
Mitochondrial oxidative phosphorylation is a downstream regulator of nitric oxide effects on...
11
ARTHRITIS & RHEUMATISM Vol. 43, No. 7, July 2000, pp 1560–1570 © 2000, American College of Rheumatology MITOCHONDRIAL OXIDATIVE PHOSPHORYLATION IS A DOWNSTREAM REGULATOR OF NITRIC OXIDE EFFECTS ON CHONDROCYTE MATRIX SYNTHESIS AND MINERALIZATION KRISTEN JOHNSON, ALEXANDER JUNG, ANNE MURPHY, ALEXANDER ANDREYEV, JAMES DYKENS, and ROBERT TERKELTAUB Objective. Increased chondrocyte nitric oxide (NO) and peroxynitrite production appears to modulate decreased matrix synthesis and increased mineraliza- tion in osteoarthritis (OA). Because NO inhibits mito- chondrial respiration, this study was undertaken to directly assess the potential role of chondrocyte mito- chondrial oxidative phosphorylation (OXPHOS) in ma- trix synthesis and mineralization. Methods. We studied cultured human articular chondrocytes and immortalized costal chondrocytes (TC28 cells). We also assessed the effects of antimycin A and oligomycin (inhibitors of mitochondrial complexes III and V, respectively) on chondrocyte mitochondrial respiration, ATP synthesis, and inorganic pyrophos- phate (PPi) generation, and the mineralizing potential of released matrix vesicles (MV). Results. Articular chondrocytes and TC28 cells respired at comparable rates. Peroxynitrite and NO donors markedly suppressed respiration and ATP gen- eration in chondrocytes. Because NO exerts multiple effects on chondrocytes, we investigated the primary functions of mitochondrial respiration and OXPHOS. To do so, we identified minimally cytotoxic doses of antimycin and oligomycin, which both induced intracell- ular ATP depletion (by 50–80%), attenuated collagen and proteoglycan synthesis, and blocked transforming growth factor b from increasing intracellular ATP and elaboration of PPi, a critical inhibitor of hydroxyapatite deposition. Antimycin and oligomycin also abrogated the ability of the ATP-hydrolyzing enzyme plasma cell membrane glycoprotein 1 (PC-1) to increase chondro- cyte PPi generation. Finally, MV from cells treated with antimycin or oligomycin contained less PPi and precip- itated >50% more 45 Ca. Conclusion. Chondrocyte mitochondrial reserve, as NO-sensitive mitochondrial respiration–mediated ATP production, appears to support matrix synthesis and PPi elaboration and to regulate MV composition and mineralizing activity. NO-induced depression of chondrocyte respiration could modulate matrix loss and secondary cartilage mineralization in OA. The pathogenesis of osteoarthritis (OA) includes elaboration of increased amounts of nitric oxide (NO), as a consequence of up-regulation of chondrocyte- inducible NO synthase induced by interleukin-1 (IL-1) and other factors (1–3). In chondrocytes, increased NO generation promotes OA (3,4) through effects that include inhibition of chondrocyte cytoskeletal actin po- lymerization and b1 integrin–dependent signaling (5,6), decreased expression of IL-1 receptor antagonist and transforming growth factor b (TGFb) (7,8), suppression of chondrocyte collagen and proteoglycan synthesis (9,10), activation of metalloproteinases (11), and sup- pression of proliferation as well as promotion of apo- ptosis (4,12). Cytokine-induced and NO-induced oxida- tive stress, the potentiation of injury by other oxidants, the formation of peroxynitrite from NO and O 2 2 , and the nitration of proteins mediate several of the afore- mentioned effects of NO on chondrocytes (3,4,10,13,14). Presented in part at the 63rd Annual Scientific Meeting of the American College of Rheumatology, Boston, MA, November 1999. Supported in part by a Veterans Affairs Medical Service Merit Review award and by NIH grant P01-AG-07996. Mr. Jung is the recipient of an NIH Predoctoral Fellowship award (HL-07491). Dr. Terkeltaub is the recipient of an Arthritis Foundation Biomedical Sciences Research Award. Kristen Johnson, BA, Alexander Jung, BS, Robert Ter- keltaub, MD: Department of Veterans Affairs Medical Center, and University of California, San Diego; Anne Murphy, PhD, Alexander Andreyev, PhD, James Dykens, PhD: Mitokor Corporation, San Diego, California. Address reprint requests to Robert Terkeltaub, MD, Depart- ment of Veterans Affairs Medical Center, 3350 La Jolla Village Drive, San Diego, CA 92161. Submitted for publication December 17, 1999; accepted in revised form March 7, 2000. 1560
Transcript of Mitochondrial oxidative phosphorylation is a downstream regulator of nitric oxide effects on...

ARTHRITIS & RHEUMATISMVol. 43, No. 7, July 2000, pp 1560–1570© 2000, American College of Rheumatology
MITOCHONDRIAL OXIDATIVE PHOSPHORYLATION IS ADOWNSTREAM REGULATOR OF NITRIC OXIDE EFFECTS ONCHONDROCYTE MATRIX SYNTHESIS AND MINERALIZATION
KRISTEN JOHNSON, ALEXANDER JUNG, ANNE MURPHY, ALEXANDER ANDREYEV,JAMES DYKENS, and ROBERT TERKELTAUB
Objective. Increased chondrocyte nitric oxide(NO) and peroxynitrite production appears to modulatedecreased matrix synthesis and increased mineraliza-tion in osteoarthritis (OA). Because NO inhibits mito-chondrial respiration, this study was undertaken todirectly assess the potential role of chondrocyte mito-chondrial oxidative phosphorylation (OXPHOS) in ma-trix synthesis and mineralization.
Methods. We studied cultured human articularchondrocytes and immortalized costal chondrocytes(TC28 cells). We also assessed the effects of antimycin Aand oligomycin (inhibitors of mitochondrial complexesIII and V, respectively) on chondrocyte mitochondrialrespiration, ATP synthesis, and inorganic pyrophos-phate (PPi) generation, and the mineralizing potentialof released matrix vesicles (MV).
Results. Articular chondrocytes and TC28 cellsrespired at comparable rates. Peroxynitrite and NOdonors markedly suppressed respiration and ATP gen-eration in chondrocytes. Because NO exerts multipleeffects on chondrocytes, we investigated the primaryfunctions of mitochondrial respiration and OXPHOS.To do so, we identified minimally cytotoxic doses of
antimycin and oligomycin, which both induced intracell-ular ATP depletion (by 50–80%), attenuated collagenand proteoglycan synthesis, and blocked transforminggrowth factor b from increasing intracellular ATP andelaboration of PPi, a critical inhibitor of hydroxyapatitedeposition. Antimycin and oligomycin also abrogatedthe ability of the ATP-hydrolyzing enzyme plasma cellmembrane glycoprotein 1 (PC-1) to increase chondro-cyte PPi generation. Finally, MV from cells treated withantimycin or oligomycin contained less PPi and precip-itated >50% more 45Ca.
Conclusion. Chondrocyte mitochondrial reserve,as NO-sensitive mitochondrial respiration–mediatedATP production, appears to support matrix synthesisand PPi elaboration and to regulate MV compositionand mineralizing activity. NO-induced depression ofchondrocyte respiration could modulate matrix loss andsecondary cartilage mineralization in OA.
