
Loss of the VHR dual-specific phosphatase causescell-cycle arrest and senescence
14
LETTERS 524 NATURE CELL BIOLOGY VOLUME 8 | NUMBER 5 | MAY 2006 Loss of the VHR dual-specific phosphatase causes cell-cycle arrest and senescence Souad Rahmouni 1,2 , Fabio Cerignoli 1 , Andres Alonso 1,3 , Toshiya Tsutji 1 , Rachel Henkens 2 , Changjun Zhu 1 , Christine Louis-dit-Sully 1 , Michel Moutschen 2 , Wei Jiang 1 and Tomas Mustelin 1,4 Protein tyrosine phosphatases regulate important processes in eukaryotic cells and have critical functions in many human diseases including diabetes to cancer 1–3 . Here, we report that the human Vaccinia H1-related (VHR) dual-specific protein tyrosine phosphatase regulates cell-cycle progression and is itself modulated during the cell cycle. Using RNA interference (RNAi), we demonstrate that cells lacking VHR arrest at the G1–S and G2–M transitions of the cell cycle and show the initial signs of senescence, such as flattening, spreading, appearance of autophagosomes, β-galactosidase staining and decreased telomerase activity. In agreement with this notion, cells lacking VHR were found to upregulate p21 Cip–Waf1 , whereas they downregulated the expression of genes for cell- cycle regulators, DNA replication, transcription and mRNA processing. Loss of VHR also caused a several-fold increase in serum-induced activation of its substrates, the mitogen- activated protein (MAP) kinases Jnk and Erk. VHR-induced cell-cycle arrest was dependent on this hyperactivation of Jnk and Erk, and was reversed by Jnk and Erk inhibition or knock-down. We conclude that VHR is required for cell-cycle progression as it modulates MAP kinase activation in a cell- cycle phase-dependent manner. The MAP kinases Erk, Jnk, and p38 are activated by dual phosphor- ylation of a tyrosine and a threonine residue in the consensus motif Thr–X–Tyr (where X is Glu, Gly or Pro) in their activation loops. More than a dozen phosphatases specifically function to dephosphorylate these residues 4 . It has been assumed that there is a great deal of redun- dancy within this group, largely because MKP1 –/– mice were perfectly healthy and their MAP kinase responses were identical to controls 5 . One of these MAP kinase phosphatases (MKPs) is the 185-amino acid long VHR 6 , which was cloned based on its homology with the first identified dual-specific phosphatase, the Vaccinia virus H1 open reading-frame 7 . Although VHR can dephosphorylate both Erk and Jnk in vivo 8–10 , it dif- fers from the other ‘typical’ MKPs (suh as PAC-1, MKP1, MKP2, MKP3, MKP4, MKP5, MKP7, hVH3, hVH5, PYST2 and MK-STYX) in that it lacks the MAP kinase-binding CDC25-homology motif found in the other MKPs 4 . Instead, VHR is a considerably smaller enzyme (with a molecular weight (M r ) of 21 K) and consists of only a catalytic domain with no recognized targeting or docking sequences. In this respect, VHR is a member of the ‘atypical’ dual-specificity phosphatase group 1 , which consists of 19 small enzymes, most of them less than 250 amino- acid residues long and some of them more related to the CDC14 cell- cycle phosphatases than to the MKPs 11 . Unlike many MKPs, VHR is not acutely upregulated in response to activation of MAP kinases 9 . For these reasons, the physiological contribution of VHR to MAP kinase regulation in cells, which invariably also express several other MKPs, has remained unclear. Here, we report that VHR is instead regulated during the cell cycle. VHR was barely detectable in cells synchronized in G1 with a double thymidine block, but then gradually increased during the progression of the cells through S phase (Fig. 1a, b). The expression of VHR peaked at 10 h when most cells went through the first round of cell division (Fig. 1c). This was followed by a decline through the next G1 phase and another increase during the following S phase. VHR was most abundant in mitotic cells (Fig. 1a and see Supplementary Information, Fig. S1). Although VHR was mostly localized to the nucleas in interphase cells, it became dispersed throughout the cell during prometaphase and then concentrated around the chromatin during metaphase and between the daughter chromatids in telophase (Fig. 1d). A substantial portion of VHR was observed in the contractile ring, ended up in the mid- body and was readily detectable in purified midbody preparations (see Supplementary Information, Fig. S1). Immunoblots of synchronized cells treated for 30 min with the protein synthesis inhibitor cyclohex- imide and probe with anti-VHR antibodies revealed that the half-life of VHR was much shortened (<30 min) in G1 compared with the other phases of the cell cycle (Fig. 1e). To determine whether the biphasic regulation of VHR levels during the cell cycle (which was also seen in COS, U2OS, 293T, and primary T cells; see Supplementary Information, Fig. S2) is indicative of an active 1 The Burnham Institute for Medical Research, 10901 North Torrey Pines Road, La Jolla, CA 92037, USA. 2 University of Liège, B35, Avenue de l’Hôpital, 3, 4000 Liège 1, Belgium. 3 Instituto de Biologia y Genetica Molecular, C/Sanz y Forés, s/n, 47003, Valladolid , Spain. 4 Correspondence should be addressed to T.M. (r-mail: [email protected]) Received 16 September 2005; accepted 16 March 2006; published on line 9 April 2006; corrected after print 1 June 2006; DOI: 10.1038/ncb1398 © 2006 Nature Publishing Group
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Loss of the VHR dual-specific phosphatase causescell-cycle arrest and senescence

L E T T E R S
524 NATURE CELL BIOLOGY VOLUME 8 | NUMBER 5 | MAY 2006
Loss of the VHR dual-specific phosphatase causescell-cycle arrest and senescenceSouad Rahmouni1,2, Fabio Cerignoli1, Andres Alonso1,3, Toshiya Tsutji1, Rachel Henkens2, Changjun Zhu1, Christine Louis-dit-Sully1, Michel Moutschen2, Wei Jiang1 and Tomas Mustelin1,4
Protein tyrosine phosphatases regulate important processes in eukaryotic cells and have critical functions in many human diseases including diabetes to cancer1–3. Here, we report that the human Vaccinia H1-related (VHR) dual-specific protein tyrosine phosphatase regulates cell-cycle progression and is itself modulated during the cell cycle. Using RNA interference (RNAi), we demonstrate that cells lacking VHR arrest at the G1–S and G2–M transitions of the cell cycle and show the initial signs of senescence, such as flattening, spreading, appearance of autophagosomes, β-galactosidase staining and decreased telomerase activity. In agreement with this notion, cells lacking VHR were found to upregulate p21Cip–Waf1, whereas they downregulated the expression of genes for cell-cycle regulators, DNA replication, transcription and mRNA processing. Loss of VHR also caused a several-fold increase in serum-induced activation of its substrates, the mitogen-activated protein (MAP) kinases Jnk and Erk. VHR-induced cell-cycle arrest was dependent on this hyperactivation of Jnk and Erk, and was reversed by Jnk and Erk inhibition or knock-down. We conclude that VHR is required for cell-cycle progression as it modulates MAP kinase activation in a cell-cycle phase-dependent manner.
