
NEW CHALLENGES AND OPPORTUNITIES IN PRENATAL ...
178
MICROARRAY ANALYSIS oke Muys NEW CHALLENGES AND OPPORTUNITIES IN PRENATAL INVASIVE DIAGNOSIS MICROARRAY ANALYSIS Joke Muys Prof. Dr. Bettina Blaumeiser Prof. Dr. Yves Jacquemyn Dr. Katrien Janssens
-
Upload
khangminh22 -
Category
Documents
-
view
1 -
download
0
Transcript of NEW CHALLENGES AND OPPORTUNITIES IN PRENATAL ...

Proefschrift voorgelegd tot het behalen van de graad van doctor in de medische wetenschappen aan de Universiteit Antwerpen te verdedigen door: Joke MUYS | Promotors: Bettina Blaumeiser, Yves Jacquemyn Co-promotor: Katrien Janssens | Faculteit Geneeskunde en Gezondheidswetenschappen | Antwerpen, 2020
Proefschrift voorgelegd tot het behalen van de graad van doctor in de medische wetenschappen aan de Promotors: Bettina Blaumeiser, Yves Jacquemyn
Co-promotor: Katrien Janssens | Faculteit Geneeskunde en Gezondheidswetenschappen | Antwerpen, 2020
Proefschrift voorgelegd tot het behalen van de graad van doctor in de medische wetenschappen aan de Promotors: Bettina Blaumeiser, Yves Jacquemyn
Co-promotor: Katrien Janssens | Faculteit Geneeskunde en Gezondheidswetenschappen | Antwerpen, 2020
Proefschrift voorgelegd tot het behalen van de graad van doctor in de medische wetenschappen aan de Promotors: Bettina Blaumeiser, Yves Jacquemyn
Co-promotor: Katrien Janssens | Faculteit Geneeskunde en Gezondheidswetenschappen | Antwerpen, 2020
NEW CHALLENGES AND OPPORTUNITIES IN PRENATAL INVASIVE DIAGNOSISMICROARRAY ANALYSISJoke Muys
cover-boek-hand.indd 2cover-boek-hand.indd 2 07/07/2020 07:5807/07/2020 07:58
NEW CHALLENGES AND OPPORTUNITIES IN PRENATAL INVASIVE DIAGNOSISMICROARRAY ANALYSIS
Joke Muys
Prof. Dr. Bettina BlaumeiserProf. Dr. Yves JacquemynDr. Katrien Janssens
cover-boek-hand.indd 1cover-boek-hand.indd 1 07/07/2020 07:5807/07/2020 07:58
Proefschrift voorgelegd tot het behalen van de graad van doctor in de Medische Wetenschappen aan de Universiteit Antwerpen te verdedigen door:
Joke MUYS
Promotors: Prof. Dr. Bettina Blaumeiser, Prof. Dr. Yves Jacquemyn
Co-promotor: Dr. Katrien Janssens
Faculteit Geneeskunde en Gezondheidswetenschappen
Antwerpen, 2020
NEW
CHA
LLENG
ES AN
D O
PPORTU
NITIES
IN PREN
ATAL IN
VASIVE D
IAG
NO
SISJoke M
uys

New challenges and opportunities in prenatal invasive diagnosis: microarray analysis
Joke Muys

New challenges and opportunities in prenatal invasive diagnosis: microarray analysis
ISBN: 978-94-6416-046-8
The studies in this thesis were financially supported by Fonds Wetenschappelijk Onderzoek (FWO) (grant number: 1700917N )
Cover by Bart Muys Lay-out & printed by Ridderprint
Copyright © 2020, by Joke Muys

NEW CHALLENGES AND OPPORTUNITIES IN PRENATAL INVASIVE DIAGNOSIS
MICROARRAY ANALYSIS
NIEUWE UITDAGINGEN EN OPPORTUNITEITEN BIJ PRENATALE INVASIEVE DIAGNOSE
MICROARRAY ANALYSE
Proefschrift voorgelegd tot het behalen van de graad van doctor in de Medische Wetenschappen aan de Universiteit Antwerpen te verdedigen door:
Joke Muys
Promotors: Prof. Dr. Bettina Blaumeiser, Prof. Dr. Yves Jacquemyn
Co-promotor: Dr. Katrien Janssens
Faculteit Geneeskunde en Gezondheidswetenschappen
Antwerpen, 2020


5
MEMBERS OF THE JURYPromotors
Bettina Blaumeiser, M.D., PhD., Department of Medical Genetics, University of Antwerp
Yves Jacquemyn, M.D., PhD., Department of Obstetrics and Gynaecology, University of Antwerp
Co-promotor
Katrien Janssens, PhD., Department of Medical Genetics, University of Antwerp
PhD. Commission
Ludo Mahieu, M.D., PhD., Department of Neonatology, University of Antwerp
Geert Mortier, M.D., PhD., Department of Medical Genetics, University of Antwerp
Members of the jury
Lieve Page – Christiaens, M.D., PhD., Department of Obstetrics and Gynaecology, Utrecht University; Associate Director Medical Affairs, Reproductive Genetic Health, Illumina Inc., San Diego
Malgorzata Ilona Srebniak, PhD., Department of Medical Genetics, Rotterdam University

6

7
TABLE OF CONTENTS
1 GENERAL INTRODUCTION
2 THE BELGIAN PRENATAL MICROARRAY (BEMAPRE) DATABASE: A SYSTEMATIC NATIONWIDE REPOSITORY OF FETAL GENOMIC ABERRATIONS.
3 PRENATALLY DETECTED COPY NUMBER VARIANTS IN A NATIONAL COHORT: A POSTNATAL FOLLOW-UP STUDY.
4 CHROMOSOMAL MICRO-ARRAY ANALYSIS IN PRENATAL DIAGNOSIS: ETHICAL CONSIDERATIONS OF THE BELGIAN APPROACH.
5 PRENATAL HOMOZYGOSITY MAPPING DETECTS A NOVEL MUTATION IN CHST3 IN A FETUS WITH SKELETAL DYSPLASIA AND JOINT DISLOCATIONS.
6 GENERAL DISCUSSION AND FUTURE PERSPECTIVES
7 SUMMARY | SAMENVATTING
8 LIST OF ABBREVIATIONS
9 REFERENCES
10 ADDENDUM: QUESTIONNAIRES
11 CURRICULUM VITAE
12 ACKNOWLEDGEMENTS
9 19
41
65
79
89
99
103
107
117
161
169

8
LABO. EXPELLU PTATATURESTO BEATI-IS DUCID UT ERUNT, QUAMUSDAE PROVIDUS DIT OMNITIBUS.
cover-boek-hand.indd 8cover-boek-hand.indd 8 07/07/2020 07:5807/07/2020 07:58

9
1 GENERAL INTRODUCTIONInvasive prenatal diagnosis is designed to determine the health status and development of the fetus before birth, by examination of fetal (amniotic fluid or fetal blood) or placental (chorionic villi) material. The procedure for obtaining amniotic fluid from a pregnant woman, by inserting a needle through the abdominal wall and into the amniotic sac, is called amniocentesis. One of the first reports of transabdominal amniocentesis dates back to over 100 years ago.1, 2 Those first amniocenteses were performed in a “blind” manner: the puncture side was determined by external palpation of the uterus, and the needle was inserted. After implementation of ultrasound in obstetrics and the publication of the first case reports on identification of fetal and placental malformations by ultrasonography in the 1960s, ultrasonography was progressively used for placental localization before puncture,2, 3 and nowadays, ultrasound guidance during the procedure is obligatory. Genetic analysis of amniotic fluid was first reported in 1955, when Serr determined fetal sex by examining Barr bodies in amniotic fluid.2, 4 Several reports described an association between early amniocentesis (before the 15th week of pregnancy) and fetal loss or talipes equinovarus,5, 6 and called for an alternative method to obtain fetal samples in pregnancies with fetal abnormalities. In 1968, Mohr introduced the concept of first trimester chorionic villi sampling (CVS).2, 7 Chorionic villi are part of the placenta and can be obtained by ultrasound guidance through vaginal or transabdominal biopsy.8-10 The procedure can be performed from the 11th gestational week onwards, to avoid a delay in case of fetal abnormalities. The first reports of the clinical application of CVS for diagnosis of hemoglobinopathies and chromosomal abnormalities originate from the 1980s.8, 9, 11, 12
Since that time, the genetic landscape has drastically changed, as have the possibilities for analyzing samples obtained by invasive prenatal procedures. Genetic tests can be divided into cytogenetic and molecular analyses.
1 1

10
Chapter 1
1.1 CYTOGENETIC ANALYSESCytogenetics is the study of the structure and number of chromosomes using microscopy.
1.1.1 KaryotypingMitosis is the cell division by which the body grows and achieves tissue regeneration. In mitosis, especially during the stages of metaphase and prometaphase, the 23 chromosome pairs are at their most condensed state and become visible under a microscope. The first step in karyotyping involves the interruption of mitotic cell division, the so-called mitotic arrest, to withhold cells from proceeding to the next stages of mitosis. A hypotonic solution is applied to fixate the cells after which they can be stained by various staining methods, of which Giemsa staining is the most frequently used, in order to display their sequence content.13, 14 Giemsa staining results in a characteristic pattern of alternating light and dark lines, the so-called G-bands. The 23 chromosome pairs can be differentiated by a combination of length, G-banding pattern and location of their centromere, i.e. the point of attachment between 2 sister chromatids, which divides chromosome pairs into a short arm (p) and a long arm (q). Chromosomes are arranged in a karyogram (Figure 1.1, Figure 1.2, Figure 1.3), allowing for the determination of chromosome number, as well as chromosomal translocations (the ‘swapping’ of parts of the chromosomes). However, because of the maximally condensed state of the DNA at the time of mitotic arrest, a karyogram depicts chromosomes at 1/10000 of their fully extended state.13 Therefore, the karyotyping technique can only detect larger chromosomal abnormalities; it has a resolution (detection precision) of approximately 5000 to 10000 kilobases (kb). Time to diagnosis (turnaround time or TAT) is at least two weeks and because of the complicated process which relies on cell division, the test is prone to failure.
Delaunay was the first to define a karyotype as the phenotypic appearance of chromosomes in 1922.15 However, it took several more years (1955-1956) before the human karyotype was depicted, because of the high uncertainty about the number of chromosomes in a human cell. When the procedure of amniocentesis was fully established in the 1970s, karyotype analysis on cells cultured from amniotic fluid was introduced. Today, karyotyping remains one of the most frequently performed techniques in the analysis of prenatal samples.2

11
General introduction
Figure 1.1: Normal karyotype of a 46,XX female patient.
Figure 1.2: Normal karyotype of a 46,XY
male patient.
Figure 1.3: Karyotype of a 47,XY male patient with Down syndrome (trisomy 21). Note the presence of 3 copies of chromosome 21.
1 1

12
1.1.2 Fluorescence in situ hybridization (FISH)With FISH, labeled probes are hybridized to DNA within chromosomes to visualize numerical or structural chromosomal aberrations. The in-situ hybridization (ISH) techniques originally utilized in the 1970s and early 1980s applied radioactive probes that were replaced by non-radioactive probes labeled with fluorescent dyes (Figure 1.4). Throughout the years, the clinical use of FISH has remained unchanged and to date, it is still utilized to obtain rapid results on both numerical (e.g. trisomy 21) and structural aberrations (e.g. a balanced translocation) after invasive testing, and to test for deletions or duplications in regions of specific interest.2 The FISH technique, however, does not allow genome-wide DNA testing.
Figure 1.4: FISH of a XXY male patient, with probes at Yq12 (orange) and the X chromosome centromere (green).
Chapter 1

13
1.2 MOLECULAR GENETIC ANALYSESMolecular genetics is the study of DNA at the molecular level and requires the isolation of DNA from the patient material.
1.2.1 Polyrase Chain Reaction (PCR) techniqueIn 1983, the PCR technique was developed, allowing the selective amplification of a particular DNA sequence several billion-fold in a very limited time-frame.2
Basically, the technique is an enzymatic augmentation (or amplification) of a fragment of DNA located between two primers. These primers are designed so that one primer is complementary to one strand of the DNA molecule on one side of the targeted fragment and the other primer is complementary to the opposite strand at the other side of the fragment. After converting the double-stranded DNA into two single strands (a process called denaturation), DNA polymerase produces two new strands of DNA complementary to the target sequence. Next, the newly synthesized strands of DNA serve as templates for the primers to synthesize two additional strands of DNA. Every round doubles the amount of the target sequence, resulting in an abundance of copies of the same fragment. This amplified fragment of DNA is the basis of many molecular tests.13
Like FISH, the PCR technique does not allow genome-wide DNA testing and either requires a priori information on the mutation to be tested (e.g. in case of a familial mutation) or can be used to detect a defined small set of aberrations.2 PCR was initially used for the detection of cystic fibrosis in the fetus, but also for diagnosing fetal infectious diseases like congenital toxoplasmosis in amniotic fluid. Multiple variations of the basic PCR technique exist, two of which play a key role in rapid aneuploidy detection in prenatal diagnosis. The first is quantitative fluorescent PCR (QF-PCR), where polymorphic small tandem repeats of various length (so-called markers) are fluorescently labeled during the PCR. The ratio of the alleles of each of these markers, being distributed across the 5 chromosomes of interest (13, 18, 21, X and Y), can be used to detect an aneuploidy. Secondly, multiplex ligation-dependent probe amplification (MLPA) allows relative quantification of multiple DNA sequences in a single reaction. Both techniques are commonly utilized for rapid assessment of the presence of common aneuploidies.
General introduction
1 1

14
1.2.2 Chromosomal Microarray Analysis (CMA)In 1992, the development of array comparative genomic hybridization (array CGH) introduced a major leap in diagnostic technologies.2 With the array CGH technology, the difference in copy number, or amount, of a particular DNA segment in two different DNA samples can be determined. Total DNA from one sample (case) is labeled with a red fluorescent dye and the other (control) sample is labeled with a green dye. The two labeled DNA samples are mixed in equal amounts and applied on a microarray chip containing a large number of probes, each probe corresponding to a different unique fragment in the human genome. These fragments are distributed evenly throughout the genome; however, a higher probe density can be provided in loci associated with disease. Quantification of the dosage of a particular fragment of DNA present in the test sample versus the control sample is obtained by measuring the ratio of red-to-green fluorescence emitted by each probe.13 Inherent to the principle of microarray, the technique can detect the absence (deletion) or amplification (duplication) of very small parts of the chromosome (down to 100 kb), but it cannot detect balanced translocations or other aberrations that do not cause a net loss or gain of chromosomal material. The first publications on the application of array CGH in prenatal diagnosis date from 2005.16
The human genome project (1990 - 2003), generated sequence data from many hundreds of individuals worldwide, thus providing information about the natural variation in human DNA. Single Nucleotide Polymorphisms (SNPs) are the most common of all variations in human DNA. They appear in general once every 1000 base pairs. SNPs usually have two possible nucleotide types or alleles (simplified as A and B), that each occur in a substantial percentage of the human population. An individual inherits one allele from each parent for every SNP. Therefore, each individual has three possible genotypes (AA, AB and BB). In SNP arrays, each interrogated SNP is represented by a probe. A and B alleles are differentiated from each other by a single nucleotide extension step utilizing a two-dye chemistry (similar to the red and green color in array CGH). Next, signal intensity of each allele is determined. B allele frequency (BAF), which is the relative amount of the presence of one allele compared to the other, is determined by the ratio of the measured intensities from the two alleles. Homozygous SNPs have BAFs of 0 (AA) or 1 (BB), whilst heterozygous SNPs have BAFs near 0.5 (AB). Log R ratio (LRR) represents the ratio of observed versus expected intensities of the case sample (Figure 1.5). Consequently, SNP arrays, in addition to the typical array CGH technology, can be used to identify patterns of allelic imbalance, triploidy, maternal cell contamination and regions of homozygosity.17 Chapter 5 of this doctoral thesis discusses the introduction of homozygosity mapping with SNP array in a prenatal setting. Detecting regions of homozygosity is of added diagnostic value in case of fetal ultrasonographic abnormalities in consanguineous parents. The SNP array technology was introduced in prenatal testing in 2010.18
Chapter 1
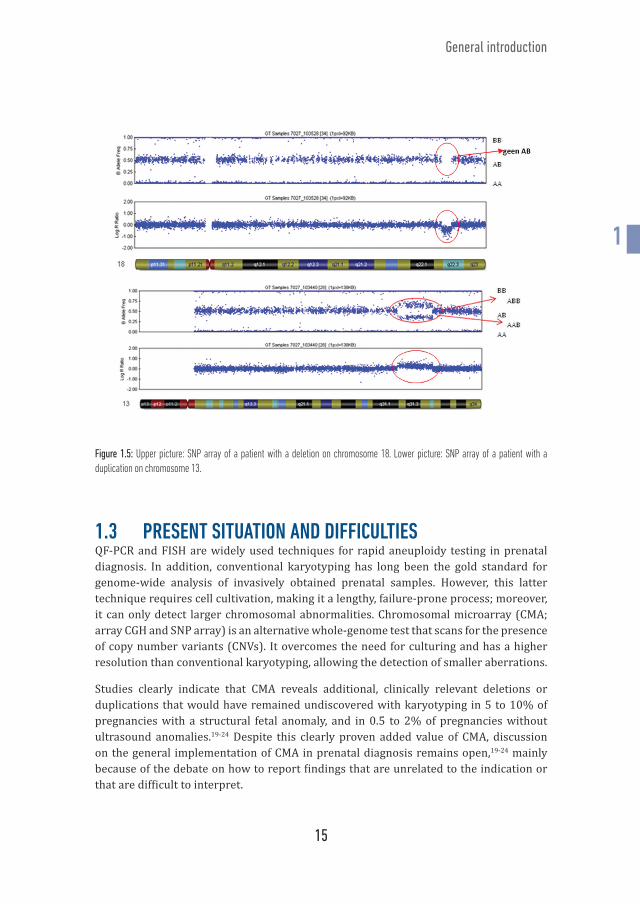
15
Figure 1.5: Upper picture: SNP array of a patient with a deletion on chromosome 18. Lower picture: SNP array of a patient with a duplication on chromosome 13.
1.3 PRESENT SITUATION AND DIFFICULTIESQF-PCR and FISH are widely used techniques for rapid aneuploidy testing in prenatal diagnosis. In addition, conventional karyotyping has long been the gold standard for genome-wide analysis of invasively obtained prenatal samples. However, this latter technique requires cell cultivation, making it a lengthy, failure-prone process; moreover, it can only detect larger chromosomal abnormalities. Chromosomal microarray (CMA; array CGH and SNP array) is an alternative whole-genome test that scans for the presence of copy number variants (CNVs). It overcomes the need for culturing and has a higher resolution than conventional karyotyping, allowing the detection of smaller aberrations.
Studies clearly indicate that CMA reveals additional, clinically relevant deletions or duplications that would have remained undiscovered with karyotyping in 5 to 10% of pregnancies with a structural fetal anomaly, and in 0.5 to 2% of pregnancies without ultrasound anomalies.19-24 Despite this clearly proven added value of CMA, discussion on the general implementation of CMA in prenatal diagnosis remains open,19-24 mainly because of the debate on how to report findings that are unrelated to the indication or that are difficult to interpret.
General introduction
1 1

16
The uncertainty concerning the significance and clinical implications of some prenatally identified CNVs by using CMA on one hand, and its obvious added clinical value on the other hand, inspired Belgian genetic centers to adopt a national consensus on how to interpret and report prenatal CMA findings.25 As of mid-2013, all samples for prenatal genetic diagnosis were no longer analyzed by karyotyping, but by CMA. (Chapter 2)
1.3.1 Towards a national databaseOriginating from this unique national consensus, the idea to create a national prenatal CNV database, linking prenatal ultrasound findings with CMA data and postnatal clinical and neurodevelopmental data, came into existence. Public databases, such as the Database of Genomic Variants, DECIPHER, Ecaruca, The International Collaboration for Clinical Genomics, and others, are useful, but consist mainly of postnatal cases, which creates ascertainment bias: this group almost certainly represents the more severe end of the phenotypic spectrum, and therefore provides an incomplete characterization of the phenotype.19 Hence, although data from symptomatic infants evaluated postnatally gives some guidance for prenatal counseling, it remains difficult to predict the phenotype based on the prenatally detected genotype. As detected CNVs can be population-dependent, a national database for prenatal microarrays in Belgium represents a necessary condition for evidence-based genetic counseling. As part of this doctoral thesis, a national prenatal database was constructed, to facilitate communication between genetic centers, increase knowledge and as a mean to answer the proposed research questions. (Chapter 2)
1.3.2 Prenatal phenotype – Genotype – Postnatal phenotypeCreation of an appropriate database is necessary for studying correlations between CNV type, CNV size, gene content, prenatal phenotype and postnatal development in children diagnosed prenatally with a non-benign CNV. The postnatal follow-up of these children is of value, first, because it increases our understanding of susceptibility and pathogenic CNVs and, secondly, because it will aid in interpreting variants where the clinical outcome is still unknown (Variants of Unknown Significance or VOUS). In this work, prenatal genotype-phenotype associations were studied, and children prenatally diagnosed with a pathogenic CNV, susceptibility CNV or VOUS were examined in a postnatal follow-up study. (Chapter 3)
1.3.3 Ethical considerationsThe publication of our Belgian approach has sparked discussions worldwide and has inspired other countries to develop guidelines.26 Current professional society guidelines suggest the application of CMA for evaluating fetuses with ultrasound anomalies.27, 28
Chapter 1

17
Recently, an online tool was introduced by the American College of Medical Genetics and Genomics (ACMG) to aid CNV classification,29 but choosing which CNVs to report and which not to in a prenatal setting remains a lab-specific decision, as no general international guidelines exist. Although there is a clear need for an international consensus on the interpretation and, more importantly, the reporting of prenatally found CNVs, the Belgian approach remains unique to date.
From an ethical perspective, reporting a CNV in a prenatal context is very different from reporting it in a postnatal setting: future parents may decide to terminate the pregnancy, even without clear proof that the child will be affected; alternatively, they may decide to continue the pregnancy, but remain fearful about their child’s health even after birth. This sparks ethical discussions on what should and should not be reported.30-32 It is the responsibility of geneticists to report findings without violating patient’s (and their future children’s) autonomy and their right “not to know”.33-36 In this doctoral thesis, cultural and ethical reflections are made regarding the Belgian reporting system. (Chapter 4)
1.4 AIMS OF THE THESISThe overall aim of this thesis was to determine/refine genotype-phenotype correlations between prenatally detected pathogenic, susceptibility and unclassified CNVs.
The first aim was to create a large national database of prenatally identified CNVs as an essential asset for the clinical geneticist and (cyto)geneticist with regards to interpretation of CNVs.
The second aim was to determine the most frequently found pathogenic CNVs, susceptibility CNVS and VOUS in a prenatal setting in Belgium and to evaluate their associated prenatal phenotypes.
The third aim was to assess postnatal clinical outcome of children diagnosed prenatally with a non-benign CNV.
The fourth aim was to reflect ethically about the Belgian prenatal reporting system. The fifth aim was to investigate other opportunities for implementing CMA in prenatal diagnosis.
General introduction
1 1

18
LABO. EXPELLU PTATATURESTO BEATI-IS DUCID UT ERUNT, QUAMUSDAE PROVIDUS DIT OMNITIBUS.
cover-boek-hand.indd 10 cover-boek-hand.indd 1007/07/2020 07:58 07/07/2020 07:58

19
2 THE BELGIAN PRENATAL MICROARRAY (BEMAPRE) DATABASE: A systematic nationwide repository of fetal genomic aberrations
Joke Muys, Bettina Blaumeiser, Yves Jacquemyn, Claude Bandelier, Nathalie Brison, Saskia Bulk, Patrizia Chiarappa, Winnie Courtens, Anne De Leener, Marjan De Rademaeker, Julie Désir, Anne Destree, Koenraad Devriendt, Annelies Dheedene, Annelies Fieuw, Erik Fransen, Jean-Stéphane Gatot, Philip Holmgren, Mauricette Jamar, Sandra Janssens, Kathelijn Keymolen, Damien Lederer, Björn Menten, Marije Meuwissen, Benoit Parmentier, Bruno Pichon, Sonia Rombout, Yves Sznajer, Ann Van Den Bogaert, Kris Van Den Bogaert, Olivier Vanakker, Joris Vermeesch, Katrien Janssens
Prenat Diagn. 2018;38(13):1120–1128. doi:10.1002/pd.5373
2 1

20
Chapter 2
2.1 ABSTRACT
2.1.1 ObjectiveWith the replacement of karyotyping by chromosomal microarray (CMA) in invasive prenatal diagnosis, new challenges have arisen. By building a national database, we standardize the classification and reporting of prenatally detected copy number variants (CNVs) across Belgian genetic centers. This database, which will link genetic and ultrasound findings with postnatal development, forms a unique resource to investigate the pathogenicity of variants of uncertain significance and to refine the phenotypic spectrum of pathogenic and susceptibility CNVs.
2.1.2 Methods The BElgian PREnatal MicroArray (BEMAPRE) consortium is a collaboration of all genetic centers in Belgium. We collected data from all invasive prenatal procedures performed between May 2013 and July 2016.
2.1.3 Results In this three-year period, 13 266 prenatal CMAs were performed. By national agreement, a limited number of susceptibility CNVs and no variants of uncertain significance were reported. Added values for using CMA versus conventional karyotyping were 1.8% in the general invasive population and 2.7% in cases with an ultrasound anomaly. Of the reported CNVs 31.5% would have remained undetected with NIPT as the first-tier test.
2.1.4 Conclusion The establishment of a national database for prenatal CNV data allows for a uniform reporting policy and the investigation of the prenatal and postnatal genotype- phenotype correlation.