The pathogenesis of osteoarthritis (OA) includeselaboration of increased amounts of nitric oxide (NO),as a consequence of up-regulation of chondrocyte-inducible NO synthase induced by interleukin-1 (IL-1)and other factors (1–3). In chondrocytes, increased NOgeneration promotes OA (3,4) through effects thatinclude inhibition of chondrocyte cytoskeletal actin po-lymerization and b1 integrin–dependent signaling (5,6),decreased expression of IL-1 receptor antagonist andtransforming growth factor b (TGFb) (7,8), suppressionof chondrocyte collagen and proteoglycan synthesis(9,10), activation of metalloproteinases (11), and sup-pression of proliferation as well as promotion of apo-ptosis (4,12). Cytokine-induced and NO-induced oxida-tive stress, the potentiation of injury by other oxidants,the formation of peroxynitrite from NO and O2
2, andthe nitration of proteins mediate several of the afore-mentioned effects of NO on chondrocytes (3,4,10,13,14).
Presented in part at the 63rd Annual Scientific Meeting of theAmerican College of Rheumatology, Boston, MA, November 1999.
Supported in part by a Veterans Affairs Medical ServiceMerit Review award and by NIH grant P01-AG-07996. Mr. Jung is therecipient of an NIH Predoctoral Fellowship award (HL-07491). Dr.Terkeltaub is the recipient of an Arthritis Foundation BiomedicalSciences Research Award.
Kristen Johnson, BA, Alexander Jung, BS, Robert Ter-keltaub, MD: Department of Veterans Affairs Medical Center, andUniversity of California, San Diego; Anne Murphy, PhD, AlexanderAndreyev, PhD, James Dykens, PhD: Mitokor Corporation, SanDiego, California.
Address reprint requests to Robert Terkeltaub, MD, Depart-ment of Veterans Affairs Medical Center, 3350 La Jolla Village Drive,San Diego, CA 92161.
Submitted for publication December 17, 1999; accepted inrevised form March 7, 2000.
1560

Pathologic hydroxyapatite (HA) crystal deposi-tion and calcium pyrophosphate dihydrate (CPPD) crys-tal deposition are both common in OA (15,16). Signifi-cantly, the NO donor sodium nitroprusside directlyinduces calcification of articular cartilage tissue slices(17). In this context, NO-induced chondrocyte apoptosiscan be associated with the release of apoptotic bodies(18,19) that function as pro-mineralizing matrix vesicles(MV) able to provide a sheltered milieu for crystalnucleation (20).
A variety of NO donors have been demonstratedto suppress energy production by mitochondrial respira-tion in different cell types (21,22), an effect enhanced atlow O2 tensions (23). However, studies on chondrocyteshave not been previously reported. Because chondrocytematrix synthesis and mineralization are modulated bythe balance between ATP generation and consumption(24,25), the mechanisms by which chondrocytes generateenergy have been a topic of interest. In this regard,articular chondrocytes are specialized to survive in anavascular environment and to rely on long diffusionpathways to obtain O2 (26,27). Consistent with thisspecialization, cultured articular chondrocytes have aparticularly active rate of anaerobic glycolysis, which is apathway that produces ATP (28,29). Moreover, previoushistochemical study of articular cartilage has demon-strated that the anaerobic glycolysis–facilitating isoen-zymes lactate dehydrogenase 4 (LDH4) and LDH5 areparticularly prominent in chondrocytes in situ (26).However, the superficial and middle zones of articularcartilage are not anoxic (29), and in this context, mito-chondrial oxidative phosphorylation (OXPHOS) is 18times as efficient in ATP generation as is glycolysis(21,30). Furthermore, OXPHOS may account for up toone-fourth of total steady-state ATP production withinarticular cartilage, and possibly more under conditionsof increased energy demands associated with cartilagestress (27).
In OXPHOS, electron transport is coupled along4 enzyme complexes (I–IV) in the mitochondrial innermembrane, and ATP is synthesized from ADP at com-plex V (ATP synthase) (21,30). Electrons initially pro-vided to the respiratory chain by the carriers NADH andFADH2 are ultimately used to reduce O2. The move-ment of electrons between the first 4 complexes of thechain produces a proton gradient across the inner mito-chondrial membrane, which is one of the factors em-ployed by complex V to generate ATP (21,30).
One clue to potential functions of OXPHOS inarticular chondrocytes is provided by the finding that ingrowth plate cartilage, a change in redox potential isassociated with a marked drop in mitochondrial respira-
tion that sharply demarcates the onset of HA crystaldeposition by terminally differentiated chondrocytes(31,32). Importantly, a relatively copious elaboration ofinorganic pyrophosphate (PPi) is a constitutive charac-teristic of chondrocytes (25), and physiologically servesas a potent suppressor of HA crystal nucleation andpropagation (25,33–35). A major substrate for PPi gen-eration is ATP, via hydrolysis by nucleoside triphosphatepyrophosphohydrolases (NTPPPH) (25). The NTPPPHplasma cell membrane glycoprotein 1 (PC-1) serves as acritical regulator of extracellular PPi generation bychondrocytes (25), and PC-1 is a physiologic constituentof MV and apoptotic bodies in mineralizing cells (19,33).Moreover, defective PC-1 activity in vivo is associatedwith pathologic HA deposition in articular cartilage inttw/ttw mice (34). In this study, we tested the hypothesisthat mitochondrial reserve, as NO donor–suppressibleOXPHOS-induced ATP generation, could be a down-stream mediator of both chondrocyte matrix synthesisand mineralization.
MATERIALS AND METHODS
Reagents. Human recombinant TGFb1 was obtainedfrom R&D Systems (Minneapolis, MN). NOC-12 (N-ethyl2-[1-ethyl-2 hydroxy-2 nitrosohydramine] ethaneamine) wasfrom Calbiochem (La Jolla, CA). Sin-1 and all other reagentswere obtained from Sigma (St. Louis, MO), unless otherwiseindicated.
Chondrocytes, culture conditions, and assays of viabil-ity. TC28 cells (immortalized human primary juvenile costalchondrocytes established after immortalization of rib chondro-cytes [36] using a retrovirus expressing SV40-TAg and theneomycin-resistance selection marker [25]) were originallyobtained from Dr. M. Goldring (Harvard Medical School,Boston, MA). TC28 cells were cultured in Dulbecco’s modifiedEagle’s medium (DMEM)/Ham’s F-12 (1:1; Gibco BRL,Grand Island, NY) supplemented with 10% fetal calf serum(FCS), 1% glutamine, 100 units/ml penicillin, and 50 mg/mlstreptomycin (Omega Scientific, Tarzana, CA) and maintainedat 37°C at normal oxygen tension in the presence of 5% CO2.
Primary human knee articular chondrocytes were ob-tained from normal donors at the time of autopsy, as previ-ously described (25). Individual donors were studied in tripli-cate; cells from different donors were not pooled in any singleexperiment. Articular chondrocytes were cultured in high-glucose DMEM supplemented with 1% glutamine, 10% FCS,100 units/ml penicillin, and 50 mg/ml streptomycin and main-tained at 37°C with 5% CO2. All studies involving cell culturewere performed in complete medium (as described above)unless otherwise stated, and we verified type II collagen andaggrecan expression by reverse transcriptase–polymerase chainreaction, as previously described (25).
Cell viability was assessed by trypan blue dye exclusionand by LDH release (cytotoxicity assay; Promega, Madison,WI). Apoptosis was assessed by gross morphology in addition
OXIDATIVE PHOSPHORYLATION AND CHONDROCYTE FUNCTION 1561

to TUNEL assay (Promega) performed according to theprotocol recommended by the manufacturer.
