples from four persons in different age were collected and prepared foranalysis. To perform the analysis of glutathione P/ACE MDQ CE Beckman system wasused. Quantitative determination of reduced and oxidized glutathione form was obtained.The analysis last about 10 min what shows that MEKC is a quiet fast and useful methodfor glutathione determination. However the blood samples must be fresh so they have tobe analyzed quickly after collecting them.
1. Z. Glatz, H. Mašlanova, Specific thiol determination by micellar electrokinetic chromatography and on-column detection reaction with 2,2’-dipyridyldisulfide, J. Chromatogr., A 895 (2000) 179–187.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-122-TU
Application of Microemulsion Electrokinetic Chromatographyfor Quantitative Analysis of αα-Tocopherol Acetate
in Pharmaceutical Preparations
Dariusz Wideł, Jerzy Oszczudlowski, Zygfryd WitkiewiczInstitute of Chemistry, Jan Kochanowski University,
15 Swietokrzyska Str., 25-406 Kielce, Poland
Microemulsion electrokinetic chromatography (MEEKC) method was modified and ap-plied for quantitative analysis of α-tocopherol acetate in five different vitamin E phar-maceuticals. Alfa-tocopherol acetate also known as vitamin E acetate, is a common vita-min supplement with the molecular formula C31H52O3. Alfa-tocopherol acetate is notoxidized and can penetrate through the skin to the living cells, where about 5% is con-verted to free tocopherol and provides beneficial antioxidant effects. Naturalα-tocopherol, occurs only as d-α-tocopherol. Synthetic α-tocopherol, used as food preser-vative, is a mixture of eight diastereoisomers, and is known as d, l-α-tocopherol.
The final microemulsion used for analysis of α-tocopherol acetate was prepared in thefollowing way: 10% (w/w) sodium dodecyl sulphate (SDS), 6.6% (w/w) 1-butanol, 0,8%(w/w) n-octane, 12.5% (w/w) 2-propanol, 40% (w/w) phosphate buffer pH = 2.5 and30.1% (w/w) water. The achieved migration time of α-tocopherol acetate is about 7 min.Different concentrations and amounts of SDS were used to obtain the best results of theanalysis. P/ACETM MDQ Capillary Electrophoresis System with diode array detector wasapplied to perform analysis.
A review of the literature revealed that there have been a few methods for the determi-nation of α-tocopherol acetate, including gas chromatography with flame-ionization detec-tion, methods based on high-performance liquid chromatography (HPLC) with fluorometricdetection and microemulsion electrokinetic chromatography with diode array detector [1].A simple and rapid voltammetric method has also been developed for the quantitativedetermination of α-tocopherol acetate in pharmaceutical preparations [2].
The analysis by HPLC method with UV-VIS detector was also performed in order tocompare the results with MEEKC method for quantitative analysis of α-tocopherol acetate.
1. S. Pedersen-Bjergaard, Ø. Næss, S. Moestue, K.E. Rasmussen, Microemulsion electrokinetic chromatogra-phy in suppressed electroosmotic flow environment: Separation of fat-soluble vitamins, J. Chromatogr., A876 (2000) 201.
2. S. Michałkiewicz, M. Pryciak, J. Małyszko, J. Oszczudłowski, Voltammetric Determination of α-TocopherylAcetate in Pharmaceutical Dosage Forms, Electroanalysis 16 (2004) 961.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-123-MO
Capillary Electrophoresis Analysis of Carbohydratesin Post-Blast Residue Extracts
Cédric Sarazin1,2, Nathalie Delaunay2, Christine Costanza1,Véronique Eudes1, Pierre Gareil1
1Central Laboratory of the Prefecture de Police,39 bis, rue de Dantzig, 75015 Paris, France
2Laboratory of Physicochemistry of Electrolytes, Colloids and Analytical Sciences (PECSA), Chimie ParisTech,UPMC Paris 06, CNRS, 11 rue Pierre et Marie Curie, 75231 Paris Cedex 05, France
Chlorate, perchlorate or nitrate salts are widely used in improvised explosive composi-tions. The addition of reducing agents, as commercial carbohydrates (saccharose, lac-tose, glucose, fructose…), in solution containing oxidant salts creates an importantgaseous release (CO or CO2) and thus amplifies the explosive properties of the mixture.The study of carbohydrate contents in extract is thus very important when chlorate,perchlorate, or nitrate anions were detected. Traditionally, carbohydrate compoundswere analyzed by TLC, LC or GC, but with long analysis times and ofter poor limits ofdetection around 100 mg L−1. In order to dispose of a faster, cheaper, and powerfulalternative technique, capillary electrophoresis (CE) was then tested. However, direct CEanalysis of carbohydrates under their anionic form turns out to be tricky due to theirhigh pKa values (comprised between 12 and 13.5) and their lack of UV absorbance. Thatis why, CE methods using in-situ chelation by borate anion are nowadays the preferredmethods.
However, Rovio et al. [1], recently demonstrated that in a highly alkaline backgroundelectrolyte (BGE) composed by sodium hydroxide and phosphate buffer, carbohydratescould be separated and detected under the enolate ion form at 270 nm. Preliminarystudies show that similar results were obtained when NaOH/phosphate buffer was com-pared to pure NaOH BGE and that cations present in BGE (Na+, K+, Li+, tetrabutyl-ammonium) played an important role in the enolate formation. In order to simplify theBGE composition, BGE composed with only NaOH was selected for the end of the study.The CE method was then optimized for BGE pH and conductivity and temperature by amultivariate approach. Thus, a separation of the 4 carbohydrate of interest and 7 carbo-hydrates potentially present in real samples (ribose, xylose, mannose, maltose, galactose,mannitol, and sorbitol) was obtained in less than 15 min. Obtained limits of detectionwere almost 15 mg L−1 and intermediate precisions were calculated at less than 5%. Thismethod appeared to be sensitive enough and reproducible.
It is important to notice that unusual phenomena were observed during the detectionby direct UV absorbance of the carbohydrates at 270 nm, bringing the presence of aphotochemical reaction in the separation window into play. In fact, the influence of

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
parameters as lamp intensity, capillary diameter, and residence time in detection window,were highlighted by comparing corrected areas and absorbances for carbohydrates andfor an internal standard.
Finally, real extracts issued from real samples collected after explosions were analyzedand the presence of carbohydrates was proved.
1. Rovio S., Yli-Kauhaluoma J., Siren H., Electrophoresis 2007, 28, 3129–3135.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-124-TU
Capillary Zone Electrophoresis for Caco-2 Cell Cytotoxicity Analysis
Lu Zhang1, Feng Qu1, Meiling Hu1, Jinmei Ding2, Beilei Lou1
1School of Life Science, Beijing Institute of Technology,Beijing, 100081, China
2 National Institute of Biological Sciences,Beijing, 102206, China
A new method to evaluate the cytotoxicity of substances by capillary zone electrophoresis(CZE) was established. The estimation of the injected cell number and the effect of injec-tion conditions on cytotoxicity determination were investigated. Caco-2 cell, the bestmodel of the intestinal absorptive epithelium, was treated by substances and then stainedby Trypan Blue and fixed by paraformaldehyde. The treated Caco-2 cell samples wereanalyzed by CZE, the simultaneous detection at 590 nm and 214 nm indicated the sub-stances cytotoxicity. The method reflected simultaneously the loss of cell membraneintegrity and the degradation / leak of intracellular components by two absorbance values(the ratio of R590/214), which was more accurate and sensitive. The cytotoxicity of foursubstances Na2SO3, MeHg, Paclitaxel and CdCl2 were determined and compared. Theresults were in good agreement with the references and the conventional Trypan Blueexclusion counting assay.
Acknowledgment: This work was supported by the National Natural Science Foundation of China (No.20875009), and the Key Laboratory of Environmental Chemistry and Ecotoxicology (KF2009-09), ResearchCenter for Eco-Environmental Sciences, Chinese Academy of Sciences.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-125-MO
Covalent Binding of Phospholipids on Fused Silica Capillariesfor Electrochromatography
Jana Lokajova1, Heidi Tiala1, Tapani Viitala2,Marja-Liisa Riekkola1, Susanne K. Wiedmer1
1Laboratory of Analytical Chemistry, Department of Chemistry,POB 55, 00014 University of Helsinki, Finland
2Division of Biopharmaceutics and Pharmacokinetics,Viikinkaari 5 (P.O. Box 56), Faculty of Pharmacy, 00014 University of Helsinki, Finland
The octanol-water partition coefficient (logP) is still today the most common measure forevaluating of drug-lipid membrane interactions. Even though it provides the descriptionof hydrophobic behavior, it fails in mimicking the biomembrane environment, mostly dueto considering only non-polar interaction. The big disadvantage of the commercial immo-bilized artificial membrane chromatography phases is that currently there is only phos-phatidylcholine (PC)-based stationary phases available and they are very expensive. PC isby far the most common lipid in biological membranes, however, it poorly representsbiological membranes because of its zwitterionic character. Instead, most biologicalmembranes are negatively charged and have complex lipid compositions.
We have developed a rapid method for covalent binding of phospholipids containingprimary amino groups to silica capillary. The procedure consists of three steps. Firstly, thecapillary is aminosilanized, then it is covalently bound with aldehyde, and the final stepconsists of nucleophilic reaction of the lipid primary amino group with aldehyde groupsattached to the silica capillary. Various types of liposomes with a broad range of composi-tions and concentrations were tested and the need for primary amino group was studied.The thickness and other properties of the attached phospholipid layer were measuredusing quartz crystal microbalance technique. The capillary surface with covalently at-tached phospholipids was tested as a high throughput assay for the determination ofinteraction between the phospholipid bilayer and the aqueous phase. A set of structurallydiverse drugs was applied into the phospholipid coated capillary and the interaction ofdrugs with phospholipid layers was quantified in means of retention factors and distribu-tion constants. Since only 2.5% of lipid containing primary amino group was needed forstable covalent binding of phosholipids to the silica capillary wall, the method offers fine-tuning of the lipid composition to mimic in vivo conditions.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-126-TU
Determination of Acetylcholinesterase Inhibitorsusing Capillary Electrophoresis – Mass Spectrometry
Irene N. Nicolaou, Constantina P. Kapnissi-ChristodoulouUniversity of Cyprus,
P.O. Box 20537, 1678, Nicosia, Cyprus
The goal of this research is the development of a Capillary Zone Electrophoresis (CZE) –Electrospray Mass Spectrometry (ESI-MS) method for the determination of the Acetylcho-linesterase Inhibitor (AChEI) rivastigmine. Rivastigmine is a pseudo-irreversible carbamateinhibitor of actylcholinesterase, and it is clinically used for the symptomatic treatment ofmild to moderate Alzheimer’s Disease (AD) patients. AD is one of the most commoncauses of mental deterioration in people of advanced age.
Among all the separation techniques used for pharmaceutical analysis, such as HighPerformance Liquid Chromatography (HPLC) and Gas Chromatography (GC), CE hasbeen established as a powerful analytical tool that provides superior resolution capability,reduced operating cost and low consumption of sample and solvents. The online couplingof CE with a powerful MS detector can improve detection sensitivity. Thereby, CE-ESI-MSis a promising alternative for the identification of pharmaceutical compounds in biologicalfluids.
In the present study, a CE-ESI-MS method was developed for the analysis ofrivastigmine in human plasma. Several electrophoretic and spectrometric parameters wereexamined in order to improve the signal-to-noise ratio and consequently the limit ofdetection. These parameters were classified into three categories: the background electro-lyte parameters, such as the concentration, the pH and the use of organic modifier, thesheath liquid parameters, such as the composition, the methanol content and the flowrate, and the spray chamber parameters, such as the temperature and the flow rate of thedrying gas and the nebulizer gas pressure. In addition, the optimized parameters werethen used for the determination of rivastigmine in human plasma. Several sample prepa-ration procedures were investigated for the extraction of rivastigmine from plasma, andthey were compared, in order to determine the most effective sample pre-treatmentmethod.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-127-MO
Determination of Acidity Constants by the CE Internal StandardMethod: Polyprotic Compounds
Joan Marc Cabot, Elisabet Fuguet, Clara Ràfols, Martí RosésDepartament de Química Analítica, Universitat de Barcelona,
Martí i Franquès 1-11, E-08028 Barcelona, Spain
The determination of acidity constants is of main interest, especially in the pharmaceuticalindustry. Many potential drugs are weak acids or bases, and their use in further studiesstrongly depend on the value of certain physicochemical parameters such as the acidityconstant, hydrophobicity, and solubility.
Several methods to determine pKa values have been described in the literature. Amongthem, capillary electrophoresis (CE) offers several advantages the presence of impurities isnot so important and only low amounts of sample are required. However, the classical CEmethod implies the measurement of the mobility of the substance of interest at severalpH values by the preparation of adequate buffers at constant ionic strength in differentpH ranges. The internal standard method is based on the use of an internal standard ofsimilar pKa value to the analyte. It has all the advantages of CE as method for pKa determi-nation, but in this case the number of measured points is lower and no pH measurementof the buffer solutions is required. With appropriate standards, the method minimizes theinteractions of analyte with the buffers.
In this work this fast CE method is developed for the routinary determination of acidityconstants of polyprotic compounds. In this case, several monoprotic internal standardsare needed (one for each acidity constant of the analyte), or only one unique standardwith the same number and type of acid-base groups than the analyte, i.e. a congenericcompound. The performance of both modes is tested.
1. E. Fuguet, C. Ràfols, E. Bosch, M. Rosés, J. Chromatogr. A 1216, 2009, 3646–36512. J.M. Cabot, E. Fuguet, C. Ràfols, M. Rosés, J. Chromatogr. A 1217, 2010, 8340–83453. E. Fuguet, C. Ràfols, E. Bosch, M. Rosés, Chemistry & Biodiversity 6, 2009, 1822–1827

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-128-TU
Determination of Alcaline Cation Mobilityin Background Electrolytes Containing Micelles
Ludmila Müllerová, Pavel Dubský, Jana Svobodová, Bohuslav GašCharles University in Prague, Faculty of Science, Department of Physical and Macromolecular Chemistry,
Albertov 2030, 128 43 Prague 2, Czech Republic
Background electrolytes (BGEs) containing micelles are widely used in CE. Mobility ofhydrophobic analytes, which are more or less incorporated in the core of the micelles, butalso mobilities of small anorganic ions are influenced by the presence of micelles in theBGE. Understanding interactions between anorganic ions and micelles can simplify predic-tion of the behavior of the micellar BGEs.
Determination of accurate effective mobilities in micellar BGEs can be complicatedbecause neutral EOF markers can interact with micelles. In this work a new method formobility determination in the presence of interacting agents in the BGE was used toensure that determination of EOF mobility is not influenced by interaction of the neutralmarker (thiourea) with micelles.
This method is based on the same principle as method developed by Williams and Vigh[1] (marker zone is situated in interacting agent free BGE, while analyte zone is in BGEcontaining interacting agent, pressure mobilization of capillary content is used). The twodetector setup is utilized for the method and the distance between them is elongated bya capillary loop. Mobility determination is based on known distances between the capil-lary inlet and the first detector and between the first and the second detector.Complexation of the marker by the interacting agent is prevented by surrounding themarker zone with interacting agent free BGE.
Mobility of sodium cation was measured by this two-detector method. Comparing theresults with classical CE measurements, thiourea was found to be suitable EOF marker forBGEs containing lithium dodecylsulphate micelles. Dependences of effective mobilities ofalkaline cations (Cs+, K+, Na+) on lithium dodecylsulphate concentration were measuredand evaluated.
1. Williams B. A., Vigh G.: Anal. Chem. 1997, 69, 4445.
This work was financially supported by the Grant Agency of the Charles University grant number 51009, theMinistry of Education, Youth and Sports, Long Term Research Plan MSM0021620857 and the Grant Agencyof the Czech Republic, grant number 203/08/1428.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-129-MO
Development of a Generic MEKC Method for the Separationof 15 Antimalarial Drugs by a Design Space Approach
Caroline Lamalle1, Roland Marini2, Benjamin Debrus2, Pierre Lebrun2,Anne-Catherine Servais1, Jacques Crommen1, Philippe Hubert2, Marianne Fillet*1
1Laboratory of Analytical Pharmaceutical Chemistry, Department of Pharmacy, University of Liège, Belgium2Laboratory of Analytical Chemistry, Department of Pharmacy, University of Liège, Belgium
Malaria is one of the most important parasitic diseases affecting people especially inAfrica. As counterfeit is largely present in the African market, it is important to develop asimple method which can control the conformity of medicines used against this disease.The interest of a generic method is to permit the quality control of pharmaceutical formu-lations whose quantitative and qualitative composition is not well known. For the mo-ment no capillary electrophoresis (CE) method has been developed to analyse simulta-neously the major antimalarials but, due to its low consumption of solvent, this techniquehas an economical advantage for an implementation in Africa.
This project consists in the analysis of 15 antimalarials (artesunate, artemether, amodia-quine, chloroquine, piperaquine, primaquine, quinine, cinchonine, mefloquine, halofantrine,sulfadoxine, sulfalen, atovaquone, proguanil and pyrimethamine). In this particular applica-tion, capillary zone electrophoresis (CZE) is not recommanded as all these molecules couldnot be charged at the same pH. Micellar electrokinetic chromatography (MEKC) was thenpreferred because it allows separation of charged and neutral compounds.
Preliminary experiments were first realised to select the most crucial factors for anti-malarials separation. Several conditions were tested and four parameters as well as theirinvestigation domain were then chosen: pH (5–10), SDS concentration (20–90 mM), aceto-nitrile proportion (10–40%) and temperature (20–35 °C). Then, an experimental designmethodology using a central composite design (CCD) was carried out. Twenty five experi-ments were defined by CCD and tested. The experiments performed at low pH could notlead to the detection of all analytes. Therefore, a custom design was performed by theremoval of 5 conditions and the addition of 8 conditions at high pH. Migration times (at thebeginning, the apex and the end) were extracted for each molecule in each condition.
Mathematical modelling allowed the prediction of the optimal condition in terms of analyteseparation. This prediction was verified experimentally and could lead to the separation of 13compounds in 7 minutes. As this condition did not lead to a complete separation of the 15molecules, the buffer concentration and the voltage were modified to enlarge the migrationwindow. Finally the 15 antimalarial drugs were nicely separated within 30 minutes.
* Mailing address: University of Liege, Laboratory of Analytical Pharmaceutical Chemistry, B36, tower 4,Avenue de l’Hopital 1, B-4000 Liege, Belgium

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-130-TU
Drug Quantification in the Presence of Matrix Proteins:A MEKC Method with Direct Sample Injection
Sascha Kühne1, Christopher Untucht2, Michael Steinert2, Hermann Wätzig1
1TU Braunschweig, Inst. f. Pharmazeutische Chemie,Beethovenstraße 55, 38106 Braunschweig, Germany
2TU Braunschweig, Inst. f. Mikrobiologie,Spielmannstraße 7, 38106 Braunschweig, Germany
In order to adopt a general workflow for complex biological matrices with respect to anew blood-brain barrier (BBB) model, a micellar electrokinetic chromatography methodhas been developed and optimised. For this purpose, a set of six drugs (acetaminophen,caffeine, carbamazepine, cimetidine, indometacin and propranolol) with various, knownpermeation characteristics has been investigated. An aqueous stock solution of each drugwas diluted with the growth medium Quantum 286 (PAA Laboratories, Pasching, Austria)to a final concentration of 0.1 mg/mL and transferred to the BBB model. The drug con-centrations of both sides of the BBB model were determined after six hours of incubation.As a result of method optimisation, separations were carried out using a 60 mM boratebuffer containing 200 mM of SDS at 30 kV voltage, leading to an analysis time of lessthan 10 min. Between runs the capillary (fused silica, ID: 50 μm, L: 50 cm, l: 42 cm) wasrinsed with a mixture of equal parts of running buffer and isopropanol (70% v/v) whichproved to be very effective to remove matrix compounds. Drug-protein interactions werereliably suppressed by the high SDS concentration; hence proteins from the growthmedium did not impede the analysis. The LOD ranged from 10 to 30 ng/mL, the typicalRSD% from 0.8% to 4% for the migration times and from 2% to 6% for the correctedpeak areas. The logarithm of the brain/blood ratio (logbb) has been calculated, showinggood to very good accordance to the reference values.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-131-MO
Electrochromatography Separation of Tryptic Digests of Enzymesusing Lauryl Methacrylate Monolithic Columns
Miriam Beneito-Cambra, Pascuala Vizcaino-Milla, Iván Esteve-Adell,Guillermo Ramis-Ramos, José Manuel Herrero-Martínez
Department of Analytical Chemistry, University of Valencia,Doctor Moliner 50, E-46100 Burjassot, València, Spain
Lauryl methacrylate monolithic columns with cationic functionalities have been designedfor capillary electrochromatography (CEC) have been prepared by UV irradiation usingtwo initiators, azobisisobutironitrile (AIBN) and lauroyl peroxide (LPO). The application ofthis type of columns to the separation of complex peptide mixtures was studied. Peptidemixtures were obtained by tryptic digestion of several enzymes commonly used in house-hold cleaning products (proteases, amylases, lipases and cellulases). Thus, the synthesis ofthe monolith was optimized to separate the resulting peptide mixtures. Particularly, themobile phase composition and the ratio of porogenic solvents in the polymerizationmixture were adjusted to obtain highly permeable rigid monoliths with adequate columnefficiency. Samples of the different classes of enzymes were analyzed, and the CEC datawere used for multivariate identification and classification of the enzyme raw materials.
Acknowledgment: CTQ2010-15335 (MEC and FEDER) and V-Segles-Empresa grant (M. B.-C., Univ. of Valen-cia and Químicas Oro).

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-132-TU
High Resolution Intact Glycoprotein Analysis by Sheathless CapillaryElectrophoresis – Electrospray Ionization-Mass Spectrometry
Hans Dewald, Scott Mack, Gina Donvito, Kimberly Kwong, Chitra RatnayakeBeckman Coulter, Inc., Discovery Solutions Business Center,
Brea, CA, USA
The analysis of intact proteins by Electrospray Ionization – Mass Spectrometry (ESI-MS)provides the most direct route for the identification and characterization of these highlycomplex molecules. With the potential presence of multiple isoforms, it is also widelyaccepted that the presence of a liquid-phase separation technique upstream from the MSdetection step would greatly contribute to a complete deciphering of the structure ofthese molecular entities. Capillary electrophoresis (CE), compatible with the preservationof delicate protein structure, post-translational and other modifications, while simplifyingsample complexity through high resolution separation capabilities, appears as a veryvaluable alternative prior to introduction into the mass spectrometer. To achieve thegreatest advantage of coupling CE with ESI-MS for intact protein analysis, a porous,sheathless interface currently in development, was implemented for the analysis of amodel glycoprotein, Ribonuclease B. The efficient coupling between these liquid and gasphase separation systems allowed the rapid, sensitive and high resolution detection of thefive known Ribonuclease B glycoforms. Further deconvolution of the resulting Time ofFlight (ToF) mass spectra by the Maximum Entropy Algorithm identified differences in theattached high mannose structure as the source of the electrophoretic heterogeneity.Produced on multiple sheathless sprayers, the data confirm that these separations arehighly repeatable and reproducible.
The High Sensitivity Porous Sprayer interface is for Laboratory Use Only; not for use in diagnostic proce-dures.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-133-MO
Highly Emissive Metal Probes Suitable for Ultratrace Detectionof Lanthanide and Actinide Ions by
Capillary Electrophoresis-Laser-Induced Fluorescence
Shingo Saito1, Tomoko Haraga2, Yuta Nakano1, Yoshiyuki Sato1, Yutaka Kameo2,Kuniaki Takahashi2, Masami Shibukawa1
1Graduate School of Science and Engineering, Saitama University,255 Shimo-Okubo, Sakura-ku, Saitama 338-8570, Japan
2Japan Atomic Energy Agency,2-4 Shirakata-Shirane, Tokai-mura, Ibaraki, Japan, 319-1195
Capillary electrophoresis with laser-induced fluorescence detection (CE-LIF) is one of themost powerful separation and detection methods. It is, however, difficult to apply toheavy metal ion analysis due to the quenching effect accompanied by complexation witha fluorescent ligand. Recently, we have developed several fluorescent probes of transitionmetal ions for CE-LIF. The probes are composed of a chelating moiety (acyclic or macro-cyclic polyaminocarboxylate with hexa-octadentate), a fluorophore (fluorescein) and aspacer between these moieties. Using these probes in CE-LIF, ultrasensitive detection oftransition metal ions could be achieved without quenching.
In particular, this study documents the use of new metal probes for lanthanide (Ln) andactinide (An) ion analysis by CE-LIF. Determination of total concentrations of trace Ln andAn ions with rapid separation and low sample volume in radioactive wastes is of impor-tance with respect to monitoring of burnup and safe disposal.
Results in this study show that the hexadentate acyclic and macrocyclic probes providehigh selectivity and sensitivity for Ln ions in combination with dynamic ternary complexreactions in the capillary. Auxiliary ligands were added to the separation buffer in order toform such ternary complexes with the mother Ln-probe complexes. The ternarycomplexation gives different mobilities to the Ln ions, thus allowing their separation.
Furthermore, the An ions, Am3+, Th4+, UO22+ and NpO2
2+, were also investigated. Am3+
was detected for all probe complexes, while UO22+ was not. Acyclic octadentates provided
selective detection of Th4+. A signal for NpO22+ was obtained when using acyclic and
macrocyclic hexadentate or heptadentate structures, although a detailed relationshipbetween detectability and chelating structure is not yet clear. Nonetheless, highly sensi-tive detection of Ln and An was achieved with detection limits in the range of sub nM-pMlevels (mid-lower ppt).

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-134-TU
Highly-Sensitive Electrophoretic Analysis of Biomolecules by LVSEP
Saeko Kinami, Takayuki Kawai, Kenji Sueyoshi, Fumihiko Kitagawa, Koji OtsukaDepartment of Material Chemistry, Graduate School of Engineering, Kyoto University,
Kyoto 615-8510, Japan
Microchip electrophoresis has great advantages such as a low consumption of samplesand rapid analysis, while a low concentration sensitivity and complicated experimentalprocedures are still regarded as major problems. To overcome these drawbacks, large-volume sample stacking with electroosmotic flow (EOF) pump (LVSEP) was combinedwith microchip zone electrophoresis (MCZE), providing a highly sensitive analysis ofoligosaccharides on a single straight channel microchip [1]. In LVSEP-MCZE, the use of apoly(vinyl alcohol) (PVA) coated channel is important to suppress the non-specific adsorp-tion of oligosaccharides. In this paper, to extend the applicability of the LVSEP technique,the LVSEP-capillary zone electrophoresis (CZE) of peptides was investigated in the PVAcoated capillary as a preliminary study of the peptide analysis.
In the LVSEP-CZE analysis, the PVA-coated capillary is filled with a low ionic strengthsolution containing anionic analytes. After filling a background solution (BGS) of the highionic strength into both the anodic and cathodic vials, a constant voltage is applied. Theanalytes are concentrated to a narrow band at the cathodic side of the boundary betweenthe sample matrix and the BGS by the difference in the electric field strength. The con-centrated band migrates toward the cathode by a temporarily enhanced EOF, which isgenerated in the low-ionic strength sample matrix. After the sample matrix is removed,the EOF is suppressed due to the high-ionic strength BGS. The analytes then start migrat-ing toward the anode, and separated by CZE.
In LVSEP-CZE, the PVA-coated capillary was effective for suppressing not only the EOFbut also a non-specific adsorption of peptides. In the conventional CZE analysis of 50 ppbFITC-labeled Angiotensin I, II, and III, a baseline separation was achieved, but the ob-tained peak heights were low. In LVSEP-CZE, on the other hand, both higher peaks andgood resolutions were obtained in spite of the 100-fold dilution of the sample solution.Comparing with conventional CZE, a 175-fold sensitivity enhancement was attained inthe LVSEP-CZE analysis of FITC-Angiotensin II. Therefore, these results revealed that theLVSEP technique is useful for the peptide analysis. The application of LVSEP to MCZE isexpected to realize highly sensitive and high-throughput analysis of peptides, and furtherinvestigation is now in progress. (368 words)
1. Kawai, T. et al. Anal. Chem. 2010, 82, 6504–6511.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-135-MO
Infrared-Based Temperature Measurements for thorough AnalyticalInstrument Qualification in Capillary Electrophoresis
Claudia Cianciulli, Hermann WätzigInstitute of Pharmaceutical Chemistry, TU Braunschweig,
Beethovenstr. 55, 38106 Braunschweig, Germany
In Capillary Electrophoresis (CE), the temperature inside the capillary is one of the crucialparameters. Therefore the accuracy and stability of the temperature has to be included ina concept for Analytical Instrument Qualification (AIQ) of CE systems. It is thus required toaccurately consider the temperature generated by the applied electrical power.
Using an infrared (IR) thermometer the generation of Joule heating can be measuredon the outside of the capillary. The strictly linear relationship between applied power perunit length (P/L) and the obtained temperature is established over a wide range of variouselectrical field strengths, buffer systems and different capillary inner diameters. Moreover,the temperature can be calculated from different electrical parameters, for example fromthe conductance [1] or the increase in current [2]. Knowing the slope of this linear rela-tionship as being 6.3 °C m W−1, the temperature can be determined just from P/L, too. Inparticular, this relationship can be used for the AIQ. A set of buffers is defined whichshould give a certain power per unit length. If the later obtained results comply with thecorresponding specifications, it can be concluded that the thermostating systems workssufficiently.
1. Evenhuis, C. J., Guijt, R. M., Macka, M., Marriott, P. J., Haddad, P. R., Electrophoresis 2005, 26, 4333–4344.2. Knox, J. H., McCormack, K. A., Chromatographia 1994, 38, 207–214.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-136-TU
Investigation of Migration Behavior and Separation Mechanismof Coumarins in Capillary Electrophoresis
Shu-Ping Wang, Ruo-Shuo Yang, Ying Yuan ChangDepartment of Applied Chemistry, Providence University
200 Chungchi Road, Shalu, Taichung, 43301, Taiwan
Coumarins are widely spread in plants and essential oils, and have been used to treatmentof cancer and oedemas. In past years, lots of attention was paid on analysis of coumarins,but few of them were dealt with the effect of substituent on migration behavior in capil-lary zone electrophoresis. Thus, for investigating the separation mechanisms, we selectedten compounds with identical skeleton of coumarin in this study.
Through the discussion on the mechanisms, 4-hydroxycoumarin possesses the smallerpKa than others with various position of hydroxyl group. We supposed resonance may beformed within lactone ring and hydroxyl group that caused the proton was dissociatedfacilely. The value of pKa of acidic coumarins and the used pH of running buffer bothwould affect the migration behavior directly. Furthermore, micelle of SDS may havevarious partition interactions with neutral species and enhance the selectivity. Moreover,the substituent of coumarin would affect the polarity and be the key factor of the separa-tion mechanism. Finally, the adequate methanol (20%) would modify the interactionbetween coumarin and micelles and provide a suitable peak shape.
There are two methods, algebraic method and differential method, to determinate thevalue of pKa by the CZE. Through the algebraic method, the pKa values of 6, 7-dihydroxy-coumarin, 7-hydroxycoumarin, coumarin-3-carboxylic acid and 4-hydroxycoumarin wereobtained successfully (6.40/7.83, 6.42, 4.54, 4.22). This would be valuable to developmedicine and relative products as well.
The proposed method of CZE was simple, fast and simultaneous for separation, and italso could be applied to analysis of food and urine with good precision. The correlationcoefficient were above 0.9955, and the RSD of migration time and peak area were under2.6% and 5.4%, respectively.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-137-MO
Label-Free Electrophoretic Analysis of Sugars using Complexationwith Aryl Boronic Acids. 2
Risa Kusumoto1, Philip Britz-McKibbin2, Takayuki Kawai1, Kenji Sueyoshi1,Fumihiko Kitagawa1, Koji Otsuka1
1Department of Material Chemistry, Graduate School of Engineering, Kyoto University,Kyoto 615-8510, Japan
2Department of Chemistry, McMaster University,Hamilton, ON, L8S 4M1, Canada
The analysis of sugars is one of the important issues in biological/clinical diagnoses sincesugars play important roles in biological functions. Among various techniques for analyz-ing sugars, capillary electrophoresis (CE) is advantageous because it provides a rapidanalysis with a low consumption of the sample. In conventional CE of sugars, however, atroublesome derivatization is often required for the detection and/or separation sincemany sugars have no UV absorbance and no charge.
To overcome this drawback, we focused on the affinity CE (ACE) analysis using arylboronic acids which form complexes with cis-diol compounds like most sugars. In a back-ground solution (BGS) containing aryl boronic acids, the complexation provides thevariations of both the wavelength of the absorption maximum and apparent electropho-retic mobility of the complex from those of native sugar, resulting in both the label-freedetection and selective separation of sugars [1].
In this study, five boronic acids, such as 3-nitrophenyl boronic acid (NPBA), 3-naphthaleneboronic acid, quinoline-3-boronic acid, 3-prydine boronic acid, and 4-methylpyridine boronicacid, were tested as the affinity ligands for the ACE analysis of sugars. Prior to the ACE analysis,the formation constants of the sugar-aryl boronic acid complexes were estimated from thechanges in the UV absorption spectra of the ligands in phosphate buffer (pH 7.0) upon addingsorbitol. Among the five boronic acids, NPBA showed the most suitable spectral changes forthe label-free ACE, i.e., Δλmax of 7 nm and Δε of 900 M−1cm−1.
The ACE analysis of the mixture of sugars, including fructose, glucose, sorbitol andmannitol, was conducted in phosphate buffer (pH 7.0) containing 3 mM NPBA. As aresult, both the detection and separation of sorbitol, mannitol, and fructose were attainedwithout any derivatization. The separation of sorbitol from stereoisomeric mannitol indi-cated the stereo-selectivity of NPBA to the difference in the cis-diol structures of thesugars. On the other hand, only glucose could not be detected since almost all of glucosewere in pyranose form in aqueous solutions, interfering sufficient complexation withNPBA. Consequently, the label-free and stereo-selective analysis of neutral sugars wassuccessfully achieved by the ACE analysis employing aryl boronic acids.
1. Kaiser, C. et al. Chem. Comm. 2008, 3, 338–340.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-138-TU
New Avenue for Mid UV-Range Detection of UnderivatizedCarbohydrates and Aminoacids in Capillary Electrophoresis
Cédric Sarazin1,2, Nathalie Delaunay2, Christine Costanza1, Véronique Eudes1,Jean-Maurice Mallet3, Pierre Gareil21Central Laboratory of the Prefecture de Police,
39 bis, rue de Dantzig, 75015 Paris, France2Laboratory of Physicochemistry of Electrolytes, Colloids and Analytical Sciences (PECSA), Chimie ParisTech,
UPMC Univ Paris 06, CNRS, 11 rue Pierre et Marie Curie, 75231 Paris Cedex 05, France3Laboratory of Biomolecules (LBM), ENS, UPMC Univ Paris 06, CNRS,
24 rue Lhomond, 75231 Paris Cedex 05, France
Classically, methods used for the separation of carbohydrates include thin-layer chroma-tography (TLC), gas chromatography (GC), and liquid chromatography (LC). In mostcases, a tedious derivatization step is needed, either to acquire appropriate volatility(GC) or detectability (LC), since direct detection modes used in LC as absorptiometry,refractometry or polarimetry do not provide enough sensitivity. More recently, impor-tant developments were carried out with the coupling between high performanceanion-exchange chromatography (HPAEC) and pulsed-amperometric detection (PAD).Nowadays, capillary electrophoresis (CE), with its high separation efficiency, low re-agent consumption, and speed, appeared as an interesting alternative to chromato-graphic methods, especially for sulfated and carboxylated carbohydrates, but the analy-sis of so-called neutral carbohydrates can be more difficult to implement, due to theabsence of easily ionizable functions (other than anomeric hydroxyl group) andchromophore groups, and the high hydrophilic character of these compounds, whichmakes the use of micelles inoperable. Recently, a promising method was proposed byRovio et al. for the CE separation under extremely high alkaline condiditions of neutralcarbohydrates under their alcoholate form and their direct UV detection, which isclaimed to rely on the absorption of enediolate at 270 nm. Even more questionable,most of the detected compounds in this paper (for example sucrose) cannot give suchenediolate, lacking a carbonyl group. In basic media, no hydrolysis of the glycosidicbond can occur; so, unsaturated group cannot originate from hydrolysis but must comefrom an oxidative process.
In this work, we thoroughly revisited the experimental conditions of this approach withrespect to both separation and detection performances. In effect, unusual detectionphenomena were observed in comparing reducing and non-reducing carbohydrate be-haviors, which pointed to the existence of photochemical reactions in the detectionwindow. A more systematic study of the influence of a number of parameter, includinglamp intensity, carbohydrate nature, BGE composition (pH, cation nature), residence time

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
in the detection window, capillary diameter, was undertaken. This study brings a deeperinsight into the understanding of the physico-chemical mechanisms involved in this newdirect UV detection mode and opens up new avenues for the detection in mid-UV rangeof non-UV absorbing compounds bearing reducing moieties, such as aminoacids andpeptides.
1. Rovio S., Yli-Kauhaluoma J., Siren H., Electrophoresis 2007, 28, 3129–3135.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-139-MO
Pre-Separation of High Volume Biological Samplesusing Divergent Flow Isoelectric Focusing
Filip Duša1,2, Jana Krenková1, Dana Moravcová1, Vladislav Kahle1, Karel Šlais1
1Institute of Analytical Chemistry of the AVCR,v.v. i., Veverí 97, 602 00 Brno, Czech Republic
2Department of Biochemistry, Faculty of Science, Masaryk University,Brno, Kotlárská 2, 611 37, Czech Republic
Biological samples are very complex mixtures differing in species of substances as well asin their concentration range. Due to difficulties in their direct analyses, efficient pre-sepa-ration steps are very often required in order to simplify the complexity of samples andenhance detection and identification of individual components.
We present an application of newly developed divergent flow isoelectric focusing (DF-IEF) instrument for fractionation of protein digests and its comparison with currently usedinstrumentation. Model protein mixture consisting of bovine serum albumin, myoglobin,and cytochrome c was digested with trypsin and resulting peptide fragments were pre-separated by DF-IEF in autofocusing mode in which the sample was desalted and separat-ed without addition of carriers forming pH gradient. The obtained fractions were analyzedusing μ-LC coupled with UV/Vis or ESI/MS detector.
Obtained chromatograms and MS spectra show well separated and focused peptides inIEF fractions covering their isoelectric points. Only some peptides were focused poorlydue to the lack of charged residues in their sequences. The DF-IEF instrument proved verygood pre-separation efficiency and ability to process a high volume throughput of lowconcentrated samples.
Acknowledgement: This work was supported by the Grant Agency of the Academy of Sciences of the CzechRepublic No. IAAX00310701 and by the Institutional research plan AVO Z40310501. The financial support ofthe Ministry of Education, Youth and Sports of the Czech Republic by grant No. LC06023 is also gratefullyacknowledged.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-140-TU
Protein-Ion Interactions Detectedby Affinity Capillary Electrophoresis
Sabine Redweik, Yuanhong Xu, Hermann WätzigTU Braunschweig, Institute of Pharmaceutical Chemistry,
Beethovenstr. 55, 38106 Braunschweig, Germany
Affinity Capillary Electrophoresis (ACE) is a precise and versatile tool to measure influenceson proteins. ACE is able to detect conformational changes as well as changes of theprotein charge. Moreover, ACE allows measuring of unspecific and specific interactions onproteins without special reagents or kits.
The influence on proteins is detected by the mobility changes which occur under theinfluence of various ligands. Therefore the migration time of the protein is measuredwithout any addition of the tested ligand and, in a further step, in presence of variousdefined ligand concentrations [1]. In order to compensate for changes on the migrationtime, which are not due to binding, the mobility ratio of an EOF-marker and the proteinis used [2]. Six repeats were done with a very good precision due to the use of a specialrinsing procedure [3]. The RSDs [%] of the migration times and the mobility ratios weretypically below 2%, very often below 0.2%. The influence of various charged species e.g.metal ions, such as Cu2+, Mn2+ and Mg2+ as well as pharmaceutical cations as ephedrinehydrochloride and pirenzepine dihydrochloride was tested on the migration behavior ofproteins, e.g. BSA, β-lactoglobulin and ovalbumin. Organic cations and metal ionsshowed clearly different interactions with the proteins. For metal ions, specific and unspe-cific interactions could be distinguished.
1. Carels, L.; Boelens, H., Journal of Chromatography A, 1997, 311–3282. Bose, S.; Yang, J.; Hage, D. S., Journal of Chromatography B, 1997, 77–883. El-Hady, D.; Kühne, S.; El-Maali, N.; Wätzig, H., Journal of Pharmaceutical and Biomedical Analysis, 2010,
232–241

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-141-MO
Quantification of Aspartate and Glutamatein Brain Microdialysates with CE-LIF
Zsolt Wagner1, Tamás Tábi1, Gergely Zachar2, András Csillag2, Éva Szöko1
1Semmelweis University, Department of Pharmacodynamics, Budapest, Hungary2Semmelweis Egyetem, Department of Anatomy, Histology and Embryology, Budapest, Hungary
Glutamate and aspartate are the two major excitatory neurotransmitters in the CNS,playing central role in learning processes and memory formation, although their excessrelease leads to excitotoxicity, thus can be responsible for neurodegenerative disorders. Inorder to elucidate the various functions of these neurotransmitters numerous samplingmethods and analytical techniques have been developed so far. Microdialysis is a valuablesampling tool for in vivo monitoring the extracellular level of these neurotransmitters. Toachieve the proper temporal resolution short sampling time combined with low flow ratewere used in our experiments, which resulted in large number of samples and only acouple of microliter sample volume. Due to the achievable short analysis time, the highsensitivity and the small required sample volume capillary electrophoresis is the mostappropriate analytical tool for analyzing brain microdialysates. Because of the lack ofeasily detectable moiety, sample derivatization providing either fluorophore or chromo-phore is to be used. However, LIF offers considerably higher sensitivity compared to UVdetections. Derivatizations in submicromolar range might be problematic for some rea-sons including the incomplete labeling reaction or the rapid hydrolysis of the reagent.
In our work the analytical performance of three commonly used fluorescent labelingtags (NBD-F, FITC. CFSE) have been compared. Separation conditions have been opti-mized to achieve short analysis time using reversed polarity separation in coated capillary.Validation data including accuracy, precision and stability have been determined accord-ing to the latest analytical guidance. Method validation has revealed that there is almosttwo orders of magnitude difference between the quantification and the detection limit,likely due to the unreliable derivatization reaction. It is also confirmed, that using a com-mon wide concentration range of calibration cannot provide accurate quantification atthe low concentration range even in the case of superior R2 value (> 0.99). In case of allthe three labeling reagents similar quantification limits were found, although there wereremarkable differences in sample stability. Based on the superior stability, FITC derivatiza-tion was used for the analysis of biological samples. The analytical performance has alsobeen tested by analyzing pooled microdialysis samples spiked with known concentrationsof analytes.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-142-TU
Searching for Ampholytes Suitable for Oscillating Electrolytes in CZE
Martina Riesová, Lucie Malináková, Bohuslav GašCharles University in Prague, Faculty of Science, Department of Physical and Macromolecular Chemistry,
Albertov 2030, 128 43 Prague 2, Czech Republic
After the first oscillating electrolyte composed of sebacic acid and imidazole was discov-ered [1], searching for other compounds capable to provide oscillating behavior startedimmediately. The criterion for electrolyte’s ability to oscillate is the existence of the com-plex system eigenmobility. System eigenmobilities can be calculated utilizing the lineartheory of electromigration which is implemented in our program PeakMaster [2]. Theknowledge of dissociation constants and limiting mobilities of the substances is essentialfor these calculations. As the first step we investigated chemicals included in PeakMaster’sdatabase where some oscillating electrolytes were found [3]. Further we revealed thatampholytes had a promising potential in terms of their oscillating behavior. The calcula-tions in PeakMaster resulted in searching for ampholyte capable to fulfill these criterions:i) both dissociation constants (of protonated and dissociated group) have mutually closevalues in acidic region of pH, ii) absolute value of the limiting mobility of negativelycharged species is higher than the value of limiting mobility of the positively chargedones. On the basis of the estimated values of dissociation constants [4] we chose severalsuitable commercially available acidic ampholytes. We determined dissociation constantsand limiting mobilities from the dependencies of effective mobilities on pH utilizing CZE.The obtained data were evaluated as described in [5]. While the dissociation constants ofall the selected ampholytes were in agreement with their estimations and their values laidin acidic pH region, only one ampholyte had appropriate values of limiting mobilities anda theoretical ability to provide oscillations. These predictions obtained by PeakMasterwere investigated experimentally.
1. V. Hruška, M. Jaroš, B. Gaš; Electrophoresis 27 (2006) 5132. http://www.natur.cuni.cz/gas3. M. Riesová, V. Hruška, E. Kenndler, B. Gaš; J. Phys. Chem. B 113 (2009) 124394. http://sparc.chem.uga.edu/sparc/5. K. Vceláková, I. Zusková, E. Kenndler, B. Gaš; Electrophoresis 25 (2004) 309
This work was financially supported by the Grant Agency of the Charles University grant number 51009, theMinistry of Education, Youth and Sports, Long Term Research Plan MSM0021620857 and the Grant Agencyof the Czech Republic, grant number 203/08/1428.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-143-MO
Simultaneous Determination of Aminothiols in Human Plasmaby Layer-by-Layer Assembly of Silica Nanoparticles
with Amperometric Detection
Lin Zhou1, Corinna Zambardi2, Mila Pravda1, Erika Scavetta2,John H.T. Luong3, Jeremy D. Glennon1
1Irish Separation Science Cluster (ISSC), Department of Chemistry and Analytical,Biological Chemistry Research Facility (ABCRE), University College Cork, Ireland
2Department of Physical and Inorganic Chemistry, Faculty of Industrial Chemistry,University of Bologna, Italy
3NRC Institute for Biological Sciences,Montreal, Quebec, Canada
Plasma aromatic and sulfur containing amino acids are good indicators of protein anabo-lism and catabolism, while blood reduced(GSH) and oxidized glutathione (GSSG) reflectoxidative status in an organism as a useful indicator of disease risk in humans [1]. GSSG isusually found in very low concentrations in comparison to GSH in normal individuals. Amethod is developed by combining silica nanoparticles modified capillary electrophoresiswith amperometric detection for the separation of GSH,GSSG and other amino thiols.Fused silica capillary was layer-by-layer assembled with poly (diallydimethylammoniumchloride) (PDDA) and silica nanoparticle (SiNPs) and PDDA again to generate a cationicsurface on the capillary wall. The positively charged PDDA and negatively charged SiNPsformed a stable coating and increased the surface charge on the capillary wall whichsignificantly improved the selectivity of the solutes via ionic interaction. Boron-dopeddiamond (BDD) electrode has been proven to be more sensitive and effective for detec-tion of overoxidized amino thiols [2]. In general, carbon electrodes, as represented byglassy carbon, show no signal corresponding to disulfides and methionine. In the presentwork, we report the possibility of the separation and detection of GSH and GSSG at BDDelectrode in acidic media. This assay is well suited for quantitative determination ofaminothiols in clinical studies and can be used to support investigations of oxidative stressin patients.
1. Maeso, N.; García-Martínez, D.; Rupérez, F. J.; Cifuentes, A.; Barbas, C., Capillary electrophoresis of gluta-thione to monitor oxidative stress and response to antioxidant treatments in an animal model. Journal ofChromatography B 2005, 822 (1–2), 61–69.
2. Terashima, C.; Rao, T. N.; Sarada, B. V.; Kubota, Y.; Fujishima, A., Direct Electrochemical Oxidation ofDisulfides at Anodically Pretreated Boron-Doped Diamond Electrodes. Analytical Chemistry 2003, 75 (7),1564–1572.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-144-TU
Speciation Analysis of Aluminium (III)-Dopamineusing Capillary Electrophoresis with Amperometric Detection
Una Barrett1, Fengjun Shang1, John, H. T. Loung2, Jeremy D. Glennon1
1Innovative Chromatography Group, Irish Separation Science Cluster (ISSC),Department of Chemistry and the Analytical and Biological Chemistry Research Facility (ABCRF),
University College Cork, Cork, Ireland2Biotechnology Research Institute, National Research Council Canada,
Montreal, Quebec, H4P2R2, Canada
Aluminium (Al) is known neurotoxic agent which may interfere with a large number ofneurochemical reactions [1]. Some neurodementia such as Parkinson’s disease (PD),Alzheimer’s disease (AD) and dialysis encephalopathy (DE) are believed to relate to theabnormally high level of Al in the brain [2]. Al has been found to hinder the rat brainsnaptosomal amine-uptake system to take in dopamine (DA) in vitro and to complex withDA moderately. In this work, a capillary electrophoretic method with end-columnamperometric detection for the rapid analysis of Al (III) dopamine complex was investigat-ed and developed. Method development included studies of the effect of applied poten-tial, buffer concentration, buffer pH, and hydrodynamic injection time on the electropho-retic separation. The method was validated with regard to linearity, precision, specificity,LOD, and LOQ. Once the separation was optimised on capillary, it was further studied ona miniaturised microchip platform (MCE).
1. Kiss, T. Archives of Gerontology and Geriatrics, 1995, 21, 99–112.2. Zhang, F.; Bi, S.; Liu, J., Yang, X.; Wang, X.; Yang, L.; Yu, T.; Chen, Y.; Dai, L.; Yang, T. Analytical Sciences,
2002, 18, 293–299.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-145-MO
Stability of Linear Polyacrylamide Coated Capillaries in Acidic Mediain the Presence of Organic Modifiers and Surfactants
Philippe Anres, Nathalie Delaunay, Pierre GareilLaboratory of Physicochemistry of Electrolytes, Colloids and Analytical Sciences (PECSA),
Chimie ParisTech, CNRS, 11 rue Pierre et Marie Curie, 75231 Paris Cedex 05, France
Since its first introduction in 1985 by Hjerten [1], linear polyacrylamide (LPA) coatedcapillaries have been widely used because of the unique properties of LPA (i) to suppresselectroosmotic flow (EOF) almost entirely and (ii) to reduce considerably adsorption ofhydrophobic compounds such as proteins or surfactants (SDS, ionic liquids). Both of theseproperties are mainly linked to its neutral and very hydrophilic character. These propertiesand the simplicity of the coating procedure made LPA-coated capillaries a prime choicefor in-situ preconcentration techniques in capillary electrophoresis involving stacking andsweeping, where EOF suppression is required to enhance performances.
Usually, with this kind of capillary, pH of background electrolytes (BGEs) are restrictedto the 4–8 range because it appears the stability of the coating is an issue for long lifeutilisation at pH below 4 and higher that 8. Indeed, in high acidic or basic conditions,acrylamide moieties can be hydrolysed into carboxylic groups, which alter capillary prop-erties regarding EOF and wall adsorption. However, to the best of our knowledge nostudies on the stability of capillaries in this pH range have been reported. Moreover, inMEKC and sweeping techniques organic modifiers are often used to tailor selectivity andno report is available on the stability of LPA-coated capillaries under such conditions.
The aim of this work was to study the stability of LPA coated capillary with a view totheir use with acidic background electrolytes (pH 2–4) in the presence of organic modifi-ers (acetonitrile and methanol), anionic surfactants (SDS) and long chain ionic liquid(dodecyldimethylimidazolium bromide). To this end, capillaries were coated followingHjerten’s procedure and allowed to be submitted for 70 hours to the different conditionsstudied, while Vigh’s tests were performed every hour to follow the evolution of EOF.
The results obtained allow concluding the coating procedure gives capillaries whereEOF is efficiently reduced and stable over 70 hours of utilisation. Consequently, this kindof capillaries can be used for preconcentration techniques in acidic media with organicmodifiers and surfactants. This opens up new possibilities to implement field enhancedsample injection (FESI)-Sweep-MEKC to preconcentrate compounds and next to separatethem in acidic media [2] and the utilisation of such capillaries will, for sure, gives betterresults in term of reproducibility and sensitivity enhancement, because the EOF is morestable and reduced comparing to non coated capillaries at pH in the range 2–4.
1. S. Hjerten, M-D. Zhu, J. Chromatogr. 1985, 346, 265.2. J.P. Quirino, S. Terabe, Anal. Chem. 2000, 72, 1023.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-146-TU
The Effect of pH of the Background Electrolyteson CIEF Applying Sequential Injection Protocol
Csilla Páger1, Andrea Vargová2, Ferenc Kilár1,2
1Institute of Bioanalysis, Faculty of Medicine, University of Pécs,Szigeti út 12, H-7624 Pécs, Hungary
2Department of Analytical and Environmental Chemistry, Faculty of Sciences, University of Pécs,Ifjúság útja 6, H-7624 Pécs, Hungary
Capillary isoelectric focusing (CIEF) in the presence of electroosmosis after the sequentialinjection of carrier ampholytes and sample, offers a highly efficient separation for ampho-teric compounds having isoelectric points outside the pH range of the appliedampholytes. The analytes (such as peptides or proteins) are concentrated in the pH gradi-ent because each component migrates to a specific position where the pH is equal to itsisoelectric point (pI).
CIEF coupled to mass spectrometry (CIEF-MS) provides an efficient identification andgives opportunity for structural characterization of the analyte. Several effects mightinfluence the effectivity of the MS detection, e.g., ion suppression by the ampholytecomponents, which would hinder the ionization of the sample components. After thesequential injection of ampholytes, of which the pH range does not cover the pI of thesamples, this problem can be avoided, because after separation, the sample compoundscan migrate out from the ampholyte zone.
Ampholytes with narrow pH range (covering only 2 pH units) were applied in theexperiments. The lengths of the ampholyte zones and the pH of the anolyte and catholytesolutions were varied in order to determine their influence on IEF. A controlled change ofthe pH of the anolyte and the catholyte solutions may cause that the analytes having pIsoutside the pH range of the applied ampholyte migrate out from the zone of theampholytes (from the pH gradient). This phenomenon will help to obtain more efficientMS detection, since the undesirable ion suppression by the ampholytes can be avoided.
The work was supported by the grants GVOP-3.2.1-0168, OTKA-K75717 and OTKA-NKTH-NI-68863.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-147-MO
Transient Isotachophoresis of Trace Metalsin a Zn Overloaded Saline Sample
Kihwan Choi, Asif Riaz, Ju Rung Park, Eun Jae Shim, Jihye Kim, Doo Soo ChungDepartment of Chemistry, Seoul National University,
Seoul 151-747, Korea
In capillary electrophoresis (CE), trace metal analysis combined with online preconcentra-tion often receives interference from concentration disparity of the analytes in the sample.Here we demonstrate that saline samples overloaded with Zn2+ could be masked com-pletely and selectively by adding thioglycolic acid (TGA) in the background electrolyte(BGE). A selective sweeping mechanism could be described as a means of masking in CE,which includes dynamic complexation and sweeping of Zn2+ selectively from the sampleby TGA. While trace metals such as Fe2+, and Ni2+ could be analyzed simultaneously bytransient isotachophoresis (TITP) using 4-(2-pyridylazo) resorcinol (PAR) as a complexingagent. Chloride and PAR in the sample acted as the leading electrolyte and the terminat-ing electrolyte, respectively. The optimized BGE was composed of 95 mM N-tris(hydroxy-methyl)methyl-3-aminopropanesulfonic acid and 80 mM triethylamine, 0.1 mM PAR, 2mM TGA and 0.02% FC-PN (a fluorocarbon neutral polymeric surfactant) at pH 9.0. Thelimits of detection (S/N = 3) were in the sub-ppb range (Fe2+ 0.3 ppb, Ni2+ 0.2 ppb). Themethod was successfully applied to determine the trace amounts of Fe2+ and Ni2+ in thestandard reference urine and human urine samples containing excessive amounts of Zn2+.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-148-TU
Transient Trapping in Enantioseparationfor High-Sensitive Detection. 2
Hiroshi Koino, Kota Hashiba, Kenji Sueyoshi, Fumihiko Kitagawa, Koji OtsukaDepartment of Material Chemistry, Graduate School of Engineering, Kyoto University,
Kyoto 615-8510, Japan
Cyclodextrin electrokinetic chromatography (CDEKC) is one of the versatile methods forthe enantioseparation in capillary electrophoresis. CDEKC has great advantages such asthe rapid separation, high resolution and low consumption of samples/reagents, while alow concentration sensitivity due to a short optical pathlength is often problematic. Toimprove the sensitivity, various online and offline preconcentration techniques have beenapplied to CDEKC [1]. In this study, a novel online sample enrichment technique namedtransient trapping (tr-trapping) [2] was combined with CDEKC to achieve a rapid andhighly sensitive enantioseparation.
In tr-trapping-CDEKC, a sample solution containing no sodium dodecyl sulfate (SDS) iselectrokinetically introduced into the capillary filled with a phosphate buffer (pH 7.0)containing cyclodextrin as a background solution (BGS) after a partial injection of a micel-lar solution plug. During the sample injection, hydrophobic analytes are trapped andenriched around the sample/micell boundary. After exchanging the inlet vial from thesample solution to the BGS, the focused analytes are consecutively released from theboundary according to the decrease in the micellar concentration by the diffusion. Afterpassing through the micellar zone, the enantioseparation of the focused analytes pro-ceeds on the basis of CDEKC in the BGS zone.
To evaluate the efficiency of tr-trapping-CDEKC, 1-aminoindan (AI) and sulfatedβ-cyclodextrin (S-β-CD) were used as model enantiomers and a chiral selector, respectively.When the sample solution was electrokinetically introduced for 420 s after the 10-s injectionof the micellar solution by pressure, a baseline separation of AI enantiomers was achievedwith a 230-fold sensitivity enhancement relative to the conventional CDEKC analysis. Thelimit of detection of AI was improved from 2.5 ppm to 25 ppb by applying the tr-trappingtechnique. When sweeping, a conventional preconcentration technique in EKC, was appliedto the enantioseparation by CDEKC and cyclodextrin modified micellar EKC, either theeffective preconcentration or enantioseparation was achieved, but not simultaneously. Theseresults demonstrated that the combination of the tr-trapping technique with CDEKC provid-ed both the effective preconcentration and adequate enantioseparation.
1. Sánchez-Hernández, L. et al. Electrophoresis. 2010, 31, 28–43.2. Sueyoshi, K.; Kitagawa, F.; Otsuka, K. Anal. Chem. 2008, 80, 1255–1262.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-149-MO
Use of Ligand Step Gradient Focusing in Combinationwith Isotachophoresis (LSGF-ITP) for the Effective Pre-Concentration
and Analysis of Heavy Metals
Eliška Glovinová, Jan PospíchalMendel University,
Brno, Czech Republic
The new method was developed for the concentration and separation of metal ions calledligand step gradient focusing-LSGF. The principle of the method is a stationary pH discon-tinuity-neutralization reaction boundary, inside an ITP capillary which creates distinctlydifferent complexing regions when a ligand is placed throughout the capillary. By select-ing conditions such that the metal is uncomplexed at low pH and complexed at high pHin such a way that it forms a negative complex, the metals can be focused around the pHchange.
The analytical procedure consisted of pre-concentration, mobilization and detection ofanalytes. The low-concentrated metal ions in the cationic form were electrokineticallycontinuously dosed into the column where they were selectively trapped on the stationaryligand step gradient in the form of unmoving zones of chelate complexes with effectivelyzero charge. After a detectable amount of analyte was accumulated, the accumulatedzones were mobilized to the analytical column, where they were analyzed by the conven-tional ITP method using conductivity detection.
The trapping selectivity was set-up by proper choice of pH and complexing agents. Amixture of heavy metals – Pb and Cd were used as model analytes, citrate were used ascomplexing agents. Using a 2000 sec. dosing time, the proposed method improved thedetection limit by 100–150 times in comparison to analysis by ITP with classical injection.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-150-TU
Comonomer Distribution across Molar Mass Distributionin Polyolefins by GPC using a Filter-Based IR Detector
Alberto Ortín1, Benjamín Monrabal1, María Celia García-Álvarez-Coque2,José Ramón Torres-Lapasió2
1Polymer Characterization, S.A.,c/ Gustave Eiffel 8, València Parc Tecnològic, 46980 Paterna, Spain
2Departament de Química Analítica, Universitat de València,c/ Dr. Moliner 50, 46100 Burjassot, Spain
Many polyolefins of industrial interest are ethylene copolymers with a-olefins containingfour to eight carbons, or ethylene-propylene copolymers. The controlled addition ofcomonomer in the polymer chains allows a balance of end-use properties in these prod-ucts, not achievable by ethylene or propylene homopolymers. Gel permeation chroma-tography (GPC) is the technique routinely used to analyse the molar mass distribution inmany natural and synthetic polymers, including polyolefins. Knowledge of the distributionof comonomer across the molar mass distribution in a copolymer is also needed, since itis a key microstructural feature, which correlates with the macroscopic properties of thematerial, and thus, its range of possible applications and performance.
In polyolefin applications, the use of infrared (IR) detectors has been extended to GPC(Macromol. Symp. 257, 29 (2007)), and more recently, to high temperature liquid chro-matography (Macromolecules 43, 3710 (2010)). The performance of the direct couplingof a filter-based IR detector to a high temperature GPC instrument, by means of a heatedflow-through cell, is here described. The detector was equipped with a broad band IRsource and two narrow bandpass interference filters. These filters allowed selecting spec-tral bands in the mid-IR region (2800–3000 cm−1), with different sensitivities to the copol-ymer components. Two signals, proportional to the transmittance, were generated, whichwere transformed to absorbance units (Abs), taking the pure solvent as reference. Thefilter-based IR detector resulted in a very simple and practical setup for the analysis ofpolyolefins against FTIR detection.
Continuous IR absorbance GPC chromatograms were collected at the two selectedbands. Their slice-by-slice AbsCH3/AbsCH ratio allowed the determination of the copolymercomposition at every molar mass fraction. The use of the dual band IR detector in theanalysis of comonomer distribution, across the molar mass distribution in polyolefincopolymers, is discussed. The high sensitivity of the detection allows the injection of lowconcentrations of the polyolefin materials, and the use of standard analysis conditions sothat chromatographic quality is not compromised, even in cases when very high molarmass fractions are present. Selected applications of the method to the analysis of polyeth-ylene and polypropylene copolymers are presented and discussed.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-151-MO
High Temperatures as a Tool to Increase Solvent Compatibility inTwo-Dimensional Liquid Chromatography
Jakob Haun1, Thorsten Teutenberg1, Torsten C. Schmidt2
1Institut für Energie- und Umwelttechnik e. V. (Institute of Energy and Environmental Technology, IUTA)Bliersheimer Strasse 60, 47229 Duisburg, Germany
2University of Duisburg-Essen, Instrumental Analytical Chemistry,45141 Essen, Germany
Whenever two-dimensional liquid chromatographic techniques are used, there are twoindependent solvent systems, one for each respective separation dimension. Duringmodulation by valve switching, not only the solutes but also considerable amounts of firstdimension solvents are injected to the second dimension. Depending on the chosensolvent systems and on the stationary phase of the second dimension, different solventeffects can negatively affect or destroy the second dimension separation. Besides miscibili-ty, solvent strength and volume overload, also effects based on viscosity differences likeviscous fingering (VF) are well described in literature [1].
As it is known from literature [2], viscosity and differences in viscosity can be effectivelyreduced by the application of high-temperature high-performance liquid chromatography(HT-HPLC). Therefore, a conventional one-dimensional HPLC system in combination witha high temperature oven was used to simulate an isothermal high-temperature seconddimension. For these experiments, a typical situation in two-dimensional liquid chroma-tography was chosen. The sample plug with high organic modifier content (from a possi-ble first separation at the end of a solvent gradient) is injected to the second dimensionwith a watery eluent. Peak shapes of the different solutes were monitored for differentinjection volumes, solute concentrations, oven temperatures, eluent compositions andtwo different column diameters in order to critically examine the nature of occurring peakdistorting effects. Moreover, some of the experiments were transferred to a capillary HPLCsystem.
1. R.A. Shalliker, G. Guiochon, Analyst 135 (2010) 222.2. T. Teutenberg, High-Temperature Liquid Chromatography – A User’s Guide for Method Development,
Royal Society of Chemistry, Cambridge, 2010.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-152-TU
The Prospect of using Sintered Nanodiamond as Stationary Phasefor High-Performance Liquid Chromatography
at Elevated Temperatures
Olga N. Fedyanina1, Pavel N. Nesterenko2
1Department of Chemistry, Moscow State University,Moscow, 109028, Russia
2University of Tasmania, Australian Centre for Research on Separation Science (ACROSS),Private Bag 75, Hobart, Tasmania, 7001, Australia
A number of stationary phases used in high-temperature liquid chromatography (HTLC)at elevated temperatures is still very limited because of lack of hydrolytic and mechani-cal stability, poor thermal conductivity of adsorbents in packed bed, difference in ther-mal expansion between column housing and adsorbents matrix. The use of zirconia-and titania, porous graphitic carbon and some organic polymers as stationary phase inHTLC has been reported. Diamond, other carbon allotrope has excellent mechanical,chemical and thermal stability as well as excellent thermal conductivity (150 times aslarge as stainless steel) and no thermal expansion at elevated temperatures. Thesephysico-chemical properties make diamond one of the most promising adsorbents forHTLC [1].
Detonation nanodiamond is inexpensive material with particles of average size 5 nm.The structure of particles is not completely proved but it is accepted now that a singleparticle consists of central diamond core covered with graphene monolayer and with a setof functional groups at outer layer. The distinguishing feature of detonation nanodiamondis unusual adsorption properties, which are associates with the presence of big number ofvarious functional groups connected with both sp2 (graphite) and sp3 (diamond) phases.The fine size of and strong aggregation of particles make difficult their direct use as adsor-bent, so sintering, milling of sintered agglomerates and size fractionation is required toget particles of microdiperse sintered nanodiamond (MSND) suitable for the use as col-umn packing.
MSND fraction with average particle size 3–4 microns, specific surface area 190 m2/gand bimodal pore size distribution was used as a column packing after additional purifica-tion and fractionation on size. Recently these columns were tested in different chromato-graphic modes such us IC, HILIC, NP and RP HPLC. The preliminary results of investiga-tion of the adsorption properties of MSND show the distinctive ability of this adsorbent toretain polar organic molecules with labile proton in functional groups (phenols,nucleotides, aromatic acids). The retention of series of phenols and nucleonic compoundson MSND is investigated at elevated temperature up to 200 °C. Effect of concentration of

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
methanol on retention of some phenols at different temperature is also studied. Thepotential application for the use this adsorbents at elevated temperatures is demonstrat-ed. It is shown that MSND thermal stability even after 720 hours column work in chemi-cally aggressive eluents.
1. P.N. Nesterenko and P.R. Haddad. Diamond-related materials as potential new separation media in separa-tion science. Anal. Bioanal. Chem. 2010. V.396. No. 1. P.205–211.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-153-MO
Fast, UHPLC-like Separation of Basic Analytes at Elevated ColumnTemperature on 3 μμm Particles under Elevated Temperature
and Isocratic Conditions
Rainer W. Schmid, Goran MitulovicClinical Department of Laboratory Medicine, Medical University of Vienna,
Währinger Gürtel 18-20, A-1090 Vienna, Austria
The aim of this study was to evaluate the importance of operating micro separation col-umns (ID 2mm) at higher temperatures and high flow rates to improve peak performancein isocratic hydrophilic interaction liquid chromatography (HILIC), using low molecularweight analytes as model compounds. We have tested different HILIC separation columnspacked with conventional silica materials (Phenomenex, ACE Silica, and TOSOH-TSK-Gel80) and core shell particles (Phenomenex Kinetex). Various solvents were tested forseparation including water, acetonitrile, methanol, and ethanol with flow rates rangingfrom 0.1 to 1.1 ml/min and columns were operated at temperatures from 55 °C to 95 °Cutilizing standard medium pressure HPLC equipment.
The separations in this study using the basic drug substances cocaine, mcPP, amphet-amine, methamphetamine, and MDPP as test analytes were carried out on three differentsilica type HILIC columns with an ID of 2 mm and 250 mm length. Influence of operatingtemperature on separation efficiency was investigated while keeping flow rates constant:While increasing the temperature had the expected effect of reduced retention indices forall analytes (and thus decreasing organic solvent consumption and significant increasedseparation speed), also selectivity shifts were observed with improved resolution betweensome pairs with alternated retention order were observed.
With decreased peak widths and successful reduction of overall separation time frominitially 8 minutes down to 1 minute nevertheless no increase of the slope of the van’tHoff curves was detected.
To find the optimal separation temperature and flow rates for the separation of furthersmall basic compounds additional experiments are needed. However, the alreadyachieved results have a significant impact on sample throughput and selectivity for reallife separations of drug samples containing amphetamine type drugs.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-154-TU
Use of Porous Graphitic Carbon in the Studyof Thermal Gradient HPLC
Anthony Edge1, Luisa Pereira1, James Heaton2, Monica Dolci1,Harald Ritchie1, Norman W. Smith2
1Thermo Fisher Scientific,Manor Park, Runcorn, WA7 1T, UK
2Pharmaceutical Science and Research Division, School of Biomedical and Health Sciences,Kings’s College London, SE1 9NH, UK
The use of porous graphitic carbon as a stationary phase for liquid chromatography willbe investigated with the use of thermal gradients to demonstrate the similarities betweenthermal gradient elution and organic gradient elution. The effect of the gradient will beinvestigated using a set of test compounds which shows the dramatic effect that usingthermal gradients can have on the separation mechanism. This will be discussed in rela-tion to solvent gradients with the differences and similarities between the two approacheshighlighted.
A further study investigates the effects of thermal lag in a liquid chromatography sys-tem, in particular the effect of the wall thickness will be discussed in relation to potentialhysteresis effects that can be observed using such systems. Decreasing the wall thicknesscan have an effect on the radial thermal heat lag and this effect will be examined withexperimental data which will demonstrate reducing the wall thickness can have alter theefficiency in isocratic and isothermal systems due to a reduction in thermal lag within thechromatographic system, and the effect on the application of thermal gradients will alsothen be discussed.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-155-MO
1-D Liquid Chromatography – ESI – Tandem MS Couplingfor Chemoselective and Enantioselective Separationsof Chiral Amino Acid Derivatives and Its Application
to Biologically Relevant Samples
Roland J. Reischl, Wolfgang LindnerUniversity of Vienna, Department of Analytical Chemistry,
Währingerstrasse 38, 1090 Vienna, Austria
The separation and identification of traces of D – amino acids (D-AAs) in the presence oflarge excesses of L – amino acids is of ever increasing importance in the field of molecularbiology. It has been shown that certain D-AAs play key roles in cellular processes of higherdeveloped species. They act as biochemical switches for insulin production [1], as neuro-modulators in the human cortex [2, 3] and are signal transmitters for the production andexcretion of hormones [4]. For these purposes they are endogenously synthesizedthrough enzymatic racemization [5]. However, many functions and inflictions of D-AAs inmammalian cellular processes still remain to be clarified. Therefore there is a strong needfor powerful highly selective and highly sensitive methods in order to precisely analyzethese molecules. We herein want to present a method that is based on the generation ofiron (II) ferrocenyl propionamides of amino acids [6] and the subsequent separation on acinchona alkloid based anion exchanger type chiral stationary phase. The combination ofLC separations with the chemoselectivity of the highly sensitive triple quadrupole MSdetection profits from great synergism and allows us to simultaneously detect the D –enantiomers of most chiral proteinogenic amino acids. We herein want to present theapplication of our method to biological samples like urine, serum and cell lysate.
1. Morikawa, A., et al., Biochemical and Biophysical Research Communications, 2007. 355(4): p. 872–876.2. Hashimoto, K., et al., Archives of General Psychiatry, 2003. 60(6): p. 572–576.3. Hashimoto, K., et al., Progress in Neuro-Psychopharmacology and Biological Psychiatry, 2004. 28(2): p.
385–388.4. Wolosker, H., A. D’Aniello, and S.H. Snyder, Neuroscience, 2000. 100(1): p. 183–189.5. Wolosker, H., S. Blackshaw, and S.H. Snyder, Proceedings of the National Academy of Sciences of the
United States of America, 1999. 96(23): p. 13409–13414.6. Bomke, S., et al., Analytical and Bioanalytical Chemistry, 2009. 393(1): p. 247–256.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-156-TU
Analysis of Decomposition Products of Hexafluorophosphate Saltsin Aqueous Solution
Lydia Terborg1,2,3, Sascha Nowak2, Paul R. Haddad3, Pavel N. Nesterenko3,Martin Winter2, Uwe Karst1
1Westfälische Wilhelms-Universität Münster, Institut für Anorganische und Analytische Chemie,Corrensstraße 30, 48149 Münster, Germany
2Westfälische Wilhelms-Universität Münster, Institut für Physikalische Chemie,Corrensstraße 28/30, 48149 Münster, Germany
3Australian Centre for Research on Separation Science (ACROSS), University of Tasmania,Private Bag 75, Hobart, Tasmania 7001, Australia
Due to its favourable properties, hexafluorophosphate (PF6−) is a common and a widely
used anion in battery research. On the one hand, PF6− is used in room temperature mol-
ten salts (ionic liquids) based novel electrolytes. Typically, ionic liquids consist of relativelysmall inorganic anions (e.g. bromide, chloride, tetrafluoroborate, hexafluorophosphate,trifluoromethylsulfonylimid) and a bulky and asymmetric nitrogen-containing organiccation (e.g. alkylpyrrolidinium, alkylimidazolium, alkylpyridinium). These compounds arecombined with lithium hexafluorophosphate (LiPF6) to obtain chemically stable andhighly conductive electrolyte systems for lithium ion batteries.
On the other hand, lithium hexafluorophosphate (LiPF6) is established as the conduct-ing salt for most organic electrolyte mixtures/systems used in commercial available energystorage systems.
Based on the high hygroscopicity of LiPF6, such systems are always contaminated witha certain amount of water that accelerates the decomposition of the conducting salt to LiFand PF5, which subsequently may release hydrofluoric acid (HF). The decompositionproducts, especially HF and PF5, have a negative influence on the performance of thelithium ion batteries, such a deterioration of the solid electrolyte interface (SEI) and canfurther act as catalysts for the decomposition of the electrolyte.
Due to the fact that the reaction mechanism of the hydrolysis of PF6− in these systems is still
unknown, this work focuses on the decomposition of different hexafluorophosphate salts.Therefore, hydrolysis of different hexafluorophosphate salts has been investigated in
purified water. For analysis, ion chromatography with UV as well as non suppressed andsuppressed conductivity detection was used. Identification was carried out by ion chroma-tography coupled with inductively coupled plasma optical emission spectrometry (IC/ICP-OES) and validated by ion chromatography coupled with electrospray ionization massspectrometry (IC/ESI-MS).

