
New disc-based technologies for diagnostic and research applications
14
New disc-based technologies for diagnostic and research applications Raj Barathur, Jack Bookout, Srinand Sreevatsan, John Gordon, Martina Werner, Gautam Thor and Mark Worthington Burstein Technologies Inc., Irvine, California, USA Correspondence to Raj Barathur, Burstein Technologies Inc., 163 Technology Drive, Irvine, California 92618, USA. E-mail: rajbarathur@bursteinlabs.com Received 22 January 2002; accepted 19 June 2002 The role of genotypic analysis in disease diagnostics and drug response assessment is continually expanding. New genomic discoveries combined with new, novel technologies may provide a greater range of testing capabilities in the near future. We describe the application of nanotechnology, in which DNA microarrays have been placed in a microchannel environment that can be read and analyzed in an optical (CD/DVD) disc drive system. The potential exists to combine molecular and immunological applications together into a rapid, low-cost, high-capacity screening platform. The relevance of this technology is discussed in respect to infectious agent detection, pharmacogenomics, neurogenomics and genetic variations associated with neurologic diseases. Psychiatr Genet 12:193–206 r 2002 Lippincott Williams & Wilkins. Keywords: CD/DVD discs, genotyping, gene variants, MEMS, microarrays, nanotechnology, neurogenomics, neurologic disorders, optical discs, pathogen detection, pharmacogenomics INTRODUCTION The past decade has seen the rapid development of BioMEMS (MicroElectroMechanical Systems) and nanotechnology, driven by ever-increasing screening requirements of the pharmaceutical/pharmaco- genomics industry and by long-held interests by the medical community for cost-effective point of care diagnostic instrumentation (Weber, 1997; Kleyn and Wesell, 1998; Kost, 2000). To meet these demands, many new plat- form technologies have been created, of which a number are reviewed in this volume. The basis for many of these advancements is the adaptation of micro-electronic technologies developed for the com- puting industry. For example, two major contribu- tions from this source are the microchip manufacturing (from which MEMS fabrication technologies evolved) and optical information and data storage devices (providing advances in instrument miniaturization with increased analy- tical capacities) (Fodor et al., 1993; Kido et al., 2000). Microchip and micro-array based platforms have received recent attention due to their relatively high-capacity sequence interrogation capabilities, and a number of research groups and companies have utilized this approach (e.g. Affymetrix Inc., Genetic MicroSystems, Gene Logic Inc., Incyte Pharmaceuticals Inc.) (Bowtell, 1999). Relatively less attention has been given to the parallel development of a smaller group of companies and researchers adapting optical disc technologies for biological and biochemical analysis (Kiddo et al., 2000) (Gyros AB, Tecan-Gamera Bioscience, Abaxis Inc., Burstein Technologies Inc.). All of these groups have seen in compact disc (CD) technology the potential for developing rapid, low-cost, high-capacity screening systems by adapting existing industry standard technology. By packaging different assay formats on disc, the goal of achieving a generic format capable of suppor- ting multiple applications would be realized. The present paper describes progress towards this end by Burstein Technologies Inc. (BTI) on molecular and immunological assays, and the future potential of this technology. The platform described en- ables physicians/laboratories to use specially designed CD discs and a CD/DVD reader attached to an ordinary personal computer to deliver a wide range of tests including clinical laboratory diagnostics, biological warfare agent detection, for- ensic DNA tests, and food and water contamination tests. 0955-8829 r 2002 Lippincott Williams & Wilkins Psychiatric Genetics 2002, Vol 12 No 4 193 Psychiatric Genetics 12:193–206 Copyright © Lippincott Williams & Wilkins. Unauthorized reproduction of this article is prohibited.
-
Upload
spanalumni -
Category
Documents
-
view
1 -
download
0
Transcript of New disc-based technologies for diagnostic and research applications

New disc-based technologies for diagnostic and researchapplications
Raj Barathur, Jack Bookout, Srinand Sreevatsan, John Gordon, Martina Werner,GautamThor and Mark Worthington
Burstein Technologies Inc., Irvine, California, USA
Correspondence to Raj Barathur, Burstein Technologies Inc., 163 Technology Drive, Irvine,California 92618, USA. E-mail: [email protected]
Received 22 January 2002; accepted 19 June 2002
The role of genotypic analysis in disease diagnostics and drug response assessment is continually expanding. New genomicdiscoveries combined with new, novel technologies may provide a greater range of testing capabilities in the near future. Wedescribe the application of nanotechnology, in which DNA microarrays have been placed in a microchannel environment thatcan be read and analyzed in an optical (CD/DVD) disc drive system. The potential exists to combine molecular andimmunological applications together into a rapid, low-cost, high-capacity screening platform. The relevance of this technologyis discussed in respect to infectious agent detection, pharmacogenomics, neurogenomics and genetic variations associated withneurologic diseases. Psychiatr Genet 12:193–206 r 2002 Lippincott Williams & Wilkins.
Keywords: CD/DVD discs, genotyping, gene variants, MEMS, microarrays, nanotechnology, neurogenomics, neurologicdisorders, optical discs, pathogen detection, pharmacogenomics
INTRODUCTION
The past decade has seen the rapid development ofBioMEMS (MicroElectroMechanical Systems) andnanotechnology, driven by ever-increasing screeningrequirements of the pharmaceutical/pharmaco-genomics industry and by long-held interests by themedical community for cost-effective point of carediagnostic instrumentation (Weber, 1997; Kleyn andWesell, 1998; Kost, 2000).
To meet these demands, many new plat-form technologies have been created, of whicha number are reviewed in this volume. The basisfor many of these advancements is the adaptation ofmicro-electronic technologies developed for the com-puting industry. For example, two major contribu-tions from this source are the microchipmanufacturing (from which MEMS fabricationtechnologies evolved) and optical informationand data storage devices (providing advances ininstrument miniaturization with increased analy-tical capacities) (Fodor et al., 1993; Kido et al.,2000).
Microchip and micro-array based platforms havereceived recent attention due to their relativelyhigh-capacity sequence interrogation capabilities,and a number of research groups and companies
have utilized this approach (e.g. Affymetrix Inc.,Genetic MicroSystems, Gene Logic Inc., IncytePharmaceuticals Inc.) (Bowtell, 1999). Relatively lessattention has been given to the parallel developmentof a smaller group of companies and researchersadapting optical disc technologies for biological andbiochemical analysis (Kiddo et al., 2000) (Gyros AB,Tecan-Gamera Bioscience, Abaxis Inc., BursteinTechnologies Inc.).
All of these groups have seen in compact disc(CD) technology the potential for developing rapid,low-cost, high-capacity screening systems byadapting existing industry standard technology. Bypackaging different assay formats on disc, the goalof achieving a generic format capable of suppor-ting multiple applications would be realized. Thepresent paper describes progress towards this endby Burstein Technologies Inc. (BTI) on molecularand immunological assays, and the future potentialof this technology. The platform described en-ables physicians/laboratories to use specially designedCD discs and a CD/DVD reader attached toan ordinary personal computer to deliver awide range of tests including clinical laboratorydiagnostics, biological warfare agent detection, for-ensic DNA tests, and food and water contaminationtests.
0955-8829 r 2002 Lippincott Williams & Wilkins Psychiatric Genetics 2002, Vol 12 No 4 193
Psychiatric Genetics 12:193–206
Copyright © Lippincott Williams & Wilkins. Unauthorized reproduction of this article is prohibited.

TECHNOLOGY
All of the BTI assays described are carried out withinan enclosed disc format read by a modified CD drive.The platform uses standard or semi-transmissive CDor DVD discs to deliver a wide range of tests using aCD reader attached to a personal computer. Thesystem will identify and quantify biochemical inter-actions inside a specially designed CD disc, theBioCompact Disc (BCD) (Table 1). A stepwisedepiction of the array utilization from hybridizedDNA at the molecular level to its detection in theBioCompact Disc reader (Figure 1) and a flowdiagram of the BCD drive function (Figure 2) areshown. The key features of this system to note is thatall analysis is carried out in the liquid phase within anenclosed chamber on disc, and that the analysis of the
biologic sample is carried out concurrently withoptical tracking of information features on disc.The BCD system is the testing platform of choice Fit contains all of the necessary elements.
