
The Fifth Congress of the Bund - Resolutions (Russian version)

Tetrahedron: Asymmetry xxx (2011) xxx–xxx
Contents lists available at ScienceDirect
Tetrahedron: Asymmetry
journal homepage: www.elsevier .com/locate / tetasy
Lipase-catalyzed kinetic resolutions of racemic 1-(10-ethyl-10H-phenothiazin-1,2, and 4-yl)ethanols and their acetates
Jürgen Brem a, Sarolta Pilbák b, Csaba Paizs a, Gergely Bánóczi b, Florin-Dan Irimie a,Monica-Ioana Tos�a a,⇑, László Poppe b,⇑a Department of Biochemistry and Biochemical Engineering, Babes�-Bolyai University of Cluj-Napoca, Ro-400028 Cluj-Napoca, Arany János 11, Romaniab Department of Organic Chemistry and Technology and Research Group for Alkaloid Chemistry HAS, Budapest University of Technology and Economics,H-1111 Budapest, M}uegyetem rkp. 3, Hungary
a r t i c l e i n f o
Article history:Received 27 April 2011Accepted 17 May 2011Available online xxxx
0957-4166/$ - see front matter � 2011 Elsevier Ltd. Adoi:10.1016/j.tetasy.2011.05.009
⇑ Corresponding authors. Tel.: +36 1 463 3299; faxE-mail address: [email protected] (L. Poppe).
Please cite this article in press as: Brem, J.;
a b s t r a c t
The synthesis of both enantiomers of 1-(10-ethyl-10H-phenothiazin-1,2, and 4-yl)ethanols 1a–c andtheir acetates via enantioselective methanolysis of the corresponding racemic esters rac 2a–c with lipaseB from Candida antarctica (CaL-B) or/and by acylation of the racemic alcohols with the lipase A or lipase Bfrom C. antarctica (CaL-A and CaL-B) is described. The absolute configuration of enantiopure 1-(10-ethyl-10H-phenothiazin-1-yl)ethyl acetate 2a was assigned as (R) by using QM/MM(hf/3–21g:uff) calculationswithin the CaL-B (1LBT crystal structure) enzymic environment.
� 2011 Elsevier Ltd. All rights reserved.
1. Introduction
Phenothiazine is one of the most valuable templates for a largenumber of organic compounds, exhibiting a wide variety of biolog-ical activities and which are used as antihistaminic, antipsychotic,anticholinergic, antiemetic, anti-inflammatory, sedative, or cito-static drugs.1 Recently the synthesis of 2-substituted N-acylpheno-thiazines and their antibacterial and antifungal activities wasreported.2
Especially in the case of central nervous system drugs,3 a closerelationship between the chemical structure and the therapeuticeffects of phenothiazine derivatives has been demonstrated. Theinteraction of the drug-pharmacological receptors strongly de-pends on the nature, position, and number of functional groupspresent in the drug molecule.4
Due to the chiral nature of biomacromolecules which are builtby chiral units such as amino acids and carbohydrates, most ofthe biologically active compounds controlling the physiologicalfunctions of living organisms are chiral. Moreover, the stereocom-plementary recognition between chiral drugs and drug-receptorhas been demonstrated.
The increased interest in the understanding of how phenothia-zine-based drugs act, explains the necessity of the development ofnovel synthetic methods for the preparation of variously substi-tuted phenothiazines in enantiopure form.
Hydrolases are frequently used as biocatalysts for the kineticresolution of chiral organic compounds. Lipases A and B from
ll rights reserved.
: +36 1 463 3697 (L.P.).
et al. Tetrahedron: Asymmet
Candida antarctica were found to be efficient catalysts for KRand DKR,5 and also for the enantioselective resolution of variousN-alkylated phenothiazines, such as phenothiazin-3-yl-cyano-hydrins,6 -3-hydroxy-propanoic acids,7 or -1-ethanols.8 In orderto investigate how the position of the asymmetric moiety at-tached to the phenothiazine could influence the activity andselectivity of lipases, the kinetic resolution of 1-(10-ethyl-10H-phenothiazin-1-,2- and 4-yl)ethanols rac-1a–c and of their acet-ylated derivatives rac-2a–c was studied. The resulting optimumconditions for the analytical scale reactions were successfullyapplied for the preparative scale synthesis of the highlyenantiomerically enriched (R)- and (S)-1-(10-ethyl-10H-phenothiazinyl)ethanols (R)- and (S)-1a–c.
Moreover, the absolute configurations of the enantiopure prod-ucts obtained were assigned using the earlier published data forenantiomerically pure enantiomers of 1d8 and non alkylated 1b9
and by QM/MM calculations within the CaL-B10 structure for 2aand 2d.
2. Results and discussion
2.1. Chemical synthesis
As shown in Scheme 1 racemic 1-(10-ethyl-10H-phenothiazi-nyl)ethanols rac-1a–c were prepared using specific methods foreach individual compound in accordance with the literature.
As starting compounds for the syntheses of alcohols rac-1a–c,the corresponding 10-ethyl-10H-phenothiazinyl-carbaldehydes(for 1a,c) or the commercially available 1-(10H-phenothiazin-2-yl)ethanone (for 1b) were used (Scheme 1).
ry (2011), doi:10.1016/j.tetasy.2011.05.009

NH
S
N
S
N
S
N
S
O OH
NH
S
O
N
S
O
N
S
OH
NH
S
O N
S
ON
S
OH
I. II. III.
II. IV.
III.II.
1a
1b
1c
V.
Scheme 1. Synthesis of the racemic 1-(10-ethyl-10H-phenothiazinyl)ethanols rac-1a–c. Reagents and conditions: I. (a) 2.5 equiv n-BuLi,�78 �C, 1 h, rt, 48 h; (b) DMF,�78 �C,rt, 2 h; II. NaH, EtI, DMF; III. MeMgI, Et2O; IV. NaBH4, methanol; V. (a) BuLi, TMEDA, �78 �C, rt 1 h; (b) DMF, �78 �C, rt, 1 h.
2 J. Brem et al. / Tetrahedron: Asymmetry xxx (2011) xxx–xxx
The direct lithiation and subsequent formylation with DMF is afacile procedure to regioselectively prepare phenothiazine deriva-tives with the formyl group adjacent to the heteroatom.11 The di-rect metallation of phenothiazine with 2.5 equiv of n-BuLifollowed by treatment with DMF resulted in selective formylationat the 1-position. The N-alkylation of the resulting aldehyde withethyl iodide followed by Grignard reaction with methyl iodideyielded almost quantitatively racemic 1-(10-ethyl-10H-phenothia-zin-1-yl)ethanol rac–1a.
The 1-(10-ethyl-10H-phenothiazin-2-yl)ethanol rac-1b was ob-tained in high overall yield by N-alkylation of the commerciallyavailable 2-acetylphenothiazine with ethyl iodide followed by sub-sequent reduction with NaBH4 in methanol.
For the synthesis of racemic 1-(10-ethyl-10H-phenothiazin-4-yl)ethanol rac-1c, lithiation of N-ethyl phenotiazine was performedin the presence of a stoichiometric amount of tetramethylethyl-ene-diamine (TMEDA) followed by formylation with DMF.12 Theforming aldehyde was transformed into the desired alcohol rac-1c via Grignard reaction with methyl iodide.