21
Prenatal database for chromosomal microarray results
2.2 INTRODUCTIONChromosomal microarray analysis (CMA) scans for the genome-wide presence of microdeletions and microduplications or copy number variants (CNVs). Recent years have seen a steady rise of CMA at the expense of karyotyping in the analysis of invasively obtained prenatal samples (amniotic fluid or chorion villi). With the use of CMA, the requirement for cell culturing, which is a lengthy and failure-prone process, is overcome. Moreover, current array designs allow for a higher resolution than conventional karyotyping (100-400 kb versus 5-10 Mb), enabling the detection of smaller CNVs.37
In 5 to 10% of pregnancies with a fetal structural anomaly and in 0.5-2% of pregnancies without, CMA reveals cryptic, clinically relevant CNVs.19-24
With the introduction of this new technique, new challenges arose. Due to the higher resolution, genetic variants causing late-onset disorders (e.g., Charcot-Marie-Tooth disease), variants with a reduced penetrance/variable expression (susceptibility CNVs), and variants for which there is no information on possible consequences (Variants Of Unknown Significance (VOUS)) can be detected.38 There is no international consensus on policy for the reporting of these findings to future parents. Reporting a CNV in a prenatal setting is ethically very different from the postnatal setting: future parents may decide to discontinue the pregnancy, even without ‘hard’ evidence that the child will be affected; alternatively, when continuing the pregnancy, they may remain anxious about the child’s development. In addition, parents may obtain knowledge about their own personal health.
In Belgium, all samples for prenatal genetic diagnosis have been analyzed by CMA since 2013.25 Despite the use of different types of genomic array platforms (SNP array and array CGH) in the eight genetic centers, a cut-off resolution of 400 kb for both deletions and duplications was agreed upon in order to maximize the detection of pathogenic variants while minimizing the number of VOUS. In the case that the genomic platform allowed for detection of clearly pathogenic CNVs smaller than 400 kb, these variants were of course reported as well.
CNVs are classified as benign, pathogenic, susceptibility or VOUS. Benign CNVs are repeatedly found in the normal population and are not associated with pathological phenotypes; they are never reported. Pathogenic CNVs are recurrent genomic rearrangements with a well-defined congenital phenotype or aberrations resulting in a known effect on the function of a gene that correlates with a known phenotype (e.g., haploinsufficiency). These CNVs are generally reported. When the finding is unrelated to the indication of the CMA (incidental finding),39 the following reporting policy is applied: dominant late-onset diseases with clinical utility (therapeutic options, preventive measures, termination of pregnancy) are reported to future parents; carriership for autosomal recessive diseases is reported if the carrier frequency is >1/50; and X-linked carrier status is always reported.
Susceptibility CNVs are genetic risk factors with reduced penetrance and/or variable
2 1

22
expression, often associated with a highly unpredictable phenotype that does not present prenatally (e.g., intellectual disability, autism spectrum disorder, epilepsy, psychiatric disorder). A limited number of susceptibility CNVs are reported in the prenatal setting. This list (Table 2.1), which was composed by geneticists from all of the Belgian genetic centers, takes into account penetrance and severity 40-43 and is updated on a yearly basis. All CNVs that cannot be classified as benign, pathogenic or susceptibility are designated VOUS.
Table 2.1: List of susceptibility CNVs reported in the prenatal setting (version February 2018)
Chr. Region del/ dup† start in Mb (hg19)
stop in Mb (hg 19) size in kb gene Phenotype morphological
anomaly OMIM ClinGen score
distal 1q21.1 dup 146,57 147,39 820 GJA5 (CX40) ID‡ , DD§, ASD¶, SZ†† macrocephaly, CHD 612475 3distal 1q21.1 del 146,57 147,39 820 GJA5 (CX40) ID, DD, ASD, SZ, facial
dysmorphismmicrocephaly, CHD, renal and urinary tract anomalies
612474 3
1q24.3 del 171,81 172,38 57 DNM3 ID IUGR, microcephaly, brachydactyly
Awaiting Review
15q13.3 del 31,13 32,48 1350 CHRNA7 DD, ID, ASD, epilepsy, SZ microcephaly, CHD 612001 315q26 del 99,36 102,52 3160 IGF1R ID IUGR 3Distal 16p11.2 del 28,74 28,96 220 SH2B1 obesity, DD, ID, SZ none 613444 2
16p11.2 proximal dup 29,59 30,19 600 TBX6 ASD, ID, DD, SZ, anorexia
microcephaly 614671 3
16p11.2 proximal del 29,59 30,19 600 TBX6 ID, DD, ASD, obesity, SZ, speech delay
macrocephaly, vertebra
611913 3
17q12 del 34,82 36,21 1390 HNF1B facial dysmorphy, gen-ital abnormalities, ID, DD, ASD, MODY‡‡
renal anomalies 614527 3
22q11.2 dup 19,02 20,29 1270 TBX1 DD, epilepsy, dysmorphic features
Microcephaly, CHD 608363 3
The ClinGen score refers to the evidence for a haploinsufficiency phenotype (deletion) or a triplosensitive phenotype (duplication). 3 = sufficient evidence; 2 = some evidence. Abbreviations: †: del/dup: deletion/duplication, ‡: ID: Intellectual Disability, §: DD: Developmental Delay, ¶: ASD: Autism Spectrum Disorder, ††: SZ: Schizophrenia, ‡‡: MODY: maturity onset diabetes of the young
Chapter 2

23
expression, often associated with a highly unpredictable phenotype that does not present prenatally (e.g., intellectual disability, autism spectrum disorder, epilepsy, psychiatric disorder). A limited number of susceptibility CNVs are reported in the prenatal setting. This list (Table 2.1), which was composed by geneticists from all of the Belgian genetic centers, takes into account penetrance and severity 40-43 and is updated on a yearly basis. All CNVs that cannot be classified as benign, pathogenic or susceptibility are designated VOUS.
Table 2.1: List of susceptibility CNVs reported in the prenatal setting (version February 2018)
Chr. Region del/ dup† start in Mb (hg19)
stop in Mb (hg 19) size in kb gene Phenotype morphological
anomaly OMIM ClinGen score
distal 1q21.1 dup 146,57 147,39 820 GJA5 (CX40) ID‡ , DD§, ASD¶, SZ†† macrocephaly, CHD 612475 3distal 1q21.1 del 146,57 147,39 820 GJA5 (CX40) ID, DD, ASD, SZ, facial
dysmorphismmicrocephaly, CHD, renal and urinary tract anomalies
612474 3
1q24.3 del 171,81 172,38 57 DNM3 ID IUGR, microcephaly, brachydactyly
Awaiting Review
15q13.3 del 31,13 32,48 1350 CHRNA7 DD, ID, ASD, epilepsy, SZ microcephaly, CHD 612001 315q26 del 99,36 102,52 3160 IGF1R ID IUGR 3Distal 16p11.2 del 28,74 28,96 220 SH2B1 obesity, DD, ID, SZ none 613444 2
16p11.2 proximal dup 29,59 30,19 600 TBX6 ASD, ID, DD, SZ, anorexia
microcephaly 614671 3
16p11.2 proximal del 29,59 30,19 600 TBX6 ID, DD, ASD, obesity, SZ, speech delay
macrocephaly, vertebra
611913 3
17q12 del 34,82 36,21 1390 HNF1B facial dysmorphy, gen-ital abnormalities, ID, DD, ASD, MODY‡‡
renal anomalies 614527 3
22q11.2 dup 19,02 20,29 1270 TBX1 DD, epilepsy, dysmorphic features
Microcephaly, CHD 608363 3
The ClinGen score refers to the evidence for a haploinsufficiency phenotype (deletion) or a triplosensitive phenotype (duplication). 3 = sufficient evidence; 2 = some evidence. Abbreviations: †: del/dup: deletion/duplication, ‡: ID: Intellectual Disability, §: DD: Developmental Delay, ¶: ASD: Autism Spectrum Disorder, ††: SZ: Schizophrenia, ‡‡: MODY: maturity onset diabetes of the young
Prenatal database for chromosomal microarray results
the first nation-wide database collecting prenatal genetic results and structural findings on ultrasound as the basis for longitudinal studies of the developmental effect of CNVs. The database, furthermore, ensures unanimous reporting and counseling policy.
Despite these guidelines, ambiguous situations still occur, which are tackled by a committee of experts. To guide their decisions, an appropriate database relating prenatal genetic and ultrasound findings to postnatal clinical and neurodevelopmental data had to be built. Here we report on the resulting BElgian PREnatal MicroArray (BEMAPRE) database, which contains the data from all Belgian invasive tests performed in a three-year period (May 2013–July 2016). This database allows the identification of the most frequent pathogenic CNVs, susceptibility CNVs and VOUS in Belgium and to calculate added values for the use of CMA versus karyotyping. To the best of our knowledge, this is
2 1

24
2.3 METHODS
2.3.1 Study conductThe BEMAPRE consortium is a collaboration of clinical and laboratory geneticists from every genetic center in Belgium (http://www.beshg.be/index.php?page=centers). It aims to collect data on all invasive procedures performed in Belgium. Approval of the central ethical committee and of the College for Human Genetics of the Federal Ministry of Public Health in Belgium has been granted for this project. Data are stored in a coded manner in the Bench Lab CNV 5.0 platform provided by Agilent Technologies (Cartagenia NV). Agilent Technologies was not involved in this research in any other way.
2.3.2 Data collectionWe collected data from invasive prenatal procedures performed between May 2013 and July 2016. The centers provided the indications for the invasive tests and the CMA results obtained. These indications comprised: an aberrant Down syndrome screening test; advanced maternal age; a structural fetal abnormality on ultrasound (including increased nuchal translucency); a familial genetic disorder; an abnormal result for a Non-Invasive Prenatal Test (NIPT); other (including maternal seroconversion for Cytomegalovirus (CMV) or Toxoplasmosis and anxiety).
Possible CMA outcomes were: no or only benign CNV(s); aneuploidy; pathogenic CNV; VOUS; susceptibility CNV reported; susceptibility CNV not reported. Note that pathogenic CNVs also include incidental findings, because a syndromic genomic disorder can be viewed as such a finding if the reason for the CMA did not relate to the syndrome. To determine the added value of CMA over karyotyping, CNVs were grouped on the basis of their size (larger/smaller than 10 Mb). All VOUS were reanalyzed in September 2017 for a possible class-change to benign or pathogenic, based on recent literature and information in publicly available CNV databases.
For all prenatal cases with a non-benign CNV (pathogenic CNV, susceptibility CNV or VOUS, with the exclusion of aneuploidies and unbalanced translocations), the following information was obtained: chromosome number, start and stop position of the CNV (hg19), size of the CNV, copy number, class, gender, clinical information (Human Phenotype Ontology (HPO)) and (whenever available) mode of inheritance. For 6,660 of 13,266 cases (50.2%), information on the indication for the invasive procedure was acquired.
Chapter 2

25
2.3.3 Data analysisRecurrence of the following CNVs was evaluated: VOUS deletion, VOUS duplication, pathogenic deletion, pathogenic duplication and susceptibility CNVs. CNVs were labeled recurrent if appearing at least five times in our population and when presenting with a smallest overlapping region of at least 80% to account for platform-specific differences. The percentage overlap takes into account the size of the regions and is calculated as follows: 2 times the overlap between 2 CNVs divided by the sum of lengths of both CNVs.
2.3.4 Statistical analysisDescriptive statistics were used to describe population, patient and CNV characteristics. SPSS 24 (IBM Corp. Released 2016. IBM SPSS Statistics for Windows, Version 24.0. Armonk, NY: IBM Corp.) was applied to analyze data. Frequency tables describing the association between indication and mutation type were visualized using correspondence analysis. The correspondence plots were generated using the ca package from the software package R, version 3.1.2.44, 45
Prenatal database for chromosomal microarray results
2 1

26
2.4 RESULTSBetween May 2013 and July 2016, 13 266 prenatal CMAs were performed in Belgium. The principal indications were a structural fetal abnormality (including increased nuchal translucency) (30.2%) and an aberrant Down syndrome screening test (30.4%). Further indications included advanced maternal age (13.1%), familial genetic disorder (10.8%), positive NIPT (2.0%), and other (13.5%).
1,347 of the 13,266 cases (10.2%) carried an aneuploidy. In 54% of these, at least one structural abnormality was visible on ultrasound investigation. Conversely, in the presence of ultrasound anomalies, 18.1% of cases demonstrated aneuploidy or an unbalanced translocation. As expected, aneuploidies were particularly common in the positive NIPT group (69.6%) (Figure 2.1; Table 2.2)
Figure 2.1: CMA results in prenatal cases subdivided according to indication for invasive prenatal testing. CMA results are classified as normal (no or only benign CNVs), aneuploidy, pathogenic CNV > 10 Mb, pathogenic CNV < 10 Mb, reported susceptibility CNV, unreported susceptibility CNV and VOUS.
Chapter 2

27
Prenatal database for chromosomal microarray results
Table 2.2: CMA results in prenatal cases subdivided according to indication for invasive prenatal testing
USA† 2013 (100)
1444 (71,7)
364 (18,1)
26 (1,3)
41 (2)
14 (0,7)
20 (1)
104 (5,2)
FTS‡ 2022 (100)
1770 (87,5)
111 (5,5)
5 (0,2)
9 (0,5)
16 (0,8)
15 (0,8)
96 (4,7)
Fam gen disorder§
720 (100)
625 (86,8)
29 (4)
1 (0,1)
8 (1,1)
9 (1,3)
4 (0,6)
44 (6,1)
NIPT ¶ 135 (100)
33 (24,5)
94 (69,6)
3 (2,2)
1 (0,7)
0 (0)
0 (0)
4 (3)
AMA†† 874 (100)
796 (91,1)
22 (2,5)
1 (0,1)
7 (0,8)
2 (0,2)
4 (0,5)
42 (4,8)
Other 896 (100)
828 (92,4)
21 (2,3)
1 (0,1)
7 (0,8)
4 (0,4)
3 (0,4)
32 (3,6)
Total 6660 (100)
5496 (82,5)
641 (9,6)
37 (0,6)
73 (1,1)
45 (0,7)
46 (0,7)
322 (4,8)
CMA results are classified as normal (no or only benign CNVs), aneuploidy, pathogenic CNV > 10 Mb, pathogenic CNV < 10 Mb, reported susceptibility CNV, unreported susceptibility CNV and VOUS. Table view. Please note that numbers and percentages are based on 6660 cases (50.2% of the population). Abbreviations: †: USA: Ultrasound anomaly, ‡: FTS: an aberrant Down screening test, §: Fam gen disorder: Known genetic disorder in the family, ¶: NIPT: abnormal result on Non-Invasive Prenatal Test, ††: AMA: Advanced Maternal Age, ‡‡: Path: Pathogenic CNV, §§: VOUS: Variant of Unknown Significance, Other: CMV, toxoplasmosis, anxiety and remaining indications.
Indi
catio
ns
Tota
l in
dica
tion
(%)
Norm
al (%
)
Aneu
ploi
dy (%
)
Path
‡‡
>10
Mb(%
)
Path
<1
0 Mb
(%)
Susc
eptib
ility
re
porte
d (%
)
Susc
eptib
ility
no
t rep
orte
d (%
)
VOUS
§§ (%
)
2 1

28
In 1.9% of cases (246/13,266), a pathogenic CNV was detected; 175 of those (71.1%) had a CNV smaller than 10 Mb that presumably would have escaped detection by karyotyping (Supplementary Table 1 available on request). More than half of the fetuses (63.0% or 155/246) with a pathogenic CNV had a structural abnormality on ultrasound investigation; 39 (25.2%) of those carried multiple structural anomalies. Figure 2.2 and Table 2.3 show the distribution of CNV classes in cases with ultrasound anomalies. In the category of ‘Positive NIPT’, a pathogenic CNV was detected in four (2.9%) cases (Figure 2.1), three of which were larger than 10 Mb (2.2%). Correspondence plots did not show an association between the indication for the invasive procedure and finding a pathogenic CNV (data not shown).
Figure 2.2: Distribution of CNV classes in cases with ultrasound anomalies, sorted according to the organ system involved. The following subcategories are defined: multiple anomalies, increased nuchal translucency (NT), cardiac anomaly, facial anomaly, anomalies of the nervous system, intrathoracic anomaly, hernia diaphragmatica, skeletal anomaly, growth anomaly, anomalies of the abdomen (including gastroschisis and omphalocoele), anomaly of the amniotic fluid, genito-urinary anomaly, twin-to-twin transfusion syndrome and unknown anomaly. Cases are classified as multiple if more than one compartment is affected. Graphic view.
Chapter 2

29
Table 2.3: Distribution of CNV classes in cases with ultrasound anomalies, sorted according to the organ system involved
Pathogenic CNV
Susceptibility reported
Susceptibility not reported VOUS§
Multiple 39 3 12 67NT¶ 28 4 11 66Heart 25 3 3 29Facial 5 0 6 19Central nervous system
15 2 7 37
Thoracic cavity 2 0 0 2Hernia diaphragmatica
6 1 1 4
Skeleton 7 3 2 6Growth 9 1 2 6Abdomen 4 4 3 6Amniotic fluid 6 0 3 3Genito-urinary 4 4 1 13Twin to twin 0 0 1 11Unspecified 7 1 2 18Total 155 26 53 287
Table view. Abbreviations: §: VOUS: Variant of Unknown Significance, ¶: NT: Increased Nuchal Translucency
Table 2.4 lists the most frequent syndromic genomic disorders in our population. The 22q11.2 deletion syndrome (OMIM #188400) is by far the most common: we detected 41 cases, accounting for 0.31% of all invasive samples. The most common incidental findings were X-Linked Ichtyosis (OMIM #308100; 13 cases, 6 female and 7 male), Hereditary Neuropathy with liability to Pressure Palsies (OMIM #162500; 6 cases) and Charcot-Marie-Tooth type 1A (OMIM #118200; 5 cases) (Table 2.4; Supplementary Table 2 available on request).
Prenatal database for chromosomal microarray results
2 1

30
Table 2.4: Most frequent syndromic disorders, susceptibility CNVs (reported and unreported), incidental findings and VOUS in our prenatal population
Syndromic disorders Location n
% of total invasive population
cases with an ultrasound anomaly % (n)
22q11 del (OMIM 188400)
22q11 41 0,31 80 (33)
X-Linked Ichtyosis (OMIM #308100)
Xp22.3 13 0,10 23 (3)
Hereditary Neurop-athy with liability to Pressure Palsies (OMIM #162500)
17p12 6 0,05 50 (3)
Wolf-Hirschhorn (OMIM 194190)
4p16.3 5 0,04 100 (5)
Charcot-Marie-Tooth type 1A (OMIM #118200)
17p12 5 0,04 40 (2)
Williams Beuren (OMIM 194050)
7q11.23 5 0,04 100 (5)
Susceptibility CNVs (reported) Location n
% of total invasive population
cases with an ultrasound anomaly % (n)
22q11.2 dup (OMIM 608363)
chr22:19.020.000-20.290.000
24 0,18 44 (11)
GJA5 dup (OMIM 612475)
chr1:146.570.000-147.390.000
14 0,11 21 (3)
CHRNA7 del (OMIM 612001)
chr15:31.130.000-32.480.000
8 0,06 14 (1)
GJA5 del (OMIM 612474)
chr1:146.570.000-147.390.000
7 0,05 50 (4)
TBX6 dup (OMIM 614671)
chr16:29.590.000-30.190.000
5 0,04 40 (2)
TBX6 del (OMIM 611913)
chr16:29.650.000-30.200.000
5 0,04 40 (2)
HNF1B del (OMIM 614527)
chr17:34.820.000-36.210.000
5 0,04 80 (4)
Chapter 2

31
Susceptibility CNVs(unreported) Location n
% of total invasive population
cases with an ultrasound anomaly % (n)
15q11.2 dup chr15:22.800.000-23.090.000
32 0,24 34 (11)
15q11.2 del (OMIM 615656)
chr15:22.800.000-23.090.000
25 0,19 56 (14)
CHRNA7 dup chr15:31.130.000-32.480.000
21 0,16 10 (2)
MYH11 dup chr16:14.980.000-16.480.000
16 0,12 56 (9)
NPHP1 dup chr2:110.870.000-110.980.000
13 0,10 46 (6)
HFE2 dup chr1:144.970.000-146.100.000
10 0,08 20 (2)
MYH11 del chr16:14.980.000-16.480.000
9 0,07 44 (4)
VOUS Location n% of total invasive population
cases with an ultrasound anomaly % (n)
6q22.31 dup chr6: 123.539.625 - 124.328.531
10 0,07 50 (5)
17p13.3 dup chr17: 148.092 - 597.702
6 0,05 0 (0)
9p23 dup chr9: 10.164.926 - 11.868.588
6 0,05 33 (2)
10q23.31 del chr10: 91.626.482 - 92.035.457
6 0,05 33 (2)
22q11.23 dup chr22: 23.720.181 - 24.959.827
6 0,05 17 (1)
14q11.2 dup chr14: 22.323.879 – 22.964.864
5 0.04 40 (2)
3p14.2 dup chr3: 59.666.501 – 60.993.079
5 0.04 20 (1)
The table shows the genomic location, the number of cases with this CNV, the frequency in our prenatal population and the percentage and number of cases with an ultrasound anomaly.
Prenatal database for chromosomal microarray results
2 1

32
Susceptibility CNVs were diagnosed in 1.6% (210/13266) of our population; based on our national guidelines,25 one third of those (71/210 or 33.8%; 0.5% of the total population) were reported (Table 2.1). In cases with an ultrasound anomaly, 0.7% carried a reported susceptibility CNV; this was not significantly different compared to the prevalence in the entire prenatal population, in accordance with the fact that susceptibility CNVs are rarely associated with ultrasound anomalies. Table 2.4 shows the most frequent susceptibility CNVs: the 22q11.2 duplication syndrome (OMIM #608363; 24 cases) and the 15q11.2 BP1-BP2 duplication46 (32 cases) are respectively the most common reported and unreported susceptibility CNV. Susceptibility CNVs were all cryptic.
The overall added diagnostic value of using CMA compared to karyotyping was 1.8%. Added values were calculated by taking into account all reported CNVs (pathogenic CNVs and reported susceptibility CNVs). Table 2.5 shows the added diagnostic value of CMA per indication. In cases with versus without an ultrasound anomaly, CMA had an added diagnostic value of respectively 2.7% and 1.5%.
Table 2.5: Yield of karyotyping, CMA and NIPT in prenatal samples subdivided according to indication
IndicationsYield karyotyping in %
Yield CMA‡‡ in%
Yield NIPT (all aneuploidies) in%
Added value CMA vs karyotyping in %
USA† 19,4 22,1 18,1 2,7FTS‡ 5,7 7 5,5 1,3Fam gen disorder§
4,2 6,7 4 2,5
NIPT ¶ 71,9 72,6 69,6 0,7AMA †† 2,6 3,7 2,5 1,1Other§§ 2,6 4,2 2,3 1,6Total 10,1 11,9 9,6 1,8
The added value of using CMA vs karyotyping is shown in the last column. Yield is the percentage of diagnoses detected by using a particular test compared to not testing at all. Abbreviations: †: USA: Ultrasound anomaly, ‡: FTS: an aberrant Down screening test , §: Fam gen disorder: Known genetic disorder in the family, ¶: NIPT: abnormal result on Non-Invasive Prenatal Test, ††: AMA: Advanced Maternal Age, ‡‡: CMA: Chromosomal Microarray Analysis, §§: Other (including CMV, toxoplasmosis, anxiety)
Chapter 2

33
Of all the cases, 5.6% (746/13,266) carried a VOUS: a deletion in 23.6% of the cases (176/746), a duplication in 72.9% (544/746), and both in 3.5% (26/746) (Supplementary Table 1 available on request). In 38.5% (287/746) of these, structural fetal abnormalities were present on ultrasound; this percentage increased to 46.8% in cases with more than one VOUS. VOUS were distributed evenly among the different indications, as revealed by correspondence analysis (data not shown).
Seven recurrent VOUS were detected in our population, one deletion and six duplications (Table 2.4). The most frequent recurrent VOUS was a duplication on chromosome 6q22.31 (ten cases). The common region (chr6:123.539.625-124.328.531; 789 kb) contains the genes TRDN (Triadin) and NKAIN2 (NA+/K+ Transporting ATPase- interacting 2). As described by Srebniak et al., this may represent a private variant that is benign when present alone, but may act as a second hit in carriers of an additional VOUS.47 In all our cases, this was an isolated finding. Moreover, indications for invasive testing and fetal phenotype were different, supporting Srebniak’s conclusion that this variant is benign when occurring privately, although a common postnatal phenotype cannot be excluded. The only recurrent deletion is located on chromosome 10q23.31 and was diagnosed in six cases (common region: chr10: 91.626.482-92.035.457; 409 kb). This region encompasses only one pseudogene. In three cases, the deletion was inherited from a phenotypically normal parent, arguing against its pathogenicity.
To explore the effect of CNV load, we examined the phenotype of children with more than one reported CNV (excluding cases with an aneuploidy or unbalanced translocation) or with one reported CNV and one VOUS, versus those with an isolated reported CNV. Of 317 cases with a reported CNV (246 with a pathogenic CNV and 71 with a susceptibility CNV), 33 cases (10.4%) had more than one reported CNV. Of those, 20 (60.6%) had structural abnormalities. Another 27 of 317 cases (8.5%) had both a reported CNV and a VOUS. Of those, 18 (66.7%) had structural abnormalities. Of the remaining 257 cases with a reported CNV, structural abnormalities were found in 143 cases (55.6%). There was no significant difference in the presence of ultrasound anomalies between groups (p = 0.497).
With the implementation of NIPT, invasive prenatal testing will increasingly become restricted to pregnancies with ultrasound anomalies and those with a known genetic defect in the family. If NIPT becomes the first-tier test for all other indications, subchromosomal aberrations will be missed. Presuming a NIPT technology that can detect all aneuploidies, this would account for 31.5% (100/317) of reported CNVs in our study population. This percentage decreases slightly to 26.2% (83/317) in case of “genome-wide NIPT” (detecting all aberrations above 10Mb) (Table 2.6). For the added values of using CMA versus karyotyping and NIPT, see Table 2.5.
Prenatal database for chromosomal microarray results
2 1

34
Table 2.6: CMA results in prenatal cases subdivided according to indication for invasive prenatal testing (13266 invasive procedures)
Pathogenic CNVs Susceptibility
n path >10Mb
n path <10Mb
n susc reported
n susc not reported
USA 51 104 26 53
FTS 6 16 21 36
Fam gen disorder 3 21 12 9
AMA 1 8 4 14
NIPT 4 1 0 1
Other Indications 3 19 5 21
- Anxiety 0 1 2 3
- CMV 1 6 2 9
- Toxoplasmosis 0 5 1 4
- Remaining 2 7 0 5
Unknown 3 6 3 5
TOTAL 71 175 71 139
CMA results are classified as pathogenic CNV > 10 Mb, pathogenic CNV < 10 Mb, reported susceptibility CNV, and unreported susceptibility CNV. USA: Ultrasound anomaly; FTS: an aberrant Down syndrome screening test; Fam gen disorder: Known genetic disorder in the family; NIPT: abnormal result on Non-Invasive Prenatal Test; AMA: Advanced Maternal Age; Other: CMV, toxoplasmosis, anxiety and remaining indications. This table shows the diagnoses that can be missed if NIPT becomes the first-tier test for all other indications.
2.5 DISCUSSIONIn Belgium, approximately 125,000 children are born every year. Over a three-year period (May 2013-July 2016), 13 266 invasive prenatal procedures were performed.
The most frequently detected syndromic genomic disorder was the 22q11.2 deletion syndrome. We encountered this deletion in 0.31% of our population (41 cases). In their prospective study analyzing 9,500 prenatal samples, Grati et al. found a comparable prevalence (0.3%).48 The reported postnatal prevalence of the syndrome is much lower: in a large population-based study involving 255,849 babies, 0.017% carried the deletion.49 We can discern several reasons for this discrepancy. First, the phenotypic spectrum of the 22q11.2 deletion syndrome is broad, causing underdiagnosis of this syndrome in the postnatal setting. Second, prenatal cases with ultrasound anomalies are more likely to be terminated. Finally, 22q11.2 pregnancies are thought to be more prone to end in a miscarriage: in a recent study in which the incidence of 22q11.2 deletions in 26,101 products of conception was examined,50 12/9,398 (0.13%) samples which were normal at karyotype resolution had an isolated 22q11.2 deletion, approaching the prevalence in our prenatal population. Of our 41 cases, 53.7% had an ultrasound anomaly
Chapter 2

35
that was clearly related to the genetic finding.
The 22q11.2 duplication syndrome was the most frequently reported susceptibility CNV in our prenatal population (24 cases or 0.18%). In a control population, the frequency is 0.05%.41 The variant has a broad phenotypic spectrum. The most common symptoms are intellectual disability/learning difficulties (97%), delayed psychomotor development (67%), growth retardation (63%), muscular hypotonia (43%), and cardiac anomalies (20%).51, 52 Patients with a 22q11.2 duplication are 4.1 to 10 times more at risk of developing a neurodevelopmental disorder.53 Although in the majority of cases (69%), the duplication is inherited from a normal parent,51 this susceptibility CNV is nevertheless reported prenatally because of its possible association with fetal structural anomalies and the importance of ultrasonographic follow-up. In this study, 11/24 (45.8%) of fetuses with a 22q11.2 duplication syndrome had ultrasonographic abnormalities (short femora (2), transposition of the great arteries (1), increased nuchal translucency (4)).
The 15q11.2 duplication (chr15:22800000–23090000, minimal size 290 kb) is the most frequently found unreported susceptibility CNV in our population (32 cases). The phenotypic spectrum of developmental delay is highly variable, from motor coordination problems to autism spectrum disorder and obsessive compulsive disorder.54 Although initially described as a susceptibility region for neurological dysfunction,46 several more recent reports failed to show a clear genotype-phenotype association. Cooper described 64/15,767 patients with developmental delay versus 36/8,329 healthy controls (penetrance 0.64),42 Coe detected the 15q11.2 duplication in 128/29,085 patients with developmental delay versus 60/19,584 healthy controls, resulting in a likelihood ratio of 1.44.40 In a study of 2,521 autism spectrum disorder families, Chaste found no difference in frequency between patients and healthy siblings.55 The highly variable and often mild phenotype and the low penetrance and likelihood ratio justify our reporting policy.
The phenotype resulting from a susceptibility CNV is highly unpredictable. Belgian geneticists compiled a limited list of susceptibility loci that should be reported and a non-exhaustive list of those that are not reported (Table 2.1 and Table 2.7), based on the clinical spectrum, expected severity, and published odds ratios or penetrance values.40-43 The fetal and parental phenotype is also taken into account. These lists are re-evaluated on a yearly basis. We observe a strong correlation between our reporting policy and the dosage sensitivity score given by ClinGen (https://www.ncbi.nlm.nih.gov/projects/dbvar/clingen/). All reported loci have a score of 3 (sufficient evidence), with the exception of the 16p11.2 distal deletion (score 2; some evidence). Conversely, unreported susceptibility CNVs have a score of 0 (no evidence), 1 (little evidence) or 2. The 2p16.3 deletion has been given a score of 3 by ClinGen; at last evaluation, we concluded that penetrance had not been sufficiently determined for this CNV. The rationale behind this strict reporting policy is to avoid anxiety in and stigmatization of future parents over a CNV for which the outcome is highly uncertain.56, 57 Nonetheless, one might still reflect on the ethical consequences of not reporting a variant that unexpectedly does cause disease. Thus, elaborate pretest and posttest genetic counseling remain crucial when using CMA in prenatal diagnosis.
Prenatal database for chromosomal microarray results
2 1

36
Table 2.7: List of susceptibility CNVs that are not reported in the prenatal setting. (Version February 2018)
CNV del/dup† start in Mb (hg19)
stop in Mb (hg 19)
size in kb Gene phenotype morphological anomaly OMIM ClinGen score
1q21.1 dup 144,97 146,61 1640 HFE2/HJV DD‡, ASD§ CHD§§ absent
2p16.3 del 50 51,11 1110 NRXN1 ID¶, ASD, SZ††, DD, dysmorphic features
none 614332 3
2q13 dup 110,87 110,98 110 NPHP1 ASD, ID none 0
3q29 dup 197,2 198,84 1600 ID, DD none unknown
13q12 dup 20,81 21,01 1200 CRYL1 unknown unknown awaiting review
15q11.2 dup 22,8 23,09 290 NIPA1 DD, motor delay, speech delay, ASD
none unlikely
15q11.2 del 22,8 23,09 290 NIPA1 ID, DD, epilepsy CHD 615656 2
15q13.3 dup 31,13 32,48 1350 CHRNA7 ADHD‡‡, ID, DD, ASD
none 1
16p13.11 dup 14,98 16,48 1500 MYH11 ID, ASD, SZ, ADHD
aorta dilatation absent
16p13.11 del 14,98 16,48 1500 MYH11 ID, DD, ASD, epilepsy
microcephaly absent
16p12.2 dup 21,94 22,46 520 EEF2K, CDR2 unknown unknown 0
16p12.2 del 21,94 22,46 520 EEF2K, CDR2 DD, speech delay
cranofacial and skeletal abnormalities, CHD
136570 2
Distal 16p11.2 dup 28,74 28,96 220 SH2B1 anorexia, ID, DD, ASD, SZ
none 1
17q12 dup 34,73 36,22 1500 HNF1B DD none 0
Distal 22q11.2 dup 21,91 23,65 1740 DD, epilepsy, dysmorphic features
none unknown
The ClinGen score refers to the evidence for a haploinsufficiency phenotype (deletion) or a triplosensitive phenotype (duplication). 3 = sufficient evidence; 2 = some evidence; 1 = little evidence; 0 = no evidence. Abbreviations: †: del/dup: deletion/duplication, ‡: DD: Developmental Delay, §: ASD: Autism Spectrum Disorder , ¶: ID: Intellectual Disability, ††: SZ: Schizophrenia, ‡‡: ADHD: Attention Deficit/Hyperactivity disorder, §§: CHD: Congenital Heart Disease
Chapter 2

