
A Critical Elaboration on the Pedagogy of Rizal's La Liga Filipina
Upload
independentCategory
view
1download
0
Chemical Physics Letters 598 (2014) 10–16
Contents lists available at ScienceDirect
Chemical Physics Letters
journal homepage: www.elsevier .com/locate /cplet t
Noncovalent functionalization of single-walled carbon nanotubesby aromatic diisocyanate molecules: A computational study
http://dx.doi.org/10.1016/j.cplett.2014.02.0420009-2614/� 2014 Elsevier B.V. All rights reserved.
⇑ Corresponding author.E-mail address: [email protected] (J. Goclon).
Jakub Goclon a,⇑, Mariana Kozlowska b, Pawel Rodziewicz b
a Interdisciplinary Center for Molecular Materials (ICMM) and Computer-Chemistry-Center (CCC), Friedrich-Alexander-University Erlangen-Nürnberg, Nägelsbachstr. 25, 91052Erlangen, Germanyb Institute of Chemistry, University of Bialystok, Hurtowa 1, 15-399 Bialystok, Poland
a r t i c l e i n f o
Article history:Received 20 December 2013In final form 17 February 2014Available online 3 March 2014
a b s t r a c t
We investigate the noncovalent functionalization of metallic single-walled carbon nanotubes (SWCNT)(6,0) by 4,40-methylene diphenyl diisocyanate (MDI) and toluene-2,4-diisocyanate (TDI) molecules usingthe density functional theory (DFT) method with van der Waals dispersion correction. The obtained localminima show the dependence between the molecular arrangement of the adsorbates on SWCNT surfaceand their binding energies. We analyze the interplay between the p–p stacking interactions and isocya-nate functional groups. For the analysis of the changes in the electronic structure we calculate the densityof states (DOS) and charge density plots.
� 2014 Elsevier B.V. All rights reserved.
1. Introduction
Carbon nanotubes (CNTs) are important compounds with com-mon applications in many areas of material science owing to theirunique mechanical and electronic properties [1]. They are used assensors [2,3], energy conversion materials to construct solar cells[4], hydrogen storage materials [5], and molecular electronic de-vices [6].
The modification of the surface of carbon nanotubes via cova-lent and noncovalent functionalization is essential for their appli-cations, since pristine CNTs are chemically inert, and suchmodifications make them soluble in various solvents [7,8]. Onthe one hand, the attachment of molecules to the surface by cova-lent bonding deforms the local structure and directly influences onthe properties of CNTs [9]. On the other hand, the noncovalentfunctionalization has only a small influence on the electronic prop-erties of the nanotubes [10] due to the weak van der Waals inter-actions between the CNT and the adsorbate.
The binding of polymers to the surface of CNTs opens new tech-nological areas. In such cases the individual properties of thesematerials can be combined giving a new hybrid material with bet-ter properties. Technically, the binding of a polymer to a CNT isusually achieved by the grafting to [11] and grafting from [12] ap-proaches. In the first case, a direct reaction occurs between theexisting polymer and CNT functional groups. In the second case,small monomer units diffuse to the surface instead of the long
polymer chains, like in the grafting to method. In both cases themain polimerization reaction is usually proceeded by the sidewallcovalent attachment of functional groups to the pristine CNT.
Polyurethanes (PUs) are segmented polymers which are usuallyproduced in the polyaddition process of diisocyanates and polyols[13]. The production of polyurethanes is one of the most importantbranches of the global chemical industry with numerous applica-tions [13,14]. Various types of diisocyanates can be used to synthe-size PUs. They can be either aromatic or aliphatic. The former onesare more reactive than the latter ones. In our work, we focus ontwo aromatic diisocyanates, namely: 4,40-methylene diphenyl diis-ocyanate (MDI) and toluene-2,4-diisocyanate (TDI), which are themost frequently used in the production of polyurethanes [15].
The functionalization of CNTs proceeds via the reaction ofamide bonds formation between the hydroxyl groups on the side-walls of the tubes and isocyanate groups of the aromatic mono-mers [9,16]. Such covalent functionalization of CNTs competeswith noncovalent one and is strictly dependent on the initial con-centration of the hydroxyl groups on the CNTs sidewalls. The fulldescription of this process needs consideration of both cases.