Chemical acetylation of alcohols rac-1a–c by acetyl chloride inthe presence of triethylamine and a catalytic amount of DMAP gavethe racemic acetates rac-2a–c (Scheme 2).
2.2. Enzymatic synthesis
To investigate the stereoselectivity of the reactions involvingracemic 1-(10-alkyl-10H-phenothiazinyl)ethanols rac-1a–c and
R
OAc
R
OH+
lipase
rac-1a-c(R)-2a-c(S)-1a-c
solvent R
OH
N
SR=
OAcCH3-COCl
Et3N, DMAPCH2Cl2
Scheme 2. Biotransformation of the racemic 1-(10-ethyl-10H-ph
Please cite this article in press as: Brem, J.; et al. Tetrahedron: Asymmet
their acetates rac-2a–c, the chromatographic separation of theenantiomers was first established (Table 7). The base-line separa-tion of the enantiomers of all rac-1,2a–c was performed using var-ious enantioselective HPLC columns.
Efficient enzymatic procedures for the synthesis of enantiomeri-cally pure N-alkyl-phenothiazin-3-yl-ethanols (R)-1d and (S)-1dand their butanoates has been already described, using L-AK andCaL-B mediated acylations of racemic 1-(10-alkyl-10H-phenothia-zin-3-yl)ethanol rac-1d and by CaL-B catalyzed methanolysis of theracemic butanoate of 1d.8 Since the nature of the alkyl group attachedto the heterocyclic nitrogen exhibited no influence on the selectivityof the enzymatic reactions, we decided to investigate only the lipasecatalyzed kinetic resolution of the racemic N-ethyl substituted1-(phenothiazinyl)ethanols rac-1a–c and their acetates rac-2a–c.
2.2.1. Analytical scale biotransformations2.2.1.1. Analytical scale enzymatic acetylation of rac-1a–c. First,the analytical scale enantioselective lipase catalyzed acetylation ofracemic 1-(10-ethyl-10H-phenothiazinyl)-ethanols rac-1a–c wasstudied in neat vinyl acetate using 14 free and immobilized com-mercial lipases (listed in the Experimental parts) (Scheme 2). Mostof them exhibited low enantioselectivities and activities, or wereactive in a nonselective manner. The best results were obtainedwith lipases A and B from C. antarctica (CaL-A and CaL-B) and withthe lipase from Pseudomonas fluorescens (L-AK), which were activeand selective biocatalysts for the kinetic resolutions of rac-1a–c asshown in Table 1.
MeOH
(S)-2a-c (R)-1a-crac-2a-c
lipase
solventR
OAc
R
OAc
R
OH+,
enothiazinyl)ethanols rac-1a–c and their acetates rac-2a–c.
ry (2011), doi:10.1016/j.tetasy.2011.05.009

Table 1Acylation of rac-1a–c (0.1 M) with lipases CaL-A, CaL-B or L-AK (50 mg/mL) in neat vinyl acetate at room temperature
Substrate Enzyme Time (h) c (%) ee(R)-2a–c (%) ee(S)-1a–c (%) E
rac-1a CaL-A 14 24 97 30 77CaL-B 85 11 94 12 36L-AK 85 7 67 6 5
rac-1b CaL-A 24 50 97 99 >200CaL-B 24 49 99 99 �200L-AK 24 50 99 97 >200
rac-1c CaL-A 15 46 91 79 47CaL-B 24 31 95 42 61L-AK 40 40 94 32 46
J. Brem et al. / Tetrahedron: Asymmetry xxx (2011) xxx–xxx 3
The most efficient lipase for the acetylation of the racemic alco-hols rac-1b,c in terms of selectivity proved to be CaL-B. In the caseof rac-1a, which is the most sterically hindered secondary alcohol,lipase A from C. antarctica cross-linked with glutaraldehyde (CaL-A-CLEA) was the most active and selective (c = 24% after 14 h,E = 77, Table 1.).
Based on the crystal structure of CaL-A and molecular modeling,it has been proposed that upon substrate binding, CaL-A undergoesa conformational change whereby the active-site ‘flap’ (Gly426-Gly440) moves around the active site, widening the available spaceessentially infinite.13 This can explain why CaL-A is active andshows enantioselectivity in the O-acylation of highly sterically hin-dered secondary alcohols.
It is known that the nature of the solvent can significantly influ-ence the selectivity of this reaction.8 Therefore, the acylation ofrac-1a–c with vinyl acetate in the presence of the most appropriateenzymes (CaL-B for rac-1b,c and CaL-A for 1a) was tested in severalorganic solvents (Table 2).
While the CaL-A (CLEA form) catalyzed acylation of rac-1a withvinyl acetate as the acyl donor in polar solvents, such as dichloro-methane, chloroform, THF, or dioxane proceeded with low activity(c <5% after 110 h), the reaction was efficient in ethers such as DIIP-E, MTBE, or diethyl ether and other non-polar solvents (n-hexaneand toluene). Acetonitrile was the only polar solvent in whichCaL-A showed good activity but low stereoselectivity, is in agree-ment with our previously reported results.14 As presented inTable 2, n-hexane was found to be an appropriate reaction media(E = 55, c = 22% after 110 h) for the CaL-A mediated acetylation ofrac-1a. For rac-1b,c CaL-B was the most active and selective whenusing chloroform as the solvent (E >200 at approx 50% conversion).
As previously found for phenotiazin-3-yl ethanols,8 during theenzymatic reactions and especially during the work-up of the reac-tion mixture, a moderate to strong instability of the ethanolic sub-strates was observed. The stability decrease in the series was asfollows: rac-1a > rac-1b > rac-1d > rac-1c. This is probably the rea-son as to why the chemical hydrolysis of the obtained (R)-2a–c wasaffected when the chemical hydrolysis of these compounds wastested. Consequently, for the synthesis (R)-1a–c the lipase cata-lyzed kinetic resolution of rac-2a–c was investigated further.
2.2.1.2. Analytical scale enzymatic alcoholysis of rac-2a–c. It isknown that lipases usually retain their enantiomeric preferencein hydrolysis or alcoholysis.11 Consequently, such reactions shouldresult in the opposite enantiomeric forms of the reaction counter-parts. For obtaining the opposite enantiomers of 1,2a–c, the kineticresolution of racemic acetates rac-a–c was performed in the nextstep.
First the enzyme mediated hydrolysis of racemic acetates rac-2a–c was tested using CaL-B, which was found to be the appropri-ate biocatalyst for the enzymatic kinetic resolution of racemic1-(10-alkyl-10H-phenothiazin-3-yl)-ethyl butanoates.8 Due to the
Please cite this article in press as: Brem, J.; et al. Tetrahedron: Asymmet
poor solubility of all the ester substrates in water, various water–organic solvent mixtures were used as reaction media. For all sub-strates in all of the reaction media used, the appearance of largeamounts of side products, which decreased the global yield for(R)-1a–c, was observed. In this way the use of water as nucleophilefor the enzymatic resolution of rac-2a–c practically was ruled out.
