
Molecular dynamics simulations of lectin domain of FimH and immunoinformatics for the design of...
7
Molecular dynamics simulations of lectin domain of FimH and immunoinformatics for the design of potential vaccine candidates Muthukumar Singaravelu a, b , Anitha Selvan a , Sharmila Anishetty a, * a Centre for Biotechnology, Anna University, Chennai 600 025, India b National Institute of Ocean Technology, Chennai 600 100, India A R T I C L E I N F O Article history: Received 2 April 2014 Received in revised form 11 August 2014 Accepted 11 August 2014 Available online xxx Keywords: FimH adhesin Molecular dynamics simulation Rigidity Motif Immunoinformatics Vaccine A B S T R A C T Adhesion of uropathogenic E. coli (UPEC) to uroepithelial cell receptors is facilitated through the lectin domain of FimH adhesin. In the current study, Molecular Dynamics (MD) simulations were performed for the lectin domain of FimH from UPEC J96. The high affinity state lectin domain was found to be stable and rigid during the simulations. Further, based on conserved subsequences around one of the disulfide forming cysteines, two sequence motifs were designed. An immunoinformatics approach was utilized to identify linear and discontinuous epitopes for the lectin domain of FimH. We propose that the accessibility of predicted epitopes should also be assessed in a dynamic aqueous environment to evaluate the potential of vaccine candidates. Since MD simulation data enables assessing the accessibility in a dynamic environment, we evaluated the accessibility of the top ranked discontinuous and linear epitopes using structures obtained at every nanosecond (ns) in the 1–20 ns MD simulation timeframe. Knowledge gained in this study has a potential utility in the design of vaccine candidates for Urinary Tract Infection (UTI). ã 2014 Elsevier Ltd. All rights reserved. 1. Introduction Bacterial adhesins are virulence factors important for patho- genesis. Urinary Tract Infection (UTI) is a disease more commonly seen in women. They have a 50% chance of an infection during their lifetime (Bouckaert et al., 2006). Uropathogenic Escherichia coli (UPEC) are implicated in 90% of UTI. 95% of UTI causing E. coli possess type1 fimbriae (Tchesnokova et al., 2011). Adhesion of UPEC to the high mannose glycoprotein uroplakin Ia receptor in the bladder epithelium is mediated by FimH of the filamentous type I pilus system (Zhou et al., 2001). The type I pilus of UPEC has four structural subunits FimA, FimF, FimG and FimH. Each structural subunit of the pilus has a single pilin domain while FimH has an additional lectin domain through which it attaches to the mannose based receptors of the host. The pilus rod is able to stretch and unwind under mechanical stress without dissociating from the host cell receptors (Puorger et al., 2011). UPEC has to withstand the shear stress while establishing an infection. Although UTI can be treated with antibiotics, recurrence is a major problem. This is compounded with the emergence of multidrug resistant UTI causing strains. Invasion of bladder epithelial cells by E. coli is also mediated by FimH (Martinez et al., 2000). When UPEC gets into the host cytosol, it can grow and form intracellular bacterial communities (IBC) (Anderson et al., 2003). The FimH lectin domain is an elongated 11 stranded beta barrel with an immunoglobulin fold (Bouckaert et al., 2005). FimH forms mechanical force enhanced catch bonds due to an interdomain allosteric regulation wherein it switches over to a high affinity state which shows stronger binding to mannose (Yakovenko et al., 2008). Experimental and Molecular Dynamics (MD) simulation studies have addressed FimH adhesin (Thomas et al., 2002; Nilsson et al., 2007; Aprikian et al., 2011). Steered Molecular Dynamics (SMD) simulation of the lectin domain of FimH was performed wherein shear induced force was simulated (Thomas et al., 2002). The importance of cysteine bond of FimH in the adaptation to bind surfaces under increased shear stress was also evaluated through experiments and MD simulations (Nilsson et al., 2007). Single molecule force spectroscopy experiments show that the fimbrial tip mediates shear dependent adhesion of bacteria to uroepithelial cells and the force enhances interaction with mannose (Aprikian et al., 2011). Further, MD simulations of the quaternary structure of the fimbrial tip was used to evaluate inter domain flexibility in the presence and absence of force and a hook chain model was proposed for the fimbrial tip complex (Aprikian et al., 2011). * Corresponding author. Tel.: +91 44 2235 0772; fax: +91 44 2235 0299. E-mail addresses: [email protected], [email protected] (S. Anishetty). http://dx.doi.org/10.1016/j.compbiolchem.2014.08.002 1476-9271/ ã 2014 Elsevier Ltd. All rights reserved. Computational Biology and Chemistry xxx (2014) 18–24 Contents lists available at ScienceDirect Computational Biology and Chemistry journal homepa ge: www.elsev ier.com/locate/compbiolchem
Transcript of Molecular dynamics simulations of lectin domain of FimH and immunoinformatics for the design of...

Computational Biology and Chemistry xxx (2014) 18–24
Molecular dynamics simulations of lectin domain of FimH andimmunoinformatics for the design of potential vaccine candidates
Muthukumar Singaravelu a,b, Anitha Selvan a, Sharmila Anishetty a,*aCentre for Biotechnology, Anna University, Chennai 600 025, IndiabNational Institute of Ocean Technology, Chennai 600 100, India
A R T I C L E I N F O
Article history:Received 2 April 2014Received in revised form 11 August 2014Accepted 11 August 2014Available online xxx
Keywords:FimH adhesinMolecular dynamics simulationRigidityMotifImmunoinformaticsVaccine
A B S T R A C T
Adhesion of uropathogenic E. coli (UPEC) to uroepithelial cell receptors is facilitated through the lectindomain of FimH adhesin. In the current study, Molecular Dynamics (MD) simulations were performed forthe lectin domain of FimH from UPEC J96. The high affinity state lectin domain was found to be stable andrigid during the simulations. Further, based on conserved subsequences around one of the disulfideforming cysteines, two sequence motifs were designed. An immunoinformatics approach was utilized toidentify linear and discontinuous epitopes for the lectin domain of FimH. We propose that the accessibilityof predicted epitopes should also be assessed in a dynamic aqueous environment to evaluate the potentialof vaccine candidates. Since MD simulation data enables assessing the accessibility in a dynamicenvironment, we evaluated the accessibility of the top ranked discontinuous and linear epitopes usingstructures obtained at every nanosecond (ns) in the 1–20 ns MD simulation timeframe. Knowledge gainedin this study has a potential utility in the design of vaccine candidates for Urinary Tract Infection (UTI).
