
Mechanical characterization of fine grained silicon carbide consolidated using polymer pyrolysis and...
11
CERAMICS INTERNATIONAL Available online at www.sciencedirect.com Ceramics International 40 (2014) 12081–12091 Mechanical characterization of fine grained silicon carbide consolidated using polymer pyrolysis and spark plasma sintering Arif Rahman, Ashish Singh, Sandip P. Harimkar, Raman P. Singh n School of Mechanical and Aerospace Engineering, Oklahoma State University, 218 Engineering North, Stillwater, OK 74078, United States Received 23 February 2014; accepted 7 April 2014 Available online 16 April 2014 Abstract In this study fine grained bulk silicon carbide ceramics are processed using a novel approach that involves pyrolysis of a preceramic polymer followed by spark plasma sintering (SPS). Allylhydridopolycarbosilane (AHPCS) is used as the preceramic polymer that is pyrolyzed under inert conditions to produce SiC powder. Fourier transform infrared spectroscopy (FTIR) is used to confirm complete conversion of the preceramic polymer to amorphous SiC at 1400 1C. Subsequently, spark plasma sintering (SPS) is used to compact the SiC powder at temperatures ranging from 1600 to 2100 1C at a uni-axial pressure of 70 MPa and a soak time of 10 min. In situ crystallization of amorphous SiC using SPS results in fine grained structure with grain size ranging from 97 to 540 nm. Close to theoretical density materials are obtained, at the higher sintering temperatures, with mechanical properties that exceed those of commercially processed sintered silicon carbide. & 2014 Elsevier Ltd and Techna Group S.r.l. All rights reserved. Keywords: Mechanical characterization; Polymer pyrolysis; Spark plasma sintering; Fracture toughness; SiC 1. Introduction Significant enhancements in mechanical properties are expected for nanostructured silicon carbide (SiC) as compared to conventional microcrystalline (micro-grained) microstructure, primarily due to the very high grain boundary area-to-volume ratio present in nanostructured materials [1,2]. Nonetheless, bulk processing of nanostructured SiC is challenging due to covalent nature of Si–C bonds and low self-diffusion coefficients. Commonly used techniques for bulk processing of SiC are pressureless sintering, hot pressing, and hot isostatic pressing using commercial powder feedstock. Bulk polycrystalline sili- con carbide consolidated using these methods exhibits coarse- grained microstructure due to the application of higher proces- sing temperatures (2100–2500 1C) for longer time (1–24 h) [3,4]. As a result, various approaches have been pursued to restrict grain growth during sintering of ceramics. One approach has involved the use of higher sintering pressures, without increasing sintering temperature or time. Shinoda et al. have used nanocrystalline SiC powder with 3.5 wt% of free carbon to consolidate bulk sample of 97.1% relative density with the use of 980 MPa pressure and 1600 1C temperature while restricting the grain size to 30 nm [5]. However, usage of ultra-high pressure makes this method unsuitable for the fabrication of large ceramic parts for commercial applications [2]. Alternatively, liquid phase sintering (LPS) with the use of a second phase allows sintering of SiC at a relatively lower temperature (1900 1C) without the use of external pressures. Gomez et al. obtained more than 95% relative density with the addition of different proportions of additives (Al 2 O 3 , Y 2 O 3 , and C) at a temperature below 1900 1C using LPS [6]. The use of such additives during sintering aids in the densifica- tion process by segregating at the grain boundaries and enhancing mass transport and diffusion through the grain boundaries [7]. While such additives effectively enhance sinter- ing, they have deleterious effects on the microstructure and properties of ceramics, especially with regards to high- temperature creep [8]. To avoid the negative effects of sintering additives, spark plasma sintering technique has recently been used to con- solidate bulk SiC. In spark plasma sintering the combined www.elsevier.com/locate/ceramint http://dx.doi.org/10.1016/j.ceramint.2014.04.048 0272-8842/& 2014 Elsevier Ltd and Techna Group S.r.l. All rights reserved. n Corresponding author. Tel.: þ1 405 744 5140; fax: þ 1 405 744 3189. E-mail address: [email protected] (R.P. Singh).
-
Upload
independent -
Category
Documents
-
view
2 -
download
0
Transcript of Mechanical characterization of fine grained silicon carbide consolidated using polymer pyrolysis and...

CERAMICSINTERNATIONAL
Available online at www.sciencedirect.com
http://dx.doi.org/0272-8842/& 20
nCorrespondinE-mail addre
(2014) 12081–12091
Ceramics International 40 www.elsevier.com/locate/ceramintMechanical characterization of fine grained silicon carbide consolidatedusing polymer pyrolysis and spark plasma sintering
Arif Rahman, Ashish Singh, Sandip P. Harimkar, Raman P. Singhn
School of Mechanical and Aerospace Engineering, Oklahoma State University, 218 Engineering North, Stillwater, OK 74078, United States
Received 23 February 2014; accepted 7 April 2014Available online 16 April 2014
Abstract
In this study fine grained bulk silicon carbide ceramics are processed using a novel approach that involves pyrolysis of a preceramic polymerfollowed by spark plasma sintering (SPS). Allylhydridopolycarbosilane (AHPCS) is used as the preceramic polymer that is pyrolyzed under inertconditions to produce SiC powder. Fourier transform infrared spectroscopy (FTIR) is used to confirm complete conversion of the preceramicpolymer to amorphous SiC at 1400 1C. Subsequently, spark plasma sintering (SPS) is used to compact the SiC powder at temperatures rangingfrom 1600 to 2100 1C at a uni-axial pressure of 70 MPa and a soak time of 10 min. In situ crystallization of amorphous SiC using SPS results infine grained structure with grain size ranging from 97 to 540 nm. Close to theoretical density materials are obtained, at the higher sinteringtemperatures, with mechanical properties that exceed those of commercially processed sintered silicon carbide.& 2014 Elsevier Ltd and Techna Group S.r.l. All rights reserved.
Keywords: Mechanical characterization; Polymer pyrolysis; Spark plasma sintering; Fracture toughness; SiC
1. Introduction
Significant enhancements in mechanical properties areexpected for nanostructured silicon carbide (SiC) as comparedto conventional microcrystalline (micro-grained) microstructure,primarily due to the very high grain boundary area-to-volumeratio present in nanostructured materials [1,2]. Nonetheless, bulkprocessing of nanostructured SiC is challenging due to covalentnature of Si–C bonds and low self-diffusion coefficients.