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-157-MO
Analysis of Fentanyl and its Metabolite, Norfentanylby Capillary Electrophoresis Coupled to Mass Spectrometry
with a Sheathless Porous Electrospray Interface
John C. HudsonBeckman Coulter, Inc., Discovery Solutions Business Center,
Brea, CA, USA
A sheathless electrospray interface for capillary electrophoresis-mass spectrometry (CE-MS), currently in development, was evaluated in a forensic application requiring highsensitivity. Fentanyl is a potent analgesic commonly administered at low dosages, 25micrograms/patch, resulting in very low therapeutic levels of 0.3 to 1.2 ng/mL of serumafter 24 hours [1].
There is a need in forensic casework to detect and quantify low levels of fentanyl afterextended periods of time between administration of the drug and collection of the sam-ple for analysis. These challenging types of cases include Drug Facilitated Sexual Assault(DFSA) and Driving Under the Influence of Drugs (DUID) where there may be a substan-tial delay in the assault being reported or samples from an impaired driver being ob-tained.
In this work, fentanyl, along with its main metabolite, norfentanyl, were analyzed inextracts of spiked serum. Samples were externally prepared along with blind controls andanalyzed in duplicate in an automated process. The results for both fentanyl andnorfentanyl showed excellent linearity (R > 0.995) over a range of 0.1 to 50 ng/mL ofserum with LOD and LOQ values of 0.1 ng/mL. Analysis of controls prepared by an exter-nal agency was within acceptable precision over the entire concentration range.
These results confirm that the interface is capable of providing the necessary sensitivity,robustness and precision for a challenging, qualitative and quantitative forensic druganalysis. Very low sample volumes (6 nL in this work) are required compared to existingtechnology such as LC-MS, making CE-MS using this sheathless interface virtually a non-destructive sampling technique. This is significant in forensic cases because analysis canthen be done on extracts of very small samples of the matrix of interest (50 to 100 μL ofserum).
1. Baselt, R.C. (2008) Disposition of Toxic Drugs and Chemicals in Man, pp. 616–619, 8th Edition, BiomedicalPublications, Foster City, California, 94404, USA.
The High Sensitivity Porous Sprayer interface is for Laboratory Use Only; not for use in diagnostic proce-dures.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-159-MO
Analysis of Multi-Component Samples by LC××LC-MS/MS
S. Nakanishi, T. Noguchi, H. Miyamoto, T. SekiAnalysis Research Department, Chemical Research Laboratories, Nissan Chemical Industries, Ltd.
2-10-1 Tsuboi-nishi, Funabashi-shi, Chiba, 274-8507, Japan
Recently, comprehensive two-dimensional liquid chromatography (LCxLC) attracts a lot ofattention for the analysis of multi-component samples, such as foods, environmentalsamples and biomaterials.
In LCxLC, two columns that have different retention mechanisms are used for two-dimensional separation. All eluted components from 1st column are divided to fractionsat regular intervals by switching valve. Each fraction is sent to 2nd column and is separat-ed again. In this way, overlapped peaks in 1st column can be separated for each peak.Thus it achieves higher peak capacity compared to conventional LC.
For an analysis of multi-component samples that contain a few hundred of compo-nents, we combined LCxLC with tandem mass spectrometry (MS/MS). LCxLC-MS/MS hasmade it possible to visualize properties of components (polarity, molecular size, functionalgroup and so on) on two-dimensional chromatogram (grouping). It also enables to figureout the characteristics of samples (sample imaging).
We applied LCxLC-MS/MS to an analysis of multi-component natural products(propolis extract). Propolis extract contains a lot of components such as phenolic acids,flavonoids, terpenes etc. A conventional LC could not separate those peaks completely ordetect imperceptible peaks hidden by major peaks. Using LCxLC-MS/MS, however, weachieved two-dimensional separation of the propolis extract with high efficiency anddetailed structural analysis with MS/MS fragmentation data. Moreover we succeeded inidentifying some components of propolis extracts using two- dimensional chromato-grams. In addition, this technique enabled to analogize and group similar structure com-ponents such as phenolic acids and flavonoids by MS/MS fragmentation data.
LCxLC-MS/MS are very powerful technique not only for the separation of multi-compo-nent samples but also for grouping and sample imaging.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-160-TU
Approaches to LC-MS Hyphenation for Simultaneous Quantificationin Biological Matrices of Xenobiotics and Metabolites
with Wide pKa and Polarity Ranges
Zsuzsanna Kuklenyik, Antonia M. Calafat, John R. Barr, James L. PirkleCenters for Disease Control and Prevention,
4770 Buford Hghwy, Atlanta, GA 30341, USA
LC-MS method optimization for analysis of biological matrices is a triangulation taskamong the physicochemical properties of the biological matrix, the chromatographicbehavior of the analytes and their optimal ionization in the LC-MS interface. This taskbecomes further complicated when the target analyte groups are not chosen based onphysicochemical similarities but rather based on application, occurrence in the samebiological matrix, and type of metabolism. In addition, limited sample amounts, economi-cal laboratory workflow and data management needs of large public health cohort studiespresent challenging situations when a group of compounds encompassing a wide pKa andhydrophobicity range has to be determined simultaneously from the same HPLC injectionwhile preserving selectivity and precision. The purpose of this presentation is to share oursuccess stories where we ventured out of the comfortable realm of single column lineargradient methods and looked deeper into the modern HPLC “tool box”. Principles andapproaches will be discussed that had key importance in the development and robustapplication of our LC-MS and LC-LC-MS hyphenated methods. For example, operationnear analyte pKa reverse phase HPLC conditions where the complex simultaneous effectsof organic modifiers on analyte and buffer pKa had to be considered (analysis of HIVreverse transcriptase inhibitor drug metabolites in serum). Our two dimensional LC-LCcolumn switching setups allowed us to use very different chromatography conditions (pH,buffer concentration, organic content) for analyte separation from the injection matrix inthe first LC dimension, and optimal analytical separation and MS detection in the secondLC dimension. For example, coupling buffer/organic LC with water/organic LC conditions(alkyl phenols in urine); coupling anion exchange LC with ion-pairing LC (intracellularnucleotide phosphates); and coupling cation exchange LC with reverse phase LC (atrazinemetabolites in urine). Our LC-MS methods incorporate many automated functions that amodern LC-MS equipment and software engineering can offer: overlapping run mode,reverse flow switching, T mixing, multiple column switching valves and multiple gradientpump programming. The presentation will include method validation results and applica-tion to public health projects at the Centers for Disease control and Prevention.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-161-MO
Challenges in the Structure Elucidation of New Bis-Indole Alkaloids:LC-FTICR-MS/MS and NMR Analysis of Vinblastine
and Vincristine Impurities
Viktor Háda1, Zsófia Dubrovay1, Zoltán Gulyás2, Ágnes Lakó Futó3
1Spectroscopic Research2Development Chemistry
3Synthetic I.,Chemical Works of Gedeon Richter Plc., Gyömroi u. 19–21., Budapest, Hungary
The bis-indole alkaloids vinblastine (VLB) and vincristine (VCR) are naturally present in theplant Catharanthus roseus, and are widely used in cancer chemotherapy. VLB is routinelyproduced by the extraction of Catharanthus roseus and subsequent industrial-scale col-umn-chromatographic purification. Since the same process is uneconomical for VCR, onan industrial scale the latter is produced via the synthetic oxidation of VLB. Recently,efforts have been directed by our company toward finding a more economical procedurefor the production of VLB. A newly developed method enables a more effective separationof VLB from the known impurities. Although the VLB and VCR drug substances obtainedare highly pure, novel minor related impurities were detected in them by analytical HPLCat the production site. Repeated attempts to isolate and purify by preparative liquidchromatography these unknown impurities for structural identification yielded smallamounts of materials whose main components were the pertinent impurities, but whichwere still contaminated with other VLB/VCR-related compounds.
In principle the most detailed and accurate structural information of an organic mole-cule is provided by NMR spectroscopy. In practice, however, VLB and VCR moleculesoften exhibit broad NMR peaks and crowded spectra, which can make structural interpre-tation difficult. Moreover, the required sophisticated NMR investigations may take severalhours or days even on ultra-sensitive instruments, which makes it impossible to performon-line LC-NMR measurements for bis-indole alkaloids. In such cases the MS supportinginformation can be indispensable. For all of the above-mentioned VLB/VCR-related impuri-ties the structures could ultimately be verified only by the complementary and iterativeuse of NMR and high-resolution tandem mass spectrometry. In most cases the classicalapproach was followed: detection by analytical HPLC, isolation and purification bypreparative liquid chromatography followed by off-line MS and NMR analysis. Neverthe-less, this strategy did not work for all the impurities: for one of the trace-level impurities inthe plant extraction the existence and natural origin could be confirmed by LC-FTICR-MS/MS measurements of several production batches. For the accurate LC-MS/MS basedidentification and structure elucidation of these complex molecules the specific MS frag-mentation mechanism of related compounds should be studied and explored in advance.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
During that process a new ESI-MS fragmentation mechanism characteristic of bis-indolealkaloids was discovered, which can facilitate the LC-MS/MS based identification andstructure elucidation of other VLB/VCR-related compounds.
The cooperation of different analytical techniques, such as HPLC, preparative liquidchromatography, ultra high-field NMR and high-resolution (LC-)MS/MS allowed thestructural identification and complete spectral characterization of several hitherto unpub-lished VLB/VCR-analogue impurities, some of which are natural products.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-162-TU
Comprehensive Investigation of the Influence of using Acidicand Basic Mobile Phases on Bioanalysis Assay Sensitivity
in Positive Ion ESI Mass Spectrometry
Paul D. Rainville1,2, Joanne Mather1, Norman W. Smith2, Robert S. Plumb1,2
1Waters Corporation34 Maple Street Milford, MA USA
2Kings College London150 Stamford Street London SE1 9NH UK
The sensitivity of a bioanalytical method is critical is defining the PK parameters of apotential new medicine. Low exposure compounds provide one of the most significantanalytical challenges due to their low systemic concentration. The sensitivity of a bio-analytical LC/MS/MS based assay can be influenced by different parameters including: thedetector, chromatographic resolution from endogenous matix compounds and ionizationefficiency of the compound being quantified. In this work, we show the influence ofaqueous mobile phase pH (acidic (pH 3), and basic (pH 10)) in conjunction with metha-nol and acetonitrile on the sensitivity of twenty-five probe pharmaceuticals in biologicalfluid extracts. The probe pharmaceuticals varied in pKa, molecular weight, hydrophobicity,and functional groups. Each compound was spiked into rat plasma,protein precipitatedand detected under ESI positive mass spectrometry. Signal-to-noise, chromatographicpeak area, retention time, and peak width where all calculated and compared for eachcompound run under differing pH and organic mobile phase conditions. Six replicatesinjections where carried out for each mobile phases combination. The results of thisinvestigation showed that tradition acidic aqueous mobile phases did not always result inthe best assay sensitivity even under electropspray positive ionization mode conditions.The observations made while utilizing the basic aqueous mobile phase were: A significantimprovement in peak shape for basic compounds resulting in sharper peaks and subse-quent higher signal-to-noise values. An increase in peak area for greater than 80 percentof the compounds tested compared to traditional acidic aqueous conditions. An increasedin retention for some poorly retained compounds. An increase in MS signal for com-pounds whose retention time was unaffected by the mobile phase conditions. Finally, theaffect of the mobile phase combinations on matrix interference resolution, such as thephospholipid fraction will be discussed. The results of these studies indicate that basicmobile phases are an effective option in order to increase the sensitivity of LC/MS/MSbioanalytical assays run under ESI positive ionization mode.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-163-MO
Coupling of HT-HPLC with Enzymatic Assays– The Issue of Organic Solvent
Romy K. Scheerle1, Thorsten Teutenberg2, Johanna Graßmann1, Thomas Letzel11TU München, Lehrstuhl für Chemisch-Technische Analyse und Chemische Lebensmitteltechnologie,
Weihenstephaner Steig 23, 85354 Freising, Germany2Institut für Energie- und Umwelttechnik e. V. (IUTA),
Bliersheimer Str. 60, 47229 Duisburg, Germany
The fast and cost-efficient screening for functional components in complex mixtures is anemerging research area. Thus, the presented project aims on the development of onlinecoupling of high performance liquid chromatography (HPLC) to diverse enzymatic assayswith mass spectrometric (MS) detection. The system provides versatile options to screenfor regulatory compounds in complex mixtures.
Samples are separated by HPLC and subsequently analyzed by an online continuousflow enzymatic assay. The use of common organic solvents is required for chromato-graphic separation. However, to maintain the enzymatic activity it is important to keepthe organic part of the mobile phase as low as possible. High-temperature liquid chroma-tography (HT-HPLC) is a possibility for the effective coupling of chromatographic separa-tion with enzymatic screening methods [1]. Compared to conventional HPLC the amountof organic solvent for the separation can be decreased markedly and often 10% organicphase is sufficient for chromatographic separation. This improves the compatibility of thechromatographic separation with the biochemical detection by enzymatic assays.
In the current study the influence of several common organic solvents (methanol,ethanol, isopropanol, acetone, acetonitrile) on enzymatic activity (hen egg white lyso-zyme, chitinase, α-chymotrypsin, elastase from human neutrophils and porcine pancreas,acetylcholinesterase) was tested systematically by electrospray ionization mass spectrome-try. The results provide an overview about the susceptibility of the enzymes towardsorganic solvents and a reference point for many other applications in analytical chemistryand biotechnology. The obtained information was applied to conduct measurements withthe online coupled setup and first results are shown.
1. de Boer, A.R., et al., High-temperature liquid chromatography coupled on-line to a continuous-flow bio-chemical screening assay with electrospray ionization mass spectrometric detection. Analytical Chemistry,2005. 77, 7894–7900.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-164-TU
Determination of Serotonin and its Precursors by Capillary LC-MS.Application to Chocolate Samples
Vanesa Guillén-Casla, Noelia Rosales-Conrado, María Eugenia de León-González,Luis Vicente Pérez-Arribas, Luis María Polo-Díez
Department of Analytical Chemistry, Faculty of Chemistry, Complutense University of Madrid,E-28040 Madrid, Spain
Serotonin (5-HT) is a neurotransmitter in the central nervous system (CNS) which issynthesised from the L-tryptophan amino acid through short a metabolic pathway involv-ing 5-hydroxytryptophan (5-HTP) as an intermediate. Serotonin has a broad activity inhuman brain, playing an important role in the modulation of anger, aggression, bodytemperature, mood, sleep, sexuality, appetite, and metabolism, as well as stimulatingvomiting.
Chocolate is a mixture of cocoa paste, cocoa butter, and sugar having well knownnutritional qualities; it also contains tryptophan, an essential amino acid which initiatethe production of the serotonin. Moreover, although serotonin could be found only insmall amounts, it plays an important role during certain nervous or depression states.Consequently, detection of serotonin and its precursors, L-tryptophan and 5-HTP, inchocolate samples is particularly important because they can be altered during cocoaprocessing.
Since concentration of these compounds is normally in the low μg g−1 range, sensitiveand selective analytical methods have to be applied for their determination. In the lastfew years, miniaturized LC has received special attention, offering the possibility toachieve good resolution and high efficiency in a short analysis time using small volumesof both samples and reagents.
The aim of the present study was to develop and optimize a capillary liquid chroma-tography cLC-MS method and a sample preparation procedure for the determination ofserotonin and its precursors in commercial chocolate samples. Using a Zorbax SB-C18column (150 x 0.5 mm I.D., 5 μm) a personalized multifactorial experimental designwas applied to optimize pH and buffer concentration of the mobile phase, injectionvolume and focusing conditions. Best results were obtained by gradient elution at 20 μLmin−1, using a mixture of ACN/ammonium formiate 5 mM pH 4 (3:97 v/v) as mobilephase. The injection volume was set at 10 μL and pure water was used as injectionsolution. Working in selected ion mode (m/z 177 for 5-HT, m/z 205 for L-tryptophanand m/z 221 for 5-HTP) detection limits were between 0.06 and 1.0 μg L−1. Linearitywas in the concentration range 0.25–500 μg L−1. Intra- and inter-day precision for thewhole cLC-MS system, expressed as relative standard deviation (RSD), were between0.10 and 6.0%.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
Additionally, a fast and simple extraction method based on two steps, the first oneinvolving fat extraction and the other one for extraction of the analytes, was developed todetermine 5-HT, 5-HTP and L-tryptophan in chocolate samples.
The optimised cLC-MS method and the sample preparation procedure were applied tothe analysis of target compounds in chocolate samples with different cocoa content (70–100%). Recoveries higher than 80% with RSD lower than 5% were obtained in spikedsamples for all analytes, showing the effectiveness of the proposed methodology. In non-spiked samples, serotonin was found in the concentration range between 13–28 μg g−1
and L-tryptophan between 86–190 μg g−1, while hydroxytryptophan was only detected inchocolate samples with the lowest cocoa content.
V. Guillén thanks the Comunidad de Madrid for financial support through a “Personal Investigador de Apoyo”grant.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-165-MO
Development of a Pre-Column Derivatization Strategyfor the Measurement of the Tricarboxylic Acid Cycle Intermediates
by Means of Reversed Phase LC-MS with ESI+ Ionization
D. Kloos1, M. Giera1, M. Wijtmans2, R. J. E. Derks3, O. A. Mayboroda3,A. M. Deelder3, W. M. A. Niessen1
1LACDR division of BioMolecular Analysis, VU Amsterdam,De Boelelaan 1083, 1081 HV Amsterdam, The Nederlands2LACDR division of Medicinal Chemistry, VU Amsterdam,
De Boelelaan 1083, 1081 HV Amsterdam, The Nederlands3Department of Parasitology, LUMC Leiden,
Albinusdreef 2, 2333 ZA Leiden, The Nederlands
The analysis of metabolic processes is of fundamental biological interest. The abundanceof small cellular metabolites, such as the intermediates of the tricarboxylic acid (TCA)cycle, contains essential information about the metabolic state of a cell. Not only is theTCA cycle key factor in the energy regulation within aerobic cells, the intermediates of theTCA cycle also play a role in cell signaling.
Several analytical techniques are available for the measurement of the TCA cycle inter-mediates, which are mainly mono- and di-carboxylic acids. Still the most prominenttechnique for their determination is gas chromatography mass spectrometry (GC/MS)after silylation of the analytes. Other liquid chromatography mass spectrometry (LC/MS)based methods involve hydrophilic interaction chromatography (HILIC) separations, ionpairing and ion chromatography techniques. However, these techniques suffer fromdifficulties with aqueous samples, MS compatibility, sensitivity or separation strength.
Modifying the carboxylic acids into RP-LC/ESI(+)-MS compatible analytes successfullyaddresses these limitations. We have developed a pre-column derivatization strategybased on the coupling of the acid function to a secondary amine via activation by acarbodiimide reagent. The applied label, N-methyl-2-phenylethanamine, was empiricallyselected because of several of its properties: secondary amines proved to prevent multipleundesired side reactions, an ethyl-spacer group between the reactive amine function andthe chromatographically important phenyl group prevented sterical hindrance and al-lowed the full conversion also of tricarboxylic acids. The presented protocol was opti-mized by means of a response surface model experimental design approach and success-fully applied to determine the TCA cycle intermediates in hamster heart muscle samples.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-166-TU
Development of a Raman Detector for Hyphenationwith High-Temperature Liquid Chromatography
and Isotope Ratio Mass Spectrometry
Björn Fischer*, Hans BettermannHeinrich-Heine-University, Institute of Physical Chemistry,
Universitätsstraße 1, 40225 Düsseldorf, Germany
Due to criminal statistics product and trademark piracy have raised dramatically in recentyears. The critical numbers of fake articles in foodstuffs, pharmacy and cosmetics exhibitpotential risks for public health.
Product piracy can be identified by determining the origin and authenticity of the chemi-cal compounds that are part of fake articles. After applying a separation by high-temperaturehigh performance liquid chromatography (HT-HPLC) origin and authenticity of analytes arerevealable by isotope ratio mass spectrometry (IRMS). The sole linkage of HT-HLPC and IRMScan be used if the composition of samples is known. The analysis of samples with unknowncomposition additionally requires the identification of the separated compounds. Especiallyfor this experimental approach we developed a detector based on Raman spectroscopy.
Raman spectroscopy yields information about chemical structure and concentration fromthe separated analytes by analyzing their spectra of molecular vibrations and the observablesignal intensities therein. With regard to the hyphenation to IRMS and since isotope ratioanalysis of organic compounds requires water as the sole eluent, the Raman spectrum ofwater does not affect Raman-active vibrations of organic compounds. The structural analysisof the separated compounds by Raman spectroscopy moreover enables differentiation ofisomeric species that are usually not separable by HT-HPLC alone. The Raman detectortherefore provides the identification of co-elution of two or more isomeric substances.
The Raman device as an online HT-HPLC detector was set between the HT-HPLC and theIRMS. Raman signals were generated within a liquid core waveguide. The laser light wasfocused into a fused silica fibre and guided to a T-fitting which coupled the HT-HPLCcapillary tubing, the liquid core waveguide and the optic fibre. The Raman light wascollected by a second fibre that directed the scattered light to the monochromator / CCDcamera unit for the spectral analysis.
At present this hyphenation concept and the coupling of HT-HPLC with Raman spectros-copy in particular was proven by investigating mixtures of food stuff additives.
This work is part of a close cooperation with the Institute of Energy and EnvironmentalTechnology e.V., IUTA (working group of Dr. T. Teutenberg) and the University Duisburg-Essen, Instrumental Analytical Chemistry (working group of Prof. Dr. T. C. Schmidt).
* Working Group for Liquid Phase Laser Spectroscopy, Room 26.33.02.32

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-167-MO
Development of a Simple, Fast and Quantitative UHPLC-MS/MSMethod for the Determination of Organic Acids
in Fermentation Processes
Burhan Ozalp, Erwin Kaal, Lucien DuchateauDSM Biotechnology Center, R&D, DSM Delft,
Alex. Fleminglaan 1, 2613 AX Delft, The Netherlands.
Organic acids play an important role during fermentation processes. To understandand/or improve the fermentation process, the whole range of organic acids at low levelhas to be analyzed. Although many methods based on GC and HPLC have already beendescribed in literature, none of these methods meet our requirements. Because of thehuge number of samples that has to be analyzed, analysis time of the method should beshort, maximum 20 minutes. Further, sample preparation has to be reduced to a mini-mum, and detection limits at ppm level have to be reached. It should be possible to inject100% aqueous solutions. To meet these requirements, a new UHPLC method using tan-dem mass spectrometry is described in this study. With the newly developed method, it ispossible to analyze the most important organic acids including mono- and di-acids.Quantification was performed by using isotope-labeled analogues of the acids as internalstandard.
The first step in the development of the RP-UHPLC-method was the investigation of theuse of modifier in the mobile phase without having too much ion-suppression problems.Trifluoroacetic acid, formic acid and methanesulfonic acid were tested and post-columnoptions were evaluated. The tested sample matrices were broth, filtrates and yeast-ex-tracts.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-168-TU
Enabling Faster Separations and Smaller Sample Volumeswith Micro-Flow Liquid Chromatography
Steve Hobbs, Dave Neyer, Khaled MriziqEksigent, Division of AB Sciex,
5875 Arnold Road, Dublin, CA 94568, USA
We have previously reported on fast cycle times and fast analyses that are enabled bymicro-flow chromatography. Key features to such separations include low delay volumes,fast gradient generation and low dispersion ESI probes. Known benefits to micro-flowchromatography include improved sensitivity for a given injection volume, reduced sol-vent consumption and reduced contamination of the mass spectrometer. In this presenta-tion we examine the benefits of moving to 1.0, 0.5 and even 0.3 mm columns for fastand high peak capacity separations using a micro-flow LC/MS/MS system designed foruse in the 1–50 μL/min range. The system uses a new autosampler configuration thatsubstantially improves sample washing and allows for faster cycle times. The autosamplerfluidic design isolates the syringe from sample and further allows the syringe needle andinjection valve to be washed rapidly.
Probe dispersion tests were performed on four ESI electrode designs at a flow rate of 10μL/min. Flow injection was performed using a micro-flow LC and an injection valve usingtime-sliced injection to provide 20 – 30 nL injection volumes. PEEK tubing with 25 μminner diameter and 30 cm length was used between the injection valve and ESI probes.An LC/MS/MS was used to measure peak widths. The width at half height was 2.5 sec-onds for a commercially available conventional probe and 2.0, 0.75 and 0.64 seconds forprobes designed specifically for micro-flow.
For investigating improvements to separation speed and cycle time, propanolol,verapamil and reserpine were separated on 1.0, 0.5 and 0.3 mm id columns. The columnswere packed with Halo Fused Core 2.7 mm diameter C18 particles and were 50 mm long.Baseline separations were possible with cycle times well under 1 minute.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-169-MO
Evaluation of SFC-MS Configurations for the Analysis of Lipids,Sterols and Polycyclic Aromatic Hydrocarbons
Melissa Dunkle1, Alberto dos Santos Pereira1, Nobukazu Higashi2,Kazuhisa Mitsui2, Hirooki Kanda3, Frank David1, Pat Sandra1
1Research Institute for Chromatography,Kennedypark 26, 8500 Kortrijk, Belgium
2Japan Tobacco Inc. Tobacco Science Research Center6-2, Umegaoka, Aoba-ku, 227-8512 Yokohama, Japan
3Gerstel K.K.,2-13-18 Nakane, Meguro-ku, 152-0031 Tokyo, Japan
A commercially available modular SFC system (1260 Infinity SFC, Agilent Technologies)was combined with single quadrupole and time-of-flight mass spectroscopy using atmo-spheric pressure chemical ionization (APCI). Different configurations, including flowsplitting before the backpressure regulator, flow splitting after the backpressure regulatorand using liquid make-up flow, were compared in terms of chromatographic resolution,repeatability and spectral quality. Mass spectra (fragmentation, isotopic ratios, etc) werecompared to data obtained by LC-MS.
Tests were performed using semi-volatile to non-volatile apolar solutes including lipids(glycerides), sterols, tocopherols and polycyclic aromatic hydrocarbons.
The data demonstrate that direct connection with additional make-up flow and inter-face heating results in superior sensitivity, repeatability and spectral quality compared toclassical configurations involving flow splitting [1]. Supercritical fluid chromatography alsoenabled the use of coupled columns, increasing column length and plate number andresulting in high resolution separations at moderate pressure drop. Interestingly, somevery apolar solutes, such as polycyclic aromatic hydrocarbons, showed better ionizationunder carbon dioxide/methanol or carbon dioxide/acetonitrile mobile phase conditionscompared to LC/MS with a water/methanol or water/acetonitrile gradient.
1. C. Aurigemma, W. Farrell, J. Chromatogr. A, 1217 (2010) 6110–6114

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-170-TU
How to Select Orthogonal Conditions in Reverse Phase to AchieveLCxLC Separations According to the Nature of Compounds
Ramia Al Bakain1, Isabelle Rivals2, Patrick Sassiat1, Didier Thiebaut1,Marie-Claire Hennion1, Guillaume Euvrard2, Jérome Vial1
1Laboratoire des Sciences Analytiques, Bioanalytiques et Miniaturisation UMR-CNRS-UPMC- PECSA,ESPCI ParisTech, 10 rue Vauquelin, 75005 Paris, France
2Équipe de Statistique Appliquée, ESPCI ParisTech,10 rue Vauquelin, 75005 Paris, France
The demand for characterization of complex samples, i.e. containing several hundreds ofcompounds, requires analytical tools able to answer this matter of increasing difficulty.Among these tools, two-dimensional liquid chromatography is a very attractive techniquedue to its separation power obtained via the coupling of two separation modes exhibitingdifferent mechanisms, i.e. orthogonal separation.
In a previous study carried out on a wide set of probe solutes, including neutral, basicand acidic compounds, it has been established that the optimal tool to evaluate theorthogonality of RPLC systems was Kendall’s correlation coefficient [1]. The factors influ-encing orthogonality are the nature of the stationary phases, the organic modifiers andthe pH values of the two systems. Experimentally, eight reverse phase stationary phaseswere considered, each of them at two different pH values (2.5 and 7) and with two or-ganic modifiers: (methanol and acetonitrile). Experiments were carried out in gradientmode.
In this presentation, the influence of the composition of the probe solute set will bestudied. In practice, subsets of probe solutes built on the basis on their acido-basic prop-erties will be used. Chromatographic behaviors will be then evaluated and Analysis OfVariance will be used to establish which factors have a significant effect and to rank thefactors according to the intensity of their effect on orthogonality, as a function of thenature of the tested compounds. Then, examples of orthogonal separations for eachsubset of compounds will be presented.
Finally, comprehensive 2D chromatograms obtained with a Time of Flight (TOF) spec-trometer detector will be shown.
1. R. Al Bakain et al., J. Chromatogr. A, under revision.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-171-MO
HPLC-MS-MS Analysis of Pteridines in Graphosoma Lineatumby Hydrophilic Interaction Liquid Chromatography
P. Kozlík1, J. Krajícek1, E. Tesarová2, R. Cabala1, Z. Bosáková1
1Department of Analytical Chemistry, Faculty of Science, Charles University in Prague,Albertov 2030, 128 43 Prague 2, Czech Republic
2Department of Physical and Macromolecular Chemistry, Faculty of Science, Charles University in Prague,Albertov 2030, 128 43 Prague 2, Czech Republic
Pteridines belong to the family of pigmentary colours of insect cuticle, but some of themare also important eye pigments. They are widely distributed in microorganisms, plants,and animals as dyes. The aim of this work was to develop a high performance liquidchromatographic method with ESI-MS-MS detection for separation of selected pteridines,namely leucopterin, isoxanthopterin, xanthopterin, erythropterin, biopterin and neopterinin insect cuticle. Several organic solvents and their mixtures with water or buffers weretested to obtain sufficient solubility of standards. Mass spectrometric detection was per-formed in reaction monitoring mode where the precursor and product ions were selectedfor each analyte individually. Considering the polar character of the studied analytes andthe applied mass spectrometric detection, hydrophilic interaction liquid chromatography(HILIC) was used. Two chromatographic columns, Atlantis HILIC Silica (Waters, 4.6 x 150mm, 3 μm) and ZIC®-HILIC (Merck, 4.6 x 150 mm, 3.5 μm), were tested in combinationwith binary mobile phases consisting of acetonitrile and various buffers. The effects ofmobile phase composition, buffer type, pH and concentration on the retention and sepa-ration of pteridines were examined in detail. The results obtained on the both columnswere critically compared and discussed. Under the optimized separation and MS condi-tions, the insect integuments were analysed after their extraction by dimethylsulfoxide.Optimization of the extraction time for achieving the equilibrium concentration wasperformed by monitoring the extraction of pteridines in the period of one week. Underthe optimized extraction and separation conditions the integuments of Graphosomalineatum, obtained from various localities (Czech Republic, Cyprus, Sweden), wereanalysed, and pteridines were separated and identified.
Acknowledgements: Financial support from the Grant Agency of the Czech Republic, grant No.P505/11/1459, Grant Agency of the Charles University, grant No. SVV 261204 and the long-term researchplan of the Ministry of Education of the Czech Republic, No. MSM0021620857, and RP 14/63 is gratefullyacknowledged.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-172-TU
Hydrophilic Interaction Liquid Chromatography (HILIC) Coupledto Mass Spectrometry for the Characterization
of Prebiotic Galactooligosaccharides
O. Hernández-Hernández1, I. Calvillo1, R. Lebrón-Aguilar2,F. J. Moreno3, M. L. Sanz1
1Instituto de Química Orgánica General (CSIC), Madrid, Spain2Instituto de Química-Física “Rocasolano” (CSIC), Madrid, Spain
3Instituto de Investigación en Ciencias de la Alimentación (CSIC-UAM), Madrid, Spain
Galactooligosaccharides (GOS) are carbohydrates with recognised prebiotic properties,produced by the action of β-galactosidases using lactose as substrate. The resulting mix-ture is composed by oligosaccharides of different glycosidic linkages and molecularweights. Depending on the enzymatic source, GOS structures can be different [1] and,consequently, their effect on gut microflora can change. Therefore, the determination oftheir chemical composition becomes essential.
Currently, hydrophilic interaction liquid chromatography (HILIC) is gaining importancein the analysis of these compounds and many HILIC stationary phases are commerciallyavailable. The use of mass spectrometric (MS) coupled to HPLC systems has considerablyenriched the field of oligosaccharide analysis, providing structural information.
To the best of our knowledge, scarce information is reported about characterization ofprebiotic carbohydrates, such as GOS, using different HILIC stationary phases coupled toMS. Therefore, in this work the separation of GOS using three different HILIC stationaryphases was evaluated and the developed HILIC-ESI-MSn method was used to characterizedifferent commercial GOS mixtures.
Analyses were carried out using ZIC®-HILIC, PolyhydroxyethylTM A and XbridgeTM Amidecolumns coupled to an ion trap mass spectrometer at positive ion mode. In each column,an optimization of the LC method was performed and multi-stage MS (MSn) was used tocharacterize structurally prebiotic GOS. The best results for the separation of GOS wereachieved using the XbridgeTM column with a gradient of ACN:H2O with 0.1% of ammoni-um hydroxide. Elucidation of molecular weights and glycosidic linkages was carried outby MSn using different standards as reference.
Different qualitative (glycosidic linkages and degree polymerization) and quantitativecarbohydrates composition was found for the three different commercial GOS samples.Mainly galactosyl-galactoses were detected in all DP fractions, prevailing the glycosidiclinkages 1–6 and 1–4 over the others.
1. Cardelle-Cobas et al., J. Agric. Food Chem. 2008, 56, 3328–3333.
This work was financed by projects AGL2009-11909 funded by Ministerio de Ciencia e Innovación andPOII10-0178-4685 funded by Junta de Comunidades de Castilla-La Mancha-FEDER. O. Hernández-Hernándezthanks CSIC for a JAE Predoc grant.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-173-MO
Improved Interface for Connecting SFC to MS and Movingtoward Open Access SFC/MS
Xu Zhang, Mark J. HaywardChemical and Pharmacokinetic Science, Lundbeck Research USA,
Paramus, NJ, USA
In an effort to set up a robust and efficient open access SFC/MS system for MedicinalChemistry support as well as achiral and chiral method screening, an improved interfacehas been designed and successfully utilized to connect the SFC to the MS. The interface isconsists of two fix restrictors made of PEEK tubing and a heater after the column. Postcolumn heating (prior to expansion to atmosphere pressure) maintains the 100 bar col-umn pressure, homogenizes the eluent, and results in more efficient (sensitive) ESI ioniza-tion and a more stable MS signal. With Masslynx 4.1 SCN798 software control, OASFC/MS provides an efficient and useful tool for normal phase Med Chem support as wellas achiral and chiral analysis and separation method screening. Preliminary data presentedhere demonstrates both the feasibility of a SFC/MS system in an open-access fashion, aswell as the high quality of data produced using the above-mentioned SFC/MS interface.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-174-TU
Improving Sensitivity in Bioanalytical LC/MS/MSthrough Efficient Management of Mobile Phase pH
Paul D. Rainville, Robert S. Plumb, Thomas WheatWaters Corporation
34 Maple Street, Milford, MA 01757, USA
Chromatography coupled with tandem quadrupole mass spectrometry is the preferredanalytical method for the quantitative analysis of compounds in biological fluids. Thebasis for this is in the ability of the technique to provide very selective and sensitive detec-tion with relatively high throughput. Endogenous matrix components in biological fluids,including: metabolites, lipids and concomitant-dosed medications can influence theionization of analytes of interest if co-elution occurs with the compound undergoingquantification. Thus elimination of these matrix effects is key in maximizing sensitivity. Inorder to overcome this problem, various LC/MS/MS method parameters can be manipu-lated in order to promote better selectivity of the compound of interest from the matrixcomponents. In this work, we show the use of efficient mobile phase pH control throughthe use of a programmable algorithm to both assess the best mobile phase pH for bothanalyte ionization efficiency and selectivity from endogenous matrix components.
All experiments were carried out using mass spectrometric detection in electrospraypositive ionization mode. A six component test mix consisting of various physical, chemi-cal properties and spiked into protein precipitated rat plasma was chosen in order toassess the overall success or failure of the approach. In addition to monitoring the testanalytes, the effect of mobile phase pH was also monitored for the choline containinglipids fraction from the biological matrix. The pH of the mobile phase was changedthrough use of an algorithm which combined different volumes of acidic and basic mobilephase in order to deliver a requested final mobile phase pH for which the analysis was tobe evaluated.
The results from the study showed mobile phase pH provided increased resolution forcomponents from biological interferences present in the matrix. That major matrix com-ponents such as choline containing lipids can be affected by mobile phase pH. Further thealgorithm provided and automated approach to effectively screen mobile phase pH toidentify the ideal mobile phase pH conditions for a bioanalytical assay.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-175-MO
In-Capillary Screening of Matrix Metalloproteinase Inhibitorsby Capillary Electrophoresis for Coupling with ESI Mass Spectrometry
Xu Wang, Erwin Adams, Ann Van SchepdaelLaboratory for Pharmaceutical Analysis, Faculty of Pharmaceutical Sciences, K. U. Leuven,
Herestraat 49 PB 923, B-3000 Leuven, Belgium
Matrix metalloproteinases (MMPs) have been considered as a novel biomarker and poten-tial therapeutic target in human cancer. The MMP-2 and MMP-9 which have been partic-ularly implicated in tumor invasion and metastasis formation were selected as modelenzymes. A capillary electrophoresis (CE) method with fluorescence detection and enzy-matic reaction inside the capillary for screening of MMP inhibitors was firstly developed inour lab. In order to develop a label free method for the in-capillary screening of MMPinhibitors, the applicability of CE-MS for this purpose is investigated. In this project, avolatile system was firstly developed with CE-fluorescence detector. The buffer concentra-tion and pH have been optimized in this system. Finally, 20 mM ammonium acetate (pH7.0) was selected for both enzyme reaction and electrophoretic separation. Then, theinline incubation was performed using this method. The whole reaction, separation anddetection were achieved within one minute. For the mass spectrometry analysis, the iontrap MS conditions were optimized using the reaction product. The negative ESI detectionmode was selected. The sheath liquid composition was also optimized. Acetonitrile-water(80:20) was finally selected for further CE-MS investigation.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-176-TU
LC-Ion Exchange SPE-NMR
Cristina Daolio, Markus Godejohann, Ulrich Braumann, Manfred SpraulBruker BioSpin GmbH,Rheinstetten, Germany
Nowadays, the technique LC-SPE-NMR is the method of choice for structure elucidation,especially for unknowns in complex mixtures. The setup provides the possibility of devel-oping the LC part under normal reversed-phase conditions and trapping individual peaksfor posterior NMR experiments. The concept presents a main advantage of multiple LCruns were the peaks are trapped post column, consecutively, on SPE cartridges increasingtheir concentration and the signal-to-noise ratio for the further NMR experiments.
The exchange of LC solvents for deuterated solvents during the elution process, afterremoval of buffers and salts by a nitrogen stream, shows that the chemical shifts areidentical to those published already. As a result we can suppose that secondary process,like oxidation or isomerisation due to the constant manipulation of the sample can beavoided delivering fast dereplication for some studies.
Although, LC-SPE-NMR is subjected to the same restrictions as reversed phase chroma-tography concerning the very polar analytes which break through the SPE cartridges. Inthese circumstances only traditional LC-NMR can be used, either loop storage or directstop flow, losing the ability of sample concentration taking the NMR measurements backto the range of milligrams per peak on column.
This work shows a solution for the peaks that break through the SPE cartridges by usingthe ion-ion interaction concept where anions are retained by positive charge immobilizedon SPE material and cations are retained by negative charge. The later elution in the NMRflow probe can be performed in a very small solvent volume by removing charge fromanalyte or SPE material.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-177-MO
Measurement of Zwitterionic Osmolytes by LC-MS/MS
Chris McEntyre1, Crystal Lenky2, Michael Lever3
1Department of Chemistry, University of Canterbury,Private Bag 4800, Christchurch 8140, New Zealand
2Gateway Antarctica, University of Canterbury,Private Bag 4800, Christchurch 8140, New Zealand
3Canterbury Health Laboratories,PO Box 151, Christchurch 8140, New Zealand
Betaine-related osmolytes are highly polar water-soluble zwitterionic compounds. Theycontain a positively charged functional group at all pHs, and a negatively charged func-tional group that can be protonated at low pH. As a result the molecules have a netpositive charge at low pH. These osmolytes can therefore be conveniently measured byliquid chromatography tandem mass spectrometry (LC-MS/MS) in the positive modeusing an acidic buffer in the mobile phase. We describe an LC-MS/MS method for mea-suring the zwitterionic osmolytes: glycine betaine, arsenobetaine, dimethylsulfonio-propionate (DMSP), homarine, trimethylamine-N-oxide. This assay will be used to mea-sure osmolytes in Antarctic Weddell seals. Samples were extracted by diluting 50 μl ofplasma into 250 μl of extraction solvent, containing 10 μmol/l of each internal standard in10% methanol, and 90% acetonitrile. Samples were analysed using an MDS Sciex API4000 (Applied Biosystems, VIC, Australia) tandem mass spectrometer with a turbo ionspray (electrospray ionisation) probe, connected to a Shimadzu Prominence HPLC system.A binary gradient was used. The flow rate was 400 μl/min. The injection volume was 10μl. Separation was performed on a Cogent 100 mm × 2.1 mm, 4 μm Diamond Hydride(Eatontown, NJ, USA) silica column, and the oven temperature was 40 °C. Osmolyteswere measured in positive ion mode using multiple reaction monitoring (MRM). Whilethe isotopic internal standards: D9-glycine betaine, 13C2 arsenobetaine, and D9-TMAOcould be purchased, D6-DMSP and D3-homarine required custom synthesis. Withoutusing appropriate compound specific isotopic standards, the recoveries were often eitherbelow or above 100% with a variation of around 10%. Future work will involve an evalua-tion of different columns, including HILIC phases and sub 2-micron particle sized columnsfor improved sample throughput. The method can easily be adapted to include otherzwitterionic metabolites (such as carnitine) for biomedical applications.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-179-MO
Optimization and Evaluation of a Sheathless CapillaryElectrophoresis-Electrospray Ionization-Mass Spectrometry(CE-ESI-MS) Platform for the Analysis of Posttranslationally
Modified Peptides: Comparison to LC-MS
Herbert Lindner, Klaus Faserl, Leopold Kremser, Bettina SargDivision of Clinical Biochemistry, Biocenter,
Innsbruck Medical University, Austria
A sheathless capillary electrophoresis-electrospray ionization-mass spectrometry (CE-ESI-MS) interface with a porous tip as nanospray emitter was evaluated for fast, sensitive andreproducible analyses of posttranslationally modified peptides. We analyzed a linkerhistone sample, consisting of multiply modified proteins as well as microsequence variantsdiffering only slightly in mass and charge. A positively charged capillary coating and 0.1%formic acid as background electrolyte were used for separation upstream from massspectrometry characterization. Challenges exist due to the large dynamic range of modifi-cations at different residues, and the different nature of amino acid modifications, e.g.,phosphorylation, deamidation, methylation and acetylation.
The sheathless CE-ESI-MS method developed was compared with nanoLC-ESI-MS forthe analysis of ArgC-digested rat linker histones. With comparable amounts of sampleapplied to both systems, the number of identified peptides increased by more than 60%when using CE-ESI-MS, and the analysis time was significantly reduced. We found thatlow molecular mass peptides (below 1400 Da) were preferentially identified by CE-ESI-MS, since this group of peptides poorly interacted with the reversed phase material in thenanoLC system. Based on the results obtained, CE-ESI-MS can be considered as a comple-mentary technique to conventional LC-ESI-MS and as an alternative approach for modernprotein/peptide analysis. Principles, advantages and disadvantages of both techniques arediscussed in detail.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-180-TU
Optimization of Lipidomic Analysis using Off-Line Two-DimensionalHILIC x RP HPLC/MS
Eva Cífková, Miroslav Lísa, Michal HolcapekUniversity of Pardubice, Faculty of Chemical Technology, Department of Analytical Chemistry,
Studentská 573, 53210 Pardubice, Czech Republic
Lipids are important components in all biological tissues having many essential functionsassociated with the proper function of organisms. The disruption of lipid metabolism canlead to serious diseases (i.e., obesity, atherosclerosis, cancer, cardiovascular problems,etc.) and the analytical characterization of the lipidome is essential to understand theirrole. For this purpose, lipidomic analysis using off-line two-dimensional coupling of hydro-philic interaction liquid chromatography (HILIC) and reversed phase (RP) high-perfor-mance liquid chromatography mode with mass spectrometry detection was optimized. Inthe first dimension, total liquid extract was fractioned using HILIC into individual lipidclasses. Chromatographic conditions have been carefully optimized to achieve the separa-tion of most lipid classes in biological tissues. Effects of column packing, mobile phasecomposition (i.e., type of organic solvent, concentration of water, concentration of addi-tive and pH), separation temperature and gradient steepness have been tested. OptimizedHILIC separation enables the fractionation of 18 lipid classes that cover a wide range ofpolarities. Collected fractions were separated using RP method into individual lipid speciesaccording to the acyls lengths and number of double bonds. Effects of C18 column pack-ing, mobile phase composition, separation temperature and gradient steepness on thechromatographic resolution have been studied. Individual lipid species and their fatty acidcomposition and position of fatty acyls on the glycerol skeleton was identified using massspectrometry. Electrospray was used for the identification of polar lipids and atmosphericpressure chemical ionization for non-polar lipids. Off-line coupling of HILIC and RP-HPLCmodes enables the comprehensive analysis of plant and animal samples.
Acknowledgement: This work was supported by the grant project No. MSM0021627502 sponsored by theMinistry of Education, Youth and Sports of the Czech Republic and projects Nos. 203/09/0139 sponsored bythe Czech Science Foundation.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-181-MO
Polyphenolic Fingerprint of Various Natural Matricesby RPLC Coupled to Quadrupole and Hybrid Mass Analyzers
Francesco Cacciola1, Paola Donato2,1, Marina Russo3,Paola Dugo1,2, Luigi Mondello1,2
1Dipartimento Farmaco-chimico, University of Messina,viale Annunziata, 98168 Messina, Italy
2University Campus Bio-Medico,Rome, Italy
3Baller (s.r.l.),Messina, Italy
Nowadays, the demand for improved efficiency has reached the highest point either byincreasing sample throughput, or by the use in multidimensional systems. The two mostpromising approaches to attain enhanced performance consist in the use of smaller parti-cle size packing material, or to carry out separations under elevated temperatures, bothaiming to achieve higher column performance in a broad flow-rate region. In order tofully exploit the benefits of innovative stationary phases, we evaluated the advantages ofshell-packed stationary phases (2.7 μm particle-size columns), consisting of a 1.7 μm solidcore and a 0.5 μm porous shell for the investigation of the polyphenolic profile of variousnatural matrices. For unambiguous identification of the occurring compounds we em-ployed both a quadrupole and hybrid mass analyzers. For the latter, the ability to performrapid switchover between positive and negative ionization modes allowed to collect morequalitative information about a sample in a single run, while the high resolution and highspecificity in precursor-ion selection often provided the exact mass assignment necessaryfor confident elucidation of the structures of unknown compounds.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-182-TU
Rapid Method for the Detection of Metabolite of Lewisite– 2-Chlorovinylarsonous Acid in Urine by Liquid
Chromatography-Negative Electrospray-Tandem Mass Spectrometry
Arkady Braun1, Igor Rodin1, Oleg Shpigun1,Andrey Stavrianidi1, Igor Rybalchenko2
1Department of Chemistry, Lomonosov Moscow State University,Moscow, Russia2“Lumex ltd.”,Moscow, Russia
The development, production, stockpiling and use of chemical weapons are prohibitedunder the Chemical Weapons Convention [1]. In cases of alleged use of chemical warfare(CW) agents, environmental samples may be collected and analyzed for agents and theirdegradation products presented as a supporting evidence of a CW attack. Biomedicalsamples, e.g. blood and urine, may be analyzed for biological markers of poisoning asevidence that individuals have been exposed to a CW agent [2]. Biomedical sample analy-sis also has applications in exposure monitoring, e.g. in individuals engaged in demilitar-ization activities, and for the diagnosis of poisoning prior to the administration of medicalcountermeasures.
Lewisite [dichloro(2-chlorovinyl)arsine], a highly toxic chemical warfare agent withvesicant properties, was developed during World War I. Lewisite irritates the skin and eyesand is also poisonous when inhaled; but its clinical effects appear within seconds of expo-sure. Lewisite is very reactive; in aqueous media, it rapidly hydrolyzes to a stable watersoluble derivative, 2-chlorovinylarsonous acid (CVAA), which is also toxic [3].
Several analytical methods have measured CVAA to determine lewisite exposure, etc.the authors [4] determined CVAA in urine by using GC-MS for the 1,3-propanedithiolderivative. Several methods, based on the detection of CVAA, have been reported for theidentification of lewisite in environmental [5] and biological [6] samples. These approach-es have advantages but may require derivatization that add to sample preparation time.The main purpose of our work was to develop a rapid, sensitive technique for CVAAdetermination in urine samples.
We present a method for the detection of CVAA in human and rat urines spiked withCVAA that is based on the materials used by liquid chromatography interfaced to negativeion-electrospray ionization-tandem mass spectrometry. Columns under gradient condi-tions were used. By automating and optimizing several sample cleanup steps, the methodprovides the LC-MS/MS instrument with sufficiently clean concentrated samples, at a ratecomparable to that at which it quantifies them. The use of fast, automated sample prepa-ration steps, employing separate solid-phase extraction analyte isolation, provides samples

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
that are sufficiently clean and the 1 ng/ml limit of detection to be achieved. Thus, themethod is well suited for the purpose of the biomonitoring of lewisite exposure in theevent of a mass-casualty terrorist incident or in case of an accident at CW storage unitscharacterized by high sample loads and the low concentrations to be detected.
1. Convention on the Prohibition of the Development. Production. Stockpiling and Use of Chemical Weaponsand on their Destruction. Technical Secretariat for the Organization for the Prohibition of Chemical Weap-ons. The Hague. 1997.
2. T.P. Logan, J.R. Smith, E.M. Jakubowski, R.E. Nielson, Toxicol. Methods 9 (1999) 275.3. W.A. Waters, J.H. Williams, J. Chem. Soc. (1950) 18.4. J.V. Wooten, D.L. Ashley, and A.M. Calafat, J. Chromatogr. B 772 (2002) 142.5. B. Szostek, J.H. Aldstadt, J. Chromatogr. A 807 (1998) 253.6. A. Fidder, D. Noort, A.G. Hulst, L.P.A. de Jong, H.P. Benschop, Arch. Toxicol. 74 (2000) 207.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-183-MO
Robustness and Metal Release Testing of the New Agilent 1260Infinity Bio-Inert LC with Agilent 7700 ICP-MS
Nicole Fellner, Jochen Strassner, Katja KornetzkyAgilent Technologies, R&D and Marketing GmbH,
Hewlett Packard-Str. 8, 76337 Waldbronn, Germany
Many analytical applications such as analysis of large bio-molecules or speciation in ultralow-levels of metals with ICP-MS require dedicated analytical systems. A stainless steel freeHPLC system is the prerequisite for such applications when inertness is crucial and metalrelease should be reduced to zero. The new 1260 Infinity Bio-inert HPLC has a titaniumbased pump and metal free sample path from autosampler to detector. The metal releaseof this new system was tested with the Agilent 7700 ICP-MS system and data comparedwith an earlier Technical Note from Agilent where metal release from an Agilent 1100HPLC system was measured with ICP-MS as well. Elements of interest were Fe, Cd, Cr, Cu,Au, Zr etc. Mobile phases tested during these experiments were common eluents like0.1% formic acid, sodium phosphate buffer and 100 mM NaOH. Furthermore, the robust-ness of the system is shown by running the system in 0.1 M molar HCl (pH 1) for elevendays where data that pumping performance was not affected.
Obtained data show that the new 1260 Infinity Bio-inert HPLC shows very high chemi-cal resistance against low and high pH solvents and high ionic strength buffers. RegardingICP-MS analysis, most elements could not be detected after treatment because the valueis below the detection limit. Others like Ti and Zr showed concentrations around 1 μg/land lower. This is due to the fact that all wetted stainless steel parts of the pump havebeen replaced by corrosion resistant materials like titanium, platin, ceramic and inertpolymers.
Therefore the 1260 Infinity Bio-inert HPLC is not just a solution for typical Bio-HPLCapplications, it is also an extremely robust and suitable front-end solution for HPLC-ICP-MS where very low background of metals is essential.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-184-TU
Separation and Structural Identification of Lipid A Moleculesby Capillary Electrophoresis – Electrospray Mass Spectrometry
Viktor Sándor1, Ágnes Dörnyei2, Anikó Kilár1, Béla Kocsis3, Ferenc Kilár1,2
1University of Pécs, Faculty of Sciences, Department of Analytical and Environmental Chemistry,Ifjúság útja 6., 7624 Pécs, Hungary
2University of Pécs, Faculty of Medicine, Institute of Bioanalysis,Szigeti út 12., 7624 Pécs, Hungary
3University of Pécs, Faculty of Medicine, Department of Medical Microbiology and Immunology,Szigeti út 12., 7624 Pécs, Hungary
Lipopolysaccharides (LPSs, or endotoxins) are the main components of the externalmembrane of Gram-negative bacteria. LPS is composed of three distinct structuralregions: the O-chain polysaccharide, the core oligosaccharide and the lipid A moiety.Lipid A serves as the hydrophobic anchor of LPS in the outer membrane and is responsi-ble for bioactivity. It generally consists of a β-1,6-linked glucosamine disaccharide back-bone that is acylated by up to seven C10-C18 fatty acids or β-hydroxy-fatty acids linked asester at C3 and C3’ positions and as amides at C2 and C2’ positions. The hydroxylgroups of these β-hydroxy-fatty acid chains can be further esterified by additional fattyacids. Phosphates, with or without other substituents (such as phosphate, ethanol-amine, or monosaccharides), are linked at C1 and/or C4’ positions. The number andtype of the acyl chains and the state of the phosphorylation of the glycolipid are funda-mental determinants of the toxicity of LPS.
The structure of lipid A is relatively conserved, compared to the highly variable O-chainpolysaccharide, however, a bacterium may contain more than one lipid A structural type,since the environmental conditions can affect the number and types of lipid A speciesfound in a single bacterial population. The moderate variability of lipid A molecules canserve as the basis for the rapid identification of bacterial strains.
The need for a fast and small-scale analysis of lipid A inspired us to develop a methodthat combines the high resolution efficiency of capillary electrophoresis and the highsensitivity and specificity of mass spectrometry. According to the chemical nature of thelipid A molecules we applied non-aqueous capillary electrophoresis coupled to electro-spray ionization mass spectrometry (NACE/ESI-MS), which might enable the separationand identification of these structurally very closely related hydrophobic compounds.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-185-MO
Separation of Polar Polycyclic-Aromatic Compounds by OnlineComprehensive Two-Dimensional Liquid Chromatography (LC××LC)
Rune Græsbøll, Nikoline Juul Nielsen, Jan H. ChristensenDepartment of Basic Science and Environment, Faculty of Life Sciences, University of Copenhagen,
Thorvaldsensvej 40, 1871 Frederiksberg C, Denmark
Comprehensive two-dimensional LC (LC×LC) systems are far from routinely used but havethe capabilities to significantly increase the chromatographic separation of single compo-nents in complex samples and/or to reduce the analysis time. This is achieved by a largeincrease in peak capacity, since a fully orthogonal LC×LC system ideally have a peak capacityequal to the product of the peak capacities of the individual LC-systems (Bedani et al.,2009). Two-dimensional LC has previously been used for analysis of a wide range of com-pounds e.g. phenolic compounds in wine (Kivilompolo et al., 2008), lipids in rice oil (Mon-delle et al., 2005) and compounds in traditional Chinese medicine (Chen et al., 2004). Herewe demonstrate that online comprehensive two-dimensional liquid chromatography can beused for the separation of polar polycyclic-aromatic compounds (PACs) and in particular oxyPACs, e.g.: dihydroxynaphthalene, anthracene-9,10-dione, 1,2-acenaphthenequinone. PACsare found in crude oils, petroleum products and are produced during burning of fossil fuels.Polar PACs are among others formed when PACs are metabolised, and are found to haveequal or higher toxicity than the parent PACs. (Lundstedt et al., 2007).
We use a system similar to the one described by Stoll et al. (2006) in which high tem-perature is used in the second dimension to allow very rapid separation, which enablesreducing the size of the fraction transferred to the second dimension. The system is fullyautomated so that the fractions from the first LC-system are automatically transferred tothe second LC-system (online and comprehensive LC×LC). We used a standard silica-based C18 column as first dimension column, while the second dimension column was ashort 5 cm zirconia-based column which allowed for the high temperature required forvery rapid separation without column bleed. The compounds are detected by a photo-diode array detector after the second dimension.
ReferencesBedani F., Kok W., and Janssen H.-G.: Optimal gradient operation in comprehensive liquid chromatography ×
liquid chromatography systems with limited orthogonality. Analytica Chemica Acta, 654, 77–84 (2009).Kivilompolo M., Oburka V., and Hyötyläinen T.: Comprehensive two-dimensionel liquid chromatography in
the analysis of antioxidant phenolic compounds in wines and juices. Analytical and bioanalytical chemistry,391, 373–380 (2008).
Mondello L., Tranchida P.Q., Stanek V., Jandera P., and Dugo P.: Silver-ion reversed-phase comprehensivetwo-dimensional liquid chromatography combined with mass spectrometric detection in lipidic food anal-ysis. Journal of Chromatography A, 1086, 91–98 (2005).

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
Chen X., Kong L., Su X., Fu H., Ni J., Zhao R., and Zou H.: Separation and identification of compounds inRhizoma chuanxiong by comprehensive two-dimensional liquid chromatography coupled to mass spec-trometry. Journal of Chromatography A, 1040, 169–178 (2004).
Lundstedt S., White P., Lemieux C., Lynes K., Öberg L., Haglund P., and Tysklind M.: Sources, Fate and ToxicHazards of Oxygenated Polycyclic Aromatic Hydrocarbon (PAHs) at PAH-contaminated Sited. AMBIO,36(6), 475–485 (2007).
Stoll D.R., Cohen J.D. and Carr P.W.: Fast, comprehensive online two-dimensional high performance liquidchromatography through the use of high temperature ultra-fast gradient elution reversed-phase liquidchromatography. Journal of Chromatography A, 112, 123–137 (2006).

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-186-TU
Sheathless Capillary Electrophoresis-Electrospray Ionization MassSpectrometry for the Analysis of Samples Available
in Minute Amounts
Jean-Marc Busnel1,2, Jeff D. Chapman1, Jerald S. Feitelson1,André M. Deelder2, Oleg A. Mayboroda2
1Beckman Coulter, Inc., Discovery Solutions Business Center,Brea, CA, USA
2Biomolecular Mass Spectrometry Unit, Department of Parasitology, Leiden University Medical Center,Leiden, The Netherlands
This presentation will discuss recent advancements of a novel sheathless interface thatcombines the resolving power and low flow characteristics of capillary electrophoresis(CE) with electrospray ionization mass spectrometry (ESI-MS) using a porous sheathlessinterface currently in development.
Recently, the analytical properties of this new interface were extensively characterizedand its capabilities, in terms of sensitivity, resolution as well as mass loading capacity,were further evaluated for the separation of complex peptide mixtures. We have demon-strated with this platform that very high sensitivities can be achieved (low picomolarrange) allowing the detection of very low amounts (< 20 attomoles analyzed) of materialswith a very high resolving power [1].
As a follow-up to this publication, an explorative study further assessed the potential ofthe proposed strategy for the profiling of very minute amounts of biological samples. Tothis end, as a typical example of cell material that would only be available in minutequantities, the malaria hypnozoïte model was chosen. More precisely, an immortalizedhepatocyte cell line derived from human hepatocytes was studied. After cell lysis, thesample was directly submitted to trypsin digestion, lyophilized and further reconstituted,only 250 nL of which were sampled and analyzed by sheathless CE-ESI-MS. It was foundthat profiling was achievable with this injected quantity accounting for the protein con-tents of approximately 10 cells, and allowed the detection of more than 2,500 molecularfeatures (average number of 4 replicates) in less than 1 hour. This confirmed the ability ofthis sheathless CE-ESI-MS platform to profile very small amounts of complex peptidemixtures.
1. Busnel, J.M.; Schoenmaker, B.; Ramautar, R.; Carrasco-Pancorbo, A.; Ratnayake, C.; Feitelson, J.S.;Chapman, J.D.; Deelder, A.M.; Mayboroda, O.A. Analytical Chemistry 2010, 82, 9476–9483.
The High Sensitivity Porous Sprayer interface is for Laboratory Use Only; not for use in diagnostic proce-dures.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-187-MO
Towards Standard-Free Quantitative and Qualitative Analysisin Liquid Chromatography
Markus M. Martin1, Frank Steiner1, Michael Heidorn1,Marc Plante2, Fraser McLeod1
1Dionex-Softron GmbH,Dornierstr. 4, 82110 Germering, Germany
2Dionex Corporation,22 Alpha Road, Chelmsford, MA 01824, USA
Since the start of analytical chemistry there have been two fundamental questions: Whatanalytes are in my sample, and what are their quantities? Answering these questions hasalways been a tedious and time-consuming job. A common approach is to use separationtechniques such as high performance liquid chromatography (HPLC), and increasingly thenew UHPLC technology (Ultra-HPLC). These technologies separate mixtures of even thehighest complexity into single zones of pure compounds; nevertheless, the biggest con-straint for drawing the full picture of an unknown sample is the applied detection tech-nique, and the biggest constraint for defining the quantities is the lack of reference stan-dards for unknown analytes. The most widely used detection principles are based onoptical methods, which implies that quantification can only be done by relative measure-ments against reference compounds. As these detectors do not provide mass or structuralinformation, compounds are only assigned by retention time comparison, again by meansof standards. To save costs, time and work, an analytical approach for identification andquantification without any reference materials would be highly desirable.
In this presentation, we demonstrate the feasibility of a solution that fulfills most re-quirements for a reference-standard-free analytical method. Detection techniques basedon nebulization of the mobile phase and formation of aerosol particles, such as thecharged-aerosol detector, show an analyte-independent response that approaches “uni-versal”; this is required for reference-free quantification. In addition, mass spectrometersin tandem-MS modes, which are also hyphenated to LC by mobile phase removal throughnebulization, give strong evidence for the identity of a molecule. As both detector typesshow a dependence of the response on the mobile phase composition, the influence ofgradient elution is compensated by an inverse gradient, ensuring a uniform responsethroughout the whole chromatographic elution window. The combined use of a tandem-MS for compound identification and a charged-aerosol detector for quantification withconsistent response allows for the rapid and comprehensive investigation of unknownsamples. The scope and suitability of this approach will be discussed by investigating theanalysis of edible oils and confirming the results by comparison with conventional refer-ence-based LC analysis.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-188-TU
Using a Sheathless MS Interface Leads to Higher Resolutionsand Sensitivities in the CE Analysis of Peptides
René Kuijpers, Peter Wierenga, Henk Schols, Harry GruppenWageningen University, Laboratory of Food Chemistry,Bomenweg 2, 6703HD Wageningen, The Nederlands
Capillary electrophoresis has been proven to be a very sensitive and powerful tool with ahigh separation efficiency for the analysis of peptides and proteins. Several detectors arecommonly used for the detection of the proteins and peptides, like the ultraviolet detec-tor(UV), contactless conductivity detector (CCD), laser-induced fluorescence (LIF) andmass-spectrometry (Electrospray ionisation (ESI-)MS).
Depending on the detection techniques, different sample concentrations will be need-ed. In this study we compare two detection methods, the UV and MS detection, wherefor the latter, two different MS-interfaces were used: a sheath-liquid ESI-interface and asheathless ESI-interface with a porous tip (under development by Beckman Coulter). Allthese detection methods were used in combination with CE – isotachoforesis.
Three different peptide samples were used: a standard mix containing 5 differentpeptides, a beta-lactoglobuline hydrolysate (degree of hydrolysis of 6.8%) and a caseinphosphopeptide hydrolysate. The different samples were all analysed in 7 different con-centrations varying between 0.125 μg/ml till 250 μg/ml covering the range 29 pg – 58ng sample per injection.
In all cases we observed a better separation when concentration decreases. With theporous sheathless ESI-MS interface we observed good electropherograms, even for thelowest concentration (29 pg) per injection. Both the sheath-liquid-ESI-MS interface andUV detection were far less sensitive with a minimum of 0,78 ng and 19 ng per injectionrespectively. Even analysis of MS-MS we observed in all mentioned results good spectra.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-189-MO
Advantages of Multiple Time-Slice Injections in Sensitive,High-Throughput LC-MS Analyses
Remco van Soest, David W. Neyer, Don W. ArnoldEksigent Technologies,Dublin, CA 94568, USA
Microflow LC-MS with column diameters ≤ 1 mm is proving to be a valuable approach forhighthroughput LC-MS in bioanalytical development, including ADME-Tox measure-ments, since it can offer a significant improvement in sensitivity over high flow applica-tions. We recently described a chromatography system designed for this application thatallows very fast separations (1 to 1.5 minute cycle times). The system can deliver a widerange of injection volumes by using a time-slice injection approach. We will present datashowing how multiple, rapid separations can be conducted on a single small samplevolume. Such an approach can provide expanded dynamic range and improvedquantitation. In addition, since such cycle times are not limited by the overhead of theautosampler, one can gain this additional data in a shorter time. Metered injections withover 10× range of volumes have been demonstrated using samples extracted from biolog-ical matrix (plasma), showing improvement in dynamic range owing to the ability toinject smaller sample volumes. This approach has potential opportunities for increasedquantitation range without additional sample preparation in DMPK and ADME/Tox workf-lows.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-190-TU
A Comparison between Ultraviolet Detectionand Pulsed Amperometric Detection for the Simultaneous Analysis
of Saikosaponins, Glycyrrhizinic Acid, Poncirin, and Naringinin Caihu-Dayuan-Yin
Ha-Jeong Kwon1, Hee-Jung Sim2, Sa-Im Lee2, Min-Jung Gu2,Seon-Pyo Hong2, Yong-Duk Park1
1Department of Preventive and Social Dentistry, Graduate School, Kyung Hee University,Hoegi-dong, Dongdaemoon-gu, Seoul 130-701, South Korea
2Department of Oriental Pharmaceutical Sciences, Kyung Hee East-West Pharmaceutical Research Institute,College of Pharmacy, Kyung Hee University,
Hoegi-dong, Dongdaemoon-gu, Seoul 130-701, South Korea
In this study, we compared between ultraviolet detection (UV) and pulsed amperometricdetection (PAD) for the HPLC analysis of saikosaponins, glycyrrhizhinic acid, poncirin andnaringin in traditional Chinese medicine, Caihi-Dayuan-Yin. The glycosides were separat-ed by using a Kinetex C18 column under 0.01% phosphoric acid-acetonitrile gradientconditions in 60 min and detected by UV at 210 nm or PAD under alkaline condition.
The coefficients of linear regression were 0.9998–1.0000 at UV method, and 0.9969–0.9996 at PAD method. In order to compare the sensitivities of the methods, limits ofdetection (LODs) and limits of qnautitation (LOQs) were investigated in both method. Incase of UV detection, flavonoid glycosides has 40–60 times higher sensitivity thantriterpenoid saponins because they have strong chromophore. The PAD detection showedbetter sensitivies than UV detection. The sensitivity of glycyryrrizinic acid was about 30times poorer than those of the other compounds.
When analyzing Caihu-Dayuan-Yin by RP-HPLC-PAD method, the analytes peaks werecompletely separated without overlapping with background peaks. In case of using RP-HPLC-UV method, determination of Saikosaponins was impossible because they containedunder the LOQ, and was overlapped with noise peak or background peak. As a result,only naringin, poncirin and glycyrrhizinic acid could be determined by RP-HPLC-UVmethod, and the obtained contents were agreed with those by RP-HPLC-PAD method.
The robustness of this method was also evaluated by intra- and inter-day validation.The UV detection (RSDs ≤ 8.34%) was more precise than PAD detection (RSDs ≤ 15.15%)when determining Caihu-Dayuan-Yin. Although UV detection showed better linearity andprecision, the PAD method were more useful when micro-analyzing compounds whichhave poor chromophore for UV detection or requiring high selectivity in complex matrix.This method would be useful for the quality control of TCMs.
This research was supported by Basic Science Research Program through the National Research Foundation ofKorea (NRF) funded by the Ministry of Education, Science and Technology (2010-0002694).

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-191-MO
Alcoholic Fraction Composition in Essential Fragrances:a Comparative Study between CG-MS and HPLC-UV-MS
with Pre-Column Derivatization
Miriam Beneito-Cambra, Tamara Cortell-Barberá, Guillermo Ramis-Ramos, JoséManuel Herrero-Martínez
Department of Analytical Chemistry, University of Valencia,Doctor Moliner 50, E-46100 Burjassot, València, Spain
The alcoholic fraction of essential fragrances of Mentha and Rose was investigated byHPLC-UV-MS previous derivatization with a cyclic aromatic anhydride; this method wascompared to the GC-MS method. For this purpose, the samples were diluted with 1,4-dioxane and diphenic anhydride was added. The mixture was maintained at 105 °C for90 min. After cooling, the residue was dissolved in a 2:1 methanol-water mixture contain-ing ammonia. Separation of the derivatized alcohols was carried out by gradient elutionon a C8 column with water/ acetonitrile in the presence of 0.1% acetic acid. In order toperform a reliable identification of different alcohols presents, HPLC-MS studies were alsodone. The results obtained with this method were compared to that obtained by GC-MS.For this purpose, the samples were diluted with acetone and injected in the gas chro-matograph. Gradient elution with an optimized temperature program was performed.The alcoholic components were tentatively identified by comparison to linear retentionindices and mass spectrum library data. The results obtained by the proposed HPLC-UV-MS method were in rough agreement with those found by GC-MS. In addition to thedifferent sensitivities, the differences found were due to the thermal decomposition ofsome analytes in the GC column. Thus, the HPLC-UV-MS method provides a reliable, easyand sensitive method of to obtain alcoholic fraction profiles of essential fragrances, withthe advantages of low LODs for some analytes, and lack of thermal destruction of labilecompounds.
Acknowledgment: CTQ2010-15335 (MEC and FEDER) and V-Segles-Empresa grant (M. B.-C., Univ. of Valen-cia and Químicas Oro).