The functionalized CNTs can be used for further applications, forexample as polymer–CNTs composites and coatings. An in situpolycondensation approach was applied to functionalized multi-walled carbon nanotube (MWCNTs) resulting in polyurea-,polyurethane- and poly(urea–urethane)-bonded CNTs [17]. Suchhybrid systems have much better properties than the polymer it-self due to the effect of the reinforcement of the polymer matrixby creating covalent bonds with the MWCNTs surface. It was foundthat CNTs based nanocomposites were much more resistant to

J. Goclon et al. / Chemical Physics Letters 598 (2014) 10–16 11
wear than the pure PUs [18]. MWCNTs-reinforced thermoplasticpolyurethane nanocomposites have better thermal stability andmechanical properties [19].
In this work, we study the energetic and electronic aspects ofthe noncovalent sidewall functionalization of metallic single-walled carbon nanotube (SWCNT) (6,0). We particularly focus onmolecules used in experiments involving polyurethane functional-ization of CNTs [17–19]. We investigate the noncovalent interac-tion between 4,40-methylene diphenyl diisocyanate (MDI) andtoluene-2,4-diisocyanate (TDI) molecules with SWCNT (6,0), utiliz-ing density functional theory (DFT) methods. Since the SWCNT are1D nanostructured materials the natural way of the computationaldescription of such system is the usage of plane waves (PW) ap-proach. We also examine the modification of the electronic struc-ture of the CNTs after the adsorption where the periodicapproach seems to be the most reliable method. Our calculationspredict that both MDI and TDI interact with the nanotube andthe calculated adsorption energies are typical for physisorptionprocesses.
2. Computational methods
Periodic ab initio DFT calculations utilizing Grimme’s correction(D2) for van der Waals interactions [20] with a plane wave basisset, as implemented in PWscf [21] program, were carried out.The electron–ion interactions were described by the Vanderbiltultrasoft pseudopotentials [22] with a kinetic energy cutoff of 30Ry. The electronic structure was calculated with the generalizedgradient approximation (GGA) using exchange–correlation Per-dew–Burke–Ernzerhof (PBE) functional [23]. All adsorption pro-cesses were considered with nonspin polarized calculations.
Conventional DFT (LDA and GGA) approach does not properlydescribe the p–p stacking interactions [24]. The accurate descrip-tion of such interactions is given by all electron many-body wave-function calculations including higher order correlation correctionsto the Hartree–Fock (HF) ground state [25]. Unfortunately, thepractical usage of these methods is limited to rather small systemsbecause of their large computational requirements.
Apart from the post-HF methods, the local density approxima-tion (LDA) has been found to give underestimated adsorption ener-gies and relatively good equilibrium distances of small aromaticmolecules adsorbed on the surface of graphene sheets [26] andof CNTs sidewalls [27]. The DFT (GGA)–vdW method has alreadybeen demonstrated to reliably depict p–p stacking interactions[26,28]. Detailed comparison of the adsorption energies and equi-librium distances within LDA (parametrized by Perdew and Zunger[29]) and GGA (PBE) with and without vdW correction is presentedin the next section on the example of benzene adsorption onSWCNT (6,0).
The nanotube was fully relaxed with an optimized length of17.065 Å (96 atoms), what corresponds to four unit cells alongthe tube axis. The lateral size of the supercell has been set up to22 Å to minimize the interactions between analyzed molecularsystem and its periodic images. Additionally, in one specific case(see Figure 2c), the supercell was extended along the tube axis to21.331 Å (five unit cells) in order to avoid the adsorbate–adsorbate
Hollow (H) Bridg
Figure 1. Three different adsorption sites of benzene molecule on SWCNT (6,0): hollowatoms are pink. (For interpretation of the references to color in this figure legend, the r
interactions. For the description of both types of representedsupercells, a 1 � 1 � 6 Monkhorst–Pack grid for the sampling ofthe Brillouin zone was taken [30]. A k-point mesh of 1 � 1 � 20sampling was performed in the case of density of states (DOS) cal-culations. All configurations were relaxed by minimizing the atom-ic forces. The total convergence was achieved when the maximumcomponent of the residual forces on the ions was less then3 � 10�3 eV/Å.
The adsorption energy (Eads) was calculated using the followingequation:
Eads ¼ Eadsorbate þ ESWCNT � Eadsorbate=SWCNT; ð1Þ
where Eadsorbate is the energy of the isolated adsorbate in its equilib-rium configuration, ESWCNT is the total energy of supercell for thecarbon nanotube, and Eadsorbate=SWCNT is the total energy of supercellfor the adsorbate/SWCNT system. Positive adsorption energy de-notes an exothermic adsorption process.
The figures were made using VESTA [31] and XCrySDen [32]programs.