The MAP kinases Erk, Jnk, and p38 are activated by dual phosphor-ylation of a tyrosine and a threonine residue in the consensus motif Thr–X–Tyr (where X is Glu, Gly or Pro) in their activation loops. More than a dozen phosphatases specifically function to dephosphorylate these residues4. It has been assumed that there is a great deal of redun-dancy within this group, largely because MKP1–/– mice were perfectly healthy and their MAP kinase responses were identical to controls5. One of these MAP kinase phosphatases (MKPs) is the 185-amino acid long VHR6, which was cloned based on its homology with the first identified dual-specific phosphatase, the Vaccinia virus H1 open reading-frame7. Although VHR can dephosphorylate both Erk and Jnk in vivo8–10, it dif-fers from the other ‘typical’ MKPs (suh as PAC-1, MKP1, MKP2, MKP3,
MKP4, MKP5, MKP7, hVH3, hVH5, PYST2 and MK-STYX) in that it lacks the MAP kinase-binding CDC25-homology motif found in the other MKPs4. Instead, VHR is a considerably smaller enzyme (with a molecular weight (Mr ) of 21 K) and consists of only a catalytic domain with no recognized targeting or docking sequences. In this respect, VHR is a member of the ‘atypical’ dual-specificity phosphatase group1, which consists of 19 small enzymes, most of them less than 250 amino-acid residues long and some of them more related to the CDC14 cell-cycle phosphatases than to the MKPs11. Unlike many MKPs, VHR is not acutely upregulated in response to activation of MAP kinases9. For these reasons, the physiological contribution of VHR to MAP kinase regulation in cells, which invariably also express several other MKPs, has remained unclear.
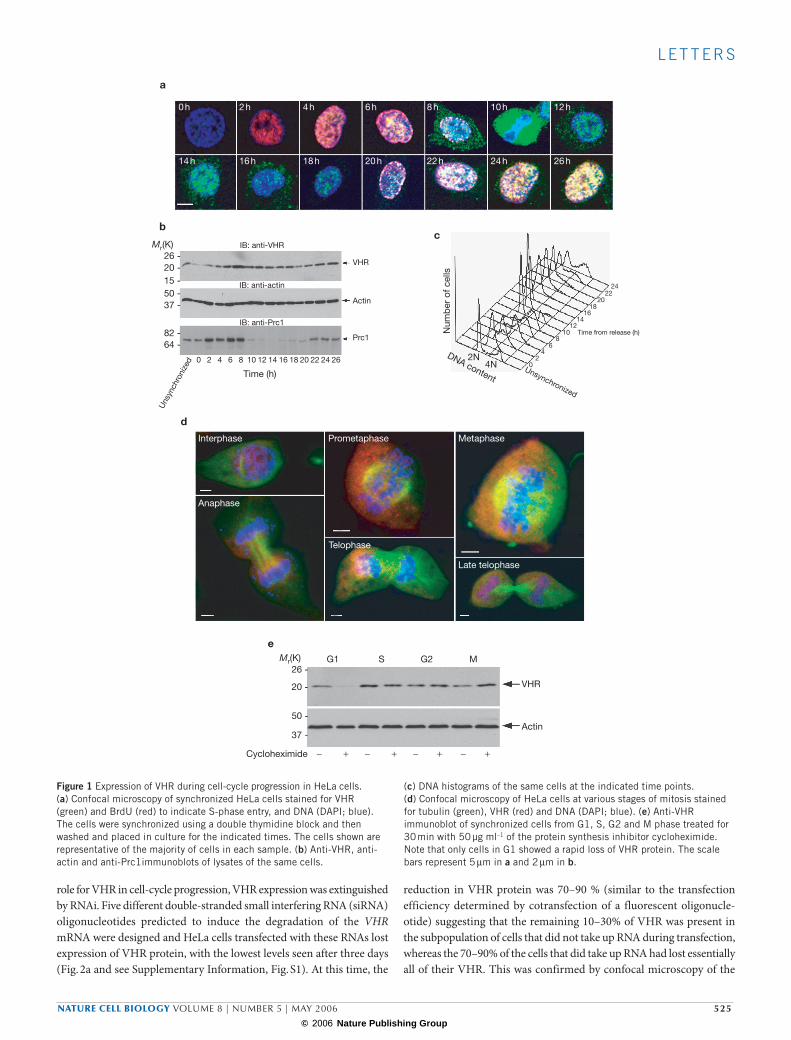
Here, we report that VHR is instead regulated during the cell cycle. VHR was barely detectable in cells synchronized in G1 with a double thymidine block, but then gradually increased during the progression of the cells through S phase (Fig. 1a, b). The expression of VHR peaked at 10 h when most cells went through the first round of cell division (Fig. 1c). This was followed by a decline through the next G1 phase and another increase during the following S phase. VHR was most abundant in mitotic cells (Fig. 1a and see Supplementary Information, Fig. S1). Although VHR was mostly localized to the nucleas in interphase cells, it became dispersed throughout the cell during prometaphase and then concentrated around the chromatin during metaphase and between the daughter chromatids in telophase (Fig. 1d). A substantial portion of VHR was observed in the contractile ring, ended up in the mid-body and was readily detectable in purified midbody preparations (see Supplementary Information, Fig. S1). Immunoblots of synchronized cells treated for 30 min with the protein synthesis inhibitor cyclohex-imide and probe with anti-VHR antibodies revealed that the half-life of VHR was much shortened (<30 min) in G1 compared with the other phases of the cell cycle (Fig. 1e).
To determine whether the biphasic regulation of VHR levels during the cell cycle (which was also seen in COS, U2OS, 293T, and primary T cells; see Supplementary Information, Fig. S2) is indicative of an active
1The Burnham Institute for Medical Research, 10901 North Torrey Pines Road, La Jolla, CA 92037, USA. 2University of Liège, B35, Avenue de l’Hôpital, 3, 4000 Liège 1, Belgium. 3Instituto de Biologia y Genetica Molecular, C/Sanz y Forés, s/n, 47003, Valladolid , Spain.4Correspondence should be addressed to T.M. (r-mail: [email protected])
Received 16 September 2005; accepted 16 March 2006; published on line 9 April 2006; corrected after print 1 June 2006; DOI: 10.1038/ncb1398
© 2006 Nature Publishing Group
NATURE CELL BIOLOGY VOLUME 8 | NUMBER 5 | MAY 2006 525
L E T T E R S
role for VHR in cell-cycle progression, VHR expression was extinguished by RNAi. Five different double-stranded small interfering RNA (siRNA) oligonucleotides predicted to induce the degradation of the VHR mRNA were designed and HeLa cells transfected with these RNAs lost expression of VHR protein, with the lowest levels seen after three days (Fig. 2a and see Supplementary Information, Fig. S1). At this time, the
reduction in VHR protein was 70–90 % (similar to the transfection efficiency determined by cotransfection of a fluorescent oligonucle-otide) suggesting that the remaining 10–30% of VHR was present in the subpopulation of cells that did not take up RNA during transfection, whereas the 70–90% of the cells that did take up RNA had lost essentially all of their VHR. This was confirmed by confocal microscopy of the
a
b
d
e
c
0 h 2 h 4 h 6 h 8 h 10 h 12 h
14 h 16 h 18 h 20 h 22 h 24 h 26 h
IB: anti-VHR
IB: anti-actin
IB: anti-Prc1
VHR
Actin
Prc1
26 -20 -15 -50 -37 -
82 -64 -
0 2 4 6 8 10 12 14 16 18 20 22 24 26
Uns
ynch
roni
zed
Num
ber
of c
ells
2N4N
DNA contentUnsynchronized
02
46
810
1214
1618
2022
24
Time from release (h)
Interphase
Anaphase
Telophase
Prometaphase Metaphase
Late telophase
G1 G2S M
VHR
Actin
26 -
20 -
50 -
37 -
Cycloheximide − + − + − + − +
Mr(K)
Mr(K)
Time (h)
Figure 1 Expression of VHR during cell-cycle progression in HeLa cells. (a) Confocal microscopy of synchronized HeLa cells stained for VHR (green) and BrdU (red) to indicate S-phase entry, and DNA (DAPI; blue). The cells were synchronized using a double thymidine block and then washed and placed in culture for the indicated times. The cells shown are representative of the majority of cells in each sample. (b) Anti-VHR, anti-actin and anti-Prc1immunoblots of lysates of the same cells.