Commonly used techniques for bulk processing of SiC arepressureless sintering, hot pressing, and hot isostatic pressingusing commercial powder feedstock. Bulk polycrystalline sili-con carbide consolidated using these methods exhibits coarse-grained microstructure due to the application of higher proces-sing temperatures (2100–2500 1C) for longer time (1–24 h)[3,4]. As a result, various approaches have been pursued torestrict grain growth during sintering of ceramics. One approachhas involved the use of higher sintering pressures, withoutincreasing sintering temperature or time. Shinoda et al. have
10.1016/j.ceramint.2014.04.04814 Elsevier Ltd and Techna Group S.r.l. All rights reserved.
g author. Tel.: þ1 405 744 5140; fax: þ1 405 744 3189.ss: [email protected] (R.P. Singh).
used nanocrystalline SiC powder with 3.5 wt% of free carbon toconsolidate bulk sample of 97.1% relative density with the useof 980 MPa pressure and 1600 1C temperature while restrictingthe grain size to 30 nm [5]. However, usage of ultra-highpressure makes this method unsuitable for the fabrication oflarge ceramic parts for commercial applications [2].Alternatively, liquid phase sintering (LPS) with the use of a
second phase allows sintering of SiC at a relatively lowertemperature (1900 1C) without the use of external pressures.Gomez et al. obtained more than 95% relative densitywith the addition of different proportions of additives (Al2O3,Y2O3, and C) at a temperature below 1900 1C using LPS [6].The use of such additives during sintering aids in the densifica-tion process by segregating at the grain boundaries andenhancing mass transport and diffusion through the grainboundaries [7]. While such additives effectively enhance sinter-ing, they have deleterious effects on the microstructure andproperties of ceramics, especially with regards to high-temperature creep [8].To avoid the negative effects of sintering additives, spark
plasma sintering technique has recently been used to con-solidate bulk SiC. In spark plasma sintering the combined

A. Rahman et al. / Ceramics International 40 (2014) 12081–1209112082
effects of pulse direct current and uni-axial pressure are usedto sinter the material at comparatively low sintering tempera-tures (200–300 1C lower than conventional sintering) andsignificantly reduced sintering time (few minutes as com-pared to hours) [9,10]. Since very high heating rates (up to600 1C/min) are achievable using SPS due to the passage ofhigh amount of pulsed direct current through graphite diesand punches, grain growth during densification of nanostruc-tured SiC can be inhibited due to exposure to high tempera-tures for very short time [11]. Yamamoto et al. used SPS toconsolidate SiC powder prepared using mechanical alloyingof commercially available Si and C powder [12]. Althoughdense SiC ceramics were prepared using this method,mechanical properties were lower compared to commerciallyavailable SiC powder consolidated with additives using SPS.More recently, Guillard et al. and Lara et al. have carried outparametric study to understand the effect of time, temperatureand pressure on densification of SiC without the use ofadditives during SPS [13,14]. While temperature and pressureplayed a vital role in densification process, grain growth wasfound to be restricted to �100 nm with the use of higherpressure (150 MPa) at the same sintering temperature. Zhanget al. investigated the effect of granulation process on themicrostructure and mechanical properties of SiC sinteredusing SPS [15]. Binderless granulation process resulting in80 μm granules was considered as the key factor in accel-erating the densification process reaching a relative density of98.5% at 1860 1C. However, this process led to coarse-grained (2:05 μm) microstructure even at a sintering tempera-ture of 1860 1C. Lomello et al. used laser pyrolysis to prepareβ-SiC nanopowders as initial powder for SPS [16]. Althoughuse of this technique to prepare initial powder seems to bebeneficial as the mean particle size was 16.6 nm, the presenceof 5 wt% oxygen and 1 wt% free carbon as a by-product inthe initial powder hinders the densification process, moreimportantly, has detrimental grain coarsening effect. Lorretteet al. reported SPS sintering of β-SiC using SiC powderproduced by laser synthesis with or without the use of boronadditives [17]. In their study, material without boron additiveand 80 nm grain size showed similar flexural strengthcompared to dense commercial reference material at highertesting temperature. However, fracture toughness values werelower compared to the reference material even at roomtemperature for nano-sized SiC.
In order to obtain nanograined microstructure in bulk SiC,it is important to have control over the microstructure of theinitial powder that is used for bulk processing. Using commer-cially available powder as starting material does not allow thatcontrol. Pyrolysis of preceramic precursors allows that controlover the microstructure of the initial powder. In this process,organometallic polymeric compounds are pyrolytically decom-posed to produce the desired ceramic [18]. While the potentialof pyrolysis of preceramic precursors to produce amorphousand nanostructured ceramics is well recognized, the approachis often associated with high porosity in the processed bulkceramics rendering this method ineffective to be used for densebulk ceramic [18,19].
In the current investigation, a novel approach is presentedthat combines the techniques of polymer precursor processingand spark plasma sintering (SPS). This process can yield bulknanostructured SiC with the possibilities of retaining grainsizes down to the sub-500 nm range while still being able tofabricate net-shape dense forms. The aim of the study is toestablish a novel processing technique to enable fabrication offine-grained SiC, investigate the microstructure and themechanical properties in comparison to commercially availablesintered SiC (Hexoloy SA, Saint-Gobain Ceramics), and SiCsintered from commercially available powder with or withoutsintering additives.
2. Experimental methods
2.1. Powder preparation
As a first step amorphous–nanocrystalline SiC powder wasprepared from a preceramic polymer, allylhydridopolycarbosi-lane (AHPCS) obtained from Starfire Systems Inc. (Malta,NY). The powder preparation process was started by heatingthe liquid polymer precursor to 650 1C, at 1 1C/min, under aninert atmosphere and then holding it at 650 1C for 10 min. Thisinitiated the cross-linking of the polymer precursor. Forcomplete conversion to amorphous–nanocrystalline SiC, theheating was continued till 1400 1C, at 3 1C/min. The materialwas held at the final temperature for 1 h to ensure thermalequilibrium and complete processing. Finally, the material wascooled down to room temperature, at 5 1C/min.Due to the release of hydrogen gas during the polymer to
ceramic conversion, the final material contained large voids.This material was ground using a hand grinder until theparticles passed through a colander of mesh size 12 followedby subsequent milling into fine powder ð � 0:5 μmÞ using ahigh energy ball mill (Pulverisette, Fritsch GmbH). Ballmilling was performed using a ball-to-powder mass ratio of5:1 with tungsten carbide (WC) balls as grinding media for15 min with 750 rpm. Scanning electron microscope (SEM)images were used to measure the particle size. Five differentimages were used and a total of �300 particles wereconsidered for the measurement. Microstructure of SiC parti-cles after ball milling can be seen in Fig. 1 which shows thevariation of particle size over a range of 0.2–1.25 μm with87% of the particles falling within a 0:5 μm range. It should beemphasized, however, that this particle size does not indicategrain size of the material after pyrolysis, which is typically inthe 1–5 nm range as reported in an earlier work [20].For comparison commercially available SiC powder from
Alfa Aesar was also used. This powder was also ball milledusing the same parameters mentioned above.