Using the aforementioned optimal conditions for the resolutionof 1-(10-alkyl-10H-phenothiazin-3-yl)-ethyl butanoates,8 themethanolysis of acetates rac-2a–c in the presence of CaL-B in ace-tonitrile was further investigated. The rate of methanolysis wasfound to be higher than those of other alcoholyses. Moreover 1-(10-alkyl-10H-phenothiazin-3-yl)-ethanol displayed a higher sta-bility in the presence of methanol than in the presence of otheralcohols and the reaction rate was highest when methanol wasused as the nucleophile. Finally, methanol enhanced the solubilityof all substrates and products. While methanolysis occured effi-ciently for rac-2b,c under these conditions, CaL-B was totally inac-tive toward rac-2a in acetonitrile. The enzyme catalyzedmethanolysis of racemic 1-(10-ethyl-10H-phenothiazin-1-yl)ethylacetate rac-2a was studied in several organic solvents. The reactiononly took place in non-polar solvents and the best results were ob-tained in n-hexane when at 50% conversion the enantiopurity ofboth products was high (ee >99%) (Table 3).
With the aim of developing a general methodology, the meth-anolysis in the same solvent was also tested for rac-1b,c. Whilethe selectivities remained high (E >200), the reaction times de-creased significantly.
2.2.2. Preparative scale synthesis of both (R)- and (S)-1,2a–cUsing the optimal conditions found for the analytical scale
enzymatic reactions, the preparative scale enzymatic synthesis ofboth (S)- and (R)-1,2a–c was performed (Table 4). To demonstratethe usefulness of these enzymatic procedures 1 g of the substratewas used for each enzymatic kinetic resolution.
The opposite enantiomeric forms of products (S)-2a–c and (R)-1a–c can be prepared by methanolysis of the corresponding race-mic acetates rac-2a–c assisted by CaL-B. The ee for both (S)-2a–cand (R)-1a–c were >99, c = 50% and E�200 when the reaction oc-curs in hexane in the presence of 10 equiv of methanol.
2.3. The absolute configurations of (R)-1b,d
The absolute configurations of the (R)-(+)-1-(10H-phenothia-zin-2-yl)ethanol [non-ethylated analogue of (R)-1b] obtained byreduction of 2-acetyl-phenothiazine in the presence of a chiral spi-roborate ester catalyst has already been described.15 Since the signof the specific rotation proved to be independent of the nature ofthe N-substitution within a series of (10-substituted-10H-pheno-thiazin-3-yl)ethanols,8 the absolute configuration of the N-ethylat-ed 1-(10H-phenothiazin-2-yl)ethanol (+)-1b could be assigned as(R). On the other hand, in the case of (R)-(+)-1-(10-ethyl-10H-phe-
ry (2011), doi:10.1016/j.tetasy.2011.05.009
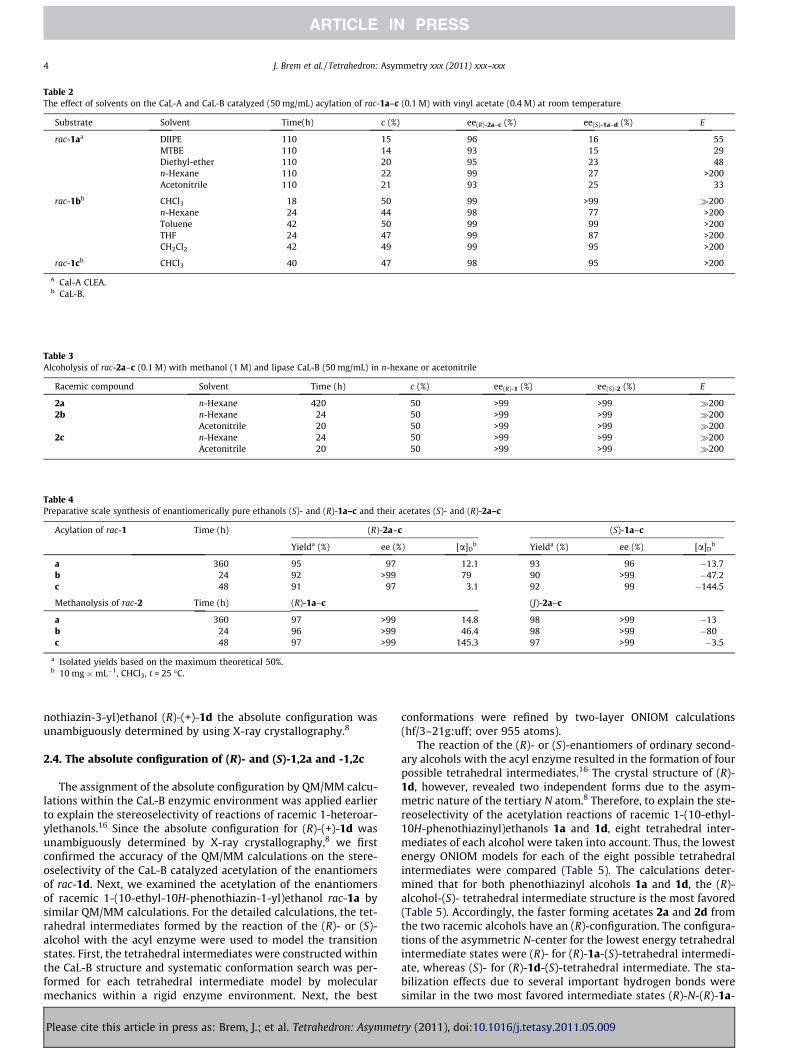
Table 2The effect of solvents on the CaL-A and CaL-B catalyzed (50 mg/mL) acylation of rac-1a–c (0.1 M) with vinyl acetate (0.4 M) at room temperature
Substrate Solvent Time(h) c (%) ee(R)-2a–c (%) ee(S)-1a–d (%) E
rac-1aa DIIPE 110 15 96 16 55MTBE 110 14 93 15 29Diethyl-ether 110 20 95 23 48n-Hexane 110 22 99 27 >200Acetonitrile 110 21 93 25 33
rac-1bb CHCl3 18 50 99 >99 �200n-Hexane 24 44 98 77 >200Toluene 42 50 99 99 >200THF 24 47 99 87 >200CH2Cl2 42 49 99 95 >200
rac-1cb CHCl3 40 47 98 95 >200
A Cal-A CLEA.b CaL-B.
Table 3Alcoholysis of rac-2a–c (0.1 M) with methanol (1 M) and lipase CaL-B (50 mg/mL) in n-hexane or acetonitrile
Racemic compound Solvent Time (h) c (%) ee(R)-1 (%) ee(S)-2 (%) E
2a n-Hexane 420 50 >99 >99 �2002b n-Hexane 24 50 >99 >99 �200
Acetonitrile 20 50 >99 >99 �2002c n-Hexane 24 50 >99 >99 �200
Acetonitrile 20 50 >99 >99 �200
Table 4Preparative scale synthesis of enantiomerically pure ethanols (S)- and (R)-1a–c and their acetates (S)- and (R)-2a–c
Acylation of rac-1 Time (h) (R)-2a–c (S)-1a–c
Yielda (%) ee (%) [a]Db Yielda (%) ee (%) [a]D
b
a 360 95 97 12.1 93 96 �13.7b 24 92 >99 79 90 >99 �47.2c 48 91 97 3.1 92 99 �144.5
Methanolysis of rac-2 Time (h) (R)-1a–c (J)-2a–c
a 360 97 >99 14.8 98 >99 �13b 24 96 >99 46.4 98 >99 �80c 48 97 >99 145.3 97 >99 �3.5
a Isolated yields based on the maximum theoretical 50%.b 10 mg �mL�1, CHCl3, t = 25 �C.