(c) DNA histograms of the same cells at the indicated time points. (d) Confocal microscopy of HeLa cells at various stages of mitosis stained for tubulin (green), VHR (red) and DNA (DAPI; blue). (e) Anti-VHR immunoblot of synchronized cells from G1, S, G2 and M phase treated for 30 min with 50 µg ml–1 of the protein synthesis inhibitor cycloheximide. Note that only cells in G1 showed a rapid loss of VHR protein. The scale bars represent 5 µm in a and 2 µm in b.
© 2006 Nature Publishing Group

526 NATURE CELL BIOLOGY VOLUME 8 | NUMBER 5 | MAY 2006
L E T T E R S
transfected cell populations (Fig. 3). All five siRNAs reduced the expres-sion of VHR and gave very similar results in subsequent experiments (see Supplementary Information, Figs S1 and S2).
HeLa cells normally double in number in approximately 24 h, but the cells treated with VHR siRNA ceased to increase in number and remained at a constant level from the fourth day (Fig. 2b and see Supplementary Information, Fig. S1). This observation was confirmed in tens of experi-ments and with all siRNAs. In contrast, MKP1 siRNA did not affect cell-number doubling time (see Supplementary Information, Fig. S3). Light microscopy also revealed that the loss of VHR was accompanied by mor-phological changes compatible with cell-cycle arrest (Fig. 2c) and much less thymidine was incorporated (Fig. 2d). The cells became flat, large and had more filopodia than control cells. Many cells stained positive for β-galac-tosidase (Fig. 2e) three days after transfection, and even more strongly after
5 or 8 days. As the morphological changes and β-galactosidase staining are indicative of the beginning cell senescence12, telomerase activity was also measured and shown to decrease after 3–5 days of VHR siRNA treatment (Fig. 2f) in a manner that correlated with loss of the VHR protein (Fig. 2g). Although this modest, but reproducible, decrease in telomerase activity supports the notion of cell-cycle arrest and the beginning senescence, it is unlikely to be responsible for these changes. Electron microscopy revealed that most VHR siRNA-treated cells contained increased numbers of membrane-enclosed autolysosomal or autophagosomal, multivesicular and sometimes ‘onion skin-like’ vacuoles (see Supplementary Information, Fig. S3). Control cells occasionally contained these structures, but never more than 1–2 per ultrathin section, whereas VHR siRNA-treated cells often had 5–10 per section. All these changes are compatible with cell-cycle arrest and the beginning senescence in the cells lacking VHR.
a
d e f
g
h
b c
Mr(K)
Mr(K)
Mr(K)
IB: anti-VHR
IB: anti-actin
26 -20 -15 -
50 -37 -
VHR
Actin
Days 0 1 2 3 4 5 0 1 2 3 4 5Control siRNA VHR siRNA
1600
1400
1200
1000
800
600
400
200
16000
14000
12000
10000
8000
6000
4000
2000
0
0
Control siRNAVHR siRNA
0 3(Day)
Control siRNA VHR siRNA3 H
-thy
mid
ine
inco
rpor
atio
n Control siRNA
VHR siRNA
Control siRNA
VHR siRNA
2
1.8
1.6
1.4
1.2
1
0.8
0.6
0.4
0.2
0
Telomerase activity
+ RN
Ase
+ RN
Ase
+ RN
Ase
0 100 300
VHR siRNA (nM)
26 -
20 -
15 -
50 -
37 -
1 2 3
0 100 300
IB: anti-VHR
IB: anti-actin
VHR
Actin
Cel
l num
ber
(tho
usan
ds)
Ab
sorb
ance
(450
−690
nm
)
VHR siRNA (nM)
Figure 2 Loss of VHR results in altered morphology and beginning cell senescence. (a) Anti-VHR and anti-actin immunoblots of lysates of HeLa cells transfected with a control luciferase siRNA or VHR siRNA and cultured for the indicated number of days. (b) Number of cells at the start of the experiment (200,000) and on day 3 for HeLa cells transfected with a control luciferase siRNA (black bars) or with VHR siRNA (white bars). (c) Light microscopy of HeLa cells transfected with a control luciferase siRNA or VHR siRNA and cultured for three days. (d) Thymidine incorporation by HeLa cells transfected with a control
luciferase siRNA or with VHR siRNA. The data represent the mean ± sd. (n = 3). (e) β-galactosidase staining of HeLa cells transfected with a control luciferase siRNA or VHR siRNA and cultured for three days. Note the increased number of blue-staining granules in many cells. (f) Telomerase activity of cells treated with the indicated concentrations of VHR siRNA. Each sample was analysed in triplicate, with RNAse added to a fourth sample. (g) Anti-VHR immunoblot of the same cells as in f. (h) Anti-actin control immunoblot of the same cells as in f. The scale bars represent 10 µm in c and 25 µm in e.
© 2006 Nature Publishing Group

NATURE CELL BIOLOGY VOLUME 8 | NUMBER 5 | MAY 2006 527
L E T T E R S
Cell-cycle phase analysis showed a loss of cells in S phase, an increased proportion of cells in G1 phase and an unchanged percentage of cells in G2 and M phases (Fig. 3a). There was no change in the very low number of cells with a sub-2N (apoptotic) DNA content. The lack of S-phase entry in VHR siRNA-treated cells was verified by a 24 h bromo-deoxyuridine
(BrdU) labelling, which showed that only 30% of the VHR siRNA-treated cells had gone through S phase during this time, compared with >96% of control cells (Fig. 3b, c and see Supplementary Information, Fig. S3). This percentage is similar to the fraction of cells that retain detectable VHR expression (Fig. 3b) and were BrdU-positive (albeit often weaker
a
e
f
g
b c
d
Control siRNA
VHR siRNA
G1
G2−MS
80 120
0
20
40
60
80
100
14
024
86
1012
70
60
50
40
30
20
10
0Cel
l-cy
cle
pha
se d
istr
ibut
ion
0
0 1 2 3 4 5
3 4 3 4
1400
1200
1000
800
600
400
200
0
(Day)
Cel
l num
ber
(tho
usan
ds)
n = 48
n = 103
n = 116
n = 60
ControlsiRNA
VHRsiRNA
ControlsiRNA
VHRsiRNA
Brd
U-p
ositi
ve c
ells
(per
cent
age)
Cel
ls in
M p
hase
(per
cent
age)
Control siRNA VHR siRNA
Anti-VHR
Anti-VHR
Anti-phosphoH3
DAPI
Merge
Anti-VHR
Anti-BrdU Anti-BrdU
DAPI DAPI
Merge Merge
Mr(K)
Mr(K)
Mr(K) Mr(K)
20 -
50 - 50 -
50 -50 -
26 -
26 - 26 -
26 -
50 -
37 -
VHR
Actin
Cdc2
Cdk2
Cdk4
Cdk6
Con
trol s
iRN
AVH
R s
iRN
A
Con
trol s
iRN
A
Con
trol s
iRN
A
VHR
siR
NA
VHR
siR
NA
#1VH
R s
iRN
A #2
KCTD
5 si
RN
A
Con
trol s
iRN
AVH
R s
iRN
A
1 2 3 4 5 6 7 8
p21Cip1−WAF126 -
20 -15 -
(Day)
Control siRNA VHR siRNA
Figure 3 Loss of VHR results in absence of cells in S phase and M phase. (a) Cell-cycle distribution and cell number count of cells transfected with control or VHR siRNA and observed for 4–5 days. The cells were stained with propidium iodide for DNA content and analysed by FACS after the indicated number of days in culture. (b) Confocal microscopy of the same HeLa cells stained for VHR (green) and BrdU (red) to indicate S-phase entry, and DNA (DAPI, blue). Control siRNA-treated cells and cells treated with VHR are shown. The bottom panels represent merges of the 3 images above. (c) Quantification of the BrdU positive cells in b. The indicated numbers of cells were counted and the data are given as percentage of BrdU positive cells of this total number. (d) Quantification of cells staining for phosphoH3.
