
Newly recognized cellular abnormalities in the gray platelet syndrome
11
doi:10.1182/blood.V98.5.1382 2001 98: 1382-1391 Mircea Adam, Marie-Anne Gougerot-Pocidalo and Elisabeth M. Cramer Arnaud Drouin, Rémi Favier, Jean-Marc Massé, Najet Debili, Alain Schmitt, Carole Elbim, Josette Guichard, Newly recognized cellular abnormalities in the gray platelet syndrome http://bloodjournal.hematologylibrary.org/content/98/5/1382.full.html Updated information and services can be found at: (3202 articles) Hematopoiesis and Stem Cells Articles on similar topics can be found in the following Blood collections http://bloodjournal.hematologylibrary.org/site/misc/rights.xhtml#repub_requests Information about reproducing this article in parts or in its entirety may be found online at: http://bloodjournal.hematologylibrary.org/site/misc/rights.xhtml#reprints Information about ordering reprints may be found online at: http://bloodjournal.hematologylibrary.org/site/subscriptions/index.xhtml Information about subscriptions and ASH membership may be found online at: Copyright 2011 by The American Society of Hematology; all rights reserved. American Society of Hematology, 2021 L St, NW, Suite 900, Washington DC 20036. Blood (print ISSN 0006-4971, online ISSN 1528-0020), is published weekly by the For personal use only. by guest on February 1, 2014. bloodjournal.hematologylibrary.org From For personal use only. by guest on February 1, 2014. bloodjournal.hematologylibrary.org From
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Newly recognized cellular abnormalities in the gray platelet syndrome

doi:10.1182/blood.V98.5.13822001 98: 1382-1391
Mircea Adam, Marie-Anne Gougerot-Pocidalo and Elisabeth M. CramerArnaud Drouin, Rémi Favier, Jean-Marc Massé, Najet Debili, Alain Schmitt, Carole Elbim, Josette Guichard, Newly recognized cellular abnormalities in the gray platelet syndrome
http://bloodjournal.hematologylibrary.org/content/98/5/1382.full.htmlUpdated information and services can be found at:
(3202 articles)Hematopoiesis and Stem Cells �Articles on similar topics can be found in the following Blood collections
http://bloodjournal.hematologylibrary.org/site/misc/rights.xhtml#repub_requestsInformation about reproducing this article in parts or in its entirety may be found online at:
http://bloodjournal.hematologylibrary.org/site/misc/rights.xhtml#reprintsInformation about ordering reprints may be found online at:
http://bloodjournal.hematologylibrary.org/site/subscriptions/index.xhtmlInformation about subscriptions and ASH membership may be found online at:
Copyright 2011 by The American Society of Hematology; all rights reserved.American Society of Hematology, 2021 L St, NW, Suite 900, Washington DC 20036.Blood (print ISSN 0006-4971, online ISSN 1528-0020), is published weekly by the
For personal use only. by guest on February 1, 2014. bloodjournal.hematologylibrary.orgFrom For personal use only. by guest on February 1, 2014. bloodjournal.hematologylibrary.orgFrom

HEMATOPOIESIS
Newly recognized cellular abnormalities in the gray platelet syndromeArnaud Drouin, Remi Favier, Jean-Marc Masse, Najet Debili, Alain Schmitt, Carole Elbim, Josette Guichard,Mircea Adam, Marie-Anne Gougerot-Pocidalo, and Elisabeth M. Cramer
The gray platelet syndrome (GPS) is arare congenital bleeding disorder inwhich thrombocytopenia is associatedwith increased platelet size and de-creased a-granule content. This reportdescribes 3 new pediatric cases present-ing with the classical platelet abnormali-ties of GPS within one family with nor-mal parents. Examination of bloodsmears of the 3 patients demonstratednot only gray platelets, but also graypolymorphonuclear neutrophils (PMNs)with decreased or abnormally distrib-uted components of secretory compart-ments (alkaline phosphatase, CD35,CD11b/CD18). Secondary granules were
also decreased in number as assayedby immunoelectron microscopy. Thesedata confirm that the secretory compart-ments in neutrophils were also deficientin this family. Megakaryocytes (MKs)were cultured from the peripheral bloodCD341 cells of the 3 patients for 14days, in the presence of thrombopoietinand processed for immunoelectron mi-croscopy. Although von Willebrand fac-tor (vWF) was virtually undetectable inplatelets, vWF immunolabeling was con-spicuous in cultured maturing MKs, par-ticularly within Golgi saccules, but in-stead of being packaged in a-granules,it was released into the demarcation
membrane system. In contrast, P-selec-tin followed a more classical pathway.Double-labeling experiments confirmedthat vWF was following an intracellularpathway distinct from the one of P-selectin. In these 3 new cases of GPS, theMKs appeared to abnormally processvWF, with secretion into the extracellularspace instead of normal a-granule pack-aging. Furthermore, the secretory com-partment of another blood cell line, theneutrophil, was also affected in this fam-ily of GPS. (Blood. 2001;98:1382-1391)
© 2001 by The American Society of Hematology
Introduction
The gray platelet syndrome (GPS) is an inherited disorder ofprimary hemostasis with associated bleeding tendency, throm-bocytopenia, and classical abnormal platelet morphology.1 Theplatelets appear characteristically gray after Romanovsky stain-ing as a result of an abnormally lowa-granule content.Immunoelectron microscopic analysis of gray platelets con-firms that thea-granule contents appear absent.2 In contrast, thegranule-limiting membranes appear present with normally ex-pressed protein components.3 This syndrome is distinct fromother inherited disorders that also affect plateleta-granules,such asa-d storage pool deficiency and the Quebec plateletdisorder. The former is combined with dense granule deficiency4
and the latter with an abnormal protease activity responsiblefor a-granule protein degradation.5 The a-granules are theprincipal storage site for hemostatic proteins such as fibrinogen,von Willebrand factor (vWF), thrombospondin, and factorV and for growth factors such as platelet-derived growth factorand transforming growth factor-b.6 In addition, plasma proteinssuch as albumin and immunoglobulins are endocytosed byplatelets and stored ina-granules.7 The a-granule proteinsare released into the open canalicular system on plateletactivation and play a key role both in hemostasis and woundhealing. In this report, we describe a new family presenting withGPS with a detailed description of their platelet, megakaryo-cytic, and polymorphonuclear neutrophil (PMN) ultrastructuralabnormalities.
Patients, materials, and methods
Patients
Approval for this study was obtained from the INSERM institutional reviewboard. Infomed consent was obtained from the patients and the parents,according to the Declaration of Helsinki.
The patients are the 3 children of a family with healthy parents. Thechildren are 2 heterozygous twins, aged 8 years at the time of diagnosis, andtheir elder sister, aged 10 years. The older sister exhibited thombocytopenia(703 109/L) with recurrent ecchymosis and epistaxis since the age of 5years. Up to the age of 2 years, she had many pulmonary infections.Peripheral blood smears showed that her platelets were large, agranular, andgray. In addition her PMNs also displayed a gray cytoplasmic appearanceinstead of their usual beige and granulous texture. On bone marrow smears,erythroblastic and granulocytic lineages were normally represented butonly rare megakaryocytes (MKs) were present. The marrow specimenobtained by biopsy showed normal cellularity and confirmed that the MKswere reduced in number. A diffuse reticular fibrosis without increasedcollagen was noticed. Kinetic studies of autologous platelets revealed anormal life span but platelet production was apparently only one third ofnormal. Other investigations revealed similar biologic platelet and PMNpatterns in her brother and her sister, both exhibiting thombocytopenia andprolonged bleeding times (Ivy tests:. 20 minutes).
