
Memory B cells from a subset of treatment naïve relapsing remitting multiple sclerosis patients...
24
Memory B cells from a subset of treatment naïve relapsing remitting multiple sclerosis patients elicit CD4+ T cell proliferation and IFN-γ production in response to MBP and MOG Christopher T. Harp 1 , Sara Ireland 1 , Laurie S. Davis 2 , Gina Remington 1 , Bonnie Cassidy 1 , Petra D. Cravens 1 , Olaf Stuve 1,5 , Amy E. Lovett-Racke 6 , Todd N. Eagar 1 , Benjamin M. Greenberg 1 , Michael K. Racke 7 , Lindsay G. Cowell 8 , Nitin J. Karandikar 1,3,4 , Elliot M. Frohman 1 , and Nancy L. Monson 1,3 1 Department of Neurology, University of Texas Southwestern Medical Center, Dallas, Texas 75390, USA 2 Department of Internal Medicine, University of Texas Southwestern Medical Center, Dallas, Texas 75390, USA 3 Department of Pathology, University of Texas Southwestern Medical Center, Dallas, Texas 75390, USA 4 Department of Immunology, University of Texas Southwestern Medical Center, Dallas, Texas 75390, USA 5 VA North Texas Health Care Systems, Dallas, TX 75216, USA 6 Departments of Molecular Virology, Immunology and Medical Genetics, Ohio State University Medical Center, Columbus, OH 43210, USA 7 Department of Neurology, Ohio State University Medical Center, Columbus, OH 43210, USA 8 Department of Biostatistics, Duke University, Durham, North Carolina, 27708, USA Abstract Recent evidence suggests that B and T cell interactions may be paramount in relapsing remitting multiple sclerosis (RRMS) disease pathogenesis. We hypothesized that memory B cell pools from RRMS patients may specifically harbor a subset of potent neuro-antigen presenting cells that support neuro-antigen reactive T cell proliferation and cytokine secretion. To test this hypothesis, we compared CD80 and HLA-DR expression, IL-10 and LTα secretion, neuro-antigen binding capacity, and neuro-antigen presentation by memory B cells from RRMS patients to naïve B cells from RRMS patients and to memory and naïve B cells from healthy donors (HD). We identified memory B cells from some RRMS patients that elicited CD4+ T cell proliferation and IFN-γ secretion in response to myelin basic protein (MBP) and myelin oligodendrocyte glycoprotein (MOG). Notwithstanding the fact that the phenotypic parameters that promote efficient antigen presentation were observed to be similar between RRMS and HD memory B cells, a corresponding capability to elicit CD4+ T cell proliferation in response to MBP and MOG was not observed in HD memory B cells. Our results demonstrate for the first time that the memory B cell pool in RRMS harbors neuro-antigen specific B cells that can activate T cells. Corresponding Author: Nancy L. Monson; Department of Neurology, University of Texas Southwestern Medical Center, 5323 Harry Hines Blvd., Dallas, TX 75390.Fax: +1-214-648-9129. [email protected]. Conflict of interest The authors declare no financial or commercial conflict of interest. NIH Public Access Author Manuscript Eur J Immunol. Author manuscript; available in PMC 2011 October 1. Published in final edited form as: Eur J Immunol. 2010 October ; 40(10): 2942–2956. doi:10.1002/eji.201040516. NIH-PA Author Manuscript NIH-PA Author Manuscript NIH-PA Author Manuscript
-
Upload
independent -
Category
Documents
-
view
2 -
download
0
Transcript of Memory B cells from a subset of treatment naïve relapsing remitting multiple sclerosis patients...

Memory B cells from a subset of treatment naïve relapsingremitting multiple sclerosis patients elicit CD4+ T cellproliferation and IFN-γ production in response to MBP and MOG
Christopher T. Harp1, Sara Ireland1, Laurie S. Davis2, Gina Remington1, Bonnie Cassidy1,Petra D. Cravens1, Olaf Stuve1,5, Amy E. Lovett-Racke6, Todd N. Eagar1, Benjamin M.Greenberg1, Michael K. Racke7, Lindsay G. Cowell8, Nitin J. Karandikar1,3,4, Elliot M.Frohman1, and Nancy L. Monson1,31 Department of Neurology, University of Texas Southwestern Medical Center, Dallas, Texas75390, USA2 Department of Internal Medicine, University of Texas Southwestern Medical Center, Dallas,Texas 75390, USA3 Department of Pathology, University of Texas Southwestern Medical Center, Dallas, Texas75390, USA4 Department of Immunology, University of Texas Southwestern Medical Center, Dallas, Texas75390, USA5 VA North Texas Health Care Systems, Dallas, TX 75216, USA6 Departments of Molecular Virology, Immunology and Medical Genetics, Ohio State UniversityMedical Center, Columbus, OH 43210, USA7 Department of Neurology, Ohio State University Medical Center, Columbus, OH 43210, USA8 Department of Biostatistics, Duke University, Durham, North Carolina, 27708, USA
AbstractRecent evidence suggests that B and T cell interactions may be paramount in relapsing remittingmultiple sclerosis (RRMS) disease pathogenesis. We hypothesized that memory B cell pools fromRRMS patients may specifically harbor a subset of potent neuro-antigen presenting cells thatsupport neuro-antigen reactive T cell proliferation and cytokine secretion. To test this hypothesis,we compared CD80 and HLA-DR expression, IL-10 and LTα secretion, neuro-antigen bindingcapacity, and neuro-antigen presentation by memory B cells from RRMS patients to naïve B cellsfrom RRMS patients and to memory and naïve B cells from healthy donors (HD). We identifiedmemory B cells from some RRMS patients that elicited CD4+ T cell proliferation and IFN-γsecretion in response to myelin basic protein (MBP) and myelin oligodendrocyte glycoprotein(MOG). Notwithstanding the fact that the phenotypic parameters that promote efficient antigenpresentation were observed to be similar between RRMS and HD memory B cells, acorresponding capability to elicit CD4+ T cell proliferation in response to MBP and MOG was notobserved in HD memory B cells. Our results demonstrate for the first time that the memory B cellpool in RRMS harbors neuro-antigen specific B cells that can activate T cells.
Corresponding Author: Nancy L. Monson; Department of Neurology, University of Texas Southwestern Medical Center, 5323 HarryHines Blvd., Dallas, TX 75390.Fax: +1-214-648-9129. [email protected] of interest The authors declare no financial or commercial conflict of interest.
NIH Public AccessAuthor ManuscriptEur J Immunol. Author manuscript; available in PMC 2011 October 1.
Published in final edited form as:Eur J Immunol. 2010 October ; 40(10): 2942–2956. doi:10.1002/eji.201040516.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

Keywordsmultiple sclerosis; B cells; autoimmunity; antigen presentation
INTRODUCTIONA role of B cells in the pathogenesis of relapsing remitting multiple sclerosis (RRMS) wasrecently substantiated, as therapeutic depletion of B cells with Rituximab resulted inreduction in disease activity as measured by gadolinium enhancing lesions on magneticresonance imaging [1]; diminution of pro-inflammatory cytokine secretion by T cells fromMS patients receiving Rituximab therapy [2] and reappearance of B cells in the peripherythat correlated with a return in RRMS symptoms [3]. Preventing B cells from entering thecentral nervous system (CNS) may be an important target of disease modifying therapy,since RRMS patients treated with the selective adhesion molecule inhibitor natalizumab, amonoclonal antibody to very late antigen-4 (VLA-4) present on a variety of leukocytes,show a disproportionate increase in the number of circulating B cells [4].
Although B cells serve varied functions in the immune system including the production ofantibodies, lymphoneogenesis, cytokine secretion and antigen presentation [5-7], which ofthese functions play a central role in the pathogenesis of RRMS remains unclear. Forinstance, it does not appear likely that antibody secretion is central to RRMS diseaseactivity, given that treatment with Rituximab confers benefit in reducing both clinical andradiographic disease manifestations without concomitant reduction of circulating antibodysince CD20, the target of Rituximab, is not present on plasma cells [1]. However, Rituximabtreatment of RRMS patients does result in a significant depletion of B cells within thecerebrospinal fluid (CSF) [8] and cerebral perivascular spaces [9]. A marked decrease in Tcell frequency in the CSF has also been observed [8], indicating a potential role of B cells inregulating T cells within the CNS. This finding was similarly observed in the mouse modelof MS, experimental autoimmune encephalomyelitis (EAE), in which depletion of B cellsafter EAE induction resulted in a reduction of antigen-specific effector T cells within theCNS [10]. Interestingly, a decline in T cell frequency in the CSF of pediatric opsoclonus-myoclonus patients receiving Rituximab was not observed [11], indicating that T cellregulation by B cells may be a unique feature of MS patients.
At first glance, these results are surprising since the most extensively studied consequenceof B-T cell interactions is the induction of T cell tolerance or expansion of regulatory T cells[12-14]. For example in mice, antigen-specific naïve B cells induce naïve T cells toproliferate and differentiate into regulatory T cells, unlike dendritic cells, which induceeffector T cell subset development [15]. In addition, hen egg lysozyme (HEL) specific Bcells fail to initiate significant T cell proliferation or IL-2 and IFN-γ secretion [16]. Further,on a per cell basis, human B cells are less potent antigen presenting cells (APC) thandendritic cells in vitro [17]. Ultimately, these findings have suggested that B cells areunlikely to play a significant role as APC in the induction of effector T cell responses.Nevertheless, these studies did not characterize APC function in the memory B cell pool.Importantly, other in vitro investigations have demonstrated that human memory B cells arepotent APC in the context of both allo-antigen [18] and exogenous foreign antigen [19].Interestingly, circulating memory B cells are also reduced in RRMS patients duringmitoxantrone [20] and IFN-β therapy [21]. The relationship of these reductions totherapeutic efficacy remains unknown.
Our studies have recently focused on the potential impact of memory B cells from RRMSpatients that dominate the B cell pool in the CNS of RRMS patients [22,23], and whether
Harp et al. Page 2
Eur J Immunol. Author manuscript; available in PMC 2011 October 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