The indicated number of cells were counted and the data are given as percentage of phosphoH3-positive cells of this total number. (e) Confocal microscopy of control or VHR siRNA-transfected HeLa cells stained for VHR (green) and phospho H3 (red) to indicate M-phase entry, and DNA (DAPI, blue). The bottom panels represent merges of the images above. (f) Immunoblots of total lysates of control siRNA transfected cells and VHR siRNA cells with the indicated antibodies. Note that VHR was downregulated by approximately 80%, wheras actin levels were unchanged. (g) Immunoblot with anti-p21Cip1–WAF1 of total lysates of cells transfected with control siRNA (lanes 1–2), two different VHR siRNAs cells (lanes 3–6) or another control siRNA (lanes 7–8). The scale bars represent 10 µm in b and e.
© 2006 Nature Publishing Group

528 NATURE CELL BIOLOGY VOLUME 8 | NUMBER 5 | MAY 2006
L E T T E R S
than controls). Thus, loss of VHR correlated very strongly with loss of entry to S phase.
Very similar results were obtained with an antibody against phos-phohistone 3 (phosphoH3), an M phase marker (Fig. 3d, e). Although control cells behaved as expected with fluctuating VHR levels and 12% of the cells in M phase, the VHR knock-down cells were mostly nega-tive for VHR and had a much reduced content of phosphoH3 (only 4% of cells stained with this antibody). Furthermore, the cells that con-tained phosphoH3 were invariably among those that also contained some VHR. Live-cell imaging also showed that VHR siRNA-treated cells, in contrast with control cells, did not divide during a 72 h-period (data not shown). Taken together, these data indicate that loss of VHR prevents cell division.
To assess the consequences of VHR loss by another approach, RNA was isolated from HeLa cells treated for 3 days with control siRNAs or VHR siRNA, and three independent comparative cDNA expression analysis experiments were performed using Affymetrix DNA arrays.
These experiments showed that of the 22,000 genes analysed, some 200 genes were downregulated and some 100 genes upregulated more than twofold in each experiment (see Supplementary Information, Table SI). Very similar data were also obtained using two different VHR siRNAs and the Illumina instrument for expression analysis. In agreement with the cell-cycle arrest, senescence13 and the morphological changes seen by light and electron microscopy, the most upregulated gene was p21Cip–Waf1 (see Supplementary Information, Table SI; and 3.29–4.42-fold with the Illumina instrument), whereas the downregulated genes included cell-cycle regulators (CDK2, CDC2, cyclins A2 and E2), general transcription factors, RNA polymerase I and II, DNA replication machinery and DNA repair proteins. Many of the same changes in gene expression were seen in U2OS cells, although the changes tended to be slightly smaller due to lower transfection efficiencies for these cells.
To verify that the DNA array results also translate into meaningful changes in protein expression, lysates of the VHR siRNA-treated cells were immunoblotted with antibodies for cyclins, cyclin-dependent kinases and other regulators of cell-cycle progression (Fig. 3f, g and see Supplementary Information, Fig. S2). Loss of VHR caused a decrease in CDC2, CDK2 and CDK4 expression, as previously reported for cells entering senescence14, whereas the level of p21 was increased (Fig. 3g). As a control for specificity, CDC2 levels were found to be reduced in cells transfected with any of the five VHR siRNA oligos (see Supplementary Information, Fig. S2). The kinase activities of CDC2, CDK2 and CDK4 were also much reduced (see Supplementary Information, Fig. S2), largely reflecting their reduced expression. Together with the expression profiling, these results confirmed that loss of VHR caused a profound cell-cycle arrest, which correlates with p21 upregulation and deficient production and activation of critical CDKs.
Only two substrates have been identified for VHR: the MAP kinases Erk8–10 and Jnk9,10. Furthermore, stimulation of VHR siRNA-treated HeLa cells (Fig. 4) with serum caused a much stronger activation of Erk and Jnk than in control RNA-treated cells. Although the time-courses of Erk and Jnk activation were not significantly altered, the peak activi-ties of both kinases were much higher in the absence of VHR and there was a clear elevation of the basal activity of Erk. The augmented Jnk activation is also notable as serum normally has only a minor effect on this kinase in HeLa cells. Activation of Mek, the kinase upstream of Erk, was similar in cells with or without VHR, whereas activation of Rsk, a kinase at least partly downstream of Erk, was elevated and prolonged. These results demonstrate, for the first time, that endogenous VHR sig-nificantly contributes to the dephosphorylation of Erk and Jnk following serum stimulation of cells.
Although MAP kinase activation is generally thought to have a growth-promoting role during the G1 phase of the cell cycle (for exam-ple in signalling pathways initiated by growth factor receptors) there are numerous reports that constitutive or prolonged activation of the Ras–Raf–Mek–Erk pathway promotes cell-cycle arrest and cell senes-cence15–17. Many growth-arresting stimuli (such as phorbol esters18, DNA damage19 or leukemia inhibitory factor20) also induce an obligatory, and often prolonged, Erk2 activation21 and elevated Erk2 activity has been shown to directly block the G2–M phase transition and exit from M phase22. Similarly, Jnk has been implicated in cell-cycle checkpoint acti-vation in response to growth inhibitory stimuli23,24. Jnk activation also leads to p53 activation and induction of p21Cip–Waf1 and Jnk has been shown to be physically associated with these two proteins25.
a
b
c
d
e
Control siRNA VHR siRNA
26 -
20 -
15 -
61 -
50 -
37 -
61 -
50 -
37 -
61 -
50 -
37 -
61 -
115 -
80 -
61 -
115 -
80 -
61 -
50 -
37 -
61 -
50 -
37 -
61 -50 -
37 -
61 -
50 -
37 -
50 -
37 -
50 -
37 -
61 -
50 -
37 -
26 -
20 -
15 -
IB: anti-VHR
IB: anti-phosphoERK
IB: anti-phosphoJnk
IB: anti-phosphoMek
IB: anti-phospho-Rsk IB: anti-phospho-Rsk
IB: anti-phosphoMek
IB: anti-phosphoJnk
IB: anti-ERK
IB: anti-Mek IB: anti-Mek
IB: anti-ERK
IB: anti-phosphoERK
IB: anti-VHR
Serum (min) Serum (min)
Mr(K) Mr(K)
0 1 5 15 30 60 0 1 5 15 30 60
Figure 4 Loss of VHR augments activation of Erk and Jnk. (a) Anti-VHR immunoblots of the same cell lysates used for assessment of MAP kinase activation in b–e. The cells were stimulated with 10% serum for 0, 1, 5, 15, 30 and 60 min, as indicated at the bottom of the figure. Equal amounts of total protein were loaded into each lane. (b–e) Immunoblots of the same lysates with the indicated antibodies for assement of MAP kinase activation.
© 2006 Nature Publishing Group
NATURE CELL BIOLOGY VOLUME 8 | NUMBER 5 | MAY 2006 529
L E T T E R S
Based on these reports, we asked whether loss of VHR leads to growth arrest because MAP kinases are not properly downregulated in the absence of VHR and therefore exert some of their growth-inhibitory actions. Perhaps the purpose of the biphasic VHR expression during cell-cycle progression is to allow full MAP kinase activation only in early G1 phase and then to suppress MAP kinase activation with increas-ing potency during S, G2 and M phases to prevent the growth-arrest-ing effects of MAP kinases. Interestingly, phosphoErk is more readily detected in cells during G1 phase (T.M., unpublished observations). To directly test whether constitutively elevated Erk and/or Jnk activ-ity caused cell-cycle arrest in our system, constitutively active Mek1 (Mek1-DD) was expressed in HeLa cells. They cells became flattened, spread out and stopped proliferating within 24–48 h of transfection (see Supplementary Information, Fig. S4). These cells were mostly in G2 phase, supporting the notion that constitutively elevated Erk activity arrests HeLa cells in this phase22.