Peripheral blood cells (platelets and PMNs) and cultured MKs from the3 children were also studied. Ultrastructural examination of plateletsdemonstrated the absence of normala-granules and prominent vacuoliza-tion. Fibrinogen as well as thrombospondin content were reduced (20%of normal).
From INSERM U. 474, Hopital de Port-Royal; INSERM U. 479, Hopital Bichat;Laboratoire d’hematologie, Hopital Trousseau, Paris, France; and INSERM U.362, Institut Gustave Roussy, Villejuif, France.
Submitted July 13, 2000; accepted April 24, 2001.
Reprints: Elisabeth M. Cramer. INSERM U.474, Maternite Port-Royal, 5eme
etage, 123 Boulevard de Port-Royal, Paris 75014, France; e-mail:[email protected].
The publication costs of this article were defrayed in part by page chargepayment. Therefore, and solely to indicate this fact, this article is herebymarked ‘‘advertisement’’ in accordance with 18 U.S.C. section 1734.
© 2001 by The American Society of Hematology
1382 BLOOD, 1 SEPTEMBER 2001 z VOLUME 98, NUMBER 5
For personal use only. by guest on February 1, 2014. bloodjournal.hematologylibrary.orgFrom

Platelet number and morphology were normal in the parents and thegrandparents of these children. The karyotypes, studied by conventionalcytogenetic analysis, were normal.
Cells
Blood samples were taken from the 3 GPS children by venipuncture intoplastic tubes containing ACD-C buffer (6.8 mM citric acid, 11.2 mMglucose, pH 4.2) or lithium heparin. Platelet-rich plasma (PRP) wasobtained by centrifugation of PRP for 10 minutes at 180g and 22°C. Theisolated platelets were obtained by centrifugation of PRP for 10 minutes at1100gand washed 3 times with Tyrode buffer (360 mM citric acid, 5 mMKCl, 2 mM CaCl2, 1 mM MgCl2, 103 mM glucose, pH 7.4) containing 3.5mg/mL bovine serum albumin (BSA; Sigma Chemical, St Louis, MO). Thewashed platelets were resuspended and fixed with 1% glutaraldehyde (LaddResearch, Burlington, VT) in phosphate buffer (0.1 M, pH 7.4). MKs weregrown in liquid culture from circulating CD341 blood precursors. CD341
cells were isolated from heparin-anticoagulated blood, using a Ficollgradient followed by a purification on magnetic beads of the Miltenyitechnique. The cells were then cultured for 12 days in serum-free liquidmedium containing recombinant human stem cell factor and thrombopoi-etin (Rhu PEG-MGDF; Amgen, Thousand Oaks, CA) at a final concentra-tion of 10 ng/mL. Cells were fixed at day 12 of culture in 1% glutaraldehyde(Ladd Research, Burlington, VT) in phosphate buffer (0.1 mol/L, pH 7.4),and further processed for electron microscopy.8
For functional tests, PMNs were rapidly isolated at 4°C (to avoidactivation) on 2% dextran T 500 (Pharmacia LKB, Uppsala, Sweden) inphosphate-buffered saline (PBS), then on a Ficoll-Isopaque gradient. Afterred blood cell lysis and washing with cold PBS, PMNs were adjusted to106/mL in PBS lacking Ca11 and Mg11.9 Viability was always more than99% in the trypan blue dye exclusion test.
Antibodies
Polyclonal rabbit antibodies were used for immunoelectron microscopy;anti-CD35 (Santa Cruz Biotechnology, Santa Cruz, CA), anti-vWF (Dako-patts, Glostrup, Denmark), antiglycocalicin (anti-GPIb),10 anti-P-selectin.11
(kind gifts from Dr Michael Berndt, Victoria, Australia) and antilactofer-rin (Cappel Lab, Downington, PA) were used at 10mg/mL. Goat antirabbitIgG coupled to 10 nm gold was purchased from British Biocell Interna-tional (Cardiff, United Kingdom).
Electron microscopy
Normal PMNs, platelets, and MKs were prepared for immunoelectronmicroscopy as follows. After fixation, cells were washed 3 times withphosphate buffer, embedded in sucrose, and frozen in liquid nitrogen.Immunochemical reactions were performed on thin sections collected ongrids according to the method of Bendayan.12After a brief incubation of thesections with 0.1% BSA and 15% normal goat serum, they were labeledwith the polyclonal rabbit antibodies diluted in Tris-buffered saline (TBS)containing 1% BSA and 4% normal goat serum for 2 hours at 22°C, washed3 times with TBS containing 0.1% BSA, and then incubated with goatantirabbit-gold (10 nm) for 1 hour at 22°C. Double-labeling experimentswere performed according to Slot and Geuze.13 The sections werecounterstained with uranyl acetate and lead citrate. Samples were observedon a Philips 450 CM 10 electron microscope (Eindoven, The Netherlands).
Leukocyte alkaline phosphatase cytochemistry
Freshly prepared blood smears from the 3 patients, from their parents, andfrom a control subject were fixed in a solution of formol and methanol (1:9)for 1 minute, then covered with a mixture containinga-naphtyl-phosphateNa (Sigma Chemical), 0.1 g and Fast Garnet (Sigma Chemical) 0.1 g, inpropanediol buffer (25 mL) and HCl N/10 (5 mL) added to distilledwater (70 mL) for 25 minutes. In each sample, at least 100 PMNs wereexamined and the staining intensity of each of them was scored from 0 to4 (0, no staining; 1, light beige cytoplasm; 2, deep beige cytoplasm; 3,brown staining in part of the cytoplasm; 4, homogeneously dark browncytoplasm).
Expression of CD11b/CD18 and CD35 at the PMN surface
Heparinized blood was placed on ice and analyzed immediately. Wholeblood samples were either kept on ice or incubated with PBS orN-formyl-methyonyl-leucyl-phenylalanine (fMLP; 1026 M), at 37°C for 5minutes. To study CD11b, CD18, and CD35 expression, samples (100mL)from each patient were then incubated with phycoerythrin (PE)-conjugatedmonoclonal mouse antihuman CD11b antibody (Dakopatts), fluoresceinisothiocyanate (FITC)-conjugated monoclonal mouse antihuman CD18antibody (Becton Dickinson, Immunocytometry Systems, San Jose, CA), orFITC-conjugated monoclonal mouse antihuman CD35 antibody (Immuno-tech, Marseilles, France) for 30 minutes at 4°C. Red cells were lysed withFACS lysing solution. After one wash with ice-cold PBS, the cells wereresuspended in 1% paraformaldehyde-PBS and kept on ice until analysis.Nonspecific antibody binding was determined on cells incubated with thesame concentration of an irrelevant antibody of the same isotype.
PMN functions
Activity of reduced nicotinamide adenine dinucleotide phosphate (NADPH)-oxidase was tested as follows. The nitroblue tetrazolium reduction assaywas applied to whole blood in the presence of endotoxin (lipopolysaccha-rides,Escherichia coliO 55:B5, 10mg/mL for 15 minutes) orStaphylococ-cus epidermidis(107 colony-forming units (CFUs)/mL for 15 minutes).After red blood cell lysis, leukocytes were cytospun and stained withfuchsin, and the percentage of PMNs containing dark blue formazanprecipitates was counted microscopically.
The luminol-amplified chemiluminescence assay was performed afterstimulation of isolated PMNs with either fMLP (1026 M for 5 minutes) orphorbol-myristate acetate (PMA; 100 ng/mL for 5 minutes).