Treatment with mitochondrial inhibitors and mea-surement of O2 consumption. Oligomycin or antimycin A wasadded to cells (confirmed to be .90% viable by trypan bluedye exclusion) at 0, 24, 48, and 72 hours in culture, as indicated.To measure the rate of O2 consumption, we used a Clarkelectrode (Rank Brothers, Bottisham, Cambridge, UK). Forstudies involving intact cell respiration, cells were cultured to;90% confluence, washed once, and then incubated withbuffer alone, 0.025 mM antimycin, or 0.02 mg/ml (0.025 mM)oligomycin in complete DMEM growth medium (as describedabove) for 24 hours. The cells were trypsinized, counted, andresuspended in the complete growth medium with constantstirring and stabilized at 37°C. O2 consumption was thenmeasured in cells in complete growth medium and designatedas follows: 1) state 3/4 (describes basal respiration with nosubstrate addition in intact cells, conditions under whichrespiration is intermediate between state 3 and state 4); 2)state 4 (caused by the addition of 5 mg/ml of oligomycin); 3)state 3U (maximal state 3: uncoupled respiration due totitrated addition of 10-ng/ml aliquots of carbonylcyanide-P–trifluoromethoxyphenylhydrazone).
For digitonin-permeabilized cell respiration studies,cells were grown to 90% confluence, washed once, and incu-bated with or without the peroxynitrite donor Sin-1 (500 mM)or the NO donor NOC-12 (250 mM) for 2 hours, with .98%viability confirmed by trypan blue dye exclusion. In each study,2.5 3 106 cells were resuspended in 0.25 ml of KCl medium(125 mM KCl, 2 mM K2HPO4, 20 mM HEPES, pH 7.0) withthe addition of 0.967 mM MgCl2, 5 mM potassium glutamate,5 mM potassium malate, 0.01% digitonin, and 250 mM EGTA.With cells undergoing constant stirring, the rate of O2 con-sumption was measured for state 3 respiration (by the additionof 0.4 mM ADP), and state 4 and 3U respiration as above.
Transfection and adenoviral infection of chondrocytes.Transient transfections were performed on 5 3 105 TC28 cellsin a 35-mm tissue culture dish using Lipofectamine Plus (LifeTechnologies, Grand Island, NY). Briefly, 2 mg of emptyplasmid DNA or wild-type human PC-1 in pcDNA3.1 (25) wasadded to the lipid reagent following a complex formation ofthe DNA to the “Plus” reagent. The cells were then incubatedwith plasmid DNA for 3 hours at 37°C in DMEM/F-12 mediumcontaining 1% FCS and then washed with phosphate bufferedsaline (PBS). Complete medium was added to the cells, whichwere then incubated for an additional 72 hours.
For adenoviral gene transfer, we followed a previouslydescribed protocol (25). Briefly, 5 3 105 cells were plated inculture dishes and incubated for 18 hours at 37oC. The cellswere washed twice with PBS, and then incubated for 6–8 hourswith 5 3 103 plaque-forming units/ml of empty virus orrecombinant virus containing wild-type human PC-1 in high-glucose DMEM containing 1% serum. Cells were then washedagain and cultured for an additional 72 hours.
Assays of ATP and lactate. To assay ATP, washed cells(5 3 105) were centrifuged for 15 seconds, and the cell pelletwas frozen immediately in a dry ice/ethanol bath. ATP wasmeasured by a luciferase method that employed a biolumines-cence assay kit (Sigma) (25). To assay lactate, cells werecultured in colorless, serum-free DMEM/F-12 or DMEMsupplemented with 1% glutamine and penicillin/streptomycinas above. Where indicated, 0.025 mM antimycin or 0.02 mg/ml
oligomycin was added for 72 hours. The medium was thentreated with an equal volume of 0.4M trichloroacetic acid(TCA) at 4oC for 10 minutes, centrifuged for 5 minutes at14,000g, and the supernatant measured for lactate content byreduction of NAD to NADH (measured at 340 nm in thepresence of excess NAD), using the Sigma Diagnostics Lactatekit.
PPi metabolism and cellular DNA assays. PPi wasmeasured and equalized for DNA concentrations, as previ-ously described (25). Specific activities of NTPPPH and alka-line phosphatase (AP), and their translocation to the plasmamembrane, were measured as described (25).
Assays of collagen and proteoglycan synthesis. Tomeasure proteoglycan synthesis, 3 3 106 cells were plated in a10-cm culture dish and the mitochondrial inhibitors addedafter 18 hours. After a further 48 hours in culture, 20 mCi/ml of35S–sodium sulfate was added to each plate in serum-freemedium. After 24 hours, the medium was collected and thecells were washed once with PBS, placed into 0.5M NaOH, andmixed for 48 hours at 4°C. The media and the cell extract wereeluted from Sephadex G-25M PD-10 columns (AmershamPharmacia, Piscataway, NJ) with 4M GuHCl. The elutedportion was added to scintillation fluid and counted.
To assay collagen production, 5 3 105 adherent cellswere treated with mitochondrial inhibitors, as indicated, for 48hours. The medium was replaced with serum-free mediumcontaining 5 ml/ml of 3H proline, 50 mg/ml ascorbate, and 100mg/ml of b-aminopropionitrile for 8 hours, and was thencollected and precipitated with 15% TCA. Cells were extractedin 1M NaCl, 1 mM N-ethylmaleimide, 0.2 mM phenylmethyl-sulfonyl fluoride, 0.75 mM EDTA and then precipitated with15% TCA. The precipitate was washed 3 times with 5% TCAand once with 5% TCA containing 0.5% tannic acid. The pelletwas dissolved and resuspended in 0.05M Tris HCl, pH 7.6, 5mM CaCl2, and 2.5 mM N-ethylmaleimide. Collagenase-sensitive protein per mg of total protein was determined byincubation for 2 hours with 80 units/ml of collagenase, followed
Figure 1. Rates of oxygen consumption by articular chondrocytes andTC28 cells in monolayer culture. Cells (2.5 3 106) were resuspended in0.25 ml of complete growth medium as described in Materials andMethods. These nonpermeabilized cells were monitored for oxygenconsumption using a Clark electrode in which state 3/4 rates, state 4 rates(with 5 mg/ml oligomycin added), and state 3 uncoupled (3U) rates (with200 ng/ml carbonylcyanide-P–trifluoromethoxyphenylhydrazone added)were determined as described in Materials and Methods (n 5 9 for TC28cells; n 5 9 replicates from 3 donors for articular chondrocytes [for eachchondrocyte experiment indicated here and in the other figures, chondro-cytes from individual donors were studied in triplicate]).
1562 JOHNSON ET AL

by addition of 10% TCA and 0.5% tannic acid, and addition ofthe supernatant to scintillation fluid.
MV isolation and 45Ca precipitation assay. Condi-tioned media from cultured cells were collected at 72 hoursafter transfection or adenoviral gene transfer as indicated, andMV collected by differential ultracentrifugation as previouslydescribed (33). MV fractions (0.04 mg protein in 0.025 ml)were added in triplicate to 0.5 ml calcifying medium (2.2 mMCaCl2, [1 mCi/ml 45Ca], 1.6 mM KH2PO4, with or without 1mM ATP disodium salt, 15 mM KCl, 10 mM NaHCO3, 50 mMN-Tris [hydroxymethyl] methyl-2 aminoethanesulfonic acid,pH 7.6), vortexed, and incubated at 37°C for 24 hours. Washedprecipitates were then prepared and counted (33).
Statistical analysis. Values are presented as themean 6 SD. Statistical analysis was performed using Student’st-test (paired 2-sample testing for means).
