
M13 Bacteriophage and Adeno-Associated Virus Hybrid for Novel Tissue Engineering Material with Gene...
6
© 2015 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim wileyonlinelibrary.com 1 COMMUNICATION M13 Bacteriophage and Adeno-Associated Virus Hybrid for Novel Tissue Engineering Material with Gene Delivery Functions So Young Yoo,* Hyo-Eon Jin, Dong Shin Choi, Masae Kobayashi, Yohan Farouz, Sky Wang, and Seung-Wuk Lee* Dr. S. Y. Yoo, Dr. H.-E. Jin, D. S. Choi, M. Kobayashi, Y. Farouz, S. Wang, Prof. S.-W. Lee Department of Bioengineering University of California, Berkeley Physical Biosciences Division Lawrence Berkeley National Laboratory Berkeley Nanoscience and Nanoengineering Institute Berkeley, CA 94720, USA E-mail: [email protected]; [email protected] Dr. S. Y. Yoo BIO-IT Foundry Technology Institute Pusan National University Busan 609-735, and Research Institute for Convergence of Biomedical Science and Technology Yangsan 626-770, Republic of Korea Y. Farouz Biology Department Ecole Polytechnique Route de Saclay 91128, Palaiseau, Cedex, France DOI: 10.1002/adhm.201500179 infect even by these virus-based approaches. [10] (2) Viruses often cause host immune responses that eliminate the infecting virus and its side effects on the surrounding cells. Gene delivery vec- tors must be able to escape the body's natural surveillance sys- tems, otherwise a resulting adverse immune reaction can cause serious illness or even death. [11] (3) Random insertion of viral genome into host genome can cause unexpected mutations that can give rise to abnormal cell formation such as cancer. [12] Therefore, selective, safe, stable, and predictable gene delivery is still challenging in terms of developing targeted gene delivery. Recent advances in the phage biotechnology provide remark- able pathways to develop novel biomaterials. Phages possess many desirable features that make them attractive as versatile gene delivery materials. Phage have little harmful effects and are easily removed from the body through lysosomal degrada- tion processes, causing few known side-effects. [13] Phage can be modified to display functional peptide motifs on their minor (pIII, pIX) and major (pVIII) coat proteins. [14–18] Large quanti- ties of identical phage building blocks can be easily prepared through bacterial amplification. Due to their long-filamentous shape (aspect ratio: 130), phage can self-assemble into nanofi- brous tissue-like matrix structures. Recently, we engineered various biochemical cues (i.e., RGD (Arg-Gly-Asp), IKVAV (Ile- Lys-Val-Ala-Val), and DGEA (Asp-Gly-Glu-Ala)) on major coat proteins (pVIII) of the phage and developed self-assembled tissue engineering materials for regulating cellular behaviors of desired cells. [15,16,19–22] Using this phage-based tissue matrix system, we demonstrated that engineered phages could regulate various cellular behaviors such as proliferation and differentia- tion. [14–16,19,20,23] Here, we developed novel tissue engineering materials with gene delivery functions through the hybridiza- tion of M13 bacteriophage (phage) and AAV. We engineered the M13 phage with RGD peptide on the major coat proteins. We then fused its gene with AAV gene with eukaryotic invading functional gene, ITR. The resulting M13–AAV hybrid phage exhibited enhanced internalization into target cells as compared to minor coat engineered phage with RGD or wild-type phage. The resulting hybrid phage formed nanofibrous matrices that could support the cellular growth and deliver desired gene information into the target tissues. Our novel M13–AAV hybrid phage matrices can provide selective, stable, and safe gene delivery ( Figure 1). We believe that it can be also developed as a topical therapeutic tissue patch in the future. In order to construct a novel phage tissue engineering mate- rial with gene delivery function ( Figure 2), we constructed the hybrid phage (M13 RGD8 –AAV GFP ) carrying both RGD major coat Reprogramming of cellular functions through gene delivery is critical in treating cellular/tissue abnormalities as well as in studying the mechanism of cellular behavior of many cells. [1] Many researchers have utilized nanosized natural [2] or syn- thetic materials such as surfactants, [3] peptide or protein nano- spheres, [4] and polymersomes for gene delivery. [5] Recently, virus-based gene delivery vehicles, such as retrovirus or adeno- virus, are considered as promising delivery vehicles for transfer of genetic information into target cells. [6] Among them, adeno- associated virus (AAV) has been largely adapted for therapeutic gene transfer due to its innocuousness and high resistance to extreme conditions. [7] The AAV is a nonpathogenic virus with a linear single-stranded DNA genome that contains eukaryotic gene integrating functional genes, named inverted terminal repeats (ITRs; 145 bp in length for AAV2), [8] at each end of the AAV viral genome terminus. The ITR possesses a T-shaped hairpin structure with self-complementary guanine-cyto- sine rich sequences; the gene enables integration of the viral genome into as well as rescue from a specific section of the host genome (19th chromosome in humans). [9] Despite the develop- ment of various viral gene delivery vehicles, their therapeutic application still remains challenging due to several reasons. (1) Viruses can hardly reach their desired targets. Viruses have lim- ited ranges of the type of host cells they can infect, although adenoviruses and AAV are able to infect a relatively broad range of cells efficiently. Still, some cell types are unmanageable to Adv. Healthcare Mater. 2015, DOI: 10.1002/adhm.201500179 www.advhealthmat.de www.MaterialsViews.com
-
Upload
univ-paris5 -
Category
Documents
-
view
1 -
download
0
Transcript of M13 Bacteriophage and Adeno-Associated Virus Hybrid for Novel Tissue Engineering Material with Gene...