We also measured H2O2 production using a flow cytometric assayaccording to Bass and coworkers.14,15 This technique is based on theoxidation of the nonfluorescent 29,79-dichlorofluorescin into highly fluores-cent dichlorofluorescein (DCF), by H2O2 produced by PMA-stimulatedPMNs.
Spontaneous and directed migration under agarose, toward fMLP (1027
M) and activated serum, was measured as previously described.16
Flow cytometry analysis
Flow cytometry analysis was performed using a Becton Dickinson FACScan(Becton Dickinson Immunocytometry Systems) with a 15-mW, 488-nmargon laser. Forward and side scatters were used to identify the granulocytepopulation and to gate out other cells and debris. The purity of the gatedcells was assessed by using FITC- or PE-conjugated CD3, CD45, CD14,and CD15 antibodies (Becton Dickinson). The green fluorescence ofFITC-conjugated antibodies and DCF was recorded from 515 to 545 nm;the red fluorescence of PE-conjugated anti-CD11b was recorded from 563to 607 nm. In all cases, unstained cells were used and the photomultipliersettings were adjusted so that the unstained cell population appeared in thelower left-hand corner of the fluorescence display. All the results wereobtained with a constant photomultiplier gain. The data were analyzedusing LYSIS II software (Becton Dickinson) and the mean fluorescenceintensity (MFI) was used to quantify the responses. The results werecompared to the normal ranges obtained from members of the laboratorystaff as controls.
Results
Blood smears
Light microscopic examination of the blood smears of the 3children was performed. Platelets displayed a characteristic graycolor confirming the diagnosis of GPS. Furthermore, the PMNcytoplasm background staining was also gray with a degranulatedappearance compared with normal PMNs (Figure 1A,B). Theblood smears from the 2 parents were normal and platelets andPMNs displayed normal color.
GRAY PLATELET SYNDROME 1383BLOOD, 1 SEPTEMBER 2001 z VOLUME 98, NUMBER 5
For personal use only. by guest on February 1, 2014. bloodjournal.hematologylibrary.orgFrom

PMN cytochemical and ultrastructural examination
Because of thegray appearance on the smears of the PMN cytoplasm,the different neutrophil granule compartments were explored inthese patients. The cytochemical detection of the leukocyte enzymealkaline phosphatase (a specific component of the membrane ofsecretory vesicles) was consistently and strongly decreased com-pared to normal PMNs (Figure 1C,D), with a mean score of 5, 7,and 5, respectively, compared against a control mean of 62.
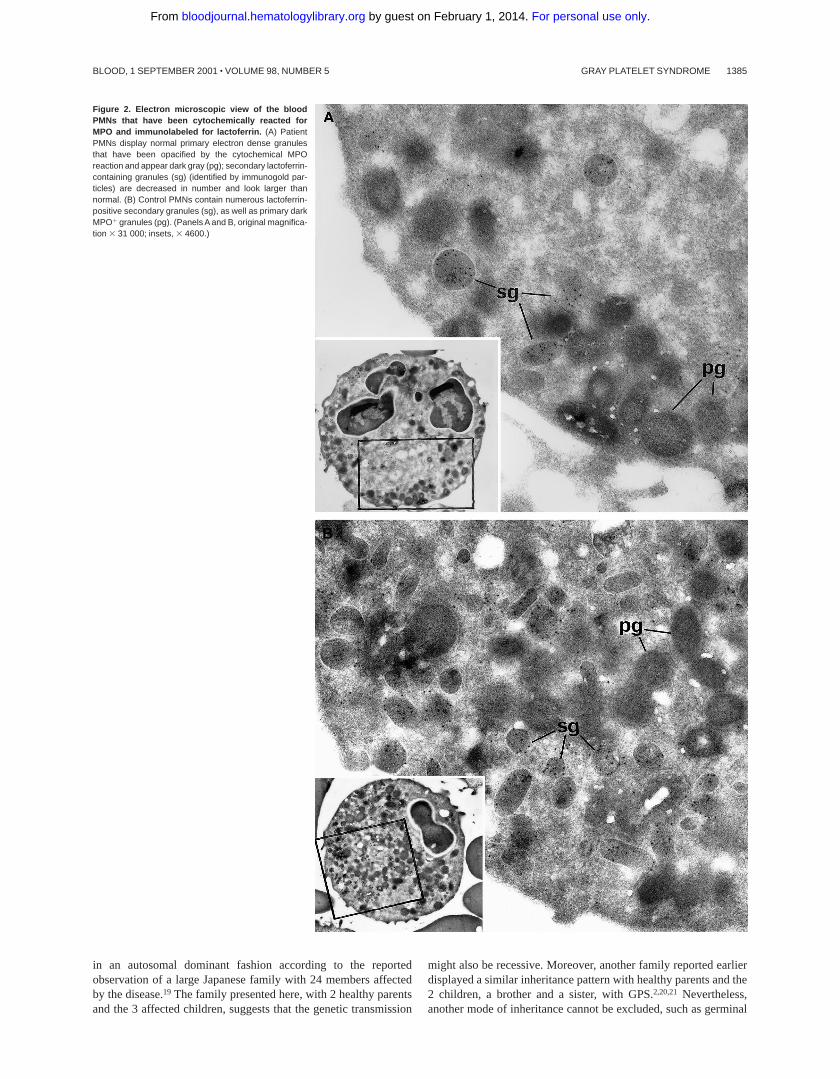
Myeloperoxidase (MPO)-positive primary granules were nor-mally present, as attested by cytochemical reactions observed byelectron microscopy. Immunolabeling for lactoferrin showed astrong decrease in secondary granules in the 2 patients whosePMNs could be studied by electron microscopy (Figure 2). Theratio of secondary/primary granules was diminished in thesepatients: control5 60%, patient Ju5 26%, patient Me5 21%.Control immunolabeling was performed by omitting the primaryantibody in the reaction that led to the absence of gold particles onthe structures.
PMN flow cytometry and functional studies
Because alkaline phosphatase is a component of the membrane ofsecretory vesicles, which was undetectable in the 3 patients’PMNs,17,18 we investigated the distribution of the other specificcomponents of secretory vesicles—the complement receptor CD35,which is normally restricted to the membrane of secretory vesicles;the integrins CD11b/CD18, which are expressed within bothsecretory vesicles and specific granules. The expression of CD35,CD11b, and CD18 (Table 1) was increased in the patients at thebasal state when compared to controls. The membrane expressionfor those proteins was maximal on unstimulated neutrophils anddid not increase on stimulation by fMLP as is the case in normalneutrophils. Interestingly, even if CD11b and CD18 expressionswere highly expressed at the membrane in the PMN basal state, andbarely increased after fMLP stimulation, the level of CD11b andCD18 at the membrane did not reach more than 30% of the control
levels. The high basal expression of these receptors in the patientscould not be explained by in vivo or in vitro PMN activation,because L-selectin expression remained in the normal range:controls5 166616; patient Me5 137; patient Ju5 145.
Functional studies of PMN leukocytes were normal. Oxidaseactivity after PMA, fMLP, endotoxin, and bacterial stimulation aswell as spontaneous and oriented migration were studied in the 3children and did not differ from normal controls (data not shown).
