
Manuale d'uso
76
Assurity™, Assurity MRI™, Endurity™, Endurity™ Core, Endurity MRI™, Zenex™, Zenex™ MRI, Zenus™, Zenus™ MRI Generatore di impulsi Allure™ RF, Allure Quadra™ RF, Quadra Allure™, Quadra Allure MP™, Quadra Allure MP™ RF Generatore di impulsi per terapia di resincronizzazione cardiaca Manuale d'uso
-
Upload
khangminh22 -
Category
Documents
-
view
1 -
download
0
Transcript of Manuale d'uso

Assurity™, Assurity MRI™, Endurity™, Endurity™ Core, Endurity MRI™, Zenex™, Zenex™ MRI, Zenus™, Zenus™ MRI Generatore di impulsi Allure™ RF, Allure Quadra™ RF, Quadra Allure™, Quadra Allure MP™, Quadra Allure MP™ RF Generatore di impulsi per terapia di resincronizzazione cardiaca
Manuale d'uso

Salvo diversa indicazione, il simbolo ™ indica che il nome è un marchio commerciale di St. Jude Medical e/o delle sue consociate o concesso in licenza alle stesse. ST. JUDE MEDICAL e il simbolo dei nove quadrati sono marchi commerciali e marchi di servizio di St. Jude Medical, LLC e delle sue consociate.
Pat. http://patents.sjm.com
© 2019 St. Jude Medical, LLC. Tutti i diritti riservati.

1
Descrizione del dispositivo In questo manuale vengono descritti i generatori di impulsi St. Jude Medical™ elencati nella tabella1.
Questi dispositivi possono essere programmati con il Sistema per la cura del paziente Merlin™ (Merlin PCS) dotato di software Modello 3330 versione 21.2.2 (o successiva). Per informazioni sulla programmazione, fare riferimento all’help in linea del programmatore.
ATTENZIONE: Non tutti i modelli di dispositivo sono disponibili in tutti i paesi.
Non tutti i prodotti elencati come dispositivi a compatibilità condizionata alla RM sono approvati per l’uso a compatibilità condizionata alla RM in tutti i paesi o in tutte le regioni.
Prima di eseguire una scansione MRI sui pazienti ai quali è stato impiantato uno di questi dispositivi, contattare St. Jude Medical o consultare le autorità di regolazione di competenza per verificare se i prodotti sono stati certificati come dispositivi a compatibilità condizionata alla RM.
1 Non tutti i modelli di dispositivo sono disponibili in tutti i paesi.

2
Tabella 1. Descrizione dei generatori di impulsi
Nome Numero di modello
Descrizione Tipo di connettore
Stato MRI
Endurity™ Core PM1140 Generatore di impulsi monocamerale
IS-1 Compatibilità condizionata alla RM
Endurity™ Core PM1152 Generatore di impulsi monocamerale
IS-1 Compatibilità condizionata alla RM
Endurity™ PM1160 Generatore di impulsi monocamerale
IS-1 Non testato
Endurity™ PM1162 Generatore di impulsi monocamerale
IS-1 Compatibilità condizionata alla RM
Zenus™ PM1170 Generatore di impulsi monocamerale
IS-1 Non testato

3
Tabella 1. Descrizione dei generatori di impulsi
Nome Numero di modello
Descrizione Tipo di connettore
Stato MRI
Endurity MRI™ PM1172 Generatore di impulsi monocamerale
IS-1 Compatibilità condizionata alla RM
Zenus™ MRI PM1182 Generatore di impulsi monocamerale
IS-1 Compatibilità condizionata alla RM
Assurity™ PM1240 Generatore di impulsi monocamerale con telemetria RF
IS-1 Non testato
Zenex™ PM1250 Generatore di impulsi monocamerale con telemetria RF
IS-1 Non testato
Assurity MRI™ PM1272 Generatore di impulsi monocamerale con telemetria RF
IS-1 Compatibilità condizionata alla RM

4
Tabella 1. Descrizione dei generatori di impulsi
Nome Numero di modello
Descrizione Tipo di connettore
Stato MRI
Zenex™ MRI PM1282 Generatore di impulsi monocamerale con telemetria RF
IS-1 Compatibilità condizionata alla RM
Endurity™ Core PM2140 Generatore di impulsi bicamerale IS-1 Compatibilità condizionata alla RM
Endurity™ Core PM2152 Generatore di impulsi bicamerale IS-1 Compatibilità condizionata alla RM
Endurity™ PM2160 Generatore di impulsi bicamerale IS-1 Non testato
Endurity™ PM2162 Generatore di impulsi bicamerale IS-1 Compatibilità condizionata alla RM
Zenus™ PM2170 Generatore di impulsi bicamerale IS-1 Non testato

5
Tabella 1. Descrizione dei generatori di impulsi
Nome Numero di modello
Descrizione Tipo di connettore
Stato MRI
Endurity MRI™ PM2172 Generatore di impulsi bicamerale IS-1 Compatibilità condizionata alla RM
Zenus™ MRI PM2182 Generatore di impulsi bicamerale IS-1 Compatibilità condizionata alla RM
Assurity™ PM2240 Generatore di impulsi bicamerale con telemetria RF
IS-1 Non testato
Zenex™ PM2250 Generatore di impulsi bicamerale con telemetria RF
IS-1 Non testato
Assurity MRI™ PM2272 Generatore di impulsi bicamerale con telemetria RF
IS-1 Compatibilità condizionata alla RM

6
Tabella 1. Descrizione dei generatori di impulsi
Nome Numero di modello
Descrizione Tipo di connettore
Stato MRI
Zenex™ MRI PM2282 Generatore di impulsi bicamerale con telemetria RF
IS-1 Compatibilità condizionata alla RM
Allure™ PM3120 CRT-P IS-1 Non testato
Allure™ RF PM3222 CRT-P con telemetria RF IS-1 Non testato
Allure Quadra™ RF PM3242 CRT-P con telemetria RF IS-1/IS4-LLLL Non testato
Quadra Allure MP™ RF
PM3262 CRT-P con telemetria RF IS-1/IS4-LLLL Non testato
Quadra Allure™ PM3542 CRT-P IS-1/IS4-LLLL Compatibilità condizionata alla RM
Quadra Allure MP™ PM3562 CRT-P IS-1/IS4-LLLL Compatibilità condizionata alla RM

7
Indicazioni L’impianto di un generatore di impulsi monocamerale, di un generatore di impulsi bicamerale o di un CRT-P (Pacemaker per terapia di resincronizzazione cardiaca) è indicato in una o più delle seguenti condizioni permanenti o in una combinazione di questi sintomi2,3,4: Sincope
Presincope
Spossatezza
Disorientamento dovuto ad aritmia/bradicardia
L’impianto di un dispositivo CRT-P è indicato nei pazienti che: trarrebbero beneficio da una resincronizzazione tra il ventricolo destro e il sinistro.
presentano una o più indicazioni convenzionali per l’impianto di un pacemaker.
Un generatore di impulsi a compatibilità condizionata alla RM è sicuro per l’uso nell’ambiente MRI in maniera condizionata quando viene utilizzato in un sistema di stimolazione a compatibilità condizionata alla RM completo e in conformità alle istruzioni contenute nel manuale del sistema di stimolazione MRI Ready per il sistema di stimolazione a compatibilità condizionata alla RM di St. Jude Medical™. 2 Tracy CM, Epstein AE, Darbar D, et al. ACCF/AHA/HR Focused Updated of the 2008 Guidelines for Device Based Therapy of Cardiac Rhythm Abnormalities. Circulation 2012;126:1784-1800. 3 Brignole M, Auricchio A, Baron-Esquivias G. et al. 2013 ESC Guidelines on cardiac pacing and cardiac resynchronization therapy: The Task Force on cardiac pacing and resynchronization therapy of the European Society of Cardiology (ESC). Developed in collaboration with the European Heart Rhythm Association (EHRA). Eur Heart J. 2013;34(29):2281-329. 4 Epstein AE, DiMarco JP, Ellenborgen KA, et al. ACC/AHA/HRS 2008 Guidelines for Device-Based Therapy of Cardiac Rhythm Abnormalities. J Am Coll Cardiol 2008;51:e1-62.

8
La stimolazione con modulazione della frequenza è indicata nei pazienti con incompetenza cronotropa e per quelli che, in concomitanza con l’attività fisica, potrebbero trarre beneficio da frequenze di stimolazione più alte.
La stimolazione bicamerale (generatori di impulsi bicamerali, CRT-P) è indicata nei pazienti che evidenziano: malattia del nodo del seno
blocco AV cronico di secondo e terzo grado, sintomatico
sindrome di Adams-Stokes ricorrente
blocco di branca sintomatico bilaterale, se sono state escluse tachiaritmia ed altre cause
La stimolazione atriale è indicata per pazienti con una disfunzione del nodo del seno e normali sistemi di conduzione AV e intraventricolare.
La stimolazione ventricolare è indicata per pazienti con bradicardia significativa e: ritmo sinusale normale con soltanto rari episodi di blocco A-V o arresto sinusale
fibrillazione atriale cronica (AF)
grave invalidità fisica.
La stimolazione AF Suppression™ (generatori di impulsi bicamerali, CRT-P) è indicata per la soppressione di episodi di fibrillazione atriale parossistica o persistente in pazienti con una o più delle summenzionate indicazioni per la stimolazione.