K Centrifugation capability (variation in speeds andtiming enable sample processing within micro-fluidic chambers).
K The laser serves as the analytical tool used to readthe test results. The diagram in Figure 2 depictsthe basics of laser optics used for diagnostics. Thisis the ‘heart’ of the BTI platform.
K Software included on each BCD disc sends testdata securely and instantaneously via the Internetto a central data repository and authorizedindividuals who need the information.
Disc readerThe optical disc drive is fast becoming more thanjust a data storage and retrieval device. It is asophisticated laser-scanning device with inherentcapabilities in:
K physical process/measurementK image recognitionK laser scanning microscopy.
Just like a scanning microscope can be used tocharacterize microscopic objects, the optical disc driveis designed to identify micrometer-sized features at arate of a million per second. This feature can be used as
FIGURE 1. Micro-array technology in the BCD disc system. Clockwise from top right: atomic force microscopic imageof probe-captured DNA in the spot zone; three-dimensional presentation of DNA array data as analysed in the reader;amicro-array grid analysis screen of a BCD disc; a BCD discwith amicro-array cassette; and a readerdevice for readingBCD discs.
TABLE1. Principalof the technology: highlights
K The optical disc drive is a sophisticated laser-scanningmicroscope.
K The optical disc drive is also a centrifuge.K The CD (DVD,HD-DVD) is a biocompatible ‘solid phase’,
which can substitute for standard consumablesincluding slides, micro-wells, centrifuge tubes.
K Real-timeanalysispossible: forexample, a1mm2
micro-array canbe scanned in 20^30 swith a totaldata reduction time of 5 min.A customalgorithmperforms interpretations in real time.The image showsanoverall scheme of BTImicro-arraydesignandanalysis.
BARATHUR ET AL.
194 Psychiatric Genetics 2002, Vol 12 No 4
Copyright © Lippincott Williams & Wilkins. Unauthorized reproduction of this article is prohibited.

indicated in Figure 2 to identify objects by theirelectronic signatures, and even to quantitate thepresence of these signatures within a given field.
Micro£uidicsA key feature to adapting assays to the disc isnanotechnology (i.e. the miniaturization of systemsthrough nanofabrication processes). This is done insuch a way that assays can be scaled down withoutsacrificing performance. Nanotechnology also lowerstest cost by reducing reagent requirements. The BCDdisc utilizes systems of microchannels to deliver smallbut exact amounts of reagents and sample to theappropriate zones on the disc. In essence, themicrofluidic engineering is married with the high-resolution, optical capabilities of the CD/DVD read-er. Fluid flow has to be controlled to provide specialconditions where mixing is desired, or uniform flowover assay zones where homogeneous assay perfor-mance must be assured. As indicated earlier, the driveacts as a centrifuge, controlling sequencing of fluidaliquots into channels and flow rates over assayzones, etc., with highly calculated accuracy.
Micro-array technologyMicro-array technology is a very important platformtool, and its application is just as important inimmunoassays as it is in genetic analysis (Joos et al.,2000; Kim et al., 2000). Micro-arrays can provide
huge amounts of biological data, which can beapplied to answer many questions at one time. Figure1 provides an indication of how arrays are integratedinto the BCD disc. Arrays of 30–75 mm diameterspots can be densely grouped on the disc to readilyachieve from 150 to 1500 spots/mm2 (Kiddo et al.,2000). Several formats of data analysis can be used.Figure 3 shows a simple array of three spots, withwhich only the determination of the presence orabsence of the probed sequence is desired. The arrayspot is analysed for the overall concentration (ordensity) of beads bound to hybridized complexes.Further a quantitative relationship of reporter beadsand the amount of probed sequence present can beassessed, and the copy number can be defined inrelation to internal controls within the testing system.This provides both a qualitative and a quantitativeformat for data analysis.
The principle for generation of signals for the formatgiven in Figure 3 is shown schematically in Figure 4.The capture probe zone, in which target DNA iscaptured, can be designed into a microline format (forgenetic fingerprint visualization and display) or intomicrodot arrays (for analysis by the reader). Further-more, the multiple probe combinations can be usedwithin the same capture spot. As previously noted,each object provides a unique electronic signature(Figure 2). Because of this attribute, reporter micro-spheres of different characteristics (size, light transmis-sive or light absorbing, etc.) can be used together. Each
FIGURE 2. Schematic of a BCD disc readerand a picture of the internal drive components.Reportermicrospheres on thedisc result in electronic signatures to be generated that are characteristic of the microspheres. Such signatures can beutilized to provide eitherqualitative orquantitative results.
NEW DISC-BASED TECHNOLOGIES FOR DIAGNOSTIC AND RESEARCH APPLICATIONS
Psychiatric Genetics 2002, Vol 12 No 4 195
Copyright © Lippincott Williams & Wilkins. Unauthorized reproduction of this article is prohibited.

reporter can be coated with different receptors thatcorrespond with specific ligands, which in turn arepresent on the amplified target DNA (e.g. biotinylatedtarget DNA will attract strepavidin-coated micro-spheres, while digoxigenin-labelled target DNA wouldattract microspheres labelled with anti-digoxigenin).Such combinations become useful in analysis of mixedallotypes or for quantitating target DNA againstinternal control DNA copies.
By integrating previously separate steps of stan-dard assay formats into a seamless, simplifiedprocess, this new technology enables rapid, inexpen-sive and unambiguous evaluation of clinical samples.
What previously required many steps and up to24 h before a physician could be given test results, will
now take only a few steps and just minutes. The keyadvantage of the BTI system is that it dramaticallysimplifies the testing process and makes it moreconvenient by replacing complex (and expensive) testequipment with an ordinary PC and a CD/DVDreader. Thus, testing can be done at the point of careas easily as it could be done in a laboratory setting.
APPLICATIONS IN INFECTIOUSDISEASE
An area of practical application of the BCD disc is inthe diagnostics and characterization of infectiousdisease. The disc can be utilized to perform immu-noassays for detection of antibodies or antigens thatmight be associated with infection by a particular
Sample Addition Port Fluidic Channel To Disc Margin
FIGURE 3. A qualitative result for a simple triplet array.Biotinylated target DNA is added and hybridized to capture probe.Strepavidin-Reporter microspheres are added.The disc is spun for 5min, at 3,000 rpm to remove free microspheres.Thereader thenanalyses event signatures in the capture zone.
BARATHUR ET AL.
196 Psychiatric Genetics 2002, Vol 12 No 4
Copyright © Lippincott Williams & Wilkins. Unauthorized reproduction of this article is prohibited.

pathogen. But for this discussion, utilization of thedisc for detection DNA or RNA of the pathogen willbe emphasized. Based on similar reporter systems asthose already described, micro-arrays for proprietarytarget sequences are being prepared to identifyspecific pathogens. The process begins with a sample(e.g. body fluids such as blood, urine, saliva or milk,
tissue, etc.), followed by extraction of nucleic acid,amplification of targets and, finally, hybridizationand detection using micro-arrays (the last step beingperformed in the BCD disc). Diagnostics currentlybeing developed will enable multiplex identificationof specific pathogen types such as Mycobacteriumparatuberculosis, Staphylococcus aureus, coliformorganisms, streptococcal species, Salmonellae, Bru-cellae, Mycobacterium bovis, Campylobacter, HIV,hepatitis type B and type C, and other pathogens.
One such example would be for use with brucellosisinfections. Brucellosis, a disease of humans andanimals, has both health and economic importancein many developing countries (Nicoltti, 1980; Achaand Szyfres, 1987). Six species of Brucella arecurrently known to cause brucellosis: Brucella abor-tus, Brucella suis, Brucella melitensis, Brucella ovis,Brucella neotomae, and Brucella canis (Nicoltti, 1980;Acha and Szyfres, 1987). Infection begins as septice-mia with sudden insidious onset characterized bycontinued, intermittent or irregular fever (Acha andSzyfres, 1987). Symptomatology is similar to that ofother febrile illnesses, but with a marked musculo-skeletal involvement (severe fatigue, aches and pains)(Acha and Szyfres, 1987). Severe complicationsinclude meningitis, encephalitis, spondylitis, suppura-tive arthritis, and vegetative endocarditis (Acha andSzyfres, 1987). The infection is treatable with tetra-
FIGURE 4. Use of reporter microspheres in signal analy-sis. Capture probes within the array spot hybridize to thetarget DNA, which is biotinylated (free biotinylated primersare not complementary to the capture probe sequences).Streptavidin-coated microspheres then bind to the spotzones in which the target DNA is hybridized. Unbound mi-crospheres are removed through spinning the disc.