© 2015 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim wileyonlinelibrary.com 1
CO
MM
UN
ICATIO
N
M13 Bacteriophage and Adeno-Associated Virus Hybrid for Novel Tissue Engineering Material with Gene Delivery Functions
So Young Yoo , * Hyo-Eon Jin , Dong Shin Choi , Masae Kobayashi , Yohan Farouz , Sky Wang , and Seung-Wuk Lee *
Dr. S. Y. Yoo, Dr. H.-E. Jin, D. S. Choi, M. Kobayashi, Y. Farouz, S. Wang, Prof. S.-W. Lee Department of Bioengineering University of California, Berkeley Physical Biosciences Division Lawrence Berkeley National Laboratory Berkeley Nanoscience and Nanoengineering Institute Berkeley , CA 94720 , USAE-mail: [email protected]; [email protected] Dr. S. Y. Yoo BIO-IT Foundry Technology Institute Pusan National University Busan 609-735, and Research Institute for Convergence of Biomedical Science and Technology Yangsan 626-770 , Republic of Korea Y. Farouz Biology Department Ecole Polytechnique Route de Saclay 91128 , Palaiseau , Cedex , France
DOI: 10.1002/adhm.201500179
infect even by these virus-based approaches. [ 10 ] (2) Viruses often cause host immune responses that eliminate the infecting virus and its side effects on the surrounding cells. Gene delivery vec-tors must be able to escape the body's natural surveillance sys-tems, otherwise a resulting adverse immune reaction can cause serious illness or even death. [ 11 ] (3) Random insertion of viral genome into host genome can cause unexpected mutations that can give rise to abnormal cell formation such as cancer. [ 12 ] Therefore, selective, safe, stable, and predictable gene delivery is still challenging in terms of developing targeted gene delivery.
Recent advances in the phage biotechnology provide remark-able pathways to develop novel biomaterials. Phages possess many desirable features that make them attractive as versatile gene delivery materials. Phage have little harmful effects and are easily removed from the body through lysosomal degrada-tion processes, causing few known side-effects. [ 13 ] Phage can be modifi ed to display functional peptide motifs on their minor (pIII, pIX) and major (pVIII) coat proteins. [ 14–18 ] Large quanti-ties of identical phage building blocks can be easily prepared through bacterial amplifi cation. Due to their long-fi lamentous shape (aspect ratio: 130), phage can self-assemble into nanofi -brous tissue-like matrix structures. Recently, we engineered various biochemical cues (i.e., RGD (Arg-Gly-Asp), IKVAV (Ile-Lys-Val-Ala-Val), and DGEA (Asp-Gly-Glu-Ala)) on major coat proteins (pVIII) of the phage and developed self-assembled tissue engineering materials for regulating cellular behaviors of desired cells. [ 15,16,19–22 ] Using this phage-based tissue matrix system, we demonstrated that engineered phages could regulate various cellular behaviors such as proliferation and differentia-tion. [ 14–16,19,20,23 ] Here, we developed novel tissue engineering materials with gene delivery functions through the hybridiza-tion of M13 bacteriophage (phage) and AAV. We engineered the M13 phage with RGD peptide on the major coat proteins. We then fused its gene with AAV gene with eukaryotic invading functional gene, ITR. The resulting M13–AAV hybrid phage exhibited enhanced internalization into target cells as compared to minor coat engineered phage with RGD or wild-type phage. The resulting hybrid phage formed nanofi brous matrices that could support the cellular growth and deliver desired gene information into the target tissues. Our novel M13–AAV hybrid phage matrices can provide selective, stable, and safe gene delivery ( Figure 1 ). We believe that it can be also developed as a topical therapeutic tissue patch in the future.
In order to construct a novel phage tissue engineering mate-rial with gene delivery function ( Figure 2 ), we constructed the hybrid phage (M13 RGD8 –AAV GFP ) carrying both RGD major coat
Reprogramming of cellular functions through gene delivery is critical in treating cellular/tissue abnormalities as well as in studying the mechanism of cellular behavior of many cells. [ 1 ] Many researchers have utilized nanosized natural [ 2 ] or syn-thetic materials such as surfactants, [ 3 ] peptide or protein nano-spheres, [ 4 ] and polymersomes for gene delivery. [ 5 ] Recently, virus-based gene delivery vehicles, such as retrovirus or adeno-virus, are considered as promising delivery vehicles for transfer of genetic information into target cells. [ 6 ] Among them, adeno-associated virus (AAV) has been largely adapted for therapeutic gene transfer due to its innocuousness and high resistance to extreme conditions. [ 7 ] The AAV is a nonpathogenic virus with a linear single-stranded DNA genome that contains eukaryotic gene integrating functional genes, named inverted terminal repeats (ITRs; 145 bp in length for AAV2), [ 8 ] at each end of the AAV viral genome terminus. The ITR possesses a T-shaped hairpin structure with self-complementary guanine-cyto-sine rich sequences; the gene enables integration of the viral genome into as well as rescue from a specifi c section of the host genome (19th chromosome in humans). [ 9 ] Despite the develop-ment of various viral gene delivery vehicles, their therapeutic application still remains challenging due to several reasons. (1) Viruses can hardly reach their desired targets. Viruses have lim-ited ranges of the type of host cells they can infect, although adenoviruses and AAV are able to infect a relatively broad range of cells effi ciently. Still, some cell types are unmanageable to
Adv. Healthcare Mater. 2015, DOI: 10.1002/adhm.201500179
www.advhealthmat.dewww.MaterialsViews.com

© 2015 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheimwileyonlinelibrary.com2
CO
MM
UN
ICATI
ON
engineered M13 phage (M13 RGD8 ) gene and ITR-fl anked cas-sette containing the human recombinant green fl uorescent pro-tein (GFP) gene using recombinant DNA technology. Previously, this similar approach was developed for biomedical imaging. [ 24 ]
Briefl y, M13 phage was engineered to display the RGD pep-tide on pVIII major coat protein (M13 RGD8 ) by inverted poly-merase chain reaction (PCR) cloning method. [ 15,20,25,26 ] Human recombinant GFP (hrGFP) in AAV-hrGFP (stratagene) was
Adv. Healthcare Mater. 2015, DOI: 10.1002/adhm.201500179
www.advhealthmat.dewww.MaterialsViews.com
Figure 1. Schematic illustration of our approach to construct M13 RGD8 –AAV GFP hybrid phage and its application in tissue engineering with gene delivery functions. A) We genetically engineered M13 phage and AAV hybrid virus. Constructed hybrid virus (M13 RGD8 –AAV GFP ) has longer DNA resulting longer phage (1.4 µm) than M13 RGD8 (880 nm). B) The resulting hybrid phage could form tissue-like nanofi brous structures. The resulting phage-tissue-like materials can transduce desired genetic information to express GFP proteins.