9
Algoritmo di rilevamento AT/AF. L’algoritmo per il rilevamento di AT/AF è indicato per rilevare le tachiaritmie atriali che sono risultate associate a un rischio aumentato di ictus nei pazienti anziani, ipertesi, portatori di pacemaker senza precedente storia clinica di AF.
Per specifiche indicazioni associate alle singole modalità, consultare l’help in linea del programmatore.
Accessori, Uso designato Con i generatori di impulsi descritti in questo manuale è autorizzato esclusivamente l’uso degli accessori qui elencati.
Tabella 2. Accessori e usi previsti
Accessorio Uso previsto
Chiave torsiometrica Fissare i connettori dell’elettrocatetere e gli otturatori all’interno del connettore del dispositivo.
Tappo del connettore dell’elettrocatetere IS-1
Isolare e proteggere le prese dell’elettrocatetere inutilizzate.
Otturatore IS4/DF4 Sigillare i fori per gli elettrocateteri non utilizzati

10
Controindicazioni Dispositivo di cardioversione-defibrillazione impiantato (ICD). I generatori di impulsi monocamerali, i generatori di impulsi bicamerali e i dispositivi CRT-P sono controindicati nei pazienti con un dispositivo di cardioversione-defibrillazione impiantato.
La stimolazione a frequenza adattiva potrebbe essere non appropriata per i pazienti che soffrono di angina o che accusano altri sintomi di disfunzione del miocardio alle frequenze superiori guidate da sensore. Per selezionare il valore appropriato di Massima Frequenza del Sensore occorre basarsi sulla valutazione della massima frequenza di stimolazione tollerata dal paziente.
La stimolazione AF Suppression (generatori di impulsi bicamerali, dispositivi CRT-P) è sconsigliata nei pazienti non in grado di tollerare una frequenza di stimolazione atriale elevata.
La stimolazione bicamerale (generatori di impulsi bicamerali, dispositivi CRT-P), sebbene non controindicata nei pazienti con flutter atriale cronico, fibrillazione atriale cronica o atri silenti, potrebbe limitarsi ad offrire a questi pazienti benefici non superiori a quelli della stimolazione monocamerale.
La stimolazione monocamerale ventricolare (demand) è relativamente controindicata nei pazienti affetti da sindrome da pacemaker, che hanno una conduzione VA retrograda o che accusano una caduta della pressione arteriosa all’inizio della stimolazione ventricolare.
La stimolazione monocamerale atriale è relativamente controindicata nei pazienti che presentano una conduzione AV compromessa.

11
Scansione RM. I pazienti che non hanno un sistema completo di stimolazione a compatibilità condizionata alla RM St. Jude Medical™ che comprende un generatore di impulsi a compatibilità condizionata alla RM St. Jude Medical ed elettrocateteri a compatibilità condizionata alla RM St. Jude Medical sono controindicati per una scansione MRI.
Per specifiche controindicazioni associate alle singole modalità, consultare l’help in linea del programmatore.
Avvertenze Per evitare danni irreparabili al dispositivo e lesioni del tessuto nel punto di interfaccia elettrodo-tessuto, evitare l’esposizione a quanto segue: Elettrochirurgia. Non usare dispositivi elettrochirurgici in prossimità di un dispositivo impiantato.
Se è necessaria elettrocauterizzazione, usare un cauterizzatore bipolare o allontanare il più possibile l’elettrodo indifferente dal dispositivo.
Litotripsia. Non focalizzare un raggio da litotripsia entro 16 cm dal dispositivo. Prima della litotripsia, programmare il dispositivo su Sensore Off per evitare aumenti inappropriati della frequenza di stimolazione. Dopo l’eventuale esposizione alla litotripsia, occorre controllare accuratamente il funzionamento del dispositivo, prestando particolare attenzione al sensore.
Radioterapia. Non usare radiazioni ionizzanti a scopo terapeutico in prossimità di un dispositivo impiantato, poiché la radioterapia potrebbe danneggiarne i circuiti elettronici. Eventuali danni al

12
dispositivo causati da radiazioni ionizzanti utilizzate a scopo terapeutico potrebbero non essere immediatamente rilevabili.
Terapia a ultrasuoni. Non esporre il dispositivo a livelli terapeutici di energia ultrasonica, poiché il dispositivo potrebbe accidentalmente concentrare il campo degli ultrasuoni e provocare danni non immediatamente rilevabili. Non è stato accertato se il trattamento a ultrasuoni a fini diagnostici alteri o meno il corretto funzionamento del dispositivo.
Sensing ventricolare. Nei dispositivi CRT-P, la sensibilità ventricolare deve essere programmata sull’impostazione più alta (sensibilità minima) in grado di assicurare un adeguato margine per il sensing ventricolare. La dislocazione dell’elettrocatetere ventricolare sinistro in una posizione prossima agli atri può determinare oversensing atriale e inibizione ventricolare.
Dopo l’eventuale esposizione a uno dei trattamenti precedenti, verificare accuratamente il funzionamento del dispositivo.
Risonanza magnetica per immagini (MRI). Generatori di impulsi a compatibilità condizionata alla RM. I test hanno dimostrato che il
sistema di stimolazione a compatibilità condizionata alla RM St. Jude Medical™ è sicuro in ambiente di risonanza magnetica se utilizzato in conformità alle istruzioni contenute nel manuale del sistema di stimolazione MRI-Ready. Il sistema di stimolazione St. Jude Medical a compatibilità condizionata alla RM include un generatore di impulsi St. Jude Medical a compatibilità condizionata alla RM collegato a uno o più elettrocateteri St. Jude Medical a compatibilità condizionata alla RM.

13
Generatori di impulsi non testati per la RM. “Non testato” indica che il dispositivo non è stato testato e il suo utilizzo in un ambiente di RM non è stato determinato. Per ulteriori informazioni, consultare il manuale del sistema di stimolazione MRI-Ready.
Funzionamento in modalità di Backup VVI. In rari casi, il dispositivo può ripristinare il funzionamento in modalità di Backup VVI alle impostazioni elencate nella tabella seguente. Queste impostazioni non sono programmabili.
Quando il dispositivo ha ripristinato il funzionamento in modalità Backup VVI, il programmatore visualizza un messaggio a comparsa a indicare che il dispositivo è operativo secondo i valori di Backup VVI. Premere il pulsante Continua e seguire le istruzioni sullo schermo.
Tabella 3. Impostazioni Backup VVI
Parametro Impostazione
Generatori di impulsi monocamerali Generatori di impulsi bicamerali
CRT-P
Modalità VVI VVI
Frequenza Base 67 min§ 67 min§
Stimolazione ventricolo n/d VS —> VD

14
Tabella 3. Impostazioni Backup VVI
Parametro Impostazione
Generatori di impulsi monocamerali Generatori di impulsi bicamerali
CRT-P
Configurazione impulso Unipolare Punta Unipolare VD Punta Unipolare VS
Configurazione Sensing Punta Unipolare Punta unipolare VD
Ampiezza dell’impulso 5,0 V 5,0 V
Durata dell’impulso 0,6 ms 0,6 ms
Periodo refrattario 337 ms 337 ms
Sensibilità 2,0 mV 2,0 mV
Ritardo interventricolare n/d 16 ms
Indicatore di sostituzione elettiva (ERI) (pagina 47). All’ERI, la durata nominale del dispositivo è di tre o sei mesi. Quando il dispositivo manifesta segni di ERI, deve essere prontamente sostituito.

15
Programmare le visite di follow-up del paziente con una frequenza appropriata, in modo da rilevare l’ERI prima della fine della durata operativa (EOL).
Stimolazione programmata non invasiva (NIPS). Nel corso della NIPS, possono verificarsi episodi di tachicardia o fibrillazione ventricolari potenzialmente fatali, pertanto: (1) monitorare attentamente il paziente e (2) mettere a pronta disposizione del personale specializzato in queste procedure le apparecchiature per defibrillazione e rianimazione. La NIPS deve essere utilizzata esclusivamente da medici specializzati nell’applicazione dei protocolli di induzione e reversione della tachicardia. Per ulteriori informazioni sulla NIPS, consultare la guida in linea del programmatore.
La Stimolazione di supporto ventricolare durante i test NIPS (generatori di impulsi bicamerali, CRT-P) viene erogata in modalità VOO. Le indicazioni e le controindicazioni specifiche per la modalità VOO sono reperibili nella guida in linea del programmatore.
Precauzioni Esclusivamente monouso. Comunicazione con il dispositivo. La comunicazione con il dispositivo può essere alterata da interferenze elettriche e da forti campi magnetici. In tal caso, spegnere le apparecchiature elettriche presenti nelle immediate vicinanze oppure allontanarle dal paziente e dal programmatore. Se il problema persiste, rivolgersi a St. Jude Medical.