Base Pairs
300
200
100
B. melitensis
B. melitensis
B. suis
B. suis
B. abortis
B. abortis
PCR generated amplicons stained with ethidium bromide
Multiplexed (3 species) Probes
FIGURE 5. Multiplexampli¢cationanddetection: agarosegeland CDreader results.Gel results showmultiplex-ampli¢edDNA from three species of Brucella.These productswere thenhybridized to probe capture zones on the disc and analysed(after addition of reporter microspheres) for the presence of target DNA.Each capture zone provides speci¢c detection ofDNAof the species of interest or in the presence of DNAofanyof the species of interest (multiplexedprobezone).
NEW DISC-BASED TECHNOLOGIES FOR DIAGNOSTIC AND RESEARCH APPLICATIONS
Psychiatric Genetics 2002, Vol 12 No 4 197
Copyright © Lippincott Williams & Wilkins. Unauthorized reproduction of this article is prohibited.

cyclines, but can become established chronically insome cases.
Figure 5 indicates how amplified DNA fromdifferent species of Brucella can be detected usingthe BCD disc. The region amplified codes for aportion of 16s RNA (Dorsch et al., 1989; Fox et al.,1998). One primer (forward) is common for allspecies, while the second primer (reverse) differs foreach species. With this design, the amplified DNAproduct from each pathogen differed in number ofbase pairs, but also each product had sequences thatare specific for that pathogen. The results show thatDNA from each organism could be detected usingarray spots with probes specific for that species, orthe presence of DNA from any of these organisms ina sample could be detected in an array spot contain-ing probes for all three organisms (a multiplexedprobe spot or zone). The detection process is highlyspecific, since removal of B. suis DNA (or DNA ofanother species) from the test sample results in anegative result from the array analysis for thatorganism (data not shown). It should be noted fromthis that the specific pathogen could be identifiedusing species-specific probe spots or, if desired toscreen for multiple potential pathogens, arrays ofmultiplexed probe spots could be used to searchthrough to multiple possible pathogens. A similardetection format has been used for detection of B.abortus and M. bovis in milk collected from infectedherds of cattle (Sreevatsan et al., 2000). That assaysensitivity was found to detect as few as 10–100genomic DNA equivalents per milliliter of specimen(Sreevatsan et al., 2000).
Viral load analysis is a standard of practicefor HIV-infected and hepatitis C virus-infectedpatients currently, not only in monitoring clinicalstatus of patients, but also (and more importantly)in monitoring efficacy of drugs on patients (Zeinet al., 1996; Bell et al., 1997). Since the technologyrequires operators with skill and experience, andvirtually mandates that molecular biologists managelaboratories performing these assays, testing islimited to those hospitals and clinical laboratoriesthat can provide this type of support (Krajden, 1995).Adaptation to the BCD disc platform for the virusdetection (and quantifying viral RNA copiesper milliliter of plasma) is currently underway.A third infectious disease, molecular applicationunder study for the BCD disc utilization is genotypicfingerprinting for drug resistance in HIV, hepatitis Cvirus and Mycobacterium tuberculosis. These lastapplications entail the utilization of extensive arraysthat probe for the specific known mutations ofinterest.
APPLICATIONS IN NEUROGENOMICSANDPHARMACOGENOMICS
To most in the field of human genomics, the post-genomic era is the close of just the beginning. The nextstep is to mine through the millions of pieces ofinformation that comprise the genome and make senseof its composition. In so doing, a better understandingof gene associations with disease, drug response andother phenotypic expression will be gained for use byphysicians and pharmaceutical companies.
It is now estimated that the human genome iscomposed of about 50 000–90 000 genes, but thatutilization of those genes results in more than 1million proteins (of which fewer than 8000 genefunctions are known) (Kennedy, 2000; Roest et al.,2000; Venter et al., 2001). One of the first tasks in thisnew era is to catalogue the most common types ofgenetic differences. Exons and regulatory sequencesconstitute only about 1–3% of the genome (Venteret al., 2001). About 75% of the genome lacks truegene composition, as it consists mainly of random,nonsense and repetitive sequence regions (Venteret al., 2001). Single nucleotide polymorphisms (SNPs)occur at about one in every 1250 base pairs (clusteredor dispersed and in coding or non-coding regions),and constitute about 80% of all sequence-defineddifferences in populations (Altshuler et al., 2000;Sachidanandam et al., 2001; Venter et al., 2001). Theshort tandem repeats, usually of five to nine basesequence repeats, are also present in the genome atthe frequency of about one in 3 million base pairs(Edwards et al., 1991, 1992; Warne et al., 1991;Kupferschmid et al., 1999).
While other technologies may currently be requiredfor analysing genotypic differences resulting fromshort tandem repeats, the BCD disc technologies arevery useful for detecting SNPs and many knowngenetic deletions and insertions. As already notedwith the previous examples, specific genetic sequencescan be detected using arrays of various compositionson the BCD disc. It is now possible to identifydisease-validated targets and use them to aid in bothdiagnostic and therapeutic decisions. To illustratethis, the rapidly unfolding advances in two importantareas of gene discovery will be discussed: pharmaco-genomics and neurogenomics.
Pharmacogenomics is a segment of genome re-search that emphasizes discovery of genes that affecthow drugs work in different individuals (Weber, 1997;Kleyn and Wesell, 1998). Population differences anddisease associations with genetic variation are criticalfor this process, as pharmacogenomics is an efforttoward a systematic approach to elucidating gene
BARATHUR ET AL.
198 Psychiatric Genetics 2002, Vol 12 No 4
Copyright © Lippincott Williams & Wilkins. Unauthorized reproduction of this article is prohibited.

function and validating drug targets (Miller et al.,1997; Ingelman-Sundberd, 1998; Kleyn and Vesell,1998). Arrays can be used to provide gene expressionprofiles that help define the effect of therapeuticcompounds of relevant genes. Genetic variations canaffect expression patterns, can impair or enhance drugeffects and can even contribute to drug toxicity (Milleret al., 1997; Ingelman-Sundberd, 1998).
Several examples of genotypic polymorphisms andtheir effect on drug utility are presented in Table 2. Itis estimated that only 30–60% of patients with thesame disease will give an effective response to manyof the commonly used drugs (Shi et al., 2001). But avariable number will also have a range of side effects.Both efficacy and adverse reactions have foundationsin genetic make-up. CYP3A4, for example, is knownto affect the metabolism of a large number of drugs(Evans and Relling, 1999). Several altering mutationsare known, including a common promoter poly-morphism (Evans and Relling, 1999). Anotherexample is ApoE4, an apolipoprotein E locusallotype that not only correlates with elevated riskfor Alzheimer’s disease, but serves as a negativepredictive marker for response to cholinesteraseinhibitor treatment (Farlow et al., 1996, 1998). Manymutations known to affect drug activity are SNPs.About 2–10% of the population are homozygous fornon-functional CYP2D6 alleles (Evans and Relling,1999). This correlates with reduced codeine responseand less pain tolerance (Evans and Relling, 1999).Known CYP2D6 mutations include splice site muta-tions causing exon skipping, deletions and geneduplications, and early stop codon point mutations(Evans and Relling, 1999). Many diagnostic groupsare using or developing the array technology as aformat for looking at the various pharmacogenomicmarkers such as those indicated.
Another segment of genome research is neuroge-netics, which emphasizes exploring genetic variation
and the effects on neurologic diseases. Table 3illustrates (by the selected diseases listed) the diversityof genes affected or contributing to diseases withneurologic symptoms.