Figure 2. Constructing M13 RGD8 –AAV GFP hybrid phage. A) We used genetically engineered M13 phage modifi ed with RGD on its major coat protein (M13 RGD8 ) as backbone and introduced AAV ITR cassette harboring ITR-CMV-eGFP-ITR into the M13 phage gene. B) After construction, we obtained the mixture of the M13 RGD8 –AAV GFP and M13 RGD8 . After ultracentrifugation, we obtained purifi ed hybrid phage (M13 RGD8 –AAV GFP ). C) AFM image showed that the constructed phage have ≈1400 nm in length. D) The resulting hybrid phage could form nanofi brous phage fi lms. Scale bars in C and D are 1 µm.

© 2015 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim wileyonlinelibrary.com 3
CO
MM
UN
ICATIO
N
replaced with enhanced GFP (eGFP), and ITR cassette (ITR–CMV–eGFP–ITR) regions were cut by PvuII sites ( ≈3.4 kb) and inserted into M13 RGD8 gene backbone (≈7.1 kb) (Figure 2 A). The sequence of cloned hybrid phage vector was confi rmed by DNA sequencing using sequencing primers listed in Table S1 (Supporting Information). The combination of M13 (≈7.1 kb) and ITR cassette from the AAV (≈3.4 kb) resulted in a longer hybrid ssDNA (M13 RGD8 –AAV GFP phage; ≈10.5 kb, Figure 2 B) (detailed procedures are available in the Experimental Sec-tion). Therefore, the resulting hybrid phage exhibited longer phage than wild-type phage. [ 27 ] Atomic force microscopy (AFM) images of the constructed hybrid phage show that the M13 RGD8 –AAV GFP phage exhibited ≈1400 nm phage (compared with the M13 RGD8 (≈880 nm) (Figure 2 C). Due to the elongated nanofi brous structure of the phage, we can easily fabricate self-assembled phage matrix structures by making liquid crystalline phage fi lms. [ 19,20,23,25,28 ] The AFM image of the drop-cast fi lms show that the engineered phage could form fi brous matrices (Figure 2 D).
The phage engineered with RGD-peptides on major coat protein (pVIII) exhibited enhanced RGD-induced endocy-tosis to the target cells. Previously, we characterized the tissue engineering functions of the major coat engineered phage which exhibited good biocompatibility and cell viability. [ 15,25 ] Through the chemical and biological cue control, the phage-based tissue engineering materials could guide the directional growth of the desired cells. [ 15,23 ] During this investigation, we also found that many of the phages were internalized into the target cells that we utilized (mouse neural progenitor cells and MC3T3 fi broblasts) [ 16,22,25 ] with little effect on their cell viability. Because the effect of the phage coat protein engi-neering on the internalization process to the target cells has been rarely characterized previously, we characterized phage coat engineering effect on internalization processes here by quantifying bound phage on target cell surfaces and subse-quent internalized phage to the target cells. First, we coated
the engineered phage on substrates to form tissue scaffold-like structures (Figure 2 D). The target cells (1 000 cells well −1 ) were then applied onto engineered-phage-coated surface to deter-mine the phage binding on the target cells, their internaliza-tion, and subsequent gene expression. [ 29 ] In order to compare the internalization effi ciency, we characterize uptakes of M13 phage modifi ed with RGD peptide on the pVIII major coat pro-tein (M13 RGD8 ) or on the pIII minor coat protein (M13 RGD3 ). We also used wild-type M13 phage without any modifi cation (M13 WT ) as a control ( Table 1 ). We used HeLa cells, which are known to express large numbers of integrin proteins on the cell surface, and used MC3T3 cells to test possible topical gene delivery application. Using fl uorescent microscopy techniques after the immunostaining, we quantifi ed and visualized the phage after phage treatment (see details in the Experimental Section). We also quantifi ed the amount of the phage bound on the target cell surfaces and internalized phage through the phage washing assays followed by the phage titering in HeLa ( Figure 3 ) and MC3T3 cells (Figure S2, Supporting Informa-tion). The major coat engineered phage (the M13 RGD8 ) with the RGD peptide exhibited enhanced uptake into target eukar-yotic cells and the internalization effi ciency of the phage was dependent on the copy number of the displayed RGD peptide. The fl uorescent images and their quantifi cation (Figure 3 A,B) showed that the major coat engineered phage with the RGD peptide (M13 RGD8 ; red) were internalized more in HeLa cells
Adv. Healthcare Mater. 2015, DOI: 10.1002/adhm.201500179
www.advhealthmat.dewww.MaterialsViews.com
Table 1. Different phage types used in this study.