3. Preliminary results on model p–p system
The main subject of our study is the interaction between TDIand MDI molecules with the surface of SWCNT. Both adsorbatemolecules possess the common structural element, namely thearomatic ring. Since the molecular aggregation and structuralchanges are driven by the p–p type of interactions, we firstly studythe benzene molecule adsorption on SWCNT as the model process.Secondly, the adsorption energies of benzene molecule are studiedin order to compare the trustworthiness of the GGA+D2 methodwith conventional ones: LDA and GGA. We analyze the magnitudeof the adsorption energy and the types of adsorption sites for thenoncovalent interactions of the benzene molecule with SWCNT(6,0). Previous calculations showed the perpendicular arrangementof the plane of the benzene molecule with respect to the SWCNTsurface is less stable than the parallel one [33].
Here we consider three symmetric adsorption sites of the paral-lel orientation of benzene on SWCNT sidewall [34]. They aremarked in Figure 1 as hollow (H), bridge (B) and top (T). The calcu-lated energetic and structural parameters are presented in Table 1.Several studies of the noncovalent interactions between benzeneand different types of SWCNTs have been already done[27,33,34]. Those studies predict a wide range of the binding ener-gies depending on the method or the type of exchange–correlationfunctionals used within DFT calculations.
Our results of binding energies obtained with LDA functional(147–228 meV (3.39–5.26 kcal/mol)) for the system of benzeneand SWCNT (6,0) are close to those presented by Woods et al.[34] (151–260 meV) for SWCNT (8,0) with the same order of thestability of adsorption sites. Our studies show almost no bindingenergy (20–28 meV) for the calculations within GGA approach.However, when we included the D2 correction for PBE functionalcalculations, the values of the adsorption energy increased aboutten times, see Table 1. The results obtained at the MP2 level [33]for benzene adsorption on SWCNT (10,0) are equal to 201 and254 meV for hollow and bridge sites, respectively, and rather
e (B) Top (T)
(H), bridge (B) and top (T). Carbon atoms of the adsorbate are green and hydrogeneader is referred to the web version of this article.)

Table 1Adsorption energies [meV] and C–C distances [Å] calculated within LDA, GGA (PBE)and GGA (PBE)+D2 for the adsorption of benzene on SWCNT (6,0) at three differentsites: hollow (H), bridge (B) and top (T), see Figure 1. The C–C denotes the shortestdistance between the carbon atom from the benzene ring and that from the SWCNT.
Site Energy[meV]
C–C[Å]
LDA PBE PBE+D2 LDA PBE PBE+D2
Hollow (H) 147 20 216 3.33 4.20 3.39Bridge (B) 228 28 287 3.18 3.99 3.22Top (T) 220 27 278 3.23 4.00 3.25
12 J. Goclon et al. / Chemical Physics Letters 598 (2014) 10–16
slightly change than our PBE+D2 results (lower by 15 and 33 meV,respectively). Additional test calculations showed that increasingthe cutoff energy value from 30 to 35 Ry for PBE+D2 calculationscaused the increase of the adsorption energy only by 1 meV. Itshows that van der Waals forces are mainly responsible for thebinding of the benzene molecule to the SWCNT. The equilibriumdistances between benzene molecule and the SWCNT calculatedusing GGA (PBE) functionals are the largest compared to the corre-sponding ones for LDA and GGA (PBE)+D2, see Table 1. The calcu-lated values for LDA functional are slightly smaller than theseobtained with GGA (PBE)+D2. Similar energetic dependencies areobserved for adenine adsorption on graphite [28] with the adsorp-tion energies equal to 0.46, 0.07 and 1.09 eV within LDA, GGA andGGA+vdW, respectively. The last value is very close to that ex-tracted from the experiment (1.01 eV [35]).
4. Results and discussion
The calculations of the strength of interactions between ben-zene molecule and SWCNT (6,0) show the importance of van derWaals forces and the need of the usage of the dispersion correctionin our DFT calculations. Our main goal is the theoretical descriptionof the adsorption process of larger molecules, such as 4,40-methy-lene diphenyl diisocyanate (MDI) and toluene-2,4-diisocyanate
(a)
(c) (d)
I
III IV
Figure 2. Optimized structures for physisorption of 4,40-methylene diphenyl diisocyanatatoms are blue, oxygen atoms are red and hydrogen atoms are pink. (For interpretation ofof this article.)
