
Hybrid Materials from Intermolecular Associations between Cationic Lipid and Polymers
Upload
independentCategory
view
0download
0
PAPER www.rsc.org/crystengcomm | CrystEngComm
MMH-2 as a new approach for the prediction of intermolecularinteractions: the crystal packing of acetamide†
Edelsys Codorniu-Hern�andez,‡*a A. Daniel Boese,b Carsten Schauerte,b Alberto Rolo-Naranjo,a
Ram�on Miranda-Quintana,a Luis A. Montero-Cabrerac and Roland Boeseb
Received 23rd March 2009, Accepted 8th June 2009
First published as an Advance Article on the web 23rd July 2009
DOI: 10.1039/b905779j
A new approach (MMH-2) was applied and tested for the prediction of intermolecular interactions in
the crystal packing of acetamide. In MMH-2, energies of random molecular interaction configurations
are computed. It uses molecular association quantities from statistical thermodynamics in order to
obtain intermolecular interaction motifs that follow a ranking process. The most important motifs are
optimized. Here, the AM1 semiempirical Hamiltonian was applied for the calculation and optimization
of each obtained configuration and a comparison to MP2 results is provided. Such a stepwise procedure
follows the assumed genesis of crystal growth without using experimental input. For evaluation
purposes, graph set analysis was used to classify the structural patterns of both acetamide polymorphs.
It was also necessary to introduce a new geometrical similarity index for the comparison of calculated
and experimental motifs. As a result, all experimental hydrogen bond patterns were found and
molecular synthons in both polymorphic acetamide structures were predicted as local minima. This
suggests a new strategy for crystal structure prediction of flexible molecules with a possible subsequent
progress in crystal engineering in silico.
1. Introduction
Most of the neat solids contain crystals or crystalline regions
which influence the respective solid state properties. All funda-
mental chemical and physical properties such as solubility,
melting temperature, hardness or hygroscopicity are therefore
determined not only by molecular structure but also by the
packing of the molecules in the crystals. This becomes particu-
larly evident with polymorphs which have the same molecules
arranged with different crystal packing. For active pharmaceu-
tical ingredients polymorphism has become an important issue
not only for the obvious reasons of modulated properties which
influence the bioavailability and other features but also for legal
issues in the context of intellectual property protection.1 This
caused a boom in research activities but the possibility to predict
a crystals structure starting from a molecular structure is still in
its infancy. Even though more than 450 000 crystal structures of
aDepartment of Molecular Design and Synthesis, Higher Institute ofTechnologies and Applied Sciences, Ave Salvador Allende y Luaces,Quinta de los Molinos, Plaza de la Revoluci�on, Ciudad Habana, CP10600 AP 6163, CubabDepartment of Chemistry, University of Duisburg-Essen,Universit€atsstrasse 5-7, 45117 Essen, GermanycLaboratory of Computational and Theoretical Chemistry, Faculty ofChemistry, University of Havana, Zapata e G y Maz�on, CP 10400Ciudad Habana, Cuba
† Electronic supplementary information (ESI) available: A brief outlineof MMH procedures; results of MMH-1/AM1 for acetamide dimers(Fig. S1–S4); additional results comparing MMH-2/AM1 with MP2and DFT calculations (Tables S1–S12, Fig. S5–S6). See DOI:10.1039/b905779j
‡ Current address: Department of Chemistry. University of Calgary.2500 University Drive N.W., Calgary, Alberta, Canada, T2N 1N4.E-mail: [email protected]; Tel.: +1(403)220-7561.
2358 | CrystEngComm, 2009, 11, 2358–2370
organic molecules are precisely described in literature
(Cambridge Structural Database (CSD)),2 the open problem of
‘crystal structure prediction’ (CSP) has not been conclusively
solved.3–7
The Cambridge Crystallographic Data Centre (CCDC) orga-
nized regularly blind tests in CSP since 2000 with the participa-
tion of different computational laboratories, but the problem
turned out to be much more subtle than originally anticipated.
There were severe restrictions for the candidates selected for the
tests in respect of the size of the molecules, the number of
molecules in the asymmetric unit, the kind of elements, flexibility,
space groups etc. which were thought to reduce the problem and
computational times. However the restrictions had a significant
influence on the approaches, in spite of major advances8 there
still seems to be no general solution in sight.9–11
The restriction of space groups and the number of independent
molecules in the asymmetric unit favoured approaches which
start with a symmetry relationship between two molecules and by
adding further molecules ending in a pattern that corresponds to
one of the most favourable space groups for organic molecules as
found in the CSD.
Amongst different strategies there is one which assembles
molecules according to the most frequent and robust patterns
found in the CSD, called supramolecular synthons.12 Such
preferred motifs went through different steps of crystal growth
and therefore they represent an experimental result which has all
the prerequisites of an existing crystal.
Other approaches follow much more the genesis of a crystal
and do not apply symmetry restrictions when assembling clus-
ters. Two molecules meeting each other do not have symmetry in
mind, their goal is to minimize their relative energy followed by
adding stepwise an increasing number of molecules.
This journal is ª The Royal Society of Chemistry 2009

A strategy which seems to be most promising, combines the
two approaches which consider the genesis of a crystal by ana-
lysing the energy for clusters of increasing size and then selecting
motifs which proved to be stable synthons for existing crystals.
The first step is therefore the formation of clusters without
employing any symmetry restrictions. The second step consists of
finding motifs which can be localised with the help of the less
specific graph-set analysis for hydrogen bonds and the third step
is allocating the supramolecular synthons. The final step should
be generally constituted as a selective aid used to rule out less
favourable patterns which are less likely to exist according to
statistical searches in the CSD.
The importance of the first step is undisputed and consists of
the development and application of reliable methods for the
correct estimation of intermolecular interactions. Our aim is to
introduce and test a new approach (MMH-2) in order to provide
the packing motifs. MMH-2 is based on a previous methodology
‘‘Multiple minima hypersurface procedure’’ (MMH, now to be
called MMH-1) published ten years ago by one of the authors
(LMC)13 as an alternative method for the study of explicit solvent
effects. MMH-1 has been extensively applied in the last years for
the study of intermolecular associations.13–18 In recent papers,
MMH-1 was also recognized as a very reliable procedure for
searching minima in weakly interacting complexes.16–18 The new
approach keeps the conception of the original MMH-1
combining quantum mechanical methods and statistical ther-
modynamics in order to obtain thermodynamic magnitudes
related with molecular association processes.
The remainder of this article is organized as follows: In section
2 we describe the general steps of the proposed approach (MMH-
2) and the consecutive procedures for the application of MMH-2
and MMH-1 in the elucidation of acetamide hydrogen bond
motifs. In this section we also discuss our computational setup
with the application of different simulation packages. In section
3 we present the graph set results and supramolecular synthons
of acetamide polymorphs in order to have the experimental
patterns for comparison with the theoretical results. Finally, in
section 4 we summarize our findings and discuss the possible
application of MMH-2 for CSP.