2.2. Sintering parameters
Spark plasma sintering (SPS) was used to consolidatenanostructured SiC obtained through pyrolysis of preceramicprecursor using an SPS system (Model 10-3, Thermal Tech-nology, LLC., Santa Rosa, California, USA). The DC pulse
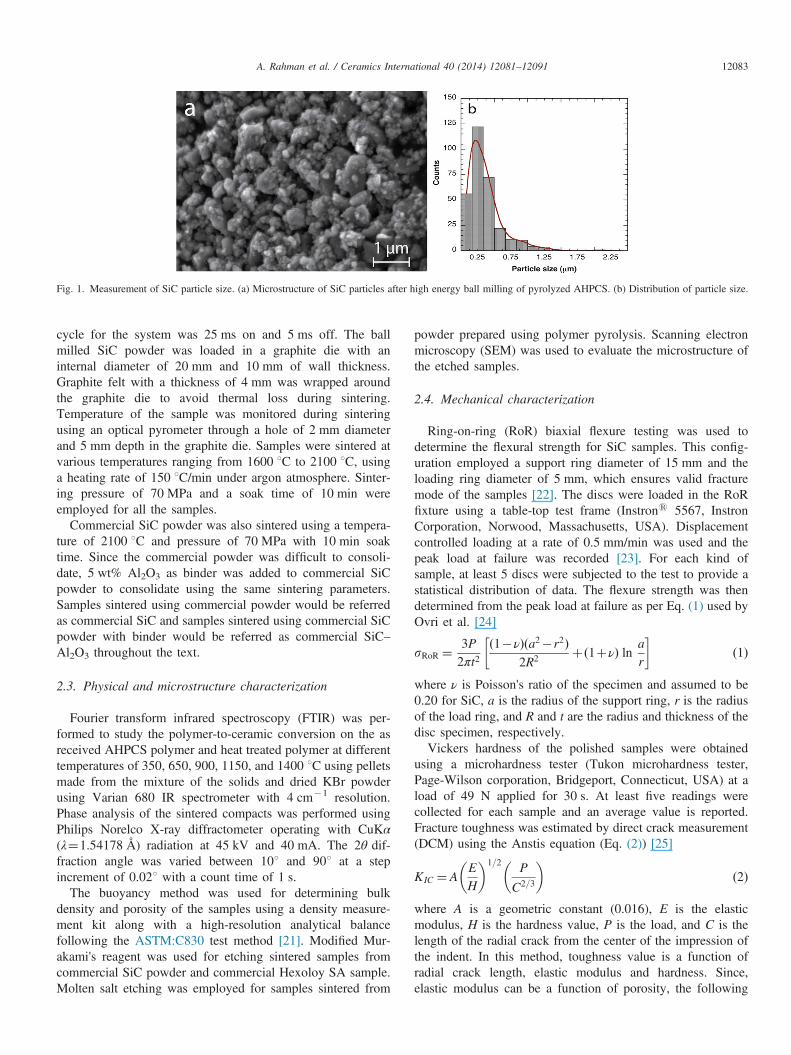
Fig. 1. Measurement of SiC particle size. (a) Microstructure of SiC particles after high energy ball milling of pyrolyzed AHPCS. (b) Distribution of particle size.
A. Rahman et al. / Ceramics International 40 (2014) 12081–12091 12083
cycle for the system was 25 ms on and 5 ms off. The ballmilled SiC powder was loaded in a graphite die with aninternal diameter of 20 mm and 10 mm of wall thickness.Graphite felt with a thickness of 4 mm was wrapped aroundthe graphite die to avoid thermal loss during sintering.Temperature of the sample was monitored during sinteringusing an optical pyrometer through a hole of 2 mm diameterand 5 mm depth in the graphite die. Samples were sintered atvarious temperatures ranging from 1600 1C to 2100 1C, usinga heating rate of 150 1C/min under argon atmosphere. Sinter-ing pressure of 70 MPa and a soak time of 10 min wereemployed for all the samples.
Commercial SiC powder was also sintered using a tempera-ture of 2100 1C and pressure of 70 MPa with 10 min soaktime. Since the commercial powder was difficult to consoli-date, 5 wt% Al2O3 as binder was added to commercial SiCpowder to consolidate using the same sintering parameters.Samples sintered using commercial powder would be referredas commercial SiC and samples sintered using commercial SiCpowder with binder would be referred as commercial SiC–Al2O3 throughout the text.
2.3. Physical and microstructure characterization
Fourier transform infrared spectroscopy (FTIR) was per-formed to study the polymer-to-ceramic conversion on the asreceived AHPCS polymer and heat treated polymer at differenttemperatures of 350, 650, 900, 1150, and 1400 1C using pelletsmade from the mixture of the solids and dried KBr powderusing Varian 680 IR spectrometer with 4 cm�1 resolution.Phase analysis of the sintered compacts was performed usingPhilips Norelco X-ray diffractometer operating with CuKα(λ¼1.54178 Å) radiation at 45 kV and 40 mA. The 2θ dif-fraction angle was varied between 101 and 901 at a stepincrement of 0.021 with a count time of 1 s.
The buoyancy method was used for determining bulkdensity and porosity of the samples using a density measure-ment kit along with a high-resolution analytical balancefollowing the ASTM:C830 test method [21]. Modified Mur-akami's reagent was used for etching sintered samples fromcommercial SiC powder and commercial Hexoloy SA sample.Molten salt etching was employed for samples sintered from
powder prepared using polymer pyrolysis. Scanning electronmicroscopy (SEM) was used to evaluate the microstructure ofthe etched samples.
2.4. Mechanical characterization
Ring-on-ring (RoR) biaxial flexure testing was used todetermine the flexural strength for SiC samples. This config-uration employed a support ring diameter of 15 mm and theloading ring diameter of 5 mm, which ensures valid fracturemode of the samples [22]. The discs were loaded in the RoRfixture using a table-top test frame (Instrons 5567, InstronCorporation, Norwood, Massachusetts, USA). Displacementcontrolled loading at a rate of 0.5 mm/min was used and thepeak load at failure was recorded [23]. For each kind ofsample, at least 5 discs were subjected to the test to provide astatistical distribution of data. The flexure strength was thendetermined from the peak load at failure as per Eq. (1) used byOvri et al. [24]
sRoR ¼ 3P2πt2
ð1�νÞða2�r2Þ2R2 þ 1þνð Þ ln a
r
� �ð1Þ
where ν is Poisson's ratio of the specimen and assumed to be0.20 for SiC, a is the radius of the support ring, r is the radiusof the load ring, and R and t are the radius and thickness of thedisc specimen, respectively.Vickers hardness of the polished samples were obtained
using a microhardness tester (Tukon microhardness tester,Page-Wilson corporation, Bridgeport, Connecticut, USA) at aload of 49 N applied for 30 s. At least five readings werecollected for each sample and an average value is reported.Fracture toughness was estimated by direct crack measurement(DCM) using the Anstis equation (Eq. (2)) [25]
KIC ¼ AE
H
� �1=2 P
C2=3
� �ð2Þ
where A is a geometric constant (0.016), E is the elasticmodulus, H is the hardness value, P is the load, and C is thelength of the radial crack from the center of the impression ofthe indent. In this method, toughness value is a function ofradial crack length, elastic modulus and hardness. Since,elastic modulus can be a function of porosity, the following

(a. u
.)
(iv)
(v)
A. Rahman et al. / Ceramics International 40 (2014) 12081–1209112084
equation proposed by Snead et al. was used for calculatingelastic modulus [26]
E¼ E0 expð�CVpÞ ð3Þwhere E0 ¼ 460 GPa is the modulus of non-porous SiC, andC¼3.57 is a constant.