RESULTSMitochondrial respiration and ATP generation
by chondrocytes in culture. Human articular chondro-cytes and immortalized costal chondrocytes (TC28 cells)respired at a comparable rate in culture (Figure 1). We
Figure 2. Effects of the NO and O22 donor SIN-1 on chondrocytic
respiration. TC28 cells were incubated for 2 hours at 37°C with orwithout 500 mM SIN-1. The cells were then digitonin permeabilizedand respiration measured as described in Materials and Methods (n 512 replicates). p 5 P , 0.01.
Figure 3. Dose-dependent effects of antimycin and oligomycin on articular chondrocyte and TC28 cell viability andintracellular ATP concentration. A, Cells (5 3 105) in 5.0 ml were studied in triplicate in the presence of antimycinor oligomycin at the indicated concentrations, administered every 24 hours for a total of 72 hours. Total intracellularATP concentration was assayed as described in Materials and Methods. B, Cells (2 3 104) in 0.2 ml were studiedin triplicate in the presence of antimycin or oligomycin at the indicated concentrations, administered every 24 hoursfor a total of 72 hours, as described in Materials and Methods. The proportion of cells viable at 72 hours wasassessed by trypan blue dye exclusion (.200 cells counted for each replicate). Results obtained using the Promegacytotoxicity assay (lactate dehydrogenase release assay) were essentially identical.
OXIDATIVE PHOSPHORYLATION AND CHONDROCYTE FUNCTION 1563

treated TC28 cells for 2 hours with the NO and O22
donor Sin-1 (500 mM), which markedly inhibited the rateof respiration (Figure 2). Comparable results were ob-tained by treating with 250 mM of the NO donorNOC-12, which donates 2 equivalents of NO per mole-cule (data not shown). Furthermore, extension of theSin-1 or NOC-12 treatment of TC28 cells and articularchondrocytes for 6 hours and 24 hours decreased thelevels of intracellular ATP by ;50% (data not shown).Because of the broad variety of effects of NO onchondrocyte function cited above, we investigated thepotential primary significance of NO-induced suppres-sion of respiration in chondrocyte function. To do so, wetreated intact cells with the OXPHOS complex III
inhibitor antimycin A and the mitochondrial ATP syn-thase (complex V) inhibitor oligomycin (30).
Dose-response studies first identified, for furtherstudy, minimally cytotoxic daily doses of antimycin(0.025 mM) and oligomycin (0.02 mg/ml), which inducedintracellular ATP depletion (by 50–80% at 72 hours) inTC28 cells and articular chondrocytes (Figure 3). States3/4 and 3U respiration of intact (nonpermeabilized)chondrocytes were significantly inhibited by 0.025 mMantimycin (Figure 4). Furthermore, state 3/4 respirationin the intact cells was inhibited by 0.02 mg/ml oligomycin(Figure 4). Though depression of intracellular ATP wasobserved at these doses, neither antimycin nor oligomy-cin decreased lactate generation (data not shown), whichwas active at a robust basal level (mean 6 SD 100.5 63.8 mg/ml and 58.2 6 4.4 mg/ml lactate generated per 48hours by TC28 cells [n 5 8] and articular chondrocytes[n 5 15], respectively).
TGFb (10 ng/ml), which is known to augmentchondrocyte anaerobic glycolysis (24), increased intra-cellular ATP 2–3 fold in TC28 cells and articular chon-drocytes (Table 1). TGFb did not increase chondrocyterespiration (data not shown), and in oligomycin-treatedcells, TGFb remained able to double intracellular ATP(Table 1). However, TGFb raised the ATP concentra-tion only back to resting baseline levels (not to theTGFb-induced levels of ATP achieved in the absence ofoligomycin). Additional results with antimycin (Table 1)further indicated that integrity of OXPHOS was neededto support intracellular ATP levels significantly in both
Figure 4. Effects of selected doses of antimycin A and oligomycin onO2 consumption in articular chondrocytes and TC28 cells. Articularchondrocytes and TC28 cells were incubated for 24 hours with 0.025mM antimycin and 0.02 mg/ml oligomycin. Intact cell respirationstudies were performed as described in Materials and Methods, afterresuspension of 2.5 3 106 cells in 0.25 ml of growth media. State 3/4respiration was measured, and then 5 mg/ml of oligomycin and 500ng/ml of carbonylcyanide-P–trifluoromethoxyphenylhydrazone wereadded to monitor state 4 and state 3U respiration, respectively. A,Articular chondrocytes (n 5 6 [as 2 donors, each run in triplicate]).p 5 P , 0.05. B, TC28 cells (n 5 6 replicates). p 5 P , 0.01.
Table 1. Effects of selected doses of oligomycin and antimycin onintracellular ATP levels*
Condition
ATP, pmoles/mg DNA
Articularchondrocytes TC28 cells
Buffer control 435.2 6 4.2 393.8 6 29.8Oligomycin 221.2 6 14.0† 198.6 6 3.7†TGFb 1,158 6 6.9† 801.6 6 4.9†Oligomycin 1 TGFb 398.3 6 3.6 369.6 6 5.0Antimycin 93.73 6 5.9† 69.78 6 4.0†Antimycin 1 TGFb 456.6 6 9.6 299.6 6 2.0
* Articular chondrocytes or TC28 cells (5 3 105 cells) were cultured in5 ml of complete medium and treated with 10 ng/ml transforminggrowth factor b (TGFb), 0.025 mM antimycin, or 0.02 mg/ml oligomy-cin. The cells were incubated at 37°C for 48 hours, with antimycin andoligomycin replenished after the first 24 hours. The cells were thenwashed with phosphate buffered saline, frozen on dry ice, and the ATPassay performed as described in Materials and Methods (n 5 9).Values are the mean 6 SD.† P , 0.01 versus control.
1564 JOHNSON ET AL

resting and growth factor–stimulated chondrocytes inculture.
Mitochondrial functions, including cytochrome crelease, are central to apoptosis (37). However, apopto-sis requires ATP-dependent caspase activation and isgenerally inhibited following primary suppression of themitochondrial electron transport chain (38–41). Underthe conditions and doses chosen for 48–72 hours ofantimycin and oligomycin treatment, which included thepresence of 10% calf serum to provide peptide growthfactors, we saw no increase in apoptotic changes, such asrounding up of cells or blebbing, by gross morphologicassessment. Approximately 1% of untreated cells, 8% ofantimycin-treated cells, and 15% of oligomycin-treated
cells developed positive TUNEL staining (n 5 300each). In contrast, ;85% of the cells treated with apositive control for apoptosis induction (100 nM stauro-sporine) developed TUNEL staining, and gross mor-phology was consistent with widespread apoptoticchanges in the positive control cells. Thus, the effects ofantimycin and oligomycin on ATP generation by chon-drocytes were not simply attributable to apoptosis.
Effects of inhibition of mitochondrial respirationand ATP generation on chondrocyte functions. Chon-drocyte mineralization is modulated by production of analtered extracellular matrix and the release of MV intothis matrix (16,20). Thus, we assessed the potential roleof OXPHOS in regulating chondrocyte matrix synthesis,PPi elaboration, and the mineralizing potential of MV.We observed that antimycin and oligomycin treatmentboth markedly decreased proteoglycan and collagensynthesis (Figures 5 and 6).