Electron microscopy of platelets and MKs
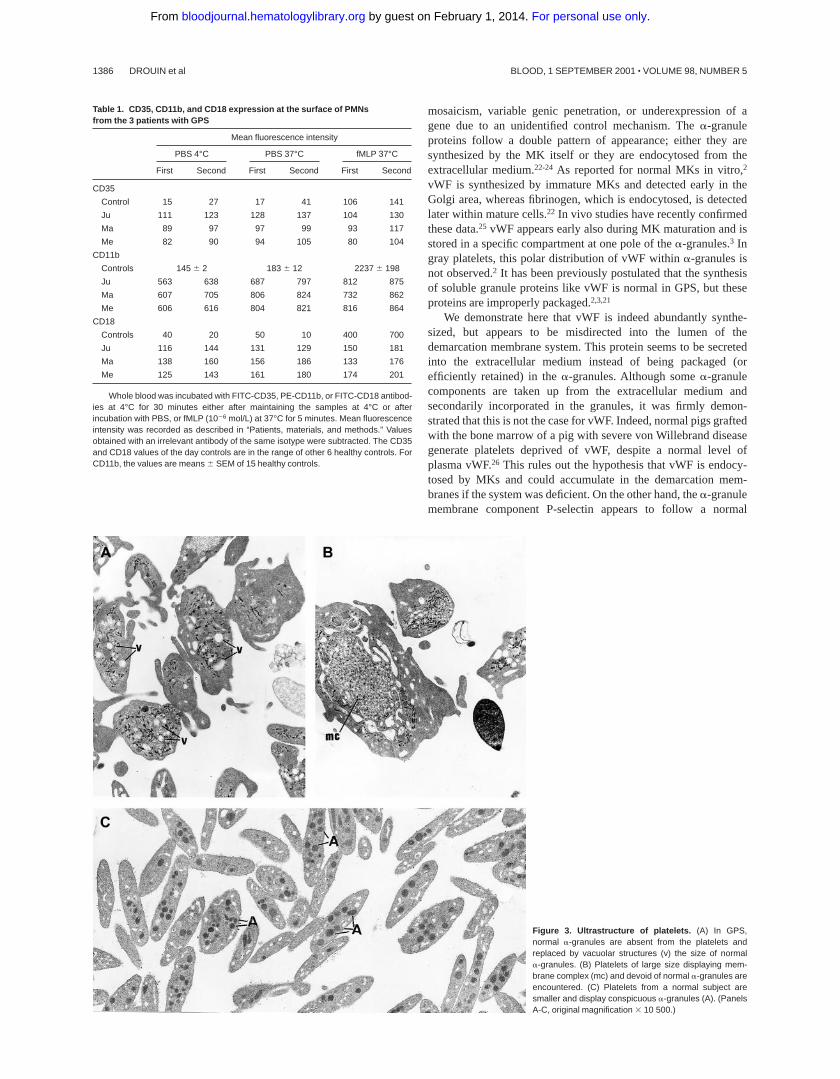
The platelets from the 3 children displayed similar ultrastructure.Platelet size was heterogeneous, and many platelets were largerthan a red blood cell or a small lymphocyte. The most strikingmorphologic abnormality was the lack of normala-granules andprominent vacuolization of the cytoplasm (Figure 3A). Membranecomplexes, which represent abnormally distributed intracellularmembrane, were also observed (Figure 3B).
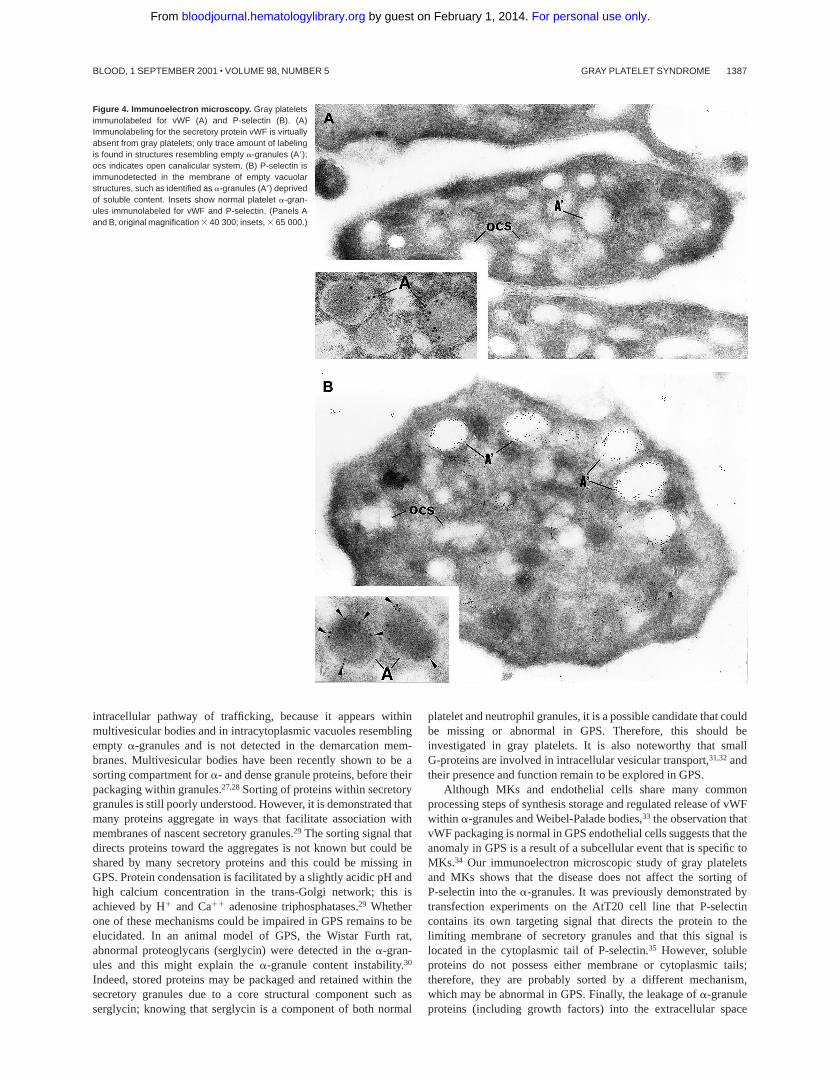
Intracellular trafficking ofa-granule proteins was studied byimmunoelectron microscopy in the 3 patients. First, gray plateletswere examined for the secretory protein vWF; it was virtuallyabsent from all putative emptya-granules (Figure 4A). In contrast,in gray platelets, P-selectin was immunodetected in the membraneof empty vacuolar structures, thus identified asa-granules deprivedof soluble content (Figure 4B).
Then, intracellular trafficking ofa-granule proteins was studiedby immunoelectron microscopy in the 3 patients. In maturing GPSMKs, vWF immunolabeling appeared to be strongly detectedwithin the Golgi complex, associated vesicles, and small granules(not shown), and heavy labeling was found within the lumen of thedemarcation membrane system (Figure 5A). This characteristiclabeling has also been observed in the cultured MKs from the 3patients suggesting that in GPS, vWF is abundantly synthesized,not packaged or not retained ina-granules, misdirected toward thedemarcation membrane system, and constitutively secreted into theextracellular medium. In maturing GPS MKs, P-selectin wasdetected within some vesicles arising from the Golgi complex (notshown) and nascenta-granules, which became electron lucentduring maturation (Figure 5B). It was also abundantly detected inthe multivesicular bodies and in the membrane of occasionalvacuoles (Figure 5B). Thus, and in contrast to vWF, P-selectinappeared to be normally targeted toward the membrane of vacuolarstructures previously identified as emptya-granules. Doubleimmunolabeling confirmed that P-selectin and vWF were follow-ing a distinct pattern of intracellular trafficking; P-selectin wasfound in the multivesicular bodies, where no GPIb was present andwere thus distinct from demarcation membranes (Figure 6A). vWFwas detected in the lumen of demarcation membranes, identified bytheir GPIb content (Figure 6B); multivesicular bodies were virtu-ally deprived of vWF. Double labeling for vWF and P-selectinconfirmed that the 2 proteins had followed divergent pathwaysbecause P-selectin was detected in multivesicular bodies, whereasvWF was found in the elongated channels of the demarcationmembrane system (Figure 6C).