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-193-MO
Analysis of Native Carotenoid Composition in Sweet Bell Peppersusing C30 Columns in Tandem
Paola Dugo1,2, Daniele Giuffrida3, Germana Torre1, Francesco Cacciola1,Paola Donato2,1, Luigi Mondello1,2
1Dipartimento Farmaco-chimico, University of Messina,viale Annunziata, 98168 Messina, Italy
2University Campus Bio-Medico,Rome, Italy
3Dipartimento di Scienze degli Alimenti e dell’Ambiente,University of Messina, Italy
Serial coupled columns reversed-phase separations in high-performance liquid chroma-tography can be an useful tool for the analysis of complex real samples. The great difficul-ties that are found when analyzing complex carotenoid samples, due to the high naturalvariability of these compounds as well as to the presence of carotenoid esters are welldocumented. In the present contribution, the applicability of connecting two C30 columnsto significantly increase the separation power, resolution and peak capacity for the analy-sis of carotenoids in a complex carotenoids sample like sweet bell pepper has been shownfor the first time. By using LC coupled to PDA/APCI-MS detectors, 56 different carotenoidshave been detected in red sweet bell peppers. By using two serial coupled C30 columns apeak capacity of 95.4 was obtained, compared to 73 achieved using a single column.Moreover, resolution greatly improved between different critical peaks when using twoserial coupled C30 columns, compared to a single column. Interestingly, free carotenoids,mono-esters and di-esters were quantitatively equally represented (around 33% for eachdifferent class) in red sweet bell pepper. Free β-carotene (12.68%), capsanthin-C14:0(8.47%), and capsanthin-C12:0-C14:0 (8.95%) were the most abundant carotenoids inthe three different classes of red sweet bell pepper. No carotenoids esters were detectedin yellow or green sweet bell peppers. The application of such methodology in the analy-sis of other complex carotenoids matrices could be a future objective of research.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-194-TU
Analysis of Plant Hormones by HPLC-MS Basedon New Pretreatment Methods
Lan Zhang, Qiaomei LuMinistry of Education Key Laboratory of Analysis and Detection for Food Safety (Fuzhou University),
Department of Chemistry, Fuzhou University, Fuzhou 350002, Fujian, China
Plant hormones (PHs) are a series of low-molecular-weight organic compounds synthe-sized in plants, regulating almost every aspect of plant life. There are five classical kinds ofhormones, including auxins, gibberellins, abscissic acid, cytokines and ethylene [1]. PHsare difficult to detect due to their low amounts and serious interfering substances coexist-ing in plants, thus, sample preparation method with high preconcentration factors andhigh sensitivity are quite welcome. Liquid chromatography-mass spectrometry (LC-MS)combines the efficient separation capability of HPLC and structural characterization of MS,so it is a robust tool to analyze PHs.
Traditional sample pretreatment methods involving liquid-liquid extraction (LLE) andsolid phase extraction (SPE) were firstly studied. For instance, identification and quantita-tion of four auxins was realized by LLE-LC-MS [2]. Additionally, three endogenous PHsunder salt stress were investigated using SPE-LC-MS assay [3].
Then, some new pretreatment methods were used to study PHs. Combined with amolecularly imprinted solid-phase microextraction (MISPME) technique, a selective fiberusing indole-acetic acid as template molecules was prepared and characterized in ourlaboratory. All the results demonstrated that MISPME-HPLC-MS method was potential todetect low amounts auxin in real sample.
As a good alternative to MISPME, dispersive liquid-liquid microextraction (DLLME) intro-duced in 2006 has become popular for its merits including high enrichment ability and simpleoperation. Two PHs in peach were investigated by DLLME-HPLC-MS system. And another newsample pretreatment method named solidified floating organic drop microextraction(SFODME) combining with LC-MS was tested to four PHs. Results showed that SFODME wasa powerful preconcentration method, with the enrichment factors about 200-fold.
In short, LC-MS is very suitable for PHs analyses considering its powerful separation andqualitative ability. How to extract and preconcentrate PHs effectively from plant matrix isstill the bottleneck in PHs research. It is predicted that new pretreatment techniques willhave wider application in future.
1. H. Li. Modern Plant Physiology, 2nd ed.; Higher Education Press: Beijing, 2006.2. Q. Lu, L. Zhang, et al. Rapid Commun. Mass Spectrom. 2008; 22: 2565–2572.3. Q. Lu, L. Zhang, et al. Sci. in China (Series B: Chemistry) 2009, 39 (8): 785–792.
The authors are grateful for the National Nature Sciences Foundation of China (21075016), the CultivationFund of the Key Scientific and Technical Innovation Project, Ministry of Education of China (708056), China.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-195-MO
Analysis of Polysaccharides from Chinese Herbsusing Saccharide Mapping
Jing Zhao, Jing Xie, Yiwen Chen, Jia Guan, Shao-ping LiState Key Laboratory for Quality Research in Chinese Medicine and Institute of Chinese Medical Sciences,
University of Macau, Macau SAR, China
Polysaccharides from Traditional herbs exhibit multiple pharmacological activities. Howev-er, quality control of polysaccharides is a challenge because of their complex. In thispresentation, saccharide mapping based on specific enzymatic digestion of poly-saccharides and HPLC analysis was introduced, and the application in qualitative analysisof polysaccharides from different Chinese herbs were also reported. The results showedthat saccharide mapping is helpful to improve the quality control of polysaccharides fromChinese herbs.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-196-TU
Analysis of Radix Angelicae Sinensis by ComprehensiveTwo-Dimensional Chromatographic Separation Techniques
Roman Dück1, Alexandra von Trotha1, Margit Geißler2, Oliver J. Schmitz1
1Department of Analytical Chemistry, University of Wuppertal,Gauss-Str. 20, 42119 Wuppertal, Germany
2Shimadzu Europa GmbH,Albert-Hahn-Str. 6-10, 47269 Duisburg, Germany
The use of traditional Chinese medicine (TCM) has attracted a lot of attention not onlyin the East but also worldwide. Scientists are interested in the chemical constituents ofTCMs and the origins of their pharmacological and thus therapeutic activities. RadixAngelicae sinensis, one of the most widely used TCMs, has proven to have a variety ofbioactivities.
Samples with complex matrices such as herbal extracts represent a major challenge forseparation scientists. Radix Angelicae sinensis as a natural product represents a very com-plex matrix. Only with high-resolution separation techniques it is possible to identify theconstituents. One-dimensional techniques are not sufficient in most cases. Therefore, inthis work comprehensive two-dimensional chromatographic techniques such as GCxGC-quadrupole(MS) for volatile GC-accessible compounds and LCxLC-qTOF(MS) for polarcompounds are used. For the LCxLC analysis, two possibilities, off-line and on-line LCxLC,are compared. In off-line method, the eluate of the first dimension was collected in frac-tions, and each fraction was separated in the second dimension. With the on-line method,the collected eluates of the first dimension were analyzed simultaneously in the seconddimension. In the two methods the same columns were used. The methods have beenoptimized separately to yield the best possible results. To show the advantages and disad-vantages of the two methods, the following parameters have been compared: peak ca-pacity, duration of the analysis, consumption of solvent and the quality of the MS spectraobtained.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-197-MO
Application of Acetylcholinesterase Enzyme Silica CapillaryTube Reactor for Online Ligands and Extracts Screening
Joyce I. da Silva1 (PG), Marcela C. de Morais2 (PG), Cláudio Viegas Jr.3 (PQ),Antonia Q. L Souza4 (PQ), Afonso D.L. de Souza4 (PQ), Quezia B. Cass2 (PQ),
Carmen Lúcia Cardoso1 (PQ)1Departamento de Química, Faculdade de Filosofia,
Ciências e Letras de Ribeirão Preto, USP. Brazil2Departamento de Química, Universidade Federal de São Carlos-
UFSCar- São Carlos, SP, Brazil3Laboratório de Fitoquímica e Química Medicinal, Universidade Federal de Alfenas,
UNIFAL-MG, Brazil4Laboratório de Produtos de Origem Microbiana – Universidade Federal do Amazonas-ICB-DFCA/UFAM
– Manaus, Brazil
The cholinesterases (ChEs) are key enzymes in a wide range of important areas such asneurobiology, toxicology, and pharmacology. Among the ChEs, the enzyme acetylcho-linesterase (AChE) represents a widely studied target enzyme especially in the realm ofdrug discovery programs concerning Alzheimer’s disease (AD) [1]. Considering theseimportant applications, a promising approach is the use of immobilized enzymes ontosolid support creating immobilized enzyme reactors (IMERs), which can be used as anLC column to select specific inhibitors by employing rapid on-line screening of naturaland combinatorial libraries. To prepare AChE-IMERs, AChE from electric fish (Electro-phorus electricus) or AChE from human erythrocytes, was covalently immobilized onfused silica capillary using the homobifuncional agent glutaraldehyde as spacer, whichresulted in two bioreactors: AChE-ee-IMER and AChE-Hu-IMER. The IMERs were coupledto a high performance liquid chromatography system with UV-vis detection. The analyt-ical method was validated for both IMERs, which were then used in a study for determi-nation of the inhibitory potency (IC50) of standard AChE inhibitors. In addition, a total of12 3-acetyl-N-benzylpiperidine derivatives and 48 natural extracts were screened. Re-sults showed lower inhibition activity for these compounds with the best values beingaround 53% inhibition in 200 μM. Screening of 48 microbiological or plant extractsidentified 11 promising active extracts of Amazonian endophytic fungi with more than50% inhibition. The sample that afforded 81% inhibition in ACHe-ee-IMER and 70% inAche-Hu-IMER were selected for the purification work. Samples were also evaluated byanticholinesterase solution assay and false positive assay for comparison purposes [2–4]which evidenced high similarity. Results revealed that this method is suited to the iden-tification of the inhibition activity of more complex mixtures without the need for addi-tional pre-fractionation. This is a fast and useful approach for the accurate and repro-

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
ducible automated screening of synthetic or natural ligands. Moreover, these IMERs area great alternative for the preservation of the enzymatic activity (retention of 78% initialenzymatic activity for 10 months for IMER-AChE-ee and 60 days for IMER-AChE-Hu).
1. Sugimoto H, (2008) Chemico-Biological Interactions 175: 204–208.2. Ellman GL, Courtney KD, Andres Jr. V, Featherstone M. (1961) Biochem. Pharmacol 7: 88–95.3. Hosttetman, K.; Marston, A.; Kissling, J. Phytochem. Anal. 13:51–54, 2002.4. Verpoorte, R.; Rhee, I. K.; Van Rijin, R. M. Phytochem. Anal. 14: 127, 2003.
Acknowledgments: FAPESP, CNPq, and CAPES

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-198-TU
Application of an HPLC Method on Analysis of Bioactive Compoundsof Niuchangchih (Antrodia Camphorata) on Hepatoprotection
against Alcoholic Injury
Analyses of Bioactive Compounds in Niuchangchih(Antrodia Camphorata) via an HPLC and Its Hepatoprotection
against Chronic Alcohol Injury
Yi-Chen Chen1, Min-Tze Wu2, Chia-Hsin Huang2, Yuan-Yen Chang3
1Department of Animal Science and Technology, National Taiwan University,No. 50, Ln. 155, Sec. 3, Keelung Rd., Taipei 106, Taiwan
2Biotechnology Division, Taiwan Agricultural Research Institute, Council of Agriculture,Taichung County 413, Taiwan
3Department of Microbiology and Immunology, School of Medicine, Chung Shan Medical University,Taichung 402, Taiwan
Alcoholic fatty liver disease (AFLD) is due to excessive consumption of alcohol.Niuchangchih (Antrodia camphorata) is a fungus and contains polysaccharides, tri-terpenoids, steroids, benzenoids, and maleic/succinic acid derivatives. HPLC chromato-gram revealed there are five major compounds of our wild Niuchangchih: 2,4,5-tri-methoxybenzaldehyde, zhankuic acid C, dehydrosulphurenic acid, dehydroeburicoic acid,and ergostatrien-3β-ol. Those major compounds in wild Niuchangchih are zhankuic acidC (12.05%) most, followed by dehydrosulphurenic acid (3.55%), ergostatrien-3β-ol(0.7%), 2,4,5-trimethoxybenzaldehyde (0.03%), and dehydroeburicoic acid (0.03%).Because 2,4,5-trimethoxybenzaldehyde, zhankuic acid C, and dehydroeburicoic acidshowed some benefits in cell and animal model previously, we would like to study if wildNiuchangchih can protect liver from a chronic alcohol injury. Nine male Wistar rats pergroup were randomly assigned to one of the following drinking treatments: a 20% (w/w)alcohol solution (ALC); a 20% (w/w) alcohol solution co-treated with 0.25 g silymarin /kgBW/day (ALC_Sil); a 20% alcohol solution co-treated with 0.025 g Niuchangchih /kgBW/day for 4 weeks. Rats with co-treatments of silymarin or Niuchangchih had smaller (p< 0.05) relative liver size, less (p < 0.05) liver lipid accumulation, and lower (p < 0.05)liver damage indices [aspartate aminotransferase (AST) and alkaline phosphatase (ALP)values]. In the regulation of alcohol metabolism, the lower serum alcohol level was onlyobserved in alcohol-fed rats supplemented with Niuchangchih. Meanwhile, co-treatmentof silymarin or Niuchangchih increased (p < 0.05) catalase (CAT), and aldehyde dehydro-genase (ALDH) activities but did not (p > 0.05) affect alcohol dehydrogenase (ADH) andcytochrome P450, family 2, subfamily e, polypeptide 1 (CYP2E1) expressions, whichaccelerate the alcohol metabolism in the body. Additionally, silymarin or Niuchangchih

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
did not (p > 0.05) influence serum/hepatic matrix metalloproteinase-2 (MMP-2) activities,and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), activatingprotein-1 (AP1), and alpha-smooth muscle actin (α-SMA) gene expressions, butserum/hepatic MMP-9 activities and tumor necrosis factor-α (TNF-α), Kruppel-like factor 6(KLF-6), and transforming growth factor-beta 1 (TGF-β1) gene expressions of alcohol-fedrats were downregulated (p < 0.05) by silymarin or Niuchangchih, which also couldexplain lower liver damage observed in chronic-alcohol fed rats.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-199-MO
Application of Chromatographic Methods in the Studyand the Characterization of Stem Bark Extracts
from Phyllanthus muellerianus (Kuntze) Excell
Caterina Temporini1, Gloria Brusotti1,2, Ilaria Cesari1,2,Giorgio Frassà1,2, Gabriele Caccialanza1,2
1Department of Drug Sciences, University of Pavia,Pavia, Italy
1Center for Studies and Researches in Ethnopharmacy (C.I.St.R.E.), University of Pavia,Pavia, Italy
The plants of the genus Phyllanthus (Euphorbiaceae) are widely distributed in most tropi-cal and subtropical countries, and have long been used in folk medicine to treat kidneyand urinary bladder disturbance, intestinal infections, diabetes and hepatitis B [1].Phyllanthus muellerianus (Kuntze) Excell is a medicinal plant widespread in the tropicalregion of West Africa and the Baka pygmies from Cameroon prepare a water decoction ofPhyllanthus muellerianus stem bark which is used as a remedy for tetanus and woundinfections [2, 3]. In our previous work [4] the antimicrobial properties of P. muellerianusstem bark extracts have been demonstrated against different micro-organisms such asClostridium sporogenes Streptococcus mutans and Streptococcus pyogenes.
Bioassay-guided isolation together with preliminary phytochemical investigationsallowed to identify a basic fraction responsible for the biological activity. In order toseparate the more active fraction in each component and to collect them for furtherbiological assays an HPLC method was developed.
Considering the basic nature of the active fraction a cation-exchange chromatographicapproach has been chosen and successfully applied to the separation of the mixturecomponents. The optimal mobile phase is a binary mixture of 80% ammonium acetatebuffer (20 mM) and 20% acetonitrile. The separation procedure was carried out underisocratic elution at a flow rate of 0.7 ml/min.
These experimental conditions allowed us to obtain pure individual compounds thatwill be characterized by means of HPLC-ESI-MS and NMR.
In this work we have also analyzed, by capillary GC-MS technique, the oil obtained byhydrodistillation, of Phyllantus muellerianus air dried stem bark. Forty compounds repre-senting 83.5% were identified, while nineteen compounds accounting for 14.2% were notidentified. The major component of the essential oil (45%) was found to be (E)-isoelemicinidentified by comparison of its 1H NMR experimental data, with literature data [5].
1. J.B. Calixto, A.R.S. Santos, V.C. Filho, R.A. Yunes, 1998. Medicinal research reviews 18, 225–58.2. J.L. Betti, 2004. African study monographs 25, 1–27.3. R. Brisson, 1999. Etudes pygmées, SELAF n 376, Ed Peeters, Paris.4. G. Brusotti, I. Cesari, G. Frassà, P. Grisoli, C. Dacarro, G. Caccialanza, 2011, Journal of Ethnopharmacology,
DOI: 10.1016/j.jep.2011.03.0425. A. Kilic, H. Hafizoglu, H. Kollmannsberger, S. Nitz, 2004, J. Agri. Food Chem., 52, 1601.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-201-MO
Application of Multidimensional Chromatographic Methodusing Combination of IMAC, SEC and RP-HPLCfor Characterization of Soil Humic Substances
Róbert Góra, Radoslav Halko, Milan Hutta, Pavol Rohárik,Imrišcák Lukáš, Pociatková Veronika
Department of Analytical Chemistry, Faculty of Natural Sciences, Comenius University,Mlynská dolina CH-2, 84215 Bratislava, Slovakia
Multidimensional chromatography has a proven to be useful for the analysis of complexsamples such as humic substances or lignin samples. From the point-of-view of chemicalanalysis, characteristic feature of these analytes is diffuse non-distinct analytical signalproduced by many detection principles. This signal does not usually result in an exactnumerical physical-chemical data, but is described also by their distribution function orrange of validity. This dictates the necessity of development of automated complexseparation procedures with minimal sample pretreatment, and the use of on-line (off-line)multidimensional chromatographic techniques is a logical solution to these requirements.
The aim of this work was the introductory study for fractionation and characterizationof HS using off-line and on-line combination of IMAC with Al (III) and RP-HPLC or SEC,respectively. IMAC technique is based on the interaction between molecules in solutionand metal ions immobilized on a solid support. The molecules are separated according totheir affinity towards chelating metal ions, which depends on the coordination betweenthe chelating metal ion and electron donor groups from the ligand. The ligands areassumed to bind to the metal through ligand exchange.
With respect to the non-common approach we focused to evaluation of its potential tocreate orthogonal, i.e. on different separation principles working two dimensionalcomprehensive separation methods. Obtained results indicate, that such methods couldbe combined in a compact, automatized, orthogonal separation systems forcharacterization of such complicated natural substances as are examined humic acids andobtain so more information about their character.
This work was supported by the financial support of projects VEGA 1/0870/09, VEGA 1/0329/10, APVV-0595-07 and VVCE-0070-07.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-202-TU
Application of TLC-MS to Characterize Multifarious Plant Extracts
Árpád Könczöl1, Ágnes Alberti2, Ágnes Kéry2, Gyula Beke1, György Tibor Balogh1
1Compound Profiling Laboratory, Gedeon Richter Plc.,Gyömrôi u. 19–21., H-1475 Budapest, Hungary
2 Department of Pharmacognosy, Semmelweis University,Üllôi u. 26., H-1085 Budapest, Hungary
Thin layer chromatography (TLC), as a simple, cost-effective and easy-to-operate planarchromatographic technique, has been playing primary and fundamental role inphytochemical analysis for several decades. Recently, interfacing TLC with massspectrometry (MS) allows the identification and structure elucidation of the separatedcompounds, by extracting semi-automatically zones of interest from TLC plates anddirecting them online into the ion source of the MS instrument.
Several plant extracts with different and diverse secondary metabolite profile werechosen in order to test and demonstrate the features and applicability of TLC-MS inmodern phytochemical analysis. Extracts and marker compounds of Sempervivum tectorumand Matricaria recutita (for flavonoids) Curcuma spp. (for diarylheptanoids) Bergenia spp.(for glycosylated hydroquinones), Piper spp. and Lobelia inflata (for piperidine alkaloids),Camellia sinensis (for purine alkaloids), Ruta spp. (for coumarins), Thymus spp. andHarpagophytum spp. (for terpenoids) were investigated in terms of detectability,throughput and reliability. Moreover, the general usability of TLC-MS for semi-quantitative purposes was also tested and validated with reference compounds andstandardized herbal extracts.
Based on our experiments, advantages and limitations of this promising coupledtechnique will be discussed.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-203-MO
Catalytic Hydrolysis of Cellulose in Ionic Liquid
Kati Helmja, Merike VaherInstitute of Chemistry, Tallinn University of Technology,
Akadeemia tee 15, 12618 Tallinn, Estonia
The interest in cellulose as the major constituent of biomass has attracted due to itspotential application in sustainable energy production [1, 2]. During hydrolysis celluloseis transformed to monosaccharides of which glycose, fructose, mannose, arabinose andxylose are the most common. Cellulose is highly resistance to hydrolyse because of itstight hydrogen-bonding network and van der Waals interactions in molecule. However, ithas been reported that suitable and also environmental friendly solvents, ionic liquidshave found efficient application in the dissolution of cellulose [1–3].
The main purpose of the present study was catalytic hydrolysis of cellulose and willow(Salix viminalis clone 78183) tree in ionic liquid and subsequent analysis of the productionof di- and monosaccharides capillary electrophoretically. In the current work as ionicliquid 1-butyl-3-methyl imidazole chloride and as solid catalyst Amberlyst® Dry was used.At different reaction time interval, the samples (100 μl) were taken from the reactor andeach sample was diluted 5-folds by deionised water. The qualitative and quantitativeanalyses of di- and monosaccharide were performed by capillary electrophoresis, whichsuitable method was previously developed [4, 5]. It appeared, that the main disaccharidesdetected during cellulose hydrolyses were saccharose and cellubiose, but amongmonosaccharides glucose was determined. But during willow tree hydrolysis cellubiose,galactose, arabinose and xylose were monitored. Besides, the production of5-hydroxymethyl-furfural (5-HMF) was observed in both hydrolyzates.
1. Watanabe, H., Carbohydr. Polym. 2010, 80, 1168–1171.2. Jiang, F., Zhu, Q., Ma, D., Liu, X., Han, X., J. Mol. Catalys. A: Chem. 2011, 334, 8–12.3. Feng, L., Chen, Z.-I., J Mol. Liq. 2008, 142, 1–5.4. Rovio, S., Yli-Kauhaluoma, J., Siren, H., Electrophoresis, 2007, 28, 3129–3136.5. Vaher, M., Koel, M., Kazarjan, J., Kaljurand, M., Electrophoresis, 2011, 32, in press.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-204-TU
Characterization of Biological Adhesives – Insectsas an Inspiration for Biomimetic Applications
Lisa Steinhauser, Julius Braun, Manuela Gradl, Tim Nicholson, Klaus AlbertInstitute of Organic Chemistry, University of TübingenAuf der Morgenstelle 18, 72076 Tübingen, Germany
Many insect can climb on vertical walls or across ceilings. They have micro-structured toepads and additionally secrete a thin-layered fluid to attach strongly to surfaces. Thisbiological glue has some remarkable features:
1) The attachment is extremely fast but reversible.2) It works on a great variety of surfaces (smooth, rough, even wet).3) It is eco-friendly and biodegradable.Therefore it offers a high potential to inspire superior industrial adhesives for high-tech
applications.The main objective in this study is to gain new understandings of the composition of
the adhesives material secreted by insects. The results will help to develop new syntheticcounterparts with improved functions.
We investigate different kind of insect like black beetles (cockroaches) and locusts(grasshoppers). We improved the way collecting fluids from the toe pads. A SPME-fibercan be used or the pads are washed manually with water or non-polar solvent.Hydrophilic and hydrophopic compounds can be investigated separately.
Because of the small amount of the samples the structure elucidation is mainly donewith GC-MS. The derivatization depends on the different volatility of the substanceclasses. We also tried to use UHPLC and capillary NMR to characterize the compounds ofthe biological adhesives.
We could identify hydrocarbons in the hydrophobic extracts. The water extractcontains carbohydrates and amino acids that can be a hint for peptids. Future work willconcentrate on the exact function mechanism to develop a biomimetic model of theadhesive.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-205-MO
Chromatographic Profiles of Betalain Degradation Productsfrom Red Beet Root Extracts
Aneta Spórna-Kucab, Sławomir Wybraniec, Paweł StalicaFaculty of Analytical Chemistry, Institute C-1, Department of Chemical Engineering and Technology,
Cracow University of Technology,ul. Warszawska 24, Cracow 31-155, Poland
Red beet (Beta vulgaris L.) root extract is increasingly utilized as a natural food colorantdue to a growing interest of consumers in potential health benefits and non-toxic featuresof betalains. Betanin is the principal betacyanin present in red beet. However, applicationof these compounds is still problematic because they are sensitive to several degradationfactors such as elevated temperature, unfavorable pH or presence of metal cations [1].Increasing the stability of betalains will allow to elevate their utilization in pharmacy aswell as food and cosmetic industry. HPLC is the only technique used for comprehensiveseparation and analysis of betalains.
The stability study was performed on pure betanin which was isolated by HPLC fromred beet root and on its decarboxylated derivatives. Thermal treatment (85 °C) ofbetalains was carried out in citrate or acetate buffers (with/without EDTA) with addition ofdifferent solvents (ethanol, methanol and acetonitrile) during 60 min heating. Changes inpigment stability were assessed at a pH range of 3–8. The relationship between thechromatographic profiles of generated degradation products and the applied physico-chemical conditions was studied.
The pigments were much more stabile in acetate buffer with EDTA than acetate orcitrate buffer at pH 3–8. Influence of EDTA on the degradation was lower in alkalineconditions. The main degradation product of betanin was 17-decarboxy-betanin, whichwas formed at the whole pH range. In alkaline conditions, a formation of neobetanin andits further degradation during prolonged heating was observed. Decarboxylatedcompounds were more stabile than betanin which implicates their higher potential asstabile colourants in industry.
1. Wybraniec, S. Formation of Decarboxylated Betacyanins in Heated Purified Betacyanin Fractions from RedBeet Root (Beta vulgaris L.). Journal of Agricultural and. Food Chemistry, (53), (2005) 3483–3487.
This research was financed by Polish Ministry of Science and Higher Education for years 2007–2010 (ProjectNo. N205 2991 33).

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-206-TU
Comparison of Different Techniques to Identify Similar Strainsof Pseudomonas
Anna Kubesová1,3, Marie Horká1, Jaroslav Horký2, Hana Matoušková2, Karel Šlais1
1Institute of Analytical Chemistry of the ASCR,v. v. i., Veverí 97, 602 00 Brno, Czech Republic
2State Phytosanitary Administration, Division of Diagnostics,Šlechtitelu 23, 77900 Olomouc, Czech Republic
3 Department of Biochemistry, Faculty of Science, Masaryk University Brno,Kotlárská 267/2, 611 37 Brno, Czech Republic
Pseudomonas syringae pathovars (pv.), are economically important and the most studiedas a plant pathogen with more than 50 pathovars [1]. Therfore, they appear to besuitable candidates for a demonstration and comparison of the appropriate procedures forthe identification of microorganisms. The common identification of bacteria by gaschromatographic analysis of fatty acid methyl esters [2, 3] needs time-consumingisolation of microorganisms from the matrix and then the cultivation of isolates on specialagar media for at least 24 h. The gas chromatographic spectra are easy to evaluate andare useful for the characterization of individual species, and sometimes even subspecies.As a result, misidentifications may appear because of insufficient databases.
Our comparative study focuses on the analytical techniques potentially suitable fordiagnostic purposes in microbiology. The similar plant-associated strains of P. syringae,the collection of the strains or isolates from plant tissues, pv. lachrymans, maculicola,morsprunorum, tomato, mori and persicae, have been used as model microbes. They wereidentified by gas chromatographic analysis of fatty acid methyl esters [2–4], or separatedby capillary electrophoresis and capillary isoelectric focusing techniques [4, 5]. The lysatesfrom 30 microbial strains were also separated by gel IEF to obtain the characteristicproteomic ‘fingerprint’ of the Pseudomonas strain. MALDI-TOF MS has been used for theanalysis of intact proteins from whole cells for an acquirement of the characteristic proteinmass-spectrum fingerprints of particular pathovars (pv.). Based on our findings theoptimal differentiation procedure was designed.
1. V. Catara, Mol. Plant Pathol., 2007, 8, 233–244.2. A. M. Alvarez, Annu. Rev. Phytopathol., 2004, 42, 339–366.3. P. Dawyndt, M. Vancanneyt, C. Snauwaert, B. De Baets, H. De Meyer and J. Swings, J. Microbiol. Methods,
2006, 66, 410–433.4. M. Horká, J. Horký, H. Matoušková, K. Šlais, Anal. Chem., 2007, 79, 9539–9546.5. M. Horká, J. Horký, H. Matoušková, K. Šlais, J. Chromatogr., A,2009, 1216, 1019–1024.
Acknowledgement: This work was supported by the Grant Agency of the Academy of Sciences of the CzechRepublic No. IAAX00310701, grand No. 1740/2011/G6 from Czech Ministry of Education (FRVŠ) and by theInstitutional research plan AVO Z40310501.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-208-TU
Comprehensive 2-Dimensional Liquid Chromatographic Analysisof Grape Tannins
Kathithileni M. Kalili, André de VilliersStellenbosch University,
Private Bag X1, Matieland 7602, South Africa
Despite the vital role tannins play in many natural products, comparatively little iscurrently know about the exact chemical composition of this diverse group ofcompounds. This shortcoming can primarily be ascribed to the lack of suitable analyticaltechniques required for their detailed characterization. In this work, off-line, stop-flow andon-line comprehensive 2-dimensional liquid chromatographic methods for improvedanalysis of grape tannins are reported. A diol stationary phase was used in hydrophilicinteraction chromatography (HILIC) mode in the first dimension to separate compoundson the basis of polarity, while a C18 column employed in the second dimension providedreversed phase separation based on hydrophobicity. A combination of fluorescence, UVand mass spectrometric (MS) detection methods facilitated identification of compounds.Grape seeds were found to contain primarily procyanidins and galloylated procyanidins,while the skins consisted of a mixture of procyanidins, galloylated procyanidins as well asprodelphinidins. Procyanidin oligomers up to nonamers and heptamers were identified inthe grape seeds and skins, respectively. The combination of these two orthogonal 1-Dseparations (r2 < 0.3) resulted in exceptionally high practical peak capacities, particularlythose measured for the off-line and stop-flow methods. The values were slightly reducedin the on-line system due to lower peak capacity in the second dimension, but thisapproach provided the highest peak capacity production rate. Furthermore, the stop-flowand on-line methods provided automation and less risk for sample alteration. HILIC×RP-LCwas found to provide a powerful separation method for the in-depth investigation ofcondensed tannins.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-209-MO
Comprehensive 2-Dimensional Liquid Chromatographic Analysisof Rooibos Tea (Aspalathus linearis) Phenolics
T. Beelders1, K.M. Kalili2, D. De Beer3, E. Joubert1,3, A. de Villiers2
1Department of Food Science, Stellenbosch University,Private Bag X1, Matieland 7602, South Africa
2Department of Chemistry, Stellenbosch University,Private Bag X1, Matieland 7602, South Africa
3Post-Harvest and Wine Technology Division, ARC Infruitec-Nietvoorbij,Private Bag X5026, Stellenbosch 7599, South Africa
Phenolic compounds are associated with various health-promoting benefits of Rooibostea, a beverage produced from the indigenous South African shrub Aspalathus linearis.The detailed investigation of Rooibos bio-active phenolic compounds is howeverhampered by limitations associated with high performance liquid chromatographic(HPLC) methods extensively used for Rooibos analysis. Comprehensive 2-dimensionalliquid chromatography (2D-LC) offers a powerful technique for the separation of complexnatural samples due to the exceptionally high peak capacity that may be attained. Herewe report the systematic development of comprehensive 2D-LC methods for the analysisof Rooibos phenolics. In the first dimension, compounds were separated according topolarity by hydrophilic interaction chromatography (HILIC), whilst gradient reversedphase liquid chromatography (RP-LC) using a C18 column was employed in the seconddimension. Column dimensions, flow rates, gradient profiles and sampling rates wereoptimised for both on-line and off-line HILIC×RP-LC. Following method development,both methods were applied to the detailed investigation of phenolic compounds inaqueous infusions of Rooibos. Diode array- and electrospray ionisation mass spectrometry(ESI-MS) were employed for peak identification. The combination of highly orthogonalseparations provides a significant improvement in resolution, as evidenced by practicalpeak capacities in excess of 2000 measured for off-line HILIC×RP-LC analysis of Rooibosinfusions. HILIC×RP-LC demonstrated its suitability for the analysis of a diverse group ofphenolic compounds, including hydroxybenzoic and hydroxycinnamic acids,dihydrochalcones, flavones and flavonols. The separation of all these phenolics cannot beachieved in a single analysis by conventional HPLC.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-210-TU
Detection of Panax Ginseng Extracts using ComprehensiveTwo-Dimensional LC-MS Coupled with PCA
Tatsunari Yoshida1, Ken-ichiro Tanaka1, Tairo Ogura1, Tsutomu Nishine1,Hirohisa Mikami1, Luigi Mondello2, Paola Dugo2
1Analytical Applications Department, Shimadzu Corporation,380-1, Horiyamashita, Hadano-city, Kanagawa 259-1304, Japan
2Dipartimento Farmaco-chimico, University of Messina,Viale Annunziata s.n, 98168, Messina, Italy
The comprehensive two-dimensional liquid chromatography (2D-LC) is a powerful toolfor the analysis of complex samples including pharmaceutical, biological, and naturalproducts. The favourable features of 2D-LC were already demonstrated more than thirtyyears ago; since then, a lot of efforts have been put in the development ofmultidimensional techniques of ever higher peak capacity. Recently, UHPLC (Ultra HighPerformance Liquid Chromatography) has been successfully employed to remarkablydecrease analysis time in the second dimension of comprehensive LC system.
We employed a combination of RPLC (low pH) x RPLC (high pH) for 2D-LC separation.A narrow bore column (1.0 mm ID x 75 mm length) for the first dimension of 2D-LC anda short column (2.1 mm ID x 50 mm length) packed with sub-2 μm particles for seconddimension were selected. The use of a significant different pH mobile phase gave lot ofpeaks in 2D chromatogram. All experiments have been run on a 2D-LC system (ShimadzuProminence), equipped with four high pressure pumps, and interfaced through ESI to ahybrid mass spectrometer (Shimadzu LCMS-IT-TOF), which possesses both MSn ability ofan ion trap and the excellent resolution and mass accuracy of time-of-flight. The 2D-LCdata were arranged to the array data set by Profiling Solution Software (MSS) (Shimadzu)and the array data set were analyzed by SIMCA-P+ (Umertrice) for Principal ComponentAnalysis (PCA). Furthermore, the 2D-LC data were visualized and elaborated into two andthree dimensions using Chromsquare ver. 1.3 software (Chromaleont, Messina, Italy).
In this study, in order to find the similarities and differences of the four differentproducing of Panax ginseng, a popular Chinese herbal medicine, we employedcomprehensive 2D-LC for the analysis of their extracts of row forms. The chromatographicbehavior of some major ginsenosides (ginsenoside Rg1, Re, Rb1, Rc, and etc) and otherminor compounds was monitored. The hundreds array data of retention time and m/zpairs were produced from the 2D chromatographic data by MSS and analyzed by PCAmethod. The PCA scores plot could be largely divided into two clusters.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-211-MO
Determination of Astragalin and Astragalosides I–IV in RadixAstragali using High-Performance Liquid Chromatography Method
with Pulsed Amperometric Detection
Ha-Jeong Kwon, Yong-Duk ParkDepartment of Preventive and Social Dentistry, Graduate School, Kyung Hee University,
Hoegi-dong, Dongdaemoon-gu, Seoul 130-701, South Korea
We developed a highly sensitive reversed-phase HPLC-pulsed amperometric detectionmethod for the determination of astragalin and astragaloside I–IV in Radix Astragali.
Pulsed amperometry permits detection of carbohydrates with excellent signal-to-noiseratios without requiring derivatization. Carbohydrates are detected by measuring theelectrical current generated by their oxidation at the surface of a gold electrode. Theastragalin and astragalosides are glycosides which have carbohydrates moiety in theirchemical structures. Therefore, glycosides can be also detected at PAD with highsensitivity. The glycosides were completely separated within 20 min on a reversed-phasecolumn using a water-acetonitrile gradient, and were detected using a PAD under NaOHalkaline conditions. Sodium hydroxide solution is added to post-column reagents becausesugar is converted to anions, which can be detected by PAD under strongly alkalineconditions.
Absolute methanol was used as dissolution and extraction solvent becauseastragaloside I–III are decomposed in the aqueous solution. Radix Astragali were extractedby sonication in 100% MeOH for 30 min. Digitoxin (10 μg/mL) was used as an internalstandard.
The detection (S/N = 3) and quantification (S/N = 10) limits for the compounds were0.03–0.15 ng and 0.10–0.45 ng, respectively. The linear regression coefficient was0.9982–1.0000 for concentrations of LOQs-25 μg/mL.
The contents of astragalin and astragalosides are successfully determined in main roots,lateral roots and rhizome in the 4-year-old, 5-year-old, and 6-year-old were determined.This analytical method showed good precision and accuracy.
This research was supported by Basic Science Research Program through the National Research Foundation ofKorea (NRF) funded by the Ministry of Education, Science and Technology (2010-0002694).

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-212-TU
Determination of Carotenoids in Commonly Consumed Vegetables
Zora Kotíková, Alena Hejtmánková, Jaromír LachmanDepartment of Chemistry, Faculty of Agrobiology, Food and Natural Resources,
Czech University of Life Sciences, Prague, Czech Republic
This study was carried out to determine the concentrations of the main carotenoids inseven types of vegetables, which are commonly consumed in the Czech diet, namelylettuce (Lactuca sativa, L.), broccoli (Brassica oleracea, L.), carrot (Daucus carota, L.), greenand yellow pepper (Capsicum annuum, L.), tomato (Solanum lycopersicum, L.) and potato(Solanum tuberosum, L.). The vegetables were purchased from Prague supermarkets.Carotenoids were extracted into a mixture of ethanol:acetone:hexan (1:1:2, by volume)with addition of 0.2% BHT. Determination of samples was carried out in triplicates usinga HPLC system (ULTIMATE 3000, DIONEX) with PDA detection. For separation ofcarotenoids a C30 YMC Carotenoid Column (3.0 × 150 mm, 5 μm, WATERS) was used.The gradient elution operating at 30 °C for 35 up to 55 min at flow rate 0.8 ml/minconsisted of methanol, water and tert-butyl(methyl)ether. The carotenoids were detectedat 445 nm and were quantified using the external standards method.
The green vegetables, lettuce (L) and broccoli (B) showed similar elution profiles. Theyboth contained high levels of xanthophylls lutein (18.2 μg/g fw L, 21.5 μg/g B),violaxanthin (12.7 μg/g L, 5.1 μg/g B) and neoxanthin (10.9 μg/g L, 5.2 μg/g B) andrelatively high amount of β-carotene (13.5 μg/g L, 8.8 μg/g B). In addition, broccolicontained a small amount of zeaxanthin (1.2 μg/g).
The major contributor to the total carotenoid content in yellow (Y) and green (G)pepper was lutein (2.2 μg/g Y, 3.5 μg/g G) followed by β-carotene (0.9 μg/g Y, 1.3 μg/gG). Yellow pepper also contained α-carotene (0.5 μg/g). On the other hand, in greenpepper neoxanthin (0.8 μg/g) and zeaxanthin (0.4 μg/g) were found.
Carrot and tomato had relatively simple profiles. Carrot contained α-carotene (43.9μg/g) and β-carotene (86.6 μg/g) as main carotenoids and lutein (3.1 μg/g) as minorcomponent. Tomato, a rich source of lycopene (27.9 μg/g), also contained β-carotene(3.6 μg/g) and lutein (0.4 μg/g) in smaller amounts. Potato was relatively poor source ofcarotenoids. Most of the total carotenoids were xanthophylls, namely violaxanthin (0.3μg/g), neoxanthin (0.3 μg/g), lutein (1.7 μg/g) and zeaxanthin (0.1 μg/g). β-Carotenewas also found but only in trace amount (0.1 μg/g).

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-213-MO
Determination of Carotenoids, Flavonoids, Phenolic Acidsand Tocopherols in Ethanolic Extracts of Pleurotus eryngii
Fruit Bodies Harvested at Various Time by HPLCand Antioxidant Capacities of the Extracts
Deng-Jye Yang1, Jau-Tien Lin2, Lao-Dar Juang3
1School of Health Diet and Industry Management, Chung Shan Medical University,110, Section 1, Jianguo N. Road, Taichung 402, Taiwan
2Department of Applied Chemistry, Chung Shan Medical University,110, Section 1, Jianguo N. Road, Taichung 402, Taiwan
3Department of Horticulture, National Chung Hsing University,250, Kuo Kuang Road, Taichung 402, Taiwan
Pleurotus eryngii is a popular edible mushroom in Taiwan. In the study, compositions ofcarotenoids, flavonoids, phenolic acids and tocopherols in ethanolic extracts of themushroom fruit bodies harvested at the 10th, 12th and 15th days from sawdust mediumpacking bags after the full growth of the mycelia and inducing the fruit body formationwere determined by HPLC. Antioxidant properties of the extracts were also evaluated.Harvest time would influence compositions of these antioxidant components in theethanolic extracts and their antioxidant capacities. The extract from the fruit bodyharvested at the 10th day had the highest antioxidant components and antioxidantcapacities, followed by those harvested at the 12th and 15th days. The highest levels ofphenolic acid, flavonoid, tocopherol and carotenoid in the extracts were syringic acid,epicatechin, α-tocopherol and all-trans-β-carotene, respectively.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-214-TU
Determination of Certain Flavonoids by Liquid ChromatograhyEmploying Internal Standard and its Application
to Some Aromatic Plants
Ulku Dilek Uysal1, Muzaffer Tuncel2, Nilgun Ozturk3, Elif Mine Oncu-Kaya1,Sinem Türkkan1, Kubra Kurtulan1
1Anadolu University, Faculty of Science, Department of Chemistry,26470 Eskisehir, Turkey
2Anadolu University, Faculty of Pharmacy, Department of Analytical Chemistry,26470 Eskisehir, Turkey
3Anadolu University, Faculty of Pharmacy, Department of Pharmacognosy,26470 Eskisehir, Turkey
This study describes a method development for the determination of five flavonoids byusing a gradient HPLC method with a suitable internal standard and its application tocertain aromatic plants which are used as herbal tea in Turkey. The analysis wasperformed by utilizing a two solvents system [A: methanol:water:formic acid (10:88:2;v:v:v); B: methanol:water:formic acid (90:8:2; v:v:v)] on a reverse phase column. Theflow-rate and injection volume were 1 mL min−1 and 10 μL, respectively. Signals weredetected at 340 nm where all flavonoids have the most compromising rate of peaknormalization. The utilized flavonoids appeared, in turn, in the presented conditions asrutin, quercetin, luteolin, kaempferol, apigenin. An internal standard [5,5’-dithio-bis(2-nitrobenzoic acid)] technique was also applied for the analysis of flavonoids to increaseprecision. The repeatability results as RSD% were among 0.74 to 4.64 for intra-day and2.02 to 5.67 was for inter-day with the employment of (9.92x10−6 – 4.17x10−5 M)flavonoids. Linearity parameters were also examined in the range of 1.27x10−6 M –4.10x10−5 M flavonoids, and very good correlations were obtained. The limit of detectionand limit of quantification of each flavonoid were calculated according to the criterions(S/N = 3.3) and (S/N = 10), respectively. The method was applied to the extracts ofcertain Salvia species [Salvia tchihatcheffii (Fisch. & Camey.), Salvia triloba L. and Salviatomentosa Mill.] used as herbal tea in Turkish traditional medicine and reasonable resultswere obtained.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-216-TU
Determination of Phenolic Acids in Gingerby High Performance Liquid Chromatography
Elif Mine Öncü Kaya1, Nilgün Öztürk2, Ülkü Dilek Uysal1, Muzaffer Tunçel31Anadolu University, Faculty of Science, Department of Chemistry,
26470 Eskisehir, Turkey2Anadolu University, Faculty of Pharmacy, Department of Pharmacognosy,
26470, Eskisehir, Turkey3Anadolu University, Faculty of Pharmacy, Department of Analytical Chemistry,
26470, Eskisehir, Turkey
Ginger (Zingiber officinale Roscoe, Zingiberacae) is widely used around the world in foodsas a spice and for medicinal purposes. It has also been suggested for the treatment ofvarious other conditions, including atherosclerosis, migraine headaches, rheumatoidarthritis, high cholesterol, ulcers, depression, and impotence. In addition to thesemedicinal uses, is believed to help the common cold, flu-like symptoms, and even painfulmenstrual periods. Some phenolic substances present in ginger, generally, possess stronganti-inflammatory and anti-oxidative properties and exert substantial anti-carcinogenicand anti-mutagenic activities. In this study, the determination of phenolic acids of theinfusion, methanol and ethyl acetate extracts obtained from ginger roots, HPLC analysiswas performed by utilizing two solvents system [A: methanol:water:formic acid (10:88:2;v:v:v); B: methanol:water:formic acid (90:8:2; v:v:v)] on a C18 column. The flow-rate andinjection volume were 1 ml min−1 and 10 μL, respectively. Signals were detected at 280nm. Besides, internal standard technique (propylparaben) was applied for the analysis ofphenolic acids to increase the precision. Chlorogenic acid was the dominant phenolic acidfound in extracts. The other phenolic acids detected from the extracts wereprotocathechuic, caffeic, p-coumaric and o-coumaric acids.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-217-MO
Determination of Saponins in Paris formosana Hayataby High-Performance Liquid Chromatographywith Evaporative Light Scattering Detection
Jau-Tien Lin1, Yan-Zin Chang2, Shih-Chuan Liu3, Mei-Peng Lu1, Deng-Jye Yang3
1Department of Applied Chemistry, Chung Shan Medical University,110, Section 1, Jianguo N. Road, Taichung 402, Taiwan
2Department of Nutrition, Chung Shan Medical University Hospital,110, Section 1, Jianguo N. Road, Taichung 402, Taiwan
3School of Health Diet and Industry Management, Chung Shan Medical University,110, Section 1, Jianguo N. Road, Taichung 402, Taiwan
Nine steroidal saponins were found in Paris formosana Hayata. The saponins were threefurostanol glycosides, two pennogenyl glycosides (spirostanol glycosides) and fourdiosgenyl glycosides (spirostanol glycosides). In the study, a high performance liquidchromatographic method was developed to simultaneously determine ten steroidalsaponins including three furostanol glycosides, three pennogenyl glycosides and fourdiosgenyl glycosides which was used to quantified the steroidal saponins in Parisformosana Hayata. The stationary phase was a C18 column kept at 35 °C and the mobilephase was a gradient solvent system consisting of acetonitrile/deionized at flow rate of 1ml/min. An evaporative light scattering detector was used to detect these saponins; thelimit of detections (LODs) and limit of quantifications (LOQs) were 0.01~0.27 and0.04~0.90 μg, respectively. The separation factors (α) and resolutions (Rs) of the saponinswere larger than 1. The saponins in P. formosana Hayata gathered from various areas ofTaiwan were also determined.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-218-TU
Determination of the Composition of Natural Productsby HPLC with Charged Aerosol Detection
Ian N. Acworth, Bruce Bailey, Paul Gamache, John WaraskaESA – A Dionex Company,
22 Alpha Rd., Chelmsford, MA 01824, USA
Natural products contain a great diversity of compounds that can show extreme variationin their physicochemical properties. Analysis of active components can be challenging asnot all contain a chromophore or can ionize, thereby limiting the use of UV absorbanceand mass spectrometry, respectively. Charged aerosol detection is a sensitive, universal(nonselective) approach that can measure any nonvolatile and many semivolatilecompounds. A number of isocratic and gradient HPLC-Corona® charged aerosol detector(CAD®) methods were developed and evaluated for the measurement of analytes from avariety of natural products including black cohosh, ginkgo biloba, ginseng, soy, andstevia. Analytes showed consistent response independent of chemical structure (typically< 10% variability between compounds). All methods had a wide dynamic range (4 ordersof magnitude), good sensitivity (typically low ng levels of detection), and excellentreproducibility (RSDs typically < 2%) even at low detection levels.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-219-MO
Determination of Tocopherols in Pumpkin Seed Oils by HPLC-UV-Vis:Application to Oil Classification According to Genetic Variety
Yanelis Saucedo-Hernández1, María Jesús Lerma-García2,José Manuel Herrero-Martínez2, Guillermo Ramis Ramos2, Elisa Jorge Rodríguez1,
Ernesto F. Simó-Alfonso2
1Pharmacy Department, Faculty of Chemistry and Pharmacy, Central University “Marta Abreu” of Las Villas,C-54830 Santa Clara, Cuba
2Department of Analytical Chemistry, University of Valencia,Doctor Moliner 50, E-46100 Burjassot, Valencia, Spain
A method for the determination of tocopherols (Ts) in pumpkin seed oils by HPLC withUV-Vis detection was developed. Owing to the significant variation of the pharmaceuticaland nutritional properties of the oils with the genetic variety, the development ofauthentification methods, capable of distinguishing the genetic variety, is of interest. Oilswere extracted by pressing the seeds of three different genetic varieties of Cucurbitamoschata, RG, Inivit C-88 and Inivit C-2000, commonly grown in Cuba. Extracts of theoils in methanol were processed. Optimal separation of the Ts was achieved at 45 °C,using a C18 column (5 μm, 15 cm x 4,6 mm) and a mobile phase constituted bymethanol with a small water percentage (ca. 2%). With a flow rate of 2 mL min−1, totalanalysis time was 8 min. The method was applied to the quantification of the Ts in theoils. Identification of other minor components which are characteristic of Cucurbita oilextracts is in progress. Further, multivariate processing of the chromatographic data wasuseful to classify the pumpkin seed oils according to their respective genetic varieties. Forthis purpose, linear discriminant analysis was used. The model provided classification ofCucurbita moschata oils of different varieties with high reliability.
Acknowledgements: Projects CTQ2010-15335 (MICINN and FEDER funds) and A/031344/10 (AECI). M.J.Lerma-García thanks the Generalitat Valenciana for an FPI grant for PhD studies.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-220-TU
Development and Certification of Catechins and Alkaloidsin Green Tea Containing Reference Materials
L. C. Sander1, K. E. Sharpless1, S. A. Wise1, M. Bedner1, M. C. Tims1, R. Lieberman1,J. H. Yen2, A. NguyenPho3, M. C. Roman4, M. Payne5, J. M. Betz6
1National Institute of Standards and Technology (NIST), Analytical Chemistry Division,100 Bureau Drive, MS 8392, Gaithersburg, MD 20899-8392, USA
2National Institute of Standards and Technology (NIST), Statistical Engineering Division,Gaithersburg, MD 20899-8980, USA
3Center for Drug Evaluation and Research (CDER), Food and Drug Administration,College Park, MD 20740, USA
4Tampa Bay Analytical Research, Inc.,Largo, FL 33777, USA
5Hershey Foods Corporation,Hershey, PA, USA
6Office of Dietary Supplements, National Institutes of Health,Bethesda, MD 20892, USA
Tea (Camellia sinensis L.) is a popular beverage that has been consumed in some culturesfor centuries. Increased interest in this beverage has resulted from perceived healthbenefits that may be associated with its consumption, and it is now commonly used indietary supplement formulations. The latter materials are typically marketed as aids forweight loss and as stimulants to promote energy; they are also advertised as exhibitingantioxidant properties.
NIST is working in collaboration with the National Institutes of Health, Office of DietarySupplements (NIH-ODS) and the Food and Drug Administration (FDA) to developStandard Reference Materials (SRMs) to support the chemical analysis of dietarysupplements. These materials are being developed as suites to provide a close match fordifferent types of sample matrices that may provide similar analytical challenges thatwould be encountered by the analyst. The materials are intended primarily for use invalidating analytical methods and for quality assurance when assigning values to in-housecontrol materials. By providing SRMs with known composition, a major source ofmeasurement uncertainty is eliminated leading to improved measurement accuracy.
A suite of three green tea-containing SRMs has been issued as part of this effort: SRM3254 Camellia sinensis (Green Tea) Leaves, SRM 3255 Camellia sinensis (Green Tea)Extract, and SRM 3256 Green Tea-Containing Solid Oral Dosage Form. These referencematerials were characterized for catechins, caffeine and other xanthine alkaloids, usingmultiple methods (i.e., liquid chromatography with absorbance, fluorescence, and massspectrometry detection). These SRMs are the first certified reference materials (CRMs)designed specifically to support the measurement of catechins and alkaloids in green tea.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-221-MO
Extraction and Analyses of Chiral Isomers (Eipgoitrin/Goitrin)from Isatis Indigotica Fort Root Extract using SFE and SFC-MS
Jacquelyn Runco1, Jeff Wright1, Li Yang2, Kate Yu3, Rui Wang2, Yiming Li2,Zhengtao Wang2, Alan Millar3, Harbaksh Sidhu4, Rui Chen1
1Waters Corporation,New Castle, DE, USA
2Shanghai Traditional Chinese Medicine University,Shanghai, P. R. China3Waters Corporation,
Milford, MA, USA4Waters Corporation,Pittsburgh, PA, USA
Traditional Chinese Medicines (TCMs) have played an important role in Asia as major clinicaltherapies for thousands of years. In the past decade, the usage of TCMs has expandedglobally and gained increasing acceptance as a complement to modern Western medicine.A large number of TCMs are chiral compounds. Since optical isomers could possessconsiderable different bioactivities, it is therefore important to quantify enantiomers in orderto better understand stereo-specific bio-disposition of these enantiomers in TCM.
(R, S)-goitrin are found in Isatis indigotica Fort (known as the ban lan gen in China), butonly the R-goitrin (Epigoitrin) displays the desired antiviral activity. Currently, thequantitative analysis of (R, S)-goitrin often involves a solvent extraction step, which cantake several hours to days, followed by a RPLC based quantitation methodology.However, RPLC does not resolve R- and S-goitrin; therefore it can’t accurately quantify thebioactive R-goitrin. Recently, Lin et al. demonstrated a normal phase HPLC/UV basedchiral separation and quantitation of R- and S-goitrin. The separation time was 50 min.Here, we report a supercritical fluid based workflow for qualitative and quantitativeanalysis of (R, S)-goitrin: SFE extraction followed by SFC-PDA-MS based separation andquantitation of R- and S-goitrin with significantly reduced overall process time.
We first used the racemate to develop an SFC method for the chiral separation of (R,S)-goitrin. Multiple chiral stationary phases (CSPs) and co-solvents were screened using ageneric gradient. Next, we will explore the SFE extraction process. Parameters such aspressure, co-solvent and processing time will be varied to selectively extract (R, S)-goitrinfrom the Isatis indigotica Fort root. The extraction efficiency and specificity will beassessed and compared to the traditional solvent extraction approach. Finally, we willoptimize the chromatographic conditions for the extract analysis to reduce the run time.The optimized chromatographic conditions will then be applied to perform a series ofquantitative analyses of R- and S-goitrin using SFC-PDA-MS, including LOD, LOQ,linearity, calibration curve, precision, accuracy, reproducibility, stability and recovery.Analyses of several commercially available ban lan gen powder will also be presented.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-222-TU
Gradient HPLC with Electrochemical Array Detectionfor the Measurement of Polyphenols in Natural Products, Botanicals,
Supplements, and Animal and Human Tissues
Ian N. Acworth, John Waraska, Paul GamacheESA – A Dionex Company,
22 Alpha Rd, Chelmsford, MA 01824, USA
There continues to be great interest in the potential health benefits of polyphenoliccompounds present in fruits, vegetables and natural products. Of all the multitude ofpolyphenols present in plant materials, the most widely studied include catechins,flavonols and phytoestrogens. The biological effects of these compounds, as shown bynumerous in vitro and animal studies, suggest that they may be protective against cancer,cardiovascular, inflammation and other diseases. Although there have been great stridesin understanding the occurrence, bio-availability and biological activities of some of thesecompounds, their in vivo effects in humans is an active area of research. Analyticalmethods that are capable of measuring low levels of these compounds and theirmetabolites with a high degree of specificity in biological tissues are therefore needed.
Polyphenols are electrochemically active and can be measured using HPLC withcoulometric electrode array detection. This technique utilizes multiple electrochemicalsensors that can be optimized for more than one chemical class. Easily oxidizedcompounds can be selectively detected at upstream, low potential sensors, while higheroxidizing compounds respond at downstream higher potential sensors. Consequently,this approach is both extremely sensitive (fg levels) and provides qualitative information(voltammetric behavior) for analyte identification. It also offers a wide linear range and isthe only electrochemical approach that is fully compatible with gradient elution.Presented here are examples of its use for targeted analysis of polyphenols (e.g.,catechins, isoflavones) in a number of animal and human tissues (e.g., urine, plasma andbrain tissue) and its use in metabolomics for generation of global metabolite patterns fordetermining the content of commercially available natural product supplements.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-223-MO
High Throughput Methodology to Characterize Red WinePolyphenols using Solid Phase Extraction Combined
with Ultra-Performance Liquid Chromatography
Jorge Pereira, Catarina Luis Silva, José S. CâmaraCentro de Química da Madeira, Centro de Ciências Exactas e da Engenharia da Universidade da Madeira,
Campus Universitário da Penteada, 9000-390 Funchal, Portugal
Red wine has been extensively associated to the health protection provided by the famousMediterranean diet. In this study we present a fast, reliable and high throughputmethodology involving Ultra-Performance Liquid Chromatography (UPLC) with photodiodearray (PDA) detection that allows the simultaneous characterization of 15 importantbioactive metabolites present in red wine from different geographical origins. Themetabolites analysed were the polyphenols gallic acid, protocatechuic acid, (−)-catechin,gentisic acid, (−)-epicatechin, syringic acid, p-coumaric acid, ferulic acid, m-coumaric acid,rutin, trans-resveratrol, myricetin, quercetin, cinnamic acid and kaempferol. To reduce thecomplexity of wine extract and improve the recovery efficiency of the methodology,samples were subjected to a reverse-phase solid-phase extraction (SPE) procedure using assorbent a new macroporous copolymer made from a balanced ratio of two monomers, thelipophilic divinylbenzene and the hydrophilic N-vinylpyrrolidone (OasisTM HLB) prior toUPLC-PDA analysis. This analysis involved a 50 mm column packed with 1.7 μm particlesoperating at elevated pressure to attain ultra-fast analysis and highly efficient separations.The method was validated using a Madeira Island wine sample spiked with polyphenols atconcentration levels ranging from 0.06 (cinnamic acid) to 12 μg mL−1 ((−)-catechin). Thecalibration curves of metabolites showed good linearity within the studied range. Detection(LOD) and quantification (LOQ) limits ranged from 0.006 μg mL−1 to 0.581 μg mL−1, andfrom 0.019 μg mL−1 to 1.938 μg mL−1, for gallic and gentisic acids, respectively. The averagerecoveries (n = 6) ranged from 40% (m-coumaric acid) to 149% (myricetin), with relativestandard deviations between 2.02% ((−)-catechin) and 25.16% (kaempferol). The validatedmethod was then applied to red and white wines from different geographical origins(Azores, Canary and Madeira Islands). (−)-Epicatechin and (−)-catechin were found themajor polyphenols in the wines assayed. Red wines were found to be richer than whitewines in the polyphenols analysed.
The method proved to be sensitive, selective, and with good precision, and moreover,an ultra-fast approach allowing the separation of the 15 bioactive metabolitesinvestigated with high resolution power in within 5 min.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-224-TU
High Throughput SPE/UPLC-PDA-Based Methodologyfor the Simultaneous Determination of
Bioactive Phenolic Metabolites in Food Dietary Products
Carla Miguel, Jaime Camacho, Paulo Craveiro,Catarina L. Silva, Jorge Pereira, José S. Câmara
CQM – Centro de Química da Madeira, Centro de Ciências Exactas e da Engenharia da Universidade daMadeira, Campus Universitário da Penteada, 9000-390 Funchal, Portugal
Polyphenols are common constituents of foods of plant origin and major antioxidants ofour diet. The main dietary sources of polyphenols are fruits, vegetables, and beverages,such as tea, coffee, or wine. Several hundreds of different polyphenols have beenidentified in foods [1]. The two main types of polyphenols are (i) flavonoids (flavonols,flavones, flavan-3-ols, flavanones, isoflavones, and anthocyanidins) and (ii) nonflavonoidcompounds (phenolic acids, hydroxycinammates and stilbenes). Arising biogeneticallyfrom either, the shikimate/ phenylpropanoid pathway or ‘polyketide’ acetate/malonatepathway, or both, producing monomeric and polymeric phenols and polyphenols, aschemically defined above, and which fulfill a very broad range of physiological roles inplants. As antioxidants, polyphenols may protect cell constituents against oxidativedamage and, therefore, limit the risk of various degenerative diseases associated tooxidative stress, namely cardiovascular diseases, cancers, neurodegenerative diseases,diabetes, or osteoporosis. There is, therefore, increasing interest in the role of nutritionand specific dietary constituents in the prevention of such diseases.
Therefore in this study, a reversed-phase UPLC method that uses gradient elution andphotodiode array detection was used to quantitate several biologically active phenolicconstituents of vegetables: broccoli (Brassica oleracea L.); tomato (Lycopersicon esculentum,Mill) and carrot (Daucus carota L.). The bioactive metabolites were extracted by Ultrasoundextraction. The performance of different solvents, methanol, ethanol, water, and ethylacetate, to isolate phenolic compounds was tested and evaluated. The extract clean-up wascarried out on a macroporous copolymer made from a balanced ratio of two monomers, thelipophilic divinylbenzene and the hydrophilic N-vinylpyrrolidone (OasisTM HLB). Thebioactive phenolics were eluted by 250 μL of a mixture containing 95% methanol and 5%water, and the separation was carried out on a HSS T3 analytical column (100 mm × 2.1mm, 1.8 μm particle size) using a binary mobile phase composed of aqueous 0.1% formicacid (eluent A) and methanol (eluent B) in the gradient elution mode. Each analysis requiredan equilibration period of 3 min and a run time of 13 min for completion.
In broccoli, gentisic acid was, by far, the most abundant phenolic metabolite, followedby (−)-catechin and ferulic acid. The main components isolated from carrots werequercitin, gentisic acid and kaempferol, whereas in tomato ferulic acid, (−)-catechin andgentisic acid are the dominants bioactive phenolics.
1. A. Scalbert, G. Williamson, J. Nutr., 130 (2000) 2073S.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-225-MO
High-Performance Liquid Chromatographic Determination ofBergenin and Arbutin in Leaves of Different Bergenia Species
Borbála Boros1, Silvia Jakabová2,3, Réka Molnár4, Ágnes Farkas4, Attila Felinger2
1Institute of Viticulture and Oenology, Faculty of Sciences, University of Pécs,Pázmány Péter u. 4, H-7634 Pécs, Hungary
2Department of Analytical and Environmental Chemistry, Faculty of Science, University of Pécs,Ifjúság útja 6, H-7624, Pécs, Hungary
3Department of Chemistry, Faculty of Natural Sciences, Constantine the Philosopher University in Nitra,Tr. A. Hlinku 1, 949 01 Nitra, Slovakia
4Institute of Pharmacognosy, Medical School, University of Pécs,Rókus utca 2, H-7624, Pécs, Hungary
Bergenia species (Saxifragaceae) are important medicinal plants distributed in South andEast Asia, as well as European countries. In India these plants grow at high altitudes in theHimalayas usually in rocky areas and on cliffs. They are popularly known in the Indiansystem of medicine as Pashanbhed (meaning ‘to dissolve the stone’) because the rhizomeshave been used for centuries in herbal formulations for dissolution of kidney and bladderstones, and for treatment of leucorrhea, piles, and pulmonary infections [1].
Bergenia crassifolia L. is commonly known as Siberian tea or elephant ear. The plant isvalued for its antibacterial, anti-inflammatory and antioxidant effect, and recently theadaptogenic properties of the leaves have been described [2].
In our experimental garden at the University of Pécs, a plantation of six differentBergenia lines was established from the propagation material originating from the MTTResearch Institute, Finland. The main goal of our study was to elaborate an optimalmethod for detecting various levels of the main active components, arbutin and bergeninin leaf samples of the six Bergenia lines.
High-performance liquid chromatography (HPLC) coupled with diode array detector(DAD) method was used for the analysis. The method was evaluated for a number ofvalidation characteristics (repeatability and intermediate precision, LOD, LOQ, calibrationrange, and recovery).
Significant differences were detected in the level of active compounds in the sixinvestigated Bergenia lines, and the measured data were within the previously reportedrange of arbutin content [2, 3].
The present study highlights the importance of selecting medicinal plant lines withoptimal levels of effective substances and of elaborating suitable methods for the fast andefficient detection of these active agents in the drug samples.
1. L.V. Asolkar, K.K. Kakkar, and O.J. Chakre, Glossary of Indian Medical Plants with Active Principles. PID,CSIR, New Delhi, 1992, p. 122

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
2. A.N. Shikov, O.N. Pozharitskaya, M.N. Makarova., H.J. Damien Dorman, V.G. Makarov, R. Hiltunen, B.Galambosi, J. Functional Foods 2, 71–76 (2010)
3. C. Po p, L. Vlase L, T. Mircea, Not. Bot. Hort. Agrobot. Cluj 37, 129–132 (2009)
Acknowledgement: The work was supported by the grants OTKA K75717 and SROP-4.2.2/08/1/2008-0011.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-226-TU
Identification of Morphological Similar Species of Genus Moniliniaby Capillary and Gel Format of the Electromigration Techniques
and MALDI-TOF MS
Anna Kubesová1,3, Marie Horká1, Jirí Šalplachta1, Eva Zapletalová2, Jaroslav Horký2
1Institute of Analytical Chemistry of the ASCR,v. v. i., Veverí 97, 602 00 Brno, Czech Republic
2State Phytosanitary Administration, Division of Diagnostics,Šlechtitelu 23, 77900 Olomouc, Czech Republic
3Department of Biochemistry, Faculty of Science, Masaryk University Brno,Kotlárská 267/2, 611 37 Brno, Czech Republic
Monilinia spp. are responsible for major losses in production of, e.g., Rosaceae family gen-era. The disease may seriously reduce or even destroy a crop by affecting blossoms andfruits, either on the tree or after harvest [1]. Four closely related spp. of Monilinia havebeen identified. In North America, brown rot of fruit is mainly caused by Moniliniafructicola and to lesser extent by Monilinia laxa. In Europe, the main causal agents of thedisease are Monilinia fructigena and M. laxa. Monilie polystroma are similar to those of M.fructigena and has to date been found only in Japan [2]. However, this pathogen wasalready identified on apple shoots in Hungary [3]. The identification of the Monilinia spprelies on few morphological characteristics, and identification of the isolates in culturemedium (after 4–7 days of incubation) and mycelial growth rate is difficult, since theirappearance vary from isolate to isolate within species [4, 5]. They can not be in principlebe distinguished from other brown rot fungi except by laboratory examination.
This research will build on our experimental knowledge arising from the CIEF and CZEseparation of the hydrophobic conidia from the cultures of different strains of thefilamentous fungi, Aspergillus, Fusarium and Penicillium [6]. Now we develop rapid andreproducible methods for identification of intact spores of Monilinia spp. based on off-linecombination CZE, CIEF, gel IEF, SDS-PAGE and MALDI-TOF MS techniques. The optimizedmethods can be applied also at the identification of the Monilinia spp. from the real plantsamples and at the identification of four M. polystroma strains. The results were comparedwith those obtained by conventional microbiological identification procedures.
1. J. P. Kutter, S. C. Jacobson, J. M. Ramsey, J. Microcolumn Sep. (2000), 12, 93–97.2. M. J.Cote, M. C Tardif, A. J. Meldrum, Plant Dis. (2004), 88, 1219–1225.3. M. Petroczy, L. Palkovics, Eur. J. Plant. Pathol. (2009), 125, 343–347.4. L. J. Pernose, J. Tarran, A.-L. Wong, Aust. J. Agric. Res. (1976), 27, 547–556.5. J. Hinrichs-Berger, G. Mueller, J. Plant Dis. Protect. (2010), 117, 110–111.6. M. Horká, F. Ružicka, A. Kubesová, V. Holá, K. Šlais Anal Chem (2009), 81, 3997–4004.
Acknowledgement: This work was supported by the Grant Agency of the Academy of Sciences of the CzechRepublic No. IAAX00310701. Institutional research plan AVO Z40310501.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-227-MO
Improved Analysis of Isoquinoline Alkaloids in Goldenseal RootExtract using Charged Surface Hybrid Technology
Mia Summers, Kenneth J. FountainWaters Corporation,
34 Maple St. Milford, MA 01757, USA
Goldenseal extract from the native North American plant, Hydrastis canadensis, has longbeen used as a traditional medicine across many cultures. Goldenseal possesses anti-microbial and anti-inflammatory properties and demonstrates broad medicinal applicationincluding treatment of gastrointestinal disorders, liver disease and skin infections. Bio-active components in Goldenseal include isoquinoline alkaloids such as berberine andhydrastine, which typically show poor peak shape and loadability on traditional reversedphase columns.
Here we demonstrate the analysis of Goldenseal root extract by UPLC using a chargedsurface hybrid column, showing enhanced peak shape of the isoquinoline alkaloids andincreased loading capacity of the sample. The improved peak shape allows for greaterresolution and sensitive detection of minor components. Identification of components inGoldenseal extract is achieved using accurate mass measurement on a UPLC-ToF-MSsystem. Method scaling from UPLC to preparative HPLC is also shown, where improvedpeak shape and sample loading provide more effective preparative-scale purification ofcomponents.
The superior peak shape and loadability of alkaloids using charged surface hybridcolumns can facilitate identification and purification of components in natural productextracts such as Goldenseal root. Further, the addition of ToF-MS detection using accuratemass calibration can provide rapid identification of components in these complexmixtures.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-228-TU
Improving the Analysis of Complex Natural Productsthrough Systematic Evaluation of Different Liquid Chromatography
Stationary Phase Chemistries and Modern Particle Platforms
David S. Bell, Craig Aurand, Anders Fridstrom, Frank MichelSigma-Aldrich/Supelco
595 North Harrison Road, Bellefonte, PA 16823, USA
Natural products often consist of a highly complex mixture of both active and inactiveingredients. The development of appropriate analytical separation methods can thereforebe an arduous task. Increasing government regulations relating to the production andsale of natural products have placed even more burden on the analytical chemist todevelop effective and robust methods. To assure the quality of herbal medicinal productsthe content of either constituent(s) with known therapeutic activity, active markers oranalytical markers has got to be determined in the starting materials, and within in-process controls and batch release controls [1].
Due to the complex composition of natural products HPLC analysis time is very oftenmore than one hour. Modern particle technologies often provide the ability to reduce theanalysis time without sacrificing resolution [2, 3]. When separating the components of acomplex mixture, traditional alkyl stationary phases may also be inadequate. The choiceof alternative phases, however, is often made arbitrarily and thus may be ineffective atsolving a given issue.
In this study, the separation of active and inactive components for several popularnatural products is investigated using a variety of commercially available stationary phasechemistries based on the modern particle technologies. Parameters such as resolution,retention and selectivity are compared, contrasted and related to known retentionmechanisms exhibited by the phase chemistries. General trends and guidance will beprovided that promises to greatly facilitate the method development in this complexenvironment.
1. Guideline on Quality of Herbal Medicinal Products/Traditional Herbal Medicinal Products http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003370.pdf
2. E.R. Badman et al., J. Chromatogr. B 878 (2010) 2307–2313.3. A. Abrahim et al., Journal of Pharmaceutical and Biomedical Analysis 51 (2010) 131–137.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-229-MO
Isolation, Characterization and Qualitative Analysisof 5-Deoxyflavones in Mimosa Displotricha
Lie-Chwen Lin, Chun-Tang ChiouNational Research Institute of Chinese Medicine,
Taipei, Taiwan
The herb of Mimosa displotricha has been used as anti-cancer agent in Formosan folkmedicine. In our bioactive screening, the EtOH of M. displotricha also showed thecytotoxic activity against a small panel of human cancer lines. Bioassay-guided isolation ofMimosa displotricha led to the isolation of five new 5-deoxyflavones named displotrin A-E(1–5), together with eleven known flavonoids, flavonolignans, and triterpenoids. On thebasis of spectroscopic evidence, compounds 1–5 were characterized as 2’,5’-dihydroxy-3,7,8,4’-tetramethoxyflavone (1), 3’-hydroxy-3,7,8,4’-tetramethoxyflavone (2), 7,3’,4’-trihydroxy-3,8-dimethoxyflavone (3), 2’-hydroxy-7,4’,5’-trimethoxyflavone (4), and4-hydroxy-3,10,11-trimethoxy-[1] benzopyrano- [3,2-c] [2]-benzopyran-7(5H)-one (5).Compounds 1, 2, 9 (5’’-methoxyhydnocarpin-D), 10 (betulinic acid), and 11(luteolin)showed anti-proliferation activity against human lung cancer cell A549 with GI50 valuesrange from 2.70 to 41.70 μM; against human gastric adenocarcinoma AGS cell with GI50
values range from 1.68 to 24.78 μM; against human colorectal adenocarcinoma HT-29cell with GI50 values range from 4.10 to 28.60 μM; and against human prostate carcinomaPC3 cell with GI50 values range from 2.30 to 21.09 μM, respectively. Eight of theseisolated compounds in ethanolic extract of M. displotricha were unambiguouslycharacterized by HPLC coupled with photodiode array (DAD). The HPLC separation wasachieved on a Purosher STAR RP-18 (5 μm, 250 x 4.6 mm) column using a gradientmobile phase of actonitrile in H2O at a flow rate of 1 ml/min. The develop method wasfound to be useful for determination of 5-deoxyflavones in mimosa species.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-230-TU
Isolation, Identification and Quantification of Cytokinin Nucleotidesby High Performance Liquid Chromatography
and Capillary Electrophoresis
Tibor Béres1, Petr Tarkowski2, Miroslav Strnad1,2, Karel Doležal1,2
1Laboratory of Growth Regulators, Institute of Experimental Botany ASCR,Šlechtitelu 11, 783 71 Olomouc, Czech Republic
2Centre of the Region Haná for Biotechnological and Agricultural Research, Faculty of Science,Palacký University,
Šlechtitelu 11, 783 71 Olomouc, Czech Republic
There is very little known about cytokinin nucleotides. Although the endogenousoccurrence of these compounds in plants has been predicted a decade ago, up to date,their functions are still not fully understood. Interestingly, when applied exogenously,cytokinin nucleotides play a role in animal cell death and are of medicinal interest. Thelack of analytical methods for intact cytokinin nucleotides determination together withlack of standards are the main obstacles in understanding of their functions both in plantand animal organism. To study the biological importance of these compounds, newmethods are essential. The developed HPLC-MS and CE-UV/MS methods for identificationand quantification of intact cytokinin nucleotides were applied in analysis of thesecompounds in leukemia cells and in vitro enzymatic reaction mixtures.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-231-MO
LC/MS Separation of Natural Antioxidants in Herbsand Honey Extracts using PFP Column
Petra Dinisová, Petr Cesla, Lenka Ceslová, Jan FischerUniversity of Pardubice, Faculty of Chemical Technology, Department of Analytical Chemistry,
Studentská 573, CZ-53210 Pardubice, Czech Republic
A liquid chromatography mass spectrometry method has been developed for separationof polyphenolic compounds including phenolic acids and flavonoids in natural matrices.The composition of mobile phase, type of the column and gradient profile wereoptimized. Five different columns with C18, phenyl-hexyl and pentafluorophenyl (PFP)chemically bonded stationary phases were compared using mobile phases with methanoland acetonitrile mixtures with addition of 0,3% (v/v) formic acid. The best separation ofselected polyphenolic compounds was achieved using column with PFP chemicallybonded stationary phase. The segmented linear gradient profile, i.e. initial concentrationof acetonitrile and gradient steepness in each segment, was optimized by numericaliteration procedure using regression parameters of experimental dependencies ofretention factors of antioxidants under isocratic conditions on concentration ofacetonitrile in mobile phase. The water extracts of flower of wild chamomile, lemon balm,sage and water extract of honey were analyzed under optimal conditions.
Authors acknowledge the financial support by the Ministry of Education, Youth and Sports of Czech Republic,project No. MSM0021627502 and by the Czech Science Foundation, project No. 203/07/0641.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-232-TU
LCxLC Separation of Polyphenols from the Stems of Rubus idaeus
M. Majdan-Skóra, D. Głód, M. Krauze-BaranowskaDepartment of Pharmacognosy, Medical University of Gdansk,
Al. Gen. J. Hallera 107, 80-416 Gdansk, Poland
Rubus idaeus L (Rosaceae) (red raspberry) is a source of fruits-rich in anthocyanins andstems, which are used in traditional medicine in a treatment of cold and flulike infections.The stems are a popular remedy mainly in western regions of Poland and Lithuania. Thechemical composition of this traditional herbal remedy remains still unknown. The LCxLC“comprehensive” technique was employed to separation and identification of polyphenolspresent in the stems of 11 cultivars of red raspberry. The conditions of separation wereoptimized in both directions using a mixture of 23 standard compounds belonging to agroup of ellagitannins (1), flavonols (11), phenolic acids (7) and flavan-3-ols (5). Finally,in the first direction the resolution was performed on a Discovery HS C18 column withgradient elution in a mixture of 0,1% TFA in water and acetonitrile. In the seconddirection was employed a Chromolith Performance RP-18 column and gradient elutionwith the same mixture of solvent, but the gradient program was different (tG – 64 min,modulation time – 2 min, sampling time – 25 Hz, time constant 0,08 sec). Ellagic acidwas shown to be a major compound in all analysed cultivars. Other phenolic compounds,cinnamic and benzoic acid derivatives, procyanidins and flavonols, flavan-3-ols, weredetected in minor concentrations as free aglycones of glycosides. Among flavonols,hyperoside and isoquercetin were dominant compound. The stems of Rubus idaerus seemsto be a rich source of polyphenols possesing antioxidant activity, what can explain itstraditional use as acurative in a cold.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-233-MO
Molecularly Imprinted Polymers of Fructosazine for Extractionfrom Selected Plants
Nathaly Henry1,2, Raphaël Delepee1, Sylvie Coquerelle2,Jean-Marc Seigneuret2, Luigi A. Agrofoglio1
1Institut de Chimie Organique et Analytique (ICOA), UMR CNRS 6005, Université d’Orléans,Orléans, France
2Alban Muller, Laboratoires PRAT,Montreuil, France
Since 1990, natural products have gained of interest for both cosmetic and pharmaceuticmanufacturers. For instance, two D-glucosamine derivatives, the 2,5-deoxyfructosazineand the fructosazine exhibit biological activity against diabetic’s diseases [1, 2]. They arealso very useful as flavoring additives [3] or light stabilizers [4] for foods. Instead ofsynthesizing these compounds, we have developed a selective method based on themolecularly imprinted polymer solid phase extraction (MISPE) to extract thesecompounds naturally from plants extracts.
MIPSE consists in creating a complementary structural picture of a target molecule (ortemplate), inside a polymer. In our case, the polymer contains specific cavities of 2,5-deoxyfructosazine and fructosazine and can rebind theses molecules from plants extracts.An original polymerization method was developed as the template is obtained, in situ,under the polymerization conditions. Indeed, the monomer is acting as a catalyst of theformation of the template in basic aqueous solution. Thus, it’s not necessary to start with2,5-deoxyfructosazine or fructosazine to create a specific molecularly imprinted polymer.
Moreover, a “green chemistry” approach has been used in order to reduceenvironmental impact. Consequently, water as polymerization solvent and ethyl alcoholas rebinding solvent were preferred to more toxic acetonitrile or chloroform. Severalparameters such as the choice of reactants’ amount, duration and type of polymerizationand conditions of rebinding were optimized.
These optimized conditions allowed us to extract fructosazine and 2,5-deoxyfructosazine from chicory leaves with an important selectivity. These compoundsare present in this plant in significant amounts (0.2% and 1%, respectively). All polymeroptimization and compounds origin will be discussed.
1. H. Bouchard, A. Commercon, J.F. Peyronel, T. Corinne, Médicaments contenant en tant que principe actifau moins un dérivé substitué de polyhydroxyalkylpyrazines, les nouveaux dérivéspolyhydroxyalkylpyrazines et leur préparation, French Patent Nº 97 09186, 18.07.97
2. M. Evers, Association de déoxyfructosazine et d’un antidiabétique agoniste du récepteur gamma-active parle proliférateur de peroxisome, French Patent Nº 99 16359, 23.12.99
3. P. Li, Y. Zong, B. Lu, Z. Qu, C-Q. Yang, Y-B. Song, S-H. Sun, Application of deoxyfructosazine in cigaretteflavoring, Yancao Keji (2008) 11: 30–35.
4. R. Van Der Ark, P. Blokker, L. Bolshaw, E. Brouwer, P. Hughes, H. Kessels, F. Olierook, M. Van Veen, Pyrazinesas light stabilizers for beer and other foods and beverages, PCT Int. Appl. WO 2005030920 07.04.2005

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-234-TU
Monitoring of Opium Alkaloids from Papaver somniferum L. Collectedin the Czech Republic Using LC-ESI-MS/MS Method
Alena Hejtmánková1, Irena Stránská2, Katerina Hejtmánková1,Vladimír Pivec1, Kristina Jíru1, Jan Novák2
1Department of Chemistry,2Department of Botany and Plant Physiology, Faculty of Agrobiology, Food and Natural Resources,
Czech University of Life Sciences Prague, Czech Republic
Analytical method including reliable solid phase extraction and liquid chromatography –electrospray ionisation – mass spectrometry (LC-ESI-MS/MS) has been developed fordetermination of 5 pharmaceutically important alkaloids (morphine, codeine, thebaine,papaverine and noscapine) in opium poppy.
The Czech Republic is the most important European grower and exporter of opiumpoppy. Poppy, grown as an oil plant, foodstuff, pharmaceutical and ornamental plant isan important cultural species. Poppy seed is suitable for human consumption, inparticular for the excellent dietetic properties. Poppy capsule (a mixture of crushed drycapsules with up to 15 cm long stems under capsule) is a coveted raw material for thepharmaceutical industry, used for the isolation of the alkaloids. For the pharmaceuticalindustry are interesting variety with higher content of morphine in the poppy capsules(ca. > 0.8%), for food use are appropriate to the contrary low-morphine variety. Theaim of this study is to determine the contents of the 5 main alkaloids in poppy capsulesof 39 selected agro-technically interesting varieties having various colour seeds for thepurposes of the renewal of poppy breeding in the Czech Republic with a view to itsfurther usage.
A powdered ripe capsule without seeds was extracted with 5% acetic acid undersonication. The obtained suspension was centrifuged and the supernatant was purified bysolid-phase extraction cartridges (Oasis MCX). The eluate was evaporated to drynessusing rotary vacuum evaporator. The residue was dissolved in 50% aqueous methanol,filtered through a membrane PVDF filter (0.45 μm) and analyzed by HPLC-MS/MS.
Reversed phase chromatography (HyPURITY AQUASTAR column) using gradient elutionwith A: (0.1% formic acid in methanol, v/v) and B: (0.1% formic acid in deionized water,v/v) as a mobile phase was employed. The detection was carried out using 3200 QTRAPdetector with electrospray ionisation in positive ion mode.
The significant differences of the total quantity and content of individual alkaloidsamong certain poppy varieties was demonstrated. The average content of morphine in allvarieties 6006 μg/g (range 2793–17175 μg/g), codeine 650 mg/g (range 70–1994 μg/g),noscapine 728 μg/g (range < 1,3–4269 μg/g), papaverine 158 mg/g (range < 1,1–1050mg/g), and thebaine 223 μg/g (range < 52,5–1701 μg/g) was determined. Higher

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
morphine content of above 0.8% was found only in 3 varieties The total maximumcontent of alkaloids 17.3 ± 0.7 mg/g as well as morphine 16,4 ± 0.7 mg/g wasdetermined in the Buddha variety, the lowest total content of alkaloids 3.9 ± 0.5 mg/gwas found in the Lori variety.
This study was supported by the Ministry of Education, Youth and Sports of the Czech Republic, (ResearchProject No. MSM 6046070901).