Extracellular PPi critically regulates cartilage HAand CPPD crystal deposition (25,33–35), and TGFb,which is produced in abundance by hypertrophic chon-drocytes, is relatively unique among chondrocyte growthfactors in causing a marked increase of PPi elaboration(25). Antimycin and oligomycin both decreased basallevels of intracellular PPi and extracellular PPi (Figure
Figure 5. Effects of mitochondrial inhibitors on proteoglycan synthe-sis by cultured chondrocytes (A) and TC28 cells (B), as measured by35S incorporation. Cells (3 3 106) were plated in a 10-cm dish in 30.0ml medium, and 0.02 mg/ml oligomycin or 0.025 mM antimycin wasadded every 24 hours for 72 hours. After 48 hours of culture, 20 mCi/mlof 35S-sodium sulfate was added to each plate in serum-free medium.After a total of 72 hours, the medium was collected and the cells werewashed once with phosphate buffered saline and then scraped into0.5M NaOH and mixed for 48 hours at 4°C. The medium and the cellextract were eluted from Sephadex G-25M PD-10 columns with 4MGuHCl. The eluted portion was added to scintillation fluid andcounted (n 5 9 replicates in A and 9 replicates in B). p 5 P , 0.05.
Figure 6. Effects of mitochondrial inhibitors on collagen synthesis bycultured chondrocytes, as measured by 3H incorporation. Cells (5 3105) were plated in 5 ml medium, and 0.02 mg/ml of oligomycin or0.025 mM of antimycin was added every 24 hours. After the mitochon-drial inhibitors were added, the cells were cultured for a further 48hours. The medium was then removed from the plates and replacedwith serum-free medium containing 5 ml/ml of 3H-proline, 50 mg/mlascorbate, and 100 mg/ml of b-aminopropionitrile, and incubationcontinued for 8 hours. The medium was collected and immediatelyprecipitated with 15% trichloroacetic acid. The cells were lysed asdescribed in Materials and Methods. Collagenase-sensitive protein wasdetermined by digesting the total protein with 80 units/ml of collage-nase as described in Materials and Methods (n 5 9 replicates). p 5 P ,0.05.
OXIDATIVE PHOSPHORYLATION AND CHONDROCYTE FUNCTION 1565
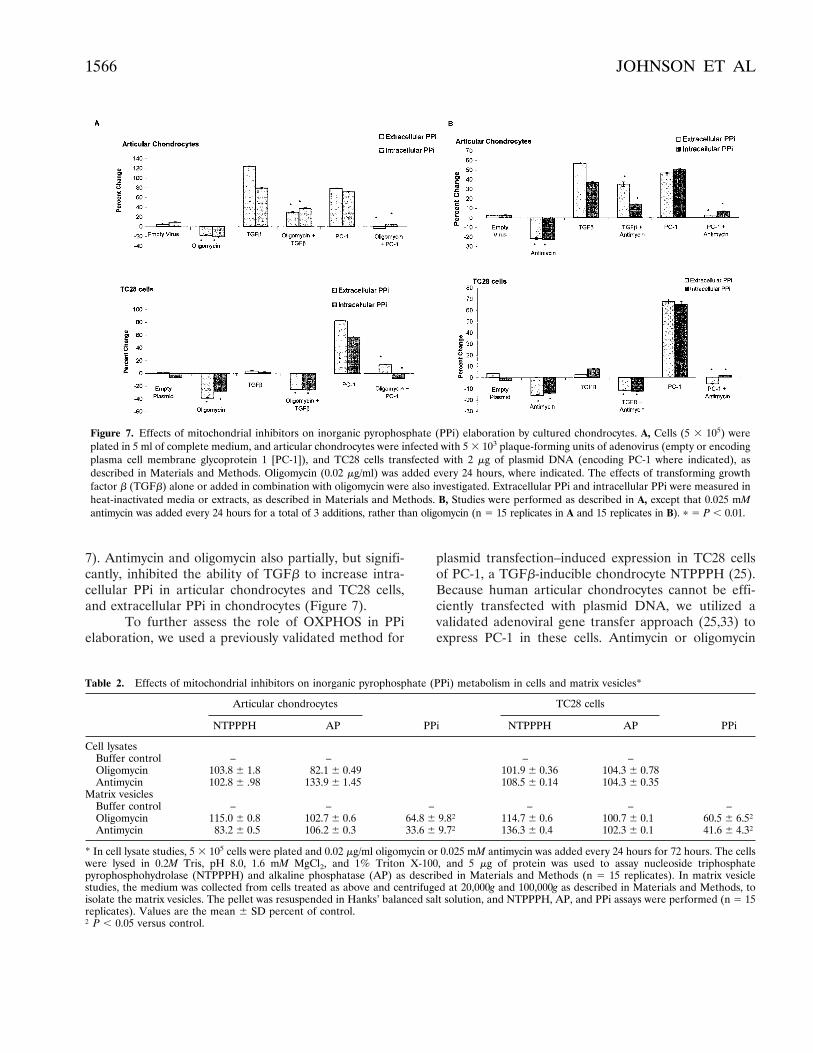
7). Antimycin and oligomycin also partially, but signifi-cantly, inhibited the ability of TGFb to increase intra-cellular PPi in articular chondrocytes and TC28 cells,and extracellular PPi in chondrocytes (Figure 7).
To further assess the role of OXPHOS in PPielaboration, we used a previously validated method for
plasmid transfection–induced expression in TC28 cellsof PC-1, a TGFb-inducible chondrocyte NTPPPH (25).Because human articular chondrocytes cannot be effi-ciently transfected with plasmid DNA, we utilized avalidated adenoviral gene transfer approach (25,33) toexpress PC-1 in these cells. Antimycin or oligomycin
Figure 7. Effects of mitochondrial inhibitors on inorganic pyrophosphate (PPi) elaboration by cultured chondrocytes. A, Cells (5 3 105) wereplated in 5 ml of complete medium, and articular chondrocytes were infected with 5 3 103 plaque-forming units of adenovirus (empty or encodingplasma cell membrane glycoprotein 1 [PC-1]), and TC28 cells transfected with 2 mg of plasmid DNA (encoding PC-1 where indicated), asdescribed in Materials and Methods. Oligomycin (0.02 mg/ml) was added every 24 hours, where indicated. The effects of transforming growthfactor b (TGFb) alone or added in combination with oligomycin were also investigated. Extracellular PPi and intracellular PPi were measured inheat-inactivated media or extracts, as described in Materials and Methods. B, Studies were performed as described in A, except that 0.025 mMantimycin was added every 24 hours for a total of 3 additions, rather than oligomycin (n 5 15 replicates in A and 15 replicates in B). p 5 P , 0.01.
Table 2. Effects of mitochondrial inhibitors on inorganic pyrophosphate (PPi) metabolism in cells and matrix vesicles*
Articular chondrocytes
PPi
TC28 cells
PPiNTPPPH AP NTPPPH AP
Cell lysatesBuffer control – – – –Oligomycin 103.8 6 1.8 82.1 6 0.49 101.9 6 0.36 104.3 6 0.78Antimycin 102.8 6 .98 133.9 6 1.45 108.5 6 0.14 104.3 6 0.35
Matrix vesiclesBuffer control – – – – – –Oligomycin 115.0 6 0.8 102.7 6 0.6 64.8 6 9.8† 114.7 6 0.6 100.7 6 0.1 60.5 6 6.5†Antimycin 83.2 6 0.5 106.2 6 0.3 33.6 6 9.7† 136.3 6 0.4 102.3 6 0.1 41.6 6 4.3†
* In cell lysate studies, 5 3 105 cells were plated and 0.02 mg/ml oligomycin or 0.025 mM antimycin was added every 24 hours for 72 hours. The cellswere lysed in 0.2M Tris, pH 8.0, 1.6 mM MgCl2, and 1% Triton X-100, and 5 mg of protein was used to assay nucleoside triphosphatepyrophosphohydrolase (NTPPPH) and alkaline phosphatase (AP) as described in Materials and Methods (n 5 15 replicates). In matrix vesiclestudies, the medium was collected from cells treated as above and centrifuged at 20,000g and 100,000g as described in Materials and Methods, toisolate the matrix vesicles. The pellet was resuspended in Hanks’ balanced salt solution, and NTPPPH, AP, and PPi assays were performed (n 5 15replicates). Values are the mean 6 SD percent of control.† P , 0.05 versus control.