Control immunolabeling was performed by omitting the pri-mary antibody in the reaction, which led to the absence of goldparticles on the structures.
Discussion
This study describes a new family of 3 children with GPS. GPS is arare congenital disorder, which can be either sporadic or inherited
Figure 1. Light microscopy of blood smears. (A) In Romanovsky-stained smearsfrom a normal subject, the PMNs have a granular cytoplasm and a platelet displaysgranules in the cytoplasm (arrow). (B) In similarly stained smears from a patient withGPS, the PMN cytoplasm appears agranular and platelets are also degranulated(arrow). (C,D) Cytochemical detection of leukocyte alkaline phosphatase. PMNstaining intensity is easily detected in the normal state (C) but strongly decreased inGPS compared to normal neutrophils (D).
1384 DROUIN et al BLOOD, 1 SEPTEMBER 2001 z VOLUME 98, NUMBER 5
For personal use only. by guest on February 1, 2014. bloodjournal.hematologylibrary.orgFrom
in an autosomal dominant fashion according to the reportedobservation of a large Japanese family with 24 members affectedby the disease.19 The family presented here, with 2 healthy parentsand the 3 affected children, suggests that the genetic transmission
might also be recessive. Moreover, another family reported earlierdisplayed a similar inheritance pattern with healthy parents and the2 children, a brother and a sister, with GPS.2,20,21 Nevertheless,another mode of inheritance cannot be excluded, such as germinal
Figure 2. Electron microscopic view of the bloodPMNs that have been cytochemically reacted forMPO and immunolabeled for lactoferrin. (A) PatientPMNs display normal primary electron dense granulesthat have been opacified by the cytochemical MPOreaction and appear dark gray (pg); secondary lactoferrin-containing granules (sg) (identified by immunogold par-ticles) are decreased in number and look larger thannormal. (B) Control PMNs contain numerous lactoferrin-positive secondary granules (sg), as well as primary darkMPO1 granules (pg). (Panels A and B, original magnifica-tion 3 31 000; insets, 3 4600.)
GRAY PLATELET SYNDROME 1385BLOOD, 1 SEPTEMBER 2001 z VOLUME 98, NUMBER 5
For personal use only. by guest on February 1, 2014. bloodjournal.hematologylibrary.orgFrom
mosaicism, variable genic penetration, or underexpression of agene due to an unidentified control mechanism. Thea-granuleproteins follow a double pattern of appearance; either they aresynthesized by the MK itself or they are endocytosed from theextracellular medium.22-24 As reported for normal MKs in vitro,2
vWF is synthesized by immature MKs and detected early in theGolgi area, whereas fibrinogen, which is endocytosed, is detectedlater within mature cells.22 In vivo studies have recently confirmedthese data.25 vWF appears early also during MK maturation and isstored in a specific compartment at one pole of thea-granules.3 Ingray platelets, this polar distribution of vWF withina-granules isnot observed.2 It has been previously postulated that the synthesisof soluble granule proteins like vWF is normal in GPS, but theseproteins are improperly packaged.2,3,21
We demonstrate here that vWF is indeed abundantly synthe-sized, but appears to be misdirected into the lumen of thedemarcation membrane system. This protein seems to be secretedinto the extracellular medium instead of being packaged (orefficiently retained) in thea-granules. Although somea-granulecomponents are taken up from the extracellular medium andsecondarily incorporated in the granules, it was firmly demon-strated that this is not the case for vWF. Indeed, normal pigs graftedwith the bone marrow of a pig with severe von Willebrand diseasegenerate platelets deprived of vWF, despite a normal level ofplasma vWF.26 This rules out the hypothesis that vWF is endocy-tosed by MKs and could accumulate in the demarcation mem-branes if the system was deficient. On the other hand, thea-granulemembrane component P-selectin appears to follow a normal
Table 1. CD35, CD11b, and CD18 expression at the surface of PMNsfrom the 3 patients with GPS
Mean fluorescence intensity
PBS 4°C PBS 37°C fMLP 37°C
First Second First Second First Second
CD35
Control 15 27 17 41 106 141
Ju 111 123 128 137 104 130
Ma 89 97 97 99 93 117
Me 82 90 94 105 80 104
CD11b
Controls 145 6 2 183 6 12 2237 6 198
Ju 563 638 687 797 812 875
Ma 607 705 806 824 732 862
Me 606 616 804 821 816 864
CD18
Controls 40 20 50 10 400 700
Ju 116 144 131 129 150 181
Ma 138 160 156 186 133 176
Me 125 143 161 180 174 201
Whole blood was incubated with FITC-CD35, PE-CD11b, or FITC-CD18 antibod-ies at 4°C for 30 minutes either after maintaining the samples at 4°C or afterincubation with PBS, or fMLP (1026 mol/L) at 37°C for 5 minutes. Mean fluorescenceintensity was recorded as described in “Patients, materials, and methods.” Valuesobtained with an irrelevant antibody of the same isotype were subtracted. The CD35and CD18 values of the day controls are in the range of other 6 healthy controls. ForCD11b, the values are means 6 SEM of 15 healthy controls.
Figure 3. Ultrastructure of platelets. (A) In GPS,normal a-granules are absent from the platelets andreplaced by vacuolar structures (v) the size of normala-granules. (B) Platelets of large size displaying mem-brane complex (mc) and devoid of normal a-granules areencountered. (C) Platelets from a normal subject aresmaller and display conspicuous a-granules (A). (PanelsA-C, original magnification 3 10 500.)
1386 DROUIN et al BLOOD, 1 SEPTEMBER 2001 z VOLUME 98, NUMBER 5
For personal use only. by guest on February 1, 2014. bloodjournal.hematologylibrary.orgFrom
intracellular pathway of trafficking, because it appears withinmultivesicular bodies and in intracytoplasmic vacuoles resemblingempty a-granules and is not detected in the demarcation mem-branes. Multivesicular bodies have been recently shown to be asorting compartment fora- and dense granule proteins, before theirpackaging within granules.27,28Sorting of proteins within secretorygranules is still poorly understood. However, it is demonstrated thatmany proteins aggregate in ways that facilitate association withmembranes of nascent secretory granules.29 The sorting signal thatdirects proteins toward the aggregates is not known but could beshared by many secretory proteins and this could be missing inGPS. Protein condensation is facilitated by a slightly acidic pH andhigh calcium concentration in the trans-Golgi network; this isachieved by H1 and Ca11 adenosine triphosphatases.29 Whetherone of these mechanisms could be impaired in GPS remains to beelucidated. In an animal model of GPS, the Wistar Furth rat,abnormal proteoglycans (serglycin) were detected in thea-gran-ules and this might explain thea-granule content instability.30
Indeed, stored proteins may be packaged and retained within thesecretory granules due to a core structural component such asserglycin; knowing that serglycin is a component of both normal
platelet and neutrophil granules, it is a possible candidate that couldbe missing or abnormal in GPS. Therefore, this should beinvestigated in gray platelets. It is also noteworthy that smallG-proteins are involved in intracellular vesicular transport,31,32andtheir presence and function remain to be explored in GPS.