37
Table 2.7: List of susceptibility CNVs that are not reported in the prenatal setting. (Version February 2018)
CNV del/dup† start in Mb (hg19)
stop in Mb (hg 19)
size in kb Gene phenotype morphological anomaly OMIM ClinGen score
1q21.1 dup 144,97 146,61 1640 HFE2/HJV DD‡, ASD§ CHD§§ absent
2p16.3 del 50 51,11 1110 NRXN1 ID¶, ASD, SZ††, DD, dysmorphic features
none 614332 3
2q13 dup 110,87 110,98 110 NPHP1 ASD, ID none 0
3q29 dup 197,2 198,84 1600 ID, DD none unknown
13q12 dup 20,81 21,01 1200 CRYL1 unknown unknown awaiting review
15q11.2 dup 22,8 23,09 290 NIPA1 DD, motor delay, speech delay, ASD
none unlikely
15q11.2 del 22,8 23,09 290 NIPA1 ID, DD, epilepsy CHD 615656 2
15q13.3 dup 31,13 32,48 1350 CHRNA7 ADHD‡‡, ID, DD, ASD
none 1
16p13.11 dup 14,98 16,48 1500 MYH11 ID, ASD, SZ, ADHD
aorta dilatation absent
16p13.11 del 14,98 16,48 1500 MYH11 ID, DD, ASD, epilepsy
microcephaly absent
16p12.2 dup 21,94 22,46 520 EEF2K, CDR2 unknown unknown 0
16p12.2 del 21,94 22,46 520 EEF2K, CDR2 DD, speech delay
cranofacial and skeletal abnormalities, CHD
136570 2
Distal 16p11.2 dup 28,74 28,96 220 SH2B1 anorexia, ID, DD, ASD, SZ
none 1
17q12 dup 34,73 36,22 1500 HNF1B DD none 0
Distal 22q11.2 dup 21,91 23,65 1740 DD, epilepsy, dysmorphic features
none unknown
The ClinGen score refers to the evidence for a haploinsufficiency phenotype (deletion) or a triplosensitive phenotype (duplication). 3 = sufficient evidence; 2 = some evidence; 1 = little evidence; 0 = no evidence. Abbreviations: †: del/dup: deletion/duplication, ‡: DD: Developmental Delay, §: ASD: Autism Spectrum Disorder , ¶: ID: Intellectual Disability, ††: SZ: Schizophrenia, ‡‡: ADHD: Attention Deficit/Hyperactivity disorder, §§: CHD: Congenital Heart Disease
Prenatal database for chromosomal microarray results
2 1

38
The added value of using CMA rather than conventional karyotyping was 1.8% in the general invasive population and increased to 2.7% in cases with an ultrasound anomaly. Upon inclusion of unreported susceptibility CNVs, the added values rose to 2.5% and 3.7%, respectively. In 2014, De Wit and colleagues performed a systematic review of the added value of prenatal CMA in fetuses with an isolated structural anomaly.20
They found that in 5.6% of these pregnancies a pathogenic, cryptic CNV could be detected. Discrepancies in added values between different studies, even after homo- genizing cohorts, were explained by small samples sizes, differences in cohort selection and differences between array platforms applied. Our study data show that in addition, the classification and reporting policy of the laboratory strongly affects the added values. In the absence of structural anomalies on ultrasound, the added value of using CMA was 1.5% in our prenatal population; this further decreased to 1.1% when taking only uneventful pregnancies (advanced maternal age or maternal anxiety) into account. In a recent systematic review of the literature and meta-analysis, a similar risk figure of 0.86% for a submicroscopic pathogenic CNV was found for uneventful pregnancies.58
CNV load is known to contribute to the severity of neurodevelopmental and psychiatric disorders, but evidence of an association of CNV load and ultrasound anomalies is lacking.59 In this study, having a higher CNV load (2 vs 1 pathogenic CNV) was not associated with a higher incidence of ultrasound anomalies. (p=0.497)
Knowing the inheritance pattern of a VOUS can be powerful information: a de novo VOUS is more likely to be pathogenic than a VOUS inherited from an unaffected parent. As our reporting policy dictates not to communicate VOUS, examining inheritance is not obligatory. Consequently, the inheritance pattern was investigated for only 27.1% of our cases. Of the de novo cases (3.9% or 29 cases), 65.5% had ultrasound anomalies versus 30.6% in cases with a parentally inherited VOUS (173 cases or 23.2% of the population). We acknowledge that knowledge on the inheritance mode of all VOUS would have strengthened the paper and will reconsider our policy for future cases.
Worldwide, the number of invasive procedures is declining rapidly with the growing implementation of NIPT.60 As of July 1, 2017, Belgium became the first country in the world to fully reimburse NIPT for all pregnancies, resulting in an even steeper increase in NIPT uptake. Our study population (invasive prenatal testing between May 2013 and July 2016) was given the opportunity for a non-reimbursed NIPT for all indications. In the case of ultrasound anomalies, we observed a 4% difference (18.1% versus 22.1%) in the diagnostic yield of NIPT versus CMA (Table 2.5), clearly demonstrating that NIPT cannot replace CMA for this indication. With respect to the implementation of NIPT for pregnancies without ultrasound anomalies, concerns have also been raised, as subchromosomal pathogenic CNVs will be missed.61, 62 In our population, 26.2% (83/317) of reported CNVs below 10 Mb were found in cases with the indication ‘an aberrant Down syndrome screening test’, ‘advanced maternal age’ or ‘other indications’, all of which would have remained undetected with NIPT as the first-tier test, even when assuming a resolution similar to that of karyotyping.
Chapter 2

39
Prenatal database for chromosomal microarray results
2
Extensive pretest counseling is and will remain absolutely crucial to inform patients about the pros and cons of NIPT versus invasive prenatal testing and to help them choose the prenatal test that is most appropriate for their situation.
Publicly available CNV databases such as the Database of Genomic Variants, DECIPHER, Ecaruca and The International Collaboration for Clinical Genomics are valuable, but mainly consist of postnatal cases. As a consequence, these databases contain cases at the more severe end of the phenotypic spectrum, providing an incomplete characterization of the phenotype associated with a particular CNV. To increase our knowledge of the phenotypic spectrum of CNVs, we embarked on a postnatal follow-up project, the aim of which is to determine the relationship between the genetic result, prenatal findings and postnatal development, to reclassify VOUS and improve our comprehension about both susceptibility and pathogenic CNVs. On January 2017, postnatal clinical and neurodevelopmental follow-up at the age of 3 years for all children included in the BEMAPRE database was launched.
2.6 CONCLUSIONIn Belgium, a uniform reporting system facilitates the national registration of all non-benign CNVs. Our prenatal strategy is unique, as we are the only country with a nationwide uniform approach to prenatal CMA analysis, reporting and communal CNV data storage. In this paper, we reported on our national prenatal data. This large and unique dataset provides us with insights into the incidence of CNVs, possible associations with the indication for the invasive procedure and the fetal phenotype. The content of the database is made publicly available to researchers and clinicians worldwide through the website of the Belgian Society of Human Genetics (http://www.beshg.be/index.php?page=guidelines) and will be updated on a regular basis. Postnatal follow-up has been initiated and will be extremely valuable, as it will facilitate the association between prenatally detected CNVs and postnatal phenotypes.
1

40
LABO. EXPELLU PTATATURESTO BEATI-IS DUCID UT ERUNT, QUAMUSDAE PROVIDUS DIT OMNITIBUS.
cover-boek-hand.indd 8cover-boek-hand.indd 8 07/07/2020 07:5807/07/2020 07:58

41
3. PRENATALLY DETECTED COPY NUMBER VARIANTS IN A NATIONAL COHORT: a postnatal follow-up study
Joke Muys, Yves Jacquemyn, Bettina Blaumeiser, Laura Bourlard, Nathalie Brison, Saskia Bulk, Patrizia Chiarappa, Anne De Leener, Marjan De Rademaeker, Julie Désir, Anne Destrée, Koenraad Devriendt, Annelies Dheedene, Armelle Duquenne, Annelies Fieuw, Erik Fransen, Jean-Stéphane Gatot, Mauricette Jamar, Sandra Janssens, Jorien Kerstjens, Kathelijn Keymolen, Damien Lederer, Björn Menten, Bruno Pichon, Sonia Rombout, Yves Sznajer, Ann Van Den Bogaert, Kris Van Den Bogaert, Joris Vermeesch, Katrien Janssens
Prenat Diagn. 2020;10.1002/pd.5751. doi:10.1002/pd.5751
3 1

42
3.1 ABSTRACT
3.1.1 ObjectiveBelgian genetic centers established a database containing data on all chromosomal microarrays (CMA) performed in a prenatal context. A study was initiated to evaluate postnatal development in children diagnosed prenatally with a non-benign copy number variant (CNV).
3.1.2 MethodsAll children diagnosed with a prenatally detected non-benign CNV in a Belgian genetic center between May 2013 and February 2015 were included in the patient population. The control population consisted of children who had undergone an invasive procedure during pregnancy, with no or only benign CNVs. Child development was evaluated at 36 months using three (3) questionnaires: Ages and Stages Questionnaire Third edition, Ages and Stages Questionnaire Social-Emotional Second Edition and a general questionnaire.
3.1.3 ResultsA significant difference in communication and personal-social development was detected between children with a reported susceptibility CNV and both children with an unreported susceptibility CNV and the control population. The outcome of children with a particular CNV is discussed in a case-by-case manner.
3.1.4 ConclusionOur postnatal follow-up project of children with a prenatally detected non-benign CNV is the first nationwide project of its kind. A higher number of cases for each CNV category is however needed to confirm our findings.
Chapter 3

43
3.2 INTRODUCTIONFollowing the introduction of chromosomal microarray (CMA) in prenatal invasive diagnosis, difficulties arose concerning the interpretation and reporting of prenatally detected copy number variants (CNVs) to future parents.23, 25, 57, 63-66 Although the added value of using CMA over conventional karyotyping for the analysis of invasively obtained prenatal samples is extensively proven,19-22, 38, 67, 68 the higher resolution of the test not only increases detection of clinically relevant CNVs, but also reveals a higher number of variants of unknown significance (VOUS), incidental findings or variants with a variable expression or incomplete penetrance (susceptibility variants).
Publicly available CNV databases are valuable, but mainly rely on postnatal results and contain cases at the more severe end of the phenotypic spectrum, providing an incomplete characterization of the phenotype associated with a particular CNV, thus complicating the interpretation of prenatally detected CNVs. Additionally, upon reporting a CNV in a prenatal setting, future parents could consider discontinuing the pregnancy, even when only limited information exists on the variant found,63 or they may choose to continue the pregnancy, but remain anxious about the future health of their baby.56, 57
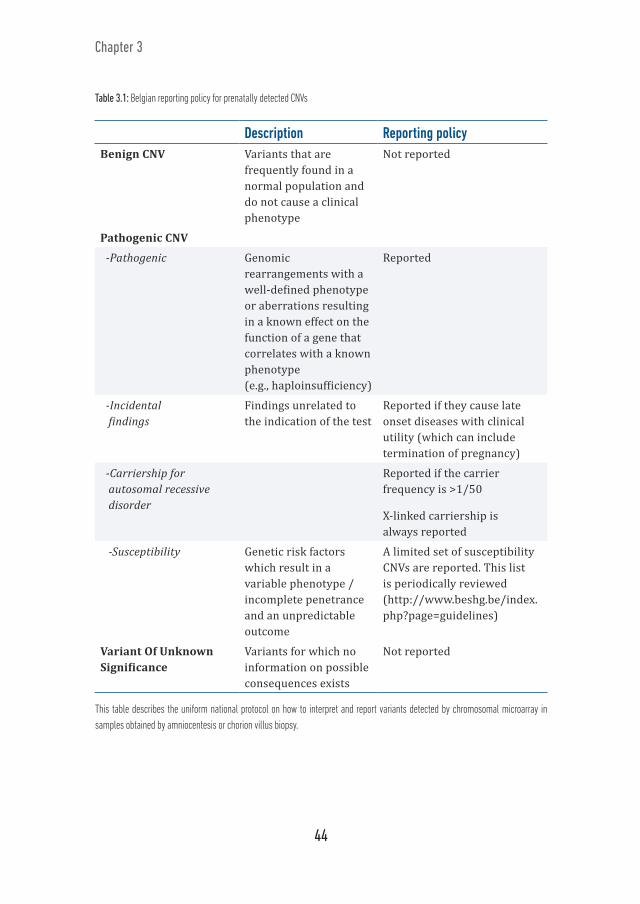
In Belgium, all genetic centers embarked on a unique national project.25 They agreed to use CMA for all indications for invasive prenatal testing. As previously published, a uniform national protocol on how to interpret and report variants was developed (Table 3.1).25, 65
A postnatal follow up-study of prenatally detected CNVs
3 1
44
Table 3.1: Belgian reporting policy for prenatally detected CNVs
Description Reporting policyBenign CNV Variants that are
frequently found in a normal population and do not cause a clinical phenotype
Not reported
Pathogenic CNV
-Pathogenic Genomic rearrangements with a well-defined phenotype or aberrations resulting in a known effect on the function of a gene that correlates with a known phenotype (e.g., haploinsufficiency)
Reported
-Incidental findings
Findings unrelated to the indication of the test
Reported if they cause late onset diseases with clinical utility (which can include termination of pregnancy)
-Carriership for autosomal recessive disorder
Reported if the carrier frequency is >1/50
X-linked carriership is always reported
-Susceptibility Genetic risk factors which result in a variable phenotype / incomplete penetrance and an unpredictable outcome
A limited set of susceptibility CNVs are reported. This list is periodically reviewed (http://www.beshg.be/index.php?page=guidelines)
Variant Of Unknown Significance
Variants for which no information on possible consequences exists
Not reported
This table describes the uniform national protocol on how to interpret and report variants detected by chromosomal microarray in samples obtained by amniocentesis or chorion villus biopsy.
Chapter 3

45
Furthermore, Belgian genetic centers established a shared prenatal database, gathering data on all prenatal CMAs performed since the switch from conventional karyotyping to CMA in 2013. This database facilitates data sharing and communication. In a recent study,65 analysis of the prenatal data gathered over a 3-year period showed pathogenic variants in 1.9% of cases; 71% of these cases were cryptic. The 22q11.2 deletion syndrome was the most frequently found genomic disorder. Of all cases, 1.6% carried a susceptibility CNV of which one-third (33.8%) was reported. The 22q11.2 duplication syndrome was the most frequent reported susceptibility CNV (SR for ‘susceptibility reported’), and the 15q11.2 BP1-BP2 duplication the most frequent unreported susceptibility CNV (SNR for ‘susceptibility not reported’). VOUS were detected in 5.6% of cases. The overall added value for using CMA instead of conventional karyotyping in all pregnancies where an invasive procedure was performed was 1.8%. The added value increased to 2.7% when anomalies were present in fetal ultrasound.
Since publicly available CNV databases do not provide a complete characterization of the phenotypic spectrum of a CNV, we initiated a national postnatal follow-up project to look at development in children diagnosed prenatally with a non-benign CNV in an unbiased manner. To the best of our knowledge, this is the first nationwide project initialized to follow up on children with prenatally detected CNVs.
3.3 METHODSThe central ethical committee and the College for Human Genetics of the Federal Ministry of Public Health in Belgium approved this project.
Human reference genome GRCh37 – hg19 was used for indicating start and stop positions of the CNVs.
The patient population was defined as: all children diagnosed in a Belgian genetic center with a prenatally detected pathogenic CNV (including incidental findings, but excluding aneuploidies and unbalanced translocations), susceptibility CNV (SR or SNR) or VOUS, collectively termed ‘non-benign CNVs’ between May 2013 and February 2015. The control population consisted of an equal number of children who had undergone an invasive procedure during pregnancy in the same study period, but had only benign CNVs or no CNVs. The goal was to create a similar distribution of indications for the invasive procedure compared to the patient population. Unless clear identification of each of the twin members was possible, twin pregnancies were excluded, as well as pregnancies that were known to be discontinued. After parental approval, child development was evaluated using 3 questionnaires when the child reached the age of 36 months.
The first questionnaire was the “Ages and Stages Questionnaire: a Parent-Completed Child Monitoring System, Third edition (ASQ-3)”.69 This questionnaire contains 30 developmental items, organized in five areas: communication, gross motor, fine motor,
A postnatal follow up-study of prenatally detected CNVs
3 1

46
problem solving and personal-social development and one overall section that focuses on general parental concerns. The scores are compared to the mean for each area of development, based on more than 18000 completed questionnaires. Children who score 1-2 standard deviations (SD) below the mean are considered to be in the monitoring zone and require close attention, specialized activities and/or repeat screening. If a child scores ≥ 2 SD below the mean, further diagnostic assessment is recommended for that specific area. Inclusion was allowed between 34 months 16 days and 38 months 30 days.
The second survey used was the “Ages and Stages Questionnaire: Social-Emotional Second Edition (ASQ-SE2)”,70 developed to complement the ASQ-3 and which focuses exclusively on the child’s social-emotional behavior. If the child scores within the monitoring zone (close to the referral cutoff point), follow-up actions for items of concern are required. Children scoring below the referral cutoff point are identified as needing further attention. The ASQ-SE2 has a permitted age range between 33 months 0 days and 41 months 30 days.
The last survey was a general questionnaire, enquiring about parental age, parental education, ethnicity, course of pregnancy, delivery etc. This questionnaire was composed in collaboration with the Children’s Neurodevelopmental Unit of the University Hospital in Antwerp, Belgium.
Patient and control samples were encoded. In each genetic center, only one researcher was granted authority to decode the center’s samples and contact the parents. Only the encoded data were used for all further data processing.
3.3.1 StatisticsTo test if cases versus controls and responders versus non-responders differed with regard to the indications, a Monte Carlo Chi-square test was carried out. The association between variant type and ASQ-3 and ASQ-SE2 results was tested using a one-way ANOVA, followed by a Posthoc analysis with Tukey correction for multiple testing. The effect of covariates on ASQ-3 and ASQ-SE2 scores was studied using multiple linear regression models with the scores as dependent variables, and parental level of education, multiple languages, surgical interventions, pre-term birth and age of the father as covariates. The model was simplified using stepwise backward elimination. Lastly, the association between pregnancy termination and variant type was investigated using a Monte-Carlo Chi-square test.
Statistical analyses were carried out using SPSS 24 (IBM Corp. Released 2016. IBM SPSS Statistics for Windows, Version 24.0. Armonk, NY.) and R, version 3.5.1.71
Chapter 3

47
3.4 RESULTSA non-benign CNV was detected in 757 cases. These children are referred to as the patient group. The control population was composed of 793 random samples. Indications for performing an invasive procedure on these samples are described in Table 3.2. There was no statistical difference in indications between both groups (p=0.23).
Table 3.2: Indications for invasive procedure in the patient population versus control group.
Indication Patient group (% of n=757)
Control group (% of n=793)
Fetal abnormality 37,6 28,1An aberrant down syndrome screening test 27,7 29,4Advanced maternal age 12,2 12,6A familial genetic disorder 11 8,7Toxoplasmosis or cmv† seroconversion 6,1 6,3An abnormal result for a NIPT‡ 0,5 0,5Other 3,7 3,7Unknown 1,2 10,7
Indications include: a fetal abnormality, including increased nuchal translucency; an aberrant Down syndrome screening test: first trimester combined test [ultrasound measurement of nuchal translucency + pregnancy-associated plasma protein A (PAPP-A) + free beta human chorionic gonadotrophin (hCG)] or second trimester triple test [alpha-fetoprotein (AFP) + hCG (total or free-β ) + unconjugated estriol]; advanced maternal age: 35 years or older at the time of conception; a familial genetic disorder: known cytogenetic or molecular aberration for which a prenatal test is warranted; toxoplasmosis or CMV† (cytomegalovirus) seroconversion; an abnormal result for NIPT‡ (non invasive prenatal test); other indications: this includes e.g. parental anxiety; unknown indication. There is no statistical difference in indications between both groups. CMV†: cytomegalovirus; NIPT‡: non invasive prenatal test.
After excluding unidentifiable members of twin pregnancies, known discontinued pregnancies and patients whose addresses were unavailable, 616 and 719 questionnaires were sent to patients and controls, respectively. Ninety-three parents (93/616, 15.1%) from the patient population and one hundred and thirty-eight parents (138/719, 19.2%) from the control population participated in the study (Figure 3.1). A statistical difference between indications for performing an invasive procedure between responders and non-responders could be detected (p=0.026). Parents were more likely to participate if the indication for the invasive procedure was ‘advanced maternal age’ or an ‘abnormal result for the Non-Invasive Prenatal Test (NIPT)’. Parents were less likely to participate if the indication for the invasive procedure was ‘other indications’, which mainly encompasses parental anxiety.
A postnatal follow up-study of prenatally detected CNVs
3 1

48
757 children with a non-benign CNV
616 questionnaires sent
93 responders
85 responders
41 appropriate age for ASQ-3
75 appropriate age for ASQ-SE2
33 VOUS4 SNR4 SR
62 VOUS4 SNR6 SR
3 Path
111 twin/discontinued pregnancies
30 unreachable
8 pregnancies discontinued
793 children with no or only benign CNV
719 questionnaires sent
138 responders
123 responders
84 appropriate age for ASQ-3
109 appropriate age for ASQ-SE2
29 twin/discontinued pregnancies 43 unreachable
2 participation refusals
13 pregnancies discontinued2 neonatal deaths
Figure 3.1: Inclusion flow chart of the patient and control groups. Inclusions and exclusions in the patient population and control group. Abbreviations: CNV: Copy Number Variant; VOUS: Variant Of Unknown Significance; SNR: unreported susceptibility CNV; SR: reported susceptibility CNV; Path: Pathogenic CNV.
In the patient population, eight parents (8/93) indicated that the pregnancy was terminated: four because of the genetic result, as these fetuses carried a pathogenic variant, and four because of an ultrasound anomaly. None of the 93 patient responders indicated a neonatal death. Of the responders in the control population, thirteen parents (13/138) indicated that the pregnancy was discontinued because of an ultrasound anomaly. Two responders in the control population indicated their child died after birth; in both cases, the child had severe anomalies.
In total, questionnaires were completed for 208 children (85 patients and 123 controls) (Supplementary Table 3 available on request). However, since not all parents completed the questionnaires at the required age, only 125 children were scored for ASQ-3 (41 patients and 84 controls) and 184 children for ASQ-SE2 (75 patients and 109 controls) (Figure 3.1). Characteristics of these two groups are summarized in Table 3.3 and Table 3.4.
Chapter 3

49
Table 3.3: Patient description for ASQ-3 participants per CNV type.
VOUS† n = 33
SR‡ n = 4
SNR§ n = 4
Control n = 84
Age mother (years) 37 ± 5 38 ± 5 32 ± 4 35 ± 6Age father (years) 40 ± 8 40 ± 15 34 ± 5 39 ± 6Parental education
Primary school 2 0 1 1Secondary school 9 1 1 20Higher education 22 3 2 63
Birth weight (grams) 3197 ± 692 3063 ± 358 3019 ± 344 3275 ± 525Prematurity (n) 4 0 2 8Indication test
Aberrant Down screening test (n)
10 1 2 25
Advanced maternal age (n)
9 1 0 14
Ultrasound anomaly (n) 6 0 1 23Multiple 1 0 0 0Nuchal translucency
2 0 0 10
Facial 1 0 1 1Central nervous system
0 0 0 2
Hernia diaphragmatica
0 0 0 1
Skeletal 0 0 0 1Growth 0 0 0 3Abdominal 0 0 0 2Amniotic fluid 0 0 0 1Unspecified 1 0 0 1
Familial genetic disorder (n)
5 0 0 9
Toxoplasmosis (n) 0 1 0 4CMV (n) 3 1 0 4 NIPT (n) 0 0 0 3Other (n) 0 0 1 2
The table provides information about characteristics of 125 children, scored for ASQ-3 (41 patients and 84 controls). VOUS†: Variant of Unknown Significance (VOUS); SR‡: reported susceptibility CNV; SNR§: unreported susceptibility CNV; Path¶: Pathogenic CNV; Control: no or only benign CNVs.
A postnatal follow up-study of prenatally detected CNVs
3 1

50
Table 3.4: Patient description for ASQ-SE-2 participants per CNV type.
VOUS† n = 62
PATH¶ n = 3
SR‡ n = 6
SNR§ n = 4
Control n = 109
Age mother (years) 36 ± 5 32 ± 4 38 ± 6 32 ± 4 36 ± 6Age father (years) 38 ± 7 33 ± 3 42 ± 13 34 ± 5 38 ± 6Parental education
Primary school 3 0 0 1 1Secondary school 14 2 1 1 25Higher education 45 1 5 2 83
Birth weight (grams) 3252 ± 639
2583 ± 1152
2987 ± 689
3019 ± 344
3286 ± 531
Prematurity (n) 6 2 0 2 9Indication test
Aberrant Down screening test (n)
20 0 1 2 33
Adv. maternal age (n) 13 0 1 0 19Ultrasound anomaly(n) 13 2 2 1 30
Multiple 1 1 0 0 0Nuchal translucency 4 0 1 0 15Cardiac 1 1 0 0 0Facial 1 0 0 1 2Central nervous system 1 0 0 0 2Hernia diaphragmatica 0 0 0 0 1Skeletal 0 0 0 0 1Growth 1 0 1 0 4Abdominal 1 0 0 0 2Amniotic fluid 0 0 0 0 1Unspecified 1 0 0 0 1
Familial genetic dis-order (n) 11 0 0 0 13Toxoplasmosis (n) 1 0 1 0 5CMV (n) 4 1 1 0 4NIPT (n) 0 0 0 0 3Other (n) 0 0 0 1 2
The table provides information about characteristics of 184 children, scored for ASQ-SE2 (75 patients and 109 controls). VOUS†: Variant of Unknown Significance (VOUS); SR‡: reported susceptibility CNV; SNR§: unreported susceptibility CNV; Path¶: Pathogenic CNV; Control: no or only benign CNVs.
Chapter 3

51
To determine the relationship between the genetic result and postnatal clinical and neurological development, the association between variant type and ASQ-3/ASQ-SE2 results was analyzed. Boxplots of ASQ-3 subcategories versus variant group are shown in Figure 3.2. Note that there were no children of appropriate age for the ASQ-3 who had a pathogenic CNV. Also, since our reporting policy states not to communicate VOUS, inheritance was known in less than half of the cases (44.3%), and the VOUS population was excluded from all statistical analysis. To study the association between variant type and the five subcategories of the ASQ-3, a one-way ANOVA comparing children with an SR, an SNR and the control population was carried out. Significant differences between the groups were detected for communication and personal-social skills: a one-way ANOVA followed by a posthoc analysis showed that for both outcomes, children with an SR scored worse compared to the control and SNR categories, whereas there was no significant difference in mean outcome between the SNR and control groups. P-values for the different tests and differences in mean outcomes between the groups are shown in Table 3.5. Multiple linear regression analysis showed that the covariates parental level of education, multiple languages, neonatal surgery, pre-term birth and age of the father had no significant effect on the ASQ-3 sub-scores. However, given the small inclusion numbers, no conclusions could be made regarding the covariables tested.
F igure 3.2: Boxplots of ASQ-3 subcategories versus variant group. Children with an SR scored worse in the categories communications skills and personal-social skills in comparison to children in the control population and children with an SNR. ASQ-3: Ages and Stages Questionnaire: a Parent-Completed Child Monitoring System, Third edition; SNR: unreported susceptibility CNV; SR: reported Susceptibility CNV.
A postnatal follow up-study of prenatally detected CNVs
3 1

52
Chapter 3
Tabl
e 3.5:
Stati
stica
l ana
lysis
of AS
Q-3 r
esult
s
P-va
lue
ANOV
ACo
mpa
red
grou
psDi
ffere
nce
in m
ean
95%
CI†
Lo
wer l
imit
95%
CI
Uppe
r lim
itP-
valu
e Tu
key
corr
ecte
d
Com
mun
icat
ion
0,00
012
SNR‡ -
Cont
rol
1.96
-5.1
79.
090.
85
SR¶ -
Cont
rol
-15.
79-2
2.92
-8.6
6<0
.001
SR –
SN
R-1
7.75
-27.
60-7
.90
0.00
19
Gros
s m
otor
0,31
8
Fine
mot
or0,
586
Prob
lem
-sol
ving
0,24
88
Pers
onal
-soc
ial
0,00
3079
SNR
- Con
trol
-0
.27
-6.1
15.
560.
995
SR -
Cont
rol
-10.
27-1
6.11
-4.4
40.
0022
SR -
SNR
-10
-18.
06-1
.94
0.04
This
table
desc
ribes
the s
tatist
ical a
nalys
is of
ASQ-
3 res
ults,
subd
ivide
d in
the fi
ve d
iffere
nt ca
tegor
ies: c
omm
unica
tion
skills
, gros
s moto
r dev
elopm
ent, fi
ne m
otor
deve
lopm
ent, p
roblem
-solv
ing sk
ills an
d pers
onal-
socia
l dev
elopm
ent. S
ignific
ant d
iffere
nces
betw
een t
he gr
oups
were
detec
ted fo
r two
deve
lopm
ental
area
s: ch
ildren
wi
th an
SR sc
ored w
orse
in th
e cate
gorie
s com
mun
icatio
ns sk
ills (p
=0.00
01) a
nd pe
rsona
l-soc
ial sk
ills (p
=0.00
3), c
ompa
red to
child
ren in
the c
ontro
l pop
ulatio
n and
ch
ildren
with
a SN
R. CI†
: Con
fiden
ce In
terva
l; SNR
‡: un
repor
ted su
scep
tibilit
y CNV
; SR¶
: repo
rted s
usce
ptibi
lity CN
V.

