
Past, Present and Future of Targeted Therapy in Solid Tumors
29
Current Cancer Drug Targets, 2010, 10, ???-??? 1 1568-0096/10 $55.00+.00 © 2010 Bentham Science Publishers Ltd. Past, Present and Future of Targeted Therapy in Solid Tumors A. Palazzo* ,# , R. Iacovelli # and E. Cortesi Division of Medical Oncology, Sapienza University of Rome, Italy Abstract: Targeted therapies affecting specific molecular target, expressed preferentially by neoplastic cells, block cancer growth. Current targets are represented by cell-surface trans-membrane proteins, intracellular proteins, and by growth fac- tors. Today a targeted therapy exists for most commonly diagnosed types of human cancer often combined with chemo- therapy or sometimes as monotherapy option. The epidermal growth factor receptors (EGFR) and vascular endothelial growth factors (VEGF) are known as the two main control key intracellular pathways, governing fundamental processes in cancer cells. The concept of using anti-EGFR and anti-VEGF strategies, as cancer treatment, has been soon developed and exploited extensively. We review targeted drugs currently available for routine treatment of lung, breast, colorectal and renal cell cancers, summarizing the history of identification and molecular characterization of targets or signaling pathways responsible for abnormal cell growth. We also focus on new targeted strategies, still under investigation, able to affect simultaneously tightly interconnected biological pathways or directed against new molecular targets. Keywords: Target therapy, breast cancer, lung cancer, colorectal cancer, kidney cancer. INTRODUCTION “Molecular targets,” “molecularly targeted drugs,” or “molecularly targeted therapies,” are currently used to define drugs that block cancer growth by interfering with specific molecules involved in tumor growth and progression. Though both interfere with cancer cell division, prolifera- tion and spread, traditional chemotherapies are directed against all proliferating cells, while targeted therapies affect specific molecular targets preferentially expressed by neo- plastic cells, thus resulting in considerable reduced toxicities. This point of view represents, undoubtedly, a revolution in cancer treatment by leading to a shift from non specific use of antineoplastic agents to the use of specific, targeted ones. The “ideal target” could be preferentially considered as one expressed in high copies, on the membrane or within tumor cells, genetically stable, not shed or secreted, and playing a causal role in tumor development and/or progres- sion. Current targets are cell-surface trans-membrane pro- teins, as tyrosine kinases receptors, intracellular proteins, as transcription factors, that play a role in the cytoplasmatic or nuclear signaling, and growth factors, that are serum proteins that stimulate their specific pathway through binding to their cell-surface receptors. The best model of targeting a receptor tyrosine kinase is represented by the treatment gastrointestinal stromal tumors (GIST). This neoplasia is a rare disease of the gastrointesti- nal tract driven by the expression of a cell-surface trans- membrane protein, that is a product of the KIT proto- oncogene with tyrosine kinase activity. The constitutive acti- vation of KIT signaling leads to uncontrolled cell prolifera- tion and resistance to apoptosis. In 2002 George Demetri and colleagues reported that imatinib mesylate, a selective inhibi- tor of protein tyrosine kinases, including the transmembrane *Address correspondence to this author at the Divisione di Oncologia Medica B, Sapienza Università di Roma, Viale Regina Elena, 324 00161 Roma, Italy; Tel: 39-6-4462982; Fax: 39-6-4463686; E-mail: [email protected] # These authors have contributed equally. receptor KIT, induce a response in 98.5% of patients with unresectable or metastatic GIST [1]. The first established possibility to target intracellular nu- clear receptors is represented by the approval of synthetic oestrogen-blocker tamoxifen for the treatment of metastatic breast cancer (MBC) in the 1970s [2]. This agent targets the oestrogen receptor (ER), a steroid hormone nuclear receptor which, when bound to oestrogen, modulates the transcrip- tional activity of the genes involved in proliferation and sur- vival of breast cancer cells. Since the pioneering observation of Beatson of a remission in premenopausal women with advanced breast carcinoma, following a surgical oophorec- tomy, the hormonal manipulation remains, to date, an old concept of targeted therapeutic approach in medical history [3]. Circulating growth factors and their transmembrane re- ceptors became quickly an interesting and attractive target for monoclonal antibody (mAb), that have higher specificity to bind specific targets, sparing normal tissues and causing fewer side-effects than conventional cytotoxic agents. The first proof that a monoclonal antibody therapy could improve clinical outcome was obtained with trastuzumab, a mono- clonal antibody that blocks the human epidermal growth factor receptor 2 (HER2) protein, that is overexpressed in around 25% of breast cancer patients conferring a worse prognosis [4]. In 2001 Dennis Slamon and colleagues found that women with advanced breast cancer overexpressing HER2, who received trastuzumab plus chemotherapy had an increase survival compared to those who received chemo- therapy alone [5]. Although trastuzumab in breast cancer and imatinib me- sylate, in chronic myeloid leukemia and GIST, have pro- vided early success story in the field of targeted agents, modern biology identified new potential targets for cancer therapy and new crucial pathways at the basis of tumor growth. During last years, considerable efforts have been spent in order to translate the growing molecular knowledge in targeted agents. Therefore, from pioneering trastuzumab and Imatinib mesylate to the wide range of targeted mole- cules now available many years and a hard work have been
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Past, Present and Future of Targeted Therapy in Solid Tumors

Current Cancer Drug Targets, 2010, 10, ???-??? 1
1568-0096/10 $55.00+.00 © 2010 Bentham Science Publishers Ltd.
Past, Present and Future of Targeted Therapy in Solid Tumors
A. Palazzo*,#, R. Iacovelli
# and E. Cortesi
Division of Medical Oncology, Sapienza University of Rome, Italy
Abstract: Targeted therapies affecting specific molecular target, expressed preferentially by neoplastic cells, block cancer
growth. Current targets are represented by cell-surface trans-membrane proteins, intracellular proteins, and by growth fac-
tors. Today a targeted therapy exists for most commonly diagnosed types of human cancer often combined with chemo-
therapy or sometimes as monotherapy option. The epidermal growth factor receptors (EGFR) and vascular endothelial
growth factors (VEGF) are known as the two main control key intracellular pathways, governing fundamental processes
in cancer cells. The concept of using anti-EGFR and anti-VEGF strategies, as cancer treatment, has been soon developed
and exploited extensively. We review targeted drugs currently available for routine treatment of lung, breast, colorectal
and renal cell cancers, summarizing the history of identification and molecular characterization of targets or signaling
pathways responsible for abnormal cell growth. We also focus on new targeted strategies, still under investigation, able to
affect simultaneously tightly interconnected biological pathways or directed against new molecular targets.
Keywords: Target therapy, breast cancer, lung cancer, colorectal cancer, kidney cancer.
INTRODUCTION
“Molecular targets,” “molecularly targeted drugs,” or “molecularly targeted therapies,” are currently used to define drugs that block cancer growth by interfering with specific molecules involved in tumor growth and progression.
Though both interfere with cancer cell division, prolifera-tion and spread, traditional chemotherapies are directed against all proliferating cells, while targeted therapies affect specific molecular targets preferentially expressed by neo-plastic cells, thus resulting in considerable reduced toxicities.
This point of view represents, undoubtedly, a revolution in cancer treatment by leading to a shift from non specific use of antineoplastic agents to the use of specific, targeted ones. The “ideal target” could be preferentially considered as one expressed in high copies, on the membrane or within tumor cells, genetically stable, not shed or secreted, and playing a causal role in tumor development and/or progres-sion. Current targets are cell-surface trans-membrane pro-teins, as tyrosine kinases receptors, intracellular proteins, as transcription factors, that play a role in the cytoplasmatic or nuclear signaling, and growth factors, that are serum proteins that stimulate their specific pathway through binding to their cell-surface receptors.
The best model of targeting a receptor tyrosine kinase is represented by the treatment gastrointestinal stromal tumors (GIST). This neoplasia is a rare disease of the gastrointesti-nal tract driven by the expression of a cell-surface trans-membrane protein, that is a product of the KIT proto-oncogene with tyrosine kinase activity. The constitutive acti-vation of KIT signaling leads to uncontrolled cell prolifera-tion and resistance to apoptosis. In 2002 George Demetri and colleagues reported that imatinib mesylate, a selective inhibi-tor of protein tyrosine kinases, including the transmembrane
*Address correspondence to this author at the Divisione di Oncologia
Medica B, Sapienza Università di Roma, Viale Regina Elena, 324 00161
Roma, Italy; Tel: 39-6-4462982; Fax: 39-6-4463686;
E-mail: [email protected] #These authors have contributed equally.
receptor KIT, induce a response in 98.5% of patients with unresectable or metastatic GIST [1].
The first established possibility to target intracellular nu-clear receptors is represented by the approval of synthetic oestrogen-blocker tamoxifen for the treatment of metastatic breast cancer (MBC) in the 1970s [2]. This agent targets the oestrogen receptor (ER), a steroid hormone nuclear receptor which, when bound to oestrogen, modulates the transcrip-tional activity of the genes involved in proliferation and sur-vival of breast cancer cells. Since the pioneering observation of Beatson of a remission in premenopausal women with advanced breast carcinoma, following a surgical oophorec-tomy, the hormonal manipulation remains, to date, an old concept of targeted therapeutic approach in medical history [3].
Circulating growth factors and their transmembrane re-ceptors became quickly an interesting and attractive target for monoclonal antibody (mAb), that have higher specificity to bind specific targets, sparing normal tissues and causing fewer side-effects than conventional cytotoxic agents. The first proof that a monoclonal antibody therapy could improve clinical outcome was obtained with trastuzumab, a mono-clonal antibody that blocks the human epidermal growth factor receptor 2 (HER2) protein, that is overexpressed in around 25% of breast cancer patients conferring a worse prognosis [4]. In 2001 Dennis Slamon and colleagues found that women with advanced breast cancer overexpressing HER2, who received trastuzumab plus chemotherapy had an increase survival compared to those who received chemo-therapy alone [5].
Although trastuzumab in breast cancer and imatinib me-sylate, in chronic myeloid leukemia and GIST, have pro-vided early success story in the field of targeted agents, modern biology identified new potential targets for cancer therapy and new crucial pathways at the basis of tumor growth. During last years, considerable efforts have been spent in order to translate the growing molecular knowledge in targeted agents. Therefore, from pioneering trastuzumab and Imatinib mesylate to the wide range of targeted mole-cules now available many years and a hard work have been

2 Current Cancer Drug Targets, 2010, Vol. 10, No. 4 Palazzo et al.
necessary. Fig. (1). Today all the “big killers” have at least one target therapy registered, for the use in a specific phase of cancer treatment, often combined with chemotherapy or sometimes as monotherapy option. These therapies allowed to treat some cancers that, until yesterday, were named “or-phan diseases” as GIST, renal cell carcinoma (RCC) or hepa-tocellular carcinoma (HCC). Despite the great possibilities offered by the use of target therapy in clinical practice, can-cers of lung and bronchus, colon and rectum and kidney ac-count for about 30%, of all newly diagnosed cancers among men and for about 27% among women in 2009. Breast can-cer alone is expected to account for 27% of all new cancer cases among women. The expected rate of death from these cancers projected for 2009 is of 42% for men and about 50% for women [6]. Among all known pathways (Table 1) the epidermal growth factor receptors (EGFR) and vascular en-dothelial growth factors (VEGF) act as two main control key intracellular pathways, that govern fundamental cellular processes and for more than two decades both represent the focus of attention of cancer research. Epidermal growth fac-tor (EGF) was first discovered in 1962 from new-born mice by Stanley Cohen, and the human forms were isolated later [7]. The receptor for human EGF was purified two decades after the EGF discovery and its role in malignant transforma-tion [8] was later established. Preliminary data hypothesized in 1971 that tumour growth is dependent on angiogenesis and is mainly mediated by vascular endothelial growth fac-tor. VEGF is expressed by up to 60% of human tumours. No angiogenesis inhibitors existed before 1980, and the first one was reported from the Folkman laboratory [9]. The concept of using anti-EGFR and anti-VEGF strategies in cancer treatment was soon developed and extensively exploited. This review will focus on the target therapy found to be sig-
nificantly efficacious and the novel approaches with clinical promise.
NON SMALL CELL LUNG CANCER
Lung cancer is classified histologically as squamous-cell carcinoma, small cell carcinoma, adenocarcinoma, large cell carcinoma; and clinically as small cell lung cancer (SCLC; 14%) or non-small cell (NSCLC; 85%) for treatment pur-pose. Current treatment options include surgical resection, platinum-based chemotherapy and radiation therapy alone or in combination. Unfortunately, despite these therapies, the disease is rarely curable and prognosis is poor, with an over-all 5-year survival rate of only 15% . Due to the poor prog-nosis of the advanced disease, much effort has been applied in the comprehension of the molecular pathogenesis aimed to find targets to design new therapies. In this search, special attention is paid to identifying single or multiple genes that the lung cancer cells absolutely require for their malignant phenotype and survival. In fact, recent advances in molecular medicine describe a pathogenetic development of lung can-cer through a stepwise process from normal lung epithelial cells towards frank malignancy. This process can be differ-ent in smokers and never smoker’s patients and a different molecular profile proved this difference [10]. Biological drugs specifically target important molecules and pathways involved in lung cancer cells proliferation, inhibition of apoptosis, angiogenesis, and invasion and are currently un-der investigation in clinical trials for lung cancer. These in-clude agents specifically inhibiting components of EGFR and other family members (such as ERBB2/Her2) and/or VEGFR pathways (with monoclonal antibodies and receptor TKIs or with inhibitors of key downstream pathway
Fig. (1). Highlights in the approval of target therapies for human solid tumors by Food and Drug Administration (FDA).
Targeted Therapy in Cancer Current Cancer Drug Targets, 2010, Vol. 10, No. 4 3
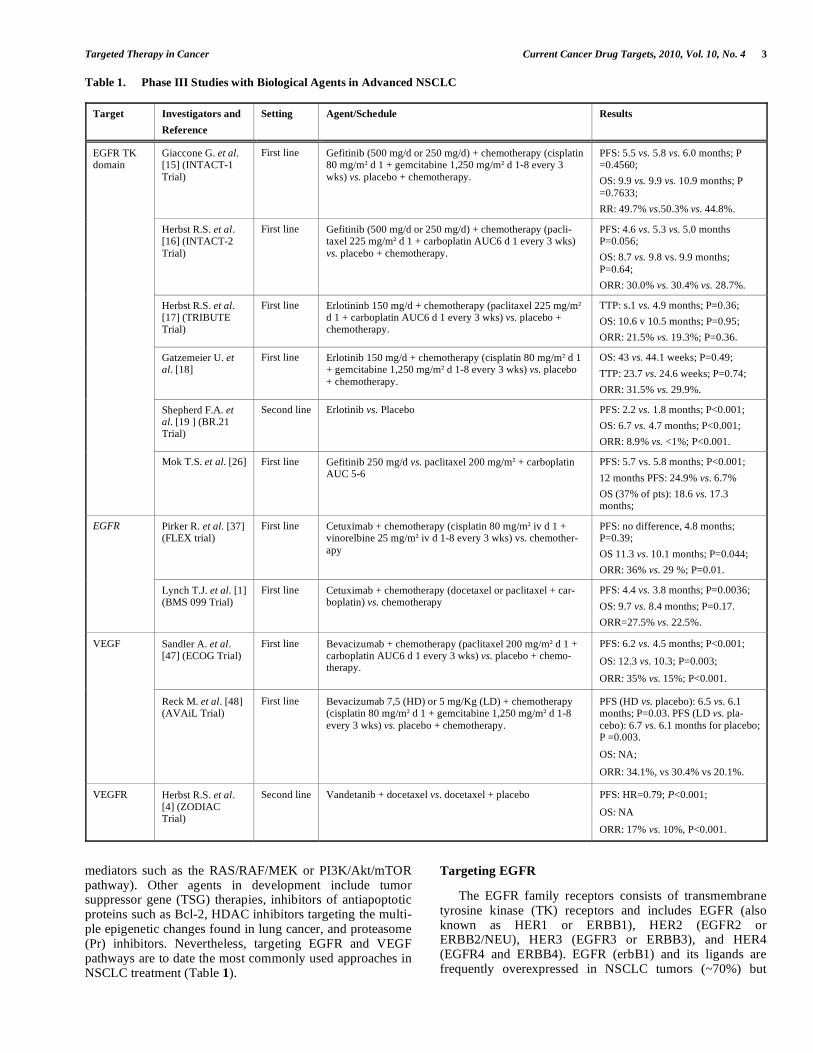
Table 1. Phase III Studies with Biological Agents in Advanced NSCLC
Target Investigators and
Reference
Setting Agent/Schedule Results
Giaccone G. et al. [15] (INTACT-1
Trial)
First line Gefitinib (500 mg/d or 250 mg/d) + chemotherapy (cisplatin 80 mg/m d 1 + gemcitabine 1,250 mg/m d 1-8 every 3
wks) vs. placebo + chemotherapy.
PFS: 5.5 vs. 5.8 vs. 6.0 months; P =0.4560;
OS: 9.9 vs. 9.9 vs. 10.9 months; P
=0.7633;
RR: 49.7% vs.50.3% vs. 44.8%.
Herbst R.S. et al. [16] (INTACT-2
Trial)
First line Gefitinib (500 mg/d or 250 mg/d) + chemotherapy (pacli-taxel 225 mg/m d 1 + carboplatin AUC6 d 1 every 3 wks)
vs. placebo + chemotherapy.
PFS: 4.6 vs. 5.3 vs. 5.0 months P=0.056;
OS: 8.7 vs. 9.8 vs. 9.9 months;
P=0.64;
ORR: 30.0% vs. 30.4% vs. 28.7%.
Herbst R.S. et al. [17] (TRIBUTE Trial)
First line Erlotininb 150 mg/d + chemotherapy (paclitaxel 225 mg/m d 1 + carboplatin AUC6 d 1 every 3 wks) vs. placebo + chemotherapy.
TTP: s.1 vs. 4.9 months; P=0.36;
OS: 10.6 v 10.5 months; P=0.95;
ORR: 21.5% vs. 19.3%; P=0.36.
Gatzemeier U. et al. [18]
First line Erlotinib 150 mg/d + chemotherapy (cisplatin 80 mg/m d 1 + gemcitabine 1,250 mg/m d 1-8 every 3 wks) vs. placebo
+ chemotherapy.
OS: 43 vs. 44.1 weeks; P=0.49;
TTP: 23.7 vs. 24.6 weeks; P=0.74;
ORR: 31.5% vs. 29.9%.
Shepherd F.A. et al. [19 ] (BR.21 Trial)
Second line Erlotinib vs. Placebo PFS: 2.2 vs. 1.8 months; P<0.001;
OS: 6.7 vs. 4.7 months; P<0.001;
ORR: 8.9% vs. <1%; P<0.001.
EGFR TK domain
Mok T.S. et al. [26] First line Gefitinib 250 mg/d vs. paclitaxel 200 mg/m + carboplatin AUC 5-6
PFS: 5.7 vs. 5.8 months; P<0.001;
12 months PFS: 24.9% vs. 6.7%
OS (37% of pts): 18.6 vs. 17.3 months;
Pirker R. et al. [37] (FLEX trial)
First line Cetuximab + chemotherapy (cisplatin 80 mg/m iv d 1 + vinorelbine 25 mg/m iv d 1-8 every 3 wks) vs. chemother-
apy
PFS: no difference, 4.8 months; P=0.39;
OS 11.3 vs. 10.1 months; P=0.044;
ORR: 36% vs. 29 %; P=0.01.
EGFR
Lynch T.J. et al. [1] (BMS 099 Trial)
First line Cetuximab + chemotherapy (docetaxel or paclitaxel + car-boplatin) vs. chemotherapy
PFS: 4.4 vs. 3.8 months; P=0.0036;
OS: 9.7 vs. 8.4 months; P=0.17.
ORR=27.5% vs. 22.5%.
Sandler A. et al. [47] (ECOG Trial)
First line Bevacizumab + chemotherapy (paclitaxel 200 mg/m d 1 + carboplatin AUC6 d 1 every 3 wks) vs. placebo + chemo-therapy.
PFS: 6.2 vs. 4.5 months; P<0.001;
OS: 12.3 vs. 10.3; P=0.003;
ORR: 35% vs. 15%; P<0.001.
VEGF
Reck M. et al. [48] (AVAiL Trial)
First line Bevacizumab 7,5 (HD) or 5 mg/Kg (LD) + chemotherapy (cisplatin 80 mg/m d 1 + gemcitabine 1,250 mg/m d 1-8
every 3 wks) vs. placebo + chemotherapy.
PFS (HD vs. placebo): 6.5 vs. 6.1 months; P=0.03. PFS (LD vs. pla-
cebo): 6.7 vs. 6.1 months for placebo; P =0.003.
OS: NA;
ORR: 34.1%, vs 30.4% vs 20.1%.
VEGFR Herbst R.S. et al. [4] (ZODIAC Trial)
Second line Vandetanib + docetaxel vs. docetaxel + placebo PFS: HR=0.79; P<0.001;
OS: NA
ORR: 17% vs. 10%, P<0.001.
mediators such as the RAS/RAF/MEK or PI3K/Akt/mTOR pathway). Other agents in development include tumor suppressor gene (TSG) therapies, inhibitors of antiapoptotic proteins such as Bcl-2, HDAC inhibitors targeting the multi-ple epigenetic changes found in lung cancer, and proteasome (Pr) inhibitors. Nevertheless, targeting EGFR and VEGF pathways are to date the most commonly used approaches in NSCLC treatment (Table 1).
Targeting EGFR
The EGFR family receptors consists of transmembrane tyrosine kinase (TK) receptors and includes EGFR (also known as HER1 or ERBB1), HER2 (EGFR2 or ERBB2/NEU), HER3 (EGFR3 or ERBB3), and HER4 (EGFR4 and ERBB4). EGFR (erbB1) and its ligands are frequently overexpressed in NSCLC tumors (~70%) but

4 Current Cancer Drug Targets, 2010, Vol. 10, No. 4 Palazzo et al.
rarely expressed in SCLCs. Binding of ligand to the EGFR causes dimerization of the receptor, which in turn activates the intracellular TK domain of the receptor, leading to its autophosphorylation and further activating a cascade of in-tracellular events leading to cell proliferation, inhibition of apoptosis, angiogenesis, and invasion, all resulting in tumor growth and spread.The overexpression of epidermal growth factor receptor (EGFR) was pronounced in virtually all squamous carcinomas and in more than 65% of large cell and adenocarcinomas [11]. Though the immunohistochemi-cal expression of EGFR was not directly related to the prog-nosis, the co-overexpression with other proteins such as HER2-neu, carbonic anhydrase (CA) IX or matrix metallo-proteinase-9 (MMP-9) correlated with shorter time to recur-rence and overall survival. The expression of these proteins as well as the presence of a phosphorilated EGFR could rep-resent the expression of an activated receptor able to turn on the intracellular pathway [12-14]. These data make of EGFR an attractive target in NSCLC. Actually several molecules that inhibit EGFR are available: the small-molecule tyrosine kinase inhibitors (TKi) as gefitinib and erlotinib, which tar-get the intracellular domains, and monoclonal antibodies as cetuximab, targeting the extracellular domain.
The INTACT 1 and 2 phase III trials reported as the addi-tion of gefitinib to conventional first line regimens of che-motherapy (cisplatin-gemcitabine or carboplatin-paclitaxel) did not provide clinical benefit over chemotherapy alone [15, 16]. Similarly erlotinib, another TK inhibitor acting with a very similar mechanism as gefitinib, when combined with chemotherapy, did not confer any survival advantage over chemotherapy alone, in a subset of patients similar to that of the INTACT trials [17, 18].
Divergent results have been obtained in the BR-21 phase III trial, reporting a positive role of erlotinib compared to placebo when administered to patients who progressed after one or two chemotherapy lines. Specifically, Erlotinib in-creased response rate, progression-free survival and overall survival, with a 5% of patients that discontinued the treat-ment because of side effects [19].
These data provides evidence for the FDA to approve the use of erlotinib for the treatment of locally advanced or me-tastatic NSCLC, after failure of at least one prior chemother-apy regimen.
The originally defined “obscure” reason for the unsuc-cessful combination of these molecules with chemotherapy, has recently been clarified through a better molecular knowl-edge of the EGFR transduction pathway. Indeed, molecular predictive markers such as EGFR expression or molecules involved into the transduction of signal pathway down-streaming the EGFR activation, as the PI3K/Akt, Ras/Raf/Erk, and Jak/STAT pathways, have been investi-gated, even though with controversial result in the correla-tion
with response [20, 21].
A retrospective mutational analysis on tumor samples of patients treated with a single agent gefitinib, at Massachu-setts General Hospital of Boston, sequenced the entire cod-ing region of the EGFR gene, using PCR amplification of individual exons. The study described heterozygous muta-tions in the tyrosine kinase domain of receptor, that were
able to determine continued activation of the mutant recep-tors. Moreover, mutant receptors were more sensitive than the wild-type receptor to inhibition by gefitinib [22].
A second analysis performed on tumor samples of Japa-nese and American patients confirmed the presence of so-matic mutations of the EGFR which were most commonly characterized by deletions in exon 19 (E19del) and mutation in exon 21 (L858R), clustering around the active kinasic domain. The BR-21 multivariate analysis of response rate reported more frequently association among these mutations and adenocarcinoma hystotype, femal sex, and Asian origin [23]. By the contrary, additional retrospective mutational analysis, performed by DNA amplification and sequencing of the exons 18 through 21, did not show any significant difference on survival with erlotinib, compared with placebo, among patients with exon 19 deletions or exon 21 (L858R) mutations, and no correlation between the presence of mu-tated or overexpressed EGFR was found [24]. Recently, a large retrospective analysis of 223 patients with known EGFR mutation status treated with erlotinib or gefitinib monotherapy showed that EGFR mutations were associated with a 67% response rate, a time to progression of 11.8 months and an overall survival of 23.9 months. In particular, TTP and OS were longer in patient with exon 19 deletions compared with L858R mutations, confirming that EGFR mutation status is associated with sensitivity to treatment with an EGFR-TKI in patients with advanced NSCLC [25].
Although gefitinib in combination with chemotherapy did not increase TTP or OS, its use as single agent in first line setting was effective, when compared to chemotherapy, in a selected population. A phase III randomized clinical trial was conducted in East Asia to compare gefitinib (250 mg per day) with carboplatin plus paclitaxel in clinically selected patients with NSCLC [26]. Patients were all chemonaïve, with histological features of adenocarcinoma (including bronchoalveolar carcinoma), nonsmokers or former light smokers. The median progression-free survival was 5.7 months in the gefitinib group and 5.8 months in the car-boplatin-paclitaxel group, but the 12-month rates of PFS were 24.9% with gefitinib and 6.7% with carboplatin-paclitaxel. The overall survival in the early analysis was similar between the two groups in the overall population but toxicity was lower in the gefitinib group. In this study, 35.9% of patients were evaluated for EGFR mutations and of all these the 59.7% were positive for a mutation of the exon 19 through 21. In this mutation-positive group, progression-free survival and objective response rate was significantly longer among patients receiving gefitinib than among those receiving carboplatin–paclitaxel. This study also showed as ethnic origin (Asian), smoking status, and histologic findings help to identify patients with a high likelihood of having an EGFR mutation (59.7%), suggesting that, whenever possible, mutational status should be determined before starting treat-ment of pulmonary adenocarcinoma. Recently the feasibility of large-scale screening for EGFR mutations in patients with the same disease characteristics was also recommended in non-Asian women with lung cancer [27].
Another important question is represented by the investi-gation about the role of K-RAS status in the response to TKi in NSCLC. The importance of Ras signaling in cell growth