7001400210028003500
I R a
bsor
banc
e
Wavenumber (cm-1)
(i)
(ii)
(iii)
Fig. 3. IR Spectra for AHPCS heated to 350 1C (i), 650 1C (ii), 900 1C (iii),1150 1C (iv), and 1400 1C (v).
20 30 40 50 60 70 80 90
Inte
nsit
y (a
.u.)
2θθθθ (degree)
Pyrolysis at 1400ºC SiC
SPS at 1600ºC SiC
SPS at 1700ºC SiC
SPS at 1800ºC SiC
SPS at 1900ºC SiC
SPS at 2000ºC SiC
SPS at 2100ºC SiC
SiC(01-073-1665)WC(01-072-0097)
3. Results and discussion
3.1. Polymer–ceramic formation and microstructuralevolution
To identify the functional groups present in AHPCS and tounderstand the conversion from polymer to ceramic FTIR wasperformed. Fig. 2 shows the FTIR spectra of the as-receivedpolymer precursor. The peaks of interest are located at 840,1045, 1250, 1355, 1630, 2120, 2920, 2950, and 3073 cm�1.The backbone of the polymer is the Si–CH2–Si chain asconfirmed by the three peaks at around 1045 cm�1 corre-sponding to CH2 bending, at 1355 cm�1 corresponding toSi–CH2–Si deformation, and at 2920 cm�1 corresponding toC–H stretching. The presence of Si–CH3 is also identified bythree peaks; at around 840 cm�1 corresponding to Si–CH3
rocking and Si–C stretching, at around 1250 cm�1 correspond-ing to Si–CH3 stretching, and at around 2950 cm�1 corre-sponding to C–H stretching in Si–CH3. Peaks indicatingCH¼CH2 are at 1630 cm�1 corresponding to C¼C stretchingand 3073 cm�1 corresponding to C–H vibration. A strongpeak at 2120 cm�1 corresponds to Si–H. A peak around3400 cm�1 is due to the water absorbed in the KBr duringFTIR experiment [27–29].
After pyrolysis to 350 1C (i), intensity of most of the peaksis greatly decreased, specially for Si–H and Si–CH3, as shownin Fig. 3. The intensity of the C¼C stretching at 1630 cm�1
and C–H vibration at 3073 cm�1 almost disappears from thespectra indicating evolution of CH¼CH2. At higher pyrolysistemperature (ii), gradually peak intensity of Si–H decreases
7001400210028003500
I R a
bsor
banc
e (a
. u.)
Wavenumber (cm-1)
Si-CH3Si-CH2-Si
C=C
Si-H
Si-CH3
C-H-CH
2-
Si-CH3
Si-CH2-Si
Fig. 2. IR Spectra for as received allylhydridopolycarbosilane.
Fig. 4. XRD patterns from pyrolyzed SiC powder and spark plasma sinteredSiC compacts processed at various temperatures.
which ensures cross-linking by hydrosilylation and dehydro-coupling reactions [27,28]. Even at 900 1C (iii) small peak forSi–H is visible, which disappears at 1150 1C (iv) completely ashydrogen is expelled from the structure and polymer toceramic conversion is completed. At 1400 1C (v) only a broadpeak corresponding to SiC is present in the IR spectra. Thisindicates that pyrolyzing the polymer precursor to 1400 1Cresults in complete removal of hydrogen from the structure andconversion to SiC.X-ray diffraction (XRD) was used to investigate the evolu-
tion of microstructure in the starting SiC powder as it wassubjected to further processing by SPS. Fig. 4 shows XRDpatterns of starting amorphous powder as well as samplescompacted using SPS. The XRD pattern of the initial SiCpowder, obtained through pyrolysis at 1400 1C, exhibited

Table 1Estimated grain size from full pattern analysis of XRD and SEM micrograph ofetched samples.
Material Sinteringtemperature (1C)
Estimated grain size
XRD SEM
SiC 1600 97 nm –
SiC 1700 187 nm –
SiC 1800 197 nm –
SiC 1900 271 nm 260 nmSiC 2000 370 nm 345 nmSiC 2100 540 nm 470 nmCommercial SiC 2100 4:1 μm 5:0 μmCommercial SiC–5%Al2O3 2100 3:0 μm 3:9 μmHexoloy SA – 4:2 μm 4:4 μm
20 30 40 50 60 70 80 90
Inte
nsity
(a.u
.)
2θθθθ (degree)
Hexoloy, SA SiC
Commercial SiC SPS sintered
Commercial SiC–5%Al2O3 SPS sintered
6H-SiC (01-073-1663)
C (00-008-0415)4H-SiC (01-073-1664)
15R-SiC (01-073-1662)
6H
4H, 15R 4H, 6H, 15R4H, 6H
C 6H 4H 6H
4H, 6H, 15R
4H, 6H4H, 6H, 15R
4H, 6H4H
4H
Fig. 5. XRD patterns from SPS sintered commercial SiC, commercial SiC–Al2O3, and commercial Hexoloy SA.
A. Rahman et al. / Ceramics International 40 (2014) 12081–12091 12085
characteristic broad halo peaks with diffused intensity indicat-ing an amorphous/nanocrystalline structure. SPS of amor-phous/nanocrystalline powder at various temperaturesranging from 1600 1C to 2100 1C resulted in crystallizationof amorphous powder during sintering. Peaks at 361, 421, 601,and 721 correspond to (111), (200), (220) and (311) planes ofβ-SiC (ICCD: 29–1129). Two small peaks correspond toresidual tungsten carbide (WC) which was used as a grindingmedia for ball milling. Full pattern analysis was performedusing XFIT program utilizing fundamental parameters (FP)approach [30]. Calculated average crystallite size for all thesamples is presented in Table 1. With the increase in sinteringtemperature from 1600 1C to 2100 1C crystallite size increasedfrom 97 nm to 540 nm.
XRD patterns for Hexoloy SA, SPS sintered commercialSiC and SPS sintered commercial SiC with binder show peaksfor α-SiC as shown in Fig. 5. Note that samples sintered fromcommercial powder showed the presence of graphite peak at26.431 indicating excess carbon in the structure. Full patternanalysis was employed in a manner mentioned earlier to
estimate average crystal size. Estimated grain sizes were inthe range of 3:0–4:0 μm as shown in Table 1.For comparison, grain sizes were also estimated from SEM
images of etched samples. Molten salt etching was used for β-SiC samples sintered at 1900, 2000, and 2100 1C and modifiedMurakami's reagent was used for etching Hexoloy SA,commercial SiC, and commercial SiC–Al2O3 samples. In theetching process, etchant selectively etches the grain boundarydelineating the microstructure. Note that samples sinteredat 1600, 1700, and 1800 1C were difficult to etch due tohigher porosity content and were not used for grain sizeestimation from SEM micrographs. Fig. 6 shows variation ofmicrostructure based on sintering temperature and source ofinitial powder. Linear intercept method was used to estimategrain sizes from the SEM images and the shape correctionfactor employed was 1.22 considering equiaxed grains. Forsamples sintered using pyrolyzed powder, grain sizesvaried between 260 and 470 nm for sintering temperature1900, 2000, and 2100 1C (Table 1). For samples sintered usingcommercial powder, and Hexoloy SA grain sizes were in4:0–5:0 μm range.Estimated grain sizes clearly indicate that preparing amor-
phous/nanocrystalline SiC powder using pyrolysis of precera-mic precursor and then preparing bulk SiC using SPS can beused effectively to control the grain size without the use of anygrain growth inhibiting additives. Using same processingparameters for commercial SiC powder resulted in a signifi-cantly larger (650%) grain size for samples sintered fromcommercial SiC powder due to crystalline nature of initialpowder as compared to pyrolyzed powder that was mostlyamorphous in nature. However, the use of Al2O3 as binder hasshown to be effective in restricting grain growth for thecommercial SiC sample by 37%. Al2O3 restricts grain growthby hindering grain boundary diffusion as reported previously[31]. Nonetheless, the fabrication approach utilized in thecurrent investigation allows better control over evolution ofmicrostructure during fabrication without the use of anyadditive.