(TDI), which is a much more complex task. We will refer, however,to the results obtained for the benzene molecule, in particular tothe stability of its different binding sites. Apart from the aromaticring, the MDI and TDI molecules interact with SWCNTs via flexibleisocyanate groups. The influence of this important structural ele-ment to the binding energy will be also analyzed.
We started from different nonequivalent positions of the MDIand TDI molecules adsorbed on the SWCNT (6,0) surface. Thegeometry of the MDI and TDI structures are shown in Figures 2and 3, respectively. The corresponding adsorption energies and se-lected equilibrium distances, calculated using PBE+D2, are dis-played in Tables 2 and 3, respectively. Due to the geometriccomplexity of the MDI molecule [36] its adsorption pattern onthe SWCNT surface is more complicated than that for TDI. Our pre-vious work [36] presents strong structural flexibility of 4,40-meth-ylene diphenyl diisocyanate (MDI) molecule in the gas phase,therefore easy geometrical adaptation of the MDI molecule to dif-ferent positions on the CNTs sidewalls is expected.
4.1. MDI adsorption
We considered five different structures (I–V) for the MDI mole-cule adsorbed on the SWCNT surface. The structures I and II pre-sented in Figure 2(a and b) correspond to the adsorption of theMDI molecule across the SWCNT. Calculated adsorption energiesfor these configurations are very close: 671 and 660 meV, respec-tively. Structural flexibility of MDI molecule is steered by the –CH2– group, which can not be freely displaced in every direction[36]. Hence, there is no possibility to cover uniformly the SWCNTsidewall by two rings of the MDI molecule. The preferred adsorp-tion sites for such arrangement of MDI are near to the bridge andthe top ones (see the previous section). The shortest distance be-tween the carbon atom from the aromatic ring of MDI and SWCNTis in the range of 3.13–3.22 Å (for both aromatic rings of the MDImolecule, see Supplementary Material for I structure), see Table 2.
The angle between two rings in the MDI molecule (\(CCC)MDI)only slightly changes after the adsorption (maximum decrease by
(b)
(e)
II
V
e (MDI) on pristine SWCNT (6,0). Carbon atoms of the adsorbate are green, nitrogenthe references to color in this figure legend, the reader is referred to the web version

(a) (b) (c)
(d) (e)
(g) (h)
I-(H) II-(H-6 )o0 oIII-(H-120 )
IV-(B) V-(B-6 )o0 VI-(B-12 )o0
VII-(T) VIII-(T-6 )o0
(f)
Figure 3. Optimized structures for physisorption of toluene-2,4-diisocyanate (TDI) on pristine SWCNT (6,0). Carbon atoms of the adsorbate are green, nitrogen atoms areblue, oxygen atoms are red and hydrogen atoms are pink. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of thisarticle.)
Table 2PBE+D2 adsorption energies [meV] and selected distances [Å] for different configu-rations of MDI/SWCNT (6,0). The Ctop–C and Cdown–C represent the shortest distancebetween the carbon atom from the top and the bottom ring with that from the CNT,respectively, see Figure 2. The \(CCC)MDI denotes the angle between two rings of theMDI molecule.
Structure Eads
[meV]Ctop–C
[Å]Cdown–C
[Å]\(CCC)MDI
[�]
I 671 3.14 3.13 111.6II 660 3.13 3.22 112.6III 555 3.29 3.23 117.4IV 448 3.29 – 112.4V 361 – – 112.5
Table 3PBE+D2 adsorption energies [meV] and C–C distances [Å] for the different adsorptionconfigurations of TDI/SWCNT (6,0), see Figure 3. The C–C represents the shortestdistance between the carbon atom from the TDI ring and that from the CNT.