2. Theory and procedures
The original MMH-1 approach has already been explained in
details13–15 and is also explained in the supplementary
information.‡ It was created with the original purpose of eval-
uating the thermodynamic association functions of various
molecular clusters (using the partition function) and to randomly
explore the multiple minima hypersurface of a supermolecular
system. The exploration is made by generating several sets of
initial random geometries. This is followed by a gradient
pathway search to the local minima that could be statistically
significant.
MMH-1 outlines as:
(1) Random generation of different molecular configurations
starting from the optimized fragment geometries.
(2) Geometry optimization of the selected configurations by
standard gradient path minimization.
(3) Selection of the most important configurations through
molecular association magnitudes obtained by statistical
This journal is ª The Royal Society of Chemistry 2009
thermodynamic formulas (association energies and entropies,
population of states, See Supplementary Information).
This scheme is modified in MMH-2 in the following way:
(1) Random generation of different molecular configurations
starting from the optimized fragment geometries.
(2) Single-point energy calculations of each different configu-
ration.
(3) Selection of the most important configurations through
molecular association magnitudes obtained by statistical ther-
modynamic formulas (association energies and entropies,
population of states, See Supplementary Information).
(4) Geometry optimization of the selected configurations by
standard gradient path minimization.
(5) Ranking order of the structural motifs by the configuration
energies (Scheme 1).
Thus, the MMH-1 method consists of steps 1, 4, and 3 from
the MMH-2 scheme.
If we have to compare with experimental patterns, one extra
point should be added:
(6) Selection of the best superimposed structures (comparing
theoretical and experimental patterns) through the similarity
index values. Graph set analysis was used to classify the struc-
tural patterns of acetamide.
After the last step in this scheme, the simulation is stopped
after a yet undefined halting criterion has been met. In our case,
the calculation finishes after the intermolecular associations of
the experimental structures for acetamide that favour the growth
of both polymorphs and the three-dimensional behaviour of the
crystal have been found. Association complexes with many
monomers are likely to show the building blocks when predicting
crystal structures. Here, another criterion to stop the simulation
can be found when a large increase in the dissociation energy is
observed.
For this scheme, two crucial choices have to be made in each
particular test applying MMH-2 for CSP:
—The appropriate selection of the underlying methodology
for the single point energy calculation (step 2) and the optimi-
zation (step 4) of each supermolecule.
—The way to compare calculated and experimental structural
motifs (step 6)
Concerning the first choice, our aim herein is to find those
crystal structures favoured by nature, which can become
a formidable task. In some cases, the experimentally obtained
structures are not related to the computed ‘‘global minimum’’ of
either the condensed structure or the fragments. Here, kinetic
effects might become important. Thus, previous attempts to
apply quantum chemical methods (ab initio or DFT) to find the
global minimum for CSP did not succeed in all cases.8–11
We believe that a statistical thermodynamic approach to find
the most important local minima will prove more successful.
Following a recent experience with MMH-1,13–15 the structural
analysis shows that various ‘‘local minima’’ may contribute
significantly to the stabilization of the system. However, in the
case of acetamide, MMH-1 was unable to reproduce the exper-
imental patterns found in the crystal structure.
MMH-2 is much faster and as we shall see later, much more
robust in the prediction of the experimental patterns. The AM1
semiempirical Hamiltonian is used to calculate the preliminary
energies of all the generated molecular arrangements.
CrystEngComm, 2009, 11, 2358–2370 | 2359

Scheme 1 Graphic representation of the novel MMH-2 approach
Since several hundred supermolecular geometries are gener-
ated during the first step, the use of ab initio methods for struc-
tures larger than dimers is computationally not feasible.
semiempirical methods have been confirmed as a good choice for
the discrimination of geometries.16–19 Here, the computational
process is fast enough to treat a huge number of different
structures that could also include large molecular systems (such
as proteins and nanoclusters). For acetamide, semiempirical
results can be tested and complemented with more sophisticated
and accurate calculations. This provides a useful test for the
reliability of this kind of Hamiltonian. In this work, we compare
the results of the AM1 Hamiltonian with the experimental
patterns as well as other methods (DFT and MP2).
For the geometrical comparison of the calculated and experi-
mental structural motifs, an overlap matrix was calculated
through TGSA2001 program,20 with the maximum overlap
among all the structures. Some quantum similarity indices were
tested in order to select the best overlap structures.21–24 Unfortu-
nately, none of them was useful for our approach. Here, the
geometric comparison was not explicitly considered. This possibly
caused some failures in the recognition of the best overlap between
calculated and experimental structures. We introduced a new
Positional similarity index (PSI). PSI ˛ [0, 1] while PSI ¼ 1
corresponds to an ideal structural overlap (eqn (1)). PSI is based
on the geometric coincidences among the atoms from two mole-
cules i and i0. Two atoms are considered coincident if the distance
between them is lower than a pre-defined thereshold (3).
PSIMOL ¼
PNATOM
i¼1
Ci;i0 ð3Þ
NATOM
(1)
The coincidence Ci,i0 is defined as:l
2360 | CrystEngComm, 2009, 11, 2358–2370
Ci;i0 ð3Þ ¼
8>><>>:
1 if dði; i0Þ# 3
5%ðdMAX � dði; i0ÞÞ if 3\dði; i0Þ# dMAX
0 if dði; i0Þ. dMAX
9>>=>>;(2)
dMAX is the maximum distance value considered in the specific
acetamide atomic comparison process. In the case of acetamide
dMAX has a the value of 3 �A. For d(i,i0 0) values higher than dMAX
the calculated molecules do not have geometrical proximity.
d (i,i0) represents the Euclidean norm between atoms i and i0. The
set of those superimposed structures having a higher overlap
between the experimental and predicted pairs of structures is an
automatic output of the methodology.
Computational details
Different calculations were carried out in order to compare the
results of MMH-2/AM1 and other ab initio and DFT methods.
The latter methods were used to reoptimize the local minima
found by the new approach.
semiempirical Hamiltonian. For obtaining minimal energy
structures AM1 was applied, using the MOPAC25 program. In all
calculations, the eigenvector following the routine was used and
all convergence thresholds have been tightened by a factor of
1000. The MMOK option for optimizing peptidic bonds was
used by means of a molecular mechanical field. This is better
suited to reproduce the planar geometry of the peptidic bond and
the experimental value of the Z/E interconversion barriers.
Ab initio methods. The lowest structures found by the AM1
Hamiltonian were optimized using different levels of calculation:
This journal is ª The Royal Society of Chemistry 2009

Fig. 1 A portion of the crystal structures of acetamide polymorphs
(ACEMID03 and ACEMID06).
The dimers were fully optimized with HF/6-311G(d,p)26 and
MP2/6-311G(d,p)27 using the Gaussian 0328 program. Further-
more, MP2/aug0-cc-pVTZ29,30 (denoting that we used diffuse
functions only on the nitrogen and oxygen atoms) values were
obtained using the Turbomole31 program. Here, MP2 was
calculated within the RI approximation. We used MP2 rather
than e.g. SCS-MP2 because of its good prediction of hydrogen
bonds, being still unsurpassed in accuracy by other methods of
this speed.32 Using these structures, single-point energies were
calculated using the MP2/aug0-cc-pVQZ29,30 and MP2/aug0-cc-
pV5Z29,30 basis sets. The trimers and tetramers from MMH-2/
AM1 were optimized by MP2/aug0-cc-pVTZ, the pentamers and
hexamers by MP2/aug0-cc-pVDZ.