Targeted Therapy in Cancer Current Cancer Drug Targets, 2010, Vol. 10, No. 4 5
and survival is supported by the importance of Ras in onco-genesis. K-RAS was identified as a transforming protein in human tumors that was analogous to the transforming pro-tein of the Kirsten murine sarcoma virus. Several K-RAS point mutations resulting in constitutive activation are found at high frequency in a variety of human tumors. Recent stud-ies indicate that patients with mutant K-RAS tumors fail to benefit from adjuvant chemotherapy and do not respond to EGFR inhibitors. K-RAS mutations were found in about 10% of patients affected by NSCLC, in 15% of patients en-rolled in the BR-21 trial and 21% of patients enrolled in the TRIBUTE trial, in which the mutational status was associ-ated with significantly decreased TTP and survival in er-lotinib plus chemotherapy–treated patients [28-30].
Together, EGFR and K-RAS appear two main targets in NSCLC treatment. Considering that mutations of both genes in individual tumors are almost mutually exclusive, it is pos-sible to distinguish two different subpopulations affected by EGFR or K-RAS mutations. Clinically, EGFR mutations affect female sex, never-smoker status, and East Asian eth-nicity, while K-RAS mutations affect male sex, ever-smoker status, and non–East Asian ethnicity. Molecular EGFR muta-tions selectively activate the Akt survival associated pathway in contrast to the K-RAS mediated cell proliferation pathway [28, 31-33].
Another EGFR targeting approach is the monoclonal an-tibody cetuximab. Many phase II studies on cetuximab com-bined with various chemotherapy schedules tested the toler-ability and the clinical activity of this molecule in patients affected by NSCLC, but only one considered the preliminary evaluation of the immunohistochemistry expression of EGFR [34-36].
In the phase III FLEX trial, 1125 patients with stage IIIB or IV, EGFR positive NSCLC were randomly assigned to receive standard platinum-based chemotherapy alone or chemotherapy plus cetuximab as first line treatment. The median overall survival was 11.3 months in the chemother-apy plus-cetuximab group and 10.1 months in the chemo-therapy alone group. The subgroup analyses reported sur-vival benefit in all histological type of non-small-cell lung cancer [37]. By the contrary, the results of a similar phase III trial, the BMS-099, did not reach statistical significance in overall survival analysis
1.
Based on these data, the Committee for Medicinal Prod-ucts for Human Use (CHMP) of the European Medicine Agency (EMEA) judged the benefits of adding cetuximab to standard platinum-based chemotherapy as modest in terms of survival times. Since it failed to have convincing effect on survival prolongation, the approval of cetuximab in patients affected by NSCLC was thus refused.
Targeting Angiogenesis
The growth and development of solid tumors is critically dependent on a functional vascular supply in the absence of which tumors remain unable to metastasize. Initiation of new
1Lynch, T. J.; Patel, T.; Dreisbach, L.; McCleod, M.; Heim, W.; Hermann, R. C.;
Paschold, E.; Pautret, V.; Weber, M. R.; Hart, L. L. Overall survival results from the phase III trial BMS 099: cetuximab+taxane/carboplatin as 1st-line treatment for ad-
vanced NSCLC. J. Thorac. Oncol. 2008, 3, abstract S305.
blood vessel formation, more currently called neoangiogene-sis, is believed to be reliant on an angiogenic "switch," which leads to a complex series of events, starting with the release of tumor-related proangiogenic factors, endothelial cell activation, and the release of proteolytic enzymes, fol-lowed by endothelial cell migration, proliferation, and capil-lary tube formation. Angiogenic factors affect vasculature formation and vascular permeability, modulate host response and influence tumor invasion, metastasis, and prognosis. Among the most important angiogenic cytokines are the VEGFs. Tumor angiogenesis, quantified in terms of mi-crovessel counts, was related with a number of parameters as tumor stage, regional lymph node involvement, disease-free interval and reduced overall survival in NSCLC, in both pro-spective and retrospective case series [38-42].
Although the prognostic and predictive role of circulating levels of VEGF, platelet-derived endothelial cell growth fac-tor (PD-ECGF) and basic fibroblast growth factor (bFGF) remains contradictory [43-45], actually the VEGF is the most promising target, either by preventing VEGF-receptor binding or by inhibiting downstream receptor signaling. However, many other approaches against tumor vasculature are also in development.
Bevacizumab is a humanized monoclonal antibody that acts by binding and neutralizing all VEGF-A isoforms. The efficacy and safety of bevacizumab in combination with car-boplatin and paclitaxel, as first line therapy, has been as-sessed in a phase II study in patients with advanced or recur-rent NSCLC [46]. The Eastern Cooperative Oncology Group (ECOG) conducted a randomized phase III trial comparing carboplatin and paclitaxel with or without bevacizumab in a similar setting of disease (stage IIIB or IV). The results re-ported a significant improvement in response rate, overall survival and progression-free survival for patients treated with bevacizumab plus chemotherapy compared with che-motherapy alone [47].
A very similar second phase III randomized trial, con-ducted in Europe and Canada (AVAiL trial), compared cis-platin and gemcitabine with or without Bevacizumab. The study confirms the data of the ECOG trial, reporting an in-crease of PFS, both for low and high dose of bevacizumab, compared to chemotherapy alone. The objective response rate were 20.1%, 34.1%, and 30.4% for the placebo, low-dose bevacizumab and high-dose bevacizumab arms respec-tively, while OS data were immature because of limited fol-low-up [48].
ECOG trial performed an exploratory analysis on the baseline levels and on the serial changes in ICAM, E-selectin, and bFGF. Authors found that ICAM expression significantly correlated with clinical response and baseline ICAM seemed to be a significant prognostic factor for sur-vival .
The primary end point of the ECOG correlative study, which was to determine whether baseline VEGF levels were predictive of response to chemotherapy ± bevacizumab, was met. Patients with high baseline VEGF levels were more likely to have an increased probability of response with the chemotherapy plus bevacizumab versus chemotherapy alone, than those with low VEGF levels. Patients with a low VEGF

6 Current Cancer Drug Targets, 2010, Vol. 10, No. 4 Palazzo et al.
level, on the contrary, had a better PFS compared with pa-tients with a high level.
Although the basal level of ICAM appears to be a strong independent prognostic and predictive factor, the research on predictive markers of response and of benefit from antiangi-ogenic agents is still ongoing [49].
To target angiogenesis in NSLC patients, small molecule receptor tyrosine kinase inhibitors (TKi) as sorafenib, sunit-inib, cediranib, and vandetanib are available.
In phase I-II studies, sorafenib shown preliminary activ-ity in combination with chemotherapy and with epidermal growth factor receptor inhibitors (erlotinib or gefitinib), al-though there were no confirmed partial response as single agent [50, 51].
At the same manner, sunitinib was tested as single agent in patients with stage IIIB or IV NSCLC, which had pro-gressed during or after treatment with at least one platinum-based combination chemotherapy regimen. 11.1% of patients had confirmed partial responses and 28.6% had a best re-sponse of stable disease for 8 weeks or longer. Discontinua-tion of the treatment because of an adverse event was re-ported in 29% of patients. Median PFS was 12.0 weeks, and median OS was 23.4 weeks, with a 1-year survival rate of 20.2% [52]. Two phase I studies evaluated sunitinib (37.5 mg or 50 mg) in combination with cisplatin and gemcitabine or docetaxel in untreated or previous treated patients respec-tively with advanced NSCLC. The combination of oral SU 37.5 mg/day with chemotherapy was judged as safe and manageable for further study in combination with other treat-ments for NSCLC
2, 3.
Cediranib, in preclinical models of human lung tumor xenografts, induced rapid onset of vessel regression, within 52 hours, that became progressively greater with the duration of treatment. In particular, the inhibition of tumor growth was probably due to a direct effect on tumor endothelium, which was likely to be derived from potent inhibition of VEGF signalling [53]. Two phase I trial of National Cancer Institute of Canada clinical trials group evaluated escalating doses of cediranib in combination with standard chemother-apy (carboplatin and paclitaxel or cisplatin and gemcitabine) in patients with advanced NSCLC. No dose-limiting tox-icities were observed during cycle 1 at each dose and fatigue, nausea, diarrhoea, anorexia and granulocytopenia were the most common adverse events. The studies encourage the assessment of antitumor activity of cediranib combined with standard doses of chemotherapy in phase II-III trials [54, 55].
The activity of vandetanib, as single-agent in patients with NSCLC progressive on first- or second line platinum-based therapy, was tested in a randomized phase II trial and compared with gefitinib. Vandetanib showed a significant prolongation of PFS compared with those randomly assigned
2Robert, F., Sandler, A.; Schiller, J. H.; Ilagan, J.; VerMeulen, W.; Harper, K.; Liu, G.;
Tye, L.; Chao, R., Traynor, A. A phase I dose-escalation and pharmacokinetic study of sunitinib plus docetaxel in patients with advanced solid tumors. J. Clin. Oncol. 2008,
26, abstact 3564. 3Reck, M.; Frickhofen, N.; Gatzemeier, U.; Fuhr, H.; Lanzalone, S.; Lechuga, M. J.;
Wang, E.; Chao, R.; Felip, E. A phase 1 dose escalation study of sunitinib in combina-tion with gemcitabine 1 cisplatin for advanced non-small cell lung cancer (NSCLC). J.
Clin. Oncol. 2007, 25, abstact 18057.
to gefitinib (11.0 and 8.1 weeks respectively), moreover the trend toward advantage in PFS was independent to the sex of patients and histology of the tumor [56].
An increase in median PFS for the combination therapy of vandetanib with chemotherapy (docetaxel) compared with chemotherapy alone was showed in a phase II study enroll-ing patients with locally advanced or metastatic (stage IIIB/IV) NSCLC after failure of first-line platinum-based chemotherapy [57]. This advantage in PFS was confirmed by another phase II study investigating a combination therapy with carboplatin and paclitaxel. This trial randomized pa-tients affected by all NSCLC histologies, including patients with previously treated CNS metastases [58]. Unexpectedly, an antitumor activity was observed when vandetanib was administered, in combination regimen, at 100 mg dose rather than at 300 mg , although the reasons of these findings are unclear.
The results of a randomized phase III trial (ZODIAC) comparing vandetanib plus docetaxel versus docetaxel plus placebo as second-line treatment for patients with advanced NSCLC have been recently presented (ASCO 2009). These data revealed, after a median duration of follow-up of 12.8 months, a statistically significant improvement in PFS and overall response rate for vandetanib plus docetaxel. A posi-tive trend for combined therapy was showed in overall sur-vival although the data was not statistically significant. De-spite the increase of adverse event reported in combined therapy arm, the study concludes that vandetanib is the first oral targeted drug in phase III trials with significant evidence of clinical benefit when added to standard chemotherapy in NSCLC
4.
Future Considerations
Since a multitude of studies have reported that a poli-chemotherapy regimen (such as triplet combination) did not confer an increase in survival, great hopes have been di-rected to the target therapies [59]. In first line setting, bevacizumab added to conventional chemotherapy achieved a 2.0 months increase in OS, but this advantage was limited to patients affected by a non squamous histology or a not-centrally located disease [47]. The same improvement in survival has been observed for erlotinib as single agent, after platinum based chemotherapy progression, in a population unselected for EGFR mutational status. To date, the role of the erlotinib after chemotherapy regimens containing bevaci-zumab and the benefit deriving from its use in EGFR mu-tated patients in first line setting is unclear.
Erlotinib efficacy is under evaluation by fourteen phase III open studies registered at clinicaltrial.gov, comparing cisplatin-gemcitabine or pemetrexed ± docetaxel in the first and second line respectively. Similarly, gefitinib is compared to platinum-based chemotherapy in EGFR FISH positive advanced NSCLC patients in ONC-2008-001 trial. The Most relevant studies in this context were two trials which evalu-ated the role of Erlotinib, as maintenance after adjuvant
4Herbst, R. S.; Sun, Y.; Korfee, S.; Germonpré, P.; Saijo, N.; Zhou, C., Wang, J.; Langmuir, P.; Kennedy, S. J.; Johnson, B. E. Vandetanib plus docetaxel versus do-
cetaxel as second-line treatment for patients with advanced non-small cell lung cancer (NSCLC): A randomized, double-blind phase III trial (ZODIAC). J. Clin. Oncol. 2009,
27, abstract CRA8003.

Targeted Therapy in Cancer Current Cancer Drug Targets, 2010, Vol. 10, No. 4 7
standard chemotherapy, in patients with stage II or IIIA NSCLC respectively.
Among anti-angiogenetic strategies, there are two ongo-ing trials (AVAPERL1 and NCT00762034) evaluating the role of a maintenance sequential therapy with bevacizumab, with or without pemetrexed, after a response to a first line chemotherapy containing bevacizumab respectively with or without pemetrexed, in patients with advanced NSCLC. In the adjuvant setting, the ECOG-E1505 phase III randomized trial is going to evaluate the benefit from bevacizumab com-bined to chemotherapy in patients with completely resected stage IB-IIIA NSCLC. Another phase III trial (SWOG-S0819) will evaluate the efficacy of a combined target ther-apy with cetuximab plus bevacizumab added to a standard chemotherapy (carboplatin-paclitaxel) as first line treatment of EGFR FISH-positive patients with NSCLC.
Future approaches to NSCLC include the discovery of new targets to increase the survival, to overcome the resis-tance to target therapies or to target subgroups of patients who did not benefit from standard therapy. To this purpose, the efficacy of figitumumab a fully human, IgG2 monoclonal antibody against the insulin-like growth factor type I recep-tor (IGF-IR), has been proven in advanced treatment-naïve NSCLC with a squamous cell histology.
The presence of multiple targets may thus allow to de-velop new drugs able to selectively interfere with different steps in tumorigenesis, with the hope to achieve a significant improvement in lung cancer therapy.
BREAST CANCER (BC)
Breast cancer is considered as a molecularly heterogene-ous disease and the large number of genes potentially in-volved in controlling cell growth, death, and differentiation emphasize the importance of studying multiple genetic al-terations in concert. To date, the natural history and the re-sponsiveness to treatments of breast tumor is reflected by a number of classical prognostic variables such as nodal status, tumor size, histological grade, age and hormone receptor status, protooncogenes like ERBB2 and mutations in the p53 gene [60].
Women with hormone receptor-positive disease generally have a better prognosis than hormone-negative, mainly de-pending on their response to estrogen deprivation. Similarly, women with human epidermal growth factor receptor 2 (HER2)-amplified disease have a more aggressive phenotype but are candidates for trastuzumab, a monoclonal antibody against HER2, which dramatically changed the natural his-tory of this type of breast cancer. Among breast cancer types there are also tumors so-called ‘triple negative’, defined by the estrogen / progesterone receptors (ER and PgR), and HER2 absence, usually associated to a poor prognosis and lack of ‘specific’ treatment options.
Studies of gene profiling have been proposed in order to explain the variations of breast tumors in growth rate, in the activity of specific signaling pathways, and in the cellular composition, with the aim to better predict the outcome of disease. The gene expression analysis identified four sub-groups (ER+/luminal-like A and B, epidermal growth factor
receptor-2 positive and basal-like) and all the molecular al-terations driving these subtype of disease are still to be de-fined. Major known pathways include the epidermal growth factor signaling cascade, with many important down stream-ing key proteins identified as potential targets such as PI3K, PTEN, mTOR, Src , the DNA-repair pathway and the vascu-lar endothelial growth factor (VEGF). These data are now being used to drive therapeutic development in breast cancer by guiding the use of existing therapies and identifying new targets.
Targeting EGFR Signaling Pathway
The ERBB2 amplification was first described twenty
years ago as consistent alteration found in breast cancer [4,
61, 62]. HER2 is overexpressed and/or amplified in 25% of
breast tumours and confers a more aggressive clinical course and a worse survival [63, 64].
Women with BC overexpressing HER2 are at greater risk
for disease progression and death than women whose tumors do not. [4]
The oncogenic potential of HER-2 activation results in an
increased cell proliferation, cell motility, tumor invasiveness,
progressive regional and distant metastases, accelerated an-giogenesis, and reduced apoptosis [65].
To trigger signaling cascades it is necessary, beyond a
ligand receptor binding, a hetero-dimerization between re-
ceptors. In contrast to the other members of the family,
HER2 is considered an ‘orphan’ receptor, since no natural
ligand is known to date [66]. However, it is well described
that HER2/neu may be activated by ligand-dependent and
ligand-independent manner, maintaining an active role in
ligand-mediated signaling through hetero-dimerization with other erbB family members [67, 68].
Furthermore, this receptor may be activated through a
ligand-independent pathway in presence of HER2 overex-
pression, with a spontaneous dimerization/oligodimerization
induced when a critical threshold level of HER2 expression
is reached [69]. Evidences suggest that overexpressed ErbB2
is constitutively phosphorylated in breast cancer cell lines as
well as in human tumors and that the extracellular domain of
HER2 can adopt a fixed conformation resulting in a ligand-
activated state, thus allowing to dimerize in the absence of a ligand [70].
Upon dimerization, intracellular tyrosine kinase (TK)
domains are phosphorylated, which in turn provide docking
sites for several adaptor proteins and signaling enzymes in-volved in a wide variety of cellular processes.
It has been observed that targeting overexpressed active
ErbB2 results in efficient inhibition of breast cancer cell pro-
liferation, which proceeds via inhibition of these intracellular
signaling pathways and directly targets various members of the cell cycle machinery [71-74].
All these biological consequences encouraged the devel-
opment of anti-HER2 therapies such as monoclonal antibod-
ies (mAbs) and small molecules inhibitors of tyrosine kinase (TKi) activity.
8 Current Cancer Drug Targets, 2010, Vol. 10, No. 4 Palazzo et al.
Trastuzumab is a recombinant humanized monoclonal antibody. The treatment of BC cells with ErbB2-specific antagonistic antibodies or with kinase inhibitors blocks tu-mor cells in the G1 phase of the cell cycle [69]. In vitro stud-ies show that trastuzumab is synergistic with a variety of chemotherapies [75] and that the synergy with DNA-damaging agents is due to trastuzumab-mediated inhibition of DNA repair by promoting increase in DNA strand breaks [76].
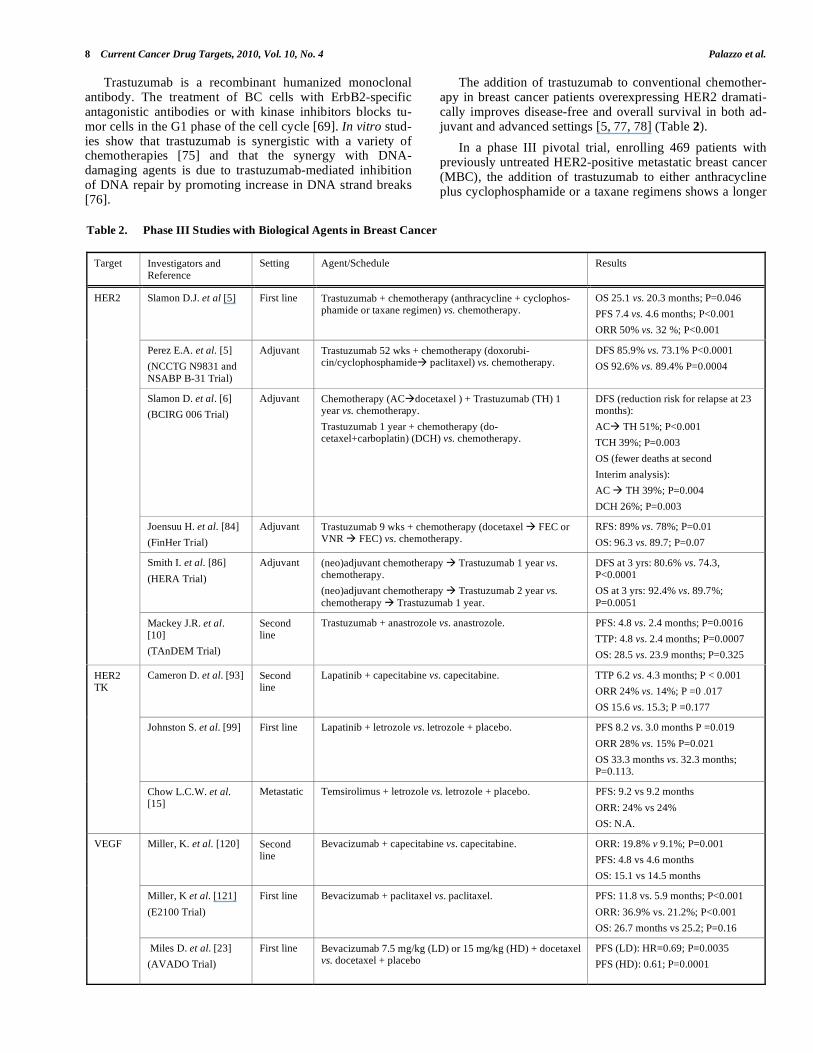
The addition of trastuzumab to conventional chemother-apy in breast cancer patients overexpressing HER2 dramati-cally improves disease-free and overall survival in both ad-juvant and advanced settings [5, 77, 78] (Table 2).
In a phase III pivotal trial, enrolling 469 patients with previously untreated HER2-positive metastatic breast cancer (MBC), the addition of trastuzumab to either anthracycline plus cyclophosphamide or a taxane regimens shows a longer
Table 2. Phase III Studies with Biological Agents in Breast Cancer
Target Investigators and Reference
Setting Agent/Schedule Results
Slamon D.J. et al [5] First line Trastuzumab + chemotherapy (anthracycline + cyclophos-phamide or taxane regimen) vs. chemotherapy.
OS 25.1 vs. 20.3 months; P=0.046
PFS 7.4 vs. 4.6 months; P<0.001
ORR 50% vs. 32 %; P<0.001
Perez E.A. et al. [5]
(NCCTG N9831 and
NSABP B-31 Trial)
Adjuvant Trastuzumab 52 wks + chemotherapy (doxorubi-cin/cyclophosphamide paclitaxel) vs. chemotherapy.
DFS 85.9% vs. 73.1% P<0.0001
OS 92.6% vs. 89.4% P=0.0004
Slamon D. et al. [6]
(BCIRG 006 Trial)
Adjuvant Chemotherapy (AC docetaxel ) + Trastuzumab (TH) 1 year vs. chemotherapy.
Trastuzumab 1 year + chemotherapy (do-cetaxel+carboplatin) (DCH) vs. chemotherapy.
DFS (reduction risk for relapse at 23 months):
AC TH 51%; P<0.001
TCH 39%; P=0.003
OS (fewer deaths at second
Interim analysis):
AC TH 39%; P=0.004
DCH 26%; P=0.003
Joensuu H. et al. [84]
(FinHer Trial)
Adjuvant Trastuzumab 9 wks + chemotherapy (docetaxel FEC or VNR FEC) vs. chemotherapy.
RFS: 89% vs. 78%; P=0.01
OS: 96.3 vs. 89.7; P=0.07
Smith I. et al. [86]
(HERA Trial)
Adjuvant (neo)adjuvant chemotherapy Trastuzumab 1 year vs. chemotherapy.
(neo)adjuvant chemotherapy Trastuzumab 2 year vs.
chemotherapy Trastuzumab 1 year.
DFS at 3 yrs: 80.6% vs. 74.3, P<0.0001
OS at 3 yrs: 92.4% vs. 89.7%;
P=0.0051
HER2
Mackey J.R. et al. [10]
(TAnDEM Trial)
Second line
Trastuzumab + anastrozole vs. anastrozole. PFS: 4.8 vs. 2.4 months; P=0.0016
TTP: 4.8 vs. 2.4 months; P=0.0007
OS: 28.5 vs. 23.9 months; P=0.325
Cameron D. et al. [93] Second line
Lapatinib + capecitabine vs. capecitabine. TTP 6.2 vs. 4.3 months; P < 0.001
ORR 24% vs. 14%; P =0 .017
OS 15.6 vs. 15.3; P =0.177
Johnston S. et al. [99] First line Lapatinib + letrozole vs. letrozole + placebo. PFS 8.2 vs. 3.0 months P =0.019
ORR 28% vs. 15% P=0.021
OS 33.3 months vs. 32.3 months; P=0.113.
HER2 TK
Chow L.C.W. et al. [15]
Metastatic Temsirolimus + letrozole vs. letrozole + placebo. PFS: 9.2 vs 9.2 months
ORR: 24% vs 24%
OS: N.A.
Miller, K. et al. [120] Second line
Bevacizumab + capecitabine vs. capecitabine. ORR: 19.8% v 9.1%; P=0.001
PFS: 4.8 vs 4.6 months
OS: 15.1 vs 14.5 months
Miller, K et al. [121]
(E2100 Trial)
First line Bevacizumab + paclitaxel vs. paclitaxel. PFS: 11.8 vs. 5.9 months; P<0.001
ORR: 36.9% vs. 21.2%; P<0.001
OS: 26.7 months vs 25.2; P=0.16
VEGF
Miles D. et al. [23]
(AVADO Trial)
First line Bevacizumab 7.5 mg/kg (LD) or 15 mg/kg (HD) + docetaxel vs. docetaxel + placebo
PFS (LD): HR=0.69; P=0.0035
PFS (HD): 0.61; P=0.0001