16
Comunicazione RF non ottimale. Il Sistema per la cura del paziente Merlin™ (Merlin PCS) segnala la qualità della comunicazione a radiofrequenza (RF) mediante gli appositi indicatori a LED dell’intensità della telemetria presenti sia sul programmatore sia sull’Antenna Merlin™. Di seguito è riportato un elenco di potenziali cause di radiocomunicazione non ottimale:
Tabella 4. Possibili cause e soluzioni per una comunicazione in radiofrequenza (RF) non ottimale
Possibili cause Soluzioni
L’orientamento/posizione dell’antenna Merlin non è ottimale.
Spostare o riorientare leggermente l’antenna Merlin. Controllare che la parte anteriore dell’antenna Merlin sia rivolta verso il dispositivo impiantabile.
Persone o oggetti interferiscono nella comunicazione tra l’antenna Merlin ed il dispositivo.
Accertarsi che nello spazio tra l’antenna Merlin e il dispositivo non vi siano oggetti e/o persone la cui presenza provochi interferenze.
L’antenna Merlin è troppo lontana dal dispositivo. Avvicinare l’antenna Merlin al dispositivo.
Qualcuno sta tenendo l’antenna Merlin. Posizionare l’antenna Merlin su una superficie piatta. Non tenere l’antenna Merlin.

17
Tabella 4. Possibili cause e soluzioni per una comunicazione in radiofrequenza (RF) non ottimale
Possibili cause Soluzioni
Altri apparecchi posizionati nelle vicinanze provocano interferenze elettromagnetiche (EMI).
Spegnere o rimuovere l’apparecchiatura che potrebbe causare le interferenze elettromagnetiche (EMI).
Il cavo dell’antenna Merlin è avvolto attorno all’antenna stessa.
Verificare che il cavo dell’antenna Merlin non sia avvolto attorno all’antenna stessa.
Scansioni TC. A causa degli aumentati livelli di potenza e dei lunghi tempi di esposizione, vi è la remota possibilità che le scansioni TC (tomografia computerizzata) possano interferire con i dispositivi impiantati. La potenziale interferenza è transitoria e comunque si verifica soltanto in presenza del segnale prodotto dai raggi-X. Un’esposizione continua può indurre un temporaneo aumento della frequenza del sensore. Esiste inoltre una remota possibilità di oversensing intermittente del dispositivo mentre il fascio della scansione TC si trova direttamente sopra il dispositivo impiantato.

18
Sterilizzazione Il contenuto della confezione è stato sterilizzato con ossido di etilene prima della spedizione. Questo dispositivo è esclusivamente monouso e non deve essere risterilizzato.
Se la sterilità della confezione è stata compromessa, contattare, St. Jude Medical.
Conservazione e manipolazione Shock meccanico. I generatori di impulsi St. Jude Medical™ sono di solida costruzione. Tuttavia, se si sospetta che il dispositivo sia stato danneggiato, non impiantarlo e restituirlo a St. Jude Medical.
Temperatura. Conservare il dispositivo a temperature comprese tra -5 °C (23 °F) e 50 °C (122 °F). Non sottoporre il dispositivo a temperature inferiori a -20° C (-4 °F) e superiori a 55 °C (131 °F). Se il dispositivo è stato immagazzinato in un luogo freddo, prima di programmarlo o impiantarlo attendere che ritorni alla temperatura ambiente dato che le temperature basse possono influire sul suo funzionamento iniziale.
Incenerimento. Non incenerire il dispositivo. Restituire i generatori espiantati a St. Jude Medical.

19
Preparativi per l'impianto Etichetta della confezione. Prima di aprire la confezione sterile, leggere attentamente l’etichetta e verificare che la confezione contenga il dispositivo desiderato. Non impiantare il generatore di impulsi nelle seguenti circostanze: Confezione danneggiata o umida.
Colorazione viola del puntino sull’etichetta dell’ossido di etilene, condizione che denota mancanza di sterilizzazione.
Superamento della data di scadenza riportata sulla confezione esterna e sul supporto. La data di scadenza coincide con la tensione minima della batteria necessaria per raggiungere la longevità della batteria prevista e mostrata nell’help in linea del programmatore.
Verifica del funzionamento. Prima di aprire la confezione sterile, verificare che il generatore di impulsi funzioni correttamente interrogandolo nella confezione. Rimuovere il magnete dalla sonda telemetrica del programmatore e stabilire la comunicazione: Comunicazione induttiva. Posizionare la sonda telemetrica del Sistema per la cura del paziente
Merlin™ (Merlin PCS) sopra la confezione e selezionare “Interroga”.

20
Comunicazione RF. Per stabilire una comunicazione RF fra il dispositivo e il programmatore e risolvere i problemi di comunicazione, collegare prima l’antenna Merlin al programmatore. Fare riferimento al manuale d’uso del Sistema per la Cura del Paziente Merlin PCS fornito in dotazione con il programmatore e con l’antenna Merlin. Utilizzare gli indicatori di intensità della telemetria per valutare la comunicazione. Se il dispositivo è RF-compatibile, un’icona nell’angolo superiore sinistro dello schermo durante la sessione di programmazione indica lo stato del link di comunicazione RF. Se l’icona RF non compare sullo schermo nel corso della sessione, il dispositivo non è RF-compatibile. Una volta stabilita la telemetria, selezionare “Interroga”.
I dati misurati dell’unità verranno visualizzati nella schermata Sommario FastPath™ dove dovrebbero indicare una tensione e uno stato della batteria nella norma e i parametri programmati dovrebbero corrispondere alle impostazioni alla consegna visualizzate nell’help in linea del programmatore.
Integrità della confezione. Accertarsi che la confezione non sia stata aperta o in alcun modo manomessa. Se si sospetta un danno, restituirla al produttore.
Data di scadenza. Non impiantare il dispositivo successivamente alla data di scadenza stampata sull’etichetta.
Apertura della confezione. Se l’interrogazione del dispositivo ancora all’interno della sua confezione sterile ne indica il normale funzionamento, estrarlo dalla confezione. La vaschetta esterna può essere aperta in ambiente non sterile. Tuttavia, quando si apre la vaschetta interna, occorre osservare integralmente la procedura sterile.

21
Test prima dell'impianto Analizzatore di Soglia di Stimolazione. Prima dell’impianto, il medico potrebbe ritenere opportuno testare il dispositivo utilizzando un sistema analizzatore di soglia di stimolazione (PSA) compatibile con impostazioni di sensibilità e di uscita calibrate. Quando si collega la sonda al connettore del dispositivo, i parametri programmati devono essere identici alle impostazioni alla consegna visualizzate nell’help in linea del programmatore.
Connessione cavi. Per il test del dispositivo utilizzare solo sonde PSA con cavo IS-1. Cavi diversi da quelli specificati potrebbero danneggiare il connettore. Non usare sonde con cavo IS-1 nel connettore IS4-LLLL.
Elettrocateteri di stimolazione compatibili. I dispositivi con connettore IS-1 supportano elettrocateteri unipolari o bipolari IS-1 con spinotto terminale corto. I dispositivi con connettore IS4-LLLL sono compatibili con elettrocateteri quadripolari IS4-LLLL. Prima dell’impianto accertarsi che gli elettrocateteri entrino agevolmente nel connettore del dispositivo.
Soglie di cattura/sensing. Le soglie di cattura e di sensing devono essere determinate con un PSA prima dell’impianto del dispositivo. Il terminale negativo del PSA (nero) va collegato al segmento dello spinotto terminale dell’elettrocatetere corrispondente all’elettrodo distale. Collegare il terminale positivo (rosso) alla parte dello spinotto corrispondente all’elettrodo ad anello per gli elettrocateteri bipolari o ad un elettrodo indifferente. Per ulteriori informazioni sulla conduzione dei test di soglia di cattura e di sensing, consultare il manuale tecnico del PSA.

22
Determinazione delle soglie di cattura/sensing basali. Successivamente all’impianto degli elettrocateteri e prima del collegamento degli stessi al dispositivo, stabilire e documentare la morfologia basale per le soglie di cattura e sensing di ogni singolo elettrocatetere utilizzando un sistema di registrazione idoneo, quale un ECG a 12 derivazioni o un elettrogramma intracardiaco (IEGM).
Impianto Preparazione del personale medico. Prima di intraprendere tale procedura il medico deve conoscere bene tutti i componenti del sistema e il contenuto del presente manuale.
Defibrillatore esterno. Tenere sempre immediatamente disponibile un defibrillatore esterno di riserva.
Trasmissione dei dati. Per una trasmissione dei dati affidabile con la sonda telemetrica induttiva del Merlin™ PCS, il generatore d’impulsi deve essere impiantato ad una profondità non superiore ai 5 cm. Per i generatori di impulsi a compatibilità condizionata alla RM, impiantare il generatore di impulsi a una profondità non superiore a 4 cm per garantire una trasmissione dei dati affidabile con il dispositivo portatile SJM MRI Activator™.
Comfort del paziente. Per un maggior comfort del paziente, impiantare il generatore di impulsi ad almeno 1,25 cm dall’osso, fatto salvo nei casi in cui ciò sia inevitabile.
Marcature sulla cassa. Controllare le marcature sulla cassa del dispositivo e verificare il corretto collegamento atriale e ventricolare.