ã 2014 Elsevier Ltd. All rights reserved.
Contents lists available at ScienceDirect
Computational Biology and Chemistry
journal homepa ge: www.elsev ier .com/locate /compbiolchem
1. Introduction
Bacterial adhesins are virulence factors important for patho-genesis. Urinary Tract Infection (UTI) is a disease more commonlyseen in women. They have a 50% chance of an infection during theirlifetime (Bouckaert et al., 2006). Uropathogenic Escherichia coli(UPEC) are implicated in 90% of UTI. 95% of UTI causing E. colipossess type1 fimbriae (Tchesnokova et al., 2011).
Adhesion of UPEC to the high mannose glycoprotein uroplakinIa receptor in the bladder epithelium is mediated by FimH of thefilamentous type I pilus system (Zhou et al., 2001). The type I pilusof UPEC has four structural subunits FimA, FimF, FimG and FimH.Each structural subunit of the pilus has a single pilin domain whileFimH has an additional lectin domain through which it attaches tothe mannose based receptors of the host. The pilus rod is able tostretch and unwind under mechanical stress without dissociatingfrom the host cell receptors (Puorger et al., 2011). UPEC has towithstand the shear stress while establishing an infection.
Although UTI can be treated with antibiotics, recurrence is amajor problem. This is compounded with the emergence of
* Corresponding author. Tel.: +91 44 2235 0772; fax: +91 44 2235 0299.E-mail addresses: [email protected], [email protected]
(S. Anishetty).
http://dx.doi.org/10.1016/j.compbiolchem.2014.08.0021476-9271/ã 2014 Elsevier Ltd. All rights reserved.
multidrug resistant UTI causing strains. Invasion of bladderepithelial cells by E. coli is also mediated by FimH (Martinezet al., 2000). When UPEC gets into the host cytosol, it can grow andform intracellular bacterial communities (IBC) (Anderson et al.,2003).
The FimH lectin domain is an elongated 11 stranded beta barrelwith an immunoglobulin fold (Bouckaert et al., 2005). FimH formsmechanical force enhanced catch bonds due to an interdomainallosteric regulation wherein it switches over to a high affinitystate which shows stronger binding to mannose (Yakovenko et al.,2008). Experimental and Molecular Dynamics (MD) simulationstudies have addressed FimH adhesin (Thomas et al., 2002; Nilssonet al., 2007; Aprikian et al., 2011). Steered Molecular Dynamics(SMD) simulation of the lectin domain of FimH was performedwherein shear induced force was simulated (Thomas et al., 2002).The importance of cysteine bond of FimH in the adaptation to bindsurfaces under increased shear stress was also evaluated throughexperiments and MD simulations (Nilsson et al., 2007). Singlemolecule force spectroscopy experiments show that the fimbrialtip mediates shear dependent adhesion of bacteria to uroepithelialcells and the force enhances interaction with mannose (Aprikianet al., 2011). Further, MD simulations of the quaternary structure ofthe fimbrial tip was used to evaluate inter domain flexibility in thepresence and absence of force and a hook chain model wasproposed for the fimbrial tip complex (Aprikian et al., 2011).

M. Singaravelu et al. / Computational Biology and Chemistry xxx (2014) 18–24 19
Crystal structures of FimH show an allosteric conformationalchange, but in order to assess if all the other observed structuraldifferences (beta bulge, alpha switch and pocket zipper) betweenthe two states are also part of the allosteric mechanism, Rodriguezet al. used experimental and computational strategies. Usingcrystal structures of low and high affinity states of FimH, theypredicted 10 point mutations in four regions that could stabilizeone of the two states: high or low affinity states. Further, usingexperimental techniques, they validated that these regions have arole in the allosteric pathway. They also identified low affinity stateFimH variants (Rodriguez et al., 2013).
In the current study, an extended conformation of FimH lectindomain from a pathogenic strain UPEC J96 has been used for MDsimulations with the objective of finding the stability andflexibility of the high affinity elongated conformation of thelectin domain and for design of potential vaccine candidates. Theavailability of immune related experimental data and develop-ment of bioinformatics tools and techniques have made an impacton immunology research (Brusic and Petrovsky, 2005). Immu-noinformatics deals with in silico analysis and modeling ofimmunological data (Patronov and Doytchinova, 2013). It offersseveral tools for computational identification of epitopes. It morespecifically deals with algorithms for mapping B and T cellepitopes which aids in vaccine research. MD simulations havebeen used in the past to study MHC protein – peptide complexes,for testing immunogenicity of peptide vaccine candidates amongstothers (Rognan et al., 1994; Oomen et al., 2003; Flower et al., 2010).Most epitope prediction strategies are data driven. MD simulationshave an advantage in the context of epitope prediction as they arenot data driven and they can also account for explicit solvation andintrinsic flexibility of the receptor and ligand (Flower et al., 2010).In the current study, we have used an immunoinformaticstechnique for identifying epitopes and further assessed theiraccessibility using MD simulations. It is essential to know if theepitopes predicted through the crystal structures are stillaccessible in a dynamic system. Our approach of following theaccessibility of the epitopes from structures obtained at differenttimeframes of the simulation in an aqueous environmentfacilitates this.