Targeted Therapy in Cancer Current Cancer Drug Targets, 2010, Vol. 10, No. 4 9
time to disease progression, an higher rate of objective re-sponse , a longer duration of response, and a longer survival duration [5].
In 1998, the United States Food and Drug Administration (FDA) approved the humanized monoclonal antibody trastuzumab, a humanized mAb of the immunoglobulin G1 type directed against the extracellular portion of HER2. It was the first HER2-targeted agent approved for clinical use in breast cancer patients overexpressing HER2.
Several mechanisms of action of trastuzumab have been proposed to date. In vivo breast cancer models and clinical trials have demonstrated that trastuzumab has not only cy-tostatic but also cytotoxic properties. At least in part, these properties may be due to the activation of antibody-dependent cellular cytotoxicity (ADCC), through the activa-tion of natural killer cells (NK), expressing the Fc gamma receptor, which can be bound by the Fc domain of trastuzu-mab. This event activates the lysis of cancer cells bound to trastuzumab. When overexpressed, HER2 undergoes prote-olytic cleavage which results in the release of the extracellu-lar domain and in the production of a truncated membrane-bound fragment (p95) .
In HER2-overexpressing breast cancer cell lines it has been demonstrated that trastuzumab can block the shedding of the extracellular domain of HER2 by inhibiting metallo-proteinase activity. A further proposed mechanism of action of trastuzumab is the reduction of the signalling from PI3K pathways, thus promoting apoptosis and the arrest of prolif-eration.
The randomized phase II study M77001 that was de-signed to compare the efficacy of trastuzumab plus docetaxel versus docetaxel alone, confirmed the superiority in terms of overall response rate time to disease progression , time to treatment failure and a significant survival benefit for trastu-zumab in combination with docetaxel, providing for an addi-tional valuable first-line treatment option in routine clinical practice for HER2-positive MBC [79].
The optimal combination of trastuzumab plus chemo-therapy for first line MBC as well as the optional timing and sequence of combination therapy are still under investiga-tion. Preclinical studies had suggested additive and synergis-tic effects from these combinations [80, 81].
Various non randomized phase II trials have shown the efficacy and relative safety of trastuzumab in combination with many other chemotherapeutic agents routinely used as
vinorelbine, paclitaxel, docetaxel, gemcitabine, capecitabine, cisplatin and carboplatin. In general, vinorelbine and taxane-containing regimens seem to be the most active with RR ranging from 45% to 86% with a median duration of TTP of 7–17 months [82] .
Moreover it is not clear whether the use of more than one cytostatic agent may be associated with increased efficacy.
Since the FDA approval in 1998 for use of trastuzumab in HER-2 positive MBC, many large trials combining trastu-zumab with chemotherapy in the adjuvant setting have been conducted and all together demonstrated that trastuzumab significantly improves disease-free survival and overall sur-vival in early stage breast cancer [83]. To date, trastuzumab
represents the only target therapy used and for cancer treat-ment in adjuvant setting.
Updated results, after a median follow-up of 3 years, from a joint analysis of two adjuvant studies (NCCTG N9831 and NSABP B-31) were presented at the ASCO meeting 2007. Both similar studies, performed in node posi-tive or in high risk node negative patients, compared four cycles of doxorubicin-cyclophosphamide followed by pacli-taxel with the same chemotherapy plus 52 weeks of trastu-zumab (H), beginning on day 1 of paclitaxel therapy. The NSABPB-31 study added a third group, in which trastuzu-mab was given after the completion of paclitaxel to evaluate the use of sequential trastuzumab
5.
The other major phase III trastuzumab based adjuvant trials (Breast Cancer International Research Group [BCIRG] 006 trials and The Finland Herceptin [FIN Her] trial) used several combinations of cytotoxic agents with monoclonal antibody and confirmed the improvement in outcome with the addition of trastuzumab to chemotherapy
6 [84].
Although the addition of trastuzumab (H) to anthracy-cline-based adjuvant regimens has been proven effective, it has also been associated with increased cardiac toxicity.
Evidences from clinical trials limit the use of trastuzu-mab in association with chemotherapy. HER2-positive tu-mours less than 1 cm in size without axillary nodal involve-ment and without other features indicating increased metas-tatic potential (e.g. vascular invasion) might not need adju-vant chemotherapy and the limited evidence of increased risk among these patients does not allow definitive recommenda-tion regarding anti-HER2 therapy. Nevertheless, the major part of the expert panel at last St Gallen International Expert Consensus Conference, despite the absence of definitive clinical trial evidence, was ready to use trastuzumab in asso-ciation with endocrine therapy in adjuvant setting [85]
The optimal duration of trastuzumab therapy in the adju-vant or neoadjuvant setting is still to be defined. In adjuvant trials, trastuzumab was studied for a short duration of 9 weeks, for 52 weeks or for 2 years. To date only the results from the comparison of one year trastuzumab versus no tras-tuzumab have been published. [78, 86].
Despite several phase II trials evaluating the use of tras-tuzumab in the preoperative setting, results from only two randomized trials were reported. The primary endpoint in these studies was pathological complete response (pCR) rate and event free survival (EFS).
Buzdar et al. reported on 42 of a planned 165 patients a significant 39% gain in the pCR by adding trastuzumab to neoadjuvant chemotherapy. Patient’s accrual was early
5Perez, E. A.; Romond, E. H.; Suman, V. J.; Jeong, J.; Davidson, N. E.; Geyer, C. E.;
Martino, S.; Mamounas, E. P.; Kauffman, P. A.; Wolmark N. Updated results of the combined analysis of NCCTG N9831 and NSABP B-31 adjuvant chemotherapy
with/without trastuzumab in patients with HER2-positive breast cancer. J. Clin. Oncol.
2007, 25, 512. 6Slamon, D.; Eiermann, W.; Robert, N.; Pienkowski, T.; Martin, M.; Pawlicki, M.;
Chan, A.; Smylie, M.; Liu, M.; Falkson, C.; Pinter, T.; Fornander, T.; Shiftan, T.; Valero, V.; Mackey, J.; Tabah-Fisch, I.; Buyse, M.; Lindsay, M.; Riva, A.; Bee, V.;
Pegram, M.; Press, M.; Crown, J. BCIRG 006: 2nd interim analysis phase III random-ized trial comparing doxorubicin and cyclophosphamide followed by docetaxel with
doxorubicin and cyclophosphamide followed by docetaxel and trastuzumab with do-cetaxel, carboplatin and trastuzumab in Her2neu positive early breast cancer patients.
San Antonio Breast Cancer Symposium 2006, abstract 52.

10 Current Cancer Drug Targets, 2010, Vol. 10, No. 4 Palazzo et al.
stopped because of this evident statistical advantage for tras-tuzumab [87].
The analysis of results of the larger NeOAdjuvant Her-ceptin (NOAH) trial presented at San Antonio Breast Cancer Symposium 2008 establishes neoadjuvant trastuzumab (H) with chemotherapy (CT) as a standard treatment option in women with HER2-positive locally advanced breast cancer. Event free survival rate at 3 years was significantly better in the H + CT arm compared with CT alone: 70.1% versus 53.3%, respectively. Both ORR and pCR were significantly higher in the H + CT arm compared to CT alone
7.
More recently, the GeparQuattro study reported a similar doubling in the observed pCR rate using always a schedule of trastuzumab concurrently with anthracycline regimen.
One of the major drawbacks in the use of trastuzumab is the development of drug resistance, although it is not clear whether tumor cells are resistant ab initio or become resis-tant during treatment [88]. In vitro studies have demon-strated that, in sensitive cells, trastuzumab causes a disrup-tion of the binding of Src to HER2, allowing PTEN to inhibit AKT and induce growth arrest. When PTEN levels are low, however, AKT remains active and trastuzumab efficacy is impaired. Other mechanisms have been proposed to explain resistance to trastuzumab. In SKBR3 cell line, resistant to trastuzumab, the presence of IGF1R/HER2 heterodimers has been observed and correlated to the inability of trastuzumab to block cell proliferation. These results point to IGF-IR as a possible mediator of trastuzumab resistance and a hypotheti-cal therapeutic target in patients with trastuzumab-resistant disease. Further studies suggested that lack of accessibility of the epitope to trastuzumab may limit the activity of this antibody.
Among all the proposed mechanisms, loss of PTEN was found in about 15% to 35% of patients with breast cancer [89]. Furthermore PTEN loss has been associated with poor prognosis in patients with ER-positive breast cancer treated with tamoxifen [90]
Since HER2 and EGFR coexpression occurs in 30% of breast cancers, blockage of both receptors is a rational strat-egy which may improve response rates to trastuzumab
Lapatinib is the first orally administered dual inhibitor of epidermal growth factor receptor (EGFR) and human epi-dermal growth factor receptor 2 (HER2) tyrosine kinases. In addition to inhibiting wild-type HER receptors, lapatinib is able to inhibit truncated forms of these receptors [91].
According to recent studies, the benefit from lapatinib in women with HER2+
MBC appears to be limited to patients
with positive FISH or IHC3+ intensity, and EGFR determi-
nation did not contribute to improved patient selection
8.
The pivotal registrative phase 3 study comparing la-patinib plus capecitabine with capecitabine alone in women
7Gianni, L.; Eiermann, W.; Semiglazov, V.; Manikhas, G. M.; Lluch, A.; Tjulandin, S.;
Feyereislova, A.; Valagussa, P.; Baselga, J. Neoadjuvant trastuzumab in patients with HER2-positive locally advanced breast cancer: Primary efficacy analysis of the NOAH
trial. San Antonio Breast Cancer Symposium 2008, abstract 31. 8Press, M. F.; Finn, R. S.; Di Leo, A.; Cameron, D. A.; Geyer, C. E.; Martin, A.; New-
stat, B.; Gagnon, R.; Arbushites, M.; Koehler, M. Correlation of HER2 gene amplifica-tion, HER2 and EGFR expression (protein and mRNA) with lapatinib efficacy in
women with metastatic breast cancer. J. Clin. Oncol. 2008, 26, abstract 1007.
with HER2-positive, locally advanced or MBC, progressive after anthracycline-taxane and trastuzumab therapy demon-strated, in 324 patients, that lapatinib plus capecitabine was superior to capecitabine alone in time-to disease progression (TTP) [92].
In 2007 the FDA approved lapatinib in combination with capecitabine in the treatment of HER-2 positive MBC pro-gressed under standard regimens.
In an update of the registrative study, recently published on a total of 399 women, the significant reduction in the rela-tive risk of progression when lapatinib is added to capecit-abine corresponded to an improvement in median TTP from 4.3 to 6.2 months. [93].
Since lapatinib is able to cross the blood-brain barrier in patient with CNS metastases [94] an exploratory analysis, performed in the pivotal trial, showed a symptomatic CNS progression in four (2%) patients in the combination-therapy group compared with 13 (6%) patients in the monotherapy group.
Based on the activity in metastatic disease (Table 2), la-patinib in the adjuvant and neoadjuvant settings is currently being explored. The Tykerb Evaluation After Chemotherapy (TEACH) trial is a phase III randomized trial comparing adjuvant lapatinib with placebo in women affected by early-stage HER-2-positive BC who have completed adjuvant chemotherapy and who have not received trastuzumab. This trial planned to enroll 3,000 patients and was recently closed [95].
The Adjuvant Lapatinib and/or Trastuzumab Treatment Optimization (ALTTO) trial has been designed with four arms in order to compare lapatinib for one year alone, versus one year of trastuzumab, versus trastuzumab followed by lapatinib versus lapatinib concomitantly with trastuzumab and it is actually recruiting. The Neoadjuvant Lapatinib and/or Trastuzumab Treatment Optimisation (Neo-ALTTO) trial is a very similar randomized, open-label, multicenter, phase III study, with three arms of comparison for the effi-cacy of lapatinib/trastuzumab plus paclitaxel in HER-2 posi-tive primary breast cancer without arms of sequential treat-ment with lapatinib followed by trastuzumab.
The integration of trastuzumab or lapatinib into current neoadjuvant chemotherapy regimens is currently under in-vestigation in GeparQuinto trial.
Targeting EGFR Pathway in Combination with Endo-
crine Therapies
An inverse association has been described between HER2 amplification/overexpression and steroid hormones estrogen and progesterone receptors in both experimental and clinical studies. However data are not entirely clear, as in some studies up to 50% of HER2+ tumors are also ER+.
There is some experimental evidence to support a role for growth factor receptor pathways, such as HER1 and HER2 together in the resistance to antiestrogen therapy. Transfec-tion of c-erbB2 into the human breast cancer cell line MCF-7 results in cells that are estrogen independent and tamoxifen resistant both in vitro and as xenografts in nude mice [96]. ErbB receptors enhance ER signaling either by directly acti-

Targeted Therapy in Cancer Current Cancer Drug Targets, 2010, Vol. 10, No. 4 11
vating ER or through activation of MAPK and Akt. So far, preclinical data have shown that a combining approach with agents blocking HER-1 or HER-2 driven signaling pathways can restore hormone sensitivity in endocrine-resistant, HER-2 overexpressing breast tumors, improving the therapeutic efficacy of currently available endocrine options [97].
Targeting HER2 in hormone-receptor positive breast cancer may involve re-expression of silenced ER
9. In the
randomized, controlled, open-label, multicenter, phase III TAnDEM trial, the efficacy of trastuzumab plus anastrozole was compared with anastrozole alone in 207 postmenopausal women with HER2 and ER positive MBC. The combination therapy regimen produced significantly greater improve-ments in response rate, time to progression, and clinical benefit rate than anastrozole alone; overall survival (OS) was also longer in patients receiving trastuzumab plus anastro-zole, despite the crossover of more than 50% of patients from the anastrozole monotherapy group to the combination therapy group following disease progression
10.
The growing body of evidence to support the role of EGFR and HER2 in cross-talk activation of estrogen recep-tor signaling has led to studies aimed to identify whether small-molecule tyrosine kinase inhibitors targeted against these receptors, such as lapatinib and gefitinib, give additive or synergistic effects when combined with endocrine agents.
In vitro data demonstrate that estrogen deprivation sig-nificantly enhances the anti-proliferative effects of lapatinib in HER2-amplified breast cancer cell lines. Preclinical evi-dence suggests that lapatinib can significantly enhance sensi-tivity to tamoxifen in cell lines with acquired tamoxifen re-sistance [98]. A phase III trial, recently published, involved patients affected by ER-positive MBC who were randomly assigned to receive either letrozole alone or letrozole com-bined with lapatinib. This trial demonstrated in 219 patients coexpressing estrogen receptor and HER2 that the combined targeted strategy significantly enhanced PFS and clinical benefit rates in patients with MBC [99].
A number of phase II studies are actually ongoing to bet-ter investigate on lapatinib and chemotherapy or endocrine therapy combination in the neoadjuvant setting (CHERLOB and LETLOB phase II trials).
Besides HER2, enhanced expression of HER1 has been found in BC cells that become resistant over time to endo-crine therapy either with tamoxifen or long-term estrogen deprivation. The HER1 expression rate in BC is in the range of 14–91%, depending on the method of assessment, and it is almost always caused by increased receptor synthesis. HER1 mutations have been reported in 78% of BC cases by RT–PCR and 27% by IHC [97]
Breast cancer cells with high expression of HER1or HER2 were most sensitive to gefitinib, which induced a sig-nificant G1-S cell cycle arrest, together with induction of apoptosis. ER-positive tumors that overexpress HER2 and
9Johnston, S. R. D. Integration of endocrine therapy with targeted agents. Breast Can-
cer Research 2008, 10, S20. 10Mackey, J. R.; Kaufman, B.; Clemens, M.; Bapsy, P. P.; Vaid, A.; Wardley, A.;
Tjulandin, S., Jahn, M.; Lehle, M.; Jones, A. Trastuzumab prolongs progression-free survival in hormone-dependent and HER2-positive metastatic breast cancer. San Anto-
nio Breast Cancer Symposium 2006, Abstract 3.
become resistant to tamoxifen can be growth-inhibited by gefitinib, which targets HER1, due to disruption of het-erodimerization between HER1 with HER2, which abrogates HER2-dependent growth [100].
Up to date some randomized studies in ER-positive MBC have been reported. A double-blind, placebo-controlled phase II trial of tamoxifen with/without gefitinib as first line endocrine therapy was conducted in 290 postmenopausal women
11. In endocrine naïve patients an advantage in PFS
was observed.
The first results of a second randomized trial of gefitinib and anastrozole versus anastrozole alone, conducted in a similar first-line patients population were reported at the ASCO meeting in 2008
12. Enrollment was stopped early due
to slow recruitment and hence limited statistical analyses were performed. There was a significant prolongation of PFS from a median of 8.2 months with anastrozole to 14.6 months with the combination.
New Target Agents for HER Family Pathway
Pertuzumab represents the first in a new class of agents known as HER dimerization inhibitors. It is a fully recombi-nant humanized monoclonal antibody that binds extracellu-larly to the dimerization arm near the junctions of domains I–III of ErbB2 and thus sterically blocking heterodimeriza-tion of HER2 with HER1 and HER3 thereby inhibiting intra-cellular signaling.
Pertuzumab, as trastuzumab, mediates ADCC, but it does not block ErbB2 shedding. Unlike in the case of trastuzu-mab, the effect of pertuzumab does not strictly require ErbB2 overexpression. In cells where ErbB2 was activated, but not overexpressed, the Fab fragment of pertuzumab was just as effective in inhibiting ErbB2 mediated signaling as the intact antibody [101, 102].
Pertuzumab is currently under early clinical evaluation, and results from phase I trials have shown that the drug is well tolerated and clinically active [103]. Pertuzumab mono-therapy has been shown activity against HER2-positive breast cancer which has progressed during trastuzumab-based therapy. The combination of the two antibodies ap-pears to be more active than either antibody alone, also in patients that had failed both antibodies given separately
13.
Neratinib is a potent, low molecular weight, orally active, pan erbB receptor tyrosine kinase inhibitor that blocks signal transduction through three receptors, erbB-1, erbB2, and erbB4 by irreversible covalent binding to their respective intracellular tyrosine kinase domains.
11Osborne, K.; Neven, P.; Dirix, L.; Mackey, J.; Robert, J.; Underhill, C.; Gutierrez, C.;
Magill, P.; Hargreaves, L. Randomized Phase II study of gefitinib (IRESSA) or pla-cebo in combination with tamoxifen in patients with hormone receptor positive metas-
tatic breast cancer. San Antonio breast Cancer Symposium 2007, abstract 2067. 12Cristofanilli, M.; Valero, V.; Mangalik, A.; Rabinowitz, I.; Arena, F. P.; Kroener, J.
F.; Curcio, E.; Watkins, C.; Magill, P. A phase II multicenter, double-blind, random-
ised trial to compare anastrozole plus gefitinib with anastrozole plus placebo in post-menopausal women with hormone recptor positive metastatic breast cancer. J. Clin.
Oncol. 2008, 26, abstract 1012. 13Cortés, J.; Baselga, J.; Petrella, T.; Gelmon, K.; Fumoleau, P.; Verma, S.; Pivot, X.;
Ross, G.; Szado, T.; Gianni, L. Pertuzumab monotherapy following trastuzumab-based treatment: Activity and tolerability in patients with advanced HER2- positive breast
cancer. J. Clin. Oncol. 2009, 27, abstract 1022.

12 Current Cancer Drug Targets, 2010, Vol. 10, No. 4 Palazzo et al.
Both preclinical and human studies have shown that neratinib has promising activity in both advanced breast can-cer and NSCLC with an acceptable safety profile.
In a phase I study performed on EGFR or HER-2 ex-pressing tumors, neratinib is generally well tolerated and shows activity in pretreated BC, previously exposed to tras-tuzumab, and in tumors with a baseline ErbB-2 immunohis-tochemical staining intensity of 2+ or 3+ [104]. Neratinib demonstrates in a phase II MBC study a robust antitumor activity in pts trastuzumab-naive compared to pts who re-ceived prior treatment
14.
Targeting Intracellular HER Family Pathway
PI3K/Akt is the most important pathway activated down-stream of ErbB2 in breast cancer.
Somatic activating mutations in Akt and the p110a subunit of the PI3K have been detected in 3–5% and 20–25% of primary breast tumors, respectively [105].
mTOR is a key growth factor-mediated signal transduc-tion pathway that regulates cell growth, closely related to the PI3K/Akt pathway [106]. Akt acts blocking the tuberous sclerosis protein complex (TSC1/2) that is a negative regula-tor of mTOR complex (mTORC1). The final effect of the inhibition on TSC1/2, by Akt, is the release of its specific inhibition of mTORC1, so that it (mTORC1) can phosphory-late some regulatory proteins as translational regulator 4E-BP1 (eukaryotic initiation factor 4E-binding protein) and the ribosomal protein p70s6k (the 70-kDa S6 kinase). [107].
The inhibition of mTOR directly prevents protein transla-tion via these two regulatory proteins, blocks the progression of the cell cycle at the G1 phase and so inhibits all mTOR-dependent growth factor signaling include estrogen, HER-2/neu and IGF-1 [107] .
The original inhibitor of mTOR is rapamycin with a poor solubility and chemical stability that limited its development as anticancer agent. So Rapamycin analogs with a more fa-vorable pharmacologic profile were synthesized, including CCI-779 (temsirolimus) and RAD-001 (everolimus). Temsi-rolimus was the first mTOR inhibitor to be investigated for the breast cancer [108].
Considering the cross-talk between the estrogen receptor and the PI3K/Akt/mTOR pathways, clinical trials exploring combined therapy of mTOR inhibitors with endocrine ther-apy were undertaken.
There is experimental evidence supporting the existence of a cross talk between ER and the PI3K/Akt pathway, that potentiate survival signals and accounts for survival of cells despite the presence of continued endocrine blockade. [109] Two ways of potentiating ER are described: a growth factor-mediated activation of mitogen-activated protein kinase (MAPK) or Akt, which directly phosphorylates ER within AF-1 and potentiates its transcriptional activity. In the other way there is a non genomic effect by which ER can interact
14Burstein, H. J.; Sun, Y.; Tan, A. R.; Dirix, L.; Vermette, J. J.; Powell, C.; Zachar-
chuk, C.; Badwe, R. A. Neratinib (HKI-272), an irreversible pan erbB receptor tyrosine kinase inhibitor: phase 2 results in patients with advanced HER2+ breast cancer. San
Antonio Breast Cancer Symposium 2008, abstract 37.
with growth factor receptors at the cell surface thus facilitat-ing PI3K/Akt signaling.
Such ER cross-talk with the PI3K/Akt pathway has pro-vided a rationale for exploring the combination of mTOR antagonists with endocrine therapy with the aim to overcome resistance [107].
A large-scale, multicenter phase III clinical trial com-pared temsirolimus–letrozole combinations to letrozole alone in postmenopausal patients with ER-positive, locally ad-vanced or MBC, suitable for first-line hormone therapy
15.
Although the early phase II data seemed to suggest that PFS may be prolonged for the temsirolimus–letrozole combina-tions
16, the phase III trial was terminated early, after the
demonstration, by an interim analysis, of a complete lack of benefit for this combination in terms of PFS and ORR (Table 2).
A potential explanation for the limited effectiveness of mTOR inhibitors in breast cancer and other cancer could be the presence of two key regulatory loops of the signaling pathway.
The first loop consisted in a negative feedback loop ex-ists downstream in the PI3K/Akt/mTOR pathway. The mTOR-activated kinase, called as S6K1, phosphorylates and destabilizes the IRS1 and IRS2 proteins that are present in insulin-like growth factor (IGF) responsive cells. mTOR inhibitors can lead to a reduction in S6K1 activity, with a consequent loss of its negative feedback on IRS1/2 proteins allowing to an increased activation of Akt activity dependent from insulin growth factor stimuli .
The second loop that could limit the efficacy of mTOR inhibitors is a positive regulatory loop involving the mTORC2 complex that is differently regulated in contrast to mTORC1. The mTORC2 complex can be activated by growth factors and phosphorylates Akt directly. Rapamycin analogs that target mTOR proteins appear to specifically block only the mTORC1 without affecting mTORC2 com-plex.[110]
In order to select patients who may benefit from mTOR inhibition when combined with endocrine therapy many studies in neoadjuvant setting have been started.
In a randomized phase II study conducted in 270 post-menopausal women with ER-positive primary operable breast cancer, the combination of letrozole and everolimus (RAD001) for 4 months before surgery resulted in signifi-cantly greater tumor shrinkage with regard to both clinical and biological response (reduction in Ki67)
17.
15Chow, L. W. C.; Sun, Y.; Jassem, J.; Baselga, J.; Hayes, D. F.; Wolff, A. C.;
Hachemi, S.; Cincotta, M.; Yu, B. W.; Kong, S.; Moore, L. Phase 3 study of temsi-rolimus with letrozole or letrozole alone in postmenopausal women with locally ad-
vanced or metastatic breast cancer. San Antonio Breast Cancer Symposium 2006, abstract 6091. 16Baselga, J.; Roche, H.; Fumoleau, P.; Campone, M.; Colomer, R.; Cortes-Funes, H.; Gil, M.; Chan, S.; Boni, J.; Kong, S.; Cincotta, M.; Moore, L. Treatment of postmeno-
pausal women with locally advanced or metastatic breast cancer with letrozole alone or
in combination with temsirolimus; a randomized 3-arm, phase 2 study. Breast Cancer
Res Treat. 2005; 94, abstract A1068. 17Baselga, J.; van Dam, P. A.; Greil, R.; Gardner, H.; Bandaru, R.; Molloy, B.; Stein-seifer, J.; Phillips, P.; Dixon, J. M.; Rugo, H. S. Improved clinical and cell cycle re-
sponse with an mTOR inhibitor, daily oral RAD001 (everolimus) plus letrozole versus placebo plus letrozole in a randomized phase II neoadjuvant trial in ER+ breast cancer.
J. Clin. Oncol. 2008, 26, abstract 530

Targeted Therapy in Cancer Current Cancer Drug Targets, 2010, Vol. 10, No. 4 13
The GeparQuinto trial is investigating in HER2 negative
patients the incorporation into neoadjuvant regimens of bevacizumab (B), RAD001 (everolimus).
The combination therapy with RAD001 (everolimus) and
trastuzumab is currently in clinical trials for HER-2–positive
BC. In an ongoing clinical trial, early efficacy data suggests
the possibility of significant synergism from the addition of
everolimus to a trastuzumab-taxane regimen in the metastatic setting
18.
New Target Agents for Intracellular HER Family Path-
way
Src is a non-receptor protein tyrosine kinase, known as a
member of the Src family kinases, initially identified by Pey-
ton Rous in 1911 as the transforming agent in chicken sar-
comas. It is downstream in signaling from a number of
growth factor receptors including PDGF receptor (PDGFR),
epidermal growth factor receptor (EGFR), and insulin-like
growth factor-1 receptor (IGF-1R) playing a role of key mes-
senger in important cellular pathways. It could be also acti-
vated by cytoplasmic proteins involved in cellular adhesion signaling.
Recent studies suggest an association between Src TK
and the development, progression and metastasis of BC,
even if, after transfection, it does not have transforming abil-
ity. The mitogenic and tumorigenic effect of Src may be ex-
plained by the cross talk between growth factors and down-
stream proteins: the overexpression of upstream growth fac-
tors such as EGFR, HER2, PDGFR and VEGFR cause a
deregulation with a subsequent increased activity of Src,
confirmed by in vitro studies on human breast cancer cells
[111, 112].
Currently, small-molecule Src inhibitors are in early
phases of clinical development either as single agents and in
combination with cytotoxic agents or in combination with
hormonal treatment. Dasatinib (BMS-354825) is a potent
orally-available inhibitor of Src-family kinases and other
kinases with anti-proliferative, anti-osteoclastic and anti-
metastatic activity, approved for the treatment of patients
with imatinib-refractory chronic myelogenous leukaemia and
Philadelphia chromosome-positive acute lymphoid leukae-
mia, due to its ability to inhibit other protein kinases: brc-abl,
c-Kit, EphA2, and PDGF-beta.
Dasatinib is currently being studied in clinical trials for the treatment of solid tumours, including breast cancer. A recent phase II study demonstrated an encouraging single-agent activity of dasatinib in patients with ER positive and/or HER2 amplified MBC
19. A modest single agent activity was,
instead, observed in patients with advanced triple-negative
18André, F.; Campone, M.; Hurvitz, S. A.; André, F.; Vittori, L.; Pylvaenaeinen, I.;
Sahmoud, T.; O'Regan, R. M. Multicentre phase I clinical trial of daily and weekly
RAD001 in combination with weekly paclitaxel and trastuzumab in patients with HER2-overexpressing metastatic breast cancer with prior resistance to trastuzumab. J.
Clin. Oncol. 2008, 26, abstract 1003. 19Mayer, E.; Baurain, J.; Sparano, J.; Strauss, L.; Campone, M.; Fumoleau, P.; Rugo,
H.; Awada, A.; Sy, O.; Llombart, A. Dasatinib in advanced HER2/neu amplified and ER/PR-positive breast cancer: Phase II study CA180088. J. Clin. Oncol. 2009, 27,
abstract 1011.
breast cancers, with clinical benefit rate of 9.3%20
. Future studies will address the combination of dasatinib with hor-monal or cytotoxic therapies.
Tipifarnib (R115777) is an oral farnesyl transferase in-hibitor (FTI) active against human tumor cell lines and ex-hibiting in phase II studies, modest single agent activity and in combination with chemotherapy or hormone-therapy in patients with previously treated metastatic breast cancer. Farnesyl transferases are proteins catalyzing activating post translational modification of Ras. Despite the absence of stimulation, Ras proteins are activated in 30% of human can-cers, including BC [70] and convey uncontrolled prolifera-tive signals. More studies are required in order to determine the efficacy of tipifarnib in BC [113-115].
Targeting DNA Repair
Targeted drugs have shown considerable success in the treatment of hormone-receptor-positive and ERBB2-overexpressing breast cancers. However, no agents are avail-able for BC lacking these characteristics, such as ‘triple-negative’, which account for almost 15% of breast cancers.
Triple negative BC are often characterized by mutations in the genes encoding breast cancer type 1 susceptibility pro-tein BRCA1 and BRCA2 [115], which are both essential for the repair of double strand DNA breaks (DSBs) and col-lapsed replication forks by the process of homologous re-combination (HR). This is a conservative form of DNA re-pair by restoring of original DNA sequence at damaging site. In the absence of HR, cells that have lost wild type BRCA1 or BRCA2 increase their usage of non-conservative mecha-nisms to repair DSBs/replication forks. These non-conservative mechanisms contribute to the tumor cell pheno-type being a ready source of genetic variation.
Inheriting a mutation in either the BRCA1 or the BRCA2 gene, renders the individual at high lifetime risk of develop-ing breast and ovarian cancer .
The lifetime risk of breast cancer in female BRCA1 mu-tation carriers has been reported to be as high as 84% and between 60–80% in BRCA2 mutation carriers [116].
The sensitivity of BRCA1- and BRCA2-deficient cell lines to different inhibitors of the DNA repair enzymes, PARP1 and PARP2 has been demonstrated.
The inhibition of PARPs causes the accumulation of DNA single-strand breaks by lacking of DNA single-strand breaks (SSBs) repair. It has been observed that SSBs ,when encountered by replication forks, cause fork collapse and lead to the formation of DNA double- strand breaks (DSBs). Normally, these breaks are repaired by means of the error-free homologous-recombination (HR) double-stranded DNA repair pathway. So in BRCA deficient cells, that give rise to a defective HR pathway, the use of PARP inhibitors seems likely a target therapy for tumors harboring defects in DNA repair. Theodosius Dobzhansky in the 1940s first described the concept of synthetic lethality as an event that occurs be-
20Finn, R. S.; Bengala, C.; Ibrahim, N.; Strauss, L. C.; Fairchild, J.; Sy, O.; Roche, H.;
Sparano, J.; Goldstein, L. J. Phase II trial of dasatinib in triple-negative breast cancer: results of study CA180059. San Antonio Breast Cancer Symposium 2008, abstract
3118.