Similarities with the pharmacogenomic researchmay be noted. Several of the metabolic diseases,particularly Lesch–Nyhan syndrome and atypicalphenylketonuria (PKU) (Type II), appear to involvethe dopaminergic pathways in the brain (pathwaysinvolved in the pathogenesis of many other neurolo-gic and psychiatric diseases including Parkinson’sdisease and schizophrenia). Investigations into howthese in-born errors of metabolism produce seizuresand abnormal behaviour may provide insight intohow these same systems malfunction in other morewidespread diseases.
Mutational analysis can also reveal patients withhomocystinuria who are cobalamin (B6 vitamin)responsive or who are unresponsive. Responsive pa-tients usually have residual cystathionine b-synthetaseactivity, which can be further stimulated by large dosesof the B6 vitamin, while non-responsive patients havemutations that completely abolish the enzyme activity.
Some diseases are caused in a defect of a singlegene, yet the location of the mutation affects thetypes or degree of symptoms expressed, while otherdiseases may result from mutations in different genes.And again, for example, with Maple Syrup UrineDisease, the mutation may result in expression ofdifferent clinical symptoms, suggesting contributoryinfluences not well defined to date. Yet specificmutations identified in Table 3 are known.
The use of micro-arrays that identify specificmutations of a patient could greatly benefit theunderstanding of underlying effects and influences ofgene interactions. For example, the number ofpossible gene variants makes array technologypotentially very useful in prenatal genetic diagnosisof PKU. Over 70 different mutations of the gene have
TABLE 2. Genotypingdrugmetabolism: drugexamples anddruge¡ect alteringmarkers (Farlow et al.,1998; KleynandWesell,1998; Emilein et al.,1999; Evans and Relling,1999; Shiet al., 2001)
Drug Druge¡ect alteringmarkers
Acetaminophen CYP1A2,CYP2E1,CYP3A4 (genesof cytochrome P450 superfamily)Beta blockers CYP2D6 (Debrisoquinhydrolase)AntidepressantsAntipsychoticsNeuroleptics Dopamine receptors D2,D3,D4,D5Cholinergic agents ApoEFallotype E4(Alzheimer’s disease,Tourette syndrome)Dexamethasone Glutathione S-transferasesFYb1,Yb2 and Yb3Lovastatin CYP2B1/2,CYP3A1/2,CYP3A4Thiopurine Thiopurinemethyl transferase,CYP2D6
NEW DISC-BASED TECHNOLOGIES FOR DIAGNOSTIC AND RESEARCH APPLICATIONS
Psychiatric Genetics 2002, Vol 12 No 4 199
Copyright © Lippincott Williams & Wilkins. Unauthorized reproduction of this article is prohibited.

TABLE 3. Selected reviewof neurologic diseasesandgenetic features
Disease Symptoms/neurologicmanifestations
Mutations(chromosome site)
Array requirements(challenges)
Additional comments References
Alzheimer’s disease Mainlyold-age onset ofmemoryandpersonalityloss.Severe degenerationof brainwith proteinplaques (beta-amyloid)andneuro¢brillarytangles.Progressivedementia andmemoryimpairment with at leastone of: aphasia, apraxia,agnosia or lossofexecutive functioning
Amyloid beta A4precursor proteinAPP (21q21)
Presenilin1 (14q24.3)Presenilin 2 (1q31^q42)Apolipoprotein
type E (19q13.2)
12þmutations72þmutationsAt least sixmutationsFourallotypesFE4
allotype carries risk
Otherassociatedmarkers still underinvestigation.Apo4is considered to bea susceptibility riskmarker.Mutationsin APPor Presenilinsfound in 30^40% ofautosomaldominantAlzheimer’s diseaseand 5% of casesin general
Goate et al.,1991;Corder et al.,1993;Hutton et al.,1996;De Jonghe et al.,1999;Finckh et al., 2000;Nilsberth et al., 2000
Carbohydrate-de¢ciencyglyco-proteinsyndrome
Only type Ia hasprominent neurologicalmanifestations.Severepsychomotor retardation(‘£oppybaby’syndrome).Multi-organ involvement
Phosphomannomutase(16p13.3^p13.12)
230þmutations anddeletions
R141H,N216IandF119Lde¢ne 88%of cases.Commonmutationsnoted forScandinavians
Hagberg et al.,1993;Jaeken et al.,1993;Van SchaftingenandJaeken,1995; Matthijset al.,1997,1999, 2000;Bjursellet al.,1998,2000; Kjaergaardet al.,1998,1999;Kondo et al.,1999
Galactosemia Symptomatic triad:hepatomegaly, cataractsandmental retardationfailure to thrivewithincreased intracranialpressureand cerebraledema.De¢cient speechand language skills.Cerebralatrophyandwhitematter lesions
Galactose1-phosphateuridyltransferase(9p13)
130þmutations Q188Rand K285Nde¢ne 70% ofCaucasian cases(absent inJapanese).S134Lcharacteristicof 62% of blackethnic cases.Duartevariant (N314D/N314D) gives 50%reducedenzymeactivity.Los Angelesvariant of Duartegives increasedactivity
Hsia,1967; ReichardtandWoo,1990,1991;Schweitzer et al.,1993;Langley et al.,1997;Elsas and Lai,1998
Homocytinuria Thromboembolicdisease; multi-organsa¡ected.Glaucoma,
Cystathionine b-synthetase (21q22.3)
Array to address492mutations
Mostlymissensemutations; mostcommon I278T
Mudd et al.,1985;Visy et al.,1991;Tsaiet al.,1996;
BARATHUR
ETAL.
200Psych
iatricGenetics
2002,Vol12
No4
Copyright © Lippincott Williams & Wilkins. Unauthorized reproduction of this article is prohibited.

osteoporosis.Mentalretardation (IQ 50^70)but one-third havenormal IQ.Many (not all)with psychiatricdisturbances (depression,obsessive-compulsive,etc.)
(vitamin B6responsive) andG307S (vitamin B6non-responsive)
Chasse et al.,1997;Kraus et al.,1999
Hyperphenyl-alaninemia
Type1 [classicalphenylketonuria (PKU)]:centralnervous systeminsult withmentalretardation, epilepsy,myelination defects.Treatment ismainlydietary
Phenylalaninehydroxylase (12q34.1)
70þ haplotypesFsixmutationsmakeup 80%of PKU.For Caucasians,most eithera singlebase change in the12th intronora R408Wmissensemutation
Variousethnicpatternsnoted.Mutation correlateswith type, residualenzymeactivityandPKUseverity
Paine,1957; DiLellaet al.,1986; GuttlerandWoo,1986; Daigeret al., 1989; Hofman et al.,1989, 1991; Scriver et al.,1989; Konecki and Lichter-Konecki,1991; Degioanniet al.,1994; Kaufman,1999;Gri⁄ths et al., 2000
Type II (atypical PKU):neurologicaldefectmore severe than type I.SymptomslikeParkinson’s disease
Dihydropteridinereductase (4p15.31)leads to a dysfunctionof tetrahydrobipterin(BH4)
50þmutationsFmissense, nonsense,insertions anddeletions
Inadequate levels of BH4:(a) six PTPS de¢ciency:Parkinsonian-likesymptoms, hypotonia,swallowingdi⁄culties,neuropsychologicaldevelopmentimpariments, seizures.(b) GTPcyclohydrolasede¢ciency
6-pyruvovyltetrahydropterinsynthetase(11q22.3^q23.3)
GTPcyclohydrolase(14q22.1^q22.2)
10þmutations
Only six patientsreported to date
Strongethnicpatternsnoted
Hypertyrosinemia Type1: pain in extremities(peripheralneurologicalcrisis) with autonomicdysfunction andprogressive paralysis.Bizarre postures torelievepain.Convulsions.Normal IQ.Treatablewithmedication
Furarylacetoacetatehydrolase (15q23^q25)
26mutationsFmissense, nonsensesplicing defects
Genotypeandphenotypecorrelationnotwell de¢nedto date
Natt et al.,1986,1992;Mitchellet al.,1990;Hahn et al.,1995;Timmers and Grompe,1996; Louis and Tanguay,1997
NEW
DISC-BASED
TECHNOLOGIE
SFOR
DIA
GNOSTIC
AND
RESEARCH
APPLIC
ATIO
NS
Psych
iatricGenetics
2002,Vol12
No4
201
Copyright © Lippincott Williams & Wilkins. Unauthorized reproduction of this article is prohibited.