Name PVIII PIII
M13 RGD8 A- DSG RGD TE -DP SHS-AET
M13 RGD3 A-EGD-DP SHS- ACG RGD SCGGGS -AET
M13 WT A-EGD-DP SHS-AET
Peptide inserts are shown in italic.
Figure 3. Cellular uptake of engineered phage according to different coat protein modifi cations. A) Immunofl uorescent staining results after phage treatment. Cells were incubated with different engineered phages, M13 RGD8 versus M13 RGD3 (10 11 pfu well −1 ) in cell culture media for 16 h at 37 °C. An acid wash was done with a low pH elution buffer. Permeabilization with Triton-X can lead phage antibodies inside cells to stain internalized phages. Then, only internalized phages were successfully stained. (red: phage, green: actin phalloidin, blue: DAPI; Scale bar = 100 µm). B) Relative internaliza-tion was calculated with phage stained area of (A) with Image J software. The graphs show relative internalization according to different coat modifi ca-tions. It shows that the pVIII-modifi ed phage (M13 RGD8 ) can be internalized into cells more than the pIII modifi cation (M13 RGD3 ) or no modifi cation (M13 WT ). C) After cell culture, membrane bound phages were taken after elution with an acid buffer (“elution”), then internalized phages were taken from cell lysates (“lysis”). The phages from each fraction were titrated. (* p < 0.01, t -test, and n = 3)
© 2015 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheimwileyonlinelibrary.com4
CO
MM
UN
ICATI
ON
than those of the M13 RGD3 (Figure 3 A). The M13 RGD3 also exhibited better internalization than the M13 WT . The MC3T3 cells also exhibited a similar internalization pattern (Figure S2, Supporting Information). We quantifi ed number of phages bound on the target cell surfaces through phage titering after acid washing elution of the bound phage. We also quanti-fi ed the titer of the internalized phage after lysis of the target cells (detailed procedures are available in the Experimental Section). [ 29,30 ] The phage quantifi cation assays from the cell surface bound phage showed that the major coat engineered phage (M13 RGD8 ) could bind to the cellular membrane better than the minor coat engineered phage (M13 RGD3 ) (Figure 3 C). We believe that the major coat engineered phage (M13 RGD8 ) possessed more proteins (2700 copies) than that of the minor coat engineered phage (M13 RGD3 ; fi ve copies). We could also observe more phages were internalized on the major coat engineered phage than that of the minor coat. These trends are similar both in the HeLa cells and the MC3T3 cells (Figure 3 C and Figure S2C, Supporting Information). However, due to the large number of the integrin expressed on the HeLa cell surfaces, we could see more phage are bound and internalized on the HeLa cells than that of the MC3T3 cells.
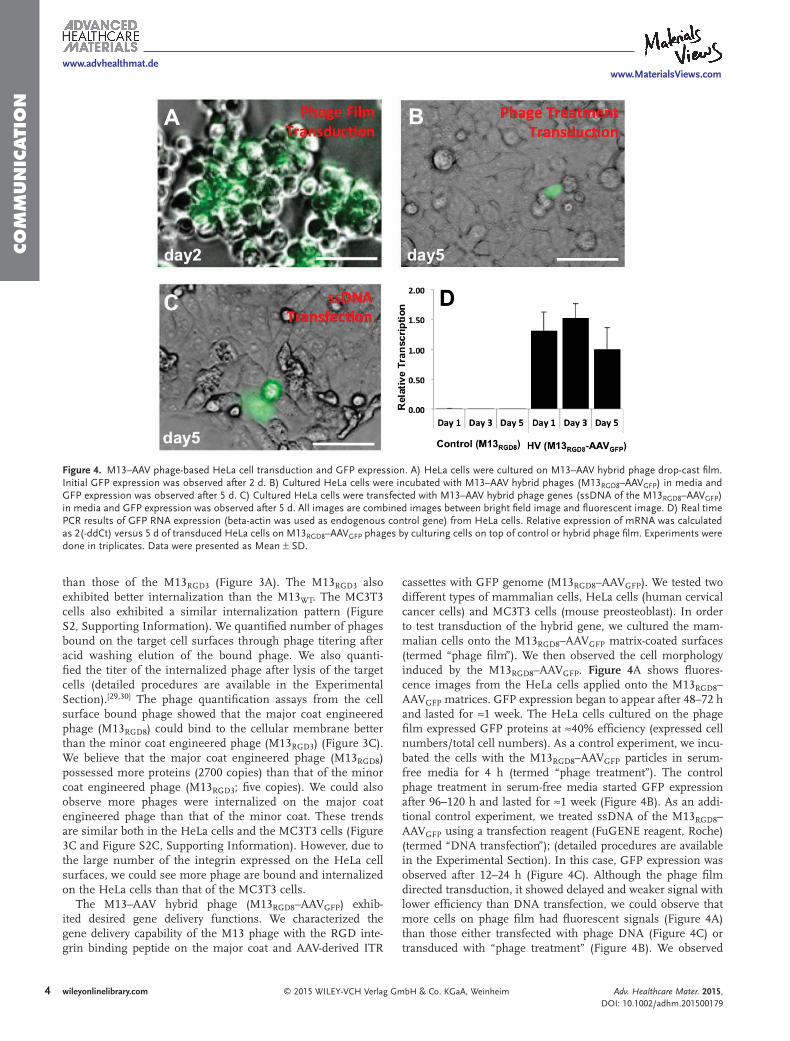
The M13–AAV hybrid phage (M13 RGD8 –AAV GFP ) exhib-ited desired gene delivery functions. We characterized the gene delivery capability of the M13 phage with the RGD inte-grin binding peptide on the major coat and AAV-derived ITR
cassettes with GFP genome (M13 RGD8 –AAV GFP ). We tested two different types of mammalian cells, HeLa cells (human cervical cancer cells) and MC3T3 cells (mouse preosteoblast). In order to test transduction of the hybrid gene, we cultured the mam-malian cells onto the M13 RGD8 –AAV GFP matrix-coated surfaces (termed “phage fi lm”). We then observed the cell morphology induced by the M13 RGD8 –AAV GFP . Figure 4 A shows fl uores-cence images from the HeLa cells applied onto the M13 RGD8 –AAV GFP matrices. GFP expression began to appear after 48–72 h and lasted for ≈1 week. The HeLa cells cultured on the phage fi lm expressed GFP proteins at ≈40% effi ciency (expressed cell numbers/total cell numbers). As a control experiment, we incu-bated the cells with the M13 RGD8 –AAV GFP particles in serum-free media for 4 h (termed “phage treatment”). The control phage treatment in serum-free media started GFP expression after 96–120 h and lasted for ≈1 week (Figure 4 B). As an addi-tional control experiment, we treated ssDNA of the M13 RGD8 –AAV GFP using a transfection reagent (FuGENE reagent, Roche) (termed “DNA transfection”); (detailed procedures are available in the Experimental Section). In this case, GFP expression was observed after 12–24 h (Figure 4 C). Although the phage fi lm directed transduction, it showed delayed and weaker signal with lower effi ciency than DNA transfection, we could observe that more cells on phage fi lm had fl uorescent signals (Figure 4 A) than those either transfected with phage DNA (Figure 4 C) or transduced with “phage treatment” (Figure 4 B). We observed