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-235-MO
Monitoring of Vitamine E Stability in Milk and Creamusing RSLC-ESI-MS Method
Katerina Hejtmánková1, Alena Hejtmánková1, Vladimír Pivec1,Tereza Michlová1, Ondrej Elich2
1Department of Chemistry, Faculty of Agrobiology, Food and Natural Resources,Czech University of Life Sciences Prague, Czech Republic
2Dairy Research Institute,MILCOM a.s., Prague, Czech Republic
Analytical method including saponification and rapid separation liquid chromatography –electrospray ionisation – mass spectromethry (RSLC-ESI-MS) has been developed fordetermination of tocopherols and tocotrienols in milk and cream.
Saponification was carried out with methanolic potassium hydroxide with parallelprotection of vitamine E by adding antioxidant pyrocatechol. After saponification vitamin Ewas extracted to hexane. Aliquot of hexane extract was evaporated under gently stream ofnitrogen and redisolved in methanol. Reversed phase chromatography with 6 mM NH3 in97% methanol and 3% deionized water as a mobil phase was employed. 6 mM NH3 has tobe used for support of deprotonation of analytes in electrospray negative ion mode. Goodseparation of α-, β-/γ- and δ-tocopherols and tocotrienols was achived in 5 minutes. Itshould to be noted that positional isomers β- and γ-tocopherol and β- and γ-tocotrienolcannot be resolved by reversed phase liquid chromatography.
The aim of our investigation is to evaluate internal reference standard of milksamples. α-tocopherol was chosen as a marker of stability. In the year 2010 was studiedstability of α-tocopherol in differently modified milk (original, skimmed milk, milk withadded lactose, protein, water and cream) and cream (20, 30, 40% fat content) afterchilling in the period of 3 month. All of samples were chilled in 3 different ways: byoperating undercooled ethanol, liquid carbon dioxide and liquid nitrogen.Consequently samples were stored in refrigerator at −20 °C.
The assumed positive correlation between content of α-tocopherol and fat content wasconfirmed. Coefficient of determination was 0.96. The lowest content of α-tocopherol wasdetermined in skimmed milk, on average 38 μg/100 g and the highest content wasdetermined in the fattest cream, on average 1043 μg/100 g. The way of freezing and thetime of storing were the main parameters studied. The mostly vitamine – friendly freezingmethod seems to be the application of liquid nitrogen vs. the undercooled ethanol andthe liquid carbon dioxide (α-tocopherol content on average 340 μg/100 g vs. 294 and297 μg/100 g). During the time of storage the decrease of vitamine E has not been noted.Furthermore no influence of added nutrient on vitamin E stability was found.
This study was supported by the Ministry of Education, Youth and Sports of the Czech Republic, Project No.MSMT 2B08072.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-237-MO
Novel HPLC-Based Approach for the Global Measurement of Lipids
Fraser McLeod, Marc Plante, Ian N. Acworth, Frank SteinerESA – A Dionex Company,
22 Alpha Rd., Chelmsford, MA 01824, USA
Lipids are a structurally diverse group of compounds that can be challenging to measure.Typically, the sample is first extracted using organic solvents, prior to derivatization toeither render the lipid more volatile for gas chromatography (GC) determination or tointroduce a chromophore for UV detection. Sometimes a combination of techniques areused to more fully characterize the sample, including GC with flame ionization detection,high performance liquid chromatography (HPLC) with evaporative light scatteringdetection (ELSD), and LC-mass spectrometry. Each form of detection has benefits andlimitations. Sample preparation for GC often requires the addition of carefully choseninternal standards, extraction, and derivatization for lipids. Errors in accuracy can result, aswell as analytes not being detected, due to non-reactivity. MS requires expensiveinstrumentation and maintenance costs can be high. The charged aerosol detector(CAD®) is a mass-sensitive, detector capable of directly measuring any non-volatile andmany semi-volatile analytes. Unlike ELSD it shows high sensitivity (low ng), wide dynamicrange (> 4 orders), high precision, and more consistent inter-analyte responseindependent of chemical structure, thus making it an ideal detector for simultaneouslymeasuring different lipid classes. Several HPLC methods are presented here that illustratethe determination of different lipid classes, including a universal, reversed-phase methodthat can resolve steroids, free fatty acids, free fatty alcohols, phytosterols, monoglycerides,diglycerides, triglycerides, phospholipids, and paraffins in a single run. For an example ofnormal-phase LC, a method for single-peak phospholipid quantification is shown. Practicalexamples are also presented, including total glycerides in biodiesel by normal-phase LC,phytosterols in natural oils, and fat soluble vitamins found in commercially availablesupplements.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-238-TU
Phenolic Antioxidant Tyrosol in Czech Wines by HPLC
Matyáš Orsák, Katerina Hejtmánková, Vladimír Pivec, Jaromír LachmanDepartment of Chemistry, Faculty of Agrobiology, Food and Natural Resources,
Czech University of Life Sciences Prague, Czech Republic
Tyrosol [4-(2-Hydroxyethyl)phenol] is a phenolic antioxidant which is formed duringfermentation by deamination of amino acid tyrosine. The principal source of tyrosol inhuman diet is olive oil; nevertheless tyrosol has been shown to possess cardio protectiveeffects mainly in white wines. Previously, higher amounts of tyrosol have been found inred wines. Tyrosol content in wine is influenced, as many other phenolic compounds too,by grape variety, weather, geographic area, yeast’s strain and used wine makingtechnology. Alcohol eases the extraction of tyrosol from the grape into the wine. 30Czech wine samples were analyzed in total. Samples comprised of white and red wine ofvarious producers, years and grape varieties.
A modified method described in Cabrita et al., 2007 was used. Briefly, wine sample wasextracted by diethyl ether, residual water removed by a drying agent, sample wassubsequently evaporated, reconstituted in methanol (10% v/v) and finally analyzed byHPLC-DAD at 280 nm. A Superpher® 100, C18 (5 μm, 250 mm x 4,6 mm) column hasbeen used for separation. Mobile phase A consisted of water acidified by formic acid (pH3) and B of methanol. Gradient elution was used. Tyrosol has been confirmed byretention time and UV spectra.
Tyrosol in white wines ranged from 1.6 to 8.2 mg/L and from 6.1 to 12.7 mg/L in redwines respectively. It has been found that the tyrosol content is dependent on grapevariety, years and producers.
This study was supported by the Ministry of Education, Youth and Sports of the Czech Republic, (ResearchProject No. MSM 6046070901) and by The National Agency for Agriculture Research of the Czech Republic,(Research Project No. NAZV QI111B107).

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-239-MO
Polyphenol Composition and Antioxidant Potentialof Certain Salvia Species
Nilgun Ozturk1, Hasibe Ozcan2, Muzaffer Tuncel3,Ismuhan Potoglu-Erkara2, Onur Koyuncu2
1Department of Pharmacognosy, Faculty of Pharmacy, Anadolu University,26470 Eskisehir, Turkey
2Department of Biology, Faculty of Art and Sciences, Eskisehir Osmangazi University,26480 Turkey
3Department of Analytical Chemistry, Faculty of Pharmacy, Anadolu University,26470 Eskisehir, Turkey
The determination of phenolic acids as well as investigation of antioxidant activity ofmethanol and ethyl acetate extracts from the herbs of Salvia candidisima Vahl. subsp.occidentalis Hedge, S. cryptantha Montbret & Aucher and S. forskahlei L. (Lamiaceae)which are growing in Turkey is described. Total phenolic contents of the extracts wereinvestigated by the Folin–Ciocalteau test. Antioxidant activity of the plants were alsoexamined by 2,2-diphenyl-1-picrylhydrazyl free radical scavenging and β-carotene-linoleicacid system method and the results were compared to those of BHT as syntheticantioxidant. Total phenolic contents of the extracts ranged from 39.28 ± 1.18 to 164.91± 1.53 mg GAE/g extract. The turn of the antioxidant capacities of the extracts fromSalvia species was found to be BHT > S. forskahlei > S. cryptantha > S. candidisima subsp.occidentalis. The methanol extracts of the studied species possessed the highest antiradicaland antioxidant activities and the highest total phenol content. For the determination ofphenolic acids from methanol and ethyl acetate extracts, HPLC analysis was performed byutilizing two solvents system [A: methanol:water:formic acid (10:88:2; v:v:v); B:methanol:water:formic acid (90:8:2; v:v:v)] on a C18 column. The flow-rate and injectionvolume were 1 ml min−1 and 10 μL, respectively. Signals were detected at 280 nm.Besides, internal standard technique (propylparaben) was applied for the analysis ofphenolic acids to increase the precision. Rosmarinic acid was the dominant phenolic acidfound in extracts and ranged from 1.90 to 136.13 mg/g of dry weight. The otherphenolic acids detected from the extracts were tr-cinnamic, ferulic, vanillic andp-hydroxybenzoic acids.
The results indicate us Salvia species are promising plants because of their potentialantioxidant activity.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-240-TU
Post-Column pH Buffered Electric Conductivity Detectionof Organic Acids with Ultra High Performance
Liquid Chromatography
Kyoko Watanabe, Makoto Ogaito, Atsushi Ieuji, Hirohisa MikamiAnalytical Applications Department, Shimadzu Co.,
1, Nishinokyo-Kuwabaracho, Nakagyo-ku, Kyoto, 604-8511, Japan
For the analysis of organic acids by HPLC, UV detection at around 210 nm has beenconventionally used as a convenient and simple technique. The UV detection method,however, has poor sensitivity and/or selectivity when analyzing organic acids in food,environmental or biological samples which include many kinds of UV absorbing co-existing compounds.
Conductivity detection is one of the most popular techniques to detect the ioniccompounds with high selectivity, because sensitivity for unionized compounds is prettylow. On the other hand, this feature means that it is difficult to gain good sensitivity foracidic compounds in acidic mobile phase. For highly sensitive determination of organicacids by the conductivity detector, “Post-column pH-buffered electric conductivitydetection” was developed by Shimadzu. After separating organic acids by ion-exclusionchromatography, pH-buffered solution is added to the column eluate constantly in orderto ionize the ionic compounds. This technique makes the sensitivity of organic acids byconductivity detector pretty better.
In this study, we applied the post-column pH buffered electric conductivity detectionto reversed phase ultra high-performance liquid chromatography (UHPLC) for the highspeed analysis of organic acids. Using Nexera system by Shimadzu, analytical conditionsfor separation (mobile phase and columns) and detection (Bis-Tris and EDTAconcentration in pH buffering solution) were optimized. Under optimized conditions,organic acids were separated within a few minutes. Good linearity and repeatability wereobtained and LOD of formic acid was 16.3 pmol.
This method was applied to the analysis of organic acids in some brewed foods. Theresults suggested that this method would be useful for the rapid determination of organicacids in samples which contain low amount of organic acids and/or large number of co-existing compounds.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-241-MO
Rapid Analytical Approach for Quantification the Total L-AscorbicAcid Content in Fruits and Vegetables by Ultra-Performance
Liquid Chromatography
Vitor Spinola, Berta Mendes, Jorge Pereira, José S. Câmara, Paula CastilhoCentro de Química da Madeira, Centro de Ciências Exactas e da Engenharia da Universidade da Madeira,
Campus Universitário da Penteada, 9000-390 Funchal, Portugal
Vitamin C, including ascorbic acid (AA) and dehydroascorbic acid (DHA), is one of themost important vitamins for human nutrition, supplied by fruits and vegetables. It isinvolved in several biochemical mechanisms, such as, collagen synthesis, immuneresponse, iron absorption and cell protection against oxidative stress. There are manyanalytical methods to measure ascorbic acid content in food, the liquid chromatographymethods being the most used and reliable [1]. The objective of the present work was todevelop and validate a method in order to determine the total content of Vitamin C (AA+ DHA) in passion fruits, strawberries, watercress and green peppers using UPLC-PDAsystem. DHA was determined indirectly after its reduction with dithiothreitol (DTT) beforeanalysis. The chromatographic system was equipped with a Acquity HSS T3 analyticalcolumn (100 mm × 2.1 mm, 1.8 μm particle size) using a isocratic mobile phasecomposed of aqueous 0.1% formic acid at a flow rate of 250 μL/min and the injectionvolume was 2 μL. The detection wavelength for the photo-diode detector was set at 245nm and the analytical column was kept at room temperature. The performance of themethod was evaluated in terms of sensitivity, limits of detection (LOD), limit ofquantification (LOQ), accuracy and precision. The method validation gave satisfactoryresults: good linearity with r2-values > 0.999, the sensitivity based on LOD values rangedbetween 0,9 and 1,3 ng/mL; LOQ values varied between 3 and 15 ng/mL; the accuracyof the method (evaluated at low, medium and high concentration levels) ranged between95 and 99.8%, and the intermediate precision between 0,2 and 0,8%. Samples wereextracted with a 3% metaphosporic-8% acetic acid-1 mM EDTA solution after 24, 48, 72,96 and 144 hours. The extracts were prepared and stored at −80 °C immediately afterextraction until analysis (within 1 week). Moreover, the stability of Vitamin C onrefrigerated extracts (4, −20 and −80 °C) has also been studied. This UPLC-PDAmethodology proves to be an improved, simple and ultra-fast approach for determinationthe total content of vitamin C in various food commodities, with high sensitivity,selectivity and resolving power within only 3 min of run analysis.
1. Novakova, L., Solich, P., & Solichova, D. (2008). HPLC methods for simultaneous determination ofascorbic and dehydroascorbic acids. Trac-Trends in Analytical Chemistry, 27(10), 942–958.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-242-TU
Rapid Approach by Ultra Performance Liquid ChromatographyCombined with Microextraction by Packed Sorbent
for Determination of Trans-Resveratrol in Wines
João Gonçalves, Jorge Pereira, José S. CâmaraCentro de Química da Madeira, Centro de Ciências Exactas e da Engenharia da Universidade da Madeira,
Campus Universitário da Penteada, 9000-390 Funchal, Portugal
The current research study is dedicated towards the development and validation of anovel sensitive, fast and accurate approach for determination of trans-resveratrol inwines based on microextraction by packed sorbent (MEPS), combined with ultra-performance liquid chromatography (UPLC) equipped with a photodiode array (PDA)detection system. Important factors affecting the performance of MEPS such as the typeof sorbent material (C2, C8, C18, SIL and M1), number of extraction cycles, and samplevolume, were studied. The method was fully validated in terms of linearity, detection(LOD) and quantification (LOQ) limits, extraction yield, accuracy and inter/intra-dayprecision, using a Madeira wine sample (ET) spiked with trans-resveratrol atconcentration levels ranging from 10 to 60 μg mL−1. The method gave satisfactoryresults in terms of linearity (r2 = 0.998) within the studied concentration range.Detection and quantification limits ranged from 1.28 μg mL−1 and 4.28 μg mL−1,respectively. The extraction yields of the MEPSC8/UPLC-PDA methodology was higherthan 84.2%, and the inter-day precision (n = 3 days), expressed as the relative standarddeviation (RSD%), varied between 0.5% and 2.6%. On the basis of analytical validation,the MEPSC8/UPLC-PDA methodology proves to be sensitive, selective, and with goodprecision, and moreover, an ultra-fast approach allowing the determination of trans-resveratrol with high resolution power in within 7 min.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-243-MO
Rapid Determination of Anthocyanins in Pomegranate Juiceand Bilberries
Pranathi P. Perati, Brian M. De Borba, Jeffrey S. RohrerDionex Corporation
1214 Oakmead Parkway, Sunnyvale, CA 94085-4024, USA
Anthocyanins are a subclass of flavanoid polyphenols responsible for the brilliant red,orange, and blue colors of most fruits, vegetables and flowers. The chemical structures ofanthocyanins are naturally electron deficient and therefore are very reactive towards freeradicals, and are consequently powerful natural antioxidants. Major sources ofanthocyanins include bilberries, strawberries, black currants, purple grapes, andpomegranates. Pomegranate juice (PJ) and bilberries have been reported to containhigher antioxidant activity than most commercially available fruit juices and fruit extracts.Due to the increased health consciousness of consumers combined with the potentialhealth benefits of PJ, the demand for PJ and pomegranate-related products has grownrapidly in the past several years. However, the high demand and short supply ofpomegranates has caused increased adulteration of PJ. Consumption of bilberry basednutritional supplements has increased globally due to its reported health benefits.Analytical methods are therefore required to quantify anthocyanins in both sources. Thispaper describes two sensitive, fast, and accurate UHPLC methods to determine sixanthocyanins in pomegranate beverages and 15 anthocyanins and 5 anthocyanidins(anthocyanins aglycones) in bilberry-based nutritional supplements. The methodsdemonstrated good sensitivity, enabling the detection and quantification of even lowconcentrations of anthocyanins found in pomegranate-related juice products. Methodsensitivity was also important for detecting the wide range of anthocyanins in bilberrysupplements. The linearity, LODs, LOQs, and accuracy of each method for determiningthe target compounds in these products will be reported.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-244-TU
Separation and Quantification of Different Flavonoids Foundin Natural Therapeutics and Functional Foods by RP-HPLC
on a Stationary Phase Providing ππ-ππ Interactions
Robert Fredriksson, Joakim Högblom, Sylvia Winkel-PetterssonSeparation Products, Eka Chemicals AB,
SE 445 80 Bohus, Sweden
Today, the modern pharmaceutical industry frequently utilizes reversed phase HPLC forthe purification of peptides and synthetic molecules, as well as for analyzing new drugcandidates. The use of smaller particles, packed in columns < 100 mm, supports the useof higher flow rates, leading to short cycle times, which increases the throughput in thescreening process, as well as for routine analysis.
Many new drug candidates are found in nature. A group of water soluble aromaticcompounds, the flavonoids, shows promising anti-inflammatory and anti-cancerproperties. Most flavonoids are found in widely available plants such as commonvegetables or fruit, like citrus fruits and onions, or in beverages, like green tea or red wine.From these common products, functional food can be made, or the flavonoids can beextracted and purified further use in natural therapeutics.
To guarantee the quality, clinical effects, and safety of the natural therapeutics, it is ofhighest importance that relevant and efficient analytical tools are used, and this posterpresents both how the flavonoids can be extracted and purified from natural products,and how the flavonoids are quantified and analyzed on a Kromasil Eternity-2.5 μm-PhenylHexyl column. The Kromasil Eternity Phenylhexyl stationary phase is an excellentchose when it comes to the detection and quantification of aromatic compounds, basedon HPLC techniques.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-245-MO
Separation of Betalains from Red Beet (Beta vulgaris L.)by High-Performance Countercurrent Chromatography (HPCCC)
using High Salt-Solvent Systems
Aneta Spórna-Kucab1, Svetlana Ignatova2, Ian Garrard2,Sławomir Wybraniec1, Paweł Stalica1
1Faculty of Analytical Chemistry, Institute C-1, Department of Chemical Engineering and Technology,Cracow University of Technology,
ul. Warszawska 24, Cracow 31-155, Poland2Brunel Institute for Bioengineering, Brunel University,
Uxbridge, West London, UK
High Performance and High Speed Counter Current Chromatography (HP & HSCCC) havebeen shown to be important techniques for the separation of a wide range of importantnatural plant pigments, due to their versatility [1]. CCC is an all liquid chromatographictechnique which has many advantages over the conventional column chromatography. Theliquid-liquid nature of CCC eliminates the problem of irreversible adsorption of the sampleon supports such as silica gel. When using a high G-force HPCCC instrument, further advan-tages are that higher amounts of sample can be introduced into the HPCCC apparatus, anda shorter separation time is possible with lower consumption of solvents [2].
Red beet root (Beta vulgaris L.) is a source of betalains – natural pigments which havepotential health benefits (antioxidative and anticarcinogenic) and non-toxic features.Betanin, the main betacyanin of red beet, is sensitive to several degradation factors suchas elevated temperature, pH or the presence of metal cations. As a result of low stability,betanin undergoes degradation and transformation reactions leading to the formation ofcomplex mixtures of derivatives [3].
Therefore, HPCCC is a suitable technique for the separation of labile betalain mixtures. Inthis study, the pigments were separated using highly polar solvent systems with high saltconcentrations: 1-propanol-acetonitrile-saturated ammonium sulphate-water (v/v/v/v,1:0.5:1.2:1); ethanol-acetonitrile-1-propanol-saturated ammonium sulphate-water (v/v/v/v,0.5:0.5:0.5:1.2:1) and ethanol-1-butanol-acetonitrile-saturated ammonium sulphate-water(v/v/v/v, 0.5:0.5:0.5:1.2:1) using an HPCCC chromatograph (Dynamic Extractions Ltd., UK)with 18.4 ml capacity, coil length 37 m and i.d. 0.8 mm, running at a flow rate 0.15 ml/minin normal phase mode.
The applied solvent systems separated the pigments with high efficiencywith the morehydrophobic compounds eluting first as expected. Moreover, for the first time, the sepa-ration of 2-decarboxy-betanin from 17- and 2,17-decarboxy-betanin,as well as fromneobetanin and betanin, was achieved.
1. P. Winterhalter, Trends Food Sci. Tech. 18 (2007) 507–513.2. S. Wybraniec, P. Stalica, G. Jerz, B. Klose, N. Gebers, P. Winterhalter, A. Spórna, M. Szaleniec, Y. Mizrahi, J.
Chromatogr. A, 1216 (2009) 6890–6899.3. S. Wybraniec, J. Agric. Food Chem. 53 (2005) 3483–3487.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-246-TU
Separation of Phenolic Acids in Marine Plants by MolecularImprinting Anion-Exchangeable Polymer Confined Ionic Liquids
Wentao Bi, Minglei Tian, Kyung Ho RowDepartment of Chemical Engineering, Inha University,
253 Yonghyun-Dong, Nam-Ku, Incheon 402-751, Korea
Polymer confined ionic liquid have been investigated for it to become a kind of effectiveseparation media for isolating bioactive compounds from interference. Due to the specialproperties of ionic liquids, ionic liquid-based materials enabled separation of organic acidsby anion-exchange mechanism. However, the non-directional ion-ion interaction ofanion-exchange and other interactions of interference/ionic liquids cause decrease ofselectivity. In order to overcome the shortage, molecular imprinting technique wasinvolved to synthesize the polymer confined ionic liquids. This proposed method wasthoroughly evaluated by solid-phase extraction of three phenol acids (protocatechuicacid, ferulic acid and caffeic acid) from Saliconia herbaces L. extract. By comparing theadsorption behavior of these three phenolic acids on different polymer confined ionicliquids, the suitable sorbent was selected with the highest adsorption capacity of phenolicacids. After solid-phase extraction, the phenolic acids (94.7% for protocatechuic acid,80.1% for ferulic acid and 89.3% for caffeic acid) were successfully recovered fromextract.
Acknowledgements: This research was supported by the National Natural Science Foundation of China(21011140338) and the International Research & Development Program of the National Research Foundationof Korea (NRF) funded by the Ministry of Education, Science and Technology (MEST) of Korea (2010-D00016).

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-247-MO
Separation of Triacylglycerol Enantiomers and Regioisomersusing Chiral HPLC/MS
Miroslav Lísa, Michal HolcapekUniversity of Pardubice, Faculty of Chemical Technology, Department of Analytical Chemistry,
Studentská 573, 532 10 Pardubice, Czech Republic
Triacylglycerols are triesters of fatty acids and glycerol having many important functionsin human organism, i.e., source of energy, essential fatty acids and lipophilic vitamins,thermal and mechanical protective layer around some organs, etc. They can differ by thecomposition of esterified fatty acids, i.e., acyl chain lengths, number of double bonds andtheir position and configuration (cis-/trans-). Fatty acids can be esterified in differentpositions on the glycerol skeleton (sn-1, 2 or 3) providing regio (R1R2R2 vs. R2R1R2) orchiral (R1R2R2 vs. R2R2R1) isomers, which are not resolved using reversed-phase HPLCcommonly used for the analysis of triacylglycerol samples. The knowledge ofstereochemical position of fatty acids on the glycerol skeleton is important from thenutrition point of view due to the stereospecific environment in the organism. In thiswork, method for the analysis of both types of isomers in one run have been developedbased on the separation of triacylglycerols using chiral stationary phase in the normal-phase mode. In this system, the retention behavior of triacylglycerols is governed by thenumber of double bonds, i.e., retention times of triacylglycerols increase with increasingnumber of double bonds. Separation of triacylglycerol enantiomers and regioisomersusing optimized HPLC conditions is demonstrated on the analysis of mixtures oftriacylglycerol standards prepared from selected mono-acyl triacylglycerols (R1R1R1)containing fatty acids commonly presented in the natural samples (i.e., palmitic, stearic,oleic, linoleic, linolenic and arachidic acids) by the randomization reaction providingmixtures of all isomers. Triacylglycerol regioisomers are identified based on different ratiosof fragment ions formed by losses of fatty acids from the glycerol skeleton in positive-ionatmospheric pressure chemical ionization mass spectra, while optically pure triacylglycerolstandards are prepared for the identification of retention order of enantiomers.
This work was supported by the project MSM0021627502 sponsored by the Ministry of Education, Youth andSports of the Czech Republic and by the project 203/09/P249 sponsored by the Czech Science Foundation.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-248-TU
Simultaneous Analysis Method for Polar and Non-Polar Ginsenosidesin White and Red Ginseng by Reversed-Phase HPLC-PAD
Sa-Im Lee1, Ha-Jeong Kwon2, Seon-Pyo Hong1
1Department of Oriental Pharmaceutical Sciences, Kyung Hee East-West Pharmaceutical Research Institute,College of Pharmacy, Kyung Hee University,
Hoegi-dong, Dongdaemoon-gu, Seoul 130-701, South Korea2Department of Preventive and Social Dentistry, College of Dentistry, Kyung Hee University,
Hoegi-dong, Dongdaemoon-gu, Seoul 130-701, South Korea
The paper describes a simultaneous determination method for polar and non-polarginsenosides in red ginseng with a reversed-phase high-performance liquidchromatography-pulsed amperometric detection. Furthermore, we could establishdetermination of polar and non-polar ginsenodsides in white ginseng in accordance withdifferent parts, regions, and ages. This method could be applied directly without anypretreatment steps and enabled the performance of highly sensitive analysis within 1 h.The detection (S/N = 3) and quantification (S/N = 10) limits for the ginsenosides ranged0.02–0.10 ng and 0.1–0.3 ng, respectively. The linear regression coefficients ranged0.9975–0.9998. Intra- and inter-day precisions were < 9.91%. The mean recoveriesranged 98.08–103.06%. The total amount of ginsenosides in the hairy root of redginseng was higher than that in the main root. Besides, more kinds of non-polarginsenosides are founded in the hairy root of white ginseng than that of main root.
This study was supported by a Korea Research Foundation (KRF) grant funded by the Korea government(MEST) (No. 2009-0073818).

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-249-MO
Simultaneous Determination of Eight Bioactive Compoundsin Polygonum Multiflorum using Pressurized Hot Water Extraction
and High Performance Liquid Chromatography
Dong-qi Han, Jing Zhao, Shao-ping LiState Key Laboratory for Quality Research in Chinese Medicine, and Institute of Chinese Medical Sciences,
University of Macau,Macao SAR, P. R. China
Polygonum multiflorum, He-Shou-Wu in Chinese, is one of commonly used traditionalChinese herbal medicines, which has considered being beneficial to health, such aspreventing premature greying of hair, in China and other Asian countries for manycenturies. In this study, a rapid and reliable pressurized hot water extraction (PHWE) andHPLC method was developed for simultaneous determination eitht compounds, includinggallic acid, catechin, proanthocyanidin B1, epicatechin, proanthocyanidin B2, emodin-8-O-β-D-glucopyranoside, hypaphorine and 2,3,5,4’-tetrahydroxystilbene-2-O-β-D-glucopyranoside in P. multiflorum. Besides, their contents in samples of P. multiflorum fromdifferent locations of China and the different parts of plant were also compared usinghierarchical clustering analysis, principle component analysis.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-250-TU
Simultaneous Determination of Eleven Intact Glucosinolatesin Fresh and Controlled Atmospheres Stored Broccoli Plants
by HPLC-MS/MS
M. F. Fernández-León1, M. Lozano1, M.C. Ayuso2, D. González-Gómez1
1Instituto Agroalimentario de Extremadura (INTAEX)Ctra. San Vicente s/n, 06071 Badajoz, Spain
2Agricultural Engineering School, University of ExtremaduraCtra. de Cáceres s/n, 06007, Badajoz, Spain
A number of epidemiological studies indicate that a regular consumption of brassicafamily vegetables helps decrease the risk of development of different type of cancer. Inthis family, broccoli (Brassica oleracea L. var. italica) is distinguished for the high numberof health-promoting compounds such as glucosinolates, phenolics, carotenoids,chlorophylls and different vitamins making broccoli an interesting commodity forproducers and consumers.
Intact glucosinolates were extracted from broccoli plants and the extracts were analyzedin a HPLC-MS/MS instrumentation. HPLC separation was performed at 40 °C on a 150 x 2.0mm (3 μm) C18 reversed-phase column and compounds were indentified and quantified bymeans of an ion trap mass spectrometer. The following transitions were used to assay 11individual glucosinolates: glucoraphanin (436 > 97), glucoiberin (422 > 97), progoitrin (388> 97), sinigrin (358 > 97), glucobrassicin (447 > 97), glucoalysin (450 > 97), gluconasturtiin(422 > 97), neoglucobrassicin (477 > 97), 4-methoxyglucobrassicin (477 > 97), gluconapin(372 > 97) and glucobrassicanapin (386 > 97).
This methodology was applied to study how postharvest conditions influenced theabundance of intact glucosinolates in broccoli plants. The analyses were performed afterharvest and after the storage under refrigeration at atmospheric conditions and withcontrolled atmospheres conditions (gas composition 10% O2 and 5% CO2).
Authors thanks Junta de Extremadura of Spain (D.G. de Ciencia y Tecnología) and FEDER’s funds for thefinancial support (Project GR10006).
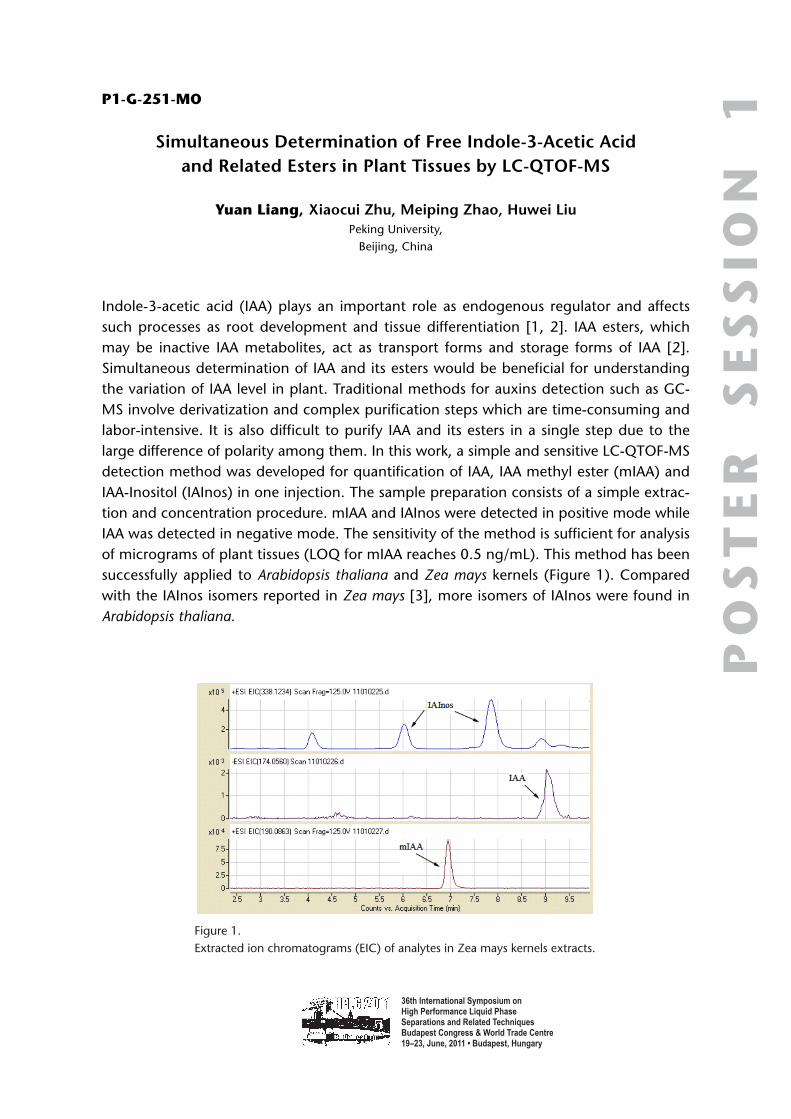
36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-251-MO
Simultaneous Determination of Free Indole-3-Acetic Acidand Related Esters in Plant Tissues by LC-QTOF-MS
Yuan Liang, Xiaocui Zhu, Meiping Zhao, Huwei LiuPeking University,
Beijing, China
Indole-3-acetic acid (IAA) plays an important role as endogenous regulator and affectssuch processes as root development and tissue differentiation [1, 2]. IAA esters, whichmay be inactive IAA metabolites, act as transport forms and storage forms of IAA [2].Simultaneous determination of IAA and its esters would be beneficial for understandingthe variation of IAA level in plant. Traditional methods for auxins detection such as GC-MS involve derivatization and complex purification steps which are time-consuming andlabor-intensive. It is also difficult to purify IAA and its esters in a single step due to thelarge difference of polarity among them. In this work, a simple and sensitive LC-QTOF-MSdetection method was developed for quantification of IAA, IAA methyl ester (mIAA) andIAA-Inositol (IAInos) in one injection. The sample preparation consists of a simple extrac-tion and concentration procedure. mIAA and IAInos were detected in positive mode whileIAA was detected in negative mode. The sensitivity of the method is sufficient for analysisof micrograms of plant tissues (LOQ for mIAA reaches 0.5 ng/mL). This method has beensuccessfully applied to Arabidopsis thaliana and Zea mays kernels (Figure 1). Comparedwith the IAInos isomers reported in Zea mays [3], more isomers of IAInos were found inArabidopsis thaliana.
Figure 1.
Extracted ion chromatograms (EIC) of analytes in Zea mays kernels extracts.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
1. Sandberg G, Crozier A, Ernstsen A, in: Rivier L, Crozier A (Eds). Principles and practice of plant hormoneanalysis, Volumne 2, London: Academic Press, 1987: 169.
2. Ribnicky D M, Cooke T J, Cohen J D. Planta, 1998, 204: 1–7.3. Nicholls P B. Planta, 1967, 72: 258–264.
Acknowledgement: National Natural Science Foundation of China (Grant No. 90717002).

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-252-TU
Simultaneous Identification of Dyes and Bindingin Graphic Documents by Capillary Electrophoresis
Ana Mª López-Montes1, Cristina Prats Henares2, Mª Esther Castillo Valdivia2,Teresa Espejo Arias1, José Luis Vílchez Quero2, Mª Rosario Blanc García2
1Department of Painting, Faculty of Fine Arts, University of Granada,Avda. Andalucía s/n, 18071 Granada, Spain
2Department of Analytical Chemistry, Faculty of Sciences, University of Granada,Avda. Fuentenueva s/n, 18071 Granada, Spain
Chemical analysis of graphic documents is a common procedure for their care, conserva-tion and restoration, and the amount of analytical work done in this area has increasedrecently with the development of new analytical techniques.
Most graphic documents on parchment and paper had been written and illustratedusing water-based techniques such as watercolour or gouache where arabic gum wasused as binding and dyes or pigments were the elements that provided colour to the inks.
Analytical methods for the identification of these components must be highly selectiveand sensitive because of the complexity of the matrix and the limited amount and size ofsample available in the case of graphic documents and archival material. For this reason,in this work capillary electrophoresis with diode array UV-Vis spectrophotometric detec-tion (CE-DAD) has been selected for developing a simple and rapid method for the identi-fication of dyes and gums. The dyes used like reference substances were obtained byextraction from different sources: red carmine from Coccus cacti insect, madder lake fromRubia tinctorum root and brazilwood from Caesalpinia echinata tree. The binding chosenas a reference has been arabic gum (from Acacia senegal tree). The method has beenapplied to the samples prepared at the laboratory composed by inks applied on paper.The sampling was carried out using a brush impregnated with adequate solvent rubbingdirectly onto the surface of paper.
The main advantages of the developed method are that only small amount of sampleis required, sample treatment is simple and analysis time is short. In fact gums and dyescan be identified by a single analysis of one single sample, which means that sampling isreduced considerably in order to identify the principal compounds. Also the proposedsampling technique does not damage the artefact.
The knowledge of the elements that constitute graphic documents facilitates dating, identi-fying and also help to understand the evolution of the techniques used in their production.Moreover, from the point of view of documental heritage conservation, analysis and scientificknowledge of materials and techniques used in the documents are of paramount importanceto determine the best conservation conditions and restoration treatments.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-253-MO
The Development and Validation of an LC-MS/MS Methodsfor the Determination of Tetrodotoxin (TTX) in Trumpet Shell,
Charonia lampas
Sharon Hutchinson1, Paul Barnes2, Joana Azevedo3,5, Vitor Vasconcelos3,4,Cowan Higgins6, Judith Nzoughet7, Chris Elliott7, Ambrose Furey1
1Department of Chemistry, Cork Institute of Technology,Rossa Avenue, Cork, Ireland
2Agri-Food and Biosciences Institute – Stormont,Stoney Road, Belfast, County Antrim, BT4 3SD, UK
3Centre of Marine and Environmental Research – CIMAR / CIIMAR – U.P.,Rua dos Bragas, 289, 4050-123 Porto, Portugal
4Department of Biology, Faculty of Sciences, University of Porto,Rua do Campo Alegre, 4169-007 Porto, Portugal
5School of Health Technology of Porto,Rua Valente Perfeito, 322, 4400-330 Vila Nova de Gaia, Portugal
6Marine Biotoxin Unit, Chemical Surveillance Branch, Agri-Food and Biosciences Institute,Stormont, Belfast BT4 3SD, UK
7Institute of Agri Food and Land Use, School of Biological Sciences, Queen’s University Belfast,Belfast, UK
Tetrodotoxin (TTX) is a marine toxin and it is reported to have caused numerous deathsin Japan following the consumption of fish and seafood containing TTX. It is a neurotoxinthat blocks voltage-gated sodium channels, resulting in respiratory paralysis and death.Therefore, simple and reliable analytical methods are required to monitor TTX. As part ofthe EU Atlantox project, this paper describes an extraction method combined with an LC-MS/MS for the quantitative detection of TTX in Charonia lampas (Trumpet shell) samples.The mass spectrometer used is a Thermo Quantum triple quadrupole (Discovery MAX)coupled to a Thermo Accela HPLC pump. The LC separation was performed on a RPAmide C16 column (150 mm x 4.6 mm i.d.) using a gradient elution with mobile phases10% Acetonitrile/90% 2 mM Ammonium Formate, 50 mM Formic acid in water and 90%Acetonitrile/10% 2 mM Ammonium Formate, 50 mM Formic acid in water, at a flow rateof 0.6 mL/min. The mass spectral acquisition was done in the positive ion mode by apply-ing selected reaction monitoring (SRM). Three MRM transitions were observed at m/z =320/302, 320/162 and 320/149 for TTX. The validated method has a limit of quantitation(LOQ) of 0.01 μg/ml and linearity correlation coefficient 0.999 for solvent based standardsolutions. Twenty four samples of Charonia lampas (Trumpet shell) were assessed for thepresence of TTX using the developed LC-MS method. Positive samples will be confirmedusing a hybrid linear ion-trap Orbitrap mass spectrometer with high mass resolution andhigh mass accuracy capabilities.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-254-TU
UHPLC Determination of Glycoside Sweeteners:Steviol Glycosides and Mogroside V
Deanna C. Hurum, Brian M. De Borba, Deepali Mohindra, Jeffrey S. RohrerDionex Corporation,
1214 Oakmead Parkway, Sunnyvale, CA 94085, USA
Terpene glycoside extracts of plants are gaining popularity as sweeteners and some; suchhave stevia extracts, have been successfully commercialized as off-the-shelf sugar substi-tutes. These compounds are non-caloric, yet are greater than 300 times sweeter thanequivalent sucrose solutions, making them attractive sugar replacements. Both purifiedforms of the glycosides from plant extracts, and whole fruit or leaf extracts are commer-cially available as purified sweeteners, herbal teas, or as nutritional supplements.
Many glycosides are present in stevia (Stevia reabaudiana (Bertoni)) extracts, withrebaudioside A of primary commercial interest. Another example terpene glycoside sweet-ener, Mogroside V is extracted from the fruit of the lo han guo plant (Siraitia grosvenorii).Due to the structural similarity of these terpene glycosides, chromatographic separationcan be challenging.
In this work, an isocratic method using a tri-mode column for terpene glycoside analy-sis is discussed. The high volatility of the mobile phase make the method ideal for anaerosolizing detection technique, such as charged aerosol detection (CAD), following UVdetection. This dual detection method was used to determine the relative proportion ofglycosides in two commercial stevia-based sweeteners and a lo han guo tea with calibra-tion ranges between 70–280 μg/mL. Retention time and peak area precisions (RSDs) were< 0.1 and < 2.0, respectively, with consistent retention times over several days of analysis.Advantages of using two detection techniques are illustrated using off-the-shelf sampleswith detection sensitivity by CAD improved 3–4 fold for glycosides that do not stronglyabsorb in the UV, such as rebaudioside B and mogroside V.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-255-MO
Ultrafast-High Performance Liquid Chromatographyas a Suitable Methodology for the Analysis of Carotenoids
in Honey-Bee Pollen
D. González-Gómez, D. Bohoyo-Gil, S. Nogales-Delgado,M. Izquierdo Rey, D. Domínguez-Valhondo
Instituto Tecnológico Agroalimentario,Carretera de Cáceres s/n. 06071, Badajoz, Spain
High Performance Liquid Chromatography (HPLC) has been widely used for the determi-nation of bioactive compounds in food products. In this work, in order to save up re-agents and time, a new technique called Ultra High Performance Liquid Chromatography(UHPLC) is optimized for the separation of carotenoid pigments. Honey-bee collectedpollen is a very valuable product due to its high content of functional compounds such asantioxidants or vitamins. Therefore, directly and as a supplement consume is quite de-manded in healthy diets.
The aim of this work was to compare both HPLC and UHPLC methodologies in terms ofresolution, selectivity, sensitivity and time and reagent saving. No significant differenceswere observed between both methodologies except in reagent and time consumption,where an important saving is achieved. A total of six carotenoids were separated andquantified in less than 8.5 minutes. For all components the resolution was higher than 1.5and LOD were below 1.0 μg mL−1 when using UHPLC. To determine the intraday andreproducibility assays were conducted for method validation.
The proposed methodology was applied for the determination of the carotene contentsto a batch of honey bee pollen samples in order to determine the differences among thepollen floral origin and the influence of the industrial processing.
Authors thanks Junta de Extremadura of Spain (D.G. de Ciencia y Tecnología) and FEDER’s funds for thefinancial support (Project GR10006 and PDT09B002)

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-256-TU
Advantage of a New Generation of Evaporative Light-ScatteringDetectors: Universality, Higher Sensitivities and Cost-Effectiveness
for Multi-Element Analyses in Liquid Chromatography– An Application Review
Eric VeretteSEDERE SAS,
841 blvd Duhamel du Monceau, 45160 Olivet, France
Among the detectors available in Liquid Chromatography, Evaporative Light-ScatteringDetector (ELSD) became in recent years a well established instrument thanks to severaltheoretical studies based on fundamental investigations and numerous applications pro-vided during the last thirty years. Indeed, ELSD is considered as a nearly Universal, power-ful and cost-effective technique, and is ideally appropriate for the majority of the liquidchromatography applications. Today, the power of this detection mode is further extend-ed with a new model which proposes a genuine and efficient Low-Temperature technolo-gy (LT-ELSDTM) combined with an innovative detection chamber, thus providing thehighest sensitivities with all compounds including semi-volatile and thermo-labile ones.
To show the strength and the versatility of this new ELSD model, several liquid chroma-tography applications are thoroughly developed in this work. These applications use themost recent liquid chromatography media, such as multi-mode, HILIC and sub-two-mi-cron particle phases, allowing outstanding separations and simultaneous analyses of awide range of compounds belonging to the major chemical and biochemical classes.
This work proposes several generic LC-ELSD methods and includes the analyses of thefollowing groups of compounds:
– Carbohydrates (mono-, di- and oligosaccharides) and polyols obtained with a gradi-ent elution.
– Polar and non-polar lipids, which represents a very convenient and efficient alterna-tive to GC analyses.
– Amino acids without any tedious derivation step before detection.– Inorganic ions without the use of any additional equipment such as ion suppressors.– Water-soluble and fat-soluble vitamins analyzed simultaneously.– Organic and phenolic acids.Validation data are provided and include low limits of detection (down to the sub-
nanogram levels) to emphasize the universality and the strength of this outstanding,highly sensitive and cost-effective detection mode.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-257-MO
Assessment of a New Generation of Evaporative Light-ScatteringDetectors for Liquid Chromatography: Sensitivity, Linearity,
Dynamic Range, Analyte Dispersionand Response Variation with Eluent Composition
Eric VeretteSEDERE SAS, 841 blvd Duhamel du Monceau, 45160 Olivet, France
Among the detectors available in Liquid Chromatography, Evaporative Light-ScatteringDetector (ELSD) became in recent years a well established instrument thanks to severaltheoretical studies based on fundamental investigations and numerous applications pro-vided during the last thirty years. Indeed, ELSD is considered as a nearly universal, powerfuland cost-effective technique, and is ideally appropriate for a great majority of liquid chroma-tography applications containing chromophoric and non-chromophoric compounds.
However, some limitations have been attributed to this type of detection in the past,such as low sensitivities, non-linearity, reduced dynamic range, large analyte dispersionand wide response variation with the mobile phase composition.
Today, a new ELSD model is proposed which offers a genuine and efficient Low-Tem-perature technology (LT-ELSDTM) combined with an innovative detection chamber. Theoverall design of this new detector results in a significant increase of sensitivity providinglimits of detection down to the sub-nanogram levels even for semi-volatile compounds.Moreover, it provides an improved direct linearity with correlation coefficients reaching0.999 and an extended overall dynamic range exceeding the four decades. Also, thismodel is optimized for the recent U-HPLC technique giving peak widths below 1 second.Finally, a study on the response variation with the mobile phase composition showed asmaller effect compared to other aerosol-based detectors.
In this work, several examples and data are provided on these topics to emphasize thegreat advancement in this technology, thus bringing to the analyst a relevant and cost-effective solution to their chromatography challenges.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-258-TU
Behavior of Glassy Carbon Paste Electrodein HPLC with Amperometric Detection
Hana Dejmkova, Jan Mika, Jiri Barek, Jiri ZimaCharles University in Prague, Faculty of Science, Department of Analytical Chemistry,
UNESCO Laboratory of Environmental Electrochemistry,Albertov 6, CZ-12843 Prague 2, Czech Republic
Application of carbon paste electrodes in HPLC with amperometric detection is generallydifficult, as the composition of the carbon paste is not resistant towards solutions withhigh content of organic solvents. However, carbon paste prepared of glassy carbon micro-particles is stable even in solutions containing 80% of methanol and was successfully usedin such media.
This contribution deals with the changes in the electrode surface after its contact withmethanol-containing solution. Dependence of the electrode behavior on the methanolcontent and on the time period of the contact with methanolic solution is investigated.Microscopic examination of the surface revealed, that the coverage of the surface bypasting liquid does not change, but the electrode roughness increases. Both batch andflow measurements confirm the related increase in the electroactive area of the electrode,which is expressed in the distinct increase of the background current and slight increaseof the signal. Growing density of the paste suggests also general change in the pastecomposition.
Applicability of the glassy carbon paste electrode in flow measurements with highcontent of organic solvents, particularly HPLC, was confirmed, although measurement insuch media is less advantageous than in aqueous media.
Acknowledgement: Financial support of the Czech Ministry of Education, Youth and Sports (projects MSM0021620857, RP14/63 and LC06035), KONTAKT (AMVIS) project ME10004 and of the grant SVV-2011-263204 is gratefully acknowledged.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-259-MO
A Complete Solution for Method Linearity in HPLC and UHPLC
Frank Steiner, Tobias Fehrenbach, Andreas Brunner, Fraser McLeodDionex Corporation,
Dornierstraße 4, 82110 Germering, Germany
Validation for linearity requires the preparation and analysis of a set of several indepen-dently prepared solutions. HPLC method linearity according to ICH guidelines, as anexample, is based on five concentration levels between 70% and 130% of the nominalconcentration, each to be injected three times. The laboratory effort for this process canbe quite substantial, especially if the method has to quantify several substances simulta-neously. To save time, it is common to prepare only one stock solution and subsequentlydilute this to the different concentration levels. This procedure means that any error in thestock solution preparation is carried through all subsequent dilutions. In addition, itmeans that the preparation of the standards differs from the process that would be usedin day-to-day operation of the method, thus making comparison of the two processesdifficult.
We present a fully automated gravimetrical solution to prepare all solutions for a cali-bration or linearity test independently and without error prone manual interference. Allsolutions prepared are immediately ready for injection onto the UHPLC analysis system. Acomprehensive software package, as an important part of the solution, sets up the se-quence for linearity validation automatically. As soon as the HPLC results are available,linearity according to ICH is calculated instantaneously by the software and all results areassessed against specification requirements. This complete solution speeds up the processsignificantly, excludes typical operator errors that arise during manual sample preparationand result calculation, and produces results of full statistical integrity.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-260-TU
Complex Rapid Temperature Programming for Capillaryand Microscale Liquid Chromatography through the Application
of Direct Contact Thermoelectric Module Arrays
David Collins1, Ekaterina Nesterenko1, Damian Connolly1, Mirek Macka2,Dermot Brabazon1, Brett Paull1,2
1Irish Separation Science Cluster (ISSC), National Centre for Sensor Research, Dublin City University,Glasnevin, Dublin 9, Ireland
2Australian Centre for Research on Separation Science (ACROSS), University of Tasmania,Hobart, Australia
Column temperature is an important, yet under utilised, parameter in liquid chromatogra-phy, which can be controlled to effect drastic changes in run times, peak efficiency andresolution, sensitivity, and mobile phase consumption. Furthermore, rapid response tem-perature programming can be used for the focusing peaks, particularly in emergingmodes of chromatography, such as those involving thermally responsive phases, or toimprove the overall resolution of complex mixtures. These advantages can be achievedthrough the application of temperature gradients over specific zones of the column, orwhich may need to be applied very rapidly. The problem with the application of suchrapid gradients and high temperature is that most commercially available column ovenscannot provide the high ramp rates or the high temperatures that may be required. Themajority of LC column ovens can only achieve a maximum ramp rate of 30 °C/min, whilethe upper temperature limit for most of these ovens is in the region of 80 – 90 °C. Inaddition, there are currently no commercially available ovens that offer distinct, isolatedtemperature zones, enabling the application of heated or cooled zones at specific andvariable locations along the length of the column.
In the work presented herein, a novel column heater based on thermoelectric moduleswith rapid temperature response for capillary and micro-bore liquid chromatography isdescribed. The operating temperature range of the novel modular system is between 15to 200 °C, with a ramp rate of approximately 400 °C/min, allowing for accurate andrapid temperature programming, with precise and isolated column temperature zonalcontrol. The potential of the approach is demonstrated with chromatographic separationsin reversed-phase mode with dual temperature/flow rate gradients, showing substantialimprovements in chromatographic performance and reduced run-times. Due to thespecific features of thermoelectric modules (precise temperature control, fast response)further application of the system to the fabrication of thermally initiated polymer mono-liths with gradients of functionality or porosity, in both fused silica capillary housing andmicro-fluidic chips (where the ability to precisely control temperature during polymerisa-tion is essential) is also demonstrated.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-261-MO
Fast LC using a New Generation Micro-UHPLC Systemwith Array Based UV Detection
Khaled S. Mriziq, David Neyer, Steve Hobbs, Remco van Soest,Phillip Paul, Don Arnold
Eksigent Technologies,Dublin, CA 94568, USA
With the availability of new stationary phase particles there are high demands for instru-ments for faster LC separations, high resolution and throughput. Existing systems canyield satisfactory performance with standard 4.6 mm ID analytical columns but not withsmaller IDs columns where system volume has a clear impact on peak width. Low-volumeseparations require maximizing the separation efficiency to provide greater quality infor-mation. Systems using 2.1 mm ID columns are required to run at flow rates beyond theoptimum linear velocity to be suited for fast LC, this result in high solvent consumptionand generating frictional heating through the column. In this presentation we discuss theadvantages of using micro-flow rates with 0.5 mm to 1 mm IDs columns. The experi-ments to demonstrate that were carried on a newly developed ultra-high pressure systemcapable of operating at 10,000 psi column pressure allowing the use of columns packedwith particles < 3 μm. The LC system is a low dispersion system, and ideally suited for fastLC, the later results in more than 90% solvent consumption saving compared to thestandard analytical columns. The LC system utilizes an optimized and highly sensitivemicro-fabricated UV flow cell and CCD-based spectrometer. Applications that demon-strate the impact of the LC design on the quality of the LC separation, in particular high-resolution and low volume separations, and the capabilities of the system in increasingefficiency, reducing analysis time, high sensitivity and minimum band broadening arediscussed.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-262-TU
Non-Discriminatory, Universal and Sensitive Detection Technologiesfor Fluid Based Separation Techniques
Joseph P. Hutchinson1, Manish Khandagale1, Jianfeng Li1, William Farrell2,Elizabeth Groeber3, Roman Szucs4, Greg Dicinoski1, Paul R. Haddad1
1Australian Centre for Research on Separation Science (ACROSS), School of Chemistry, Faculty of Science,Engineering and Technology, University of Tasmania,
Private Bag 75, Tas., 7001, Australia2Pfizer Global R&D,
La Jolla, CA, USA3Pfizer Global R&D,
Groton, CT, USA4Pfizer Global R&D,
Sandwich, Kent, CT13 9NJ, UK
Universal detection response in chromatographic analysis remains an attractive, but asyet unfulfilled, goal. Many areas of application of liquid chromatography, such as thepharmaceutical industry, would benefit from a detector which provides uniform re-sponse towards all analytes. This would allow well characterized reference standards tobe used to obtain purity results for unknown compounds within a sample. While massspectrometry (MS) has achieved prominence as a universal detector, many species arenot ionizable or suffer from variable ionization. Similarly, whilst photometric detection(UV/visible) is still widely used, UV detection requires that the analytes contain a suit-able chromophore. The simultaneous use of two or more detectors can partly overcomethe above limitations, however, this often leads to reduced sensitivity due to flow split-ting and uncertainties in determining the exact splitting ratio for quantitative purposes.Furthermore, as external calibrants are required for both MS and photometric tech-niques, quantifying unknown compounds is often not feasible. Therefore, there is aneed to develop and characterise non-discriminatory, universal detection technologiesfor fluid based separation techniques which provide the sensitivity necessary for con-temporary applications.
Aerosol detectors constitute an emergent class of mass-sensitive detectors. In thesedetectors, the HPLC column effluent is nebulized and then dried, producing analyteparticles. This process accommodates a large variety of different compound classes, pro-vided they are less volatile than the mobile phase. These dried particles are then detectedoptically in the case of the evaporative light scattering detector (ELSD) and the moresensitive condensation nucleation light scattering detector (CNLSD), or by charge transferin the case of the charged analyte detectors (Corona CAD, Corona Ultra). These detectorsprovide superior relative response factors in comparison to other types of detectors.Another option is to use the flame ionisation detector (FID) in combination with liquid

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
chromatography. This detector has predominantly been used with gas chromatography,where it has been shown to provide excellent sensitivity and linearity for the universaldetection of molecules containing carbon.
This presentation will compare the different universal detectors and highlight thefactors which contribute to variation in analyte response. These detectors have beenhyphenated with ultra high pressure liquid chromatography (UHPLC) to perform re-versed-phase gradient separations of a mixture of pharmaceuticals chosen to provide awide range of physicochemical characteristics.
ReferencesJ. P. Hutchinson, J. Li, W. Farrell, E. Groeber, R. Szucs, G. Dicinoski, P.R. Haddad. Universal response model for
a corona charged aerosol detector. J. Chromatogr. A. (2010), 1217, 7418–7427.J. P. Hutchinson, J. Li, W. Farrell, E. Groeber, R. Szucs, G. Dicinoski, P.R. Haddad. Comparison of the response of
four aerosol detectors used with ultra high pressure liquid chromatography. J. Chromatogr. A. (accepted).

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-264-TU
The Impact of Solvent Mixingon Liquid Chromatographic Performance
Christian Schmidt, Verena Jendreizik, Wulff Niedner, Fraser McLeodDionex Corporation,Germering, Germany
Gradient Elution – increasing the organic content over analysis time – nowadays is themost common technique in reversed phase HPLC. There are two different technical solu-tions for on-line (dynamic) gradient formation: high-pressure gradient proportioning(HPG) and low-pressure gradient proportioning (LPG). In both solutions compositionalfluctuation occur during gradient formation causing a deterioration of chromatographicperformance. This can be reduced by the use of a highly efficient mixer to homogenizethe compositional fluctuation. The experimental determination of mixer performance isdescribed and the impact on chromatographic performance is discussed for selectedapplication examples.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-265-MO
Total Solutions for High-Throughput Analysis and Purifications
Steve Zulli, Ziqiang Wang, Dan Rolle, Jon Jones, Tim Martin, Harbaksh SidhuSFC Division, Waters Corporation
130 Executive dr., Suite 2A, Newark, DE 19702, USA
Chromatographic tool has taken the major role in high throughput analysis and purifica-tions for its sophistications, high efficiency and fast turnaround to productivity.
With integration of high end mass spectrometry, mass-directed LC-platform technique,with its well-designed Boolean detection and collection logarithm, has shown the excel-lent performance to handle the complicated and diversified nature of drug discoverydiscipline.
The success rate for such single chromatographic platform (HPLC) based strategy hasbeen reported in the range of 50% to 70% across the industrial practices. While thisbenchmark confirms the effectiveness of the technique, it is also obvious that it will bebeneficial to have complementary and alternative technique for further improvement foroverall success rate.
In this study, we will demonstrate a complementary technique by integrating theversatile detection and collection functionalities through a supercritical fluid chromatogra-phy (SFC) platform, to work together to offer the total solution package.
The implemented functionalities on this SFC platform include hyphenated detectiontools such as MS-UV-PDA-ELSD for sophisticated determination and triggering; stackedinjections and collections to improve throughput/productivity, automatic column switch-ing for versatility, and added wasteline detection and collection to prevent sample loss.Implementation of these functionalities across several different scales of platforms will alsobe discussed.
The industrial data has shown this combined strategy can increase the success rate ofhigh throughput analysis to 85%–90%, which is significant.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-266-TU
Improvement in the Sensitivity of RT-PCR Assaysby using RP-HPLC Probe Purification
Alena G. Mosina1, Andrey N. Chuvilin2, Igor P. Smirnov2,Galina E. Pozmogova2, Ekaterina F. Kislina2
1Moscow State Academy of Fine Chemical Technology,Vernadskogo pr., 86, Moscow, the Russian Federation 119571, Russia
2Research Institute of Physical-Chemical Medicine, Moscow, Russia
Progress in the development of microanalytical methods of medical diagnostics isachieved to the large extend due to the use of DNA and its fragments, as well as oligo-nucleotides with fluorescent labels. For the diagnosis of socially important diseases it isimportant, that these methods provide timely and qualitative results.
The sensitivity and reliability of the real-time polymerase chain reaction (RT-PCR) islargely depend on the stability of oligonucleotide, which chemical behavior is not neces-sarily studied in details.
We have shown that many probes labeled with widely used hexachlorofluorescein(HEX) fluorofore contain up to 10–30% impurities with altered spectral characteristics. Wehave studied conditions of their formation, their structure and properties. It was foundthat heterocyclic oxygen in the structure of HEX is replaced nitrogen with the formationof acridine derivatives during the standard ammonolysis step [1].
In this paper, the chromatographic behavior of synthetic probes containing differentfluorophores and the fluorescence quenchers were described. The conditions of reversed-phase high performance liquid chromatography (RP HPLC) to separate the previouslyundetected impurities has been optimized, including temperature, gradient modes andtypes of columns (C16 and C18). After such purification the total sensitivity of RT-PCRassays was increased by more than 20%.
Thus, use of RP HPLC to the probe purification enhances the reliability and accuracy ofDNA identification. Therefore we may conclude, that post synthetic HPLC purification ofHEX-labeled oligonucleotides is essential for demanding applications, where sensitivityand/or fluorescent signal differentiation (e.g., in quantitative and multiplexed assays) arecritical and the possibility of false positive response must be minimized.
1. A. N. Chuvilin, M. V. Serebryakova, I. P. Smirnov, G. E. Pozmogova. Byproduct with Altered FluorescentProperties Is Formed during Standard Deprotection Step of Hexachlorofluorescein Labeled Oligo-nucleotides // Bioconjugate Chem. 2009, 20, 1441–1443

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-267-MO
UPLC System Optimization and Solid Phase Extraction MethodDevelopment Strategies for the Analysis for Oligonucleotides
Xin Zhang, Pamela Iraneta, Frank Marszalkowski, Darryl Brousmiche,Kevin Wyndham, Raymond P. Fisk, Tom Walter
Waters Corporation,34 Maple St., Milford, MA 01757, USA
Oligonucleotides are a class of biomolecule with great potential for therapeutic applica-tions. ELISA methods are considered the gold standard for quantitative oligonucleotideanalysis, but their development is time consuming and metabolite information is lost. Asthe sensitivity of mass spectrometers continues to improve, the future of quantitativeanalysis of oligonucleotides and their metabolites is expected to move in this direction.
However, complex biological samples that contain matrix specific salts, phospholipids,proteins and other endogenous compounds can potentially interfere with the quantitativedetermination of oligonucleotides and their metabolites making sample preparation acritical step. We will present a strategy for optimizing a Waters ACQUITY UPLC system forthe analysis of oligonucleotides. Factors to be considered are solubility, non-specificbinding, stability, materials (of needles, plates, and vials), and strong and weak solventwashes. These choices influence the sensitivity and the linearity of the analysis.
The systematic approach presented here for the analysis of oligonucleotides can besimilarly useful for any difficult to handle analytes, sometimes referred to as “sticky” or“labile.” Once the analytical system as been optimized then developing a sample prepara-tion method is possible. For the oligonucleotides, we chose a solid phase extraction (SPE)method using a mixed mode weak anion exchange sorbent. The method developmentsteps involved selecting appropriate loading and elution solvent systems prior to optimiz-ing the SPE methods for a variety of 15 mer to 35 mer oligonucleotides (phosphorylatedand phosphorothioated).

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-268-TU
Comparing Reversed-Phase and Micellar Liquid Chromatographyto Study Lipophylicity of 1,2,4-Triazoles
Małgorzata Janicka1, Katarzyna Stepnik1, Anna Pachuta-Stec2
1Department of Physical Chemistry, Faculty of Chemistry, Maria Curie-Skłodowska University,Maria Curie-Skłodowska Sq. 3, 20-031 Lublin, Poland
2Department of Organic Chemistry, Faculty of Pharmacy, Medical University,6 Staszica St., 20-081 Lublin, Poland
Micellar Liquid Chromatography MLC is a mode of reversed-phase liquid chromatographyusing a surfactant solution above the critical micelle concentration (cmc) as mobile phase.Retention of a solute in this technique depends on the types of interactions with themicelles and with the surfactant-modified stationary phase and is affected by hydrophobicinteractions. It should give rise to acceptable linear correlations between the chromato-graphic logk and the partition logP properties considered as lipophylicity indices. Fordifferent groups of organic solutes, linear relationships are observed between logk valuesmeasured with different pure aqueous or mixed micellar effluents and logP values. As aresult MLC becomes more and more popular in studying lipophylic properties of different,especially newly synthesized substances.
In our investigations three chromatographic techniques (HPLC, OPLC and TLC) withmicellar effluents and reversed-phase TLC were used to study lipophylic properties ofnewly synthesized 1,2,4-triazoles, potential fungicides. In experiments RP-CN, RP-8 andRP-18 stationary phases ware applied as well as different organic solvents (acetone, aceto-nitrile, dioxane and tetrahydrofuran) and surfactants (SDS and Brij-35) were used asmobile phase components. As lipophylicity descriptors there were proposed micellar logkm
and reverse-phase logkw chromatographic parameters. These values were compared withpartitioning parameters logP calculated according to molecular structures of solutestested. Linear correlations between chromatographic and partitioning indices obtained inthe studies allow determine MLC as a very good technique to study lipophylic propertiesof new organic substances.
ReferencesM.J. Ruiz-Angel, S. Carda-Broch, M.C. Garcia-Alvarez-Coque, A. Berthod, J. Chromatogr. A 1030 (2004) 279–
288M. Janicka, D. Pietras-Ozga, J. Planar Chromatogr. 23 (2010) 396–399

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-269-MO
Isolation and Characterization of Antimicrobial Compoundsfrom Plant Extracts by means of OPLC, GC-MS and BioArena
Ágnes M. Móricz1, Szabolcs Szarka2, Péter G. Ott1, Éva B. Héthelyi2,Éva Szôke2, Ernô Tyihák1
1Plant Protection Institute, Hungarian Academy of Sciences,Herman O. Str. 15, 1022 Budapest, Hungary
2Department of Pharmacognosy, Faculty of Pharmacy, Semmelweis University,Üllôi Str. 26, 1085 Budapest, Hungary
There is a more and more increased demand to the discovery of new, efficient antibioticsbecause the pathogens are resistant against the majority of presently known and usedantibiotics. Searching for bioactive natural products requires appropriate bioassays, fo-cused on the desired activity (e.g. antifungal, antibacterial). Different versions of layerliquid chromatography are ideal technical solutions for the chemical and biologicalscreening of drug ingredients, because of high-throughput, low cost, easy maintenanceand selectivity of detection reagents. Direct bioautography, linking the separation andbiological detection, potentiates the fast and easy sorting of effective components inmatrixes like plant extracts. In direct bioautography, the developed chromatoplates con-taining separated components in spots are dipped into an indicator cell suspension and,after a required incubation time, the inhibition zones are visualized using a vital dye, orsuitable luminescent indicator cells. So the evaluation of the activity is performed directlyon the adsorbent layer, at the chromatographic spots.
BioArena as a complex bioautographic system enables an unlimited possibility of inter-actions between microbes and biologically active small and big compounds in the adsor-bent layer of a chromatoplate. To study the influence of different substances on theantibacterial activity of components separated, they were dissolved in the cell suspensionimmediately before chromatoplate inoculation.
Overpressured-layer chromatography (OPLC), as an efficient forced flow planar layerliquid chromatographic technique, results in more compact chromatographic spots andbetter separation efficiency than TLC-HPTLC. OPLC is also well suited for fractionationusing analytical chromatoplates providing on-line detection and subsequent peak collec-tion. This system can be applied for efficient isolation of various substance types in gener-al, but in this case the potential antimicrobial components from plant extracts.
The OPLC separation and isolation of antibacterial components, found previously activein a TLC/OPLC-bioautographic study, of different plant extracts from species of Asteraceae,their re-chromatography, and identification by means of GC/LC-MS as well as bioauto-graphic and BioArena studies of concentrated collected peaks are demonstrated. In bioassayinvestigations Gram positive soil bacterium Bacillus subtilis (antibiotics generate white spots)and chromosomally tagged luminescence Gram negative Arabidopsis pathogen Pseudomo-nas savastanoi pv. maculicola (antibiotics generate black spots) were used.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-270-TU
Micellar HPLC, OPLC and TLC in Studying Lipophylicityof 1,2,4-Triazoles
Małgorzata Janicka1, Katarzyna Stepnik1, Anna Pachuta-Stec2
1Department of Physical Chemistry, Faculty of Chemistry, Maria Curie-Skłodowska University,Maria Curie-Skłodowska Sq. 3, 20-031 Lublin, Poland
2Department of Organic Chemistry, Faculty of Pharmacy, Medical University,6 Staszica St., 20-081 Lublin, Poland
Physicochemical properties of new compounds such as solubility, lipophylicity (hydropho-bicity), stability and acid-base character, as factor affecting bioactivity, should be deter-mined in the early stages of development. Lipophylicity is commonly measured by solutedistribution in biphasic liquid system and universal lipophylicity scale is formed by thelogarithms of partition coefficients logP. Since octanol-water was proposed as referencesystem for lipophylicity measurements, logP parameter remains as a standard for experi-mental and theoretical investigations. Due to experimental limitations connected with directmeasurements of logP parameters by shake-flask method, chromatographic techniques aregetting more and more popular in studying lipophilic properties of different potentiallybioactive substances. Though partition parameters produce more universal scale of lipo-phylicity, chromatographic approach is much more convenient, reproducible, fast andinexpensive. Recently Micellar Liquid Chromatography, MLC, is more and more popular inlipophylicity investigations. MLC is a mode of conventional reversed-phase chromatographyusing a surfactant solution above the critical micelle concentration (cmc) as mobile phase. Inthis technique solute retention depends on the types of interactions with the micelles andwith the surfactant-modified stationary phase. Nonionic solutes affected by hydrophobicinteractions should give rise to acceptable linear correlations between the chromatographicand the partition property, i.e. logk and logP parameters.
In our investigations three liquid chromatography techniques, i.e. column HPLC, over-pressured planar OPLC and thin-layer TLC, with micellar mobile phases were applied tostudy lipophylic properties of newly synthesized 1,2,4-triazoles. Different organic modifiers(acetone, dioxane, acetonitrile and tetrahydrofuran) and two surfactants (SDS and Brij35)were used as mobile phase components. In planar techniques (OPLC and TLC) RP-CNstationary phase while in HPLC RP-8 column were applied. Micellar lipophylicity parameterslogkm were compared with logP values calculated from molecular structures of solutes test-ed. Very good linear correlations observed between micellar and partitioning indices con-firm the abilities of MLC in studying lipophylic properties of organic substances.
ReferencesM.J. Ruiz-Angel, S. Carda-Broch, M.C. Garcia-Alvarez-Coque, A. Berthod, J. Chromatogr. A 1030 (2004) 279–288M. Janicka, D. Pietras-Ozga, J. Planar Chromatogr. 23 (2010) 396-399

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-271-MO
Planar Chromatography with Different Detection Techniques:the Ultrafast Quantitation of 5-Hydroxymethylfurfural in Honey
Elena S. Chernetsova1,2, Gertrud E. Morlock3
1Russian Research Centre Kurchatov Institute,Akademika Kurchatova sq. 1, 123182 Moscow, Russia
2People’s Friendship University of Russia,Miklucho-Maklaya str. 6, 117198 Moscow, Russia
3University of Hohenheim, Institute of Food Chemistry,Garbenstrasse 28, 70599 Stuttgart, Germany
During the last 20 years planar chromatography has changed, from a method with a poorreputation to a standardized, reproducible, and quantitative method, which is completelyautomated and very flexible with a variety of possibilities of analyte detection [1]. Compo-nents of a sample can be detected on a TLC or HPTLC plate using multiple detectiontechniques. Planar chromatography approaches can be successfully applied to the analysisof complex samples, especially foods and drinks, biological samples and natural products.However, the number of publications in this field is still very limited. For example, nopublications on quantitation of 5-hydroxymethylfurfural (HMF) in honey using HPTLCwere found.
HMF concentration is a very important factor reflecting the quality of honey. Nowadaysquantitation of HMF in honey is traditionally performed using spectrophotometric deter-mination after White or after Winkler or using HPLC methods [2]. However, the Winklermethod is not recommended anymore because of carcinogenic reagents and low preci-sion. The White method was also characterized with a high uncertainty for some sorts ofhoney. So, HPLC is today very often used for the quantitation of HMF in honey, and theduration of analysis is usually 30 minutes or even more. Other, more prompt, reliable andcost-effective methods for HMF quantitation are needed. To our opinion, modern planarchromatography approaches are perspective for prompt and cost-effective analysis ofhoney.
The aim of our recent studies was quantitation of HMF in honey using different planarchromatography approaches. We propose the ultrafast determination of HMF in honeyusing HPTLC instead of the traditional HPLC. The HPTLC separation lasts only 5 minutes,and up to 23 honey samples can be analyzed simultaneously on the same plate, providingthe increase of the analysis throughput in more than 20 times as compared to HPLC-based approach. Using the simplest sample preparation (just dissolving 1 g honey in 10mL water), performing a 5-minute separation and scanning at 288 nm, it was possible toquantify HMF in honey at the level of 8 mg/kg or even lower, while the concentration of5-hydroxymethylfurfural in honey should not exceed 15–80 mg/kg, according to differentregulations. The fast screening of HMF content in honey is possible using paper chroma-

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
tography as the cheapest possible alternative. Different detection techniques, such as ESI-MS and DART-MS, were applied for the quantitation of HMF in honey as well, and themethod comparison was performed. The respective results will be presented and dis-cussed.
1. G. Morlock, W. Schwack. The Contribution of Planar Chromatography to Food Analysis. Journal of PlanarChromatography (2007), 20, 399.
2. M. Zappala, B. Fallico, E. Arena, A. Verzera. Methods for the determination of HMF in honey: a compari-son. Food Control 16 (2005) 273–277.
This work was partially supported by the Council of the President of Russia (grant MK-594.2010.3).