2. Materials and methods
The crystal structure (PDB ID: 1UWF) of the lectin domain ofFimH adhesin from UPEC J96 was obtained from Protein Data Bank(PDB) (Berman et al., 2000). We chose this crystal structure since itis a good resolution (1.7 Å) PDB structure from a pathogenic strainUPEC J96, a causative organism for UTI. GROMACS software(Berendsen et al., 1995; Lindahl et al., 2001) was used for MDsimulations. The ligand in the crystal structure was removed beforeperforming the simulation. The protein was solvated with anexplicit solvent: SPC water, in a cubic box which left 2.5 nm spacearound the solute. Total number of water molecules was 45,868.One Na+ counter ion was added for neutralizing the charge.Steepest descents method was used for energy minimization. Thesystem was weakly coupled to an external bath using Berendsen’smethod. The reference temperature was fixed at 300 K. All bondswere constrained with LINCS (Miyamoto and Kollman, 1992; Hesset al., 1997). G43a1 force field in GROMACS was used for energycalculations. Periodic boundary conditions were imposed. Duringthe simulation, grid type neighbor searching was done and longrange electrostatics was handled using PME (Essmann et al., 1995).The energy minimized structure was subjected to positionrestrained MD simulation for 50 ps. This was followed by full allatom MD simulation for 20 ns. A time step of 2 fs was used.Coordinates were written every 0.5 ps to the trajectory. GROMACSbuilt in tools were used to compute Root Mean Square Deviation
(RMSD), residue wise fluctuations: Root Mean Square Fluctuations(RMSF) and distances between residues. Xmgrace (http://www.plasma-gate.weizmann.ac.il/Grace/) was used to visualize all thegraphs. Residues with RMSF of 0.2 nm and above were consideredto be mobile. 3D structural alignment was performed usingPDBeFold (http://www.ebi.ac.uk/msd-srv/ssm/cgi-bin/ssmserver)(Krissinel and Henrick, 2005). Interatomic distances from PDBstructures obtained at every ns interval were calculated using UCSFChimera (Pettersen et al., 2004).
For the sequence analysis and motif generation, we haveincluded representative sequences of FimH from various E. colistrains, E. coli K12, enteroaggregative E. coli (EAEC), enterohemor-rhagic E. coli (EHEC), enteropathogenic E. coli (EPEC), Diarrheal E.coli (DEC), a multidrug resistant environmental isolate SMS 3–5SECEC, Avian Pathogenic E. coli (APEC) and shiga toxin producing E.coli (STEC). Representative sequences of FimH from different E. colistrains bearing UniProt IDs P08191 (E. coli K12), E7H828 (EPECa14),B1LDQ6 (SMS 3–5 SECEC), H4VQ48 (DEC6C), A1AJI7 (APEC),D3GSD8 (EAEC), D5D2M2 (ExPEC), B5Z3Q8 (EHEC) and G1ZYI1(STEC_94C) were obtained from UniprotKB (release 2014_01) (TheUniProt Consortium, 2014). Sequence motifs were generatedaround Cys44 by performing multiple sequence alignment usingClustalX software (Thompson et al., 2002). The motifs werevalidated using ScanProsite tool (De Castro et al., 2006) byscanning against UniProtKB/TrEMBL databases (release 2014_01)(Boeckmann et al., 2003). From the ScanProsite results, corre-sponding UniProt protein entries matching the motif wereretrieved and their Pfam (Punta et al., 2012) domain and InterPro(Hunter et al., 2012) domain annotation noted. Linear anddiscontinuous epitopes were predicted using ElliPro server(Ponomarenko et al., 2008).
3. Results and discussion
3.1. MD simulation of lectin domain of FimH of UPEC J96
A 20 ns MD simulation was performed for the high affinitystate lectin domain of FimH. The protein stabilized around 2 nsand remained stable till 20 ns. The RMSD and RMSF graphs arepresented in Fig. 1a and b, respectively. From the RMSF data,residues with fluctuations of 0.2 nm and above were consideredmobile. Only 18 out of the 158 residues were mobile. Theseinclude Asn7, Gly8, Thr9, Pro49, Glu50, Thr51, Gly79, Thr87, Ser97,Ser114, Ala115, Gly116, Gly117, Tyr137, Asn138, Ser139,Asp140 and Thr158. Four out of these 18 residues are glycines.The rest of the protein was very rigid with fluctuations below0.2 nm with about 48 residues showing very low fluctuationsbelow 0.1 nm indicating that these residues are extremely rigid.Amongst these extremely rigid regions are beta strand 7 (residues70–76 barring residue 75), part of beta strand 11 (108–111), betastrand 13 (125–134) and beta strand 14 (142–150). The structureobtained at the end of 20 ns was superimposed with the initialstructure and is presented as Fig. 1c. When FimH is not bound tothe mannose receptor, the lectin domain is in a compressed and atwisted conformation because of the pilin domain which exertsan allosteric auto inhibition. Upon activation, it shifts from thislow affinity state to an elongated high affinity state which canpossibly remain as such without re-docking of the pilin domainon application of force to the elongated conformation (Le Tronget al., 2010).