14 Current Cancer Drug Targets, 2010, Vol. 10, No. 4 Palazzo et al.
tween two genes, when loss of both gene function (BRCA and PARP) is lethal for cellular viability [117].
PARP inhibitors were initially proposed as chemosensi-tisers, but they may also be effective as single agents in those patients whose tumours exhibit an HR defect but no BRCA mutation, the so called “ BRCAness”.
The first clinical evidence that BRCA mutated cancers may be sensitive to PARP inhibitor monotherapy was pre-sented at the 2007 ASCO meeting by Yap and co-workers with preliminary data on olaparib, a small-molecule PARP inhibitor. The follow-on phase II trial demonstrated that ola-parib is well tolerated and highly active in advanced chemo-therapy-refractory BRCA-deficient breast cancer
21.
Based on hypothesis that PARP inhibitors could enhance the efficacy of radiation therapy and chemotherapies, such as alkylating agents and platinum-based drugs, by preventing cancer cells from repairing DNA damage and promoting apoptosis, the objectives of awaited phase II study, presented at the ASCO meeting in 2009, were to evaluate BSI-201, another potent PARP1 inhibitor, in combination with gem-citabine/carboplatin (G/C) in subjects with triple negative MBC. Analyses of the first 86 of a planned 120 patients showed that BSI-201 + G/C significantly improves clinical benefit rate, median PFS, and median OS, compared with G/C alone
22.
Targeting VEGF Pathway
VEGF is often overexpressed in BC both at RNA and protein levels. Higher microvessel densities have been ob-served in BC patients and were associated with poor clinical outcome and reduced survival. Notably, this profile is corre-lated with expression of mutant p53 and with HER2 overex-pression suggesting that the induction of angiogenesis may
contribute to cancer progression [118, 119]. These data have supported the exploration of the use of anti-VEGF therapy in BC (Table 2).
In a randomized phase III trial, bevacizumab combined with capecitabine, in pretreated MBC patients, produced a significantly greater ORR compared to capecitabine alone, but had no impact on PFS or OS [120].
Conversely, the Eastern Cooperative Oncology Group (ECOG) E2100 phase III clinical trial, which provided the basis for FDA approval, investigated the use of combination therapy with bevacizumab plus paclitaxel given until disease progression. The combination compared with paclitaxel alone showed approximately doubled median progression-free survival and increased the objective response rate; con-versely, there was not a statistically significant improvement in terms of overall survival [121]. Based on these data, the EMEA and FDA have approved bevacizumab in combina-tion with paclitaxel for the first-line treatment of patients with metastatic breast cancer in March 2007.
21Tutt, A.; Robson, M.; Garber, J. E.; Domchek, S.; Audeh, M. W.; Weitzel, J. N.;
Friedlander, M.; Carmichael, J. Phase II trial of the oral PARP inhibitor olaparib in BRCA-deficient advanced breast cancer. J. Clin. Oncol. 2009, 27, abstract CRA501. 22O'Shaughnessy, J.; Osborne, C.; Pippen, J.; Yoffe, M.; Patt, D.; Monaghan, G.; Ro-cha, C.; Ossovskaya, V.; Sherman, B.; Bradley, C. Efficacy of BSI-201, a poly (ADP-
ribose) polymerase-1 (PARP1) inhibitor, in combination with gemcitabine/carboplatin in patients with metastatic triple-negative breast cancer: Results of a randomized phase
II trial. J. Clin. Oncol. 2009, 27, abstract 3.
The favorable clinical results observed in untreated MBC patients suggest that antiangiogenic agents should be given early in the course of disease. In fact, in experimental studies after VEGF suppression, the expression of additional pro-angiogenic factors such as basic fibroblast growth factor (bFGF) and transforming growth factor alpha (TGF-a) was strictly dependent on the tumour size. It was therefore sug-gested that an increased number of pro-angiogenic factors come into play at disease progression, thus making VEGF inhibition alone a less effective anti-angiogenic strategy in progressed disease [118].
The AVADO multicentre phase III trial investigates the safety and efficacy of bevacizumab in combination with do-cetaxel for first-line metastatic breast cancer. A total of 736 patients were enrolled and treatment arms containing bevaci-zumab confirmed a significant improvement in DFS
23.
Based on these results, the European Commission ap-proved on July 2009 a broader label for bevacizumab in ad-vanced breast cancer in combination with docetaxel.
An ongoing phase III trial, RIBBON 1, starts to definitely assess the clinical benefit and toxicity when bevacizumab is added to three different classes standard first line chemother-apy regimens (anthracycline-based, single-agent taxane, or single-agent capecitabine chemotherapy) for MBC. After a median follow-up of 15.6 months in the capecitabine cohort and 19.2 months in the taxane and anthracycline cohort the combination with bevacizumab confirms its statistically-significant improvement in PFS compared with placebo combination
24.
In adjuvant setting the safety and the feasibility of bevacizumab have been explored by a two arm phase II trial incorporating bevacizumab into an anthracycline regimen
25.
Neoadjuvant setting is currently under investigation and other studies are ongoing to test (BEtTH and AVEREL stud-ies) the combination of bevacizumab and trastuzumab in adjuvant and metastatic setting. The rationale of these studies relies upon the observation that VEGF, EGFR and HER-2 pathways are strictly connected [122].
Apart from bevacizumab other VEGF inhibiting drugs are currently in development for the treatment of breast can-cer , and, to date, sunitinib and pazopanib are the only anti-VEGF agents that have reached phase III trials in this tumor type.
COLORECTAL CANCER (CRC)
Combined regimens of cytotoxic agents with biological target therapies have increased response rate (RR) and im-
23Miles, D.; Chan, A.; Romieu, G.; Dirix, L.Y.; Cortes, J.; Pivot, X.; Tomczak, P.;
Taran, T.; Harbeck, N.; Steger, G.G. Randomized, double-blind, placebo-controlled, phase III study of bevacizumab with docetaxel or docetaxel with placebo as first-line
therapy for patients with locally recurrent or metastatic breast cancer: AVADO. J. Clin.
Oncol. 2008; 26, abstract LBA1011. 24Robert, N. J.; Dieras, V.; Glaspy, J.; Brufsky, A.; Bondarenko, I.; Lipatov, O.; Perez, E.; Yardley, D.; Zhou, X,; Phan, S. RIBBON-1: Randomized, double-blind, placebo-
controlled, phase III trial of chemotherapy with or without bevacizumab for first-line
treatment of HER2-negative locally recurrent or metastatic breast cancer. J. Clin.
Oncol. 2009, 27, abstract 1005. 25Miller, K. D.; O’Neill, A.; Perez, E. A.; Seidman, A. D.; Sledge, G. W. Phase II feasibility trial incorporating bevacizumab into dose-dense doxorubicin and cyclophos-
phamide followed by paclitaxel in patients with lymph node-positive breast cancer: A trial of the Eastern Cooperative Oncology Group (E2104). J. Clin. Oncol. 2008, 26,
abstract 520.

Targeted Therapy in Cancer Current Cancer Drug Targets, 2010, Vol. 10, No. 4 15
proved both progression free survival (PFS) and overall sur-vival (OS) in patient with advanced colorectal cancer dis-ease. The most recent experimental studies in this patients have been focused on the use of targeted biologic therapies. The knowledge of the molecular alterations needed to trans-form an healthy cells allowed the development of targeted agents. In colon cancer many growth pathway, when consti-tutively activated on specific genes, might initiate and/or sustain its malignant transformation and angiogenesis:
- growth factors (VEGF, PDGF, EGF, etc.);
- intracellular protein kinases (Ras, Raf, APC, etc.);
- transcription factors.
Angiogenesis and EGFR pathways represent today the main targets in the medical treatment of colorectal cancer (Table 3).
Targeting Angiogenesis
VEGF and angiogenesis appear to be as a predominant pathways in human colon cancer and are strongly associated
with metastatic disease and poor prognosis. The evaluation of vascular density in potential malignant primary colon can-cers was correlated with the development of distant metasta-sis (P<0.001): a low vessel count was found to be associated with a favourable prognosis, while a high vessel counts was observed in recurrent disease [123]. A direct correlation was also seen between VEGF expression and the development of metastatic stage of disease (P<0.001) [124-127] and with survival [128, 129]. These data show that angiogenesis with its effectors (VEGF), and its receptor (VEGFR), are the ideal targets to inhibit the vascularity, the proliferation, and the growth of colon cancer.
VEGF-A gene is a member of the PDGF/VEGF growth factor family and acts, as a glycosylated mitogen, specifi-cally on endothelial cells with various effects, including the mediation of increased vascular permeability, angiogenesis, vasculogenesis and endothelial cell growth, promoting cell migration, and inhibiting apoptosis. It is overexpressed both in some premalignant lesions of breast, cervix, and colon cancers and in invasive
cancer cells. Activation of VEGFR-1
on CRC cells with VEGF-A or -B induces downstream sig-nal, which include the activation of the Erk-1/ 2 and JNK
Table 3. Phase III Trials with Biological Agents in Colorectal Cancer
Target Investigators and
Reference
Stage Setting Agent/Schedule Results
Hurwitz H. et al. [131] IV First line Bevacizumab 5 mg/Kg every two wks + IFL vs. Placebo + IFL
PFS: 10.6 vs. 6.2 months; P<0.001;
OS: 20.3 vs. 15.6 months; P<0.001;
ORR: 44.8% vs. 34.8%; P=0.004.
Giantonio B.J. et al. [132]
IV Second line
Bevacizumab 10 mg/Kg every two wks + FOLFOX-4 chemotherapy vs. FOLFOX-4 chemotherapy vs. Bevacizumab alone
PFS: 7.3 vs. 4.7 (P<0.0001) vs. 2.7 months;
OS: 12.9 vs. 10.8 (P=0.0011) vs. 10.2 months;
ORR: 22.7% vs. 8.6% (P<0.0001) vs. 3.3%.
VEGF
Wolmark N. et al. [26] II-III Adjuvant Bevacizumab 5 mg/kg every 2 weeks x 26 + FOLFOX-6 vs. FOLFOX-6
DFS at 36 months: HR=0.89; 95% CI (0.76-1.04); P=0.15.
VEGFR Hecht J.R. et al. [27] IV First line Vatalanib + FOLFOX-4 vs. Placebo +
FOLFOX-4 PFS: 9.1 vs. 7.7 months; P=0.1;
OS: 21.4 vs. 20.5 months; P=0.28;
Van Cutsem E. et al.
[143] IV Firs line Cetuximab 400 mg/m2 initial, after 250
mg/m2 wekly + FOLFIRI vs. FOLFIRI PFS: 8.9 vs. 8.0 months; P=0.048
OS: 19.9 vs. 18.6 months; P=0.31
ORR: 46.9% vs. 38.7%; P=0.004.
Cunningham D. et al.
[137] IV Third line Cetuximab 400 mg/m2 initial, after 250
mg/m2 wekly + Irinotecan vs. Cetuximab PFS: 4.1 vs. 1.5 months, P<0.001;
OS: 8.6 vs. 6.9 months, P=0.48
ORR: 22.9% vs. 10.8%; P=0.007.
Jonker D.J. et al. [138] IV Third line Cetuximab 400 mg/m2 initial, after 250
mg/m2 wekly vs. Best supportive care.
PFS: HR=0.68; 95% CI, 0.57 to 0.80;
P<0.001;
OS: 6.1 vs. 4.6 months:
ORR: 8% vs. 0%.
EGFR
Van Cutsem E. et al.
[144] IV Third line Panitumumab 6 mg/kg every two wks vs.
Best supportive care. PFS: 8 vs. 7.3 weeks; P<0.0001;
OS: No difference; P=0.81
ORR: 10% vs. 0%; P<0.0001;
Hecht J.R. et al [147]
(PACCE Trial)
IV First line
Panitumumab 6 mg/kg every two wks +
Bevacizumab + chemotherapy vs. Bevaci-
zumab + chemotherapy.
NOTE: Chemotherapy: Oxaliplatin (Ox-T)
or Irinotecan (Iri-T) based
PFS Ox-T: 10.0 vs. 11.4 months;
PFS Iri-T: 10.1 vs. 11.7 months;
OS: 19.3 vs. 20.6 months;
ORR Ox-T: 46% vs. 48%;
ORR Iri-T: 43% vs. 40%.
EGFR,
VEGF
Tol J. et al. [146]
(CAIRO-2 Trial)
IV First line Cetuximab 400 mg/m2 initial, after 250
mg/m2 wekly + Bevacizumab 7.5 mg/Kg + XELOX vs. Bevacizumab + Xelox
PFS: 9.4 vs. 10.7 months; P=0.01;
OS: 19.4 vs. 20.3 months; P=0.16
ORR: 52.7% vs. 50.0%; P=0.49.

16 Current Cancer Drug Targets, 2010, Vol. 10, No. 4 Palazzo et al.
MAPK pathways. The addition of these cytokines led to functional changes, including increased cell migration, inva-sion, and colony formation [130].
The role of VEGF-A in tumor angiogenesis made it to become a target of therapies designed to reduce the tumor vasculature. In this setting, the first targeted agent to show a significant benefit in the treatment of metastatic colorectal cancer was bevacizumab: a humanized monoclonal antibody directed against the vascular endothelial growth factor (VEGF). Bevacizumab was tested in a phase III randomised trial comparing irinotecan, fluorouracil, and leucovorin (IFL) plus bevacizumab (5 mg/Kg) or placebo. This trial showed an increase in overall survival (20.3 vs. 15.6 months; P<0.001), rates of response (44.8% vs. 34.8%; P=0.004), and progression- free survival (10.6 vs. 6.2 months; P<0.001) [131]. In 2004, on the basis of these evidences bevacizumab was approved by FDA as first line treatment combined to chemotherapy. The addition of bevacizumab at a dose of 10 mg/m2 per week in the second-line setting (irinotecan-refractory subjects) to palliative FOLFOX-4 chemotherapy has been shown to improve survival in an ECOG random-ized phase III study [132]. A recent metanalysis of the Coch-rane Collaboration confirmed the significant benefit for both PFS and OS for the addition of bevacizumab to chemother-apy in patients with metastatic colorectal cancer [133].
In the adjuvant setting, unexpectedly, bevacizumab did not showed any advantage in disease free survival (DFS) when combined to six months of fluorouracil and oxaliplatin based chemotherapy (FOLFOX-6) and continued for one year of total treatment
26.
Bevacizumab is actually approved for the treatment of metastatic CRC in both the first and second-line, and its role of Bevacizumab in neoadjuvant and adjuvant setting is sub-ject of ongoing trials.
The inhibition of vascular endothelial growth factor re-ceptor (VEGFR) tyrosine kinase activity, represents another pathway to inhibit angiogenesis; two drugs, cediranib and vatalanib, are actually ongoing in phase III trials. Although phase I study have been demonstrated the feasibility and tolerability of cediranib as single agent or combined to ox-aliplatin and 5-fluorouracil based chemotherapy [134, 135], a phase III trial, comparing FOLFOX in combination with cediranib or bevacizumab on first line treatment is ongoing.
The role of vatalanib in colorectal cancer was studied in a phase III trial comparing FOLFOX regimen with or without vatalanib. The study was completed and the results reported no benefit in overall survival (21.4 vs. 20.5 months; p=0.28), and no statistically significant improvement in progression-free survival (9.1 vs. 7.7 months; p=0.1) from the addition of vatalanib to first-line chemotherapy
27.
26Wolmark, N.; Yothers, G.; O'Connell, M. J.; Sharif, S.; Atkins, J. N.; Seay, T. E.; Feherenbacher, L.; O'Reilly, S.; Allegra, C. J. A phase III trial comparing mFOLFOX6
to mFOLFOX6 plus bevacizumab in stage II or III carcinoma of the colon: Results of
NSABP Protocol C-08. J. Clin. Oncol. 2009, 27, abstract LBA4. 27Hecht, J. R.; Trarbach, T.; Jaeger, E.; Hainsworth, J.; Wolff, R.A.; Lloyd, K.;
Bodoky, G.; Borner, M.; Laurent, D.; Jacques C. Final overall survival results of CONFIRM1, a randomized, double–blind, placebo– controlled phase III trial in pa-
tients with metastatic adenocarcinoma of the colon or (mCRC) recieving first–line chemotherapy with oxaliplatin/5–FU/Leucovorin (FOLFOX 4) and PTK/ZK 22584
(PTK/ZK) or placebo (PBO). Eur. J. Cancer Supp. 2007, 5, 238.
Although angiogenesis pathway represents a main target in CRC treatment, actually only the addition of bevacizumab to chemotherapy has shown a significant advantage in terms of prolongation of survival and disease control in metastatic patients and received the approval for both FDA and EMEA.
Targeting EGFR
Inhibition of the EGFR is a promising concept for the treatment of CRC considering that expression of EGFR has been reported in 60–75% of colorectal cancers and has been correlated with poor prognosis [136]. From the therapeutic point of view, there are two possible strategies to target EGFR: a monoclonal antibodies (cetuximab, panitumumab) or a TK-inhibitors (TKi; erlotinib, gefitinib).
The first phase III study with a monoclonal antibody against EGFR (cetuximab) in colorectal cancer has been the BOND trial: patients, whose disease had progressed during or within 3 months after treatment with an irinotecan-based regimen, were randomized to receive cetuximab alone or in combination with irinotecan. Published results showed an increase for both the median time to progression (4.1 vs. 1.5 months, P<0.001), and response rate (22.9% vs. 10.8%; P=0.007) but not for overall survival (8.6 vs. 6.9 months; P=0.48) [137]. The BOND study also showed the capability of cetuximab to revert the resistance to irinotecan in irinote-can-previous treated tumors. These data albeit minimal, al-lowed FDA and EMEA to approve in 2004 cetuximab for treatment of patients with EGFR positive immunohistochem-istry (IHC) metastatic colorectal cancer, after failure of iri-notecan-including cytotoxic therapy. Although the BOND trial was criticized for the absence of control arm, another study, CO.17 trial, showed a significant improvement in overall survival (6.1 vs. 4.6 months; P = 0.005) and in pro-gression-free survival (P<0.001) for cetuximab treatment, compared to best supportive care, in patients with IHC posi-tive EGFR colorectal cancer previously treated with a fluoropyrimidine, irinotecan, and oxaliplatin [138].
Even if the BOND trial required the immunohistochemi-cal expression of EGFR, neither the percentage of EGFR-positive tumor cells or the maximal staining intensity per cell, did significantly correlate with the clinical response rate (P=0.87 and P=0.64, respectively). This means that, there is not an established level of EGFR expression possibly related to obtain a clinical benefit from cetuximab: in fact response rates were comparable in patients expressing 1+, 2+, or 3+ levels of EGFR [137, 139].
Moreover, a small retrospective study in advanced CRC patients treated with cetuximab at Memorial Sloan-Kettering Cancer Center (MSKCC), showed a partial response in 25% of patients EGFR negative [140]. Two hypotheses have been made to explain this data: the first one is that there could be some differences in IHC EGFR expression, between primary tumor and metastatic tissue specimens. In fact metastatic tumor acquires additional mutations conferring growth ad-vantage and the cumulative effect of clonal selection, but because of tissue availability the EGFR IHC is made mainly in primary tumor. The second explanation for the lack of correlation between EGFR expression and antitumor activity could be explained by the potential inaccuracies in IHC tests to detect different membrane levels of EGFR. A study re-

Targeted Therapy in Cancer Current Cancer Drug Targets, 2010, Vol. 10, No. 4 17
ports, in fact, the existence of two EGFR binding sites, based on a lower or higher affinity for the ligand, with a marked variability in the tumors expression [141]. The current anti-bodies anti-EGFR IHC detection systems, commercially available, do not distinguish between these two different EGFR. Thus, one could speculate that tumors carrying few EGFRs, below the threshold of IHC detection, but with an high affinity for binding site, might result to be responsive to an EGFR-targeted agent.
Although the IHC expression of the EGFR is not strictly necessary to predict a sensitivity to monoclonal antibody therapies, the non-mutated (wild-type) status of K-RAS pro-tein is mandatory for its efficacy. Mutations of K-RAS rep-resent an early event in the colorectal adenoma–carcinoma sequence and are more frequently sited in exon 2 (codons 12 and 13), and to a lesser extent in exon 3 (codon 61); mutated status [142], that occur for about 40% of colorectal cancers, results in a constitutive activation of the MAPK pathway downstream of the EGFR and, therefore, resulting in a lack of sensititivity to anti-EGFR therapy.
Several studies have recently reported the role of K-RAS mutation status to predict response to anti-EGFR monoclonal antibodies in first, second and third line of chemotherapy. The phase II OPUS study evaluated the combination of FOLFOX-4 and cetuximab in first-line, K-RAS wild type, patients. A benefit from the combination therapy, compared to FOLFOX-4 alone, was obtained both in terms of response rate (61% v 37%; P=0.01) and PFS (7.7 v 7.2 months; P=0.02)
28. The overall survival analysis for patients with K-
RAS wild type (57.1%) showed a non significant improve-ment in median OS of approximately 3 months (22.8 vs 19.5 months; P = 0.58) for patients in the combination arm, possi-bly due to the small number of patients available for this analysis
29.
A phase III trial (CRYSTAL) investigated the efficacy and safety of irinotecan in combination with a simplified regimen of fluorouracil and leucovorin (FOLFIRI) plus cetuximab, as first line treatment, in EGFR positive patients with advanced colorectal cancer. cetuximab statistically in-creased the median progression-free survival times (8.9 vs 8.0 months; P=0.048) and response rate (46.9% vs 38.7%; P=0.004) but the improvement in median overall survival times, evaluated in about 70% of patients, was not statically significant (19.9 vs 18.6 months; P=0.31) [143]. The retro-spective analysis about K-RAS status performed on ap-proximately half of the sample size reported an incidence of mutations in 36% of patients. This analysis showed that the benefit of patients treated with FOLFIRI plus cetuximab appeared to be restricted to patients without mutations in the K-RAS gene that receive targeted therapy combination with an improvement on TTP (9.9 vs. 8.7 months) and RR (59.3% vs. 43.2%) compared to chemotherapy alone. The OS among the 348 patients with wild-type–KRAS tumors showed an
28Bokemeyer, C.; Bondarenko, I.; Hartmann, J. T.; De Braud, F. G.; Volovat, C.;
Nippgen, J.; Stroh, C.; Celik, I.; Koralewski P. KRAS status and efficacy of first-line treatment of patients with metastatic colorectal cancer (mCRC) with FOLFOX with or
without cetuximab: The OPUS experience. J Clin Oncol 2008, 26, abstract 4000. 29Bokemeyer, C.; Bondarenko, I.; Hartmann, J. T.; De Braud, F.; Schuch, G.; Zubel,
A.; Celik, I.; Koralewski, P. Overall survival of patients with KRAS wild-type tumours treated with FOLFOX4 ± cetuximab as 1st-line treatment for metastatic colorectal
cancer: The OPUS study. Eur. J. Cancer. Sup. 2009, 7, 345.
hazard ratio for death of 0.84 and a median overall survival of 24.9 versus 21.0 months in the cetuximab + FOLFIRI group compared with the FOLFIRI group respectively
28, 30.
Very recently a new targeted antibody, similar to cetuxi-mab, was added to the therapeutic arsenal in CRC to a great extent. Panitumumab, a fully human antibody, showed a comparable efficacy and toxicity as single agent and in com-bination with chemotherapy in chemorefractory patients with a more rarely anaphylactic reactions.
A phase III trial randomised 1183 patient with advanced CRC to receive FOLFOX-4 with or without panitumumab 6 mg/kg every two weeks as first line therapy. The study re-ported that for patients with K-RAS wild type status, the median PFS was higher for the combination arm (9.6 vs 8.0 months; P = 0.0234) and RR was 55% and 48% respectively. In patients with mutated K-RAS, the median PFS was de-creased in those receiving panitumumab (7.3 vs. 8.8 months; P = 0.0227)
31. Second trial reported the results of the asso-
ciation of FOLFIRI chemotherapy schedule with panitumu-mab as a second line for advanced colorectal cancer. In pa-tients with K-RAS wild type status, the median PFS was 5.9 months for panitumumab + FOLFIRI and 3.9 months for FOLFIRI alone (P = 0.004); median OS was 14.5 and 12.5 months respectively (P = 0.115), while response rate was 35% and 10%
32.
In patients progressing after irinotecan and oxaliplatin combination, third-line panitumumab compared to BSC, showed a benefit for an increased PFS of 8 weeks (P<0.0001) and a RR after a 12-month minimum follow-up (10% vs 0%; P<0.0001). On the contrary no significant dif-ference regarding overall survival was observed between those groups [144]. In the subset analysis, K-RAS mutations were found in 43% of patients. The treatment effect on PFS for K-RAS wild type patients was significantly greater with a median PFS of 12.3 and 7.3 weeks and a RR of 17% and 0%, for panitumumab and BSC respectively. Even if a statis-tically significant OS difference was not observed between treatment arms among all patients analyzed (92% of sample) in the K-RAS wild type group, the OS was longer among the mutant group, adjusted for stratification factors and random-ized treatment (HR=0.67). The multivariate analysis showed that the K-RAS status was a predictor for OS in both treated and not treated patients with median OS time of 6.8 months for wild type vs. 4.5 months for mutant. A survival advan-tage was reported also for patient randomized to BSC that receive panitumumab after progression but not for patient that not receive treatment [145]. Based on these data, pani-tumumab obtained the EMEA approval, as monotherapy, for the treatment of patients with advanced colorectal cancer and wild type K-RAS status.
30Van Cutsem. E.; Lanf, I.; D’haens, G.; Moiseyenko, V.; Zaluski, J.; Folprecht, G.;
Tejpar, S.; Kisker, O.; Stroh, C.; Rougier P. KRAS status and efficacy in the first-line treatment of patients with metastatic colorectal cancer (mCRC) treated with FOLFIRI
with or without cetuximab: The CRYSTAL experience. J. Clin. Oncol. 2008, 26, abstract 2. 31Douillard, J.; Siena, S.; Cassidy, J.; Tabernero, J.; Burkes, R.; Barugel, M.E.; Hum-
blet, Y.; Cunningham, D.; Wolf, M.; Gansert, J.L.; Randomized phase 3 study of panitumumab with FOLFOX4 compared to FOLFOX4 alone as 1st-line treatment for
metastatic colorectal cancer (mCRC): the PRIME trial. Eur. J. Cancer Sup. 2009, 3, 6. 32Peeters, M.; Price, T.; Hotko, Y.; Cervantes, A.; Ducreux, M.; André, T.; Chan, E.;
Lordick, F.; Rong, A.; Gansert, J.; Randomized phase 3 study of panitumumab with FOLFIRI vs FOLFIRI alone as second-line treatment (tx) in patients (pts) with metas-
tatic colorectal cancer. Eur. J. Cancer Sup. 2009, 3, 9.