23
Vite di fissaggio. Procedere con cautela quando si gira la vite di fissaggio, la quale, se ruotata in senso antiorario per più di due giri, potrebbe uscire dal connettore.
Programmazione Programmatore. Questi dispositivi possono essere interrogati e programmati con il Sistema per la cura del paziente Merlin™ (Merlin PCS) dotato di software Modello 3330 versione 21.2.2 (o successiva).
Per l’elenco completo dei parametri e dei relativi valori programmabili, consultare la guida in linea del programmatore.
Impostazione del tipo di elettrocatetere. Quando si interroga per la prima volta il dispositivo, il programmatore richiede di impostare il parametro Tipo Elettrocatetere (nei dispositivi CRT-P, i tipi di elettrocatetere ventricolare destro e sinistro vengono impostati in modo indipendente). Poiché alcuni parametri sono condizionati dal parametro Tipo Elettrocatetere (per esempio, Configurazione impulso), occorre impostare questo parametro in sede di impianto del dispositivo. I dispositivi provvisti della funzione di rilevamento automatico della polarità dell’elettrocatetere (ALPD) rileveranno e programmeranno automaticamente le configurazioni di Tipo Elettrocatetere e Sensing. Questa funzione viene sospesa durante la telemetria e per un minuto dopo il suo completamento.
Valori di impedenza degli elettrocateteri. Nei dispositivi CRT-P, vengono visualizzati valori di impedenza degli elettrocateteri indipendenti per gli elettrocateteri VD e VS.
Ampiezze e durate dell’impulso ventricolare. Nei dispositivi CRT-P, le ampiezze e le durate dell’impulso ventricolare destro e sinistro sono programmabili in modo indipendente. Di conseguenza,

24
occorre valutare correttamente l’ampiezza dell’impulso in ciascuna camera. Normalmente, le soglie di cattura sono più elevate nel ventricolo sinistro.
Misurazioni della soglia di cattura ventricolare di follow-up. Nei dispositivi CRT-P, le misurazioni della soglia di cattura VD e VS vengono valutate in modo indipendente. Nel corso di un test di cattura VD o VS, è possibile stabilire quando si verifica la cattura annotando le variazioni morfologiche dell’ECG. I test di cattura non vengono eseguiti nelle modalità con stimolazione ventricolare triggerata. All’avvio, la modalità di stimolazione è programmata temporaneamente sulla corrispondente modalità inibita. Per ulteriori informazioni, consultare la guida in linea del programmatore.
Le modalità AOO(R), VOO(R) e DOO(R) sono principalmente destinate ad un uso diagnostico provvisorio. L’uso prolungato di queste modalità può determinare una stimolazione competitiva, inducendo aritmie potenzialmente pericolose.
La modalità Off è sconsigliata per i pazienti che potrebbero subire conseguenze negative da un’interruzione, anche di breve durata, del funzionamento del dispositivo.
Ampiezza dell’impulso. Se i sistemi di stimolazione AutoCapture™ o Cap Confirm non sono in uso, stabilire la soglia di cattura prima di programmare l’Ampiezza Impulso. Programmare il parametro Ampiezza Impulso per avere un margine di sicurezza adeguato per una cattura a lungo termine affidabile. Riesaminare periodicamente le soglie di cattura.

25
Stimolazione programmata non invasiva (NIPS). Nel corso della NIPS, possono verificarsi episodi di tachicardia o fibrillazione atriale o ventricolare, pertanto: (1) monitorare attentamente il paziente e (2) avere a pronta disposizione l’apparecchiatura di emergenza per l’esecuzione di cardioversione/defibrillazione.
Impostazioni ad Alta uscita. La programmazione di impostazioni ad alta uscita o di una Frequenza Base elevata può ridurre il tempo con cui il dispositivo raggiunge l’ERI.
Protezione dal runaway. La circuiteria hardware del dispositivo gli impedisce di stimolare a frequenze superiori alla frequenza di protezione dal runaway di seguito illustrata.
Tabella 5. Protezione dal runaway per tutti i dispositivi
Dispositivo Frequenza di protezione dal runaway
PM1140, PM1152, PM1160, PM1162, PM1170, PM1172, PM1182, PM1240, PM1250, PM1272, PM1282, PM2140, PM2152, PM2160, PM2162, PM2170, PM2172, PM2182, PM2240, PM2250, PM2272, PM2282, PM3120, PM3222, PM3242, PM3262, PM3542, PM3562
220 min§ (± 10 min§)

26
Configurazione Sensing. I test di sensing devono essere eseguiti ogni volta che vengono apportate modifiche alla configurazione del sensing.
Avviso paziente. Prima di attivare l’Avviso paziente, testare e verificare la consapevolezza del paziente di tale funzione. Per i generatori di impulsi a compatibilità condizionata alla RM dotati della funzionalità Avviso paziente, il programmatore Merlin PCS disabilita in maniera permanente l’Avviso paziente quando il generatore di impulsi è programmato con impostazioni MRI.
NOTA: per l’elenco dei dispositivi dotati della funzionalità Avviso paziente, consultare la guida in linea del programmatore.
Rischi ambientali e associati alla terapia medica
Rischi ambientali e associati alla terapia medica I dispositivi St. Jude Medical™ sono dotati di schermatura e filtri speciali che riducono notevolmente gli effetti dannosi delle interferenze elettromagnetiche (EMI) sul funzionamento del dispositivo.
I pazienti devono essere invitati a prestare la massima attenzione a evitare forti campi elettrici o magnetici. Se il dispositivo di stimolazione viene inibito o avvia il funzionamento asincrono in presenza di interferenze elettromagnetiche (EMI), il paziente dovrà allontanarsi dalla fonte delle interferenze oppure disattivare il dispositivo che le emette.

27
Chiedere ai pazienti di informarsi dal proprio medico su come comportarsi prima di entrare in ambienti che potrebbero compromettere il funzionamento del dispositivo, come le zone con cartelli di divieto d’accesso alle persone dotate di pacemaker.
Procedure ed ambienti medici In generale, i pazienti ai quali è stato impiantato un pacemaker non devono essere esposti ad attrezzature ospedaliere che generano forti segnali di campo elettromagnetico, come gli apparecchi per diatermia e le unità elettrochirurgiche. Defibrillazione esterna. I circuiti elettronici del dispositivo forniscono protezione contro le scariche
di defibrillazione. Tuttavia, occorre evitare di collocare le piastre del defibrillatore direttamente sopra il dispositivo o l’elettrocatetere di stimolazione. Dopo una defibrillazione, accertarsi che il dispositivo funzioni correttamente.
Radiazione ionizzante. La radiazione ionizzante a scopo terapeutico (usata, per esempio, negli acceleratori lineari e nelle macchine per cobaltoterapia) può danneggiare irreparabilmente i circuiti del dispositivo. L’effetto della radiazione ionizzante è cumulativo; la possibilità di danni al dispositivo è proporzionale alla dose complessiva di radiazioni assorbita dal paziente. Se il paziente deve essere esposto a radiazione ionizzante, durante la procedura proteggere il dispositivo con una schermatura locale. Qualora occorresse irradiare il tessuto in prossimità della sede di impianto, potrebbe rendersi necessario lo spostamento del dispositivo in un sito differente. Prima e dopo l’esposizione alle radiazioni, controllare il funzionamento del dispositivo per individuare eventuali effetti negativi.

28
Stimolazione elettrica transcutanea nervosa (TENS). Per ridurre la possibilità di interferenze con il funzionamento del dispositivo, posizionare gli elettrodi TENS l’uno vicino all’altro e il più distante possibile dal dispositivo. Prima di autorizzare l’uso illimitato della stimolazione elettrica nervosa transcutanea (TENS) a domicilio o in altro contesto, tenere sotto osservazione il paziente in un ambiente monitorato per escludere il verificarsi di eventuali interazioni.
Diatermia terapeutica. Evitare la diatermia, anche se il dispositivo è programmato su off, in quanto può danneggiare i tessuti adiacenti agli elettrodi impiantati o arrecare danni permanenti al dispositivo.
L’elettrocauterio può indurre aritmie e/o fibrillazione ventricolari o provocare una stimolazione asincrona o inibire il funzionamento del generatore di impulsi. Se è necessario ricorrere all’elettrocauterio, il percorso della corrente e la piastra di terra dovrebbero essere tenuti alla massima distanza possibile dal dispositivo e dagli elettrocateteri. Uno strumento per elettrocauterio bipolare può ridurre al minimo questi effetti. Dopo l’elettrocauterio, verificare accuratamente che il dispositivo funzioni normalmente.
Ablazione a radiofrequenza (RF). L’ablazione a radiofrequenza (RF) su pazienti portatori del dispositivo può indurre una stimolazione asincrona superiore o inferiore alla frequenza programmata, l’avvio di un funzionamento asincrono, un reset elettrico del dispositivo oppure un’attivazione prematura dell’indicatore di sostituzione elettiva. I rischi dell’ablazione a RF possono essere ridotti al minimo programmando una modalità di stimolazione asincrona non-rate responsive prima di intraprendere la procedura di ablazione a RF,

29
evitando il contatto diretto fra il catetere per ablazione e l’elettrocatetere impiantato o il dispositivo, posizionando la piastra di terra in modo che il percorso della corrente non attraversi o passi vicino al sistema del dispositivo, ad esempio ponendola sotto i glutei o sotto le gambe del paziente, tenendo a disposizione un programmatore oppure un’apparecchiatura per la defibrillazione esterna.
Ambiente domestico e di lavoro del paziente Linee di trasmissione ed attrezzature ad alta tensione, saldatori ad arco o a resistenza, forni a induzione ed altri apparecchi simili possono generare forti campi elettromagnetici che potrebbero interferire con il funzionamento del dispositivo.
Apparecchi di comunicazione, come trasmettitori a microonde5, amplificatori o trasmettitori ad alta potenza potrebbero generare interferenze elettromagnetiche sufficienti a pregiudicare il buon funzionamento del dispositivo. Raccomandare ai pazienti di allontanarsi da questi apparecchi per ripristinare il normale funzionamento del dispositivo.
Elettrodomestici in buone condizioni operative e dotati di opportuna messa a terra in genere non producono interferenze elettromagnetiche tali da compromettere il funzionamento del dispositivo. Vibratori, rasoi ed altri apparecchi elettrici a mano, tenuti direttamente sopra il dispositivo, potrebbero disturbarne il normale funzionamento.
Sindrome di Twiddler. Sconsigliare ai pazienti di manipolare il dispositivo impiantato poiché potrebbero derivarne danni agli elettrocateteri o un loro spostamento.
5 I forni a microonde per uso domestico non interferiscono con il funzionamento del dispositivo.