The FimH PDB structure, 1UWF used in the current study is thatof a purified lectin domain bound to a carbohydrate. It is in anelongated conformation and is considered as a high affinity state.Analyses of the structure obtained at the end of the simulationshows that the high affinity state is extremely stable and rigid withabout 90% of the protein showing very low flexibility. Few regions
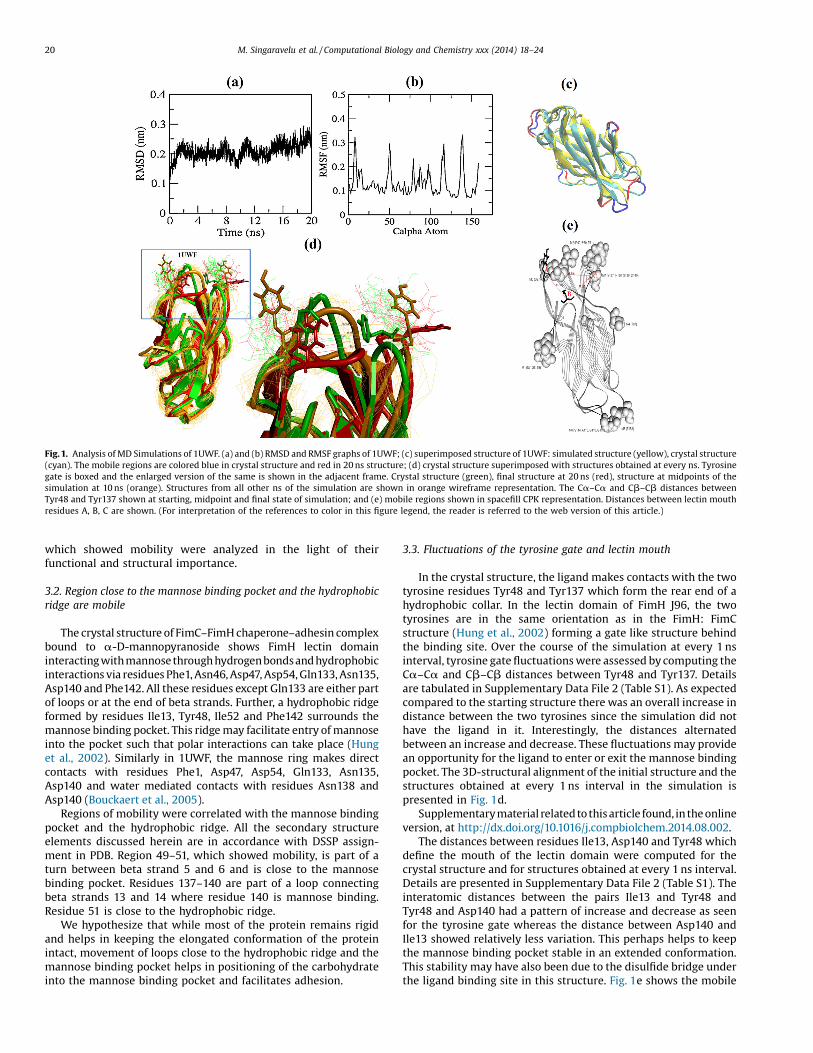
Fig. 1. Analysis of MD Simulations of 1UWF. (a) and (b) RMSD and RMSF graphs of 1UWF; (c) superimposed structure of 1UWF: simulated structure (yellow), crystal structure(cyan). The mobile regions are colored blue in crystal structure and red in 20 ns structure; (d) crystal structure superimposed with structures obtained at every ns. Tyrosinegate is boxed and the enlarged version of the same is shown in the adjacent frame. Crystal structure (green), final structure at 20 ns (red), structure at midpoints of thesimulation at 10 ns (orange). Structures from all other ns of the simulation are shown in orange wireframe representation. The Ca–Ca and Cb–Cb distances betweenTyr48 and Tyr137 shown at starting, midpoint and final state of simulation; and (e) mobile regions shown in spacefill CPK representation. Distances between lectin mouthresidues A, B, C are shown. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
20 M. Singaravelu et al. / Computational Biology and Chemistry xxx (2014) 18–24
which showed mobility were analyzed in the light of theirfunctional and structural importance.
3.2. Region close to the mannose binding pocket and the hydrophobicridge are mobile
The crystal structure of FimC–FimH chaperone–adhesin complexbound to a-D-mannopyranoside shows FimH lectin domaininteracting with mannose through hydrogen bondsand hydrophobicinteractions via residues Phe1, Asn46, Asp47, Asp54, Gln133, Asn135,Asp140 and Phe142. All these residues except Gln133 are either partof loops or at the end of beta strands. Further, a hydrophobic ridgeformed by residues Ile13, Tyr48, Ile52 and Phe142 surrounds themannose binding pocket. This ridge may facilitate entry of mannoseinto the pocket such that polar interactions can take place (Hunget al., 2002). Similarly in 1UWF, the mannose ring makes directcontacts with residues Phe1, Asp47, Asp54, Gln133, Asn135,Asp140 and water mediated contacts with residues Asn138 andAsp140 (Bouckaert et al., 2005).
Regions of mobility were correlated with the mannose bindingpocket and the hydrophobic ridge. All the secondary structureelements discussed herein are in accordance with DSSP assign-ment in PDB. Region 49–51, which showed mobility, is part of aturn between beta strand 5 and 6 and is close to the mannosebinding pocket. Residues 137–140 are part of a loop connectingbeta strands 13 and 14 where residue 140 is mannose binding.Residue 51 is close to the hydrophobic ridge.
We hypothesize that while most of the protein remains rigidand helps in keeping the elongated conformation of the proteinintact, movement of loops close to the hydrophobic ridge and themannose binding pocket helps in positioning of the carbohydrateinto the mannose binding pocket and facilitates adhesion.
3.3. Fluctuations of the tyrosine gate and lectin mouth
In the crystal structure, the ligand makes contacts with the twotyrosine residues Tyr48 and Tyr137 which form the rear end of ahydrophobic collar. In the lectin domain of FimH J96, the twotyrosines are in the same orientation as in the FimH: FimCstructure (Hung et al., 2002) forming a gate like structure behindthe binding site. Over the course of the simulation at every 1 nsinterval, tyrosine gate fluctuations were assessed by computing theCa–Ca and Cb–Cb distances between Tyr48 and Tyr137. Detailsare tabulated in Supplementary Data File 2 (Table S1). As expectedcompared to the starting structure there was an overall increase indistance between the two tyrosines since the simulation did nothave the ligand in it. Interestingly, the distances alternatedbetween an increase and decrease. These fluctuations may providean opportunity for the ligand to enter or exit the mannose bindingpocket. The 3D-structural alignment of the initial structure and thestructures obtained at every 1 ns interval in the simulation ispresented in Fig. 1d.