4 J. Brem et al. / Tetrahedron: Asymmetry xxx (2011) xxx–xxx
nothiazin-3-yl)ethanol (R)-(+)-1d the absolute configuration wasunambiguously determined by using X-ray crystallography.8
2.4. The absolute configuration of (R)- and (S)-1,2a and -1,2c
The assignment of the absolute configuration by QM/MM calcu-lations within the CaL-B enzymic environment was applied earlierto explain the stereoselectivity of reactions of racemic 1-heteroar-ylethanols.16 Since the absolute configuration for (R)-(+)-1d wasunambiguously determined by X-ray crystallography,8 we firstconfirmed the accuracy of the QM/MM calculations on the stere-oselectivity of the CaL-B catalyzed acetylation of the enantiomersof rac-1d. Next, we examined the acetylation of the enantiomersof racemic 1-(10-ethyl-10H-phenothiazin-1-yl)ethanol rac-1a bysimilar QM/MM calculations. For the detailed calculations, the tet-rahedral intermediates formed by the reaction of the (R)- or (S)-alcohol with the acyl enzyme were used to model the transitionstates. First, the tetrahedral intermediates were constructed withinthe CaL-B structure and systematic conformation search was per-formed for each tetrahedral intermediate model by molecularmechanics within a rigid enzyme environment. Next, the best
Please cite this article in press as: Brem, J.; et al. Tetrahedron: Asymmet
conformations were refined by two-layer ONIOM calculations(hf/3–21g:uff; over 955 atoms).
The reaction of the (R)- or (S)-enantiomers of ordinary second-ary alcohols with the acyl enzyme resulted in the formation of fourpossible tetrahedral intermediates.16 The crystal structure of (R)-1d, however, revealed two independent forms due to the asym-metric nature of the tertiary N atom.8 Therefore, to explain the ste-reoselectivity of the acetylation reactions of racemic 1-(10-ethyl-10H-phenothiazinyl)ethanols 1a and 1d, eight tetrahedral inter-mediates of each alcohol were taken into account. Thus, the lowestenergy ONIOM models for each of the eight possible tetrahedralintermediates were compared (Table 5). The calculations deter-mined that for both phenothiazinyl alcohols 1a and 1d, the (R)-alcohol-(S)- tetrahedral intermediate structure is the most favored(Table 5). Accordingly, the faster forming acetates 2a and 2d fromthe two racemic alcohols have an (R)-configuration. The configura-tions of the asymmetric N-center for the lowest energy tetrahedralintermediate states were (R)- for (R)-1a-(S)-tetrahedral intermedi-ate, whereas (S)- for (R)-1d-(S)-tetrahedral intermediate. The sta-bilization effects due to several important hydrogen bonds weresimilar in the two most favored intermediate states (R)-N-(R)-1a-
ry (2011), doi:10.1016/j.tetasy.2011.05.009

Table 5Comparison of the tetrahedral intermediate states of acylation of rac-1a and rac-1d within the active site of CaL-B determined by QM/MM (hf/3–21g:uff) method
N
S
OH
rac-1a:N
Srac-1d:
OH
SubstrateDE (kcal/mol)
(R)-1-(R)-Tetrahedral intermediate (R)-1-(S)-Tetrahedral intermediates (S)-1-(R)-Tetrahedral intermediate (S)-1-(S)-Tetrahedral intermediate
(S)-N (R)-N (S)-N (R)-N (S)-N (R)-N (S)-N (R)-N
rac-1a 28.6 33.3 — 0.0 — 15.3 31.9 7.9rac-1d 58.8 40.0 0.0 — 35.3 43.0 14.5a
a The N-atom of the 1-(10-ethyl-10H-phenothiazin-3-yl)ethanol moiety was nearly planar in the optimized structure.
J. Brem et al. / Tetrahedron: Asymmetry xxx (2011) xxx–xxx 5
(S)-tetrahedral intermediate and (S)-N-(R)-1d-(S)-tetrahedralintermediate (Table 6 and Fig. 1A). Irrespective of the position ofthe substituents on the phenothiazine ring in (R)-1a or (R)-1d,the alcohol moieties occupied approximately the same space ofthe active site in the most favored intermediate states (Fig. 1B).
Based on these data and on the consistent (+)-signs of the spe-cific rotations for (R)-1a,b and (R)-1d,8 the (R)-(+)-absolute config-uration was assigned to be the faster reacting enantiomer of 1-(10-ethyl-10H-phenothiazin-4-yl)ethanol 1c as well.
3. Conclusion
Efficient enzymatic procedures for the synthesis of enantiome-rically pure 1-(10-ethyl-10H-phenothiazinyl)ethanols 1a–c andtheir acetates 2a–c has been described. Due to the low stabilityof these compounds, the preparative scale biotransformationswere followed by careful isolation and purification.
While the lipase mediated acetylation afforded the enantiopure(S)-ethanols (S)-1a–c and (R)-acetates (R)-2a–c, the CaL-B cata-lyzed methanolysis of the racemic acetates rac-2a–c yielded theopposite (R)-1a–c and (S)-2a–c enantiomeric forms of the targetcompounds.
The absolute configuration of the faster reacting enantiomer of2a was determined by QM/MM calculations on all possible forms
Table 6Comparison of the H–O distances between Gln106, Thr40 and the oxoanion (d1, d2, d3), HisNs-ester-O) in the two lower energy states of (R)-1-(S)-tetrahedral intermediates
Tetrahedral intermediate structure d1 (Å) d2 (Å)
(R)-N-(R)-1a-(S)-Tetrahedral intermediate 2.16 2.77(S)-N-(R)-1d-(S)-Tetrahedral intermediate 1.93 2.00
Figure 1. Comparison of the two lower energy tetrahedral intermediate states withinacetylation of racemic 1-(10-ethyl-10H phenothiazin-1-yl)ethanol rac-1a and 1-(10-ethdiate from rac-1a; B, overlay of (R)-N-(R)-1-(S)- tetrahedral intermediate from rac-1a an
Please cite this article in press as: Brem, J.; et al. Tetrahedron: Asymmet
of the tetrahedral intermediates for the racemic alcohol rac-1awithin the active site of C. antarctica lipase B structure.