53
A boxplot of ASQ SE-2 results versus variant group is shown in Figure 3.3. A one-way ANOVA comparing the pathogenic, SR, SNR and control groups was performed. No differences in social-emotional development were observed between variant groups (p=0.069). Although the boxplot suggests a worse outcome for children diagnosed with a pathogenic CNV, the results were not statistically significant, due to the small study population and the large standard deviation within groups (group means: control 45.825 ±2.481; pathogenic 78.333±14.956; SNR 43.75±12.952; SR 64.167±10.575). The association between the ASQ-SE2 and the variant type was not influenced by accounting for the covariates parental level of education, multiple languages, possible operations, pre-term birth or age of the father. In addition, none of these covariates showed a significant effect on the ASQ-SE2. However, given the small inclusion numbers, no conclusions could be made regarding the covariables tested.
F igure 3.3: Boxplots of ASQ-SE2 versus variant group. ASQ-SE2: Ages and Stages Questionnaire: Social-Emotional Second Edition; SNR: unreported susceptibility CNV; SR: reported Susceptibility CNV.
In conclusion, w e found a statistical difference in performance between children with an SR and children with an SNR or the control population in the categories of communication skills and personal-social skills.
A second aspect that this dataset allows to investigate is the development of children with a specific CNV (susceptibility CNV, VOUS or pathogenic variant).
A postnatal follow up-study of prenatally detected CNVs
3 1

54
3.4.1 Susceptibility CNVsSusceptibility CNVs have a highly unpredictable and often prenatally undetectable phenotype. Following the Belgian reporting system, only a defined number of susceptibility CNVs are reported. Postnatal follow-up of all children with a susceptibility CNV, either reported or not, provides an unbiased insight into the development of these children and helps us to assess the value of our policy. Table 3.6 and Table 3.7 show the outcome of children with a reported versus not reported susceptibility CNV, provided that the questionnaire was completed within the correct age range.
Chapter 3
Tabl
e 3.6:
Out
com
e of t
he AS
Q-3 a
nd AS
Q-SE
2 que
stion
naire
s in c
hildr
en w
ith a
repor
ted su
scep
tibilit
y CNV
(SR)
ASQ-
3 su
bsca
leAS
Q-SE
2 sc
ore
CNV
Phen
otyp
e of
m
icrod
elet
ion
/ m
icro-
du
plica
tion
Case
nu
mbe
rAg
e ch
ild
(mon
ths)
Com
mu-
nica
tion
Gros
s m
otor
Fine
m
otor
Prob
lem
so
lvin
gPe
rson
al
Socia
l
1q21
.1
dup
ID† , D
D‡ , A
SD§ ,
SZ¶
207
36Fa
ilFa
ilFa
ilFa
ilFa
ilFa
il
1q21
.1
del
ID, D
D, A
SD,
SZ, f
acia
l dy
smor
phis
m
2935
Pass
Pass
Pass
Pass
Pass
Pass
4740
--
--
-Pa
ss
15q2
6
del
ID86
35Pa
ssPa
ssPa
ssPa
ssPa
ssPa
ss
22q1
1.2
dup
DD,
epi
leps
y, dy
smor
phic
fe
atur
es
1535
Pass
Pass
Pass
Pass
Pass
Pass
5240
--
--
-Pa
ss
Abbr
eviat
ions:
ID†:
intel
lectu
al dis
abilit
y; DD
‡: D
evelo
pmen
tal di
sord
er; A
SD§:
autis
m sp
ectru
m di
sord
er; S
Z¶: s
chizo
phren
ia

55
Tabl
e 3.7:
Outco
me o
f chil
dren
with
an un
repor
ted su
scep
tibilit
y CNV
(SNR
)
ASQ-
3 su
bsca
leAS
Q-SE
2 sc
ore
CNV
Phen
otyp
e of
m
icrod
elet
ion
/ micr
o-
dupl
icatio
n
Case
nu
mbe
rAg
e ch
ild
(mon
ths)
Com
mun
i-ca
tion
Gros
s m
otor
Fine
m
otor
Prob
lem
so
lvin
gPe
rson
al
Socia
l
15q1
1.2
BP
1-BP
2 du
p
DD
‡ , mot
or
dela
y, sp
eech
de
lay,
ASD
§
131
38Pa
ssPa
ssPa
ssPa
ssPa
ssPa
ss
16p1
3.11
de
lID
† , DD,
ASD
, ep
ileps
y16
235
Pass
Fail
Mon
itor
Pass
Pass
Pass
199
36Pa
ssPa
ssPa
ssPa
ssPa
ssM
onito
r
22q1
1.2
dist
al
dup
DD,
epi
leps
y, dy
smor
phic
fe
atur
es
197
37Pa
ssPa
ssPa
ssPa
ssPa
ssPa
ss
Abbr
eviat
ions:
ID†:
intel
lectu
al dis
abilit
y; DD
‡: D
evelo
pmen
tal di
sord
er; A
SD§:
autis
m sp
ectru
m di
sord
er; S
Z¶: s
chizo
phren
ia
A postnatal follow up-study of prenatally detected CNVs
3 1

56
3.4.1.1 Reported susceptibility CNVs
3.4.1.1.1 1q21.1 duplication syndromeThe chromosome 1q21.1 duplication syndrome, including GJA5 (OMIM 612475), is a genetic risk factor for intellectual disability, developmental delay, autism spectrum disorder, schizophrenia, macrocephaly and coronary heart disease.72, 73 The ClinGen dosage sensitivity score for this duplication is 3. This score (https://www.ncbi.nlm.nih.gov/projects/dbvar/clingen/) refers to the evidence for pathogenicity for a haploinsufficiency phenotype (deletion) or a triplosensitive phenotype (duplication), ranging from 3 (sufficient evidence) to 0 (no evidence). Mean verbal and nonverbal IQ scores are in the low average range and motor function is nearly 2 S.D below age norms.73 Another study showed that microduplications of 1q21.1 cause a range of developmental delays, neuropsychiatric abnormalities, dysmorphic features and a variety of other congenital anomalies.74 The phenotype seems to be subject to incomplete penetrance, as Coe et al. reported 48/29085 cases with developmental delay and 5/19584 healthy controls, resulting in a likelihood ratio of 6.46.40 Because of the severity of the phenotype and the relatively high likelihood ratio, this duplication is reported in the Belgian prenatal setting. Invasive testing for case 207 in our study was performed because of toxoplasmosis seroconversion, but the fetus was not affected. A 1q21.1 duplication (chr1:145.899.339-147.887.735) was reported. The parents reported a normal pregnancy and delivery, no intervention of the pediatrician and a normal birthweight. However, the child failed all five subcategories of the ASQ-3 as well as the ASQ-SE2.
3.4.1.1.2 1q21.1 deletion syndromeIndividuals with the chromosome 1q21.1 deletion syndrome, including GJA5 (OMIM 612474, ClinGen score 3), are susceptible to intellectual disability, developmental delay, autism spectrum disorder, schizophrenia, facial dysmorphism, microcephaly, coronary heart disease, renal and urinary tract anomalies.73 Coe et al. recorded a likelihood ratio of 7.63 for developmental delay in patients carrying this variant.40 Two children with a 1q21.1 deletion syndrome participated in our study cohort. In the first case, case 29, the indication for the invasive procedure was intra-uterine growth restriction (IUGR). IUGR is not typically associated with deletion of this region.74 The child passed all tests provided, although the child was one month too old for the ASQ-3 survey. The second case, case 47, had an uneventful pregnancy and delivery and passed all tests as well. In conclusion, the two cases with a 1q21.1 deletion in our study performed within the normal range for all developmental areas tested.
3.4.1.1.3 15q26 deletion Coe detected the 15q26 deletion syndrome (chr15:99.360.000-102.520.000), containing IGF1R (ClinGen score 3), in 11/29085 pediatric patients with intellectual disability, developmental delay or autism spectrum disorder versus 1/19584 healthy controls,
Chapter 3

57
A postnatal follow up-study of prenatally detected CNVs
3
resulting in a likelihood ratio of 7.41 and a penetrance of 28.6%.40 These findings, together with the importance of prenatal ultrasonographic follow-up, justify reporting this variant in a prenatal setting. Case 86 underwent invasive testing because of advanced maternal age and showed an intragenic 15q26 deletion in the IGF1R gene (chr.15:99396694-99465285). Veenma described a similar deletion of the IGF1R gene in a Dutch family75 with pre- and postnatal growth retardation, mild to moderate small head circumference, minor facial dysmorphia and mild skeletal anomalies, but without mental retardation. The child in our study was carried to term and had a birth weight of 2630gr (5th percentile). Pregnancy and delivery were uneventful, and the child passed all tests.
3.4.1.1.4 22q11.2 duplicationThe proximal (A-B) 22q11.2 duplication syndrome is the most frequently reported susceptibility CNV in our Belgian prenatal population.65 It has a ClinGen score of 3 and is a susceptibility factor for developmental delay, epilepsy and dysmorphic features and can also cause microcephaly and coronary heart disease.40 Although in the majority of cases the duplication is inherited from a normal parent,51 this susceptibility CNV is nevertheless reported prenatally in Belgium because of its possible association with fetal structural anomalies and the importance of ultrasonographic follow-up. Two cases with such a CNV participated in the study. Case 15 had an uneventful pregnancy and delivery and passed all tests. Case 52 had an increased nuchal translucency at prenatal ultrasound and was born after an instrumental delivery. Parents completed the questionnaire at 40 weeks, hence too late for the ASQ-3. The child passed all tests, including the ASQ-SE2, but scored within the monitoring zone for communications skills. Since the child was too old for the test, the results suggest communicative development is delayed. Speech delay is described in children diagnosed with a 22q11.2 duplication.51, 76, 77
3.4.1.2 Unreported Susceptibility CNVs
3.4.1.2.1 15q11.2 BP1-BP2 duplication The 15q11.2 BP1-BP2 duplication syndrome, containing NIPA1 (chr.15: 22.832.519-23.090.897), is the most frequently found SNR in the Belgian prenatal population. It is described as a susceptibility factor for developmental delay, motor delay, speech delay and autism spectrum disorder,54 but its ClinGen score is “unlikely”, indicating that its pathogenicity is at present doubtful. The highly variable, often mild phenotype and the low penetrance and likelihood ratio justify our decision not to report this CNV.40, 42, 55 In our study cohort, case 131 with this duplication (chr15:22.652.047-23.300.313) passed all tests provided.
1

58
3.4.1.2.2 16p13.11 deletionDeletion of 16p13.11 containing MYH11 is a susceptibility factor for intellectual disability, developmental delay, autism spectrum disorder, epilepsy and microcephaly.78, 79 Coe et al. detected 36/29085 cases and 7/19584 controls with this deletion, resulting in a likelihood ratio of 3.45 and a penetrance of 15.7%.40 The deletion has a ClinGen score of 3, signifying its likelihood for pathogenicity, which is confirmed in a more recent study (odds ratio 9.85).53 This deletion is currently not reported in the Belgian prenatal setting since in the majority of cases, it is inherited from an unaffected parent. Case 162, who carries the deletion, failed the ASQ-3 gross motor skills test, scored in the monitoring zone for fine motor skills and passed all other tests; no epilepsy, intellectual disability or autism spectrum disorder was reported. Invasive diagnosis in this twin pregnancy was performed because of increased nuchal translucency in the other member. Delivery was 4 weeks early, but birthweight was within the normal range. The head circumference at birth is unknown; however, prenatal ultrasound indicated a head circumference at percentile 50. No non-benign CNVs were detected in the sibling (case 163), who scored within the monitoring zone for gross motor development, suggesting that other factors may have influenced the children’s development. Case 199 was delivered at 36 weeks after a normal pregnancy. Head circumference at birth was within normal range (percentile 50). The child passed all ASQ-3 subcategories and scored within the monitoring zone for the ASQ-SE2. Based on literature78, 80, 81 and current ClinGen scores, reevaluation of the 16p13.11 deletion in our Belgian reporting system is required.
3.4.1.2.3 22q11.2 distal duplicationThe 22q11.2 distal duplication syndrome (distal type I, D-E/F) has a ClinGen score of 3 and is a susceptibility factor for developmental delay, epilepsy and dysmorphic features,82-84 but is not reported in the Belgian prenatal setting. Case 197 was diagnosed with a 22q11.2 distal duplication LCR E-H (Chr22:22.998.284-24.988.402); this particular duplication is awaiting ClinGen review. A study in 2011 describes six out of 10 patients with an LCR E/F–H duplication with speech delay and seven with mild to moderate developmental delay.84 The child in our study underwent invasive testing because of a cleft lip. The mother indicated a delay in speech, but the child passed all tests provided, including the communication test.
Chapter 3

59
3.4.2 Variants of Unknown SignificanceVOUS are variants with hitherto unknown clinical significance. Inheritance status is one of the predictors for pathogenicity of a VOUS. However, since VOUS are not reported in the Belgian prenatal setting, inheritance was tested in less than half of the cases (44.3%). Therefore, we decided to omit children with a VOUS from all statistical analyses. Still, investigation of children with a VOUS, regardless of inheritance, can help to reclassify them as (possibly) pathogenic or (possibly) benign.
3.4.2.1 Recurrent Variants of Unknown SignificanceWe detected 7 recurrent VOUS in 44 children.65 In this postnatal follow-up study, 8 children participated (cases 23, 62, 73, 129, 155, 181, 184, 191) (Table 3.8). Six of them (62, 73, 129, 155, 184, 191) passed all tests, indicating that these VOUS probably do not affect the developmental domains tested by our surveys.
A postnatal follow up-study of prenatally detected CNVs
3 1

60
Tabl
e 3.8:
Out
com
e of t
he AS
Q-3 a
nd AS
Q-SE
2 que
stion
naire
s in c
hildr
en w
ith a
recur
rent V
arian
t Of U
nkno
wn Si
gnific
ance
(VOU
S)
ASQ-
3 su
bsca
le sc
ore
ASQ-
SE2
scor
e
CNV
Locu
s (hg
19)
Case
nu
mbe
rMo
de o
f in
herit
ance
Age
child
(m
onth
s)Co
mm
u-ni
catio
nGr
oss
mot
orFi
ne
mot
orPr
oble
m
solv
ing
Pers
onal
So
cial
3p14
. 2d
up
chr3
: 59
.666
.501
–
60.9
93.0
7962
unkn
own
39Pa
ssPa
ssPa
ssPa
ssPa
ssPa
ss
191
mat
erna
l38
Pass
Pass
Pass
Pass
Pass
Pass
6q22
. 31
dup
chr6
: 12
3.53
9.62
5 -
124.
328.
531
23m
ater
nal
39Pa
ssPa
ssM
oni-
tor
Mon
itor
Pass
Pass
9p23
du
p
chr9
: 10
.164
.926
- 11
.868
.588
129
unkn
own
34Pa
ssPa
ssPa
ssPa
ssPa
ssPa
ss
155
unkn
own
38Pa
ssPa
ssPa
ssPa
ssPa
ssPa
ss
10q2
3.
31 d
el
chr1
0:
91.6
26.4
82 -
92.0
35.4
5718
1m
ater
nal
40Pa
ssFa
il-
Pass
Pass
Fail
17p1
3.
3dup
chr1
7: 1
48.0
92
- 597
.702
73m
ater
nal
9Pa
ssPa
ssPa
ssPa
ssPa
ssPa
ss
22q1
1.
23du
p
chr2
2:
23.7
20.1
81 -
24.9
59.8
2718
4m
ater
nal
35Pa
ssPa
ssPa
ssPa
ssPa
ssPa
ss
Chapter 3

61
For case 23, who carries the most frequently found recurrent VOUS (a duplication on 6q22.31 (123.539.625-124.166.602)), the questionnaire was completed at the age of 39 months. Amniocentesis was performed because of a Toxoplasmosis seroconversion, but the fetus tested negative. The child inherited the VOUS from the healthy mother, negating its pathogenicity. Although one month too old for the ASQ-3, the child scored within the monitoring zone for fine motor skills and problem-solving skills. The child passed the ASQ-SE2. As described by Srebniak et al., this CNV may represent a variant that is benign when present alone, but acts as a second hit in carriers of an additional VOUS.47 The VOUS was an isolated finding in this particular child.
Despite being one month too old for the ASQ-3 (40 months), case 181, carrying the recurrent 10q23.31 deletion (chr10:91.626.482-92.035.457), failed the gross motor skills subcategory, as well as the ASQ-SE2. The VOUS was maternally inherited, negating its pathogenicity, although reduced penetrance and variable expressivity cannot be excluded. Amniocentesis was performed because of a Toxoplasmosis seroconversion, but again the fetus was not affected. No ultrasound anomalies were detected, and pregnancy and delivery were uneventful. The deleted region encompasses only one pseudogene.
3.4.2.2 Non-recurrent Variants of Unknown SignificanceWe identified 30 children with a non-recurrent VOUS and of appropriate age for ASQ-3, and 54 children for ASQ-SE2. Four children with a non-recurrent VOUS failed one or more tests (Supplementary Table 3 available on request). However, for none of the cases, the information was sufficient to reclassify the VOUS to either benign or pathogenic.
3.4.3 Pathogenic CNVsThree children with a pathogenic variant were included in the statistical analysis of ASQ-SE2 results. Case 30 underwent an invasive procedure because of cardiac anomalies on prenatal ultrasound and carries a 9 Mb duplication on chromosome 16 (chr16:12.061.688-21.301.937). The child was born at 35 weeks with a dysmature birthweight and underwent a cardiac operation after birth. This case scored within the monitoring zone for the ASQ-SE2. Although too old for the ASQ-3 (39 months), the child only passed the communication skills test. The CNV has been discussed at a national level and was reported as pathogenic because of its size.
Case 169 was diagnosed with the 22q11.2 deletion syndrome (OMIM 188400). Ultrasound investigation indicated multiple anomalies, among which a ventricular septal defect and IUGR. The child passed the ASQ-SE2 test. Despite being too old for the ASQ-3 (41 months), the child failed the communication, problem-solving and personal-social skills test, confirming developmental delay in these areas.
Case 142 was diagnosed with a small deletion in the SHOX gene (chrX:594.241-597.792). Haploinsufficiency of this gene results in a short stature, shortening of the medial
A postnatal follow up-study of prenatally detected CNVs
3 1

62
Chapter 3
segments of the limbs, with a progressive decline in the height SD score from birth onwards. The child scored within the monitoring zone for the ASQ-SE2 test. The child was born at full term with a birthweight of 4200 grams and a birth length of 50 centimeters; current length and weight remain within normal range.
3.5 DiscussionThe landscape of prenatal diagnosis is changing drastically. Over the last 5 years, CMA has increasingly replaced conventional karyotyping for the analysis of invasively obtained samples. The Non-Invasive Prenatal Test (NIPT) has a very high uptake in both average and low risk pregnancies, and whole exome sequencing is slowly being introduced in the prenatal setting for certain ultrasonographic anomalies.85 While invasive prenatal testing suffered a steep decline with the introduction of NIPT, the expansion of NIPT to karyotype resolution has partially reversed this trend, as NIPT-positive cases need to obtain a confirmatory invasive test.
In Belgium, approximately 125,000 children are born every year. Over a 22-month period (May 2013-February 2015), circa 9200 invasive procedures were performed nationwide. Of these, 14.5% were invited to participate in this national research project. The overall response rate was 17.3%. Response rates for postal questionnaires vary from 8-9% when there is no reminder; rates increase to 31.1-32.1% after a single reminder and up to 63% after 3 reminders.86 Although 1 reminder was sent in this study, the response rate was lower than the expected rate; this could be due to the length of the questionnaire, which had three sections or 20 pages of questions.87 While a shorter questionnaire might have improved response rates, we opted for a more complete overview of the child’s development.
A statistical difference was detected in indications for the invasive procedure between responders and non-responders (p=0.026). Parents were more likely to answer questionnaires if the indication was ‘advanced maternal age’ or ‘an abnormal result for NIPT’. They were less likely to participate if the indication for the procedure was ‘other’, a category including mostly cases in which the amniocentesis was performed because of parental anxiety (e.g. because of a previous pregnancy with an aberrant genetic result.) There was no statistical difference in response rate for the indication ‘fetal ultrasonographic anomaly. It is unclear why the response rate differed for some categories. It also remains uncertain whether a worse outcome of the child influences participation rate of the parent.
We detected a significant difference in the development of children with an SR in the categories of communications skills (p=0.0001) and personal social skills (p=0.003), when compared to children with no non-benign CNV or an SNR. The phenotype of a susceptibility variant is highly unpredictable. Belgian geneticists compiled a limited list of susceptibility loci that should be reported and a non-exhaustive list of those that are not reported (http://www.beshg.be/index.php?page=guidelines).25, 65 This list is based on

63
recent literature, describing the clinical spectrum, odds ratios and penetrance values and takes into account the expected severity and the fetal and parental phenotype.25, 40-43, 65 The rationale behind this strict reporting policy is to avoid anxiety in and stigmatization of future parents over a CNV for which the outcome is highly uncertain.56, 57 This approach has been subject to international discussion88 and concerns have been raised about its legal implications. However, the Belgian genetic centers believe it to be a valuable strategy as it prevents inconsistencies in reporting between genetic centers27, 28, 64, 89-91 and reduces parental anxiety and needless terminations of pregnancy.31, 92 The differences we found between children with an SR versus children with an SNR support our choice of susceptibility CNVs to report. However, due to the low participation grade, there is currently insufficient data to validate our policy to not report other susceptibility CNVs.
We identified one child who failed the survey in the SNR category: a child with a 16p13.11 deletion. The deletion including MYH11 was assigned a ClinGen score of 3. In our study, the child failed the gross motor skills test and scored within the monitoring zone for fine motor skills. The child’s twin sibling, who did not have the deletion, also needed close attention for gross motor skills, indicating the possibility that other factors besides the CNV influenced the child’s development. A second child with the 16p13.11 deletion succeeded all tests. However, based on current literature, the 16p13.11 deletion will be reevaluated in our Belgian reporting system because current literature and ClinGen scores suggest a pathogenic nature.
Postnatal follow-up of children with a prenatally detected non-benign CNV is of importance since it allows complete phenotypical characterization of a particular CNV, as it is not dependent on cases at the more severe end of the phenotypical spectrum. Conversely, since our study population is small, our results do not necessarily reflect the results in the whole SNR population, nor can it be concluded that the developmental issues in the SR population are indeed related to the presence of a particular CNV. Hence, elaborate pre- and post-test counseling remain crucial, just as it is important to continuously review and adapt guidelines based on the most recent literature and current evidence. This is why we recently initiated a study investigating the opinion of Belgian gynecologists, general practitioners and future parents on the Belgian approach.
3.6 ConclusionIn this paper, we reported a national postnatal follow-up project, initiated to determine the relationship between the prenatal genetic results, prenatal phenotypic findings and postnatal clinical and neurological development. This study is, to the best of our knowledge, the first of its kind. Postnatal follow-up of children with a prenatally detected non-benign CNV is of great value in determining the full phenotypic spectrum of CNVs. Despite the small inclusion numbers, we could detect a significant difference in communicative and personal-social development between cases with a reported susceptibility CNV and cases with an unreported susceptibility CNV. However, a higher number of cases for each CNV category is needed to confirm our findings and we hope our study will be followed by many others worldwide.
A postnatal follow up-study of prenatally detected CNVs
3 1

64
LABO. EXPELLU PTATATURESTO BEATI-IS DUCID UT ERUNT, QUAMUSDAE PROVIDUS DIT OMNITIBUS.
cover-boek-hand.indd 6cover-boek-hand.indd 6 07/07/2020 07:5807/07/2020 07:58

65
4. CHROMOSOMAL MICRO-ARRAY ANALYSIS IN PRENATAL DIAGNOSIS: ethical considerations of the Belgian approach
Joke Muys, Bettina Blaumeiser, Katrien Janssens, Loobuyck Patrick, Yves Jacquemyn
J Med Ethics. 2020;46(2):104-109. doi:10.1136/medethics-2018-105186
4 1

66
4.1 ABSTRACTDetection of genetic aberrations in prenatal samples, obtained through amniocentesis or chorion villus biopsy, is increasingly performed using chromosomal microarray (CMA), a technique that can uncover both aneuploidies and copy number variants throughout the genome. Despite the obvious benefits of CMA, the decision on implementing the technology is complicated by ethical issues concerning variant interpretation and reporting. In Belgium, uniform guidelines were composed and a shared database for prenatal CMA findings was established. This Belgian approach sparks discussion: it is evidence-based, prevents inconsistencies and avoids parental anxiety, but can be considered paternalistic. Here, we reflect on the cultural and moral bases of the Belgian reporting system of prenatally detected variants.
4.2 INTRODUCTIONSamples obtained by prenatal invasive diagnosis (amniocentesis or chorion villi biopsy) can be investigated in multiple ways. Until recently, conventional karyotyping, which detects chromosomal aberrations of at least 5-10 Mb in size, was the preferred technology. Nowadays, chromosomal microarray analysis (CMA) is increasingly replacing karyotyping for the analysis of these samples. CMA is a whole genome test that scans for the presence of copy number variants (CNVs) and aneuploidies in DNA isolated directly from the prenatal sample. It has a higher resolution than conventional karyotyping, detecting deletions or duplications down to 100 kb.37 When compared to karyotyping, CMA detects additional clinically relevant genomic abnormalities in 5-10% of pregnancies with a fetal structural anomaly and in 0.5-2% of pregnancies without ultrasound abnormalities.19-24 In the near future, implementation of Whole Exome Sequencing (WES) and Whole Genome Sequencing (WGS) in invasive prenatal diagnosis will reveal even more detailed information to future parents.
Despite the obvious benefits of speed, higher success rate and increased diagnostic yield of CMA, the decision on implementing this procedure is complicated by ethical issues. Besides pathogenic variants related to the indication of the invasive test, the higher resolution of CMA increases the chances of revealing variants causing late-onset disorders, variants with a variable phenotype (susceptibility CNVs) and variants for which no information on possible consequences exists (Variants Of Unknown Significance (VOUS)).38 Reporting variants in a prenatal setting is very different from reporting them postnatally: future parents can decide to discontinue the pregnancy, even without (sufficient) evidence that the baby will be affected. On the other hand, they can choose to continue the pregnancy, but remain anxious about the future health of their baby, even after an uncomplicated delivery. Every health or behavioral issue of the child could be interpreted as related to the genetic variation. Furthermore, in case an inherited variant is found, parents can unsolicited obtain information about their own health, possibly leading to changes in health experience.
Chapter 4

