
Journal of Biomolecular Structure and Dynamics Modeling the interactions between MC2R and ACTH...
20
This article was downloaded by: [122.167.6.57] On: 04 May 2014, At: 22:55 Publisher: Taylor & Francis Informa Ltd Registered in England and Wales Registered Number: 1072954 Registered office: Mortimer House, 37-41 Mortimer Street, London W1T 3JH, UK Journal of Biomolecular Structure and Dynamics Publication details, including instructions for authors and subscription information: http://www.tandfonline.com/loi/tbsd20 Modeling the interactions between MC2R and ACTH models from human Mutangana Dieudonné a & K.V. Ramesh a a Department of Biotechnology, Centre for Post Graduate Studies, Jain University, 18/3, 9th Main, Jayanagar 3rd Block, Bangalore 560 011, India Accepted author version posted online: 07 Apr 2014.Published online: 28 Apr 2014. To cite this article: Mutangana Dieudonné & K.V. Ramesh (2014): Modeling the interactions between MC2R and ACTH models from human, Journal of Biomolecular Structure and Dynamics, DOI: 10.1080/07391102.2014.910475 To link to this article: http://dx.doi.org/10.1080/07391102.2014.910475 PLEASE SCROLL DOWN FOR ARTICLE Taylor & Francis makes every effort to ensure the accuracy of all the information (the “Content”) contained in the publications on our platform. However, Taylor & Francis, our agents, and our licensors make no representations or warranties whatsoever as to the accuracy, completeness, or suitability for any purpose of the Content. Any opinions and views expressed in this publication are the opinions and views of the authors, and are not the views of or endorsed by Taylor & Francis. The accuracy of the Content should not be relied upon and should be independently verified with primary sources of information. Taylor and Francis shall not be liable for any losses, actions, claims, proceedings, demands, costs, expenses, damages, and other liabilities whatsoever or howsoever caused arising directly or indirectly in connection with, in relation to or arising out of the use of the Content. This article may be used for research, teaching, and private study purposes. Any substantial or systematic reproduction, redistribution, reselling, loan, sub-licensing, systematic supply, or distribution in any form to anyone is expressly forbidden. Terms & Conditions of access and use can be found at http:// www.tandfonline.com/page/terms-and-conditions
-
Upload
jainuniversity -
Category
Documents
-
view
0 -
download
0
Transcript of Journal of Biomolecular Structure and Dynamics Modeling the interactions between MC2R and ACTH...

This article was downloaded by: [122.167.6.57]On: 04 May 2014, At: 22:55Publisher: Taylor & FrancisInforma Ltd Registered in England and Wales Registered Number: 1072954 Registered office: Mortimer House,37-41 Mortimer Street, London W1T 3JH, UK
Journal of Biomolecular Structure and DynamicsPublication details, including instructions for authors and subscription information:http://www.tandfonline.com/loi/tbsd20
Modeling the interactions between MC2R and ACTHmodels from humanMutangana Dieudonnéa & K.V. Ramesha
a Department of Biotechnology, Centre for Post Graduate Studies, Jain University, 18/3, 9thMain, Jayanagar 3rd Block, Bangalore 560 011, IndiaAccepted author version posted online: 07 Apr 2014.Published online: 28 Apr 2014.
To cite this article: Mutangana Dieudonné & K.V. Ramesh (2014): Modeling the interactions between MC2R and ACTH modelsfrom human, Journal of Biomolecular Structure and Dynamics, DOI: 10.1080/07391102.2014.910475
To link to this article: http://dx.doi.org/10.1080/07391102.2014.910475
PLEASE SCROLL DOWN FOR ARTICLE
Taylor & Francis makes every effort to ensure the accuracy of all the information (the “Content”) containedin the publications on our platform. However, Taylor & Francis, our agents, and our licensors make norepresentations or warranties whatsoever as to the accuracy, completeness, or suitability for any purpose of theContent. Any opinions and views expressed in this publication are the opinions and views of the authors, andare not the views of or endorsed by Taylor & Francis. The accuracy of the Content should not be relied upon andshould be independently verified with primary sources of information. Taylor and Francis shall not be liable forany losses, actions, claims, proceedings, demands, costs, expenses, damages, and other liabilities whatsoeveror howsoever caused arising directly or indirectly in connection with, in relation to or arising out of the use ofthe Content.
This article may be used for research, teaching, and private study purposes. Any substantial or systematicreproduction, redistribution, reselling, loan, sub-licensing, systematic supply, or distribution in anyform to anyone is expressly forbidden. Terms & Conditions of access and use can be found at http://www.tandfonline.com/page/terms-and-conditions

Modeling the interactions between MC2R and ACTH models from human
Mutangana Dieudonné and K.V. Ramesh*
Department of Biotechnology, Centre for Post Graduate Studies, Jain University, 18/3, 9th Main, Jayanagar 3rd Block, Bangalore560 011, India
Communicated by Ramaswamy H. Sarma
(Received 8 August 2013; accepted 28 March 2014)
Melanocortin system is composed of four peptide hormones namely α-, β-, -γ, and adrenocorticotropic hormone (ACTH),derived from post-translational cleavage of a polypeptide precursor ‘proopiomelanocortin (POMC).’ Among these hor-mones, ACTH, a 38 amino acid residue peptide fragment is an important hormone as it is involved in steroid secretion.In addition to this, to cite a few, this hormone is also known to induce variety of other effects, such as alterations inmotor/sexual behavior, improvement in memory, and anti-inflammatory effects. To date, five melanocortin receptors(MC1R–MC5R) have been characterized with tissue-specific expression patterns and different binding affinities for eachof the melanocortin hormones to regulate various biological functions. In the present work, three-dimensional (3D)models of MC2R and ACTH from human have been predicted, followed by docking and molecular dynamics simula-tion. While the 3D model of MC2R receptor has been predicted through threading approach, structure of ACTH wasbuilt based on ab initio technique. The MC2R model was later successfully docked onto the ACTH structure. Moleculardynamics (MD) simulation for 20 ns was used to compute the binding free energy of MC2R with ACTH model underimplicit solvent conditions.
Keywords: molecular dynamics; melanocortin; adrenocorticotropic hormone; melanocortin 2 receptor; binding energy
Introduction
Melanocortin system is composed of α-, β-, γ- melano-cyte-stimulating hormones and adrenocorticotropin hor-mone (ACTH). These melanocortins are derived from thepost-translational cleavage of a polypeptide precursor, theproopiomelanocortin (POMC) gene product (Castro &Morrison, 1997). All these hormones share core aminoacid sequence ‘HFRW’, a key pharmacophore, essentialfor biological activity. Structure activity and computa-tional studies of α-MSH peptides revealed the presence ofmotif ‘HFRW’ having a β-turn (Haskell-Luevano et al.,1996). ACTH, which is identical to α-MSH in its first 13amino acids, exerts its effects on the adrenal cortexthrough the receptor ‘MC2R’ (Bernard & Keit, 2006). Todate, five melanocortin receptors (MC1R–MC5R) havebeen characterized; these are G protein-coupled receptorswith tissue-specific expression patterns and differentbinding affinities for each of the melanocortin hormones(Ira & Tung, 2003).
The MC1R is a ‘classical’ melanocyte α-MSH recep-tor expressed by cutaneous melanocytes, where it has akey role in determining skin and hair pigmentation.However, other cell types in the skin also expressMC1R, including keratinocytes, fibroblasts, endothelialcells, and antigen-presenting cells (Luger, Schwarz,Scholzen, Schwarz, & Brazoska, 1999). Other tissues
and cell types have also been found to express MC1R(Chhajlani, 1996). In this respect, it is notable thatMC1R is expressed by leukocytes, where it mediatesanti-inflammatory and immune-modulatory properties ofmelanocortins. Chhajlani and Wikberg (1992) andSuzuki, Cone, Im, Nordlund, and Abdel-Malek (1996)have investigated the affinity of melanocortins towardsMC2R and their results suggest the affinity between themelanocortins with the receptors in the following order:α-MSH = ACTH > β-MSH > γ-MSH. MC2R is a classicalreceptor for ACTH expressed in the adrenal cortex medi-ating the effect of the hormone on steroid secretion. Thereceptor MC2R is distinguished pharmacologically fromthe other MCR subtypes as it is activated only by ACTHand has no affinity for α-, β-, or γ-MSH (Ira & Tung,2003). Although other four melanocortin receptors couldbe activated by this hormone, the studies conducted byCone et al. (1996) and Schiöth, Chhajlani, Muceniece,Klusa, and Wikberg (1996) have shown exclusive affin-ity between ACTH and MC2R. ACTH upon binding toMC2R activates the G protein Gαs to stimulate adenylylcyclase, increase intracellular cyclic adenosine mono-phosphate (cAMP) content, and activate protein kinaseA. cAMP is an obligatory second messenger for most ofthe effects of ACTH on steroidogenesis (Bernard & Keit,2006). Tsigo, Hung, and Chrousos (1993) have reported
*Corresponding author. Email: [email protected]
© 2014 Taylor & Francis
Journal of Biomolecular Structure and Dynamics, 2014http://dx.doi.org/10.1080/07391102.2014.910475
Dow
nloa
ded
by [
122.
167.
6.57
] at
22:
55 0
4 M
ay 2
014
mature
Highlight

that isolated glucocorticoid deficiency (IGD) is associ-ated with the abnormalities of adrenocorticotrophinreceptor gene. There are studies which report that MC2Ris also expressed by adipose tissue in mice and humans(Wikberg, 1999). Although ACTH is lipolytic in mice, itis not so in humans and the function of MC2R in humanadipose tissue is presently unclear.
Normal level of ACTH in the body results in theproduction of corticosteroids required for energy; how-ever, abnormal level of ACTH (increase or decrease) canlead to multiple disorders. Primary adrenal insufficiencycalled as ‘Addison’s disease’ occurs when adrenal glandproduction of cortisol is chronically deficient, resultingin chronically elevated ACTH levels (Arlt & Allolio,2003). Also, elevated ACTH level can lead to pituitarytumor, referred to as ‘Cushing’s Disease.’ On the con-trary, the signs and symptoms of excess cortisol produc-tion (hypercortisolism) is correlated with low level ofACTH secretion and the disease is known as ’Cushing’ssyndrome’ (Kumar, Abbas, Fausto, & Aster, 2005). Inaddition to these, altered level of ACTH is associatedwith various other disorders: hypopituitarism, small cellcarcinoma (Nasu et al., 2011), congenital adrenalhyperplasia (Warrell, Timothy, Firth, & Benz, 2005),Nelson’s syndrome (Biller, Grossman, & Stewart, 2008),adrenoleukodystrophy, and West syndrome (infantilespasms). In a recent review by Liang, Angleson, andDores (2013), studies using analog of human ACTH(1–24) revealed two critical amino acid motifs namely‘HFR’ and ‘KKRRP’ which are required for activationof melanocortin-2 receptor. Based on modeling and site-directed mutagenesis of human MC4R, binding site ofthis receptor has been characterized (Pogozheva et al.,2005). Outcome from this study reveals that there are sixcritical amino acids positions distributed among TM2(transmembrane) TM3, TM6, and TM7 domains whichin fact are conserved in MC2R too. However, this hasbeen elucidated without evidences based on experimen-tally determined 3D structures.