pathogenic mechanisms are mediated through the process of neuro-antigen presentation toeffector T cells. Here we demonstrate for the first time that memory B cells from theperipheral blood of some RRMS patients are able to induce significant neuro-antigenspecific T cell proliferation and IFN-γ secretion in comparison to peripheral memory B cellsfrom healthy donors. Our findings may be germane to advancing our understanding of therelationship of compartment specific memory B cell responses and the pathobiologicunderpinnings of MS.
RESULTSNaïve and memory B cell subsets from RRMS patients are phenotypically similar to thosefrom HD
Recent studies on an untreated RRMS patient cohort similar to ours found no significantdifferences in the percentages of circulating naïve and memory B cells between HD andRRMS patients [21]. Nevertheless, we wanted to confirm this phenotype in our cohorts aspatients with other autoimmune disorders have a decrease in the total percentage ofcirculating memory B cells in the peripheral blood (PB) compared to HD [24], supportingthe idea that these cells are at the site of inflammation during the course of autoimmuneinflammation, and not in circulation. The HD and RRMS patients in our cohort showedsimilar percentages of memory B cells (4.42±0.56% vs. 4.07±0.56%, p=0.12 Figure 1B) andnaïve B cells (13.70±2.01% vs. 9.66±1.20%, p=0.12, Figure 1B) within the PB, similar towhat was previously reported [21]. Also, as previously observed [23], naïve B lymphocytesdominate the PB of both HD (13.70±2.01% naïve B cells vs. 4.42±0.56% memory B cells,p<0.001, Figure 1B), and RRMS patients (9.66±1.20% naïve B cells vs. 4.07±0.56%memory B cells, p<0.001, Figure 1B), with average naïve:memory ratios of 3.27±0.37 and2.68±0.40 in HD and RRMS patients, respectively.
Costimulatory molecule expression can influence the potency of B cell antigen presentation,and is differentially regulated between naïve and memory B cells in HD [18,19]. In addition,one study had demonstrated that CD80+ B cells expand in the peripheral blood of MSpatients undergoing exacerbation [25]. Therefore, we examined memory and naive B cellsubsets for their relative expression of CD80 (B7.1) in our RRMS cohort and comparedsubset expression to HD using mean fluorescence intensity (MFI) of CD80 expression byflow cytometry. Memory B cells expressed significantly higher levels of CD80 than naïve Bcells in both HD (6.51±0.49 vs. 3.99±0.44, p<0.001, Figure 1D) and RRMS patients(6.88±0.45 vs. 4.03±0.25, p<0.001, Figure 1D). CD80 MFI naïve:memory ratios in HD andRRMS patients were similar (0.60±0.02 and 0.59±0.02, respectively).
HLA-DR is a major component of human MHCII and is constitutively expressed on restingB cells and other APC, but is highly upregulated upon activation [26]. Given the increasedexpression of CD80 on memory B cells in our cohorts, we hypothesized that HLA-DRexpression may also be significantly increased on memory B cells as compared to naïve Bcells in both HD and RRMS. Instead, we observed that the memory B cell compartmentexpressed significantly lower levels of HLA-DR compared to the naïve B cell compartmentin both HD (217.6±38.7 vs. 394.2±65.4, p<0.001, Figure 1F) and RRMS patients (98.2±16.9vs. 198.8±33.7 p<0.01, Figure 1F). However, the relative ratio of naïve:memory B cellHLA-DR expression was similar in the two cohorts (2.00±0.10 vs. 2.12±0.14) indicatingthat even though naïve and memory B cells from the HD cohort in general had higher HLA-DR expression than RRMS, the relative ratio of expression between naïve and memory Bcells in both cohorts was similar.
Harp et al. Page 3
Eur J Immunol. Author manuscript; available in PMC 2011 October 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

Memory B cells from both HD and RRMS secrete pro-inflammatory Lymphotoxin-alphaMemory and naïve B cells from HD secrete pro- and anti-inflammatory cytokines,respectively, when polyclonally stimulated in vitro [20], potentially indicating a reciprocalrole in modulating T cell responses through antigen presentation during the course ofRRMS. LTα is a prototypic inflammatory cytokine critical in the development of some EAEmodels of MS [27], and IL-10 is a prototypic anti-inflammatory cytokine that inhibitshuman T cell proliferation [28]. Therefore, we chose to examine LTα secretion as a measureof pro-inflammatory activity and IL-10 secretion as a measure of anti-inflammatory/regulatory activity in both our HD and RRMS patient cohort. Accordingly, CD40Lstimulation served as a model of T cell dependent bystander activation, and BCR + CD40Lstimulation a model of cognate antigen recognition followed by T cell help.
Memory B cells produced significantly more LTα than naïve B cells when stimulated withCD40L alone in both HD (483±49 pg/mL vs. 125±36 pg/mL, p<0.001, paired t-test, Figure2A) and RRMS patients (314±72 pg/mL vs. 78±20 pg/mL p<0.01, paired t-test, Figure 2A).There was no significant difference between the HD and RRMS cohorts when we comparedLTα production in the memory B cell compartment or the naïve B cell compartment(p=0.05). Similar response patterns were observed with the combination of BCR and CD40Lstimulation, such that LTα secretion by memory B cells was greater than LTα secretion bynaïve B cells in both HD (455±36 vs. 56±18 pg/mL, p<0.001, paired t-test, Figure 2B) andRRMS patients (325±61 vs. 80±22, p<0.001, paired t-test, Figure 2B). No significantdifferences in LTα production between memory or naïve B cells from HD or RRMS patients(Figure 2B) were observed (p=0.29). These data suggest that the addition of BCRstimulation at this dose does not induce greater LTα secretion than what is observed withCD40L alone in either the naïve or memory B cell compartment in HD and RRMS patients.
Although naïve B cells from HD and RRMS patients secreted similar amounts of LTα, naïveB cells from RRMS patients stimulated with CD40L alone produced significantly moreIL-10 than naïve B cells from HD stimulated with CD40L alone (89±30 pg/mL vs. 9±9 pg/mL, p<0.01, Figure 2C). In fact, naïve B cells from RRMS patients stimulated with CD40Lalone produced significantly more IL-10 than memory B cells from the same RRMS patients(89±30 pg/mL vs. 13±10 pg/mL, p<0.01, Figure 2C), suggesting that a subset of naïve Bcells from RRMS patients may be functioning as regulatory cells not present in HD. Uponaddition of BCR stimulation, we observed an insignificant decrease in the amount of IL-10produced by RRMS naïve B cells stimulated with BCR and CD40L stimulation incombination versus CD40L stimulation alone (89±30 pg/mL vs. 28±9 pg/mL, p=0.09, un-paired t-test, Figure 2C and 2D). Others have demonstrated that naïve B cells from HDsecrete significant concentrations of IL-10 [20, 29], but we did not observe any significantIL-10 production by naïve or memory B cells from HD in our cohort stimulated with eitherCD40L alone or BCR and CD40L stimulation in combination. This discrepancy is likelyattributed to differences in culturing systems, and not a feature peculiar to our HD cohort.
Memory B Cells from RRMS patients elicit more neuro-antigen specific CD4+ T cellproliferation than memory B cells from HD
In vivo depletion of B cells in the context of RRMS results in a reduction of gadoliniumenhancing lesions and a reduction in the number of T cells within the CSF [1], whichsubstantiates a role for B cells in MS as mediators of T cell expansion, activation andpathogenicity. Since the majority of B cells found in the CSF of RRMS patients are short-lived plasma blasts and memory B cells [22,23], we hypothesized that within the memory Bcell pool of RRMS patients, there is a sub-population of memory B cells that operate aspotent neuro-antigen presenting cells that supports neuro-antigen reactive T cell expansionand activation. To test this hypothesis, we investigated the potential of primary ex vivo
Harp et al. Page 4
Eur J Immunol. Author manuscript; available in PMC 2011 October 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

memory and naïve B cells to elicit autologous T cell proliferation to one control antigen(TT), two candidate neuro-antigens (MBP and MOG), and GA, which is an FDA approvedtherapeutic agent in the treatment of MS that has similar biochemical properties to MBP[30]. Purified memory or naïve B cells were incubated with purified autologous T cells, andT cell proliferation was measured by CFSE dilution as described in the materials andmethods. Two examples (MS-10 and MS-1) of the output data highlighting the variance inresponses between patients are provided in Figure 3. Memory and naïve B cells from MS-10(Figure 3A) and MS-1 (Figure 3B) did not induce CD4+ T cell proliferation in the absenceof antigen. Both naïve and memory B cells from MS-10 and MS-1 induced CD4+ T cellproliferation in response to TT. Memory B cells from MS-10, but not MS-1, induced CD4+T cell proliferation in response to MBP. Memory and naïve B cells from MS-10, but notMS-1, induced CD4+ T cell proliferation in response to MOG. Naïve and memory B cellsfrom MS-10 and memory B cells from MS-1 induced CD4+ T cell proliferation in responseto GA. These same assays were performed on 11 MS patients and 10 HD and the resultsdepicted in Figure 4.
Because HD and RRMS are presumably immunized to tetanus at the same frequency andhave similar frequencies of residual memory B and T cells, we expected that CD4+ T cellproliferation in response to TT when memory B cells were included in the cultures would besimilar in HD and RRMS patients. Indeed, memory B cells from HD and RRMS patientselicited similar CD4+ T cell proliferation in response to TT (p=0.16, χ2-test, Figure 4A).Also as expected, memory B cells elicited greater mean CD4+ T cell proliferation inresponse to TT than naïve B cells in HD (6.3±2.4% vs. 3.4±1.6%, p<0.05, Figure 4A).However, in the RRMS patient cohort, CD4+ T cell proliferation in response to TT wassimilar in co-cultures containing memory B or naïve B cells (5.3±2.3% vs. 3.4±1.7%,p>0.05, Figure 4A).
In contrast, memory B cells from some of the RRMS patients, but not from any of the HD,elicited CD4+ T cell proliferation in response to MBP (4/9 vs. 0/10 responders, respectively,p=0.018, χ2-test, Figure 4B). Memory B cells from RRMS patients were also more likely toelicit CD4+ T cell proliferation than memory B cells from HD in response to MOG,although this trend did not meet statistical significance (5/10 vs. 1/9 responders respectively,p=0.069, χ2-test, Figure 4D). In contrast, memory B cells from HD and RRMS patients hadenhanced capacities to elicit CD4+ T cell proliferation in response to GA in comparison tonaïve B cells (3.3±1.0% vs. 1.02±0.4%, p<0.01 and 3.6±0.7 vs. 0.86±0.32, p<0.01,respectively, Figure 4C). Interestingly, naïve B cells from RRMS patients or HD could notelicit CD4+ T cell proliferation in response to MBP or GA, but naïve B cells from RRMSpatients could elicit CD4+ T cell proliferation in response to MOG more readily than naïveB cells from HD (p=0.02, χ2-test, Figure 4D). The frequency of MOG specific CD4+ T cellsis increased in the RRMS patient cohort in comparison to HD (Supplemental Figure 1),which may explain this finding. The frequency of TT, MBP and GA specific CD4+ T cellswas similar in the HD and RRMS patient cohorts (Supplemental Figure 1).
Memory B cells from RRMS patients induce greater antigen-specific IFN-γ secretion thanmemory B cells from HD
CD4+ T cells proliferated in response to both neuro-antigen and control antigens in vitro,and so our next goal was to determine whether IFN-γ secretion (as a measure of Th1responses) was affected when memory or naïve B cells were the source APC in these cultureconditions (Figure 5). In whole PBMC cultures from HD, TT does not typically induce asignificant IFN-γ response [31,32]. In agreement with these data, both memory and naïve Bcells from our HD cohort induced little IFN-γ production in B-T cell co-culture supernatantsin response to TT (85±33 pg/mL vs. 68±40 pg/mL, p=0.86, Figure 5A). However, bothmemory and naïve B cells from the RRMS patient cohort induced levels of IFN-γ above the
Harp et al. Page 5
Eur J Immunol. Author manuscript; available in PMC 2011 October 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