67
In practice, prenatal experts are expected to adopt a position on which CNV’s, detected by a prenatal invasive procedure, are to be communicated to expecting parents and which are not. First, data transfer can be regulated and thereby, the amount of information given to the expecting couple is fixed. One can choose to reveal only known pathogenic CNV’s to all clients, or, in contrast, unfiltered information of all CNV types (including VOUS). Although impartial, this approach rises some questions: who decides on the access to information as the genetic material is belonging to the mother as long as the fetus is intrauterine, what happens at a later moment when more knowledge is available and a VOUS is recognized as pathogenic, what happens when a child is born with health problems and it can, at a later stage of science, be proven that these are related to a CNV that was not revealed? In case all CNV’s are communicated to future parents, the presence of a VOUS or susceptibility CNV can provoke demand for termination of pregnancy (TOP), which then leads to the still unresolved question about acceptability of TOP.
Secondly, data transfer to expecting parents can be individualized. Personalized medicine is considered more and more relevant and future-oriented. This personalization can include individualized levels of information transfer, stated as “how detailed do you want to know?”. Such an attitude can rapidly become highly problematic. On one side, every genetic counseling will involve, consciously or unconsciously, communicative elements such as emotional and intellectual differences between clients. The discussion on who decides what to say and what to detect is subjective and can easily develop towards a paternalistic attitude. In most real-life clinical situations, geneticists and other workers in prenatal diagnosis struggle daily with the equilibrium between their clients right to know and the professional limitations of insufficient knowledge and scientific uncertainty. Professionals would welcome some kind of guidance.27, 28, 64, 89-91
Because of the ongoing debate on how and for which indications to implement CMA in a prenatal setting, all eight Belgian genetic centers embarked on a unique project.25 Clinical and laboratory geneticists agreed upon using CMA with a resolution of 400 kb for all indications of invasive prenatal testing. Uniform guidelines on how to interpret and report variants were composed and a shared database for prenatal CMA findings was established.65
In Belgium, CNVs are classified as benign, pathogenic, susceptibility or VOUS. Benign variants are variants that do not cause a phenotype and are frequent in a normal population; they are not reported.
Pathogenic CNVs cause a well-defined phenotype and are reported when causing a congenital anomaly e.g. 22q11.2 deletion syndrome. When the phenotype is unrelated to the indication of the CMA (incidental finding), the following reporting policy is applied: dominant late-onset diseases with clinical utility (PMP22 duplication causing Charcot-Marie-Tooth disease), carriership for autosomal recessive diseases with a carrier frequency >1/50 (e.g. GJB6 deletions causing autosomal recessive deafness) and X-linked carrier status (e.g. STS deletions causing ichthyosis, a skin disorder) are reported to future parents. Susceptibility CNVs are genetic risk factors with reduced
Ethical considerations of the Belgian approach
4 1

68
penetrance and/or variable expression, often associated with a highly unpredictable phenotype that does not present prenatally (e.g., intellectual disability, autism spectrum disorder, epilepsy); only a limited number are reported in the prenatal setting. The 22q11.2 duplication syndrome (OMIM #608363) is the most frequently reported susceptibility CNV in our Belgian prenatal population. The variant has a broad phenotypic spectrum. The most common symptoms are intellectual disability/learning difficulties (97%), delayed psychomotor development (67%), growth retardation (63%), muscular hypotonia (43%), and cardiac anomalies (20%).51, 52 The 15q11.2 duplication (GRCh37 chr15:22800000–23090000, minimal size 290 kb) is the most frequently found unreported susceptibility CNV in our Belgian prenatal population. The phenotypic spectrum of developmental delay is highly variable, from motor coordination problems to autism spectrum disorder and obsessive compulsive disorder.54 The highly variable and often mild phenotype as well as the low penetrance and likelihood ratio justify our reporting policy.40, 42, 55
All CNVs that cannot be classified as benign, pathogenic or susceptibility are designated VOUS and are not reported. CNVs are communicated both to clinicians and future parents.
Decision on the classification is reached within the Belgian Society for Human Genetics (BeSHG) Prenatal Committee. The committee is a collaboration of clinical and laboratory geneticists from every genetic center in Belgium (http://www.beshg.be/index.php?page=centers). Guidelines are approved by the Belgian college for Medical Genetics.
Despite Belgian guidelines, ambiguous situations still occur, which are tackled by a committee of experts, an ad hoc committee, who will give advice within 48 hours on how to classify and whether or not to report the CNV found.
The Belgian approach sparks worldwide discussion. It has the advantage to prevent inconsistencies between genetic centers within the same country27, 28, 64, 89-91 and avoids parental anxiety and needless terminations of pregnancy in case of VOUS.31, 92 On the other hand, this approach can be viewed upon as paternalistic, raising concerns about the legal implications and ignoring the personal wishes of the patient and with progressing knowledge non-communicated VOUS can become relevant later, leading to inconsistent counseling over several years.
In this paper, we reflect on the cultural and moral basis of the Belgian reporting system of prenatally detected variants.
Chapter 4

69
4.3. CULTURAL BASIS OF THE BELGIAN REPORTING SYSTEMIt could be said that medical communication inevitably finds its basis in culture. In this text we use “culture” in a general but also limited meaning as the symbols, rituals, values that seem to distinguish people in different groups. As we report a “Belgian” approach we define groups as countries. We do realize that this mode of operandi is limiting for that every individual is multiculturally layered. However, we use this simplification for the ease of discussing a national guideline. Several studies suggest that a small country like Belgium, lying on the dynamic border between northern and southern Europe, has a distinctive medical culture.93-96 Even in such a seemingly clear forward diagnosis as uncomplicated cystitis, it has been shown that Belgian guideline authors are more concerned by safety in their diagnosis and therapeutic approach and less by “the evidence”. Safety necessitates also good guidance on what to do in doubtful cases.97 A key aspect of the Belgian health system is the emphasis on highly individualized care and the freedom to choose one’s own doctor(s). Medical care givers in Belgium tend to have long- standing trustful relationships with their patients.98 This individualized face-to-face type of care facilitates interpersonal communication, but will also put more pressure on the caregivers as the client/patient wants all information from his/her personal doctor and not from e.g. an anonymous geneticist. These cultural modifications in medical communication have not been thoroughly studied.93 In the non-medical community one can find several approaches. A frequently used approach is the model proposed by Hofstede.94, 99-102 In his manuscript dating from 1991 and extended in 2005, Hofstede discusses the international differences in the five dimensions of cultural variability: Individualism-collectivism, uncertainty avoidance, power distance, masculinity-femininity and long-term versus short-term orientation. In individualistic cultures, the needs, values and goals of the individual are prioritized over those of the group. If a culture shows a high level of uncertainty avoidance, there is a low tolerance for uncertainty and ambiguity and a greater need for formal rules and absolute truth. Power distance indicates the extent to which less powerful members of society accept that power is distributed unequally. Cultural systems high on “masculinity” emphasize differentiated sex roles, performance, ambition and independence, while more “feminine” cultures value fluid sex roles, quality of life, service and interdependence. Long-term orientation indicates a strong orientation towards future rewards (perseverance, adapting to changing circumstances), while short-term orientation is more related to the past and presence (e.g. respect for tradition, fulfilling social obligations).99 More recently, a sixth dimension has been added, namely indulgence versus self-restraint, concerning the degree of freedom to develop one’s personal human desires.
In a review of literature, Schouten reveals major differences in doctor-patient communication as a consequence of patients’ ethnic background.96 One of five key predictors of culture-related communication problems are cultural values e.g. individualism-collectivism. Predominantly people in individualistic cultures see themselves as independent, whereas in collectivistic cultures, people consider
Ethical considerations of the Belgian approach
4 1

70
themselves as part of the group. For example, Americans, who are part of a very individualistic culture, are more assertive and direct than Asians, who are more collectivistically oriented. Another key factor, proposed in the paper as an answer to differences in doctor-patient communication with different ethnic backgrounds, are patient preferences. In the western world, the past decades, a shift was noticeable from the paternalistic way of treating a patient towards an interactive doctor-patient relationship, with shared responsibility. However, shared decision making is not desired by every patient and will depend from one’s personality, beliefs about to which extent powerful others [for example, doctors] should be responsible for making decisions about patients’ health and the ethnic background.96
Meeuwesen focused on the five original dimensions in the context of medical communication in European countries, including Belgium, in 2009.95 For this study, 307 general practitioners and 5820 patients in 10 European countries (Belgium, Estonia, Germany, Great Britain, The Netherlands, Poland, Romania, Spain, Sweden and Switzerland) were interviewed. Belgium has a high power distance (as do Romania, Poland and Spain), the highest uncertainty avoidance (together with Romania and Spain), a very individualistic culture (comparable to Great Britain and the Netherlands), profiles itself in the middle when it comes to masculinity-femininity and, like Switzerland and the Netherlands, is known for its long-term orientation. Meeuwesen concluded that the five dimensions of cultural variability could predict cross-national variation in medical communication and that, unexpectedly, the more “feminine” a country, the more biomedical information exchange occurred. From this study, Belgium stood out as a country in which patient’s and doctor’s roles are fixed and patients accept an unequal distribution of power. There is little room for unexpected information and both doctors and patients feel threatened by ambiguous or unknown situations. Although Belgian patients want to be involved and informed about their situation, they consider it the medical professional’s responsibility to take the final decision; a professional unable to do so is distrusted and considered incapable. Furthermore, Belgian doctors attach importance to the psychosocial status of their patient by creating the possibility to talk about his worries and concerns. In contrast, the Netherlands, despite being our neighbour, have a very “feminine” communication style. Patients and doctors strive for equality in communication and do not avoid uncertainty, both parties ask a lot of questions and there is ample bio-medical information exchange. A Dutch study on parent opinions in the context of reporting prenatally detected variants showed that 79% of parents wanted to decide themselves about the type of test performed and the variants reported; a mere 1% of parents wanted the doctor to decide.35 On the downside, 8% of future parents to whom a susceptibility CNV was reported, felt stigmatized and 19% worried about the possible consequences of their decision.57
Outside Europe, we notice that North American doctors are considered to be ‘cold experts’103 as they ask more questions, give more biomedical information and are more disease-centered. A survey by the American College of Obstetricians and Gynecologists
Chapter 4

71
(ACOG) demonstrated that more than three quarters (77.3%) of the questioned fellows had been sued in the past.104 Although it is not known whether the high number of law suits is related to this attitude (as a cause or as a result), this medical liability evokes stress and is bound to affect medical communication: most prenatally found CNVs are reported, regardless of proven pathogenicity. In an American study inquiring about parental concerns, patients indicated that they would question their child’s development after birth in case a susceptibility CNV would be returned.105
It is clear that a worldwide “one-size-fits-all” model of medical/genetic information sharing between professionals and patients will never agree with expectations and demands in our varied cultural landscape. Knowledge about the five dimensions of cultural variability in communication will help mutual international understanding and cooperation.
Belgian guidelines on how to classify and how to report results obtained by invasive prenatal diagnosis are designed to give the needed guidance to genetic counsellors and geneticists. Evaluating these guidelines in a retrospective manner, one can conclude that they do comply with the Belgian culture, since the final decision is made by the professional and uncertainty is avoided by adopting guidelines. They even imply the introduction of a failure procedure for when guidelines are not applicable (the adhoc committee).
4.4 ETHICAL REFLECTIONS CONCERNING THE BELGIAN REPORTING SYSTEMAlthough Belgian guidelines give the guidance that geneticists and health professionals desire, they are also subject to discussion for they can be viewed upon as paternalistic and they do not accommodate an individualized approach. Therefore, it is interesting to scrutinize the Belgian approach from an ethical point of view.
Players to take into account in the setting of prenatal genetic diagnosis are the parents, fetus, physician, parental family and society. In medical ethics generally, reflections are to be made about non-maleficence (principle of doing no harm), beneficence (principle of doing good), autonomy (an individual’s right to self-determination) and empowerment (participating actively and autonomously in policies that affect one’s health or well-being).
4.4.1 Parental perspectiveBelgian geneticists attach great value to the principle of non-maleficence. In a prenatal setting, not reporting a VOUS or susceptibility CNV above all serves to avoid unnecessary anxiety for the pregnant mother. However, this involves an inherent risk of paternalism:106,
107 an expectant mother has every right to a “healthy” child, but how can anyone else
Ethical considerations of the Belgian approach
4 1

72
decide what defines healthy? On the other hand, can a pregnant woman be expected to be knowledgeable enough to evaluate the meaning of a certain CNV, even after having received pretest counseling? Future parents can feel stigmatized after receiving the diagnosis of e.g. a susceptibility CNV and might worry about the consequences of their decision.57 Some parents will terminate the pregnancy, but might feel guilty about having aborted a possibly healthy baby. The uncertainty about the meaning of a result complicates both professional counseling and the decision-making process for parents-to-be. Communication with the parents-to-be about these uncertainties can vary between paternalistic and directive on how to proceed, to placing all responsibility on the parents after disclosing any known information. Brabbing-Goldstein found that 18% of pregnancies with a low penetrance neuro-susceptibility CNV and without fetal anomalies were terminated,63 illustrating possible unnecessary concerns and terminations of pregnancy. Others will carry the pregnancy to term but might find it more difficult to bond with their child or will consider any developmental delay proof of the causality of the variant. However, in specific cases, for example in disorders like autism spectrum disorder, in which outcome is improved by early intervention, knowing about such variants could be beneficial.108, 109 Parental concern is determined by five factors: inheritance, possible phenotype, manageability of outcome, availability and strength of evidence as well as the providers’ messages, and it is key that the counselor provides all available information on each of these topics.105 Nevertheless, even with today’s broad genetic knowledge, the clinical implications of certain variants remain elusive, complicating counseling; should projecting the burden to the pregnant woman be viewed upon as medical escapism or as patient empowering, offering the future mother the opportunity to decide what to do upon detection of a VOUS? Reporting a VOUS will lead to parental genetic testing. If the VOUS is inherited, this can damage self-esteem, cause guilt in the carrier and reproach by the non-carrier, compromising the couple’s relationship. On the other hand, inheritance of a CNV from a healthy parent might as well decrease parental concern.105
At the least, counselors should make sure that the pregnant woman pays attention, absorbs and recalls the provided information, understands what applies to her, before she can be expected to consider all alternative outcomes.106, 107
Non- maleficence in case of the other, non-pregnant parent could mean avoidance of substantial emotional stress and fear, which can in turn cause relational distress. Their self- esteem could be affected when they are no longer capable to protect their family from the agony inflicted by reporting a CNV in a prenatal setting. Their anxiety will also be affected by the degree of their right to co-decide about the fate of the unborn child. When asked a few weeks after diagnosis of a pathogenic CNV or VOUS in their unborn child, future fathers remembered less than future mothers about the type of variant found and about the possible consequences for their child’s development; future mothers were more involved in the conversations with the counselor while fathers received a lot of information through their partner, rather than directly from the counselor.105 Both parents can consider wrongful birth in case a child is born with congenital
Chapter 4

73
health problems. Indeed, the main reason for prenatal genetic testing is to enhance the autonomy of the couple concerning preventing the birth of a child with a serious disorder or disability. Future parents expect to receive accurate information about the health of their child. The term ‘wrongful birth’ refers to claims for negligence where an opportunity has been lost to parents to terminate a pregnancy and involve a claim for damages by the parents of a child for, most importantly, the costs of bringing up the child.110 If prenatal diagnosis was performed, but the CNV responsible for the disability was not disclosed to the parents, because, at the time it was deemed that evidence for the pathogenicity of the CNV was not sufficient or that the penetrance of the disease was considered not high enough, parents are more likely to sue for wrongful birth.
4.4.2 Some aspects concerning the fetusAt the fetal level, ethical and legal issues mix up, since the concept of the fetus as a patient is not without difficulty. It can be, but is not by default, dependent on pregnancy duration. Even in Europe, abortion laws differ between countries. In some countries, a fetus is legally considered a person when viability is reached, even if the fetus is not yet born (enabling “murder” on an unborn child). In Belgium, an unborn child is not considered a legal entity, but becomes a rightful person at birth (“murder” on an unborn is impossible, legally allowing for intra-uterine feticide irrespective of gestational age). This special legal status of an unborn fetus in Belgium has major consequences in daily clinical practice. Termination of pregnancy on medical grounds (danger for the mother’s life or a disease in the fetus considered (by consensus) serious and non-curable) is allowed at any gestational age without upper limit, even in the viable ages after 24 weeks. But, in case the fetus is born it becomes legally obligatory to give medical care and support to this child, even if it has a serious and non-curable disease. Consequently, a termination of pregnancy for fetal anomalies or genetic disease at a later gestational age is in Belgium necessarily accompanied by intrauterine feticide.
Ethically, the status of the fetus as a patient is defined by its possibility to become an individual, but this possibility is intimately linked to the fact that the fetus can, biologically, not demonstrate autonomy, being completely dependent on his mother to survive. Consequently it is the pregnant woman who decides on the status of her unborn child.32
In case of the fetus, non-maleficence can be either interpreted as its right to exist or the avoidance of future illness. Is it in the best interest of the fetus to perform a termination of pregnancy to avoid disability? The idea that we decide for the unborn child can be interpreted as paternalistic, and this is true both for the medical professional and the parents. Even those who call themselves “lawyers or defenders of the fetus” cannot but act paternalistic as one will only be able to ask the opinion of the individual that will develop from the fetus many years later. Consequently, autonomy and empowerment are not characteristics of the fetus but of the imaginary person that can develop from the fetus.
If parents were not informed about CNV’s detected during invasive prenatal testing and they are causative for disability, the disabled child (or his/her representative), can
Ethical considerations of the Belgian approach
4 1

74
claim for ‘wrongful life’. These claims, for having to live a life full of suffering because of a disability, are brought against a doctor or obstetrician. In these cases, the claim is that, overall, living is considered to bring more harm than good.110 This raises the question: has the child a right to its own abortion? Although there is no alternative. Or the child exists, or the child does not.
4.4.3 Doctor’s perspectiveFrom a medicolegal point of view, communicating all genetic variants found in the newborn, hence avoiding all responsibility, is the best choice. Additionally, it could be argued that a clinician should not be put in the position to withhold medical information, as this is ethically challenging and jeopardizes the doctor-patient relationship. Because of the increased patient autonomy, the physician progressively escapes deciding alone.111,
112 While this sharing of responsibility can be viewed upon as an improvement, it can also cause a mental burden when helping or referring a patient desiring a termination of pregnancy for reasons the doctor cannot support. Oppositely, should doctors not have the opportunity to determine for which variants they offer a termination of pregnancy, because of the physician’s right to self-determination?
A medical professional can feel frustrated, when after extensive counselling a couple reaches the opposite conclusion from what was expected (e.g. ‘we want to terminate this pregnancy anyhow’ for a variant for which evidence of pathogenicity is lacking). It is unavoidable that even the most experienced “non-directive” counsellor still has a personal opinion that can conflict with the parents’ view.
4.4.4 Parental family’s perspectiveSince the genetic variant found prenatally can be inherited, communication of a clinically relevant variant can be of interest for the extended family. Reporting a VOUS can cause unnecessary anxiety and problems with self-esteem within a family. Medicolegally, the family does not have the autonomy and is not empowered to decide about which variants should be communicated, because of privacy issues of the index patient, the expecting mother.
4.4.5 Society’s perspectiveThe society’s perspective can be surprisingly different from the parents’ viewpoint. Do no harm could be interpreted as the desire to reduce costs to society by limiting the number of mentally or physically impaired children. Genetic laboratory costs can be high, but these costs are always less than the treatment of and/or care for dependent individuals. Parents could be judged for choosing to pursue a pregnancy when a certain genetic variant is found. In a group of 46 parents with a prenatal diagnosis of a child with Down’s syndrome who decided to continue the pregnancy, 9% reported a negative experience
Chapter 4

75
with family or friends and 35% felt unsupported by their medical professional, who had advised them to terminate the pregnancy.113 Knowledge about genetic variation within a population can also improve health care by providing the necessary medical support from birth onwards, and members of society should decide if they want to invest in this knowledge. It is in the society’s interest to collect as much information as possible about uncertain variants, for example via national databases,65 since it increases knowledge and can reduce anxiety, as more variants will move from VOUS to benign or pathogenic.
Guidelines, as those discussed in this paper, are a pragmatic response to the consequences, in this case the consequences of technology evolving faster than in-depth knowledge. In an ideal situation, before performing any (prenatal) test, we consider all alternatives, reflect on their consequences for all parties involved in an impartial manner and thereafter act on those reflections. In this paper’s context, all alternatives to consider are: applying microarray for all indications for prenatal testing, applying microarray only for certain indications (e.g. ultrasound anomalies) or not applying microarray at all. The same possibilities apply for WGS/WES. With regard to reporting, the following options are available: reporting all genetic variants to parents, reporting only variants deemed clinically relevant or letting parents decide themselves about the variants they want to be informed about.
Applying microarray for all indications detects 0.5-10% additional relevant diagnoses (depending on the indication of the invasive procedure) as compared to karyotyping and a VOUS in up to 7.3% of patients.19-24 Implementation only in case of ultrasound anomalies would miss 0.5-2% of relevant diagnoses. When reporting all non-benign variants, up to 7.3% of pregnant women might remain anxious about their child’s health during pregnancy and even after birth, although elaborate counseling might be able to partially prevent/reduce anxiety. If a variant is not considered to be pathogenic based on current literature, it remains ethically difficult for a physician to abort a pregnancy solely based on the genetic result in case of parental request. This could lead to more terminations of pregnancy but would relieve physicians of the burden of not reporting certain variants. If, like in Belgium, a committee of genetic experts decides which variants are reported and which are withheld, it relieves the burden of the treating physician. However, if a variant with a highly variable expression or unknown significance at the time of the prenatal test unexpectedly does cause disease, discussion rises about the legal consequences, effectively shifting liability from the physician to the geneticist. If parents reach a decision based on all detected variants, responsibilities of the geneticist and physician are in turn shifted to the parents. Some parents would appreciate this, others would remain anxious, worrying about the decision they made.57
After careful consideration of the consequences for the stakeholders and the possibilities Belgian geneticists decided to apply microarray for all indications, whilst a committee of experts decides, based on current literature, which variants to report and which not, since they consider it to be the option causing the least anxiety for parents and physicians. Pre- and post-test counseling are considered of high importance and support is provided for all patients in need for it.
Ethical considerations of the Belgian approach
4 1

76
4.5 CONCLUSIONIn Belgium, geneticists agreed upon using microarray for all indications of invasive prenatal testing. They decided to compile uniform guidelines on how to interpret and report variants. In this paper, we reflect upon this reporting policy, using Belgium’s cultural background and using ethical statements. Our current approach is consistent with Belgium’s high power distance and high uncertainty avoidance. To reinforce this conviction, we recently initiated a study investigating the opinion of Belgian gynecologists, general practitioners and future parents on the Belgian approach.
Chapter 4

77
Chromosomal micro-array analysis in prenatal diagnosis: ethical considerations of the Belgian approach
4 1

78
LABO. EXPELLU PTATATURESTO BEATI-IS DUCID UT ERUNT, QUAMUSDAE PROVIDUS DIT OMNITIBUS.
cover-boek-hand.indd 10cover-boek-hand.indd 10 07/07/2020 07:5807/07/2020 07:58

79
5. PRENATAL HOMOZYGOSITY MAPPING DETECTS A NOVEL MUTATION IN CHST3 IN A FETUS WITH SKELETAL DYSPLASIA AND JOINT DISLOCATIONS
Joke Muys, Bettina Blaumeiser, Yves Jacquemyn, Katrien Janssens
Clin Case Rep. 2017;5(4):440–445. Published 2017 Mar 1. doi:10.1002/ccr3.800
LABO. EXPELLU PTATATURESTO BEATI-IS DUCID UT ERUNT, QUAMUSDAE PROVIDUS DIT OMNITIBUS.
cover-boek-hand.indd 10cover-boek-hand.indd 10 07/07/2020 07:5807/07/2020 07:58
5 1

80
5.1 INTRODUCTIONIn 1987, Lander et al published the first paper on homozygosity mapping for human genes that cause recessive traits. He describes the usefulness of the method to map recessive diseases for which it is impractical or impossible to collect adequate numbers of families with multiple affected offspring.114 Single-nucleotide polymorphism (SNP) microarray has been documented to have clinical utility in the detection of regions of homozygosity.115 In recent years, homozygosity mapping has demonstrated its effectiveness in the detection of disease loci in affected children from consanguineous couples in the neonatal setting, as well as after pregnancy termination for prenatally diagnosed malformations.116-119 To our knowledge, the use of homozygosity mapping has only been reported once in the prenatal setting (on DNA isolated from amniotic fluid or a chorion villi biopsy),120 although prenatal homozygosity mapping shows additional value because it can help parents to reach an underpinned decision on whether or not to terminate the pregnancy.
5.2 CLINICAL PRESENTATIONThe case presented here involved a G3P2 woman, originating from Afghanistan, at 17 weeks gestational age. Her first two pregnancies were uncomplicated and resulted in two healthy boys. The couple was consanguineous (first cousins). In the current pregnancy, the gynecologist referred the couple for a suspicion of fetal limb malformation on routine ultrasound. Advanced ultrasound showed an abnormal morphology of the fetal lower limbs, short femora and humeri, abnormal position of the knee joints and bilateral pes equinovarus, suggestive of arthrogryposis (Figure 5.1). Family history showed that the brother of the proband’s mother had had clinical arthrogryposis and had died during pregnancy (Figure 5.2). Based upon the combination of ultrasonographic anomalies, differential diagnosis included: autosomal dominant Larsen syndrome, caused by mutations in FLNB,121 autosomal recessive Larsen syndrome, caused by mutations in B3GAT3,122 Diastrophic dysplasia, caused by homozygous or compound heterozygous mutations in SLC26A2,123 Desbusquois dysplasia, caused by homozygous or compound heterozygous mutations in CANT1,124 Chondrodysplasia with joint dislocations, caused by homozygous mutations in IMPAD1,125 and spondyloepiphyseal dysplasia with congenital joint dislocations, caused by homozygous or compound heterozygous mutations in CHST3.126
Chapter 5

81
Figure 5.1: Advanced prenatal ultrasound showed an abnormal morphology of the fetal lower limbs. This figure shows the abnormal position of the knee joints and bilateral pes equinovarus on 3D ultrasound examination.
Prenatal homozygosity mapping in a fetus with skeletal dysplasia
5 1

82
Figure 5.2: Family history. The unborn child is indicated as the proband (P-IV:3). Note the consanguinity of the parents (III:5 and III:6) (first cousins). Family history shows that the brother of the proband’s mother (III:10) had had clinical arthrogryposis and had died during pregnancy.
Given the anomalies found on prenatal ultrasound, amniocentesis was performed. Rapid aneuploidy testing (QF-PCR) did not reveal any aneuploidies. SNP array analysis to detect deletions and duplications was performed using a HumanCyto-SNP-12 v2.1 BeadChip on an iScan system following standard protocols as provided by the manufacturer (Illumina). Copy number variant (CNV) analysis was performed with CNV-WebStore.127 SNP array analysis showed no pathogenic CNVs.
The pregnancy was terminated two weeks after the ultrasonographic findings because of the severity of the phenotype and after genetic counselling regarding future prospects for the unborn child. Clinical genetic inspection of the fetus confirmed the joint and feet anomalies (Figure 5.3). Radiographic examination confirmed the abnormal position of the left and right knee joints (Figure 5.4). The ribs, upper limbs and skull were normal. Autopsy report described a male fetus with biometry in the lower normal range for 17 weeks gestational age. Elbows showed a fixed flexion and knees had a fixed forward flexion as well. Macroscopically, there were no other remarkable skeletal anomalies. Facial features were normal. All organs in neck, thorax, abdomen and pelvis were histologically and anatomically normal.
Chapter 5

83
Fi gure 5.3: Postnatal clinical inspection and autopsy confi rmed the joint and feet anomalies.
As the parents are first cousins, autozygosity, the presence of 2 alleles at a locus originating from a common ancestor as a result of inbreeding (identical by descent), might explain the phenotype; the fetus could have inherited the same mutant allele responsible for an autosomal recessive trait from both parents. To detect regions of autozygosity, possibly harbouring a mutation, homozygosity mapping was performed on the sample obtained by amniocentesis by using CNV-WebStore for data analysis.127 Twenty seven homozygous regions larger than 1 Mb were identified, accounting for 8.43% of the genome (Table 5.1). Among the 2507 genes present in these regions, we carefully inspected all 362 MORBID genes. CHST3, located on chromosome 10q22.1, was considered to be the strongest candidate gene since homozygous and compound heterozygous mutations in this gene have been identified in Spondyloephiphyseal Dysplasia with Congenital Joint Dislocations (OMIM #143095). This disorder is characterised by short stature (both prenatally and postnatally), a broad forehead, long philtrum, small ears, hypertelorism, high-arched palate, variable cardiac anomalies, broad chest, joint dislocations in knee, hip and shoulder, limb malformations (e.g. fixed elbow flexion and knee dislocations), brachydactyly, campodactyly and feet malformations. Affected individuals have a normal intelligence, but a delayed gross motor development. Our case presented with several characteristics representative for this disorder. DNA of the fetus and his parents was analysed via PCR amplification and direct sequencing of CHST3. A hitherto unknown variant, c.491C>T (p.P164L), in exon 3 of the CHST3 gene was detected in a homozygous state in the fetus. This variant was classified as a likely pathogenic variant based on in silico predictions by Alamut (Alamut Visual version 2.7 (Interactive Biosoftware, Rouen, France)). This variant lies in the so-called P-loop containing nucleoside triphosphate
Figure 5.4: Radiographic examination confi rmed the abnormal position of the left and right knee joints.
Prenatal homozygosity mapping in a fetus with skeletal dysplasia
5 1