Structure Eads
[meV]C–C[Å]
I-(H) 490 3.08II-(H-60�) 458 3.19III-(H-120�) 450 3.27IV-(B) 435 3.25V-(B-60�) 455 3.06VI-(B-120�) 510 3.17VII-(T) 488 3.07VIII-(T-60�) 460 3.17
J. Goclon et al. / Chemical Physics Letters 598 (2014) 10–16 13
-7 -6 -5 -4 -3 -2 -1 0 1
-7 -6 -5 -4 -3 -2 -1 0 1
-7 -6 -5 -4 -3 -2 -1 0 1Energy [eV]
CNT
MDI/CNT
TDI/CNT
(a)
(b)
(c)
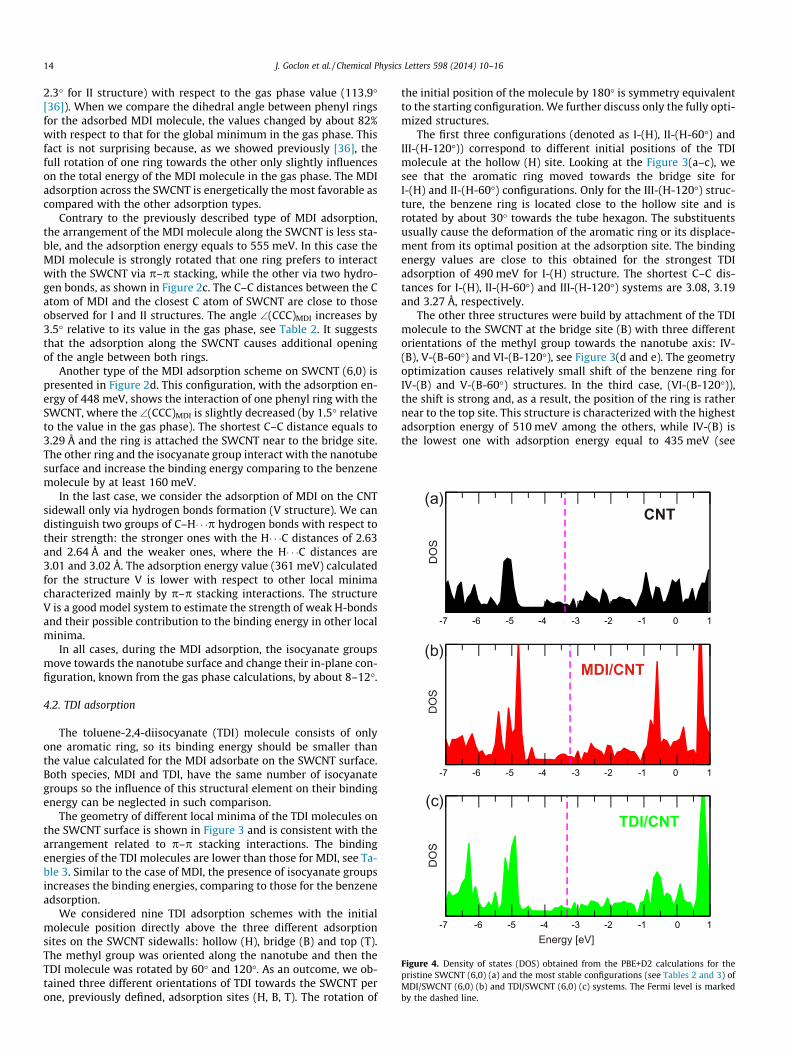
Figure 4. Density of states (DOS) obtained from the PBE+D2 calculations for thepristine SWCNT (6,0) (a) and the most stable configurations (see Tables 2 and 3) ofMDI/SWCNT (6,0) (b) and TDI/SWCNT (6,0) (c) systems. The Fermi level is markedby the dashed line.
14 J. Goclon et al. / Chemical Physics Letters 598 (2014) 10–16
2.3� for II structure) with respect to the gas phase value (113.9�[36]). When we compare the dihedral angle between phenyl ringsfor the adsorbed MDI molecule, the values changed by about 82%with respect to that for the global minimum in the gas phase. Thisfact is not surprising because, as we showed previously [36], thefull rotation of one ring towards the other only slightly influenceson the total energy of the MDI molecule in the gas phase. The MDIadsorption across the SWCNT is energetically the most favorable ascompared with the other adsorption types.
Contrary to the previously described type of MDI adsorption,the arrangement of the MDI molecule along the SWCNT is less sta-ble, and the adsorption energy equals to 555 meV. In this case theMDI molecule is strongly rotated that one ring prefers to interactwith the SWCNT via p–p stacking, while the other via two hydro-gen bonds, as shown in Figure 2c. The C–C distances between the Catom of MDI and the closest C atom of SWCNT are close to thoseobserved for I and II structures. The angle \(CCC)MDI increases by3.5� relative to its value in the gas phase, see Table 2. It suggeststhat the adsorption along the SWCNT causes additional openingof the angle between both rings.
Another type of the MDI adsorption scheme on SWCNT (6,0) ispresented in Figure 2d. This configuration, with the adsorption en-ergy of 448 meV, shows the interaction of one phenyl ring with theSWCNT, where the \(CCC)MDI is slightly decreased (by 1.5� relativeto the value in the gas phase). The shortest C–C distance equals to3.29 Å and the ring is attached the SWCNT near to the bridge site.The other ring and the isocyanate group interact with the nanotubesurface and increase the binding energy comparing to the benzenemolecule by at least 160 meV.