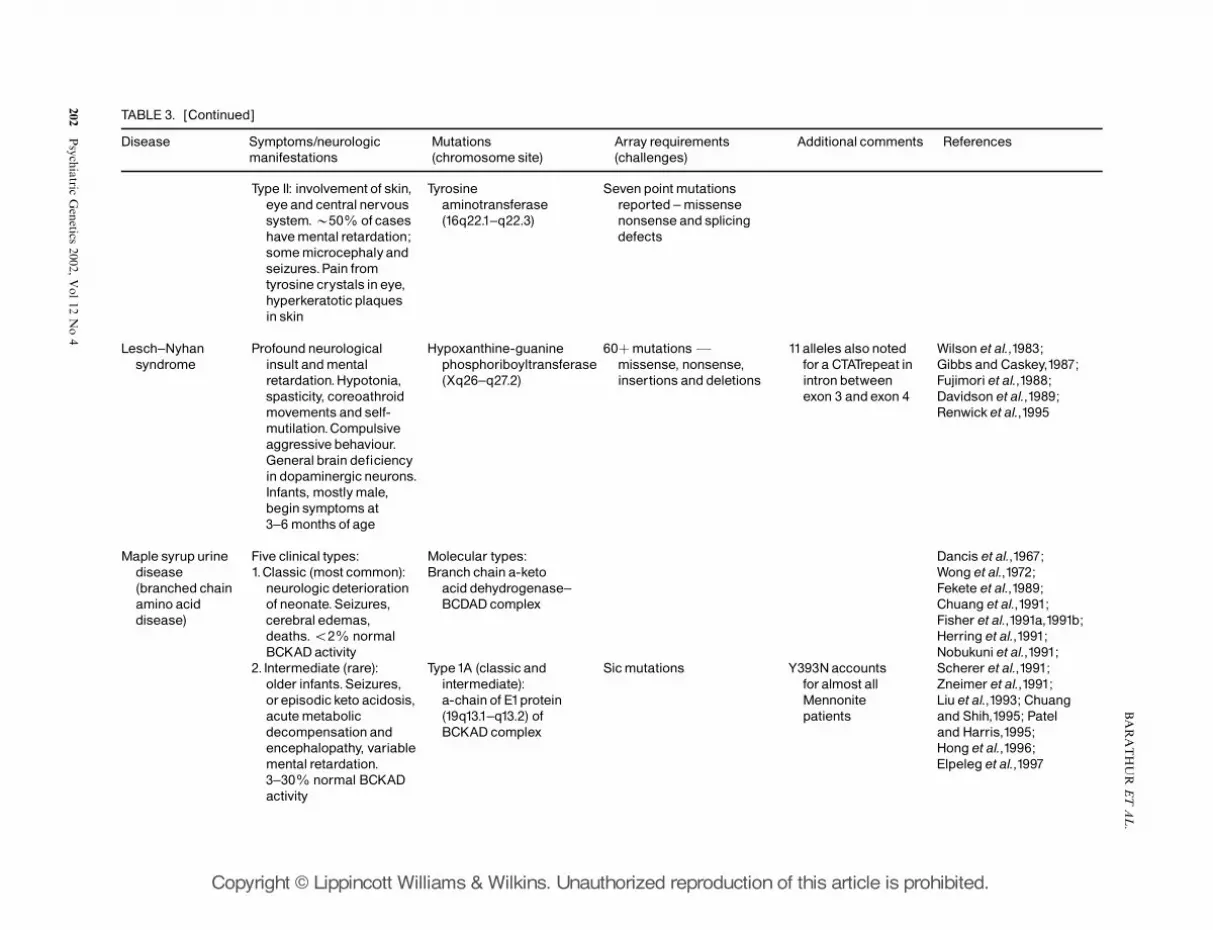
Type II: involvement of skin,eyeand centralnervoussystem.B50% of caseshavemental retardation;somemicrocephalyandseizures.Pain fromtyrosine crystals in eye,hyperkeratotic plaquesin skin
Tyrosineaminotransferase(16q22.1^q22.3)
Sevenpoint mutationsreported ^missensenonsenseand splicingdefects
Lesch^Nyhansyndrome
Profoundneurologicalinsult andmentalretardation.Hypotonia,spasticity, coreoathroidmovements and self-mutilation.Compulsiveaggressive behaviour.Generalbrain de¢ciencyin dopaminergic neurons.Infants, mostlymale,begin symptomsat3^6monthsof age
Hypoxanthine-guaninephosphoriboyltransferase(Xq26^q27.2)
60þmutationsFmissense, nonsense,insertions and deletions
11alleles alsonotedfora CTATrepeat inintronbetweenexon 3 andexon 4
Wilson et al.,1983;Gibbs and Caskey,1987;Fujimoriet al.,1988;Davidson et al.,1989;Renwicket al.,1995
Maple syrup urinedisease(branched chainamino aciddisease)
Five clinical types:1.Classic (most common):
neurologic deteriorationof neonate.Seizures,cerebraledemas,deaths.o2% normalBCKADactivity
2. Intermediate (rare):older infants.Seizures,orepisodic keto acidosis,acutemetabolicdecompensationandencephalopathy, variablemental retardation.3^30% normal BCKADactivity
Molecular types:Branch chain a-keto
acid dehydrogenase^BCDAD complex
Type1A (classic andintermediate):a-chain of E1protein(19q13.1^q13.2) ofBCKAD complex
Sicmutations Y393Naccountsforalmost allMennonitepatients
Dancis et al.,1967;Wong et al.,1972;Fekete et al.,1989;Chuang et al.,1991;Fisher et al.,1991a,1991b;Herring et al.,1991;Nobukuniet al.,1991;Scherer et al.,1991;Zneimer et al.,1991;Liu et al.,1993; Chuangand Shih,1995; Pateland Harris,1995;Hong et al.,1996;Elpeleg et al.,1997
TABLE 3. [Continued]
Disease Symptoms/neurologicmanifestations
Mutations(chromosome site)
Array requirements(challenges)
Additional comments References
BARATHUR
ETAL.
202Psych
iatricGenetics
2002,Vol12
No4
Copyright © Lippincott Williams & Wilkins. Unauthorized reproduction of this article is prohibited.

been identified (Guttler and Woo, 1986; Scriver et al.,1989; Konecki and Lichter-Konecki, 1991), and six ofthese account for over 80% of PKU (Konecki andLichter-Konecki, 1991). Ethnic distributions of mu-tations have been shown in the Chinese, YemeniteJewish, and Afro-American populations (Daiger etal., 1989; Hofman et al., 1991; Degioanni et al.,1994). There is also good correlation between thetype of mutation, the amount of residual enzymeactivity, and the severity of the PKU (Daiger et al.,1989; Hofman et al., 1991; Degioanni et al., 1994).Future diagnostic tests should detect most possiblemutations in the phenylalanine hydroxylase gene andprovide phenotype/genotype correlations strong en-ough to predict which mutations will lead to a severePKU phenotype. Until that can be done, diagnosiswill continue to depend on screening newborn bloodfor phenylalanine levels.
Molecular diagnostics may become more routineas these technologies become available. In thisrespect, while numerous mutations are described foreach disease, because many are defined, arrays can bedeveloped to screen for these mutations. In so doing,a disease mutation could be detected in most cases,with only a small frequency not identified due to thepresence of a new mutation.
THECHANGES IN PROGRESS
The technological advances and molecular applica-tions, which we have noted, hold the potential toallow physicians and researchers to diagnose orfurther characterize the complexities of many dis-eases. Furthermore, these advances may soon aid intherapeutics: first, in the design of medications, basedon patient genotypes, that are more effective and lesstoxic; and second, by providing the medical practicewith better tools for therapeutic response assess-ments. It is anticipated that much of the guessworkcurrently associated with patient care will be reduced.