Density functional theory. DFT calculations have been per-
formed using the CP2K33 simulation package for the optimiza-
tion of the acetamide trimers in isolated conditions. The density
functional theory implementation in CP2K (Quickstep)34 is
based on the hybrid Gaussian plane wave (GPW) scheme.35 In
this scheme, an efficient algorithm for the calculation of the
Kohn–Sham matrix is obtained through a dual representation of
the electron density. The Goedecker–Teter–Hutter (GTH)
pseudopotential36–38 has been employed for all DFT calculations
using the HCTH/120 functional.39 A 350 Ryd plane wave density
cut-off has been applied. Carbon, hydrogen and nitrogen of the
acetamide molecules were described by standard triple-z basis
with one set of polarization functions (TZVP).
In the paper we are presenting the results of MMH-2/AM1 and
MMH-1/AM1 in the prediction of structural motifs of acetamide
crystals, providing a clear comparison between both methodol-
ogies for the case of dimer configurations. The details in these
calculations are summarized as follows:
Details in the application of MMH-1/AM1 methodology.
Acetamide dimers and trimers calculations were performed
through the generation of 100 different random geometries,
starting from the optimized geometries. These 100 configurations
were optimized and final geometries and energies were processed
by statistical thermodynamic procedures in order to obtain
a reduced set of configurations that represent the most important
contributions to the whole system. The selected configurations
were used for the comparison with acetamide experimental
patterns. As a simultaneous step we also analyzed all the opti-
mized configurations created and optimized by MMH-1, in order
to look for the structural patterns in non-associated configura-
tions (higher energies).
Details in the application of MMH-2/AM1 methodology.
Acetamide dimer to hexamer calculations were performed
through the generation of 200 different random geometries of
acetamide clusters as the first step. Then, all consecutive steps
presented in Scheme 1 were followed. The most stable dimer
configurations were used as building blocks for the simulation of
trimers, those of trimers for tetramers, and so on. In addition,
different dimers–hexamers configurations were constructed
starting from the isolated acetamide molecules. In order to
compare all the structural patterns and theoretical results, the
best overlapped structures served as input file for calculating PSI
(eqn (1)).
This journal is ª The Royal Society of Chemistry 2009
3. The system under study
We investigated the structure of acetamide, which was described
by Bernstein et al. in 199540 concerning its hydrogen bond
patterns and its respective graph sets. Solid acetamide exists in
two crystal forms under ambient conditions.41 The most stable
modification is rhombohedral42,43 which contains one molecule
in the asymmetric unit and the metastable is an orthorhombic
form with two molecules in the asymmetric unit.44,45 The
crystal structures of the two forms were taken from the crystal
structures database, which we refer to as ACEMID06 and
ACEMID03 henceforth. Fig. 1 shows a part of the crystal
structures of both polymorphs. The hydrogen bond patterns
presented in both acetamide polymorphs were obtained
through a graph set analysis. Therefore, each acetamide poly-
morph was analyzed and catalogued in a readily recognizable
notation (Fig. 2 and Fig. 3).40 All these structures were used as
experimental patterns for comparison with the theoretical
predictions.
4. Results and discussion
MMH-1/AM1 is unable to reproduce the experimental patterns
of acetamide polymorphs. This can be deduced from the
behaviour of the thermodynamic association magnitudes of
acetamide dimers which is shown in the supplementary materi-
al.‡ The MMH-1/AM1 method predicts the true global minimum
corresponding to the R22(8) pattern shown in Fig. 2. This
CrystEngComm, 2009, 11, 2358–2370 | 2361

Fig. 2 Hydrogen bond motifs from ACEMID03 polymorph.
Fig. 3 Hydrogen bond motifs from ACEMID06 polymorph.
2362 | CrystEngComm, 2009, 11, 2358–2370
structure coincides with the structural pattern present in the
asymmetric unit of one of the acetamide polymorphs (ACE-
MID06). However, none of the other structural motifs are found
with MMH-1/AM1. Only the C11(4) pattern is present if we
consider energies more than 3 kcal mol�1 above the global
minimum, which is one of the structural patterns presented in
ACEMID03. The other experimental patterns found are not
predicted as stable structures. Thus, the information provided by
MMH-1/AM1 is incomplete, especially if several patterns and
polymorphs are present. MMH-1/AM1 was designed to predict
clusters in the gas phase to condense without any other
interaction than those among the considered molecules. Those
interactions could be exaggerated because they are focused
exclusively on the gradient path optimization after the random
cell generation.
Hence, an extension of this approach is needed, where the most
important local minima are considered and calculated, arriving
at the new MMH-2/AM1 method. Here, Scheme 1 is used for the
prediction of crystal structures.
4.1 MMH-2/AM1 results
4.1.1 Acetamide dimer configurations. Fig. 4 shows the
behaviour of the association thermodynamic magnitudes
obtained by MMH-2/AM1 for the acetamide dimers. It is evident
that other possibilities of acetamide associations are now
provided by the new approach, rather than just finding one
minimum structure. MMH-2/AM1 yields many dimer molecular
associations within 3 kcal mol�1 of the global minimum. In the
graphical representation of the population of the states of
MMH-2/AM1 (Fig. 5) there are several structures with
This journal is ª The Royal Society of Chemistry 2009

Fig. 4 Thermodynamic association magnitudes of acetamide dimer configurations obtained by MMH-2/AM1.
Fig. 5 Population of states and energies of each acetamide dimer
configuration obtained by MMH-2/AM1. Note that the structures with
any significant population correspond to the four local minima also
found in the experimental patterns.
Fig. 6 Superimposed experimental and predicted acetamide dimer
structures obtained by MMH-2/AM1. d1, d2 and d4 correspond to
ACEMID06, whereas d3 and d4 can be found in ACEMID03.
important statistical contributions. All these structures lead to
the local minima reported in Fig. 6. These structures were
collected and superimposed with all the dimer acetamide patterns
in order to obtain the similarity indices.
The four predicted MMH-2/AM1 structures with the
lowest energies coincide with the four experimental patterns
found. Hence, all experimentally found dimer patterns were
predicted by MMH-2/AM1 as minima (Fig.6). MMH-2/AM1
is thus able to reproduce the most important hydrogen bond
motifs.
In order to provide a comparison among the structures
obtained by MMH-2/AM1 and more sophisticated ab initio
methods, the energies are presented in Table 1.
For MMH-2/AM1, all energies obtained by the calculated
dimer structures of acetamide are basically equivalent to MP2. d2
is the most important dimer, followed by d3, with d1 and d4
having the smallest interaction energies. Note that for MP2
without diffuse functions, d3, d1 and d4 are basically equivalent,
and it is imperative to include diffuse basis functions in such
a study.