18 Current Cancer Drug Targets, 2010, Vol. 10, No. 4 Palazzo et al.
To evaluate an increase of PFS, response rate and overall survival of advanced patients, the CAIRO2 and PACCE tri-als studied the role of bevacizumab based chemotherapy combined with cetuximab or panitumumab respectively [146, 147]. Both trial confirmed the effectiveness of anti-EGFR monoclonal antibodies in wild type K-RAS patients, but at the same time reported a worsening of the PFS (and OS in case of panitumumab) and an increase number of seri-ous toxicity events when combined with bevacizumab based chemotherapy, independently by the schedule agent. The explanation of these side effects have not been identified and authors speculated about a possible and unexpected interac-tion between the two monoclonal antibodies suggesting that the combination of targeted therapies may not be analogous to the combination of different cytotoxic chemotherapy drugs. This is due presumably because EGFR mediates al-terations of downstream targets required for the activity of bevacizumab and/or of chemotherapy or because the induc-tion of EGFR-mediated cell-cycle arrest leads to resistance to cytotoxic therapies. These inefficacy and toxicity of com-bined targeted therapies were also confirmed in analogous phase II study [148], combining a small EGFR tyrosine kinase inhibitor, erlotinib, with bevacizumab.
These trials signed the end of the combined target ther-apy with anti EGFR and anti VEGF monoclonal antibody in colorectal cancer. More possibilities are any how coming out by new targets useful to select the best patient’s category for an existing therapy o for the development of new molecules against CRC.
Future Considerations
Routine use of monoclonal antibody in the treatment of mCRC changed dramatically the history of this disease offer-ing different therapeutic options that should be evaluated at the time of the therapeutic choice. The clinicians have to consider, at the same time, the patient characteristics (age, performance status, symptoms, co-morbidity), the status of disease (resectable or unresectable disease) and, now, the genomic profile of cancer.
Is it possible to ameliorate the survival and the control of disease of these patients?
While biochemistry can be useful to study new molecules with higher affinity to known biological target, a better knowledge of cancer molecular pathways might represents the key to found new targets.
Aflibercept, is an interesting alternative to target the cir-culating VEGF. It is an engineering protein comprised of segments of the extracellular domains of VEGFR1 and 2 fused to constant region (Fc) of human IgG1 with a potential antiangiogenic effect. Aflibercept acts as a soluble decoy receptor that binds to pro-angiogenic vascular endothelial growth factors (VEGFs), preventing VEGF/receptor binding event. Phase II and III trials with different schedules of che-motherapy combined with aflibercept as first and second line of treatment are ongoing.
Recently the downstream proteins of the K-RAS path-way, particularly B-RAF, represent an interesting point of investigation. This protein, belonging to the raf/mil family of serine/threonine protein kinases, plays a role in regulating
the MAP kinase/ERKs signaling pathway, which affects cell division, differentiation, and secretion. Mutations in this gene have also been associated with many tumors included 10% of colon cancer, but were rare in rectal cancers, and was independently associated with increasing age, female gender and right-sided colon cancer, but not with stage of disease. Moreover, a lower overall survival in B-RAF mutated pa-tients with a Duke’s stage-D was reported (P=0.0003; HR 0.38) [149]
33. B-RAF and K-RAS mutations are mutually
exclusive and were both related to inefficacy of cetuximab or panitumumab therapies [150, 151]. A potential pharmacol-ogical option to inhibit B-RAF mutated forms, with the in-tent to restore the sensitivity to cetuximab or panitumumab, could be represented by sorafenib. Moreover, this theoreti-cally efficacious combination is limited by a great toxicity and the possibility to revert the resistance to monoclonal antibody against EGFR, in B-RAF mutated patients, seems to be far off
34. A recent phase III trial randomised chemo-
naïve patients, to receive an oxaliplatin fluorouracil chemo-therapy with the addition of sorafenib or placebo. This study did not provide any patient selection based on molecular profile and probably this will be made retrospectively.
RENAL CELL CARCINOMA (RCC)
A multidisciplinary approach is also needed in RCC with integration by surgeon, radiotherapist and medical oncolo-gist. Since both cytotoxic and cytokine therapy were not able to improve the poor prognosis of advanced RCC, in the last decade this tumor has been a good model for the develop-ment of new biological agents.
Many target have been identified as cell cycle regulators of cancer progression in renal cancer (VHL, VEGF, TGF-a, HIF-a and mTOR) but preclinical studies have emphasize two relevant pathways: the vascular endothelial growth fac-tor (VEGF) and the mammalian target of rapamicin (mTOR) (Table 4). The availability of more than one agent for each of these targets complicated so far the management of patients in clinical practice.
However, even if recent studies suggested that some of the predictive biomarkers currently under investigation in clear cell renal carcinoma are promising, actually we have not any molecular parameters able to predict the efficacy of therapy, and the choice for the target therapies is based only on clinical parameters as the Memorial Sloan-Kettering Can-cer Center (MSKCC) score and its last modified version [152, 153]. This systems is founded on six clinical and bio-chemical parameters (ECOG performance status, serum he-moglobin, time from diagnosis to treatment, the value of corrected serum calcium, lactate dehydrogenase, and number of metastatic sites) in order to categorize patients on three prognosis groups: favorable ( 1 risk factor), intermediate (2
33Tie, J.; Sieber, O. M.; Gibbs, P.; Lipton, L.; Jorissen, R. N.; Langland, R.; Kosmider,
S.; McKay, D.; Nolop, K. B.; Desai. J. Selecting subjects for a therapeutic target in
colorectal cancer (CRC): Using a clinical database to enrich for patients harboring the BRAFV600E mutation. J. Clin. Oncol. 2009, 27, abstract 11003. 34Messersmith, W. A.; Jimeno, A.; Laheru, D.; Suárez-Gauthier, A.; Ma, W. W.; Rudek, M.; Khan, Y.; Donehower, R. C.; López-Ríos, F.; Wright, J.: Hidalgo. M. A
phase I biologic study of sorafenib (S) combined with irinotecan (I)/cetuximab (C) in previously treated patients with advanced colorectal cancer (CRC). Gastrointestinal
Cancers Symposium 2008 Abstract 439.

Targeted Therapy in Cancer Current Cancer Drug Targets, 2010, Vol. 10, No. 4 19
risk factors), and poor (>3 risk factors) with a median sur-vival of 29.6, 13.8 and 4.9 months respectively.
Targeting Hypoxia: VHL and HIF Function and Path-
ways in RCC
The pathogenesis of RCC is elucidated by the study on rare families exhibiting an inherited predisposition to de-velop RCC and by the discovery of the von Hippel-Lindau (VHL) gene, that give its name to the familial cancer syn-drome in which it is implicated [154].
The VHL is a tumor suppressor gene which loss of het-erozygosity predispose individuals to visceral cysts (espe-cially in the kidney and pancreas) and to a variety of tu-mours, including haemangioblastomas of the retina and cen-tral nervous system, clear cell renal carcinomas, pheochro-mocytomas, endolymphatic sac tumours, pancreatic islet cell tumours, and epididymal (male) and broad ligament (female) papillary cyst adenomas. The VHL loss of the heterozygosity is also frequently observed (about 90%) in sporadic RCC with a clear cell histological subtype [154].
VHL has multiple functions and therefore it is involved in the ubiquitination and degradation of hypoxia-inducible-factor (HIF). Biochemical studies reported the evidence be-
tween the lacking of the wild type VHL and the overexpres-sion of HIF-1alpha and HIF-2alpha proteins under normoxic conditions, suggesting as the deregulation of HIF-alpha, and especially HIF-2alpha, contributes to VHL-defective renal carcinogenesis [155-157].
Hypoxic cells, or cells lacking pVHL, accumulate high
levels of HIF, which activates the transcription of a variety of
genes, including vascular endothelial growth factor (VEGF), platelet-derived
growth factor B (PDGF-B), and transform-
ing growth factor alpha (TGF-alpha). RNA polymerase II subunit POLR2G/RPB7 is also reported to be a target of VHL protein with the effect of stabilizing functions on vari-ous mRNA coding for proteins important in malignant trans-formation including VEGF and TGF-beta [157-159]. Moreo-ver, the expression of VHL gene is not statistically related to anti vascular endothelial growth factor (VEGF) target therapy effect (p=0.34). Conversely, patient with VHL inactivation had a greater response rate to VEGF target ther-apy (41% vs. 31% for those wild-type VHL), enabled to identify loss of function of VHL gene as an independent pre-dictive factor [160]. Moreover, overexpression of VEGF-A and cytoplasmic HIF-1 in tumor cells highlights a more aggressive subtype of RCC associated to clinicopathological characteristics as higher nuclear grade, larger tumor size, higher stage and shorter survival [161].
Table 4. Phase III Studies in mRCC
Target Investigators
and Reference
MSKCC
Risk Score
Setting Agent/Schedule Results
VEGFR, PDGFR, Raf.
Escudier B. et al. [179, 180]
Low and intermediate
Second line
(after IFN or IL2)
Sorafenib 400 mg BID vs. Placebo PFS: 5.5 vs. 2.8 months; P=0.000001.
OS: 17.8 vs. 15.2 months; P=0.146
[x]
ORR: 10% vs. 2%: P<0.001.
VEGFR, PDGFR,
Kit, CSF-1R, Flt-3.
Motzer RJ. et al. [182 ,183]
All First line Sunitinib 50 mg/d 4 wks on + 2 wks off vs. IFN-alpha 9MUI three times
weekly.
PFS: 11 vs. 5 months; P<0.001.
ORR: 31% vs. 6%; P<0.001
OS: 26.4 vs. 21.8 months; P=0.051 [x]
Escudier B. et al. [185]
(AVOREN)
All First line Bevacizumab 10 mg/Kg every two wks + IFN-alpha 9MUI three times
weekly vs. Placebo + IFN-alpha 9MUI three times weekly
PFS: 10.2 vs. 5.4 months; P<0.0001
OS: 23.3 vs. 21.3 months; P=0.129;
ORR: 31% vs. 13%; P<0.0001
VEGF
Rini BI. et al. [186] (CALGB),
All First line Bevacizumab 10 mg/Kg every two wks + IFN-alpha 9MUI three times
weekly vs. IFN-alpha 9MUI three times weekly
PFS: 8.5 vs. 5.2 months; P<0.0001.
OS: 18.3 vs. 17.4 months; P=0.069.
ORR: 25% vs. 13%; P<0.0001
Hudes G. et al. [172]
Poor First line Temsirolimus 25mg/ weekly vs. IFN-alpha 18 MUI three times weekly vs.
Temsirolimus 15mg/weekly + IFN-alpha 6 MUI three times weekly.
PFS: 5.5 vs. 3.1 v 4.7 months;
OS: 10.9 vs. 7.3 v 8.4 months;
ORR: 8.6% vs. 4.8% v 8.1%; P=0.002
mTOR
Motzer RJ. et al. [173]
All Second line
(after sorafenib, sunitinib, bevaci-
zumab, IL-2 or IFN)
Everolimus 10 mg/d vs. Placebo PFS: 4 vs. 1.9 months, P<0.0001;
OS: NA vs. 8.8 months
ORR: 1% vs. 0%
VEGFR, PDGFR, c-
kit
Sternberg CN. et al.37
All First/second line
(after one prior cytokine-based
therapy)
Pazopanib 800 mg/d vs. Placebo PFS overall: 9.2 vs. 4.2 months; P < 0.0001
OS: NA
ORR: 30% vs 3%

20 Current Cancer Drug Targets, 2010, Vol. 10, No. 4 Palazzo et al.
In the presence of high levels of oxygen, HIF-alpha is hydroxylated on one (or both) of two proline (Pro) residues by prolyl hydroxlase 2 (PHD2), generating a binding site for the von Hippel–Lindau protein (VHL). The recruitment of the VHL ubiquitin (Ub) ligase complex containing elongin C (ELC), elongin B (ELB), cullin 2 (CUL2) and RING-box protein 1 (RBX1) leads to the polyubiquitylation and protea-somal degradation of HIF-alpha. By contrast, with low oxy-gen, HIF-alpha subunit remain unmodified at the prolyl resi-dues, escape pVHL recognition and increase its intracellular values. Therefore, HIF-alpha binds HIF-beta subunits form-ing a heterodimer that translocate to the nucleus inducing changes in gene expression [162].
Considering the central role of HIF-1 transcription factor, it is clear that modulation of this activity could be a potent mechanism for the treatment of a wide range of hypoxia-related pathologies. Current approaches to inhibiting HIF-1alpha include the prevention of hypoxia-inducible factor (HIF)-1alpha from interaction with proteins that might modulate its activity, or inhibiting signal transduction path-ways. The inhibition of the activity of a HIF-1alpha deubiq-uitylation enzyme that controls the removal of ubiquitin would be another new approach to inhibiting HIF-1 [163]. Many HIF-1alpha inhibitor as antineoplastic antibiotics (gel-danamycin, tanespimycin), estradiol metabolites (2-methoxyestradiol), or chemotherapy agents (vincristine sul-fate) are under clinical investigation or were tested in pa-tients with RCC without relevant results [164], and actually there are not ongoing phase III trials.
The mTOR Function and Pathway in RCC
Several evidences have implicated mTOR (mammalian target of rapamycin) in the development of renal-cell carci-noma. The protein encoded by this gene belongs to a family of phosphatidylinositol kinase-related kinases that mediate cellular responses to stress, such as the DNA damage and the nutrient deprivation.
mTOR forms at least two distinct multi-protein com-plexes: mTORC1 and mTORC2. The increased expression of its efferctors (eIF4E and S6K1) occurs in many cancers, and makes a fundamental contributions to carcinogenesis by stimulating the expression of cancer-related genes at post-transcriptional levels and may adversely affect the prognosis of patients [165-167].
Rapamycin (also known as sirolimus) was originally found as an antifungal agent, and even today, it is not com-pletely understood how rapamycin perturbs mTOR function. The complex of rapamycin with its intracellular receptor FKBP12 binds directly to mTORC1 and, at least in vitro, suppresses mTORC1-mediated phosphorylation of the sub-strates S6K1 and 4EBP1. Therefore rapamycin and its ana-logues (temsirolimus and everolimus) inhibit several proc-esses that are relevant to the antitumor properties and exiting a modulating effect on cell proliferation, cell survival and angiogenesis [168-170].
The temsirolimus phase II trial for the treatment of renal cell carcinoma showed a major activity in a “poor risk” group of patient as defined by the Memorial Sloan-Kettering Cancer Center (MSKCC) parameters [171]. A phase III ran-
domized trial was conducted in this subset of patients ran-domized to receive IFN-alpha (18 x10^6 IU 3 times weekly) or temsirolimus (25 mg intravenous weekly), or IFN-alpha (6 x10^6 IU 3 times weekly) + temsirolimus (15 mg intrave-nous weekly). The median overall survival in the group treated with temsirolimus alone was 10.9 months, as com-pared with 7.3 and 8.4 months in the groups treated with IFN-alpha or combination therapy respectively. Median pro-gression-free survival times in these three groups were 5.5 vs. 3.1 vs. 4.7 months respectively. Combined therapy not improves OS or PFS, but increased the amount of toxic ad-verse events [172]. These data permitted the approvation of temsirolimus by FDA and EMEA in poor risk patients with advanced RCC.
The everolimus activity profile was shown in a small phase II trial involving patients who received two previous lines of therapies and who had progressed on tyrosine kinase inhibition therapy, reporting an interesting 10% of partial response rate with 74% of stable disease
35.
The subsequent phase III trial reported activity of ever-olimus in patient progressed to sunitinib, sorafenib or both, with a statistically and clinically significant improvement in PFS over placebo (4 vs. 1.9 months, P<0.0001) [173]. Based on this data everolimus was approved by the FDA and EMEA for the treatment of advanced RCC in patients who fail to respond to sunitinib or sorafenib.
Targeting Angiogenesis: The VEGF and PDGF-Beta
Functions and Pathways in Renal Cell Carcinoma
Kidney cancers are highly angiogenic and in the last dec-ade, many angiogenic factors with their receptors have been found to be expressed in RCC [174]. Vascular endothelial growth factor receptor (VEGFR) and platelet-derived growth factor receptor (PDGFR) play a key role in the pathogenesis of clear-cell carcinoma and therapeutic targeting of VEGF and related pathways has been studied [175, 176].
Expression, availability, and activity of VEGF are modu-lated by several signals product by cellular stress conditions or alteration such as hypoxia, oncogene and tumor suppres-sor dysregulation, transcription factors, inflammatory media-tors, and mechanical forces. Increased levels of HIF-alpha promote increased expression of vascular endothelial growth factor (VEGF) and platelet-derived growth factor (PDGF).
Substantial clinical activity has been reported for multi-ple tyrosin kinases inhibitors agents, leading to several clini-cal questions regarding their optimal use of them.
A recent final analysis of a large phase III trial about the value of plasmatic VEGF level before the treatment with sorafenib showed how VEGF act as a prognostic factor in a multivariate analyses including Memorial Sloan-Kettering Cancer Centre (MSKCC) score and ECOG PS, suggesting that patients with an high level of VEGF have a more ag-gressive tumor, but at the same time benefit from sorafenib therapy than patient with low-VEGF level (p=0.096) [177].
35Jac, J.; Amato, J.;, Giessinger, S.; Saxena, S.; Willis, J. P. A phase II study with a
daily regimen of the oral mTOR inhibitor RAD001 (everolimus) in patients with me-tastatic renal cell carcinoma which has progressed on tyrosine kinase inhibition ther-
apy. J. Clin. Oncol. 2008, 26, abstract LBA5026.

Targeted Therapy in Cancer Current Cancer Drug Targets, 2010, Vol. 10, No. 4 21
Sorafenib and sunitinib are both oral multitarget tyrosine kinase inhibitors (TKI) but each of then has a distinct target with different affinities and pharmacokinetics.
In a phase II randomized discontinuation trial, sorafenib showed a high progression free survival (PFS) on RCC pa-tients [178]. In a large phase III randomized trial, conducted on 903 patients refractory to a previous therapy by interferon alpha (IFN-alpha) or interleukin 2 (IL-2), it showed a sig-nificantly improvement PFS compared with patients ran-domly assigned to placebo (5.5 v 2.8 months, respectively; P=0.000001) [179]. The overall survival benefit was missed due to the crossover permitted for patients treated in the pla-cebo arm. Recent publication of the data regarding the over-all survival data showed that no statistically advantage was present in the analysis performed before and after the cross-over. The final analysis reported that overall survival was not increased by sorafenib therapy (17.8 v 15.2 months; P=0.146) [177].
Although sunitinib showed a response rate of 45% on re-nal cell carcinoma after a first line therapy with interferon-alpha or interleukin-2 [180], a major interest about this tyro-sine kinase inhibitor came from a randomized up-front, phase III trial comparing sunitinib with IFN-alpha [181]. This trial showed a higher response rate (31% vs. 6% respec-tively; P<0.001), and an increased PFS (11 vs. 5 months re-spectively HR=0.42; 95% CI, 0.32 to 0.54; P<0.001) over IFN-alpha arm. The updated results shows as sunitinib in-crease also the overall survival (26.4 vs. 21.8 months; P=0.051) representing, by now, the only drugs that meet this end point [182].
The management and treatment of non–clear cell renal cancer (papillary and chromophobe histological subtype), remain an open question. The clinical activity of both soraf-enib and sunitinib in this subset was supported by results from the expanded-access trials, although the molecular mechanisms of pathogenesis, between clear and non-clear cell, seem to be rather different [183]. Both sorafenib and sunitinib have been approved by FDA and EMEA for treat-ment of advanced RCC.
The results of a phase III trial on pazopanib were recently presented at ASCO 2009. A previous phase II trial, which enrolled cytokine naïve and refractory
(failed one prior cyto-
kine or bevacizumab-containing regimens) RCC
patients showed partial response in 40% and stable disease in 42% of patient evaluated by independent review after 12 weeks of therapy
36. In the phase III trial patients with clear cell ad-
vanced RCC and measurable disease, with no prior treatment or one prior cytokine-based treatment, were randomized to receive pazopanib 800 mg or placebo. PFS was significantly longer with pazopanib in the overall study population (9.2 vs. 4.2 months; P < 0.0000001), in treatment naïve (11.1 vs. 2.8 months; P < 0.0000001), and in cytokine-pretreated (7.4 vs. 4.2 months; P < 0.001) patients. The response rate was higher for pazopanib (30% vs 3% with placebo) and median duration of response was 58.7 weeks. The overall survival
3649- Hutson, T. E.; Davis, I. D.; Machiels, J. P.; de Souza, P. L.; Hong, B. F.; Rottey,
S.; Baker, K. L.; Crofts, T.; Pandite, L.; Figlin R. Pazopanib (GW786034) is active in metastatic renal cell carcinoma (RCC): Interim results of a phase II randomized discon-
tinuation trial (RDT). J. Clin. Oncol. 2007, 25, abstract 5031
results have not been presented yet37
. Pazopanib was ap-proved by FDA for treatment of patients with advanced RCC in October 2009.
Another strategy to inhibit angiogenesis is the possibility to target the circulating VEGF with bevacizumab. The activ-ity of this drug in metastatic RCC was proved by two multi-center randomized phase III trials. The AVOREN trial ran-domized patients to receive IFN-alpha 2a (9 x10^6 UI) and bevacizumab (10 mg/Kg IV every two weeks) versus IFN alpha 2a plus placebo. This trial reported a statistically sig-nificant difference in favor of the combination arm for PFS (10.2 vs. 5.4 months, P<0.0001) and for objective response rate (31% vs. 13%, P<0.0001). The overall survival, that was the primary endpoint of the study, was not reached for the early interruption after the interim analysis reporting the ad-vantage for bevacizumab and IFN-alpha arm [184]. Final overall survival data presented at ASCO 2009 did not show a statistically significant advantage for the bevacizumab over IFN-alpha arm (23.3 vs. 21.3 months P= 0.129). The second trial conducted by Cancer And Leukemia Group B (CALGB), randomized only North American patients to re-ceive IFN-alpha 2a (9 x10^6 IU 3 times weekly) and bevaci-zumab (10 mg/Kg IV every two weeks) versus IFN alpha 2a without placebo. This trial showed a statistically significant difference in favor of the combined treatment for PFS (8.5 vs. 5.2 months, P<0.0001) and response rate (25% vs. 13%, P<0.0001) [185] but, as for AVOREN trial, the combined treatment did not increased the overall survival (18.3 vs. 17.4 months, P=0.069)
38. An interesting observation from the
AVOREN trial is that patients requiring IFN-alpha dose re-duction showed no differences in terms of PFS compared to patients receiving full-dose of treatment [186]. Based on these findings combined therapy with bevacizumab and IFN alpha has been approved by FDA and EMEA in advanced renal cell carcinoma.
Future Considerations
RCC treatment accomplished great progress since the ap-proval of the first biological target drugs in 2006. Contrary to the past, today many drugs are available to treat RCC but the number of target agents, the improvement in survival and quality of life open some interesting questions: “have target agents a role in the prevention of the recurrence after nephrectomy and would be useful as adjuvant therapy in RCC?”, and “New molecules or combined target agent could increase the response rate and survival without worsening the toxicity?”, and “What is the better sequence to treat these patients?”
Recurrence rates in patients with localised RCC range from 35%
to 65% [187]. Clinical data about target therapy
based adjuvant therapy in RCC are still not available; but this area offers interesting question about the type, the timing
37Sternberg, C. N.; Szczylik, C.; Lee, E.; Salman, P. V.; Mardiak, J.; Davis, I. D.;
Pandite, L.; Chen, M.; McCann, L.; Hawkins R. A randomized, double-blind phase III
study of pazopanib in treatment-naive and cytokine-pretreated patients with advanced renal cell carcinoma. J. Clin. Oncol. 2009, 27, abstract 5021 38Rini, B. I.; Halabi, S.; Rosenberg, J.; Stadler, W. M.; Vaena, D. A.; Atkins, J. N.; Picus, J.; Czaykowski, P.; Dutcher, J.; Small, E. J. Bevacizumab plus interferon-alpha
versus interferon-alpha monotherapy in patients with metastatic renal cell carcinoma: Results of overall survival for CALGB 90206. J. Clin. Oncol. 2009, 27, abstract
LBA5019.