30
Attività del paziente. Qualsiasi attività del paziente che comporti ripetute scosse o vibrazioni (come l’equitazione, l’uso di una perforatrice, ecc.) potrebbe aumentare la frequenza di stimolazione, se l’opzione Sensore del dispositivo è programmata su On. Sconsigliare ai pazienti di svolgere queste attività e programmare i parametri del sensore tenendo conto di tali attività.
Dispositivi per la sorveglianza elettronica degli articoli (EAS). Informare i pazienti che i Sistemi per la sorveglianza elettronica degli articoli (EAS) come quelli in uso presso i punti vendita e all’entrata/uscita di negozi, biblioteche, banche, ecc., emettono segnali che possono influire negativamente sul funzionamento dei pacemaker e dei dispositivi CRT-P. È altamente improbabile che questi sistemi interagiscano in maniera significativa con il dispositivo. Per ridurre al minimo le possibilità di interazione, consigliare ai pazienti di camminare semplicemente a un’andatura normale durante l’attraversamento di queste aree e di non indugiare o trattenersi più del necessario.
Simbolo di divieto d’accesso ai portatori di stimolatori cardiaci. Indica ai pazienti ai quali è stato impiantato questo dispositivo di evitare le zone contrassegnate con il simbolo di divieto d’accesso ai portatori di stimolatori cardiaci.

31
Figura 1. Simbolo di divieto d’accesso ai portatori di stimolatori cardiaci
Telefoni cellulari. Un filtro protettivo di progettazione St. Jude Medical installato nel dispositivo impedisce che i segnali elettromagnetici emessi dai telefoni cellulari interferiscano con il funzionamento del dispositivo.6
Il dispositivo è inoltre stato sottoposto alle prove di compatibilità con le ricetrasmittenti senza fili, conformemente ai requisiti di AAMI PC69. Questo test copre le frequenze operative (450 MHz-3 GHz) e tecniche di modulazione pulsata di tutte le tecnologie della telefonia cellulare digitale attualmente in uso nel mondo. Per ulteriori informazioni, i medici e i pazienti possono contattare il servizio di assistenza tecnica (pagina 51).
Espianto e smaltimento Non riutilizzare dispositivi ed elettrocateteri espiantati. 6 Carrillo R, Williams DB, Traad EA, Schor JS. Electromagnetic filters impeded adverse interference of pacemakers by digital cellular telephones. JACC 1996; 27(2A):15A Abstract 901-22.

32
Pulire i dispositivi espiantati con una soluzione all’1% di ipoclorito di sodio, risciacquarli con acqua e asciugarli.
Restituire il dispositivo espiantato al costruttore.
Espiantare il dispositivo utilizzando strumenti chirurgici standard.
Espiantare il dispositivo prima della cremazione di un paziente deceduto.
Per scollegare un dispositivo precedentemente impiantato dagli elettrocateteri di stimolazione a permanenza vengono fornite in dotazione apposite chiavi esagonali che possono essere richieste al rappresentante St. Jude Medical locale.
Informazioni relative alla sicurezza RM I pacemaker e i dispositivi CRT-P a compatibilità condizionata alla RM sono sicuri in ambiente di risonanza magnetica quando utilizzati in un sistema completo a compatibilità condizionata alla RM e in conformità alle istruzioni contenute nel manuale dei sistemi di stimolazione MRI-Ready St. Jude Medical™. L’esame eseguito in condizioni diverse può causare gravi lesioni o morte del paziente o malfunzionamento del dispositivo.
Potenziali effetti indesiderati Le complicanze elencate di seguito potrebbero verificarsi usando qualsiasi sistema di stimolazione cardiaca:

33
Ulteriori interventi chirurgici Reazione allergica Aritmia Lesione cardiovascolare come una lesione termica Insufficienza cardiaca scompensata Morte Embolia Erosione Stimolazione extracardiaca Ematomi Infezione Perdita di stimolazione cardiaca Perdita di sincronia Pneumotorace
Connettore del generatore di impulsi I connettori del generatore di impulsi sono illustrati nella tabella seguente, seguita dalla legenda relativa ai fori dell’elettrocatetere. (pagina 36)

34
Tabella 6. Connettori del generatore di impulsi. Consultare la legenda dei fori dell’elettrocatetere (pagina 36)
Assurity PM2240 Assurity MRI PM2272 Endurity PM2160, PM2162 Endurity MRI PM2172 Endurity Core PM2140, PM2152 Zenex PM2250 Zenex MRI PM2282 Zenus PM2170 Zenus MRI PM2182
Assurity MRI PM1272 Assurity PM1240 Endurity PM1160 Endurity MRI PM1172, PM1162 Endurity Core PM1140, PM1152 Zenex PM1250 Zenex MRI PM1282 Zenus PM1170 Zenus MRI PM1182

35
Tabella 6. Connettori del generatore di impulsi. Consultare la legenda dei fori dell’elettrocatetere (pagina 36)
Allure PM3120 Allure RF PM3222
Allure Quadra RF PM3242 Quadra Allure PM3542 Quadra Allure MP PM3562 Quadra Allure MP RF PM3262

36
Tipi di connettori della porta dell'elettrocatetere
Tabella 7. Porte per elettrocateteri
Legenda Porta Tipo di elettrocatetere Connettore
1 V (IS-1 BI) SENSING/STIMOLAZIONE
Endocardico bipolare IS-1 bipolare in linea
2 A (IS-1 BI) SENSING/STIMOLAZIONE O TAPPO
Bipolare endocardico; tappo IS-1 (quando non viene utilizzato un elettrocatetere atriale)
IS-1 bipolare in linea
3 VD (IS-1 BI) SENSING/STIMOLAZIONE
Bipolare endocardico nel ventricolo destro (CRT-P)
IS-1 bipolare in linea
4 VS (IS-1 BI) SENSING/STIMOLAZIONE O TAPPO
Bipolare endocardico nel ventricolo sinistro (CRT-P); tappo IS-1 (quando non viene utilizzato un elettrocatetere VS)
IS-1 bipolare in linea
5 S (A/V) (IS-1 BI) SENSING/STIMOLAZIONE
Bipolare endocardico; monocamerale, designato A o V in base alla programmazione
IS-1 bipolare in linea

37
Tabella 7. Porte per elettrocateteri
Legenda Porta Tipo di elettrocatetere Connettore
6 VS (IS-4 Quad) SENSING/STIMOLAZIONE O TAPPO
Quadripolare endocardico; tappo IS-4/DF4 (quando non viene utilizzato un elettrocatetere VS)
IS4-LLLL
NOTE:
Quando si eseguono i collegamenti degli elettrocateteri al generatore d’impulsi, assicurarsi che il connettore dell’elettrocatetere sia inserito nella porta per elettrocatetere appropriata. Per il sensing e la stimolazione, è importante assicurarsi che i segnali atriali e ventricolari siano correttamente rilevati e che gli impulsi di stimolazione siano erogati nella camera desiderata.
La porta dell’elettrocatetere IS4-LLLL può essere utilizzata soltanto con elettrocateteri cardiaci sinistri IS4-LLLL.

38
Indicazioni per la programmazione
Informazioni generali Per l'elenco completo dei parametri programmabili e delle loro impostazioni, consultare l'help in linea del programmatore.
Uso del magnete Per interrogare il dispositivo, rimuovere il magnete dalla sonda telemetrica del programmatore. Il magnete interferisce con il corretto funzionamento della telemetria.
Programmazione temporanea Questi dispositivi sono dotati della funzione di programmazione temporanea per assistere il medico nella diagnosi e nel trattamento del paziente. Il medico può programmare temporaneamente i parametri per valutarne gli effetti e quindi annullare rapidamente l’impostazione o programmarla in modo permanente. Per maggiori informazioni, fare riferimento all'help in linea del programmatore.