This journal is ª The Royal Society of Chemistry 2009
The d2 structure has two strong hydrogen bonds (Fig. 6). This
corresponds to the experimental findings: d2 is the dimer present
in tACEMID06 form. From the geometry optimizations
obtained by MP2 (supplementary material‡), d2 and d3 do
not change much when the AM1 optimized structure is provided.
For the other two structures d1 and d4, which are the
starting blocks for the growing of linear chains in the crystal, the
deviations from the experiments and AM1 are larger. They
do not distort completely, however, so that the building
blocks are still visible. These results can be confirmed when using
AM1 to optimize the MP2 structures. The deviation between
MP2 and AM1 is thus likely to be caused by the AM1
parametrization.
Hydrogen bond distances from MP2 results are smaller than
those found in the crystal patterns (Table 2). This is because of
the lack of other acetamide molecules in the calculations in the
CrystEngComm, 2009, 11, 2358–2370 | 2363

Table 1 Energy differences (kcal mol�1) of fully optimized acetamide dimers by different methodologies
Acetamide dimer configuration DEMMH-2/AM1 DEMP2/6-311G** DEMP2/aug0-cc-pVTZ DEMP2/aug0-cc-pVQZ (SP)
d1 3.89 5.86 7.03 5.74d2 0 0 0 0d3 2.35 5.05 5.49 4.01d4 4.04 5.78 6.93 5.89
Table 2 Geometrical parameters experimental and theoretical of acetamide dimers
Conf d(H-bond)/�A Exp. MMH-2/AM1 MP2/aug0-cc-pVTZ MP2/6-311G**
d1 CO–NH2 1.86 2.19 1.94 1.99NH2–CH3 3.08 3.11 4.05 3.89
d2 CO–NH2 2.02 2.05 1.82 1.882.05 2.07 1.82 1.88
d3 NH2–CO 1.89 2.09 1.88 1.94CO–CH3 2.72 2.23 2.29 2.30
d4 CO–NH2 2.01 2.21 1.88 1.99
Fig. 7 Superimposed experimental and predicted structures by MMH-2/
AM1 of acetamide trimers. t2, t3, t4, t5 and t6 correspond to ACEMID06,
whereas t1 and t6 can be found in ACEMID03.
gas phase, as normally occur in the crystal. Since the hydrogen
bond interaction energies are not additive, a hydrogen bond in
the crystal will be somewhat weaker. This phenomenon is also
present in AM1 optimizations. Here, the parameterization of
AM1 probably yields worse structures for the gas phase, but
better structures for the crystal, giving the right answer for the
wrong reason. As shown in Table 1 both d1 and d4, which are
found in the experimental patterns, have the largest energies for
all methods. d2 and d3 correspond to each polymorph, which
means that by MP2, both polymorph building blocks are pre-
dicted. However, d1 and d4 are needed for predicting the full
crystal of both polymorphs, since these interactions are impor-
tant for the interactions of the individual d2 and d3 building
blocks.
The structures of acetamide trimers, tetramers, pentamers
and hexamers were built using the dimer configurations as
building blocks for larger simulations. For comparison,
MMH-1/AM1 was applied as well, but the methodology
failed completely, giving none of the experimentally found
structures.
In MMH-2/AM1, two main pathways were followed to
construct the corresponding structures: (1) generation of acet-
amide trimers, tetramers, pentamers and hexamers starting from
isolated acetamide molecules and (2) generation of acetamide
trimers, tetramers, pentamers and hexamers starting from the
most important dimer (or n � 1 order) motifs as building blocks
(Scheme 1). The results are summarized as follows:
4.1.2 Acetamide trimer configurations. MMH-2/AM1
provided all the trimer acetamide patterns obtained from the
graph set analysis of the experimental patterns. Here, the six
lowest energy structures correspond to the experimental findings.
For trimers, the populations of states of MMH-2/AM1 and their
consequent optimizations only lead to these local minima. As we
can see in Fig. 7, the superimposed structures between calculated
and experimental patterns are not always exactly the same, but
the approach reproduces the structural motifs.
Table 3 shows the energetic ranking of the trimers by different
methodologies. In this case, MMH-2/AM1 finds a difference
2364 | CrystEngComm, 2009, 11, 2358–2370
between linear trimer patterns and the more associated ones. The
MP2 and DFT methods basically yield the same energetic
pattern but the optimized geometries of the MP2 calculations
again deviate from both the experimental patterns and MMH-2/
AM1, especially in the cases of t5 and t6 (ESI‡). Those corre-
spond to the two linear structures found in the experimental
patterns. Again, these two structures are highest in energy, as
predicted by all methods used. In contrast to d2 and d3, t5 and t6
do not exist in the gas phase and get completely distorted by MP2
(as shown in Fig. 8). DFT, lacking a good performance to van
der Waals interactions, interestingly yields the same results as
AM1. However, they correspond to experimentally important
This journal is ª The Royal Society of Chemistry 2009

Table 3 Energetic differences (kcal mol�1) of acetamide trimers among different methods
Acetamide trimer configuration DEMMH-2/AM1 DEMP2/aug0-cc-pVTZ DE CP2K HCTH120/TZVP
t1 3.35 3.44t2 0 1.57 0t3 2.67 0 0.23t4 2.66 0.11 0.85t5 5.29 4.24 5.08t6 5.44 4.64 3.76
Fig. 8 Geometrical orientations of t6 by different methods.
geometrical patterns. Since we are looking for the most impor-
tant interactions that favour the growth of the crystal, a majority
of them are linear structures. The success of the MMH-2/AM1
approach is the combinations of several steps (Scheme 1) that
includes a selection of representative clusters by statistical ther-
modynamics, a good strategy for the generation of the random
configurations and the use of a semiempirical Hamiltonian for
the final optimization. The potential energy surface of AM1 is in
some cases quite flat, providing geometries of several local
minima very close to the experimental patterns, without moving
the structures to the global minimum. In the case of trimers, the
found structures do not always correspond to the minimum in
the gas phase, but model the crystal structure with a surprising
Table 4 Geometrical parameters of acetamide trimers by different methodo
Configuration d(H-bond)/�A Exp. MMH-2/A
t1 CO–CH3 (1) 2.01 2.24CO–NH2 (1) 1.89 2.09CO–CH3 (2) 2.73 2.28CO–NH2 (2) 1.89 2.08
t2 CO–NH2 (1) 2.09 2.15CO–NH2 (2) 2.05 2.09CO–NH2 (3) 2.02 2.05
t3 CO–NH2 (1) 2.09 2.15CO–NH2 (2) 2.05 2.09CO–NH2 (3) 2.02 2.05
t4 CO–NH2 (1) 2.01 2.20CO–NH2 (2) 2.02 2.10CO–NH2 (3) 2.05 2.05
t5 CO–NH2 (1) 2.01 2.17CO–NH2 (2) 2.01 2.14
t6 CO–NH2 (1) 1.86 2.13CO–NH2 (2) 1.86 2.37
This journal is ª The Royal Society of Chemistry 2009
accuracy. A similar behaviour is presented in the results of
tetramers–hexamers.