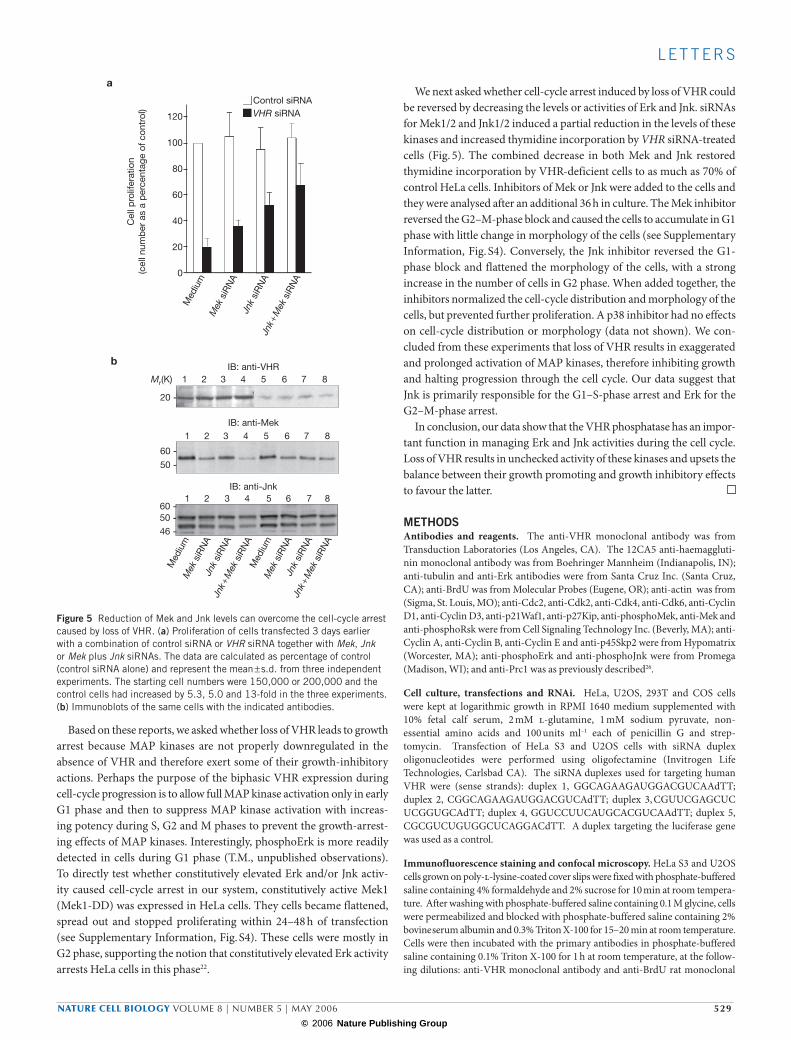
We next asked whether cell-cycle arrest induced by loss of VHR could be reversed by decreasing the levels or activities of Erk and Jnk. siRNAs for Mek1/2 and Jnk1/2 induced a partial reduction in the levels of these kinases and increased thymidine incorporation by VHR siRNA-treated cells (Fig. 5). The combined decrease in both Mek and Jnk restored thymidine incorporation by VHR-deficient cells to as much as 70% of control HeLa cells. Inhibitors of Mek or Jnk were added to the cells and they were analysed after an additional 36 h in culture. The Mek inhibitor reversed the G2–M-phase block and caused the cells to accumulate in G1 phase with little change in morphology of the cells (see Supplementary Information, Fig. S4). Conversely, the Jnk inhibitor reversed the G1-phase block and flattened the morphology of the cells, with a strong increase in the number of cells in G2 phase. When added together, the inhibitors normalized the cell-cycle distribution and morphology of the cells, but prevented further proliferation. A p38 inhibitor had no effects on cell-cycle distribution or morphology (data not shown). We con-cluded from these experiments that loss of VHR results in exaggerated and prolonged activation of MAP kinases, therefore inhibiting growth and halting progression through the cell cycle. Our data suggest that Jnk is primarily responsible for the G1–S-phase arrest and Erk for the G2–M-phase arrest.
In conclusion, our data show that the VHR phosphatase has an impor-tant function in managing Erk and Jnk activities during the cell cycle. Loss of VHR results in unchecked activity of these kinases and upsets the balance between their growth promoting and growth inhibitory effects to favour the latter.
METHODSAntibodies and reagents. The anti-VHR monoclonal antibody was from Transduction Laboratories (Los Angeles, CA). The 12CA5 anti-haemaggluti-nin monoclonal antibody was from Boehringer Mannheim (Indianapolis, IN); anti-tubulin and anti-Erk antibodies were from Santa Cruz Inc. (Santa Cruz, CA); anti-BrdU was from Molecular Probes (Eugene, OR); anti-actin was from (Sigma, St. Louis, MO); anti-Cdc2, anti-Cdk2, anti-Cdk4, anti-Cdk6, anti-Cyclin D1, anti-Cyclin D3, anti-p21Waf1, anti-p27Kip, anti-phosphoMek, anti-Mek and anti-phosphoRsk were from Cell Signaling Technology Inc. (Beverly, MA); anti-Cyclin A, anti-Cyclin B, anti-Cyclin E and anti-p45Skp2 were from Hypomatrix (Worcester, MA); anti-phosphoErk and anti-phosphoJnk were from Promega (Madison, WI); and anti-Prc1 was as previously described26.
Cell culture, transfections and RNAi. HeLa, U2OS, 293T and COS cells were kept at logarithmic growth in RPMI 1640 medium supplemented with 10% fetal calf serum, 2 mM l-glutamine, 1 mM sodium pyruvate, non-essential amino acids and 100 units ml–1 each of penicillin G and strep-tomycin. Transfection of HeLa S3 and U2OS cells with siRNA duplex oligonucleotides were performed using oligofectamine (Invitrogen Life Technologies, Carlsbad CA). The siRNA duplexes used for targeting human VHR were (sense strands): duplex 1, GGCAGAAGAUGGACGUCAAdTT; duplex 2, CGGCAGAAGAUGGACGUCAdTT; duplex 3, CGUUCGAGCUCUCGGUGCAdTT; duplex 4, GGUCCUUCAUGCACGUCAAdTT; duplex 5, CGCGUCUGUGGCUCAGGACdTT. A duplex targeting the luciferase gene was used as a control.
Immunofluorescence staining and confocal microscopy. HeLa S3 and U2OS cells grown on poly-l-lysine-coated cover slips were fixed with phosphate-buffered saline containing 4% formaldehyde and 2% sucrose for 10 min at room tempera-ture. After washing with phosphate-buffered saline containing 0.1 M glycine, cells were permeabilized and blocked with phosphate-buffered saline containing 2% bovine serum albumin and 0.3% Triton X-100 for 15–20 min at room temperature. Cells were then incubated with the primary antibodies in phosphate-buffered saline containing 0.1% Triton X-100 for 1 h at room temperature, at the follow-ing dilutions: anti-VHR monoclonal antibody and anti-BrdU rat monoclonal
Control siRNAVHR siRNA120
100
80
60
40
20
0
Med
ium
Mek
siR
NA
Jnk
siR
NA
Jnk +
Mek
siR
NA
a
Cel
l pro
lifer
atio
n(c
ell n
umb
er a
s a
per
cent
age
of c
ontr
ol)
Med
ium
Mek
siR
NA
Jnk
siR
NA
Jnk +
Mek
siR
NA
Med
ium
Mek
siR
NA
Jnk
siR
NA
Jnk +
Mek
siR
NA
b
Mr(K)
20 -
60 -
60 -50 -
46 -
50 -
1 2 3 4 5 6 7 8
1 2 3 4 5 6 7 8
1 2 3 4 5 6 7 8
IB: anti-VHR
IB: anti-Mek
IB: anti-Jnk
Figure 5 Reduction of Mek and Jnk levels can overcome the cell-cycle arrest caused by loss of VHR. (a) Proliferation of cells transfected 3 days earlier with a combination of control siRNA or VHR siRNA together with Mek, Jnk or Mek plus Jnk siRNAs. The data are calculated as percentage of control (control siRNA alone) and represent the mean ± s.d. from three independent experiments. The starting cell numbers were 150,000 or 200,000 and the control cells had increased by 5.3, 5.0 and 13-fold in the three experiments. (b) Immunoblots of the same cells with the indicated antibodies.