set threshold in response to TT, and memory B cells from RRMS patients were more likelyto induce IFN-γ secretion than memory B cells from HD (0/7 vs. 6/9, respectively, p=0.006,χ2-test, Figure 5A).
Because we observed a significantly greater number of memory B-T cell co-cultures fromRRMS patients that resulted in CD4+ T cell proliferation to MBP compared to HD, weexpected to find that IFN-γ secretion would also be concomitantly increased in response tomemory B cells from RRMS patients presenting MBP to autologous T cells. Indeed, weobserved that memory B cells from 3 of 9 RRMS patients induced IFN-γ secretion in B-Tcell co-cultures with MBP, while memory B cells from 0 of 7 HD did not (Figure 5B). NaiveB cells from either HD or RRMS patients did not induce significant IFN-γ secretion inresponse to MBP (Figure 5B). These results would indicate that in at least a subset of RRMSpatients, IFN-γ secretion in response to MBP is induced by memory B cells but not naïve Bcells. However, it should be noted that the amount of IFN-γ secretion did not consistentlycorrelate with the frequency of CD4+ T cells that proliferated in these culture conditions(Supplementary Table 1), and statistical significance was not reached when comparing thenumber of responders in each cohort (p=0.09, χ2-test).
Glatiramer acetate induces both IFN-γ and IL-5 cytokine secretion in T cell lines derivedfrom HD naïve to GA [33]. Therefore, we hypothesized that primary ex vivo T cells fromHD and our GA-treatment naïve RRMS patient cohort would also secrete IFN-γ and IL-5 inresponse to both memory and naïve B cells presenting GA. Instead, no significant IFN-γ wasproduced in response to GA when either naïve or memory B cells from HD were APC (0/8vs. 0/8 responders respectively, p=1, χ2-test, Figure 5C). Only 2/11 donors from the RRMScohort produced significant amounts of IFN-γ (Figure 5C) and IL-5 (data not shown) inresponse to GA presented by either memory or naïve B cells.
In a previous study, IFN-γ secreting T cells from untreated MS patients were increased inresponse to purified whole MOG in comparison to control patients [34]. Similar to thatobservation, memory B cells from 4 of 10 RRMS patients, and 0 of 6 HD, induced IFN-γsecretion in the B-T cell co-cultures in response to MOG (Figure 5D). Naïve B cells from 3of 10 RRMS patients and 0 of 6 HD induced IFN-γ secretion in the B-T cell cultures inresponse to MOG (Figure 5D). However, statistical significance was not reached whencomparing the number of RRMS and HD responders that secreted IFN-γ in response toMOG presented by memory or naïve B cells (p=0.09 and p=0.13, respectively, χ2-test).There was also no significant difference in the mean concentration of IFN-γ detected in B-Tcell co-cultures containing memory or naïve B cells presenting MOG (460±230 vs. 532±414 pg/mL, p=0.9).
MBP and GA binding B cells are enriched in the memory B cell compartment of both HDand RRMS patients
In order to determine if we could correlate the extent of CD4+ T cell proliferation thatoccurred through B cell antigen presentation and IFN-γ secretion to the frequency of antigenspecific B cells present in memory and naïve B cell populations in our donors, we chose tomake use of a technique that has been utilized by us [26] and others [35] to identify antigenspecific B cells by flow cytometry. An example of the gating strategy used to identify OVAand GA antigen binding memory and naïve B cells is shown in Figure 6.
The number of naïve and memory B cells that bound TT was similar in the HD and RRMSpatient cohorts (Figure 6C). However, MBP binding B cells were significantly enriched inthe memory B cell compartment in comparison to the relative number of naïve B cells thatbound MBP in both HD (2342±678 vs. 329±90 per 1×106 cells, p<0.01, Figure 6D) andRRMS patients (3931±652 vs. 1255±309 per 1×106 cells, p<0.001, Figure 6D). The
Harp et al. Page 6
Eur J Immunol. Author manuscript; available in PMC 2011 October 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

frequency of MBP binding memory B cells was similar between HD and RRMS patients(p=0.053, Figure 6D), although only memory B cells from RRMS patients were able to elicitCD4+ T cell proliferation (Figure 4B). In contrast to the relative enrichment of MBP bindingB cells in the memory B cell compartment in both HD and RRMS patients, no such relativeenrichment of MOG binding B cells in the memory B cell compartment of HD and RRMSpatients was observed (Figure 6F), even though B cells from RRMS patients and not HDwere able to elicit CD4T cell proliferation in response to MOG.
GA has many similar biochemical properties to MBP including undefined secondarystructure in solution and overall positive charge, so we expected to find that the number of Bcells that bound GA would be enriched in the memory B cell compartment as well. Indeed,GA binding B cells were also significantly enriched in the memory B cell compartment incomparison to the relative number of naïve B cells that bound GA in both HD (3095±579 vs.517±166 per 1×106 cells, p<0.05, Figure 6E) and RRMS patients (5010±1250 vs. 735±235per 1×106 cells, n=11, p<0.001, Figure 6E). Interestingly, in addition to memory B cellscontaining higher frequencies of GA binding, memory B cells were also able to elicit CD4+T cell proliferation more readily in response to GA than their naïve B cell counterparts.
DISCUSSIONIn this study, we investigated the phenotypic attributes and antigen-presenting role ofmemory vs. naïve B cells from MS patients. While B cells from MS patients werephenotypically similar to those from healthy subjects and also showed similar patterns ofLTα secretion (as a representative of pro-inflammatory cytokine release potential) inresponse to polyclonal stimuli, we found that memory B cells from a subset of MS patients,but not HD, elicited autologous CD4+ T cell proliferation in response to MBP and MOG. Atthe same time, both MS patients and HD elicited autologous CD4+ T cell proliferation inresponse to TT and GA. Thus, the differences were significantly centered on CNS-specific Tcell responses, which are relevant to the pathogenesis of MS.
Our findings cannot be attributed to an enrichment of neuro-antigen reactive memory B cellsin RRMS patients, since the neuro-antigen binding assay indicated that the frequencies ofneuro-antigen binding memory B cells are similar in HD and RRMS patients. We cannotrule out, however, that other features of memory B cells from RRMS patients render themmore capable of inducing neuro-antigen specific CD4+ T cell proliferation and cytokinesecretion than their HD counterparts, including higher production of other inflammatorycytokines such as IL-1, IL-6, IL-8 and TNF, which are also readily produced by human Bcells [36], but were not measured in this series of experiments. It should also be noted thatthe ability of memory B cells from RRMS patients, but not HD to elicit CD4+ T cellproliferation in response to MBP was not attributable to general enrichment of MBP specificT cells in the RRMS patients, since the frequency of MBP specific T cells was similar in theHD and RRMS patient cohorts for this study. Others have also observed that the overallfrequency of MBP specific T cells is similar between HD and RRMS patients [37-39].
In contrast, the frequency of MOG specific T cells in the RRMS cohort was enriched incomparison to the HD cohort used for this study. Others have also observed that thefrequency of MOG specific T cells is enriched in RRMS patients compared to HD [40,41]. Itis possible then, that the enhanced ability of memory and naïve B cells from RRMS patients,but not HD, to elicit CD4+ T cell proliferation and IFN-γ secretion in response to MOG isnot due to enrichment of MOG-binding B cells in RRMS patients compared to HD, but mayinstead be due to enrichment of MOG specific T cells in the RRMS patients. Prior reports byothers indicate that neuro-antigen-reactive memory T cells in particular are more prominentin RRMS patients than in HD [38,42]. Thus, it is possible that B cells preferentially elicit
Harp et al. Page 7
Eur J Immunol. Author manuscript; available in PMC 2011 October 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

proliferation and cytokine secretion by memory T cells, which may be most relevant to theunderlying disease process. Determining the type of CD4+ T cell that is engaged by naïveand memory B cells in a MOG-specific manner is the focus of future studies by ourlaboratory.
The finding that MBP specific memory B cells are enriched in the peripheral blood ofRRMS patients (compared to naïve) suggests that perhaps MBP specific B cells were primedin the periphery and have an effector function in the periphery as well. B cells couldinfluence the pathogenesis of MS by priming T cells in secondary peripheral lymphoidorgans, which then migrate to the CNS. B cells residing in the CNS may further promoteCNS T cell activation, although this has not been formally tested. However, it should benoted that MBP specific memory B cells were also enriched in the peripheral blood of HDwhen compared to naïve B cells. In this scenario, HD peripheral memory B cells do notelicit CD4+ T cell proliferation in response to MBP. What impact removal of MBP-bindingmemory B cells has on the ability of the remaining B cell pool to elicit effector T cellfunction in response to MBP is under investigation in our laboratory.
GA-specific memory B cells, which are cross-reactive with MBP, may elicit the activationof MBP specific CD4+ T regulatory cells (Treg). In vitro treated CD4+CD25high Treg showincreased IL-10 production in response GA treatment [43]. In addition, it was recentlydemonstrated that while untreated MS patients have reduced numbers of peripheral CD4+Treg in comparison to HD, GA treatment restores both the number and functionality of theseTreg [44]. Perhaps this would suggest that memory B cells contribute most significantly tothe expansion of CD4+ Treg in patients undergoing GA therapy.
Our data demonstrated that memory B cells from a subset of RRMS patients elicited IFN-γsecretion in response to MBP and MOG, but that memory B cells from HD did not elicitIFN-γ secretion in response to MBP and MOG. This data suggests that memory B cells fromRRMS patients are more likely to promote activation of Th1 specific clones in response toMBP and MOG than memory B cells from HD. Of note, this response did not correlate withCD4+ T cell proliferation in individual patients (Supplementary Table 1), and a comparisonof MBP and MOG responders (defined as those patients whose B cells elicited IFN-γsecretion in response to MBP or MOG) in the RRMS and HD cohorts did not reachstatistical significance. Other factors, such as the influence of CD8 T cells [45-47] mayexplain the dichotomy in responses. Naïve B cells from 3 of 10 RRMS patients were alsoable to elicit IFN-γ secretion in response to MOG, and we cannot rule out that a smallpopulation of memory B cells that are CD27- [48] may be present in our naïve B cell pools.Thus, it is possible that the IFN-γ secretion elicited by the naïve B cells from these 3 RRMSpatients was mediated by CD27- memory B cells.
Interestingly, significant IFN-γ and IL-5 secretion was consistently observed in cultureswhere memory B cells from RRMS patients were presenting Tetanus Toxoid (TT) to T cells,suggesting that memory B cells are capable of stimulating both Th1 and Th2 TT reactiveclones. Also of note, IFN-γ secretion in response to memory B cells from RRMS patientspresenting TT was enhanced compared to memory B cells from HD presenting TT (Figure5A). This is significant as it would suggest that TT specific T cell clones which do nottypically secrete IFN-γ or show a predominant Th1 profile in HD [32, 49-51] have beenskewed towards a Th1 phenotype in some RRMS patients. In fact, it was recentlydemonstrated that female RRMS patients show an exaggerated IFN-γ response to TT inPBMC as compared to control females [52]. Our data may suggest that memory B cells fromRRMS patients are likely candidates influencing this phenomenon.
Harp et al. Page 8
Eur J Immunol. Author manuscript; available in PMC 2011 October 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

In contrast to the increase in IFN-γ secretion in response to memory B cells presentingneuro-antigens or TT, IFN-γ secretion in response to memory B cells presenting GA wasrarely observed (Figure 5C)(compare 6/9 TT responders to 2/11 GA responders, p=0.03).Yet CD4+ T cell proliferation in the presence of memory B cells and GA was robust in themajority of RRMS patients (Figure 4C). CD4+ T cell proliferation in the presence ofmemory B cells and GA was also robust in the majority of HD, yet IFN-γ was not observedin the majority of these cultures either. This data would suggest that memory B cells fromRRMS patients and HD naïve to GA therapy maintain their capacity to elicit CD4+ T cellproliferation in response to GA, but that in most cases, are not able to drive T cell effectorfunctions such as IFN-γ secretion. This was somewhat unexpected as the majority of GA-specific T cell lines derived from GA-naïve MS patients secrete IFN-γ readily [53, 54].However, GA-specific T cell lines generated from MS patients that have been treated forseveral months with GA secrete significantly less IFN-γ [53]. We would not expect thatIFN-γ reactive T cell clones were absent in the MS patient cohort samples used in thesestudies since the patients were treatment naïve and in early stages of disease. It would beinteresting to determine whether memory B cells from RRMS patients benefiting from GAtherapy would additionally lose their capacity to elicit CD4+ T cell proliferation in responseto GA, and the duration of GA therapy that is required to initiate this effect.
Of note, it has been documented that GA may not need to be processed since it bindsdirectly to HLA-DR on the surface of APC [55]. We were able to purify sufficient numbersof memory B cells from one patient to test whether GA processing by memory B cells wasrequired to induce CD4+ T cell proliferation. Indeed, pretreatment of memory B cells withchloroquine, a lysosomotropic agent that prevents antigen processing, prior to co-culturewith T cells and GA, reduced CD4+ T cell proliferation below the detection threshold inresponse to GA (data not shown).
Since sera from a subset of RRMS patients and HD contain MBP and MOG reactiveantibodies [56], we predicted that we would be able to detect memory B cells from RRMSpatients and HD that would bind to MBP and MOG. Indeed, we detected memory B cellsfrom RRMS patients and HD that bound MBP and MOG, yet only memory B cells fromRRMS patients, but not from HD, elicited CD4+ T cell proliferation in response to MBP orMOG. As mentioned earlier in the discussion, this finding may be attributable to features ofmemory B cells from RRMS patients that are not present on memory B cells from HD orsome differential enrichment of MBP- or MOG-specific memory T cells in the RRMSpatients in comparison to HD. Thus, neuro-antigen binding memory B cells may have meritas a new target for immunotherapy.
However, naïve B cells from some RRMS patients, which bind to MOG at similarfrequencies as memory B cells from RRMS patients, are also able to elicit CD4+ T cellproliferation in response to MOG. This finding is unique to MOG-reactive naïve B cellsfrom RRMS patients since MBP-reactive naïve B cells from the same patients do not elicitappreciable CD4+ T cell proliferation in response to MBP. One possibility is that MOG-specific memory T cells enriched in RRMS patients can be reactivated by both memory andnaïve B cells. Previous studies indicate that naïve B cells can elicit memory T cell activation[12]. However, this hypothesis would apply only to MOG-specific responses, since naïve Bcells do not elicit CD4+ T cell proliferation in response to MBP. It is also possible that if wehad pre-activated naïve B cells from the RRMS patients, we may have also observed CD4+T cell proliferation in response to MBP.
A second possibility is that MOG may be serving as a molecular mimic for an antigenrecognized equally well by both the memory and naïve B cell repertoire, explaining theability of both of these subtypes of B cells to bind and present MOG to T cells. For example,
Harp et al. Page 9
Eur J Immunol. Author manuscript; available in PMC 2011 October 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