1566 JOHNSON ET AL

treatment abrogated PC-1–induced increases in intra-cellular and extracellular PPi in articular chondrocytesand TC28 cells (Figure 7). Neither oligomycin norantimycin treatment suppressed cell-associatedNTPPPH activity, and neither agent altered PPi-degrading AP activity (Table 2). Furthermore, neitheroligomycin nor antimycin suppressed PC-1 translocationto the plasma membrane (data not shown), a TGFb-stimulated event critical for PC-1 to promote increasedextracellular PPi (25).
MV-induced cartilage matrix mineralization isgenerally enhanced by exogenous ATP (20). We ob-served that oligomycin and antimycin treatment of botharticular chondrocytes and TC28 cells induced a signif-icantly decreased concentration of PPi associated withcell-derived MV (Table 2). In addition, ATP-treatedMV derived from chondrocytes treated with oligomycinor antimycin precipitated .50% more 45Ca (Figure 8).Thus, direct suppression of mitochondrial respirationpromoted MV-mediated mineralization in chondro-cytes.
DISCUSSION
NO and peroxynitrite generation appear to mod-ulate the pathogenesis of matrix loss and articularcartilage calcification in OA (3,4,17). NO reversiblysuppresses and peroxynitrite can irreversibly suppressmitochondrial respiration in cells other than chondro-cytes (21–23). In this study, we observed that the per-oxynitrite generator Sin-1 (42) and the NO donorNOC-12 markedly suppressed mitochondrial respiration
and ATP levels in chondrocytes. Therefore, we focusedon the potential for mitochondrial OXPHOS to functionboth in the maintenance of intracellular energy (as ATP)and in the regulation of ATP-mediated chondrocytemineralization. In doing so, we first identified doses ofantimycin and oligomycin that inhibited respiration andinduced intracellular ATP depletion. Our results sug-gested a significant supportive role of OXPHOS-mediated ATP generation in collagen and proteoglycansynthesis.
A previous study of rabbit articular chondrocytes,which used citrate and isocitrate as substrates in stan-dard aerobic culture, demonstrated respiration at a basalrate that was ,10% of the rate of respiration of culturedhepatocytes, despite comparable rates of glycolysis (24).Findings in the current study indicated that intact humanarticular chondrocytes respired in aerobic culture.Moreover, cultured immortalized endochondral and pri-mary articular chondrocytes carried out respiration atcomparable rates. Significantly, the present study wasperformed under conditions that allowed detection of an;5-fold higher rate of chondrocyte respiration than inthe earlier study of rabbit chondrocytes (24). To accom-plish this, we used respiration substrates (glutamate andmalate) that presumably were less subject to transportlimitations for access to OXPHOS complexes in theinner mitochondrial membrane (30).
In this study, we cultured chondrocytes in thepresence of 10% serum to lessen cell death. Inhibition ofmitochondrial ATP generation generally inhibits apop-tosis (38–41). There was no morphologic evidence for anincrease in apoptosis in antimycin- and oligomycin-treated chondrocytes. Furthermore, the marked effectsof antimycin and oligomycin on the mitochondrial re-serve for ATP production, and on collagen and proteo-glycan synthesis, were substantially greater than thelimited effects of the mitochondrial inhibitors on thedevelopment of TUNEL staining. Thus, the short-termeffects on chondrocyte functions observed in response todepressed mitochondrial ATP synthesis were not attrib-utable to either cytotoxicity or apoptosis. However, thisstudy was limited to short-term cultures. Because attach-ment to the pericellular matrix is a central factor regu-lating apoptosis (37), dysregulated chondrocyte OX-PHOS, as well as reactive oxygen species generated fromdisruption of mitochondrial electron transport (14),have the potential to contribute to the enhancement ofapoptosis noted in OA chondrocytes in situ (43).
Intraarticular TGFb activity is significantly in-creased in OA (44), and TGFb increases glycolysis andATP levels in chondrocytes (24,25). We observed that
Figure 8. Effects of mitochondrial inhibitors on matrix vesicle (MV)–mediated mineralization. Articular chondrocytes (5 3 105) were platedin 5 ml of complete medium, and 0.02 mg/ml of oligomycin or 0.025mM of antimycin was added every 24 hours for a total of 72 hours. TheMV were collected as described in Figure 7 and resuspended in Hanks’balanced slat solution. Aliquots of 40 mg of MV protein were added tocalcifying media and incubated with or without ATP for 24 hours.Precipitated calcium per each 40-mg sample of MV protein wasdetermined as described in Materials and Methods (n 5 9 replicates).p 5 P , 0.05.
OXIDATIVE PHOSPHORYLATION AND CHONDROCYTE FUNCTION 1567

functional integrity of OXPHOS was required not onlyfor basal intracellular generation of ATP, but also forTGFb to drive maximal intracellular ATP generation.This finding suggests the potential for an increased roleof OXPHOS in chondrocyte function under conditionsof cartilage stress and increased energy demands, asmight be associated with OA and cartilage repair. Be-cause TGFb did not by itself directly stimulate chondro-cyte respiration, it is possible that OXPHOS may beneeded to support TGFb-induced changes in chondro-cyte differentiation.
Our findings demonstrated that OXPHOS pro-vided critical support for not only basal and TGFb-induced changes in the level of the NTPPPH substrateATP, but also TGFb-induced and PC-1/NTPPPH–induced generation of PPi by cultured chondrocytes.The mechanisms of PPi elaboration by TC28 cells andarticular chondrocytes exhibit certain distinctions (25),reinforcing the importance of the central supportive roleof OXPHOS in PPi elaboration by both chondrocytetypes used in this study. Significantly, increased cartilageNTPPPH activity, increased ambient extracellular ATPlevels in the joint space, and excess PPi elaboration arestrongly linked to idiopathic CPPD crystal depositiondisease in aging (16,25,45). In degenerating articularcartilage, a close physical association has been observedbetween CPPD crystal deposits, which concentrate inthe cartilage midzone, and chondrocytes that progressedfrom resting to hypertrophic differentiation (46). More-over, hypertrophic chondrocytes are more metabolicallyactive than resting cells, are responsive to TGFb, elab-orate increased PPi, and shed more MV (47,48). OX-PHOS could provide a reserve for both increased met-abolic activity and PPi elaboration by hypertrophicchondrocytes.
HA crystal deposition, like CPPD crystal deposi-tion, is common in articular cartilage affected by OA,particularly in advanced disease (15). Significantly, theonset of HA deposition around hypertrophic endochon-dral cartilage chondrocytes coincides with sharply de-fined changes in the redox and metabolic states ofchondrocytes, in association with focal loss of cell respi-ration (31). Moreover, a marked decrease in MV PPi isobligatory for HA crystal deposition to be initiated inthe interior of MV in vitro (33,35). In addition, mainte-nance of a threshold level of PPi elaboration, partly bythe action of PC-1 (25), also prevents normal articularcartilage from mineralizing with HA in vivo (34).