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-272-TU
Potential of OPLC System with Multi-Inlet/Outlet
Emil Mincsovics1,2
1OPLC-NIT Ltd.,Andor Street 60, 1119 Budapest, Hungary
2Department of Genetics and Plant Breading, Corvinus University, Faculty of Horticultural Sciences,Villányi Street 29–43, 1118 Budapest, Hungary
OPLC system with one inlet and one outlet has a high flexibility and the principal opera-tion steps – sample application, separation, detection and isolation – can be freely com-bined. The fully off-line process corresponds to TLC and the separation and starts withdry non-segmented adsorbent layer having single and/or parallel samples spot-ted/streaked onto the localized areas of the bed. The separation can be fulfilled with orwithout overrun and the detection can be carried out in-situ on the layer or the impor-tant component bands can be isolated from the layer. The fully on-line OPLC is analo-gous with HPLC and at the same time single sample can be processed. OPLC with themulti-inlet/outlet configuration makes it possible to overcome this limitation and, withthe appropriate cassettes, results in high flexibility. In flowing eluent wall (FEW) technol-ogy only one pump is used for mobile phase introduction and the samples can be in-jected in parallel onto the localized areas of the wet non-segmented adsorbent layerdetermined by the inlet troughs. To prevent mixing of neighboring sample lanes, mo-bile phase, only, is introduced to the layer between the samples and at the sides, divid-ing the bed into active regions, where separation is performed, and non-active regions,where there is no separation; both mobile phase and sample are introduced to theactive separation regions. At the outlet the components eluted from a given lane can becollected and detected separately, with the corresponding FEW lines. The number ofinjection and collection troughs and the separation lanes is the same. The number ofFEW lines (n+1) is tailored to the number of separation channels (n). This correspondsto the number of chamber inlet/outlet as well.
Without using the FEW a new solution of injection on to specific region of the layerwill be introduced. In this solution the number of inlet and outlet of the chamber andalso the separation lanes is the same. OPLC system with one inlet and one outlet hasbeen used for these experiments; however the 5-inlet/outlet OPLC system with one-channel arrangement and the appropriate cassette results in the same results using onlyone inlet and one outlet. To enter the sample into the localized area of the wet beddetermined by the inlet trough an air bubble restricted process has been used. Theinjected air come ends of the channel where the access of the layer for the sample istemporarily inhibited; resulting in a shorter sample band than the trough. This processcan be used for fully on-line isolation.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-273-MO
The Investigations of the Chromatographic Properties of Selectedd- and 4f-electron Ion Element Complexes with Organic Ligands
by TLC and Magneto-TLC Methods
I. Malinowska, W. Ferenc, A. WronkaFaculty of Chemistry, Maria Curie-Skłodowska University,
Pl. M. Curie-Skłodowskiej 3, 20-031 Lublin, Poland
The carboxylates of d- and 4f-ion elements play an important role in inorganic and bio-inorganic chemistry because many of them are components of vitamins and drugs. Thecomplexes of carboxylic acids with various metal ions may be used as electric materials inthe modern branches of techniques and technology or they may have applications asprecursors in superconducting ceramic and magnetic field productions. These type ofcompounds are also used as catalysts, pigments, solvents, food preservatives, plastics andcosmetics.
Metal carboxylates are applied for the production of high degree purity of metal oxidesand polycarboxylic acids are used in supramolecular compound synthesis, which in manycases, form with metal ions the molecular polymers containing in their structures poresand channels owning to them they appear catalytic and adsorption properties. Thereforethey may be used for the adsorption of gases such as: argon,nitrogen and hydrocarbonsor small molecules of another inorganic compounds. Polycarboxylic acids may also formthe molecules with two- and three-dimensional structures, that have the special magneticand luminescence properties which let them be used in optical and electric industries.
Because the potential applications of the metal carboxylates are very wide especially inmedicine and pharmacy it is interesting to study the chromatographic properties of thesecompounds.
In the presented investigations the retention of 10 complexes of d- and 12 of 4f- elec-tron metals with different organic ligands in RP and NP chromatographic systems weredemonstrated. Analysis of the retention of the compounds may give us an informationabout the affinity of these compounds to the different stationary phase surface and aboutthe influence of the kind of organic ligand or central ion on the retention of the com-pounds.
Taking into account this fact that now magnetic fields being different than terrestrial inmany places, it is interesting to examine how the magnetic field influences the propertiesof the investigated compounds. Therefore the chromatograms were developed simulta-neously in two same chambers and one of them was situated in external magnetic fieldwith two B values (0.2 and 0.4 T). In magnetic field a retention of the some complexeschanges, it means that magnetic field influences the properties of the analyzed com-pounds and their interactions with surface of the stationary phase.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
Chromatographic investigations in RP systems can give us a preliminary informationabout biological activity of the compounds by logkw values. In RP systems the logkw weredetermined for investigated compounds in and outside of magnetic filed. Magnetic fieldchanges the logkw values. This is an important information because some of the analysedcompounds may be applied in medicine and beauty care.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-274-TU
TLC of Some 1,3,4 – Thiadiazole Derivatives in Magnetic Field
M. Studzinski1, I. Malinowska1, H. Malinowski21Department of Planar Chomatography, of Faculty of Chemistry, Maria Curie-Skłodowska University,
Lublin, Poland2Veksler and Baldin Laboratory of High Energy Physics, of Joint Institute For Nuclear Research Dubna,
Russia
Thin Layer Chromatography is one of the most popular method applied in life sciences. Itis also widely used in organic chemistry for screening of reaction kinetics and monitoringlevel of main products and contaminants in post reaction mixture. TLC is also used as aninstrument for controlling level of pollution in natural environment. It is fast reliable andaccurate, very robust for impurities present in biological samples but, in spite of thisadvantages, it must be constantly developed in order to “keep up” with other analyticalmethods. One path of improving of performance is changing properties of stationaryphase, which effected invention of HPTLC plates and monolithic silica gel plates (forexample). The other path of making TLC more competitive to other chromatographicmethods, was to combine it with different separation methods, and in the result, methodssuch as Overpressured Layer Chromatography (OPLC) and Planar Electrochromatography(PEC) were born.
Another way to enhance the abilities of chromatography, not only in analytical but alsoin physicochemical area as well, is application of magnetic field.
The magnetic field, different than terrestrial can influence on results of the chromato-graphic separation. On the other hand, comparison of retention of the chromatographedsolutes in magnetic field and without it can give us some information about influence ofthe magnetic filed on properties of the solutes and their interactions with surface ofstationary phase.
In presented studies TLC chromatography in magnetic field some derivatives of 1,3,4 –thiadiazoles have been demonstrated. Thin-layer chromatography was chosen as experi-mental method, due the fact that the modernization of the equipment for TLC for thework in the magnetic field is quite easy and cheap operation.
In our research, two kinds of the magnetic fields were used:Perpendicular – which lines was perpendicular to the sorbent layer and mobile phase
migration direction. The magnetic field was induced using a pair of neodymium perma-nent magnets. Maximum inductivity of the field was approximately 0,4 T.
Parallel – which direction was parallel to sorbent layer and mobile phase direction. Themagnetic field was generated by a superconducting electromagnet. The induction of thefield can be adjustable and B values was from 0.56 T to 3.1 T.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
NP and RP chromatogratography systems were used. It seems that, magnetic field notonly changes retention of the examined solutes (RF values) but, also spots width, and inconsequence, efficiency of the chromatographic systems.
In case of the reversed phase experiment, the influence of the magnetic field on logkw
– one of the crucial QSAR parameters – was investigated. Because logkW values of investi-gated compounds obtained in magnetic field and outside are different, it may suggest,that presence of external magnetic field may also amplify or otherwise modify biologicalactivity of examined compounds.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-275-MO
A Modified SVM Method for Analyzing Metabonomics Datafrom HPLC-MS
Xiaohui Lin1, Qiang Ruan1, Kang Yan1, Shili Chen2, Xinjie Zhao2,Yang Zhang1, Guowang Xu2
1School of Computer Science & Technology, Dalian University of Technology,Dalian 116024, China
2CAS Key Laboratory of Separation Science for Analytical Chemistry, Dalian Institute of Chemical Physics,Chinese Academy of Sciences,
Dalian 116023, China
Liquid chromatography-mass spectrometry (HPLC-MS) is an effective analytical techniquewhich has been used in many applications, such as proteomics and metabolomics. Sincethe data produced by HPLC-MS usually contain hundreds (or even more) of variablesincluding noisy and nonrelated information, selecting meaningful information from thedata becomes quite critic.
Support vector machine recursive feature elimination (SVM-RFE) is a very popular featureselection technique which is based on support vector machine (SVM). It has been successful-ly applied in analyzing biological data. In SVM-RFE, Filter-out-Factor (m), the number of thebottom ranked features to be deleted in each loop, can influence the performance of thealgorithm. Different m results in the different selected feature subsets, hence the perfor-mances of the corresponding SVM classification models are quite different. In order toproduce a stable result in processing high dimensional HPLC-MS data, we proposed animproved SVM-RFE method based on the dynamic Filter-out-Factor (SVM-RFE-DFF). In eachloop, only the features lying in a specific window and having no contribution to improvingthe classification performance are eliminated. To show the usefulness of our new SVM-RFE-DFF method we applied it to process metabonomics data of metabolic syndrome and liverdiseases from UPLC/Q-TOF MS platform. Results showed that the SVM-RFE-DFF outperformsSVM-RFE in discriminating the patients from healthy controls.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-276-TU
A New High Capacity MALDI Target Format for ImprovedLC-MALDI Analysis of Complex Proteomics Samples
Zoltan Czentnar, Arndt Asperger, Martin Schuerenberg,Andrea Kiehne, Detlev Suckau, Marcus Macht
Bruker Daltonik GmbH,Fahrenheitstr. 4, 28359 Bremen, Germany
High speed MALDI-TOF/TOF instruments are perfectly suited for LC-MALDI analysis ofcomplex proteome samples. To fully explore the benefits offered by these mass spectrom-eters, target plates are required that can accommodate large spot numbers. Such targetsallow the use of longer LC gradients enabling better separation and achieving improveddepth of analysis due to more comprehensive MS/MS analysis.
We describe a new pre-structured MALDI target that accomodates 1536 spots. Forbenchmarking, the target was applied to the LC-MALDI analysis of 500 ng E.coli trypsindigest using an extended LC gradient.
The new MALDI plate allowed to acquire significantly more high quality MS/MS dataper spot within a reduced time period, resulting in a clearly improved protein identifica-tion rate.
500 ng of E.coli cell lysate digested with trypsin was analyzed by LC-MALDI-TOF/TOF.The nanoLC system in use was equipped with a trap column (C18, I.D. 100 μm, 2 cmlong, 5 μm particles) and a standard analytical column (C18, I.D. 75 μm, 15 cm long, 3μm particles). LC separation was performed applying a 192 min gradient ranging from 2to 40% acetonitrile.
LC fractions were automatically deposited on a pre-structured MALDI target with a1536 spot format. 1152 fractions, 10s each, were spotted. HCCA matrix was mixed withthe LC eluate automatically.
All MALDI-MS and MS/MS data were acquired at a laser repetition rate of 1000 Hz inpositive ion mode.
For benchmarking of the new target format, 500 ng of E.coli trypsin digest was ana-lyzed by LC-MALDI. All 1152 fractions collected from a 192 min gradient were depositedon one plate, which allowed the MALDI-MS and MS/MS acquisition to be completedwithin a single overnight run without the need of target exchange.
The pre-structured target surface, characterized by hydrophilic sample spots surround-ed by a hydrophobic surface, enables optimum spot alignment, which results in 100%spot finding efficiency of the laser and allows for complete utilization of the sample spotarea during MALDI acquisition.
Furthermore, the new target facilitates improved HCCA crystalization homogeneity andenhanced robustness of the matrix preparation spots against sample consumption by the

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
laser. This enabled an increased number of MS/MS spectra to be acquired from individualtarget positions resulting in a significantly expanded depth of analysis.The complexity ofthe E.coli digest analyzed here required more than 30 MS/MS precursors to be fragment-ed from individual fraction spots. However, as the resulting MS/MS data reveals, highlysignificant peptide identifications were obtained even from those MS/MS spectra that hadbeen acquired when a spot had already seen far more than 100,000 laser shots. As anexample, the MS/MS spectrum, which was recorded at position 29 in the acquisitionorder of the fraction spot corresponding to a retention time 104.3 min, identified peptideHLWLEVK from Aspartyl-tRNA synthetase (E.coli). This clearly illustrates the outstandingMS/MS capacity offered by the new MALDI plate.
Overall, LC-MALDI analysis of 500 ng E.coli digest when using the 1536 format targetin combination with an expanded LC gradient resulted in 939 identified E.coli proteins,which represents a 20% increase compared to a previous approach based on a 2 hourgradient sampled onto two 384 format target plates.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-277-MO
An Improved nanoLC/MS/MS Ion Trap Setup for the Identificationof 800 Proteins from a Cell Line Lysate
Andrea Schneider, Andrea Kiehne, Markus Meyer, Arnd IngendohBruker Daltonik GmbH,
Bremen, Germany
One distinct goal in proteomics is still the survey of an entire cell lysate with regard to themaximum number of unambiguously identified proteins. While high resolution instru-ments are favored for this application, recent improvements in both nanoLC separation aswell as in ion trap technology lead to a much higher number of identified proteins fromsuch a sample in single LC/MS/MS experiments. Concepts and methods are demonstratedon a simple human cell line lysate.
The human cell line HT 29 was digested by trypsin and injected in different amountsranging from 200 ng to 1 μg sample into a Dionex RSLCnano coupled to a Bruker ama-Zon ETD ion trap. Separation was done with a 25 cm PepMap 75 μm column using anACN/H2O gradient of 100 or 180 min. The amaZon ion trap was set to data-dependentCID-MS/MS on 10 precursors from each respective MS spectra with dynamic exclusion ofalready fragmented peptides. A novel software algorithm reduced the time for eachMS/MS spectrum by about a factor of 2 to increase the MS/MS spectral rate. The data-base search was done in the NCBI database using Mascot (Matrix Science). Data valida-tion was done by ProteinScape (Bruker).
Very restrictive criteria were applied in the database search to largely avoid false posi-tive hits. The peptide identity threshold was set to be at least 35, the peptide false discov-ery rate to FDR < 1%. The list of hits was further screened for multiple or redundantproteins found for the same peptides which were eliminated.
Best results in terms of long protein lists were achieved, as expected, with the 180 mingradient. Injecting 1 μg of sample, more than 800 proteins could be unambiguouslyidentified (FDR = 0.94%, p = 0.04, peptide identity threshold > 35) with this relativelysimple instrumental setup. However, even for the low sample amount of 200 ng in a 100min gradient, 533 proteins could be found. This proves the high usefulness and sensitivityof the ion trap for a fast survey screening of cell lysates.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-278-TU
Analysis of Single Amino Acid Mutations in Intact Proteins
H. J. Wirth1, D. Steer2, X. Yan3, T. Bannam3, J. Rood3, A. Gooley1
1SGE Analytical Science,7 Argent Place, Ringwood, Vic 3134 Australia
2Monash Biomedical Proteomics Facility, Monash University,Clayton, Vic. 3128, Australia
3Department of Microbiology, Monash University,Clayton, Vic. 3128, Australia
With the increasing availability of high resolution mass spectrometers the analysis of intactproteins becomes a feasible alternative to the more commonly employed shot-gun tech-niques in proteomics. High resolution separations of proteins are hindered by the size ofthe analyte molecules and the resulting low diffusion rates especially inside the poresystem. The stationary phase of the column employed in these studies is a 3 μm C8 silicawith 1000 Å pore size.
By using a dedicated protein separation column (ProteCol C8 HQ1003) we demon-strate that it is possible to separate single amino acid mutants in a 36 kDa protein.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-279-MO
Characterization of the In Vitro and In Vivo Metabolitesof the Novel Thiosemicarbazone Anti-Tumour Agents
using HPLC-MS/MS
Jan Stariat1, Petra Kovarikova1, Milan Nobilis1, Vit Sestak1,Zuzana Kollarova1, Jiri Klimes1, R. Des Richardson2
1Department of Pharmaceutical Chemistry and Drug Control, Faculty of Pharmacy in Hradec Kralove,Charles University in Prague,
Heyrovskeho 1203, 500 05 Hradec Kralove, Czech Republic2Iron Metabolism and Chelation Program, Department of Pathology and Bosch Institute, University of Sydney,
Sydney, New South Wales 2006, Australia
Selective targeting of the essential nutrient iron (Fe) using Fe chelators is a promising newstrategy for anti-cancer therapy. Thiosemicarbazone Fe chelators – 2-benzoylpyridine-4-ethyl-3-thiosemicarbazone (Bp4eT) and di-2-pyridylketone-4,4-dimethyl-3-thiosemi-carbazone (Dp44mT) are among the most promising anti-proliferative agents currentlyunder development.
Investigation of the fate of these drugs in an organism is essential for further progress intheir development. Biotransformation knowledge serves as the basis for the rationale struc-ture modification in order to improve their pharmacokinetic/toxicological profile.
The aim of this project is to identify the phase I metabolites of both compounds thatare formed both in vitro and in vivo.
Initially, both drugs were incubated with human and rat hepatocyte microsomes/cyto-sol and after simple treatment, samples were analysed using LC-MS/MS on Supelco Dis-covery HS C18 column. The mobile phase was composed of the mixure of ammoniumformate and acetonitrile.
The structures of putative metabolites were suggested according to MSn experiments.In the case of Bp4eT, oxidative substitution of the sulphur by oxygen was proposed, forDp44mT hydrolysis and demethylation were suggested. The standards of the putativemetabolites were synthesized and the structures were confirmed by NMR.
Subsequently, preliminary in vivo experiments in rats were performed in order tosearch for in vivo phase I metabolites. In the case of Bp4eT, oxidative metabolite wasobserved in plasma, urine, intestinal content and faeces. Furthermore, a new metabolitewith the unknown structure was detected in these materials. The product of hydrolyticcleavage of the thiosemicarbazone bond was found for Dp44mT in plasma and faeces,while demethylated metabolite was revealed in plasma and urine.
The further effort will be focused on searching of the possible phase II metabolites aswell as the structure identification of the unknown Bp4eT metabolite.
This work was supported by the grant GAUK FaF/B-CH/85510, by the Charles University in Prague (ProjectSVV 263 001) and by research project MSM 0021620822.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-280-TU
Chromatographic Separation of Phosphorylated Neuropeptidesby Applying 3D- ERLIC-HILIC-Reversed Phase Separation.
Evaluation of Separation Conditions
Goran Mitulovic1, Verena Tretter2
1Proteomics Core Facility, Clinical Department of Laboratory Medicine, Medical University of Vienna,Währinger Gürtel 18–20, A-1090 Vienna, Austria
2Department for Biochemistry & Molecular Biology, Center for Brain Research Medical University of Vienna,Spitalgasse 4, A-1090 Vienna, Austria
The analysis of neuropeptides is highly complicated due to the complexity of the brainand because the compounds are often present in very low concentrations, and require apowerful separation technique. Usually, the reversed phase chromatography is applied forseparating peptides extracted from brain tissue; however, in most cases it does not pro-vide sufficient chromatographic resolution, making the sample characterization difficultand prude to errors.
The most used 2-D LC approach in neuropeptidomics is the combination of strongcation exchange (SCX) chromatography for first dimension separation and RP chroma-tography for second dimension separation. However, the orthogonality of this combina-tion is limited by the fact that peptides are mostly doubly or triply charged, and thusthe majority of them will cluster within few and relatively narrow SCX elution windows.In addition, the phosphorylated peptides, which are of extreme importance, are routine-ly eluted in early fractions without proper separation. Further, we have previously ob-served contamination originating from salts and column bleeding when employing SCX-RP-MS/ MS.
Therefore, we have developed a three-dimensional separation approach for phosphory-lated neuropeptides by applying ERLIC (Electrostatic Repulsion Liquid Interaction Chroma-tography) as the first separation dimension followed by parallel separation of collectedfractions on nano HILIC (Hydrophilic Interaction Liquid Chromatography) and RP columnsand MS/MS detection.
Although a powerful separation mode, a major limitation of RPLC is the lack of ade-quate retention of polar molecules. Normal Phase Liquid Chromatography (NPLC) is auseful separation technique as it provides a different selectivity than RPLC.
Following the fractionation on ERLIC phase, it is possible to inject the fraction directlyonto the non-polar HILIC stationary phase and provide a fully orthogonal separation incomparison to the s the separation with the reversed phase chromatography.
Here, we have evaluated the impact of different separation parameters such as percent-age and type of organic modifier for ERLIC and HILIC separations, the impact of salt-freeERLIC fractionation and the salt containing separation. Further, we have evaluated the

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
impact of the type of the HILIC stationary phase (polar or non-polar) on amount of identi-fied peptides in samples from rat brain.
Initial results show that, the use of ERLIC and HILIC in combination with RP separationleads to a higher number of identified peptides, and that the number of identified phos-phorylations was also substantially increased compared to SCX-RP separation and the RP-only separation.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-281-MO
Comparison of Glycoprotein Enrichment Methodson Human Plasma Samples
Lilla Turiák, Oliver Ozohanics, László Drahos, Károly VékeyHungarian Academy of Sciences, Chemical Research Center
H-1025, Pusztaszeri 59–67., Budapest, Hungary
Glycosylation is one of the most frequent and most important post translational modifica-tions. It is involved in various processes such as basic protein folding and systemic im-mune response. Analytical techniques are intensively beeing developed in order to pro-mote the description of glycosylation. Nevertheless, analysis of glycosylation is still in itsinfancy. Determinations of glycosylation patterns (glycan heterogenity for one glyco-sylation site of a protein) of proteins present in the blood plasma are currently a hugechallenge as these studies require extensive (and expensive) sample preparation. Thedifficulties lie in the low blood plasma level and in the multitude of glycoforms of theglycoproteins.
Our group has recently developed in-house built software (GlycoMiner, www.chemres.hu/ms/glycominer) to easily evaluate glycosylation patterns of glycoproteins and glyco-protein mixtures. Our present aim was to compare sample enrichment techniques ofglycoproteins; and test the influence of the methods on the glycosylation pattern ob-served. We investigated whether the commonly observed sample preparation techniquesresult in biased or unbiased glycosylation patterns.
We compared several solid phase extraction (SPE) based methods, as these can beeasily automated: strong anion-exchange on a preequilibrated SPE coloumn, phenyl-boronic acid affinity SPE and lectin affinity using wheat germ agglutinin (WGA). Thesemethods are common steps of sample enrichment or clean-up. Following gycoproteinenrichment the samples were digested by trypsin. After the digestion nano-LC-MS andMS/MS were performed. The glycosylation pattern of processed and control samples werestudied and will be presented.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-282-TU
Comparison of Microparticulate and Monolithic Reversed-PhaseColumns for HPLC Analysis of Tryptic Digestsfrom Industrial Enzymes in Cleaning Products
Miriam Beneito-Cambra1, José Manuel Herrero-Martínez1,Guillermo Ramis-Ramos1, Wolfgang Lindner2, Michael Lämmerhofer2
1Department of Analytical Chemistry, University of Valencia,Doctor Moliner 50, E-46100 Burjassot, València, Spain
2Department of Analytical Chemistry, University of Vienna,Waehinger Strasse 38, A-1090 Vienna, Austria
Enzymes for cleaning products constitute the largest segment of the world market forindustrial enzymes. Today, enzymes are important components of many laundry, dish-washing and other cleaning products. Cleaning power enhancement by enzymes leads toa reduction of washing times and temperatures, with the subsequent savings of water andenergy. Further environmental advantages arise from the lower consumption of aggres-sive chemicals, with the additional benefit of a better care of fabrics during washing. Inthis work, tryptic digests of industrial enzyme concentrates of several proteases, lipases,amylases and celluloses classes were analyzed. For this purpose, several reversed-phasestationary supports including microparticulate and monolithic columns for HPLC com-pared. The monolithic columns employed were: ProSwift (polystyrene-divinylbenzene)from Dionex, and Chromolith (silica-C18) from Merck. On the other hand, the micro-particulate silica-C18 columns: Gemini (5 and 3 μm), and Kinetex (shell-core technology,2.6 μm) from Phenomenex were used. Separations were performed by gradient elutionwith water/acetonitrile containing 0.1% trifluoroacetic acid, using UV detection at 214and 280 nm. The flow rate was modified to obtain the same linear flow velocity. Takinginto account the number of resolved peaks, peak capacity and global resolution, the bestseparations were obtained with the Kinetex column. Using this column in the optimizedconditions, to the enzymes present in detergent bases and laundry cleaning productswere analyzed. For this purpose, the enzymes were precipitated from the cleaners, usingacetone, and the precipitate was dissolved in water, precipitated again and dissolved inthe trypsine digestion buffer.
Acknowledgements: Project CTQ2010-15335 (MEC and FEDER funds) and V-Segles-Empresa grant for PhDstudies (M. Beneito-Cambra, University of Valencia and Químicas Oro).

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-283-MO
Data Mining of an Untargeted LCMS Metabolomics Study Performedusing Aqueous Compatible Reverse Phase Column
Syed Salman Lateef1, Sudha Rajagopalan1, Siji Joseph1, Nilanjan Guha1,Yugandhar Reddy1, Michael Frank2
1Agilent Technologies India Pvt. Ltd,RMZ Centennial, “C” Block, ITPL Road, Mahadevapura Post, Bangalore, 560048, India
2Agilent Technologies Ltd,Waldbronn, Germany
IntroductionRapamycin is an immunosuppressant drug that has been shown to inhibit cancer
growth. The effect of Rapamycin on the metabalome of HEK 293 Human cancer cells isinvestigated in this study by interrogating the extracted metabolites by LC/QTOFMS. Theextracted metabolites were separated on a SB-Aq column, which is a reverse phase col-umn compatible with an aqueous mobile phase. This untargeted data is mined using adatabase of compounds found in a specific pathway. Such a database is created by ex-tracting the accurate mass of compounds found in selected specific pathways. Such anapproach helps to derive information from a metabolomics study about differential ex-pression of compounds found in specific pathways. The differential expression analysis isperformed by a chemometric software that also allows searching for biological pathways.
MethodHEK 293 cells are treated with Rapamycin or vehicle only for 16 hrs. Metabolites are
extracted twice from drug treated and control cells using a 2:1:2 (Methanol:water:chloro-form) mixture. The aqueous layer is collected for LC-MS analysis. The metabolites are sepa-rated using a methanol/ water gradient on a SQ-Aq column and identified using a QTOFmass spectrometer. The gradient runs from 2% methanol to 98% methanol in 13 min.Databases of metabolites found in specific pathways are created using a custom softwarepackage written to extract and create an accurate mass database from selected KEGG path-ways. This database is used for data mining of untargeted LC-MS data.
Preliminary DataA database of metabolites found in ABS transporter pathways is created and is used to
mine data from rapamycin treated and untreated cells. The results show that 48 and 42entities respectively are found in rapamycin and non treated cells. 41 of these entities areshared commonly by the control and treated cells. There are seven entities found only inthe treated samples. In this experiment, an untargeted metabolomics data is mined usinga database of compounds found in specific pathways.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-284-TU
Development of an Immunoaffinity Sorbent for the Analysisof Opioid Peptides by IA-SPE-CE-MS
Fernando Benavente, Silvia Medina-Casanellas,José Barbosa, Victoria Sanz-Nebot
Departament de Química Analítica, Universitat de Barcelona,Avinguda Diagonal 647, (08028), Barcelona, Spain
Over the last decade immunoaffinity capillary electrophoresis (IACE) has become a usefultool for the analysis of different compounds in biological samples. IACE is carried out inseveral formats that differ in the set-up to establish the specific interaction for the comp-lexation of the antigenic analyte and the antibody before the electrophoretic separation.Approaches based on immobilized antibodies onto the inner wall or onto an appropriatesolid support as a small binding area integrated into the separation capillary can be regard-ed as a variant of on-line solid-phase extraction capillary electrophoresis (SPE-CE), which hasbeen extensively used with typical chromatographic sorbents for sample clean-up andanalyte preconcentration. In this work, we explored a procedure for the preparation of an IAsorbent for the analysis of opioid peptides by on-line immunoaffinity solid-phase extractioncapillary electrophoresis-mass spectrometry (IA-SPE-CE-MS). We followed a site-specificantibody immobilization approach based on the covalent attachment of the oxidized anti-bodies through their carbohydrate moieties to hydrazide silica particles, using a polyclonalantibody against Endomorphin 1 and 2 (End1 and End2). The main features of the IAsorbent were studied, such as the amount of hydrazide groups and antibodies attachedonto oxidized diol silica particles. Once the procedure was optimized, standard solutions ofEnd1 and End2 were used in order to establish the IA-CE-MS methodology. Acceptablerepeatability, reproducibility and linearity range values were obtained for the proposedmethodology. The limits of detection (LODs) of 1 ng mL−1 were approximately 100-foldbetter than those obtained by CE-MS. Selectivity of the IA sorbent was good but some cross-reactivity against Dynorphin A (1–7) was observed when a mixture of several opioidpeptides was analyzed. Human plasma samples spiked with End1 and End2 were also ana-lyzed and both peptides could be detected until 100 ng mL−1.
1. A. C. Moser, D. S. Hage. Electrophoresis, 2008, 29, 3279–3295.2. N. A. Guzman, T. Blanc, T. M. Phillips. Electrophoresis, 2008, 29, 3259–3278.3. E. Hernandez, F. Benavente, V. Sanz-Nebot, J. Barbosa. Electrophoresis, 2008, 29, 3366–3376.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-285-MO
Efficient In-Line Digestion Methods to Support LC-MS Workflows
K. Flook1, E. J. Sneekes2, Y. Agroskin1, R. Swart2, C. Pohl11Dionex Corporation,
1228 Titan Way, Sunnyvale, CA, USA2Dionex Corporation,
114 Aberdaan, Amsterdam, The Netherlands
The objective of bioanalytical workflows is generally to detect, identify and sometimesquantify proteins. Despite tremendous advances in instrument capabilities, the analysisand detection of intact proteins remains a challenging task. Consequently, the majority ofworkflows center on the analysis of peptides, whether these are in proteomics or peptidemapping of biopharmaceuticals.
Trypsin is a commonly used protease that cleaves at the carboxyl side of the aminoacids lysine and arginine; the generated peptides are easily separated and detected by LC-MS techniques and the predictable cleavage sites can facilitate data analysis. Usually thedigestion is performed off-line, in solution and is a time consuming and laborious process.Immobilizing the protease on a monolithic column creates several advantages. It enablesan automated, in-line process, speeds up the digestion and opens up new LC-MS work-flows, relevant to all fields that face the challenges of intact protein analysis. The use of amonolithic carrier for immobilization provides open flow through channels for fast effi-cient digestion.
Using a monolithic column with immobilized trypsin we demonstrate a simple methodto determine digestion efficiency as well as digestion capability for different proteins ofvarious complexities. Protein standards have been chosen to represent samples fromresearch fields, such as proteomics and biopharmaceutical analysis. The intact proteins areeither directly applied to the digestion column or prefractionated at the intact proteinlevel. The digestion efficiency is evaluated in terms of sequence coverage and missedcleavages. Reversed phase nanoLC is used after the immobilized trypsin column to deter-mine efficient and complete digestion of the protein samples Workflows are evaluated interms of performance, MS compatibility and efficiency.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-286-TU
Evaluation of Engineered AMPs Production in Transgenic Plantsby MRM
Esther Izquierdo Alegre1, Sonia Hem1, Claude Nespoulous1,Maria Montero2, Maria Pla2, Michel Rossignol1
1Laboratoire de Proteomique Fonctionnelle,UR1199, INRA, 2, Place Pierre Viala-Bat. 13, 34060 Montpellier, France2Institut de Tecnologia Agroalimentària (INTEA), Universitat de Girona,
Campus Montilivi, EPS-I, 17071 Girona, Spain
Antimicrobial peptides (AMPs) are short sequence peptides, around less than 50 amino acidresidues, reported to constitute a first line of defense in plants and animals. They offer greatperspectives as a novel class of therapeutics, especially against fungal infections and antibi-otic resistant bacterial pathogens. The knowledge of the structure-function relationships inAMPs have provided the rationale for the design of novel molecules based on the naturalstructure of defensins, bacteriocins, antifungal proteins, peptaibols and cyclic peptides ofanimal plant and microbial origin. The objective of our project is to address and develop thesustainable production of small rationally designed AMPs using plants as biofactories.
Genetically modified (GM) plants (rice, potato and tobacco) expressing engineeredAMPs have been generated with different types of AMP transgenes and engineered ver-sions of them (tandem, enlarged, with processing elements). The efficiency of AMPsexpression in these plants must be evaluated and, for this purpose, purification and detec-tion of AMPs is clearly a critical aspect.
In this work we present the extraction, purification and analysis of AMPs from GM plants.Targeted proteomics by MRM (multiple reaction monitoring) has been used for the detectionof expression products using a hybrid Triple Quadrupole/Ion Trap mass spectrometer coupledto nano-LC chromatography. This technique provides stable, sensitive and reproducible assaysto specifically detect and quantify proteins of interest in complex mixtures. The optimalMS/MS transitions and mass parameters (DP, CE and DT) have been determined both fortrypsin digested AMPs and non-digested peptides, since sensitivity varies significantly depend-ing on selected peptides. For example, for some AMPs, MRM signal can be up to 100 timesmore intense when working with non-digested peptides compared to tryptic fragments.
Extraction and purification of AMPs expressed by the GM plants constitute a challengedue to the amphipathic and “sticking” nature of the AMPs. We have observed that whenworking with this kind of peptides, more than 90% of AMPs contained in a standardsolution can be lost by adhesion on tube walls or during chromatography. Specific proce-dures must be set up at all the purification steps in order to allow robust quantification ofsuch peptides. Presented data will cover both the technological development and resultsobtained in GM plants overproducing the peptides.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-287-MO
Evaluation of Fully Porous and Superficially Porous Particlesin Metabolomics Applications
Ruben t’Kindt1, Gerd Vanhoenacker2, Frank David2, Pat Sandra2, Koen Sandra1
1Metablys,President Kennedypark 26, 8500 Kortrijk, Belgium
2Research Institute for Chromatography,President Kennedypark 26, 8500 Kortrijk, Belgium
In HPLC based separations, researchers are constantly striving for higher separation effi-ciency and faster speed of analysis. Especially in the field of metabolomics, where one isconfronted with highly complex metabolite samples, the benefit of higher separationefficiency is indisputable. Metabolomics analyses also imply the use of large samplegroups, next to quality control samples and control standard metabolite mixtures, whichlengthen the overall analysis time. The introduction of sub-2 μm particles definitely ful-filled the requirements of improved column efficiency and speed of analysis. Nowadays,column manufacturers are boosting the use of superficially porous particles as a worthyalternative for these sub-2 μm particles. These columns provide similar performance atreduced backpressure, thereby avoiding the use of ultra-high pressure equipment.
Here we critically investigate the benefit of both fully and superficially porous particlesin a typical metabolomics analysis setting. The primary goal was to obtain the highestseparation efficiency, with optimal MS results. A comparison is made between sub-2 μmfully porous columns (Waters Acquity HSS T3 C18, Agilent Zorbax SB-Aq) and core-shellparticle columns (Phenomenex Kinetex C18, Agilent Poroshell EC-C18), in identical exper-imental conditions, using gradient elution chromatography at two different flow rates.Urine samples, next to a standard metabolite mixture, were analyzed on the differentcolumn types. Column performance was evaluated at the chromatography level (reten-tion time, repeatability, peak width, peak capacity) as well as at the mass spectrometrylevel (number of detected features, number of identified metabolites).

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-288-TU
Identification and Characterization of Wheat High-Molecular-WeightGlutenin Subunits by Dynamic Capillary Isoelectric Focusing
Boleslaw P. SalmanowiczInstitute of Plant Genetics, Polish Academy of Sciences,
Strzeszynska Str. 34, PL 60-479 Poznan, Poland
A capillary isoelectric focusing (cIEF) method for the evolution of charge heterogeneity ofwheat high-molecular-weight glutenin subunits (HMW-GS) was developed using dynami-cally coated fused silica capillaries. The wheat HMW-GS are major class of wheat storageproteins and primarily responsible for the strength and elasticity of four dough. TheHMW-GS are determined by genes at Glu-A1 loci, with multiple alleles present in thisspecies. Simultaneous focusing and mobilization were successfully carried out using themixture of hydrophilic polymers [in particular polyethylene oxide (PEO) and polyvinylpyr-rolidone (PVP)], which were added during sample preparation. Resolution between partic-ular HMW glutenin isoforms was increased by incorporating urea and neutral detergentsinto the carrier ampholyte solution. In order to optimize major analytical parameters foreffective mobilization, experimental responses from five pI markers were selected. Per-forming rinse capillary with high 2M concentration of hydrochloric acid for five minutesafter each run, followed by water for 15 minutes improved the reproducibility of separa-tion parameters. In addition, it should be noticed that non-linear pH gradient within theperformed ranges, from pI 10.4 to 3.8, was observed, and the pH gradient caused theinconsistency of estimated pI ranges between CIEF and gel IEF. The optimization of focus-ing time, and ampholyte and anodic stabilizer concentrations were critical for the devel-opment of method with high reproducibility in the acid pH range (pI 3.8 – 6.0). Theoptimized method gave a good repeatability and intermediate precision in estimated pIvalues and peak areas of charge isoforms with relative standard deviations of not morethan 2,8% and 4,4%, respectively. Obtained data indicate that cIEF and CZE are comple-mentary systems for separation of wheat HMW glutenin subunits.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-289-MO
Improving Quality to Improve Throughputin Quantitative Neurotransmitter Analysis
David P. Budac, Xu Zhang, Mark J. HaywardLundbeck Research USA,
215 College Rd, Paramus, NJ 07652, USA
Biomarkers are endogenous compounds whose quantities are indicative of disease /symptom / treatment state and/or change in that state. We are specifically interested inanalysis of neurotransmitter biomarkers associated with central nervous system disorderssuch as monoamines, amino acids, acetylcholine and neuropeptides. Many of theseneurotransmitters are present at low pg/mL concentrations in biological samples andtherefore the greatest attention to detail in all aspects of the sample collection and in theanalytical techniques utilized must be practiced in order to successfully analyse them. Thebiggest challenges are the small sample volumes available and sensitivity.
Monoamine analyses were performed on a Waters Alliance 2795 attached to an AntecLeyden electrochemical detector. Amino acid analyses were performed on a WatersAcquity equipped with a fluorescence detector utilizing OPA derivatization. Acetylcholinewas analysed on a Waters Acquity equipped with a Waters Quattro Premier XE (triplequad mass spectrometer). Neuropeptides were analysed using an Eksigent 2D nano-LCattached to a Waters Quattro Premier XE. In vivo microdialysis samples were supplied byour neuroscience group.
Miniaturization of the components of the Alliance autosampler was performed toincrease the sensitivity, robustness and throughput of the monoamine analyses (norepi-nephrine, dopamine and serotonin). The sample loop was reduced down to either 2 or 5μL which along with other modifications facilitated lower sample consumption.
High throughput analysis of glutamate has been achieved using the Acquity UPLCsetup. Sample usage is minimal (< 5 μL) and therefore this analysis can be readily per-formed along side the monoamine analysis. Similarly, a high throughput analysis of ace-tylcholine has been implemented requiring < 5 μL of sample.
Neuropeptides (met and leu-enkephaline & angiotensin II) are presently under exami-nation utilizing a nano-LC setup attached to a triple quad mass spectrometer. To date wehave successfully measured low pg/mL (< 10 pg/mL) levels of peptides in plasma and CSFsamples. Work is underway to observe the indicated neuropeptides in microdialysis samples.
In parallel to the methods described above efforts are underway to further improve themonoamine analysis by utilizing gradient LC/MS/MS methodologies. One of the immedi-ately recognizable benefits achieved is the ability to measure multiple analytes from onesample which was previously not possible.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-290-TU
Integration of Protein Digestion Microreactors in a ComprehensiveTwo-Dimensional Liquid Chromatography System
Filippo Bedani, Huiming Yuan, Lihua Zhang, Yukui ZhangKey Laboratory of Separation Science for Analytical Chemistry,
National Chromatographic Research and Analysis Center, Dalian Institute of Chemical Physics,Chinese Academy of Science,
457 Zhongshan Road, Dalian 116023, China
Proteins are an extremely important class of (bio)macromolecules as virtually no processwithin cells occurs without their direct involvement. As a consequence, their analysis is ofparamount relevance in fields, such as proteomics, food chemistry, pharmaceutical andlife-sciences. One of the most common approaches to determine protein structure, as wellas to identify them, consists in (1) digesting them and (2) analyzing the resultingpeptides. This idea is at the heart of the “bottom-up” approach to proteomics. Protocolsinclude proteolysis (digestion) in a solution containing both a proteolytic enzyme, e.g.trypsin, and the protein(s) of interest. When done in solution, the proteolysis must gener-ally be carried out with only a small amount of the soluble enzyme to avoid autodigestionof the exogenous enzyme. Therefore, the process is slow, requiring several hours to com-plete. Some of the problems typical of protein digestion in solution can be overcome withuse of the enzyme immobilized on a solid support. This approach enables a high localizedconcentration of the enzyme and a significant acceleration of the digestion without anyappreciable autodigestion due to the site isolation effect [1].
One of the most appealing aspects of on-line two-dimensional liquid chromatography(2D-LC) systems is the possibility of automating the whole analysis. When this is done,operator involvement is greatly reduced and the process is speeded-up. Typically, proteindigestion occurs as a preliminary step followed by peptide analysis. As an alternative, thewhole protein analysis – including digestion – can be automated. For example, Yuan et al.[2] have recently developed a 2D-LC system for high-throughput analysis of intact proteins,in which on-line tryptic digestion and peptide analysis are combined. In this work, wepropose to implement 2D-LC systems which integrate digestion microreactors immobilizingdifferent enzymes. The microreactors are set in parallel after the first dimension proteinseparation. The advantage of this strategy is that separated proteins are subjected to differ-ent proteolytic digestion, hence improving sequence coverage. A second separation con-ducted on the peptides results in valuable information which can be used to more reliablyidentify protein sequences. The proposed integrated platform will provide a promising toolfor high-throughput, high accuracy and high sequence coverage protein analysis.
1. J. Krenkova, N. A. Lacher, F. Svec, Anal. Chem. 81 (2009) 2004.2. H. Yuan, L. Zhang, C. Hou, G. Zhu, D. Tao, Z. Liang, Y. Zhang, Anal. Chem. 81 (2009) 8708.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-292-TU
Metabolomic Approach with LC-QTOF-MS to Investigate PlasmaSamples from Patients with Abdominal Aortic Aneurysm
Joanna Teul1,2, Francisco Javier Rupérez2, Michal Ciborowski2,3,Jose Luis Martin-Ventura4,5, Jesus Egido4,5), Coral Barbas2
1Department of Pharmaceutical Analysis, Medical University in Bialystok,Kilinskiego 1, 15-089 Bialystok, Poland
2CEMBIO (Center for Metabolomics and Bioanlysis), Pharmacy Faculty, San Pablo-CEU University,Campus Monteprincipe, Boadilla del Monte 28668, Madrid, Spain3Department of Physical Chemistry, Medical University in Bialystok,
Kilinskiego 1, 15-089 Bialystok, Poland4Vascular Research Laboratory, Fundación Jiménez Díaz,
Madrid, Spain5Autónoma University,
Madrid, Spain
Metabolomics is a global non-target analysis of all the changes in metabolite concentra-tions in biological samples and recently has become very popular in clinical research fornovel biomarker discovery.
Abdominal aortic aneurysm (AAA) is an multifactoral inflammatory disorder character-ized by loss of elastin and fibrillar collagen, leading to aortic dilatation and rupture. It isa common cause of mortality in older people in developed countries. The disease isusually asymptomatic and mainly detected by unrelated physical examination Althoughits development is associated with many factors the origin of the pathology still remainto be defined. The risk of aorta rupture depends on the size of an aneurysm diameter. Itincreases dramatically when AAA > 5 cm. Therefore in this study plasma from twogroups of patients with AAA: small aneurysm (AAA < 5 cm) and big aneurysm (AAA > 5cm) was analysed and compared to healthy subjects, without significant differences inage and sex.
Plasma profiles were obtained with liquid chromatography coupled to accurate massquadruple time-of-flight mass spectrometry detector (LC-QTOF-MS). The resulting datawas evaluated in tree steps. Firstly chemometric tools were applied to reduce multivariatedata matrices, discard background noise and create molecular features. Then combinationof several well-established statistical methods was applied for processing data set in orderto detect differences in metabolic profiles in combination with a large biological variation.Finally accurate masses of features representing significant differences were searchedagainst the METLIN, KEGG, LIPIDMAPS, and HMDB databases. Significant changes wereobserved in concentrations of lysophosphatidylcholines, lysophosphatidylethanolamines,carnitines, sphingolipds, fatty acids amides and some others metabolites.
The research proved that metabolic fingerprinting by LC-QTOF appears as notablypowerful in obtaining detailed information of metabolic changes in pathological state andenables to discover potential markers of AAA.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-294-TU
Monolithic Silica Based Columns for Fast Peptide Separationsin Proteomics
Magnus Røgeberg1, Steven Ray Wilson1, Helle Malerød1, Elsa Lundanes1,Nobuo Tanaka2, Tyge Greibrokk1
1Department of Chemistry, University of Oslo,Post Box 1033, Blindern N-0315 Oslo, Norway
2Kyoto Institute of Technology,Kyoto, Japan
The greatest challenges in proteomics are the sample complexity and the large dynamicrange. Mass spectrometry (MS) is the most used method for detection in proteomics, butcan only tolerate a certain amount of sample, and the dynamic range is limited in a singlespectrum [1]. Hence peptide overlap should be minimized, by separation, for identifica-tion and quantification of both the low and high abundant peptides.
In the ongoing quest for better peptide separations, monolithic columns appear to bepromising. Monolithic columns, consisting of a single piece of separation medium, arecharacterized by a high permeability (low backpressure) and low mass transfer resistance.
In the present study, we have examined the potential of silica based monolithic col-umns for fast separation of peptides. High flow rates, short gradient times and elevatedtemperatures were explored. An optimized method based on maximizing peak capacitywas used to show the potential of this separation technology for analysis of tryptic digest-ed gel-spots. The optimized method has a great potential as the second dimension in a 2dimensional liquid chromatography (2D-LC) system.
1. J.M. Jacobs, J.N. Adkins, W.-J. Qian, T. Liu, Y. Shen, D.G. Camp, II, R.D. Smith, J. Proteome Res. 4 (2005)1073.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-295-MO
N-Glycosylation of Antibodies Characterized by LC-MALDIin an Integrated Software Approach
Zoltan Czentnar, Ulrike Schweiger-Hufnagel, Arndt Asperger,Anja Resemann, Detlev Suckau
Bruker Daltonik GmbH,Fahrenheitstraße 4, 28359 Bremen, Germany
Antibodies represent one of the most important classes of glycoproteins playing a centralrole in the immune response of living organisms. Furthermore, there is a growing interestin recombinant antibodies as potential biotherapeutic agents. The analysis of theN-glycosylation pattern present on antibodies is challenging due to its heterogeneousstructure. The glycan profile is highly dependent on the process by which a recombinantglycoprotein is generated, such as host organism and growth conditions. Changes to theglycosylation pattern can significantly alter biological function.
To characterize the N-glycosylation pattern of a recombinant antibody, a bottom-upapproach was chosen. Tryptically digested antibody samples were separated by nano LCand analyzed by MALDI mass spectrometry. An integrated software approach allowed adetailed characterization of the glycosylation pattern and visualization of the relevantmass spectra.
LC-MALDI-TOF/TOF analysis of the digested antibody provided, in addition to a nearlycomplete sequence coverage of the non-glycosylated peptides, a detail-rich picture of thehighly complex pattern of N-linked glycans in form of the respective N-glycopeptides.Targeted analysis of potential glycopeptide compounds significantly increased the infor-mation content. Our integrated glycoanalytics software allowed identification of glycanmodifications and interactive result validation. In this way it facilitates the initial character-ization of the antibody as well as the subsequent quality control tasks.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-296-TU
Non-Invasive Assessment of Embryo Viability in AssistedReproduction using Capillary Electrophoresis
Ales Madr1, Katerina Foltova1, Jana Zakova2, Eva Lousova2,Igor Crha2, Zdenek Glatz1
1Department of Biochemistry, Faculty of Science, Masaryk University,Kamenice 5, 625 00, Brno, Czech Republic
2Centre of Assisted Reproduction, Faculty Hospital Brno,Obilni trh 11, 602 00, Brno, Czech Republic
Multiple gestations are the one of the main problems associated with human assisted con-ception which originate from higher number of embryos implanted to increase the chanceof successful implantation. Multiple births are a public health concern, as the higher rate ofpreterm delivery in these neonates compromises their survival chances. Therefore there areattempts to select embryos with the highest developmental potential thus reduce number ofembryos implanted, pregnancy risks and increase survival chances of infants. Nowadays is ina common praxis to assess developmental potential of embryos using morphological evalua-tions and cleavage rates. Nevertheless more than four decades ago there were graduallyrevealed correlations between glucose, lactate, pyruvate and amino acids uptake and em-bryo viability. There were published many articles which offering different analytical tools tomeasure carboxylic and amino acids concentration thus provide addition information forselection of the most suitable embryos for implantation, namely LC and GC coupled withMS or MS/MS, 1H-NMR, Raman and near-infrared spectroscopy.
Since carboxylic and amino acids are highly polar molecules they can be analyzed bycapillary electrophoresis (CE). Mentioned molecules lack of chromophores, thus opticaldetection is applicable only after suitable derivatization. This time-consuming step can beoverrun by use of capacitively coupled contactless conductivity detection (C4D). Separationof amino acids by CE-C4D was demonstrated many times by Tuma’s research group ondifferent biological samples [1–3]. The goal of our study is to establish simple, reliable andfast analysis of carboxylic and amino acids based on CE-C4D. Our progress in an analysis ofthe of-the-shelf embryo culture medium G1TM (Vitrolife, Göteborg, Sweden) is presented.Using capillary with 50 μm internal diameter and total length 33.0 cm (18.3 cm to detec-tor), separation voltage +20 kV and cassette temperature controlled to constant 25 °C, theanalysis time take no longer than 6 minutes. The worst relative standard deviation in migra-tion times and peak areas of all relevant analytes are 1.6% and 4%, respectively.
1. Tuma, P. et al., Electrophoresis 2009, 30, 3436–3441.2. Samcova, E., Tuma, P., Electroanalysis 2006, 18, 152–157.3. Tuma, P., Samcova, E., Andelova, K., J. Chromatogr. B 2006, 839, 12–18.
This work was supported by the Research Project of the Grant Agency of Czech Republic No. P206/11/0009and by Research Centre LC06023 and Research Project MSM 0021622413 granted by Ministry of Education,Youth and Sports of the Czech Republic.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-297-MO
Novel Aqueous Two-Phase Systems for Extraction of Biomolecules
Morteza G. Khaledi, Samuel I. Jenkins, Christopher Collins, Mahboubeh Nejati,Nathaniel Weisner, Shuang Liang
North Carolina State University, Department of Chemistry,Raleigh, NC 27695-8204, USA
Purification, separation and analysis of protein samples pose a great challenge due to theenormous complexity and diversity of molecular structure that incorporate a wide range ofsize, charge, hydrophobicity, conformation, and modification of the genetically codedcomposition. In addition, other issues such as abundance, interaction or binding to othermolecules like lipids, and self aggregation are important considerations in development ofeffective strategies in separation and analysis. Furthermore, their delicate tertiary and quater-nary structures could be easily disturbed by various parameters such as pH, ionic strength,temperature, etc. that might limit selection of optimum conditions for separation.
Aqueous Two Phase Systems (ATPS) are among the most widely used for the extractionof biomolecules, especially proteins. The popular ATPS for proteins extractions are com-posed of neutrally charged polymers such as dextran-PEG or nonionic surfactants abovetheir cloud points (Cloud Point Systems). These ATPS offer mild and non-denaturingconditions for proteins, however, offer limited selectivity for extraction.
We will present a unique and nearly universal methodology for creating phase separa-tion in the aqueous solutions of a wide range of charged amphiphillic molecules such assurfactants and polyelectrolytes. These ATPS offer unique advantages over the existingmethods in enhancing selectivity for the extraction of biomolecules such as proteins,peptides, DNA, as well small molecules in pharmaceutical and clinical applications. This isdue to the existence of sites for electrostatic and hydrogen bonding interactions withbiomolecules. These novel ATPS are stable over a wide range of temperatures and longtime periods. In general, ATPS offer a number of advantages over other liquid-liquid andsolid phase extractions such as high water content in both phases that would make themsuitable for biological molecules, low interfacial tension that would facilitate mass transfer,feasibility for scaling up, high capacity, low cost and low toxicity. In this presentation,special attention will be paid to extraction of soluble as well as hydrophobic and mem-brane proteins and peptides.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-298-TU
On the Specificity of Selected Reaction Monitoring Coupledwith Chromatographic Separations for Proteomic Studies
Konstantinos Petritis, Ashoka Polpitiya, Jian LiuCenter for Proteomics, The Translational Genomics Research Institute (TGen),
445 N. Fifth St., Phoenix, AZ 85004, USA
Selected Reaction Monitoring (SRM) assays have been widely adopted for the absolutequantitation of bio-molecules. In the analysis of complex proteomic samples, several SRMtransitions are normally required to provide specificities high enough to exclude all pep-tides with redundant transitions. Monitoring of multiple transitions, however, not onlyincreases SRM cycle time but requires productions of multiple characteristic product ions,which might not always be available or abundant enough. With a limited number oftransitions, the required specificity can also be met with the addition of extra specificitiesprovided from other peptide properties such as their LC retention times (RT). The value ofpeptide RT on improving specificities associated with SRM experiments was simulatedhere in the analysis of complex proteomes.
The present work extends the work by Sherman et al (MCP,8,(2009),2051) who simu-lated the SRM specificity, albeit without using the chromatographic separation (which isa common practice in proteomics) as an additional filter. In this work, we simulate theincrease in specificity in SRM measurements when reversed phase (RP) and/or strongcation exchange (SCX) chromatography is used to separate those peptides. Peptideretention times for reversed phase chromatography were predicted by the algorithms ofPetritis et al. (Anal.Chem.78,(2006),5026). A novel artificial neural network algorithm forthe peptide retention time prediction in strong cation exchange is described in this paperand used for the specificity simulations. Peptides from each database are encoded in 1803vectors and their uniqueness is calculated by m/z (Q1), in-silico calculated fragmentation(Q3), RP and SCX. Peptides were fragmented in silico into b/y ions with m/z between300–2000 and a charge state of +1 or +2 or +3. Uniqueness tolerances for Q1, Q3, RPand SCX were ±1 Th, ±0.8 Th, ±20 sec and ±60 sec, respectively. The chromatographicseparation greatly increased the uniqueness of identifications even in the cases of largeproteomes. Although SCX offered lower peak capacities (i.e. higher RT tolerances) itgreatly improved the SRM uniqueness as unlike the RP, there is no correlation betweenthe peptide mass and it’s retention time in SCX.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-299-MO
Optimizing Particle Size and Column Length, what is the Best Wayto Utilize Nano UHPLC in Proteomics?
W. Decrop1, E. J. Sneekes1, Thomas Köcher2, K. Dekker1,B. de Haan1, Karl Mechtler2, R. Swart1
1Dionex CorporationAmsterdam, The Netherlands
2Research Institute of Molecular Pathology (IMP),Vienna, Austria
The development of UHPLC capabilities on nano LC scale has opened up new ways toprovide proteomics researchers with the peak capacity they need for their work. UHPLCprinciples involve the use of smaller particles allowing faster analysis and/or better resolu-tion; it is usually focused on throughput in small molecule analysis. However inproteomics analysis speed is of lesser concern compared to peak capacity. The increasedpressuresin UHPLC can be used to support increased column lengths as well. With 25 andeven 50 cm columns being implemented in routine applications, the 1 meter barrier hasbecome visible. Here we utilize the full potential of nano UHPLC to determine the effectsof smaller particles and column length on protein identification.
In this study we useddifferent nano columns at the highest possible pressure to sepa-rate a complex tryptic digest. The particle size and the length of the column were variedto create columns that operate at upper end of the system pressure limits. All columnswere operated at typical nano flowrates of 200–300 nl/min and directly interfaced (ESI)with a mass spectrometer. The gradient length was varied to determine the optimalgradient for each column length. Peak capacity, sequence coverage and protein identifi-cations were used to determine the efficiency of the separation.We identified severalthousand proteins in the trypticdigest of 1 μg of HeLa lysate using different columns.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-300-TU
Phosphoglycolipid Profiling of Bacterial Endotoxins
Ágnes Dörnyei1, Anikó Kilár2, Viktor Sándor2, Béla Kocsis3, Ferenc Kilár1,2
1Institute of Bioanalysis, Faculty of Medicine, University of Pécs,Szigeti út 12., H-7624 Pécs, Hungary
2Department of Analytical and Environmental Chemistry, Faculty of Sciences, University of Pécs,Ifjúság útja 6., H-7624 Pécs, Hungary
3Department of Medical Microbiology and Immunology, Faculty of Medicine, University of Pécs,Szigeti út 12., H-7624 Pécs, Hungary
Most Gram-negative bacteria have lipopolysaccharide (LPS) on their outer membrane. Ingeneral a LPS consists of a lipid anchor (lipid A) and two variable oligosaccharide regions(the core and the O-antigen), meanwhile it is the lipid A component that is responsiblefor the endotoxic activity. At low levels, endotoxin-mediated activation of Toll-like recep-tor 4 is beneficial, because downstream signaling pathways mobilize defense mechanismsagainst the invading bacteria. Severe infections can however cause excessive endotoxinexposure, which can lead to shock, multiple organ failure and death.
The endotoxic lipid A is an amphiphilic molecule belonging to the group of phospho-glycolipids. Generally, it is composed of a β-D-GlcN-(1’-6)-α-D-GlcN disaccharide bis-phosphorylated at C1 and C4’ positions. This backbone is acylated with β-hydroxylatedfatty acids (primary fatty acids), linked as ester at C3 and C3’ positions and as amides atC2 and C2’ positions. Further nonhydroxylated or, less frequently, β-hydroxylated fattyacids (secondary fatty acids) can be connected in ester linkage at C3 position of primaryfatty acids. Lipid A diversity is observed both in the nature, number, and length of fatty-acid side chains and also in the phosphorylation configuration of the disaccharide back-bone. The acylation and the phosphorylation state strongly influence the secondarystructure of the lipid A and consequently, its toxicity properties.
The structural analysis of endotoxin extracted from bacteria is a complex task, since theLPS or the lipid A, as well, usually possesses a considerable proportion of partial or incom-plete structures as a result of incomplete biosynthesis. A comprehensive study was contin-ued to develop phosphoglycolipid profiling techniques determining the acylation andphosphorylation pattern of lipid A extracted from several bacterial strains. Mass spectrom-etry itself, and hyphenated with online separation techniques (e.g. HPLC, CE) are usefultools in the structural elucidation of intact LPSs or the lipid A moiety. With tandem massspectrometry both the degree of acylation and the positions of the fatty acids on thedisaccharide backbone could be determined.
The work was supported by the grant OTKA-NKTH-NI-68863 and Á.D. acknowledges the support of the JánosBolyai Research Scholarship (Hungarian Academy of Sciences).

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-301-MO
Evaluating Accelerating Methods for Trypsin Digestionin LC-MS Based Proteomics
Helle Malerod1, Hanne Kolsrud Hustoft1, Steven Ray Wilson1, Jan Leo Reubsaet2,Elsa Lundanes1, Tyge Greibrokk1
1Department of Chemistry, University of Oslo,P.O. Box 1033 Blindern, 0315 Oslo, Norway
2School of Pharmacy, University of Oslo,Norway
There is a jungle of different sample preparation methods for LC-MS/MS based pro-teomics and most steps are carried out differently from lab to lab. The main step in thesample preparation procedure includes enzymatic digestion of proteins to peptides.Protein digestion can be very time consuming and different methods have been suggest-ed in order to accelerate the digestion time, such as heating, microspin columns, ultra-sonic energy, infrared energy (IR), microwave energy and microreactors where the en-zyme is immobilized on a solid support. However, do these methods provide a moreefficient digestion than the conventional method at 37 °C, and will these potential accel-erating methods digest all proteins in a shot-gun analysis? To answer these questions, thedigestion efficiency of accelerating digestion methods has been evaluated. An optimalsample preparation in LC-MS based proteomics using trypsin for protein digestion issuggested.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-302-TU
Selectivity of Mixed-Mode Chromatography for Structural Isomersof Phosphorylated Carbohydrate Metabolites
H. Hinterwirth, A. Gargano, M. Lämmerhofer, W. LindnerUniversity of Vienna,
Währingerstrasse 38, 1090 Vienna, Austria
Carbohydrates and sugar phosphates are central compounds in metabolism (such asglycolysis, glycogenolysis pathways as well as pentosephospate cycle) and in signal path-ways. Despite the prime interest in metabolomics research, their analysis is still a chal-lenge due to their hydrophilicity (not suitable for common RP methods), low sensitivity(poor UV absorbance, non-volatility and lack of specific fragmentations in MS/MS) andtheir distribution among structural isomers.
Recently, an HPLC-MS/MS based method platform for the quantitative metabolicprofiling of about 250 intra- and extra-cellular metabolites in fermentation broths wasdeveloped in our working group based on two pre-validated HILIC and two RP-LC assays[1, 2]. While one of the HILIC assays (zwitterionic ZIC-HILIC as stationary phase, pH 7.5)enabled to measure some phosphorylated carbohydrates as the sum of all isomers, dis-tinction between individual isomers was not possible.
The objective of our study [3] was the development of a new separation method for thedetermination of phosphorylated carbohydrates such as hexose and pentose phosphates, 2-and 3-phosphoglycerate, dihydroxyacetone phosphate and glyceraldehyde 3-phosphate, aswell as glucosamine 1- and 6-phosphate utilizing mixed-mode chromatography with re-versed-phase/weak anion-exchangers (RP/WAX) [4] and charged aerosol detector (CAD).The influence on the separation performance of pH, organic modifier (WAX vs. HILIC/WAX),counterions and temperature was studied. Particular attention was also paid on the separa-tion of anomers and on the on-column interconversion, so called mutarotation.
Employed acidic conditions have led to the complete separation of α- and β-anomers ofglucose 6-phosphate at low temperature. The anomers coeluted in a single peak at elevatedtemperatures (> 40 °C) (peak coalescence), while at intermediate temperatures on-columninterconversion with a plateau in-between resolved anomer peaks was observed with appar-ent reaction rate constants between 0.1 and 27.8×10−4 s−1. Dynamic HPLC under specifiedconditions enabled to investigate mutarotation of phosphorylated carbohydrates, theirinterconversion kinetics, and energy barriers for interconversion.
A complex mixture of six hexose phosphate structural isomers could be resolved almostcompletely.
1. B. Preinerstorfer, S. Schiesel, M. Lämmerhofer, W. Lindner, J. Chromatogr. A, 2010, 1217, 312–328.2. S. Schiesel, M. Lämmerhofer, W. Lindner, Anal. Bioanal. Chem., 2010, 396, 1655–1679.3. H. Hinterwirth, M. Lämmerhofer, B. Preinerstorfer, A. Gargano, R. Reischl, W. Bicker, O. Trapp, L. Brecker,
W. Lindner, J.Sep.Sci, 2010, 33, 3273–32824. M. Lämmerhofer, M. Richter, J. Wu, R. Nogueira, W. Bicker, W. Lindner J. Sep. Sci., 2008, 31, 2572–2588.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-303-MO
Study on a Rapid and Highly Selective Colorimetric Methodfor the Detection of Tryptophan in a Mixture of Amino Acids
using Liquid Chromatography-Mass Spectrometry
Yanyan Huang, Guoquan Liu, Rui ZhaoCAS Key Laboratory of Analytical Chemistry for Living Biosystems, Institute of Chemistry,
Chinese Academy of Sciences, Beijing, 100190, China
Tryptophan, an essential amino acid, is well known as the building block for proteinbiosynthesis and important in determining protein activity, hydrophobicity, and diversity.Highly specific detection of tryptophan is urgently needed for studying its diverse biologi-cal functions and applications.
A rapid and highly selective method has been developed for the colorimetric detectionof tryptophan in a mixture of amino acids without any pretreatment or separation proce-dures (Figure 1). Simply by adding different amount of DMSO into the reaction mixture,the color generating rate can be either accelerated or inhibited. The dual function ofDMSO on the color reaction of tryptophan was extensively studied by LC-MS as well asphoto-diode array detection. Two new chromatographic peaks having the same m/z wasseparated and characterized as tautomer, which was closely related with the content ofDMSO (Figure 2). The mechanism study demonstrates that DMSO acts both as a rate-enhancing solvent and an oxidizing reagent. By controlling the amount, the catalytic
Figure 1.
Photograph of reacting solutions containing different
compounds. A) Reagent blank; B) tryptophan without
DMSO; C) tryptophan with 0.2% DMSO; D) a mixture of
amino acids in the absence of tryptophan; D) amino acid
mixture in the presence of tryptophan.
Figure 2:
RP-HPLC separation of reacting solutions with
different amount of DMSO. Peak 1 and peak 2
are the oxidized products of tryptophan. Peak 3
is the precursor of the colored compound.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
effect of DMSO can take the leading role and greatly enhancing the efficiency and thesensitivity of the color reaction.
1. Z. Bao, S. Sun, J. Li, X. Chen, S. Dong, H. Ma. Angew. Chem. Int. Ed. 2006, 45, 6723–6725.
2. A. J. Parker. Chem. Rev. 1969, 69, 1–32.
Financial supports from NSFC and CAS are appreciated.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-304-TU
The Hormonome of Developing Wheat and Bean Seedsas Revealed by LC/MS
Petre I. Dobrev1, Marie Trcková2, Miroslav Kamínek1
1Institute of Experimental Botany AS CR v.v.i.,Rozvojová 263, 165 02, Prague, Czech Republic
2Crop Research Institute,Drnovská 507, 161 00, Prague, Czech Republic
Plant hormones play crucial role in control of growth and development of plants. Amongplant organs the developing seeds are the richest in some types of plant hormones. Hor-mone concentrations during seed development show distinct and reproducible patterning,which correlates well with important developmental stages like cell multiplication andenlargement, and seed filling and desiccation. Whereas the research on plant hormones wasmainly concentrated on their biologically active forms, the hormone metabolites weremostly ignored. Accumulating knowledge about the potential roles of predecessors andmetabolites of plant hormones in control of hormonal homeostasis has increased demandson analysis the whole spectrum of compounds involved in hormone biosynthesis and me-tabolism. This is in concert with the recently increasing interest on more “holistic” view ofthe complex interactions in the biological systems leading to emerging of the genomics,transcriptomics, proteomics and metabolomics. Understanding of metabolome of planthormones (hormonome) could provide powerful tool for control of plant development. Weanalyzed the hormonome of wheat and bean seeds during their development. We utilizedbroad, nonselective extraction and purification procedure, which separated the extractedhormones into two fractions, 1) fraction A – containing the hormones of acidic and neutralcharacter, and 2) fraction B – containing the hormones of basic character. Hormonome wasquantified using HPLC coupled to tandem mass spectrometer set at selected reaction moni-toring mode. The following hormones and their metabolites were analyzed: cytokinins,auxins, abscissic acid, jasmonate, gibberellins and salicylic acid. During the initial stages ofseed development associated with active cell divisions and increase of seed weight therewere peaks of some hormones like cytokinins and salicylic acid. Later, during seed matura-tion, the levels of auxin, jasmonate and some gibberellins were increased. At the last stage,during desiccation of seeds, overall decrease of hormones was observed. The dynamics ofthe metabolites of hormones behaved differently, with some metabolites preceding themain peak of active hormone, whereas others increased afterwards. Transient accumulationof some inactive metabolites, as e.g. of auxin, indicates their functioning in suppression ofendogenous levels of active hormone at certain stages of seed development.
This research was supported by grants from the Ministry of Education, Youth and Sports CR (1M06030) andthe Grant Agency of the Academy of Sciences CR (IAA600380805).