Acknowledgements
The authors would like to acknowledge the efforts of VitoDelveccio in providing sequence information and samplesfor the work in Brucellae speciation. Special thanks alsoto Ramon Valencia and Ryan McCrae for their con-tributions to the studies presented in this publication.
REFERENCES
Acha PN, Szyfres B (1987). Zoonoses and CommunicableDiseases Common to Man and Animals, Scientificpublication series No. 503, 2nd ed. Washington, DC:Pan American Health Organization. pp. 24–45.
3.Interm
ittan
t:olde
rinfants.Normal
grow
thbu
tcan
trigge
rmetab
olic
deco
mpe
nsation,
lethargy,dea
th.
5^20
%BCKAD
activity
Type
1B:(clas
sican
dinterm
ediate)b
-cha
inof
E1protein(6p2
2-p21)
ofBCKADco
mplex
MainmutationF
11bp
deletio
n,exon
1Type
1BF
mos
tlyJapa
nese
origin
4.Th
iamine-resp
onsive:
symptom
ssimilarto
interm
ediate
Type
II(classican
dthiamine-respon
sive):
E2protein(1p31)
ofBCKADco
mplex
25þmissense,
nonsen
se,insertio
nsde
letio
ns,and
splice
mutations
Array
matrix
toad
dres
sea
chse
quen
ceof
interest
5.Dihydrolipoyldeh
ydro-
gena
se(DHD)d
e¢cien
cy:
norm
alinfanc
ywith
interm
ediatesymptom
splus
severe
metab
olic
lacticacidosis.N
eurologic
insult(dem
yelination,
cavitatio
n)with
acidosis
Type
III(DHDde
¢cienc
y):
E3protein(7q31^
q32)
ofBCKADco
mplex
6þrepo
rted
mutations
Fno
nsen
sean
dmisssen
se
NEW DISC-BASED TECHNOLOGIES FOR DIAGNOSTIC AND RESEARCH APPLICATIONS
Psychiatric Genetics 2002, Vol 12 No 4 203
Copyright © Lippincott Williams & Wilkins. Unauthorized reproduction of this article is prohibited.

Altshuler D, Pollara VJ, Cowles CR, Van Etten WJ,Baldwin J, Linton L, Lander ES (2000). An SNP map ofthe human genome generated by reduced representationshotgun sequencing. Nature 407:513–516.
Bell H, Hellum K, Harthug S, Maeland A, Ritland S,Myrvang B, et al. (1997). Genotype, viral load and age asindependent predictors of treatment outcome of inter-feron-a 2a treatment in patients with chronic hepatitis C.Scand J Infect Dis 29:17–22.
Bjursell C, Wahlstrom J, Berg K, Stibler H, Kristiansson B,Matthijs G, Martinsson T (1998). Detailed mapping ofthe phosphomannomutase 2 (PMM2) gene and mutationdetection enable improved analysis for ScandinavianCDG type I families. Eur J Hum Genet 6:603–611.
Bjursell C, Erlandson A, Nordling M, Nilsson S, Wahl-strom J, Stibler H, Kristiansson B, Martinsson T (2000).PMM2 mutation spectrum, including 10 novel muta-tions, in a large CDG type 1A family material with afocus on Scandinavian families. Hum Mutat 16:395–400.
Bowtell DDL (1999). Options available-from start to finish-for obtaining expression data by microarray. Nat GenetSuppl 21:25–31.
Chasse JF, Paul V, Escanez R, Kamoun P, London J(1997). Human cystathionine beta-synthase: gene orga-nization and expression of different 5-prime alternativesplicing. Mammalian Genome 8:917–921.
Chuang DT, Fisher CW, Lau KS, Griffin TA, Wynn RM,Cox RP (1991). Maple syrup urine disease: domainstructure, mutations and exon skipping in the dihydro-lipoyl transacylase (E2) component of the branched-chain alpha-keto acid dehydrogenase complex. MolecBiol Med 8:49–63.
Chuang DT, Shih VE (1995). Disorders of branched chainamino acid and keto acid metabolism. In: Metabolic andMolecular Bases of Inherited Disease, 7th ed., ScriverCR, Beaudet AL, Sly WS, Valle D (editors). New York:McGraw-Hill. pp. 1239–1277.
Corder EH, Saunders AM, Strittmatter WJ, SchmechelDE, Gaskell PC, Small GW, et al. (1993). Gene dose ofapolipoprotein E type 4 allele and the risk of Alzheimer’sdisease in late onset families. Science 261:921–923.
Daiger SP, Reel L, Huong SS, Zeng YT, Low H, Okano Y,et al. (1989). Polymorphic DNA haplotypes at thephenylalanine hydroxylase (PAH) locus in Asianfamilies with phenylketonuria (PKU). Am J Hum Genet45:319–324.
Dancis J, Hutzler J, Rokkones T (1967). Intermittentbranched-chain ketonuria. Variant of maple-syrup-urinedisease. N Engl J Med 276:84–89.
Davidson BL, Pashmforoush M, Kelley WN, Palella TD(1989). Human hypoxanthine-guanine phosphoribosyl-transferase deficiency: the molecular defect in a patientwith gout (HPRT-Ashville). J Biol Chem 264:520–525.
Degioanni A, Darlu P (1994). Analysis of the molecularvariance at the phenylalanine hydroxylase (PAH) locus.Eur J Hum Genet 2:166–176.
De Jonghe C, Cruts M, Rogaeva EA, Tysoe C, Singleton A,Vanderstichele H, et al. (1999). Aberrant splicing in thepresenilin-1 intron 4 mutation causes presenile Alzhei-mer’s disease by increased Abeta42 secretion. HumMolec Genet 8:1539–1540.
DiLella AG, Kwok SCM, Ledley FD, Marvit J, Woo SLC(1986). Molecular structure and polymorphic map of the
human phenylalanine hydroxylase gene. Biochemistry25:743–749.
Dorsch M, Moreno E, Stackebrandt E (1989). Nucleotidesequence of the 16S rRNA from Brucella abortus.Nucleic Acids Res 17:1765–1765.
Edwards A, Civitello A, Hammond HA, Caskey CT (1991).DNA typing and genetic mapping with trimeric andtetrameric tandem repeats. Am J Hum Genet 49:746–756.
Edwards A, Hammond HA, Jin L, Caskey CT, Chakra-borty R (1992). Genetic variation at five trimeric andtetrameric tandem repeat loci in four human populationgroups. Genomics 12:241–253.
Elpeleg ON, Shaag A, Glustein JZ, Anikster Y, Joseph A,Saada A (1997). Lipoamide dehydrogenase deficiency inAshkenazi Jews: an insertion mutation in the mitochon-drial leader sequence. Hum Mutat 10:256–257.
Elsas LJ II, Lai K (1998). The molecular biology ofgalactosemia. Genet Med 1:40–48.
Emilein G, Maloteaux JM, Geurts M, Owen J (1999).Dopamine receptors and schizophrenia: contribution ofmolecular genetics and clinical neuropsychology.J Neuropsychol 2:197–227.
Evans WE, Relling MV (1999). Pharmacogenomics: trans-lating functional genomics into rational therapeutics.Science 286:487–491.
Farlow MR, Lahiri DK, Poirer J (1996). Apolipoprotein Egenotype and gender influence response to tarcrinetherapy. Ann NY Acad Sci 802:101–110.
Farlow MR, Lahiri DK, Poirer J, Davignan J, Hui S(1998). Treatment outcome of tacrine therapy dependson apolipoprotein genotype and gender of the subjectswith Alzheimer’s disease. Neurology 50:669–677.
Fekete G, Plattner R, Crabb DW, Zhang B, Harris RA,Heerema N, Palmer CG (1989). Localization of thehuman gene for the El-alpha subunit of branched chainketo acid dehydrogenase (BCKDHA) to chromosome19q13.1-q13.2. Cytogenet Cell Genet 50:236–237.
Finckh U, Muller-Thomsen T, Mann U, Eggers C,Marksteiner J, Meins W, et al. (2000). High prevalenceof pathogenic mutations in patients with early-onsetdementia detected by sequence analyses of four differentgenes. Am J Hum Genet 66:110–117.