Previously described changes in mitochondrialfunction that directly modulate mineralization includeregulation of intramitochondrial calcium accumulation
and release of intramitochondrial calcium stores (49).Results of this study suggest additional models in whichattenuation of OXPHOS serves to help remove physio-logic suppression of HA deposition by chondrocytes, inboth articular and endochondral cartilage, via primaryregulatory effects on ATP availability and PPi genera-tion. A model based purely on mitochondrial ATPgeneration is supported by evidence for a critical effectof extracellular ATP availability on the differentialdeposition of HA and CPPD crystals ex vivo by MVisolated from OA articular cartilage, where exogenousATP favored MV CPPD deposition over hydroxyapatitedeposition (50). Therefore, chondrocyte OXPHOScould partly regulate differential deposition of HA andCPPD.
Chondrocyte mitochondrial OXPHOS also regu-lated mineralization at the levels of the MV content ofPPi and the calcium-precipitating ability associated withMV. Our findings on MV PPi content reinforce previousevidence that the composition (and function) of MV canreflect the underlying metabolic activity and PPi metab-olism of cells that give rise to these structures (19,33).Significantly, the NO donor sodium nitroprusside, whichinduced calcification of articular cartilage slices (17), candonate cyanide, which inhibits OXPHOS at complex IV(30). Moreover, sodium nitroprusside induced the re-lease from cultured chondrocytes of apoptotic bodiesand MV which precipitated more calcium than MVderived from untreated chondrocytes (17). Thus, it willbe of interest to directly test in vivo whether bothprimary dysregulation of OXPHOS and secondary OX-PHOS suppression by NO modulates cartilage mineral-ization at the level of MV in OA. NO production bystimulated chondrocytes appears to decrease with ad-vanced age and is lower in the middle than in thesuperficial zone of articular cartilage (51). Thus, wepropose a model in which it is speculated that dimin-ished suppressive effects of NO on ATP and PPi-supported CPPD deposition may contribute to the in-creased frequency of idiopathic CPPD deposition withadvanced age, and the general focus of CPPD deposits inthe articular cartilage midzone (16).
We conclude that regulation of OXPHOS may beone of the signaling pathways by which NO modulatesarticular cartilage matrix biosynthesis and pathologicmineralization. Moreover, direct suppression of articularchondrocyte OXPHOS induces a decrease in matrixsynthesis and an increase in chondrocyte MV–mediatedmineralizing potential, events common to OA cartilage.Though the prevalence of OA and cartilage calcificationincrease with each decade in aging, the primary factors
1568 JOHNSON ET AL

that initiate and exacerbate these pathologic changes arenot completely understood. Oxidant-induced mitochon-drial damage, through aging, chronic cell stress, andinflammation (14,15), may account for varying rates ofcellular senescence among tissues, and the pathogenesisof several degenerative disorders (52,53). Thus, furtherinvestigation is indicated to assess the integrity of mito-chondrial electron transport in OA cartilage and thepotential for preservation of OXPHOS as a therapeutictarget in OA.
ACKNOWLEDGMENTS
We are grateful to J. Quach and Dr. M. Lotz (ScrippsResearch Institute, La Jolla, CA) for generously providingnormal human knee articular cartilage samples.
REFERENCES
1. Pelletier J, Jovanovic D, Fernandes JC, Manning P, Connor JR,Currie MG, et al. Reduction in the structural changes of experi-mental osteoarthritis by a nitric oxide inhibitor. OsteoarthritisCartilage 1999;7:416–8.
2. Hashimoto S, Takahashi K, Amiel D, Coutts RD, Lotz M. Chondro-cyte apoptosis and nitric oxide production during experimentallyinduced osteoarthritis. Arthritis Rheum 1998;41:1266–74.
3. Studer R, Jaffurs D, Stefanovic-Racic M, Robbins PD, Evans CH.Nitric oxide in osteoarthritis. Osteoarthritis Cartilage 1999;7:377–9.
4. Lotz M. The role of nitric oxide in articular cartilage damage.Rheum Dis Clin North Am 1999;25:269–82.
5. Frenkel SR, Clancy RM, Ricci JL, Di Cesare PE, Rediske JJ,Abramson SB. Effects of nitric oxide on chondrocyte migration,adhesion, and cytoskeletal assembly. Arthritis Rheum 1996;39:1905–12.
6. Clancy RM, Rediske J, Tang X, Nijher N, Frenkel S, Philips M, etal. Outside-in signaling in the chondrocyte: nitric oxide disruptsfibronectin-induced assembly of a subplasmalemmal actin/rhoA/focal adhesion kinase signaling complex. J Clin Invest 1997;100:1789–96.
7. Pelletier JP, Mineau F, Ranger P, Tardif G, Martel-Pelletier J. Theincreased synthesis of inducible nitric oxide inhibits IL-1ra synthesisby human articular chondrocytes: possible role in osteoarthriticcartilage degradation. Osteoarthritis Cartilage 1996;4:77–84.
8. Studer RK, Georgescu HI, Miller LA, Evans CH. Inhibition oftransforming growth factor b production by nitric oxide-treatedchondrocytes: implications for matrix synthesis. Arthritis Rheum1999;42:248–57.
9. Stefanovic-Racic M, Morales TI, Taskiran D, McIntyre LA, EvansCH. The role of nitric oxide in proteoglycan turnover by bovinearticular cartilage organ cultures. J Immunol 1996;156:1213–20.
10. Oh M, Fukuda K, Asada S, Yasuda Y, Tanaka S. Concurrentgeneration of nitric oxide and superoxide inhibits proteoglycansynthesis in bovine articular chondrocytes: involvement of per-oxynitrite. J Rheumatol 1998;25:2169–74.
11. Murrell GA, Jang D, Williams RJ. Nitric oxide activates metallo-protease enzymes in articular cartilage. Biochem Biophys ResCommun 1995;206:15–21.
12. Blanco FJ, Lotz M. IL-1-induced nitric oxide inhibits chondrocyteproliferation via PGE2. Exp Cell Res 1995;218:319–25.
13. Clancy RM, Abramson SB, Kohne C, Rediske J. Nitric oxideattenuates cellular hexose monophosphate shunt response to
oxidants in articular chondrocytes and acts to promote oxidantinjury. J Cell Physiol 1997;172:183–91.
14. Rediske J, Koehne C, Stoyanovsky S, Abramson S, Clancy B.TNFa sensitizes articular chondrocytes to oxidant stress [abstract].Arthritis Rheum 1999;42 Suppl 9:S404.
15. Halverson PB, McCarty D. Basic calcium phosphate (apatite,octacalcium phosphate, tricalcium phosphate) crystal depositiondiseases; calcinosis. In: Koopman W, editor. Arthritis and alliedconditions: a textbook of rheumatology. 13th ed. Baltimore:Williams and Wilkins; 1997. p. 2127–46.
16. Ryan LM, McCarty DJ. Calcium pyrophosphate crystal depositiondisease, pseudogout, and articular chondrocalcinosis. In: Koop-man W, editor. Arthritis and allied conditions: a textbook ofrheumatology. 13th ed. Baltimore: Williams and Wilkins; 1997. p.2103–26.
17. Cheung HS, Ryan LM. Phosphocitrate blocks nitric oxide-inducedcalcification of cartilage and chondrocyte-derived apoptotic bod-ies. Osteoarthritis Cartilage 1999;7:409–12.