Although MKs and endothelial cells share many commonprocessing steps of synthesis storage and regulated release of vWFwithin a-granules and Weibel-Palade bodies,33 the observation thatvWF packaging is normal in GPS endothelial cells suggests that theanomaly in GPS is a result of a subcellular event that is specific toMKs.34 Our immunoelectron microscopic study of gray plateletsand MKs shows that the disease does not affect the sorting ofP-selectin into thea-granules. It was previously demonstrated bytransfection experiments on the AtT20 cell line that P-selectincontains its own targeting signal that directs the protein to thelimiting membrane of secretory granules and that this signal islocated in the cytoplasmic tail of P-selectin.35 However, solubleproteins do not possess either membrane or cytoplasmic tails;therefore, they are probably sorted by a different mechanism,which may be abnormal in GPS. Finally, the leakage ofa-granuleproteins (including growth factors) into the extracellular space
Figure 4. Immunoelectron microscopy. Gray plateletsimmunolabeled for vWF (A) and P-selectin (B). (A)Immunolabeling for the secretory protein vWF is virtuallyabsent from gray platelets; only trace amount of labelingis found in structures resembling empty a-granules (A9);ocs indicates open canalicular system. (B) P-selectin isimmunodetected in the membrane of empty vacuolarstructures, such as identified as a-granules (A9) deprivedof soluble content. Insets show normal platelet a-gran-ules immunolabeled for vWF and P-selectin. (Panels Aand B, original magnification 3 40 300; insets, 3 65 000.)
GRAY PLATELET SYNDROME 1387BLOOD, 1 SEPTEMBER 2001 z VOLUME 98, NUMBER 5
For personal use only. by guest on February 1, 2014. bloodjournal.hematologylibrary.orgFrom
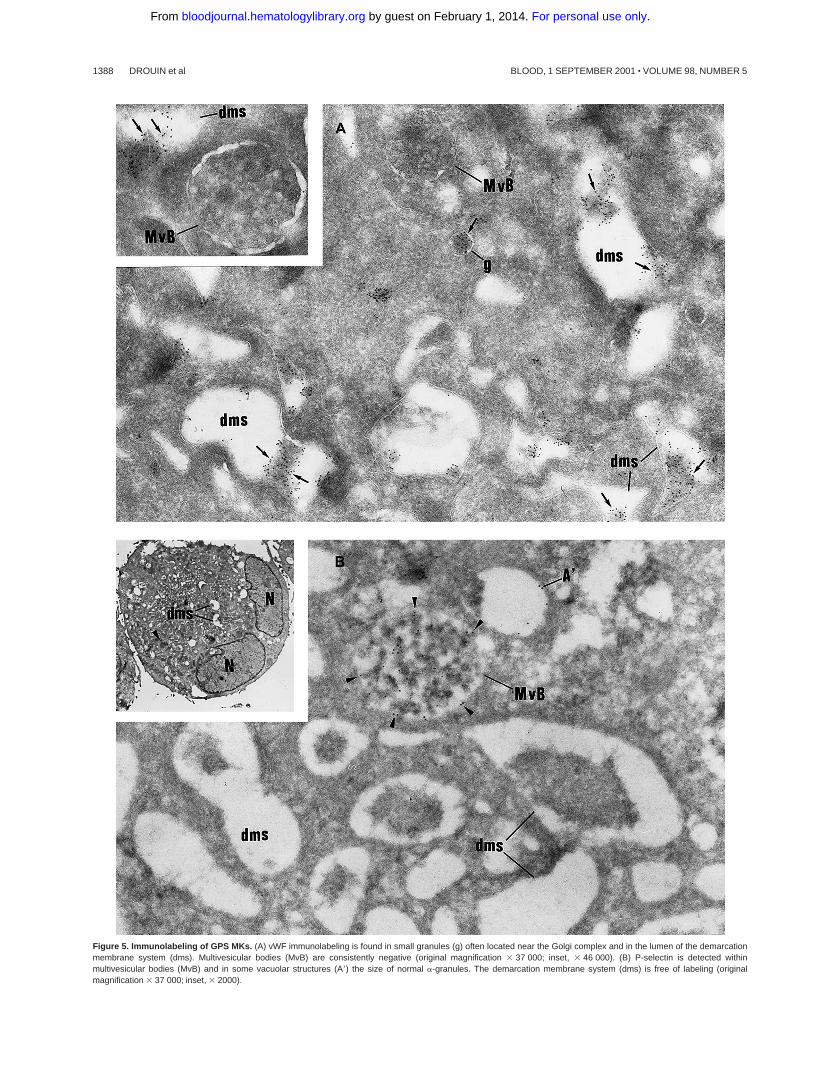
Figure 5. Immunolabeling of GPS MKs. (A) vWF immunolabeling is found in small granules (g) often located near the Golgi complex and in the lumen of the demarcationmembrane system (dms). Multivesicular bodies (MvB) are consistently negative (original magnification 3 37 000; inset, 3 46 000). (B) P-selectin is detected withinmultivesicular bodies (MvB) and in some vacuolar structures (A9) the size of normal a-granules. The demarcation membrane system (dms) is free of labeling (originalmagnification 3 37 000; inset, 3 2000).
1388 DROUIN et al BLOOD, 1 SEPTEMBER 2001 z VOLUME 98, NUMBER 5
For personal use only. by guest on February 1, 2014. bloodjournal.hematologylibrary.orgFrom
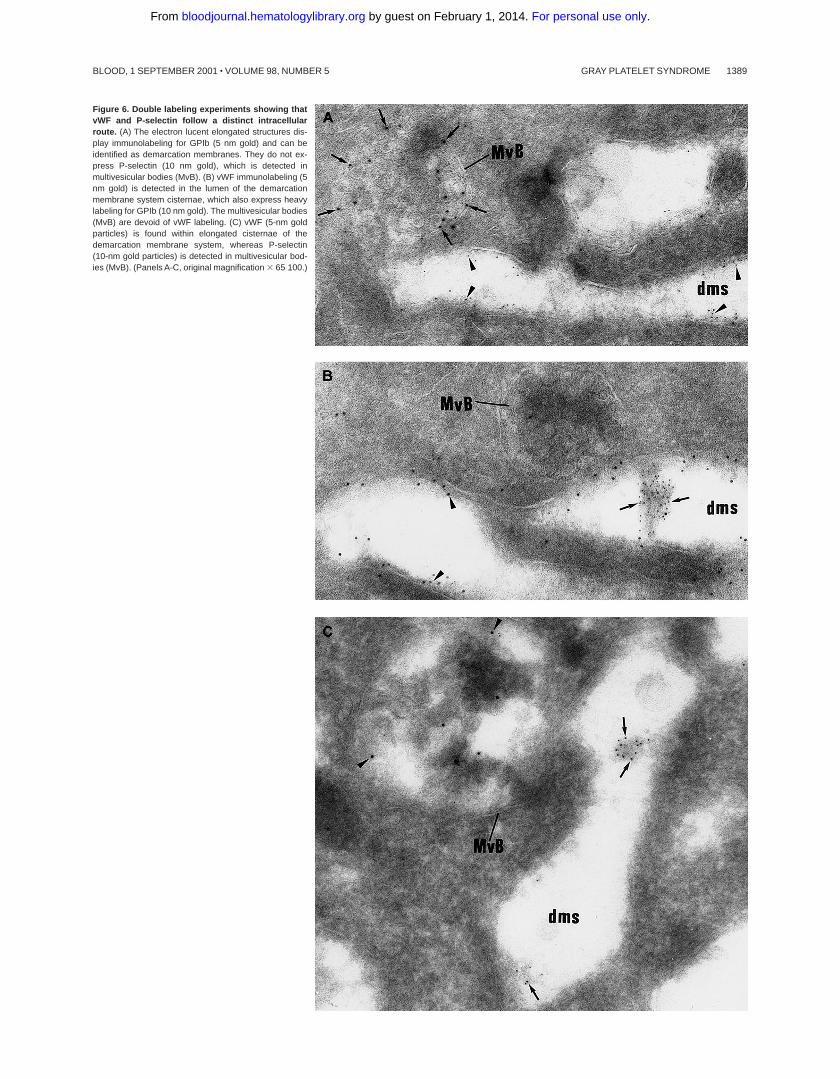
Figure 6. Double labeling experiments showing thatvWF and P-selectin follow a distinct intracellularroute. (A) The electron lucent elongated structures dis-play immunolabeling for GPIb (5 nm gold) and can beidentified as demarcation membranes. They do not ex-press P-selectin (10 nm gold), which is detected inmultivesicular bodies (MvB). (B) vWF immunolabeling (5nm gold) is detected in the lumen of the demarcationmembrane system cisternae, which also express heavylabeling for GPIb (10 nm gold). The multivesicular bodies(MvB) are devoid of vWF labeling. (C) vWF (5-nm goldparticles) is found within elongated cisternae of thedemarcation membrane system, whereas P-selectin(10-nm gold particles) is detected in multivesicular bod-ies (MvB). (Panels A-C, original magnification 3 65 100.)