it has been demonstrated that MOG serum antibodies cross-react with epitopes present in themilk protein, butyrophilin [57]. Additionally mice transgenic for IgH and IgL recognizing aconformationally specific epitope of MOG undergo receptor editing to endogenous lightchains on both MOG sufficient and MOG deficient genetic backgrounds indicating someform of cross-reactivity of the MOG reactive antibodies to a distinct self protein [58]. MOGcould also potentially act as an auto-stimulatory TLR agonist in the context of RRMS asribonucleoprotein particles do in the context of SLE [59].
Speculation as to the development of neuro-antigen specific memory B cells in the contextof RRMS is of great interest given the fact that autoreactive B cells are typically negativelyselected during development [60], or are prevented from accessing a germinal centerreaction [61,62]. Our results suggest that in this subset of RRMS patients whose memory Bcells promoted CD4+ T cell expansion to MBP or MOG in vitro, either peripheralmechanisms of B cell tolerance failed or tolerance has been overcome through sensitization.Tolerance for example can be overcome by sensitization in a mouse model where B cellsthat express BCR specific for an endogenous neo-antigen are negatively selected during Bcell development but none-the-less can develop a memory B cell repertoire that recognizesthis neo-antigen after immunization [63]. In addition, it was demonstrated that inherentlyautoreactive VH4-34 expressing B cells are not excluded from a germinal center reactionand can develop into memory B cells in the context of SLE [61,64]. Whether this sameprinciple explains the enrichment of neuro-antigen specific B cells in RRMS patientsremains unexplored.
In conclusion, these studies identify MBP and MOG specific memory B cells as potentactivators of neuro-antigen specific T cells from RRMS patients, but not HD. Thus, memoryB cells may promote the exacerbation of RRMS by activating T cells in the periphery. Suchstudies provide a mechanistic explanation for why specific depletion of B cells from RRMSpatients is beneficial for some MS patients and may indicate the need to investigatedepletion of specific subsets (memory B cells) as a therapeutic strategy for patients whosememory B cells elicit CD4+ T cell proliferation and IFN-γ secretion in response to neuro-antigens.
MATERIALS AND METHODSHUMAN SAMPLES
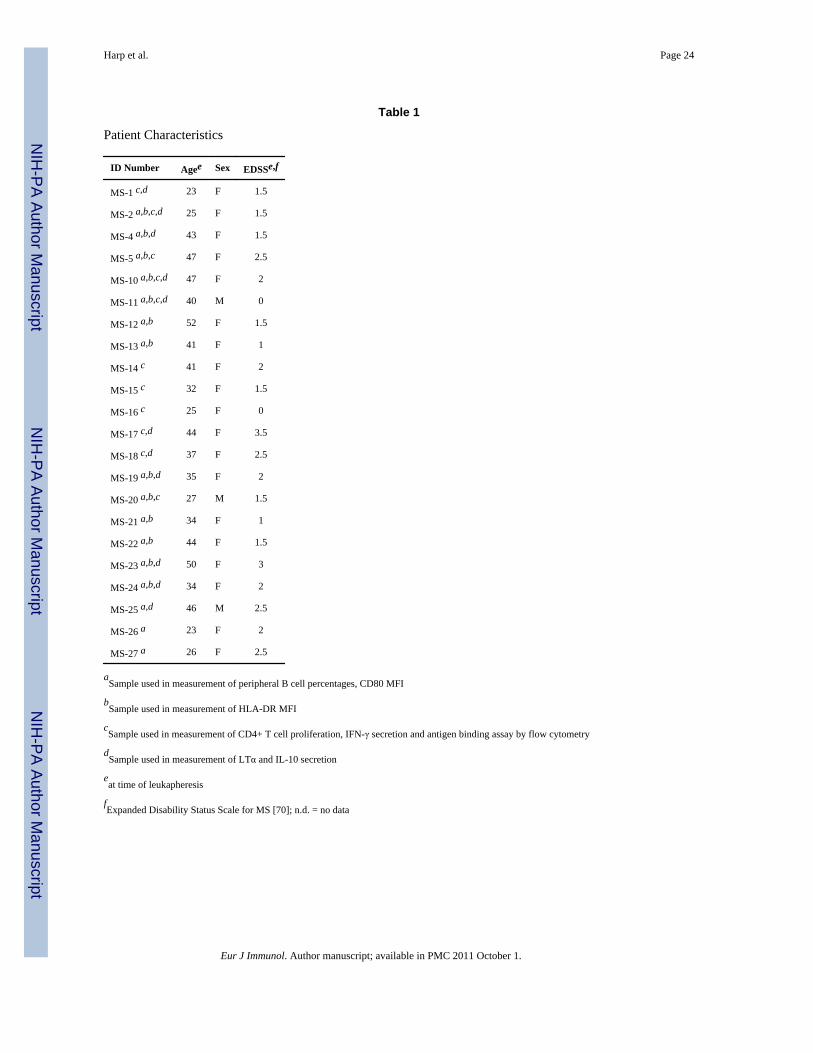
MS patients were recruited at the University of Texas Southwestern Medical Centeraccording to Institutional Review Board approved criteria and underwent leukapheresis. MSpatients included in this study had been diagnosed with clinically definite relapsingremitting MS within two years of leukapheresis, or had monosymptomatic multiple sclerosismeeting the controlled criteria for the high-risk subjects Avonex multiple sclerosisprevention study (CHAMPS) [65]. MS patient characteristics are listed in Table 1. All MSpatients had at least one exacerbation in the preceding two years but not within 60 days priorto leukapheresis sampling. None of the MS patients included in this study had primaryprogressive, secondary progressive or progressive relapsing MS. MS patients did not usecorticosteroids 60 days prior to leukapheresis and were treatment naïve to disease modifyingimmunomodulatory therapies including interferons, monoclonal antibodies, glatirameracetate, or methotrexate. MS patient peripheral blood mononuclear cells (PBMC) wereobtained from leukapheresis through ficoll density separation and cryopreserved on the sameday of leukapheresis. Healthy donor (HD) buffy coats (white blood cell filter bags) werepurchased from Carter Blood Care (Bedford, TX). Buffy coats from HD were processed viaficoll density separation to obtain PBMC, which were cryopreserved on the same day as theblood donation.
Harp et al. Page 10
Eur J Immunol. Author manuscript; available in PMC 2011 October 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

B CELL PURIFICATION AND SORTINGB cells from both RRMS and HD were enriched from cryopreserved PBMC through CD19magnetic positive selection using the CD19-DM system (BD Biosciences, San Jose, CA)according to manufacturers instructions. Enriched B cells were then stained with CD19-PECy5 and CD27-PE (BD Biosciences) and sorted on a FACS Aria (BD Biosciences). LiveB cells based on FSC vs. SSC characteristics were sorted to purity (≥99%) into naïve(CD19+CD27-) and memory (CD19+CD27+) B cell populations. The naïve B cellpopulations used for these studies may have included a small subset of memory B cells thatare CD27- [48].
INDUCTION OF B CELL CYTOKINE PRODUCTION IN RESPONSE TO CD40L STIMULATIONCytokine production was measured in sorted naïve and memory B cell culture supernatantsthat had been stimulated with CD40L or CD40L plus anti-IgM/IgG using modifications ofmethods described previously [20]. Briefly, sorted naïve and memory B cells were platedinto 96-well flat-bottom plates (Costar) at 1.5×105 cells per well in 200 μL of human culturemedium (10% human serum AB, 100 μg/mL Penicillin, 100 IU Streptomycin, 2 mM l-glutamine, 90% IMDM). For CD40L stimulation, NIH-3T3 cells expressing human CD40L(a gift from Gordon Freeman, [66]) were irradiated with 96 Gy and were added to B cells ata ratio of 15:1 B cells to NIH-3T3 cells. For BCR crosslinking plus CD40L stimulation,1.5×105 naïve or memory B cells were incubated with 0.5 μg/mL of F(ab)2 anti-human IgGand IgM (Jackson Immunoresearch) for 24 hours before being transferred to a pre-adheredmonolayer of irradiated CD40L+ NIH-3T3 cells at a ratio of 15:1 B cells to NIH-3T3 cells.
B-T CULTURE SET UPPurified T cells were obtained by negative magnetic selection from autologous donorcryopreserved PBMC using the T cell enrichment-DM system (BD Biosciences) accordingto manufacturers instructions. Purified T cells were diluted to 50×106 cells per milliliter(mL) and stained with 10 μM carboxyfluorescein diacetate, succinimidyl ester (CFSE;Invitrogen) for 5 minutes in PBS. CFSE labeled T cells were washed twice and resuspendedin human culture medium (10% human serum AB, 100 μg/mL Penicillin, 100 IUStreptomycin, 2 mM l-glutamine, 90% IMDM). 1×106 CFSE labeled T cells were culturedwith no additional cells, 1×105 naïve B cells, or 1×105 memory B cells in 1 mL of humanculture media in round bottom polystyrene tubes (Becton Dickinson). Neither the memory ornaïve B cells were pre-activated for this series of experiments. For whole PBMC cultures,PBMC were cultured at a density of 1×106 cells in 1 mL of human culture media in roundbottom polystyrene tubes. PBMC and B-T cell co-cultures contained either no exogenousantigen, 5 μg/mL Tetanus Toxoid (TT; Massachusetts Biological Laboratories) 10 μg/mLnative bovine myelin basic protein (MBP; Genway Bio-Products), 10 μg/mL glatirameracetate (GA; Teva Pharmaceuticals), or 10 μg/mL of the recombinant extracellular domainof human oligodendrocyte glycoprotein (MOG; a kind gift from Jeri Anne Lyons,Milwaukee, WI), and were cultured for 5 days at 37°C in a humidified chamber.
EXAMINATION OF T CELL SUBSETS FOR ACTIVATION AND PROLIFERATIVERESPONSES IN B-T CELL CO-CULTURES
B-T cell co-culture supernatants were harvested after 5 days in culture and frozen at −20°Cfor batch analysis of cytokine secretion. PBMC or cells in the B-T cell co-cultures werestained with optimal amounts of CD4-PE/CY7, and CD3-APC. At least 100,000 events inthe live cell gate (based on FSC×SSC characteristics) were collected for each condition on aFACS LSRII (BD Biosciences). CD4+ T helper cells were considered CD4+CD3+. CD4+ Tcell percent viability was estimated by dividing the number of CD4+ T cells in the live cellgates by the total number of CD4+ T cells collected. Proliferation was quantified as a
Harp et al. Page 11
Eur J Immunol. Author manuscript; available in PMC 2011 October 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