Both, MC3R as well as MC4R, are expressed inmany areas of central nervous system (CNS); however,MC3R is also expressed in several other peripheral tis-sues, including the gastrointestinal tract and placenta(Chhajlani, 1996). All the four melanocortins (α-, β-, γ-MSH and ACTH) show equal affinity to MC3R (Ira &Tung, 2003). On the contrary, Versteeg et al. (1998) havenoticed γ-MSH having greatest affinity to MC3R, anobservation that is assumed to be of physiological signifi-cance in energy homeostasis. As is the case with MC3R,MC4R is also involved in energy homeostasis and playsa role in sexual function too. Van der Ploeg et al. (2002)have shown that MC4R is involved in mediating theperipheral actions of melanocortin effects on erectilefunction and copulatory behavior in male rodents.Potency of melanocortins on the activation of MC4R was
shown : α-MSH = ACTH > β-MSH > γ-MSH (Ira & Tung,2003). MC5R is expressed in numerous human peripheraltissues, including the adrenal gland, adipocytes, leuko-cytes, and many others (Chhajlani, 1996). It has also avery limited distribution in the CNS. The only firmlyestablished function of MC5R, which was discovered bytargeting deletion of that receptor, is its participation inexocrine function, particularly sebaceous gland secretion(Chen et al., 1997). The role of MC5R in exocrine secre-tion has the potential to be exploited for the treatment ofskin disorders such as acne and dermatitis. The agonistpotency of melanocortins is believed to be α-MSH >ACTH > β-MSH > γ-MSH (Ira & Tung, 2003). While bio-logical functions of MCRs and of their respective ligandshave been explained by many researchers, little data areavailable regarding their NMR or X-ray crystallographystructures. Therefore, an attempt has been made in thisresearch (i) to predict the three-dimensional (3D) struc-tures of MC2R and ACTH using computational tech-niques, (ii) to analyze the interaction between MC2R andACTH through docking approach followed by (iii)molecular dynamics simulation to analyze the effect ofthe solvent environment on the binding affinity underimplicit solvent conditions.
Material and methods
Molecular modeling of MC2R and ACTH from human
Sequence information of MC2R protein was retrievedfrom UniProt database (ID: Q01718). Prediction of trans-membrane helices for this protein was done by submittingthe sequence information to TMHMM server. This is amembrane protein topology prediction method based on ahidden Markov model (Krogh, Larsson, Von Heijne, &Sonnhammer, 2001). Search for potential templatestructures for this sequence was performed usingPSI-BLAST tool with PDB as the database (Altschulet al., 1997). Based on the sequence identity, theoreticalstructure of MC2R was constructed either by homologymodeling (SWISS MODEL server) (Torsten, Jurgen,Nicolas, & Manuel, 2003) or threading approaches(I-TASSER server) (Zhang, 2008). The initial 3D modelof the receptor was subjected to loop refinement based onthe reports generated by ERRAT (Colovos & Yeates,1993) and PROCHECK (Laskowski, MacArthur, &Thornton, 1993). Loop regions of the model showing ahigh percentage of error were refined through ModLoopserver (Fiser, Gian Do, & Šali, 2000). After re-validatingthe refined structure once again, it was subjected to energyminimization using the Deep View package. The coordi-nates of the energy minimized structure were submitted tothe DaliLite server (Holm & Rosenström, 2010) whichcompares it against the structural homologs deposited inthe Protein Data Bank.
2 M. Dieudonné and K.V. Ramesh
Dow
nloa
ded
by [
122.
167.
6.57
] at
22:
55 0
4 M
ay 2
014

The PSI blast tool (Altschul et al., 1997) could notretrieve any good template structures from PDB forACTH sequence (UniProt database ID P01189 |138-176)having a sequence identity of more than 20%. Since theminimum sequence identity between the query with thetemplate sequence for homology modeling and threadingshould be not less than 30% (Johnson & Overington,1993), tertiary structure of ACTH was predicted usingab initio strategy followed by molecular dynamics simu-lations (MD) as implemented in AMBER 10 package(Case et al., 2008). This was performed on a computercluster system (18 CPUs). The initial structure of ACTHin the linear polypeptide form was constructed usingLEaP program with ‘ff99SB’ as the force field (Hornaket al., 2006) with Poison Boltzmann (PB) radii set todefault value 2 (set default PBradii mbindi2) and slowlyvarying terms in the force field were evaluated less fre-quently. Using the same program, initial topology andcoordinates files were generated for ACTH structure. MDsimulations were fully unrestrained and carried out withno periodic boundary conditions in the NPT ensembleusing SANDER module of AMBER package. SHAKEalgorithm was applied to constrain bonds involvinghydrogen as proposed by Padhi, Kumar, Vasaikar,Jayaram, and Gomes (2012) and Ryckaert, Ciccotti, andBerendsen (1997). During this exercise, no cut-off wasused to truncate the non-bonded interactions betweenatom pairs. Generalized Born surface area (gbsa) wascomputed using the Linear Combinations of PairwiseOverlaps (LCPO) model (Weiser, Shenkin, & Still, 1999).Surface tension used to calculate the non-polar contribu-tion to the free energy of solvation was computed bysetting gbsa = 1, wherein the default value for surfacetension is 0.005 kcal/mol/A2 (Sitkoff, Sharp, & Honig,1994).
Ab initio calculations were started by energy minimi-zation of the starting linear ACTH polypeptide (639atoms) in a total number of 1000 cycles with a switchfrom steepest descent to conjugate gradient algorithmafter 500 cycles. Implicit solvation effect was introducedusing the generalized Born model as suggested by Still,Tempczyk, and Hawley (1990). Born radii set as pro-posed by Onufriev, Bashford, and Case (2004) fromAMBER10 force field were rescaled to account the inter-stitial space between atom spheres. The solvent is con-sidered as a dielectric continuum. Both the interior andexterior dielectric constants were set to 1 and 80, respec-tively. Salt concentration was set to default physiologicalionic strength. In the heating stage, the system was grad-ually heated from 0 to 300 K, spread over six stages (anincrease of 50 K per stage), with time set to 5 ps for eachof the first five stages and to 25 ps for the last stage.Weak-coupling algorithm was used to control the temper-ature (Berendsen, Postma, van Gunsteren, DiNola, &Haak, 1984). The heat bath coupling for the system was
set to default during the entire heating stage. This wasfollowed by equilibration spread over 30 stages. Eachstage was run for 5 ns with the temperature being main-tained at 300 K, with total simulation time of 150 ns.During this stage, Langevin dynamics were used withthe collision frequency controlled by the Leapfrog inte-grator to propagate the dynamics (Pastor, Brooks, &Szabo, 1988). Output from MD simulation results, afterprocessing using the PTRAJ command of AMBER, wassubsequently plotted using the GRACE software package(Stambulchik, 1997). Conformational stability of the pre-dicted structures was analyzed by exploring the forma-tion of hydrogen bonds using PTRAJ command, polarcontacts and salt bridges using VMD package (Hum-phrey, Dalke, & Schulten, 1996). The ab initio predictedmodel was compared with a short peptide (12 residues)NMR resolved α-MSH synthetic analog (PDB id 1BOQ;181atoms) (Giblin, Wang, Hoffman, Jurisson, & Quinn,1998) by subjecting it to MD simulation (150 ns) usingthe same set of parameters. This NMR-derived structure,despite sharing the same sequence motif ‘HFRW’ withACTH, did not display any reverse β strand when visual-ized using any visualization tool.
Docking studies
Docking of ACTH model onto its MC2R receptor modelwas carried out using HADDOCK (High AmbiguityDriven protein-protein docking) server (de Vries,van Dijik, & Bonvin, 2010). It is an information-drivenflexible docking approach for the modeling of bimolecularcomplexes. Docking was performed using the easy inter-face program. The active and passive residues of bothmodels were obtained using C-PORT (de Vries & Bonvin,2011) server, an algorithm for the prediction of protein–protein interface residues. Predicted active and passiveresidues were assigned for both the ACTH and MC2R inthe easy interface program. After docking was completed,docking output was recorded and the complexMC2R_ACTH was loaded into Deep View package (Guex& Peitsch, 1997) or PyMOL (DeLano, 2002) package foranalyzing the interacting residues.
Molecular dynamics simulation of MC2R_ACTHcomplex, MC2R and ACTH
Preparation of starting structures
Molecular dynamics simulations of the docked‘MC2R_ACTH’ complex as well as the predicted struc-tures of MC2R (receptor) and ACTH (ligand) were per-formed separately for calculating binding free energy(ΔG) using a combination of quantum mechanics andmolecular mechanics approach as implemented inAMBER 10 package installed on SGI Altix UV 10. Allthe three structures: MC2R_ACTH complex, MC2R, and
Human adrenocorticotropic hormone 3
Dow
nloa
ded
by [
122.
167.
6.57
] at
22:
55 0
4 M
ay 2
014

ACTH were loaded using LEaP tool with ‘ff99SB’ asthe force field. Upon addition of hydrogen atoms, theinitial topology and coordinates files were generated forall the three models. Implicit solvation effects using gen-eralized Born model (Still, Tempczyk, & Hawley, 1990)were introduced for all the three simulations as explainedearlier for ACTH model.
Heating and equilibration
All the three 3D models: MC2R_ACTH complex (5515atoms), MC2R (4856 atoms), and ACTH (659 atoms)were subjected to energy minimization, heating, andequilibration for a total time of 20 ns spread over fourstages for the receptor as well as the complex and sevenstages for the ligand using SANDER program followingthe same parameters as explained earlier with minormodification introduced at equilibration stage. While thecut-off to truncate the non-bonded interactions betweenatom pairs was set to 16 Å, relative geometrical tolerancefor coordinate resetting in SHAKE algorithm was set todefault (0.00001). AMBER package must have takenentropy contribution into consideration while simulatingthe molecule using implicit condition. That is to say, byNOT including nscm flag in all the input script files,translational and rotational motions of the atoms werecompletely removed while writing to the output files atthe end of every simulation step. In other words, valuesfor various variables (bond, angles, and electrostatic,etc.) in the output files are shown after subtracting theentropy energy. MD simulation output from all the threemodels were separately analyzed using the PTRAJ com-mand as well as perl script. Energy and RMSD plotswere obtained using the GRACE software package.Summary of salt bridges formed by MC2R model andMC2R_ACTH complex before and after MD simulationwas computed by VMD package.