GRAY PLATELET SYNDROME 1389BLOOD, 1 SEPTEMBER 2001 z VOLUME 98, NUMBER 5
For personal use only. by guest on February 1, 2014. bloodjournal.hematologylibrary.orgFrom

seems to be responsible for the development of myelofibrosis,which is a virtually constant finding in GPS.36,37
We have also extensively studied PMNs of the 3 childrenbecause of the gray staining with Romanovsky stain. Interestingly,a retrospective study of the blood smears from 2 previouslypublished cases indicated the same gray color of PMNs. Due to thesimilar staining properties between platelets and PMNs, we haveinvestigated the PMN granules. At least 3 well-defined secretoryorganelles have been described in PMNs: primary granules thatcontain MPO, secondary granules that contain lactoferrin, andsecretory vesicles that contain the alkaline phosphatase and CD35receptor in their membrane and plasma proteins (albumin) in theirlumen.17,38,39
The 3 children with GPS presented an impairment of thesecretory vesicle and secondary granule compartments, whereasMPO-containing primary granules were normally present. Thecytochemical activity of the leukocyte alkaline phosphatase wasexamined in the 3 GPS children, their 2 parents, and the 2previously reported patients.2 In the 5 patients this enzymaticcytochemical activity was very low, only barely detectable, whereasit was normal in the parents. In addition, secretory vesicles providean intracellular CD35 reservoir, from which this membrane proteinmay be recruited to the cell surface after fMLP stimulation.17 Ourflow cytometric studies showed a maximal CD35 expression at thesurface of the patient PMNs, in their resting state. This expressiondid not increase after fMLP stimulation. The high basal expressionof CD35 without up-regulation suggests that during granulopoiesis,newly synthesized CD35 was not retained in the secretory vesicles,but was routed to the plasma membrane. This is in accordance withthe findings that a promyelocytic cell line, NB4 cells, forced todifferentiate into neutrophils with dimethyl sulfoxide or retinoicacid, lacks specific granules, whereas some of the cell line’sendogenous protein content localizes to the plasma membrane.40,41
The integrin CD11b/CD18 is present in both secretory vesicles andspecific granules. It is also up-regulated by degranulation in a
hierarchical manner, playing a key role in the different stepsleading to PMN infiltration of inflammatory sites. In the childrenwith GPS, CD11b/CD18 is also highly expressed at the membranein the basal resting state. This is in accordance with the ultrastruc-tural observation of a decrease in specific granules.17 The highbasal membrane expression of CD35, CD11b/CD18 was not due toPMN activation because L-selectin was not decreased. Indeed, thisadhesion molecule is constitutively expressed at the PMN plasmamembrane, and on activation, it is shed from the membrane byproteolysis.
The importance of the specific granules in neutrophil functionhas been shown in patients who lack specific granules.42 Thesepatients are susceptible to repeated skin and respiratory infectionsand have defective neutrophil chemotaxis and adhesion. To ourknowledge, the clinical consequences of secretory vesicle defect inpatients has not been described. In the present cases, one of thechildren has suffered many respiratory infections but not the others.
This study describes a new association between thrombocytope-nia and PMN abnormalities. Noteworthy, such an association ispresent in the May-Hegglin syndrome,43 Fechtner syndrome (avariant of Alport syndrome with leukocyte inclusions and macro-thrombocytopenia),44 and Chediak-Higashi syndrome.45 The obser-vation that the secretory compartments of 2 different lineages areaffected by the same pathologic process in GPS strongly suggests asimilar molecular defect in the common hematopoietic precursor.
Acknowledgments
The authors gratefully acknowledge Dr Fadila Souni for the lightmicroscopic work on PMNs, Pr Jacques Caen for allowing theretrospective study of the PMNs of 2 GPS patients, Dr PaulHarrison for improving the manuscript, and Dr Paul-Henri Romeofor his constant support.
References
1. Raccuglia G. Gray platelet syndrome: a variety ofqualitative platelet disorders. Am J Med. 1971;51:818-828.
2. Cramer EM, Vainchenker W, Vinci G, Guichard J,Breton-Gorius J. Gray platelet syndrome: immu-noelectron microscopic localisation of fibrinogenand von Willebrand factor in platelets andmegakaryocytes. Blood. 1985;66:1309-1316.
3. Rosa JP, George JN, Bainton DF, Nurden AT,Caen JP, McEver RP. Gray platelet syndrome:demonstration of a-granule membranes that canfuse with the cell surface. J Clin Invest. 1987;80:1138-1146.
4. Weiss HJ, Witte LD, Kaplan KL, et al. Heteroge-neity in storage pool deficiency: studies on gran-ule bound substances in 18 patients includingvariants deficient in alpha granules, platelet factor4, beta-thromboglobulin and platelet derivedgrowth factor. Blood. 1979;54:1296-1319.
5. Hayward CP, Cramer EM, Kane WH, et al. Stud-ies of a second family with the Quebec plateletdisorder: evidence that the degradation of thea-granule membrane and its soluble contents arenot secondary to a defect in targeting proteins toa-granules. Blood. 1997;89:1243-1253.
6. Harrison P, Cramer EM. Platelet a-granules.Blood Rev. 1993;7:52-62.
7. Harrison P, Savidge GF, Cramer EM. Annotation:the origin and physiological relevance of a-gran-ule adhesive proteins. Br J Haematol. 1990;74:125-130.
8. Cramer EM, Norol F, Guichard J, et al. Ultrastruc-
ture of platelet formation by human megakaryo-cytes cultured with the Mpl ligand. Blood. 1997;89:2336-2346.
9. Dang PM, Dewas C, Gaudry M, et al. Priming ofhuman neutrophil respiratory burst by granulo-cyte/macrophage colony-stimulating factor (GM-CSF) involves partial phosphorylation ofp47(phox). J Biol Chem. 1999;274:20704-20708.
10. Berger G, Masse JM, Cramer EM. Alpha-granulemembrane mirrors the platelet plasma membraneand contains the glycoproteins Ib, IX, and V.Blood. 1996;87:1385-1396.
11. Gamble JR, Skinner MP, Berndt MC, Vadas MA.Prevention of activated neutrophil adhesion toendothelium by soluble adhesion proteinGMP140. Science. 1990;249:414-417.
12. Bendayan M, Roth J, Perrelet A, Orci L. Quantita-tive immunocytochemical localization of pancre-atic secretory proteins in subcellular compart-ments of the rat acinar cell. J HistochemCytochem. 1980;28:149-160.
13. Slot JW, Geuze HJ. Immunoelectron microscopicexploration of the Golgi complex. J HistochemCytochem. 1983;31:1049-1056.