3.2. Relative density and densification
Variation of relative density as a function of sinteringtemperature is shown in Fig. 7, indicating influence ofsintering temperature on densification behavior of SPS sinteredSiC. At 1600 1C sintering temperature, relative density of thecompact is �60% and increase in sintering temperature to2000 1C showed sudden increase in relative density reaching92%. The maximum relative density obtained in this investiga-tion is about 95% at 2100 1C sintering temperature. Tounderstand the densification process, temperature profile,punch displacement, D and displacement rate, dD=dt as afunction of time for 2100 1C sintering temperature is plotted asshown in Fig. 8. Note that sintering pressure of 70 MPa wasapplied such a way that in each case the pressure reached70 MPa when the temperature reached desired sinteringtemperature. Initially applied pressure starts to rearrange theparticles, however, rise in temperature results in expansion of

Fig. 6. Scanning electron micrographs of etched (a) 1900 1C SiC, (b) 2000 1C SiC, (c) 2100 1C SiC, (d) commercial SiC, (e) commercial SiC–Al2O3, and(f) Hexoloy SA.
A. Rahman et al. / Ceramics International 40 (2014) 12081–1209112086
the punch as shown in case of displacement till �12.5 min.During this period densification of the compact is much lowercompared to expansion of the punch, hence decrease indisplacement is observed. This is also evident in the displace-ment rate curve. After that initial period at around 1875 1C,displacement starts to increase and displacement rate ismaximum at 2000 1C. Temperature dependent densificationmechanism such as diffusion is activated after 1875 1C. Hence,sintering temperature below 1900 1C results in lower relativedensity of the compacts.
Commercial SiC powder was difficult to sinter withoutsintering additives even at 2100 1C reaching a relative densityof 82%. Addition of Al2O3 as binder improved sinterability ofthe commercial powder increasing the relative density to 96%.Relative density of conventionally hot pressed Hexoloy, SAsample was 98.4%.
Although densification of SiC without binder is difficult,proper initial powder and optimization of processing para-meters can lead to densification in bulk SiC samples. In thisstudy, pyrolysis of preceramic polymer lead to initial SiCpowder that was sintered to 95% relative density at 2100 1C.However, commercial SiC powder was difficult to sinter usingthe same processing parameters. This shows that the source ofinitial powder plays an important role in densification process.
3.3. Mechanical properties
3.3.1. Flexural strengthIn the current study, flexural strength was determined using
a bi-axial flexure setup that utilizes ring-on-ring configuration.Flexural strengths of all the samples are presented in Table 2.Flexural strength increased from 46 MPa to 302 MPa as

A. Rahman et al. / Ceramics International 40 (2014) 12081–12091 12087
porosity content was reduced with the increase in sinteringtemperature from 1600 to 2100 1C. The ring-on-ring config-uration gives the most accurate measure of strength among all
50
60
70
80
90
100
1500 1600 1700 1800 1900 2000 2100 2200
Rel
ativ
e D
esni
ty (%
TD)
Sintering Temperature (ºC)
Fig. 7. Variation of density as a function of sintering temperature for SPSsintered SiC samples.
600
800
1000
1200
1400
1600
1800
2000
2200
0
1
2
3
4
5 10 15 20 25
Tem
pera
ture
(ºC
)
Displacem
ent , D (m
m)
Time (min)
dD/dt (m
m/m
in)
Fig. 8. Variation of temperature profile, punch displacement, D and displace-ment rate, dD=dt as a function of time for sample sintered at 2100 1C.
Table 2A summary of properties of sintered SiC from AHPCS, powder from commercial
Material Sinteringtemperature (1C)
Relativedensity (%)
SiC 1600 60.0SiC 1700 65.0SiC 1800 71.0SiC 1900 82.0SiC 2000 92.3SiC 2100 95.0Commercial SiC 2100 82.0Commercial SiC–5%Al2O3 2100 96.0Hexoloy SA – 98.7
the bi-axial setup that are being used [32]. Moreover, bi-axialflexure condition is considered to be more reliable compared touni-axial flexure because of maximum tensile stresses occur-ring within the central loading area, and failure is independentof edge condition.Strength of ceramics depends on relative density, flaw size
and grain size. However, for porous ceramics, strength isaffected more by porosity than grain size. In general, porosityreduces strength of ceramics by reducing the effective loadbearing area [33]. Using the relationship proposed by Ryshke-witch for dependence of strength on porosity content, a curvefitted to the strength values (Fig. 9) produced a good fit (R-squared value �0.96) [34]. The highest value of flexuralstrength of 302 MPa was observed for sample sintered at2100 1C. This strength value may seem to be a little lowercompared to usually published uni-axial strength data for SiC,but it should be noted that equibiaxial strength of a brittlematerial is usually lower than uni-axial strength because ofmore flaws being subjected to high stress at bi-axial testconfiguration [35]. Several authors have found bi-axialstrength to be 20–35% less than uni-axial strength whenfour-point bending configuration was used [22,36].
source and Hexoloy SA determined in this study.
Flexuralstrength (MPa)
Vickershardness (GPa)
Fracture toughness(MPa m1/2)
46720 4.370.4 –
72719 6.070.3 –
117724 9.070.3 –
195730 14.570.5 3.070.1242721 22.671.2 3.470.1302729 25.170.9 3.570.1104715 8.070.5 –
276754 20.571.4 4.070.4289756 25.270.8 2.570.1
0
50
100
150
200
250
300
350
0.1 0.2 0.3 0.4
Flex
ural
str
engt
h (M
Pa)
Porosity content
σ = σ0∗exp(-4.4∗V
p)
R2 = 0.96193
Fig. 9. Flexural strength as a function of porosity content for SPS sintered SiCsamples.

Fig. 10. SEM fracture surface images of (a) SiC sintered at 2100 1C, (b) Hexoloy, SA, (c) commercial SiC sintered at 2100 1C and (d) commercial SiC–Al2O3
sintered at 2100 1C showing different fracture modes.