Adv. Healthcare Mater. 2015, DOI: 10.1002/adhm.201500179
www.advhealthmat.dewww.MaterialsViews.com
Figure 4. M13–AAV phage-based HeLa cell transduction and GFP expression. A) HeLa cells were cultured on M13–AAV hybrid phage drop-cast fi lm. Initial GFP expression was observed after 2 d. B) Cultured HeLa cells were incubated with M13–AAV hybrid phages (M13 RGD8 –AAV GFP ) in media and GFP expression was observed after 5 d. C) Cultured HeLa cells were transfected with M13–AAV hybrid phage genes (ssDNA of the M13 RGD8 –AAV GFP ) in media and GFP expression was observed after 5 d. All images are combined images between bright fi eld image and fl uorescent image. D) Real time PCR results of GFP RNA expression (beta-actin was used as endogenous control gene) from HeLa cells. Relative expression of mRNA was calculated as 2(-ddCt) versus 5 d of transduced HeLa cells on M13 RGD8 –AAV GFP phages by culturing cells on top of control or hybrid phage fi lm. Experiments were done in triplicates. Data were presented as Mean ± SD.

© 2015 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim wileyonlinelibrary.com 5
CO
MM
UN
ICATIO
N
similar trends in the MC3T3 cells characterization (Figure S3, Supporting Information). When we characterized the relative gene transcription of the hybrid gene using real time (RT)-PCR of the mRNA, the results from the phage fi lm showed that the hybrid gene (M13 RGD8 –AAV GFP ) was transcribed from the fi rst day (day 1) in HeLa cell, whereas MC3T3 cells exhibited the gradual uptake in day 1 and 3, but the maximum uptakes happened on day 5 (Figure S3, Supporting Information). Trans-fection effi ciency differs between different cell lines, but trans-duction effi ciency of phage matrices varied based on receptor–ligand binding. We believe that it is because transduction occurs via a ligand–receptor-mediated mechanism as shown in Figure S2 (Supporting Information). Since RGD-peptide dis-played on M13 RGD8 –AAV GFP is specifi c to α v β 1 integrin receptor on the cell surface, [ 29,31 ] the transduction effi ciency would be more enhanced if we express target cell-specifi c ligands on phage coat proteins by genetic engineering, and if we use the specifi c dosage optimized for the target cells.
In our work, immobilized phage-based tissue engineering materials stably provide and expose the RGD-integrin binding peptide to the cells and gradually enter cells via the RGD-integrin-mediated endocytosis. The effi ciency of the resulting engineered phage can be controlled by major coat modifi ca-tion. [ 22 ] The ITR cassette released from internalized phages can be successfully inserted into the host genome and can induce transgene expression. In addition, our successful demonstration of the delivery of GFP reporter genes shows that it is possible to deliver therapeutic genes using a similar method demonstrated here. For example, the tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) gene, an apoptotic gene showing signifi cant results against tumor cells, can be easily integrated into our modularly modifi able phage vector. [ 32 ] Therefore, delivering TRAIL to specifi c cancer cells could be very effi cient when combined with the specifi city of the RGD motif (the αv-integrin overexpressed in tumor cells). Furthermore, as we previously demonstrated, the M13 phage has the ability to serve as a nanomaterial to direct cellular growth and differentiation. [ 15,20,23,25 ] If this feature is com-bined with the ability of the phage to deliver specifi c genes or proteins, it could lead to the production of multifunctional nanomaterials.