14. Bass DA, Parce JW, Dechatelet LR, Szejda P,Seeds MC, Thomas M. Flow cytometric studies ofoxidative product formation by neutrophils: agraded response to membrane stimulation. J Im-munol. 1983;130:1910-1917.
15. Elbim C, Prevot MH, Bouscarat F, et al. PMNfrom HIV-infected patients show enhanced acti-vation, diminished fMLP-induced L-selectin shed-
ding and an impaired oxidative burst after cyto-kine priming. Blood. 1994;84:2759-2766.
16. Amar M, Amit N, Pham Huu T, et al. Productionby K562 cells of an inhibitor of adherence-relatedfunctions of human neutrophils. J Immunol. 1990;144:4749-4756.
17. Sengelov H, Kjeldsen L, Kroeze W, Berger M,Borregaard N. Secretory vesicles are the intracel-lular reservoir of complement receptor 1 in humanneutrophils. J Immunol. 1994;153:804-810.
18. Witko-Sarsat V, Cramer EM, Hieblot C, et al.Presence of proteinase 3 in secretory vesicles:evidence of a novel, highly mobilizable intracellu-lar pool distinct from azurophil granules. Blood.1999;94:2487-2496.
19. Mori K, Suzuki S, Sugai K. Electron microscopicand functional studies on platelets in gray plateletsyndrome. Tohoku J Exp Med. 1984;143:261-287.
20. Levy-Toledano S, Caen JP, Breton-Gorius J, et al.Gray platelet syndrome: a-granule deficiency. Itsinfluence on platelet function. J Lab Clin Med.1981;98:831-848.
21. Breton-Gorius J, Vainchenker W, Nurden A, Levy-Toledano S, Caen J. Defective a-granule produc-tion in megakaryocytes from gray platelet syn-drome. Am J Pathol. 1981;102:10-19.
22. Cramer EM, Debili N, Martin JF, et al. Uncoordi-nated expression of fibrinogen compared withthrombospondin and von Willebrand factor in ma-turing human megakaryocytes. Blood. 1989;73:1123-1129.
1390 DROUIN et al BLOOD, 1 SEPTEMBER 2001 z VOLUME 98, NUMBER 5
For personal use only. by guest on February 1, 2014. bloodjournal.hematologylibrary.orgFrom

23. Harrison P, Wilbourn BR, Debili N, et al. Uptake ofplasma fibrinogen into the a-granules of humanmegakaryocytes. J Clin Invest. 1989;84:1320-1324.
24. Handagama P, Rappolee DA, Werb Z, Levin J,Bainton DF. Platelet a-granule fibrinogen, albu-min, and immunoglobulin G are not synthesizedby rat and mouse megakaryocytes. J Clin Invest.1990;86:1364-1368.
25. de Larouziere V, Brouland JP, Souni F, Drouet L,Cramer E. Inverse immunostaining pattern forsynthesized versus endocytosed alpha-granuleproteins in human bone marrow megakaryocytes.Br J Haematol. 1998;101:118-125.
26. Roussi J, Drouet L, Sigman J, et al. Absence ofincorporation of plasma von Willebrand factor intoporcine platelet alpha-granules. Br J Haematol.1995;90:661-668.
27. Heijnen HF, Debili N, Vainchencker W, Breton-Gorius J, Geuze HJ, Sixma JJ. Multivesicularbodies are an intermediate stage in the formationof platelet alpha-granules. Blood. 1998;91:2313-2325.
28. Youssefian T, Cramer EM. Multivesicular bodiesas a sorting compartment for alpha and densegranule components. Blood. 2000;95:4004-4007.
29. Rindler MJ. Biogenesis of storage granules andvesicles. Curr Opin Cell Biol. 1992;4:616-622.
30. Schick BP, Pestina TI, San Antonio JD, StenbergPE, Jackson CW. Decreased serglycin proteogly-can size is associated with the platelet alphagranule storage defect in Wistar Furth hereditarymacrothrombocytopenic rats: serglycin binding
affinity to type I collagen is unaltered. J CellPhysiol. 1997;172:87-93.
31. Balch WB. Small GTP binding proteins in vesicu-lar transport. Trends Biochem Sci. 1990;15:473-477.
32. Chavrier P, Goud B. The role of ARF and RabGTPases in membrane transport. Curr Opin CellBiol. 1999;11:466-475.
33. Wagner D, Marder V. Biosynthesis of von Wille-brand protein by human endothelial cells: pro-cessing steps and their intracellular localizationJ Cell Biol. 1984;99:2123-2130.
34. Gebrane-Younes J, Cramer EM, Orcel J, CaenJP. Gray platelet syndrome: dissociation betweenabnormal sorting in megakaryocyte a-granulesand normal sorting in Weibel-Palade bodies ofendothelial cells. J Clin Invest. 1993;92:3023-3028.
35. Disdier M, Morrissey JH, Fugate RD, Bainton DF,McEver RP. Cytoplasmic domain of P-selectin(CD62) contains the signal for sorting into theregulated secretory pathway. Mol Biol Cell. 1992;3:309-321.
36. Castro-Malaspina H. Pathogenesis of myelofibro-sis: role of ineffective megakaryopoiesis andmegakaryocyte components. Prog Clin Biol Res.1984;154:427-454.
37. Schmitt A, Jouault H, Drouin A, Guichard J, Wen-dling F, Cramer EM. Pathological interaction be-tween megakaryocytes and PMN leukocytes inmyelofibrosis. Blood. 2000;96:1342-1347.
38. Borregaard N, Lollike K, Kjeldsen L, et al. Human
neutrophil granules and secretory vesicles. Eur JHaematol. 1993;4:187-198.
39. Borregaard N, Cowland JB. Granules of the hu-man neutrophilic polymorphonuclear leukocyte.Blood. 1997;89:3503-3521.
40. Gregoire C, Welch H, Astarie-Dequeker C, Mari-donneau-Parini I. Expression of azurophil andspecific granule proteins during differentiation ofNB4 cells in neutrophils. J Cell Physiol. 1998;175:203-210.
41. Gianni M, Terao M, Zanotta S, Barbui T, Ram-baldi A, Garattini E. Retinoic acid and granulo-cyte colony-stimulating factor synergisticallyinduce leukocyte alkaline phosphatase in acutepromyelocytic leukemia cells. Blood. 1994;83:1909-1921.
42. Ganz T, Metcalf JA, Gallin JI, Boxer LA, LehrerRI. Microbicidal/cytotoxic proteins of neutrophilsare deficient in two disorders: Chediak-Higashisyndrome and “specific” granule deficiency. J ClinInvest. 1988;82:552-556.
43. Noris P, Spedini P, Belletti S, Magrini U, BalduiniCL. Thrombocytopenia, giant platelets, and leu-kocyte inclusion bodies (May-Hegglin anomaly):clinical and laboratory findings. Am J Med. 1998;104:355-360.
44. Peterson LC, Rao KV, Crosson JT, White JG.Fechtner syndrome, a variant of Alport’s syn-drome with leukocyte inclusions and macrothrom-bocytopenia. Blood. 1985;65:397-406.
45. Introne W, Boissy RE, Gahl WA. Clinical, molecu-lar, and cell biological aspects of Chediak-Higashisyndrome. Mol Genet Metab. 1999;68:283-303.
GRAY PLATELET SYNDROME 1391BLOOD, 1 SEPTEMBER 2001 z VOLUME 98, NUMBER 5
For personal use only. by guest on February 1, 2014. bloodjournal.hematologylibrary.orgFrom




