84
hydrolase fold (amino acids 132-168). The Exome Aggregation Consortium (ExAC)128 reports the heterozygous change of the same amino acid to Arg (Pro164Arg; rs771866012) in one of their 60700 individuals. The variant is classified as probably damaging by several in silico prediction programs, but no further studies have been performed.
Table 5.1: Regions of homozygosity in the fetus
Chromo-some Start End Size (bp) Nr.
Probes %Hom %Het Nr. Genes
1 213827795 216840371 3012577 407 0.995 0.002 92 86023398 87052934 1029537 163 0.994 0.006 173 206714121 221239894 14525774 1017 0.999 0.001 1364 96162805 97346618 1183814 151 1.000 0.000 15 31661814 40255501 8593688 553 0.996 0.004 326 75771799 157116213 81344415 5134 1.000 0.000 3627 171324289 175220428 3896140 603 0.998 0.002 278 47921405 49039681 1118277 141 0.986 0.014 68 140733440 142600093 1866654 290 1.000 0.000 109 101297144 102540372 1243229 199 0.995 0.005 810 3239252 19471726 16232475 2045 1.000 0.000 9910 37608337 38685231 1076895 137 1.000 0.000 1010 71056150 95747476 24691327 2845 1.000 0.000 20110 121922699 135430043 13507345 1950 0.999 0.001 10511 2348778 11644920 9296143 1277 0.998 0.001 19011 46196053 47246397 1050345 151 0.987 0.013 2212 87738065 88801020 1062956 159 1.000 0.000 512 111768973 113025901 1256929 170 0.982 0.018 1712 120822453 121874019 1051567 160 0.981 0.019 2516 30172627 31383304 1210678 241 1.000 0.000 6716 82678897 87111021 4432125 697 0.997 0.003 4317 7314216 18747176 11432961 1698 0.999 0.000 18417 37265378 75271787 38006410 3837 0.999 0.001 65017 77291311 81047565 3756255 555 0.998 0.002 9418 18540853 19601717 1060865 179 0.989 0.011 919 9247389 13355633 4108245 565 0.995 0.004 14922 40731134 42196467 1465334 208 0.995 0.005 29
Indicated in the table are number of the chromosome; start and stop position of the region of homozygosity; size of the region in base pairs (bp); the number of probes in this region; percentage of homozygosity (% Hom) and heterozygosity (% Het) as well as the number of genes involved, accounting in total for 8.43% of the genome. Genome Build: GRCh37 (hg19).
Chapter 5

85
Mutations in three other amino acids in this domain have been described.129, 130 Functional analysis of one of these, T141M, shows it to have a dramatically decreased sulfotransferase activity.129 The proband’s mother and father carry the mutation in a heterozygous state.
The proband’s parents returned for further discussion eight weeks after delivery. They received extensive genetic counselling and were informed about the possibility of preimplantation genetic diagnosis (PGD) or invasive testing in future pregnancies. The importance of further family testing was stressed. An informed consent for publication of the case was obtained after carefully informing the parents about the clinical significance of the case.
5.3 DISCUSSIONSince 2013, genome-wide array – SNP array or array CGH (comparative genomic hybridisation) – has replaced karyotyping in the prenatal setting in Belgium. At the time of introduction of this novel technology in the prenatal field, all Belgian genetic centres agreed on a resolution of 400kb to maximize the detection of pathogenic CNVs whilst minimalizing the detection of Variants of Unknown Significance (VOUS) and on a list of susceptibility loci to report.25 The increase in resolution from 5-10 Mb to 400 kb has led to an average increase of 6.0 % of diagnoses in fetuses with ultrasound anomalies,19 but smaller deletions/duplications and single point mutations stay well below the radar. The arrival of high-density SNP array however,131, 132 as utilized in our center, allows the simultaneous determination of regions of homozygosity, which can lead to the detection of autosomal recessive mutations in offspring of consanguineous partners.
We could only identify one publication on the use of homozygosity mapping in the prenatal setting,120 making this report the second in its kind. The advantage of prenatal homozygosity mapping lies mainly in the short-term interval between recognition of the problem (ultrasound anomalies) and the definite diagnosis (a recessive mutation). In several countries, termination of pregnancy can only be legally performed until 24 weeks gestation, since the fetus becomes viable around that age. With the workflow described here, complete results can be obtained within approximately 6 weeks: if amniocentesis is performed at 15 weeks gestational age, SNP array and homozygosity mapping results are known at 17 weeks gestational age, and targeted analysis results at 21 weeks gestational age, well before 24 weeks. The result will guide gynaecologists in counselling the parents correctly and can aid parents in reaching a well-informed decision on whether or not to terminate the pregnancy. Moreover, since the parents can be screened immediately for carrier state of the recessive trait, they know whether or not they are at risk of having an affected fetus in a future pregnancy and can be counselled for the different options to avoid this, including PGD and invasive prenatal testing.
Prenatal homozygosity mapping in a fetus with skeletal dysplasia
5 1

86
In this particular case, with the parents being first cousins, on average 6.25% of the genome is expected to be autozygous. Consequently, hundreds to thousands of putative candidate genes could be identified and prioritization of the genes, based on information regarding their function is crucial. If the phenotype of the fetus can be clearly linked to one or a few of the genes, Sanger sequencing is the preferred method of choice. However, this approach precludes the identification of less obvious candidate genes. Therefore, in most cases successful autozygosity mapping further narrows the number of regions of interest, whenever it is impossible to investigate multiple affected siblings. When no other affected family members are available, a combination of autozygosity mapping and next-generation sequencing (NGS) is recommended, as it allows either massive parallel sequencing of all autozygous regions or filtering of variants found by whole exome or whole genome sequencing based on autozygosity mapping. 133 One can argue that autozygosity mapping can be performed directly on the NGS data, but cost and technical limitations of NGS (e.g. low read depth in some positions) preclude this for the time being. Moreover, if, based on the examination of the autozygome, a strong candidate gene is identified and sequenced, as was the case here, this is much more cost effective than performing exome/whole genome sequencing.
5.4 CONCLUSIONWe conclude that in selected cases, homozygosity mapping followed by direct sequencing of one or a few carefully selected candidate genes in a prenatal setting can be beneficial to obtain diagnosis in consanguineous families.

87
5 1

88
Ehenitate od uta sim et aut ilictur, sita doluptibus cusantibeat.Licit fugit, od mi, conse nosse delloru mquidebist, conse volenih iliaepta volupis elendandae consed et escillaut arita consequos volorum aut ex et mincta quae vel eariasped ut exces minte pel endici-mintur aut velest inciisimo il ilic tet adigenissima seque poremqu isquunt od etur mi, que volecabore sedias sima nates voluptatur, optaqua musdam, esto quo tem aceribus, aperum quis aut et labor a vel molesequid quat peligniam aliquatur, seque minim voluptiscid maximet prepelecte pa dem alicips apicaep erspelest omnihil etur sape enimo ea quam, offi ci aut magnis et autaspit facest ute cuptae. Ut unt atem. Itaepud ionsed unt voluptati blam, odio dit ad maximo quunt as que lam, om-moluptur, voluptu reruntus alit latur, sum, culluptae. Ugiam sum ipsa simus volorum laccabo ribus-aped mi, ist iumet que sum sit, que nonsenitam, sum qui sunt rerum is dunderchit laborep tatecae nonsequas earcid que nat.
Uditatet fugia nonsequi consent magniae. Et mo blamus, ut re, que laccae volorenditis et eum, om-molup taquide lenimpe rchitatet verum rernatus vel ius autat quam volor sunt ea dolutempores re vo-lupta temperi oreius dolupta eribus etur? Quia sim nobis rem sa ipsaes atem voluptur maiore volorat.Parchit dolent. Cum eossunt quo tenis autempori alitior ad et ex expelignam quiatem fuga. Tioribus eossitatium ex ea ius, odia sequid ut es minverest modicie nisque vid mint iscil in re nimincia pratur archiciis dolore, offi ci aut fugit, serem ipsam, inciusc ilibus doluptaqui aligenis dolorro rempor modi cum quatecus dolesti usaera simpel ipici dolupie ntust, cum sum et, aut oditium ipsam earibusam in ex est dolesedio cuptas aturis aut as eatur?Aquo con peres por aut lam, offi cium rem num abo
cover-boek-hand.indd 7 cover-boek-hand.indd 707/07/2020 07:58 07/07/2020 07:58

89
6 GENERAL DISCUSSION AND FUTURE PERSPECTIVES
The landscape of prenatal invasive testing is subject to continuous innovation. Over the last 5 years, chromosomal microarray analysis (CMA) has increasingly replaced conventional karyotyping for the analysis of invasively obtained samples. As described in the general introduction (Chapter 1), conventional karyotyping has long been the gold standard for analyzing prenatal invasive samples. The value of CMA fundamentally lies in its higher resolution, revealing additional clinically relevant diagnoses in the presence and absence of ultrasound anomalies when compared to karyotyping alone. However, inherent to the higher resolution of CMA, it also detects variants with an unknown phenotype (variants of unknown significance (VOUS)), variants for which the clinical outcome is highly variable (susceptibility variants), and incidental findings, findings unrelated to the indication of the invasive test. These issues impede international consensus on how to implement CMA in daily invasive prenatal practice, how to interpret its results and whether and how to report results to future parents.19-24 Interpretation of prenatally detected copy number variants (CNVs) is challenging because knowledge on variants is mainly based on examination of affected individuals, instigating a bias to the more severe end of the phenotypic spectrum. Understandably, reporting a CNV in a prenatal setting differs from reporting it postnatally, since it can lead to interruption of pregnancy and parental anxiety. Therefore, in Belgium, with the introduction of CMA in prenatal invasive diagnosis in 2013, all 8 genetic centers developed uniform guidelines on how to classify, interpret and report variants.25 Belgian genetic centers also agreed on the construction of a prenatal database (Chapter 2), in which data of children who underwent prenatal invasive testing during pregnancy are gathered and linked to their prenatal and postnatal phenotype. The overall aim of this thesis was to determine genotype-phenotype correlations of prenatally detected pathogenic, susceptibility and unclassified CNVs.
6 1

90
Chapter 6
In Chapter 2, the most frequently found genomic disorders, reported and unreported susceptibility CNVs, and recurrent VOUS in the Belgian prenatal population are described. To enable an establishment of prenatal phenotype and genotype associations, correspondence analysis was performed, examining the relationship between the indication of the invasive test and the variant detected. This analysis failed to detect such an association. In cases with an ultrasound anomaly, 0.7% carried a reported susceptibility CNV. As expected, this was not significantly different compared to the prevalence in the entire prenatal population, as susceptibility CNVs are rarely associated with ultrasound anomalies. VOUS were distributed evenly among the different indications. Since genomic variants can be population-specific, knowing which variants are recurrent in our population is of great value, particularly if the genotype can be associated with a certain prenatal and postnatal phenotype. The in-depth comprehension of population-specific variants is only feasible when regionwide or nationwide cooperation is achieved, including uniform classification of CNVs. To the best of our knowledge, this national research project was the first of its kind, the gathered data and research learnings are unique till date.
To allow complete phenotyping of the variants found, postnatal follow-up of children prenatally diagnosed with a non-benign CNV was undertaken (Chapter 3). This study indicated that, for the categories of communication skills and personal-social skills, children with a reported susceptibility CNV performed worse than children with an unreported susceptibility CNV or children with no CNVs or only benign CNVs. Furthermore, development of children with an unreported susceptibility variant did not differ from that of the control population. These results indicate that our classification system can differentiate between children who will perform well and those who will not, helping geneticists to report variants accordingly. Obviously, a child’s development is not exclusively influenced by genetics, but also by many other covariables. We tested the effect of the covariates parental education, multilingualism, a surgical history, pre-term birth and paternal age on test scores but found no significant effect.
On an international level, the Belgian approach to implement CMA in prenatal diagnosis has been the subject of discussion. In Chapter 4, we elaborate on the cultural and ethical background of the Belgian guidelines. Several studies suggest that Belgium has a distinctive medical culture.93-96 In Belgium, there is a low tolerance for uncertainty and ambiguity. Belgian patients demand clear advice from their medical doctors. This poses an additional need for formal rules and absolute truth, and is in line with the Belgian prenatal guidelines. Moreover, chapter 4 clarifies differences between countries regarding their reporting strategy and the lack of an international uniform reporting system. A one-size-fits-all policy will be very difficult to reach, because patients and doctors’ preferences differ based on cultural and ethical background.
In Chapter 5, we describe how, in the search for new opportunities for using CMA in prenatal diagnosis, application of homozygosity mapping using SNP array in prenatal invasive testing leads to a timely diagnosis, enabling a more informed pregnancy

91
General discussion and future perspectives
termination. The case described is only the second published using this technique. Out of eight Belgian academic genetic centers, Antwerp University is the only one analyzing prenatal samples with SNP array. SNP array has the additional benefits over array CGH of detecting triploidy, maternal cell contamination and regions of homozygosity, properties that are all very valuable in the analysis of prenatal samples.
6.1 METHODOLOGICAL CONSIDERATIONSAs previously stated, the objective of this doctoral thesis was to determine genotype-phenotype correlations of prenatally detected pathogenic, susceptibility and unclassified CNVs. First, we focused on examining the association between genotype and prenatal phenotype. Notwithstanding the inclusion of 13,266 samples in the database, the number of cases with a pathogenic CNV was limited (1.9% or 246) and many of those were unique in our dataset. More than half of the fetuses with a pathogenic CNV (63.0% or 155/246) had a structural abnormality on ultrasound investigation. Classifying those cases per affected organ system decreased the number of cases per category even further. In conclusion, it was challenging to draw any clear conclusions on genotype-prenatal phenotype correlations. Despite this being a nationwide project, the number of cases will need to be increased drastically to perform statistical analyses, which can be obtained by collecting data over a longer period of time and/or by participating in international collaborations.
In the postnatal follow-up project, difficulties of reaching sufficiently large numbers for statistical analysis recurred. Postnatal follow-up of children is even more demanding than prenatal evaluation, since it requires sending out questionnaires, motivating parents to participate and analyzing data. In this work, neither the researchers nor the parents received any personal benefit, and this may have influenced the willingness to collaborate. Delays in sending out the questionnaires by dedicated researchers in the different genetic centers, as well as delays in completing the questionnaires by parents led to a significant percentage of questionnaires being completed outside the appropriate age window. Additionally, the elaborate questionnaire, containing 20 pages of questions, was probably responsible for the lower than expected response rate, which reached 17.3% after one reminder. A shorter survey might have improved response rates, but would preclude obtaining a complete overview of the child’s development. We chose to include the ASQ-3, a validated questionnaire that looks at communication skills, gross motor, fine motor, problem solving and personal-social development in a child and the ASQ-SE2 for evaluation of social and emotional health, since CNVs can also influence the psycho-social development of the child. ASQ-3 personal-social development mostly assesses whether a child can self-help itself in an age-appropriate manner, while ASQ-SE2 addresses the broad and complex nature of social-emotional development: self-regulation, compliance, social communication, adaptive functioning, autonomy and interaction with people. Finally, we added a general questionnaire to allow investigation of covariates possibly influencing the child’s development.
6 1

92
Although we detected a significantly worse outcome for the categories communication skills and personal-social skills in children with a reported susceptibility CNV, which emphasizes the value of our reporting system, it has to be noted that only four children with a reported susceptibility variant were included. Despite the very small numbers, this research project is of great value, since it is the first of its kind and underscores the value of our reporting system. As is the case for our prenatal dataset, statistical analysis of the correlation between genotype and postnatal development would highly benefit from larger datasets.
Children were evaluated at the age of 36 months. Because of the restricted timeframe of this project, a single evaluation moment was chosen. However, a child’s development cannot be fully evaluated using a single time-point measurement. Therefore, future research should include timely re-evaluation of children until adulthood.
Based on our prenatal data, the added value of using CMA compared to karyotyping was calculated; CMA had an added diagnostic value of 2.7% and 1.5%, respectively, in cases with an ultrasound anomaly versus cases without. A wide range of added values are reported in literature: 5.2-10% in cases with an anomaly versus 0.5-2% in cases without ultrasonographic abnormalities.20, 23 Discrepancies can be explained by differences in cohort size, cohort selection and array platforms; no international agreement exists on the resolution of the array platform to be used for prenatal diagnosis, which also influences the number of CNVs detected and, therefore, the added value of the CMA performed. In Belgium, a resolution of 400 kb was chosen to maximize detection rate while minimizing the amount of VOUS detected.25
As shown by our results, the classification and reporting policy of the laboratory also affects added values. For example, a systematic review in 2014 describes an added value of 6.8% for CMA in fetuses with an ultrasound anomaly, taking into account all pathogenic variants.20 Hillman reports an added value of 10% in cases with an ultrasonographic abnormality, but likely pathogenic variants were also included in this calculation.21 In our study cohort, for calculating added values of using CMA rather than conventional karyotyping, we considered all pathogenic variants and reported susceptibility variants, according to Belgian guidelines. Obviously, added values increase if unreported susceptibility variants and VOUS are included in the calculation (e.g. for fetuses with ultrasound anomalies, added values increase to respectively 3.7% and 8.9%). Uniform guidelines for classifying and communicating variants are imperative to enable researchers to compare study results.
Chapter 6

93
6.2 FUTURE POSSIBILITIES AND CHALLENGES IN PRENATAL TESTING
6.2.1 Worldwide data sharingThe need for large datasets to study genotype-phenotype correlations of rare CNVs warrants worldwide data sharing. Next to the Belgian prenatal database, medical investigators in New Zealand and Australia have initiated a prospective research project to centralize their data together with Belgian prenatal data. To avoid ascertainment bias, data on non-affected carriers (e.g. as is achieved by investigating development of children with a prenatally detected CNV) must be included in databases containing information on affected individuals. The following issues complicate worldwide data sharing:First, as described earlier, analyzing data from large datasets is labor-intensive and time-consuming. Genomic data are too complex to be analyzed by manual inspection alone. Machine learning, which is based on the development of computer algorithms that improve with experience, is increasingly applied in genetics and genomics and can be used to help analyzing huge data sets.134, 135 High performance of a machine learning method depends on theoretical and practical knowledge of both machine learning methodology as well as genomics. Two kinds of machine learning exist. In supervised machine learning, a researcher creates a computer algorithm, provides a reference set with negative and positive cases to the computer from which it can learn and thereafter provides a new, large set of data to be analyzed. Unsupervised machine learning is feeding the computer raw data, without a reference set and prior learning. In general, if one can compile a list of CNVs, together with the genes involved and possible phenotypical properties, a machine learning system can probably be trained to search for genotype-phenotype associations. Whilst a dysmorphologist can utilize the Human Phenotype Ontology (HPO) to describe human phenotypic malformations in a standardized manner, a machine learning tool can be taught to search for phenotypic features in even more detail, by extracting them from photographs, for example, and linking them to genomic data.136 In the future, the rapidly developing field of machine learning will allow for more accurate analysis of genetic data, incorporating genotype-phenotype associations.137
Second, adding information about non-affected carriers to genomic databases is essential, as it decreases bias towards the more severe end of the phenotypical spectrum. In this thesis, developing a prenatal database and performing postnatal follow-up for children with a prenatally detected non-benign CNV, is proposed as a tool to avoid such a bias. Timely re-evaluation of these children until adulthood is of importance to accurately determine their development. However, as discussed in Chapter 4, there are ethical considerations to be made. One can question whether the unborn child has a “right not to know” he or she is carrying a CNV that could possibly predispose him/her to a genetic disease. Moreover, if such sensitive, genomic information is stored in a database, the question arises about ownership of the archived data, which is of societal,
General discussion and future perspectives
6 1

94
economical and legal interest to patients. A currently healthy person can be considered at a high-risk for future medical costs, prolonged absence at work etc. based on the genomic information stored. However, fully anonymizing data is not possible if children need to be re-evaluated at given time points. With the recent introduction of General Data Protection Rules (GDPR) in the European Union, gathering such sensitive genetic data is even more challenging. Another means to obtain unbiased genotype-phenotype information is to make a genomic passport for every individual in society by determining their genomic data and registering their health status. Although commercial institutions increasingly offer genome-wide testing to individuals, it is a highly disputed step to perform such testing globally and gather it in an open-access database.
6.2.2 Whole exome and whole genome sequencing (WES and WGS) in prenatal diagnosisTo date, invasive prenatal testing strategies include quantitative fluorescent polymerase chain reaction (QF-PCR) or fluorescence in-situ hybridization (FISH) for rapid aneuploidy testing on the one hand, and conventional karyotyping and CMA for genome-wide testing on the other hand. In a postnatal setting, WES and WGS are slowly but surely being introduced, as they offer the advantage of a resolution down to the single base-pair level. Understandably, implementation of WES/WGS in a prenatal setting will pose even more challenges concerning interpretation of findings, reporting to future parents and ethical considerations. A systematic review states a wide range from 6.2 to 80% in diagnostic yield for WES in fetuses with ultrasound anomalies;138 the diagnostic rate was higher in fetuses with multiple anomalies. Currently, the American College of Medical Genetics and Genomics (ACMG) recommends the use of WES/WGS when other genetic tests fail to determine a diagnosis in a fetus with multiple anomalies139 and the Committee on Genetics and the Society for Maternal-Fetal Medicine additionally advice consultation with a geneticist because of the difficulty of the analysis.27 In Belgium, the eight genetic centers are working on national guidelines for the implementation of WES/WGS in ongoing pregnancies and a shared database for variants, similar to what has been done for CMA.
6.2.3 Towards a prenatal genome ID cart for every fetus using non-invasive prenatal testing?Non-invasive prenatal testing (NIPT) is increasingly available to pregnant women worldwide. Most NIPT methodologies are genome-wide, but in general, protocols only report on aneuploidies of chromosomes 13, 18 and 21. In a very recent national study in the Netherlands, NIPT had a positive predictive value (PPV) of 96% for trisomy 21, 98% for trisomy 18, 53% for trisomy 13 and 6% for other trisomies; the low PPV for the latter trisomies can be largely explained by confined placental mosaicism. For structural
Chapter 6

95
chromosomal aberrations, the PPV was 32%, and 64% for complex abnormal profiles indicative of maternal malignancies.140 In case of ultrasound anomalies, we observed a 4% higher diagnostic yield of CMA compared to NIPT (Chapter 2), clearly demonstrating that NIPT cannot replace CMA when ultrasonographic anomalies are present. The resolution of NIPT can become similar to that of CMA or even that of WES/WGS, but that requires a much higher sequencing depth, which comes at a cost that is currently too high for a population screening. Moreover, there are several other biological factors that hinder the expansion of NIPT resolution and implementation.
First, the current NIPT methodology is based on the analysis of cell-free DNA (cfDNA). cfDNA contains only 5-20% fetal DNA (cffDNA), which impedes the resolution, specificity and utility of NIPT – the fetal profile is always ‘clouded’ by the much stronger maternal profile. Second, cffDNA originates from circulating placental cells in maternal blood: in cases of confined placental mosaicism, where the aneuploidy or other genetic aberration is confined to the placenta and does not affect the fetus, this can lead to a false positive NIPT result. Vice versa, fetal mosaicism that does not affect the cells of the placenta could lead to a false negative NIPT result. Both problems could be overcome by analyzing circulating fetal cells, but the numbers of these cells in maternal blood are extremely low and their collection and purification for use in a cell-based NIPT are still in a research phase.141 It is to be expected though that methods to analyze sources of pure fetal DNA will eventually facilitate NIPT to become a diagnostic test instead of a screening test.
With the increasing implementation of NIPT for pregnancies without ultrasound anomalies, concerns have been raised that sub-chromosomal aberrations (which are distributed evenly between indications), will be missed.61, 62 In low-risk pregnancies, the complication risk of an invasive procedure explains the success of NIPT. Although systematic review and meta-analysis of the literature indicates a negligible procedure-related risk for miscarriage of 0.12% (95% CI, −0.05 to 0.30%) for amniocentesis and −0.11% (95% CI, −0.29 to 0.08%) for chorion villi sampling when compared to control groups with a similar risk profile,142 these results are strongly dependent on operator experience. As of July 1, 2017, Belgium became the first country in the world to fully reimburse NIPT for all pregnancies, making it a first-tier method which resulted in a steep increase in NIPT uptake and a rapid decline in the number of invasive procedures. To minimize procedure-related complications of amniocentesis and chorion villi biopsy due to lack of operator experience, centralization of invasive prenatal procedures in tertiary centers should be recommended.
It goes without saying that with the general implementation of a genome-wide NIPT, issues concerning classification of variants and their reporting in a prenatal setting, as discussed in this thesis, remain relevant.143
General discussion and future perspectives
6 1

96
6.3 GENERAL CONCLUSIONThe replacement of classical, cytogenetic karyotyping with molecular karyotyping, as CMA is also called, for the analysis of invasively obtained prenatal samples, was revolutionary, but new technologies like NIPT and WES/WGS are being implemented at an ever-increasing pace. The discussions on classification and reporting of variants to future parents remain unresolved until this day. This thesis contributes to the classification of variants, since it describes genotype-phenotype correlations of prenatally detected pathogenic, susceptibility and unclassified CNVs in a national cohort. Prenatal and postnatal phenotyping of prenatally detected CNVs, as described in this thesis, is of high importance, since it allows complete phenotypical characterization of CNVs, without ascertainment bias. Furthermore, this thesis describes differences in acceptability of reporting strategies, and their cultural and ethical backgrounds. In future research, international cooperation is warranted, because larger prenatal datasets are required to continue accurate phenotyping of children diagnosed with a prenatally detected non-benign CNV and hence, to enable the best possible counseling of future parents.
6.4 FUTURE RESEARCH OPPORTUNITIESThe findings of this doctoral thesis are of importance for the design and focus of future research.
1. Expansion and continuation of the Belgian prenatal database should be pursued to enable accurate phenotyping of children prenatally detected with a non-benign CNV. For the purpose of increasing available data, international cooperation is necessary.
2. The introduction of WES and WGS in prenatal diagnosis necessitates similar studies on prenatal WES and WGS data and the creation of an appropriate database.
3. The preferences of parents and other health care professionals with regard to the ethical issues of the Belgian reporting system should be investigated.
Chapter 6

97
General discussion and future perspectives
6 1

98
LABO. EXPELLU PTATATURESTO BEATI-IS DUCID UT ERUNT, QUAMUSDAE PROVIDUS DIT OMNITIBUS.
cover-boek-hand.indd 8cover-boek-hand.indd 8 07/07/2020 07:5807/07/2020 07:58

99
Summary / Samenvatting
7 SUMMARY / SAMENVATTING
7.1 SUMMARYThe genetic landscape is constantly changing, as are the possibilities for analyzing samples obtained by an invasive prenatal procedure. For over a decade, conventional karyotyping has been the gold standard for the analysis of amniotic fluid or chorionic villi. Chromosomal microarray (CMA) is a whole genome test with a lower failure rate and a higher resolution than conventional karyotyping. Accordingly, CMA allows the detection of smaller aberrations; however, this means it can also detect variants with a variable clinical phenotype (susceptibility variants) or variants of unknown significance, as well as pathogenic variants unrelated to the indication of the invasive procedure. Because of the lack of international guidelines on how to classify, interpret and report these findings to future parents, Belgian genetic centers developed their own uniform guidelines when starting the analysis of all invasively obtained samples for prenatal genetic diagnosis by CMA in 2013.
As part of this doctoral thesis, a national prenatal database assembling prenatal phenotypic data, genotype and postnatal developmental data of children who underwent invasive prenatal sampling during pregnancy was created. The overall aim was to determine/refine genotype-phenotype correlations between prenatally detected pathogenic, susceptibility and unclassified variants. This thesis describes an overall added diagnostic value of using CMA compared to karyotyping of 2.7% in cases with ultrasound anomalies and 1.5% in cases without ultrasound anomalies. Recurrent variants in the Belgian population were determined. Associations between the prenatal phenotype and genotype were investigated. A postnatal follow-up study demonstrated that, for the categories communication skills and personal-social skills, children with a reported susceptibility variant performed worse than children with an unreported susceptibility variant and children of the control population. The development of children with an unreported variant did not differ from those of the control population. Subsequently, this thesis outlines Belgium’s cultural background and the ethical considerations of Belgian healthcare workers for choosing a well-defined reporting system that is not tailored to the individual preferences of parents involved. Finally, this doctoral thesis introduces the application of homozygosity mapping using SNP array in prenatal invasive testing.
7 1

100
In conclusion, this thesis contributes to the knowledge of genotype-phenotype correlations between prenatally detected pathogenic, susceptibility and unclassified variants, by investigating pre- and postnatal phenotype of children prenatally diagnosed with a non-benign variant in a national cohort. Finally, it underscores the new challenges and opportunities in prenatal invasive diagnosis using chromosomal microarray analysis.
7.2 SAMENVATTINGHet genetisch landschap evolueert snel, en zo ook de testen gebruikt voor het analyseren van invasief bekomen prenatale stalen. Gedurende decennia was conventionele karyotypering de standaard genetische test voor het onderzoeken van vruchtwater of placenta-vlokken. Chromosomale microarray analyse (CMA) is een nieuwere genoomwijde test met een veel lager falingsrisico en een hogere resolutie in vergelijking met karyotypering, wat wil zeggen dat CMA kleinere afwijkingen kan detecteren. Hoewel een grotere detectieratio een voordeel is, kunnen tijdens de analyse ook varianten worden ontdekt die een variabel fenotype veroorzaken (susceptibiliteitsvarianten), varianten waarvan de klinische impact niet gekend is of toevallige vondsten die niet verwant zijn aan de reden van de invasieve test. Internationaal bestaat geen consensus over de classificatie, interpretatie en vooral de rapportering van deze bevindingen aan toekomstige ouders. Om die reden werd in België in 2013 een uniek project opgezet, waarbij alle genetische centra beslisten om invasieve prenatale stalen bekomen voor genetische analyse exclusief met CMA te analyseren en voor de classificatie, interpretatie en rapportering uniforme richtlijnen uit te schrijven en toe te passen.
Als onderdeel van deze doctoraatsthesis werd een nationale databank gecreëerd, waarin prenatale fenotypische informatie, genetische informatie en postnatale ontwikkelings-informatie van kinderen die een invasief onderzoek ondergingen tijdens de zwangerschap werd verzameld, met als doel genotype/fenotype relaties te onderzoeken van prenataal gediagnostiseerde pathogene, susceptibiliteits- en ongeklasseerde varianten. De toegevoegde waarde van het gebruik van CMA in plaats van karyotypering bedroeg respectievelijk 2,7% en 1,5% in de aan- en afwezigheid van echografische afwijkingen. Deze thesis beschrijft recurrente varianten in de Belgische populatie. Associaties tussen prenataal fenotype en genotype werden onderzocht. Er werd aangetoond dat kinderen met een gerapporteerde susceptibiliteitsvariant minder goed scoorden betreft communicatieve en sociale vaardigheden dan kinderen uit de controlepopulatie of kinderen met een susceptibiliteitsvariant die niet werd gerapporteerd. De ontwikkeling van kinderen gediagnostiseerd met een variant die niet werd gerapporteerd was vergelijkbaar met deze van kinderen uit de controlepopulatie. Aansluitend wordt in deze thesis ingegaan op de Belgische culturele achtergrond en de ethische overwegingen die geleid hebben tot het invoeren van het huidige rapporteringssysteem.