In the last case, we consider the adsorption of MDI on the CNTsidewall only via hydrogen bonds formation (V structure). We candistinguish two groups of C–H� � �p hydrogen bonds with respect totheir strength: the stronger ones with the H� � �C distances of 2.63and 2.64 Å and the weaker ones, where the H� � �C distances are3.01 and 3.02 Å. The adsorption energy value (361 meV) calculatedfor the structure V is lower with respect to other local minimacharacterized mainly by p–p stacking interactions. The structureV is a good model system to estimate the strength of weak H-bondsand their possible contribution to the binding energy in other localminima.
In all cases, during the MDI adsorption, the isocyanate groupsmove towards the nanotube surface and change their in-plane con-figuration, known from the gas phase calculations, by about 8–12�.
4.2. TDI adsorption
The toluene-2,4-diisocyanate (TDI) molecule consists of onlyone aromatic ring, so its binding energy should be smaller thanthe value calculated for the MDI adsorbate on the SWCNT surface.Both species, MDI and TDI, have the same number of isocyanategroups so the influence of this structural element on their bindingenergy can be neglected in such comparison.
The geometry of different local minima of the TDI molecules onthe SWCNT surface is shown in Figure 3 and is consistent with thearrangement related to p–p stacking interactions. The bindingenergies of the TDI molecules are lower than those for MDI, see Ta-ble 3. Similar to the case of MDI, the presence of isocyanate groupsincreases the binding energies, comparing to those for the benzeneadsorption.
We considered nine TDI adsorption schemes with the initialmolecule position directly above the three different adsorptionsites on the SWCNT sidewalls: hollow (H), bridge (B) and top (T).The methyl group was oriented along the nanotube and then theTDI molecule was rotated by 60� and 120�. As an outcome, we ob-tained three different orientations of TDI towards the SWCNT perone, previously defined, adsorption sites (H, B, T). The rotation of
the initial position of the molecule by 180� is symmetry equivalentto the starting configuration. We further discuss only the fully opti-mized structures.
The first three configurations (denoted as I-(H), II-(H-60�) andIII-(H-120�)) correspond to different initial positions of the TDImolecule at the hollow (H) site. Looking at the Figure 3(a–c), wesee that the aromatic ring moved towards the bridge site forI-(H) and II-(H-60�) configurations. Only for the III-(H-120�) struc-ture, the benzene ring is located close to the hollow site and isrotated by about 30� towards the tube hexagon. The substituentsusually cause the deformation of the aromatic ring or its displace-ment from its optimal position at the adsorption site. The bindingenergy values are close to this obtained for the strongest TDIadsorption of 490 meV for I-(H) structure. The shortest C–C dis-tances for I-(H), II-(H-60�) and III-(H-120�) systems are 3.08, 3.19and 3.27 Å, respectively.
The other three structures were build by attachment of the TDImolecule to the SWCNT at the bridge site (B) with three differentorientations of the methyl group towards the nanotube axis: IV-(B), V-(B-60�) and VI-(B-120�), see Figure 3(d and e). The geometryoptimization causes relatively small shift of the benzene ring forIV-(B) and V-(B-60�) structures. In the third case, (VI-(B-120�)),the shift is strong and, as a result, the position of the ring is rathernear to the top site. This structure is characterized with the highestadsorption energy of 510 meV among the others, while IV-(B) isthe lowest one with adsorption energy equal to 435 meV (see

(a) (b)
0.0000.0020.0050.0100.0230.050
Figure 5. Isosurface and contour plot of the charge density for the most stable configurations of MDI/SWCNT (6,0) (a) and TDI/SWCNT (6,0) (b). Charge distribution is plottedin a logarithmic scale. The isosurface corresponds to the charge density of 0.05 e/Å3.
J. Goclon et al. / Chemical Physics Letters 598 (2014) 10–16 15
Table 3). The C–C distances are equal to 3.25, 3.06 and 3.17 Å forIV-(B), V-(B-60�) and VI-(B-120�) structures, respectively.
The TDI adsorption at the top site (T) gives two new structures:VII-(T) and VIII-(T-60�). The structure IX-(T-120�) after geometryoptimization is the same like VI-(B-120�), which is the most stableone (see Table 3). We observe the small shift of the benzene ring ofthe TDI molecule on the tube sidewall for the VIII-(T-60�) localminimum and the large one for VII-(T). The binding energies andthe C–C distances are in the range of the results presented for otherstructures (see Table 3).