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-305-MO
The New Panacea in Metabolomics, Proteomics and Genomics– Electrochemistry / MS
Jean-Pierre Chervet1, Agnieszka Kraj1, Martin Eysberg1; Herbert Oberacher2
1Antec,Industrieweg 12, 2382NV Zoeterwoude, The Netherlands2Institute of Legal Medicine, Innsbruck Medical University,
Innsbruck, Austria
Recently, the scope of Electrochemistry (EC) upfront MS has been extended from mimick-ing drug metabolism towards new applications such as: protein/peptide cleavage, disul-fide bonds reduction in proteins/peptides, covalent DNA adduct formation, covalentdrug-protein binding, etc. In this poster we will show the application of on-line EC/MS asa powerful tool to simulate various oxidation and reduction processes in life sciences.
Electrochemistry up front MS can be applied for protein and peptide cleavage (as apromising new approach to enzymatic digestion). Electrochemical cleavage of proteinsand peptides occurs very specifically at C-terminal of the Tyrosine and Tryptophan pep-tide bonds. Examples of oxidative cleavage will be presented.
Disulfide bonds are one of the most important post-translational modifications forproteins. In this poster we present the structural analysis of biologically active peptidesand proteins containing disulfide bonds (e.g., GSSG, insulin, etc) using electrochemistry(EC) combined with mass spectrometry. Therefore the sample undergoes electrolyticdisulfide cleavage in the electrochemical flow cell followed by online MS analysis.
Furthermore, quick electrochemical activation of electrode and new scanning methodfor efficient metabolite synthesis will be presented. New scanning method was applied foroxidation of the highly concentrated samples (mM range) to achieve high yield in themetabolites formation. Stable oxidation conditions were obtained without the need ofany cell maintenance for a prolonged period of time.
EC was used to initiate adduct formation with DNA. The obtained reaction productswere separated by LC and detected by MS. Tandem MS experiments were used for struc-tural confirmation. In a proof of principle study acetaminophen was selected as modelcompound. Covalent adduct formation was observed for electrochemical activated mix-tures of acetaminophen and guanosine.
All these applications illustrate the tremendous power and broad applicability of elec-trochemistry as a tool to mimic nature’s Redox reactions within a few seconds or minutes.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-306-TU
Top-Down LC-MALDI Identification of Protein in Mixturesof Moderate Complexity and N- and C-Terminal Assignments
Sven Meyer1, Andrea Kiehne1, Anja Resemann1, Shannon Cornett2,Eckhardt Belau1, Detlev Suckau1
1Bruker Daltonik GmbH,Bremen, Germany
2Bruker Daltonics Inc.,Fairview, TN, USA
Mass spectrometric top-down sequence analysis of undigested proteins by MALDI in-source decay (MALDI-TDS) can provide identification as well as terminal sequence analysisin one step. MALDI-TDS is therefore very useful for side product characterization of re-combinant proteins, which typically involve protein truncation variants. However, MALDI-TDS requires pure proteins for analysis, as the in-source decay process does not permit forprecursor ion selection. Therefore, protein separation is required in order to increase thesample compatibility with MALDI-TDS. We developed a method that provides proteinidentification from moderately complex protein mixtures.
His-tagged α-casein, β-casein (technical grade) and carbonic anhydrase were obtainedfrom commercial sources. These proteins were dissolved at 100 pmol/μL in 0.1% TFA andseparated on a 500 μm PepSwift column (Dionex) using a 30 min acetonitrile gradient.Eluting fractions were collected in 20 sec fractions and directly spotted onto a MALDItarget. Spotted fractions were then covered either by sDHB or 1,5-DAN matrix and ana-lyzed using a MALDI-TOF/TOF instrument either in linear MS mode or by ISD, in bothpositive and negative ion mode. Proteins were identified from MALDI-TDS spectra withstandard Mascot searches (Matrix Science) and sequences were assigned to the MALDI-TDS spectra for manual validation.
Despite the proteins being purchased from commercial sources, all investigated pro-teins were protein mixtures, mimicking the typical situation of pseudo-pure proteinsthat are to be analyzed in mass spectrometry laboratories that cause various difficultiesin their characterization. Proteins were successfully separated with total column loadingsbetween 50 and 500 pmoles. More than 10 proteins were detected in each analysis byMS; individual proteins were identified by their N- and C-terminal sequences derivedfrom MALDI-TDS. In a bovine carbonic anhydrase lot, proteins in the mass range of 8 to29 kDa were detected. Aside from carbonic anhydrase, superoxide dismutase, andubiquitin (all bovine) were identified by MALDI-TDS as high abundant contaminants.Two low abundant proteins with 18 and 21 kDa respectively could also be analyzed.Technical grade beta casein separated into 3 main components; identified as α-S1-, α-S2- and β-casein. Successive phosphorylation of N-terminal serine residues increasingly

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
impaired the formation of positive mode c-ions but promoted the formation of negativemode c-ions.
The developed method can be used for the characterization of side products in bio-therapeutics development and quality control. Due to the high loading capacity of thecolumns this methodology can also be applied to moderately complex protein mixturessuch as natural protein isolates and with proteomics experiments subsequent to upstreamprotein chromatographic separation with orthogonal methods such as gel-free IEF tech-niques, ion exchange or GPC.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-307-MO
Two Dimensional SEC // RP Capillary LCfor Top-Down Proteomics Analysis
Evert-Jan Sneekes, Macro Karsten, Wim Decrop, Remco SwartDionex Corporation,
Amsterdam, The Nederlands
Top-down proteomic LC-MS aims at investigating protein structure and post-translationalmodifications (PTM) through liquid phase separation and MS analysis of intact proteins.The advantage of top-down approaches over bottom-up approaches is the absence of aproteolytic cleavage step. This keeps all protein information within one molecule and doesnot multiply sample complexity.However, working with intact proteins introduces chal-lenges for the analytical technology used. Proteins are more difficult to separate thanpeptides using standard LC methods and more difficult to study by MS. Recently, differ-ent electron based dissociation techniques, such as ECD and ETD have been successfullyappliedto intact proteins to obtain sequence and PTM information.The successful applica-tion of these techniques in tandem mass spectrometry for protein mixtures requires aseparation step butpowerful liquid chromatographic methods for proteins are scarcelypublished and need to be developed and optimized for direct interfacing with massspectrometry.In this study we have developeda two dimensional capillary scale LC meth-od for the separation of intact proteins. Proteins are separated and fractionated on acapillary size exclusion column. Next, proteins contained in the size fractions are separat-ed on reversed phase capillary monolithic columns prior to UV and MS detection. Themethod has been optimized with respect to SEC fractionation and RP gradient conditionsfor standard proteins and complex mixtures.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-308-TU
UHR-Q-TOF Analysis can Address Common Challengesin Targeted and Untargeted Metabolomics
Zoltan Czentnar1, Aiko Barsch1, G. Zurek1, D. Krug2, R. Müller2
1Bruker Daltonics,Bremen, Germany
2Universität des Saarlandes,Saarbrücken, Germany
Here, we present an ESI-UHR-Q-TOF based analysis of myxobacterial secondary metabo-lites, which permits to solve several challenges frequently encountered in metaboliteprofiling studies. Myxobacteria are promising producers of natural products exhibitingpotent biological activities, and several myxobacterial metabolites are currently underinvestigation as potential leads for novel drugs. However, the myxobacteria are also astriking example for the divergence between the genetic capacity for the production ofsecondary metabolites and the number of compounds that could be characterised todate. Wildtype and mutant strains were analyzed concerning the production patterns ofknown metabolites and with regard to the discovery of new metabolites.
Extracts from Myxococcus xanthus wildtype and mutant strains (4 biological replicates) wereseparated on a UHPLC RP-C18 column (50x2 mm, 1.7 μm particle size). MS analysis wascarried out using an ultra-high resolution TOF mass spectrometer. The data was processedusing a compound finding algorithm prior to statistical interpretation by principal componentanalysis (PCA) and t-test. Additionally, a targeted search for known metabolites was carriedout using the retention time, mass accuracy and isotopic pattern as identification criteria.
Since mass accuracy and resolution of TOF instruments are independent of the acquisitionrate, they are perfectly suited for a coupling to UHPLC separations. These hyphenations enablea reduction of analysis time in combination with a high chromatographic resolution andtherefore permit an increased sample throughput. The UHR-TOF analysis revealed that anacquisition rate of up to 20 Hz did not compromise the achieved mass accuracy or resolution.
Acquisition of full scan accurate mass spectra enable the targeted screening for knowncompounds e.g. from the class of DKxanthenes based on very selective high resolutionEIC (hrEIC) traces with small mass windows of 1.0 – 0.5 mDa.
A comparison of several datasets following a “comprehensive feature extraction” com-bined with a statistical analysis permits an untargeted discovery of novel biomarkers usingthe same data files as for the targeted analysis.
Even a mass accuracy of 0.1 ppm is not sufficient for an unambiguous formula identifi-cation for m/z values above 500. A combination of accurate mass data and isotopic pat-tern information in MS and MS/MS spectra can extend this m/z range for reliable formulasuggestions. Examples for novel metabolites from Myxobacteria will be shown.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-309-MO
Urinary Metabolic Profiling Analysis to DiscoverPotential Diagnostic Biomarkers Based
on LTQ-Orbitrap High Resolution Mass Spectrometry
Lina Zhou, Peiyuan Yin, Xin Lu, Guowang XuCAS Key Laboratory of Separation Science for Analytical Chemistry, National Chromatographic R&A Center,
Dalian Institute of Chemical Physics, Chinese Academy of Sciences,Dalian 116023, China
Urine is a very useful clinical sample and can be collected in a non-invasive form. Informa-tion on specific diseases or physiological variables can be taken from the appearance,smell, and taste of urine. Especially, the urinary metabolites are correlated with manylevels of metabolic activity. Therefore, in metabolomics, study urine will play a very im-portant role. Unfortunately, because of so many polar metabolites included in urine, itscomprehensive analysis is not so easy. In this study, we coupled liquid chromatography toLTQ-Orbitrap MS as a metabolic profiling platform. To get as much metabolite informa-tion as possible in a single run, a Waters HSS T3 column was used to increase the reten-tion of polar compounds.
Hepatocellular carcinoma (HCC) is known as one of the most common malignancy forits high mortality rate. The exploration of early diagnostic biomarker for HCC is veryimportant for a better surgery outcome. To find the potential early metabolic markers,paired urine samples from the same HCC patient at the diagnosis time and two yearsbefore diagnosis (TYBD) were collected. Urine samples from healthy volunteers were usedas the control.
After peak alignment from metabolic profiling, 550 and 1220 ions, respectively in thepositive and negative modes, were imported to SIMCA-P 11.0 for multivariate analysis.Some metabolites were found to be potential early diagnostic biomarkers of HCC.
In conclusion, this coupling HSS T3 LC/ LTQ-Orbitrap system is proved effective formetabolic profiling of urine samples and is especially helpful to identify potential bio-markers.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-310-TU
The Use of Dual cHiPLC Columns to Increase Throughputin Quantitative Peptide MRM Analyses
J. Bryce Young, Nicole Hebert, Erika Lin, Remco van SoestEksigent Technologies,Dublin, CA 94568, USA
The reproducibility of nanoLC analysis is critical to many proteomics workflows, particu-larly those involving analysis of replicate samples. The variability in performance betweentraditional nanoLC columns, makes proteomics methods extremely difficult to standard-ize, while the long gradients and extended equilibration times of these columns canimpose limitations on sample throughput. The cHiPLC nanoflex microfluidic platformdelivers easy to use, dead volume-free connections providing a robust solution withexcellent column-to-column reproducibility. The cHiPLC system offers much flexibility innanoLC approach, providing options for trap-elute and direct on-column workflows whichcan be switched between with relative simplicity due to the dead-volume-free connec-tions. In addition to this, the described two-column switching approach offers significantimprovement in sample throughput, since sample gradients can be performed on a sec-ond column during the equilibration of the first column used in the previous analysis, thusmaximizing the analysis time of the system. High-throughput analysis spanning 5 days ofcontinuous operation is described with maximum analysis time and <1% RSD retentiontime stability across all analyses.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-311-MO
On-Line cHiPLC Based Digestion in NanoLC-MSfor Increased Reproducibility
Remco van Soest, J. Bryce Young, Don Arnold, Nicole HebertEksigent Technologies,Dublin, CA 94568, USA
The reproducibility of sample preparation and nanoLC analysis is critical to manyproteomics workflows, particularly those involving analysis of replicate samples. ThecHiPLC nanoflex microfluidic platform delivers easy to use, dead volume-free connectionsin addition to excellent column-to-column reproducibility.The addition of an immobilizedtrypsin digestion column to the cHiPLC workflow to increase overall proteomics workflowreproducibility is described. Completion of nanoLC analyses immediately after on-chipprotein digestion has several advantages including reduced sample handling and verylittle time lag between digestion and analysis. Both improvements reduce loss of peptidesassociated with adsorption to the sample vial – a problem that is enhanced for low quan-tity peptides when there is a long time between (in solution) digestion and analysis. On-line digestion in the easy to use cHiPLC format shows good potential as a replacement forbatch processed in-solution digestion, especially when processing larger numbers ofsamples.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-312-TU
High Performance at Highest Speeds for Protein Identificationin Complex Matrices
Christie L. Hunter1, Sean L Seymour1, Doug Simmons2, Matthias Glueckmann3,Thomas Knapman3, Henri Snijders3
1AB SCIEX, USA2AB SCIEX, Canada3AB SCIEX, Europe
A major challenge in proteomics research and biomarker discovery by mass spectrometry isthe analysis of complex samples. As sample complexity increases, so does the need forpowerful hardware with high sensitivity and large dynamic range capabilities. Equally impor-tant for a thorough analysis of complex samples is the ability to acquire the maximumnumber of MS/MS spectra without loss of spectral quality during an LC/MS/MS run. Howev-er with most high performance mass spectrometer instruments available today, high speedMS/MS acquisition is performed at the expense of high quality data. This is detrimental ashigher mass accuracy and higher resolution improves the specificity of peptide identifica-tion. The AB SCIEX TripleTOFTM 5600 system is capable of achieving high acquisition speedswhile maintaining high-resolution and high mass accuracy in both MS and MS/MS mode.The system can acquire up to 50 MS/MS spectra in a second using powerful IDA (informa-tion-dependent acquisition) workflows, and its performance was investigated to understandthe advantages for protein identification from complex samples.
Obtaining the most in-depth interrogation of a proteome also depends on the qualityof the LC separation and the power of the database search engine. Here, we use theEksigent nanoLC Ultra system with the cHiPLCTM-nanoflex to obtain the highest qualityseparations with the highest reproducibility. Digging deeper into a proteomic samperequires a database search algorithm that is able to broadly identify many different PTMsand other unexpected peptides that will now be observed. ProteinPilotTM Software with itsunique ParagonTM Algorithm enables the simultaneous identification of hundreds of modi-fications, substitutions, and unexpected cleavages.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-313-MO
Application of Modern Stationary Phases for Determinationof Retinol and αα-Tocopherol in Biological Material by UHPLC
Barbora Kucerová1,2, Lenka Krcmová1,2, Jirí Plíšek1,2, Markéta Kašparová1,2,Dagmar Solichová2, Petr Solich1
1Department of Analytical Chemistry, Faculty of Pharmacy, Charles University,Heyrovského 1203, 500 05 Hradec Králové, Czech Republic
2Department of Metabolic Care and Gerontology, Teaching Hospital,Sokolská 581, 500 05 Hradec Králové, Czech Republic
Retinol and α-Tocopherol are liposoluble vitamins which play a role as antioxidants thathelp to remove free radicals away from the human body and thus they can prevent tu-morous transformation of cells. Retinol also participates in many other functions throughthe body, such as gene expression, vision, embry development and reproduction, bonemetabolism or immune functions. α-Tocopherol is the major liposoluble antioxidant inhuman body that protects lipid parts of cell membrane, blood fats, cholesterol and otherfatty substances against oxidation. Decrease of α-Tocopherol levels causes higher possibili-ty of generation of oxidative stress manifested in all organs.
Retinol and α-Tocopherol were determinated using Ultra High Performance LiquidChromatography (UHPLC) which is a new system of liquid chromatography that uses veryhigh pressure (18 000 psi) for better effectivity of separation process. Utilization of UHPLCprovides faster analysis with lower consumption of mobile phase and amount of samplecompared with HPLC system.
Monolithic and core – shell technology columns (Chromolith, Kinetex, Ascentis) ofdifferent length, particle size and diameter were tested under various conditions such astemperature, injection volume, flow rate and composition of mobile phase. Validationparameters were evaluated with regard to time of analysis and consumption of mobilephase. The best results for simultaneous analysis of retinol and α-Tocopherol wereachieved with column Ascentis Express C18, 150 x 3.0 mm, filled with 2.7 μm fused-coreparticles. Optimal temperature during analysis was 50 °C. The mixture consisted of 70%of methanol and 30% of propanol at the flow rate 1.5 ml/min was used as the mobilephase. The fluorescent detection of retinol and α-Tocopherol was carried out at 325 and295 nm, respectively.
Supported by: MSM0021620822, MZO00179906, MSM0021620820, GAUK124809/2011

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-314-TU
Application of Whelk O-1 and DACH-DNB Selectorson Sub-2 Micron Particles for Enantioselective UHPLC Separations
in the Ultra-Fast Regime
Alessia Ciogli1, Ilaria D’Acquarica1, Francesco Gasparrini1, Carmela Molinaro1,Marco Pierini1, Claudio Villani1, Jelena Kocergin2, Ted Szczerba2, Harald Ritchie3
1Dipartimento di Chimica e Tecnologie del Farmaco, Sapienza Università di Roma,Piazzale Aldo Moro 5, 00185 Rome, Italy
2Regis Technologies, Inc.,8210 Austin Avenue, Morton Grove, IL 60053, USA
3 Thermo Fisher Scientific,93–96 Chadwick Road, Runcorn, UK
In the last decade, the Ultra Performance High Liquid Chromatography (UHPLC) repre-sented the new trend in liquid chromatography providing better solutions in terms ofspeed, resolution and sensitivity respect to the conventional HPLC analysis [1, 2]. TheUHPLC advantages, based on the use of sub-2 micron particle packed columns, could bein principle combined with the capability of chiral selector to realize the enantioselective-UHPLC. In this context, brush-type selectors represent the preferred choice for high speedanalysis owing to their excellent kinetic performances. Recently, in order to shift the HPLCchiral separations in ultra-fast regime, we reported the development of DACH-DNB chiralstationary phase on 1.9 μm silica particles [3].
Herein, we report the application of two examples of brush-type CSPs, based on theDACH-DNB and Whelk O-1 selectors grafted on 1.7 μm silica particles together with areproducible procedures for their synthesis.
The transition of enantioseparations from HPLC to UHPLC was efficiently performedover a broad variety of samples, typically resolved with these selectors: sulfoxides; phos-phine oxides; aryl propanoic acids; benzodiazepines and N-protected amino acids, etc.
Finally, the optimization of separation conditions in e-UHPLC afforded very high speedseparations in sub-minute time-scale. These conditions were exploited for the enantiosepara-tion of both stereo-stable and stereo-labile chiral compounds by dynamic e-UHPLC [4].
1. N. Wu, J. A. Lippert, M. L. Lee, J. Chromatogr. A, 911 (2001) 1–122. P. W. Carr, X. Wang, Anal. Chem., 81 (2009) 5342–53533. G. Cancelliere, A. Ciogli, I. D’Acquarica, F. Gasparrini, J Kocergin, D. Misiti, M. Pierini, H. Ritchie, P.
Simone, C. Villani, J. Chromatogr. A, 1217 (2010) 990–9994. I. D’Acquarica, F. Gasparrini, M. Pierini, C. Villani, G. Zappia J. Sep. Sci., 29 (2006) 1508–1516.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-315-MO
Characterization of System Dispersion and its Impacton Chromatographic Separations
Aparna Chavali, Peyton Beals, Richard W. Andrews,Tanya Jenkins, Patricia McConville
Waters Corporation,34 Maple Street, Milford, MA 01757, USA
Liquid chromatography separations run using sub-2 μm columns provide higher resolu-tion due to reduced on-column dispersion effects. To maximize the performance ofthese columns, extra-column band spread effects must also be effectively controlled.Sources of extra-column band spread can include injection mechanisms, tubing internaldiameters, connections, and flow cell design and volume. The goal was to develop asystematic way to independently measure these sources of extra-column band spreadthat effect resolution. To evaluate the contributions from each of these components, theeffect on the band spread measurement was assessed. The impact of each of thesevariables on the ½ height, 5σ and 6σ peak widths as well as peak tailing factors wasrelated to the chromatographic resolution. Additionally, the impact of injection volume,matched to these measurable attributes, changes the weighting of the effects of theextra-column band spread variables. The relative impact of these variables is different inisocratic and gradient separations. The result is a simplified matrix which allows foreasier identification of the sources of extra-column dispersion when optimizing highresolution chromatographic separations.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-316-TU
Chromatographic Time Scale Transition from Minutes to Secondsin Chiral UHPLC with the Sub-2 Micron Whelk O-1 Stationary Phase
Alessia Ciogli1, Ilaria D’Acquarica1, Francesco Gasparrini1, Carmela Molinaro1,Claudio Villani1, Jelena Kocergin2, Ted Szczerba2, Harald Ritchie3
1Dipartimento di Chimica e Tecnologie del Farmaco, Sapienza Università di Roma,Piazzale Aldo Moro 5, 00185 Rome, Italy
2Regis Technologies, Inc.,8210 Austin Avenue, Morton Grove, IL 60053, USA
3 Thermo Fisher Scientific,93–96 Chadwick Road, Runcorn, UK
Chiral chromatography has nowadays become a preferred method for rapid separation ofenantiomers in the lab-scale compound screening and compounds isolation as well as inbiochemical and pharmaceutical industries. Research continuously promotes the design ofnew chiral selectors capable of separating a wide variety of chiral compounds. In thiscontext, the brush-type chiral stationary phases (CSPs) represent the more efficient sup-ports in the “CSPs world” from the kinetic point of view. Normally, they are employed inthe direct chromatographic resolution of racemates using standard analytical geometrycolumns (L: 250 or 150 mm, particle size: 3–5 μm).
In order to increase the kinetic performances of the separation process without loss ofselectivity, efficiency and resolution, analytical chemists moved up to Ultra-High-Pressure-LC (UHPLC), where shorter columns (50 – 100 mm length) with smaller ID (3.2 – 2.1mm) are packed with particles of smaller sizes (sub-2 μm) [1, 2].
The challenge was to amplify the availability of enantioselective UHPLC supports,already introduced in our previous study regarding the DACH-DNB selector grafted on1.9 μm silica particles [3]. Herein, we report the synthesis of a CSP using the well-knownWhelk O-1 selector grafted onto high surface area silica of 1.7 μm particle size.
Complete investigation of efficiency at wide linear velocity range (Van Deemter plots) andthe determination of permeability of the new columns confirmed the theoretical basis ofUHPLC. Remarkable results, in term of resolution, were obtained with columns of differentgeometries (length range: 30–150 mm and ID: 2.1, 3.2, 4.6 mm) over a wide number ofsamples both in normal phase (NP) and reversed phase (RP) elution. Moreover, the availabili-ty of chiral phases in both enantiomeric versions of selector allowed the application of the“ICCA approach” [4]. The very high efficiency of these systems let us to achieve also veryhigh speed separations in the sub-minute time scale with 50 mm length columns.
1. M. E. Swartz, J. Liq. Chromatogr. & Rel. Technol., 28 (2005) 12532. F. Gritti, G. Guichon, J. Chromatogr. A, 1217 (2010) 76773. G. Cancelliere, A. Ciogli, I. D’Acquarica, F. Gasparrini, J Kocergin, D. Misiti, M. Pierini, H. Ritchie, P.
Simone, C. Villani, J. Chromatogr. A, 1217 (2010) 9904. E. Badaloni, W. Cabri, A. Ciogli, R. Deias, F. Gasparrini, F. Giorgi, A. Vigevani, C. Villani, Anal. Chem., 79
(2007) 6013.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-317-MO
Comparative Study of Different UHPLC Systemsby Kinetic Plot Method
Judit Orgoványi, Erzsébet Oláh, János Horváth, Mónika BabjákGedeon Richter Plc.,
Gyömrôi út 19–21, H-1103 Budapest, Hungary
In pharmaceutical industry there is a continuous need to reduce analysis time and in-crease efficiency of the separation. For this task, ultra-high pressure liquid chromatograph-ic or ultra-high performance liquid chromatographic (UHPLC) systems can be suitable.
In the past decades column technology is developed as well, new packing materialsappeared. Monolithic columns have lower flow resistance; therefore can provide higherflow rate than packed beds [1]. Columns packed with sub-2 μm particles can be appliedat higher linear velocity without significant loss of efficiency [2]. Nowadays columnspacked with shell or fused core particles (Ascentis Express/HALO, Poroshell, Kinetex) getcomprehensive attention, studies deal with performance of these types of stationaryphases [3]. But how does performance of new columns depend on applied HPLC orUHPLC instruments?
To compare different kind of chromatographic techniques kinetic plot method (KPM)can be used, originally developed for evaluation of isocratic separations [4]. Recently, apioneer work of Broeckhoven et. al. [5] proved that KPM can be extended the practicallymore relevant case of gradient separations as well.
Aim of our study was to compare different UHPLC systems in an aspect of band broad-ening effect coming from the chromatographic systems in an example of the separationof an active pharmaceutical compound and its related substances with Kinetex PFP col-umn under both isocratic and gradient conditions. Results were evaluated with kineticplot method.
1. H. Minakuchi, K. Nakanishi, N. Soga, N. Ishizuka, N. Tanaka J. Chromatogr. 762 (1997) 135–1462. L. Nováková, H. Vlcková Anal. Chim. Acta 656 (2009) 8–353. Sz. Fekete, J. Fekete, K. Ganzler J. Pharm. Biomed. Anal. 50 (2009) 703–7094. G. Desmet, D. Clicq, Piotr Gzil.: Anal. Chem. 2005, 77, 4058–40705. K. Broeckhoven, D. Cabooter, F. Lynen, P. Sandra, G. Desmet: J. Chromatogr. A, 1217 (2010) 2787–2795

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-318-TU
Development and Validation of a UHPLC-UV Methodfor the Detection and Quantification of Erectile Dysfunction Drugs
and Some of their Analogues Found in Counterfeit Medicines
Pierre-Yves Sacré1,2, Eric Deconinck1, Patrice Chiap3, Jacques Crommen2,Eric Rozet4, Patricia Courselle1, Jacques O. De Beer1
1Laboratory of Drug Analysis, Scientific Institute of Public Health,Brussels, Belgium
2Department of Analytical Pharmaceutical Chemistry, Institute of Pharmacy, University of Liège,Liège, Belgium
3Advanced Technology Corporation (A.T.C.), University Hospital of Liège,Liège, Belgium
4Department of Analytical Chemistry, Institute of Pharmacy, University of Liège,Liège, Belgium
Due to the taboo associated with erectile dysfunction, phosphodiesterase type 5 (PDE5)inhibitors are widely sold over the internet as both counterfeited medicines and illegaladulterants in herbal dietary supplements [1].
For this study, three analogues of sildenafil (acetildenafil, hydroxyacetildenafil anddimethylsildenafil), one of vardenafil (pseudovardenafil), one of tadalafil (aminotadalafil)and the bioactive diastereoisomer of tadalafil (trans-tadalafil) have been chosen.
The method is also compatible with on-line coupling mass spectrometry and will signif-icantly reduce analysis times and solvent consumption.
The separation is performed on an AcquityTM BEH Shield RP18 (100 mm x 2.1 mm I.D.,1.7 μm particle size) column. in less than 4.5 minutes. The final method is a gradientelution with pH 3.5 formate ammonium buffer (10 mM) as aqueous phase (solvent A)and acetonitrile as organic modifier (solvent B).
The accuracy profiles were within the 95–105% range. This rapidity associated to a lowflow rate permits the analysis of a large number of samples with a reduced cost andassociated solvent consumption.
The main problem with counterfeit medicines is that their chemical composition isunknown. This is why they represent a real danger for public health. The method permitsthe detection of all PDE5 inhibitors even new ones as it covers a wide range of polarity.The elucidation of structures and the confirmation of identity may be performed byUHPLC-MS systems since the mobile phase is compatible.
The fact that the method is both applicable for the analysis of counterfeit medicinesand the routine analysis of authorised medicines is a plus in a control laboratory.
1. S. Singh, B. Prasad, A. Savaliya, R. Shah, V. Gohil, A. Kaur, Trends Analyt Chem 28 (2009) 13–282. Ph. Hubert, J.-J. Nguyen-Huu, B. Boulanger, E. Chapuzet, P. Chiap, N. Cohen, P.-A. Compagnon, W.
Dewé, M. Feinberg, M. Lallier, M. Laurentie, N. Mercier, G. Muzard, C. Nivet, L. Valat, J Pharm BiomedAnal 36 (2004) 579–586

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-319-MO
Enhanced Stationary Phase Selectivity for UHPLC SeparationsBased upon a Core-Shell Technology
Jason A. Anspach, Lawrence Y. Loo, Thuylinh Tran,A. Carl Sanchez, Tivadar Farkas
Phenomenex411 Madrid Ave, Torrance, CA 90501 USA
Over the past 10 years there has been a strong drive towards faster and more efficiencyHPLC separations. As technology has evolved, there has been a trend towards the use ofdecreasing particle size to increase separation efficiency. Some of the newest particles aresmaller than 2 μm in diameter. This decrease in particle diameter has generated columnswith higher efficiencies per unit length, allowing for the use of shorter columns for fasterseparations. The drawback, however, is that the pressure required to operate these col-umns requires the installation of new instruments. Very recently a new generation of core-shell media has been introduced. Due to the particle architecture of these materials, theyare able to provide the separation efficiencies of sub 2 μm particles, at pressures that arecompatible with conventional HPLC instrumentation. While the column efficiency increasewill increase peak resolution, the column selectivity still has a large effect on overall reso-lution. In this paper we will demonstrate how combining the increased efficiency of thelatest generation core-shell media with recent enhances in stationary selectivity can im-prove HPLC separations in a variety of applications.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-320-TU
Fast LC Method Optimisation using Kinetic Plots
L. Pereira, T. Edge, H. Ritchie, S. Luke, D. MiltonThermo Fisher Scientific,
Manor Park, Runcorn, WA7 1T, UK
There are several reasons for scaling down a method from a conventional 4.6 mm IDcolumn packed with 5 of 3 μm particles to narrowbore columns packed with sub-3 μmparticles. Sub-3 μm particle packed columns offer advantages over the more traditionalcolumns packed with 3 and 5 μm particles through shorter analysis times, improvementsin resolving power, sensitivity and peak capacity. Significant solvent savings can also beachieved by scaling down the method. When transferring methods to fast LC, severalapproaches can be taken, depending on the analytical needs. If column dimensions aremaintained and only particle size is reduced then an improvement in efficiency and,therefore, resolution and peak capacity is obtained. A second approach is to reduce notonly particle size but also column dimensions, which has the benefit of reducing analysistime. Generally, method transfer calculations only take into consideration the effect ofparticle size and flow rate on performance (Van Deemter curve), and the column dimen-sions to account for gradient and injection volume changes. The Kinetic plot method canbe used to turn each experimental point of the Van Deemter curve into the conditions ofmaximum plate-generation velocity, or in other words, the separation time and efficiencythat can be obtained at a given linear velocity on the longest possible column, at theimposed pressure limit of the system. These data points represent the best kinetic perfor-mance for the column material used. Hence, it is possible to quickly optimise the analyti-cal conditions to obtain the fastest, most efficient separation at a given pressure drop.
The work presented in this poster provides practical guidelines on scaling down separa-tions by taking into account column particle size, ID, length, and packing. Further meth-od optimization to achieve the fastest analysis time with best efficiency and resolution isalso included based on the kinetic plot method.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-321-MO
High Throughput Analysis of Model Drugs by CombiningUltra High Pressure and High Temperature Chromatography
using 1 mm ID Columns
Thorsten Teutenberg, Steffen Wiese, Paul ErmischInstitut für Energie- und Umwelttechnik e. V. (Institute of Energy and Environmental Technology, IUTA),
Bliersheimer Strasse 60, 47229 Duisburg, Germany
Ultra performance liquid chromatography is well established in routine laboratoriesaround the world. This is due to the fact that there is an ever increasing demand to speedup analysis. The upper pressure limit is continuously increased as instrument manufactur-ers present new technological developments.
In this presentation, ultra fast separations are shown using columns with an internaldiameter of 1 mm. By working at the upper pressure and temperature limit of the columnwhich is around 1,200 bars and 90 °C, extremely high linear velocities of about 4 cm s−1
are obtained. This means that the re-equilibration after a solvent gradient can be reducedto a few seconds. The chromatographic efficiency is calculated by the means of gradientkinetic plots. Although a low efficiency is obtained, this approach may be well suited forfast second dimension runs in comprehensive liquid chromatography.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-322-TU
Intelligent System Emulation: Achieving Method Compatibilitybetween HPLC and UHPLC
Monika M. Dittmann, Heike Otero-Martinez, Konstantin Choikhet,Klaus Witt, Gerhart Metzler
Agilent Technologies GmbH,Waldbronn, Germany
Next generation UHPLC instruments that have been introduced to the market in the pastfew years are typically designed for the operation of small bore columns (2.1 mm – 3 mminternal diameter) packed with small particles to achieve high separation efficiencies. Withregard to solvent delivery these systems usually differ significantly from standard HPLCsystems, in particular the dwell volumes of such systems are minimized to allow fastgradient separations.
When methods developed on standard HPLC systems are run on modern UHPLC sys-tems, changes in retention times and often also in selectivity due to the differences insystem characteristics are observed.
The authors present an approach to emulate the gradient performance of an HPLCsystem on an UHPLC system. In this way a method developed on an HPLC system systemsyields the same results in terms of retention time and resolution when run on a UHPLCsystem.
The emulation is based on a model for the system characteristics of the emulatingsystem and the system to be emulated, taking into account dwell volume, mixing proper-ties and composition offsets.
Results will be presented for various applications and a range of different gradientconditions.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-323-MO
Method Transfer from HPLC to Ultra PerformanceLiquid Chromatography (UPLCTM):
Biogenic Amines in Different Cheese Varieties
H. K. Mayer, G. FiechterBOKU – University of Natural Resources and Life Sciences Vienna,
Department of Food Science and Technology, Food Chemistry Laboratory,Muthgasse 11, A-1190 Vienna, Austria
Proteolysis is the principal and most complex biochemical event occurring during matura-tion of most cheese varieties, being of high significance for the development of textureand flavour of the final product. Biogenic amines are produced in cheese by enzymaticdecarboxylation of amino acids; and high levels can result in food poisoning.
The objective of this study was to establish a pre-column derivatization method to deter-mine biogenic amines in different cheese varieties. An existing HPLC protocol was to betransferred to ultra performance liquid chromatography (UPLCTM). AccQ-Fluor derivatizingreagent (6-Aminoquinolyl-N-hydroxysuccinimidyl carbamate) was used to analyze primaryand secondary biogenic amines. A reliable UPLCTM method was developed and optimizedfor the separation of 19 biogenic amines in cheese within nine minutes.
Commercial cheese samples from retail outlets in Vienna were analyzed for their bio-genic amine content, that varied to a great extent, depending not only on the type ofcheese, but also within a certain cheese variety. About 13.8% of samples had histamine,and 22.4% had tyramine content above 100 mg/kg. The highest concentration of hista-mine was found in Tiroler Almkäse (1.160 mg/kg) and Vorarlberger Bergkäse (397mg/kg), the highest amount of tyramine was found in Cantal, Olmützer Quargel andTiroler Almkäse (about 470 mg/kg). Moreover, 8.6% of samples had a putrescine orcadaverine content higher than 100 mg/kg. The highest concentrations were detected inOlmützer Quargel (523 mg putrescine, 748 mg cadaverine per kg). Thus, the total con-centration of biogenic amines in some samples was about 1.940 mg/kg.
In conclusion, a HPLC method for the quantitation of biogenic amines in cheese sam-ples was successfully transferred and optimized for UPLCTM. This novel UPLCTM methodenabled not only a higher analysis throughput, but achieved also a higher resolution andsensitivity.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-324-TU
New Technologies with UHPLC Improve Method Developmentand Chromatographic Results
Anne Mack1, Maureen Joseph1, William Long1, Linda Lloyd2
1Agilent Technologies,2850 Centerville Road, Wilmington, Delaware 19808, USA
2Agilent Technologies UK Limited,Essex Road, Church Stretton, SHRO SY6 6AX, UK
Advancements in UHPLC technology aid in LC method development and in improving theresults thereafter. UHPLC instruments are capable of operating at higher pressures thantraditional HPLC instruments, up to 1200 bar. This higher system pressure limit allowsusers to take full advantage of highly efficient sub-2 μm particle columns, whether it isusing long columns to enhance resolution or using fast flow rates to improve throughput.An updated packing technique makes some sub 2 μm columns stable up to 1200 bar formaximum compatibility with corresponding 1200 bar UHPLC’s. The many stationaryphases available within a family of sub 2 micron columns allow for tremendous flexibilityin selectivity during method development, providing benefits for a range of small mole-cule and bio-molecule separation needs. Additionally, UHPLC’s can be optimized for thelowest extra column volume, to ensure minimal band broadening and optimal resultsfrom the sub-2 μm RRHD columns in the smallest dimensions.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-325-MO
The Effect of Frictional Heating under Practical UHPLC Conditions
Lucie Nováková1, Jean Luc Veuthey2, Davy Guillarme2
1Department of Analytical Chemistry, Faculty of Pharmacy, Charles University,Heyrovského 1203, 500 05, Hradec Králové, Czech Republic
2School of Pharmaceutical Sciences, University of Geneva, University of Lausanne,Boulevard d’Yvoy 20, 1211 Geneva 4, Switzerland
Frictional heating phenomenon is observed when columns packed with very fine particlesare operated at high mobile phase velocities, thus generating high pressure drop [1–3]. Itis induced by the friction of the mobile phase percolating through the column bed at veryhigh pressure, which generates heat. The heat dissipates along and across the chromato-graphic column, allowing formation of axial (longitudinal) and radial temperature gradi-ents. These thermal gradients may influence retention, selectivity and column efficiency[1, 4]. Most of studies evaluating the effect of frictional heating published so far haveemployed model compounds and conditions or focused on mathematical modeling.
In the present study, we attempted to characterize the effect of frictional heating inUHPLC under real conditions (no model analytes or pure solvents as mobile phases).Then, columns packed with sub-2 μm particles were employed in the gradient mode forthe separation of a mixture containing neutral, basic and acidic drugs in both acidic andbasic pH conditions.
The evaluation of frictional heating in UHPLC system was carried out experimentallyunder gradient elution conditions (acetonitrile/ buffer at pH 3 and 9) in the pressurerange 100 – 1000 bar on both 1.0 mm and 2.1 I.D. columns using a mixture of 10 repre-sentative pharmaceutical compounds. Under adiabatic conditions (i.e. still air oven), thelongitudinal temperature gradient was estimated at +4, +8 and +16 °C at 300, 600 and1000 bar, respectively on 2.1 mm I.D column. With the 1.0 mm I. D. column, thesevalues were reduced to +3 °C, +6 °C, +12 °C, respectively. Obtained results could behighly valuable for the method transfer between regular HPLC and UHPLC.
1. F.Gritti, M. Martin, G. Guiochon, Anal. Chem. 81 (2009) 3365.2. H. Poppe, J. C. Kraak, J. F. K. Huber, H. M. van der Berg, Chromatographia 14 (1981) 515.3. I. Halasz, R. Endele, J. Asshauer, J. Chromatogr. 112 (1975) 37.4. F. Gritti, G. Guiochon, J. Chromatogr. A 1131 (2006) 151.
Acknowledgement: The authors gratefully acknowledge research project MSM002162082.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-326-TU
Quantitative Analysis of Catechins in Tea by UHPLC/UV
Guifeng Jiang, Terry ZhangThermo Fisher Scientific, San Jose, CA, USA
Tea is a complex matrix and its components are generally analyzed by high performanceliquid chromatography (HPLC) coupled with UV detection. The main drawback of con-ventional HPLC methods for the analysis of tea catechins is the compromise betweenspeed and resolution, resulting in typical analysis times of 20 minutes or longer. Ultrahigh performance liquid chromatography (UHPLC) enables faster separations and higherresolution through the use of sub-2 μm diameter particles.
The Thermo Scientific AccelaTM LC system offers the flexibility of performing both HPLCand UHPLC separations on a single platform. The Accela 1250 quaternary pump deliversaccurate and precise flows and gradients over a wide range of flow rates (0.1 – 2 mL/min)and pressures (1–1250 bar) to accelerate method development and maximize methodflexibility. Thermo Scientific 1.9 μm Hypersil GOLDTM PFP (perfluorophenyl) columnsenhance retention and selectivity for UHPLC separations of substituted aromatic com-pounds. In this application, we demonstrate fast and robust separation, detection andquantitation of sub-ppm levels of catechins and other phenolic compounds using theAccela LC platform and high performance sub-2 μm columns.
Several method parameters – mobile phase composition, flow rate and column length– were investigated in order to optimize separation speed and resolution for highthroughput and quantitation applications. Using these UHPLC methods, significant differ-ences in catechin and phenolic content in green and black tea samples were found.Although these methods were applied for the analysis of catechins and phenols in tea,they can be adapted for analysis of other foodstuffs as well as for use in studies examiningthe effects of catechin intake on human health.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-327-MO
There are Problems Associated with Gradient and Method Transferamong HPLC, and UHPLC, Systems and Columns
– are there Explanations and Usable Workarounds?
Michael Woodman, Lori SandfordAgilent Technologies,
Wilmington, Delaware, USA
UHPLC has allowed many users to explore practical examples of what was in the past onlypossible under theoretical consideration. We observe, however, that considerable difficul-ty may be found when a demanding HPLC method is transferred to UHPLC columnsand/or systems, or the reverse is attempted. Is it due to dramatic increases in operatingpressure and viscous heating, dwell volume disparities or is it that many methods aresimply not translated with sufficient calculation precision? For every remarkable exampleof perfect translation, generally prepared by an instrument or column vendor, as many asfive examples of catastrophic results may be found. The answer is no doubt complex andis more and more the subject of discussion now that it is quite clear that UHPLC conceptsand practice are here to stay and growing in use.
We have interrogated systems in the UHPLC classification, with a selection of columnsgenerally meeting the definition of UHPLC in design and performance, and a set of HPLCconditions for comparison. Separations known to be demanding for their sensitivity topH, temperature and gradient slope are put to the test in a matrix of instrument variables.All the while, very accurate calculations of gradient transfer are ensured with a consistenttranslation tool. Extra-column dispersive effects, while of considerable interest, are ig-nored as the focus of this work is in the alpha term of the resolution equation. The natureof viscous heating and how it may affect the alpha is explored and distinctions betweengradient delay, gradient dwell volume and the intriguing transition volume effect areexplored.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-328-TU
UHPLC Considerations – Demands for Ultra Pure Solvents,Additives and New Testing Methods
Rudolf KöhlingSigma-Aldrich Production GmbH,
Industriestrasse 25, CH-9470 Buchs SG, Switzerland
Since the beginning of HPLC there has always been an ambition to decrease the run-timeand increase the efficiency of a column while still maintaining or increasing resolution.The decrease in particle size together with the development of Fused CoreTM particles istoday’s result of this need. Higher pressure tolerance, very low dead-volumes and im-proved detectors are improvements to HPLC systems ending up in the newest generationof UHPLC systems. The combination of high efficiency columns and UHPLC systemsenable very fast chromatographic separations resulting in very narrow and tall peaks [1,2]. This advantage may also become a disadvantage when even the smallest impurities inthe solvent cause background and unwanted peaks. Many technical aspects are taken intoaccount for the development of UHPLC systems and methods regarding detectors such asUV, MS, etc. but only fragmentary information is documented about specifications ofmobile phase solvents/ additives and their preparation for usage in UHPLC [3, 4].
This poster describes the influence of solvent quality on UHPLC applications and thedefinition of new testing specifications. Several examples show that new and improvedtesting methods such as more sensitive gradient tests using UV and both ESI modes(pos./neg.) are needed for the growing field of UHPLC/UV/MS applications. Examples ofsuch applications are polar analytes in metabolomics, identification and quantification ofcompounds in forensics and quality control in pharmaceutical industry [5].
1. L.R. Snyder, J.J. Kirkland, J.W. Dolan, Introduction to modern liquid chromatography, 3rd ed., Wiley, 2010,pp. 6–8.
2. P. Jandera1, G. Henze, Liquid Chromatography, in: Ullmann’s Encyclopedia of Industrial Chemistry, Wiley-VCH, 2010, DOI: 10.1002/14356007.b05_237.pub2 (online).
3. V. R. Meyer, Praxis der Hochleistungs-Flüssigchromatographie, 9. Aufl., Wiley-VCH, 2004.4. K. K. Unger, E. Weber (ed.), Handbuch der HPLC, GIT Verlag, 1995.5. S. K. Chowdhury, Identification and Quantification of Drugs, Metabolites and Metabolizing Enzymes by
LC-MS, in: Progress in Pharmaceutical and Biomedical Analysis, Vol. 8, 1st ed., Elsevier, 2005.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-329-MO
UHPLC – MS of Clinical Relevant Compoundsusing a New Type of 2 μμm Silica Particles
Michael Schulz, Anita Piper, Petra Lewits, Karin CabreraMerck KGaA,
Darmstadt, Germany
Today, HPLC columns packed with 2 μm silica particles are becoming more and morepopular to follow the needs for fast separations and high separation efficiency. This trendcan be explained with a continuously increasing demand of analysing more and moresamples per time unit in order to secure the quality of products. In this context we havedeveloped a new high purity silica which can be provided as 2 μm particles packed insmall diameter UHPLC columns (2.1 mm i.d.). The columns are especially suited for thefast analysis of samples produced in the pharmaceutical, food or cosmetics industry aswell as for laboratories engaged in clinical, diagnostical, or environmental analysis. Fur-thermore, the costs of analysis can be dramatically reduced because of low solvent con-sumption.
The increasing usage of MS (mass spectrometry) detectors has led to the developmentof these columns with smaller dimensions in length and diameter. Here, we demonstratethe use of our new UHPLC columns filled with 2 μm silica particles (Purospher STAR RP-18e) in the area of clinical applications combined with MS-detection.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-330-TU
Ultra-Fast LC Analysis of Complex Samples at UHPLC Conditions
Matthias Pursch, Romina HammesDow Deutschland Anlagengesellschaft mbH, Analytical Technology Center,
77836 Rheinmuenster, Germany
The recent introduction of UHPLC instrumentation and high-efficiency columns hasopened the way to perform LC analyses on very short time scales. Potential benefits arereductions of analysis times and/or enhanced information content by using longer highefficiency columns.
This poster details the separation of pesticides at ultra-fast and high-resolution LCconditions by applying pressures of up to 1100 bar. Analysis times in ultra-fast mode areless than 2 min for a mixture of polar and non-polar pesticides. Automated screening ofdifferent stationary phase chemistries (C18, C8, Phenyl Hexyl etc.) and different columnlengths was used to optimize the selectivity for a given sample matrix. Performance dataon reproducibility and sensitivity will be presented.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-G-331-MO
Utilizing Extended Linear Velocity to Maximize Peak Capacityin Ultra-High Power Chromatography
Dawn Stickle1, Goran Rasched2, Bob Giuffre2, Dat-Phan3
1Agilent Technologies,40 Shattuck Rd, Andover, MA 01810, USA
2Agilent Technologies,550 Clark Dr, Budd Lake, NJ 07825, USA
3Agilent Technologies,770 Stanford Dr, Columbia, MD 20580, USA
Prior to the development of ultra-high power liquid chromatography systems, the upperpressure limits and flow rate ranges that could be tested were limited by the power rangeof currently available HPLC hardware. With the advent of ultra-high power chromatogra-phy systems, the performance of the columns analyzed has been explored both theoreti-cally and experimentally. Historically, plate height has been used as the measure forseparation efficiency. However, in a practical sense, this measure is best used for isocraticand isothermal based separations. Peak capacity is a more suitable measure of efficiencyin gradient separations as it considers the partitioning effects from the change in solventstrength during an analysis. For a given gradient time, peak capacity has been shown toincrease beyond the predicted optimum based on the van Deemter equation, when flowrates are extended past optimal linear velocity.
This work seeks to explore the power limits beyond traditional commercial systems(6000 psi) up to 17,000 psi at 2 mL/min and 12,000 psi up to 5 mL/min. Kinetic (Poppe)plots will be used to illustrate the efficiencies gained by extending the power envelopeabove 10,000 psi along with the relationship between flow rate and gradient duration.Data will be shown comparing particle types (totally porous versus superficially porous),particle sizes, and columns in series, along with the affects of temperature and gradientlength.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-332-TU
A Linkage Application of Multi-Dimensional Chromatography,Solid-Phase Peptide Ligand Library,
Two-Dimensional Difference Gel Electrophoresisand Mass Spectrometry towards Plasma Biomarker Development
Tatsuo Hagiwara1,2, Yumi Saito1, Yasufumi Murakami2, Tadashi Kondo1
1Division of Pharmacoproteomics, National Cancer Center Research Institute,Tokyo, Japan
2Laboratory of Genome Biology, Department of Biological Science & Technology,Tokyo University of Science, Japan
To develop the proteomics tool for plasma biomarker development, we combined threedifferent methods; peptide ligand library, multi-dimensional chromatography, two-dimen-sional difference gel electrophoresis (2D-DIGE) and mass spectrometry. High abundanceproteins such as albumin and immunoglobulin were depleted by Blue sepharose (HiTrapBlue HP, GE) and protein G sepharose (HiTrap Protein G HP, GE) chromatography, respec-tively. The resulting plasma samples were separated into six step-wise fractions by anion-exchange chromatography (Resource Q, GE), and the fractionated samples were subject-ed to the solid-phase combinatorial peptide library (ProteoMiner, BioRad). After theseserial treatments, the resulting protein samples were labeled with CyDye DIGE Fluorsaturation dye (GE) and separated by 2D-PAGE (Multiphor II and EttanDalttwelve, GE). Wecompared the 2D-DIGE images between the fractions and between the samples with andwithout ProteoMiner treatments, and the protein spots with different intensity wereexamined by mass spectrometry for protein identification (LTQ, Thermo). The multi-dimensional separation by chromatography increased the number of protein spots of 2D-DIGE from 538 in the original plasma sample to 5852 in all fractionated protein samples.The ProteoMiner treatment increased the number of protein spots from 6390 to 8030 asa total number of protein spots. Although same protein spots were redundantly observedin the neighboring fractions, the combined use of different methods contributed to morecomprehensive plasma protein studies. The mass spectrometric study identified middle tohigh abundant proteins as those enriched by the ProteoMiner; we did not obtain the datasupporting the idea that the solid-phase peptide ligand library broadly enhances detec-tion of lower abundance plasma proteins. In conclusions, we demonstrated that thecombined use of multiple separation methods increased the number of protein spots in2D-DIGE, suggesting its possible utilities for plasma proteomics. The characterization ofenriched proteins will be required before the practical application of this approach forplasma biomarker development.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-333-MO
Analysis of the Altered Glycosylation of IgG1in Rheumatoid Arthritis by Capillary Electrophoresis
Csaba Váradi, Stefan Mittermayr, Márta Kerékgyártó, Bertalan MeskóHorváth Laboratory of Bioseparation Sciences, University of Debrecen,
Hungary
Glycosylation is one of the most important posttranslational modifications, which holdsthe promise to be widely used as biological markers for various diseaes. The N-glyco-sylation on human immunoglobulins, especially on IgG1, plays a critical role in the bioac-tivity of this goup of very important proteins. Significant differences have been reportedin IgG1 glycosylation in pregnancy, aging and various diseases. The four characteristicglycans of IgG1 are biantennary-agalacto (G0), biantennary-mongalacto (G1 and G1’)and biantennary-digalacto (G2) structures, with the relative amonut of G0 showing thehighest variability in the above mentioned conditions. The aim of this study was to inves-tigate the changes in the relative amount of G0 glycan of IgG1 in rheumatoid arthritis(RA), especially in view of transcriptomics data on galactosyl transferase experession. Weanalyzed and compared the glycosylation pattern in sex and age matched samples to seethe correlation between IgG1 N-glycan profile, galactosyl transferase expression and thepatogenesis of reumatoid arthritis. IgG1 was isolated from the patient’s samples usingProtein A affinity pulldown and the N-glycans were digested by peptide-N-glycanase F(PNGase F). The released glycans were then fluorescently labeled with aminopyrene-trisulfonate (APTS) and analyzed by capillary electrophoreiss and laser induced fluores-cence detection.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-334-TU
Ceramide Profiling in Skin using LC-QTOF-MS
Ruben t’Kindt1, Lucie Jorge1, Emmie Dumont2, Pauline Couturon3,Frank David2, Pat Sandra2, Koen Sandra1
1Metablys,President Kennedypark 26, 8500 Kortrijk, Belgium
2Research Institute for Chromatography,President Kennedypark 26, 8500 Kortrijk, Belgium
3K.U. Leuven, Chemistry Department,Celestijnenlaan 200f, 3001 Heverlee, Belgium
Ceramides are important structural elements in the complex and lipid-rich matrix of thehuman skin. About 40% of the stratum corneum (SC) consists out of these polar lipidspecies. They are indispensable in the barrier function and water-holding property of ourskin and may play essential roles in many skin diseases and defects. Ceramides are theresult of the linkage of two molecules, a long amino alcoholic chain covalently bound toa fatty acid moiety. Complete profiling of ceramides is challenging, as the class ofceramide species contains an extensive structural heterogeneity. They not only vary in thecombination of sphingoid bases and fatty acyl moieties but also in the chain length andthe number, position and stereochemistry of double bonds, hydroxyl groups and otherfunctionalities of the building blocks.
The current paper describes an LC-MS based method for the elucidation of skinceramides and the potential classification of different skin types based on ceramide com-position. Ceramide samples were collected by tape stripping of skin, after which theywere selectively extracted and concentrated. The purified ceramide extracts were thenanalyzed by reversed-phase chromatography coupled to quadrupole time-of-flight massspectrometry in both positive and negative electrospray ionization mode. MS/MS experi-ments were performed in data-dependent acquisition mode to confirm accurate mass-based identifications of all ceramides and to distinguish isomeric species. An in-houselibrary was built with formula, exact mass and retention time of all possible skinceramides, providing an automated data-processing of LC-MS profiles.
Hundreds of ceramide species, including all 16 described classes of ceramides, could bereliably separated and identified. Within each ceramide species, MS/MS fragmentationspectra revealed several isomeric structures varying in carbon length of sphingoid baseand fatty acyl building blocks, confirming the structural heterogeneity of the ceramides.
Ceramide profiling can definitely be a valuable tool in skin type classification, providingcosmetic companies leads for customized skin products matching the clients’ skin profile.Furthermore it can be useful for tracking biomarkers for various skin diseases.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-336-TU
Determination of Acrylamide Metabolites in Urineby LC-MS/MS Technique
Krystyna Tyrpien, Magdalena SzumskaDepartment of Chemistry, Faculty of Medicine, Zabrze
Silesian Medical University in Katowice, Poland
Acrylamide (AA) is recognised as neurotoxic and classified as a probable human carcino-gen (IARC, 1994) quickly absorbed and metabolised in the body. It has been shown thatAA and its carcinogenic metabolite – glycidamide (GA), are excreted as mercapturic acids,their urinary conjugations with glutathione.
The preliminary research was carried out towards mercapturic acids determination inurine samples.
That is why N-acetyl-S-(2-carbamoylethyl)-L-cysteine (AAMA) as well as N-acetyl-S-(1-carbamoyl-2-hydroxyethyl)-L-cysteine (GAMA) were synthesized as described earlier(Bjellaas et al., 2005). Purity of these standards was verified by means NMR technique.
The analytes were extracted from urine by ENV+ (Isolute IST) SPE cartridges prior toESI-RP-LC-MS/MS analysis.
The analytical method using a liquid chromatograph (Dionex Ultimate 3000) with amass spectrometer (HCT Bruker Daltonics) operated in positive electrospray ionizationmode with a quadrupole ion trap was applied. The separation step was performed usingKinetex column (100 x 3.00 mm, 2.6 μm, XB-C18 100A, Phenomenex) and 1% of formicacid in water and acetonitrile (95:5, v/v), as mobile phase.
Detection limits for both AAMA and GAMA were 4,8 μg/L and 37.5 μg/L, respectively.
1. http://www.inchem.org/documents/iarc/vol60/m60-11.html2. Bjellaas, T., Janak, K., Lundanes, E., Kronberg, L., and Becher, G. (2005). Determination and quantification
of urinary metabolites after dietary exposure to acrylamide. Xenobiotica 35, 1003–1018.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-337-MO
Determination of Galanin Family Peptides in LC-MS Methodusing Fused-Core Technology
François Mansion, Jacques Crommen, Anne-Catherine Servais, Marianne FilletLaboratory of Analytical and Pharmaceutical Chemistry, Drug and Interfaculty Research Center,
University of Liege, Liege, Belgium
Nowadays, peptides are increasingly used as specific biomarkers of some diseases or astherapeutic agents. Quick and reliable evaluation of biomarkers may save valuable timeon the disease. Analytical techniques commonly used were adapted to peptides determi-nation. Indeed, the complexity of peptides in term of structure and their tendency to theadsorption require special attention when developing the method. Due to recent advanc-es in materials (sub-2 μm particles, fused-core technology), reversed-phase liquid chroma-tography (LC) and mass spectrometry (MS) detection is particularly well suited for thiskind of applications.
In comparison to classical silica particles, fused-core particles are not fully porous. Theyare made of a silica core and a homogenous porous shell. The analyte time diffusing intoand out of the pores is reduced, which decreases band broadening and increases peakefficiency. Due to lower column backpressure, fused-core columns are compatible withany LC system.
Galanin family peptides were selected as interest peptides biomarkers. Galanin, galaninlike peptide and alarin are neuropeptides involved at different levels in the regulation ofinflammatory processes. A LC-MS method was developed to separate and quantify thesepeptides. Our LC system was first adapted to the fused – core technology: the replace-ment of the conventional needle seat capillary by a lower needle seat capillary volume,the use of tubing of 0.005 inch and zero volume finger-tight fittings, … Then, chromato-graphic performances of various fused – core columns (C18, C18 XB and PFP; 100 mm x2.1 mm; 2.6 μm) were compared to a fully porous particles column (C18; 100 mm x 2.1mm; 3 μm). Among the different fused – core stationary phases, the C18 fused-core col-umn was selected as the most suited column for our peptide biomarkers determination.Moreover, particular attention was paid to prevent peptides adsorption all over the ana-lytical system.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-338-TU
Fractionation of the Human Plasma Proteome for Monoclonal Anti-body Proteomics Based Biomarker Discovery
András Kovács1, Edit Sperling1, Stefan Mittermayr1, József Lázár2, Attila Balogh2,János Kádas2, Ákos Szekrényes1, László Takács2, István Kurucz2
1Horváth Laboratory of Bioseparation Sciences, Research Centre for Molecular Medicine,Medical and Health Science Center, University of Debrecen, Debrecen, Hungary
H-4032 Debrecen, Nagyerdei körút 98., Hungary2BioSystems International Kft, Debrecen, HungaryH-4032 Debrecen, Nagyerdei körút 98., Hungary
Monoclonal antibody proteomics is a novel methodology to recognize proteins of bio-marker potential, but requires subsequent antigen identification steps. The identificationof medium or lower abundant proteins needs a large amount of sample to assure theproper amount of antigen for the ID process. Our aim was to generate a comprehensiveanalyte library representing the human plasma proteome, balancing the presently existing11–12 order of magnitude differences, which make isolation and identification of lowconcentration proteins extremely difficult. The fractions of the analyte library can then bereadily used to accommodate the protein identification process for monoclonal antibodyproteomics based biomarker discovery. First the large human serum albumin and immu-noglobulin content were removed from 500 mL normal pooled human plasma by specificchromatography based partitioning methods and the resulting depleted plasma was pre-fractionated by ammonium sulfate precipitation. Each fraction was then further separatedby size exclusion chromatography, followed by cation and anion exchange chromatogra-phy. The 20 most concentrated ion exchange chromatography fractions were furtherseparated by hydrophobic interaction chromatography. This process resulted in 783fractions with the average protein concentration of 1 mg/mL. All chromatography andprecipitation steps were carefully designed with the purpose of maintaining the nativeforms of the intact proteins throughout the fractionation process. The separation route ofvitamin D-binding protein (an antibody proteomics lead) was followed in all major frac-tionation levels by dot blot assay in order to identify the library fraction it accumulated inand the identity of the antigen was verified by Western blot.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-339-MO
Glycosylation Profile of Haptoglobin Phenotypesin Normal and Cancer Patients
Ákos Szekrényes, Csaba Váradi, Márta Kerékgyártó, Stefan MittermayrHorváth Laboratory of Bioseparation Sciences, University of Debrecen,
Hungary
Protein glycosylation is increasingly being recognized as one of the most prominentbiochemical alterations in malignant transformation and tumorigenesis. Haptoglobin is anacute phase protein with three typical phenotypes of Hp1-1, Hp2-1 and Hp2-2. Thesedifferent forms are reportedly associated with inflammatory and malignant diseases aswell as with ovarian and prostate cancer. Haptoglobin has four characteristic glycosylationsites, which are located on its β-chain. In this study, first we have examined the qualita-tive and quantitative glycosylation differences between the haptoglobin phenotypes.Haptoglobin was isolated by immunoaffinity pulldown using a Hp-specific monoclonalantibody followed by SDS-PAGE purification. The glycans were released the endo-glycosidase enzyme PNGase F using an in-gel digestion process. The liberated carbohy-drate structures were labeled with the fluorophore dye APTS followed by capillary gel-electrophoresis analysis. The haptoglobin glycosylation profiles were then comparedbetween normal and cancer patient samples.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-340-TU
High Sensitivity Protein Quantitation usinga Triple Quadrupole with a Dual Ion Funnel
Christine Miller1, Yanan Yang1, Christian Gotenfels2, Keith Waddell11Agilent Technologies,
5301 Stevens Creek Boulevard, Santa Clara, California 95051, USA2Agilent Technologies,
Hewlett-Packard Str. 8, 76337 Waldbronn, Germany
Assays that are both specific and quantitative for target proteins are critical for preclinicalvalidation of putative biomarkers. Such assays are typically multiplexed, multiple reactionmonitoring (MRM) analyses which can provide the high-throughput required. Sensitivityis a key requirement for such assays as protein biomarker concentrations may be quitelow in commonly used biofluids such as serum and plasma. Improving the sensitivity ofLC/MS can be achieved by using nanoflow LC, and by enhancing the sampling andtransmission of ions in the mass spectrometer. This study demonstrates the 5–10x sensi-tivity gain achieved for peptides using a triple quadrupole mass spectrometer modifiedwith a dual ion funnel. The sensitivity achieved using a microfluidic-based nanoflow LCsystem compared to a standard LC system will be shown discussed.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-341-MO
HPLC-MS Method for Monitoring Aldehydic Oxidative Stress Markersin Human Clinical Samples
Kamila Syslová1, Petr Kacer1, Marek Kuzma2, Beata Vilhanová2, Štepánka Vlcková3,Jindriška Lebedová3, Zdenka Fenclová3, Daniela Pelclová3
1Institute of Cheical Technology, ICT – Prague,Technická 5, 166 28 Prague 6, Czech Republic
2Laboratory of Molecular Structure Characterization, Institute of Microbiology,Vídenská 1083, 142 20 Prague 4, Czech Republic
3Department of Occupational Medicine, 1st Medical Faculty, Charles University,Na Bojišti 1, 120 00 Prague 2, Czech Republic
Oxidative stress is increasingly seen as a major upstream component in the signalizingcascade involved in many cellular functions, such as cell proliferation, inflammatory re-sponses, stimulating adhesion molecule, and chemo-attractant production. A growingbody of evidence suggests that many of the effects of cellular dysfunction under oxidativestress are mediated by products of non-enzymatic reactions, such as the peroxidativedegradation of polyunsaturated fatty acids. Aldehydic molecules generated during lipidperoxidation have been implicated as causative agents in cytotoxic processes initiated bythe exposure of biological systems to oxidizing agents. Endogenously generatedaldehydic lipid peroxidation products are malondialdehyde, α,β-unsaturated aldehydes(mainly 4-hydroxynonenal and 4-hydroxyhexenal) and saturated aldehydes (hexanal,heptanal and nonanal). Aldehydes are formed by lipid peroxidation of ϖ-6 (arachidonicacid, linoleic acid) and ϖ-3 (oleic acid) polyunsaturated fatty acids. The aldehydes (bio-markers) quantification in various body fluids (exhaled breath condensate, plasma andurine) represents an interesting tool for oxidative stress induced diseases diagnostics.
The work presents a new method for the determination of aldehyde-biomarkers presentin body fluids (exhaled breath condensate, urine, plasma) based on the LC-ESI/MS/MS.Malondialdehyde, 4-hydroxynonenal, and saturated aldehydes (n-C6 to C13 aldehydes)were quantified after derivatization with Girard’s reagent T. LC-ESI-MS/MS operated inneutral loss (NL) mode was used for its exceptionally high degree of selectivity and sensi-tivity. The combination of mass spectrometry detection with separation techniques(HPLC) enabled retention of the analytes of interest from the solvent front and avoids co-elution of salts and endogenous matrix components which can suppress the ionization ofthe analytes during the electrospray-ionization (ESI).
The developed method enabled unequivocal parallel determination of several oxida-tive-stress biomarkers at the only one analysis run. The method was optimized and vali-dated. Finally, the method was tested on real clinical samples collected from patients withdifferent oxidative stress induced disorders (silicosis, asbestosis) and compared to thecontrol group of healthy subjects.
Acknowledgement: The work was supported by the Ministry of Health of the Czech Republic (Grant NS10298-3/2009) and Grant Agency of the Czech Republic (Grant GA CR 203/08/H032).