© 2006 Nature Publishing Group

530 NATURE CELL BIOLOGY VOLUME 8 | NUMBER 5 | MAY 2006
L E T T E R S
antibody, 1:250; anti-Prc1 polyclonal antibody and anti-tubulin polyclonal anti-body, 1:1,000; anti-phosphoH3 polyclonal antibody, 1:10,000. Primary antibod-ies were visualized using rhodamine-conjugated anti-rat antibody, Alexa-488 or Alexa-594 conjugated anti-mouse or anti-rabbit antibody, and Cy5-conjugated anti-goat antibody (Molecular Probes). DNA was counterstained with 4,6-diamid-ino-2-phenylindole (DAPI; Molecular Probes). The coverslips were then mounted using the slow-fade mounting medium (Molecular Probes) and cells were viewed under a multiphoton confocal microscope (BioRad, Hercules, CA).
Cell synchronization. HeLa S3 cells were plated on poly-l-lysine coated coverslips in Dulbecco’s Modified Eagle’s medium with 10% fetal bovine serum and 100 U penicillin–streptomycin. Cells were arrested in S phase by the addition of 2 mM thy-midine for 17–18 h, released from the arrest for 9 h and then arrested a second time with thymidine. After 18 h, the cells were again released and followed for 0–26 h. To obtain populations of cells in mitosis, cells were arrested in 2 mM thymidine for 18 h, released for 8 h and blocked in 40 µg ml–1 nocodazole for 12 h. Floating mitotic cells were collected, washed twice in phosphate-buffered saline, replated on 100 mm dishes, and collected every 2 h thereafter. Synchrony was monitored by flow cytometry of propidium iodine-stained cells and by confocal microscopy.
BrdU incorporation and analysis of cell cycle by flow cytometry. Cells were kept in culture medium supplemented with 10 µM BrdU for 15 min (Fig. 2) or 24 h (Fig. 5) and then fixed with ice-cold 70% ethanol. After washing twice with phosphate-buffered saline, the cells were treated with 2 M HCl at 25 °C for 20 min, washed twice with phosphate-buffered saline followed by phosphate-buffered saline with 0.2% Tween-20. The cell pellet was resuspended in buffer and incu-bated with 2 µl of anti-BrdU antibody (BD Biosciences, La Jolla, CA) at 25 °C for 30 min. The cells were then washed twice with phosphate-buffered saline with 0.2% Tween-20 and incubated with 5 µl fluorescein isothiocyanate-conjugated
rabbit anti-mouse IgG antibodies (DAKO, Glostrup, Denmark) at 25 °C for 30 min. The cells were washed with phosphate-buffered saline with 0.2% Tween-20, and processed for propidium iodide staining and two-dimensional flow cytometry (FACScan flow cytometer; Becton Dickinson, San Jose, CA).
Midbody isolation from HeLa cells. Mitotic HeLa cells were obtained by a thy-midine–nocodazole block as described above. Cells synchronized in S phase with a double thymidine block were used as a negative control for the midbody purifi-cation. The cells were washed, pelleted at 200g for 3 min and gently resuspended in 1 M hexylene glycol(2-methy-2,4-pentandiol), 2 mM piperazine-N,N′-bis(2-ethane sulfonic acid) at pH 7.2, 20 µM MgCl2. Cells were immediately centrifuged at 200g for 3 min and resuspended in 1% NP-40, 1 M hexylene glycol(2-methy-2,4-pentandiol), 2 mM piperazine-N,N′-bis(2-ethane sulfonic acid) at pH 7.2, 1 mM EGTA, 1 mM Na3VO4 and 10 µg ml–1 of each leupeptin and aprotinin at 37 °°C and vigorously vortexed. Cells were then incubated on ice after adding 0.3 volumes of cold 1 M hexylene glycol(2-methy-2,4-pentandiol), 50 mM 2-(N-mopholino) ethane sulfonic acid at pH 6.3. After 10 min centrifugation at 250g, the supernatant was supplemented with 5 µM taxol, layered over a cushion of 40% glycerol (w/v) in 50 mM 2-(N-mopholino) ethane sulfonic acid at pH 6.3 and then centrifuged at 2,800g for 45 min. The pellet was resuspended in 50 mM 2-(N-mopholino) ethane sulfonic acid at pH 6.3 and centrifuged again through 40% glycerol. Finally, the pellet containing the purified midbody was washed in 50 mM 2-(N-mopholino) ethane sulfonic acid with 10 µM taxol and then used for electrophoresis or immunofluorescence staining.
β-galactosidase staining. Staining of cells for β-galactosidase was performed as previously described12 with minor modifications. siRNA transfected HeLa cells were washed in phosphate-buffered saline and fixed in 2% formalde-hyde at room temperature for 3 min. After washing, the cells were incubated overnight at 37 °C (without CO2) in a freshly prepared solution of 1 mg ml–1 5-bromo-4-chloro-3-indolyl β-d-galactoside in 40 mM citric acid at pH 6.0, 150 mM sodium phosphate, 5 mM potassium ferrocyanide, 5 mM potassium ferricyanide, 150 mM NaCl and 2 mM MgCl2. The cells were then visualized in bright field at ×40–100 magnification.
RNA isolation, cDNA synthesis and microarray hybridization. RNA was prepared from HeLa or U2OS cells using TRIzol (Invitrogen) and reverse-tran-scribed into Cy5–dUTP and Cy3–dUTP labelled cDNA (Amersham Pharmacia Biotech, Piscataway NJ) as previously described27. Equal amounts of Cy5- and
Cy3-labelled cDNA were hybridized to spotted cDNA microarrays containing 22,692 elements representing approximately 16,332 different human genes and scanned in the Burnham Institute’s microarray facility.
Immunoprecipitation, SDS–PAGE and immunoblotting. These procedures were performed as previously described9,10,28,29. Briefly, cells were lysed in 20 mM Tris–HCl at pH 7.5, 150 mM NaCl, 5 mM EDTA containing 1% NP-40, 1 mM Na3VO4, 10 µg ml–1 aprotinin and leupeptin, 100 µg ml–1 soybean trypsin inhibi-tor and 1 mM phenylmethylsulphonyl fluoride, and then centrifuged at 20,000g for 20 min. The cleared lysates were preadsorbed on protein G–Sepharose and then incubated with antibody for 2 h, followed by protein G–Sepharose beads. Immune complexes were washed three times in lysis buffer, once in lysis buffer with 0.5 M NaCl, again in lysis buffer and either suspended in SDS sample buffer or used for in vitro kinase assays. Proteins resolved by SDS–PAGE were trans-ferred electrophoretically onto nitrocellulose filters, which were immunoblotted with optimal dilutions of monoclonal antibodiess, followed by anti-mouse-Ig-peroxidase. The blots were developed by enhanced chemiluminescence (ECL kit, Amersham) according to the manufacturer’s instructions.