Supplementarymaterial related to this article found, in the onlineversion, at http://dx.doi.org/10.1016/j.compbiolchem.2014.08.002.
The distances between residues Ile13, Asp140 and Tyr48 whichdefine the mouth of the lectin domain were computed for thecrystal structure and for structures obtained at every 1 ns interval.Details are presented in Supplementary Data File 2 (Table S1). Theinteratomic distances between the pairs Ile13 and Tyr48 andTyr48 and Asp140 had a pattern of increase and decrease as seenfor the tyrosine gate whereas the distance between Asp140 andIle13 showed relatively less variation. This perhaps helps to keepthe mannose binding pocket stable in an extended conformation.This stability may have also been due to the disulfide bridge underthe ligand binding site in this structure. Fig. 1e shows the mobile
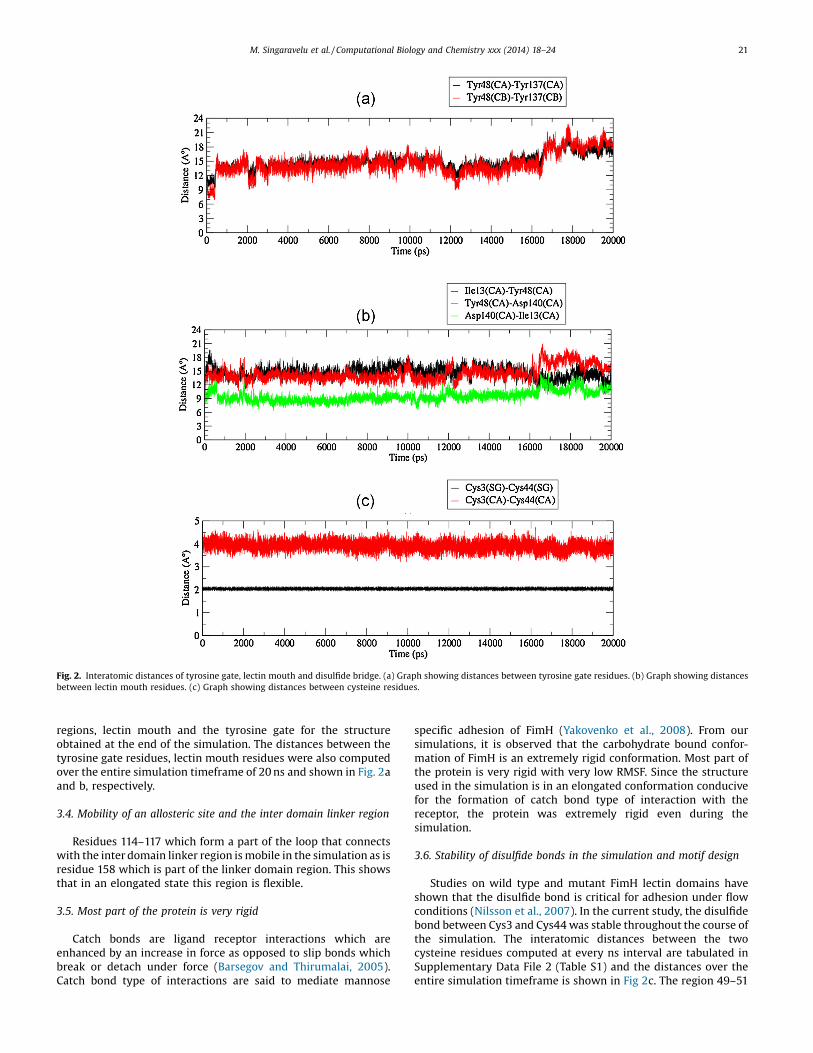
Fig. 2. Interatomic distances of tyrosine gate, lectin mouth and disulfide bridge. (a) Graph showing distances between tyrosine gate residues. (b) Graph showing distancesbetween lectin mouth residues. (c) Graph showing distances between cysteine residues.
M. Singaravelu et al. / Computational Biology and Chemistry xxx (2014) 18–24 21
regions, lectin mouth and the tyrosine gate for the structureobtained at the end of the simulation. The distances between thetyrosine gate residues, lectin mouth residues were also computedover the entire simulation timeframe of 20 ns and shown in Fig. 2aand b, respectively.
3.4. Mobility of an allosteric site and the inter domain linker region
Residues 114–117 which form a part of the loop that connectswith the inter domain linker region is mobile in the simulation as isresidue 158 which is part of the linker domain region. This showsthat in an elongated state this region is flexible.
3.5. Most part of the protein is very rigid
Catch bonds are ligand receptor interactions which areenhanced by an increase in force as opposed to slip bonds whichbreak or detach under force (Barsegov and Thirumalai, 2005).Catch bond type of interactions are said to mediate mannose
specific adhesion of FimH (Yakovenko et al., 2008). From oursimulations, it is observed that the carbohydrate bound confor-mation of FimH is an extremely rigid conformation. Most part ofthe protein is very rigid with very low RMSF. Since the structureused in the simulation is in an elongated conformation conducivefor the formation of catch bond type of interaction with thereceptor, the protein was extremely rigid even during thesimulation.
3.6. Stability of disulfide bonds in the simulation and motif design
Studies on wild type and mutant FimH lectin domains haveshown that the disulfide bond is critical for adhesion under flowconditions (Nilsson et al., 2007). In the current study, the disulfidebond between Cys3 and Cys44 was stable throughout the course ofthe simulation. The interatomic distances between the twocysteine residues computed at every ns interval are tabulated inSupplementary Data File 2 (Table S1) and the distances over theentire simulation timeframe is shown in Fig 2c. The region 49–51
22 M. Singaravelu et al. / Computational Biology and Chemistry xxx (2014) 18–24
had a low mobility probably due to the presence of a disulfide bondforming Cys44 in the vicinity. The stable disulfide bond may have arole in maintaining the overall stability and rigidity of the protein.