22 Current Cancer Drug Targets, 2010, Vol. 10, No. 4 Palazzo et al.
and the duration of target therapy. Clinical trials are ongoing to evaluate the efficacy of sorafenib (SORCE Trial) for 1 or 3 years or sunitinib (S-TRAC Trial) for 1 year versus pla-cebo, while the ASSURE trial compare the efficacy of sunit-inib or sorafenib versus placebo for one year.
In advanced/metastatic disease the possibility to increase the response rate and survival of these patients was attempt in combined target therapy as well as with new molecules. We are looking forward for the interest at the INTORACT (INvestigation of TORisel and Avastin Combination Ther-apy) phase III trial, that it comparing the combination of bevacizumab and temsirolimus to bevacizumab and Inter-feron-Alfa, but estimated date for the study completion is still far. An interesting study is also the evaluation of the new promising target therapy, pazopanib, by comparison with sunitinib in naïve patients with advanced RCC.
With the exception for sorafenib and everolimus, all other molecules currently utilized are evaluated in first line setting and actually there are no drugs able to prolong overall survival after progression to a first line. New agents that tar-get VEGF and mTOR pathways are emerging and have showed interesting clinical activity in phase II trial. Axitinib is an oral potent inhibitor of the intracellular tyrosine kinases portion of the VEGFR1, VEGFR2, PDGFR and c-KIT. A phase II study was conducted for this drugs, in patients with metastatic RCC refractory to IFN-alpha and IL-2 therapy; partial responses to treatment were reported in 44.2% of pa-tients with two complete responses [188]. Another trial re-ported its activity in 22.6% of patient who had received prior sorafenib therapy with median PFS and OS times of 7.4 and 13.6 months respectively [189]. Actually a phase III study designed to demonstrate if axitinib is superior to sorafenib in delaying tumor progression, in patients with metastatic RCC after failure of one systemic first-line regimen is ongoing.
With the availability of several target therapies on the best therapeutic is not known; is it better to target VEGF before mTOR or vice versa? Which is the better VEGF in-hibitor to start therapy? Limited retrospective experiences reported antitumor activity of sorafenib or sunitinib after a first line with antiangiogenic agents and a longer duration of disease control in patients who received sorafenib followed by sunitinib [190, 191]. A trial comparing the progression-free survival time of two the sequential therapy for first and second line therapy by sorafenib-sunitinib or by the inverse sequence (sunitinib followed by sorafenib) is ongoing. The future challenges in clinical management of RCC will be answer to these question and search other new and more ac-tive molecules, even if it is necessary to know better the po-tentiality of those molecules now available [192]. The indi-viduation of a predictive marker of response or the knowl-edge of the mechanisms of resistance to target agents, to elude them, seem to be distant goal in the future[193].
CONCLUSIONS
The rapidly expanding knowledge in tumor biology has encouraged optimism for the possibility to find and target tumor-specific mechanisms and thereby increase both effi-cacy and tolerance. A great number of ‘targeted drugs’ are being tested in clinical trials and up today many of these new
drugs are available for routine use in health care. Very en-couraging results have been obtained if administered when this target agents are used in combination chemotherapy, even if studies on combining target agents directed at poten-tially synergistic pathways are ongoing.
We might need to identify the specific target molecules in order to predict the desired effect, and it will required ad-ditional testing for validated biomarkers of targeted therapy efficacy and resistance, predictors of toxicity, and rational selection of companion cytotoxic drugs.
To optimally and efficiently develop of these drugs, we must overcome the knowledge gap on tightly interconnected biological pathways.
ABBREVIATIONS
VEGF = vascular endothelial growth factor
VEGFR = vascular endothelial growth factor receptor
EGFR = epidermal growth factor receptor
TK = tyrosine kinase
wks = weeks
PFS = progression free survival
OS = overall survival
ORR = overall response rate
NA = not applicable
d = daily
HD = high dose
LD = low dose
HER2 = epidermal growth factor receptor 2
HER2TK = epidermal growth factor receptor 2 tyrosine kinase domain
TH = docetaxel+trastuzumab
DC = docetaxel + carboplatin
AC = anthracyclin + cyclophosphamide
FEC = fluorouracil + epirubicin+ cyclophosphamide
VNR = vinorelbine
HR = hazard ratio
IFL = 5FU, leucovorin, irinotecan 4/6 weeks
FOLFOX-4 = oxaliplatin, 5FU, leucovorin days 1-2 every 2 weeks
FOLFOX-6 = oxaliplatin, 5FU, leucovorin days1 every 2 weeks
FOLFIRI = irinotecan, 5FU, leucovorin days 1-2 every 2 weeks
PDGFR = platelet derived growth factor receptor
Kit = stem cell factor receptor
Flt-3 = fms-related tyrosine kinase 3
CSF-1R = colony-stimulating factor-1 receptor

Targeted Therapy in Cancer Current Cancer Drug Targets, 2010, Vol. 10, No. 4 23
MSKCC = Memorial Sloan-Kettering Cancer Centre
IFN = interferon
IL2 = interleukin-2
MUI = million of international unit
DATABASES
Clinical Trial.gov: www.clinicaltrial.gov
National Cancer Institute: www.cancer.gov
Entrez Gene: http://www.ncbi.nlm.nih.gov/gene
REFERENCES
[1] Demetri, G. D.; von Mehren, M.; Blanke, C. D.; Van den Abbeele,
A. D.; Eisenberg, B.; Roberts, P. J.; Heinrich, M. C.; Tuveson, D.
A.; Singer, S.; Janicek, M.; Fletcher, J. A.; Silverman, S. G.; Sil-
berman, S. L.; Capdeville, R.; Kiese, B.; Peng, B.; Dimitrijevic, S.;
Druker, B. J.; Corless, C.; Fletcher, C. D.; Joensuu, H. Efficacy and
safety of imatinib mesylate in advanced gastrointestinal stromal
tumors. N. Engl. J. Med. 2002, 347, 472-480.
[2] Jordan, V. C.; Morrow, M. Tamoxifen, Raloxifene, and the Preven-
tion of Breast Cancer. Endocrine Reviews 1999, 20, 253-278.
[3] Beatson, G. T. On the treatment of inoperable cases of carcinoma
of the mamma: suggestions for a new method of treatment with il-
lustrative cases. Lancet 1896, 2, 104-107.
[4] Slamon, D. J.; Clark, G. M.; Wong, S. G.; Levin, W. J.; Ullrich, A.;
McGuire, W. L. Human breast cancer: correlation of relapse and
survival with amplification of the HER-2/neu oncogene. Science
1987, 235, 177-182.
[5] Slamon, D. J.; Leyland-Jones, B.; Shak, S.; Fuchs, H.; Paton, V.;
Bajamonde, A.; Fleming, T.; Eiermann, W.; Wolter, J.; Pegram,
M.; Baselga, J.; Norton, L. Use of chemotherapy plus a monoclonal
antibody against HER2 for metastatic breast cancer that overex-
presses HER2. N. Engl. J. Med. 2001, 344, 783-792.
[6] Jemal, A.; Siegel, R.; Ward, E.; Hao, Y.; Xu, J.; Thun, M. J. Cancer
Statistics, 2009. CA Cancer J. Clin. 2009, 59, 225-249.
[7] Lo, H. W.; Hsu, S. C.; Hung, M. C. EGFR signaling pathway in
breast cancers: from traditional signal transduction to direct nuclear
translocalization. Breast Cancer Res. Treat. 2006, 95, 211-218.
[8] Sporn, M. B.; Todaro, G. J. Autocrine secretion and malignant
transformation of cells. N. Engl. J. Med. 1980, 303, 878-880.
[9] Folkman, J. Tumor angiogenesis: therapeutic implications. N. Engl.
J. Med. 1971, 285, 1182-1186.
[10] Sun, S.; Schiller, J. H.; Gazdar, A. F. Lung cancer in never smokers
- a different disease. Nat. Rev. Cancer 2007, 7, 778-790.
[11] Franklin, W. A.; Veve, R.; Hirsch, F. R.; Helfrich, B. A.; Bunn Jr.,
P. A. Epidermal growth factor receptor family in lung cancer and
premalignancy. Semin. Oncol. 2002, 29, 3-14.
[12] Kanematsu, T.; Yano, S.; Uehara, H.; Bando, Y.; Sone, S. Phos-
phorylation, but not overexpression, of epidermal growth factor re-
ceptor is associated with poor prognosis of non-small cell lung can-
cer patients. Oncol. Res. 2003, 13, 289-298.
[13] Swinson, D. E. B.; Cox, G.; O’Byrne, K. J. Coexpression of epi-
dermal growth factor receptor with related factors is associated
with a poor prognosis in non-small-cell lung cancer. Br. J. Cancer.
2004, 91, 1301-1307.
[14] Onn, A.; Correa, A. M.; Gilcrease, M.; Isobe, T.; Massarelli, E.;
Bucana, C. D.; O'Reilly, M. S.; Hong, W. K.; Fidler, I. J.; Putnam,
J. B.; Herbst, R. S. Synchronous overexpression of epidermal
growth factor receptor and HER2-neu protein is a predictor of poor
outcome in patients with stage I non-small cell lung cancer. Clin.
Cancer Res. 2004, 10, 136-143.
[15] Giaccone, G.; Herbst, R. S.; Manegold, C.; Scagliotti, G.; Rosell,
R.; Miller, V.; Natale, R. B.; Schiller, J. H.; Von Pawel, J.; Pluzan-
ska, A.; Gatzemeier, U.; Grous, J.; Ochs, J. S.; Averbuch, S. D.;
Wolf, M. K.; Rennie, P.; Fandi, A.; Johnson, D. H. Gefitinib in
combination with gemcitabine and cisplatin in advanced non-small-
cell lung cancer: a phase III trial--INTACT 1. J. Clin. Oncol. 2004,
22, 777-784.
[16] Herbst, R. S.; Giaccone, G.; Schiller, J. H.; Natale, R. B.; Miller,
V.; Manegold, C.; Scagliotti, G.; Rosell, R.; Oliff, I.; Reeves, J. A.;
Wolf, M. K.; Krebs, A. D.; Averbuch, S. D.; Ochs, J. S.; Grous, J.;
Fandi, A.; Johnson, D. H. Gefitinib in combination with paclitaxel
and carboplatin in advanced non-small-cell lung cancer: a phase III
trial--INTACT 2. J. Clin. Oncol. 2004, 22, 785-794.
[17] Herbst, R. S.; Prager, D.; Hermann, R.; Fehrenbacher, L.; Johnson,
B. E.; Sandler, A.; Kris, M. G.; Tran, H. T.; Klein, P.; Li, X.;
Ramies, D.; Johnson, D. H., Miller, V. A. TRIBUTE: a phase III
trial of erlotinib hydrochloride (OSI-774) combined with car-
boplatin and paclitaxel chemotherapy in advanced non-small-cell
lung cancer. J. Clin. Oncol. 2005, 23, 5892-5899.
[18] Gatzemeier, U.; Pluzanska, A.; Szczesna, A.; Kaukel, E.; Roubec,
J.; De Rosa, F.; Milanowski, J.; Karnicka-Mlodkowski, H.; Pesek,
M.; Serwatowski, P.; Ramlau, R.; Janaskova, T.; Vansteenkiste, J.;
Strausz, J.; Manikhas, G. M.; Von Pawel, J. Phase III study of er-
lotinib in combination with cisplatin and gemcitabine in advanced
non-small-cell lung cancer: the Tarceva Lung Cancer Investigation
Trial. J. Clin. Oncol. 2007, 25, 1545-1552.
[19] Shepherd, F. A.; Rodrigues Pereira, J.; Ciuleanu, T.; Tan, E. H.;
Hirsh, V.; Thongprasert, S.; Campos, D.; Maoleekoonpiroj, S.;
Smylie, M.; Martins, R.; van Kooten, M.; Dediu, M.; Findlay, B.;
Tu, D.; Johnston, D.; Bezjak, A.; Clark, G.; Santabárbara, P.;
Seymour, L. Erlotinib in previously treated non-small-cell lung
cancer. N. Engl. J. Med. 2005, 353, 123-132.
[20] Cappuzzo, F.; Magrini, E.; Ceresoli, G. L.; Bartolini, S.; Rossi, E.;
Ludovini, V.; Gregorc, V.; Ligorio, C.; Cancellieri, A.; Damiani,
S.; Spreafico, A.; Paties, C. T.; Lombardo, L.; Calandri, C.;
Bellezza, G.; Tonato, M.; Crinò, L.; Akt phosphorylation and gefit-
inib efficacy in patients with advanced non-small-cell lung cancer.
J. Natl. Cancer Inst. 2004, 96, 1133-1141.
[21] Han, S. W.; Hwang, P. G.; Chung, D. H.; Kim, D. W.; Im, S. A.;
Kim, Y. T.; Kim Heo, D. S.; Bang, Y. J.; Kim, N. K. Epidermal
growth factor receptor (EGFR) downstream molecules as response
predictive markers for gefitinib (Iressa®, ZD1839) in chemother-
apy resistant non-small-cell lung cancer. Int. J. Cancer 2005, 113,
109-115.
[22] Lynch, T. J.; Bell, D. W.; Sordella, R.; Gurubhagavatula, S.; Oki-
moto, R. A.; Brannigan, B. W.; Harris, P. L.; Haserlat, S. M.;
Supko, J. G.; Haluska, F. G.; Louis, D. N.; Christiani, D. C.; Set-
tleman, J.; Haber, D. A. Activating mutations in the epidermal
growth factor receptor underlying responsiveness of non-small-cell
lung cancer to gefitinib. N. Engl. J. Med. 2004, 350, 2129-2139.
[23] Paez, J. G.; Jänne, P. A.; Lee, J. C.; Tracy, S.; Greulich, H.;
Gabriel, S.; Herman, P.; Kaye, F. J.; Lindeman, N.; Boggon, T. J.;
Naoki, K.; Sasaki, H.; Fujii, Y.; Eck, M. J.; Sellers, W. R.; John-
son, B. E.; Meyerson, M. EGFR mutations in lung cancer: correla-
tion with clinical response to gefitinib therapy. Science 2004, 304,
1497-500.
[24] Tsao, M. S.; Sakurada, A.; Cutz, J. C.; Zhu, C. Q.; Kamel-Reid, S.;
Squire, J.; Lorimer, I.; Zhang, T.; Liu, N.; Daneshmand, M.; Mar-
rano, P.; da Cunha Santos, G.; Lagarde, A.; Richardson, F.;
Seymour, L.; Whitehead, M.; Ding, K.; Pater, J.; Shepherd, F. A.
Erlotinib in lung cancer - molecular and clinical predictors of out-
come. N. Engl. J. Med. 2005, 353, 133-144.
[25] Jackman, D. M.; Miller, V. A.; Cioffredi, L. A.; Yeap, B. Y.;
Jänne, P. A.; Riely, G. J.; Ruiz, M. G.; Giaccone, G.; Sequist, L.
V.; Johnson, B. E. Impact of epidermal growth factor receptor and
KRAS mutations on clinical outcomes in previously untreated non-
small cell lung cancer patients: results of an online tumor registry
of clinical trials. Clin. Cancer Res. 2009, 15, 5267-5273.
[26] Mok, T. S.; Wu, Y. L.; Thongprasert, S.; Yang, C. H.; Chu, D. T.;
Saijo, N.; Sunpaweravong, P.; Han, B.; Margono, B.; Ichinose, Y.;
Nishiwaki, Y.; Ohe, Y.; Yang, J. J.; Chewaskulyong, B.; Jiang, H.;
Duffield, E. L.; Watkins, C. L.; Armour, A. A.; Fukuoka, M. Gefit-
inib or carboplatin–paclitaxel in pulmonary adenocarcinoma. N.
Engl. J. Med. 2009, 361, 947-957.
[27] Rosell, R.; Moran, T.; Queralt, C.; Porta, R.; Cardenal, F.; Camps,
C.; Majem, M.; Lopez-Vivanco, G.; Isla, D.; Provencio, M.; Insa,
A.; Massuti, B.; Gonzalez-Larriba, J. L.; Paz-Ares, L.; Bover, I.;
Garcia-Campelo, R.; Moreno, M. A.; Catot, S.; Rolfo, C.; Reguart,
N.; Palmero, R.; Sánchez, J. M.; Bastus, R.; Mayo, C.; Bertran-

24 Current Cancer Drug Targets, 2010, Vol. 10, No. 4 Palazzo et al.
Alamillo, J.; Molina, M. A.; Sanchez, J. J.; Taron, M. Screening for
epidermal growth factor receptor mutations in lung cancer. N. Engl.
J. Med. 2009, 361, 958-967.
[28] Shigematsu, H.; Gazdar, A. F. Somatic mutations of epidermal
growth factor receptor signaling pathway in lung cancers. Int. J.
Cancer 2006, 118, 257-262.
[29] Beau-Faller, M.; Legrain, M.; Voegeli, A. C.; Guérin, E.; Lavaux,
T.; Ruppert, A. M.; Neuville, A.; Massard, G.; Wihlm, J. M.;
Quoix, E.; Oudet, P.; Gaub, M. P. Detection of K-Ras mutations in
tumour samples of patients with non-small cell lung cancer using
PNA-mediated PCR clamping. Br. J. Cancer 2009, 100, 985-992.
[30] Eberhard, D. A.; Johnson, B. E.; Amler, L. C.; Goddard, A. D.;
Heldens, S. L.; Herbst, R. S.; Ince, W. L.; Jänne, P. A.; Januario,
T.; Johnson, D. H.; Klein, P.; Miller, V. A.; Ostland, M. A.;
Ramies, D. A.; Sebisanovic, D.; Stinson, J. A.; Zhang, Y. R.; Se-
shagiri, S.; Hillan, K. J. Mutations in the epidermal growth factor
receptor and in KRAS are predictive and prognostic indicators in
patients with non-small-cell lung cancer treated with chemotherapy
alone and in combination with erlotinib. J. Clin. Oncol. 2005, 23,
5900-5909.
[31] Gazdar, A. F. KRAS Mutations in Lung Cancer. America Society
Clinical Oncology, ASCO Educational Book 2007, pp. 433-435.
[32] Shigematsu, H.; Lin, L.; Takahashi, T.; Nomura, M.; Suzuki, M.;
Wistuba, I. I.; Fong, K. M.; Lee, H.; Toyooka, S.; Shimizu, N.; Fu-
jisawa, T.; Feng, Z.; Roth, J. A.; Herz, J.; Minna, J. D.; Gazdar, A.
F. Clinical and biological features associated with epidermal
growth factor receptor gene mutations in lung cancers. J. Natl.
Cancer Inst. 2005, 97, 339-346.
[33] Sordella, R.; Bell, D. W.; Haber, D. A.; Settleman, J. Gefitinib-
sensitizing EGFR mutations in lung cancer activate anti-apoptotic
pathways. Science 2004, 305, 1163-1167.
[34] Belani, C. P.; Schreeder, M. T.; Steis, R. G.; Guidice, R. A.;
Marsland, T. A.; Butler, E. H.; Ramalingam, S. S. Cetuximab in
combination with carboplatin and docetaxel for patients with me-
tastatic or advanced-stage nonsmall cell lung cancer: a multicenter
phase 2 study. Cancer 2008, 113, 2512-2517.
[35] Socinski, M. A.; Saleh, M. N.; Trent, D. F.; Dobbs, T. W.; Zehnge-
bot, L. M.; Levine, M. A.; Bordoni, R.; Stella, P. J. A randomized,
phase II trial of two dose schedules of car-
boplatin/paclitaxel/cetuximab in stage IIIB/IV non-small-cell lung
cancer (NSCLC). Ann. Oncol. 2009, 20, 1068-1073.
[36] Kim, E. S.; Mauer, A. M.; William, W. N., Jr.; Tran, H. T.; Liu, D.;
Lee, J. J.; Windt, P.; Hong, W. K.; Vokes, E. E.; Herbst, R. S. A
phase 2 study of cetuximab in combination with docetaxel in che-
motherapy-refractory/resistant patients with advanced nonsmall
cell lung cancer. Cancer 2009, 115, 1713-1722.
[37] Pirker, R.; Pereira, J. R.; Szczesna, A.; von Pawel, J.; Krzakowski,
M.; Ramlau, R.; Vynnychenko, I.; Park, K.; Yu, C. T.; Ganul, V.;
Roh, J. K.; Bajetta, E.; O'Byrne, K.; de Marinis, F.; Eberhardt, W.;
Goddemeier, T.; Emig, M.; Gatzemeier, U. Cetuximab plus chemo-
therapy in patients with advanced non-small-cell lung cancer
(FLEX): an open-label randomised phase III trial. Lancet 2009,
373, 1525-1531.
[38] Fontanini, G.; Lucchi, M.; Vignati, S.; Mussi, A.; Ciardiello, F.; De
Laurentiis, M.; De Placido, S.; Basolo, F.; Angeletti, C. A.;
Bevilacqua, G. Angiogenesis as a prognostic indicator of survival
in non-small-cell lung carcinoma: a prospective study. J. Natl.
Cancer Inst. 1997, 89, 881-886.
[39] Fontanini, G.; Vignati, S.; Boldrini, L.; Chine, S.; Silvestri, V.;
Lucchi, M.; Mussi, A.; Angeletti, C. A.; Bevilacqua, G. Vascular
endothelial growth factor is associated with neovascularization and
influences progression of non-small cell lung carcinoma. Clin.
Cancer Res. 1997, 3, 861-865.
[40] Macchiarini, P.; Fontanini, G.; Dulmet, E.; de Montpreville, V.;
Chapelier, A. R.; Cerrina, J.; Ladurie, F. L.; Dartevelle, P. G. An-
giogenesis: an indicator of metastasis in non-small cell lung cancer
invading the thoracic inlet. Ann. Thorac. Surg. 1994, 57, 1534-
1539.
[41] Volm, M.; Mattern, J.; Koomagi, R. Expression of platelet-derived
endothelial cell growth factor in non-small cell lung carcinomas:
relationship to various biological factors. Int. J. Oncol. 1998, 13,
975-979.
[42] Giatromanolaki, A.; Koukourakis, M.; O’Byrne, K.; Fox S.;
Whitehouse, R.; Talbot, D. C.; Harris, A. L.; Gatter, K. C. Prognos-
tic value of angiogenesis in operable non-small cell lung cancer. J.
Pathol. 1996, 179, 80-88.
[43] Chakra, M.; Pujol, J. L.; Lamy, P. J.; Bozonnat, M. C.; Quantin, X.;
Jacot, W.; Daurès, J. P. Circulating serum vascular endothelial
growth factor is not a prognostic factor of non-small cell lung can-
cer. J. Thorac. Oncol. 2008, 3, 1119-1126.
[44] O'Byrne, K. J.; Koukourakis, M. I.; Giatromanolaki, A.; Cox, G.;
Turley, H.; Steward, W. P.; Gatter, K.; Harris, A. L. Vascular endo-
thelial growth factor, platelet-derived endothelial cell growth factor
and angiogenesis in non-small-cell lung cancer. Br. J. Cancer
2000, 82, 1427-1432.
[45] Bremnes, R. M.; Camps, C.; Sirera, R. Angiogenesis in non-small
cell lung cancer: the prognostic impact of neoangiogenesis and the
cytokines VEGF and bFGF in tumours and blood. Lung Cancer
2006, 51, 143-158.
[46] Johnson, D. H.; Fehrenbacher, L.; Novotny, W. F.; Herbst, R. S.;
Nemunaitis, J. J.; Jablons, D. M.; Langer, C. J.; DeVore, R. F. 3rd.;
Gaudreault, J.; Damico, L. A.; Holmgren, E.; Kabbinavar, F.
Randomized phase II trial comparing bevacizumab plus carboplatin
and paclitaxel with carboplatin and paclitaxel alone in previously
untreated locally advanced or metastatic non-small-cell lung
cancer. J. Clin. Oncol. 2004, 22, 2184-2191.
[47] Sandler, A.; Gray, R.; Perry, M. C.; Brahmer, J.; Schiller, J. H.;
Dowlati, A.; Lilenbaum, R.; Johnson, D. H. Paclitaxel-carboplatin
alone or with bevacizumab for non-small-cell lung cancer. N. Engl.
J. Med. 2006, 355, 2542-2550.
[48] Reck, M.; von Pawel, J.; Zatloukal, P.; Ramlau, R.; Gorbounova,
V.; Hirsh, V.; Leighl, N.; Mezger, J., Archer, V.; Moore, N.;
Manegold, C. Phase III trial of cisplatin plus gemcitabine with ei-
ther placebo or bevacizumab as first-line therapy for nonsquamous
non-small-cell lung cancer: AVAil. J. Clin. Oncol. 2009, 27, 1227-
1234.
[49] Dowlati, A.; Gray, R.; Sandler, A. B.; Schiller, J. H.; Johnson, D.
H. Cell adhesion molecules, vascular endothelial growth factor, and
basic fibroblast growth factor in patients with non-small cell lung
cancer treated with chemotherapy with or without bevacizumab--an
Eastern Cooperative Oncology Group Study. Clin. Cancer Res.
2008, 14, 1407-1412.
[50] Gridelli, C.; Rossi, A.; Mongillo, F.; Bareschino, M.; Maione, P.;
Ciardiello, F. A randomized phase II study of soraf-
enib/gemcitabine or sorafenib/erlotinib for advanced non-small-cell
lung cancer in elderly patients or patients with a performance status
of 2: treatment rationale and protocol dynamics. Clin. Lung Cancer
2007, 8, 396-398.
[51] Adjei, A. A.; Molina, J. R.; Mandrekar, S. J.; Marks, R.; Reid, J.
R.; Croghan, G.; Hanson, L. J.; Jett, J. R.; Xia, C.; Lathia, C.;
Simantov, R. Phase I trial of sorafenib in combination with gefit-
inib in patients with refractory or recurrent non-small cell lung can-
cer. Clin. Cancer Res. 2007, 13, 2684-2691.
[52] Socinski, M. A.; Novello, S.; Brahmer. J. R.; Rosell, R.; Sanchez,
J. M.; Belani, C. P.; Govindan, R.; Atkins, J. N.; Gillenwater, H.
H.; Pallares, C.; Tye, L.; Selaru, P.; Chao. R. C.; Scagliotti, G. V.
Multicenter, phase II trial of sunitinib in previously treated, ad-
vanced non-small-cell lung cancer. J. Clin. Oncol. 2008, 26, 650-
656.
[53] Wedge, S. R.; Kendrew, J.; Hennequin, L. F.; Valentine, P. J.;
Barry, S. T.; Brave, S. R.; Smith, N. R.; James, N. H.; Dukes, M.;
Curwen, J. O.; Chester, R.; Jackson, J. A.; Boffey, S. J.; Kilburn, L.
L.; Barnett, S.; Richmond, G. H.; Wadsworth, P. F.; Walker, M.;
Bigley, A. L.; Taylor, S. T.; Cooper, L.; Beck, S.; Jürgensmeier, J.
M.; Ogilvie, D. J. AZD2171: A highly potent, orally bioavailable,
vascular endothelial growth factor receptor-2 tyrosine kinase in-
hibitor for the treatment of cancer. Cancer Res. 2005, 65, 4389-
4400.
[54] Goss, G.; Shepherd, F. A.; Laurie, S.; Gauthier, I.; Leighl, N.;
Chen, E.; Feld, R.; Powers, J.; Seymour, L. A phase I and pharma-
cokinetic study of daily oral cediranib, an inhibitor of vascular en-
dothelial growth factor tyrosine kinases, in combination with cis-
platin and gemcitabine in patients with advanced non-small cell
lung cancer: a study of the National Cancer Institute of Canada
Clinical Trials Group. Eur. J. Cancer 2009, 45, 782-788.
[55] Laurie, S. A.; Gauthier, I.; Arnold, A.; Shepherd, F. A.; Ellis, P.
M.; Chen, E.; Goss, G.; Powers, J.; Walsh, W.; Tu, D.; Robertson,
J.; Puchalski, T. A.; Seymour, L. Phase I and pharmacokinetic