Binding free energy (ΔGbinding) of the reaction due toconformational change was estimated based on the equa-tion proposed by Kollman et al. (2000):
DGðbindÞ ¼ DGcomplexconf � DGreceptor
conf � DGligandconf (1)
here DG�conf s the conformational free energy.
For all the three models (MC2R_ACTH complex,MC2R, and ACTH), conformational free energy wascomputed as:
Sum total of conformational energy in the gas phase(ΔGconf (g)) (also referred as vacuum) and gas to solva-tion free energy (ΔGsolv) minus entropy componentTSMM.
ΔGconf (g) is obtained from quantum mechanical cal-culation which involve sums of terms over all the inter-nal coordinates of the molecule; this includes ΔEinternal
(internal energy) arising from bond, angle, and dihedral
terms, ΔEelect (electrostatic) energy and ΔEVDW (van derWaals) energies, in the molecular mechanics (MM) forcefield.
ΔGsolv is the sum of non-polar (ΔGsolvGBSA: the non-electrostatic contribution to the solvation free energy cal-culated by solvent accessible surface area) and polar(ΔGsolvGB: the electrostatic contribution to the solvationfree energy calculated by generalized Born (GB) method.
TΔSMM the entropic cost of fixing rotational andtranslational degree of freedom. In our study, solvationentropy is included through ΔGsolv by removing entropyat the end of every simulation step of MD simulationwhile writing the trajectories to the output file. Whilesolvation entropy is included through ΔGsolv, conforma-tional entropy changes upon binding are neglected.
Statistical error
In MD simulation, samples are correlated. To accountfor this, one may compute averages for blocks of dataand use these block averages to estimate the variance.AMBER package generates root mean square fluctuation(RMSF) data, which is nothing but standard deviation, atthe end of equilibration stage (called as block, n). Thesevalues when squared give the variance.
For instance, if there are n blocks (x, y,… n),
Average varianceðzÞ ¼ ðvarðxÞ þ varðyÞ þ . . .varðnÞÞ=n
These data are later used to estimate the standard error (ε)of the mean associated with the variable of interest (A)using the equation suggested by David and Michael (2008):
�2ðAÞ � r2ðAÞn� 1
(2.1)
Alternatively,
�ðAÞ � rðAÞffiffiffiffiffiffiffiffiffiffiffi
n� 1p (2.2)
Percent relative standard error (RSE) of the variable A iscomputed using the formula
% RSEðAÞ ¼ �ðAÞ�X ðAÞ � 100 (2.3)
where �X ðAÞ is the block average of variable A.
Results and discussion
Molecular modeling studies
Melanocortin 2 receptor
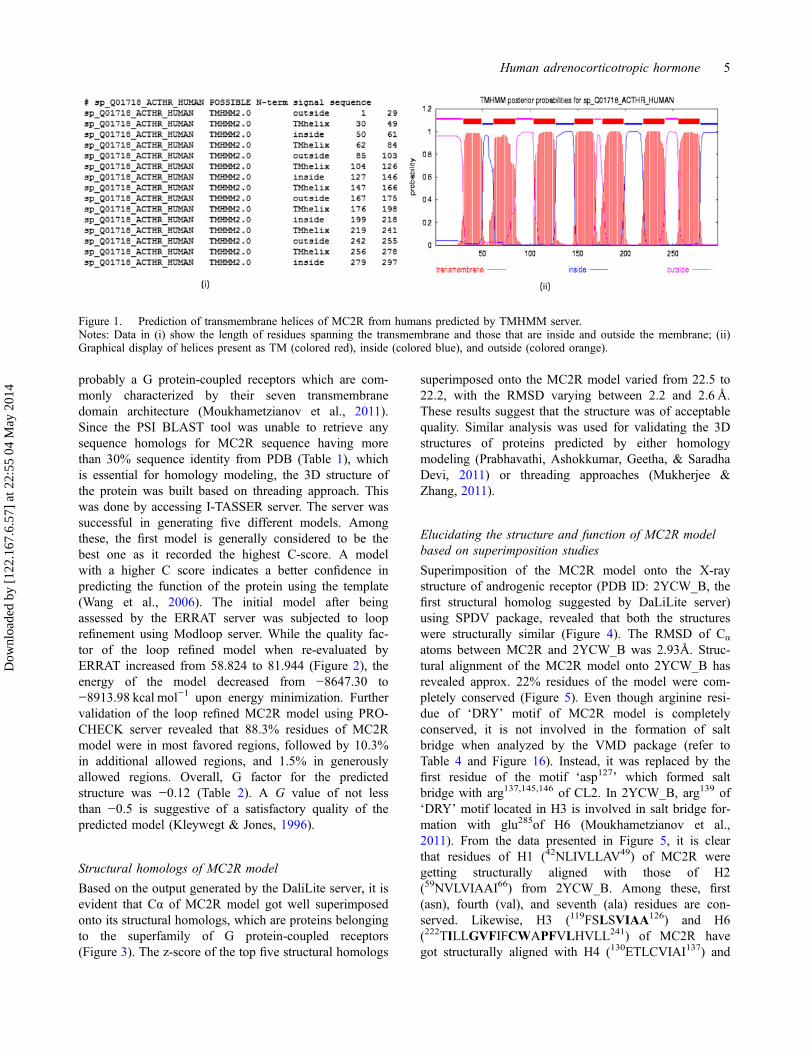
Based on MC2R sequence information, the TMHMMserver identified seven transmembrane helices, fourcytoplasmic loops, and four extracellular loops (Figure 1).These results suggest that the MC2R sequence is
4 M. Dieudonné and K.V. Ramesh
Dow
nloa
ded
by [
122.
167.
6.57
] at
22:
55 0
4 M
ay 2
014
probably a G protein-coupled receptors which are com-monly characterized by their seven transmembranedomain architecture (Moukhametzianov et al., 2011).Since the PSI BLAST tool was unable to retrieve anysequence homologs for MC2R sequence having morethan 30% sequence identity from PDB (Table 1), whichis essential for homology modeling, the 3D structure ofthe protein was built based on threading approach. Thiswas done by accessing I-TASSER server. The server wassuccessful in generating five different models. Amongthese, the first model is generally considered to be thebest one as it recorded the highest C-score. A modelwith a higher C score indicates a better confidence inpredicting the function of the protein using the template(Wang et al., 2006). The initial model after beingassessed by the ERRAT server was subjected to looprefinement using Modloop server. While the quality fac-tor of the loop refined model when re-evaluated byERRAT increased from 58.824 to 81.944 (Figure 2), theenergy of the model decreased from −8647.30 to−8913.98 kcal mol−1 upon energy minimization. Furthervalidation of the loop refined MC2R model using PRO-CHECK server revealed that 88.3% residues of MC2Rmodel were in most favored regions, followed by 10.3%in additional allowed regions, and 1.5% in generouslyallowed regions. Overall, G factor for the predictedstructure was −0.12 (Table 2). A G value of not lessthan −0.5 is suggestive of a satisfactory quality of thepredicted model (Kleywegt & Jones, 1996).
Structural homologs of MC2R model
Based on the output generated by the DaliLite server, it isevident that Cα of MC2R model got well superimposedonto its structural homologs, which are proteins belongingto the superfamily of G protein-coupled receptors(Figure 3). The z-score of the top five structural homologs
superimposed onto the MC2R model varied from 22.5 to22.2, with the RMSD varying between 2.2 and 2.6 Å.These results suggest that the structure was of acceptablequality. Similar analysis was used for validating the 3Dstructures of proteins predicted by either homologymodeling (Prabhavathi, Ashokkumar, Geetha, & SaradhaDevi, 2011) or threading approaches (Mukherjee &Zhang, 2011).
Elucidating the structure and function of MC2R modelbased on superimposition studies
Superimposition of the MC2R model onto the X-raystructure of androgenic receptor (PDB ID: 2YCW_B, thefirst structural homolog suggested by DaLiLite server)using SPDV package, revealed that both the structureswere structurally similar (Figure 4). The RMSD of Cα
atoms between MC2R and 2YCW_B was 2.93Å. Struc-tural alignment of the MC2R model onto 2YCW_B hasrevealed approx. 22% residues of the model were com-pletely conserved (Figure 5). Even though arginine resi-due of ‘DRY’ motif of MC2R model is completelyconserved, it is not involved in the formation of saltbridge when analyzed by the VMD package (refer toTable 4 and Figure 16). Instead, it was replaced by thefirst residue of the motif ‘asp127’ which formed saltbridge with arg137,145,146 of CL2. In 2YCW_B, arg139 of‘DRY’ motif located in H3 is involved in salt bridge for-mation with glu285of H6 (Moukhametzianov et al.,2011). From the data presented in Figure 5, it is clearthat residues of H1 (42NLIVLLAV49) of MC2R weregetting structurally aligned with those of H2(59NVLVIAAI66) from 2YCW_B. Among these, first(asn), fourth (val), and seventh (ala) residues are con-served. Likewise, H3 (119FSLSVIAA126) and H6(222TILLGVFIFCWAPFVLHVLL241) of MC2R havegot structurally aligned with H4 (130ETLCVIAI137) and
Figure 1. Prediction of transmembrane helices of MC2R from humans predicted by TMHMM server.Notes: Data in (i) show the length of residues spanning the transmembrane and those that are inside and outside the membrane; (ii)Graphical display of helices present as TM (colored red), inside (colored blue), and outside (colored orange).
Human adrenocorticotropic hormone 5
Dow
nloa
ded
by [
122.
167.