Further, the sequence environment around Cys44 in different E.coli FimH lectin domain sequences was analyzed since Cys44 is inclose proximity to the tyrosine gate residue Tyr48. Two sequencemotifs “FCHNDYP” and “SCWNDYGG” were designed based onmultiple sequence alignment of representative FimH sequences(Supplementary figure). On scanning for these motifs in Uni-protKB/TrEMBL it was found that the motifs are specific to FimHlectin domain sequences. While 1692 sequences match the firstmotif, 1357 sequences matched the second motif. 1493 sequencescontaining the first motif were from E. coli, 36 from Shigella,160 from Klebsiella, one each from Raoultella ornithinolytica B6,Enterococcus gallinarum and Citrobacter rodentium. Of the1357 sequences matching the second motif, 1338 were from E.coli and 19 from Shigella. Since many of the entries were fromTrEMBL, the annotation of some of the proteins as FimH was notcomplete. However, they had FimH_mannose binding domainannotation from PFam (PF09160) and InterPro (IPR015243) thus,confirming that they are FimH related sequences. Details arepresented in Supplementary File 1. Crystal structures of FimHlectin domain from PDB show that the cysteine in the first motif“FCHNDYP” is engaged in a disulfide bond except in the lectin
Table 1Linear and discontinuous epitopes of 1UWF, crystal structure of lectin domain of FimH
Linear epitopes (LE) for crystal structure 1UWF
LE no. Region
LE1 135–141
LE2 112–118
LE3 46–55
LE4 151–158
LE5 23–33
LE6 121–124
LE7 6–15
LE8 86–100
Discontinuous epitopes (DE) for crystal structure 1UWF
DE no. No. ofresidues
Residues
DE1 22 N46, D47, Y48, P49, E50, T51, I52, T53, Y55, R92, V94, N96, S9DE2 29 N23, L24, A25, P26, V27, V28, N29, V30, G31, Q32, N33, V112, S11
V155, V156, P157, T158DE3 10 A6, N7, G8, T9, A10, P12, I13, G14, G15, Q41
DE4 13 S78, G79, S80, S81, Y82, P83, P85, T86, T87, S88, E89, T90, P10
Linear epitopes (LE) for 20 ns simulated structure of 1UWF
LE no. Region
LE1 134–142
LE2 110–118
LE3 45–55
LE4 151–158
LE5 121–124
LE6 91–101
LE7 4–33
LE8 78–89
Discontinuous Epitopes (DE) for 20 ns simulated structure of 1UWF
DE no. No. ofresidues
Residues
DE1 46 K4, T5, A6, N7, G8, T9, A10, I11, P12, I13, G14, G15, G16, S17, S39, TN96, S97, R98, T99, D100, K101, T134, N135, N136, Y137, N138
DE2 36 N23, L24, A25, P26, V27, V28, N29, V30, G31, Q32, N33, G66, L68G123, S124, N151, N152, D153, V154, V155, V156, P157, T158
DE3 5 S78, G79, S80, S81, P104
The residues in DE1 of the 20 ns simulation structure mapping to DE1 of 1UWF are un
domain of FimH K-12 (1TR7). There are no representative crystalstructures for sequences bearing the second motif. With thecurrently available information, it is proposed that the first motifregion may provide a favorable environment for the formation ofdisulfide bond near the ligand binding site and contribute to thestability of the adhesin.
Supplementarymaterial related to this article found, in the onlineversion, at http://dx.doi.org/10.1016/j.compbiolchem.2014.08.002.
3.7. Immunoinformatics for linear and discontinuous epitopeprediction for lectin domain of FimH J96
B-cell epitope predictions were obtained from Ellipro server.Eight linear epitopes and four discontinuous epitopes (DE) werepredicted. Some of the DEs encompassed linear epitopes. Forexample, DE1 overlaps with linear epitopes 1, 3 and 8. Since DEsprovide information from a structural perspective, structuresavailable through the simulation were utilized to find accessibilityof these epitopes in a dynamic environment. With the structureobtained at 20 ns, eight linear epitopes and three DEs werepredicted. Details of the epitopes obtained with the crystalstructure and the 20 ns simulation structure are provided inTable 1, while epitopes for the other structures (1–19 ns) areprovided in Supplementary Data File 2 (Table S2).
J96 and its 20 ns simulation structure.
Residues Score
NNYNSDD 0.840VSSAGGV 0.815NDYPETITDY 0.734NNDVVVPT 0.721NLAPVVNVGQN 0.692KAGS 0.663ANGTAIPIGG 0.652TTSETPRVVYNSRTD 0.634
Score
7, R98, T99, D100, N135, N136, Y137, N138, S139, D140 0.7913, S114, A115, G116, G117, V118, K121, A122, G123, S124, N151, N152, D153, 0.735
0.6454 0.50
Residues Score
TNNYNSDDF 0.771TPVSSAGGV 0.761HNDYPETITDY 0.707NNDVVVPT 0.699KAGS 0.687PRVVYNSRTDK 0.651KTANGTAIPIGGGSANVYVNLAPVVNVGQN 0.602SGSSYPFPTTSE 0.593
Score
40, Q41, H45, N46, D47, Y48, P49, E50, T51, I52, T53, Y55, T90, P91, R92, V94,, S139, D140, D141, F142
0.673
, S69, N70, T110, P111, V112, S113, S114, A115, G116, G117, V118, K121, A122, 0.672
0.538
derlined.