4. Experimental
4.1. Analytical methods
The 1H and 13C-NMR spectra were recorded in CDCl3 solution ona Brucker Avance DPX-300 spectrometer operating at 300 and75 MHz, respectively. Chemical shifts on the d scale are expressedin ppm values from TMS as the internal standard. IR spectra wererecorded in KBr or CsBr plates on a Jasco FT-IR spectrometer andthe wave numbers are reported in cm�1. EI mass spectra were ta-ken on a VG 7070E mass spectrometer operating at 70 eV and arecharacterized by m/z (relative intensity %) values. Optical rotationswere determined on a Bellingham-Stanley ADP 220 polarimeter.Thin Layer Chromatography (TLC) was carried out using MerckKieselgel 60F254 sheets. Spots were visualized by treatment with5% ethanolic phosphomolybdic acid solution and heating. Prepara-tive vacuum-chromatography was performed on Merck Kieselgel60 (0.063–0.200 lm). Determination of E was based using theequation E = ln[(1 � c)(1 � eeS)]/ln[(1 � c)(1 + eeS)].17
High performance liquid chromatography analyses wereconducted with an Agilent 1200 instrument. For enantiomeric
224-Ns and tetrahedral intermediate oxygens (d4: His224-Ns-Ser105-O3; d5: His224-
d3 (Å) d4 (Å) d5 (Å) d6 (Å) d7(Å)
1.83 2.15 1.93 2.00 2.441.59 2.26 1.75 2.00 2.44
the active site of CaL-B determined by QM/MM (hf/3–21g:uff) calculations foryl-10H-phenothiazin-3-yl)ethanol rac-1d [A, (R)-N-(R)-1-(S)- tetrahedral interme-d (S)-N-(R)-1-(S)- tetrahedral intermediate from rac-1d].
ry (2011), doi:10.1016/j.tetasy.2011.05.009

Table 7Retention times for the enantiomers of 1a–c and 2a–c
Compound tR (min) Compound tR (min)
(R)-1a 6.5 (R)-2a 4.1(S)-1a 7.1 (S)-2a 3.5(R)-1b 7.9 (R)-2b 5.5(S)-1b 7.2 (S)-2b 5.3(R)-1c 6.8 (R)-2c 12.0(S)-1c 7.9 (S)-2c 9.4
6 J. Brem et al. / Tetrahedron: Asymmetry xxx (2011) xxx–xxx
separation of rac-1,2a a LiChroCART (R,R)-Whelk-O1 column(4.6 � 250 mm); for rac-1,2b Chiralpak IB column (4.6 � 250 mm)and for rac-1,2c Chiralpak IC column (4.6 � 250 mm) and a mixtureof hexane and 2-propanol, 98:2, 90:10 and 90:10, respectively, (v/v) as eluent was used, all at 1 mL/min flow rate (Table 7).
4.2. Reagents and solvents
All reagents were purchased from Aldrich or Fluka and used asreceived. Solvents and acyl donors for enzymatic reactions werestored over molecular sieves unless otherwise stated. Lipases fromAspergillus niger, P. fluorescens (L-AK), Burkholderia cepacia (L-PS),Rhizopus oryzae (L-F), Rhizopus arrhizus, Penicillium camembertiand Mucor javanicus were products of Amano. Lipases from Candidarugosa (CrL), Candida lipolytica, Mucor miehei and porcine pancreas(PPL) were purchased from Fluka. CAL-A CLEA was from Fluka. Li-pase B from C. antarctica (CAL-B, Novozym 435) and lipase fromThermomyces lanuginosus (Lipozyme TL IM) were purchased fromNovozymes, Denmark.
4.3. Synthesis of racemic alcohols rac-1,2a–c
4.3.1. Synthesis of racemic 1-(10-ethyl-10H-phenothiazin-yl)-ethanols rac-1a,c
Into a stirred solution of methyl magnesium iodide, preparedfrom magnesium (7.4 mmol, 0.177 g) and iodomethane (7.4 mmol,1.05 g, 0.460 mL) in Et2O (5 mL), a solution of a 10-ethyl-10H-phe-nothiazinyl-carbaldehyde 1a,c (6.17 mmol, 1.57 g) dissolved inEt2O (3 mL) was added slowly at 0 �C under argon. The resultingmixture was stirred at room temperature for 1.5–2 h. Afterquenching the reaction by the slow addition of saturated ammo-nium chloride solution (8 mL) and ethanol (2 mL) at t <0 �C to pro-tect the crude products, the organic layer was isolated and theaqueous layer was extracted with Et2O (2 � 10 mL). The combinedorganic layer was dried over anhydrous sodium sulfate and con-centrated in vacuo at low temperature (t <10 �C). The solid residuewas purified by vacuum-chromatography on neutral aluminumoxide (Brockmann IV) using dichloromethane, resulting in theracemic alcohol rac-1a,c as a colorless semisolid.
4.3.1.1. 1-(10-Ethyl-10H-phenothiazin-1-yl)ethanol rac-1a. Yield:91%; semisolid; 1H NMR: (300 MHz, CDCl3); d = 1.15 (t, J = 7.0 Hz,3H), 1.55 (d, J = 6.4 Hz, 3H), 3.75 (q, J = 7.0 Hz, 2H), 5.31 (q,J = 6.4 Hz, 1H), 6.97–7.20 (m, 7H); 13C NMR: (75 MHz, CDCl3):d = 14.2, 23.6, 50.8, 65.7, 114.3, 122.7; 125.1; 125.3; 125.4; 126.3,127.0, 127.2; 132.8; 141.1; 142.5, 144,1; IR (KBr): 3357, 3060,2971, 2926, 2864, 1590, 1566, 1473, 1433, 1375, 1263, 1227, 1096,1077, 787, 758, 727; HRMS: M+ found (M+ calculated forC16H17NOS): 271.1 (271.1030); MS: m/z (%) = 272 (7, M+1), 271(31, M), 242 (37), 200 (17), 199 (24), 133 (12), 91 (13), 58 (21), 56(12), 43 (100).
4.3.1.2. 1-(10-Ethyl-10H-phenothiazin-4-yl)ethanol rac-1c. Yield:90%; semisolid; 1H NMR: (300 MHz, CDCl3): d = 1.41 (t, J = 6.9 Hz,3H), 1.49 (d, J = 6.4 Hz, 3H), 3.92 (q, J = 6.9 Hz, 2H), 5.27 (q,
Please cite this article in press as: Brem, J.; et al. Tetrahedron: Asymmet
J = 6.4 Hz, 1H), 6.79–6.84 (m, 1H), 6.87–6.95 (m, 2H), 7.15–7.24(m,4H); 13C NMR: (75 MHz, CDCl3): d = 13.1, 23.5, 41.8, 67.0, 114.3,115.1, 118.8, 122.1, 122.7, 124.3, 126.9, 127.3, 127.5, 143.4, 145.1,145.7; IR (KBr): 3353, 3063, 2969, 2857, 1593, 1564, 1480, 1459,1443, 1367, 1325, 1249, 1110, 889, 785, 746; HRMS: M+ found (M+
calculated for C16H17NOS): 271.2 (271.1030); MS: m/z (%) = 273.2(4, M+2), 272.2 (14, M+1), 271.2 (86, M), 243 (17), 242 (100), 226(7), 198 (19), 191 (24), 83 (14), 43 (18).
4.3.2. Synthesis of racemic 1-(10-ethyl-10H-phenothiazin-2-yl)ethanol rac-1b
Into a stirred solution of 1-(10H-phenothiazin-2-yl)ethanone(1 mmol, 241 mg) in anhydrous DMF (20 mL), NaH (1.1 mmol,26.5 mg) was added in small portions at 0–5 �C. After 10 min, ethyliodide (1.25 mmol, 125 lL) was added and the resulting mixturewas stirred at 50 �C. After completion of the reaction (checked byTLC), the solvent from the mixture was distillated in vacuo, the res-idue was dissolved in CH2Cl2 (20 mL), filtered, and concentrated.The 1-(10-ethyl-10H-phenothiazin-2-yl)ethanone was crystallizedfrom ethanol as yellow semisolid and used in the next step withoutfurther purification.