39
Preimpostazioni di programmazione
Impostazioni alla consegna Le impostazioni dei parametri del dispositivo vengono preconfigurate al momento della fabbricazione. Per ulteriori informazioni, fare riferimento all'help in linea del programmatore.
Impostazioni di emergenza Il dispositivo è dotato di impostazioni ad alta uscita standard rapidamente programmabili mediante la funzione Emergenza VVI del programmatore. Le impostazioni della funzione Emergenza VVI sono indicate nell’help in linea del programmatore.
NOTA: se è stata selezionata la funzione di Emergenza VVI, il sistema cancella i dati diagnostici in memoria senza visualizzare alcun messaggio di avvertimento.
Identificatori radiopachi Ogni dispositivo è dotato di un marker d’assorbimento dei raggi X per l’identificazione non invasiva, il quale consiste nel logo St. Jude Medical (SJM) e in un codice modello.7
7 I modelli PM1172, PM1272, PM2172 e PM2272 potrebbero avere una targhetta SJM HM MRI a causa della distribuzione globale.

40
Tabella 8. Codici di identificazione radiologica dei modelli di dispositivi descritti nel presente manuale
Modello dispositivo Codice di identificazione radiologica del modello
PM1160, PM1170, PM1172, PM1182, PM1240, PM1250, PM1272, PM1282, PM2160, PM2170, PM2172, PM2182, PM2240, PM2250, PM2272, PM2282, PM3120, PM3222, PM3242, PM3262
HI
PM1140, PM1152, PM1162, PM2140, PM2152, PM2162 HM MRI
PM3542, PM3562 SJM HM

41
Impianto e collegamento degli elettrocateteri Contenuto della confezione I dispositivi sono forniti in una confezione sterile contenente: Un dispositivo Kit del connettore composto da:
- Chiave torsiometrica n. 2 Materiale illustrativo
Collegamento degli elettrocateteri I dispositivi con connettore IS-1 supportano elettrocateteri unipolari o bipolari IS-1 con spinotto terminale corto. I dispositivi con connettore IS4-LLLL sono compatibili con elettrocateteri quadripolari IS4-LLLL. Prima dell’impianto accertarsi che gli elettrocateteri entrino agevolmente nel connettore del dispositivo.
Questi dispositivi sono dotati di un’unica vite di fissaggio per ciascun spinotto. La vite fa contatto con lo spinotto dell’elettrocatetere fissandolo all’interno del connettore, mentre una molla anulare stabilisce il contatto con gli anelli prossimali.
NOTA: inserire il tipo di elettrocatetere per ciascun elettrocatetere nella schermata delle informazioni paziente. Per ulteriori informazioni, fare riferimento all’help in linea del programmatore.

42
ATTENZIONE: Successivamente all’impianto di tutti gli elettrocateteri e prima del collegamento degli stessi al dispositivo, stabilire e documentare la morfologia basale per le soglie di cattura e sensing di ogni singolo elettrocatetere utilizzando un sistema di registrazione idoneo, quale un ECG a 12 derivazioni o un elettrogramma intracardiaco (IEGM).
Per collegare il dispositivo agli elettrocateteri:
1. Pulire gli spinotti terminali degli elettrocateteri impiantati, liberandoli da sangue e fluidi corporei.
2. Controllare le marcature sulla cassa del dispositivo e verificare il corretto collegamento atriale e ventricolare.
ATTENZIONE: procedere con cautela quando si gira la vite di fissaggio, la quale, se ruotata in senso antiorario per più di due giri, potrebbe uscire dal connettore.
NOTA: nei dispositivi CRT-P, per ottenere un sensing e una stimolazione corretti, è della massima importanza garantire che i segnali dei ventricoli sinistro e destro vengano rilevati correttamente e che gli impulsi di stimolazione vengano erogati nella camera desiderata.
3. Utilizzare la chiave torsiometrica n. 2 in dotazione per fare arretrare le viti di fissaggio nel connettore del dispositivo in modo che gli spinotti terminali dell’elettrocatetere di stimolazione possano essere completamente inseriti.

43
4. Inserire saldamente ciascun elettrocatetere nel connettore finché lo spinotto dell’elettrocatetere è bloccato e visibile attraverso l’apposita apertura sull’altro lato del connettore.
5. Afferrare la chiave torsiometrica per il lato più grande dell’impugnatura come mostrato nella figura a sinistra di seguito. Sui modelli di chiave applicabili, non afferrare la chiave torsiometrica dalla parte più piccola e stretta dell’impugnatura come mostrato a destra.
Figura 2. Uso corretto vs. errato della chiave torsiometrica

44
6. Inserire la chiave torsiometrica n. 2 attraverso il foro sul connettore e nella vite di fissaggio posta sul lato del connettore.
7. Ruotare la chiave in senso orario finché non scatta. La chiave ha una capacità di torsione limitata e, quando utilizzata correttamente, non permette un serraggio eccessivo.
8. Ripetere la procedura sopra indicata per gli ulteriori elettrocateteri.
9. Tirare delicatamente gli elettrocateteri per accertarsi che siano ben fissati al connettore.
Per ridurre al minimo il rischio di migrazione del dispositivo, fissare il dispositivo dentro la tasca sottocutanea mediante il foro di sutura nel connettore del dispositivo.
Dopo aver impiantato il dispositivo e chiuso la tasca, interrogare il dispositivo e configurare correttamente il parametro Tipo Elettrocatetere. Le impostazioni di Tipo Elettrocatetere sono descritte nell’help in linea del programmatore. Per i generatori di impulsi a compatibilità condizionata alla RM, settare il parametro Hardware aggiuntivo sull’impostazione corretta.
NOTA:
Nei dispositivi CRT-P, le ampiezze e le durate dell’impulso ventricolare destro e sinistro sono programmabili in modo indipendente. L’ampiezza e la durata dell’impulso devono essere valutate in ciascuna camera separatamente.
Nei dispositivi CRT-P, vengono visualizzati valori di impedenza degli elettrocateteri indipendenti per gli elettrocateteri VD e VS.

45
Registrazione del dispositivo Ogni dispositivo contiene un modulo di registrazione del dispositivo impiantabile che serve da documento permanente dei dati relativi al sistema impiantato. L'originale dovrà venire rispedito, opportunamente compilato, al costruttore nell'apposita busta completa di indirizzo, già affrancata. Del suddetto modulo di registrazione vengono fornite copie destinate all'ospedale e al medico.
Durata del dispositivo Per i calcoli della durata stimata, consultare la guida in linea del programmatore.
A condizioni nominali (100% di stimolazione, 2,5 V/0,5 ms per ogni camera, 60 ppm, 500 ohm di impedenza dell’elettrocatetere), la durata stimata corrisponde a quanto segue:
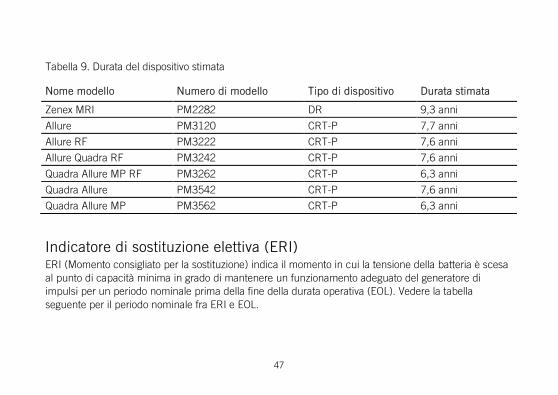
Tabella 9. Durata del dispositivo stimata
Nome modello Numero di modello Tipo di dispositivo Durata stimata
Endurity Core PM1140, PM1152 SR 14,3 anni
Endurity PM1160, PM1162 SR 14,3 anni
Zenus PM1170 SR 14,3 anni
Endurity MRI PM1172 SR 14,3 anni

46
Tabella 9. Durata del dispositivo stimata
Nome modello Numero di modello Tipo di dispositivo Durata stimata
Zenus MRI PM1182 SR 14,3 anni
Assurity PM1240 SR 13,8 anni
Zenex PM1250 SR 13,8 anni
Assurity MRI PM1272 SR 13,8 anni
Zenex MRI PM1282 SR 13,8 anni
Endurity Core PM2140, PM2152 DR 9,5 anni
Endurity PM2160, PM2162 DR 9,5 anni
Zenus PM2170 DR 9,5 anni
Endurity MRI PM2172 DR 9,5 anni
Zenus MRI PM2182 DR 9,5 anni
Assurity PM2240 DR 9,3 anni
Zenex PM2250 DR 9,3 anni
Assurity MRI PM2272 DR 9,3 anni
47
Tabella 9. Durata del dispositivo stimata
Nome modello Numero di modello Tipo di dispositivo Durata stimata
Zenex MRI PM2282 DR 9,3 anni
Allure PM3120 CRT-P 7,7 anni
Allure RF PM3222 CRT-P 7,6 anni
Allure Quadra RF PM3242 CRT-P 7,6 anni
Quadra Allure MP RF PM3262 CRT-P 6,3 anni
Quadra Allure PM3542 CRT-P 7,6 anni
Quadra Allure MP PM3562 CRT-P 6,3 anni
Indicatore di sostituzione elettiva (ERI) ERI (Momento consigliato per la sostituzione) indica il momento in cui la tensione della batteria è scesa al punto di capacità minima in grado di mantenere un funzionamento adeguato del generatore di impulsi per un periodo nominale prima della fine della durata operativa (EOL). Vedere la tabella seguente per il periodo nominale fra ERI e EOL.