6.57
] at
22:
55 0
4 M
ay 2
014
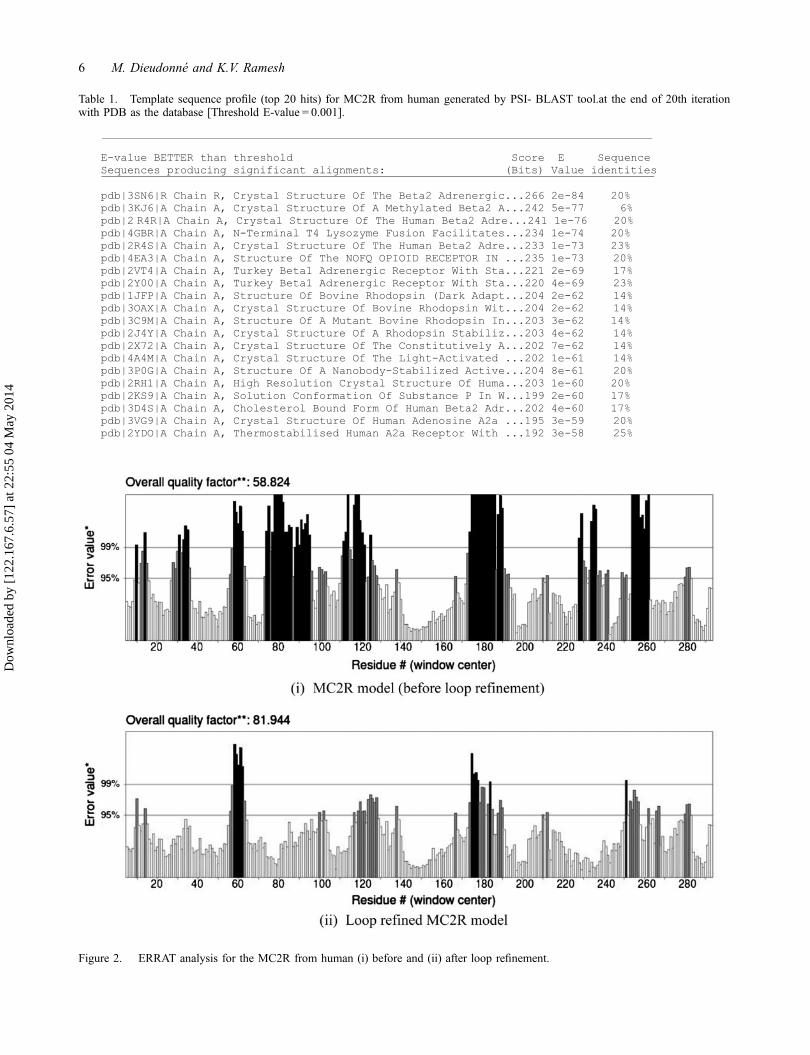
Table 1. Template sequence profile (top 20 hits) for MC2R from human generated by PSI- BLAST tool.at the end of 20th iterationwith PDB as the database [Threshold E-value = 0.001].
Figure 2. ERRAT analysis for the MC2R from human (i) before and (ii) after loop refinement.
6 M. Dieudonné and K.V. Ramesh
Dow
nloa
ded
by [
122.
167.
6.57
] at
22:
55 0
4 M
ay 2
014

H7 (264GIIMGVFTLCWLPFFLVNIV286) of 2YCW_Brespectively. Structural alignment data show that fourresidues (leu121, val123, ile124, and ala125) of H3 and nineresidues (ile223, gly226, val227, phe228, cys231, trp232,
pro234, phe235, and leu237) of H6 of MC2R model werefully conserved. From the same data we can notice thattwo residue pairs (‘174TV175’ and ‘211PR212’) have notaligned with 2YCW_B. This has resulted in the
Table 2. Summary of PROCHECK analysis for MC2R model from human.
Figure 3. Superimposition of top five structural homologs of MC2R from human generated by DaliLite server.Notes: MC2R_A: green, 2YCW_B: brick red, 4AMI_B: grey, 2YCW_A: orange, 4AMJ_B: dark blue and 2YO3_B: light blue.
Human adrenocorticotropic hormone 7
Dow
nloa
ded
by [
122.
167.
6.57
] at
22:
55 0
4 M
ay 2
014
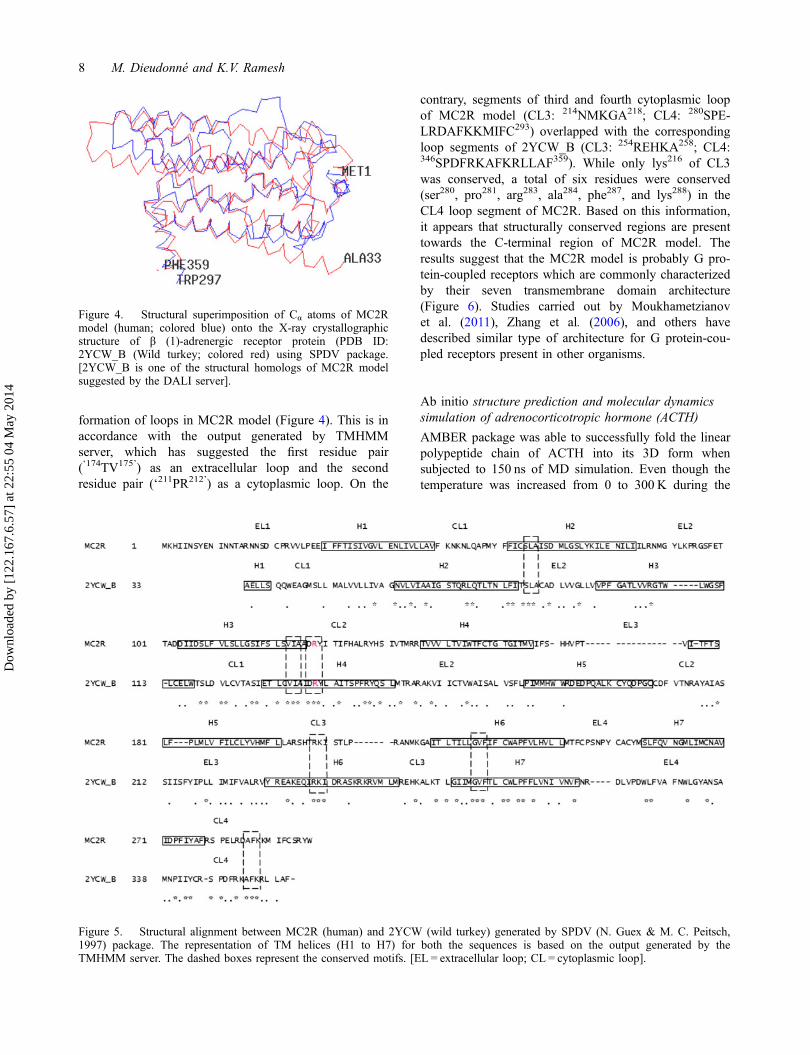
formation of loops in MC2R model (Figure 4). This is inaccordance with the output generated by TMHMMserver, which has suggested the first residue pair(‘174TV175’) as an extracellular loop and the secondresidue pair (‘211PR212’) as a cytoplasmic loop. On the
contrary, segments of third and fourth cytoplasmic loopof MC2R model (CL3: 214NMKGA218; CL4: 280SPE-LRDAFKKMIFC293) overlapped with the correspondingloop segments of 2YCW_B (CL3: 254REHKA258; CL4:346SPDFRKAFKRLLAF359). While only lys216 of CL3was conserved, a total of six residues were conserved(ser280, pro281, arg283, ala284, phe287, and lys288) in theCL4 loop segment of MC2R. Based on this information,it appears that structurally conserved regions are presenttowards the C-terminal region of MC2R model. Theresults suggest that the MC2R model is probably G pro-tein-coupled receptors which are commonly characterizedby their seven transmembrane domain architecture(Figure 6). Studies carried out by Moukhametzianovet al. (2011), Zhang et al. (2006), and others havedescribed similar type of architecture for G protein-cou-pled receptors present in other organisms.
Ab initio structure prediction and molecular dynamicssimulation of adrenocorticotropic hormone (ACTH)
AMBER package was able to successfully fold the linearpolypeptide chain of ACTH into its 3D form whensubjected to 150 ns of MD simulation. Even though thetemperature was increased from 0 to 300 K during the
Figure 4. Structural superimposition of Cα atoms of MC2Rmodel (human; colored blue) onto the X-ray crystallographicstructure of β (1)-adrenergic receptor protein (PDB ID:2YCW_B (Wild turkey; colored red) using SPDV package.[2YCW_B is one of the structural homologs of MC2R modelsuggested by the DALI server].
Figure 5. Structural alignment between MC2R (human) and 2YCW (wild turkey) generated by SPDV (N. Guex & M. C. Peitsch,1997) package. The representation of TM helices (H1 to H7) for both the sequences is based on the output generated by theTMHMM server. The dashed boxes represent the conserved motifs. [EL = extracellular loop; CL = cytoplasmic loop].
8 M. Dieudonné and K.V. Ramesh
Dow
nloa
ded
by [
122.
167.
6.57
] at
22:
55 0
4 M
ay 2
014
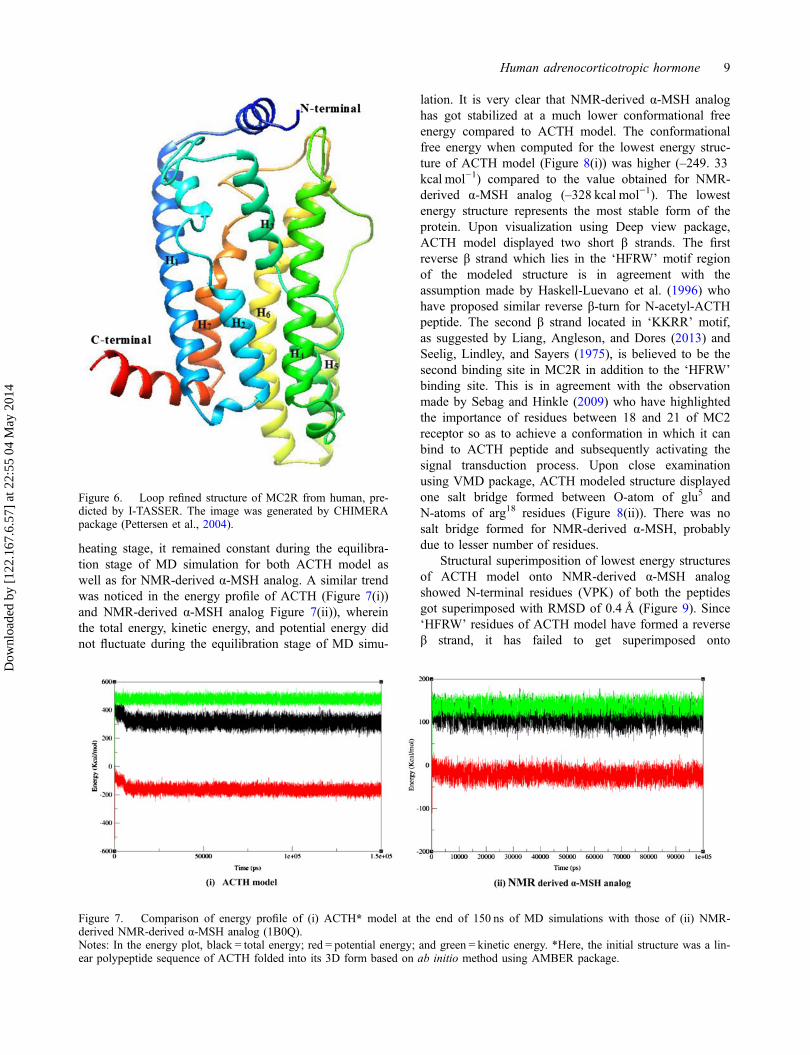
heating stage, it remained constant during the equilibra-tion stage of MD simulation for both ACTH model aswell as for NMR-derived α-MSH analog. A similar trendwas noticed in the energy profile of ACTH (Figure 7(i))and NMR-derived α-MSH analog Figure 7(ii)), whereinthe total energy, kinetic energy, and potential energy didnot fluctuate during the equilibration stage of MD simu-
lation. It is very clear that NMR-derived α-MSH analoghas got stabilized at a much lower conformational freeenergy compared to ACTH model. The conformationalfree energy when computed for the lowest energy struc-ture of ACTH model (Figure 8(i)) was higher (–249. 33kcal mol−1) compared to the value obtained for NMR-derived α-MSH analog (–328 kcal mol−1). The lowestenergy structure represents the most stable form of theprotein. Upon visualization using Deep view package,ACTH model displayed two short β strands. The firstreverse β strand which lies in the ‘HFRW’ motif regionof the modeled structure is in agreement with theassumption made by Haskell-Luevano et al. (1996) whohave proposed similar reverse β-turn for N-acetyl-ACTHpeptide. The second β strand located in ‘KKRR’ motif,as suggested by Liang, Angleson, and Dores (2013) andSeelig, Lindley, and Sayers (1975), is believed to be thesecond binding site in MC2R in addition to the ‘HFRW’binding site. This is in agreement with the observationmade by Sebag and Hinkle (2009) who have highlightedthe importance of residues between 18 and 21 of MC2receptor so as to achieve a conformation in which it canbind to ACTH peptide and subsequently activating thesignal transduction process. Upon close examinationusing VMD package, ACTH modeled structure displayedone salt bridge formed between O-atom of glu5 andN-atoms of arg18 residues (Figure 8(ii)). There was nosalt bridge formed for NMR-derived α-MSH, probablydue to lesser number of residues.