In vitro kinase assays. Immunoprecipitated kinases were included into a 20-µl reaction containing 10 µg histone or 1 µg of GST–Retinoblastoma (Rb), 50 mM Tris–HCl at pH 7.4, 10 mM MgCl2, 1 mM EGTA, 1 mM DTT, 5 mM NaF, 5 mM βglycerophosphate, 0.05 mM Na3VO4, 0.1 mM ATP and 1 µCi of γ-32PATP. After 20 min at 30 °C, reactions were stopped by the addition of 5 µl of 5× SDS sample buffer, resolved by SDS–PAGE and exposed to film.
MAP kinase activation assays. For Erk phosphorylation, cells were serum-starved by culturing in serum-free medium for 15–18 h and then were transferred to culture medium containing 10% serum for the indicated times (1–60 min). Cell lysates were subsequently immunoblotted with antibodies against the phospho-rylated forms of Erk, Jnk, Mek and Rsk. The same filters were then stripped and immunoblotted for the total amount of each kinase.
Electron microscopy. As previously described29, cells were fixed in 2% formal-dehyde and 2% glutaraldehyde in 0.1 M cacodylate at pH 7.3 for 2 h at 4 °C. Cells were washed 3 times in this buffer, followed by a secondary fixation in 2% OsO4 in the same buffer for 1 h at room temperature. Cells were again washed 3 times in buffer. Cells were then successively dehydrated in ethanol, infused with pro-pylene oxide and embedded in Epon 812. Ultrathin sections were obtained using a Reichert-Jung microtome and then double stained with uranyl-acetate and lead citrate. Specimens were examined using a Hitachi-600 electron microscope at 75 kV with magnifications ranging from ×8,000–100,000.
Note: Supplementary Information is available on the Nature Cell Biology website.
ACKNOWLEDGMENTSThis work was supported by Rotary International, the Van Beirs Foundation, Centre Anticancéreux près L’Université de Liège, the Spanish Ministry of Education and Culture, the Plan Nacional de Salud y Farmacia (SAF 2003-05194) and the National Institutes of Health (AI35603).
COMPETING FINANCIAL INTERESTSThe authors declare that they have no competing financial interests.
Published online at http://www.nature.com/naturecellbiology/Reprints and permissions information is available online at http://npg.nature.com/reprintsandpermissions/
1. Alonso, A. et al. The set of genes encoding protein tyrosine phosphatase family members in the human genome. Cell 117, 699–720 (2004).
2. Andersen, J. N. et al. A genomic perspective on protein tyrosine phosphatases: gene structure, pseudogenes, and genetic disease linkage. FASEB J. 18, 8–13 (2004).
3. Mustelin, T., Vang, T. and Bottini, N. Protein tyrosine phosphatases and the immune response. Nature Rev. Immunol. 5, 43–57 (2005).
4. Alonso, A., Rojas, A., Godzik, A. and Mustelin, T. The dual-specific protein tyrosine phosphatase family. Top. Curr. Genet. 5, 333–358 (2003).
5. Dorfman, K. et al. Disruption of the erp/mkp-1 gene does not affect mouse develop-ment: normal MAP kinase activity in ERP/MKP-1-deficient fibroblasts. Oncogene 13, 925–931 (1996).
6. Ishibashi, T., Bottaro, D. P., Chan, A., Kiki, T. and Aaronson, S. A. Expression cloning of a human dual-specificity phosphatase. Proc. Natl Acad. Sci. USA 89, 12170–12174 (1992).
© 2006 Nature Publishing Group

NATURE CELL BIOLOGY VOLUME 8 | NUMBER 5 | MAY 2006 531
L E T T E R S
7. Guan, K. L., Bryoles, S. S. and Dixon, J. E. A Tyr/Ser protein phosphatase encoded by Vaccinia virus. Nature 350, 359–362 (1991).
8. Todd, J. L., Tanner, K. G. and Denu, J. M. Extracellular regulated kinases (ERK) 1 and ERK2 are authentic substrates for the dual-specificity protein-tyrosine phosphatase VHR. A novel role in down-regulating the ERK pathway. J. Biol. Chem. 274, 13271–13280 (1999).
9. Alonso, A., Saxena, M., Williams, S. and Mustelin, T. Inhibitory role for dual-specificity phosphatase VHR in T cell antigen receptor and CD28-induced Erk and Jnk activation. J. Biol. Chem. 276, 4766–4771 (2001).
10. Alonso, A. et al. Tyrosine phosphorylation of VHR phosphatase by ZAP-70. Nature Immunol. 4, 44–48 (2003).
11. Alonso, A. et al. The minimal essential core of a cysteine-based protein tyrosine phos-phatase revealed by a novel 16-kDa VH1-like phosphatase, VHZ. J. Biol. Chem. 279, 35768–35774 (2004).
12. Dimri, G. P. et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl Acad. Sci. USA 92, 9363–9367 (1995).
13. Kang, M. K. et al. Senescence-associated genes in normal human oral keratinocytes. Exp. Cell Res. 287, 272–281 (2003).
14. Stein, G. H., Drullinger, L. F., Robetorye, R. S., Pereira-Smith, O. M. and Smith, J. R. Senescent cells fail to express cdc2, cycA, and cycB in response to mitogen stimulation. Proc. Natl Acad. Sci. USA 88, 11012–11016 (1991).
15. Serrano, M., Lin, A. W., McCurrach, M. E., Beach, D. and Lowe, S. W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 88, 593–602 (1997).
16. Pumiglia, K. M. and Decker, S. J. Cell cycle arrest mediated by the MEK/mitogen-activated protein kinase pathway. Proc. Natl Acad. Sci. USA 94, 448–452 (1997).
17. Wang, W. et al. Sequential activation of the MEK-extracellular signal-regulated kinase and MKK3/6-p38 mitogen-activated protein kinase pathways mediates oncogenic ras-induced premature senescence. Mol. Cell Biol. 22, 3389–3403 (2002).
18. Alblas, J., Slager-Davidov, R., Steenbergh, P. H., Sussenbach, J. S. and van den Burg, B. The role of MAP kinase in TPA-mediated cell cycle arrest of human breast cancer cells. Oncogene 16, 131–139 (1998).
19. Tang, D. et al. ERK activation mediates cell cycle arrest and apoptosis after DNA dam-age independently of p53. J. Biol. Chem. 277, 12710–12717 (2002).
20. Park, J.-I., Strock, C. J., Ball, D. W. and Nelkin, B. D. The Ras/Raf/MEK/extracellular signal-regulated kinase pathway induces autocrine–paracrine growth inhibition via the leukemia inhibitory factor/JAK/STAT pathway. Mol. Cell Biol. 23, 543–554 (2003).
21. Lahlou, H. et al. sst2 somatostatin receptor inhibits cell proliferation through Ras-, Rap1-, and B-Raf-dependent ERK2 activation. J. Biol. Chem. 278, 39356–39371 (2003).
22. Chau, A. S.-S. and Shibuya, E. K. Inactivation of p42 mitogen-activated protein kinase is required for exit from M-phase after cyclin destruction. J. Biol. Chem. 274, 32085–32090 (1999).
23. Tchou, W.-W., Yie, T.-A., Tan, T.-H., Rom, W. N. and Tchou-Wong, K.-M. Role of c-Jun N-terminal kinase 1 (JNK1) in cell cycle checkpoint activated by the protease inhibitor N-acetyl-leucinyl-leucinyl-norleucinal. Oncogene 18, 6974–6980 (1999).
24. Grosch, S. et al. Activation of c-Jun-N-terminal-kinase is crucial for the induction of a cell cycle arrest in human colon carcinoma cells caused by flurbiprofen enantiomers. FASEB J. 17, 1316–1318 (2003).
25. Xue, Y., Ramaswamy, N. T., Hong, X. and Pelling, J. C. Association of JNK1 with p21waf1 and p53: modulation of JNK1 activity. Mol. Carcinogen. 36, 38–44 (2003).
26. Jiang, W. et al. PRC1: a human mitotic spindle-associated CDK substrate protein required for cytokinesis. Mol. Cell 2, 877–885 (1998).