The fact that MMH-2/AM1 is able to reproduce the associated
trimers (t2, t3 and t4) as the most stable ones, gives the possibility
to see the possible interactions among different layers in the
crystal.
The hydrogen bond distances obtained by different methods
are shown in Table 4, where we obtain the same behaviour as
with the dimers. The t5 and t6 MP2 results are not comparable
because of the above mentioned changed geometries.
Correlating the theoretical results of dimers and trimers with
the crystal structures of the different acetamide polymorphs we
obtain the following conclusions: from the MMH-2/AM1
method, we obtain four dimer structures (d1–d4), in which d1 and
d4 are more thermodynamically unstable (Table 1). For the
trimers (Fig. 6), the d2 building block is combined via d1 and d4,
however not with d3. We deduce that d2, d1 and d4 belong to the
same polymorph. These results are in agreement with
the experimental crystal structures found. t2, t3 and t4 (Fig. 6) are
the most stable structures and show interactions between two
different layers in the polymorph that contain the d2, d1 and d4
building blocks of ACEMID06. We can identify the trimer
structures t5 and t6 as linear chains of one of these layers of the
ACEMID06 polymorph and t1 as linear chain of the other
polymorph ACEMID03.
Commencing from these results, we build tetramers–hexamers
starting from different combinations of d3 and t1 building blocks
in order to model the growing of ACEMID03 and different
combinations of (d2, d1 and d4) and (t2, t3, t4, t5 and t6) in order to
logies
M1 CP2K (HCTH120/pVTZ) MP2/aug0-cc-pVTZ
2.63 1.891.95 2.282.69 1.871.97 2.281.98 2.162.00 1.861.89 1.801.98 1.932.00 1.861.89 1.771.89 1.811.93 1.871.91 1.921.89 N/A1.881.87 N/A1.88
CrystEngComm, 2009, 11, 2358–2370 | 2365

model the growing of ACEMID06. Additionally, all kinds of
possible tetramers–hexamers associations starting from the iso-
lated molecules were performed. For that reason, the theoretical
results are presented now correlated with the experimental result
of each crystal packing:
4.1.3 Modelling the main intermolecular associations that
support the growing of ACEMID06 crystal structure (tetramers,
pentamers and hexamers). MMH-2/AM1 finds again four tetra-
mers as the most stable configurations (q1–q4) in perfect agreement
with the experimental results. No other local minima were found.
The superimposed experimental and theoretical structures are
provided in the supplementary material.‡ The structures q3 and q4
are more associated configurations that show the interactions
among different layers (similar to t3 and t4 in Fig. 6). The struc-
tures q1 and q2 are linear chains corresponding to the trimers t5
and t6 in Fig.6, which were no minima on the MP2 surface.
This general behaviour was found for all the MP2 optimiza-
tions of the MMH-2/AM1 geometries, going from tetramers to
hexamers: here, the linear chains reproduced by AM1 changed to
more associated clusters using the MP2 method. However, AM1
is in good agreement with the experimental crystal structures and
yield valuable information for the associated state. The
geometrical orientations of the four tetramers obtained by MP2
and MMH-2/AM1 are provided in the supplementary material.‡
Tables 5 and 6 show the energetic and geometrical parameters
of the tetramer configurations, also calculated by MP2. q1 and q2
optimized by MP2 do not even closely resemble the AM1 or
experimental linear structures, completely distorting to more
Table 5 Energetic differences of acetamide tetramers among differentmethodologies
Acetamidetetramerconfiguration DEMMH-2/AM1 DEMP2/aug0-cc-pVTZ
q1 1.85 4.72q2 1.75 4.07q3 0 0q4 0.25 1.48
Table 6 Geometrical parameters experimental and theoretical of acet-amide tetramers
Config d(H-bond)/�A Exp. MMH-2/AM1 MP2/aug0-cc-pVTZ
q1 CO–CH3 (all) 3.02 2.36 N/ACO–NH2 (1) 2.01 2.17CO–NH2 (2) 2.01 2.14CO–NH2 (3) 2.01 2.14
q2 CO–NH2 (1) 1.87 2.17 N/ACO–NH2 (2) 1.87 2.14CO–NH2 (3) 1.87 2.14CO–CH3 2.92 2.36
q3 CO–NH2 (1) 2.01 2.16 1.81CO–NH2 (2) 2.01 2.16 1.87CO–NH2 (3) 2.02 2.05 1.92CO–NH2 (4) 2.05 2.09 1.87
q4 CO–NH2 (1) 2.01 2.16 1.81CO–NH2 (2) 2.01 2.16 1.87CO–NH2 (3) 2.02 2.05 1.92CO–NH2 (4) 2.05 2.09 1.87
2366 | CrystEngComm, 2009, 11, 2358–2370
associated structures. Still, the interaction energies remain much
lower compared to q3 and q4. In these last geometries the most
stable dimer (d2) is present, probably causing this effect. This
structure has two strong hydrogen bonds that favour the stability
of acetamide associations and is responsible for the lower energy
structure. A similar energetic behaviour can be seen in the MMH-
2/AM1 results, although q1 and q2 are recognized as stable
configurations which are in good agreement with experiment.
Considering the geometries in detail, we do not report the MP2
q1 and q2 results because of the above mentioned deviation. The
MMH-2/AM1 distances are close to experiment and the orien-
tation of the molecules (supplementary material‡) is quite good
even for these simulations. The hydrogen bonds obtained by
MP2 are again smaller than the values from the experiments, as
was the case for both dimers and trimers.
Again, detailed information on the geometrical orientation
and geometrical parameters of pentamers and hexamers is
provided in the supplementary material.‡ As before, linear
chains are found by MMH-2/AM1 in good agreement with the
experimental structures. For each of the pentamers and hexam-
ers, the associated cluster is obtained as the global MP2
minimum. In these larger simulations, new information can be
gathered about the structural motifs of the interacting molecular
synthons of these polymorphs (Fig. 9). Even in MP2, a large
building block of the crystal is predicted as minimum. It is
evident that dimer d2 favours the associations between different
layers or molecular synthons in the crystal. The distances of the
calculated interactions have the same behaviour already pre-
sented for dimmers, trimers and tetramers, where MP2 obtains
smaller H-bond distances in comparison to experiment (supple-
mentary material). The energetic differences of pentamers and
hexamers will be presented at the end of the article together with
the results of the second polymorph.
Fig. 9 Representative structures obtained by MMH-2/AM1 for pen-
tamers and hexamers.