Structural superimposition of lowest energy structuresof ACTH model onto NMR-derived α-MSH analogshowed N-terminal residues (VPK) of both the peptidesgot superimposed with RMSD of 0.4 Å (Figure 9). Since‘HFRW’ residues of ACTH model have formed a reverseβ strand, it has failed to get superimposed onto
Figure 6. Loop refined structure of MC2R from human, pre-dicted by I-TASSER. The image was generated by CHIMERApackage (Pettersen et al., 2004).
Figure 7. Comparison of energy profile of (i) ACTH* model at the end of 150 ns of MD simulations with those of (ii) NMR-derived NMR-derived α-MSH analog (1B0Q).Notes: In the energy plot, black = total energy; red = potential energy; and green = kinetic energy. *Here, the initial structure was a lin-ear polypeptide sequence of ACTH folded into its 3D form based on ab initio method using AMBER package.
Human adrenocorticotropic hormone 9
Dow
nloa
ded
by [
122.
167.
6.57
] at
22:
55 0
4 M
ay 2
014

NMR-derived α-MSH analog. Even though α-MSHanalog was subjected to prolonged MD simulation (150ns), there was no formation of reverse β strand. This
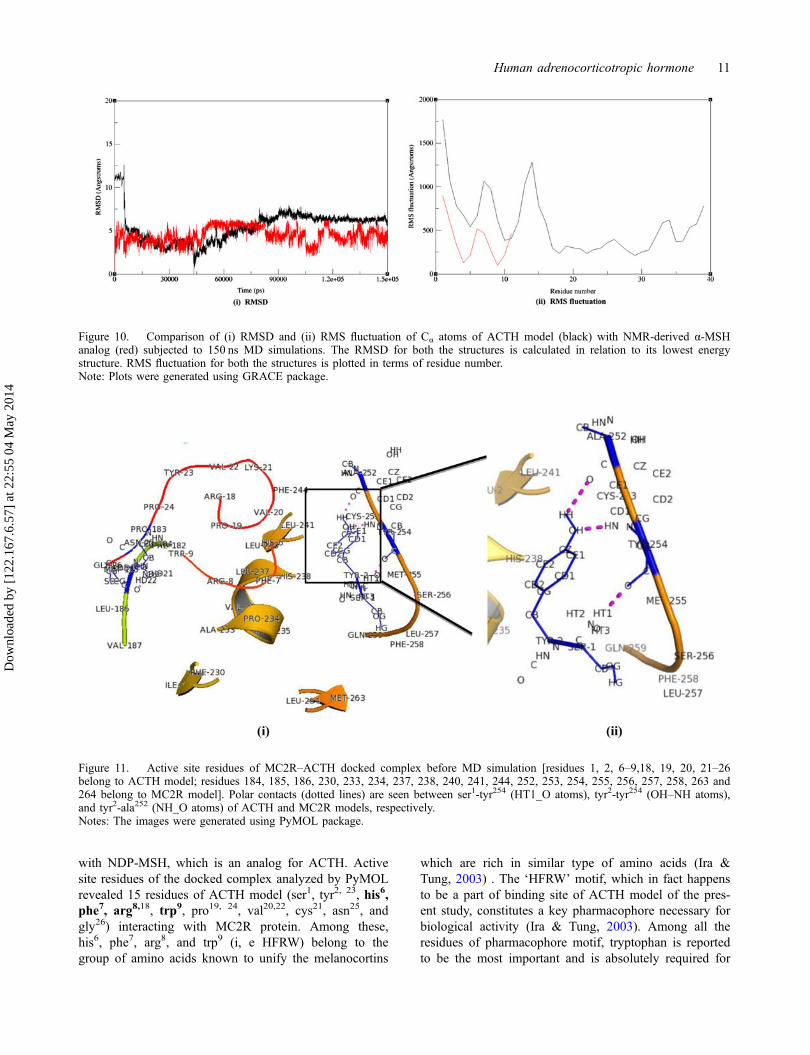
could be attributed to the lesser number of residues (12)of the peptide. On the contrary, ACTH peptide consistedof 39 residues and some of which must have assisted forthe formation of reverse β strand upon MD simulation.Further, based on RMSD plot generated after the com-pletion of MD simulation, it is very much evident thatthere is a near overlapping of Cα atoms of both ACTH-and NMR-derived α-MSH analog between 65 and 75 ns.For the rest of the simulation time, the RMSD plot ofNMR-derived α-MSH analog is higher than that ofACTH model. However, the RMSD plot of ACTHmodel is less fluctuating compared to that of NMR-derived structure after 90 ns till the end of simulationtime with the deviation of Cα atoms being within 1 Å(Figure 10(i)). Based on this, it is possible to concludethat, after an initial structural rearrangement of ACTHmodel, the structure appears to have attained conforma-tional stability after 90 ns. To bring clarity to the stabilityof ACTH model, RMSF has been compared with that ofNMR-derived α-MSH analog (Figure 10(ii)) suggestingboth of these structures do share similar type of residualfluctuation pattern. However, magnitude of these fluctua-tions in NMR-derived structure was lesser compared toACTH model. This could be due to the fact that NMR-derived structure is experimentally determined withhigher stability compared to our ACTH model.
Docking studies (before MD simulation)
Among the 10 docked clusters generated by HADDOCKserver for ACTH and MC2R, the second pose of thesecond cluster having dock energy of −89.0 kcal mol−1
(Figure 11(i)) was selected for further analysis. Thisselection is based on the binding site residues reportedby Pogozheva et al. (2005) for MC4R model interacting
Figure 8. (i) Lowest energy structure of ACTH model predicted by ab initio method (−249.33 kcal mol−1). The image was gener-ated using PyMOL package (ii) Salt bridges formed between the O atom (glu) and N atoms (arg) in the lowest energy structure ofACTH at the end of 150 ns MD simulations. Distances (Å) between the atoms are represented as dotted lines. The image was gener-ated using VMD package.
Figure 9. Superimposition of lowest energy structure ofACTH predicted by ab initio method (magenta color) ontoNMR-derived α-MSH analog structure (cyan color). Notice theoverlapping of N-terminal residues (VPK) in both the struc-tures.
10 M. Dieudonné and K.V. Ramesh
Dow
nloa
ded
by [
122.
167.
6.57
] at
22:
55 0
4 M
ay 2
014
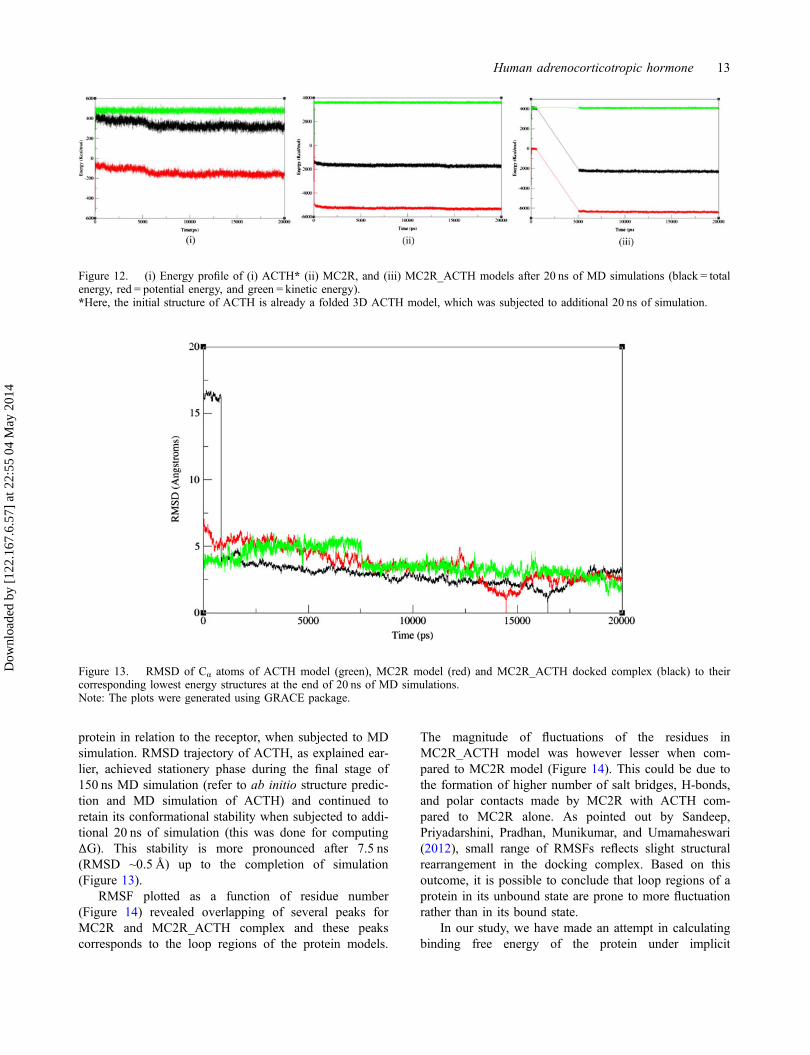
with NDP-MSH, which is an analog for ACTH. Activesite residues of the docked complex analyzed by PyMOLrevealed 15 residues of ACTH model (ser1, tyr2, 23, his6,phe7, arg8,18, trp9, pro19, 24, val20,22, cys21, asn25, andgly26) interacting with MC2R protein. Among these,his6, phe7, arg8, and trp9 (i, e HFRW) belong to thegroup of amino acids known to unify the melanocortins
which are rich in similar type of amino acids (Ira &Tung, 2003) . The ‘HFRW’ motif, which in fact happensto be a part of binding site of ACTH model of the pres-ent study, constitutes a key pharmacophore necessary forbiological activity (Ira & Tung, 2003). Among all theresidues of pharmacophore motif, tryptophan is reportedto be the most important and is absolutely required for
Figure 10. Comparison of (i) RMSD and (ii) RMS fluctuation of Cα atoms of ACTH model (black) with NMR-derived α-MSHanalog (red) subjected to 150 ns MD simulations. The RMSD for both the structures is calculated in relation to its lowest energystructure. RMS fluctuation for both the structures is plotted in terms of residue number.Note: Plots were generated using GRACE package.