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-342-TU
Levels of MDA Determined by HPLC in Patients with Leukemia
Matheus Sampaio Goveia1, Pamella Cristina Scheel1,Carlos Eduardo Coral de Oliveira2, Julie Massayo Maeda Oda2,
Maria Angélica Ehara Watanabe2, Suzana Lucy Nixdorf1
1Universidade Estadual de Londrina, Chemistry Department, DIA Lab., Brazil2Universidade Estadual de Londrina, Biology Department, DIA Lab., Brazil
Chronic myelogenous leukemia (CML) is a malignant myeloproliferative disorder originat-ed from a pluripotent stem cell, characterized by abnormal release of the expanded,malignant stem cell clone from the bone marrow into the circulation. According to Brazil-ian Ministry of Health and National Cancer Institute, by rating in 2010 arose in Brazilmore than 9500 cases of leukemia, and about 5700 deaths in 2008. Malondialdehyde(MDA) is being used as a bio-indicator, by the fact of having cytotoxic and genotoxicaction, occurring in high levels in some pathologies associated to oxidative stress. Howev-er, there aren’t studies in literature correlating its levels to leukemia stages yet. The objec-tive of this study was to verify viability of using MDA as bio-indicator to early diagnosis ofthe disease, as a faster and less invasive method, in patients from University Hospital ofLondrina, diagnosed with leukemia. The used method was described by Pilz et al.(2000)adapted and validated to analysis in HPLC-PAD. Chromatographic system used was HPLC(Alliance e2695, Waters, Milford, MA, USA) with UV/VIS-PAD 2998 detector. Mobilephases was a mixture of acetonitrile: water (0.2% of acetic acid) 35:65 (v/v) with flowrate of 0.8 mL min−1. The injection volume was 20.0 μL. All analysis was kept under con-trolled temperature of 4 °C to samples inside and 28 °C to column. Was used pre-columnand monolithic column Chromolith C-18 (150 x 3.9 mm, 5 μm, Merck, Darmstadt, Ger-many) and detection was made in λ = 310 nm. Calibration curves showed themselveslinear with r2 > 0.99 de 0.625 a 20.00 μmol L−1. MDA values determined in plasma ofhealth donors (n = 41), control samples, had an average of 1.702 ± 0.574 μmol L−1, whilepatients carrier of leukemia (n = 20) had 3.290 ± 0.822 μmol L−1, being possible to verifypositive correlation between the disease and levels of MDA, suggesting potential use of itas bio-indicator of leukemia.
ReferencePilz, J.; Meineke, I.; Gleiter, C. J. Chromatogr. B; v.742, p. 315–325, 2000.
Acknowledgements: The authors are grateful to CAPES, CNPq and Fundação Araucária for financial support.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-343-MO
Optimization and Application of Microwave-Assisted Acid Hydrolysisfor Rapid Quantification of Protein Oxidation Markers using LC-MS
Somaieh Afiuni-Zadeh1,2, Xinghua Guo1, Gholamhassan Azimi2, Ernst Lankmayr1
1Institute of Analytical Chemistry and Food Chemistry, Graz University of Technology,Stremayrgasse 9, 8010 Graz, Austria
2Department of Chemistry, University of Arak,38156 Arak, Iran
Protein oxidation is known to affect protein functionality in biological systems and foodquality. The earlier analytical methods were limited to measure total protein carbonylsformed by diverse and unspecific pathways, thus, the chemical structure and the oxida-tion mechanisms of carbonyls derived from proteins remain unknown. In medical re-search, the two products semialdehydes α-aminoadipic semialdehyde (AAS) andγ-glutamic semialdehyde (GGS) have been used as biomarkers of oxidative stress andindicators of serious age-related disorders and disease developments such as Alzheimer,rheumatoid arthritis, amyotrophic lateral sclerosis, diabetes mellitus, and Parkinson’sdisease. The aim of this work is to improve a recently published LC-MS method ofquantification by significantly reducing the total analysis time. Here simple and efficientmicrowave-assisted acid hydrolysis (MAAH) of proteins has optimized and applied forsample preparation instead of using thermal heating. The precursor amino acid residuescorresponding to AAS and GGS in oxidized proteins were derivatized by reductive amina-tion with sodium cyanoborohydride (NaCNBH3) and p-aminobenzoic acid (ABA) followedby MAAH to generate the marker derivatives AAS-ABA and GGS-ABA. The quantificationwas performed using electrospray ionization liquid chromatography-mass spectrometry(ESI LC-MS). The important parameters for hydrolysis were optimized, which include thetemperature, the reaction time, the acid concentration and volume as well as the micro-wave power. Compared to the conventional acid hydrolysis of 18–24 h using 6–12 M HClat 110 °C applied commonly in the literature, MAAH of proteins can be completed as fastas in only 2–10 minutes and, additionally, with a 3–5 times higher yield of the finalderivatization products. Furthermore, a better agreement between the ratio of the detect-ed derivatization products and the theoretical yields from the studied protein has alsobeen achieved, which indicates that MAAH may serve as a more reliable method of acidhydrolysis for this purpose than that with conventional thermal heating. The MAAH meth-od is demonstrated to be a time-saving, reproducible and efficient technique for studyingAAS and GGS as protein oxidation markers using LC-MS.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-344-TU
Quantification of Reduced Glutathione (GSH) as Biomarkerof Toxicity in Reconstructed Human Skin Models
Eric Arbey, Guillaume Léreaux, Jean-Roch Meunier, Joan Eilstein, Daniel DuchéL’Oréal Research & Innovation, Advanced Research Department,
93601 Aulnay-Sous-Bois, Cedex, France
Contact dermatitis results from the interaction between a hapten (small chemical containingelectrophilic sites) with the side chain of nucleophilic amino acids of endogenous proteins toform an antigenic complex which can induce skin sensitization. Chlorodinitrobenzene(CDNB) is a well known sensitizer that reacts with the cysteine residues of skin proteins by anaromatic nucleophilic substitution. The skin is not just a physical barrier but performs a widerange of metabolic activities and detoxification reactions as well [Gibbs et al., 2007]. Previousstudies showed that CDNB is transformed and detoxified from a normal human skin (NHS)and reconstructed human skin models (skin models) by glutathione S-transferases (GST) intoa glutathione adduct (DNP-SG) [Léreaux et al., 2010]. Then, the toxicity of CDNB couldappear after the GST becomes saturated or after cofactor is running out. Reduced glutathione(GSH), the cofactor of GST activities is a small peptide of three amino acids (glycine-cysteine-glutamic acid) that plays the role of cellular anti-oxidant and reactive intermediate scavenger[Pompella et al., 2003]. Thus, the DNP-SG and GSH concentrations were quantified in thereconstructed human skin model EpiskinTM according to increasing amounts of CDNB todetermine the apparent enzymatic parameters of the GST activity (Kmapp, Vmapp) and the skinmodel capacities to preserve the cellular GSH homeostasis. The amounts of DNP-SG formedand residual GSH (and oxidized glutathione) were measured by UV-HPLC and MS/MS-HPLCmethods, respectively. Note that before the analysis, GSH was derivatized with N-ethyl-maleimide. Results showed an apparent affinity (Kmapp) and an apparent maximal velocity(Vmaxapp) of 37.7 ± 4.1 μM and 51.0 ± 2.3 pmol of DNP-SG/min/mg of protein, respectively.Also, we observed that skin model can produce a De Novo GSH’s synthesis allowing to pro-tect a cellular homeostasis of GSH up to CDNB concentrations upper to 32 μM for which thetotality of the basal GSH pool was re-formed. So, this GSH assay and skin model can allowthe cosmetic industry to detect electrophilic compounds, determine the concentration fromwhich they become toxic and eliminate potential skin sensitizers.
ReferencesGibbs S, Van de Sandt JJM, Merk HF, Lockley DJ, Pendlington RU and Pease CK (2007) Xenobiotic metabolism
in human skin and 3D human skin reconstructs: A review. Current Drug Metab 8: 1–15Léreaux G, Eilstein J, Meunier JR, Leclaire J and Duché D (2010) Characterization of N-acetyl and glutathione
S-transferase avities in skin and reconstructed human skin models. 9th International ISSX Meeting, Turkey(abstract 211)
Pompella A, Visvikis A, Paolicchi A, De Tata V, Casini AF (2003). The changing faces of glutathione, a cellularprotagonist. Biochem. Pharmacol. 66: 1499–503

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-345-MO
Synthesis and Analysis of Neoglycoproteinsto Generate Carbohydrate Specific Antibodies
for Glycomics Based Biomarker Discovery
Márta Kerékgyártó1, Csaba Váradi1, Stefan Mittermayr1, Ákos Szekrényes1,Ildikó Olajos2, István Kurucz3
1Horváth Laboratory of Bioseparation Sciences,University of Debrecen, Hungary
2Institute of Biology and Ecology, Faculty of Sciences and Technology,University of Debrecen, Hungary
3Biosystems International Kft.,Debrecen, Hungary
Carbohydrate specific antibodies can be generated by immunizing vertebrates with specif-ic (oligo)saccharide structure containing antigens. These antibodies possess specificity forthe carbohydrate residues present as structural components of the antigen. The anti-carbohydrate antibodies are useful as diagnostic markers for detecting oligosaccharides,which might be characteristic for various diseases. For immunization purposes, thesesugar structures are usually covalently linked to macromolecular carriers such as bovine orhuman serum albumin and the resulting glycoconjugates are referred to as neoglyco-proteins. Here we report on the synthesis of maltose conjugate neoglycoproteins usingtwo reductive amination based approaches. The carbohydrate antigen (maltose) waslinked to the lysine ε-amino groups of BSA to form a Schiff base, which was consequentlyreduced with sodium cyanoborohydride to obtain a stable conjugate. In one case, theglucose at the reductive end of the maltose served as spacer (the ring opened up duringthe conjugation reaction). In the other case, an aglycon spacer of 7-(1,3-dioxan-2-yl)-heptan-1-ol was utilized conserving in this way the intact annular maltose structure ascarbohydrate antigen. The degree of sugar incorporation in the carrier protein was deter-mined by multicapillary gel electrophoresis and MALDI-TOF mass spectrometry. Thesenewly synthesized neoglycoproteins were used to immunize BALB/c-mice in order togenerate novel, sugar specific antibodies. The specificity of the polyclonal antibody re-sponse was investigated by enzyme immune-assays, where bindings of the antibodieswere demonstrated to non- and differently modified carrier proteins.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-346-TU
Universal Fluorescent Multiplex PCR and Capillary Electrophoresisfor Evaluation of Gene Conversion between SMN1/SMN2
in Spinal Muscular Atrophy
Shou-Mei Wu, Chun-Chi WangSchool of Pharmacy, College of Pharmacy, Kaohsiung Medical University,
100, Shih-Chuan 1st Road, Kaohsiung, Taiwan
In this research, we developed a capillary electrophoresis (CE) method with universalfluorescent multiplex PCR to simultaneously detect the SMN1/SMN2 genes in exon 7 and8. Spinal muscular atrophy (SMA) was a highly frequent inherited disease caused byabsence of the SMN1 gene in approximately 94% patients. Those patients were foundwith deletion of SMN1 gene or gene conversion between SMN1/SMN2. However, mostmethods were only focusing on the analysis of whole gene deletion, but ignoring geneconversion. Simultaneous quantification of SMN1/SMN2 in exon 7 and 8 is a good strate-gy for estimating SMN1 deletion or SMN1/SMN2 gene conversion. This study establisheda CE separation allowing differentiation of all copy-ratios of SMN1/SMN2 in exon 7 and 8.Among 212 detected individuals, there were 23 SMA patients, 45 carriers and 144 normalsubjects. The three individuals existed different ratios of SMN1/SMN2 in two exons, in-cluding a SMA patient having only two SMN2 copy in exon 7 but one SMN1 copy presentin exon 8. This method could provide more information about SMN1 deletion orSMN1/SMN2 gene conversion for SMA genotyping and diagnosis.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-347-MO
A New HPLC Method for the Determination of Neopterin,Kynurenine, Tryptophan and Creatinine in Exudates
and Amniotic Fluid
Lenka Krcmová1,2, Dagmar Solichová2, Bohuslav Melichar3, Petr Solich1
1Department of Analytical Chemistry, Charles University, Faculty of Pharmacy,Hradec Králové, Czech Republic
2Department of Metabolic Care and Gerontology, University Hospital,Hradec Králové, Czech Republic
3Department of Oncology, Palacký University Medical School,Olomouc, Czech Republic
Neopterin is a heterocyclic compound which is primarily produced by human macro-phages activated with Th1-type cytokine interferon-gamma. Thus, neopterin is an indica-tor of systemic immune activation. Like neopterin formation, the kynurenine/tryptophan-ratio reflects cellular immune response and IFN-γ-mediated T-cell activation and is in-creased in clinical conditions similar to those neopterin concentrations are increased.
The measurement of neopterin, kynurenine and tryptophan in exudates may be usedto assess the immune activation systemically or locally, in the microenvironment.Neopterin determination has been widely used in laboratory medicine. In addition,neopterin determination may be used to predict prognosis in cancer patients or to moni-tor the effect of therapy.
Determination of neopterin in amniotic fluid is useful for prenatal diagnostic of GTPcyclohydrolase deficiency and thus pteridine synthesis and may reflect the maturation ofpteridine metabolism in the fetus.
A new HPLC method for the simultaneous determination of neopterin, creatinine, kyn-urenine and tryptophan in various biological human fluids (amniotic fluid and exudates) wasdeveloped and validated. Monolithic stationary phase technology (two silica gel monolithiccolumns RP C18e, 50 x 4.6 mm + 100 x 3.0 mm, were connected with guard monolithiccartridge, +10 x 4.6 mm). As mobile phase 15 mmol/L phosphate buffer at pH 4.50 wasused. Neopterin and tryptophan were detected using fluorescent detection and kynurenineand creatinine were detected by diode-array detection. Using of special auto sampler formicro titration plates (samples are storage in dark cooled place protected against evapora-tion) was combined with easy sample preparation step. This method may be suitable forlarge sequences of samples in clinical research and routine practice.
Supported by: MSM0021620822, MZO00179906, MSM0021620820

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-348-TU
A Rapid and Precise HPLC-Based Lipase Assay
Astrid Wirtz1, Kathrin Klein1, Laura Schneider1, Martina Pohl2,Karl-Erich Jaeger1, Ulrich Krauss1
1Institut für Molekulare Enzymtechnologie der Heinrich-Heine-Universität Düsseldorfim Forschungszentrum Jülich, 52426 Jülich, Germany
2Institut für Bio- und Geowissenschaften, Forschungszentrum Jülich,52425 Jülich, Germany
Lipases are an important class of enzymes which catalyze the hydrolysis of a wide range offatty acid esters. In order to quantify lipolytic activity, routinely the para-nitrophenolpalmitate (pNPP) assay [1] is employed. Hereby, the physical properties of common lipasemodel substrates such as pNPP, like their low solubility in water, leads to turbid solutions(OD410 ~ 1.5). This severely compromises the photometric detection of the released chro-mogen para-nitrophenol (pNP). Moreover, the homogeneity and stability of the water/substrate emulsion may further limit the precision and accuracy of the assay. To overcomethose problems we developed a more reliable lipase assay by adapting the photometricpNPP assay. We use high-performance liquid chromatography (HPLC) to quantify thereaction product pNP formed by hydrolysis from pNPP by the action of lipolytic enzymes.Although, HPLC-based lipase assays using fully porous column materials have been de-scribed before [2], none of those methods employ next generation column materials suchas the KinetexTM Core-Shell technology.
In order to separate and quantify the reaction product pNP from residual substrate weemploy the high efficiency KinetexTM 2.6 μm Core-Shell LC-column (Phenomenex) mount-ed on an conventional HPLC system (Shimadzu, LC-2010AHT). Due to the superior proper-ties of this column material reduced separation times (3 minutes) are possible. Assayperformance was validated using lipase A from Bacillus subtilis purified as previously de-scribed [3].
The developed assay is very robust and offers good reproducibility in both intra- andinterday experiments (standard deviation below 4%). The method shows appropriatelinearity values (R2 = 0.999) and was found suitable to determine initial rate velocities. Inconclusion the assay should be applicable both in biotechnological as well as biomedicalsettings were lipase activities have to be determined routinely with high accuracy.
1. U. K. Winkler, M. Stuckmann, J Bacteriol 1979, 138, 663–670.2. V. Maurich, M. Moneghini, M. Zacchigna, A. Pitotti, E. Lencioni, J Pharm Biomed Anal 1991, 9, 427–431.3. G. van Pouderoyen, T. Eggert, K. E. Jaeger, B. W. Dijkstra, J Mol Biol 2001, 309, 215–226.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-349-MO
Advantages of UPLC in Biopharmaceutical Analysis
Yajun Jennifer Wang, Jennifer Rea, Tony G. Moreno,Tomasz Baginski, Dell Farnan
Protein Analytical Chemistry, Genentech Inc. A Member of the Roche Group,South San Francisco, CA 94080, USa
Chromatographic analyses are ubiquitous in biotechnology. The complex structures oflarge molecules including monoclonal antibodies necessitate that a wide range of chro-matographic methods are utilized to assess the product quality attributes and proteinheterogeneity. Frequently used separations include reversed phase, size exclusion, ion-exchange chromatography, and hydrophobic interaction chromatography. Increasing thethroughput and decreasing the sample requirements for these methodologies has been afocus of our research for several years. With the recent advancement in UPLC technologyand the use of sub-2 micron particle column packing, we not only can increase through-put and sensitivity, but also improve peptide recovery in some cases. Data will be present-ed to demonstrate the advantages of these methods for screening applications in formula-tion and recovery development, for product characterization using peptide map analysisvia LC-MS-MS, and for analysis of PNGase F released N-linked glycans.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-350-TU
Analysis of Antibiotics in Biological Fluidsusing Surfactant Mediated Mobile Phases
Maria Rambla-Alegre1, Julián Paños-Pérez2, Juan Peris-Vicente1,Inmaculada Casas-Breva3, Devasish Bose4, Nitasha Agrawal4,
Josep Esteve-Romero1, Samuel Carda-Broch1
1Department of Physical and Analytical Chemistry, Universitat Jaume I,12071 Castelló, Spain
2Department of Inorganic and Organic Chemistry, Universitat Jaume I3Hospital La Plana,
Vila-real, Spain4Department of Criminology & Forensic Science, Dr. H.S.Gour University,
Sagar, India
Antibiotics, or antibacterial agents, are chemicals of a low molecular weight, synthetic orproduced by microorganisms that, in small concentrations, are capable of inhibitinggrowth and of even killing certain microorganisms. Their main applications are in humanand veterinary medicine, and agriculture. In humans, they are used in the treatment ofurinary tract, respiratory and other systemic-type infections. Interest in antibiotics hasgrown in parallel with an increasingly high degree of productivity in the field of analyticalapplications. Therefore it is necessary to develop chromatographic procedures capable ofdetermining various drugs simultaneously in the shortest possible time. Micellar liquidchromatography is a reversed-phase liquid technique which offers advantages over con-ventional HPLC as far as sample preparation, selectivity and versatility are concerned. Itseluent characteristics also allow the analysis of solutes in a wide range of polarities, froma single injection to isocratic elution. It should be noted that the organic solvent concen-tration is much lower than that used in conventional RPLC. For this reason, the mobilephases in MLC are less toxic, non-flammable with a lower cost and of less environmentalimpact. The main advantage of this technique is that samples can be injected directly intothe chromatographic system with no previous step. The research work herein includedhas implied the determination of penicillins and quinolones in biological fluids usingsurfactant mediated mobile phases.
This work was supported by the projects from the Spanish Ministerio de Educación y Ciencia (MEC) CTQ200764473/BQU. Maria Rambla-Alegre also wishes to thank the MEC for her FPU grant.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-351-MO
Analysis of Biocides with the PerkinElmer Flexar FX-15 SystemEquipped with a PDA Detector
April DeAtley, Njies PedjiePerkinElmer,
Shelton, CT, USA
Biocides are chemical substances that are used to kill harmful organisms. Biocides have awide range of application s in the food industry, agriculture, cosmetic, pharmaceutical,and in construction. In the food industry methylparaben is used as an antifungal and as apreservative; benzoic acid and its salts are widely use in beverages to prevent the growthof mold, yeast and some bacteria. In cosmetic products, methylisothiazoline is used toprevent the growth of microbes. In agriculture carbendazim is used as a fungicide and iseffective in treating fungal diseases of trees. In construction benzisothiazoline is used asan antimicrobial and fungicide in adhesive and varnishes.
While undoubtedly biocides have useful applications, their use need to be regulatedbecause they are very toxic and can cause death at concentration not tolerated by hu-man. In recent years, there has been considerable concern that the indiscriminate use ofbiocides increases bacteria resistance which can cause previously treatable diseases to bebecome untreatable. In different industries where biocides are use, there is a need forsimple analytical method to constantly monitor the type and amount to insure that theirusage is safe and comply with regulations.
This application note will present a robust and sensitive reverse phase liquid chroma-tography method for the analysis of some widely used biocides. Method conditions andperformance data including precision and linearity are presented. A widely use antimicro-bial hand soap is analyzed and the biocides type and concentration confirmed.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-352-TU
Analysis of Lidocaine and its Metabolite in Plasma Samplesof Parturient Women with Gestational Diabetes Mellitus
by In-Tube Solid Phase Microextraction Coupledto Liquid Chromatography (In-Tube SPME/LC)
Bruno José Gonçalves da Silva1, Juciene Aparecida Caris1, Vera Lúcia Lanchote2,Maria Eugênia Costa Queiroz1
1Departamento de Química, Faculdade de Filosofia Ciências e Letras de Ribeirão Preto,Universidade de São Paulo,
Ribeirão Preto 14040-901, SP, Brazil2Departamento de Análises Clínicas, Toxicológicas e Bromatológicas,
Faculdade de Ciências Farmacêuticas de Ribeirão Preto, Universidade de São Paulo,Ribeirão Preto 14040-901, SP, Brazil
Lidocaine is a local anesthetic of the amide type used for anesthetic infiltration and/orregional blockade. It acts by inhibiting the ion flow necessary for the conduction of im-pulses from pain sensation fibers. However, dysfunction or changes in the activity oforgans and systems due to a diagnosed disease such as Gestational Diabetes Mellitus(GDM) as well as modifications in the maternal organism caused by pregnancy can alterthe kinetic disposition and the metabolism of lidocaine and other drugs. In such cases, alaboratory measurement of drug and metabolites plasma levels becomes mandatory forunderstanding of the pharmacology of this important anesthetic prescribed for parturientwomen.
In-tube solid-phase microextraction (in-tube SPME), an effective sample preparationtechnique, has been successfully applied to the analysis of drugs in biological fluids.Organic compounds are directly extracted and concentrated into the stationary phase ofcapillary columns by repeated draw/eject cycles of sample solution. In-tube SPME is anideal sample preparation technique because it is fast to operate, easy to automate, sol-vent-free, and inexpensive. It can be automated to perform several different tasks includ-ing continuous extraction, concentration, desorption, and injection, by using an auto-sampler that is being employed used in combination with high performance liquid chro-matography.
In this study, an in-tube SPME/LC-UV method using OV-1701 (14% cyanopropylphenylmethylpolysiloxane) capillary column was developed and validated for evaluation of theinfluence of GDM on the pharmacokinetics of lidocaine and its metabolite monoethyl-glycinexylidide (MEGX) in plasma samples from parturient women subjected to periduralanesthesia. Important factors in the optimization of in-tube SPME efficiency are discussed,including the sample draw/eject volume, number of draw/eject cycles, draw/eject flowrate, pH, and influence of plasma proteins. The limit of quantitation for both lidocaine

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
and MEGX in plasma was 50 ng mL−1, and the interassay precision had variation lowerthan 8%. The response of the in-tube SPME/LC-UV method for the drugs was linear overa dynamic ranging from 50 to 5000 ng mL−1, with correlation coefficients better than0.9985, and accuracy from 94.7 to 117.2%. After validation of the in-tube SPME/LC-UVmethod, it was successfully applied for evaluation of the kinetic disposition of lidocaine inparturient women with GDM.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-353-MO
Analysis of Protein Primary Structure using Wide PoreSub 2 μμm Particles and UHPLC
Phu T. Duong1, Brian A. Bidlingmeyer1, Linda L. Lloyd2
1Agilent Technologies,2850 Centerville Road, Wilmington, DE 19808, USA
2Agilent Technologies UK Limited,Essex Road, Church Stretton, Shropshire, SY6 6AX, UK
Due to the heterogeneity of a protein biopharmaceutical it is necessary to use a numberof chromatographic techniques to fully characterize the API. Methods include size exclu-sion chromatography for the quantitation of dimers and aggregates and ion exchange forthe identification of charge variants. Both of these techniques use aqueous eluents andnon-denaturing conditions. As part of the full characterization of a protein it is also neces-sary to look at the primary amino acid sequence and any post translational modificationsto that sequence that may have occurred during the purification or formulation steps ofmanufacture. To perform this type of analysis denaturing conditions are required and soreverse phase HPLC is normally the technique of choice.
We have evaluated a new reverse phase media, Zorbax 300SB 1.8 μm, for the reversephase separation of a typical protein biopharmaceutical, insulin. Using denaturing condi-tions we have investigated whether it is possible to use sub 2 μm particles for proteinseparations and if the advantages seen for small molecules can also be achieved for pro-tein separations.
The data presented here shows how the use of a 1.8 μm column designed for use withUHPLC systems can significantly reduced the analysis time, critical for increasing theefficiency of QC, for protein primary structure analysis. The separations we present willdemonstrate how this technology can be used to achieve resolution of various insulinisoforms.
The eluents routinely used for reverse phase analysis are acidic, contain trifluoroaceticacid or formic acid, which can reduce the lifetime of many HPLC columns. We haveshown that by using StableBond technology it is possible to produce an HPLC mediawhich is stable under the acidic conditions and will provide robust reproducible separa-tions required for protein QC.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-355-MO
Boronate Functionalised Polymer Monoliths for Identificationof New Biomarkers in Type II Diabetes Mellitus
Anikó TakátsyUniversity of Pécs, Medical School Department of Biochemistry and Medical Chemistry,
7624 Pécs, Szigeti Street 12, Hungary
Glycolipids and glycoproteins have been identified as biomarkers for a range of importantdiseases, potentially leading to new therapeutic and diagnostic techniques. Amadoricompounds (ACs), the products of nonenzymatic glycation processes (e.g. in diabetes)and advanced glycation endproducts (AGEs) are known to be responsible for the develop-ment of diabetic complications such as diabetic nephropathy and retinopathy [1]. Sincethe 1970s, the measurement of glycated hemoglobin A1c (HbA1c) has been used routine-ly as a clinical diagnostic marker for relatively long-term (4–6 weeks) glucose control indiabetic patients [2]. However, to find more specific and informative protein biomarkersfor monitoring the glycemic state and to get deeper insight into the role of glycation inthe development of diabetic complications, comprehensive proteomic studies are re-quired for identifying those glycated proteins whose altered structures may contribute topathology.
Monoliths and in particular polymer monoliths which are continuous macroporousmedia have several advantages over packed particle columns. Among others the mono-liths can be synthesised by in situ polymerisation in various geometrical formats, theirhigh permeability allows resolution to be maintained at higher flow rates, they expectedto solve the interparticular voids (typical for the packed capillary columns used in HPLC)and it is possible to accurately control the surface chemistry during polymerisation toachieve the desired surface functionality as long as beside the high specificity the sub-strate effect would be smaller than in the case of e.g. agarose.
Although affinity chromatography is a favourable technique nowadays and the syn-thesis of new monoliths is also in the focus of the analytical chemistry, there is a lack ofa new powerful specific enriching phase for glycated proteins and peptides. The aim ofthis project was to polymerise boronate coupled monolith with enhanced capacity,giving smaller substrate effect for the fast identification of new glycation sites of pro-teins and glycated peptides isolated from biological samples. Analysis of the glycatedpeptides were completed with MALDI (matrix assisted laser desorption ionisation) massspectrometer TOF (time of flight) detector for fast mass characterization in a wide massrange.
The more sensitive and exact specification of the Amadori-products can serve as poten-tial biomarkers [3].

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
1. D.R. McCance, D.G. Dyer, J.A. Dunn, K.E. Bailie, R.S. Thorpe, J.W. Baynes, T.J. Lyons, Maillard reaction
products and their relation to complication in insulin dependent diabetes mellitus, J. Clin. Invest. 91
(1993) 2470–2478.
2. L.A. Trivelli, H.M. Ranney, H.-T. Lai, Hemoglobin components in patients with diabetes mellitus, N. Engl. J.
Med. 284 (1971) 353–357.
3. A. Takátsy, K. Böddi, L. Nagy, G. Nagy, Sz. Szabó, L. Markó, I. Wittmann, R. Ohmacht, T. Ringer, G. K.
Bonn, D. Gjerde, Z. Szabó, Enrichment of Amadori products derived from the nonenzymatic glycation of
proteins using microscale boronate affinity chromatography. Analytical Biochemistry 393 (2009) 8–22.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-356-TU
By-Products in Recombinant TAU Protein Production Characterizedby LC-MALDI Top-Down Sequencing (LC-MALDI-TDS)
Sven Meyer1, Arndt Asperger1, Marcus Macht1, Verena Tellsroem1,Branislav Kovacech2, Andrej Kovac2
1Bruker Daltonik GmbH,Bremen, Germany
2Inst. of Neuroimmunol., Slovak Academy of Sciences,Bratislava, Slovakia
Compared to Edman sequencing, MALDI-TOF based top-down sequencing of intactproteins (MALDI-TDS) is much faster, can deliver significantly longer sequence readoutfrom both protein N- and C-terminus and has no limitations in case of terminally modifiedproteins. These features make MALDI-TDS an extremely powerful method for the QCanalysis of recombinant protein products, for instance biopharmaceuticals. While directMALDI-TDS analysis requires purified, homogeneous protein samples, coupling MALDI-TDS to upfront LC separation allows analyzing proteins in mixtures.
Recombinant protein products often contain unexpected by-products. As an example,we describe here the characterization by LC-MALDI-TDS of two by-products that occurredin a recombinant TAU protein expression product.
The recombinant TAU protein was dissolved in 0.1% TFA in water. Approximately 100pmol was injected onto a capillary LC system equipped with a monolithic column (sta-tionary phase PS-DVB, 200 μm I.D., 5 cm long) held at 60 °C. A linear gradient from 10to 70 percent acetonitrile was run at a flow rate of 2 μl/min. Eluate fractions were spottedevery 20 s on a MALDI target plate pre-spotted with sDHB matrix.
MALDI-MS spectra of the deposited LC fractions were collected using MALDI-TOFMS inpositive linear mode. MALDI-TDS spectra were acquired in positive reflector mode fromthose target positions yielding protein MS signals.
Direct MALDI-TOF analysis of the recombinant TAU protein product had revealed thepresence of multiple minor components of unknown identity. These components wereseparated from the main compound, the expected TAU protein, by means of capillary LC.Thus, the unknown by-products became accessible to analysis by MALDI-TDS, which usesthe in-source decomposition (ISD) of proteins in the MALDI plume, but does not providefor precursor ion selection.
sDHB was chosen as a MALDI matrix for the analysis described here as it allows toacquire intact protein MS spectra and, at the same time, is most suitable to yield bothN-terminal and C-terminal in-source decay fragments.
Identification of the pre-separated protein by-products was achieved by matching theobtained MALDI-TDS data against the amino acid sequence of the expected TAU protein.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
A 16 kDa compound was identified to represent a truncated version of TAU covering theN-terminal sequence TAU-[1–161]. The truncation site was unambiguously determined bymeans of matching series of C-terminal y- and z-type fragment ions in the MALDI-TDSspectrum. A further protein by-product was identified as TAU-[2–242] with the N-terminalmethionine residue excised. The identity of this component could be derived from match-ing series of N-terminal c-type fragment ions observed in the respective TDS spectrum.
LC-MALDI-TDS turned out to be very efficient in the analysis described above. Com-pared to alternative techniques, for example Edman sequencing or typical bottom-upmass spectrometric approaches like LC-MS/MS of enzymatic digests out of SDS-PAGE gelbands, the described LC-MALDI-TDS method was significantly faster and produced aminimum amount of data to generate the requested information.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-357-MO
Carbon Nanotubes as a Gene Delivery Vehiclefor Plant Cell Transfection
Maged F. Serag1, Noritada Kaji2, Yukihiro Okamoto1,Manabu Tokeshi1, Yoshinobu Baba1,3
1Department of Applied Chemistry, Graduate School of Engineering,FIRST Research Center for Innovative Nanobiodevice, Nagoya University,
Nagoya, Japan2Graduate School of Science, Nagoya University,
Nagoya, Japan3Health Research Institute, National Institute of Advanced Industrial Science and Technology (AIST),
Takamatsu, Japan
Carbon nanotubes are currently expected as a useful tool not only in the field of semicon-ductor but also in the field of biomolecular delivery into cells since their extraordinaryphysical, chemical, and mechanical properties. Major barriers to delivery of biomoleculesare crossing the cellular membranes and achieving a high cytoplasmic concentration bycircumventing entrapment into endosomes and other lytic organelles. In our previouswork, we proposed cellulase-modified carbon nanotubes, which have an ability to digestmajor components of cell walls, for making a small hole on the cell wall in order to en-hance DNA uptake into cytoplasm. However some processes including the modification ofthe enzyme onto the CNT and long-term preservation problem were major drawbacks tobe a conventional transfection technique. Motivated by such aim, we have investigatedthe capability of multiwalled carbon nanotubes (MWCNTs) to penetrate the cell mem-brane of plant protoplasts (plant cells made devoid of their cell walls via enzymatic treat-ment) and studied their internalization mechanism via confocal imaging and TEM tech-niques. Our results indentified an endosome-escaping uptake mode of MWCNTs by plantprotoplasts. Moreover, short MWCNTs (< 100 nm) were observed to target specific cellu-lar substructures including the nucleus, plastids, and vacuoles. These findings are expect-ed to have a significant impact on plant cell biology and transformation technologies.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-358-TU
Characterization of a Therapeutic Protein by OptimizedPeptide Mapping, SEC and IEX
Srividya Kailasam1, Siji Joseph1, Angelika Gratzfeld-Huesgen2
1 Agilent Technologies India Pvt. Ltd.,‘C’ Block, RMZ Centennial, Plot No. 8A-8D, Donddanakundi Industrial area, ITPL Road,
Mahadevapura Post, Bangalore 560048, Karnataka, India2 Agilent Technologies Waldbronn,
Germany
The physico-chemical characterization of therapeutic proteins is important to ensure drugsafety and efficacy. Identity of a protein drug can be effectively demonstrated by severalLC-based techniques, like peptide mapping, SEC and IEX. These techniques are appliedduring all the stages of the drug development. Reverse phase HPLC is used to constructthe peptide map of the therapeutic protein. The proteolytic digest of the protein is ana-lyzed and the goal of the analysis is to chromatographically resolve and detect all proteinfragments. Thus complete sequence coverage is achieved. Due to the complexity of theanalyzed sample containing hundreds of different compounds, peptide mapping processtherefore required very long analytical runs.
In addition to identification by peptide mapping, SEC is used to establish the purity ofthe NBE. This process helps discriminate between the monomeric molecular species fromthe dimeric or multimeric species that can cause allergic adverse responses. IEX is com-monly applied to demonstrate the absence or presence of acidic or basic charge variantsof the main component
In this study we demonstrate the complete characterization of a small therapeuticprotein P128 using the Agilent 1260 Infinity bio-inert LC system and the Bio-LC columns.The bio-inert LC which is completely bio-inert, corrosion resistant and high pH compati-ble is ideal for the separation and characterization of biomolecules.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-359-MO
Characterization of Amino Acid Profiles of Culture Mediausing Ultra Performance Liquid Chromatography,AQC Pre-Column Derivatization and UV Detection
Gregor Fiechter, Helmut K. MayerDepartment of Food Science & Technology, Food Chemistry Laboratory,
BOKU – University of Natural Resources & Life Sciences Vienna,Muthgasse 11, A-1190 Vienna, Austria
Considering the variety of different amino acid protocols, commercialized HPLC-designedamino acid solutions may offer some advantages as to tailor-made ready-for-use solventsystems, customized derivatization procedures, and already optimized gradient elution.However, regarding the complexity of sample matrices and the increasing demand forminimizing sampling time, conventional HPLC may lack some efficiency respective therequired sensitivity, runtime, resolution or peak capacity.
Emphasizing state-of-the-art fast LC-techniques, the Waters AccQ.TagTM method as awell-established amino acid solution designed for HPLC, 6-aminoquinolyl-N-hydroxy-succinimidyl carbamate (AQC) derivatization, and fluorescence detection, was properlytransferred and accordingly optimized for the application on ultra performance liquidchromatography (UPLCTM) and UV detection.
As compared to the original HPLC method, UPLCTM proved to be superior by facilitatingexcellent separations of 20 amino acids derivatives within 12 min only, thus demonstrat-ing significantly shortened runtimes (> 35 min for HPLC) and an enhanced chromato-graphic performance. Using UV instead of fluorescence detection, amino acid quantifica-tion after pre-column AQC derivatization yielded appropriate sensitivities with corre-sponding detection limits varying within the low pmol range.
Moreover, this newly established UPLCTM method was applied to characterize changesin the free (FAA) as well as total amino acid (TAA) profiles specific to culture media atthree typical production stages of a biotechnological compound (fresh medium, fer-mentation broth after cell mass production, and after product expression at fermenta-tion end). Amino acid profiles intrinsic to the fresh medium indicated free, hence morebioavailable, amino acids at a FAA/TAA ratio of 40%, whereas ongoing fermentationimplied a rather specific, successive decline in selective FAA concentrations. Thereby, themost distinctive variations in FAAs were highlighted by aspartic acid, serine and threo-nine, each exhibiting an almost complete uptake from the culture media (−96 to −99%)along the entire fermentation process, with remaining FAA/TAA ratios of 1%, 8%, and1% respectively. This in fact may strongly indicate arising limitations of selective aminoacids within the nutrient broth, hence showing either an insufficient supplementation ora well designed fermentation process with an efficiently calculated amino acid uptake.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
Thus, by identifying shortages in supplementation material, amino acid monitoringusing rapid techniques, such as UPLCTM, can contribute a valuable tool to properlyoptimized or re-adjust culture media to specific requirements in applied fermentationprocesses.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-360-TU
Characterization of Praziquantel and its Metabolitesby Ultra-Performance Liquid Chromatography Coupled
with Electrospray Tandem Mass Spectrometry
Veronika Jedlicková1, Robert Jirásko1, Michal Holcapek1,Ivan Vokrál2, Lenka Skálová2
1University of Pardubice, Faculty of Chemical Technology, Department of Analytical Chemistry,Studentská 573, 532 10 Pardubice, Czech Republic
2Charles University, Faculty of Pharmacy, Department of Biochemical Sciences,Heyrovského 1203, 500 05 Hradec Králové, Czech Republic
Praziquantel (PZQ) is a broad-spectrum anthelmintic drug used in cases of trematodes,cestodes and schistosomal infection. It is metabolized mainly in liver, where hydroxylatedmetabolites and glucuronide conjugates are typically formed. The excretion of thesemetabolic products is ensured by renal system. In our work, PZQ was incubated with rathepatocytes and microsomes. Subsequently, the solid-phase extraction of microsomal andhepatocyte incubation mixtures was performed. Moreover, rat urine samples were ob-tained from in vivo experiments after PZQ oral administration. The comparison of incuba-tion samples with placebo experiments simplified the identification of metabolites. Stud-ied samples were analyzed using optimized UHPLC coupled with electrospray ionizationtandem mass spectrometry method that provided information about PZQ microsomaland hepatic biotransformation pathways. Structures of particular phase I and II metabo-lites were suggested based on the high mass accuracy measurements in full scan andtandem mass spectrometry modes using QqTOF analyzer. The additional measurementsof MSn using ion trap analyzer were performed for the verification of fragmentation paths.
This work was supported by MSM0021627502 (Ministry of Education, Youth and Sports of the Czech Repub-lic) and P502/10/0217 (Czech Science Foundation).

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-361-MO
Chromatographic Determination of the AntimalarialAlpha-Beta Arteether
Josep Esteve-Romero1, Samuel Carda-Broch1, Maria Rambla-Alegre1,Adrián Martinavarro-Domínguez1, Juan Péris-Vicente1, Devasish Bose2,
Abhilasha Durgbanshi3, Nitasha Agrawal21Departament de Química Física i Analítica, Universitat Jaume I,
Castello, Spain2Department of Criminology and Forensic Science, Dr. Harisingh Gour University,
Sagar (M.P.) India3Department of Applied Chemistry, Institute of technology, Banaras Hindu University,
Varanasi (U.P.) India
A simple chromatographic procedure was developed for the determination of the anti-marial α-β arteether [1, 2]. After a methematical optimization, the recomended procedureuse a C18 column, a hybrid micellar mobile phase containing 0.15 M SDS-6% (v/v) buta-nol-0.01 M phosphate buffered at pH 7 with UV detection at 210 nm. Under these condi-tions the analysis time was less than 8 min. ICH Guideline validation studies were per-formed and calibration was linear in the range 0.25 to 5 ppm with r2 > 0.999, intra (n =10) and interday precision (n = 5) were found to be satisfactory. Method was applied tothe determination of arteether in different pharmaceutical preparation obtaining recover-ies in the 97–103% range. Compared with others chromatographic techniques, themicellar liquid chromatography has the advantages of avoiding sample pretreatment,reducing analysis time and toxicity to environment, low cost of per sample analysis.
1. Malaria vector control and personal protection: report of a WHO study group. Gene-va, World Health Organization, (2006).
2. Guidelines for the treatment of malaria. Geneva, World Health Organization, (2006).
This study was part of project CTQ2007-64473/BQU, funded by Ministeri d’Educació i Ciència (MEC). MariaRambla-Alegre also wishes to thank the MEC for her FPU grant.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-362-TU
Comparative Glycomics: Rapid and Confident IgGStructural Elucidation using a Multidimensional
Liquid Phase Separation Approach
Stefan Mittermayr1,2, Jonathan Bones1, Margaret Doherty1, Pauline M. Rudd1
1NIBRT Dublin-Oxford Glycobiology Laboratory, NIBRT– The National Institute for Bioprocessing Research and Training,
UCD Conway Institute of Biomolecular and Biomedical Research, University College Dublin,Belfield, Dublin 4, Ireland
2Horváth Laboratory of Bioseparation Sciences, Institute of Medical Chemistry, University of Debrecen,Debrecen, HU-4032, Hungary
N-glycans attached to the CH2 domains of the Fc region of IgG play an important role inboth stabilising and modulating the activity of the antibody. We compared the 1.7 μmhydrophilic interaction phase for UPLC with CE-LIF for comprehensive characterisation ofN-glycans released from polyclonal IgG from healthy human serum. A combination ofweak anion exchange based enrichment and exoglycosidase array digestions enabled theexhaustive identification of 32 IgG N-glycans using UPLC-fluorescence or CE-LIF. Combi-nation of the data demonstrated that complete structural annotation was subsequentlypossible within a twenty minute chromatographic and electrophoretic total analysis timedue to the advantageous orthogonality of the separation mechanisms. The combined useof both analytical platforms is therefore recommended for the rapid and comprehensiveanalysis of IgG N-glycosylation present on therapeutic antibodies or on antibodies ofbiomedical or pathological significance.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-363-MO
Comparison of Sialic Acid Determination in Glycoproteins:Two Liquid Chromatography Methods
Deanna C. Hurum, Jeffrey S. RohrerDionex Corporation,
1214 Oakmead Parkway, Sunnyvale, CA 94085, USA
Biopharmaceutical sialylation is important in determining glycoprotein bioavailability,function, stability, and eventual metabolism. Although over 50 natural sialic acids havebeen identified, two forms are commonly determined in glycoprotein products: N-acetyl-neuraminic acid (Neu5Ac) and N-glycolylneuraminic acid (Neu5Gc). Because humans donot generally synthesize Neu5Gc and have been shown to possess antibodies againstNeu5Gc, the presence of this sialic acid in a therapeutic agent can potentially lead to animmune response. Consequently, both total glycoprotein sialylation, and the identity ofthe sialic acids, play important roles in therapeutic protein efficacy, pharmacokinetics, andpotential immunogenicity.
This work compares two common sialic acid assays. Analyses by both high-performanceanion-exchange chromatography with pulsed amperometric detection (HPAE-PAD) andsample derivatization followed by UHPLC with fluorescence detection (UHPLC-FLD) areevaluated with comparisons of assay performance and total analysis time. Calibrationranges were chosen spanning the expected amounts of Neu5Ac and Neu5Gc in fiverepresentative glycoproteins; calf fetuin, bovine apo-transferrin, human transferrin, sheepα1-acid glycoprotein (AGP), and human α1-acid glycoprotein. For both methods, responseis linear with correlation coefficients > 0.9990 and > 0.995 for HPAE-PAD and UHPLC-FLDdetermination, respectively. In both cases, method sensitivity easily allows sialic aciddetermination in acid hydrolyzates of calf fetuin (14 μg), h. transferrin (20 μg), b. apo-transferrin (25 μg), h. AGP (13 μg), and s. APG (7 μg). Glycoprotein acid hydrolysateswere analyzed to evaluate method accuracy and precision. Retention time precision (RSDsof < 0.12) and peak area precision (RSDs of < 3.0) are good for both methods. Bothmethods investigated provided results that were consistent with previously publishedresults for the glycoproteins analyzed. As a measure of method accuracy, recoveries fromspiked samples ranged from 79–114% for Neu5Ac and 81–110% for Neu5Gc by UHPLC-FLD and from 74–107% and 72–103% for Neu5Ac and Neu5Gc by HPAE-PAD, respective-ly. Between-day assay precision (RSDs) ranged from 9.5–19% (UHPLC-FLD) and 7.9–14%(HPAE-PAD). Individual method precautions and advantages are illustrated using analysisresults from the five model glycoproteins.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-364-TU
C-terminal Processing Proteases from Pseudomonas Aeruginosa CleavePenicillin-Binding Proteins In Vitro
Astrid Wirtz1, Beatrix Santiago-Schübel2, Rien Hoge1,3,Frank Rosenau4, Diana Hofmann2
1Institut für Molekulare Enzymtechnologie, Heinrich-Heine-Universität Düsseldorf,Germany
2Forschungszentrum Jülich GmbH, ZCH/BioSpec,Jülich, Germany
3Clinical Pharmacy, Deventer Hospital,The Netherlands
4Institut für Pharmazeutische Biotechnologie, Universität Ulm,Germany
C-terminal processing proteases (CTPs) form a relatively new group of serine proteasesthat are involved in the carboxy-terminal processing of proteins [1]. The CTPs can befound in Eubacteria, Archaea, algae, plants and animals. In plants and algae one substrateis the photosynthetic protein D1, which needs to be cleaved to be functional and it there-fore essential for viability. Bacterial CTPs are involved in pathogenesis, among others, inPseudomonas aeruginosa which is an important cause of both community-acquired andhospital-acquired infections [2]. There are indications that CTPs in bacteria play a role incell wall synthesis in which they process penicillin-binding proteins (PBPs) [3]. Identifica-tion of the substrates and knowledge about the cleavage site is important for developingpossible novel antibiotic drugs. Here we present possible substrates and activities of twoCTPs from P. aeruginosa (CtpA and Prc).
Prc (E. coli, P. aeruginosa) and CtpA (P. aeruginosa) have been incubated with theC-terminal peptides of the D1-protein and PBP3 and PBP3A from P. aeruginosa overnight at 37 °C. Proteolysis products were separated on BioBasic-18 column (ThermoScientific) using gradient elution with a mobile phase of water and ACN. The HPLC wasdirectly coupled with a 4000QTRAPTM linear ion trap mass Spectrometer (Applied Bio-system/MDS SCIEX). The MS was operated in the positive EMS mode ion scanning from100 – 1800 m/z.
In contrast to most bacteria with only one CTP in their genome P. aeruginosa has twohomologous CTP proteases, namely CtpA and Prc. We could show that both, P.aeruginosa CtpA and Prc, and their homologue from E. coli are able to cleave the plantD1-peptide and one from a green algae. Cleavage occurs at a site similar to those inphotosynthetic organisms.
Furthermore, we have investigated the cleavage activity towards the penicilline-bindingproteins from P. aeruginosa PBP3 and PBP3A: CtpA and Prc were able to cleavage both

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
peptides with Prc being slightly more specific towards PBP3A, whereas CtpA showedhigher specificity towards PBP3. These results from our in vitro experiments stronglysuggest that these penicillin binding proteins are probably physiological targets of CTPs.
1. Silber KR, Keiler KC & Sauer RT (1992) Tsp: a tail-specific protease that selectively. Proc. Natl. Academ.
Sciences USA, 89, 295–299
2. Hoge R, Laschinski M, Jaeger KE, Wilhelm S & Rosenau F.(2010).The subcellular localization of a C-terminal
processing protease in Pseudomonas aeruginosa. FEMS Microbiol Lett., 316, 23–30
3. Hara H, Yamamoto Y, Higashitani A, Suzuki H & Nishumara Y (1991), Cloning, mapping, and characteriza-
tion of the E.coli prc gene, which is involved in C-terminal processing of penicillin binding protein 3. J.
Bacteriol., 173, 4799–4813

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-365-MO
Defining the Optimum Parameters for Efficient Size Separationsof Proteins
Linda Lloyd, Keeley MappAgilent Technologies UK Limited,
Essex Road, Church Stretton, Shropshire, SY6 6AX, UK
The requirement for precise and accurate information relating to protein molecularweight, size, aggregation and degradation varies according to the application. In drugdiscovery and process monitoring there is a requirement for speed but in QA/QC of finalproduct then the accuracy of the quantitation will be paramount. It is essential whenlooking at the choice of column and the method parameters that the objective of theseparation is clearly defined and the parameters that control the speed and resolution ofthe separation are fully understood.
In the work presented here we use a set of protein standards that span the typicalrange of characteristics of recombinant biopharmaceuticals – from antibodies to smallglobular proteins, to determine the influence on resolution of the selected parameters.
Parameters investigated include:1. Choice of SEC media, pore size and pore size distribution, particle size and surface
chemistry2. Columns dimensions3. Eluent pH and modifier content4. Linear velocityThe results of our experimental investigation shows the preferred options for SEC
separations based on the size and characteristics of the protein and enables recommenda-tion for the method parameters.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-366-TU
Determination of Benzophenones in Human Placental Tissue Samplesby Liquid Chromatography-Tandem Mass Spectrometry
Inmaculada Jiménez-Díaz1,2, Fernando Vela-Soria1, Rocío Rodríguez-Gómez1,Óscar Ballesteros1, Alberto Zafra-Gómez1, Alberto Navalón1,
Maria Fátima Fernández2, Nicolás Olea2
1Research Group of Analytical Chemistry and Life Sciences, Department of Analytical Chemistry,Campus of Fuentenueva, University of Granada,
E-18071 Granada, Spain2Laboratory of Medical Investigations, San Cecilio University Hospital, University of Granada,
CIBER Epidemiology and Public Health (CIBERESP),E-18071 Granada, Spain
Benzophenones (BPs) are a family of compounds widely used to protect the skin and hairfrom UV irradiation. Despite human exposure to BPs through dermal application of prod-ucts containing sunscreen agents and the increasing evidence that BPs are able to inter-fere with endocrine systems, few studies have examined the occurrence of BPs in humans.
In this work, we propose a new liquid chromatography-tandem mass spectrometry (LC-MS/MS) method to determine six BPs, namely, benzophenone-1 (BP-1), benzophenone-2(BP-2), benzophenone-3 (BP-3), benzophenone-6 (BP-6), benzophenone-8 (BP-8) and4-hydroxybenzophenone (4-OH-BP) in human placental tissue samples. The methodinvolves an extraction step of the analytes from the samples using ethyl acetate, followedby a clean-up step using centrifugation prior to their quantification by LC-MS/MS usingan atmospheric pressure chemical ionization (APCI) interface in the positive mode. Benzo-phenone-d10 (BP-d10) was used as surrogate. Found detection limits (LOD) ranged from0.07 to 0.3 ng g−1 and quantification limits (LOQ) from 0.3 to 1.0 ng g−1, while inter- andintra-day variability was under 12%. The method was validated using standard additioncalibration and a spike recovery assay. Recovery rates for spiked samples ranged from 91to 110%. This method was satisfactorily applied for the determination of BPs in 16 pla-cental tissue samples collected from women who live in Granada (Spain).

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-367-MO
Determination of Citrulline in Human Plasma Samplesusing a RP-HPLC Method
Agustín Acquaviva, Lílian Romero, Cecilia CastellsLaboratorio de Separaciones Analíticas, División Química Analítica, Facultad de Ciencias Exactas,
Universidad Nacional de La Plata47 y 115. (1900) La Plata, Buenos Aires, Argentina
Citrulline is a nonessential amino acid critical to the detoxification and elimination ofunwanted ammonia within cells; it is a precursor of arginine during metabolism and itsblood level is an important biomarker of several pathologies prognostics [1].
The amino acids concentration can be measured by reverse phase liquid chromatogra-phy using pre-column derivatization agents. Among several derivatization agents availablewe choose 9-fluorenylmethyl chloroformate (FMOC-Cl) because the reaction kinetics isvery fast at room temperature and the derivatives are very stable [2]. These features makethe automation a straightforward step. The pre-column reaction was carried out at pH 9.2conditioned by borate buffer; the reaction was stopped with β-phenylethylamine ortyramine after 1 minute of vortex shaking. Variables from this pre-column reaction wereoptimized.
Very good separation of FMOC-citrulline from other FMOC-amino acids usually presentin plasma was achieved by using an octadecylsilice column. The conditions of separationwere optimized by using an elution gradient between A solution of ACN:MeOH:Acetatebuffer (pH 4.2, 50 mM), and B solution of ACN:Acetate buffer.
The proposed methodology was applied to a human plasma sample. The methodestablished presents a short time-consuming, it is friendly green and feasible of automa-tion. Besides, this method applied to citrulline concentration determination can be usedas a diagnostic or monitoring method for a wide range of diseases fundamentally thoserelated to intestine failure.
1. Hui-ming Mao, Wei Wei, Wu-jun Xiong, Ying Lu, Bing-guan Chen, Zhongmin Liu. Clinical Biochemistry 43(2010) 1141–1147
2. Einarsson S., Josefsson B., Lagerkvist S. J. Chromatog., 282 (1983) 609–618

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-369-MO
Determination of Parabens in Human Placental Tissue Samplesby Liquid Chromatography-Tandem Mass Spectrometry
Inmaculada Jiménez-Díaz1,2, Fernando Vela-Soria1, Óscar Ballesteros1,Alberto Zafra-Gómez1, Alberto Navalón1, Maria Fátima Fernández2, Nicolás Olea2
1Research Group of Analytical Chemistry and Life Sciences, Department of Analytical Chemistry,Campus of Fuentenueva, University of Granada,
E-18071 Granada, Spain2Laboratory of Medical Investigations, San Cecilio University Hospital, University of Granada,
CIBER Epidemiology and Public Health (CIBERESP),E-18071 Granada, Spain
Endocrine disruptors are a group of organic compounds widely used, which are ubiqui-tous in the environment and in biological samples. The main effect of these compounds isassociated with their ability to mimic or block the action of natural hormones in livingorganisms, including humans. Parabens (esters of p-hydroxybenzoic acid) belong to thisgroup of compounds.
Many researchers hypothesize that exposure to EDCs during critical periods of develop-ment – in utero or early postnatal life – could cause morphologic and functional alterationsin wildlife and humans affecting growth, reproduction and also development [1, 2].
In this work, we propose a new liquid chromatography-tandem mass spectrometry(LC-MS/MS) method to asses the presence of parabens most commonly used in industrialapplications (methyl-, ethyl-, propyl- and butyl-paraben) in samples of human placentaltissue. The method involves the extraction of the analytes from the samples using ethylacetate, followed by a clean-up step using centrifugation prior to their quantification byLC-MS/MS using an atmospheric pressure chemical ionization (APCI) interface in thenegative mode. Deuterated bisphenol A (BPA-d16) was used as surrogate. Found detectionlimits (LOD) ranged from 0.03 to 0.06 ng g−1 and quantification limits (LOQ) from 0.1 to0.2 ng g−1, while inter- and intra-day variability was under 13.8%. The method was vali-dated using standard addition calibration and a spike recovery assay. Recovery rates forspiked samples ranged from 82% to 108%. This method was satisfactorily applied for thedetermination of parabens in 50 placental tissue samples collected from women who livein the province of Granada (Spain).
1. Fernández MF, Olmos B, Granada A, López-Espinosa MJ, Molina-Molina JM, Fernández JM, Cruz M, Olea-Serrano F, Olea N (2007) Environ Health Perspect 115:8–14.
2. Bosquiazzo VL, Varayoud J, Muñoz de Toro M, Luque EH, Ramos JG (2010) Biol Reprod 82(1):86–95

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-370-TU
Determination of Phytochemical Compoundsin Longan-Flower-Water-Extract (Dimocarpus Longans Lour.)
by HPLC and its Protection against Non-Alcoholic Steatohepatitis
Jung-Kai Tseng1, Yi-Chen Chen2, Deng-Jye Yang3, Chin-Lin Hsu4
1School of Optometry, Chung-Shan Medical University,110, Sec. 1, Jianguo N. Rd., Taichung 402, Taiwan
2Department of Animal Science and Technology, National Taiwan University,Taipei 106, Taiwan
3School of Health Diet and Industry Management, Chung-Shan Medical University,Taichung 402, Taiwan
4School of Nutrition, Chung-Shan Medical University,Taichung 402, Taiwan
Non-alcoholic Steatohepatitis (NASH) is primarily associated with obesity, insulin resis-tance, and the metabolic syndrome. The longan (Dimocarpus longans Lour.) is an impor-tantly economic fruit in Taiwan and the longan-flower-water extract (LFWE) contains alarge volume of antioxidant compounds, such as phenols, flavonoids, and condensedtannin. Through an HPLC analysis, in the LFWE flavonoids including (+)-catechin,epicatechin, and rutin and phenolic acids including gallic acid, gentisic acid, chlorogenicacid, vanillic acid, p-coumaric acid, ferulic acid, sinapic acid and syringic acid could befound in the LFWE. However, anthocyanins could not be detected in the extracts.Epicatechin (18.72 and 36.93 mg / 100 mL for the 1.25 and 2.5% (w/v) LFWE, respec-tively) and gentisic acid (20.07 and 39.34 mg / 100 mL for the 1.25 and 2.5% (w/v)LFWE, respectively) were the highest levels of flavonoid and phenolic acid in LFWEs,respectively. For induction of hepatosteatosis, body weight and (p < 0.05) liver size in ratswere successfully increased (p < 0.05) by a hypercaloric diet (HCD) compared to rats on anormal-caloric diet (NCD). Drinking both 1.25 and 2.5% LWFEs decreased (p < 0.05) liverlipid levels in the HCD rats and also resulted in lower (p < 0.05) hepatic malondialdehyde(MDA) levels and higher (p < 0.05) glutathione (GSH) levels, which may also result in lessliver damage (lower GOT and GPT values) in HCD rats. In addition, drinking LFWE alsodownregulated (p < 0.05) gene expressions of hepatic matrix metalloproteinases-2 and -9(MMP-2/9) in HCD rats. Finally, polyphenol-rich LFWE demonstrated hepatoprotectivecharacteristics against an energy-dense dietary habit.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-371-MO
Determination of Some Banned Aromatic Amines in Wasteusing Micellar Liquid Chromatography
Sandeep Kumar Moury1, Swati Dubey1, Devasish Bose1, Abhilasha Durgbanshi2,Josep Esteve-Romero3, Samuel Carda-Broch3, Maria Rambla Alegre3,
Monica-Ana Raviolo4
1Department of Criminology & Forensic Science, Dr. H.S.Gour University,Sagar (M.P.) India
2Department of Applied Chemistry, Banaras Hindu University,Vanarasi, India
3Química Bioanalítica, QFA, ESTCE, Universitat Jaume I,12071 Castelló, Spain
4Departamento de Química, Facultad de Farmacia, Universidad de Córdoba,Argentina
Aromatic amines are extensively used for industries but they are classified as a potentialenvironment and health hazard. In our laboratory, a new high performance liquid chro-matography method has been developed and optimized, for the determination of eightbanned aromatic amines: benzidine, o-anisidine, o-phenylenediamine, o-nitroaniline,2-methoxy-5-methylaniline (p-cresidine), o-toluidine, p-toluidine and p-chloroaniline.Conditions were: C18 column, 0.085 M SDS/3.2% (v/v) pentanol/pH 7, with detection at280 nm. Under these conditions, the eight aromatic amines were separated and quanti-fied in industrial waste waters in 16 min. Method validation studies were performedaccording to ICH Guideline [1, 2]. The possibility of direct injection using micellar liquidchromatography reduces the cost and the total time of analysis, and decreases errorsources owing to minimized risks of losses and chemical changes in the analytes. Theproposed method is a good candidate for application in the routine analysis in the area ofenvironmental monitoring.
1. Mei-Liang Chin-Chen, Samuel Carda-Broch, Devasish Bose, Josep Esteve-Romero, Food Chemistry, Volume120, Issue 3, 1 June 2010, Pages 915–920
2. Validation of Analytical Procedures: Text and Methodology Q2 (R1). ICH Harmonised Tripartite Guideline(2005). http://www.ich.org/cache/compo/363-272-1.html#Q2A (access on 27/06/09)
This study was part of project CTQ2007-64473/BQU, funded by Ministeri d’Educació i Ciència (MEC). MariaRambla-Alegre also wishes to thank the MEC for her FPU grant.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-372-TU
Determination of Pteridines and Hidropteridinesin Human Urine by HPLC-MS
Ana Jiménez Girón, Isabel Durán Martín-Merás, Anunciación Espinosa MansillaDepartment of Analytical Chemistry, University of Extremadura,
Avda. Elvas s/n, 06006 Badajoz, Spain
Non-conjugated folates (pteridines), such as biopterin (BIO), neopterin (NEO) betweenothers and its reduced forms, are of vital importance in various biochemical pathways. Thelevel of these compounds in urine and plasma is established as an important clinic criterion[1]. A significant increase in the urinary excretion of xanthopterin (XAN), NEO and pterin(PT), as well as a significant decrease in isoxanthopterin (ISO) and an NEO/BIO increment,was found in cancer patients. In mononucleosis infected children an increase of the NEOamount was also observed [2]. The relationship NEO/BIO is used as a diagnostic criterion. Itis known that alterations on the metabolic mechanism of tetrahydrobiopterin (BH4) give riseto different diseases, named hyperphenylalaninaemia (HPAs). The differential diagnostic ofthese HPAs is important in order to establish the adequate therapeutic protocol which isbased in the measurement of pteridines and other metabolites excretion. Hence, the classi-cal phenylketonuria produces increments in the urinary levels of NEO and BIO, while theBH4 deficit produces a decrease in the NEO and BIO levels.
Traditionally it has been needed to transform them into highly fluorescent aromaticforms by a previous oxidation step, in order to reach the required limit of detections inHPLC analysis. Some reduced form markers have been determined by differential meth-ods, with the loss of security that this type of analysis involves.
LC-MS is a power tool widely used for the determination of biomarkers in biologicalfluids. However, correct identification and quantification of a large number of substancesat very low concentration remains a challenge for MS. In particular, the analysis of sub-stances in biological samples, since it involves the analysis of markers and metabolites,which often are in very low concentration, being one of the main problems to carry outthe quantification of these substances the named matrix effect.
In this work, we have optimized a LC-MS method to determine several pteridinic markers inbiological fluids. Optimization of the separation step and MS detection was carried out. Aimwas to minimize sample handing and the standard addition method was applied to dilutedurine samples without a previous sample pretreatment step. Matrix effect was widely evaluated.
1. M. M. Müller, H. C. Curtius, M. Herold, C. H. Huber, Clin Chim Acta 201 (1991) 1–16.2. F. Cañada-Cañada, A. Espinosa-Mansilla, A. Muñoz de la Peña, A. Mancha de Llanos, Anal. Chim. Acta 648
(2009) 113–122.
Acknowledgements: Financial support from the Ministerio de Ciencia y Tecnología of Spain (ProjectCTQ2008-00468). Funding from the Junta de Extremadura and European Social Funds (Consolidation Projectof Research Group FQM003) reference GR10033 is also acknowledged. AJG acknowledge financial supportfrom Junta de Extremadura (ayudas para la reincorporación de doctores al Sistema de Ciencia, Tecnología eInnovación de Extremadura, expediente REI09011).

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-373-MO
Development and Validation of a Novel HPLC Methodfor the Dose Formulation Analysis of Parathyroid Hormone
and an Analog in Saline
G. Wallace1, R. O’Rielly1, A. Bartlett1, G. Hattersley2
1Charles River Laboratories, Preclinical Services,22022 Transcanadienne, Seneville (Montreal), Quebec, H9X 3R3, Canada
2Radius Health, Inc.201 Broadway, Cambridge, MA 02139, USA
Dose formulation analysis of peptides and proteins in aqueous matrices is beset withproblems of adsorption to surfaces and aggregation. Accurate analysis and LLOQs aretherefore frequently limited.
In support of preclinical studies, a reverse phase HPLC method was developed andvalidated for parathyroid hormone fragment, PTH (1–34) and a novel 34 amino acidPTH/PTHrP analog, down to 2.5 μg/mL in saline.
Optimal chromatography was acheived on a monolith column (Onyx C18, 100 x 3.0mm) with a 12 minute run time using an acetonitrile gradient with trifluoroacetic acid asan ion pairing agent. UV detection was performed at 210 nm.
Spiked samples were successfully analyzed at 10 and 50 μg/mL for PTH (1–34), and 2.5and 150 μg/mL for the analog.
Linearity was achieved over a concentration range of 1 to 25 μg/mL (r = 1.00) for PTH(1–34), and 0.75 to 25 μg/mL (r = 1.0) for the analog.
The method was validated with respect to linearity, carry-over, selectivity, precision andaccuracy, injection medium stability and matrix stability.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-374-TU
Development and Validation of Method for Retinol and αα-TocopherolAnalysis in Breast Milk by HPLC-DAD using Core-Shell Technology
Jirí Plíšek1,2, Markéta Kašparová1,2, Hana Vlcková1, Barbora Kucerová1,2,Lenka Krcmová1,2, Dagmar Solichová2, Petr Solich1
1Department of Analytical Chemistry, Faculty of Pharmacy, Charles University,Heyrovského 1203, 500 05 Hradec Králové, Czech Republic
2Department of Metabolic Care and Gerontology, Teaching Hospital,Sokolská 581, 500 05 Hradec Králové, Czech Republic
Breast milk constitutes a natural source of nutrients and water and satisfies basic nutritionrequirements for child to age 6 months. Breastfed children progress at average better and areresistant against infection, oncogenous disease and allergies. In present it is important also thatthe breastfed children are less commonly obese. Typically, major constituents such as fat andprotein can differ by two- to threefold between mothers at the same stage of lactation. More-over the concentrations of some minor constituents (e.g. vitamins) can be highly variable.
Retinol plays a role in a variety of functions throughout the body, such as vision, genetranscription, immune function, embryonic development and reproduction, bone metab-olism, haematopoiesis, skin health, antioxidant activity. α-Tocopherol is the main lipophil-ic antioxidant which inhibits peroxidation of polyunsaturated fatty acids in cell mem-branes. Preterm infants may be especially prone to develop clinical symptoms such ashemolytic anemia, retrolental fibroplasias, intraventricular hemorrhage and bronchopul-monary dysplasia as a result of Vitamin E deficiency.
Sample preparation procedure, performed before HPLC analysis, consists of de-proteination, saponification and liquid-liquid extraction steps carried out in one screw topglass tube. During development of chromatographic method mobile phase flow rate, injec-tion volume, limits of detection and quantification, peak asymmetry, resolution, efficiency,analysis time and solvent consumption were tested and optimized. The core shell technologyanalytical column Kinetex (C18, 100 Å, 2.6 μm, 100 x 4.6 mm – Phenomenex, Torrance,USA) was chosen for separation of the target analytes (retinol and α-tocopherol). As themobile phase 100% methanol was used and the detection of retinol and α-tocopherol wascarried out at 325 and 295 nm, respectively by diode array detector. The measured datashowed good linearity, repeatability of retention time and peak area. Measurement wasperformed by the chromatographic system Prominence Shimadzu (Kyoto, Japan) composedby Rack changer 1C autosampler for microtitrate plates, Degasser DGU-20A5, Column OvenCTO – 20 AC, Diode array detector SPD – M20A and Communication bus module CBM-20A.
New and fast liquid-liquid extraction and HPLC-DAD method for determination ofretinol and α-tocopherol in breast milk using core shell technology was developed andconsequently validated.
This study was supported by the Grant Agency of Charles University – GAUK No. 124809/2011 and MSMT ofCzech Republic – FRVS No. 1042/2011.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-375-MO
Development and Validation of Micellar Liquid ChromatographicMethod for Lamivudine and Derivatives
María S. Gualdesi1, Mónica A. Raviolo1, Josep Esteve-Romero2,Margarita C. Briñón1
1Departamento Farmacia. Facultad Ciencias Químicas, Universidad Nacional Córdoba,Argentina
2Química Bioanalítica, QFA-ESTCE, Universidad Jaume I,Spain
The search for an effective chemotherapeutic treatment against HIV infection has led to thedevelopment of agents that target specific and critical events in the HIV replicative cycle.Lamivudine (3TC) has been shown to be somewhat less toxic than other antiviral drugs but itstherapeutic value is limited by the resistance that is generated within a few weeks of treat-ment. To overcome this problem, we used a 5’-O-carbonate substitution strategy by linkingdifferent aliphatic alcohols on the side chains 5’-O position of 3TC. Among them, that ofassociate with methanol (3TC-Meta), ethanol (3TC-Eta), n-propanol (3TC-nPro), n-Butanol(3TC-Buta), n-Pentanol (3TC-Penta), n-Hexanol (3TC-Hexa) and n-Octanol (3TC-Octa) dem-onstrated to exhibit important biological activity against HIV and HBV [1, 2]. In the presentstudy, a simple and sensitive method by micellar liquid chromatographic (MLC) was opti-mized and validated for the simultaneous analysis of 3TC and their derivatives. Analyticalseparation was performed in less than 12.5 min, with these retention times (tR, min): 3TC: 2.4;3TC-Metha: 3.1; 3TC-Etha: 4.0; 3TC-nPro: 4.9; 3TC-Buta: 5.8; 3TC-Penta: 6.9; 3TC-Hexa: 8.1and 3TC-Octa: 10.4. For the study we using C18 ODS column with UV detection (λ = 272nm), at 30 °C, the flow rate was 1.0 mL min−1, and a micellar solution of 0.15 M sodiumdodecyl sulphate, 4% butanol buffered at pH 7 as mobile phase. The analytical parametersstudied include linearity (r > 0.9995), intra- and inter-day precision (RSD, %: 0.02–1.48 and0.07–1.66, respectively) and robustness. This method was sensitive with limit of detection(LOD) between 1.6 x 10−7 and 5.1 x 10−6 M and limit of quantification (LOQ) between 1.1 x10−6 and 1.4 x 10−5 M for all compounds, with recoveries ranging from 92.92 to 118.26%.
Zidovudine (AZT) was used (tR: 2.8 min) as internal standard for a major statistical analy-sis. Thus, the method was fully validated according to the Food and Drug Administrationguideline, and is suitable, sensitive and simple for routine analysis of 3TC derivatives.
1. Ravetti S, Gualdesi MS, Trinchero-Hernández JS, Turk G, Briñón MC. 2009. Bioorg Med Chem 17:6407–6413.
2. Gagey D, Ravetti S, Castro EF, Gualdesi MS, Briñón MC, Campos RH, Cavallaro LV. 2010. Int J of Anti-microb Agents 36:566–569.
This study was part of project CTQ2007-64473/BQU, funded by Ministeri d’Educació i Ciència (MEC, Spain)and FONCYT Préstamo BID PICT Nº 1325 (Argentina).

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-376-TU
Development of a Glycan Database for Waters Ultra PerformanceLiquid Chromatography (UPLC)
Mark Hilliard, Weston Struwe, Barbara Adamczyk, Pauline M. RuddNIBRT,
Foster’s Avenue, Mount Merrion, Blackrock, Dublin 4, Ireland
Glycosylation is the most complex post-translational protein modification. Over half of allproteins are glycosylated and to elucidate their function we also have to understand theirglycan structure. Manufacturing of protein life sciences products can be very difficult anda number of factors can have a major effect upon their glycosylation, such as dissolvedoxygen, pH, carbon source, and temperature. The correct glycosylation of protein lifescience products is absolutely required for folding and most importantly for therapeuticefficacy. Such fluctuations in the process can put product integrity at risk and thus theability to correctly monitor glycosylation is required. In collaboration with Waters, NIBRTis developing a glycan database for the ACQUITY UPLC. This system uses standard princi-ples of chromatography with columns that contain smaller particles (1.7 μm) in conjunc-tion with higher flow rates which results in overall increase in speed, sensitivity, resolutioncompared to standard HPLC systems. The BEH columns, a hybrid material utilizes bridgedethylsiloxane/silica hybrid (BEH) structure and small particle size. BEH technology ensuresthe column stability under the high pressure and through wider pH range (1–12) allowing for varied applications.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-377-MO
Development of a Sphingomyelin Biomimetic Stationary Phasefor Immobilized Artificial Membrane (IAM) Chromatography
Dieter Verzele1, Frédéric Lynen1, Adrian G. Wright2,Melissa Hanna-Brown2, Pat Sandra1
1Pfizer Analytical Research Centre, Dept. of Organic Chemistry, Ghent University,Belgium
2Pfizer European R&D Headquarters,Sandwich, UK
As innovative approach to liquid chromatography in which the stationary phase emulatesthe lipid environment of a cell membrane, the so-called immobilized artificial membrane(IAM) technology offers an in vitro HPLC model of in vivo drug partitioning [1]. High-throughput and simple, the practical assets of this chemical technique over more tradi-tional biological methods are vitiated however by oversimplification and structural ambi-guities in the sparse range of commercial, ‘phosphatidylcholine-only’ IAM columns. Con-sidering the mechanistic complexity of the in vivo uptake process [2], more sophisticatedIAM stationary phases are desired, the synthetic aspects need up-to-date revision, andassay correlations should accordingly be improved.
Attracted by its mysterious nature, we decided to focus on the development of a sphin-gomyelin [3] counterpart of the hitherto available columns. Useful as such in a broadersynthetic context, an oxidative release monitoring strategy proved invaluable to achievea straightforward, ultra-short synthesis of the desired silica material, while minimizing theaforementioned ambiguities. As such, we were able to manufacture a prototype sphingo-myelin stationary phase through an unprecedented, solid-phase inspired methodology,amenable to mixed-phase approaches towards more genuine membrane mimics.
In current communication, our efforts on both the synthesis of this column in particularand analytical applications of IAM chromatography in general will be presented.
1. Taillardat-Bertschinger, A.; Carrupt, P.; Barbato, F.; Testa, B. J. Med. Chem. 2003, 46, 655–665. Immobi-lized artificial membrane HPLC in drug research.
2. Van Meer, G.; Voelker, D. R.; Feigenson, G. W. Nature Rev. Mol. Cell Biol. 2008, 9, 112–124. Membranelipids: where they are and how they behave.
3. Liao, J.; Tao, J.; Lin, G.; Liu, D. Tetrahedron 2005, 61, 4715–4733. Chemistry and biology of sphingolipids.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-378-TU
Development of an HPLC Method to Quantify an Immunostimulantin an Emulsion-Based Vaccine Adjuvant
J.-F. Cotte1, M. Raphat1, A. Debreyer1, F. Dalençon1, P Talaga1,J. Haensler1, O. Adam1, K. Johnson2
1Research Department, Sanofi Pasteur,Marcy l’Etoile, France
23M Drug Delivery Systems Division,St. Paul, USA
New vaccines based on purified recombinant proteins are usually less immunogenicthanstandard whole-cell and virus-based vaccines. Thus, efficient and safe adjuvants arerequired to assist these new vaccines in inducing potent and persistent immune respons-es. The development of such adjuvants remains both a challenge and a necessity.
Past approaches in adjuvant research generally relied on carrier- or delivery system-typeadjuvants, such as aluminium salts or emulsions.In order to induce well-defined cell-mediated immune responses in addition to antibodies,new immunostimulants havebeen developed. Among these, synthetic toll-like receptors (TLRs) agonists, such as theResiquimod (3M company), are promising candidates [1]. However, these small molecu-lar weigh compounds are generally found most efficacious as vaccine adjuvants whencombined with an appropriate delivery system, such as an oil-in-water emulsion forinstance [2, 3].
Oil-in-water emulsions can be considered as dispersed systems, with an oily phasedispersed in an aqueous phase, and stabilized by a surfactant or a surfactant system.Quantification of Resiquimod in such a complex matrix has been an analytical challenge.
The work presented in this poster concerns the development of an HPLC method forthe quantification of Resiquimod in a complex oil-in-water emulsion. This method is rapid,sensitive and may be readily adapted for quality control of both the emulsion componentsand the immunostimulant.
1. Tomai M. A., Miller R. L., Lipson K. E., Kieper W. C., Zarraga I. E., Vasilakos J. P. Resiquimod and otherimmune response modifiers as vaccine adjuvants. Expert Rev Vaccines. 2007; 6(5):835–47.
2. Reed S. G., Bertholet S., Coler R. N., Friede M. New horizons in adjuvants for vaccine development. TrendsImmunol. 2009; 30(1):23–32.
3. Guy B. The perfect mix: recent progress in adjuvant research. Nat Rev Microbiol. 2007; 5(7):505–17.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-380-TU
Direct UPLC-ESI-MS/MS Method of Brain Microdialysates:Rapid and Easy Method for Neurotransmitters
and their Metabolites Monitoring
Petr Kacer1, Kamila Syslová1,2, Vera Najmanová1, Lukáš Rambousek1,3,4,Marek Kuzma2, Vera Bubeníková-Valešová3, Romana Šlamberová5
1Institute of Chemical Technology,Technicka 5, 166 28 Prague 6, Czech Republic
2Institute of Microbiology,Videnska 1083, 142 20 Prague 4, Czech Republic
3Prague Psychiatric Center, Department of Biochemistry and Brain Pathophysiology,Ustavni 91, 181 03 Prague 8, Czech Republic
4Institute of Physiology,Videnska 1083, 142 20 Prague 4, Czech Republic
5Department of Normal Pathological and Clinical Physiology, 3rd Faculty of Medicine, Charles University,Ruska 87, 100 00 Prague 10, Czech Republic
A sensitive assay method was developed for a parallel, rapid and precise determination ofdopamine, serotonine, γ-aminobutiric acid (GABA), glutamate and their metabolites frombrain microdialysis samples. The method consisted of 2D-UPLC method, serving thepurpose separating salts from artificial cerebrospinal fluid (ACSF) used in the microdialysis,and the detection method LC-ESI-MS/MS, where the selected reaction monitoring modewas used for its extremely high degree of selectivity and the stable-isotope-dilution assayfor its high precision of quantification. The developed method was characterized by thefollowing parameters: the precision of the developed method was ≤ 12.4% (determinedas RSD) for all the substrates; the mean accuracy was ≤ 11.8% (determined as RE). Themethod was tested on samples obtained from nucleus accumbens of rat pups after anacute methamphetamine administration. The developed assay could be applied for botha simultaneous analysis of all the four neurotransmitters (dopamine, serotonin, GABA andglutamate) and their principal metabolites in microdialysis samples acquired from the ratbrain and the time monitoring of their tenuous concentration changes on picogram levelfollowed by methamphetamine stimulus.
The developed assay method is easy to operate, robust and rapid. To best of ourknowledge, it is the first method not using the separation and pre-concentration step andalso analyzing such a large number of neurotransmitters and their metabolites in singleanalytical run. The developed method potentially expands the diagnostic/treatmentpotential in regards to neuropsychiatric and neurological disorders, where above men-tioned neurotransmitters and their metabolites concentration levels are altered in compar-ison to physiological levels. The method may well contribute to a better understanding ofthe pathophysiology and pathogenesis of many neuropsychiatric disorders (drug addic-

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
tion, schizophrenia, Parkinsonism, Alzheimer dementia) and in pharmaceutical research ofnew drugs to treat neurological diseases.
Acknowledgment: The authors wish to acknowledge with gratitude the financial support by the Grant Agen-cy of the Czech Republic (Grant GACR P303/10/0580) and the Ministry of Education of the Czech Republic(Grant CEZ: MSM 604 613 7301).