In summary, we developed novel M13–AAV hybrid phage tissue engineering matrices that can deliver programmed gene information through the combination of RGD-peptide engi-neering on major coat protein together with AAV-derived ITR gene cassette. The M13 phage system serves as a safe and con-venient backbone to support tissue growth and the ITR cassette stably delivers the target gene, and thus induces programmed protein expression. Selective delivery was achieved via integrin-mediated endocytosis through the RGD-peptide displayed on the phage major coat protein. Phage engineered with high den-sity of RGD-peptides on major coat protein exhibited higher internalization compared with that of minor coat engineered phage or wild-type phage. The resulting engineered M13–AAV hybrid phage fi lm was successfully able to induce GFP expression into mammalian cells. We believe that our M13–AAV hybrid phage matrix materials can be further applied for various biomedical applications including tropical therapeutic patches development in the future.
Experimental Section
Genetic Engineering and Purifi cation of M13 Bacteriophage : M13 phage was engineered to display the RGD peptide motif on pVIII or pIII protein by inverted PCR cloning method and a site-specifi c mutagenesis on PvuII site was performed to generate mRGD8 (M13 RGD8 ) using QuickChange site-directed mutagenesis kit as described in the manual (Figure S1A, Supporting Information). hrGFP in AAV-hrGFP (Stratagene) was replaced with eGFP and ITR cassette (ITR–CMV–eGFP–ITR) regions were cut with PvuII and inserted into M13 RGD8 backbone. R8-AAV-eGFP hybrid phages (M13 RGD8 –AAV GFP ) were fi nally generated. After genetic engineering, the successful construction of the target gene with ≈10.5 kb band using agarose gel electrophoresis was confi rmed. The mixture of M13 RGD8 (yellow arrow) and M13 RGD8 –AAV GFP (red arrow) (Figure 2 B) was found. M13 RGD8 –AAV GFP was obtained by CsCl gradient ultracentrifuge purifi cation. The ssDNA was extracted from a white band purifi ed by ultracentrifuge and loaded onto alkaline-denatured agarose gel, and it was confi rmed that the purifi ed bands were constructed hybrid phage (Figure 2 B).
Cell Culture : HeLa, human cervical cancer cells, were obtained from the cell facility at the University of California, Berkeley (Berkeley, CA). MC3T3, a nontransformed cell line established from newborn mouse calvaria and exhibits an osteoblastic phenotype, was purchased from ATCC (CRL-2593, Manassas, VA). HeLa cells were cultured in DMEM media (Invitrogen, Carlsbad, CA) with 10% heat-inactivated fetal bovine serum (FBS) and 1% antibiotics–antimicotics (AB–AM) at 37 °C, 5% CO 2, and 95% humidity. MC3T3 cells were cultured in α-MEM media with 10% heat-inactivated FBS and 1% AB–AM at 37 °C, 5% CO 2 , and 95% humidity. The growth media was changed every 2 d, and the base population of cells was passaged when near confl uency.
Atomic Force Microscopy (AFM) Analysis : Aminated surfaces were prepared to coat the phages. To characterize the length of the individual phage, 500 µL of phage solution (500 ng mL −1 ) were used. The phage deposited on the substrates was dried in a desiccator prior to analysis. The topographic images of the surfaces were acquired in air using AFM (MFP-3D AFM, Asylum Research, Santa Barbara, CA) operating on tapping mode with a silicon cantilever (AC240TS, Olympus, Tokyo, Japan).
Phage Drop-Cast Film : Phage-coated surfaces were initially coated with polyornithine to allow for better adhesion of the negatively charged phage. [ 33 ] Phage solutions in PBS (phosphate buffered saline; ≈10 12 pfu mL −1 ) were then drop-cast onto the surface and allowed to dry overnight at room temperature (30 µL well −1 , 8 well chamber slide) to form the nanofi brous phage tissue network structures. In parallel, phage solution was drop-cast onto gold substrate to analyze the nanofi brous structures on phage fi lm by AFM.
Double-Stranded DNA (dsDNA) and Single-Stranded DNA (ssDNA) Extraction : dsDNA (RF DNA) was extracted from Escherichia coli using a miniprep kit (Quiagen, Valencia, CA). ssDNA was extracted from phage solution as described by NEB (Ipswich, MA). Transfection was performed with Fugene reagent (Roche, Basel, Switzerland)
Immunostaining and Fluorescence Microscopy : Before fi xation, the cells were washed with 0.1 M glycine, pH2.0, containing 1 mg mL −1 BSA. Cells were thoroughly washed with PBS and fi xed in 3.7% formaldehyde solution for 15 min at room temperature. After four washes with PBS, cells were made permeable by incubation in 0.1% Triton X-100 for 30 min. Cells were again washed with PBS. All samples were blocked in 5% normal goat serum in PBS for 30 min. To detect internalized phages, samples were incubated with rabbit anti-fd (Sigma Aldrich, St. Louis, MO) primary antibody at 1:500 dilution/PBST(phosphate buffered saline-Tween20) and washed with PBST three times. Samples were then incubated with Alexa fl uorochrome-conjugated goat antirabbit antibody (Molecular Probes, Eugene, OR) at 1:500 dilution in PBST and washed three times with PBST. Finally, the actin fi laments and nuclei were stained with Alexa 488 phalloidin and DAPI (4',6-diamidino-2-phenylindole), respectively, as counterstaining steps. The fl uorescence images were collected using an IX71 Fluorescence Microscope (Olympus, Tokyo, Japan). Images were analyzed using NIH ImageJ (NIH, http://rsb.info.nih.gov/ij/).