Figure 11. Active site residues of MC2R–ACTH docked complex before MD simulation [residues 1, 2, 6–9,18, 19, 20, 21–26belong to ACTH model; residues 184, 185, 186, 230, 233, 234, 237, 238, 240, 241, 244, 252, 253, 254, 255, 256, 257, 258, 263 and264 belong to MC2R model]. Polar contacts (dotted lines) are seen between ser1-tyr254 (HT1_O atoms), tyr2-tyr254 (OH–NH atoms),and tyr2-ala252 (NH_O atoms) of ACTH and MC2R models, respectively.Notes: The images were generated using PyMOL package.
Human adrenocorticotropic hormone 11
Dow
nloa
ded
by [
122.
167.
6.57
] at
22:
55 0
4 M
ay 2
014

activation of the receptor (Liang, Angleson, & Dores2013). Along with HFRW motif, arg18 present in the sec-ond motif region (‘KKRR’) also required for the activa-tion of MC2R (Seelig, Lindley, & Sayers, 1975).PyMOL has also revealed 20 residues of MC2R modelinvolved in the interaction with the ACTH model(Figure 11 (ii)). Majority of the interacting residues ishydrophobic in nature and are part of transmembranehelices (H5: leu 184,186, met185; H6: phe230, ala233,pro234, leu237, his238, leu240,241, and H7: ser256, leu257,phe258, met263, and leu264). Among the 20 interactingresidues, 5 of them are localized in fourth extracellularloop regions of MC2R (phe244, ala252, cys253, tyr254 andmet255). These observations strongly suggest that ACTHmodel has got docked into the pocket occupied by theresidues belonging to H5, H6, H7, and EL4. Theseresults are in agreement with the explanations given byLiang and his co-workers (2013), who have noticed sixconserved binding sites in human MC2R (H2_glu
80,
H3_ser108, H6_cys
231, pro234, leu237, and H7_phe258). From
these observations, it may be noted that the last threeresidues (pro234, leu237 (H6), and phe258 (H7)) areappearing at the binding site interface of MC2R–ACTHdocked complex. It may also be noted from the presentstudy that none of the residues of H1–4, EL1–3, andCLs of MC2R protein were seen to be involved in inter-action with ACTH protein.
Polar contacts
Among the 15 residues of ACTH model interacting withMC2R model, only three residues formed inter-polarcontacts. The H and N atoms (HT1, NH) of ser1 and tyr2
from ACTH model were involved in forming polar con-tact with O atoms (O) of tyr254 and ala252 from MC2Rmodel, respectively (Figure 11(ii)). The O atom (OH) oftyr2 from ACTH was in contact with N atom (NH) oftyr254 residue from MC2R. Although, there are manyexperimental investigations which prove the affinity ofACTH peptide towards MC2R protein (Adan et al.,1994; Jarl, 1999; Mountjoy, Robbins, Mortrud, & Cone,1992), none of these studies provide details of structuralinteraction of the proteins. Outcome from the presentdocking exercise provides a very strong evidence ofinteraction at molecular level between ACTH and MC2Rproteins, thereby substantiating the earlier works.
MD simulation studies for computing free bindingenergy
Results of MD simulations for ACTH model, MC2Rmodel, and MC2R_ACTH docked complex were ana-lyzed for various parameters such root mean square devi-ation (RMSD), RMSF, binding free energy, polarcontacts, and salt bridge formation. Kinetic energy was
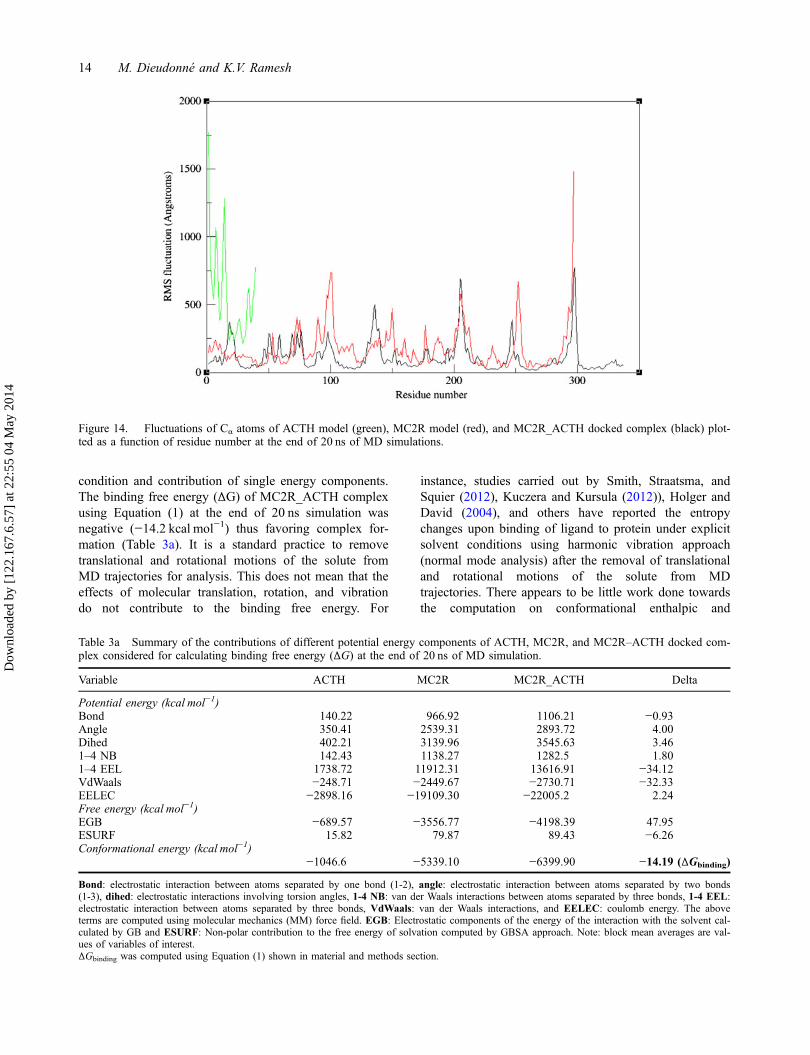
stable for all the three systems (Figure 12) as expected,since temperature which is directly proportional tokinetic energy was constant during MD simulation pro-cess. However, the first 5 ns of simulation of the com-plex shows a sudden drop in energy plot (Figure 12(iii));one of the probable reasons for this behavior could bethe conformational changes which might have takenplace while the ligand tried to locate the proper bindingpocket for the receptor. Other factors contributing for thedrastic decrease in energy level of the complex structureneed to be further explored. Nevertheless, the plot showsstability after 5 ns till the end of simulation period.
The lowest energy structure of ACTH model at theend of additional 20 ns of simulation had a conforma-tional energy of −1109.90 kcal mol−1, which is lessercompared to the value (−249.24 kcal mol−1) obtainedduring the initial 150 ns of MD simulation. Further,based on the data presented in Table 3a, it is evident thatconformational free energy computed as the sum ofpotential energy and free energy terms for the complex(MC2R_ACTH) is lesser (−6399.90 kcal mol−1) than thatof receptor (MC2R) (−5339.10 kcal mol−1) suggestingthe stability of the docked protein–protein complex. In aMD simulation study conducted by Abhinav et al.(2011) on Hsp90/Cdc37 protein docked with the ligand‘withaferin,’ energy profile of the docked complex waslesser compared to the receptor protein, suggesting thestability of the docked drug–protein complex.
The RMSD plot of MC2R and MC2R_ACTH gener-ated with reference to their corresponding lowest energystructure showed similar trend (Figure 13). However, thefact that RMSD dropped from 15A to 5A within 1 ns forMC2R_ACTH; the probable reason could be the same asexplained earlier for the complex energy fluctuation.Other factors contributing for the drastic decrease inRMSD level of the complex structure need to be furtherstudied. Nevertheless, both, MC2R and MC2R_ACTHappear to have attained structural stability after 1 ns ofsimulation which remained almost consistent for the restof the simulation time. Structural drift measured in termsof deviation of Cα atoms of MC2R and MC2R_ACTHstructures from 1 to 20 ns was about 1 Å. The results inFigure 13 show that RMSD of the complex was lowerthan the receptor protein. The extent of deviation of Cα
atoms in MC2R and MC2R_ACTH complex was higherduring the initial stages of simulation. This is becausethe initial structures taken for our simulation studieswere modeled structures. The deviation however gotdiminished during the later stages of simulation. As thesimulation progressed, ACTH in association with MC2Rtried to make interactions with the solvent (i.e., implicitcondition) and thus stabilizes the complex structurethereby resulting in lowering the RMSD trajectory incomparison to the MC2R model alone. Abhinav et al.(2011) have reported lowering of RMSD of the complex
12 M. Dieudonné and K.V. Ramesh
Dow
nloa
ded
by [
122.
167.
6.57
] at
22:
55 0
4 M
ay 2
014
protein in relation to the receptor, when subjected to MDsimulation. RMSD trajectory of ACTH, as explained ear-lier, achieved stationery phase during the final stage of150 ns MD simulation (refer to ab initio structure predic-tion and MD simulation of ACTH) and continued toretain its conformational stability when subjected to addi-tional 20 ns of simulation (this was done for computingΔG). This stability is more pronounced after 7.5 ns(RMSD ~0.5 Å) up to the completion of simulation(Figure 13).
RMSF plotted as a function of residue number(Figure 14) revealed overlapping of several peaks forMC2R and MC2R_ACTH complex and these peakscorresponds to the loop regions of the protein models.
The magnitude of fluctuations of the residues inMC2R_ACTH model was however lesser when com-pared to MC2R model (Figure 14). This could be due tothe formation of higher number of salt bridges, H-bonds,and polar contacts made by MC2R with ACTH com-pared to MC2R alone. As pointed out by Sandeep,Priyadarshini, Pradhan, Munikumar, and Umamaheswari(2012), small range of RMSFs reflects slight structuralrearrangement in the docking complex. Based on thisoutcome, it is possible to conclude that loop regions of aprotein in its unbound state are prone to more fluctuationrather than in its bound state.