percentage by dividing the number of CD4+ T cells in the CSFE low gate by the totalnumber of T cells in the live gate. In order to normalize the data between assays, thepercentage of T cell proliferation observed in the absence of antigen was subtracted from thepercentage of T cell proliferation observed in the presence of antigen (ΔProliferation). IfΔProliferation was negative, the antigen-specific proliferation was graphed as a zero value.However, for statistical comparisons, the actual normalized values were used. To clarify, theΔProliferation values represent the percent of proliferating input T cells and daughter cells.A threshold value for positive proliferation was set at 2%, similar to thresholds set forpositive proliferation documented previously [67]. All fluorescently labeled monoclonalantibodies were obtained from BD Biosciences (San Jose, CA).
MEASUREMENT OF CYTOKINE SECRETION IN B-T CELL CULTURE SUPERNATANTSInterleukin-5 (IL-5), Interferon-gamma (IFN-γ), Interleukin-10 (IL-10), or lymphotoxin-alpha (LTα) capture monoclonal antibodies were diluted to 2 μg/mL in 0.1 M NaHCO3 andcoated onto Immulon 2HB 96-well plates (Thermo-Fisher Scientific, Pittsburgh, PA)overnight at 4°C. Plates were then washed twice with wash buffer (PBS, 0.1% v/vTween-20) and incubated with blocking buffer (1% BSA in PBS) for 2 hours at roomtemperature. Plates were washed 2 times and samples which had been previously frozen at−20°C for batch analysis as well as standards were added to the plate and incubatedovernight at 4°C. Plates were washed 4 times and incubated with 1 μg/mL paired-biotinylated anti-cytokine antibodies for 1 hour. Plates were then washed 6 times andincubated with 2.5μg/mL streptavidin-HRP (Jackson Immunoresearch, West Grove, PA) for30 minutes. Signal was developed using TMB substrate (Sigma). All monoclonal antibodiesused for capture and detection were purchased from BD Biosciences (San Jose, CA). Ofnote, low levels of IFN-γ were detected by ELISA in B-T cell cultures in the absence ofantigen. Therefore, we set the threshold level of IFN-γ secretion at 237 pg/mL, which wasthe mean secretion (plus two standard deviations) observed in all B-T cell co-cultures fromHD in the absence of antigen.
CONJUGATION OF WHOLE PROTEINS WITH FITCOvalbumin (OVA; Sigma, St. Louis, MO), Tetanus Toxoid (TT; Massachusetts BiologicLaboratories, Worchester, MA), Myelin Basic Protein (MBP; Genway Biotech, San Diego,CA), Myelin Oligodendrocyte Glycoprotein (MOG; a kind gift of Jeri Lyons, Milwaukee,WI) and Glatiramer Acetate (GA; TEVA Neuroscience, Kansas City, MO) wereresuspended in PBS at a concentration of 2 mg/mL and incubated with 10 μM FITC (Pierce,Rockford, IL) for 60 min at room temperature. Unbound FITC and low molecular weightspecies of GA were removed by size exclusion gel filtration, using high performancedesalting resin with molecular weight cutoff of 7,000 daltons (Pierce, Rockford, IL). 0.1%NaN3 was added to the conjugated protein preparations as preservative. OVA-FITC wasgenerated to detect background of OVA (and FITC-) reactive B cells [68].
QUANTITATION OF ANTIGEN SPECIFIC B CELLSCryopreserved PBMC from HD and RRMS patients were assayed for their ability to bindFITC conjugated proteins as described by others [69] with modifications. Briefly, 2×106
PBMC were resuspended in 80 μL of protein binding buffer (10% FBS, 0.1% NaN3, inPBS). PBMC were incubated with either no exogenous antigen, or pre-determined amountsof FITC conjugated OVA, TT, MBP, MOG, or GA. in a final volume of 100 μL on ice for 1hour. Cells were then washed twice in ice-cold buffer and incubated with CD19-PE/CY5and CD27-PE (BD Biosciences, San Jose, CA) for 30 min on ice. Cells were then washedonce in ice-cold buffer, and 1 μL propidium iodide (PI; BD Biosciences, San Jose, CA)added to exclude dead cells. 1×106 events in the live cell gate were collected on a C6 flowcytometer (Accuri, Ann Arbor, MI) and analyzed with Flowjo Software (Treestar, Ashland,
Harp et al. Page 12
Eur J Immunol. Author manuscript; available in PMC 2011 October 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

OR). Cells were gated on live lymphocytes based on FSC×SSC characteristics and exclusionof PI, and further gated into naïve (CD19+CD27−) and memory (CD19+CD27+) B cellsubsets. The percent of naïve and memory B cells binding FITC labeled proteins werequantified as antigen specific B cells.
STATISTICSMean values are reported plus and minus the standard error (SE). Unless noted otherwise,mean naïve or memory lymphocyte percentages, CD80 MFI, HLA-DR MFI, cytokinesecretion, T cell proliferative responses, and B cell protein binding assays were comparedbetween HD and RRMS patients (between donor group) and naïve and memory B cellresponses (repeated measures within donor group) using two-way mixed model analysis ofvariance (ANOVA). When overall ANOVA p-values were found to be significant (less than0.05) a multiple comparison post-hoc analysis with Bonferroni correction was used todetermine which means differed significantly, and estimated p-values are reported. Non-significant p-values are reported as exact p values. In some cases where two-way ANOVAwas not appropriate, mean responses were compared using un-paired t-test (between donorgroup) and paired t-test (within donor group) and exact p-values are reported. Chi-Squaredanalysis was used to compare number of positive responders between HD and RRMS.
Supplementary MaterialRefer to Web version on PubMed Central for supplementary material.
AcknowledgmentsWe thank all of the patients who donated samples for use in this study. We also thank Gordon Freeman for the kindgift of CD40L+ NIH3T3 cells. This work was supported by grants from Teva Neuroscience, Kansas City, MO(NLM), Howson Funds (NLM), and the National Institute of Allergy and Immunology (NJK), and NRSA T32AI005284-28 (CTH).
Abbreviations
RRMS relapsing remitting multiple sclerosis
LTα lymphotoxin-alpha
HD healthy donor
CNS central nervous system
VLA-4 very late antigen-4
CSF cerebrospinal fluid
EAE experimental autoimmune encephalomyelitis
APC antigen presenting cell
HEL hen egg lysozyme
IFN-γ interferon-gamma
PB peripheral blood
TT tetanus toxoid
MBP myelin basic protein
MOG myelin oligodendrocyte glycoprotein
GA glatiramer acetate
Harp et al. Page 13
Eur J Immunol. Author manuscript; available in PMC 2011 October 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

MFI mean fluorescence intensity
REFERENCES1. Hauser SL, Waubant E, Arnold DL, Vollmer T, Antel J, Fox RJ, Bar-Or A, Panzara M, Sarkar N,
Agarwal S, Langer-Gould A, Smith CH. B-cell depletion with rituximab in relapsing-remittingmultiple sclerosis. N Engl J Med. 2008; 358:676–688. [PubMed: 18272891]
2. Bar-Or A, Fawaz L, Fan B, Darlington PJ, Rieger A, Ghorayeb C, Calabresi PA, Waubant E, HauserSL, Zhang J, Smith CH. Abnormal B-cell cytokine responses a trigger of T-cell-mediated disease inMS? Ann Neurol. 67:452–461. [PubMed: 20437580]
3. Stuve O, Leussink VI, Frohlich R, Hemmer B, Hartung HP, Menge T, Kieseier BC. Long-term B-lymphocyte depletion with rituximab in patients with relapsing-remitting multiple sclerosis. ArchNeurol. 2009; 66:259–261. [PubMed: 19204165]
4. Krumbholz M, Meinl I, Kumpfel T, Hohlfeld R, Meinl E. Natalizumab disproportionately increasescirculating pre-B and B cells in multiple sclerosis. Neurology. 2008; 71:1350–1354. [PubMed:18936427]
5. Gray D, Gray M, Barr T. Innate responses of B cells. Eur J Immunol. 2007; 37:3304–3310.[PubMed: 18000957]
6. Martin F, Chan AC. B cell immunobiology in disease: evolving concepts from the clinic. Annu RevImmunol. 2006; 24:467–496. [PubMed: 16551256]
7. Shlomchik MJ, Craft JE, Mamula MJ. From T to B and back again: positive feedback in systemicautoimmune disease. Nat Rev Immunol. 2001; 1:147–153. [PubMed: 11905822]
8. Cross AH, Stark JL, Lauber J, Ramsbottom MJ, Lyons JA. Rituximab reduces B cells and T cells incerebrospinal fluid of multiple sclerosis patients. J Neuroimmunol. 2006; 180:63–70. [PubMed:16904756]
9. Martin Mdel P, Cravens PD, Winger R, Kieseier BC, Cepok S, Eagar TN, Zamvil SS, Weber MS,Frohman EM, Kleinschmidt-Demasters BK, Montine TJ, Hemmer B, Marra CM, Stuve O.Depletion of B lymphocytes from cerebral perivascular spaces by rituximab. Arch Neurol. 2009;66:1016–1020. [PubMed: 19667224]
10. Matsushita T, Yanaba K, Bouaziz JD, Fujimoto M, Tedder TF. Regulatory B cells inhibit EAEinitiation in mice while other B cells promote disease progression. J Clin Invest. 2008; 118:3420–3430. [PubMed: 18802481]
11. Pranzatelli MR, Tate ED, Travelstead AL, Colliver JA. Long-Term Cerebrospinal Fluid and BloodLymphocyte Dynamics After Rituximab for Pediatric Opsoclonus-Myoclonus. J Clin Immunol.2009
12. Fuchs EJ, Matzinger P. B cells turn off virgin but not memory T cells. Science. 1992; 258:1156–1159. [PubMed: 1439825]
13. Chen LC, Delgado JC, Jensen PE, Chen X. Direct expansion of human allospecific FoxP3+CD4+regulatory T cells with allogeneic B cells for therapeutic application. J Immunol. 2009; 183:4094–4102. [PubMed: 19684083]
14. Eynon EE, Parker DC. Small B cells as antigen-presenting cells in the induction of tolerance tosoluble protein antigens. J Exp Med. 1992; 175:131–138. [PubMed: 1730913]
15. Reichardt P, Dornbach B, Rong S, Beissert S, Gueler F, Loser K, Gunzer M. Naive B cellsgenerate regulatory T cells in the presence of a mature immunologic synapse. Blood. 2007;110:1519–1529. [PubMed: 17392507]
16. Attanavanich K, Kearney JF. Marginal zone, but not follicular B cells, are potent activators ofnaive CD4 T cells. J Immunol. 2004; 172:803–811. [PubMed: 14707050]
17. Corinti S, Medaglini D, Prezzi C, Cavani A, Pozzi G, Girolomoni G. Human dendritic cells aresuperior to B cells at presenting a major histocompatibility complex class II-restrictedheterologous antigen expressed on recombinant Streptococcus gordonii. Infect Immun. 2000;68:1879–1883. [PubMed: 10722577]
Harp et al. Page 14
Eur J Immunol. Author manuscript; available in PMC 2011 October 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