Fisher CR, Chuang JL, Cox RP, Fisher CW, Star RA,Chuang DT (1991a). Maple syrup urine disease inMennonites: evidence that the Y393N mutation in E1-alpha impedes assembly of the E1 component ofbranched-chain alpha-keto acid dehydrogenase complex.J Clin Invest 88:1034–1037.
Fisher CW, Lau KS, Fisher CR, Wynn RM, Cox RP,Chuang DT (1991b). A 17-bp insertion and a phe215-to-cys missense mutation in the dihydrolipoyl transacylase(E2) mRNA from a thiamine-responsive maple syrupurine disease patient WG-34. Biochem Biophys ResCommun 174:804–809.
Fodor SP, Rava RP, Huang XC, Pease AC, Holmes CP,Adams CL (1993). Multiplexed biochemical assays withbiological chips. Nature 364:555–556.
Fox KF, Fox A, Nagpal M, Steinberg P, Heroux K (1998).Identification of Brucella by ribosomal-spacer-regionPCR and differentiation of Brucella canis from otherBrucella ssp. pathogenic for humans by carbohydrateprofiles. J Clin Microbiol 36:3217–3222.
Fujimori S, Hidaka Y, Davidson BL, Palella TD, KelleyWN (1988). Identification of a single nucleotide change
BARATHUR ET AL.
204 Psychiatric Genetics 2002, Vol 12 No 4
Copyright © Lippincott Williams & Wilkins. Unauthorized reproduction of this article is prohibited.

in a mutant gene for hypoxanthine-guanine phospho-ribosyltransferase (HPRT-Ann Arbor). Hum Genet79:39–43.
Gibbs RA, Caskey CT (1987). Identification and localiza-tion of mutations at the Lesch–Nyhan locus byribonuclease A cleavage. Science 236:303–305.
Goate A, Chartier-Harlin MC, Mullan M, Brown J,Crawford F, Fidani L, et al. (1991). Segregation of amissense mutation in the amyloid precursor protein genewith familial Alzheimer’s disease. Nature 349:704–706.
Griffiths PV, Demellweek C, Fay N, Robinson PH,Davidson DC (2000). Wechsler subscale IQ and subtestprofile in early treated phenylketonuria. Arch Dis Child82:209–215.
Guttler F, Woo SLC (1986). Molecular genetics of PKU.J Inherit Metab Dis 9(suppl 1):58–68.
Hagberg BA, Blennow G, Kristiansson B, Stibler H (1993).Carbohydrate-deficient glycoprotein syndromes: peculiargroup of new disorders. Pediatr Neurol 9:255–262.
Hahn SH, Krasnewich D, Brantly M, Kvittingen EA, GahlWA (1995). Heterozygosity for an exon 12 splicingmutation and a W234G missense mutation in anAmerican child with chronic tyrosinemia type 1. HumMutat 6:66–73.
Herring WJ, Litwer S, Weber JL, Danner DJ (1991).Molecular genetic basis of maple syrup urine disease in afamily with two defective alleles for branched chainacyltransferase and localization of the gene to humanchromosome 1. Am J Hum Genet 48:342–350.
Hofman KJ, Antonarakis SE, Missiou-Tsangaraki S,Boehm CD, Valle D (1989). Phenylketonuria in theGreek population. Haplotype analysis of the phenylala-nine hydroxylase gene and identification of a PKUmutation. Mol Biol Med 6:245–250.
Hofman KJ, Steel G, Kazazian HH, Valle D (1991).Phenylketonuria in US blacks: molecular analysis of thephenylalanine hydroxylase gene. Am J Hum Genet48:791–798.
Hong YS, Kerr DS, Craigen WJ, Tan J, Pan Y, Lusk M,Patel MS (1996). Identification of two mutations in acompound heterozygous child with dihydrolipoamidedehydrogenase deficiency. Hum Molec Genet 5:1925–1930.
Hsia D Y-Y (1967). Clinical variants of galactosemia.Metabolism 16:419–437.
Hutton M, Busfield F, Wragg M, Crook R, Perez-Tur J,Clark RF, et al. (1996). Complete analysis of thepresenilin 1 gene in early onset Alzheimer’s disease.Neuroreport 7:801–805.
Ingelman-Sundberd M (1998). Functional consequences ofpolymorphism of xenobiotic metabolizing enzymes.Toxicol Lett 102–103:155–160.
Jaeken J, De Cock P, Stibler H, Van Geet C, Kint J,Ramaekers V, Carchon H (1993). Carbohydrate-deficient glycoprotein syndrome type II. J Inherit MetabDis 16:1041.
Joos TO, Schrenk M, Hopfl P, Kroger K, Chowdhury U,Stoll D et al. (2000). A microarray enzyme-linkedimmunosorbant assay for autoimmune diagnostics.Electrophoresis 21:2641–2650.
Kaufman S (1999). A model of human phenylalaninemetabolism in normal subjects and in phenylketonuricpatients. Proc Natl Acad Sci USA 96:3160–3164.
Kennedy GC (2000). The impact of genomics on ther-apeutic drug development. EXS 89:1–10.
Kiddo H, Maquieira A, Hammock BD (2000). Disc-basedimmunoassay microarrays. Anal Chim Acta 411:1–11.
Kim S, Dougherty ER, Bittner ML, Chen Y, Sivakumar K,Metzer P (2000). General nonlinear framework for theanalysis of gene interaction via multivariate expressionarrays. J Biomed Optics 5:411–424.
Kjaergaard S, Skovby F, Schwartz M (1998). Absence ofhomozygosity for predominant mutations in PMM2 inDanish patients with carbohydrate-deficient glycopro-tein syndrome type 1. Eur J Hum Genet 6:331–336.
Kjaergaard S, Skovby F, Schwartz M (1999). Carbohy-drate-deficient glycoprotein syndrome type 1A: expres-sion and characterisation of wild type and mutantPMM2 in E. coli. Eur J Hum Genet 7:884–888.
Kleyn PW, Vesell ES (1998). Genetic variation as a guide todrug development. Science 281:1820–1821.
Kondo I, Mizugishi K, Yoneda Y, Hashimoto T, Kuwa-jima K, Yuasa L, et al. (1999). Missense mutations inphosphomannomutase 2 gene in two Japanese familieswith carbohydrate-deficient glycoprotein syndrome type1. Clin Genet 55:50–54.
Konecki DS, Lichter-Konecki U (1991). The phenylketo-nuria locus: current knowledge about alleles and muta-tions of the phenylalanine hydroxylase gene in variouspopulations. Hum Genet 87:377–388.
Kost GJ (2000). Connectivity: the millennium challenge forpoint-of-care testing. Arch Pathol Lab Med 124:1108–1110.
Krajden M (1995). Molecular detection of hepatitis C virus:impact of detection methodology on clinical andlaboratory correlations. Crit Rev Clin Lab Sci 32:41–66.
Kraus JP, Janosik M, Kozich V, Mandell R, Shih V,Sperandeo MP, et al. (1999). Cystathionine beta-synthase mutations in homocystinuria. Hum Mutat13:362–375.
Kupferschmid TD, Calicchio T, Budowle B (1999). MaineCaucasian population DNA database using twelve shorttandem repeat loci. J Forensic Sci 44:392–395.
Langley SD, Lai K, Kembrue PP, Hjelm LN, Elsas LJ(1997). Molecular basis of Duarte and Los Angelesvariant galactosemia. Am J Hum Genet 60:366–372.
Liu T-C, Kim H, Arizmendi C, Kitano A, Patel MS (1993).Identification of two missense mutations in a dihydro-lipoamide dehydrogenase-deficient patient. Proc NatlAcad Sci USA 90:5186–5190.
Louis M, Tanguay RM (1997). Mutations in the fumaryl-acetoacetate hydrolase gene causing hereditary tyrosine-mia type I: overview. Hum Mutat 9:291–299.
Matthijs G, Schollen E, Pardon E, Veiga-Da-Cunha M,Jaeken J, Cassiman J-J, Van Schaftingen E (1997).Mutations in PMM2, a phosphomannomutase gene onchromosome 16p13, in carbohydrate-deficient glycopro-tein type I syndrome (Jaeken syndrome). Nat Genet16:88–92.