In our study, we have made an attempt in calculatingbinding free energy of the protein under implicit
Figure 12. (i) Energy profile of (i) ACTH* (ii) MC2R, and (iii) MC2R_ACTH models after 20 ns of MD simulations (black = totalenergy, red = potential energy, and green = kinetic energy).*Here, the initial structure of ACTH is already a folded 3D ACTH model, which was subjected to additional 20 ns of simulation.
Figure 13. RMSD of Cα atoms of ACTH model (green), MC2R model (red) and MC2R_ACTH docked complex (black) to theircorresponding lowest energy structures at the end of 20 ns of MD simulations.Note: The plots were generated using GRACE package.
Human adrenocorticotropic hormone 13
Dow
nloa
ded
by [
122.
167.
6.57
] at
22:
55 0
4 M
ay 2
014
condition and contribution of single energy components.The binding free energy (ΔG) of MC2R_ACTH complexusing Equation (1) at the end of 20 ns simulation wasnegative (−14.2 kcal mol−1) thus favoring complex for-mation (Table 3a). It is a standard practice to removetranslational and rotational motions of the solute fromMD trajectories for analysis. This does not mean that theeffects of molecular translation, rotation, and vibrationdo not contribute to the binding free energy. For
instance, studies carried out by Smith, Straatsma, andSquier (2012), Kuczera and Kursula (2012)), Holger andDavid (2004), and others have reported the entropychanges upon binding of ligand to protein under explicitsolvent conditions using harmonic vibration approach(normal mode analysis) after the removal of translationaland rotational motions of the solute from MDtrajectories. There appears to be little work done towardsthe computation on conformational enthalpic and
Table 3a Summary of the contributions of different potential energy components of ACTH, MC2R, and MC2R–ACTH docked com-plex considered for calculating binding free energy (ΔG) at the end of 20 ns of MD simulation.
Variable ACTH MC2R MC2R_ACTH Delta
Potential energy (kcal mol−1)Bond 140.22 966.92 1106.21 −0.93Angle 350.41 2539.31 2893.72 4.00Dihed 402.21 3139.96 3545.63 3.461–4 NB 142.43 1138.27 1282.5 1.801–4 EEL 1738.72 11912.31 13616.91 −34.12VdWaals −248.71 −2449.67 −2730.71 −32.33EELEC −2898.16 −19109.30 −22005.2 2.24Free energy (kcal mol−1)EGB −689.57 −3556.77 −4198.39 47.95ESURF 15.82 79.87 89.43 −6.26Conformational energy (kcal mol−1)
−1046.6 −5339.10 −6399.90 −14.19 (ΔGbinding)
Bond: electrostatic interaction between atoms separated by one bond (1-2), angle: electrostatic interaction between atoms separated by two bonds(1-3), dihed: electrostatic interactions involving torsion angles, 1-4 NB: van der Waals interactions between atoms separated by three bonds, 1-4 EEL:electrostatic interaction between atoms separated by three bonds, VdWaals: van der Waals interactions, and EELEC: coulomb energy. The aboveterms are computed using molecular mechanics (MM) force field. EGB: Electrostatic components of the energy of the interaction with the solvent cal-culated by GB and ESURF: Non-polar contribution to the free energy of solvation computed by GBSA approach. Note: block mean averages are val-ues of variables of interest.ΔGbinding was computed using Equation (1) shown in material and methods section.
Figure 14. Fluctuations of Cα atoms of ACTH model (green), MC2R model (red), and MC2R_ACTH docked complex (black) plot-ted as a function of residue number at the end of 20 ns of MD simulations.
14 M. Dieudonné and K.V. Ramesh
Dow
nloa
ded
by [
122.
167.
6.57
] at
22:
55 0
4 M
ay 2
014

entropic changes in implicit condition. In our study,although solvation entropy is included through ΔGsolv,conformational entropy changes due to binding of ligandto the protein is ignored because as the protein goestoward its native state, it experiences entropy loss.Sciretti, Bruscolini, Pelizzola, Pretti, and Jaramillo(2009) have reported the complexity of computing theconformational entropy in a folded state of a protein.MD simulation when carried out under implicit condi-tion, however, does not allow easy way of computingentropy changes upon binding of ligand to the protein,which is one of the limitation (implicit condition), unlikenormal mode analysis (explicit condition). Since therewill be significant reduction in entropy during protein–protein interaction (Brady & Sharp, 1997), entropy con-tribution for calculating binding free energy may beassumed to be of lesser significance. This is also con-firmed by Amzel (1997), who noticed loss of transla-tional entropy associated with the formation of acomplex between two protein molecules in solution.
Analysis done with respect to single energy compo-nent (Table 3a) suggests that electrostatic interactionbetween atoms separated by three bonds strongly favorcomplex formation. This is opposed by the disfavorablecontributions calculated by the electrostatic componentsof the energy of the interaction with the solvent calculatedby GB (referred to as EGB in Table 3a). Solvation freeenergy (sum of EGB: Electrostatic components of theenergy of the interaction with the solvent calculated byGB and ESURF: Non-polar contribution to the free energyof solvation computed by GBSA approach) turns out to be
Table 3b. Summary of the contributions of different potential energy components of ACTH, MC2R, and MC2R- ACTH dockedcomplex considered for calculating Relative Standard Error (RSE) of the mean (Averaged value of all blocks).
VariableACTH MC2R MC2R_ACTH
PE(kcal mol−1) aSE�xð�Þ bRSE (%)
PE(kcal mol−1) SE�xð�Þ RSE (%)
PE(kcal mol−1) SE�xð�Þ RSE (%)
Bond 137.92 4.01 2.91 967.54 15.36 1.59 1101.63 16.23 1.47Angle 352.42 5.46 1.55 2539.09 22.28 0.88 2890.90 23.08 0.80Dihed 406.08 3.53 0.87 3145.67 13.68 0.43 3557.60 14.48 0.411–4 NB 142.71 2.011 1.41 1138.21 8.28 0.72 1284.03 8.77 0.681–4 EEL 1735.88 6.03 0.35 11897.18 26.84 0.23 13578.42 27.42 0.20VdWaals −251.82 3.64 1.45 −2392.18 17.56 0.73 −2731.64 20.47 0.75EELEC −2950.42 45.26 1.53 −19111.6 261.01 1.37 −22003.30 298.71 1.36Free energy (kcal mol−1)EGB −627.17 43.82 6.99 −3549.69 257.85 7.26 −4127.72 294.05 7.12ESURF 14.63 0.36 2.43 84.67 1.32 1.56 89.09 1.27 1.42
Note: (i) While potential energy (PE) for ACTH represents the average value calculated from seven equilibration stages, for MC2R and MC2R_ACTH,the average value is computed from four equilibration stages.(ii) In case of ACTH model, equilibration stage 1 to 5 was carried out for 1,000,000 number of steps (2 ns each) with number of snap shots = 2000 /stage, the last two stages (6 and 7) were carried out for 2,500,000 number of steps (5 ns each) with number of snap shots = 5000/stage(iii) In case of MC2R and MC2R_ACTH models, each of the equilibration stage was carried out for 2,500,000 number of steps (5 ns each) with num-ber of snap shots = 5000/stage].aSE�x = standard error of mean; bRSE = relative standard error.bSE�xð2Þ was computed using equation 2.2 shown in material and methods section.
Figure 15. Superimposition of MC2R_ACTH docked com-plex (before and after MD simulation). RMSD = 1.46Å.MC2R_ACTH before MD is shown as magenta green colorcombination and after MD is represented as cyan blue combi-nation. The image is generated using chimera package.
Human adrenocorticotropic hormone 15
Dow
nloa
ded
by [
122.
167.
6.57
] at
22:
55 0
4 M
ay 2
014

positive (41.69), thereby disfavoring the complex forma-tion. This is in general agreement with several other stud-ies (Holger, Kiel, & David, 2003; Huo, Massova, &Kollman, 2002; Massova & Kollman, 2000). On thecontrary, Sheinerman and Honig (2002) have noticed elec-trostatic interactions favoring the complex formation.Even though some internal energy terms (angle and dihed)values are positive, disfavoring the complex formation,contribution from van der Waals interaction, one of themain component favoring the complex formation, as wellas non-polar part of solvation free energy are negative(Table 3a) favoring the complex formation. According tothe studies made by Holger and David (2004) on Ras–Rafprotein, favorable contribution to binding affinity arisefrom van der Waals interactions as well as non-polar partof solvation free energy. Low-standard error values esti-mated for all the variables (Table 3b) imply that the simu-lation was reasonably accurate. Holger and David (2004)have used std. error of mean to report the accuracy ofbinding energy components for Ras–Raf complex aftersubjecting it to MD simulation.
MC2R_ACTH complex upon MD simulation under-went conformational changes (Figure 15), which resultedin bringing many of the interacting residues of these twoproteins much closer. For instance, there was break up of
existing polar contacts before simulation and formationof new ones as a result of solvent effect induced by MDsimulation. In both the models, three atoms of O (tyr2,asn25, and phe7) from ACTH were involved in non-cova-lent H bonding with two atoms of O (tyr254, lys186 andN atom of his238 from MC2R, respectively (Figure 16).
Conformational changes in MC2R_ACTH complexas a result of MD simulation were also confirmed byobserving an increase in the number of salt bridges(Table 4). Salt bridges were formed by the O atoms ofaspartate and glutamate with the N atoms of arginine,histidine as well as lysine. The data presented in Table 4show a substantial increase in the number of salt bridgesformed after the completion of simulation. The data alsoreveal the absence of salt bridge formation betweenMC2R and ACTH before the start of simulation process.However, there was a drastic increase in the number ofsalt bridges formed by the MC2R_ACTH docked com-plex at intra-molecular level. Among three salt bridgesthat were initially formed within MC2R component ofthe complex structure (asp103-lys93; glu80-arg95; andglu80-lys93), only one was retained (asp103-lys93) afterthe completion of simulation with minor variationnoticed in the distances of interacting atoms. In theACTH component of the complex structure, salt bridge
Figure 16. Polar contact (dotted lines) established between the residues of MC2R_ACTH complex after 20 ns of MD simulation. Inboth the models, three atoms of O (tyr2, asn25, and phe7) from ACTH were involved in non-covalent H bonding with two atoms ofO (tyr254 and lys186) and N atom of his238 from MC2R, respectively.Note: The figure was generated using PyMOL package.
16 M. Dieudonné and K.V. Ramesh
Dow
nloa
ded
by [
122.
167.
6.57
] at
22:
55 0
4 M
ay 2
014

that was reported earlier for unbound ACTH (glu5-arg17)was retained along with three more being formed at theend of 20 ns MD simulation (Table 4). From these obser-vations, it is possible to conclude that a protein structurewhen subjected to a prolonged period of MD simulationattains conformational stability with increased number ofpolar contacts and salt bridges. Holger, Kiel, and David(2003) have noticed the formation of polar contacts andsalt bridges in Ras–Raf protein complex attaining confor-mational stability after the completion of MD simulation.