This journal is ª The Royal Society of Chemistry 2009
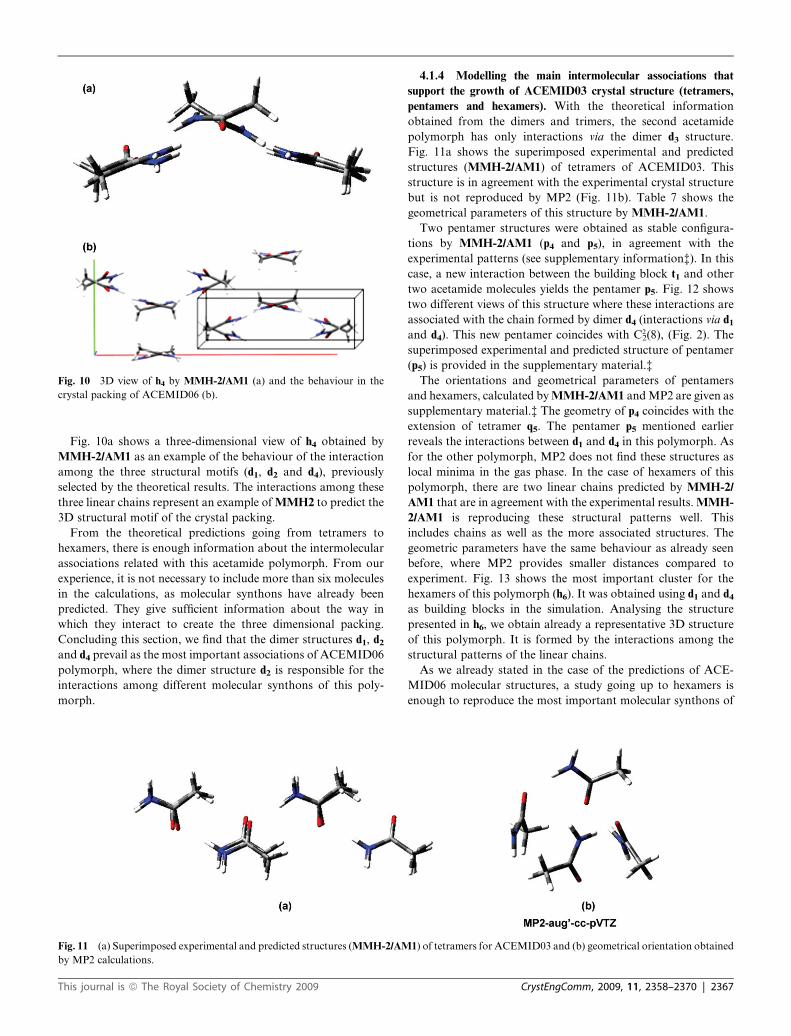
Fig. 10 3D view of h4 by MMH-2/AM1 (a) and the behaviour in the
crystal packing of ACEMID06 (b).
Fig. 10a shows a three-dimensional view of h4 obtained by
MMH-2/AM1 as an example of the behaviour of the interaction
among the three structural motifs (d1, d2 and d4), previously
selected by the theoretical results. The interactions among these
three linear chains represent an example of MMH2 to predict the
3D structural motif of the crystal packing.
From the theoretical predictions going from tetramers to
hexamers, there is enough information about the intermolecular
associations related with this acetamide polymorph. From our
experience, it is not necessary to include more than six molecules
in the calculations, as molecular synthons have already been
predicted. They give sufficient information about the way in
which they interact to create the three dimensional packing.
Concluding this section, we find that the dimer structures d1, d2
and d4 prevail as the most important associations of ACEMID06
polymorph, where the dimer structure d2 is responsible for the
interactions among different molecular synthons of this poly-
morph.
Fig. 11 (a) Superimposed experimental and predicted structures (MMH-2/AM
by MP2 calculations.
This journal is ª The Royal Society of Chemistry 2009
4.1.4 Modelling the main intermolecular associations that
support the growth of ACEMID03 crystal structure (tetramers,
pentamers and hexamers). With the theoretical information
obtained from the dimers and trimers, the second acetamide
polymorph has only interactions via the dimer d3 structure.
Fig. 11a shows the superimposed experimental and predicted
structures (MMH-2/AM1) of tetramers of ACEMID03. This
structure is in agreement with the experimental crystal structure
but is not reproduced by MP2 (Fig. 11b). Table 7 shows the
geometrical parameters of this structure by MMH-2/AM1.
Two pentamer structures were obtained as stable configura-
tions by MMH-2/AM1 (p4 and p5), in agreement with the
experimental patterns (see supplementary information‡). In this
case, a new interaction between the building block t1 and other
two acetamide molecules yields the pentamer p5. Fig. 12 shows
two different views of this structure where these interactions are
associated with the chain formed by dimer d4 (interactions via d1
and d4). This new pentamer coincides with C22(8), (Fig. 2). The
superimposed experimental and predicted structure of pentamer
(p5) is provided in the supplementary material.‡
The orientations and geometrical parameters of pentamers
and hexamers, calculated by MMH-2/AM1 and MP2 are given as
supplementary material.‡ The geometry of p4 coincides with the
extension of tetramer q5. The pentamer p5 mentioned earlier
reveals the interactions between d1 and d4 in this polymorph. As
for the other polymorph, MP2 does not find these structures as
local minima in the gas phase. In the case of hexamers of this
polymorph, there are two linear chains predicted by MMH-2/
AM1 that are in agreement with the experimental results. MMH-
2/AM1 is reproducing these structural patterns well. This
includes chains as well as the more associated structures. The
geometric parameters have the same behaviour as already seen
before, where MP2 provides smaller distances compared to
experiment. Fig. 13 shows the most important cluster for the
hexamers of this polymorph (h6). It was obtained using d1 and d4
as building blocks in the simulation. Analysing the structure
presented in h6, we obtain already a representative 3D structure
of this polymorph. It is formed by the interactions among the
structural patterns of the linear chains.
As we already stated in the case of the predictions of ACE-
MID06 molecular structures, a study going up to hexamers is
enough to reproduce the most important molecular synthons of
1) of tetramers for ACEMID03 and (b) geometrical orientation obtained
CrystEngComm, 2009, 11, 2358–2370 | 2367

Table 7 Geometrical parameters experimental and theoretical of acet-amide tetramer q5
Config d(H-bond)/�A Exp. MMH-2/AM1 MP2/aug0-cc-pVTZ
q5 CO–CH3 (all) 3.02 2.36 N/ACO–NH2 (1) 2.01 2.17CO–NH2 (2) 2.01 2.14CO–NH2 (3) 2.01 2.14
Fig. 13 A 3D view of h6 by MMH-2/AM1 (a) and the behaviour in the
crystal packing of ACEMID03 (b).
ACEMID03. This also gives some insight into the 3D crystal
packing.
4.1.5 Energetic prediction of polymorph stabilities by MMH-
2/AM1. The energetic behaviour of the predicted structures from
dimers, trimers and tetramer has been presented above when
comparing each structure. Tables 8 and 9, in contrast, show the
dissociation energy of each acetamide cluster compared to the
monomer provided by MMH-2/AM1 and MP2. For MP2, we
can compare those energies to the ones usually obtained for
hydrogen bonds.32 The range for a single hydrogen bond is
between 2 and 13 kcal mol�1. The acetamide dimer has an
interaction energy of 15 kcal mol�1 having two hydrogen bonds
in the most stable dimer. The strength of the hydrogen bond in
NH3–H2O is, in comparison, 6.5 kcal mol�1.