Targeted Therapy in Cancer Current Cancer Drug Targets, 2010, Vol. 10, No. 4 25
study of daily oral AZD2171, an inhibitor of vascular endothelial
growth factor tyrosine kinases, in combination with carboplatin and
paclitaxel in patients with advanced non-small-cell lung cancer: the
National Cancer Institute of Canada clinical trials group. J. Clin.
Oncol. 2008, 26, 1871-1878.
[56] Natale, R. B.; Bodkin, D.; Govindan, R.; Sleckman, B. G.; Rizvi,
N. A.; Capó, A.; Germonpré, P.; Eberhardt, W. E.; Stockman, P.
K.; Kennedy, S. J.; Ranson, M. Vandetanib Versus Gefitinib in Pa-
tients With Advanced Non–Small-Cell Lung Cancer: Results From
a Two-Part, Double-Blind, Randomized Phase II Study. J. Clin.
Oncol. 2009, 27, 2523-2529.
[57] Heymach, J. V.; Johnson, B. E.; Prager, D.; Csada, E.; Roubec, J.;
Pesek, M.; Spásová, I.; Belani, P.; Bodrogi, I.; Gadgeel, S.; Ken-
nedy, S. J.; Hou, J.; Herbst, R. S. Randomized, placebo-controlled
phase II study of vandetanib plus docetaxel in previously treated
non small-cell lung cancer. J. Clin. Oncol. 2007, 25, 4270-4277.
[58] Heymach, J. V.; Paz-Ares, L.; De Braud, F.; Sebastian, M.; Stew-
art, D. J.; Eberhardt, W. E.; Ranade, A. A.; Cohen, G.; Trigo, J. M.;
Sandler, A. B.; Bonomi, P. D.; Herbst, R. S.; Krebs, A. D.; Vas-
selli, J.; Johnson, B. E. Randomized phase II study of vandetanib
alone or with paclitaxel and carboplatin as first-line treatment for
advanced non–small-cell lung cancer. J. Clin. Oncol. 2008, 26,
5407-5415.
[59] Delbaldo, C.; Michiels, S.; Rolland, E.; Syz, N; Soria, J-C.; Le
Chevalier, T.; Pignon, J. P. Second or third additional chemother-
apy drug for non-small cell lung cancer in patients with advanced
disease. Cochrane Database of Systematic Reviews 2007, 4, Art.
No. CD004569.
[60] Sørlie, T.; Perou, C. M.; Tibshirani, R.; Aas, T.; Geisler, S.; John-
sen, H.; Hastie, T.; Eisen, M. B.; van de Rijn, M.; Jeffrey, S. S.;
Thorsen, T.; Quist, H.; Matese, J. C.; Brown, P. O.; Botstein, D.;
Eystein Lønning, P.; Børresen-Dale, A. L. Gene expression pat-
terns of breast carcinomas distinguish tumor subclasses with clini-
cal implications. PNAS 2001, 98, 10869-10874.
[61] Cohen, S. Isolation of a mouse submaxillary gland protein acceler-
ating incisor eruption and eyelid opening in the new-born animal. J.
Biol. Chem. 1962, 237, 1555-1562.
[62] Berger, M. S.; Locher, G. W.; Saurer, S.; Gullick, W. J.; Water-
field, M. D.; Groner, B.; Hynes, N. E. Correlation of c-erbB-2 gene
amplification and protein expression in human breast carcinoma
with nodal status and nuclear grading. Cancer Res. 1988, 48, 1238-
1243.
[63] Slamon, D. J.; Godolphin, W.; Jones, L. A.; Holt, J. A.; Wong, S.
G.; Keith, D. E.; Levin, W. J.; Stuart, S. G.; Udove, J.; Ullrich, A.;
et al. Studies of the HER-2/neu proto-oncogene in human breast
and ovarian cancer. Science 1989, 244, 707-712.
[64] Chia, S.; Norris, B.; Speers, C.; Cheang, M.; Gilks, B.; Gown, A.
M.; Huntsman, D.; Olivotto, I. A.; Nielsen, T. O.; Gelmon, K. Hu-
man epidermal growth factor receptor 2 overexpression as a prog-
nostic factor in a large tissue microarray series of node-negative
breast cancers. J. Clin. Oncol. 2008, 26, 5697-5704.
[65] Ross, J. S.; Slodkowska, E. A.; Symmans, W. F.; Pusztai, L.;
Ravdin, P. M.; Hortobagyi, G. N. The her-2 receptor and breast
cancer: ten years of targeted anti–her-2 therapy and personalized
medicine. The Oncologist 2009, 14, 320-368.
[66] Brennan, P. J.; Kumogai, T.; Berezov, A.; Murali, R.; Greene, M. I.
HER2/Neu: mechanisms of dimerization/oligomerization. Onco-
gene 2000, 19, 6093-6101.
[67] Tzahar, E.; Waterman, H.; Chen, X.; Levkowitz, G.; Karunagaran,
D.; Lavi, S.; Ratzkin, B. J.; Yarden, Y. A hierarchical network of
interreceptor interactions determines signal transduction by Neu
differentiation factor/neuregulin and epidermal growth factor. Mol.
Cell. Biol. 1996, 16, 5276-5287.
[68] Roskoski, R. Jr. The ErbB/HER receptor protein-tyrosine kinases
and cancer. Biochem. Biophys. Res. Commun. 2004, 319, 1-11.
[69] Holbro, T.; Beerli, R. R.; Maurer, F.; Koziczak, M.; Barbas, C. F.;
Hynes, N. E. The ErbB2/ErbB3 heterodimer functions as an onco-
genic unit: ErbB2 requires ErbB3 to drive breast tumor cell prolif-
eration. PNAS 2003, 100, 8933-8938.
[70] Cho, H. S.; Mason, K.; Ramyar, K. X.; Stanley, A. M.; Gabelli, S.
B.; Denney, D. W. Jr.; Leahy, D. J. Structure of the extracellular
region of HER2 alone and in complex with the Herceptin Fab. Na-
ture 2003, 421, 756-760.
[71] Neve, R. M.; Sutterlüty, H.; Pullen, N.; Lane, H. A.; Daly, J. M.;
Krek, W.; Hynes, N. E. Effects of oncogenic ErbB2 on G1 cell cy-
cle regulators in breast tumour cells. Oncogene 2000, 19, 1647-
1656.
[72] Lane, H. A.; Beuvink, I.; Motoyama, A. B.; Daly, J. M.; Neve, R.
M.; Hynes, N. E. ErbB2 potentiates breast tumor proliferation
through modulation of p27(Kip1)-Cdk2 complex formation: recep-
tor overexpression does not determine growth dependency. Mol.
Cell. Biol. 2000, 20, 3210-3223.
[73] Yakes, F. M.; Chinratanalab, W.; Ritter, C, A.; King, W.; Seelig,
S.; Arteaga, C. L. Herceptin-induced inhibition of phosphatidyli-
nositol-3 kinase and Akt Is required for antibody-mediated effects
on p27, cyclin D1, and antitumor action. Cancer Res. 2002, 62,
4132-4141.
[74] Münster, P. N.; Marchion, D. C.; Basso, A. D.; Rosen, N. Degrada-
tion of HER2 by ansamycins induces growth arrest and apoptosis in
cells with HER2 overexpression via a HER3, phosphatidylinositol
3'-kinase-AKT-dependent pathway. Cancer Res. 2002, 62, 3132-
3137.
[75] Pegram, M. D.; Konecny, G. E.; O’Callaghan, C.; Beryt, M.;
Pietras, R.; Slamon, D. J. Rational combinations of trastuzumab
with chemotherapeutic drugs used in the treatment of breast cancer.
J. Natl. Cancer Inst. 2004, 96, 739-749.
[76] Mayfield, S.; Vaughn, J. P.; Kute, T. E. DNA strand breaks and cell
cycle perturbation in Herceptin treated breast cancer cell lines.
Breast Cancer Res. Treat. 2001, 70, 123-129.
[77] Untch, M.; Rezai, M.; Loibl, S.; Fasching, P. A.; Huober, J.; Tesch,
H.; Bauerfeind, I.; Hilfrich, J.; Eidtmann, H.; Gerber, B.; Hanusch,
C.; Kühn, T.; du Bois, A.; Blohmer, J. U.; Thomssen, C.; Dan
Costa, S.; Jackisch, C.; Kaufmann, M.; Mehta, K.; von Minckwitz,
G. Neoadjuvant Treatment With Trastuzumab in HER2-Positive
Breast Cancer: Results From the GeparQuattro Study. J. Clin. On-
col. 2010, in press.
[78] Piccart-Gebhart, M. J.; Procter, M.; Leyland-Jones, B.; Goldhirsch,
A.; Untch, M.; Smith, I.; Gianni, L.; Baselga, J.; Bell, R.; Jackisch,
C.; Cameron, D.; Dowsett, M.; Barrios, C. H.; Steger, G.; Huang,
C. S.; Andersson, M.; Inbar, M.; Lichinitser, M.; Láng, I.; Nitz, U.;
Iwata, H.; Thomssen, C.; Lohrisch, C.; Suter, T. M.; Rüschoff, J,;
Suto, T.; Greatorex, V.; Ward, C.; Straehlem, C.; McFadden, E.;
Dolci, M. S.; Gelber, R. D. Trastuzumab after adjuvant chemother-
apy in HER2-positive breast cancer. N. Engl. J. Med. 2005, 353,
1659-1672.
[79] Marty, M.; Cognetti, F.; Maraninchi, D.; Snyder, R.; Mauriac, L.;
Tubiana-Hulin, M.; Chan, S.; Grimes, D.; Antón, A.; Lluch, A.;
Kennedy, J.; O'Byrne, K.; Conte, P.; Green, M.; Ward, C.; Mayne,
K.; Extra, J. M. Randomized phase II trial of the efficacy and safety
of trastuzumab combined with docetaxel in patients with human
epidermal growth factor receptor 2-positive metastatic breast can-
cer administered as first-line treatment: The M77001 study group.
J. Clin. Oncol. 2005, 23, 4265-4274.
[80] McKeage, K.; Perry, C. M. Trastuzumab: a review of its use in the
treatment of metastatic breast cancer overexpressing HER2. Drugs
2002, 62, 209-243.
[81] Pegram, M.; Hsu, S.; Lewis, G.; Pietras, R.; Beryt, M.; Sliwkowski,
M.; Coombs, D.; Baly, D.; Kabbinavar, F.; Slamon, D. Inhibitory
effects of combinations of HER-2/neu antibody and chemothera-
peutic agents used for treatment of human breast cancers. Onco-
gene 1999, 18, 2241-2251.
[82] Nielsen, D. L.; Andersson, M.; Kamby, C. HER2-targeted therapy
in breast cancer. Monoclonal antibodies and tyrosine kinase inhibi-
tors. Cancer Treat. Rev. 2009, 35, 121-136.
[83] Romond, E. H.; Perez, E. A.; Bryant, J.; Suman, V. J.; Geyer, C. E.
Jr.; Davidson, N. E.; Tan-Chiu, E.; Martino, S.; Paik, S.; Kaufman,
P. A.; Swain, S. M.; Pisansky, T. M.; Fehrenbacher, L.; Kutteh, L.
A.; Vogel, V. G.; Visscher, D. W.; Yothers, G.; Jenkins, R. B.;
Brown, A. M.; Dakhil, S. R.; Mamounas, E. P.; Lingle, W. L.;
Klein, P. M.; Ingle, J. N.; Wolmark, N. Trastuzumab plus adjuvant
chemotherapy for operable HER2-positive breast cancer. N. Engl.
J. Med. 2005, 353, 1673-1684.
[84] Joensuu, H.; Kellokumpu-Lehtinen, P. L.; Bono, P.; Alanko, T.;
Kataja, V.; Asola, R.; Utriainen, T.; Kokko, R.; Hemminki, A.;
Tarkkanen, M.; Turpeenniemi-Hujanen, T.; Jyrkkiö, S.; Flander,
M.; Helle, L.; Ingalsuo, S.; Johansson, K.; Jääskeläinen, A. S.; Pa-
junen, M.; Rauhala, M.; Kaleva-Kerola, J.; Salminen, T.; Leinonen,

26 Current Cancer Drug Targets, 2010, Vol. 10, No. 4 Palazzo et al.
M.; Elomaa, I.; Isola, J. Adjuvant docetaxel or vinorelbine with or
without trastuzumab for breast cancer. N. Engl. J. Med. 2006, 354,
809-820.
[85] Goldhirsch, A.; Ingle, J. N.; Gelber, R. D.; Coates, A. S.; Thürli-
mann, B.; Senn, H. J. Thresholds for therapies: highlights of the St
Gallen international expert consensus on the primary therapy of
early breast cancer 2009. Ann. Oncol. 2009, 20, 1319-1329.
[86] Smith, I.; Procter, M.; Gelber, R. D.; Guillaume, S.; Feyereislova,
A.; Dowsett, M.; Goldhirsch, A.; Untch, M.; Mariani, G.; Baselga,
J.; Kaufmann, M.; Cameron, D.; Bell, R.; Bergh, J.; Coleman, R.;
Wardley, A.; Harbeck, N.; Lopez, R. I.; Mallmann, P.; Gelmon, K.;
Wilcken, N.; Wist, E.; Sánchez Rovira, P.; Piccart-Gebhart, M. J.
2-year follow-up of trastuzumab after adjuvant chemotherapy in
HER2-positive breast cancer: a randomised controlled trial. Lancet
2007, 369, 29-36.
[87] Buzdar, A. U.; Valero, V.; Ibrahim, N. K.; Francis, D.; Broglio, K.
R.; Theriault, R. L.; Pusztai, L.; Green, M. C.; Singletary, S. E.;
Hunt, K. K.; Sahin, A. A.; Esteva, F.; Symmans, W. F.; Ewer, M.
S.; Buchholz, T. A.; Hortobagyi, G. N. Neoadjuvant therapy with
paclitaxel followed by 5-fluorouracil, epirubicin, and cyclophos-
phamide chemotherapy and concurrent trastuzumab in human epi-
dermal growth factor receptor 2-positive operable breast cancer: An
update of the initial randomized study population and data of addi-
tional patients treated with the same regimen. Clin. Cancer Res.
2007, 13, 228-233.
[88] Keniry, M.; Parsons, R. The role of PTEN signaling perturbations
in cancer and in targeted therapy. Oncogene 2008, 27, 5477-5485.
[89] Saal, L. H.; Holm, K.; Maurer, M.; Memeo, L.; Su, T.; Wang, X.;
Yu, J. S.; Malmström, P. O.; Mansukhani, M.; Enoksson, J.; Hib-
shoosh, H.; Borg, A.; Parsons, R. PIK3CA mutations correlate with
hormone receptors, node metastasis, and ERBB2, and are mutually
exclusive with PTEN loss in human breast carcinoma. Cancer Res.
2005, 65, 2554-2559.
[90] Simoncini, T.; Hafezi-Moghadam, A.; Brazil, D. P.; Ley, K.; Chin,
W. W.; Liao, J. K. Interaction of oestrogen receptor with the regu-
latory subunit of phosphatidylinositol-3-OH kinase. Nature 2000,
407, 538-541.
[91] Patrick, J.; Goodin, M.; Goodin, S. Lapatinib: a dual inhibitor of
human epidermal growth factor receptor tyrosine kinases. Clin.
Ther. 2008, 30, 1427-1447.
[92] Geyer, C. E.; Forster, J.; Lindquist, D.; Chan, S.; Romieu, C. G.;
Pienkowski, T.; Jagiello-Gruszfeld, A.; Crown, J.; Chan, A.;
Kaufman, B.; Skarlos, D.; Campone, M.; Davidson, N.; Berger, M.;
Oliva, C.; Rubin, S. D.; Stein, S.; Cameron, D. Lapatinib plus
capecitabine for HER2-positive advanced breast cancer. N. Engl. J.
Med. 2006, 355, 2733-2743.
[93] Cameron, D.; Casey, M.; Press, M.; Lindquist, D.; Pienkowski, T.;
Romieu, C. G.; Chan, S.; Jagiello-Gruszfeld, A.; Kaufman, B.;
Crown, J.; Chan, A.; Campone, M.; Viens, P.; Davidson, N.; Gor-
bounova, V.; Raats, J. I.; Skarlos, D.; Newstat, B.; Roychowdhury,
D.; Paoletti, P.; Oliva, C.; Rubin, S.; Stein, S.; Geyer, C. E. A
phase III randomized comparison of lapatinib plus capecitabine
versus capecitabine alone in women with advanced breast cancer
that has progressed on trastuzumab: Updated efficacy and bio-
marker analyses. Breast Cancer Res. Treat. 2008, 112, 533-543.
[94] Lin, N. U.; Carey, L. A.; Liu, M. C.; Younger, J.; Come, S. E.;
Ewend, M.; Harris, G. J.; Bullitt, E.; Van den Abbeele, A. D.; Hen-
son, J. W.; Li, X.; Gelman, R.; Burstein, H. J.; Kasparian, E.;
Kirsch D. G.; Crawford, A.; Hochberg, F.; Winer, E. P. Phase II
trial of lapatinib for brain metastases in patients with human epi-
dermal growth factor receptor 2-positive breast cancer. J. Clin. On-
col. 2008, 26, 1993-1999.
[95] Moy, B.; Goss, P. E. TEACH: Tykerb evaluation after chemother-
apy. Clin. Breast Cancer 2007, 7, 489-492.
[96] Newby, J. C.; Johnston, S. R.; Smith, I. E.; Dowsett, M. Expression
of epidermal growth factor receptor and c-erbB2 during the devel-
opment of tamoxifen resistance in human breast cancer. Clin. Can-
cer Res. 1997, 3, 1643-1651.
[97] Atalay, G.; Cardoso, F.; Awada, A.; Piccart, M. J. Novel therapeu-
tic strategies targeting the epidermal growth factor receptor
(EGFR) family and its downstream effectors in breast cancer. Ann.
Oncol. 2003, 14, 1346-1363.
[98] Chu, I.; Blackwell, K.; Chen, S.; Slingerland, J. The dual
ErbB1/ErbB2 inhibitor, lapatinib (GW572016), cooperates with
tamoxifen to inhibit both cell proliferation- and estrogen dependent
gene expression in antiestrogen-resistant breast cancer. Cancer Res.
2005, 65, 18-25.
[99] Johnston, S.; Pippen, J. Jr.; Pivot, X.; Lichinitser, M.; Sadeghi, S.;
Dieras, V.; Gomez, H. L.; Romieu, G.; Manikhas, A.; Kennedy, M.
J.; Press, M. F.; Maltzman, J.; Florance, A.; O'Rourke, L.; Oliva,
C.; Stein, S.; Pegram, M. Lapatinib Combined With Letrozole Ver-
sus Letrozole and Placebo As First-Line Therapy for Postmeno-
pausal Hormone Receptor-Positive Metastatic Breast Cancer. J.
Clin. Oncol. 2009, 27, 5538-5546.
[100] Johnston, S. R. D. Clinical Efforts to Combine Endocrine Agents
with Targeted Therapies against EpidermalGrowth FactorRecep-
tor/Human Epidermal Growth Factor Receptor 2 and Mammalian
Target of Rapamycin in Breast Cancer. Clin. Cancer Res. 2006, 12,
1061-1068.
[101] Reid, A.; Vidal, L.; Shaw, H.; De Bono, J. Dual inhibition of
ErbB1 (EGFR/HER1) and ErbB2 (HER2/neu). Eur. J. Cancer
2007, 43, 481-489.
[102] Friedländer, E.; Barok, M.; Szöllosi, J.; Vereb, G. ErbB-directed
immunotherapy: Antibodies in current practice and promising new
agents. Immunol. Lett. 2008, 116, 126-140.
[103] Agus, D. B.; Gordon, M. S.; Taylor, C.; Natale, R. B.; Karlan, B.;
Mendelson, D. S.; Press, M. F.; Allison, D. E.; Sliwkowski, M. X.;
Lieberman, G.; Kelsey, S. M.; Fyfe, G. Phase I clinical study of
pertuzumab, a novel HER dimerization inhibitor, in patients with
advanced cancer. J. Clin. Oncol. 2005, 23, 2534-2543.
[104] Wong, K. K.; Fracasso, P. M.; Bukowski, R. M.; Lynch, T. J.;
Munster, P. N.; Shapiro, G. I.; Jänne, P. A.; Eder, J. P.; Naughton,
M. J.; Ellis, M. J.; Jones, S. F.; Mekhail, T.; Zacharchuk, C.; Ver-
mette, J.; Abbas, R.; Quinn, S.; Powell, C.; Burris, H. A. A phase I
study with neratinib (HKI-272), an irreversible pan ErbB receptor
tyrosine kinase inhibitor, in patients with solid tumors. Clin. Can-
cer Res. 2009, 15, 2552-2558.
[105] Hynes, N. E.; MacDonald, G. ErbB receptors and signaling path-
ways in cancer. Curr. Opin. Cell Biology 2009, 21, 177-184.
[106] Schmelzle, T.; Hall, M. N. TOR, a central controller of cell growth.
Cell 2000, 103, 253-262.
[107] Johnston S. R. D. Role of the mTOR Pathway in Endocrine resis-
tant Breast Cancer Opportunities for Novel Combination Strategies.
Educational Book, 2009 by American Society of Clinical Oncol-
ogy.
[108] Chan, S.; Scheulen, M. E.; Johnston, S.; Mross, K.; Cardoso, F.;
Dittrich, C.; Eiermann, W.; Hess, D.; Morant, R.; Semiglazov, V.;
Borner, M.; Salzberg, M.; Ostapenko, V.; Illiger, H. J.; Behringer,
D.; Bardy-Bouxin, N.; Boni, J.; Kong, S.; Cincotta, M.; Moore, L.
Phase II study of temsirolimus (CCI-779), a novel inhibitor of
mTOR, in heavily pretreated patients with locally advanced or me-
tastatic breast cancer. J. Clin. Oncol. 2005, 23, 5314-5322.
[109] Kurokawa, H.; Arteaga, C. L. ErbB (HER) receptors can abrogate
antiestrogen action in human breast cancer by multiple signaling
mechanisms. Clin. Cancer Res. 2003, 9, 511-515S.
[110] Lu, C. H.; Wyszomierski, S. L.; Tseng, L. M.; Sun, M. H.; Lan, K.
H.; Neal, C. L.; Mills, G. B.; Hortobagyi, G. N.; Esteva, F. J.; Yu,
D. Preclinical testing of clinically applicable strategies for over-
coming trastuzumab resistance caused by PTEN deficiency. Clin.
Cancer Res. 2007, 13, 5883–5888.
[111] Finn, R. S. Targeting Src in breast cancer. Ann. Oncol. 2008, 19,
1379-1386.
[112] Di Cosimo, S.; Baselga, J. Targeted therapies in breast cancer:
Where are we now? Eur. J. Cancer 2008, 44, 2781-2790.
[113] Li, T.; Christos, P.J.; Sparano, J. A.; Hershman, D. L.; Hoschander,
S.; O'Brien, K.; Wright, J. J.; Vahdat, L. T. Phase II trial of the far-
nesyltransferase inhibitor tipifarnib plus fulvestrant in hormone re-
ceptor-positive metastatic breast cancer: New York Cancer Consor-
tium Trial P6205. Ann. Oncol. 2009, 20, 642-647.
[114] Johnston, S. R.; Semiglazov, V. F.; Manikhas, G. M.; Spaeth, D.;
Romieu, G.; Dodwell, D. J.; Wardley, A. M.; Neven, P.; Bessems,
A.; Park, Y. C.; De Porre, P. M.; Perez Ruixo, J. J.; Howes, A. J. A
phase II, randomized, blinded study of the farnesyltransferase in-
hibitor tipifarnib combined with letrozole in the treatment of ad-
vanced breast cancer after antiestrogen therapy. Breast Cancer Res.
Treat. 2008, 110, 327-335.
[115] Deann, P.; Atchley, C.; Albarracin, T.; Lopez, A.; Valero, V.;
Amos, C. I.; Gonzalez-Angulo, A. M.; Hortobagyi, G. N.; Arun, B.