A. Rahman et al. / Ceramics International 40 (2014) 12081–1209112088
In comparison, commercially available Hexoloy SA alsoshowed almost similar (�289 MPa) bi-axial strength value.Uni-axial strength of Hexoloy SA using four point bend test is380 MPa as supplied by the manufacturer. This is 24% higherthan the bi-axial strength found in the current investigation andconsistent with previous observations as mentioned earlier.Commercial SiC sample sintered at 2100 1C showed bi-axialstrength of 104 MPa that was 66% less than β-SiC samplesintered using the same parameters. Large flaws in terms ofresidual porosity in the commercial SiC due to lack ofsinterability was responsible for this low strength of thematerial. Addition of binder significantly improved densityof the sintered commercial SiC–Al2O3 to 96% and strengthalso increased to �276 MPa. With the addition of additives,14% increase in density resulted in 62% increase in strengthfor commercial SiC sample. Nonetheless, the strength ofcommercial SiC–Al2O3 was slightly less than SiC samplessintered using pyrolyzed powder. This is due to the embrittle-ment of the grain boundary as a result of impurities in the formof Al2O3 [37].
Fig. 10 shows fracture surface images of different materialsused in this study. SiC sintered from pyrolyzed powdershowed transgranular fracture. Hexoloy, SA also showedsimilar fracture mode. On the other hand, commercial SiCshowed a mix of transgranular and intergranular fracturemodes. Interestingly, addition of Al2O3 to the commercialpowder completely changed the fracture mode to intergranular.
This change is a result of grain boundary chemistry as Al andO have been shown to aggregate at the grain boundary [38,39].Recently, in an effort to sinter SiC without binders
Yamamoto et al. used SPS to consolidate SiC powder preparedusing mechanical alloying of commercially available Si and Cpowder [12]. Although dense SiC ceramics were preparedusing this method, bending strengths were lower compared tocommercially available SiC powder consolidated with addi-tives using SPS method. It is suspected that the mechanicalalloying leads to incomplete Si–C bonding in the startingpowder. This manifests itself as lower mechanical strength inthe sintered compact. The processing presented in the presentstudy, however, results in greatly improved properties as thestarting SiC powder is prepared by a chemical pyrolysis routewhich leads to proper Si–C bonding in the starting powder.
3.3.2. Vickers hardnessIn the present study, hardness increased from �4 GPa to
�25 GPa as a function of density as shown in Table 2.Increase in hardness with decreasing grain size is reported inthe literature for both oxide and non-oxide ceramics [40,41].Vickers hardness of the sintered SiC samples in the presentstudy, however, showed an increasing trend with the increasein processing temperature. This is mainly due to the higherresidual porosity in the structure at lower sintering temperatureresulting in decrease in hardness. Yamamoto et al. alsomeasured increase in Vicker's hardness with the increase in

0
5
10
15
20
25
30
0.1 0.2 0.3 0.4
Har
dnes
s (G
Pa)
Porosity content
Hv = Hv0 *exp(-5.4*V
p)
R2 = 0.98547
Fig. 11. Vickers hardness as a function of porosity content for SPS sinteredSiC samples.
A. Rahman et al. / Ceramics International 40 (2014) 12081–12091 12089
relative density reaching a hardness value of 20 GPa fornanograined-SiC consolidated using SPS [12]. Moreover,Lomello et al. reported influence of density to be strongerthan the influence of grain size when grain size of SiC wasunder 130 nm [16]. In that study, hardness increased to�25 GPa as a function of density. Ryshkewitch reported anexponential dependence of compression strength on porositycontent for different ceramic materials [34] and Snead et al.extended this relationship for hardness of ceramic materialusing the following equation [26]:
Hv¼Hv0 expð�CVpÞ ð4Þwhere Hv is Vicker's hardness in GPa, Hv0 is Vicker's hardnesswithout porosity, C is a constant (5.4), and Vp is the volumefraction of porosity. Variation of hardness as a function ofporosity content is shown in Fig. 11. The hardness valueswere fitted to a curve using Eq. (4) by least squares technique.R-squared value determined to be �0.99 ensured good fit tothe experimental data.
Hexoloy, SA with 98.4% relative density showed hardnessof 25.2 GPa which is similar to hardness of sample sinteredusing pyrolyzed powder. Dong et al. reported Vicker's hard-ness of 36 GPa for Hexoloy, SA at room temperature with anindentation load of 9.8 N [42]. This is significantly higher thanthe value observed in the current investigation. This discre-pancy is likely due to the difference in indentation load (49 N)used in the current study usually known as indentation sizeeffect [43–45]. Hardness of the commercial SiC samplesintered at 2100 1C was largely influenced by porosity of thematerial. Even at this sintering temperature lack of sintera-blility resulted in a moderate hardness of �8.1 GPa. Additionof sintering additive to the commercial powder improveddensification, hence, it was expected to have higher hardnessvalue. However, use of 49 N load resulted in deformed indentswith surface cracks making it difficult to measure hardness atthis load. Therefore, a load of 9.8 N was used to measure
hardness of the material. Even though hardness of commercialSiC improved to 20.5 GPa with the addition of binder, still itwas less than samples of similar relative density prepared usingpyrolysis.The differences in hardness could be explained based on the
fracture mode observed in respective materials. Flinders et al.previously reported decrease in hardness with change infracture mode from transgranular to intergranular [38]. Asfracture mode changes from transgranular to intergranular,displacing grains during indentation process becomes easier asthe crack propagates through the grain boundaries. Hence,drop in hardness is expected. It should be noted that both SiCsintered at 2100 1C and Hexoloy, SA showed transgranularfracture (Fig. 10a, b) that correlate with higher hardness valueobserved for these materials. Although commercial SiCsintered at 2100 1C showed a mix of transgranular andintergranular fracture, hardness value was dominated byporosity of the material. Interestingly, addition of Al2O3 tothe commercial powder completely changed the fracture modeto intergranular. This change is a result of grain boundarychemistry as Al and O have been shown to aggregate at thegrain boundary [38,39]. With this change in fracture mode,commercial SiC–Al2O3 showed less hardness even withimproved densification.
3.3.3. Fracture toughnessFracture toughness measured using direct crack measure-
ment (DCM) method is presented in Table 2. Samples withproper radial cracks were used for toughness measurement. Forsamples sintered using pyrolyzed powder, toughness increasedwith reduction in porosity. Such behavior has been observedby other authors [15]. Fracture resistance depends on crackpropagation path and impurities at grain boundary. For non-detectable impurities at grain boundary, grain boundarytoughness can be higher than grain toughness renderingtransgranular fracture [37]. This has been observed in thecurrent study for materials sintered using pyrolyzed powder asshown in Fig. 10. Interestingly, Hexoloy SA also showedtransgranular fracture as the amount of additives in the sinteredmaterial was very less [42]. Nonetheless, the fracture tough-ness of Hexoloy SA was 40% lower than the fracturetoughness of the SiC sintered at 2100 1C using pyrolyzedpowder. Similar fracture toughness of Hexoloy SA has beenreported by other authors using DCM method [46]. Since bothsamples had similar relative density and hardness, this varia-tion is only a function of crack resistance measured as radialcrack length. Finer grains can enhance toughness by impedingcrack propagation. As Hexoloy SA had much larger grain size(4:4 μm) compared to SiC sintered at 2100 1C (440 nm),difference in toughness for these materials can be observedas a result of such mechanisms. On the contrary, commercialSiC–Al2O3 showed higher fracture toughness (�4.0) com-pared to the rest of the materials. For this material, Al2O3
segregates at the grain boundary rendering the grain boundaryweaker than the SiC grains. As a result, crack propagation waschanged to intergranular mode from mixed mode in the binderless commercial SiC (Fig. 10). Intergranular fracture can

A. Rahman et al. / Ceramics International 40 (2014) 12081–1209112090
improve fracture toughness if crack driving force is decreasedby deflection of the crack along grain boundaries and notsuperseded by decrease in grain boundary toughness due to thesegregating impurities. In case of commercial SiC–Al2O3,toughness of Al2O3 is slightly less than SiC. Hence, decreasein crack driving force results in slightly higher toughness incommercial SiC–Al2O3.