To a stirred solution of the 1-(10-ethyl-10H-phenothiazin-2-yl)ethanone (0.8 mmol, 215 mg) in dry methanol (5 mL), NaBH4
(1 mmol, 38 mg) was added in small portions at room temperatureand the resulting mixture was stirred. After the reduction wascomplete (checked by TLC), the mixture was quenched by thedropwise addition of a 2 M HCl solution (1 mL) and evaporatedto a final volume of approximately 1 mL. To this residue, water(3 mL) and CH2Cl2 (6 mL) were added. After separating the two lay-ers, the aqueous layer was extracted with CH2Cl2 (6 mL). The com-bined organic layers were dried over anhydrous MgSO4 and thesolvent was removed. The residue was purified by column chroma-tography on silica gel using CH2Cl2 as eluent yielding the desiredproduct as a colorless semisolid.
4.3.2.1. 1-(10-Ethyl-10H-phenothiazin-2-yl)ethanol rac-1b. Yield:94%; semisolid; 1H NMR: (300 MHz, CDCl3): d = 1.39–1.47 (m, 6H),3.93 (q, J = 6.9 Hz, 2H), 4.81 (q, J = 6.4 Hz, 1H), 6.85–6.93 (m, 4H),7.06–7.24 (m, 3H); 13C NMR : (75 MHz, CDCl3): d = 12.9, 25.1, 41.5,70.1, 112.1, 115.0, 119.2, 122.2, 123.2, 124.3, 127.1, 127.2, 144.8,145.1, 145.2; IR (KBr): 3356, 3060, 2969, 2925, 2853, 1595, 1585,1463, 1442, 1423, 1284, 1235, 1133, 1110, 887, 815, 748; HRMS:M+ found (M+ calculated for C16H17NOS): 271.1 (271.1030); MS: m/z (%) = 271 (37, M), 242 (36), 196 (19), 135 (28), 128 (43), 119 (58),105 (25), 91 (23), 58 (39), 57 (28), 43 (100).
4.3.3. Chemical acetylation of the racemic 1-(10-ethyl-10H-phenothiazin-yl)-ethanols rac-1a–c
Into a solution of racemic 1-(10-ethyl-10H-phenothiazin-yl)ethanol rac-1a–c (5.61 mmol) in dry CH2Cl2 (15 mL), Et3N(6.16 mmol, 624 mg, 860 lL), acetyl chloride (6.17 mmol,484.3 mg, 438.7 lL) and DMAP (0.16 mmol, 20 mg) were added.The mixture was stirred at room temperature overnight and thenquenched with water (15 mL). The isolated organic layer was driedover anhydrous sodium sulfate and the solvent was distilled off byrotatory evaporation. The crude product was purified by vacuum-chromatography on neutral aluminum oxide (Brockmann IV) usingCH2Cl2 as eluent to give rac-2a–c as a semisolid.
4.3.3.1. 1-(10-Ethyl-10H-phenothiazin-1-yl)ethyl acetate rac-2a. Yield: 93%; semisolid; 1H NMR: (300 MHz, CDCl3): d = 1.07(t, J = 6.9 Hz, 3H), 1.35 (d, J = 6.6 Hz, 3H), 2.07 (s, 3H), 3.54 (q,J = 6.9 Hz, 2H), 6.34 (q, J = 6.9 Hz, 1H), 6.94–7.22 (m, 7H); 13CNMR: (75 MHz, CDCl3): d = 14.6, 21.5, 22.3, 51.2, 69.2, 124.4,124.8, 125.2, 126.0, 126.6, 127.1, 127.2, 133.0, 134.2, 138.2,142.2, 145.2, 170.5; IR (KBr): 3061, 2975, 2929, 2865, 1732,
ry (2011), doi:10.1016/j.tetasy.2011.05.009

J. Brem et al. / Tetrahedron: Asymmetry xxx (2011) xxx–xxx 7
1591, 1568, 1474, 1435, 1368, 1239, 1073, 1022, 788, 759, 728;HRMS: M+ found (M+ calculated for C18H19NO2S): 313.1(313.1136); MS: m/z (%) = 315 (5, M+2), 314 (18, M+1), 313 (100,M), 284 (100), 270 (15), 242 (35), 241 (46), 225 (26), 224 (38),223 (27), 200 (11), 199 (18), 198 (16), 43 (30).
4.3.3.2. 1-(10-Ethyl-10H-phenothiazin-2-yl)ethyl acetate rac-2b. Yield: 87%; semisolid; 1H NMR: (300 MHz, CDCl3): d = 1.41(t, J = 6.9 Hz, 3H), 1.51 (d, J = 6.6 Hz, 3H), 2.06 (s, 3H), 3.94 (q,J = 6.9 Hz, 2H), 5.80 (q, J = 6.9 Hz, 1H), 6.83–6.92 (m, 4H), 6.83–6.92 (m, 3H); 13C NMR: (75 MHz, CDCl3): d = 13.1, 21.5, 22.2,41.8, 72.3, 113.4, 115.3, 120.1, 122.5, 124.3, 124.4, 127.2, 127.4,127.5, 141.1, 144.9, 145.2, 170.4; IR (KBr): 3057, 2970, 2931,2869, 1732, 1579, 1465, 1443, 1326, 1245, 1237, 1179, 1062,816, 749; 745 HRMS: M+ found (M+ calculated for C18H19NO2S):313.2 (313.1136); MS: m/z (%) = 313 (7, M), 206 (11), 192 (6),191 (41), 149 (8), 86 (12), 84 (70), 82 (100), 71 (100), 57 (13), 47(18); 43 (20).
4.3.3.3. 1-(10-Ethyl-10H-phenothiazin-4-yl)ethyl acetate rac-2c. Yield: 81%; semisolid; 1H NMR: (300 MHz, CDCl3): d = 1.41 (t,J = 6.9 Hz, 3H), 1.55 (d, J = 6.4 Hz, 3H), 2.10 (s, 3H), 3.93 (q,J = 6.9 Hz, 2H), 6.21 (q, J = 6.4 Hz, 1H), 6.83–6.95 (m, 3H), 7.06–7.09 (m, 1H), 7.14–7.21 (m, 3H); 13C NMR: (75 MHz, CDCl3):d = 13.2, 20.9, 21.3, 42.3, 69.8, 114.9, 115.3, 119.5, 122.5, 122.6,123.8, 124.5, 127.0, 127.5, 127.8, 139.5, 145.7, 170.2; IR (KBr):3064, 2972, 2927, 2868, 1733, 1593, 1566, 1481, 1462, 1443,1370, 1240, 1072, 1058, 783, 748, 729; HRMS: M+ found (M+ calcu-lated for C18H19NO2S): 313.2 (313.1136); MS: m/z (%) = 315.2 (5,M+2), 314.2 (18, M+1), 313.2 (100, M), 285 (10), 284 (66), 226(10), 225 (15), 224 (28), 191 (8), 58 (26), 43 (72).