A detailed analysis of the geometry performed for the most sta-ble structure VI-(B-120�) (see Supplementary Material) shows onlyslight changes of the geometry of the isocyanate groups after TDIadsorption – the bond and angle changes are smaller than 0.01 Åand 2�, respectively. Only dihedral angles between the plane ofthe benzene ring and –NCO group change by 12� and 14� (with re-spect to the gas phase value) because the isocyanate groups movetowards the nanotube surface. Such behavior pattern is observedfor all studied cases.
4.3. Electronic structure analysis
The influence of the adsorption of the MDI and TDI moleculeson the electronic structure of the pristine SWCNT (6,0) is also pre-sented. The most stable adsorption configurations are considered: Ifor MDI and VI-(B-120�) for TDI. Figure 4 shows the total density ofstates (DOS) for both systems and for the pristine metallic SWCNT(6,0), as a reference system.
We found only a small shift in the Fermi level (the dashed linein Figure 4) by about 0.06 and 0.15 eV after TDI and MDI adsorp-tion on the CNT, respectively. The existence of additional bands,originating from the adsorbate molecule, is also visible in thestructure of the DOS. For MDI adsorption such bands are locatedapproximately 1.6 and 2.2 eV below the Fermi level, while forTDI adsorption �1.5 and 3.0 eV. For both cases, Figure 4(b and c)also present the appearance of additional bands located near 2.5–2.6 and 3.8–3.9 eV above the Fermi level. The highest occupiedmolecular orbital (HOMO) and the lowest unoccupied molecularorbital (LUMO) of the adsorbed molecules are located below andabove the Fermi level, far from it. When we compare the DOS inthe vicinity of the Fermi level (between �4.3 and �1.2 eV) forthe pristine and the molecule-adsorbed SWCNT, no difference isobserved.
The charge density analysis gives the information about thenature of chemical bonding. Figure 5 shows the charge densityfor two, the most stable, configurations of the MDI/SWCNT (a)and TDI/SWCNT (b) systems. It is seen that the charge density be-
tween the SWCNT and the adsorbate is small and is localizedmainly on the individual components. Thus, it is clear that thereis no bonding between adsorbates (MDI and TDI) and the SWCNT.The main interaction between the SWCNT and the molecules is ofelectrostatic nature, therefore, only minor perturbation of theSWCNT electronic structure appears.
5. Conclusions
The noncovalent adsorption of 4,40-methylene diphenyl diisocy-anate (MDI) and toluene-2,4-diisocyanate (TDI) on single-walledcarbon nanotube (SWCNT) (6,0) was studied using GGA (PBE) func-tional including dispersion energy correction for the properdescription of van der Waals interactions. Initial tests for the ben-zene adsorption on SWCNT (6,0) showed that the augmentation ofthe GGA (PBE) method by an empirical dispersion term appears tobe a good description of p–p stacking interactions. Beside the dom-inant role of the stacking interactions, the contribution to the bind-ing energy from the isocyanate groups, present in MDI and TDI,seems to be also very important.
We considered five different configurations for the MDI/SWCNTsystem and eight for the TDI/SWCNT one. Calculated binding ener-gies and electronic properties are characteristic for a physisorptionprocess. The structure with the MDI molecule adsorbed across theSWCNT was found to be energetically the most favorable. The localminimum where only hydrogen bonds stabilize the system is theleast stable. The binding energy values of TDI adsorption are closeto each other. The structure with the initial rotation of the TDI ringby 120� towards the nanotube, adsorbed near to the top site, is themost stable.
The equilibrium distances for all studied configurations showedcompetition between the aromatic ring and the isocyanate groupsand are shorter than those for the benzene adsorption. Further-more, the isocyanate groups moved towards the nanotubesidewalls.
The only slight perturbations in the density of states (DOS)plot, close to the Fermi level, and the charge density analysis indi-cate no significant charge transfer between the adsorbate and theSWCNT, unambiguously showing on the van der Waals interac-tion type.
Acknowledgments
This work was supported by the Foundation for Polish Science(FNP) within the HOMING PLUS programme. Computational re-sources were provided by RRZE (Erlangen).

16 J. Goclon et al. / Chemical Physics Letters 598 (2014) 10–16
Appendix A. Supplementary data
Supplementary data associated with this article can be found, inthe online version, at http://dx.doi.org/10.1016/j.cplett.2014.02.042.