4. Conclusion
Conventional sintering requires usage of high temperatureand pressure along with additive to consolidate bulk SiC. SPS,an alternative, has recently been explored for fabrication ofbulk SiC without binders. However, depending on the sourceof the initial powder, the SPS process can lead to bulk sampleswith varying mechanical properties. Hence, proper fabricationprocess for bulk SiC is still being investigated for dense SiCsamples with controlled microstructure without the use ofbinders. In that respect, current investigation combines the useof polymer pyrolysis and SPS to obtain dense SiC ceramicwith controlled microstructure. Based on the experimentalinvestigation the following conclusions are drawn:
1.
Chemical changes to the polymer precursor (AHPCS)during pyrolysis to different temperatures were identifiedusing IR spectroscopy. Pyrolysis of the polymer precursorgradually expelled the hydrogen from the network andresulted in hydrogen free at 1150 1C. Pyrolysis to 1400 1Cconfirmed complete polymer-to-ceramic conversion as evi-dent by the IR spectroscopy.2.
X-ray diffraction analysis revealed that the pyrolyzedpowder was amorphous/nanocrystalline in nature. Furtherprocessing of the pyrolyzed powder in terms of SPSresulted in increase in crystallite size from 97 to 540nm with increasing sintering temperature from 1600 to2100 1C, respectively. Similar processing parameters lead toexaggerated grain growth of the commercial crystallinepowder. These observations were also confirmed withscanning electron microscopy of the etched samples.3.
The sintering temperature was found to play a vital role incompacting SiC. Increase in the processing temperatureresulted in an increase in the relative bulk density from 60%to 95% through ordering of the structure. Punch displace-ment study revealed that densification started at around1875 1C and maximum densification was observed at2000 1C. In comparison, commercial SiC powder couldnot be densified without additive. Al2O3 in the form ofadditive improved densification for the commercial powder.4.
Mechanical properties were found to be dependent onporosity rather than grain size for pyrolyzed powder. Bothflexural strength and Vickers hardness showed exponentialdependence on porosity. However, mechanical properties ofpyrolyzed powder sintered at 2100 1C were comparable toother reference materials used in this study. The highestflexural strength of �302 MPa was observed for SiCsintered at 2100 1C at a relative density of 95%, whichwas similar to near theoretically dense commerciallyavailable SiC (Hexoloy SA). Moreover, SiC sintered at2100 1C showed 40% higher fracture toughness comparedto Hexoloy SA. Furthermore, commercial SiC powdersintered using an additive exhibited strength and hardnessvalues that were lower for this material due to embrittle-ment caused by the presence of impurity at the grainboundary. Thus, it is concluded that the combination ofpolymer pyrolysis with spark plasma sintering techniqueyields dense SiC with superior mechanical properties whilerestricting grain growth.
References
[1] I. Szlufarska, A. Nakano, P. Vashishta, A crossover in the mechanicalresponse of nanocrystalline ceramics, Science 309 (5736) (2005) 911–914.
[2] B. Kearand, A. Mukherjee, Far-from-equilibrium processing of nanostructured ceramics, in: Materials Processing Handbook, CRC Press,pp. 7-1–7-18, 2007.
[3] D. Liu, Oxidation of polycrystalline α-silicon carbide ceramic, Ceram.Int. 23 (5) (1997) 425–436.
[4] F. VanDijen, E. Mayer, Liquid phase sintering of silicon carbide, J. Eur.Ceram. Soc. 16 (4) (1996) 413–420.
[5] Y. Shinoda, T. Nagano, F. Wakai, Fabrication of nanograined siliconcarbide by ultrahigh-pressure hot isostatic pressing, J. Am. Ceram. Soc.82 (3) (1999) 771–773.
[6] E. Gomez, J. Echeberria, I. Iturriza, F. Castro, Liquid phase sintering ofsic with additions of Y2O3, Al2O3 and SiO2, J. Eur. Ceram. Soc. 24 (9)(2004) 2895–2903.
[7] J. Lee, Y. Ahn, T. Nishimura, H. Tanak, L. Sea-Hoon, Ultra-low-temperature sintering of nanostructured β-sic, J. Am. Ceram. Soc. 94 (2)(2011) 324–327.
[8] M. Backhausricoult, N. Mozdzierz, P. Eveno, Impurities in silicon-carbide ceramics and their role during high-temperature creep, J. Phys. III3 (December (12)) (1993) 2189–2210.
[9] G.B. Yadhukulakrishnan, A. Rahman, S. Karumuri, M.M. Stackpoole,A.K. Kalkan, R.P. Singh, S.P. Harimkar, Spark plasma sintering ofsilicon carbide and multi-walled carbon nanotube reinforced zirconiumdiboride ceramic composite, Mater. Sci. Eng.: A 552 (0) (2012) 125–133.
[10] G.B. Yadhukulakrishnan, S. Karumuri, A. Rahman, R.P. Singh,A.K. Kalkan, S.P. Harimkar, Spark plasma sintering of graphenereinforced zirconium diboride ultra-high temperature ceramic composites,Ceram. Int. 39 (6) (2013) 6637–6646.
[11] Z. Munir, U. Anselmi-Tamburini, M. Ohyanagi, The effect of electricfield and pressure on the synthesis and consolidation of materials: areview of the spark plasma sintering method, J. Mater. Sci. 41 (February(3)) (2006) 763–777.
[12] T. Yamamoto, T. Kondou, Y. Kodera, T. Ishii, M. Ohyanagi, Z. Munir,Mechanical properties of beta-SiC fabricated by spark plasma sintering,J. Mater. Eng. Perform. 14 (4) (2005) 460–466.
[13] F. Guillard, A. Allemand, J.-D. Lulewicz, J. Galy, Densification of SiCby SPS-effects of time, temperature and pressure, J. Eur. Ceram. Soc. 27(7) (2007) 2725–2728.
[14] A. Lara, A.L. Ortiz, A. Munoz, A. Dominguez-Rodriguez, Densificationof additive-free polycrystalline β-SiC by spark-plasma sintering, Ceram.Int. 38 (1) (2012) 45–53.
[15] Z.-H. Zhang, F.-C. Wang, J. Luo, S.-K. Lee, L. Wang, Processing andcharacterization of fine-grained monolithic SiC ceramic synthesized byspark plasma sintering, Mater. Sci. Eng.: A 527 (7–8) (2010) 2099–2103.