18. Good KL, Avery DT, Tangye SG. Resting human memory B cells are intrinsically programmed forenhanced survival and responsiveness to diverse stimuli compared to naive B cells. J Immunol.2009; 182:890–901. [PubMed: 19124732]
19. Bar-Or A, Oliveira EM, Anderson DE, Krieger JI, Duddy M, O’Connor KC, Hafler DA.Immunological memory: contribution of memory B cells expressing costimulatory molecules inthe resting state. J Immunol. 2001; 167:5669–5677. [PubMed: 11698439]
20. Duddy M, Niino M, Adatia F, Hebert S, Freedman M, Atkins H, Kim HJ, Bar-Or A. Distincteffector cytokine profiles of memory and naive human B cell subsets and implication in multiplesclerosis. J Immunol. 2007; 178:6092–6099. [PubMed: 17475834]
21. Niino M, Hirotani M, Miyazaki Y, Sasaki H. Memory and naive B-cell subsets in patients withmultiple sclerosis. Neurosci Lett. 2009; 464:74–78. [PubMed: 19666086]
22. Cepok S, Rosche B, Grummel V, Vogel F, Zhou D, Sayn J, Sommer N, Hartung HP, Hemmer B.Short-lived plasma blasts are the main B cell effector subset during the course of multiplesclerosis. Brain. 2005; 128:1667–1676. [PubMed: 15800022]
23. Harp C, Lee J, Lambracht-Washington D, Cameron E, Olsen G, Frohman E, Racke M, Monson N.Cerebrospinal fluid B cells from multiple sclerosis patients are subject to normal germinal centerselection. J Neuroimmunol. 2007; 183:189–199. [PubMed: 17169437]
24. Hansen A, Odendahl M, Reiter K, Jacobi AM, Feist E, Scholze J, Burmester GR, Lipsky PE,Dorner T. Diminished peripheral blood memory B cells and accumulation of memory B cells inthe salivary glands of patients with Sjogren’s syndrome. Arthritis Rheum. 2002; 46:2160–2171.[PubMed: 12209521]
25. Genc K, Dona DL, Reder AT. Increased CD80(+) B cells in active multiple sclerosis and reversalby interferon beta-1b therapy. J Clin Invest. 1997; 99:2664–2671. [PubMed: 9169496]
26. Harp CT, Lovett-Racke AE, Racke MK, Frohman EM, Monson NL. Impact of myelin-specificantigen presenting B cells on T cell activation in multiple sclerosis. Clin Immunol. 2008; 128:382–391. [PubMed: 18599355]
27. Suen WE, Bergman CM, Hjelmstrom P, Ruddle NH. A critical role for lymphotoxin inexperimental allergic encephalomyelitis. J Exp Med. 1997; 186:1233–1240. [PubMed: 9334362]
28. Taga K, Tosato G. IL-10 inhibits human T cell proliferation and IL-2 production. J Immunol.1992; 148:1143–1148. [PubMed: 1737931]
29. Correale J, Farez M, Razzitte G. Helminth infections associated with multiple sclerosis induceregulatory B cells. Ann Neurol. 2008; 64:187–199. [PubMed: 18655096]
30. Dhib-Jalbut S. Glatiramer acetate (Copaxone) therapy for multiple sclerosis. Pharmacol Ther.2003; 98:245–255. [PubMed: 12725872]
31. Barth H, Berg PA, Klein R. Methods for the in vitro determination of an individual dispositiontowards TH1- or TH2-reactivity by the application of appropriate stimulatory antigens. Clin ExpImmunol. 2003; 134:78–85. [PubMed: 12974758]
32. Lenarczyk A, Helsloot J, Farmer K, Peters L, Sturgess A, Kirkham B. Antigen-induced IL-17response in the peripheral blood mononuclear cells (PBMC) of healthy controls. Clin ExpImmunol. 2000; 122:41–48. [PubMed: 11012616]
33. Duda PW, Krieger JI, Schmied MC, Balentine C, Hafler DA. Human and murine CD4 T cellreactivity to a complex antigen: recognition of the synthetic random polypeptide glatirameracetate. J Immunol. 2000; 165:7300–7307. [PubMed: 11120865]
34. Sun J, Link H, Olsson T, Xiao BG, Andersson G, Ekre HP, Linington C, Diener P. T and B cellresponses to myelin-oligodendrocyte glycoprotein in multiple sclerosis. J Immunol. 1991;146:1490–1495. [PubMed: 1899688]
35. Julius MH, Masuda T, Herzenberg LA. Demonstration that antigen-binding cells are precursors ofantibody-producing cells after purification with a fluorescence-activated cell sorter. Proc NatlAcad Sci U S A. 1972; 69:1934–1938. [PubMed: 4114858]
36. Pistoia V. Production of cytokines by human B cells in health and disease. Immunol Today. 1997;18:343–350. [PubMed: 9238838]
37. Lindert RB, Haase CG, Brehm U, Linington C, Wekerle H, Hohlfeld R. Multiple sclerosis: B- andT-cell responses to the extracellular domain of the myelin oligodendrocyte glycoprotein. Brain.1999; 122(Pt 11):2089–2100. [PubMed: 10545394]
Harp et al. Page 15
Eur J Immunol. Author manuscript; available in PMC 2011 October 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

38. Lovett-Racke AE, Trotter JL, Lauber J, Perrin PJ, June CH, Racke MK. Decreased dependence ofmyelin basic protein-reactive T cells on CD28-mediated costimulation in multiple sclerosispatients. A marker of activated/memory T cells. J Clin Invest. 1998; 101:725–730. [PubMed:9466965]
39. Hellings N, Baree M, Verhoeven C, D’Hooghe M,B, Medaer R, Bernard CC, Raus J, Stinissen P.T-cell reactivity to multiple myelin antigens in multiple sclerosis patients and healthy controls. JNeurosci Res. 2001; 63:290–302. [PubMed: 11170179]
40. de Rosbo, N. Kerlero; Milo, R.; Lees, MB.; Burger, D.; Bernard, CC.; Ben-Nun, A. Reactivity tomyelin antigens in multiple sclerosis. Peripheral blood lymphocytes respond predominantly tomyelin oligodendrocyte glycoprotein. J Clin Invest. 1993; 92:2602–2608. [PubMed: 7504688]
41. de Rosbo, N. Kerlero; Hoffman, M.; Mendel, I.; Yust, I.; Kaye, J.; Bakimer, R.; Flechter, S.;Abramsky, O.; Milo, R.; Karni, A.; Ben-Nun, A. Predominance of the autoimmune response tomyelin oligodendrocyte glycoprotein (MOG) in multiple sclerosis: reactivity to the extracellulardomain of MOG is directed against three main regions. Eur J Immunol. 1997; 27:3059–3069.[PubMed: 9394837]
42. Scholz C, Patton KT, Anderson DE, Freeman GJ, Hafler DA. Expansion of autoreactive T cells inmultiple sclerosis is independent of exogenous B7 costimulation. J Immunol. 1998; 160:1532–1538. [PubMed: 9570577]
43. Putheti P, Soderstrom M, Link H, Huang YM. Effect of glatiramer acetate (Copaxone) onCD4+CD25high T regulatory cells and their IL-10 production in multiple sclerosis. JNeuroimmunol. 2003; 144:125–131. [PubMed: 14597106]
44. Haas J, Korporal M, Balint B, Fritzsching B, Schwarz A, Wildemann B. Glatiramer acetateimproves regulatory T-cell function by expansion of naive CD4(+)CD25(+)FOXP3(+)CD31(+) T-cells in patients with multiple sclerosis. J Neuroimmunol. 2009; 216:113–117. [PubMed:19646767]
45. Mikulkova Z, Praksova P, Stourac P, Bednarik J, Strajtova L, Pacasova R, Belobradkova J, Dite P,Michalek J. Numerical defects in CD8+CD28− T-suppressor lymphocyte population in patientswith type 1 diabetes mellitus and multiple sclerosis. Cell Immunol. 262:75–79. [PubMed:20219185]
46. Correale J, Villa A. Role of CD8+ CD25+ Foxp3+ regulatory T cells in multiple sclerosis. AnnNeurol. 67:625–638. [PubMed: 20437560]
47. Zang YC, Li S, Rivera VM, Hong J, Robinson RR, Breitbach WT, Killian J, Zhang JZ. IncreasedCD8+ cytotoxic T cell responses to myelin basic protein in multiple sclerosis. J Immunol. 2004;172:5120–5127. [PubMed: 15067096]
48. Wei C, Anolik J, Cappione A, Zheng B, Pugh-Bernard A, Brooks J, Lee EH, Milner EC, Sanz I. Anew population of cells lacking expression of CD27 represents a notable component of the B cellmemory compartment in systemic lupus erythematosus. J Immunol. 2007; 178:6624–6633.[PubMed: 17475894]
49. Barth H, Klein R, Berg PA, Wiedenmann B, Hopf U, Berg T. Analysis of the effect of IL-12therapy on immunoregulatory T-cell subsets in patients with chronic hepatitis C infection.Hepatogastroenterology. 2003; 50:201–206. [PubMed: 12630023]
50. Davis LS, Schulze-Koops H, Lipsky PE. Human CD4+ T cell differentiation and effector function:implications for autoimmunity. Immunol Res. 1999; 19:25–34. [PubMed: 10374693]
51. Rivino L, Messi M, Jarrossay D, Lanzavecchia A, Sallusto F, Geginat J. Chemokine receptorexpression identifies Pre-T helper (Th)1, Pre-Th2, and nonpolarized cells among human CD4+central memory T cells. J Exp Med. 2004; 200:725–735. [PubMed: 15381728]
52. Moldovan IR, Cotleur AC, Zamor N, Butler RS, Pelfrey CM. Multiple sclerosis patients showsexual dimorphism in cytokine responses to myelin antigens. J Neuroimmunol. 2008; 193:161–169. [PubMed: 18022700]
53. Duda PW, Schmied MC, Cook SL, Krieger JI, Hafler DA. Glatiramer acetate (Copaxone) inducesdegenerate, Th2-polarized immune responses in patients with multiple sclerosis. J Clin Invest.2000; 105:967–976. [PubMed: 10749576]
54. Neuhaus O, Farina C, Yassouridis A, Wiendl H, Bergh F. Then, Dose T, Wekerle H, Hohlfeld R.Multiple sclerosis: comparison of copolymer-1- reactive T cell lines from treated and untreated
Harp et al. Page 16
Eur J Immunol. Author manuscript; available in PMC 2011 October 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