Adv. Healthcare Mater. 2015, DOI: 10.1002/adhm.201500179
www.advhealthmat.dewww.MaterialsViews.com

© 2015 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheimwileyonlinelibrary.com6
CO
MM
UN
ICATI
ON
Adv. Healthcare Mater. 2015, DOI: 10.1002/adhm.201500179
www.advhealthmat.dewww.MaterialsViews.com
Quantitative Real Time Polymerase Chain Reaction (qRT-PCR) : qRT-PCR was conducted on iQ5 RT-PCR using total RNA extracted and individual primer sets for enhanced GFP (eGFP) and β-actin (see Table S1, Supporting Information) with iScript One-Step RT-PCR Kit (Bio-Rad, Hercules, CA). Data for all genes were characterized by the cycle threshold (Ct), defi ned as the fractional number of cycles at which the reporter fl uorescent emission reached a fi xed threshold level in the exponential region of the amplifi cation plot. Melting curves were plotted at the end of each experiment, with all products demonstrating one predominant peak. Relative expression (vs 5 d culture on HV phages) was calculated by the Δ C t method normalized to β-actin (where C t is threshold cycle).
Supporting Information Supporting Information is available from the Wiley Online Library or from the author.
Acknowledgements S.Y.Y. and H.-E.J. contributed equally to this work. The authors would like to thank Professor David Schaffer for generously gifting eGFP cDNA for cloning. This work was supported by the Hellman Family Faculty Fund (SWL); start-up funds from the Berkeley Nanoscience and Nanoengineering Institute at the University of California, Berkeley (SWL); the Laboratory Directed Research and Development fund from the Lawrence Berkeley National Laboratory; and Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT and Future Planning (Grant No. 2013R1A1A3008484) and the Korean Government (Grant No. NRF-2014S1A2A2027641).
Received: March 13, 2015 Published online:
[1] a) L. Shao , W.-S. Wu , Expert Opin. Biol. Ther. 2010 , 10 , 231 ; b) M. Ginsberg , D. James , B.-S. Ding , D. Nolan , F. Geng , Jason M. Butler , W. Schachterle , Venkat R. Pulijaal , S. Mathew , Stephen T. Chasen , J. Xiang , Z. Rosenwaks , K. Shido , O. Elemento , Sina Y. Rabbany , S. Rafi i , Cell 2012 , 151 , 559 ; c) D.-Q. Tang , L. Shun , V. Koya , Y. Sun , Q. Wang , H. Wang , S.-W. Li , Y. Sun , D. L. Purich , C. Zhang , B. Hansen , K. Qian , M. Atkinson , M. I. Phillips , L.-J. Yang , Am. J. Transl. Res. 2013 , 5 , 184 ; d) Y. Ikada , J. R. Soc. Interface 2006 , 3 , 589 .
[2] J. M. Dang , K. W. Leong , Adv. Drug Delivery Rev. 2006 , 58 , 487 . [3] a) M. Donkuru , I. Badea , S. Wettig , R. Verrall , M. Elsabahy ,
M. Foldvari , Nanomedicine 2010 , 5 , 1103 ; b) G. Candiani , D. Pezzoli , L. Ciani , R. Chiesa , S. Ristori , PLoS One 2010 , 5 , e13430 .
[4] a) S. Kommareddy , M. M. Amiji , Cold Spring Harb. Protoc. 2008 , 2008 , pdb.top30; b) K. W. Leong , H. Q. Mao , V. L. Truong-Le , K. Roy , S. M. Walsh , J. T. August , J. Controlled Release 1998 , 53 , 183 .
[5] a) J. H. Jeong , S. W. Kim , T. G. Park , Prog. Polym. Sci. 2007 , 32 , 1239 ; b) T. Niidome , L. Huang , Gene Ther. 2002 , 9 , 1647 .
[6] a) R. G. Hawley , F. H. Lieu , A. Z. Fong , T. S. Hawley , Gene Ther. 1994 , 1 , 136 ; b) T. Friedmann , R. Roblin , Science 1972 , 175 , 949 ; c) M. Ali , N. R. Lemoine , C. J. Ring , Gene Ther. 1994 , 1 , 367 .
[7] a) R. W. Atchison , B. C. Casto , W. M. Hammon , Science 1965 , 149 , 754 ; b) M. Mezzina , O.-W. Merten , 2011 , 737 , 211;
c) J. Grieger , R. Samulski , in, Gene Therapy and Gene Delivery Systems , Vol., 99 (Eds: D. Schaffer , W. Zhou ), Springer , Berlin 2005 , p. 119 .
[8] a) F. J. Koczot , B. J. Carter , C. F. Garon , J. A. Rose , Proc. Natl. Acad. Sci. U.S.A. 1973 , 70 , 215 ; b) E. Lusby , K. H. Fife , K. I. Berns , J. Virol. 1980 , 34 , 402 .