48
Quando il dispositivo raggiunge l’ERI, il medico viene avvertito della condizione da diversi indicatori. Per informazioni su queste condizioni, fare riferimento alla guida in linea del programmatore.
Tabella 10. Tempo nominale fra ERI e EOL per tutti i dispositivi
Dispositivo Periodo di tempo nominale tra ERI e EOL
PM1140, PM1152, PM1160, PM1162, PM1170, PM1172, PM1182, PM1240, PM1250, PM1272, PM1282, PM2140, PM2152, PM2160, PM2162, PM2170, PM2172, PM2182, PM2240, PM2250, PM2272, PM2282, PM3120, PM3222, PM3242, PM3262, PM3542, PM3562
6 mesi
Come cancellare l'ERI Quando il programmatore visualizza un messaggio indicante che il dispositivo ha raggiunto l’ERI, è possibile cancellare l’ERI. Per ulteriori informazioni sulla cancellazione dell’ERI, consultare la guida in linea del programmatore.

49
ATTENZIONE:
La programmazione di impostazioni ad alta uscita o di una Frequenza Base elevata può ridurre il tempo con cui il dispositivo raggiunge l’ERI. La programmazione di frequenze e uscite inferiori può ripristinare il normale stato della batteria.
Se il programmatore visualizza un messaggio di avvertimento indicante che è stato raggiunto l’ERI, il medico deve effettuare una valutazione completa del dispositivo.
AVVERTENZA: all’ERI, la durata nominale del dispositivo è di tre o sei mesi. (pagina 47) Quando manifesta segni di ERI (descritti nella guida in linea del programmatore), il dispositivo deve essere prontamente sostituito.
Fine servizio La condizione di fine della durata operativa (EOL) si verifica quando la tensione della batteria è scesa a un livello designato nella tabella seguente.
Per ulteriori informazioni, consultare la guida in linea del programmatore.

50
Tabella 11. Tensione della batteria approssimativa a fine della durata operativa per tutti i dispositivi
Dispositivo Tensione della batteria approssimativa all’EOL
PM1140, PM1152, PM1160, PM1162, PM1170, PM1172, PM1182, PM1240, PM1250, PM1272, PM1282, PM2140, PM2152, PM2160, PM2162, PM2170, PM2172, PM2182, PM2240, PM2250, PM2272, PM2282, PM3120, PM3222, PM3242, PM3262, PM3542, PM3562
2,47 V

51
Assistenza tecnica St. Jude Medical mette a disposizione linee telefoniche in funzione 24 ore su 24 per rispondere a domande tecniche e per fornire supporto: 1 818 362 6822
1 800 722 3774 (numero verde nel Nord America)
+ 46 8 474 4147 (Svezia)
+ 61 2 9936 1200 (Australia)
manuals.sjm.com
Per richiedere ulteriore assistenza, contattare il rappresentante St. Jude Medical locale.
Informazioni aggiuntive Per ulteriori informazioni su questo dispositivo, consultare l'help in linea del programmatore.
52
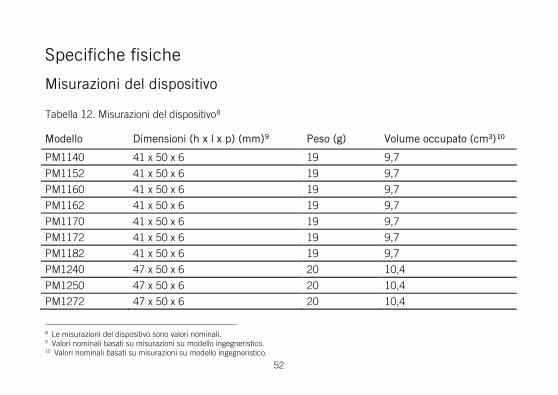
Specifiche fisiche
Misurazioni del dispositivo
Tabella 12. Misurazioni del dispositivo8
Modello Dimensioni (h x l x p) (mm)9 Peso (g) Volume occupato (cm3)10
PM1140 41 x 50 x 6 19 9,7
PM1152 41 x 50 x 6 19 9,7
PM1160 41 x 50 x 6 19 9,7
PM1162 41 x 50 x 6 19 9,7
PM1170 41 x 50 x 6 19 9,7
PM1172 41 x 50 x 6 19 9,7
PM1182 41 x 50 x 6 19 9,7
PM1240 47 x 50 x 6 20 10,4
PM1250 47 x 50 x 6 20 10,4
PM1272 47 x 50 x 6 20 10,4
8 Le misurazioni del dispositivo sono valori nominali. 9 Valori nominali basati su misurazioni su modello ingegneristico. 10 Valori nominali basati su misurazioni su modello ingegneristico.

53
Tabella 12. Misurazioni del dispositivo8
Modello Dimensioni (h x l x p) (mm)9 Peso (g) Volume occupato (cm3)10
PM1282 47 x 50 x 6 20 10,4
PM2140 46 x 50 x 6 19 10,4
PM2152 46 x 50 x 6 19 10,4
PM2160 46 x 50 x 6 19 10,4
PM2162 46 x 50 x 6 19 10,4
PM2170 46 x 50 x 6 19 10,4
PM2172 46 x 50 x 6 19 10,4
PM2182 46 x 50 x 6 19 10,4
PM2240 47 x 50 x 6 20 10,4
PM2250 47 x 50 x 6 20 10,4
PM2272 47 x 50 x 6 20 10,4
PM2282 47 x 50 x 6 20 10,4
PM3120 55 x 59 x 6 24 14

54
Tabella 12. Misurazioni del dispositivo8
Modello Dimensioni (h x l x p) (mm)9 Peso (g) Volume occupato (cm3)10
PM3222 55 x 59 x 6 24 14
PM3242 55 x 59 x 6 27 15
PM3262 56 x 59 x 6 27 15
PM3542 56 x 59 x 6 26 15
PM3562 56 x 59 x 6 26 15

55
Materiali del dispositivo
Tabella 13. Materiali del dispositivo
Modello Cassa Rivestimento cassa
Antenna RF11 Materiale del connettore
Tutti i dispositivi
Titanio Nessuna Titanio Può contenere uno o più dei seguenti: Resina epossidica, poliuretano termoplastico, polisulfone
11 Per dispositivi con funzione telemetrica RF.

56
Compatibilità degli elettrocateteri
Tabella 14. Compatibilità degli elettrocateteri
Modello Compatibilità degli elettrocateteri
PM3242 PM3262 PM3542 PM3562
IS-1 e IS4-LLLL
Tutti gli altri dispositivi IS-112
12 Compatibile con elettrocateteri IS-1 con spinotto terminale corto.

57
Dati sulla batteria
Tabella 15. Informazioni sulla batteria
Modello Alimentazione Produttore; Modello
Tensione e inizio vita (BOL)
Tensione necessaria per attivare l’indicatore di sostituzione previsto
PM3120 PM3222 PM3242 PM3262 PM3542 PM3562
1 cella QMR13
Greatbatch Medical; modello 2662
3,20 V
2,62 V
Tutti gli altri dispositivi
2,60 V
13 QMR è un marchio di Greatbatch Medical.

58
Frequenze operative RF Le apparecchiature adiacenti che emettono forti campi magnetici possono interferire con la comunicazione in radiofrequenza (RF), anche se l’altra apparecchiatura è conforme ai requisiti sulle emissioni CISPR. Le caratteristiche operative sono le seguenti:
Banda MICS: 402-405 MHz. La potenza effettiva irradiata è inferiore ai limiti specificati in: Europa: EN ETSI 301 839
USA: FCC 47 CFR Parte 95; 95.601-95.673 Sottoparte E, 95.1201-95.1221 Sottoparte I
ID FCC: RIASJMRFB
Armonizzato con la FCC
L’indicazione seguente è applicabile solo al Canada:
Il presente dispositivo non determina interferenze con stazioni che impiegano la banda 400.150-406.000 MHz per i servizi meteorologici, stazioni di trasmissione con i satelliti meteorologici e servizi di trasmissione con i satelliti per l’esplorazione terrestre e deve accettare eventuali interferenze ricevute, incluse quelle in grado di causare problemi di funzionamento.
Questo dispositivo è conforme agli standard RSS esenti da licenza di Industry Canada. Il suo utilizzo è subordinato alle due condizioni seguenti: (1) questo dispositivo non dovrebbe provocare interferenze dannose e (2) è in grado di accettare eventuali interferenze provenienti da altri dispositivi, incluse quelle che potrebbero provocare anomalie nel funzionamento.