M. Singaravelu et al. / Computational Biology and Chemistry xxx (2014) 18–24 23
3.8. Correlation of epitopes (DEs) to mannose binding site and tyrosinegate
DE1 obtained from crystal structure 1UWF had 22 residueswhich include four of the mannose binding pocket residues Asn46,Asp47, Asn135 and Asp140 and two tyrosine gate residuesTyr48 and Tyr137. This is significant since blocking of the mannosebinding site will abolish adhesion. Since the crystal structure is astatic structure, we propose that accessibility of epitopes in adynamic environment as is encountered in a physiological stateshould be assessed. The epitopes predicted from the structuresobtained through MD simulations can be used to assess theaccessibility. Additionally, any new epitopic regions which becomeaccessible in a dynamic state can be predicted from the MDsimulation structures. It was found that DE1 of crystal structureremains accessible throughout as part of atleast one of the top twoDEs predicted with the MD simulation data. The epitopes predictedat the end of the simulation timeframe 20 ns is discussed. Analysisshows that DE1 predicted from the 20 ns simulation includesresidues 4–17, 39–41, 45–53, 55, 90–94, 96–101, and 134–142. Inaddition to all the residues of DE1 of crystal structure, it includes anadditional mannose binding residue Phe142 along withjavascript:void(0) other residues. Thus, DE1 of the crystal structure remainsaccessible even in a dynamic environment. The top scoring linearepitope of 1UWF is also a part of DE1 of crystal structure andremains accessible. The epitope DE1 obtained from the crystalstructure and its overlap with epitopes obtained from structures atevery ns of the simulation is shown in Fig. 3a and b, respectively.
Previous studies have addressed the potential of FimH and FimCHas a vaccine candidate (Tchesnokova et al., 2011; Langermann et al.,1997, 2000; Thankavel et al., 1997). In one of the studies, antibodiesraised against the lectin domain of FimH increased adhesion of FimHto uroepithelial cells instead of inhibiting it. The conformationalepitopes identified in their study did not include the mannosebindingsite (Tchesnokovaetal.,2011;Kisielaetal.,2013).Kisielaetal.in a recent study proposed that in the functionally activeconformation of FimH lectin domain, the binding pocket epitopesare shielded by ligand masking. They also developed an interestingstrategy wherein they made a functionally defective FimH lectindomain which was locked in a low-binding conformation and theyfurther showed that the antibodies that were induced by this mutantlectin domain were able to block the adhesion (Kisiela et al., 2013).Other experimental studies have also shown the potential of antiFimH antibodies inhibiting adhesion (Langermann et al.,1997, 2000;Thankavel et al., 1997).
The epitopes identified in the current study have a potential inthe design of vaccine candidates against UTI, since they weregenerated from the 3D structure of the high affinity state FimHlectin domain, and they also include some of the mannose binding
Fig. 3. Discontinuous epitopes in 1UWF and its 20 ns simulation structure. (a) DE1 of1UWF in green. (b) DE1 of 20 ns structure of 1UWF; the region in green is where DE1 of1UWF overlaps with DE1 of 20 ns; rest of the DE1 of 20 ns is in red and the remainingportion of the protein is in cyan. (For interpretation of the references to color in thisfigure legend, the reader is referred to the web version of this article.)
residues and remain accessible even in a dynamic aqueousenvironment. We therefore propose that these epitopes have apotential to be immunogenic.
4. Conclusions
MD simulation of the high affinity conformation of FimH showsthat the entire lectin domain is largely rigid with very few regionsshowing mobility. The rigidity of the protein with selectivemovement of regions in the vicinity of the mannose bindingpocket and the hydrophobic ridge perhaps helps the protein inattaching to the mannose based receptors while staying stableunder shear stress. Linear and DEs were predicted for the lectindomain of FimH of UPEC J96 and their accessibility was assessedwith structures obtained from the MD simulation studies.
Contributors
Muthukumar Singaravelu and Anitha Selvan contributedequally to the work and may be considered as joint first authors.
Acknowledgements
The authors thank BTIS, Department of Biotechnology forcomputational facilities. Muthukumar Singaravelu thanks MBTNIOT for support. Anitha Selvan thanks CSIR for Senior ResearchFellowship.
References
Anderson, G., Palermo, J., Schilling, D., et al., 2003. Intracellular bacterial biofilm-likepods in urinary tract infections. Science 301, 105–107.
Aprikian, P., Interlandi, G., Kidd, B.A., et al., 2011. The bacterial fimbrial tip acts as amechanical force sensor. PLoS Biol. 9 (5), e1000617.
Barsegov, V., Thirumalai, D., 2005. Dynamics of unbinding of cell adhesionmolecules: transition from catch to slip bonds. Proc. Natl. Acad. Sci. U. S. A. 102,1835–1839.
Berendsen, H.J.C., Van der Spoel, D., Van Drunen, R., 1995. GROMACS: a message-passing parallel molecular dynamics implementation. Comput. Phys. Commun.91, 43–56.
Berman, H.M., Westbrook, J., Feng, Z., et al., 2000. The protein data bank. NucleicAcids Res. 28, 235–242.
Boeckmann, B., Bairoch, A., Apweiler, R., et al., 2003. The SWISS-PROT proteinknowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 31, 365–370.
Bouckaert, J., Berglund, J., Schembri, M., et al., 2005. Receptor binding studiesdisclose a novel class of high affinity inhibitors of the Escherichia coli FimHadhesion. Mol. Microbiol. 55, 441–455.
Bouckaert, J., Mackenzie, J., De Paz, P., et al., 2006. The affinity of the FimH fimbrialadhesin is receptor driven and quasi independent of Escherichia coli pathotypes.Mol. Microbiol. 61, 1556–1568.
Brusic, V., Petrovsky, N., 2005. Immunoinformatics and its relevance tounderstanding human immune disease. Exp. Rev. Clin. Immunol. 1, 145–157.
De Castro, E., Sigrist, C.J., Gattiker, A., et al., 2006. ScanProsite: detection of PROSITEsignature matches and ProRule-associated functional and structural residues inproteins. Nucleic Acids Res. 34, W362–W365.
Essmann, U., Perela, L., Berkowitz, M.L., et al., 1995. A smooth particle mesh ewaldmethod. J. Chem. Phys. 103, 8577–8593.