101
Tot slot introduceert deze thesis ook de toepassing van homozygositeitsmapping (door middel van SNP array technologie) in prenatale invasieve diagnostiek.
Concluderend draagt deze doctoraatsthesis bij tot de kennis over genotype en fenotype van prenataal vastgestelde pathogene, susceptibiliteits- en ongeklasseerde varianten, aan de hand van de studie van het pre- en postnatale fenotype van kinderen die prenataal werden gediagnosticeerd met een niet-benigne variant. Tot slot behandelt dit werk de uitdagingen en opportuniteiten die de implementatie van microarray analyse in prenatale invasieve diagnostiek met zich meebrengt.
7 1

102
LABO. EXPELLU PTATATURESTO BEATI-IS DUCID UT ERUNT, QUAMUSDAE PROVIDUS DIT OMNITIBUS.
cover-boek-hand.indd 6cover-boek-hand.indd 6 07/07/2020 07:5807/07/2020 07:58

103
8 LIST OF ABBREVIATIONS%Het Percentage of Heterozygosity%Hom Percentage of HomozygosityACMG American College of Medical Genetics and GenomicsACOG American College of Obstetricians and GynecologistsAFP Alpha FetoproteinAMA Advanced Maternal AgeArray CGH Array Comparative Genomic HybridizationASD Autism Spectrum DisorderASQ-3 Ages and Stages Questionnaire: a Parent-Completed Child Monitoring System, Third editionASQ-SE2 Ages and Stages Questionnaire: Social-Emotional, Second EditionB3GAT3 Beta-1,3-glucuronyltransferase 3BAF B-allele frequencyBEMAPRE BElgian MicroArray PREnatalBeSHG Belgian Society for Human Geneticsbp BasepaircfDNA cell free DNAcffDNA cell free fetal DNAChr ChromosomeCI Confidence IntervalCMA Chromosomal Microarray AnalysisCMV CytomegalovirusCNV Copy Number VariantCVS Chorionic Villi SamplingDD Developmental Delaydel/dup Deletion/DuplicationDNA Deoxyribonucleic acid
8 1

104
Fam gen disorder Known genetic disorder in the familyFISH Fluorescence In Situ HybridizationFTS First trimester screeningG3P2 Gravida 3 Para 2G-bands Giemsa-bandingGDPR General Data Protection RulesGRCh37 Genome Reference Consortium Human Build 37hCG Human chorionic gonadotropinhg19 Human genome 19HPO Human Phenotype OntologyID Intellectual DisabilityId Identification numberIQ Intelligence QuotientISH In Situ HybridizationIUGR Intra-uterine growth restrictionK+ Kaliumkb KilobasesLCR Low-copy repeatsLRR Log R ratioMb MegabasesMLPA Multiplex Ligation-dependent Probe AmplificationMODY Maturity Onset Diabetes of the Youngn NumberNa+ NatriumNGS Next-Generation SequencingNIPT Non-Invasive Prenatal TestNT Nuchal translucencyOMIM Online Mendelian Inheritance in Man PAPP-A Pregnancy-associated plasma protein APath PathogenicPCR Polymerase Chain ReactionPGD Preimplantation genetic diagnosisPPV Positive predictive valueQF-PCR Quantitative Fluorescent Polymerase Chain ReactionSD Standard deviationSNP Single Nucleotide Polymorphism
Chapter 8

105
SNR Susceptibility not reportedSR Susceptibility reportedSZ SchizophreniaTAT Turn Around Time TOP Termination of pregnancyUSA Ultrasound anomalyVOUS Variant of Unknown SignificanceVs VersusWES Whole Exome SequencingWGS Whole Genome Sequencing
8
List of abbreviations
1

106
LABO. EXPELLU PTATATURESTO BEATI-
IS DUCID UT ERUNT, QUAMUSDAE
PROVIDUS DIT OMNITIBUS.
cover-boek-hand.indd 6
cover-boek-hand.indd 6
07/07/2020 07:58
07/07/2020 07:58

107
9 REFERENCES
1. Prochownick. Beitrage zur Lehre vom Fruchtwasser und seiner Entstehung. Arch Gynakol. 1897;11:304.2. Levy B, Stosic M. Traditional Prenatal Diagnosis: Past to Present. Methods Mol Biol. 2019;1885:3-22.3. Hinselmann M, Hindemann P, Kubli F. Placental localisation by B-scan ultrasound in transabdominal amniocentesis. Ann Ostet Ginecol Med Perinat. 1970;92(8):503-6.4. Serr DM, Sachs L, Danon M. The diagnosis of sex before birth using cells from the amniotic fluid (a preliminary report). Bull Res Counc Isr. 1955;5B(2):137-8.5. Randomised trial to assess safety and fetal outcome of early and midtrimester amniocentesis. The Canadian Early and Mid-trimester Amniocentesis Trial (CEMAT) Group. Lancet. 1998;351(9098):242-7.6. Sundberg K, Bang J, Smidt-Jensen S, et al. Randomised study of risk of fetal loss related to early amniocentesis versus chorionic villus sampling. Lancet. 1997;350(9079):697-703.7. Mohr J. Foetal genetic diagnosis: development of techniques for early sampling of foetal cells. Acta Pathol Microbiol Scand. 1968;73(1):73-7.8. Brambati B. Prenatal diagnosis of genetic diseases. Eur J Obstet Gynecol Reprod Biol. 2000;90(2):165-9.9. Brambati B, Simoni G. Diagnosis of fetal trisomy 21 in first trimester. Lancet. 1983;1(8324):586.10. Smidt-Jensen S, Hahnemann N. Transabdominal fine needle biopsy from chorionic villi in the first trimester. Prenat Diagn. 1984;4(3):163-9.11. Goossens M, Dumez Y, Kaplan L, et al. Prenatal diagnosis of sickle-cell anemia in the first trimester of pregnancy. N Engl J Med. 1983;309(14):831-3.12. Old JM, Ward RH, Petrou M, et al. First-trimester fetal diagnosis for haemoglobinopathies: three cases. Lancet. 1982;2(8313):1413-6.13. Nussbaum RL, McInnes RR, Willard HF, et al. Thompson & Thompson genetics in medicine. 7th ed. / Robert 1. Nussbaum, Roderick R. McInnes, Huntington F. Willard ; with clinical case studies updated and new cases prepared by Ada Hamosh. ed. Philadelphia: Saunders/Elsevier; 2007.
9 1

108
14. Howe B, Umrigar A, Tsien F. Chromosome preparation from cultured cells. J Vis Exp. 2014(83):e50203.15. N. DL. Comparative karyological study of species Muscari Mill. and Bellevalia Lapeyr. Bulletin of the Tiflis Botanical Garden. 1922;2(1):1-32.16. Lichtenbelt KD, Knoers NV, Schuring-Blom GH. From karyotyping to array-CGH in prenatal diagnosis. Cytogenet Genome Res. 2011;135(3-4):241-50.17. Attiyeh EF, Diskin SJ, Attiyeh MA, et al. Genomic copy number determination in cancer cells from single nucleotide polymorphism microarrays based on quantitative genotyping corrected for aneuploidy. Genome Res. 2009;19(2):276-83.18. Faas BH, van der Burgt I, Kooper AJ, et al. Identification of clinically significant, submicroscopic chromosome alterations and UPD in fetuses with ultrasound anomalies using genome-wide 250k SNP array analysis. J Med Genet. 2010;47(9):586-94.19. Wapner RJ, Martin CL, Levy B, et al. Chromosomal microarray versus karyotyping for prenatal diagnosis. N Engl J Med. 2012;367(23):2175-84.20. de Wit MC, Srebniak MI, Govaerts LC, et al. Additional value of prenatal genomic array testing in fetuses with isolated structural ultrasound abnormalities and a normal karyotype: a systematic review of the literature. Ultrasound Obstet Gynecol. 2014;43(2):139-46.21. Hillman SC, McMullan DJ, Hall G, et al. Use of prenatal chromosomal microarray: prospective cohort study and systematic review and meta-analysis. Ultrasound Obstet Gynecol. 2013;41(6):610-20.22. Hillman SC, Pretlove S, Coomarasamy A, et al. Additional information from array comparative genomic hybridization technology over conventional karyotyping in prenatal diagnosis: a systematic review and meta-analysis. Ultrasound Obstet Gynecol. 2011;37(1):6-14.23. Oneda B, Rauch A. Microarrays in prenatal diagnosis. Best Pract Res Clin Obstet Gynaecol. 2017;42:53-63.24. Brady PD, Delle Chiaie B, Christenhusz G, et al. A prospective study of the clinical utility of prenatal chromosomal microarray analysis in fetuses with ultrasound abnormalities and an exploration of a framework for reporting unclassified variants and risk factors. Genet Med. 2014;16(6):469-76.25. Vanakker O, Vilain C, Janssens K, et al. Implementation of genomic arrays in prenatal diagnosis: the Belgian approach to meet the challenges. Eur J Med Genet. 2014;57(4):151-6.26. Buchanan JA, Chitayat D, Kolomietz E, et al. Prenatal genomic microarray and sequencing in Canadian medical practice: towards consensus. Journal of medical |genetics. 2015;52(9):585-6.27. Committee on G, the Society for Maternal-Fetal M. Committee Opinion No.682: Microarrays and Next-Generation Sequencing Technology: The Use of Advanced Genetic Diagnostic Tools in Obstetrics and Gynecology. Obstet Gynecol. 2016;128(6):e262-e8.28. Society for Maternal-Fetal Medicine . Electronic address pso, Dugoff L, Norton ME, et al. The use of chromosomal microarray for prenatal diagnosis. Am J Obstet Gynecol. 2016;215(4):B2-9.29. Riggs ER, Andersen EF, Cherry AM, et al. Technical standards for the
Chapter 9

109
interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet Med. 2019.30. Bringman JJ. Prenatal chromosomal microarray for the Catholic physician. Linacre Q. 2014;81(2):162-71.31. de Jong A, Dondorp WJ, Macville MV, et al. Microarrays as a diagnostic tool in prenatal screening strategies: ethical reflection. Hum Genet. 2014;133(2):163-72.32. Dondorp WJ, Page-Christiaens GC, de Wert GM. Genomic futures of prenatal screening: ethical reflection. Clin Genet. 2016;89(5):531-8.33. Hardisty EE, Vora NL. Advances in genetic prenatal diagnosis and screening. Curr Opin Pediatr. 2014;26(6):634-8.34. Srebniak MI, Van Opstal D, Joosten M, et al. Whole-genome array as a first-line cytogenetic test in prenatal diagnosis. Ultrasound Obstet Gynecol. 2015;45(4):363-72.35. van der Steen SL, Diderich KE, Riedijk SR, et al. Pregnant couples at increased risk for common aneuploidies choose maximal information from invasive genetic testing. Clin Genet. 2015;88(1):25-31.36. Walser SA, Kellom KS, Palmer SC, et al. Comparing genetic counselor’s and patient’s perceptions of needs in prenatal chromosomal microarray testing. Prenat Diagn. 2015;35(9):870-8.37. Brady PD, Vermeesch JR. Genomic microarrays: a technology overview. Prenat Diagn. 2012;32(4):336-43.38. Srebniak MI, Diderich KE, Joosten M, et al. Prenatal SNP array testing in 1000 fetuses with ultrasound anomalies: causative, unexpected and susceptibility CNVs. Eur J Hum Genet. 2016;24(5):645-51.39. Weiner C. Anticipate and communicate: Ethical management of incidental and secondary findings in the clinical, research, and direct-to-consumer contexts (December 2013 report of the Presidential Commission for the Study of Bioethical Issues). Am J Epidemiol. 2014;180(6):562-4.40. Coe BP, Witherspoon K, Rosenfeld JA, et al. Refining analyses of copy number variation identifies specific genes associated with developmental delay. Nat Genet. 2014;46(10):1063-71.41. Rosenfeld JA, Coe BP, Eichler EE, et al. Estimates of penetrance for recurrent pathogenic copy-number variations. Genet Med. 2013;15(6):478-81.42. Cooper GM, Coe BP, Girirajan S, et al. A copy number variation morbidity map of developmental delay. Nat Genet. 2011;43(9):838-46.43. Girirajan S, Rosenfeld JA, Coe BP, et al. Phenotypic heterogeneity of genomic disorders and rare copy-number variants. N Engl J Med. 2012;367(14):1321-31.44. Nenadic O, Greenacre M. Correspondence analysis in R, with two- and three-dimensional graphics: The ca package. J Stat Softw. 2007;20(3).45. Team RC. R: A language and environment for statistical computing. . R Foundation for Statistical Computing, Vienna, Austria. 2014;URL http://www.R-project.org/. 46. Burnside RD, Pasion R, Mikhail FM, et al. Microdeletion/microduplication of proximal 15q11.2 between BP1 and BP2: a susceptibility region for neurological
9
References
1

110
dysfunction including developmental and language delay. Hum Genet. 2011; 130(4):517-28.47. Srebniak MI, van Zutven LJ, Petit F, et al. Interstitial 6q21q23 duplication - variant of variable phenotype and incomplete penetrance or benign duplication? Mol Cytogenet. 2016;9:43.48. Grati FR, Molina Gomes D, Ferreira JC, et al. Prevalence of recurrent pathogenic microdeletions and microduplications in over 9500 pregnancies. Prenat Diagn. 2015;35(8):801-9.49. Botto LD, May K, Fernhoff PM, et al. A population-based study of the 22q11.2 deletion: phenotype, incidence, and contribution to major birth defects in the population. Pediatrics. 2003;112(1 Pt 1):101-7.50. Maisenbacher MK, Merrion K, Pettersen B, et al. Incidence of the 22q11.2 deletion in a large cohort of miscarriage samples. Mol Cytogenet. 2017;10:6.51. Wentzel C, Fernstrom M, Ohrner Y, et al. Clinical variability of the 22q11.2 duplication syndrome. Eur J Med Genet. 2008;51(6):501-10.52. Rosa RF, Zen PR, Ricachinevsky CP, et al. 22q11.2 duplication and congenital heart defects. Arq Bras Cardiol. 2009;93(4):e67-9, e55-7.53. Torres F, Barbosa M, Maciel P. Recurrent copy number variations as risk factors for neurodevelopmental disorders: critical overview and analysis of clinical implications. J Med Genet. 2016;53(2):73-90.54. Picinelli C, Lintas C, Piras IS, et al. Recurrent 15q11.2 BP1-BP2 microdeletions and microduplications in the etiology of neurodevelopmental disorders. Am J Med Genet B Neuropsychiatr Genet. 2016;171(8):1088-98.55. Chaste P, Sanders SJ, Mohan KN, et al. Modest impact on risk for autism spectrum disorder of rare copy number variants at 15q11.2, specifically breakpoints 1 to 2. Autism Res. 2014;7(3):355-62.56. Bernhardt BA, Soucier D, Hanson K, et al. Women’s experiences receiving abnormal prenatal chromosomal microarray testing results. Genet Med. 2013;15(2):139-45.57. van der Steen SL, Riedijk SR, Verhagen-Visser J, et al. The Psychological Impact of Prenatal Diagnosis and Disclosure of Susceptibility Loci: First Impressions of Parents’ Experiences. J Genet Couns. 2016;25(6):1227-34.58. Srebniak MI, Joosten M, Knapen M, et al. Frequency of submicroscopic chromosome aberrations in pregnancies without increased risk for structural chromosome aberrations: a systematic review of literature and meta-analysis. Ultrasound Obstet Gynecol. 2017.59. Girirajan S, Johnson RL, Tassone F, et al. Global increases in both common and rare copy number load associated with autism. Hum Mol Genet. 2013;22(14):2870-80.60. Johnson K, Kelley J, Saxton V, et al. Declining invasive prenatal diagnostic procedures: A comparison of tertiary hospital and national data from 2012 to 2015. Aust N Z J Obstet Gynaecol. 2017;57(2):152-6. 61. Srebniak MI, Knapen M, Polak M, et al. The influence of SNP-based chromosomal microarray and NIPT on the diagnostic yield in 10,000 fetuses with and without fetal ultrasound anomalies. Hum Mutat. 2017;38(7):880-8.
Chapter 9

111
62. Konialis C, Pangalos C. Dilemmas in Prenatal Chromosomal Diagnosis Revealed Through a Single Center’s 30 Years’ Experience and 90,000 Cases. Fetal Diagn Ther. 2015;38(3):218-32.63. Brabbing-Goldstein D, Reches A, Svirsky R, et al. Dilemmas in genetic counseling for low-penetrance neuro-susceptibility loci detected on prenatal chromosomal microarray analysis. Am J Obstet Gynecol. 2018;218(2):247 e1- e12.64. Malan V, Lapierre JM, Egloff M, et al. A French Approach to Test Fetuses with Ultrasound Abnormalities Using a Customized Microarray as First-Tier Genetic Test. Cytogenet Genome Res. 2015;147(2-3):103-10.65. Muys J, Blaumeiser B, Jacquemyn Y, et al. The BElgian PREnatal MicroArray (BEMAPRE) database: A systematic nationwide repository of fetal genomic aberrations. Prenat Diagn. 2018.66. Van Opstal D, de Vries F, Govaerts L, et al. Benefits and burdens of using a SNP array in pregnancies at increased risk for the common aneuploidies. Hum Mutat. 2015;36(3):319-26.67. Fiorentino F, Napoletano S, Caiazzo F, et al. Chromosomal microarray analysis as a first-line test in pregnancies with a priori low risk for the detection of submicroscopic chromosomal abnormalities. Eur J Hum Genet. 2013;21(7):725-30.68. Oneda B, Baldinger R, Reissmann R, et al. High-resolution chromosomal microarrays in prenatal diagnosis significantly increase diagnostic power. Prenat Diagn. 2014;34(6):525-33.69. Squires J. ASQ-3 user’s guide. Baltimore: Paul H. Brookes Pub.; 2009. xxiv, 227 p. p.70. Squires J, Bricker DD, Twombly E. ASQ-SE-2 user’s guide. Second edition. ed. Baltimore: Paul H. Brookes Publishing, Co.; 2015. xxiv, 291 pages p.71. Team RC. R: A Language and Environment for Statistical Computing. R Foun- dation for Statistical Computing, Vienna. 2018 [Available from: https://www.R-project.org.72. Soemedi R, Topf A, Wilson IJ, et al. Phenotype-specific effect of chromosome 1q21.1 rearrangements and GJA5 duplications in 2436 congenital heart disease patients and 6760 controls. Hum Mol Genet. 2012;21(7):1513-20.73. Bernier R, Steinman KJ, Reilly B, et al. Clinical phenotype of the recurrent 1q21.1 copy-number variant. Genet Med. 2016;18(4):341-9.74. Brunetti-Pierri N, Berg JS, Scaglia F, et al. Recurrent reciprocal 1q21.1 deletions and duplications associated with microcephaly or macrocephaly and developmental and behavioral abnormalities. Nat Genet. 2008;40(12):1466-71.75. Veenma DC, Eussen HJ, Govaerts LC, et al. Phenotype-genotype correlation in a familial IGF1R microdeletion case. J Med Genet. 2010;47(7):492-8.76. Edelmann L, Pandita RK, Spiteri E, et al. A common molecular basis for rearrangement disorders on chromosome 22q11. Hum Mol Genet. 1999;8(7):1157-67.77. Yobb TM, Somerville MJ, Willatt L, et al. Microduplication and triplication of 22q11.2: a highly variable syndrome. Am J Hum Genet. 2005;76(5):865-76.78. Hannes FD, Sharp AJ, Mefford HC, et al. Recurrent reciprocal deletions and duplications of 16p13.11: the deletion is a risk factor for MR/MCA while the duplication may be a rare benign variant. J Med Genet. 2009;46(4):223-32.
9
References
1

112
79. Ingason A, Rujescu D, Cichon S, et al. Copy number variations of chromosome 16p13.1 region associated with schizophrenia. Mol Psychiatry. 2011;16(1):17-25.80. de Kovel CG, Trucks H, Helbig I, et al. Recurrent microdeletions at 15q11.2 and 16p13.11 predispose to idiopathic generalized epilepsies. Brain. 2010;133(Pt 1):23-32.81. Jahn JA, von Spiczak S, Muhle H, et al. Iterative phenotyping of 15q11.2, 15q13.3 and 16p13.11 microdeletion carriers in pediatric epilepsies. Epilepsy Res. 2014;108(1):109-16.82. Coppinger J, McDonald-McGinn D, Zackai E, et al. Identification of familial and de novo microduplications of 22q11.21-q11.23 distal to the 22q11.21 microdeletion syndrome region. Hum Mol Genet. 2009;18(8):1377-83.83. Ou Z, Berg JS, Yonath H, et al. Microduplications of 22q11.2 are frequently inherited and are associated with variable phenotypes. Genet Med. 2008;10(4):267-77.84. Wincent J, Bruno DL, van Bon BW, et al. Sixteen New Cases Contributing to the Characterization of Patients with Distal 22q11.2 Microduplications. Mol Syndromol. 2010;1(5):246-54.85. Leung GKC, Mak CCY, Fung JLF, et al. Identifying the genetic causes for prenatally diagnosed structural congenital anomalies (SCAs) by whole-exome sequencing (WES). BMC Med Genomics. 2018;11(1):93.86. Weston D, Parsons V, Ntani G, et al. Mixed contact methods to improve response to a postal questionnaire. Occup Med (Lond). 2017;67(4):305-7.87. Edwards P, Roberts I, Clarke M, et al. Methods to increase response rates to postal questionnaires. Cochrane Database Syst Rev. 2007(2):MR000008.88. Muys J, Blaumeiser B, Janssens K, et al. Chromosomal microarray analysis in prenatal diagnosis: ethical considerations of the Belgian approach. J Med Ethics. 2019.89. Miller DT, Adam MP, Aradhya S, et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet. 2010;86(5):749-64.90. Borrell A. A new comprehensive paradigm for Prenatal Diagnosis. Ultrasound Obstet Gynecol. 2018.91. Prenatal Diagnosis Through Chromosomal Microarray Analysis (CMA). SBU Systematic Review Summaries. Stockholm2016.92. Rigter T, Henneman L, Kristoffersson U, et al. Reflecting on earlier experiences with unsolicited findings: points to consider for next-generation sequencing and informed consent in diagnostics. Hum Mutat. 2013;34(10):1322-8.93. Gudykunst WB. Cultural variability in communication. Thousand Oaks, Calif.,1997. S. 325-454. p.94. Hofstede G. Cultures and organizations : software of the mind. London: McGraw-Hill; 1991. xii, 279 s. p.95. Meeuwesen L, van den Brink-Muinen A, Hofstede G. Can dimensions of national culture predict cross-national differences in medical communication? Patient Educ Couns. 2009;75(1):58-66.96. Schouten BC, Meeuwesen L. Cultural differences in medical communication: a review of the literature. Patient Educ Couns. 2006;64(1-3):21-34.97. Christiaens T, De Backer D, Burgers J, et al. Guidelines, evidence, and cultural
Chapter 9

113
factors. Scand J Prim Health Care. 2004;22(3):141-5.98. Gysels M, Evans N, Menaca A, et al. Culture and end of life care: a scoping exercise in seven European countries. PLoS One. 2012;7(4):e34188.99. Hofstede G, Hofstede GJ. Cultures and organizations : software of the mind. Revised and expanded 2. edition. ed. London: McGraw-Hill; 2005. xii, 434 sider p.100. Hofstede GH. Masculinity and femininity : the taboo dimension of national cultures.101. Hofstede GH. Measuring hierarchical power distance in thirty-seven countries. [S.l.]: European Institute for Advanced Studies in Management; 1976.102. Hofstede GH. Culture’s consequences : comparing values, behaviors, institutions, and organizations across nations. 2nd ed. ed. Thousand Oaks, Calif. ; London: SAGE; 2001.103. Bensing JM, Roter DL, Hulsman RL. Communication patterns of primary care physicians in the United States and the Netherlands. J Gen Intern Med. 2003;18(5):335-42.104. American College of O, Gynecologists. ACOG Committee opinion no. 551: coping with the stress of medical professional liability litigation. Obstet Gynecol. 2013;121(1):220-2.105. Walser SA, Werner-Lin A, Russell A, et al. “Something Extra on Chromosome 5”: Parents’ Understanding of Positive Prenatal Chromosomal Microarray Analysis (CMA) Results. J Genet Couns. 2016;25(5):1116-26.106. Chervenak FA, McCullough LB. Ethics of research in perinatal medicine. Semin Perinatol. 2009;33(6):391-6.107. Chervenak FA, McCullough LB. Ethical issues in perinatal genetics. Semin Fetal Neonatal Med. 2011;16(2):70-3.108. McPheeters ML, Weitlauf A, Vehorn A, et al. Screening for Autism Spectrum Disorder in Young Children: A Systematic Evidence Review for the US Preventive Services Task Force. U.S. Preventive Services Task Force Evidence Syntheses, formerly Systematic Evidence Reviews. Rockville (MD)2016.109. Reichow B, Hume K, Barton EE, et al. Early intensive behavioral intervention (EIBI) for young children with autism spectrum disorders (ASD). Cochrane Database Syst Rev. 2018;5:CD009260.110. Frati P, Fineschi V, Di Sanzo M, et al. Preimplantation and prenatal diagnosis, wrongful birth and wrongful life: a global view of bioethical and legal controversies. Hum Reprod Update. 2017;23(3):338-57.111. Thompson JM. The changing patient-physician relationship. AMA J Ethics. 2015;17(5):473-6.112. Grunloh C, Myreteg G, Cajander A, et al. “Why Do They Need to Check Me?” Patient Participation Through eHealth and the Doctor-Patient Relationship: Qualitative Study. J Med Internet Res. 2018;20(1):e11.113. Nelson Goff BS, Springer N, Foote LC, et al. Receiving the initial Down syndrome diagnosis: a comparison of prenatal and postnatal parent group experiences. Intellect Dev Disabil. 2013;51(6):446-57.114. Lander ES, Botstein D. Homozygosity mapping: a way to map human recessive traits with the DNA of inbred children. Science. 1987;236(4808):1567-70.115. Sund KL, Zimmerman SL, Thomas C, et al. Regions of homozygosity identified
9
References
1

114
by SNP microarray analysis aid in the diagnosis of autosomal recessive disease and incidentally detect parental blood relationships. Genet Med. 2013;15(1):70-8.116. Capo-Chichi JM, Boissel S, Brustein E, et al. Disruption of CLPB is associated with congenital microcephaly, severe encephalopathy and 3-methylglutaconic aciduria. J Med Genet. 2015;52(5):303-11.117. Fiskerstrand T, Houge G, Sund S, et al. Identification of a gene for renal- hepatic-pancreatic dysplasia by microarray-based homozygosity mapping. J Mol Diagn. 2010;12(1):125-31.118. Marshall CR, Farrell SA, Cushing D, et al. Whole-exome analysis of foetal autopsy tissue reveals a frameshift mutation in OBSL1, consistent with a diagnosis of 3-M Syndrome. BMC Genomics. 2015;16 Suppl 1:S12.119. Xin B, Puffenberger E, Tumbush J, et al. Homozygosity for a novel splice site mutation in the cardiac myosin-binding protein C gene causes severe neonatal hypertrophic cardiomyopathy. Am J Med Genet A. 2007;143A(22):2662-7.120. Abumansour IS, Al Sulmi E, Chodirker BN, et al. Prenatal Diagnosis of Walker-Warburg Syndrome Using Single Nucleotide Polymorphism Array: A Clinical Experience from Three Related Palestinian Families with Congenital Hydrocephalus. AJP Rep. 2015;5(2):e116-20.121. Krakow D, Robertson SP, King LM, et al. Mutations in the gene encoding filamin B disrupt vertebral segmentation, joint formation and skeletogenesis. Nat Genet. 2004;36(4):405-10.122. Baasanjav S, Al-Gazali L, Hashiguchi T, et al. Faulty initiation of proteoglycan synthesis causes cardiac and joint defects. Am J Hum Genet. 2011;89(1):15-27.123. Hastbacka J, de la Chapelle A, Mahtani MM, et al. The diastrophic dysplasia gene encodes a novel sulfate transporter: positional cloning by fine-structure linkage disequilibrium mapping. Cell. 1994;78(6):1073-87.124. Faden M, Al-Zahrani F, Arafah D, et al. Mutation of CANT1 causes Desbuquois dysplasia. Am J Med Genet A. 2010;152A(5):1157-60.125. Vissers LE, Lausch E, Unger S, et al. Chondrodysplasia and abnormal joint development associated with mutations in IMPAD1, encoding the Golgi-resident nucleotide phosphatase, gPAPP. Am J Hum Genet. 2011;88(5):608-15.126. Rajab A, Kunze J, Mundlos S. Spondyloepiphyseal dysplasia Omani type: a new recessive type of SED with progressive spinal involvement. Am J Med Genet A. 2004;126A(4):413-9.127. Vandeweyer G, Reyniers E, Wuyts W, et al. CNV-WebStore: online CNV analysis, storage and interpretation. BMC Bioinformatics. 2011;12:4.128. Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536(7616):285-91.129. Tuysuz B, Mizumoto S, Sugahara K, et al. Omani-type spondyloepiphyseal dysplasia with cardiac involvement caused by a missense mutation in CHST3. Clin Genet. 2009;75(4):375-83.130. Unger S, Lausch E, Rossi A, et al. Phenotypic features of carbohydrate sulfotransferase 3 (CHST3) deficiency in 24 patients: congenital dislocations and vertebral changes as principal diagnostic features. Am J Med Genet A. 2010;152A(10):2543-9.
Chapter 9