59
Prestazioni di rilevamento in presenza di interferenza elettromagnetica Quando il parametro Configurazione Sensing è impostato su Bipolare, valori di Sensibilità Atriale di 0,2 mV o più elevati possono essere maggiormente sensibili alle interferenze elettromagnetiche (EMI). I dispositivi sono conformi ai requisiti della compatibilità elettromagnetica della clausola 27.5 della norma CENELEC EN45502-2-114 ad impostazioni della Sensibilità Atriale di 0,3 mV e impostazioni meno sensibili.
Quando il parametro Configurazione Sensing è impostato su Unipolare, i valori di Sensibilità Atriale e Ventricolare di 2,0 mV o più elevati possono essere maggiormente sensibili alle interferenze elettromagnetiche (EMI). I dispositivi sono conformi ai requisiti della compatibilità elettromagnetica della clausola 27.5 della norma CENELEC EN45502-2-1, ad impostazioni della Sensibilità Atriale e Ventricolare di 2,0 mV e impostazioni meno sensibili. (La clausola 27.5.1 della norma CENELEC EN45502-2-1 impone che il generatore di impulsi impiantabile sia costruito in modo da rendere improbabile che i segnali elettromagnetici più comuni vengano scambiati per battiti rilevati e che cambi il comportamento terapeutico del generatore di impulsi impiantabile).
Come previsto dalla clausola 27.4 della norma EN45502-2-1, la modalità di funzionamento per le interferenze del dispositivo si caratterizza come segue: La modalità di rumori atriali è “stimolazione disattivata” per le frequenze EMI inferiori a circa
30 Hz e “stimolazione a frequenza costante” per le frequenze superiori a circa 30 Hz.
14 Come specificato in questa sezione, la norma CENELEC EN45502-2-1:2003 è equivalente alla ANSI/AAMI PC69:2007.

60
La modalità per i rumori ventricolari è “stimolazione a frequenza costante” per le frequenze EMI comprese tra 16,6 Hz e-167 kHz.
Effetti della temperatura I parametri di stimolazione quali Frequenza Impulso, Durata Impulso, Ampiezza Impulso e Sensibilità soddisfano le tolleranze nominali definite nell'help in linea del programmatore oltre il range di temperature compreso fra 25 e 45°C (±2°C).
Impedenza d'ingresso
Tabella 16. Impedenza di ingresso
Misurazione Range
Impedenza di ingresso 30-75 k¬

61
Capacità di stimolazione effettiva
Tabella 17. Capacità di stimolazione effettiva
Misurazione Range
Capacità di stimolazione effettiva 4,7 µF±10%
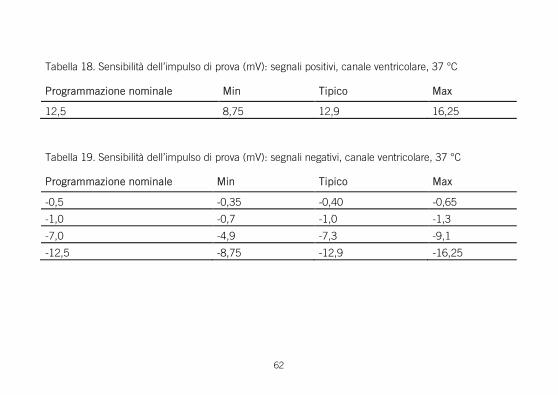
Sensibilità dell'impulso di prova Sensibilità misurata usando l’impulso di prova mostrato nella figura sotto riportata.
Tabella 18. Sensibilità dell’impulso di prova (mV): segnali positivi, canale ventricolare, 37 °C
Programmazione nominale Min Tipico Max
0,5 0,35 0,40 0,65
1,0 0,7 1,0 1,3
7,0 4,9 7,3 9,1
62
Tabella 18. Sensibilità dell’impulso di prova (mV): segnali positivi, canale ventricolare, 37 °C
Programmazione nominale Min Tipico Max
12,5 8,75 12,9 16,25
Tabella 19. Sensibilità dell’impulso di prova (mV): segnali negativi, canale ventricolare, 37 °C
Programmazione nominale Min Tipico Max
-0,5 -0,35 -0,40 -0,65
-1,0 -0,7 -1,0 -1,3
-7,0 -4,9 -7,3 -9,1
-12,5 -8,75 -12,9 -16,25

63
Figura 3. Descrizione dell’impulso di prova
1. 2,0 ms 2. 13 ms

64
Curva di scarica della batteria
Figura 4. Curva di scarica della batteria
1. Tensione (mV) 2. % di profondità di scarica (mAh)

65
Simboli I simboli seguenti e i simboli armonizzati possono essere riportati sul prodotto o sulla sua etichetta. Per i simboli armonizzati, consultare il Glossario dei simbli universali all’indirizzo https://manuals.sjm.com.
Simbolo Descrizione
Dispositivo monocamerale modulato in frequenza
Dispositivo bicamerale modulato in frequenza
Dispositivo di comunicazione monocamerale
Dispositivo di comunicazione bicamerale
Pacemaker per terapia di resincronizzazione cardiaca, atriale destro, ventricolare destro, ventricolare sinistro
Sensing bipolare/Stimolazione bipolare
Sensing/Stimolazione unipolare
DDDR NBG - stimolazione bicamerale, sensing doppio, risposta doppia, modulazione della frequenza

66
Simbolo Descrizione
DDDRV NBG - stimolazione bicamerale, sensing bicamerale, risposta bicamerale, modulazione della frequenza, sensing e stimolazione biventricolare
SSI NBG - stimolazione atriale o ventricolare, sensing atriale oventricolare, risposta inibita, nessuna modulazione della frequenza
SSIR NBG - stimolazione atriale o ventricolare, sensing atriale o ventricolare, risposta inibita, modulazione della frequenza
IS-1 Il connettore dell'elettrocatetere supporta elettrocateteri unipolari o bipolari IS-1 (Standard internazionale-1) con spinotto terminale corto.
IS4-LLLL Il connettore dell'elettrocatetere è compatibile con elettrocateteri IS4-LLLL quadripolari. SJ4-LLLL è equivalente a IS4-LLLL. Le cavità dei connettori SJ4 e IS4 St. Jude Medical sono conformi alla norma ISO27186:2010(E).
VVI NBG - stimolazione ventricolare, sensing ventricolare, inibizione della risposta
DDD NBG - stimolazione bicamerale, sensing bicamerale, risposta bicamerale, nessuna modulazione della frequenza
Seguire le istruzioni per l'uso riportate su questo sito Internet

67
Simbolo Descrizione
Stabilimento di produzione
Marchio di conformità europeo, apposto conformemente alle disposizioni pertinenti previste dalla direttiva AIMD 90/385/CEE e dalla direttiva RE 2014/53/UE Allegato II. Con il presente marchio, St. Jude Medical dichiara che il dispositivo soddisfa i requisiti essenziali e altre disposizioni pertinenti delle direttive citate. Il testo completo della dichiarazione di conformità dell’Unione Europea della direttiva RE 2014/53/UE è disponibile al seguente indirizzo Internet: www.sjmglobal.com/euconformity. Questo prodotto funziona in una banda di frequenza compresa tra 402 e -405 MHz con una potenza irradiata effettiva inferiore a 25 µW ERP. Questo prodotto funziona tra 9 e 200 kHz con un’intensità di campo H-inferiore a 25 dBµA/m a 10 m.

68
Simbolo Descrizione
Il dispositivo contiene una batteria e l'etichetta è apposta in conformità alla Direttiva 2006/66/CE del Parlamento Europeo e del Consiglio. Restituire il dispositivo a St. Jude Medical dopo l'espianto o smaltire come materiale potenzialmente a rischio biologico in conformità alla pratica medica e alle leggi e alle regolamentazioni locali, statali e federali applicabili.
Per l’uso solo su prescrizione
Questa apparecchiatura è dotata della certificazione in base al tipo ai sensi dell'Articolo 38-24 della Japan Radio Law
Marchio di certificazione coreana per i dispositivi elettrici
Marchio di conformità normativa (Regulatory Compliance Mark, RCM) dell'Australian Communications and Media Authority (ACMA) e della Radio Spectrum Management (RSM) della Nuova Zelanda

69
Simbolo Descrizione
Inizio della vita di servizio
Parametro ventricolo sinistro
Parametro atrio destro e ventricolo destro
Indicazione di intervallo o ritardo. Quando è presente un intervallo o un ritardo tra due camere, l'immagine viene modificata per includere uno spazio o un vuoto.
Intervallo rilevato da atriale a ventricolare
Ritardo AV: Stimolato/Rilevato
Ampiezza e durata impulso
Ampiezza/durata impulso, ventricolare destra, ventricolare sinistra
Sensibilità
Periodo refrattario

70
Simbolo Descrizione
Sensing
Terapia erogata
Rilevamento automatico polarità
S (A/V)- Monocamerale (A o V) Connettore IS-1
Connettore IS-1 A- Atriale; Connettore IS-1 V- Ventricolare
Connettore IS-1 A- Atriale; Connettore IS-4 VS- Ventricolare sinistro; Connettore IS-1 VD- Ventricolare destro
Accessori

71
Simbolo Descrizione
Documentazione del prodotto
Prodotto in Malesia
Fabbricato negli Stati Uniti



St. Jude Medical Cardiac Rhythm Management Division 15900 Valley View Court Sylmar, CA 91342 USA +1 818 362 6822
St. Jude Medical Coordination Center BVBA The Corporate Village Da Vincilaan 11 Box F1 1935 Zaventem Belgium +32 2 774 68 11
St. Jude Medical Australia Pty. Limited 17 Orion Road Lane Cove NSW 2066 Australia
St. Jude Medical Puerto Rico LLC Lot A Interior - #2 Rd Km. 67.5 Santana Industrial Park Arecibo, PR 00612 USA
St. Jude Medical Operations (M) Sdn. Bhd. Plot 102, Lebuhraya Kampung Jawa, Bayan Lepas Industrial Zone 11900 Penang Malaysia
sjm.com
2019-07 ARTIT100171075A




