[9] X.-S. Wang , S. Ponnazhagan , A. Srivastava , J. Mol. Biol. 1995 , 250 , 573 .
[10] a) J. Zabner , M. Seiler , R. Walters , R. M. Kotin , W. Fulgeras , B. L. Davidson , J. A. Chiorini , J. Virol. 2000 , 74 , 3852 ; b) K. Hueffer , C. R. Parrish , Curr. Opin. Microbiol. 2003 , 6 , 392 .
[11] J. Savulescu , J. Med. Ethics 2001 , 27 , 148 . [12] N.-B. Woods , V. Bottero , M. Schmidt , C. von Kalle , I. M. Verma ,
Nature 2006 , 440 , 1123 . [13] a) C. R. Merril , D. Scholl , S. L. Adhya , Cold Spring Harb. 2003 , 2 ,
489 ; b) J. Milstien , J. Walker , J. Petricciani , Science 1977 , 197 , 469 . [14] S. Y. Yoo , A. Merzlyak , S.-W. Lee , Mediators Infl ammation 2014 , 11 ,
192790 . [15] S. Y. Yoo , A. Merzlyak , S.-W. Lee , Soft Matter 2011 , 7 , 1660 . [16] S. R. Bhattarai , S. Y. Yoo , S.-W. Lee , D. Dean , Biomaterials 2012 , 33 ,
5166 . [17] C. M. Hammers , J. R. Stanley , J. Invest. Dermatol. 2014 , 134 , e17 . [18] H. E. Jin , R. Farr , S. W. Lee , Biomaterials 2014 , 35 , 9236 . [19] S. Y. Yoo , M. Kobayashi , P. P. Lee , S.-W. Lee , Biomacromolecules
2011 , 12 , 987 . [20] S. Y. Yoo , W.-J. Chung , T. H. Kim , M. Le , S.-W. Lee , Soft Matter 2011 ,
7 , 363 . [21] a) S. Y. Yoo , J.-W. Oh , S.-W. Lee , Langmuir , 2011 , 28 , 2166 ;
b) W.-J. Chung , D.-Y. Lee , S. Y. Yoo , Int. J. Nanomed. 2014 , 9 , 5825 .
[22] D. S. Choi , H.-E. Jin , S. Y. Yoo , S.-W. Lee , Bioconjugate Chem. 2013 , 25 , 216 .
[23] W.-J. Chung , A. Merzlyak , S. Y. Yoo , S.-W. Lee , Langmuir 2010 , 26 , 9885 .
[24] A. Hajitou , M. Trepel , C. E. Lilley , S. Soghomonyan , M. M. Alauddin , F. C. Marini III , B. H. Restel , M. G. Ozawa , C. A. Moya , R. Rangel , Y. Sun , K. Zaoui , M. Schmidt , C. von Kalle , M. D. Weitzman , J. G. Gelovani , R. Pasqualini , W. Arap , Cell 2006 , 125 , 385 .
[25] A. Merzlyak , S. Indrakanti , S.-W. Lee , Nano Lett. 2009 , 9 , 846 . [26] J. W. Kehoe , B. K. Kay , Chem. Rev. 2005 , 105 , 4056 . [27] a) D. A. Marvin , Curr. Opin. Structural Biol. 1998 , 8 , 150 ;
b) K. R. Purdy , S. Fraden , Phys. Rev. E 2004 , 70 , 061703 . [28] a) W.-J. Chung , J.-W. Oh , K. Kwak , B. Y. Lee , J. Meyer , E. Wang ,
A. Hexemer , S.-W. Lee , Nature 2011 , 478 , 364 ; b) C. Y. Xu , R. Inai , M. Kotaki , S. Ramakrishna , Biomaterials 2004 , 25 , 877 .
[29] S. L. Hart , A. M. Knight , R. P. Harbottle , A. Mistry , H. D. Hunger , D. F. Cutler , R. Williamson , C. Coutelle , J. Biol. Chem. 1994 , 269 , 12468 .
[30] V. V. Ivanenkov , F. Felici , A. G. Menon , Biochim. Biophys. Acta – Mol. Cell Res. 1999 , 1448 , 450 .
[31] R. P. Harbottle , R. G. Cooper , S. L. Hart , A. Ladhoff , T. McKay , A. M. Knight , E. Wagner , A. D. Miller , C. Coutelle , Human Gene Ther. 1998 , 9 , 1037 .
[32] a) W. Guo , H. Zhu , L. Zhang , J. Davis , F. Teraishi , J. A. Roth , C. Stephens , J. Fueyo , H. Jiang , C. Conrad , B. Fang , Cancer Gene Ther. 2005 , 13 , 82 ; b) R. A. Daniels , H. Turley , F. C. Kimberley , X. S. Liu , J. Mongkolsapaya , P. Ch’en , X. N. Xu , B. Jin , F. Pezzella , G. R. Screaton , Cell Res 2005 , 15 , 430 ; c) A. Mohr , G. Henderson , L. Dudus , I. Herr , T. Kuerschner , K.-M. Debatin , H. Weiher , K. Fisher , R. Zwacka , Gene Ther. 2004 , 11 , 534 ; d) J. Lee , M. Hampl , P. Albert , H. A. Fine , Neoplasia 2002 , 4 , 312 .
[33] A. P. Krueger , R. C. Ritter , S. P. Smith , J. Exp. Med. 1929 , 50 , 739 .




