Flower, D.R., Phadwal, K., Macdonald, I., et al., 2010. T-cell epitope prediction andimmune complex simulation using molecular dynamics: state of the art andpersisting challenges. Immunome Res. 6, S4.
Hess, B., Bekker, H., Berendsen, H.J.C., et al.,1997. LINCS: a linear constraint solver formolecular simulations. J. Comput. Chem. 18, 1463–1472.
Hung, C.S., Bouckaert, J., Hung, D., et al., 2002. Structural basis of tropism ofEscherichia coli to the bladder during urinary tract infection. Mol. Microbiol. 44,903–915.
Hunter, S., Jones, P., Mitchell, A., et al., 2012. InterPro in 2011: new developmentsin the family and domain prediction database. Nucleic Acids Res. 40, D306–D312.
Krissinel, E., Henrick, K., 2005. Multiple alignment of protein structures in threedimensions. CompLife 3695, 67–78.
Kisiela, D.I., Rodriguez, V.B., Tchesnokova, V., et al., 2013. Conformationalinactivation induces immunogenicity of the receptor-binding pocket of abacterial adhesin. Proc. Natl. Acad. Sci. 110 (47), 19089–19094.
Langermann, S., Palaszynski, S., Barnhart, M., et al., 1997. Prevention of mucosalEscherichia coli infection by FimH-adhesin-based systemic vaccination. Science276, 607–611.

24 M. Singaravelu et al. / Computational Biology and Chemistry xxx (2014) 18–24
Langermann, S., Möllby, R., Burlein, J.E., et al., 2000. Vaccination with FimH adhesinprotects cynomolgus monkeys from colonization and infection by uropatho-genic Escherichia coli. J. Infect. Dis. 181, 774–778.
Le Trong, P., Aprikian, B., Kidd, A., et al., 2010. Structural basis for mechanical forceregulation of the adhesin FimH via finger trap-like beta sheet twisting. Cell 141,645–655.
Lindahl, E., Hess, B., Van Der Spoel, D., 2001. GROMACS 3.0: a package for molecularsimulation and trajectory analysis. J. Mol. Model. 7, 306–317.
Martinez, J., Mulvey, A., Schilling, D., et al., 2000. Type 1 pilus-mediated bacterialinvasion of bladder epithelial cells. EMBO J. 19, 2803–2812.
Miyamoto, S., Kollman, P.A., 1992. SETTLE: an analytical version of the SHAKE andRATTLE algorithm for rigid water models. J. Comput. Chem. 13, 952–962.
Nilsson, L.M., Yakovenko, O., Tchesnokova, V., et al., 2007. The cysteine bond in theEscherichia coli FimH adhesin is critical for adhesion under flow conditions. Mol.Microbiol. 65, 1158–1169.
Oomen, C.J., Hoogerhout, P., Bonvin, A.M., et al., 2003. Immunogenicity of peptide-vaccine candidates predicted by molecular dynamics simulations. J. Mol. Biol.328 (5), 1083–1089.
Patronov, A., Doytchinova, I., 2013. T-cell epitope vaccine design by immunoinfor-matics. Open Biol. 3 (1), 120139.
Pettersen, E.F., Goddard, T.D., Huang, C.C., et al., 2004. UCSF Chimera—avisualization system for exploratory research and analysis. J. Comput. Chem.25, 1605–1612.
Ponomarenko, J., Bui, H.H., Li, W., et al., 2008. ElliPro: a new structure-based tool forthe prediction of antibody epitopes. BMC Bioinform. 9514.
Punta, M., Coggill, P.C., Eberhardt, R.Y., et al., 2012. The Pfam protein familiesdatabase. Nucleic Acids Res. 40, D290–D301.
Puorger, C., Vetsch, M., Wider, G., et al., 2011. Structure, folding and stability of FimA,the main structural subunit of type 1 pili from uropathogenic Escherichia colistrains. J. Mol. Biol. 412, 520–535.
Rodriguez, V.B., Kidd, B.A., Interlandi, G., et al., 2013. Allosteric coupling in thebacterial adhesive protein FimH. J. Biol. Chem. 288 (33), 24128–24139.
Rognan, D., Scapozza, L., Folkers, G., Daser, A., 1994. Molecular dynamics simulationof MHC-peptide complexes as a tool for predicting potential T cell epitopes.Biochemistry 33 (38), 11476–11485.
Tchesnokova, V., Aprikian, P., Kisiela, D., et al., 2011. Type 1 fimbrial adhesin FimHelicits an immune response that enhances cell adhesion of Escherichia coli.Infect. Immun. 79, 3895–3904.
Thankavel, K., Madison, B., Ikeda, T., et al.,1997. Localization of a domain in the FimHadhesin of Escherichia coli type 1 fimbriae capable of receptor recognition anduse of a domain-specific antibody to confer protection against experimentalurinary tract infection. J. Clin. Investig. 100, 1123–1136.
The UniProt Consortium, 2014. Activities at the Universal Protein Resource(UniProt). Nucleic Acids Res. 42, D191–D198.
Thomas, W.E., Trintchina, E., Forero, M., et al., 2002. Bacterial adhesion to target cellsenhanced by shear force. Cell 109, 913–923.
Thompson, J.D., Gibson, T.J., Higgins, D.G., 2002. Multiple sequence alignment usingClustalW and ClustalX. Curr. Protoc. Bioinform. 2.3.1–2.3.22.
Yakovenko, O., Sharma, S., Forero, M., et al., 2008. FimH forms catch bonds that areenhanced by mechanical force due to allosteric regulation. J. Biol. Chem. 283,11596–11605.
Zhou, G., Mo, W.J., Sebbel, P., et al., 2001. Uroplakin Ia is the urothelial receptor foruropathogenic Escherichia coli: evidence from in vitro FimH binding. J. Cell Sci.114, 4095–4103.




