Matthijs G, Schollen E, Heykants L, Grunewald S (1999).Phosphomannomutase deficiency: the molecular basis ofthe classical Jaeken syndrome (CDGS type Ia). MolecGenet Metab 68:220–226.
Matthijs G, Schollen E, Bjursell C, Erlandson A, Freeze H,Imtiaz F, et al. (2000). Mutations in PMM2 that causecongenital disorders of glycosylation, type Ia (CDG-Ia).Hum Mutat 16:386–394.
NEW DISC-BASED TECHNOLOGIES FOR DIAGNOSTIC AND RESEARCH APPLICATIONS
Psychiatric Genetics 2002, Vol 12 No 4 205
Copyright © Lippincott Williams & Wilkins. Unauthorized reproduction of this article is prohibited.

Miller MS, McCarver DG, Bell DA, Eaton DL, GoldsteinJA (1997). Genetic polymorphisms in human drugmetabolic enzymes. Fundam Appl Toxicol 40:1–14.
Mitchell G, Larochelle J, Lambert M, Michaud J, GrenierA, Ogier H, et al. (1990). Neurologic crises in hereditarytyrosinemia. N Engl J Med 322:432–437.
Mudd SH, Skovby F, Levy HL, Pettigrew KD, Wilcken B,Pyeritz RE, et al. (1985). The natural history ofhomocystinuria due to cystathionine beta-synthasedeficiency. Am J Hum Genet 37:1–31.
Natt E, Kao F-T, Rettenmeier R, Scherer G (1986).Assignment of the human tyrosine aminotransferasegene to chromosome 16. Hum Genet 72:225–228.
Natt E, Kida K, Odievre M, Di Rocco M, Scherer G(1992). Point mutations in the tyrosine aminotransferasegene in tyrosinemia type II. Proc Natl Acad Sci 89:9297–9301.
Nicoltti P (1980). The epidemiology of brucellosis. Adv VetSci Comp Med 24:69–98.
Nilsberth C, Westlind-Danielsson A, Eckman CB, ForsellC, Axelman K, Luthman J, et al. (2000). The Arctic APPmutation (E693G) causes Alzheimer’s disease through anovel mechanism: increased amyloid b protofibrilformation and decreased amyloid b levels in plasmaand conditioned media. Neurobiol Aging 21(suppl 1):S58.
Nobukuni Y, Mitsubuchi H, Akaboshi I, Indo Y, Endo F,Yoshioka A, Matsuda I (1991). Maple syrup urinedisease: complete defect of the E1-beta subunit of thebranched chain alpha-ketoacid dehydrogenase complexdue to a deletion of an 11-bp repeat sequence whichencodes a mitochondrial targeting leader peptide in afamily with the disease. J Clin Invest 87:1862–1866.
Paine RS (1957). The variability in manifestations ofuntreated patients with phenylketonuria (phenylpyruvicaciduria). Pediatrics 20:290–302.
Patel MS, Harris RA (1995). Mammalian alpha-keto aciddehydrogenase complexes: gene regulation and geneticdefects. FASEB J 9:1164–1172.
Reichardt JKV, Woo SLC (1990). Missense mutations ingalactosemia (abstract). Am J Hum Genet 47(suppl):A164.
Reichardt JKV, Woo SLC (1991). Molecular basis ofgalactosemia: mutations and polymorphisms in the geneencoding human galactose-1-phosphate uridyltransfer-ase. Proc Natl Acad Sci USA 88:2633–2637.
Renwick PJ, Birley AJ, McKeown CME, Hulten M (1995).Southern analysis reveals a large deletion at thehypoxanthine phosphoribosyltransferase locus in a pa-tient with Lesch–Nyhan syndrome. Clin Genet 48:80–84.
Roest Crollius H, Jaillon O, Bernot A, Dasilva C, BouneauL, Fischer C, et al. (2000). Estimate of human genenumber provided by genome-wide analysis using Tetra-odon nigroviridis DNA sequence. Nat Genet 25:235–238.
Sachidanandam R, Weissman D, Schmidt SC, Kakol JM,Stein LD, Marth G, et al. (2001). A map of humangenome sequence variation containing 1.42 million singlenucleotide polymorphisms. Nature 409:928–933.
Scherer SW, Otulakowski G, Robinson BH, Tsui L-C(1991). Localization of the human dihydrolipoamide
dehydrogenase gene (DLD) to 7q31-q32. Cytogenet CellGenet 56:176–177.
Schweitzer S, Shin Y, Jakobs C, Brodehl J (1993). Long-term outcome in 134 patients with galactosaemia. Eur JPediatr 152:36–43.
Scriver CR, Kaufman S, Woo SLC (1989). The hyperphe-nylalaninemias. In: The Metabolic Basis of InheritedDisease, 6th ed., Scriver CR, Beaudet AL, Sly WS, ValleD (editors). New York: McGraw-Hill. pp. 495–546.
Shi MM, Mehrens D, Dacus K (2001). Pharmacogenomics:changing the health care paradigm. Modern DrugDiscovery 4:27–32.
Sreevatsan S, Bookout JB, Ringpis F, Perumaala S, FichtTA, Adams LG, et al. (2000). A multiplex approach tomolecular detection of Brucella abortus and/or Myco-bacterium bovis infection in cattle. J Clin Microsc38:2602–2610.
Timmers C, Grompe M (1996). Six novel mutations in thefumarylacetoacetate hydrolase gene of patients withhereditary tyrosinemia type I. Hum Mutat 7:367–369.
Tsai MY, Bignell M, Schwichtenberg K, Hanson NQ(1996). High prevalence of a mutation in the cystathio-nine beta-synthase gene. Am J Hum Genet 59:1262–1267.
Van Schaftingen E, Jaeken J (1995). Phosphomannomutasedeficiency is a cause of carbohydrate-deficient glycopro-tein syndrome type I. FEBS Lett 377:318–320.
Venter JC, Adams MD, Meyers EW, Li PW, Mural RJ,Sutton GG, et al. (2001). The sequence of the humangenome. Science 291:1304–1351.
Visy JM, Le Coz P, Chadefaux B, Fressinaud C, WoimantF, Marquet J, et al. (1991). Homocystinuria due to 5,10-methylenetetra-hydrofolate reductase deficiency revealedby stroke in adult siblings. Neurology 41:1313–1315.
Warne D, Watkins C, Bodfish P, Nyberg K, Spurr NK(1991). Tetranucleotide repeat polymorphism at thehuman beta-actin related pseudogene 2 (ACTBP2)detected using the polymerase chain reaction. Nucl AcidsRes 19:6980.
Weber WW (1997). Pharmacogenomics. New York: OxfordUniversity Press.
Wilson JM, Frossard P, Nussbaum RL, Caskey CT, KelleyWN (1983). Human hypoxanthine-guanine phospho-ribosyltransferase: detection of a mutant allele byrestriction endonuclease analysis. J Clin Invest 72:767–772.
Wong PWK, Justice P, Smith GF, Hsia DY-Y (1972). Acase of classical maple syrup urine disease, ‘thiaminenon-responsive’. Clin Genet 3:27–33.
Zein NN, Rakela J, Krawitt EL, Reddy KR, Tominga T,Persing DH, Group CS (1996). Hepatitis C virusgenotypes in the United States: epidemiology, patho-genicity, and response to interferon therapy. Ann IntMed 125:634–639.
Zneimer SM, Lau KS, Eddy RL, Shows TB, Chuang JL,Chuang DT, Cox RP (1991). Regional assignment of twogenes of the human branched-chain alpha-keto aciddehydrogenase complex: the E1-beta gene (BCKDHB)to chromosome 6p21–22 and the E2 gene (DBT) tochromosome 1p31. Genomics 10:740–747.
BARATHUR ET AL.
206 Psychiatric Genetics 2002, Vol 12 No 4
Copyright © Lippincott Williams & Wilkins. Unauthorized reproduction of this article is prohibited.




