[16] F. Lomello, G. Bonnefont, Y. Leconte, N. Herlin-Boime, G. Fantozzi,Processing of nano-SiC ceramics: densification by {SPS} and mechanicalcharacterization, J. Eur. Ceram. Soc. 32 (3) (2012) 633–641.
[17] C. Lorrette, A. Reau, L. Briottet, Mechanical properties of nanostructuredsilicon carbide consolidated by spark plasma sintering, J. Eur. Ceram.Soc. 33 (1) (2013) 147–156.

A. Rahman et al. / Ceramics International 40 (2014) 12081–12091 12091
[18] R. Riedel, G. Mera, R. Hauser, A. Klonczynski, Silicon-based polymer-derived ceramics: synthesis properties and applications—a review, J.Ceram. Soc. Jpn. 114 (June (1330)) (2006) 425–444.
[19] A. Rahman, S.C. Zunjarrao, R. Singh, Comparison of bulk and nanoscaleproperties of polymer precursor derived silicon carbide with sinteredsilicon carbide, in: Processing and Properties of Advanced Ceramics andComposites, 2009, pp. 63–76.
[20] S.C. Zunjarrao, A. Rahman, R.P. Singh, Characterization of the evolutionand properties of silicon carbide derived from a preceramic polymerprecursor, J. Am. Ceram. Soc. 96 (6) (2013) 1869–1876.
[21] ASTM-C830-00, Standard Test Methods for Apparent Porosity, LiquidAbsorption, Apparent Specific Gravity, and Bulk Density of RefractoryShapes by Vacuum Pressure, American Society for Testing and Materials,West Conshohocken, PA, 2000.
[22] A.A. Wereszczak, J.J. Swab, R.H. Kraft, Effects of Machining on theUniaxial and Equibiaxial Flexure Strength of CAP3 AD-995 Al2O3,Technical Report ARL-TR-3617, 2005.
[23] ASTM-C1499-09, Standard Test Method for Monotonic EquibiaxialFlexural Strength of Advanced Ceramics at Ambient Temperature,American Society for Testing and Materials, West Conshohocken, PA,2001.
[24] J. Ovri, A parametric study of the biaxial strength test for brittle materials,Mater. Chem. Phys. 66 (1) (2000) 1–5.
[25] G. Anstis, P. Chantikul, B. Lawn, D. Marshall, A critical evaluation ofindentation techniques for measuring fracture toughness: I direct crackmeasurements, J. Am. Ceram. Soc. 64 (9) (1981) 533–538.
[26] L.L. Snead, T. Nozawa, Y. Katoh, T.-S. Byun, S. Kondo, D.A. Petti,Handbook of SiC properties for fuel performance modeling, J. Nucl.Mater. 371 (September 15 (1–3)) (2007) 329–377 (1st Symposium on Nuclear Fuels and Structural Materials for Next GenerationNuclear Reactors, Reno, NV, June 04–08, 2006).
[27] H. Li, L. Zhang, L. Cheng, Y. Wang, Z. Yu, M. Huang, H. Tu, H. Xia,Effect of the polycarbosilane structure on its final ceramic yield, J. Eur.Ceram. Soc. 28 (4) (2008) 887–891.
[28] H. Li, L. Zhang, L. Cheng, Y. Wang, Z. Yu, M. Huang, H. Tu, H. Xia,Polymer-ceramic conversion of a highly branched liquid polycarbosilanefor SiC-based ceramics, J. Mater. Sci. 43 (April (8)) (2008) 2806–2811.
[29] M. Huang, Y. Fang, R. Li, T. Huang, Z. Yu, H. Xia, Synthesis andproperties of liquid polycarbosilanes with hyperbranched structures,J. Appl. Polym. Sci. 113 (August (3)) (2009) 1611–1618.
[30] R.W. Cheary, A.A. Coelho, Programs XFIT and FOURYA, Deposited inCCP14 Powder Diffraction Library, Engineering and Physical SciencesResearch Council, Daresbury laboratory, Warrington, England, 1996.
[31] F. Lange, Hot-pressing behavior of silicon carbide powders withadditions of aluminum oxide, J. Mater. Sci. 10 (2) (1975) 314–320.
[32] J. Ritter, K. Jakus, A. Batakis, N. Bandyopadhyay, Appraisal of biaxialstrength testing, J. Non-Cryst. Solids 38–39 (Part 1) (1980) 419–424.
[33] F. Knudsen, Dependence of mechanical strength of brittle polycrystallinespecimens on porosity and grain size, J. Am. Ceram. Soc. 42 (8) (1959)376–387.
[34] E. Ryshkewitch, Compression strength of porous sintered alumina andzirconia, J. Am. Ceram. Soc. 36 (2) (1953) 65–68.
[35] M. Giovan, G. Sines, Biaxial and uniaxial data for statistical comparisonsof a ceramics strength, J. Am. Ceram. Soc. 62 (9–10) (1979) 510–515.
[36] A.K. Singh, S.C. Zunjarrao, R.P. Singh, Processing of uranium oxide andsilicon carbide based fuel using polymer infiltration and pyrolysis,J. Nucl. Mater. 378 (3) (2008) 238–243.
[37] C. Conner, K. Faber, Segregant-enhanced fracture in magnesium-oxide,J. Mater. Sci. 25 (June (6)) (1990) 2737–2742.
[38] M. Flinders, D. Ray, A. Anderson, R. Cutler, High-toughness siliconcarbide as armor, J. Am. Ceram. Soc. 88 (August (8)) (2005) 2217–2226.
[39] X. Zhang, M. Sixta, L. De Jonghe, Grain boundary evolution in hot-pressed ABC-SiC, J. Am. Ceram. Soc. 83 (November (11)) (2000)2813–2820.
[40] A. Krell, P. Blank, Grain-size dependence of hardness in dense submic-rometer alumina, J. Am. Ceram. Soc. 78 (April (4)) (1995) 1118–1120.
[41] R. Rice, C. Wu, F. Borchelt, Hardness grain-size relations in ceramics,J. Am. Ceram. Soc. 77 (October (10)) (1994) 2539–2553.
[42] X. Dong, S. Jahanmir, L. Ives, Wear transition diagram for siliconcarbide, Tribol. Int. 28 (8) (1995) 559–572.
[43] A. Mukhopadhyay, S. Datta, D. Chakraborty, On the microhardness ofsilicon nitride and sialon ceramics, J. Eur. Ceram. Soc. 6 (5) (1990)303–311.
[44] J. Gong, J. Wu, Z. Guan, Analysis of the indentation size effect on theapparent hardness for ceramics, Mater. Lett. 38 (3) (1999) 197–201.
[45] J. Gong, J. Wu, Z. Guan, Examination of the indentation size effect inlow-load Vickers hardness testing of ceramics, J. Eur. Ceram. Soc. 19(15) (1999) 2625–2631.
[46] K. Faber, A. Evans, Intergranular crack-deflection toughening in silicon-carbide, J. Am. Ceram. Soc. 66 (6) (1983) C94–C96.




