4.4. Enzyme mediated biotransformation of racemic 1-(10-ethyl-10H-phenothiazin-yl)ethanols, rac-1a–c and theiracetates racemic 1-(10-ethyl-10H-phenothiazin-yl)ethylacetates, rac-2a–c on an analytical scale
4.4.1. Enzymatic kinetic resolution of racemic 1-(10-ethyl-10H-phenothiazinyl)ethanols rac-1a–c with vinyl acetate anddifferent lipases
Into a solution of racemic 1-(10-ethyl-10H-phenothiazinyletha-nol rac-1a–c (0.1 mmol, 27.1 mg) in vinyl acetate (1 mL), lipase(50 mg) was added. The reaction mixture was shaken at 300 rpmat room temperature. For HPLC analysis, samples were taken fromthe reaction mixture (10 lL), diluted to 500 lL with 2-propanoland filtered before injection. Data on the conversion and enantio-meric composition of the products for the solvents tested are pre-sented in Table 1.
4.4.2. Enzymatic kinetic resolution of racemic 1-(10-ethyl-10H-phenothiazin-yl)ethanols rac-1a–c with vinyl acetate indifferent solvents with CaL-A/CaL-B
Into a solution of racemic 1-(10-ethyl-10H-phenothiazin-yl)ethanol rac-1a–c (0.1 mmol, 27.1 mg) in different solvents(1 mL), vinyl acetate (0.4 mmol, 34.4 mg, 36.8 lL), and lipase(50 mg, CaL-A or CaL-B) were added. The reaction mixture was sha-ken at 300 rpm at room temperature. Samples were analyzed byHPLC similarly as described in Section 4.4.1. Data on the conver-sion and enantiomeric composition of the products are presentedin Table 2.
4.4.3. Enzymatic alcoholysis of racemic 1-(10-ethyl-10H-phenothiazinyl)ethyl acetates rac-2a–c
Into a mixture of rac-2a–c (0.1 mmol, 31.3 mg) in n-hexane/ace-tonitrile (1 mL), CaL-B (50 mg) and methanol (1 mmol, 32 mg,40.4 lL) were added. The reaction mixture was shaken at
Please cite this article in press as: Brem, J.; et al. Tetrahedron: Asymmet
300 rpm at room temperature. Samples were analyzed by HPLCas described in Section 4.3.1. Data on conversion and enantiomericcomposition of the products are presented in Table 3.
4.5. Enzyme mediated biotransformation of racemic 1-(10-ethyl-10H-phenothiazinyl)ethanols, rac-1a–c and their acetatesrac-2a–c on a preparative scale
Into the solution of racemic 1-(10-ethyl-10H-phenothiazi-nyl)ethanols rac-2a–c (3.69 mmol, 1 g) in n-hexane or chloroformdepending on the enzyme used (36.9 mL), vinyl acetate(14.7 mmol, 1.27 g, 1.36 mL) and lipase (1.84 g, CaL-A or CaL-B)were added. The reaction mixture was shaken at 300 rpm at roomtemperature. The reactions were monitored by TLC and stopped atapprox. 50% conversion by removing the enzyme by filtration. Sol-vents were removed in vacuo at low temperature (t <10 �C) and thecrude product was purified by vacuum-chromatography on neutralaluminum oxide (Brockmann IV) using dichloromethane as eluent,resulting in virtually enantiopure (S)-1-(10-ethyl-10H-phenothia-zin-yl)ethanols (S)-1a–c and (R)-1-(10-ethyl-10H-phenothiazin-yl)ethyl acetate (R)-2a–c as semisolids.
Into a solution of racemic 1-(10-ethyl-10H-phenothiazi-nyl)ethyl acetates rac-2a–c (3.19 mmol, 1 g) in acetonitrile(31.9 mL), methanol (31.9 mmol, 1 g, 1.29 mL) and CaL-B (1.59 g)were added. The reaction mixture was shaken at 300 rpm at roomtemperature. The reactions were monitored by TLC and stopped atan approx. 50% conversion by removing the enzyme by filtration.Solvents were removed in vacuo at <10 �C and the crude productwas purified by vacuum-chromatography on neutral aluminumoxide (Brockmann IV) using dichloromethane as eluent, resultingin virtually enantiopure (S)-1-(10-ethyl-10H-phenothiazin-yl)ethyl acetates (S)-2a–c and (R)-1-(10-alkyl-10H-phenothiazin-yl)ethanols (R)-1a–c as semisolids.
The desired compounds were isolated in good yields, withoutaltering their enantiomeric purity (Table 7).
4.6. Computational Methods
4.6.1. Systematic conformational search within the active site ofCaLB at MM level
The calculations were performed within the active site of thecrystal structure CaL-B containing Tween80 (T80) as the substratemimic (PDB code: 1LBT).10 From this CaL-B structure, a part con-taining the residues within 10 Å spheres around the Ser105 Oc,T80 C10 and T80 C20 atoms was cut off as the active site modelfor CS and later QM/MM calculations. Next, T80 was removedand by the Hyperchem18 standard procedure, hydrogens wereadded to the amino acid residues and water oxygens. The C- andN-termini at cutting were completed to neutral aldehyde and ami-no moieties. During our calculations, His224 was protonated andAsp187 was deprotonated.
The tetrahedral intermediate states were built in the active sitemodel by constructing a covalent bond between the Oc atom ofSer105 and the carbonyl carbon of the (R)- and (S)-enantiomersof 2a and 2d acetates. In this way, starting structures were con-structed for the eight tetrahedral intermediate states [(R)-N-(R)-1-(R)-tetrahedral intermediates, (S)-N-(R)-1-(R)-tetrahedral inter-mediate, (R)-N-(R)-1-(S)-tetrahedral intermediate, (S)-N-(R)-1-(S)-tetrahedral intermediate, (R)-N-(S)-1-(R)-tetrahedral intermediate,(S)-N-(S)-1-(R)-tetrahedral intermediate, (R)-N-(S)-1-(S)-tetrahe-dral intermediate, (S)-N-(S)-1-(S)-tetrahedral intermediate] foracetylation of each racemic alcohols rac-1a and rac-1d. The initialtetrahedral intermediates including Ca and Cb (with their hydro-gens) of Ser105 (47 atoms for 2a and 2d acetates) were optimizedby the MM+ method of Hyperchem18 within the rigid enzymicenvironment (including no waters).
ry (2011), doi:10.1016/j.tetasy.2011.05.009

8 J. Brem et al. / Tetrahedron: Asymmetry xxx (2011) xxx–xxx
The conformational search within this active site model wasperformed by the conformational search module implemented inHyperchem18 at molecular mechanics (MM+) level using defaultsettings (MM+ forcefield; gradient: 0.1 kcal/mol; Polak-Ribieremethod until 0.05 kcal/mol RMS gradient or maximum 600 cycles;limits: 300 iterations, 150 optimizations, 15 conformations; testoptions: ‘skip if atoms are closer than 0.3 Å’). During the conforma-tional search, five torsion angles (starting from Ca of Ser105) of thealcohols 1a and 1d were chosen to vary in a rigid enzymic environ-ment (including no waters). The three lowest energy conforma-tions of each 1a and 1d alcohol–acyl enzyme tetrahedralintermediate systems were investigated further by QM/MMmethods.
4.6.2. QM/MM calculations within the active site of CaL-BAll QM/MM optimizations were carried out using the ONIOM
protocol as implemented in Gaussian0319 with the aid of Gauss-View20 for selecting and preparing the calculation levels.