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-381-MO
Efficient Use of pH Gradients in the Ion Exchange Analysis of Proteins
Thomas E. Wheat, Daniel S. Root, Patricia R. McConvilleWaters Corporation
34 Maple Street, Milford, MA0 1757, USA
The analysis of proteins is complicated by their large size and by the heterogeneity ofchemical properties. Because these factors affect the selectivity of the analysis, it is neces-sary to combine multiple analytical techniques that are sensitive to different physical andchemical properties to ensure complete characterization of a protein sample. Within theset of chromatographic techniques included in this approach, ion exchange can be themost discriminating. Realization of the full power of ion exchange requires optimization ofboth the column and the mobile phase. Among the parameters requiring optimization,the pH of the separation buffer has the largest effect on the selectivity of the method.Efficient techniques have been developed for screening separations with a selection ofcolumns over a series of different pH buffers. It is, in addition, recognized that pH gradi-ents can provide particularly useful protein separations. Such gradients could complementthe use of iterative experiments over small increments of pH. In practice, pH gradientshave not been widely applied because they are difficult to control. Because pH is a loga-rithmic function, simple buffer mixing does not give the desired pH profile. As the targetpH deviates further from the pK, pH changes become very large with small changes inproportions. It is, therefore, also complicated to control the pH profile when changingcolumn dimensions or flow rate. We have developed and evaluated algorithms and asoftware user interface to simplify this process. The technology is implemented on a foursolvent pump and the gradient table is programmed directly in units of pH and salt con-centration. The system uses a “buffer system” that is freely defined by the user to calcu-late the required proportions of the four mobile phase reservoirs and to recalculate thoseproportions at each pump stroke. We will show the use of this system for both anionicand cationic buffers. Several examples of protein separations, including monoclonalantibodies, will also be shown.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-382-TU
A New UHPLC Column for Polar Analyte Retention
Ken Butchart, Mark WoodruffFortis Technologies Ltd
45 Coalbrookdale Road, Clayhill Industrial Park, Neston, Cheshire, CH64 3UG, UK
Ultra high pressure liquid chromatography (UHPLC) has taken the market by storm allow-ing faster method development to be achieved in a shorter period of time. More efficien-cy and more speed are both sound parameters to be able to manipulate, however thestrongest parameter in method development is resolution.
There are many hydrophilic compounds that are difficult to retain on RP-18 stationaryphases and in order to produce robust, accurate data sufficient retention of these analytesis critical.
In this poster we discuss the use of a new stationary phase designed specifically for theretention and separation of these more hydrophilic molecules. We show the extra resolu-tion that can be achieved between analytes due to the mechanism of the stationaryphase, exhibiting both hydrophobic and hydrophilic retention profiles. We highlight theapplications that this new stationary phase can achieve, comparing it to commerciallyavailable phases to study the differences.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-383-MO
Evaluation of Size Exclusion Chromatography Packing Materialsfor the Analysis of Proteins and Higher Order Aggregates
Paula Hong, Edouard S. P. Bouvier, Kenneth J. FountainWaters Corporation,
34 Maple Street, Milford, MA 01757, USA
Size exclusion chromatography (SEC) is typically used to measure protein aggregates andother size variants present in biopharmaceuticals. Size-based HPLC separations havetraditionally been under low pressure conditions with silica-based material. These meth-ods can be unreliable due to changes in retention time, peak shape, resolution as well asirreproducibility between columns. New advances in packing materials and instrumenta-tion have allowed faster and more reproducible separations to be achieved. However, therecovery of proteins and higher order aggregates still remains a critical part of the successof an SEC method.
In this presentation, we will discuss the factors that can influence quantitation of bio-molecules on size exclusion packing materials. An extensive evaluation of the columncontribution to protein recovery is also presented for a variety of different packing materi-als. The effect of variables such as flow rate, temperature, mobile phase composition, andformulation excipients will be measured for a wide variety of proteins, including mono-clonal antibodies. Due to the increasing growth in the production of biopharmaceuticals,these studies will help establish guidelines for reliable assessment of protein purity.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-384-TU
Exploration of pH Gradient Ion-Exchange Chromatographyfor High Resolution Protein Separations in Biotechnology
and Proteomics
Wim Decrop, Marie-Jeanne Olivo, Evert-Jan Sneekes, Remco SwartDionex Corporation
Amsterdam, The Nederlands
Ion-exchange chromatography (IEC) is a versatile separation technique for biomoleculesseparated by either salt or pH gradients. Although salt gradients are more commonlyapplied, the utilization of pH gradients can provide significant advantages such as: i)improved separation resolution, ii) lower salt concentration in collected fractions, and iii)the possibility to correlatea proteinsiso-electric point(pI) data with elution profiles. Recent-ly the application of pH-gradient ion-exchange chromatography has been described forthe separation of standard proteins [1] and monoclonal antibodies [2, 3].
In this study we report on our experiences with pH-gradient ion-exchange chromatog-raphy applied for the separation of proteins from various sources. High resolution separa-tions of a monoclonal antibody and its isoforms were achieved on a new non-porousstrong cation exchange resin. Results are comparedto those obtained with salt gradiention-exchange chromatography. Complex protein mixtures typically found in proteomicswere separated with pH-gradient anion-exchange chromatography. Developed methodol-ogy is validated for pH profile shape and precision, retention time precision, peak capaci-ty, sample capacity and robustness towards sample solvent composition.
1. TangirAhamed et al., J. Chrom. A, 1194 (2008) p. 22–292. Dell Farnan and G. Tony Moreno, Anal. Chem., 2009, 81 (21), p. 8846–88573. TangirAhamedet al., J. Chrom. A, 1164 (2007) p. 181–188

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-385-MO
Forming Highly Accurate pH- and Salt-Gradientsfor Biomolecules Separation using First Principle Calculations
Vlastimil Hruska1, Bohuslav Gas1, Uwe Effelsberg2,Jochen Strassner2, Tom van deGoor2
1Charles University Prague, Faculty of Science,Czech Republic
2Agilent Technologies,Waldbronn, Germany
In bio-molecule separations e.g. ion exchange chromatography, both pH and salt gradi-ents are used to optimize resolution of proteins. Often simple binary mixing of two solu-tions with different pH or salt concentration is used, which results in significant linearitydeviations on the created pH gradient (not fully linear and containing regions with insuffi-cient buffering capacity) or non-constant pH conditions in the case of salt gradients.
A first principle model was developed that exactly calculates, based on pKa, the re-quired composition of a quaternary mixture of individual compound solutions as a func-tion of time to create fully linear pH gradients and salt gradients at constant pH. Accuracyerror calculation and predicting insufficient buffering capacity are added to assist the userin preparing optimal separation conditions. A ready to use pump time table can be ex-ported to be used with the Agilent 1260 Infinity Bio-inert LC.
Capabilities of this utility and its application to ion exchange chromatography of pro-teins will be shown.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-386-TU
Functionalised Polymeric Monolithic Sheets for Dried Bloodand Plasma Spotting
Anna Nordborg1, Esme Candish1, Pavel N. Nesterenko1, Greg W. Dicinoski1,Graeme Clark2, Paul R. Haddad1, Emily F. Hilder1
1Pfizer Analytical Research Centre (PARC), Australian Centre for Research on Separation Science (ACROSS),School of Chemistry University of Tasmania,
Hobart, Australia2Pfizer Global Research & Development,
Sandwich, UK
In clinical diagnostics, a popular tool is the use of dried blood spotting (DBS). The use ofdried blood spots has a number of advantages including a simplified collection, transportand storage of samples. In addition, the risk of infection during sample handling is re-duced. It also reduces the sample volumes needed and enables the use of less invasivesample collection techniques. This has an important beneficial effect when used in animalstudies as it reduces the number of animals needed.
The material predominantly used in dried blood spotting is paper. Paper is suitable forthe purpose and by different impregnation strategies the paper characteristics can bealtered. However, the possibility of functionalisation of the paper surface is still limited.For this reason, the introduction and use of other media in dried blood spotting is desir-able and we have demonstrated that polymeric monoliths offer a viable alternative.
Polymeric monolithic materials are porous materials that can be made in situ in differ-ent confinements such as capillaries or sheets. The wide variety of functional monomersavailable in combination with different post-polymerization modification strategies en-ables the preparation of materials with a wide variety of characteristics and functionality.The wide range of possible modification strategies of monolithic materials is therefore anadvantage over paper based materials.
In this study we present the design, synthesis and characterization of polymeric mono-lithic stationary phases prepared in sheet format for use in dried blood spotting. A fewexamples of functionalised materials are given with the desired functionality imparted inone of three ways; co-polymerization, reaction with surface group and post-polymeriza-tion grafting. Finally, we present the use of the prepared polymeric monoliths for driedblood spotting of blood and plasma spiked with a mixed cocktail of pharmaceuticals.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-387-MO
Glycopeptide Analysis by Capillary Electrophoresis– QTOF – Mass Spectrometry
Suresh Babu CV1, Ravindra Gudihal1, Syed Salman Lateef1, Ning Tang2,Tobias Preckel3, Martin Greiner3, Stephan Buckenmaier3
1Agilent Technologies India Pvt. Ltd.,Bangalore, India
2Agilent Technologies,Santa Clara, CA, USA
3Agilent Technologies R&D and Mktg. GmbH & Co.KG,Waldbronn, Germany
Glycosylation is an important post-translational modification (PTM) of proteins. Due tothe importance of monoclonal antibodies (mAb) as therapeutic agents, there is a growingdemand for monitoring the carbohydrate structures attached to antibody. For enhancedseparation efficiency, higher resolution, shorter run times, minimal sample/solvent con-sumption and flexibility, capillary electrophoresis (CE) has an enormous potential for theanalysis of biopharmaceuticals. Further, there is growing interest in exploring CE coupledto mass spectrometry (MS) for the higher sensitivity and better compound identificationwith accurate mass measurements.
In this study, we have analyzed the glycopeptide of a monoclonal antibody using CEcoupled to Accurate-Mass Q-TOF mass spectrometry with a coaxial sheath liquid inter-face. The tryptic peptide map of the monoclonal antibody was generated and the glyco-peptide was assigned using accurate mass measurement. Peptide mapping of heavy andlight chains resulted in 93% of protein sequence coverage with mass accuracy of 10 ppm.With a mass error limit of 1 ppm, the glycopeptides were confirmed using the intenseextracted ion electropherogram (EIC) at m/z 204.085 corresponding to the diagnosticsugar oxonium fragment Ions. Analysis of the MS/MS spectrum revealed the minor andmajor forms G1F/G2F and G0F modifications of this mAb.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-388-TU
High Throughput Determination of Levonorgestrel in Human Plasmausing a Sensitive LC-MS/MS Method
Cédric Hubert1, Bruno Streel2, Renilde Sibenaler3, Joëlle Widart4,Attilio Ceccato5, Philippe Hubert1
1Laboratory of Analytical Chemistry, CIRM, Institute of Pharmacy, University of Liège,B 36, B-4000 Liège, Belgium
2Galephar,39, rue du Parc Industriel, B-6900 Marche-en-Famenne, Belgium
3SMB Laboratories,26-28, rue de la Pastorale, B-1080 Bruxelles, Belgium
4Department of Pharmacy, University of Liège,B 36, B-4000 Liège, Belgium
5Odyssea Pharma,16, Rue du Travail, B-4460 Grâce-Hollogne, Belgium
The present LC method was developed for the monitoring of plasmatic concentration oflevonorgestrel (LNG) after insertion of a hormonal intra-uterine device (IUD). In thisstudy, very small plasmatic concentrations of LNG were expected, ranging from 500pg/mL to 100 pg/mL. Consequently, the present method must be as sensible as possible.For this kind of concentration the liquid chromatography coupled to tandem mass spec-trometry detection (LC-MS/MS) has proved its efficiency and usefulness. In this scope, thesolid phase extraction (SPE) is generally recommended to avoid matrix effect as well asion suppression.
Due to the hydrophobic character of the compound of interest, an end-capped octylsilica (C8(EC)) disposable extraction cartridge (DEC) was selected. The plasma samplewas first diluted with orthophosphoric acid. Methyltestosterone, the internal standard(IS), was then added and the resulting sample was treated using a GILSON MultipleProbe 215 SPE system. This system processes eight samples simultaneously allowing ahigh throughput sample preparation. The elution was made in two times using 450 μLof methanol. 100 μL of 0.1% formic acid in water were then added in order to reconsti-tute mobile phase.
The separation was carried out on a Phenomenex phenyl-hexyl column (150 x 4.6,5 μm) using a mobile phase consisting in a mixture of methanol and formic acid 0.1%in water (90/10, v/v). Using these LC conditions, a short run time was obtained. Themass transition 313.4/245.1 and 303.0/97.1 for levonorgestrel and IS was respectivelyselected.
The coupling of the GILSON Multiple Probe 215 SPE system with the LC-MS/MS condi-tions allows a high throughput determination of levonorgestrel in a complex matrix (>100 samples per day).

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
Afterwards, the validation of the method was considered using an approach based onthe accuracy profile [1, 2] allowing to manage the risk associated to the use of thesemethods in routine analysis [3].
1. PAT Initiative, FDA, http://www.fda.gov/cder/OPS/PAT.htm2. Ph. Hubert et al, Harmonization of strategies for the validation of quantitative analytical procedures: A
SFSTP proposal-part I, J. Pharm. Biomed. Anal. Vol. 36, 579 (2004).3. ICH guidelines Q9, EMEA, http://www.emea.eu.int/Inspections/docs/ICHQ9Step4QRM.pdf

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-390-TU
Hydrophilic Interaction Liquid Chromatography Coupledto Mass Spectrometry for the Separation of Dalargin
and Structurally Related Enkephalins
Ayat Abbood1, Christine Herrenknecht2, Rana Alsalim1,Myriam Taverna1, Claire Smadja1
1Université Paris-Sud, UMR 8612, Protéines et Nanotechnologies en Sciences Séparatives, 92296,Faculté de Pharmacie, Châtenay-Malabry, France
2Université de Nantes, EA2160, Mer Molécules Santé (MMS), Faculté de Pharmacie,44000 Nantes, France
Dalargin is a synthetic hexapeptide, analogue of Leu-enkephalin, involved in the regula-tion of many physiological processes including pain. Retention behavior of this peptideand its five structural analogues has been firstly studied by hydrophilic interaction liquidchromatography (HILIC) on a bare silica stationary phase. The influence of organic modifi-er content, buffer pH, and ionic strength, on peptides retention was evaluated. The varia-tion of organic content of the mobile phase shows an hydrophilic partitioning phenome-non. The pH modification suggested that electrostatic interaction between the most polarpeptides (Dalargine and Des-Tyr-LE) and the sorbent could also occur. The cation-ex-change phenomenon was demonstrated for these two peptides by varying the ionicstrength of the mobile phase. The method was applied to the analysis of non purifieddalargin, resulting from the solid-phase synthesis. The optimization of the separation ofthe target peptide from its side products has been first performed with UV detection. Thisstudy showed a rapid and efficient separation was obtained by mixing ACN and MeOH inthe mobile phase. The precision of the method was evaluated by five replicate analyses oronce per day over 5-days for intra- and inter-day repeatabilities, respectively. Excellentintra- and inter-day repeatabilities were obtained for k (RSDs < 0.9%). Robustness of theHILIC method was checked through minor changes in chromatographic conditions suchas: pH of the mobile phase (±0.2 unit) and column temperature (±2 °C). The RSDs were< 1.5% for retention factors. The HILIC method for dalargin is robust and suitable forroutine analysis. The identification of the target peptides (Dalargine) and the other impu-rities present in the synthesis mixture was performed by coupling the HILIC column withelectrospray ionization mass spectrometry in negative and positive mode.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-391-MO
Identification and Characterization of Tetracosactide Impuritiesby CE and RP-HPLC Coupled to TOF MS
Angelina Taichrib1, Gerhard K. E. Scriba2, Christian Neusüß1
1Aalen University, Chemistry Department,Beethovenstrasse 1, 73430 Aalen, Germany
2 Friedrich Schiller University of Jena, School of Biology and Pharmacy,Philosophenweg 14, 07746 Jena, Germany
The peptide tetracosactide (TCS) is a synthetic analogue to naturally-occurring humanadrenocorticotropic hormone (ACTH). Its physiologic effect is based on the stimulation ofthe production of steroid hormones like cortisol in the adrenal glands. Hence, TCS showsseveral pharmaceutical applications, in particular regarding the diagnosis of adrenalcortex dysfunction. Besides, TCS is considered to be a performance enhancing substanceand is thus on the list of prohibited substances of the World Anti-Doping Agency.
The analysis of TCS plays an important role in terms of doping control, but even moreregarding the quality control of the synthetic product for pharmaceutical applications.The standard method for routine analysis established by the European Pharmacopoeiainvolves the separation by reversed phase high performance liquid chromatography (RP-HPLC) and analyte detection by a UV-spectrometer. The corresponding chromatogramsrevealed several peaks (partially) separated from the main substance, that couldn’t beassociated with known impurities, like the oxidized form of TCS. For the identification ofthese unknown impurities both capillary electrophoresis (CE) and RP-HPLC were coupledto a quadrupole time-of-flight mass spectrometer (QTOF MS) which enabled the fastdetermination of the accurate masses and fragmentation experiments. By this means, theunknown substances were identified to be products of incomplete TCS synthesis, i.e. N-and C-terminally truncated peptides, and peptides with unreleased protecting groupsused for side chain protection during solid phase synthesis of TCS. A semi-quantitativeestimation of the relative amounts of the impurities was carried out.
The selectivity of RP-HPLC and CE regarding the separation of tetracosactide impuritiesis discussed. Both separation techniques could be used for the appropriate separation ofdifferent peptides in the tetracosactide sample. However, due to wider differences in themass to charge ratio CE showed a higher suitability for the separation of tetracosactidefragments (smaller peptides); while the larger peptides, i.e. those wearing protectinggroups, were separated more efficiently by RP-HPLC, which is due to higher differences inhydrophobicity.
Finally, different TCS sources were compared regarding the number and type of therespective impurities.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-392-TU
Internal Standard Addition for Dried Blood Spot Analysis Basedon Flow Through Desorption Solid-Phase Extraction
and Mass Spectrometry
Lena Knegt, Bert Ooms, Emile KosterSpark Holland,
P.de Keyserstraat 8, 7825VE Emmen, The Netherlands
Dried Blood Spot (DBS) analysis has many advantages with respect to sampling, ship-ment, storage. Many studies have been performed to demonstrate the benefits of DBSprocessing. However, the workload within the lab has increased because efficient automa-tion is not yet available. Another major issue in DBS analysis is the addition of the internalstandard, because adding the standard to the sample before applying it on the filterpaper is hardly possible. In this presentation, an on-line DBS extraction/analysis concept ispresented that utilizes flow through desorption of the bloodspot, coupled directly to massspectrometric analysis via online solid-phase extraction. Furthermore, various ways ofinternal standard addition are investigated and discussed.
Blank blood is spotted onto Whatman DMPK-C filter paper cards (15 μL-aliquots). Thespots were dried (> 2 hours) and stored until used. DBS analysis is performed by clampingthe blood spot directly in a special device that allows for the flow though desorption ofthe blood sample without punching. The desorbed sample is subsequently flushed to-wards a solid-phase extraction cartridge for cleanup and then directly towards a WatersAcquity TQD mass spectrometer for selective detection of the analyte(s). Internal standardsolution is either added to the desorption liquid via a loop, sprayed on the dried bloodspot before desorption or applied on the paper before applying the blood sample.
Test data proved that the developed system can be used to clamp and seal filter papercards leak tight. Samples were directly flushed from the paper onto a C18 SPE cartridge bymeans of 1mL of water (0.2% formic acid) at 2 mL/min. During transfer of the blood samplefrom the paper to the SPE cartridge, an accurate volume of internal standard solution (pro-pranolol in water) is introduced in the flow path upstream of the DBS card by means of a 20μL sample loop (size can be varied if needed). Subsequently, the disposable SPE cartridge iswashed to remove interfering matrix compounds and then directly eluted towards the MS bymeans of a gradient. The precision of the internal standard addition itself via a loop (upstreama DBS card) turned out to be about 2.5% RSD for the complete DBS-SPE-MS/MS methodolo-gy. Adding the internal standard to the DBS card by means of a sprayer either before or afterthe application of the blood sample is currently under investigation. This means the bloodspot itself contains the internal standard before flow through desorption takes place. Differenc-es in DBS desorption are assumed to be corrected this way, but obviously, this only works withadequate precision of the spray process. Preliminary tests show lower precision than the loop-approach. Results of a more thorough investigation will be shared.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-394-TU
Laboratory Diagnostics of Adenosindeaminase (ADA) Deficiencyby HPLC
J. Bártl1, P. Hornik1, J. Krijt1, S. Štastná1, I. Šebesta1,2
1Institute of Inherited Metabolic Disorders2Institute of Clinical Biochemistry and Laboratory Diagnostics
General Faculty Hospital and First Faculty of Medicine, Charles University, Prague, Czech Republic
Background:ADA deficiency is an autosomal recessive inborn error of purine metabolism. Enzyme
deficiency results in an accumulation of toxic deoxyadenosine and adenosine in biologicalfluids and consequently disturbs humoral and cell immunity.
Diagnosis of this disease relies on the analysis of adenosine and deoxyadenosine levelsin urine. We report here biochemical findings of patient suffered from ADA deficiency,who was identified by HPLC on biochemical and enzymatic level.
Method:The urinary adenosine and deoxyadenosine were analyzed on Alliance 2695 and Photo-
diode Array Detector 2998 (Waters). Before HPLC analyses, the urine samples were dilut-ed with water to give concentration of creatinine of 1 mmol/L. The determination wasaffected by reverse-phase Prontosil C18 AQ (200 mm x 4.0 mm i.d.), 3 μm particle size(Bischoff), and eluted by linear 15 min. gradient from 0 to 30% of acetonitrile in 40 mMammonium acetate buffer (pH 5), flow – rate 0.7 ml/min and peaks were detected usingUV absorbance scanned over the range of 230–305 nm.
For the ADA activity calculations, the concentration of inosine produced by the enzymereaction in erythrocytes was measured after incubation for 4 hours.
Results:Deoxyadenosine urinary excretion was elevated 236 mmol/mol creatinine (ref. value <
1 mmol/mol creatinine). Adenosine was not detected in urine. The ADA activity was 3.0nmol/h/mg of hemoglobine, the activity in control individuals (n = 50) were in the rangeof 18.0–89.0 nmol/h/mg of hemoglobine.
Diagnosis was acknowledged on molecular genetic level by foreign laboratories.
Conclusion:This method is suitable for routine quantification urinary purine and pyrimidine
nucleosides in general. It is a useful and rapid tool for diagnosis of inherited metabolicdiseases of purine and pyrimidine metabolism.
Supported by the project MZ0VFN 2005 by Ministry of Health of the Czech Republic.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-395-MO
Liquid Chromatographic Method for Simultaneous Determinationof Fungicides in Seeds, Fungicide Formulations, Plant Material,
Soil and Water Samples
Sandeep Kumar Moury1, Devasish Bose1, Abhilasha Durgbanshi2,Josep Esteve-Romero3, Samuel Carda-Broch3, Maria Rambla-Alegre3,
Sergio Marco-Peiró3, Monica-Ana Raviolo4
1Department of Criminology & Forensic Science, Dr. H.S.Gour University,Sagar (M.P.) India
2Department of Applied Chemistry, Banaras Hindu University,Vanarasi, India
3Química Bioanalítica, QFA., ESTCE, Universitat Jaume I,12071 Castelló, Spain
4Departamento de Química, Facultad de Farmacia, Universidad de Córdoba,Argentina
Thiram, carbendazim and ziram are used as fungicides, seed treating agents or as rubberaccelerators (thiram, ziram). In the present work the separation and determination ofthese fungicides was achieved on HPLC using a mobile phase containing 0.13 M sodiumdodecyl sulphate-2.5% (v/v) pentanol- 0.01 M Na2HPO4 at pH 7, a C18 column (250 mm× 4.6 mm, 5 μm particle size) coupled to a photo diode array detector set at 275 nm withan analysis time of less then 10 min. Validation studies were performed using ICH guide-line, obtaining good linearity (r2 > 0.999), limits of detection (3s criteria) of 26, 24 and 32ng/mL for thiram, carbendazim and ziram respectively, and intra- and inter-day precision(R.S.D, %) was less than 2.5. The developed method was applied for the determination ofselected fungicide in seeds, commercial formulation, plant material, soil and water withany prior pretreatment step (apart form washing, mincing or filtering) and injectingdirectly onto the chromatographic system. The micellar liquid chromatography (MLC)method herein reported is simple, sensitive, precise, robust and the samples can be direct-ly injected into the column without any pre-treatment step.
1. http://dacnet.nic.in/ppin/PDF/Seed%20Treatment%20Rabi123.html (access on March 21, 2011).2. http://www.indianexpress.com/ie/daily/20000709/ina09061.html (access on March, 21, 2011).
This study was part of project CTQ2007-64473/BQU, funded by Ministeri d’Educació i Ciència (MEC) andFONCYT Préstamo BID PICT Nº 1325. Maria Rambla-Alegre also wishes to thank the MEC for her FPU grant.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-396-TU
Micellar Liquid Cromatography in Bioanalytical Chemistry
Josep Esteve-Romero1, Samuel Carda-Broch1, Maria Rambla-Alegre1,Mei-Liang Chin-Chen1, Mónica-Ana Raviolo2, Devasish Bose3,
Abhilasha Durgbanshi4, Inmaculada Casas-Breva5
1Química Bioanalítica, QFA-ESTCE, Universitat Jaume I,Campus Riu Sec, 12071 Castelló, Spain
2Departamento Farmacia, Facultad Ciencias Químicas, Universidad Nacional Córdoba,Argentina
3Department of Criminology & Forensic Science, Dr. H.S. Gour University,Sagar (M.P.) India
4Department of Applied Chemistry, Banaras Hindu University,Vanarasi, India
5Servei de Farmàcia Hospitalaria, Hospital la Plana,Vila-real, Spain
In micellar liquid chromatography (MLC), the mobile phase is composed of a surfactantand an alcohol. The parameters that must be optimized in MLC are the type of column,the pH, the nature of the surfactant and modifier, and their concentrations. Optimizationis performed using an interpretive strategy in which the best mobile phase is selected forvalidation studies. Optimized methods in MLC provide fast, efficient separations, therebyallowing the determination of a great variety of substances in a complex matrix. MLC is auseful technique for the identification of drugs in biological fluids, which often can beinjected directly into the chromatographic system even in the presence of other com-pounds, including proteins, without any pretreatment other than filtration. In describingthe many applications of MLC, this review draws upon examples taken from the literatureas well as recent results of the authors’ own research. It describes the theoretical ap-proach, optimization strategy, and applications of MLC in the determination of differentgroups of drug substances. The compounds and matrices under discussion are: antide-pressants in pharmaceuticals, calcium-channel blocking agents in serum, anserine andcarnosine in meat samples, tyramine and tryptamine in wines and sulfonamides in milk.
1. J. Esteve-Romero, S. Carda-Broch, M Gil-Agustí, M.E. Capella-Peiró, D. Bose, Trends in Analytical Chemistry24 (2005) 75–91
2. J. Esteve-Romero, S. Carda-Broch, M. Rambla-Alegre, Contributions to Science, 6(1) (2010) 105–114
This study was part of project CTQ2007-64473/BQU, funded by Ministeri d’Educació i Ciència (MEC) andFONCYT Préstamo BID PICT Nº 1325 (Argentina). Maria Rambla-Alegre also wishes to thank the MEC for herFPU grant.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-397-MO
Mobile Phase Considerations for Improved LC-MS AmenablePeptide Separations
Anders Fridstrom1, Hillel K. Brandes2, David S. Bell2, Craig R. Aurand2
1Sigma-Aldrich Chemie GmbH,Industristrasse 25, Buchs, 9471, Switzerland
2Sigma-Aldrich/Supelco,595 North Harrison Road, Bellefonte, Pennsylvania 16823, USA
Elution of peptides on silica-based, reversed-phase alkyl phases under the common MS-compatible condition of dilute formic acid is generally not as symmetrical or efficient aswith the traditional UV-based methods with dilute trifluoroacetic acid (TFA). This is acomplex phenomenon that may involve charge-charge interactions between the peptideanalytes as well as between the peptide and silica surface. Therefore, such interactionsmay be mitigated by pH and/or mobile phase additives that may function as counterionsin an ion-exchange process. This paper describes the chromatographic behavior of re-versed-phase peptide separations as a function of pH and counterion concentration. Theimpetus to explore this is driven by rapid growth in peptide drug candidates and peptide-based active pharmaceutical ingredients.
Peptides varying in basicity were chromatographed, and peak efficiency and symmetryrecorded as a function of acidic modifier, pH and/or counterion concentration. Dataanalysis deciphered mechanistic reasons for poorer peak shape of peptides with formicacid (as compared to traditional methods with TFA). These studies were performed withdesigned synthetic peptides as well as enzymatic protein digests.
Peak shape of basic peptides was affected by the concentration of the acidic ion-pairreagent (TFA or formate), which in turn is controlled by pH relative to pKa of the ion-pairing anion. Concentration of the cation counterion, independent of pH, did not miti-gate against poor peak shape. Peptides that are not particularly basic, display satisfactorypeak shape in the presence of low concentrations of ion-pairing anion. Control of mobilephase pH not only effects peptide peak shape, but can be very significant in affectingselectivity as well.
Optimal mobile phase conditions for reversed-phase, LC-MS amenable peptide chro-matography depends in part, on the nature of the peptides being analyzed. Not only ispeak shape and selectivity varied by relatively minor changes in pH, but the results arealso affected by the peptide pI. Implications are also raised for deactivation of the silicasurface through control of the mobile phase composition. The results of this work provideinsights leading to facile development of robust and rugged LC-MS methods for peptideanalysis.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-398-TU
Newly Developed Two Types of Packing Materials Basedon Organic/Inorganic Hybrid Silica and High-Strength Silica
for Preparative HPLC of Peptides and Proteins
Noriko Shoji, Chie Yamashita, Takatomo Takai, Masakatsu Omote,Naohiro Kuriyama, Takashi Sato
YMC Co., Ltd., Ishikawa, Japan
Nowadays, reversed-phase (RP) chromatography has an important role in precise purifica-tion of high value-added compounds, such as pharmaceutical peptides or proteins. Wehave developed two types of preparative bulk packing media based on the different basematerials, high-strength silica (YMC-Exphere) and organic/inorganic hybrid silica (YMC-Triart Prep), to provide improved recovery, selectivity, and longer life time especially inpurification of peptides and proteins.
The novel high-strength silica particles for YMC-Exphere was successfully prepared byour original process, which allows the higher density and narrower particle size distribu-tion than typical silica gel. YMC-Exphere gives superior column efficiency and highermechanical strength in packing/unpacking process of dynamic axial compression col-umns. The chemical bonding method and density of C18 or C8 functional groups wereoptimized to enhance chemical stability and selectivity under the typical peptide purifica-tion conditions. Furthermore, YMC-Triart Prep which is based on multi layered organ-ic/inorganic hybrid silica particles has been developed by using microreactor technologyand novel surface modification technology for higher chemical stability especially inalkaline conditions. The pore size and pore volume of both of these new particles wereoptimized for the effective separations of peptides and proteins.
In this poster, we will evaluate pH stability and mechanical stability of these brand newpacking materials comparing existing conventional silica media and also compare thechromatographic performance for peptides and proteins.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-399-MO
Novel Derivatives of Lamivudine. Stability Studiesin Different Matrices
María S. Gualdesi1, Mónica A. Raviolo1, Josep Esteve-Romero2,Margarita C. Briñón1
1Departamento Farmacia. Facultad Ciencias Químicas. Universidad Nacional Córdoba,Argentina
2Química Bioanalítica, QFA-ESTCE, Universidad Jaume I, Spain
Aiming to develop antiviral agents (anti-HIV) with optimal pharmacokinetics properties,we have investigated novel derivatives of lamivudine (3TC), by association with differentaliphatic alcohols. Among them, that of associate with methanol (3TC-Meta), ethanol(3TC-Eta), n-propanol (3TC-nPro), n-Butanol (3TC-Buta), n-Pentanol (3TC-Penta), n-Hexa-nol (3TC-Hexa) and n-Octanol (3TC-Octa) demonstrated to exhibit important biologicalactivity against HIV and HBV [1, 2]. The Food and Drug Administration (FDA) Guidancefor Industry concerning in vivo bioavailability and bioequivalence studies for immediaterelease solid oral dosage forms includes a discussion on gastrointestinal stability. Stabilityin the gastrointestinal tract (GIT) may be confirmed by incubating the drug substance ingastric and intestinal fluids that are representative of in vivo drug exposure to these fluids;e.g., 1 h in simulated gastric fluids (SGF) and 3 h in simulated intestinal fluids (SIF). Signif-icant degradation (> 5%) of a drug assessed in this manner could suggest potential insta-bility in the GIT. In this work, we present stability studies of 3TC and their derivatives inbuffer pH’s 1.2 and 6.8; simulated gastric fluid (SGF, pH 1.2) and intestinal one (SIF, pH6.8), all as indicated in USP 32. As analytical method we have used Micellar Liquid Chro-matography (MLC), validated according to the FDA guideline [3]. Both, the disappear-ance of reagents (3TC derivatives) and the appearance of the only degradation product(3TC) were analyzed for each compound. Zidovudine was used as internal standard for amajor statistical analysis. In buffer pH’s 1.2 and 6.8 as well as in SGF, no degradation ofintact drugs was observed after 48 h of incubation, while in SIF different degradationprocess was detected, caused by the presence of pancreatin. Table 1 shows the percent-ages of degradation of each derivative in SIF at 3 h of incubation, which follows a pseudo-first-order kinetics.
Table 1. Percentage of degradation at 3 hours of study of carbonates of 3TC in SIF.
Compound 3TC-Metha
3TC-Etha
3TC-nPro
3TC-Buta
3TC-Penta
3TC-Hexa
3TC-Octa
% degradation at 3 hs 9.97 10.86 19.26 17.90 11.30 57.50 44.44
The degradation of the derivatives is independent of medium pH and the presence ofpepsin, since no degradation was observed at pH’s 1.2, 6.8 and SGF. By contrast, 3TC

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
derivatives are affected by the presence of pancreatin (Table 1), degraded at a rate > 5%after 3 h of incubation in SIF. These results indicate that the 3TC derivatives if given orallyshall be absorbed as a percentage > 10%, and if you want to form a prodrug reservoir forlater release of 3TC should be studied roads administration other than oral.
1. Ravetti S, Gualdesi MS, Trinchero-Hernández JS, Turk G, Briñón MC. 2009. Bioorg Med Chem 17:6407–6413.
2. Gagey D, Ravetti S, Castro EF, Gualdesi MS, Briñón MC, Campos RH, Cavallaro LV. 2010. Int J of Anti-microb Agents 36:566–569.
3. Development and validation of Micellar Liquid Chromatographic method for lamivudine and derivatives.Gualdesi, MS, Raviolo, MA, Esteve Romero, JS, Briñón, MC. 36th International Symposium of High-Perfor-mance Liquid Phase Separation and Related Techniques. 2011. Hungary.
This study was part of project CTQ2007-64473/BQU, funded by Ministeri d’Educació i Ciència (MEC) andFONCYT Préstamo BID PICT Nº 1325 (Argentina).

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-400-TU
On-Line Simultaneous Analysis of Negative Charged and PositiveCharged Analytes by Capillary Electrophoresis
with FASI-Sweeping and its Applications
Shao-Yun Wei, Su-Hwei ChenSchool of Pharmacy, College of Pharmacy, Kaohsiung Medical University,
Kaohsiung, Taiwan
We developed an on-line stacking method, including field-amplified sample injection(FASI) and sweeping for simultaneous separations of acid and basic drugs in commercialtablets and plasma simple by capillary electrophoresis (CE) with UV detection at 254 nm.
A sample pretreatment by liquid-liquid extraction with CH2Cl2/diethyl ester (v/v, 40:60)for plasma pretreatment and subsequent quantitation by FASI-sweeping-CE was used.Before sample loading, a rinse buffer containing 50 mM Tris/phosphate (pH 3), 41%MeOH and 0.1% polyethylene oxide (PEO) (as a dynamic coating polymer) at 50 psi for5 min in order to suppress the EOF. Sample loading of anionic drugs was achieved byelectrokinetic injection at a negative voltage of −2.5 kV for 80 s and anions turned toneutral compounds as soon as analytes entered the acid buffer (pH 3).
The basic drugs were injected at a positive voltage of +5 kV for 120 s. Finally, thesweeping with SDS micelles from BGE (25 mM Tris/phosphate and 60 mM SDS, pH 3)was performed by negative voltage of −20 kV.
Under the optimal FASI-sweeping CE condition, good separation with high efficiencyand short migration time is achieved. Several parameters affecting the sample stackingand separation of the drug were studied, including sample matrix, pH and concentrationsof the buffer and surfactant.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-401-MO
Optimization of Analytical Conditions for Amino Acidsin Urine by CE-LIF
Mª Paz Lorenzo, Carmen Fernández, Coral Barbas, Antonia GarcíaCEMBIO (Center for Metabolomics and Bioanalysis) Faculty of Pharmacy, San Pablo-CEU University,
Campus Montepríncipe, Boadilla del Monte 28668, Madrid, Spain
Amino acids play a major role in energy metabolism, neurotransmission, and lipid trans-port. Their quantitative analysis is important in disease diagnostics and, increasingly, inelucidating nutritional influences on physiology. Twenty different amino acids are used tosynthesize proteins.
There are several methods for amino acid analysis in biological samples [1] all of themwith strenghts and drawbacks. Capillary electrophoresis is a well suited technique forbiofluid analysis. It requires small sample volumes, and has high efficiency to separateconsiderable number of sample components. CE-UV may suffer from poor concentrationsensitivity, compared to UV absorbance, laser induced fluorescence detection offers bettersensitivity, besides it is regarded to be more selective.
4-Fluoro- or 4-chloro-7-nitro-2,1,3-benzoxadiazole (NBD-F) is a fluorogenic labelingagents for high speed derivatization of amino acid analysis. NBD-F labeled amines areefficiently excited using 488 nm light allowing more common lasers to be used for excita-tion than has been previously possible.
Both the derivatization process for urine samples and standards and the separationconditions were optimized. Conditions such as: temperature (50–80 °C), reaction time(5–20 min), reactants (10–125 mM) or pH (8–11) were tested for the derivatization.Regarding the separation conditions, stacking effect, BGE concentration (75–125 mM),pH, cyclodextrin concentration (5–25 mM) and internal standard were also tested, as wellas voltage, capillary temperature and stability of the treated urine samples.
The final CE-LIF method allows a baseline separation of several amino acids in less than20 min, including glutamate, glycine, taurine, L-and D-serine, aspartate, glutamine,histidine, 1-methylhistidine, alanine and β-aminoisobutyric acid with high sensitivitywhich are the main amino acids in urine. The CE-LIF method has been validated for 10representative amino acids and applied to quantify urine samples.
1. Éva Szökö, Tamás Tabi. J. Pharm. and Biom. Anal. 53 (2010) 1180–1192

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-402-TU
Optimization of Monoclonal Antibody Separation Conditionson Size Exclusion Chromatography
Hiroyuki Moriyama, Hiroyuki Yamasaki, Michiko SakataSeparation center, TOSOH Corporation,
Kaisei-cho 4560, Shu-Nan, Yamaguchi 746-8501, Japan
Monoclonal antibodies (mAbs) are widely using as a biopharmaceutical and still develop-ing new mAbs by modifying the complementarity determining regions. There are someanalysis technologies for quality control of mAbs such as centrifugation, size exclusion andion exchange chromatography, etc. Size exclusion chromatography (SEC) is a powerfuland convenient tool for determining mAb monomers and their impurities including ag-gregates, oligomers and mAb fragments. Although each mAb have different properties onhydrophobicity, hydrophilicity and electro-statics, chromatographic conditions such aspH, ionic strength, and operating temperature play an important role on obtaining betterresolutions by narrowing peak widths and enlarging elution gaps of mAbs and theirrelated fragments. Although the pore size for separating mAb monomer and its dimerrequired is estimated around 100 KDa – 500 KDa, the slope of the calibration curve oversuch range are more critical property to obtain better resolutions.
We report here the investigation of chromatographic conditions for affecting the reso-lution between monomer and dimer of mAb sample on SEC mode.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-403-MO
Optimization of Protein Separations on Weak Cation ExchangeColumns – a Study of Particle Size, Buffer Salts and Gradients
Faizy Ahmed1,2, Christina Song2, Jennifer Palladino2, Taegen Clary1
1Agilent Technologies,5301 Stevens Creek Blvd. Santa Clara, CA, USA
2Department of Pharmacology,Gillespie NRF, UC Irvine, CA, USA
Bio-therapeutic proteins, such as monoclonal antibodies (mAbs), undergo structural andchemical changes during preparation, formulation or storage. These include chemicalmodifications such as oxidation or deamidation and physical changes such as denatur-ation or the formation of aggregates. C-terminal processing of monoclonal antibodiesleads to the absence of lysine residues in the monoclonal antibodies (mAbs) derived frommammalian cells. Weak Cation Exchange (WCX) chromatography is used for the charac-terization of these charge-variant of mAbs.
We first studied the effects of different buffer salts – NaCl vs. NaPO4 for the separationof protein standards on Bio MAB WCX columns. It was found that the proteins did notelute according to their pI with one of them, RNAse, eluting after lysozyme in NaCl gradi-ent whereas the order of separation occurred according the pI in NaPO4 gradient. How-ever, when the NaCl gradient was used on a monolith column (Bio Monolith CM-15) theelution order remained according the protein pI. We studied the effect of particle size (1.7μm to 10 μm), buffer salts, pH and column lengths and gradient times to optimize theseparation of proteins from their impurities. Better separations were obtained with NaPO4compared to the NaCl gradients. Chromatographic conditions were then optimized forthe resolution of the charge-variants of mAbs. Digestion of mAbs with carboxypeptidaseB resulted in the disappearance of one of the basic peaks.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-404-TU
Performance Characteristics of Commercially Available Gelsfor Protein Analysis by Capillary Gel Electrophoresis
with UV Detection
Christian Wenz, Rainer Nitsche, Hans Brunnert, Martin GreinerAgilent Technologies R&D and Marketing GmbH & Co KG,
Hewlett-Packard-Str. 8, 76337 Waldbronn, Germany
Capillary gel electrophoresis is a widely used tool for the size-based analysis of proteins.Due to several advantages with regard to automation, reproducibility and resolution it hasreplaced the classical technique sodium dodecyl sulfate-polyacrylamide gel electrophore-sis in many labs, especially in the biotechnology industry. Capillary gel electrophoresis itis now routinely used in the quality control environment to assess purity and integrity oftherapeutic proteins including monoclonal antibody. Commonly commercially availabledextran-based separation matrices are used for these separations. Here, two separationmatrices for protein characterization by capillary gel electrophoresis with UV detection,the SDS Gel Buffer from Beckman Coulter and the recently introduced Protigels fromAdvanced Analytical, were compared in detail on the Agilent 7100 Capillary Electrophore-sis system. Two different sample sets were analyzed: first, a protein size standard and BSAas a test protein for molecular weight determination; second, a reduced antibody stan-dard for quantification of light chain, non-glycosylated heavy chain and heavy chain. Toget an estimate for batch-to-batch variability, all experiments were performed with twodifferent gel and capillary batches, respectively. Across these experiments, the gels fromboth suppliers showed a similar performance. Furthermore, impurity detection experi-ments were done with a low molecular weight protein spiked into a non-reduced anti-body standard sample. With the gels of both suppliers it was possible to detect the lowmolecular weight protein down to a level of 0.1%.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-405-MO
Proline-Hydroxyproline Dipeptide Analysis using Barium Hydroxideand Barium Acetate by High-Performance Anion-Exchange
Chromatography Coupled with Pulsed Amperometric Detection
Ha-Jeong Kwin, Hee-Jung Sim, Seon-Pyo HongDepartment of Oriental Pharmaceutical Sciences, College of Pharmacy, Kyung Hee University,
Dongdaemoon-gu Hoegi-dong #1, Seoul 130-701, Korea
We developed a simultaneous analytic method for proline (Pro), 4-hydroxyproline (Hyp)and proline-hydroxyproline dipeptide (PHP) by high-performance anion-exchange chro-matography (HPLC) with pulsed amperometric detection (PAD). Three analytes wereseparated on an anion exchange column (AminoPac PA10, 4 mm i.d. × 250 mm) using1.2 mM barium hydroxide and 1.5 mM barium acetate as a mobile phase. Resolutionswere 2.7 (PHP and Hyp) and 1.6 (Hyp and Pro) under this condition. Normalized capacityfactor (k’/k’0) of PHP was measured by 20 times injection of PHP standard solution with aforty-minute cycle for evaluating stability of retention time and finally it came out as agood stability of retention time (k’/k’0 > 0.97). Sample pretreatment steps were simplifiedwithout derivatization. The detection limit (S/N = 3) for Pro, Hyp, and PHP were 0.1,0.05, and 0.3 μM, respectively. Linear dynamic range was 1–100 μM (r2 > 0.9990) for anon-hydrolyzed rat urine sample. The intra- and inter-day precisions were < 8.6%, withsatisfactory mean recoveries (91.6–100.0%). This method was applied to non-hydrolyzedrat urine samples and differentiated osteoporosis groups from normal groups and showeda possibility to be a practical procedure for screening and follow-up monitoring of osteo-porosis and other bone diseases.
This study was supported by a Korea Research Foundation (KRF) grant funded by the Korea government(MEST) (No. 2009-0073818).

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-406-TU
Proteins and Peptides Separation with 2 μμm Non-PorousHigh Resolution ODS Column
Itaru YazawaImtakt Corporation,
Kyoto Research Park, Kyoto 600-8813, Japan
Life science research requires the separation of many different proteins and peptides, suchas monoclonal antibody (mAb), pegylated protein, proteomics, etc. For these demands,we have developed a novel 2 μm non-porous ODS column, which offers the high-perfor-mance of 75,000 plates for 250 x 4.6 mm column dimensions. Because the column im-proves peak recovery due to the absence of pores, it is particularly useful for proteinseparation.
We have tested this non-porous ODS column with protein and peptide separation inthis study.
We found that our non-porous ODS column generates much larger numbers of peptidepeaks compared to porous ODS columns. Accordingly, non-porous ODS can detect smallamounts of peptides, which may be important as bio-markers.
Also we found that this column has an excellent separation performance for mAb witha shallow gradient elution at high temperature. Using a gradient slope was very importantfor molecular recognition of the protein.
For polyclonal IgG in human serum, this novel high resolution ODS column generated1200 peak numbers with a 300 minute run time. This result demonstrates that, despite alow surface area, our non-porous ODS column has excellent molecular recognition perfor-mance. In another separation study, around 700 peaks of hemocyanin and hemoglobinwere identified.
Pegylated protein consists of a fixed MW protein and various MW of PEG. This columnseparated several peaks of different MW of pegylated protein.
We have tested other proteins with consistently excellent separation. In conclusion, thisnon-porous high resolution ODS column creates novel RP separations of proteins andpeptides.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-407-MO
Qualitative and Quantitative Determination of Brain Phospholipidsby Reversed Phase LC/MS(MS) Method
Róbert Berkecz1, Zoltán Kele1, Zoltán Szabó1, Heikki Tanila2,Daniel Michaelson3, Tamás Janáky1
1Department of Medical Chemistry, University of Szeged, Dóm tér 8,6720 Szeged, Hungary
2A. I. Virtanen Insitute, University of Eastern Finland,Neulaniementie 2, 70211 Kuopio, Finland
3Department of Neurobiochemistry, Tel-Aviv University,Ramat-Aviv, 69978 Tel-Aviv, Israel
Lipidomics, after genomics and proteomics, is a new and rapidly growing researchfield. Lipids are an important group of biomolecules that exist in great variety in higherorganisms. Glycerophospholipids (PL) as a major class of lipids are known as a mem-brane builder owing to his amphipathic properties, as cellular messengers, and asenzyme activators. PLs are a principal component of the brain. Dependence betweenchanging of level of PLs and some disease such as schizophrenia, Alzheimer’s, Farberdisease, Niemann-Pick disease, Gaucher disease and Down syndrome are intensivelyinvestigated area.
Chemically all glycerophospholipids are derivatives of phosphatidic acid where thephosphate group is bonded to the glycerol backbone at sn-3 position, additional hydroxylgroups of glycerol are esterified to the carboxyl goups of two fatty acid chains at sn-1 andsn-2 positions, moreover. The main classes can be classified the following phosphat-idylcholine (PC), phosphatidylethanolamine (PE), phosphatidylinositol (PI), phosphatidyl-serine (PS), phosphatidylglycerol (PG) and finally the phosphatidic acid (PA) where thephosphate group is not derivatised.
Analysis of membrane PL is a challenge both in identification and quantification foranalytical chemists because of wide range distribution of PLs in tissues and the diversebehavior of different PL classes during the separation (LC) and ionization process (MS).The mass spectrometry analysis has been widely used in lipidomics due to its advantagessuch as unparalleled sensitivity, specificity and versatility. The predominated soft ioniza-tion techniques are electrospray ionization, ESI; atmospheric pressure chemical ionization,APCI and matrix-assistant laser desorption ionization, MALDI in this field.
This study was undertaken with two specific goals. First was to develop an efficientreversed-phase liquid chromatography/electrospray ionization mass spectrometrymethod for analysis of glycerophospholipids from biological samples. Our second aimwas to determine the effects of fish oil, Fortasyn (a mixed chow of Danone) and phyto-sterols enriched diets on the membrane PL compositions in different mouse brainareas.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
The reverse-phased LC separation of PLs was achieved on Kinetex® C18 2.1 x 100 mmI.D. with 2.6 μm particle size 100 Å column afforded in high separation capability. Usingour LC-MSMS method we were able to identify 14 PS, 31 PE and 36 PC in positive modeand 11 PS, 30 PE, 11 PS, 8 PI and 2 PG species in negative mode on a QTOF Premier(Waters) mass spectrometer applying Lipidomics Gateway Phopholipid Database. Theidentified PLs were quantified by LC-MS measurement using the Quanlynx software.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-408-TU
Quality Control of Intact Biotherapeuticsby High-Resolution LC/MS
Arnd Ingendoh, Dirk Wunderlich, Christian AlbersBruker Daltonik GmbH,
Bremen, Germany
The production of recombinant proteins is one of the fastest growing sectors in the phar-maceutical industry as these proteins are increasingly used as drugs. With this interest innew biopharmaceuticals proper quality control is needed to ensure the use of the rightbatches in the proteins production. This includes knowledge about the correct amino acidsequence as well as characterization of modification sites. Using a high-resolution instru-ment is recommended due to the heterogeneity of those samples as well as the advan-tage of having an exact mass on the intact protein and enzymatic fragments.
We use an ultrahigh-resolution QTOF for the LC/MS analysis of various proteins, likerecombinant IgG. The IgG was obtained from Chinese Hamster Ovary (CHO) cells. Inaddition, a mixture of recombinant proteins was obtained from E.Coli cells. Proteins wereseparated with a Zorbax SBC8, Rapid Resolution Cartridge (2.1 x 30 mm, 3.5 μm) within15 minutes and analyzed by MS.
The resolution and wide mass range of the instrumental setup allowed for discrimina-tion of discrete changes in the glycosylation patterns even for proteins like IgG. Furtheranalysis of the recombinant IgG after reduction and alkylation allowed to measure theMW of the light chain at 25 kDa with 0.2 ppm mass accuracy. The MS spectra of therecombinant E. coli proteins allowed to distinguish between protein monomers anddimers, covering a mass range up to 40 kDa.
The data was processed by specially designed software modules, which compare masspatterns and accuracies with expected values ensuring a fast and reliable informationretrieval, which is mandatory for quality control. In combination with the possibility ofrunning the intact proteins directly, a high-throughput analysis is possible.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-410-TU
Reversed-Phase Performance Liquid Chromatographic Determinationof Lipophilicity of Potential Antitrypanosomal Compounds
Mónica A. Raviolo1, Margarita C. Briñón1, Maria Rambla-Alegre2,Samuel Carda-Broch2, Adrián Martinavarro-Domínguez2,
Josep Esteve-Romero2, Devasish Bose2
1Departamento Farmacia. Facultad Ciencias Químicas. Universidad Nacional Córdoba,Argentina
2Química Bioanalítica, QFA-ESTCE, Universidad Jaume I,Spain
Lipophilicity of a substance is one of the parameters which plays a basic role in manybiological processes. Fujita et al have proposed the n-octanol/water partition coeffi-cient, Po/w, as a measure of compound’s lipophilicity. The logarithm of Po/w (logPo/w) iswidely used because of its simplicity and some similarity between n-octanol and bio-logical membranes. Nowadays, the determination of the partition coefficient usuallymeasured by the “shake-flask” method, is often superseded by chromatographic meth-ods. It has been demonstrated that the capacity factor (k) of a compound by RP-HPLCmethod is a reliable indirect descriptor of lipophilicity of a compound. The capacityfactor is given by k = (tr − t0)/t0, where tr and t0 are the retention times of the solute andthe unretained compound, respectively. Moreover, some studies have shown thatlogk’w, the capacity factor which is extrapolated from the binary phase to 100% waterin RP-HPLC system, is an even better descriptor of lipophilicity than an isocratic factorbecause it is independent of any organic modifier effects, and it reflects polar – non-polar partitioning in a manner similar to the “shake-flask” measurement. This methodwas extensively evaluated and the Organization for Economic Cooperation and Devel-opment (OECD) to prepare a guideline that should be followed to produce reliablelogPo/w values using RP-HPLC. According to OECD the logPo/w value of an analyte canbe evaluated from a calibration graph constructed from the logPo/w and logk’w values ofreferences compounds. The reference substances not only should have logPo/w valueswhich encompass the logPo/w of the test substances but also it is preferable that shouldbe structurally related to them. Based on it, selected references substances were: ani-line, phenol, benzoic acid, toluene, allopurinol, didanosine. High correlation was foundbetween logPo/w (bibliography) and logk’w (experimental) values (r2 = 0.9960). The aimof this study was to determine the logPo/w of six novel derivatives of allopurinol, synthe-sized as potential antitrypanosomal drugs (etiologic agent of Chagas’ disease). For thispurpose we determined the logk’w of theses derivatives on a C-18 stationary phase at aflow rate of 1 mL/min using the mixture of methanol and water as eluent and UVdetection. The logPo/w of each derivative of allopurinol were evaluated from the calibra-

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
tion graph and as expected the derivatives were more lipophilic that allopurinol, whichgives them advantages in terms of pharmacokinetic properties such as cross mem-branes by passive diffusion.
This study was part of project CTQ2007-64473/BQU, funded by Ministeri d’Educació i Ciència (MEC) andFONCYT Préstamo BID PICT Nº 1325 (Argentina). Maria Rambla-Alegre also wishes to thank the MEC for herFPU grant.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-411-MO
Separation of Peptides and RNA/DNA Fragments in 100%Aqueous Solution on RPLC-Phases with Tuned Hydrophobicity
Joakim Högblom, Anders Törncrona, Maria Forsberg, Sylvia Winkel-PetterssonSeparation Products, Eka Chemicals AB, SE
445 80 Bohus, Sweden
Biopharmaceutical drug candidates are becoming ever more dominant players in thepharmaceutical development pipeline. To guarantee the quality and clinical efficacy andsafety, it is of uppermost importance to have access to relevant and efficient analyticaltools. Detection and quantification based on HPLC techniques has proven to be thetechnique of choice for many years and thus present focus is set on developing stationaryphases that are combining the credited HPLC technique with specific needs defined bynew classes of biopharmaceuticals.
The objective of the present work was to investigate the effect of different surfacecompositions of silica based stationary phases on their ability to separate different compo-nents in 100% aqueous solutions containing biological solutes. A silica matrix was surfacemodified by different proportions of hydrophobic and hydrophilic moieties to tune thehydrophobic/hydrophilic character of the resulting materials. Some materials were alsoprovided with cationic moieties to add ion-exchange interactions to broaden their separa-tion ability.
The resulting materials were characterized by their chemical surface composition,wettability of 100% aqueous solutions, and their chromatographic performance. Nucleicacids and enzymatic digests of proteins in 100% aqueous solutions were separated oncolumns packed with the stationary phases of different hydrophobicities.
In addition, the prepared materials were evaluated with respect to their ability toseparate fragments of DNA. It was found that relatively minor variations in the hydropho-bic/hydrophilic balance of the stationary phase enabled fine tuning of the separation ofRNA/DNA fragments, obtained by means of restriction enzymes, or oligonucleotides.Similar results were obtained with separation of enzymatic digests (endoproteases) ofproteins.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-412-TU
Simultaneous Determination of Catecholaminesfrom Rat Cerebrospinal Fluid by High-Performance
Liquid Chromatography with UV Detection
Min-Jung Gu, Hee-Jung Sim, Seon-Pyo HongDepartment of Oriental Pharmaceutical Sciences, Kyung Hee University,
Dongdaemoon-gu Hoegi-dong #1, Seoul 130-701, Korea
A simple determination method has been developed for serotonin (5-HT), 5-hydroxy-indole-3-methoxybenzeneacetic acid (5-HIAA), homovanilic acid (HVA), dopamine (DA),and 3,4-dihydroxyphenyl acetic acid (DOPAC) by reversed-phase high-performanceliquid chromatography with UV detection. Separation of the five catecholamines wasobtained on an aqueous column (Hypersil GOLD AQ, 2.1 mm i.d. × 150 mm, 5 μm)with a flow rate of 200 mL/min and 20 mM KH2PO4 with 95% phosphate buffer pH 3.0was used as a mobile phase. The sample injection volume was 10 μL and the optimal UVabsorbance wavelength was 270 nm. The column oven temperature was maintained at30 °C. All analytes were separated under these conditions and this method was appliedto determinate catecholamines from rat cerebrospinal fluid samples. Simultaneousdetermination of DA, 5-HT, DOPAC, and HVA was conducted from rat cerebrospinalfluid samples pretreated with perchloric acid. All samples were stored under −20 °Cbecause It was found that the storage of rat cerebrospinal fluid at −20 °C cause losses ofdopamine and serotonin.
This study was supported by a Korea Research Foundation (KRF) grant funded by the Korea government(MEST) (No. 2009-0073818).

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-413-MO
Simultaneous Determination of Clarithromycin and Tobramycinin Different Plasma by Sensitive LC-MS/MS
B. Cahay, B. Mertens, R. Klinkenberg, B. StreelGalephar M/F,
39 rue du Parc Industriel, B-6900 Marche-en-Famenne, Belgium
Tobramycin is an aminoglycoside used in the treatment of Pseudomonas Aeruginosa, themain opportunistic bacteria found in cysticfibrosis patients. Clarithromycin is an antibiotic(macrolide) active on gram+ and gram− bacteria.
An ion-pairing chromatographic separation coupled with a MS/MS detection wasdeveloped due to the lack of chromophor group in tobramycin and the high difference ofpolarity between the two active molecules.
A solid-phase extraction (SPE) containing cation exchange phase (Oasis® MCX) is usedto isolate the compounds from the biological matrix. The eluate was evaporated andreconstituted with mobile phase before analysis in the LC-MS/MS system. A good separa-tion was obtained on an octadecyl silica based column using a mobile phase gradientsystem composed of water, heptafluoro butiric acid and formic acid.
Deuterated clarithromycin and deuterated tobramycin were used as internal standards.Mass spectrometric detection was carried out using a triple quadrupole mass spectrom-
eter apparatus equipped with a TurboIonSpray interface operating in the positive ionmode.
The lower limits of quantification obtained were 50 ng/ml for tobramycin and 5 ng/mlclarithromycin.
The method was validated using the total error concept and the accuracy profiles as adecision tool [1].
1. Ph Hubert and Al, STP Pharma Pratiques 13 (2003); 101–138

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-414-TU
Simultaneous Determination of Doxorubicin and Epirubicinin Plasma by Capillary Electrophoresis with Field Amplified
Sample Injection and its Application
Su-Hwei Chen, Ping-Chih LinSchool of Pharmacy, College of Pharmacy, Kaohsiung Medical University,
Kaohsiung, Taiwan
Doxorubicin and epirubicin are anthracycline antibiotic epimers widely used in treatmentsarcomas and a variety of carcinomas. They are usually used in combination with otheragents such as cyclophosphamide, vincristine, procarbazine, etc. Myelosuppression is amajor dose-limiting complication. On the other hand, cumulative dose-related chroniccardiotoxicity is a serious clinical complication of anthracycline therapy. The clinical valueof doxorubicin and epirubicin is limited by the dose-dependent cardiotoxicity. Therefore,it is very necessary to monitor the plasma concentration levels of doxorubicin andepirubicin during chemotherapy for preventing the cardiotoxicity.
A capillary electrophoresis using field amplified sample injection with UV detection (CE-UV) is described for the simultaneous determination of doxorubucin and epirubicin inhuman plasma. After sample pretreatment, Field-amplified sample stacking method isapplied for sensitive improvment. The sample was empolyed with an electrokinetic injec-tion of 10 kV for 30 sec. CE separation of doxorubicin and epirubicin from human plasmawas performed at 25 °C using a background electrolyte consisting of phosphate buffer(75 mM, pH 5) containing 0.02% PVP(55000) and 45% organic modifier (methanol 40%and 5% acetonitrile).

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-415-MO
Simultaneous Determination of Drugs in Human Autopsy Material
R. Oertel1, J. Pietsch2, N. Arenz1, S. G. Zeitz1, L. Goltz1, W. Kirch1
1Institute of Clinical Pharmacology, Medical Faculty Carl Gustav Carus, Technical University Dresden,Fiedlerstr. 27, 01307 Dresden, Germany
2Institute of Legal Medicine, Medical Faculty Carl Gustav Carus, Technical University Dresden, Germany
This study describes a cooperation between legal medicine and clinical pharmacology. Inthe legal medicine autopsy material is normally investigated to find out the cause ofdeath. But in many cases drugs were detected without connection to the cause of deathand up to now no further investigations have taken place. In our study it was possible tomeasure more than twenty drugs in human directly in several compartments. The de-ceased had a continual therapeutic treatment, a treatment during an operation or anunsuccessful urgent therapy.
A liquid/liquid extraction and a LC/MS/MS method were developed for the determina-tion of these drug concentrations. Using LC/MS/MS a complete chromatographic separa-tion of all analytes is not necessary. But when measuring many transitions in a biologicalmatrix two problems should be excluded: ion suppression and a too small number ofmeasurement points per peak. A relatively short operation time and a sufficient separationwere reached by column, eluent, and gradient optimization with POPLC (Phase Opti-mized Liquid Chromatography).
Different autopsy materials of about 150 cases were investigated. Especially in caseswith four or more simultaneously given drugs their distribution in the compartments isvery interesting for pharmacokinetic examinations. In the autopsy material of seven de-ceased four or more drugs could be proved and determined simultaneously. So the distri-bution patterns of the drugs in the compartments of one individual deceased were com-pared. This means that the great differences between the subjects like age, gender,weight, disease, and cause of death you normally meet in those studies could be exclud-ed. This is a great advantage of our study. Mostly the dosage of the drugs was unknownand only sometimes the time between drug application and death was documented.
Measurements of drug concentrations in human autopsy material deepen the knowl-edge of the respective drugs’ pharmacokinetic.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-416-TU
Strategies for Coupling of IEC and SEC to MS Detectionfor the Separation and Characterization of Related Substances
in Biopharmaceuticals
W. Decrop, E. J. Sneekes, R. SwartDionex Corporation,
Amsterdam, The Nederlands
The identification of related substances and impurities in drug products is an importanttask during all staged of the pharmaceutical development process. For small moleculedrugs, reversed phase HPLC in combination with mass spectrometric detection is ideallysuited for this task. However biological drug compounds (biopharmaceuticals) are com-plex molecules and require additional LC methods for the separation of related substancesand their characterization. Ion-exchange chromatography is typically used for the separa-tion of charge variants; size-exclusion chromatography is used for the separation of aggre-gates and degradation products. Mobile phases applied in these LC techniques typicallycontain high concentrations of non-volatile buffer salts.
The experimental workflow includes the following steps: i) separation of protein drugand related substances on either ion-exchange or size exclusion column, ii) fractionationof peaks of interest, iii) desalting of fractions on RP SPE column, iv) elution of desaltingcompounds to MS and MS and MS2 detection of the protein.
Ion-exchange chromatography is a well established LC technique for the characteriza-tion of proteins therapeutics. For the separation of protein variants which often beardifferent charges, ion-exchange LC is more selective than reversed phase HPLC. Theapplication of ion-exchange chromatography of intact proteins was tested with the analy-sis of ribonuclease A. Application of the set-up for biopharmaceutical analysis will bedemonstrated.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-417-MO
Two-Dimensional Chromatography for Quantitation and MS Analysisof Monoclonal Antibodies in Complex Samples
Sean M. McCarthy, Thomas E. Wheat, Ying Qing Yu, Jeff R. MazzeoWaters Corporation, Biopharmaceuticals,
Milford, USA
Identification, characterization, and quantitation of monoclonal antibodies (mAbs) isrequired at many stages of biopharmaceutical research and development. The primaryanalytical tools are liquid chromatography coupled with UV or mass spectrometry (MS).Both techniques can be compromised by interferences in the sample matrix, includinghigh salt concentrations, other proteins, or the components of cell culture media. A highthroughput analytical technique should combine sample preparation and chromatograph-ic techniques to ensure accurate and robust quantitiation. Affinity chromatography onimmobilized Protein A can be used to isolate the antibody from a complex matrix whilereversed phase LC is useful for introducing a salt free, concentrated sample into an MSion-source.
Affinity chromatography is often used to determine protein titer by UV absorbanceusing peak area due to its high specificity, linearity, and reproducibility. Mass spectromet-ric analysis allows the practitioner to determine the mass, glycoprofile, and other massbased attributes of the analyte. While each of these methods can be performed veryrapidly, they are generally accomplished by using separate chromatographic systemssince the eluents used for affinity chromatography are incompatible with mass spectrome-try. In addition, each analysis requires different sample preparation protocols which canincrease the time necessary for analysis.
In this presentation we describe the use of an ACQUITY UPLC system with 2D technolo-gy. The 2D system allows for simultaneous purification and quantitation of monoclonalantibodies by Protein A affinity chromatography and determination of mass profile by MSanalysis after desalting on a short reversed phase column. The peak of interest is elutedfrom the first dimension affinity separation to a second dimension reverse phase separa-tion. Our data demonstrates the linearity and reproducibility of response for mAbs fromcomplex matricies. We also discuss carryover and chromatographic reproducibility ob-served when performing protein A affinity chromatography with the 2D system. 2D UPLCsystem optimization for peak collection, desalting, and MS detection of antibody samplesis also discussed.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-418-TU
UHPLC/MS/MS Metabolites Characterization of Flubendazole
Robert Jirásko1, Michal Holcapek1, Veronika Jedlicková1, Milan Nobilis2,Lenka Skálová2, Jirí Lamka2
1Department of Analytical Chemistry, Faculty of Chemical Technology, University of Pardubice,Studentská 573, 532 10, Pardubice, Czech Republic
2 Faculty of Pharmacy, Charles University,Heyrovského 1203, 500 05 Hradec Králové, Czech Republic
The detection and structure elucidation of Phase I and II metabolites of new drugs is animportant step for the drug research and development. Combination of our UHPLC sepa-ration with subsequent high mass accuracy measurement in full scan and tandem massspectra modes using QqTOF analyzer enables the identification even for trace metabolitesand give the reliable picture about present drug metabolites within several minutes. First,the data evaluation is performed and the elemental composition of present metabolites isdetermined based on the combination of following information: high mass accuracy,specific mass defect, characteristic neutral losses, comparison of fragmentation withparent drug or relative retention shifts. UHPLC/MS/MS detection of chromophore-con-taining metabolites in the complex mixture is improved by parallel UV detection. Ad-vanced software tools are applied for the metabolite identification using the comparisonof the blank chromatogram with the real incubation sample together with the softwareprediction and detection of possible metabolites. Moreover, the multistage mass spectrameasurement using ion trap based analyzer with direct infusion introduction of selectedmetabolites after their isolation provides useful information about their fragmentationpattern.
This work was supported by P502/10/0217 (GACR, Czech Republic), MSM0021627502 and MSM0021620822 (MŠMT, Czech Republic).

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-419-MO
Analytical Study of a Resin used as Sealing Materialin Ancient Pottery from Archaeological Site
by Light Microscopy, Vibrational Spectroscopyand Pyrolysis-Gas Chromatography-Mass Spectrometry
J. Peris-Vicente1, L. Osete-Cortina2, F. M. Valle-Algarra3, M. A. Ferrer-Eres4,J. V. Gimeno-Adelantado3, M. T. Doménech-Carbó2, R. Mateo-Castro3,
M. D. Soriano-Piñol3, M. Rambla-Alegre1, S. Carda-Broch1, J. Esteve-Romero1
1Química Bioanalítica, QFA, ESTCE, Universitat Jaume I,12071 Castelló, Spain
2Department of Conservation and Restoration of Cultural Goods, Heritage Conservation Institute,Polytechnical University of Valencia,
Camino de Vera 14, 46022 Valencia, Spain3Department of Analytical Chemistry, Faculty of Chemistry, University of Valencia,
C/Doctor Moliner 50, 46100 Burjassot, Valencia, Spain4Department of Prehistory and Archaeology, Faculty of History and Geography, University of Valencia,
Avda. Blasco Ibañez 28, 46010, Valencia, Spain
The archaeological site of Lixus situated on the meanders of the river Luckus (Morocco) iscurrently the object of archaeological study. This ancient Phoenician city was an impor-tant centre of metallurgical activity, especially bronze and iron. In fact, previous studieshave shown that high amount of iron objects have been produced in Lixus furnaces,especially in the 2nd century b.c. Once made, these finished objects were usually intro-duced in plugged pottery for keeping or trading. However, iron is easily corroded underthe influence of atmospheric oxygen and moisture. Thus, the storing had to assure a totalisolation from the environment and, therefore, a sealing substance was added arroundthe plug. The study of the materials used to close up the vessels containing metallicobjects will provide useful information about the storing up conditions of the iron objects.The sealing materials used in the Ancient Time have normally been taken from naturalsubstances, due to their accessibility and abundance. Resins are sticky and mainly water-insoluble materials, which are extracted from by-products of metabolism spontaneouslyexuded by a large number of trees and plants. Because of their properties, resins as anexcellent material to cover crack, fissures and joints, in order to hermetically close a ves-sel. Pine is a tree specie very abundant in the Mediterranean basin, and so that Pinus resinis often found in ancient historical and archaeological objects.
In a previous paper, a complete study about iron metallurgy has been reported, bymeans of the analysis of materials from several phases of metal production processesfound in a hermetically closed amphora. In this work, the study is focused on the resinousmaterial used with the plug to improve the sealing of the vessel, and its comparison witha present-day Pinus resin. These materials were analyzed by light microscopy, FTIR and

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
pyrolysis-GC-MS. This analytical technique provides useful information about the chemicalcomposition of the samples. In both materials, diterpenoid acids were detected, but inthe archaeological sample the amount of oxidized compounds was higher.
Acknowledgments: R+D+I Project CTQ2005-09339-CO3-02/BQU and E.R.D.F.

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-420-TU
Chromatographic and Mass-Spectrometric Techniquesfor Characterizing Carbene-Type Polymers
Petra Aarnoutse1, Eva Reingruber1, Erica Jellema2, Markus Finger2,Bas de Bruin2, Peter Schoenmakers1
1Analytical-Chemistry Group,2Department of Homogeneous Catalysis,
Van ‘t Hoff Institute for Molecular Sciences, University of Amsterdam,Science park 904, 1098 XH Amsterdam, The Netherlands
The polymerization of carbenes yields a unique new class of polymers, in which eachC-atom in the chain bears a functional group [1]. A variety of different carbene-typemolecules have been synthesized, using one (homopolymers) or two (copolymers) differ-ent monomeric units [2, 3].
Advanced characterization techniques are essential to guide the development of a newclass of polymers. Homopolymers feature different end groups, which provide essentialinformation on the reaction mechanism. Copolymers show various complex variationsaround an average chemical composition.
End-group (or “functionality-type”) distributions (FTDs) can be obtained from liquid-chromatographic separations at the critical conditions (CC), where retention is indepen-dent of the molecular weight and solely governed by the functional groups (or endgroups). A complete characterization of the FTD would require comprehensive two-di-mensional liquid chromatography separations, with CC conditions in one of the separa-tion stages and size-exclusion chromatography in the other (CC×SEC) [4]. A great deal ofcomplementary information can be obtained from matrix-assisted laser- desorption/ioniz-ation mass spectrometry (MALDI-MS). The information from MALDI tends to be moredetailed, whereas the LC information is generally more quantitative and comprehensive(covering a broader range of molecular weights).
For the characterization of the chemical-composition distributions (CCDs) of copoly-mers gradient-elution LC is commonly used. However, the resulting separation is con-founded by molecular-weight effects. The rigorous characterization of copolymers there-fore requires two-dimensional separations, in which gradient-elution LC is combined withSEC [3].
In this poster we will present results on the characterization of functionality-type distri-butions of homopolymers by MALDI-MS and chemical-composition distributions byLC×SEC. It will be demonstrated that indispensable information can be obtained onpolymer structures and reaction mechanisms.
1. Jellema, E. et al. C1 polymerisation and related C-C bond forming carbene insertion reactions. Chem. Soc.Rev., 2010, 39, 1706

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
2. Jellema, E. et al. Rh-mediated polymerization of carbenes: Mechanism and stereoregulation. J. Am. Chem.Soc., 2007, 129, 11631–11641
3. Jellema, E. et al. Rhodium-Mediated Stereospecific Carbene Polymerization: From Homopolymers to Ran-dom and Block Copolymers. Macromolecules, 2010, 43, 8892–8903
4. Jiang, X. et al. Comprehensive two-dimensional liquid chromatography for the characterization of func-tional acrylate polymers. J. Chrom. A., 2005, 1076 (1–2), 51–61

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-421-MO
Deformation and Degradation of Polymers in Ultra-High-PressureLiquid Chromatography
Elena Uliyanchenko1, Sjoerd van der Wal1,2, Peter Schoenmakers1
1Analytical-Chemistry Group, Van ’t Hoff Institute for Molecular Sciences (HIMS), Faculty of Science,University of Amsterdam,
Science Park 904, 1098 XH Amsterdam, The Netherlands2DSM Resolve,
P.O. Box 18, 6160 MD Geleen, The Netherlands
Ultra-high-pressure liquid chromatography (UHPLC) has become a widely used separationmethod for analyses in many fields, such as pharmaceutical, environmental and foodanalysis, life science, etc. The proliferation of UHPLC applications over the last years hasbeen triggered by the advantages it offers for the separations of complex samples. Byusing smaller (sub-2 μm) particles at higher pressures (above 100 MPa) it provides signifi-cant improvements in efficiency and / or in speed of analyses.
Although size-exclusion separations of polymers at UHPLC conditions are not yet com-mon, they have great potential [1]. However, there is a fundamental limitation for thistype of separations, which is associated with shear degradation of macromolecules. Be-cause of small particles, high pressures and narrow flow channels utilized in UHPLC sys-tems, such a limit is easier encountered at UHPLC conditions compared to common HPLCpractice.
Another phenomenon caused by the high shear forces is deformation of macro-molecules. If polymer molecules are stretched in the direction of flow, the elution from aSEC column will no longer follow the conventional SEC mechanism. So-called slalomchromatography can be observed [2]. When such conditions are achieved for polydispersesamples, this may lead to the appearance of double peaks even from polymer standardswith narrow molecular-mass distributions [3].
In the present work we study the behaviour of polymers at high shear rates generatedin a UHPLC system. We show evidence that both the deformation and the degradation ofmacromolecules may occur during analyses at common UHPLC conditions. We demon-strate that slalom chromatography of polydisperse samples may be a useful analyticaltool, when understanding the behaviour of polymers at high shear rates. We attempt tospecify the molecular weight limit and the useful range of chromatographic conditions,which allow to avoid the degradation of polymers during UHPLC analyses.
1. E. Uliyanchenko, P.J. Schoenmakers, Sj. Van der Wal J. Chromatogr. A 1218 (2011) 15092. J. Hirabayashi and K. Kasai Anal. Biochem. 178 (1989) 3363. Y. Liu, W. Radke and H. Pasch Macromolecules 38 (2005) 7476

36th International Symposium on
High Performance Liquid Phase
Separations and Related Techniques
Budapest Congress & World Trade Centre
19–23, June, 2011 • Budapest, Hungary
PO
ST
ER
SE
SS
IO
N1
P1-S-422-TU
HPLC and SFC Analyses of Trimer Fatty Acids
John Kimmel, Lisa Zang, Sue D’AntonioAgilent Technologies, Life Science and Chemical Analysis,
5301 Stevens Creek Blvd, Santa Clara, CA 95051, USA
Dimer and Trimer Fatty Acids have extensive applications in the areas of polyamide resins forinks and ddhesives, lubricants, greases, oilfield chemicals, fuel additives, corrosion inhibitors,sealants, polymer intermediates and personal skin care products. The polymerization of C18based fatty acids produces complex mixtures of dimeric, trimeric and higher molecularweight acids including their isomers. It is important to determine the distribution amongmonomers, dimers, and trimers in these fatty acid components. The conventional separationmethods for analyzing these mixtures involve lengthy normal phase HPLC gradient orderivatizing the acids into esters and then analyzing indirectly with GC. Our goal is to devel-op better and faster HPLC methods using commercially available instruments to analyzetrimers fatty acids for quality control without derivitization.
In this study, we investigated alternative HPLC methods using Agilent 1200 InfinityHPLC systems including a 1260 SFC (supercritical fluid chromatography) system. Fordetection of these highly saturated fatty acid trimers, Agilent ELSD and Single QuadrupleMS detector were used. With MSD we were able to identify dimmers, trimers and highermolecular weight polymers in the complex trimer samples. The fatty acid trimer sampleswere generously provided by Chemco Systems. Samples were dissolved in isopropylalcohol without any treatment. Numbers of reverse phase, normal phase and GPC col-umns were used in this study. The separation results from different liquid chromatograph-ic techniques are compared. Faster or better separations and identifications can beachieved using some of these methods.















![10 11 Session HR Session Seri Management[1]](https://static.fdokumen.com/doc/165x107/6314ba61fc260b71020fb0ee/10-11-session-hr-session-seri-management1.jpg)