Targeted Therapy in Cancer Current Cancer Drug Targets, 2010, Vol. 10, No. 4 27
K. Clinical and pathologic characteristics of patients with BRCA-
positive and BRCA-negative breast cancer. J. Clin. Oncol. 2008,
26, 4282-4288.
[116] Drew, Y.; Calvert, H. The Potential of PARP Inhibitors in Genetic
Breast and Ovarian Cancers. Ann. N. Y. Acad. Sci. 2008, 1138, 136-
145.
[117] Lord, C. J.; Ashworth, A. Targeted therapy for cancer using PARP
inhibitors. Curr. Opin. Pharmacol. 2008, 8, 363-369.
[118] Harbeck, N. Clinical data for anti-angiogenic agents in previously
treated advanced breast cancer. Eur. J. Cancer Suppl. 2008, 6, 14-
20.
[119] Sledge, G. W. VEGF-Targeting Therapy for Breast Cancer. J.
Mammary Gland. Biol. Neoplasia. 2005, 10, 319-323.
[120] Marty, M.; Pivot, X. The potential of anti-vascular endothelial
growth factor therapy in metastatic breast cancer: Clinical experi-
ence with anti-angiogenic agents, focusing on bevacizumab. Eur. J.
Cancer 2008, 44, 912-920.
[121] Miller, K. D.; Chap, L. I.; Holmes, F. A.; Cobleigh, M. A.; Mar-
com, P. K.; Fehrenbacher, L.; Dickler, M.; Overmoyer, B. A.; Re-
imann, J. D.; Sing, A. P.; Langmuir, V.; Rugo, H. S. Randomized
phase III trial of capecitabine compared with bevacizumab plus
capecitabine in patients with previously treated metastatic breast
cancer. J. Clin. Oncol. 2005, 23, 792-799.
[122] Miller, K.; Wang, M.; Gralow, J.; Dickler, M.; Cobleigh, M.;
Perez, E. A.; Shenkier, T.; Cella, D.; Davidson, N.E. Paclitaxel plus
bevacizumab versus paclitaxel alone for metastatic breast cancer.
N. Engl. J. Med. 2007, 357, 2666-2676.
[123] Takahashi, Y.; Tucker, S. L.; Kitadai, Y.; Koura, A. N.; Bucana, C.
D.; Cleary, K. R.; Ellis, L. M. Vessel counts and expression of vas-
cular endothelial growth factor as prognostic factors in node-
negative colon cancer. Arch. Surg. 1997, 132, 541-546.
[124] Takahashi, Y.; Kitadai, Y.; Bucana, C. D.; Cleary, K. R.; Ellis, L.
M. Expression of vascular endothelial growth factor and its recep-
tor, KDR, correlates with vascularity, metastasis, and proliferation
of human colon cancer. Cancer Res. 1995, 55, 3964-3968.
[125] Brown, L. F.; Berse, B.; Jackman, R. W.; Tognazzi, K.; Guidi, A.
J.; Dvorak H. F.; Senger, D. R.; Connolly, J. L.; Schnitt S. J. Ex-
pression of vascular permeability factor (vascular endothelial
growth factor) and its receptors in breast cancer. Hum. Pathol.
1995, 26, 86-91.
[126] Guidi, A. J.; Abu-Jawdeh, G.; Berse, B.; Jackman, R. W.; Tog-
nazzi, K.; Dvorak, H. F.; Brown, L. F. Vascular permeability factor
(vascular endothelial growth factor) expression and angiogenesis in
cervical neoplasia. J. Natl. Cancer. Inst. 1995, 87, 1237-1245.
[127] Wong, M. P.; Cheung, N.; Yuen, S. T.; Leung, S. Y.; Chung, L. P.
Vascular endothelial growth factor is up-regulated in the early pre-
malignant stage of colorectal tumour progression. Int. J. Cancer
1999, 81, 845-850.
[128] Ellis, L. M.; Takahashi, Y.; Liu, W.; Shaheen, R. M. Vascular
endothelial growth factor in human colon cancer: biology and
therapeutic implications. Oncologist 2000, 5, 11-15.
[129] Cascinu, S.; Staccioli, M. P.; Gasparini, G.; Giordani, P.; Catalano,
V.; Ghiselli, R.; Rossi, C.; Baldelli, A. M.; Graziano, F.; Saba, V.;
Muretto, P.; Catalano, G. Expression of vascular endothelial
growth factor can predict event-free survival in stage II colon can-
cer. Clin. Cancer Res. 2000, 6, 2803-2807.
[130] Fan, F.; Wey, J. S.; McCarty, M. F.; Belcheva, A.; Liu, W.; Bauer,
T. W.; Somcio, R. J.; Wu, Y.; Hooper, A.; Hicklin, D. J.; Ellis, L.
M. Expression and function of vascular endothelial growth factor
receptor-1 on human colorectal cancer cells. Oncogene 2005, 24,
2647-2653.
[131] Hurwitz, H.; Fehrenbacher, L.; Novotny, W.; Cartwright, T.;
Hainsworth, J.; Heim, W.; Berlin, J.; Baron, A.; Griffing, S.;
Holmgren, E.; Ferrara, N.; Fyfe, G.; Rogers, B.; Ross, R.; Kabbi-
navar, F. Bevacizumab plus irinotecan, fluorouracil, and leucovorin
for metastatic colorectal cancer. N. Engl. J. Med. 2004, 350, 2335-
2342.
[132] Giantonio, B. J.; Catalano, P. J.; Meropol, N. J.; O'Dwyer, P. J.;
Mitchell, E. P.; Alberts, S. R.; Schwartz, M. A.; Benson, A. B. 3rd.
Bevacizumab in combination with oxaliplatin, fluorouracil, and
leucovorin (FOLFOX4) for previously treated metastatic colorectal
cancer: results from the Eastern Cooperative Oncology Group
Study E3200. J. Clin. Oncol. 2007, 25, 1539-1544.
[133] Wagner, A. D.; Arnold, D.; Grothey, A. A.; Haerting, J.; Unver-
zagt, S. Anti-angiogenic therapies for metastatic colorectal cancer.
The Cochrane Library 2009, Issue 3, CD005392.
[134] Yamamoto, N.; Tamura, T.; Yamamoto, N.; Yamada, K.; Yamada,
Y.; Nokihara, H.; Fujiwara, Y.; Takahashi, T.; Murakami, H.;
Boku, N.; Yamazaki, K.; Puchalski, T. A.; Shin, E. Phase I, dose
escalation and pharmacokinetic study of cediranib (RECENTIN), a
highly potent and selective VEGFR signaling inhibitor, in Japanese
patients with advanced solid tumors. Cancer Chemother. Pharma-
col. 2009, 64, 1165-1172.
[135] Chen, E.; Jonker, D.; Gauthier, I.; MacLean, M.; Wells, J.; Powers,
J.; Seymour, L. Phase I study of cediranib in combination with ox-
aliplatin and infusional 5-Fluorouracil in patients with advanced
colorectal cancer. Clin. Cancer Res. 2009, 15, 1481-1486.
[136] Veronese, M. L.; O’Dwyer, P. J. Monoclonal antibodies in the
treatment of colorectal cancer. Eur. J. Cancer 2004, 40, 1292-1301.
[137] Cunningham, D.; Humblet, Y.; Siena, S.; Khayat, D.; Bleiberg, H.;
Santoro, A.; Bets, D.; Mueser, M.; Harstrick, A.; Verslype, C.;
Chau, I.; Van Cutsem, E. Cetuximab monotherapy and cetuximab
plus irinotecan in irinotecan-refractory metastatic colorectal cancer.
N. Engl. J. Med. 2004, 351, 337-345.
[138] Jonker, D. J.; O’Callaghan, C. J.; Karapetis, C. S.; Zalcberg, J. R.;
Tu, D.; Au, H. J.; Berry, S. R.; Krahn, M.; Price, T.; Simes, R. J.;
Tebbutt, N. C.; van Hazel, G.; Wierzbicki, R.; Langer, C.; Moore,
M. J. Cetuximab for the Treatment of Colorectal Cancer. N. Engl.
J. Med. 2007, 357, 2040-2048.
[139] Saltz, L. B.; Meropol, N. J.; Loehrer, P. J. Sr.; Needle, M. N.;
Kopit, J.; Mayer, R. J. Phase II trial of cetuximab in patients with
refractory colorectal cancer that expresses the epidermal growth
factor receptor. J. Clin. Oncol. 2004, 22, 1201-1208.
[140] Chung, K. Y.; Shia, J.; Kemeny, N. E.; Shah, M.; Schwartz, G. K.;
Tse, A.; Hamilton, A.; Pan, D.; Schrag, D.; Schwartz, L.; Klimstra,
D. S.; Fridman, D.; Kelsen, D. P.; Saltz, L. B. Cetuximab shows ac-
tivity in colorectal cancer patients with tumors that do not express
the epidermal growth factor receptor by immunohistochemistry. J.
Clin. Oncol. 2005, 23, 1803-1810.
[141] Francoual, M.; Etienne-Grimaldi, M.C.; Formento, J. L.; Benchi-
mol, D.; Bourgeon, A.; Chazal, M.; Letoublon, C.; André, T.;
Gilly, N.; Delpero, J. R.; Lasser, P.; Spano, J. P.; Milano, G. EGFR
in colorectal cancer: more than a simple receptor. Ann. Oncol.
2006, 17, 962-967.
[142] Andreyev, H. J.; Norman, A. R.; Cunningham, D.; Oates, J. R.;
Clarke, P. A. Kirsten ras mutations in patients with colorectal can-
cer: the multicenter “RASCAL” study. J. Natl. Cancer Inst. 1998,
90, 675-684.
[143] Van Cutsem, E.; Köhne, C. H.; Hitre, E.; Zaluski, J.; Chang Chien,
C. R.; Makhson, A.; D'Haens, G.; Pintér, T.; Lim, R.; Bodoky, G.;
Roh, J. K.; Folprecht, G.; Ruff, P.; Stroh, C.; Tejpar, S.;
Schlichting, M.; Nippgen, J.; Rougier, P. Cetuximab and chemo-
therapy as initial treatment for metastatic colorectal cancer. N.
Engl. J. Med. 2009, 360, 1408-1417.
[144] Van Cutsem, E.; Peeters, M.; Siena, S.; Humblet, Y.; Hendlisz, A.;
Neyns, B.; Canon, J. L.; Van Laethem, J. L.; Maurel, J.;
Richardson, G.; Wolf, M.; Amado, R. G. Open-label phase III Trial
of panitumumab plus best supportive care compared with best sup-
portive care alone in patients with chemotherapy-refractory metas-
tatic colorectal cancer. J. Clin. Oncol. 2007, 25, 1658-1664.
[145] Amado, R. G.; Wolf, M.; Peeters, M.; Van Cutsem, E.; Siena, S.;
Freeman, D. J.; Juan, T.; Sikorski, R.; Suggs, S.; Radinsky, R.; Pat-
terson, S. D.; Chang, D. D. Wild-type KRAS is required for pani-
tumumab efficacy in patients with metastatic colorectal cancer. J.
Clin. Oncol. 2008, 26, 1626-1634.
[146] Tol, J.; Koopman, M.; Cats, A.; Rodenburg, C. J.; Creemers, G. J.;
Schrama, J. G.; Erdkamp, F. L.; Vos, A. H.; van Groeningen, C. J.;
Sinnige, H. A.; Richel, D. J.; Voest, E. E.; Dijkstra, J. R.; Vink-
Börger, M. E.; Antonini, N. F.; Mol, L.; van Krieken, J. H.; Dale-
sio, O.; Punt, C. J. Chemotherapy, bevacizumab, and cetuximab in
metastatic colorectal cancer. N. Engl. J. Med. 2009, 360, 563-572.
[147] Hecht, J. R.; Mitchell, E.; Chidiac, T.; Scroggin, C.; Hagenstad, C.;
Spigel, D.; Marshall, J.; Cohn, A.; McCollum, D.; Stella, P.;
Deeter, R.; Shahin, S.; Amado, R. G. A randomized phase IIIb trial
of chemotherapy, bevacizumab, and panitumumab compared with
chemotherapy and bevacizumab alone for metastatic colorectal
cancer. J. Clin. Oncol. 2009, 27, 672-680.

28 Current Cancer Drug Targets, 2010, Vol. 10, No. 4 Palazzo et al.
[148] Meyerhardt, J. A.; Stuart, K.; Fuchs, C. S.; Zhu, A. X.; Earle, C. C.;
Bhargava, P.; Blaszkowsky, L.; Enzinger, P.; Mayer, R. J.; Battu,
S.; Lawrence, C.; Ryan, D. P. Phase II study of FOLFOX, bevaci-
zumab and erlotinib as first-line therapy for patients with metastas-
tic colorectal cancer. Ann. Oncol. 2007, 18, 1185-1189.
[149] Michaloglou, C.; Vredeveld, L. C. W.; Mooi, W. J.; Peeper, D. S.
BRAFE600 in benign and malignant human tumours. Oncogene
2008, 27, 877-895.
[150] Souglakos, J.; Philips, J.; Wang, R.; Marwah, S.; Silver, M.;
Tzardi, M.; Silver, J.; Ogino, S.; Hooshmand, S.; Kwak, E.; Freed,
E.; Meyerhardt, J. A.; Saridaki, Z.; Georgoulias, V.; Finkelstein,
D,; Fuchs, C. S.; Kulke, M. H.; Shivdasani, R. A. Prognostic and
predictive value of common mutations for treatment response and
survival in patients with metastatic colorectal cancer. Br. J. Cancer
2009, 101, 465-472.
[151] Di Nicolantonio, F.; Martini, M.; Molinari, F.; Sartore-Bianchi, A.;
Arena, S.; Saletti, P.; De Dosso, S.; Mazzucchelli, L.; Frattini, M.;
Siena, S.; Bardelli, A. Wild-Type BRAF Is Required for Response
to Panitumumab or Cetuximab in Metastatic Colorectal Cancer. J.
Clin. Oncol. 2008, 26, 5705-5712.
[152] Motzer, R. J.; Mazumdar, M.; Bacik, J.; Berg, W.; Amsterdam, A.;
Ferrara, J. Survival and prognostic stratification of 670 patients
with advanced renal cell carcinoma. J. Clin. Oncol. 1999, 17, 2530-
2540.
[153] Mekhail, T. M.; Abou-Jawde, R. M.; Boumerhi, G.; Malhi, S.;
Wood, L.; Elson, P.; Bukowski, R. Validation and extension of the
Memorial Sloan-Kettering prognostic factors model for survival in
patients with previously untreated metastatic renal cell carcinoma.
J. Clin. Oncol. 2005, 23, 832-840.
[154] Latif, F.; Tory, K.; Gnarra, J.; Yao, M.; Duh, F. M.; Orcutt, M. L.;
Stackhouse, T.; Kuzmin, I.; Modi, W.; Geil, L.; et al. Identification
of the von Hippel-Lindau disease tumor suppressor gene. Science
1993, 260, 1317-1320.
[155] Wang, G. L.; Jiang, B. -H.; Rue, A. E.; Semenza, G. L. Hypoxia-
inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer
regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995,
92, 5510-5514.
[156] Huang, L. E.; Arany, Z.; Livingston, D. M.; Bunn, H. F. Activation
of hypoxia-inducible transcription factor depends primarily upon
redox-sensitive stabilization of its alpha subunit. J. Biol. Chem.
1996, 271, 32253-32259.
[157] Krieg, M.; Haas, R.; Brauch, H.; Acker, T.; Flamme, I.; Plate, K.
H. Up-regulation of hypoxia-inducible factors HIF-1alpha and HIF-
2alpha under normoxic conditions in renal carcinoma cells by von
Hippel-Lindau tumor suppressor gene loss of function. Oncogene
2000, 19, 5435-5443.
[158] Iliopoulos, O.; Kibel, A.; Gray, S.; Kaelin, W. G. Tumor suppres-
sion by the human von Hippel–Lindau gene product. Nature Med.
1995, 1, 822-826.
[159] Davidowitz, E.; Schoenfeld, A.; Burk, R. VHL induces renal cell
differentiation and growth arrest through integration of cell-cell and
cell-extracellular matrix signaling. Mol. Cell. Biol. 2001, 21, 865-
874.
[160] Choueiri, T. K.; Vaziri, S. A.; Jaeger, E.; Elson, P.; Wood, L.;
Bhalla, I. P.; Small, E. J.; Weinberg, V.; Sein, N.; Simko, J.; Gol-
shayan, A. R.; Sercia, L.; Zhou, M.; Waldman, F. M.; Rini, B. I.;
Bukowski, R. M.; Ganapathi, R. von Hippel-Lindau gene status and
response to vascular endothelial growth factor targeted therapy for
metastatic clear cell renal cell carcinoma. J. Urol. 2008, 180, 860-
865.
[161] Dorevi , G.; Matusan-Ilijas, K.; Babarovi , E.; Hadzisejdi , I.;
Grahovac, M.; Grahovac, B.; Jonji , N. Hypoxia inducible factor-
1alpha correlates with vascular endothelial growth factor A and C
indicating worse prognosis in clear cell renal cell carcinoma. J.
Exp. Clin. Cancer Res. 2009, 40, 20-28.
[162] Kaelin, W. G. Jr. The von Hippel-Lindau tumor suppressor gene
and kidney cancer. Clin. Cancer Res. 2004, 10, 6290-6295S.
[163] Giaccia, A.; Siim, B. G.; Johnson, R. S. HIF-1 as a target for drug
development. Nature Rev. Drug Discov. 2003, 2, 1-9.
[164] Ronnen, E. A.; Kondagunta, G. V.; Ishill, N.; Sweeney, S. M.;
Deluca, J. K.; Schwartz, L.; Bacik, J.; Motzer, R. J. A phase II trial
of 17-(Allylamino)-17-demethoxygeldanamycin in patients with
papillary and clear cell renal cell carcinoma. Invest. New Drugs
2006, 24, 543-546.
[165] Coleman, L. J.; Peter, M. B.; Teall, T. J.; Brannan, R. A.; Hanby,
A. M.; Honarpisheh, H.; Shaaban, A. M.; Smith, L.; Speirs, V.;
Verghese, E. T.; McElwaine, J. N.; Hughes, T. A. Combined analy-
sis of eIF4E and 4E-binding protein expression predicts breast can-
cer survival and estimates eIF4E activity. Br. J. Cancer 2009, 100,
1393-1399.
[166] Khoury, T.; Alrawi, S.; Ramnath, N.; Li, Q.; Grimm, M.; Black, J.;
Tan, D. Eukaryotic initiation factor-4E and cyclin D1 expression
associated with patient survival in lung cancer. Clin Lung Cancer
2009, 10, 58-66.
[167] Castellvi, J.; Garcia, A.; Rojo, F.; Ruiz-Marcellan, C.; Gil, A.,
Baselga, J.; Ramon y Cajal, S. Phosphorylated 4E binding protein
1: a hallmark of cell signaling that correlates with survival in ovar-
ian cancer. Cancer 2006, 107, 1801-1811.
[168] Sabatini, D. M. mTOR and cancer: insights into a complex rela-
tionship. Nat. Rev. Cancer 2006, 6, 729-734.
[169] Lane, H. A.; Wood, J. M.; McSheehy, P. M.; Allegrini, P. R.; Bou-
lay, A.; Brueggen, J.; Littlewood-Evans, A.; Maira, S. M.; Martiny-
Baron, G.; Schnell, C. R.; Sini, P.; O'Reilly, T. mTOR inhibitor
RAD001 (everolimus) has antiangiogenic/vascular properties dis-
tinct from a VEGFR tyrosine kinase inhibitor. Clin. Cancer Res.
2009, 15, 1612-1622.
[170] Rini, B.; Kar, S.; Kirkpatrick, P. Temsirolimus. Nature Rev. Drug
Discov. 2007, 6, 599-600.
[171] Atkins, M. B.; Hidalgo, M.; Stadler, W. M.; Logan, T. F.; Dutcher,
J. P.; Hudes, G. R.; Park, Y.; Liou, S. H.; Marshall, B.; Boni, J. P.;
Dukart, G.; Sherman, M. L. Randomized phase II study of multiple
dose levels of CCI-779, a novel mammalian target of rapamycin
kinase inhibitor, in patients with advanced refractory renal cell car-
cinoma. J. Clin. Oncol. 2004, 22, 909-918.
[172] Hudes, G.; Carducci, M.; Tomczak, P.; Dutcher, J.; Figlin, R.;
Kapoor, A.; Staroslawska, E.; Sosman, J.; McDermott, D.; Bo-
drogi, I.; Kovacevic, Z.; Lesovoy, V.; Schmidt-Wolf, I. G.; Barba-
rash, O.; Gokmen, E.; O'Toole, T.; Lustgarten, S.; Moore, L.;
Motzer, R. J. Temsirolimus, interferon-alpha, or both for advanced
renal cell carcinoma. N. Eng. J. Med. 2007, 356, 2271-2281.
[173] Motzer, R. J.; Escudier, B.; Oudard, S.; Hutson, T. E.; Porta, C.;
Bracarda, S.; Grünwald, V.; Thompson, J. A.; Figlin, R. A.; Hol-
laender, N.; Urbanowitz, G.; Berg, W. J.; Kay, A.; Lebwohl, D.;
Ravaud, A. Efficacy of everolimus in advanced renal cell carci-
noma: a double-blind randomized placebo controlled phase III trial.
Lancet 2008, 372, 449-456.
[174] Gunningham, S. P.; Currie, M. J.; Han, C.; Turner, K.; Scott, P. A.;
Robinson, B. A.; Harris, A. L.; Fox, S. B. Vascular endothelial
growth factor-B and vascular endothelial growth factor-C expres-
sion in renal cell carcinomas: regulation by the von Hippel-Lindau
gene and hypoxia. Cancer Res. 2001, 61, 3206-3211.
[175] Mendel, D. B.; Laird, A. D.; Xin, X.; Louie, S. G.; Christensen, J.
G.; Li, G.; Schreck, R. E.; Abrams, T. J.; Ngai, T. J.; Lee, L. B.;
Murray, L. J.; Carver, J.; Chan, E.; Moss, K. G.; Haznedar, J. O.;
Sukbuntherng, J.; Blake, R. A.; Sun, L.; Tang, C.; Miller, T.; Shi-
razian, S.; McMahon, G.; Cherrington, J. M. In vivo antitumor ac-
tivity of SU11248, a novel tyrosine kinase inhibitor targeting vas-
cular endothelial growth factor and platelet derived growth factor
receptors: determination of a pharmacokinetic/pharmacodynamic
relationship. Clin. Cancer Res. 2003, 9, 327-337.
[176] O’Farrell, A. M.; Abrams, T. J.; Yuen, H. A.; Ngai, T. J.; Louie, S.
G.; Yee, K. W.; Wong, L. M.; Hong, W.; Lee, L. B.; Town, A.;
Smolich, B. D.; Manning, W. C.; Murray, L. J.; Heinrich, M. C.;
Cherrington, J. M. SU11248 is a novel FLT3 tyrosine kinase in-
hibitor with potent activity in vitro and in vivo. Blood 2003, 101,
3597-3596.
[177] Escudier, B.; Eisen, T.; Stadler, W. M.; Szczylik, C.; Oudard, S.;
Staehler, M.; Negrier, S.; Chevreau, C.; Desai, A. A.; Rolland, F.;
Demkow, T.; Hutson, T. E,; Gore, M.; Anderson, S.; Hofilena, G.;
Shan, M.; Pena, C.; Lathia, C.; Bukowski, R. M. Sorafenib for
treatment of renal cell carcinoma: final efficacy and safety results
of the phase III treatment approaches in renal cancer global evalua-
tion trial. J. Clin. Oncol. 2009, 27, 3312-3318.
[178] Ratain, M. J.; Eisen, T.; Stadler, W. M.; Flaherty, K. T.; Kaye, S.
B.; Rosner, G. L.; Gore, M.; Desai, A. A.; Patnaik, A.; Xiong, H.
Q.; Rowinsky, E.; Abbruzzese, J. L.; Xia, C.; Simantov, R.;
Schwartz, B.; O'Dwyer, P. J. Phase II placebo controlled random-

Targeted Therapy in Cancer Current Cancer Drug Targets, 2010, Vol. 10, No. 4 29
ized discontinuation trial of Sorafenib in patient with metastatic re-
nal cell carcinoma. J. Clin. Oncol. 2006, 24, 2505-2512.
[179] Escudier, B.; Eisen, T.; Stadler, W. M.; Szczylik, C.; Oudard, S.;
Siebels, M.; Negrier, S.; Chevreau, C.; Solska, E.; Desai, A. A.;
Rolland, F.; Demkow, T.; Hutson, T. E.; Gore, M.; Freeman, S.;
Schwartz, B.; Shan, M.; Simantov, R.; Bukowski, R. M, Sorafenib
in advanced clear-cell renal-cell carcinoma. N. Engl. J. Med. 2007,
356, 125-134.
[180] Motzer, R. J.; Michaelson, M. D.; Redman, B. G.; Hudes, G. R.;
Wilding, G.; Figlin, R. A.; Ginsberg, M. S.; Kim, S. T.; Baum, C.
M.; DePrimo, S. E.; Li, J. Z.; Bello, C. L.; Theuer, C. P.; George,
D. J.; Rini, B. I. Activity of SU11248, a multi-targeted inhibitor of
vascular endothelial growth factor receptor and platelet-derived
growth factor receptor, in patients with metastatic renal cell carci-
noma. J. Clin. Oncol. 2006, 24, 16-24.
[181] Motzer, R. J.; Hutson, T. E.; Tomczak, P.; Michaelson, M. D.;
Bukowski, R. M.; Rixe, O.; Oudard, S.; Negrier, S.; Szczylik, C.;
Kim, S. T.; Chen, I.; Bycott, P. W.; Baum, C. M.; Figlin, R. A.
Sunitinib versus interferon alfa in metastatic renal-cell carcinoma.
N. Engl. J. Med. 2007, 356, 115-124.
[182] Motzer, R. J.; Hutson, T. E.; Tomczak, P.; Michaelson, M. D.;
Bukowski, R. M.; Oudard, S.; Negrier, S.; Szczylik, C.; Pili, R.;
Bjarnason, G. A.; Garcia-del-Muro, X.; Sosman, J. A.; Solska, E.;
Wilding, G.; Thompson, J. A.; Kim, S. T.; Chen, I.; Huang, X.;
Figlin, R. A. Overall survival and updated results for sunitinib
compared with interferon alfa in patients with metastatic renal cell
carcinoma. J. Clin. Oncol. 2009, 27, 3584-3590.
[183] Choueiri, T. K.; Plantade, A.; Elson, P.; Negrier, S.; Ravaud, A.;
Oudard, S.; Zhou, M.; Rini, B. I.; Bukowski, R. M.; Escudier, B.
Efficacy of sunitinib and Sorafenib in metastatic papilarry and
chromophobe renal cell carcinoma. J. Clin. Oncol. 2008, 26, 127-
131.
[184] Escudier, B.; Pluzanska, A.; Koralewski, P.; Ravaud, A.; Bracarda,
S.; Szczylik, C.; Chevreau, C.; Filipek, M.; Melichar, B.; Bajetta,
E.; Gorbunova, V.; Bay, J. O.; Bodrogi, I.; Jagiello-Gruszfeld, A.;
Moore, N. Bevacizumab plus interferon- 2a for treatment of metas-
tatic renal cell carcinoma: A randomized, double-blind phase III
trial. Lancet 2007, 370, 2103-2111.
[185] Rini, B. I.; Halabi, S.; Rosenberg, J. E.; Stadler, W. M.; Vaena, D.
A.; Ou, S. S.; Archer, L.; Atkins, J. N.; Picus, J.; Czaykowski, P.;
Dutcher, J.; Small, E. J. Bevacizumab plus interferon alfa com-
pared with interferon alfa monotherapy in patients with metastatic
renal cell carcinoma: CALGB 90206. J. Clin. Oncol. 2008, 26,
5422-5428.
[186] Melichar, B.; Koralewski, P.; Ravaud, A.; Pluzanska, A.; Bracarda,
S.; Szczylik, C.; Chevreau, C.; Filipek, M.; Delva, R.; Sevin, E.;
Négrier, S.; McKendrick, J.; Santoro, A.; Pisa, P.; Escudier, B.
First-line bevacizumab combined with reduced dose interferon-
alpha2a is active in patients with metastatic renal cell carcinoma.
Ann. Oncol 2008, 19, 1470-1476.
[187] Lam, J. S.; Belldegrun, A. S.; Figlin, R. A. Adjuvant treatment for
renal cell carcinoma. Exp. Opin. Pharmacother 2006, 7, 705-720.
[188] Rixe, O.; Bukowski, R. M.; Michaelson, M. D.; Wilding, G.; Hu-
des, G. R.; Bolte, O.; Motzer, R. J.; Bycott, P.; Liau, K. F.; Freddo,
J.; Trask, P. C.; Kim, S.; Rini, B. I. Axitinib treatment in patients
with cytokine-refractory metastatic renal-cell cancer: a phase II
study. Lancet Oncol. 2007, 8, 975-984.
[189] Rini, B. I.; Wilding, G.; Hudes, G.; Stadler, W. M.; Kim, S.; Ta-
razi, J.; Rosbrook, B.; Trask, P. C.; Wood, L.; Dutcher, J. P. Phase
II Study of Axitinib in Sorafenib-Refractory Metastatic Renal Cell
Carcinoma. J. Clin. Oncol. 2009, 27, 4462-4468.
[190] Tamaskar, I.; Garcia, J. A.; Elson, P.; Wood, L.; Mekhail, T.;
Dreicer, R.; Rini, B. I.; Bukowski, R. M. Antitumor effects of
sunitinib or sorafenib in patients with metastatic renal cell carci-
noma who received prior antiangiogenic therapy. J. Urol. 2008,
179, 81-86.
[191] Dudek, A. Z.; Zolnierek, J.; Dham, A.; Lindgren, B. R.; Szczylik,
C. Sequential therapy with sorafenib and sunitinib in renal cell car-
cinoma. Cancer 2009, 115, 61-67.
[192] Huang, H.; Menefee, M.; Edgerly, M.; Zhuang, S.; Kotz, H.;
Poruchynsky, M.; Huff, L. M.; Bates, S.; Fojo, T. A phase II clini-
cal trial of ixabepilone (Ixempra; BMS-247550; NSC 710428), an
epothilone B analog, in patients with metastatic renal cell carci-
noma. Clin. Cancer Res. 2010, 16(5), 1634-1641.
[193] Choueiri, T. K.; Regan, M. M.; Rosenberg, J. E.; Oh, W. K.; Clem-
ent, J.; Amato, A. M.; McDermott, D.; Cho, D. C.; Atkins, M. B.;
Signoretti, S. Carbonic anhydrase IX and pathological features as
predictors of outcome in patients with metastatic clear-cell renal
cell carcinoma receiving vascular endothelial growth factor-
targeted therapy. BJU Int. 2010, in press.
Received: December 14, 2009 Revised: April 08, 2010 Accepted: April 10, 2010




