subjects reveals cytokine shift from T helper 1 to T helper 2 cells. Proc Natl Acad Sci U S A.2000; 97:7452–7457. [PubMed: 10861011]
55. Fridkis-Hareli M, Teitelbaum D, Gurevich E, Pecht I, Brautbar C, Kwon OJ, Brenner T, Arnon R,Sela M. Direct binding of myelin basic protein and synthetic copolymer 1 to class II majorhistocompatibility complex molecules on living antigen-presenting cells--specificity andpromiscuity. Proc Natl Acad Sci U S A. 1994; 91:4872–4876. [PubMed: 7515181]
56. Reindl M, Linington C, Brehm U, Egg R, Dilitz E, Deisenhammer F, Poewe W, Berger T.Antibodies against the myelin oligodendrocyte glycoprotein and the myelin basic protein inmultiple sclerosis and other neurological diseases: a comparative study. Brain. 1999; 122(Pt 11):2047–2056. [PubMed: 10545390]
57. Guggenmos J, Schubart AS, Ogg S, Andersson M, Olsson T, Mather IH, Linington C. Antibodycross-reactivity between myelin oligodendrocyte glycoprotein and the milk protein butyrophilin inmultiple sclerosis. J Immunol. 2004; 172:661–668. [PubMed: 14688379]
58. Litzenburger T, Bluthmann H, Morales P, Pham-Dinh D, Dautigny A, Wekerle H, Iglesias A.Development of myelin oligodendrocyte glycoprotein autoreactive transgenic B lymphocytes:receptor editing in vivo after encounter of a self-antigen distinct from myelin oligodendrocyteglycoprotein. J Immunol. 2000; 165:5360–5366. [PubMed: 11046072]
59. Vollmer J, Tluk S, Schmitz C, Hamm S, Jurk M, Forsbach A, Akira S, Kelly KM, Reeves WH,Bauer S, Krieg AM. Immune stimulation mediated by autoantigen binding sites within smallnuclear RNAs involves Toll-like receptors 7 and 8. J Exp Med. 2005; 202:1575–1585. [PubMed:16330816]
60. Wardemann H, Yurasov S, Schaefer A, Young JW, Meffre E, Nussenzweig MC. Predominantautoantibody production by early human B cell precursors. Science. 2003; 301:1374–1377.[PubMed: 12920303]
61. Cappione A 3rd, Anolik JH, Pugh-Bernard A, Barnard J, Dutcher P, Silverman G, Sanz I.Germinal center exclusion of autoreactive B cells is defective in human systemic lupuserythematosus. J Clin Invest. 2005; 115:3205–3216. [PubMed: 16211091]
62. Ekland EH, Forster R, Lipp M, Cyster JG. Requirements for follicular exclusion and competitiveelimination of autoantigen-binding B cells. J Immunol. 2004; 172:4700–4708. [PubMed:15067045]
63. Guay HM, Larkin J 3rd, Picca CC, Panarey L, Caton AJ. Spontaneous autoreactive memory B cellformation driven by a high frequency of autoreactive CD4+ T cells. J Immunol. 2007; 178:4793–4802. [PubMed: 17404260]
64. Pugh-Bernard AE, Silverman GJ, Cappione AJ, Villano ME, Ryan DH, Insel RA, Sanz I.Regulation of inherently autoreactive VH4-34 B cells in the maintenance of human B celltolerance. J Clin Invest. 2001; 108:1061–1070. [PubMed: 11581307]
65. Beck RW, Chandler DL, Cole SR, Simon JH, Jacobs LD, Kinkel RP, Selhorst JB, Rose JW,Cooper JA, Rice G, Murray TJ, Sandrock AW. Interferon beta-1a for early multiple sclerosis:CHAMPS trial subgroup analyses. Ann Neurol. 2002; 51:481–490. [PubMed: 11921054]
66. Schultze JL, Cardoso AA, Freeman GJ, Seamon MJ, Daley J, Pinkus GS, Gribben JG, Nadler LM.Follicular lymphomas can be induced to present alloantigen efficiently: a conceptual model toimprove their tumor immunogenicity. Proc Natl Acad Sci U S A. 1995; 92:8200–8204. [PubMed:7545296]
67. Crawford MP, Yan SX, Ortega SB, Mehta RS, Hewitt RE, Price DA, Stastny P, Douek DC, KoupRA, Racke MK, Karandikar NJ. High prevalence of autoreactive, neuroantigen-specific CD8+ Tcells in multiple sclerosis revealed by novel flow cytometric assay. Blood. 2004; 103:4222–4231.[PubMed: 14976054]
68. Scott DW. Antifluorescein affinity columns. Isolation and immunocompetence of lymphocytes thatbind fluoresceinated antigens in vivo or in vitro. J Exp Med. 1976; 144:69–78. [PubMed:1084410]
69. Moody MA, Haynes BF. Antigen-specific B cell detection reagents: use and quality control.Cytometry A. 2008; 73:1086–1092. [PubMed: 18613115]
70. Kurtzke JF. Rating neurologic impairment in multiple sclerosis: an expanded disability status scale(EDSS). Neurology. 1983; 33:1444–1452. [PubMed: 6685237]
Harp et al. Page 17
Eur J Immunol. Author manuscript; available in PMC 2011 October 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

FIGURE 1. Memory B cells from RRMS patients and HD exhibit an activated phenotype butexpress less HLA-DR(A) Gating strategy used to determine the percentage of peripheral blood memory(CD19+CD27+) and naïve (CD19+CD27−) B cell subsets from live lymphocytes based onforward scatter and side scatter characteristics from a single HD; percentage within eachgate indicated in corners of dot plot. (B) Percentage of naïve and memory B cell subsets inthe peripheral blood lymphocyte population of HD and RRMS patient cohorts; individualpoints represent individual subjects, and black bars represent mean values of each group.Representative histograms of (C) CD80 and (E) HLA-DR expression from one HD; isotypecontrol (grey line), naïve B cells (thin black line) and memory B cells (thick black line). Theexpression of CD80 (D) and HLA-DR (F) on memory and naïve B cell subsets in theperipheral blood lymphocyte population of HD and RRMS patient cohorts were quantifiedby mean fluorescence intensity (MFI) of the population; individual points representindividual subjects, and black bars represent mean values of each group. **p<0.01,***p<0.001 multiple comparisons post-hoc analysis with Bonferroni correction after two-way mixed-model ANOVA.
Harp et al. Page 18
Eur J Immunol. Author manuscript; available in PMC 2011 October 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
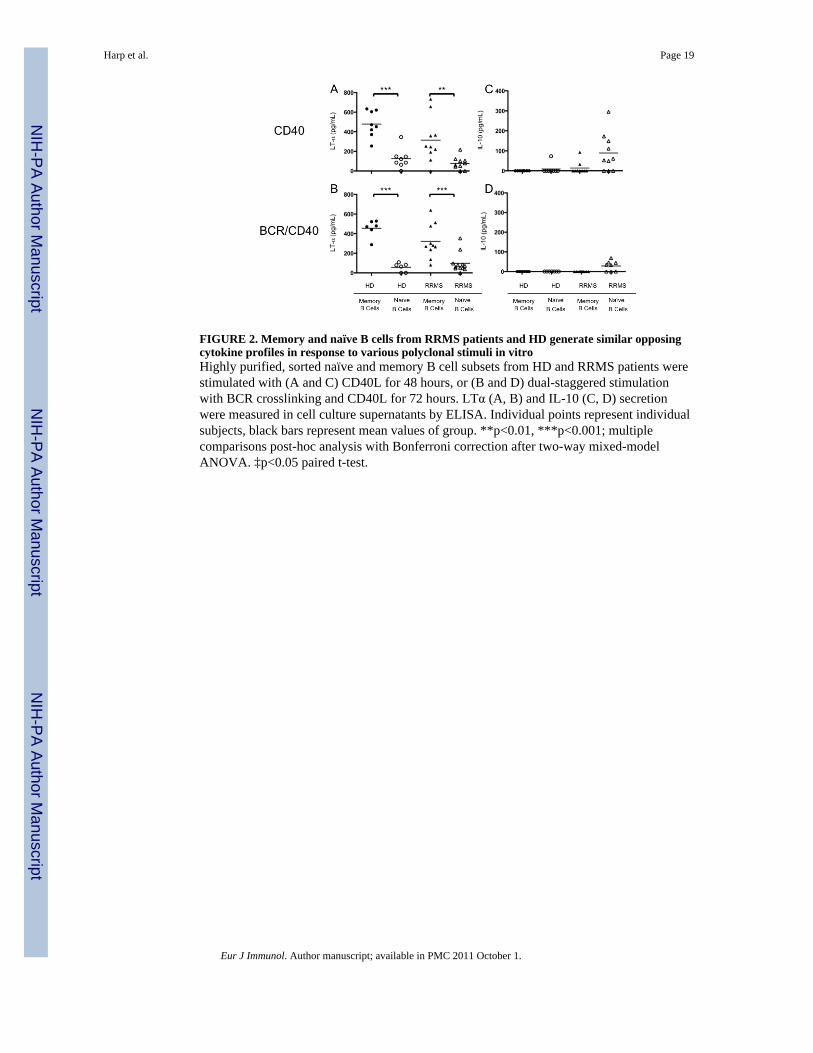
FIGURE 2. Memory and naïve B cells from RRMS patients and HD generate similar opposingcytokine profiles in response to various polyclonal stimuli in vitroHighly purified, sorted naïve and memory B cell subsets from HD and RRMS patients werestimulated with (A and C) CD40L for 48 hours, or (B and D) dual-staggered stimulationwith BCR crosslinking and CD40L for 72 hours. LTα (A, B) and IL-10 (C, D) secretionwere measured in cell culture supernatants by ELISA. Individual points represent individualsubjects, black bars represent mean values of group. **p<0.01, ***p<0.001; multiplecomparisons post-hoc analysis with Bonferroni correction after two-way mixed-modelANOVA. ‡p<0.05 paired t-test.
Harp et al. Page 19
Eur J Immunol. Author manuscript; available in PMC 2011 October 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript

FIGURE 3. CD4+ T cell proliferation from RRMS patientsA) Example of gating strategy used to obtain the raw percentage of CD3+CD4+ T cellproliferation in co-culture of purified CFSE-labeled T cells and autologous FACS purifiedCD27+CD19+ memory B cells in the presence of 5 ug/mL of TT, from HD-12. Numbersinside of gates are the percentage of cells in each gate. Example of proliferation histogramsfrom MS-10 (B), and MS-1 (C) of CD3+CD4+ T cell proliferation in cocultures of purifiedCFSE-labeled T cells and autologous highly purified FACS-sorted naïve B cells (top rows)or memory B cells (bottom rows) along with antigens where indicated (above columns).Raw percentage of CD4+ T cell proliferation was determined by gating onCFSElowCD3+CD4+ cells and listed above the histogram gates. ΔProliferation wascalculated by subtracting background (No Ag) proliferation (listed in parenthesis abovegate). MS-10 CD4+ T cell viability was 40.6±0.3% and 40.8±1.6% in naïve B-T andmemory B-T cell cultures respectively. MS-1 CD4+ T cell viability was 36.9±1.7% and38.3±2.2% in naïve and memory B-T cell cultures respectively.
Harp et al. Page 20
Eur J Immunol. Author manuscript; available in PMC 2011 October 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
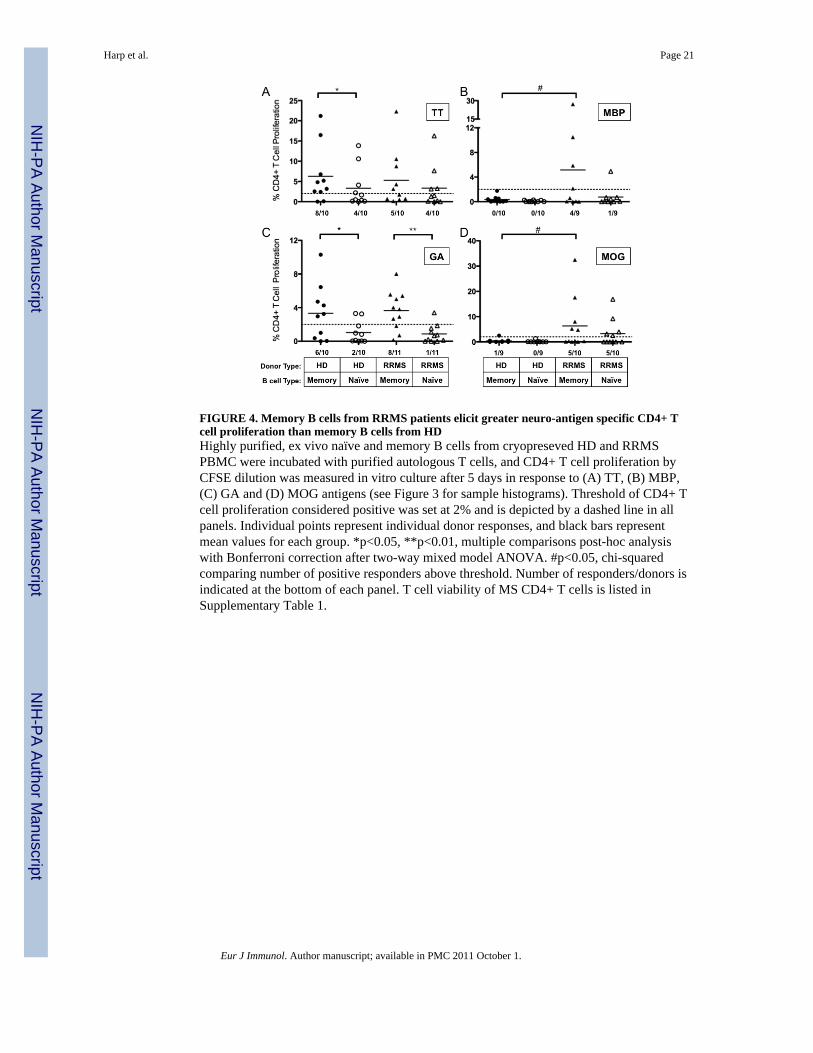
FIGURE 4. Memory B cells from RRMS patients elicit greater neuro-antigen specific CD4+ Tcell proliferation than memory B cells from HDHighly purified, ex vivo naïve and memory B cells from cryopreseved HD and RRMSPBMC were incubated with purified autologous T cells, and CD4+ T cell proliferation byCFSE dilution was measured in vitro culture after 5 days in response to (A) TT, (B) MBP,(C) GA and (D) MOG antigens (see Figure 3 for sample histograms). Threshold of CD4+ Tcell proliferation considered positive was set at 2% and is depicted by a dashed line in allpanels. Individual points represent individual donor responses, and black bars representmean values for each group. *p<0.05, **p<0.01, multiple comparisons post-hoc analysiswith Bonferroni correction after two-way mixed model ANOVA. #p<0.05, chi-squaredcomparing number of positive responders above threshold. Number of responders/donors isindicated at the bottom of each panel. T cell viability of MS CD4+ T cells is listed inSupplementary Table 1.
Harp et al. Page 21
Eur J Immunol. Author manuscript; available in PMC 2011 October 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
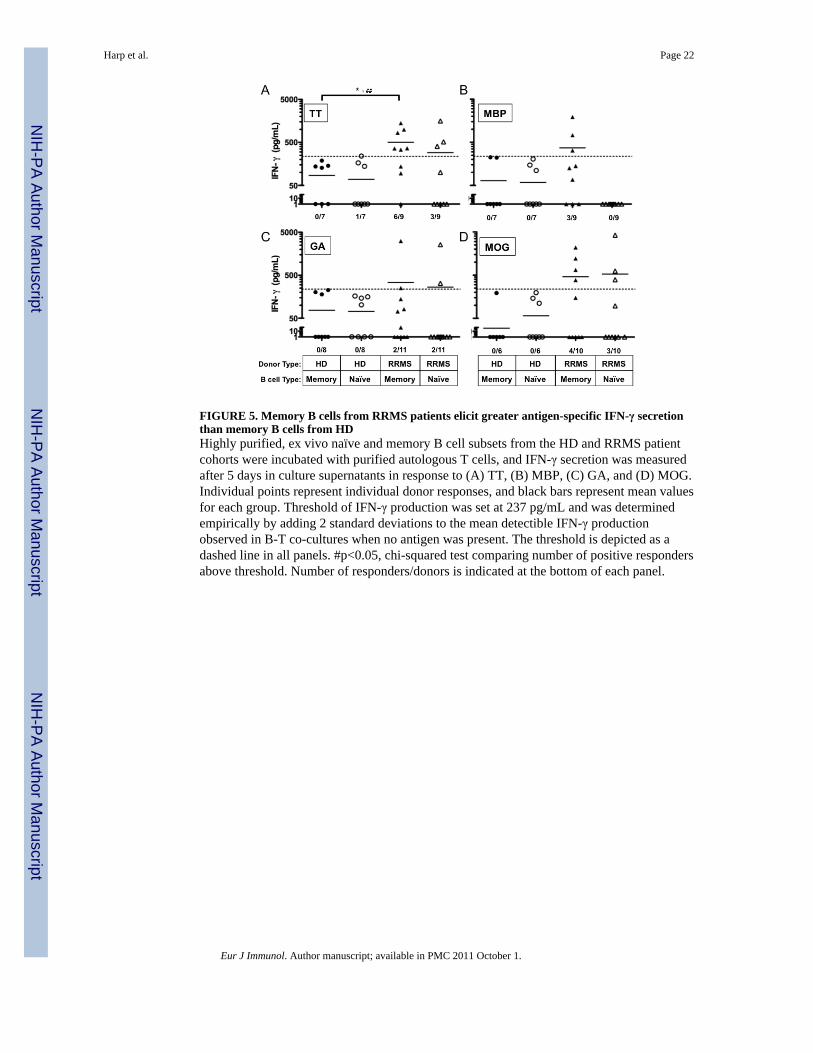
FIGURE 5. Memory B cells from RRMS patients elicit greater antigen-specific IFN-γ secretionthan memory B cells from HDHighly purified, ex vivo naïve and memory B cell subsets from the HD and RRMS patientcohorts were incubated with purified autologous T cells, and IFN-γ secretion was measuredafter 5 days in culture supernatants in response to (A) TT, (B) MBP, (C) GA, and (D) MOG.Individual points represent individual donor responses, and black bars represent mean valuesfor each group. Threshold of IFN-γ production was set at 237 pg/mL and was determinedempirically by adding 2 standard deviations to the mean detectible IFN-γ productionobserved in B-T co-cultures when no antigen was present. The threshold is depicted as adashed line in all panels. #p<0.05, chi-squared test comparing number of positive respondersabove threshold. Number of responders/donors is indicated at the bottom of each panel.
Harp et al. Page 22
Eur J Immunol. Author manuscript; available in PMC 2011 October 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
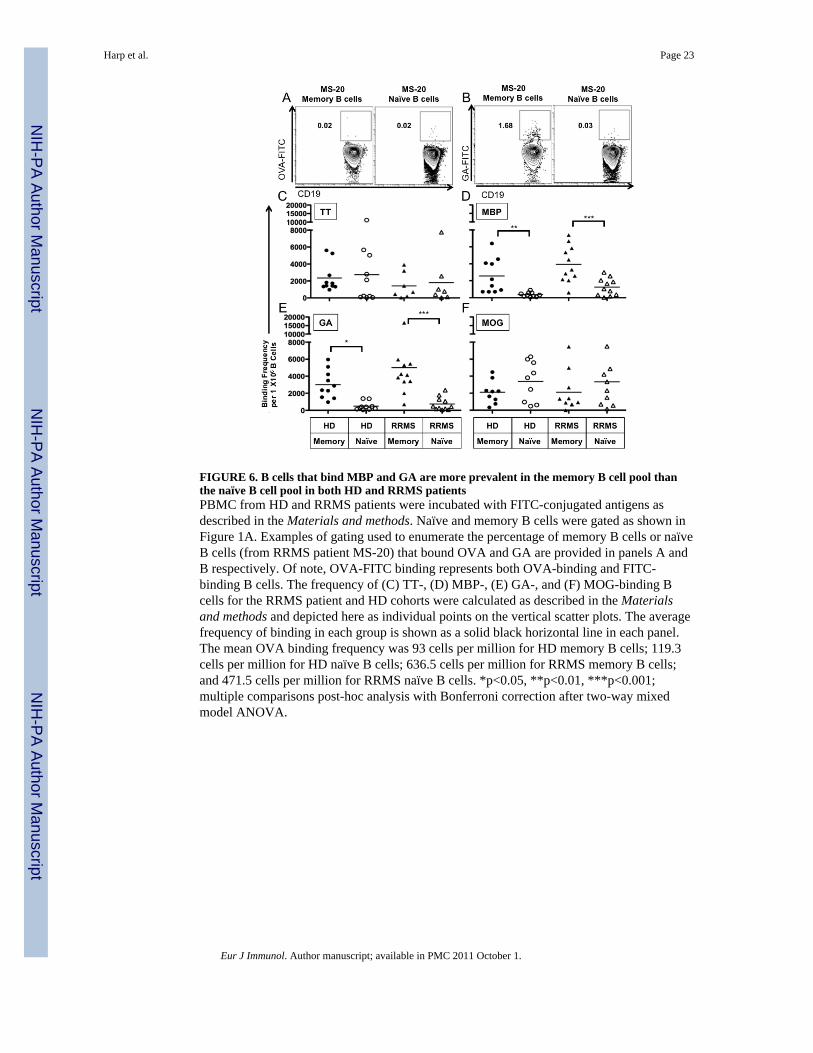
FIGURE 6. B cells that bind MBP and GA are more prevalent in the memory B cell pool thanthe naïve B cell pool in both HD and RRMS patientsPBMC from HD and RRMS patients were incubated with FITC-conjugated antigens asdescribed in the Materials and methods. Naïve and memory B cells were gated as shown inFigure 1A. Examples of gating used to enumerate the percentage of memory B cells or naïveB cells (from RRMS patient MS-20) that bound OVA and GA are provided in panels A andB respectively. Of note, OVA-FITC binding represents both OVA-binding and FITC-binding B cells. The frequency of (C) TT-, (D) MBP-, (E) GA-, and (F) MOG-binding Bcells for the RRMS patient and HD cohorts were calculated as described in the Materialsand methods and depicted here as individual points on the vertical scatter plots. The averagefrequency of binding in each group is shown as a solid black horizontal line in each panel.The mean OVA binding frequency was 93 cells per million for HD memory B cells; 119.3cells per million for HD naïve B cells; 636.5 cells per million for RRMS memory B cells;and 471.5 cells per million for RRMS naïve B cells. *p<0.05, **p<0.01, ***p<0.001;multiple comparisons post-hoc analysis with Bonferroni correction after two-way mixedmodel ANOVA.
Harp et al. Page 23
Eur J Immunol. Author manuscript; available in PMC 2011 October 1.
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
NIH
-PA Author Manuscript
Harp et al. Page 24
Table 1
Patient Characteristics
ID Number Agee Sex EDSSe,f
MS-1 c,d 23 F 1.5
MS-2 a,b,c,d 25 F 1.5
MS-4 a,b,d 43 F 1.5
MS-5 a,b,c 47 F 2.5
MS-10 a,b,c,d 47 F 2
MS-11 a,b,c,d 40 M 0
MS-12 a,b 52 F 1.5
MS-13 a,b 41 F 1
MS-14 c 41 F 2
MS-15 c 32 F 1.5
MS-16 c 25 F 0
MS-17 c,d 44 F 3.5
MS-18 c,d 37 F 2.5
MS-19 a,b,d 35 F 2
MS-20 a,b,c 27 M 1.5
MS-21 a,b 34 F 1
MS-22 a,b 44 F 1.5
MS-23 a,b,d 50 F 3
MS-24 a,b,d 34 F 2
MS-25 a,d 46 M 2.5
MS-26 a 23 F 2
MS-27 a 26 F 2.5
aSample used in measurement of peripheral B cell percentages, CD80 MFI
bSample used in measurement of HLA-DR MFI
cSample used in measurement of CD4+ T cell proliferation, IFN-γ secretion and antigen binding assay by flow cytometry
dSample used in measurement of LTα and IL-10 secretion
eat time of leukapheresis
fExpanded Disability Status Scale for MS [70]; n.d. = no data
Eur J Immunol. Author manuscript; available in PMC 2011 October 1.




