The best conformations for each tetrahedral intermediate wereoptimized using a two-layer ONIOM method (hf/3–21g:uff) over955 atoms. The high layer included the atoms of the alcohols 1aand 1d, the acetate part of tetrahedral intermediate, the side chainsof the catalytic triad (Ser105, His224, Asp187 + one water), the rel-evant parts of the residues forming the so-called oxoanion hole(Gln106, Thr40). The high layer was relaxed with Hartree-Fock(hf/3–21g) method. Residues from the low layer, except Trp104(+ one water), Asp134, Gln157, Ile189, Ala281, Ala282 and Ile285,were kept fixed during the molecular mechanics (UFF)optimization.
Acknowledgments
J.B. gives thanks for the financial support provided from pro-grams co financed by The Sectoral operational program human re-sources development, Contract POSDRU 6/1.5/S/3 – ‘Doctoralstudies: through science towards society’. This work is also relatedto the scientific program of ‘Development of quality-oriented andharmonized R+D+I strategy and functional model at BME’ (TÁMOP-4.2.1/B-09/1/KMR-2010-0002), supported by the New HungaryDevelopment Plan.
References
1. (a) Mosnaim, A. D.; Ranade, V. V.; Wolf, M. E.; Puente, J.; Antonieta, V. M. Am. J.Ther. 2006, 13, 261–273; (b) Kaiser, C.; Pavloff, A. M.; Garvey, E.; Fowler, P. J.;
Please cite this article in press as: Brem, J.; et al. Tetrahedron: Asymmet
Tedeschi, D. H.; Zirkle, C. L.; Nodiff, E. A.; Saggiomo, A. J. J. Med. Chem. 1972, 15,665–673.
2. Bansode, T. N.; Shelke, J. V.; Dongre, V. G. Eur. J. Med. Chem. 2009, 44, 5094–5098.
3. Feinberg, A. P.; Snyder, S. H. PNAS 1975, 72, 1899–1903.4. Long, J. P.; Lands, A. M.; Zenitz, B. L.; Beach, J. J. Pharmacol. Exp. Ther. 1957,
119(4), 479–484.5. (a) Bencze, L. C.; Paizs, C.; Tos�a, M. I.; Trif, M.; Irimie, F. D. Tetrahedron:
Asymmetry 2010, 21, 1999–2004; (b) Paizs, C.; Tosa, M.; Majdik, C.; Tähtinen, P.;Irimie, F. D.; Kanerva, L. T. Tetrahedron: Asymmetry 2003, 14, 619–627; (c) Kirk,O.; Christensen, M. W. Org. Process Res. Dev. 2002, 6, 446–451. De Gonzalo, G.;(d) Lavandera, I.; Brieva, R.; Gotor, V. Tetrahedron 2004, 60, 10525–10532.
6. Paizs, C.; Taehtinen, P.; Tosa, M.; Majdik, C.; Irimie, F. D.; Kanerva, L. T.Tetrahedron 2004, 60(46), 10533–10540.
7. Brem, J.; Tos�a, M. I.; Paizs, C.; Vass, E.; Irimie, F. D. Tetrahedron: Asymmetry2010, 21, 365–373.
8. Brem, J.; Tos�a, M. I.; Paizs, C.; Munceanu, A.; Matkovic-Calogovic, D.; Irimie, F.D. Tetrahedron: Asymmetry 2010, 21, 1993–1998.
9. Stepanenko, V.; De Jesus, M.; Correa, W.; Guzman, I.; Vazquez, C.; Ortiz, L.;Ortiz-Marciales, M. Tetrahedron: Asymmetry 2007, 18, 2738–2745.
10. Uppenberg, J.; Ohmer, N.; Norin, M.; Hult, K.; Kleywegt, G. J.; Patkar, S.;Waagen, V.; Anthomen, T.; Jones, T. A. Biochemistry 1995, 34, 16838–16851.
11. Katritzky, A. R.; He, H. Y.; Long, Q.; Cui, X.; Level, J.; Wilcox, A. V. ARKIVOC 2000,iii, 240–251.
12. Mortier, J.; Nguyen, T. H.; Tilly, D.; Castanet, A. S. ARKIVOC 2007, vi, 47–54.13. (a) Ericsson, D. J.; Kasrayan, A.; Johansson, P.; Bergfors, T.; Sandström, A. G.;
Bäckvall, J.-E.; Mowbray, S. L. J. Mol. Biol. 2008, 376, 109–119; (b) Engström, K.;Nyhlén, J.; Sandström, A. G.; Bäckvall, J.-E. J. Am. Chem. Soc. 2010, 132, 7038–7042; (c) Naik, S.; Basu, A. B.; Saikia, R.; Madan, B.; Paul, P.; Chaterjee, R.; Brask,J.; Svendsen, A. J. Mol. Catal. B: Enzym. 2010, 65, 18–23.
14. Brem, J.; Liljeblad, A.; Paizs, C.; Tosa, M. I.; Irimie, F. D.; Kanerva, L. T.Tetrahedron: Asymmetry 2011, 22, 315–322.
15. Stepanenko, V.; De Jesus, M.; Correa, W.; Guzman, I.; Vazquez, C.; Ortiz, L.;Ortiz-Marciales, M. Tetrahedron: Asymmetry 2007, 18(23), 2738–2745.
16. Tosa, M.; Pilbák, S.; Moldovan, P.; Paizs, C.; Szatzker, G.; Szakács, G.; Novák, L.;Irimie, F. D.; Poppe, L. Tetrahedron: Asymmetry 2008, 19, 1844–1852.
17. Chen, C. S.; Fujimoto, Y.; Girdaukas, G.; Sih, C. J. J. Am. Chem. Soc. 1982, 104,7294–7299.
18. Hyperchem 7.5, Hypercube, Inc., 2003.19. Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.;
Cheeseman, J. R.; Montgomery, J. A., Jr.; Vreven, T.; Kudin, K. N.; Burant, J. C.;Millam, J. M.; Iyengar, S. S.; Tomasi, J.; Barone, V.; Mennucci, B.; Cossi, M.;Scalmani, G.; Rega, N.; Petersson, G. A.; Nakatsuji, H.; Hada, M.; Ehara, M.;Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao,O.; Nakai, H.; Klene, M.; Li, X.; Knox, J. E.; Hratchian, H. P.; Cross, J. B.; Bakken,V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A.J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Ayala, P. Y.; Morokuma, K.; Voth, G.A.; Salvador, P.; Dannenberg, J. J.; Zakrzewski, V. G.; Dapprich, S.; Daniels, A. D.;Strain, M. C.; Farkas, O.; Malick, D. K.; Rabuck, A. D.; Raghavachari, K.;Foresman, J. B.; Ortiz, J. V.; Cui, Q.; Baboul, A. G.; Clifford, S.; Cioslowski, J.;Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Martin, R. L.;Fox, D. J.; Keith, T.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Challacombe,M.; Gill, P. M. W.; Johnson, B.; Chen, W.; Wong, M. W.; Gonzalez, C.; Pople, J. A.Gaussian 03, Revision B.05; Gaussian, Inc.: Wallingford CT, 2003.
20. Dennington, R., II; Keith, T.; Millam, J.; Eppinnett, K.; Hovell, W. L.; Gilliland, R.GaussView, Version 3.09; Semichem, Inc.: Shawnee Mission, KS, 2003.
ry (2011), doi:10.1016/j.tetasy.2011.05.009
Copyright © 2022 FDOKUMEN




