The dissociation energy values presented in Tables 8 and 9 are
sensitive in the prediction of the crystal growing. Here, we
observe quite a different behaviour for the AM1 and MP2
methods. For AM1, trimers are very stable, while the predicted
tetramers are increasing the dissociation energy of the cluster by
only 2 kcal mol�1. When going from pentamers to hexamers, we
observe a large jump in the dissociation energy by 13 kcal mol�1
using AM1 and almost 30 kcal mol�1 using MP2. This is indic-
ative of the fact that one very important building block has been
obtained (Fig. 14). Here, we have obviously arrived at a very
stable structure. Overall, this shows the success of MMH-2 to
achieve the most stable isomers for larger structures, such as
hexamers. These can later be identified by ab initio methods, and
be preserved as a building block when predicting the crystal
structures.
Fig. 12 Different views of pentamer p5 which shows the inte
2368 | CrystEngComm, 2009, 11, 2358–2370
From Tables 8 and 9, we can also compare the stability of
the clusters between each other and predict the thermo-
dynamically most stable polymorph. The more stable acetamide
clusters (d2, t2, t3 and t4, q3 and q4, p3 and p4 and h3 and h4)
belong to ACEMID06. Therefore, from our predictions
ACEMID06 should be the thermodynamically more stable
polymorph.
ractions among two dimer structural motifs (d1 and d4).
This journal is ª The Royal Society of Chemistry 2009

Table 8 Formation energy (kcal mol�1) of acetamide clusters by MMH-2/AM1
Efdimer Eftrimer Eftetramer Efpentamer Efhexamers
d1 4.42 t1 11.92 q1 15.30 p1 20.95 h1 26.67d2 8.48 t2 15.28 q2 15.40 p2 21.12 h2 26.26d3 6.14 t3 12.55 q3 17.89 p3 27.00 h3 39.44d4 4.59 t4 12.62 q4 17.63 p4 23.30 h4 38.61
t5 9.98 q5 17.55 p5 20.37 h5 26.85t6 9.83 h6 27.58
Table 9 Formation energy (kcal mol�1) of acetamide clusters by MP2/aug0-cc-pVTZ
Efdimer Eftrimer Eftetramer Efpentamer Efhexamer
d1 8.31 t1 19.44 q1 N/A p1 N/A h1 N/Ad2 15.34 t2 21.47 q2 N/A p2 N/A h2 N/Ad3 9.85 t3 22.88 q3 31.53 p3 38.25 h3 66.77d4 8.40 t4 22.83 q4 31.28 p4 N/A h4 62.37
t5 N/A q5 N/A p5 N/A h5 N/At6 N/A h6 43.39
4.1.6 Possible contributions of MMH-2 to CSP. With the
results from MMH-2/AM1 we are presenting a way of predicting
the growth of crystal structures. For organic molecules, mainly
weak interactions favour the existence of stable and metastable
polymorphs in which active pharmaceutical ingredients (API)
have a crucial importance. Most computational methods of CSP
are based on searching the global minimum in the lattice energy.
This process is challenging because of the wide range of space
Fig. 14 Structure of the most stable pattern found by MMH-2/A
This journal is ª The Royal Society of Chemistry 2009
groups and cell dimensions which need to be considered. MMH-
2/AM1 provides the possibility to predict the most stable
intermolecular associations that favour the growing of crystal
structures without requiring any experimental input or symmetry
restrictions. Correlating to the theoretical results, we can also
recognize the structures that belong to different polymorphs with
their respective energetic stabilities. MMH-2 proved to be a very
promising approach and might be also combined with some
developed genetic algorithms methods46,47 in order to improve
the structural assembly. In our opinion, MMH-2 has a wide
range of applications due to the eminent importance of inter-
molecular interactions for the structure, function, and dynamics
of a vast number of chemical and biological systems. For CSP, it
could be an alternative, fast and useful tool.
Conclusions
We have introduced a novel approach (MMH-2) for the predic-
tion of intermolecular interactions with a broad range of appli-
cations and a contribution to crystal structure prediction. Our
results indicate that it is an important tool for the elucidation of
structural motifs of all possible molecular synthons. The interac-
tions among different layers in the crystal in one-dimensional and
three-dimensional structures are also observed. Moreover, it is an
efficient way to predict the existence of various polymorphic
structures and their respective thermodynamic stabilities.
MMH-2 was tested for the case of acetamide polymorphs,
giving the correct results compared to experimental findings.
Using AM1 as an underlying theoretical model, we were able to
predict all structures found in the graph set analysis of the
experimental patterns. As a more sophisticated method, we used
M1 and MP2 in good agreement with the crystal structure.
CrystEngComm, 2009, 11, 2358–2370 | 2369

MP2 for validation. MP2 does not always yield the linear asso-
ciated motifs found by AM1 as local minima in the gas phase.
Despite this drawback, the AM1 motifs proved very useful for
the construction of further associated structures in comparison
to experiment. For the very stable hexamer forms, MP2 finally
agrees with the AM1 results, showing how small building blocks
are formed and the crystal structure might evolve.
Thus, MMH-2 may be an important tool for the development
of crystal engineering.
Acknowledgements
E.C.H. acknowledges financial support by the Deutsche For-
schungsgemeinschaft, Forschergruppe 618 for research grant in
Essen, Germany, in 2007 and 2008. Most of the calculations were
performed using the computing facilities of the group of Prof.
Georg Jansen, Essen, whom we thank for useful discussions.
Additionally E.C.H. thanks Prof. Dr Michelle Parrinello for the
financial support of a scientific research grant in 2008, Dr Clo-
tilde S. Cucinotta for useful discussions and help for the DFT
calculations, and Sean Christopher Wood (Calgary, Canada) for
his kind review of the English version of this manuscript. We
want also acknowledge Prof. Ram�on Carb�o-Dorca for giving us
the access to TGSA program.
References
1 J. Bernstein, Polymorphism in Molecular Crystals, 2002, published inthe US by Oxford University Press. Inc., New York. ISBN-10:0198506058.
2 F. H. Allen, Acta Crsytallogr., Sect. B, 2002, 58, 380–388.3 G. R. Desiraju, Angew. Chem., Int. Ed., 2007, 46, 8342–8356.4 T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48–76.5 C. B. Aaker€oy, J. Desper and M. M. Smith, Chem. Commun., 2007,
3936–3938.6 J. D. Dunitz and A. Gavezzotti, Cryst. Growth Des., 2005, 5(6), 2180–
2189.7 S. L. Price, CrystEngComm, 2004, 6(61), 344–353.8 M. A. Neumann, F. J. J. Leusen and J. Kendrich, Angew. Chem., Int.
Ed., 2008, 47, 2427–2430.9 J. P. M. Lommerse, W. D. S. Motherwell, H. L. Ammon,
J. D. Dunitz, A. Gavezzotti, D. W. M. Hofmann, F. J. J. Leusen,W. T. M. Mooij, S. L. Price, B. Schweizer, M. U. Schmidt,B. P. van Eijck, P. Verwer and D. E. Williams, Acta Crystallogr.,Sect. B: Struct. Sci., 2000, 56, 697.