18. Blanco FJ, Ochs RL, Schwarz H, Lotz M. Chondrocyte apoptosisinduced by nitric oxide. Am J Pathol 1995;146:75–85.
19. Hashimoto S, Ochs RL, Rosen F, Quach J, McCabe G, Solan J, etal. Chondrocyte-derived apoptotic bodies and calcification ofarticular cartilage. Proc Natl Acad Sci U S A 1998;95:3094–9.
20. Anderson HC. Molecular biology of matrix vesicles. Clin Orthop1995;314:266–80.
21. Brookes PS, Bolanos JP, Heales SJ. The assumption that nitricoxide inhibits mitochondrial ATP synthesis is correct. FEBS Lett1999;446:261–3.
22. Brown GC. Nitric oxide and mitochondrial respiration. BiochimBiophys Acta 1999;1411:351–69.
23. Koivisto A, Pittner J, Froelich M, Persson AE. Oxygen-dependentinhibition of respiration in isolated renal tubules by nitric oxide.Kidney Int 1999;55:2368–75.
24. Stefanovic-Racic M, Stadler J, Georgescu HI, Evans CH. Nitricoxide and energy production in articular chondrocytes. J CellPhysiol 1994;159:274–80.
25. Johnson K, Vaingankar S, Chen Y, Moffa A, Goldring MB, SanoK, et al. Differential mechanisms of inorganic pyrophosphateproduction by plasma cell membrane glycoprotein-1 and B10 inchondrocytes. Arthritis Rheum 1999;42:1986–97.
26. Tushan FS, Rodnan GP, Altman M, Robin ED. Anaerobicglycolysis and lactate dehydrogenase (LDH) isozymes in articularcartilage. J Lab Clin Med 1969;73:649–50.
27. Lee RB, Urban JP. Evidence for a negative Pasteur effect inarticular cartilage. Biochem J 1997;321:95–102.
28. Marcus RE, Srivastava VML. Effect of low oxygen tension onglucose-metabolizing enzymes in cultured articular chondrocytes.Soc Exp Biol Med Proc 1973;143:488–91.
29. Oegema TR Jr, Thompson RC. Metabolism of chondrocytesderived from normal and osteoarthritic human cartilage. In:Kuettner K, editor. Articular cartilage biochemistry. New York:Raven Press; 1986. p. 257–71.
30. Murphy AN, Fiskum G, Beal MF. Mitochondria in neurodegen-eration: bioenergetic function in cell life and death. J Cereb BloodFlow Metab 1999;19:231–45.
31. Shapiro IM, Golub EE, Kakuta S, Hazelgrove J, Harvey J, ChanceB. Initiation of endochondral calcification is related to changes inthe redox state of hypertrophic chondrocytes. Science 1982;217:950–2.
32. Matsumoto H, DeBolt K, Shapiro IM. Adenine, guanine, andinosine nucleotides of chock growth cartilage: relationship be-tween energy status and the mineralization process. J Bone MinerRes 1988;3:347–52.
33. Johnson K, Moffa A, Pritzker K, Chen Y, Goding J, Terkeltaub R.Matrix vesicle plasma cell membrane glycoprotein-1 (PC-1) regu-lates mineralization by murine osteoblastic MC3T3 cells. J BoneMiner Res 1999;14:883–92.
OXIDATIVE PHOSPHORYLATION AND CHONDROCYTE FUNCTION 1569

34. Okawa A, Nakamura I, Goto S, Moriya H, Nakamura Y, IkegawaS. Mutation in Npps in a mouse model of ossification of theposterior longitudinal ligament of the spine. Nat Genet 1998;19:271–3.
35. Meyer JL. Can biological calcification occur in the presence ofpyrophosphate? Arch Biochem Biophys 1984;231:1–8.
36. Goldring MB, Birkhead JR, Suen LF, Yamin R, Mizuno S,Glowacki J, et al. Interleukin-1 beta-modulated gene expression inimmortalized human chondrocytes. J Clin Invest 1994;94:2307–16.
37. Green DR, Reed JC. Mitochondria and apoptosis. Science 1998;281:1309–12.
38. Petit PX, Zamzami N, Vayssiere JL, Mignotte B, Kroemer G,Castedo M. Implication of mitochondria in apoptosis. Mol CellBiochem 1997;174:185–8.
39. Galitovsky VE, Gogvadze VG. Inhibitors of mitochondrial energyproduction prevent DNA internucleosomal fragmentation in thy-mocytes. Biochemistry 1998;63:1374–7.
40. Nicotera P, Leist M, Ferrando-May E. Intracellular ATP, a switchin the decision between apoptosis and necrosis. Toxicol Lett1998;102-103:139–42.
41. Leist M, Single B, Naumann H, Fava E, Simon B, Kuhnle S, et al.Nitric oxide inhibits execution of apoptosis at two distinct ATP-dependent steps upstream and downstream of mitochondrialcytochrome c release. Biochem Biophys Res Commun 1999;258:215–21.
42. Singh RJ, Hogg N, Joseph J, Konorev E, Kalyanaraman B. Theperoxynitrite generator, SIN-1, becomes a nitric oxide donor in thepresence of electron acceptors. Arch Biochem Biophys 1999;361:331–9.
43. Blanco FJ, Guitian R, Vazquez-Martul E, de Toro FJ, Galdo F.Osteoarthritis chondrocytes die by apoptosis: a possible pathwayfor osteoarthritis pathology. Arthritis Rheum 1998;41:284–9.
44. Moos V, Fickert S, Muller B, Weber U, Sieper J. Immunohisto-logical analysis of cytokine expression in human osteoarthritic andhealthy cartilage. J Rheumatol 1999;26:870–9.
45. Ryan LM, Rachow JW, McCarty DJ. Synovial fluid ATP: apotential substrate for the production of inorganic pyrophosphate.J Rheumatol 1991;18:716–20.
46. Ishikawa K, Masuda I, Ohira T, Yokoyama M. A histological studyof calcium pyrophosphate dihydrate crystal-deposition disease.J Bone Joint Surg Am 1989;71:875–86.
47. Rosenthal AK, Henry LA. Thyroid hormones induce features ofthe hypertrophic phenotype and stimulate correlates of CPPDcrystal formation in articular chondrocytes. J Rheumatol 1999;26:395–401.
48. Kirsch T, Nah HD, Shapiro IM, Pacifici M. Regulated productionof mineralization-competent matrix vesicles in hypertrophic chon-drocytes. J Cell Biol 1997;137:1149–60.
49. Brighton CT, Hunt RM. Mitochondrial calcium and its role incalcification. Clin Orthop 1974;100:406–16.
50. Derfus B, Kranendonk S, Camacho N, Mandel N, Kushnaryov V,Lynch K, et al. Human osteoarthritic cartilage matrix vesiclesgenerate both calcium pyrophosphate dihydrate and apatite invitro. Calcif Tissue Int 1998;63:258–62.
51. Hauselmann HJ, Stefanovic-Racic M, Michel BA, Evans CH.Differences in nitric oxide production by superficial and deephuman articular chondrocytes: implications for proteoglycan turn-over in inflammatory joint diseases. J Immunol 1998;160:1444–8.
52. Grishko VI, Druzhyna N, LeDoux SP, Wilson GL. Nitric oxide-induced damage to mtDNA and its subsequent repair. NucleicAcids Res 1999;27:4510–6.
53. Papa S, Skulachev VP. Reactive oxygen species, mitochondria,apoptosis and aging. Mol Cell Biochem 1997;174:305–19.
1570 JOHNSON ET AL




