27. Eisen, M. B. and Brown, P.O. DNA arrays for analysis of gene expression. Methods Enzymol. 303, 179–205 (1999).
28. Saxena, M., Williams, S., Taskén, K. and Mustelin, T. Crosstalk between cAMP-depend-ent kinase and MAP kinase through hematopoietic protein tyrosine phosphatase (HePTP). Nature Cell Biol. 1, 305–311 (1999).
29. Huynh, H. et al. Control of vesicle fusion by a tyrosine phosphatase. Nature Cell. Biol. 6, 831–839 (2004).
© 2006 Nature Publishing Group
© 2006 Nature Publishing Group

Supplementary Fig. S2
© 2006 Nature Publishing Group
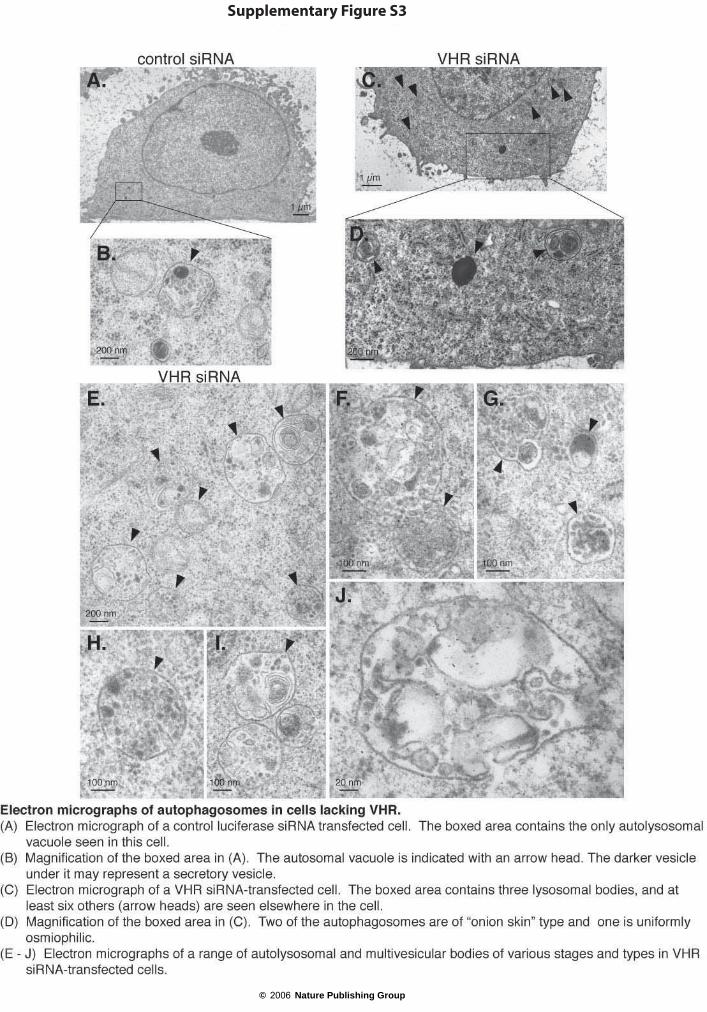
Supplementary Figure S3
© 2006 Nature Publishing Group

© 2006 Nature Publishing Group

Supplemental Table SI.
Changes in expression of genes relevant to cell cycle arrest
Downregulated ratio in HeLaa U2OSb
retinoblastoma-like 1 (p107) (NM_002895) 0.227 ± 0.156 1.009CDC2 (NM_001786) 0.248 ± 0.080 0.801cyclin E2 (NM_057749) 0.286 ± 0.049 0.502, 0.574RNA polymerase α (NM_002689) 0.367 ± 0.349 0.860PTEN (NM_000314) 0.413 ± 0.111 n.d.cyclin A2 (NM_001237) 0.443 ± 0.341 0.920, 0.940activating transcription factor 2 (NM_001880) 0.444 ± 0.182 n.d.DNA topoisomerase II α (NM_001067) 0.462 ± 0.361 0.537CDK2 (NM_001798) 0.467 ± 0.182 0.691general transcription factor IIIC, peptide 4 (NM_012204) 0.471 ± 0.229 0.453E2F transcription factor (NM_001949) 0.479 ± 0.371 0.621transcription elongation regulator 1 (CA150) (NM_006706) 0.489 ± 0.172 n.d.Mdm2, (NM_002392) 0.509 ± 0.240 1.317importin α1 (NM_002266) 0.512 ± 0.218 n.d.checkpoint suppressor 1 (NM_005197) 0.533 ± 0.252 0.594replication factor C (activator 1) (NM_002915) 0.594 ± 0.298 1.030
Upregulatedhistone 2, H2be (NM_003528) 3.180 ± 0.968 1.929MAX binding protein (NM_020310) 2.975 ± 0.106 n.d.suppression of tumorigenicity 1 (NM_005380) 2.937 ± 0.301 1.371CDKI 1A (p21, Cip1/Waf1) (NM_000389) 2.435 ± 1.502 2.196, 2.346transcription termination factor-like protein (NM_025198) 2.328 ± 1.312 n.d.MKP-1 (DUSP1) (NM_004417) 2.279 ± 0.689 0.789G-2 and S-phase expressed 1 (NM_016426) 2.145 ± 1.193 1.857CDKI3/KAP (NM_005192) 1.851 ± 0.300 n.d.__________________________________________________________________________a ratios are signal from VHR siRNA treated cells / control siRNA treated cells and values represent the mean ± SD
from 3 independent experiments.b ratios are calculated as for HeLa cells and are from a single experiment, with some values in duplicates. Note that
the transfection efficiency is lower in U2OS than in HeLa cells and that changes therefore seem to be lower.
Supplemental Table SII.
Changes in the expression of genes possibly relevant senescence
Reproducibly upregulated ratio in HeLa U2OS selenoprotein SelM (AY043487) 5.221 ± 1.054 2.062glutathione peroxidase 1 (NM_000581) 4.302 ± 0.261 1.807apolipoprotein A-I (NM_000039) 2.462 ± 0.885 1.495apolipoprotein E (NM_000041) 2.412 ± 0.532 n.d.mucolipin 1 (NM_020533) 2.346 ± 1.123 n.d.apolipoprotein B mRNA editing enzyme (NM_001644) 2.282 ± 1.687 1.260NADH dehydrogenase 1 alpha (NM_002488) 2.204 ± 1.117 1.573ceroid-lipofuscinosis (NM_000086) 2.058 ± 1.326 n.d.__________________________________________________________________________a ratios are the signal from VHR siRNA treated cells / control siRNA treated cells and values represent the mean ±
SD from 3 independent experiments.b ratios are calculated as for HeLa cells and are from a single experiment, with some values in duplicates. Note that
the transfection efficiency is lower in U2OS than in HeLa cells and that changes therefore seem to be lower.
© 2006 Nature Publishing Group

E R R AT U M
NATURE CELL BIOLOGY VOLUME 8 | NUMBER 6 | JUNE 2006 642
In the letter by Rahmouni et al. (Nature Cell Biol. 8, 524–531; 2006), the antibodies used in Fig. 5b were incorrectly labelled. This figure has been corrected online and is shown below.
Med
ium
Mek
siR
NA
Jnk
siR
NA
Jnk +
Mek
siR
NA
Med
ium
Mek
siR
NA
Jnk
siR
NA
Jnk +
Mek
siR
NA
b
Mr(K)
20 -
60 -
60 -50 -
46 -
50 -
1 2 3 4 5 6 7 8
1 2 3 4 5 6 7 8
1 2 3 4 5 6 7 8
IB: anti-VHR
IB: anti-Mek
IB: anti-Jnk
© 2006 Nature Publishing Group




