10 W. D. S. Motherwell, H. L. Ammon, J. D. Dunitz, A. Dzyabchenko,P. Erk, A. Gavezzotti, D. W. M. Hofmann, F. J. J. Leusen,J. P. M. Lommerse, W. T. M. Mooij, S. L. Price, H. Scheraga,B. Schweizer, M. U. Schmidt, B. P. van Eijck, P. Verwer andD. E. Williams, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 647.
11 G. M. Day, W. D. S. Motherwell, H. L. Ammon, S. X. M. Boerrigter,R. G. Della Valle, E. Venuti, A. Dzyabchenko, J. D. Dunitz,B. Schweizer, B. P. van Eijck, P. Erk, J. C. Facelli, V. E. Bazterra,M. B. Ferraro, D. W. M. Hofmann, F. J. J. Leusen, C. Liang,C. C. Pantelides, P. G. Karamertzanis, S. L. Price, T. C. Lewis,H. Nowell, A. Torrisi, H. A. Scheraga, Y. A. Arnautova,M. U. Schmidt and P. Verwer, Acta Crystallogr., Sect. B: Struct.Sci., 2005, 61, 511.
12 A. Dey, N. N. Pati and G. R. Desiraju, CrystEngComm, 2006, 8, 751.13 L. A. Montero, A. M. Esteva, J. Molina, A. Zapardiel, L. Hern�andez,
H. M�arquez and A. Acosta, J. Am. Chem. Soc., 1998, 120, 12023.14 E. Codorniu-Hern�andez, A. Mesa-Ibirico, R. Hern�andez-
Santiesteban, L. A. Montero-Cabrera, F. Martinez-Luzardo andW. D. Stohrer, THEOCHEM, 2005, 715(1–3), 227.
15 E. Codorniu-Hern�andez, A. Mesa-Ibirico, R. Hern�andez-Santiesteban,L. A. Montero-Cabrera, F. Martinez-Luzardo, J. L. Santana-Romero,T. Bormann and W. D. Stohrer, Int. J. Quantum Chem., 2005, 103(1), 82.
16 E. S�anchez-Garcı́a, L. A. Montero and W. Sander, J. Phys. Chem. A,2006, 110, 12613.
2370 | CrystEngComm, 2009, 11, 2358–2370
17 E. S�anchez-Garcı́a, M. Studentkowski, L. A. Montero andW. Sander, ChemPhysChem, 2005, 6, 618.
18 E. S�anchez-Garcı́a, L. George, L. A. Montero and W. Sander,J. Phys. Chem. A, 2004, 108(52), 11846.
19 W. Thiel, semiempirical Methods, Modern Methods and Algorithmsof Quantum Chemistry, Proceedings, 2nd edn, 2000, ed.J. Grotendorst, John von Neumann Institute for Computing, J€ulich,NIC Series, Vol. 3, ISBN 3-00-005834-6, pp. 261.
20 R. Carb�o-Dorca and X. Giron�es, Int. J. Quantum Chem., 2005, 101, 8.21 J. T. Tou, R. C. Gonz�alez, Pattern Recognition Principles,
Addison –Wesley, Reading, 1974.22 J. D. Petke, J. Comput. Chem., 1993, 14, 928.23 E. E. Hodgkin and W. G. Richards, Int. J. Quantum Chem., 1987, 14,
105.24 E. E. Hodgkin and W. G. Richards, Chem. Ber., 1988, 24, 1141.25 J. J. P. Stewart, MOPAC, Manual sixth edition, Frank J. Seiler.
Research Laboratory United State Air Force Academy, 1990.(MOPAC2009, James J. P. Stewart, Stewart ComputationalChemistry, Version 8.303W web: http://OpenMOPAC.net).
26 P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213.27 W. J. Hehre, L. Radom, P. V. R. Schleyer, J. A. Pople, Ab initio
Molecular Orbital Theory, Wiley, New York, 1986.28 M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria,
M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr., T. Vreven,K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi,V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega,G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota,R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda,O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian,J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts,R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli,J. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador,J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels,M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck,K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui,A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu,A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox,T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara,M. Challacombe, P. M. W. Gill, B. G. Johnson, W. Chen,M. W. Wong, C. Gonzalez and J. A. Pople, GAUSSIAN 03(Revision C.02), Gaussian, Inc., Wallingford, CT, 2004.
29 T. H. Dunning Jr., J. Chem. Phys., 1989, 90, 1007.30 R. A. Kendall, T. H. Dunning and R. J. Harrison Jr., J. Chem. Phys.,
1992, 96, 6796.31 TURBOMOLE, version 5.8, developed by R. Ahlrichs, F. Furche,
C. H€attig, W. Klopper, M. Sierka, F. Weigend, and coworkers.32 A. D. Boese, J. M. L. Martin and W. Klopper, J. Phys. Chem. A, 2007,
111, 11122.33 CP2K version 2.0.0 (development version), the CP2K developers
group 2007 freely available at http://cp2k.berlios.de/.34 J. Vande Vondele, M. Krack, F. Mohamed, M. Parrinello,
T. Chassaing and J. Hutter, Comput. Phys. Commun., 2005, 167, 103.35 G. Lippert, J. Hutter and M. Parrinello, Theor. Chem. Acc., 1999, 103,
124.36 S. Goedecker, M. Teter and J. Hutter, Phys. Rev. B: Condens. Matter
Mater. Phys., 1996, 54, 1703.37 C. Hartwigsen, S. Goedecker and J. Hutter, Phys. Rev. B: Condens.
Matter Mater. Phys., 1998, 58, 3641.38 M. Krack, Theor. Chem. Acc., 2005, 114, 145.39 A. D. Boese, N. L. Doltsinis, N. C. Handy and M. Sprik, J. Chem.
Phys., 2000, 112, 1670.40 J. Bernstein, R. E. Davis, L. Shimoni and N. Chang, Angew. Chem.,
Int. Ed. Engl., 1995, 34, 1555.41 F. P. A. Fabbiani, D. R. Allan, W. G. Marshall, S. Parsons,
C. R. Pulham and R. I. Smith, J. Cryst. Growth, 2005, 275, 185.42 I. F. Senti and D. Harker, J. Am. Chem. Soc., 1940, 62, 2008.43 G. A. Jeffrey, J. R. Ruble, R. K. McMullan, D. J. DeFrees,
J. S. Binkley and J. A. Pople, Acta Crystallogr., Sect. B: Struct.Crystallogr. Cryst. Chem., 1980, 36, 2292.
44 W. C. Hamilton, Acta Crystallogr., 1965, 18, 866.45 S. Watanabe, Y. Abe and R. Yoshizaki, J. Phys. Soc. Jpn., 1986, 55,
2400.46 A. R. Oganov and C. W. Glass, J. Chem. Phys., 2006, 124, 244704.47 E. Oger, N. R. M. Crawford, R. Kelting, P. Weis, M. M. Kappes and
R. Ahlrichs, Angew. Chem., Int. Ed., 2007, 46, 8503.
This journal is ª The Royal Society of Chemistry 2009
Copyright © 2022 FDOKUMEN




