References
[1] R. Saito, G. Dresselhaus, M.S. Dresselhaus, Physical Properties of CarbonNanotubes, Imperial College Press, London, 1998.
[2] J. Kong, N.R. Franklin, C.W. Zhou, M.G. Chapline, S. Peng, K.J. Cho, H.J. Dai,Science 287 (2000) 622.
[3] P.G. Collins, K. Bradley, M. Ishigami, A. Zettl, Science 287 (2000) 1801.[4] J. Wei et al., Nano Lett. 7 (2007) 2317.[5] C. Liu, Y.Y. Fan, M. Liu, H.T. Cong, H.M. Cheng, M.S. Dresselhaus, Science 286
(1999) 1127.[6] M.P. Anantram, F. Lonard, Rep. Prog. Phys. 69 (2006) 507.[7] K. Balasubramanian, M. Burghard, Small 1 (2005) 180.[8] W.E. Ford, A. Jung, A. Hirsch, R. Graupner, F. Scholz, A. Yasuda, J.M. Wessels,
Adv. Mater. 18 (2006) 1193.[9] Y.C. Jung, H. Muramatsu, T. Hayashi, J.H. Kim, Y.A. Kim, M. Endo, M.S.
Dresselhaus, Eur. J. Inorg. Chem. 27 (2010) 4305.[10] C.-H. Liu, J.-J. Li, H.-L. Zhang, B.-R. Li, Y. Guo, Colloids Surf., A 313 (2008) 9.[11] Y.-P. Sun, K. Fu, Y. Lin, W. Huang, Acc. Chem. Res. 35 (2002) 1096.[12] Y.-T. Shieh, G.-L. Liu, K.C. Hwang, C.-C. Chen, Polymer 46 (2005) 10945.
[13] G. Woods, The ICI Polyurethane Book, second edn., Wiley, New York, 1990.[14] G. Randall, The Polyurethane Book, Wiley-VCH, Weinheim, 2002.[15] S.A. Madbouly, J.U. Otaigbe, Prog. Polm. Sci. 34 (2009) 1283.[16] C. Zhao, L. Ji, H. Liu, G. Hu, S. Zhang, M. Yang, Z. Yang, J. Solid State Chem. 177
(2004) 4394.[17] C. Gao et al., J. Phys. Chem. B 109 (2005) 11925.[18] H.-J. Song, Z.-Z. Zhang, X.-H. Men, Eur. Polym. J. 43 (2007) 4092.[19] T.M. Madkour, F.M. Hagag, W. Mamdouh, R.A. Azzam, Polymer 53 (2012) 5788.[20] S. Grimme, J. Comput. Chem. 27 (2006) 1787.[21] The PWscf code. Avaialble from: http://www.pwscf.org.[22] D. Vanderbilt, Phys. Rev. B 41 (1990) 7892.[23] J.P. Perdew, K. Burke, M. Ernzerhof, Phys. Rev. Lett. 77 (1996) 3865. 78 (1997)
1396.[24] Y. Zhao, N.E. Schultz, D.G. Truhlar, J. Chem. Theory Comput. 2 (2006) 364.[25] M.O. Sinnokrot, E.F. Valeev, C.D. Sherrill, J. Am. Chem. Soc. 124 (2002) 10887.[26] J.D. Wuest, A. Rochefort, Chem. Commun. 46 (2010) 2923.[27] S. Lim, N. Park, Appl. Phys. Lett. 95 (2009) 243110.[28] F. Ortmann, W.G. Schmidt, F. Bechstedt, Phys. Rev. Lett. 95 (2005) 186101.[29] J.P. Perdew, A. Zunger, Phys. Rev. B 23 (1981) 5048.[30] H.J. Monkhorst, J.D. Pack, Phys. Rev. B 13 (1976) 5188.[31] K. Momma, F. Izumi, J. Appl. Crystallogr. 44 (2011) 1272.[32] A. Kokalj, Comput. Mater. Sci. 28 (2003) 155.[33] T. Kar, H.F. Bettinger, S. Scheiner, A.K. Roy, J. Phys. Chem. C 112 (2008)
20070.[34] L.M. Woods, S�.C. Badescu, T.L. Reinecke, Phys. Rev. B 75 (2007) 155415.[35] J.E. Freund, Ph. D. thesis, Ludwig-Maximilians-Universität, München, 1998.[36] P. Rodziewicz, J. Goclon, J. Mol. Model. 20 (2014) 2097.
Copyright © 2022 FDOKUMEN




