115
9
References
131. Liao C, Fu F, Li R, et al. Implementation of high-resolution SNP arrays in the investigation of fetuses with ultrasound malformations: 5 years of clinical experience. Clin Genet. 2014;86(3):264-9.132. Srebniak MI, Boter M, Oudesluijs GO, et al. Genomic SNP array as a gold standard for prenatal diagnosis of foetal ultrasound abnormalities. Mol Cytogenet. 2012;5(1):14.133. Alkuraya FS. The application of next-generation sequencing in the autozygosity mapping of human recessive diseases. Hum Genet. 2013;132(11):1197-211.134. Eraslan G, Avsec Z, Gagneur J, et al. Deep learning: new computational modelling techniques for genomics. Nat Rev Genet. 2019;20(7):389-403.135. Libbrecht MW, Noble WS. Machine learning applications in genetics and genomics. Nat Rev Genet. 2015;16(6):321-32.136. Gurovich Y, Hanani Y, Bar O, et al. Identifying facial phenotypes of genetic disorders using deep learning. Nat Med. 2019;25(1):60-4.137. Dias R, Torkamani A. Artificial intelligence in clinical and genomic diagnostics. Genome Med. 2019;11(1):70.138. Best S, Wou K, Vora N, et al. Promises, pitfalls and practicalities of prenatal whole exome sequencing. Prenat Diagn. 2018;38(1):10-9.139. Directors ABo. Points to consider for informed consent for genome/exome sequencing. Genet Med. 2013;15(9):748-9.140. van der Meij KRM, Sistermans EA, Macville MVE, et al. TRIDENT-2: National Implementation of Genome-Wide Non-Invasive Prenatal Testing as a First-Tier Screening Test in the Netherlands. Am J Hum Genet. 2019.141. Vossaert L, Wang Q, Salman R, et al. Validation Studies for Single Circulating Trophoblast Genetic Testing as a Form of Noninvasive Prenatal Diagnosis. Am J Hum Genet. 2019;105(6):1262-73.142. Salomon LJ, Sotiriadis A, Wulff CB, et al. Risk of miscarriage following amniocentesis or chorionic villus sampling: systematic review of literature and updated meta-analysis. Ultrasound Obstet Gynecol. 2019;54(4):442-51.143. Jani JC, Gil MM, Benachi A, et al. Genome-wide cfDNA testing of maternal blood. Ultrasound Obstet Gynecol. 2020;55(1):13-4.
1

116
LABO. EXPELLU PTATATURESTO BEATI-IS DUCID UT ERUNT, QUAMUSDAE PROVIDUS DIT OMNITIBUS.
LABO. EXPELLU PTATATURESTO BEATI-IS DUCID UT ERUNT, QUAMUSDAE
cover-boek-hand.indd 12cover-boek-hand.indd 12 07/07/2020 07:5807/07/2020 07:58

117
10 ADDENDUM: Questionnaires
10.1 DUTCH QUESTIONNAIRELABO. EXPELLU PTATATURESTO BEATI-IS DUCID UT ERUNT, QUAMUSDAE PROVIDUS DIT OMNITIBUS.
LABO. EXPELLU PTATATURESTO BEATI-IS DUCID UT ERUNT, QUAMUSDAE
cover-boek-hand.indd 12cover-boek-hand.indd 12 07/07/2020 07:5807/07/2020 07:58
10
119
1

118
Chapter 10
119

119
10
Addendum: Questionnaires
119
1

120
120
Chapter 10

121
120
10
Addendum: Questionnaires
1

122
121
Chapter 10

123
121
10
Addendum: Questionnaires
1

124
122
Chapter 10

125
122
10
Addendum: Questionnaires
1

126
123
Chapter 10

127
123
Addendum: Questionnaires
10 1

128
124
Chapter 10

129
124
Addendum: Questionnaires
10 1

130
125
Chapter 10

131
125
Addendum: Questionnaires
10 1

132
126
Chapter 10

133
126
Addendum: Questionnaires
10 1

134
127
Chapter 10

135
127
Addendum: Questionnaires
10 1

136
128
Chapter 10

137
128
Addendum: Questionnaires
10 1

138
129
Chapter 10

139
131
Addendum: Questionnaires
10
10.2 FRENCH QUESTIONNAIRE
1

140
Chapter 10
131

141
Addendum: Questionnaires
10
132
1

142
Chapter 10
132

143
Addendum: Questionnaires
10
133
1

144
Chapter 10
133

145
Addendum: Questionnaires
10
134
1

146
Chapter 10
134

147
Addendum: Questionnaires
10
135
1

148
Chapter 10
135

149
Addendum: Questionnaires
10
136
1

150
Chapter 10
136

151
Addendum: Questionnaires
10
137
1

152
Chapter 10
137

153
Addendum: Questionnaires
10
138
1

154
Chapter 10
138

155
Addendum: Questionnaires
10
139
1

156
Chapter 10
139

157
Addendum: Questionnaires
10
140
1

158
Chapter 10
140

159
Addendum: Questionnaires
10
141
1

160
LABO. EXPELLU PTATATURESTO BEATI-
IS DUCID UT ERUNT, QUAMUSDAE
PROVIDUS DIT OMNITIBUS.
cover-boek-hand.indd 8
cover-boek-hand.indd 8
07/07/2020 07:58
07/07/2020 07:58

161
11 CURRICULUM VITAE
11.1 GENERAL INFORMATIONSurname : MuysGiven name : Joke Anna AlfonsDate of birth : 08 January 1982Place of birth : LierNationality : BelgianE-mail : [email protected] | [email protected]
11.2 Education Secondary school: Mathematics; Science; Latin - Heilig Hart van Maria Instituut, Berlaar, 2000
University Degree: Medical Doctor - University of Antwerp, Antwerp, 2007 (Cum Laude)
Specialisation: Obstetrics and Gynaecology, Recognised by the Belgian Ministery of Health December 2014. Clinical residency training in Ziekenhuis Oost Limburg, Genk, Belgium; Klina Brasschaat, Brasschaat, Belgium; St. Augustinus, GZA Hospitals, Wilrijk, Belgium; MC Erasmus Sophia, Rotterdam, The Netherlands and University Hospital Antwerp, Edegem, Belgium
Prenatal Ultrasound and Genetics Structureel Echoscopisch Onderzoek (SEO) Erasmus MC Rotterdam, the Netherlands, 2014
Interuniversity Permanent Education course in Human Genetics, Belgium, 2016
Interuniversity Education in prenatal ultrasound and fetal medicine, Belgium, 2018
11 1

162
Chapter 11
11.3 Board membership Member of the recognition committee in Obstetrics - Gynaecology (since 2017) Board member of Studiecentrum voor Perinatale Epidemiologie (SPE) (since 2019)
Scientific committee member of Le Centre d’épidémiologie périnatale (since 2019) Coordinating Intership Supervisor Obstetrics – Gynaecology of Antwerp University and University Hospital Antwerp (since 2020)
11.4 Scientific career11.4.1 Thesis Master thesis “Quality of life after successful resuscitation” (Promotor: Prof. Dr. Leo Bossaert)
PhD thesis “New challenges and opportunities in prenatal invasive diagnosis: microarray analysis” (Promotors: Prof. Dr. Yves Jacquemyn, Prof. Dr. Bettina Blaumeiser; Co-promotor: Dr. Katrien Janssens)
11.4.2 Research grants Clinical PhD fellowship research grant from the Research Foundation Flanders - Fonds Wetenschappelijk Onderzoek (FWO) – Belgium (Grant number 1700917N). The FWO supports ground-breaking fundamental and strategic research at universities of the Flemish Community.
11.4.3 Scientific awards Best Early Career Award Finalist. International conference on prenatal diagnosis and therapy, International Society for Prenatal Diagnosis (ISPD), 2018, Antwerp, Belgium
Award Best Oral Presentation in its category: International Society of Ultrasound in Obstetrics and Gynaecology (ISUOG), “The Belgian Microarray Prenatal Consortium.”, 2016, Rome, Italy

163
Curriculum vitae
11.4.4 Co-supervisor master thesis of master in medicine. University of Antwerp Deprez V: Phenotype and pregnancy outcomes at 36 months in Belgian children who received prenatal genetic testing: a comparison between 22q11.2 microduplication and microdeletion cases, and controls. Period: 2015-2018
Geypen A and Verhaelen L: Phenotype at 36 months in Belgian children who received prenatal genetic testing: an evaluation of 15q11.2 deletion and 15q11.2 duplication cases. Period: 2016-2019
de Raedemaeker H and de Winter J: The value of postpartum ultrasound for the diagnosis of retained products of conception. Period: 2016-2019
11.4.5 Co-supervisor master thesis of master in Verpleeg- en vroedkunde. University of Antwerp Coremans S: Postpartum echografie door de vroedvrouw. Visie van de vroed- vrouw, reproduceerbaarheid tussen vroedvrouw, assistent gynaecologie en gynaecoloog, beleving door de kraamvrouw. Period: 2016-2017
Mertens K: CRP-bepaling in het postpartum, ontwikkeling van model ter predictie van postpartum infectie. Period: 2016-2017
Rousseau S and Van den Nieuwenhuyzen E: Genetic counselling in Belgium concerning microarrayanalysis in prenatal testing and NIPT: parental and physician’s preferences. Period: 2017-2018
11.4.6 Publications Muys J, Jacquemyn Y, Blaumeiser B, et al. Prenatally detected copy number variants in a national cohort: a postnatal follow-up study [published online ahead of print, 2020 May 21]. Prenat Diagn. 2020;10.1002/pd.5751. doi:10.1002/pd.5751
Jacquemyn Y, Mannaerts D, Muys J, De Bruyn Ch, Gyselaers W, Van Craenenbroeck E, Van Berendoncks A. Pre-eclampsie als venster op gezondheidspreventie: langetermijneffecten na hypertensieve zwanger- schapscomplicaties. Tijdschr. Voor Geneeskunde, 76, nr. 13, 2020. doi:10.2143/ TVG.76.13.2003091
Muys J, Blaumeiser B, Janssens K, Loobuyck P, Jacquemyn Y. Chromosomal microarray analysis in prenatal diagnosis: ethical considerations of the Belgian approach. J Med Ethics. 2019 Sep 16. pii: medethics-2018-105186. doi: 10.1136/medethics-2018-105186.
11 1

164
Muys J, Blaumeiser B, Jacquemyn Y, Bandelier C, Brison N, Bulk S, Chiarappa P, Courtens W, De Leener A, De Rademaeker M, Désir J, Destrée A, Devriendt K, Dheedene A, Fieuw A, Fransen E, Gatot JS, Holmgren P, Jamar M, Janssens S, Keymolen K, Lederer D, Menten B, Meuwissen M, Parmentier B, Pichon B, Rombout S, Sznajer Y, Van Den Bogaert A, Van Den Bogaert K, Vanakker O, Vermeesch J, Janssens K. The Belgian MicroArray Prenatal (BEMAPRE) database: A systematic nationwide repository of fetal genomic aberrations. Prenat Diagn. 2018 Dec;38(13):1120-1128. doi: 10.1002/pd.5373. Epub 2018 Nov 14.
Muys J, Blaumeiser B, Jacquemyn Y, Devriendt K, Janssens S, Keymolen K, Rom bout S, Gatot JS, Desir J, Sznajer Y, Meuwissen M, Vermeesch J, Menten B, Fieuw A, Parmentier B, Bulk S, Pichon B, Bandelier C, Fransen E, Van Den Bo gaert K, Dheedene A, De Rademaeker M, Destree A, Courtens W, Deleener A, Brison N, Vanakker O, Van Den Bogaert A, Jamar M, Chiarappa P, Lederer D, The Belgian Microarray Prenatal Consortium (BEMAPRE), Janssens K. The BElgian PREnatal MicroArray consortium: towards relating prenatally detected CNVs, prenatal phenotype and postnatal clinical data Special Issue: Abstracts of the ISPD 22nd International Conference on Prenatal Diagnosis and Therapy, Antwerp, Belgium, 8-11 July 2018. Prenat Diagn. 2018 Oct;38 Suppl 1:3-114. doi: 10.1002/pd.5300.
De Winter J, De Raedemaecker H, Muys J, Jacquemyn Y. The value of post- partum ultrasound for the diagnosis of retained products of conception: A systematic review. Facts Views Vis Obgyn. 2017 Dec;9(4):207-216. Muys J, Blaumeiser B, Janssens K, Jacquemyn Y. Wat te doen met onverwachte vondsten bij invasieve diagnostiek? Proceedings van het 21e Doelencongres Infertiliteit, Gynaecologie en Obstetrie april 2017
Muys J, Blaumeiser B, Jacquemyn Y, Janssens K. Prenatal homozygosity mapping detects a novel mutation in CHST3 in a fetus with skeletal dysplasia and joint dislocations. Clin Case Rep. 2017 Mar 1;5(4):440-445. doi: 10.1002/ ccr3.800. eCollection 2017 Apr.
Van Avermaete F, Muys J, Jacquemyn Y. Management of Hermansky-Pudlak syndrome in pregnancy and review of literature. BMJ Case Rep. 2016 Nov 17;2016. pii: bcr2016217719. doi: 10.1136/bcr-2016-217719.
Muys J, Blaumeiser B, Janssens K, Bandelier C, Gatot J, Van Den Bogaert A, Vermeesch J, Rombout S, Menten B, Pichon B, Keymolen K, Van Den Bogaert K, Janssens S, Caberg J, Désir J, Sznajer Y, Destree A, Jacquemyn Y. The Belgian approach to meet the challenge in interpreting prenatal microarray results Ab stracts of the 26th World Congress on Ultrasound in Obstetrics and Gynecology,
Chapter 11

165
Rome, Italy, 24-28 September 2016. Ultrasound Obstet Gynecol. 2016 Sep;48 Suppl 1:11. doi: 10.1002/uog.16055.
Bruwiere E, Van Roosbroeck S, Van Hal G, Muys J, Jacquemyn Y. An exploration of attitudes towards breast cancer screening in orthodox Jewish women in Antwerp-- Belgium. Eur J Gynaecol Oncol. 2016;37(3):384-7.
Sonnemans H, Schmid A, Muys J, Jacquemyn Y. Flemish obstetricians’ personal preference regarding induction of labor and mode of delivery in term births. Clin. Exp. Obstet. Gynecol. - ISSN: 0390-6663 XLIII, n. 6, 2016 doi: 10.12891/ceog3404.2016
Verbruggen M, Muys J, Mannaerts D, Lambert J, Jacquemyn Y. Acne en anticonceptie, wat kunnen wij aanraden aan onze patiënten. 2016 Jan. 72(2):111-116. DOI:10.2143/TVG.72.02.2002044,
Verbruggen M, Mannaerts D, Muys J, Jacquemyn Y. Use of ticagrelor in human pregnancy, the first experience. BMJ Case Rep. 2015 Nov 25;2015. pii: bcr2015212217. doi: 10.1136/bcr-2015-212217.
Jacquemyn Y, Mannaerts D, Muys J. Risicostratificatie in de vroege zwangerschap. Proceedings van het 20e Doelencongres Infertiliteit, Gynaecologie en Obstetrie april 2015: 37-43
Mannaerts D, Muys J, Blaumeiser B, Jacquemyn Y. A rare cause of primary amenorrhea, the XY female with gonadal dysgenesis.BMJ Case Rep. 2015 Feb 9;2015. pii: bcr2014206609. doi: 10.1136/bcr-2014-206609.
Mannaerts D, Muys J, Ramaekers P, Jacquemyn Y. Relapsing fetal bilateral hydrothorax, an isolated expression of a vein of Galen aneurysmal malfor- mation. BMJ Case Rep. 2015 Jan 27;2015. pii: bcr2014208384. doi: 10.1136/ bcr-2014- 208384.
Muys J, Van Doorn HC, Tjalma W. De follow up van Gynaecologische tumoren in Vlaanderen en in Nederland. Reproductieve geneeskunde, Gynaecologie en Obstetrie anno 2011: Proceedings van het 18e Doelencongres Infertiliteit, Gynaecologie en Obstetrie 6,7 en 8 april 2011: 555- 568
Muys J, Gyselaers W, Martens W, Ombelet W. Sectio-audit door middel van de Robsonanalyse, Gunaikeia 2010; 15(2): 52-57
Beersmans S, Muys J, Bossaert L. Are the current tools for assessing the quality of life after cardiac arrest valid?, Abstract in proceedings Resuscitation 2006, 69 (1): 37; Oral presentation by fellow researcher
Curriculum vitae
11 1

166
11.4.7 Presentations
11.4.7.1 Oral presentations Hypo- en hyperthyreoïdie, nut van foetale bewaking en echoscopie, 22e Nederlands Vlaams Congres Infertiliteit-gynaecologie – obstetrie. Invited speaker. April 2019, Rotterdam, The Netherlands
Geneeskundige Dagen Antwerpen: Genetica in de dagelijkse praktijk: nieuwe inzichten. Invited speaker. Prenatale diagnostiek. 14-15 September 2018. Antwerp. Belgium.
The BElgian PREnatal MicroArray consortium: towards relating prenatally detected CNVs, prenatal phenotype and postnatal clinical data. 22nd International Conference on Prenatal Diagnosis and Therapy, 8-11 July 2018, Antwerp, Belgium.
The BElgian PREnatal MicroArray (BEMAPRE) consortium: A systematic nation-wide database of fetal genomic aberrations and postnatal follow up study. 45th Annual Meeting on Fetal and Neonatal Physiological Society. 24-27 June 2018, Maastricht, the Netherlands.
Wat te doen met onverwachte vondsten bij invasieve diagnostiek? Invited speaker, 21e Doelencongres Infertiliteit, Gynaecologie en Obstetrie april 2017, Rotterdam, The Netherlands
The Belgian approach to meet the challenge in interpreting prenatal microarray results. 26th World Congress on Ultrasound in Obstetrics and Gynecology, Rome, Italy, 24-28 September 2016. Award best oral presentation in its category.
The Belgian Microarray Prenatal Consortium. Realization of the Belgian Prenatal Microarray (BEMAPRE) database and update of the Belgian reporting approach. International Society on Prenatal Diagnosis, 2016, Berlin, Germany
The Belgian Prenatal Microarray (BEMAPRE) consortium: sharing prenatal genomic array data in a national database. XXV European Congress on Perinatal Medicine 2016, Maastricht, The Netherlands
Arraydiagnostiek in prenatale diagnose, 20e Nederlands Vlaams Congres Infertiliteit-gynaecologie – obstetrie. Invited speaker. April 2015, Rotterdam, The Netherlands
Chapter 11

167
11.4.7.2 Poster presentations Joke Muys, Y Jacquemyn, B Blaumeiser, BEMAPRE consortium, J Kertjens, K Janssens. Ameliorating genotype–phenotype knowledge of prenatally determined CNVs. The BElgianPREnatalMicroArray (BEMAPRE) consortium. International Society for prenatal diagnosis, San Diego, United States, July 2016
Joke Muys, K. Janssens, B. Blaumeiser, O. Vanakker, C. Vilain, G. Smits, C. Bandelier, S. Bulk, J. Caberg, A. De Leener, M. De Rademaeker, T. de Ravel, J. Desir, M. Crespen, A. Dheedene, S. Gaillez, B. Parmentier, JS. Gatot, S. Janssens, K. Keymolen, B. Menten, B. Pichon, M. Ravoet, N. Revencu, S. Rombout, C. Staessens, A. Van Den Bogaert, F. Kooy, K. Van Den Bogaert, J. Vermeesch, Y. Sznajer, M. Meuwissen, K. Devriendt; Y. Jacquemyn. Belgian Prenatal MicroArray (BEMAPRE) Initiative. International Society for prenatal diagnosis, Washington, United States, July 2015
Joke Muys, K. Janssens, B. Blaumeiser, Y. Jacquemyn. The Belgian MicroArray Prenatal (BEMAPRE) database. European Society on Human Genetics, Glasgow, Scotland, June 2015
Curriculum vitae
11 1

168
LABO. EXPELLU PTATATURESTO BEATI-IS DUCID UT ERUNT, QUAMUSDAE PROVIDUS DIT OMNITIBUS.
cover-boek-hand.indd 8cover-boek-hand.indd 8 07/07/2020 07:5807/07/2020 07:58

169
12 ACKNOWLEDGEMENTS
Gedurende dit onderzoek werd ik geïnspireerd en geholpen door velen. Ik ben bijzonder blij en trots om eindelijk het resultaat met jullie te kunnen delen. Ik kan jullie niet genoeg bedanken, maar ik doe heel graag een poging om mijn gevoelens hier onder woorden te brengen. Ik hoop dat ik jullie allen genoeg recht kan doen.
Eerst en vooral een warm dankjewel aan alle kinderen en hun ouders die hebben deelgenomen aan dit onderzoek. Het invullen van de vragenlijsten kostte heel wat tijd en moeite, en de ouders werden hierbij vaak herinnerd aan een heel moeilijke periode in hun leven. Zonder hun doorzettingsvermogen was dit onderzoek onmogelijk.
To the Belgian Society for Human Genetics Workgroup on Prenatal Testing: thank you so much for spending many hours on my research, for uploading the required genetic data, for forwarding the surveys to your patients, and for the interesting discussions.
Professor Blaumeiser, beste Bettina, u vroeg me tijdens mijn stageperiode op de dienst Genetica of ik interesse had in het starten van een doctoraatsonderzoek, en u reikte me het onderwerp van deze thesis aan. Ik heb enorme bewondering voor uw kennis en toewijding aan dit onderzoeksveld. Ik waardeer heel erg uw steun, advies en inspirerende vastberadenheid. Ik hoop oprecht u trots te hebben gemaakt met het resultaat.
Professor Jacquemyn, beste Yves, al van bij de start van mijn opleiding tot gynaecoloog kon ik rekenen op uw hulp en uw vertrouwen in mij. Toen ik mijn opleiding moest verlengen omwille van redenen buiten mijn macht, zorgde u ervoor dat ik me tijdens deze periode kon verdiepen in mijn passies genetica en prenatale echografie. Dankzij uw onvoorwaardelijke steun ben ik met dit doctoraatsonderzoek kunnen starten. Uw visie en expertise hebben de koers van dit proefschrift bepaald. Bedankt voor alle kansen die u me biedt en al heeft geboden.
Doctor Janssens, beste Katrien, jij was onmiskenbaar een drijvende kracht en tilde dit proefschrift tot een hoger niveau. Bedankt voor je geduld, het delen van je kennis en je uitmuntende begeleiding. Ik hoop dat iedereen een co-promotor mag hebben zoals jij die was voor mij.
12 1

170
Chapter 12
Dank aan professor Geert Mortier en professor Ludo Mahieu voor jullie vertrouwen in dit onderzoek en jullie kritische beoordeling van mijn proefschrift.
Professor Lieve Page-Christiaens en doctor Malgorzata Srebniak, jullie zijn autoriteiten in het onderzoek naar prenatale genetische diagnostiek. Ik ben vereerd dat jullie dit proefschrift willen jureren.
Collega gynaecologen van het Universitair Ziekenhuis Antwerpen (UZA), bedankt voor alle steun en luisterende oren. Jullie zijn een geweldig team en onze patiënten zijn bij jullie in de allerbeste handen! Christine en Dominique, bedankt voor het delen van doctoraatslief en -leed. Ellen, dankjewel voor alles wat je voor mij gedaan hebt.
Aan alle assistenten, vroedvrouwen, en secretaresses van de dienst Gynaecologie van het UZA: jullie zijn kanjers!
Collega’s van de dienst Genetica, bedankt voor het warme welkom op jullie dienst, de interessante journal clubs, en de leuke babbels aan het koffietoestel.
Professor Fransen, Erik, uw hulp bij dit proefschrift was van groot belang. We delen de liefde voor de fiets. Bedankt om in de statistische analyse de kop te trekken en mij een tandje hoger te laten schakelen.
Dankjewel aan doctor Sigri Beckers en dokter Philip Loquet voor het aanleveren van beeldmateriaal.
Valerie, Antje en Lisa, gefeliciteerd met de mooie masterproef die jullie hebben afgelegd op dit onderwerp.
Ellen Vermeiren, bedankt voor je hulp bij het online plaatsen van de vragenlijsten.
Lieve vrienden en vriendinnen, heerlijk hoelang wij elkaar al kennen. Het is niet onder woorden te brengen hoeveel jullie voor me betekenen. Dankjewel ook aan de vrienden van Tria-GO!, waarbij een bijzondere sportieve uitdaging tijdens dit doctoraat heeft geleid tot extra energie, nieuwe vriendschappen en een diepe dankbaarheid voor het leven.
Paula en Jos, lieve schoonouders, jullie onophoudelijke hulp en ondersteuning tijdens mijn studies en bij de opvang van ons patatje Hugo zijn van onschatbare waarde.
Lena en Alexander, jullie zijn de beste plus-broer en -zus die ik me kan wensen. Bart, lieve broer, ik bewonder je gave om mooie dingen te maken. Dankjewel om een perfecte cover voor dit proefschrift te ontwerpen. Lieve zus Katrien, bedankt om er steeds voor ons te zijn. Lieve petekindjes Brynn en Ylian, jullie zijn schatjes!

171
Acknowledgements
Papa en Ingrid, ik kan steeds rekenen op jullie steun en waardevolle advies. Ik zie jullie graag.
Mama, dankjewel voor de grenzeloze liefde en ontelbare keren dat je me geholpen hebt.
Tom en Hugo, I see trees of green, red roses too. I see them bloom, for me and you. And I think to myself, what a wonderful world.
12 1

172
Joke Muys

173
New challenges and opportunities in prenatal invasive diagnosis

174
Joke Muys

175
New challenges and opportunities in prenatal invasive diagnosis

176

Proefschrift voorgelegd tot het behalen van de graad van doctor in de medische wetenschappen aan de Universiteit Antwerpen te verdedigen door: Joke MUYS | Promotors: Bettina Blaumeiser, Yves Jacquemyn Co-promotor: Katrien Janssens | Faculteit Geneeskunde en Gezondheidswetenschappen | Antwerpen, 2020
Proefschrift voorgelegd tot het behalen van de graad van doctor in de medische wetenschappen aan de Promotors: Bettina Blaumeiser, Yves Jacquemyn
Co-promotor: Katrien Janssens | Faculteit Geneeskunde en Gezondheidswetenschappen | Antwerpen, 2020
Proefschrift voorgelegd tot het behalen van de graad van doctor in de medische wetenschappen aan de Promotors: Bettina Blaumeiser, Yves Jacquemyn
Co-promotor: Katrien Janssens | Faculteit Geneeskunde en Gezondheidswetenschappen | Antwerpen, 2020
Proefschrift voorgelegd tot het behalen van de graad van doctor in de medische wetenschappen aan de Promotors: Bettina Blaumeiser, Yves Jacquemyn
Co-promotor: Katrien Janssens | Faculteit Geneeskunde en Gezondheidswetenschappen | Antwerpen, 2020
NEW CHALLENGES AND OPPORTUNITIES IN PRENATAL INVASIVE DIAGNOSISMICROARRAY ANALYSISJoke Muys
cover-boek-hand.indd 2cover-boek-hand.indd 2 07/07/2020 07:5807/07/2020 07:58
NEW CHALLENGES AND OPPORTUNITIES IN PRENATAL INVASIVE DIAGNOSISMICROARRAY ANALYSIS
Joke Muys
Prof. Dr. Bettina BlaumeiserProf. Dr. Yves JacquemynDr. Katrien Janssens
cover-boek-hand.indd 1cover-boek-hand.indd 1 07/07/2020 07:5807/07/2020 07:58
Proefschrift voorgelegd tot het behalen van de graad van doctor in de Medische Wetenschappen aan de Universiteit Antwerpen te verdedigen door:
Joke MUYS
Promotors: Prof. Dr. Bettina Blaumeiser, Prof. Dr. Yves Jacquemyn
Co-promotor: Dr. Katrien Janssens
Faculteit Geneeskunde en Gezondheidswetenschappen
Antwerpen, 2020
NEW
CHA
LLENG
ES AN
D O
PPORTU
NITIES
IN PREN
ATAL IN
VASIVE D
IAG
NO
SISJoke M
uys




