Output of this research has shed more insight intothe molecular interactions between MC2R and ACTHmodels and thus, may help in the development of mela-nocortin-based drugs.
Summary
In the present study, computational approaches havebeen applied for molecular modeling of MC2R andACTH from human. 3D structure prediction was fol-lowed by docking studies to predict the binding pocketsbetween MC2R and ACTH models. Finally, MD simula-tions were run to investigate the stability ofMC2R_ACTH docked complex structure under implicitsolvent environment. Analyses of the predicted MC2Rand ACTH models have revealed their secondary struc-ture information. While MC2R protein was made up ofseven transmembrane helices (H1-7), four extracellularloops (EL1-4), and four cytoplasmic loops (CL1-4),ACTH model displayed two short beta-sheets. MC2Rgot successfully docked onto ACTH with dock energy of−89.0 kcal mol−1. Energy profile, RMSD, and RMSFrecorded for MC2R_ACTH complex revealed that thestructure attained conformational stability at end of 20 nsof MD simulation. This is clear from the increased num-ber of polar contacts and salt bridges formed in the com-plex. Binding free energy being negative for
MC2R_ACTH docked complex suggests that this couldbe a thermodynamically spontaneous reaction.
AcknowledgmentsWe are grateful to Dr Chenraj Roychand, Chairman, Jain Groupof Institution, Dr Sudha Deshmukh, Dean (Sciences), CPGS,Jain U’sty, and Dr P.C. Deshmukh, Professor of Physics, IndianInstitute of Technology, Chennai for creating the computationalfacilities. We also acknowledge Dr Sunil S. More, Dr Rathore,and Dr Abhay for giving useful points during the progress ofthis work. We also thank Mr Anand and his team members ofLintechnokrat, Bangalore for the efficient maintenance of ourcomputer cluster system.
ReferencesAbhinav, G., Ashutosh, S., Vibhuti, A., Piyush, P., Virendra, S. B.,
Durai, S., & Divya, B. (2011). Hsp90/Cdc37 Chaperone/co-chaperone complex, a novel junction anticancer target eluci-dated by the mode of action of herbal drug Withaferin A.BMC Bioinformatics, 12, S30.
Adan, R. A. H., Oosterom, J., Ludvigsdottir, G., Brakkee, J. H.,Burbach, J. P. H., & Gispen, W. H. (1994). Identification ofantagonists for melanocortin MC3, MC4 and MC5 receptors.European Journal of Pharmacology: Molecular Pharmacol-ogy, 269, 331–337.
Altschul, S. F., Madden, T. L., Schäffer, A. A., Zhang, J., Zhang,Z., Miller, W., & Lipman, D. J. (1997). Gapped BLAST andPSI-BLAST: a new generation of protein database searchprograms. Nucleic Acids Research, 25, 3389–3402.
Amzel, L. M. (1997). Loss of translational entropy in binding,folding, and catalysis. Proteins: Structure, Function, andGenetics, 28, 144–149.
Arlt, W., & Allolio, B. (2003). Adrenal insufficiency. The Lan-cet, 361, 1881–1893.
Berendsen, H. J. C., Postma, J. P. M., van Gunsteren, W. F.,DiNola, A., & Haak, J. R. (1984). Molecular dynamicswith coupling to an external bath. The Journal of ChemicalPhysics, 81, 3684–3690.
Bernard, P. S., & Keit, L. P. (2006). Adrenocorticotropic hor-mone; adrenocortical steroids and their synthetic analogs;inhibitors of the synthesis and actions of adrenocortical
Table 4. Summary of salt bridges formed by MC2R_ACTH complex (before and after MD simulation) computed by VMD pack-age.
Before MD After MD
Intra Intra
MC2R ACTH MC2R ACTH
ASP103-LYS93 (3.60) GLU5-ARG17 (3.61) ASP103-LYS93 (3.91) GLU5-ARG17 (3.77)GLU80-ARG95 (3.38) ASP20-ARG23 (4.03) GLU5-ARG8 (4.38)GLU80-LYS93 (2.99) ASP104-ARG16 (3.43) ASP29-ARG18 (3.54)
ASP127-ARG146 (4.40) GLU38-LYS16 (4.17)ASP285-LYS289(3.54)GLU9-LYS93 (2.50)GLU28-ARG95 (4.00)GLU80-LYS77(2.93)GLU282-ARG212 (4.66)GLU282-LYS216 (4.07)
Notes: Average distances between O and N atoms (Å) between the ionic residue pairs are shown in parentheses
Human adrenocorticotropic hormone 17
Dow
nloa
ded
by [
122.
167.
6.57
] at
22:
55 0
4 M
ay 2
014

hormones. In L. B. Laurence, L. John, & L. P. Keith(Eds.), The pharmacological basis of therapeutics, Vol. 11(p. 1589). New York, NY: McGraw-Hill.
Biller, B. M., Grossman, A. B., & Stewart, P. M. (2008). Treat-ment of adrenocorticotropin-dependent Cushing’s syn-drome: a consensus statement. The Journal of ClinicalEndocrinology & Metabolism, 93, 2454–2462.
Brady, G. P., & Sharp, K. A. (1997). Entropy in protein foldingand in protein—protein interactions. Current Opinion inStructural Biology, 7, 215–221.
Case, D. A., Darden, T. A., Cheatham, T. E., Simmerling, C. L.,Wang, J., Duke, R. E., … Kollman, P. A. (2008).AMBER 10.
Castro, M. G., & Morrison, E. (1997). Post-translational pro-cessing of proopiomelanocortin in the pituitary and in thebrain. Critical Reviews of Neurobiology, 11, 35–57.
Chen, W., Kelly, M. A., Opitz-Araya, X., Thomas, R. E., Low,M. J., & Cone, R. D. (1997). Exocrine gland dysfunctionin MC5-R-deficient mice: Evidence for coordinated regula-tion of exocrine gland function by melanocortin peptides.Cell, 91, 789–798.
Chhajlani, V. (1996). Distribution of cDNA for melanocortinreceptor subtypes in human tissues. Biochemistry andMolecular Biology International, 38, 73–80.
Chhajlani, V., & Wikberg, J. E. (1992). Molecular cloning andexpression of the human melanocyte stimulating hormonereceptor cDNA. FEBS Letters, 309, 417–420.
Colovos, C., & Yeates, T. O. (1993). Verification of proteinstructures: Patterns of nonbonded atomic interactions. Pro-tein Science, 2, 1511–1519.
Cone, R. D., Lu, D., Koppula, S., Vage, D. I., Klungland, H.,Boston, B., Chen, W., … Pouton, C. K. R. A. (1996). Themelanocortin receptors: Agonists, antagonists, and the hor-monal control of pigmentation. Recent Progress in Hor-mone Research, 51, 287–317.
David, J. E., & Michael, W. D. (2008). Monte Carlos simula-tions. In A. Kukol (Ed.), Methods in molecular BIology,molecular modeling of proteins, Vol. 443 (pp. 25–36).Totowa, NJ: Humana Press.
DeLano, W. L. (2002). The PyMOL molecular graphics system,Version 1.5.0.4. Schrödinger: LLC.
Fiser, A., Gian Do, R. K., & Šali, A. (2000). Modeling ofloops in protein structures. Protein Sciences, 9, 753–1773.
Giblin, M. F., Wang, N., Hoffman, T. J., Jurisson, S. S., &Quinn, T. P. (1998). Design and characterization of alpha-melanotropin peptide analogs cyclized through rhenium andtechnetium metal coordination. Proceedings of the NationalAcademy of Sciences, 95, 12814–12818.
Guex, N., & Peitsch, M. C. (1997). SWISS-MODEL and theSwiss-Pdb Viewer: An environment for comparative proteinmodeling. Electrophoresis, 18, 2714–2723.
Haskell-Luevano, C., Sawyer, T. K., Trumpp-Kallmeyer, S.,Bikker, J. A., Humblet, C., Gantz, I., & Hruby, V. J.(1996). Three-dimensional molecular models of thehMC1R melanocortin receptor: complexes with melanotro-pin peptide agonists. Drug Design and Discovery, 14, 197–211.
Holger, G., & David, A. C. (2004). Converging free energyestimates_ MM_PBGBSA studies on the protein–proteincomplex ras raf. Journal of Computational Chemistry, 25,238–250.
Holger, G., Kiel, C., & David, A. C. (2003). Insights into pro-tein-protein binding by binding free energy calculation andfree energy decomposition for the ras-raf and ras–RalGDScomplexes. Journal of Molecular Biology, 330, 891–913.
Holm, L., & Rosenström, P. (2010). Dali server: Conservationmapping in 3D. Nucleic Acids Research, 38, W545–W549.
Hornak, V., Abel, R., Okur, A., Strockbine, B., Roitberg, A., &Simmerling, C. (2006). Comparison of multiple Amberforce fields and development of improved protein backboneparameters. Proteins: Structure, Function, and Bioinformat-ics, 65, 712–725.
Humphrey, W., Dalke, A., & Schulten, K. (1996). VMD:Visual molecular dynamics. Journal of Molecular Graph-ics, 14, 33–38.
Huo, S., Massova, I., & Kollman, P. A. (2002). Computationalalanine scanning of the 1:1 human growth hormone-receptor complex. Journal of Computational Chemistry, 23,15–27.
Ira, G., & Tung, M. F. (2003). The melanocortin system. Amer-ican Journal of Physiology Endocrinology Metabolism,284, E468–E474.
Jarl, E. S. W. (1999). Melanocortin receptors: Perspectives fornovel drugs. European Journal of Pharmaceutical Sciences,375, 295–310.
Johnson, M. S., & Overington, J. P. (1993). A structural basisfor sequence comparisons. Journal of Molecular Biology,233, 716–738.
Kleywegt, K. J., & Jones, T. A. (1996). Phi/Psi-chology: Rama-chandran revisited. Structure, 4, 1395–1400.
Kollman, P. A., Massova, I., Reyes, C., Kuhn, B., Huo, S.,Chong, L. … Lee, Ma. (2000). Calculating structures andfree energies of complex molecules: Combining molecularmechanics and continuum models. Accounts of ChemicalResearch, 33, 889–897.
Krogh, A., Larsson, B., Von Heijne, G., & Sonnhammer, E. L. L.(2001). Predicting transmembrane protein topology with ahidden Markov model: Application to complete genomes.Journal of Molecular Biology, 305, 567–580.
Kuczera, K., & Kursula, P. (2012). Interactions of calmodulinwith death-associated protein kinase peptides: experimentaland modeling studies. Journal of Biomolecular Structureand Dynamics, 30, 45–61.
Kumar, V., Abbas, A. K., Fausto, N., & Aster, A. (2005).Robbins and Cotran pathologic basis of disease (7th ed.).New York, NY: Elsevier-Saunders.
Laskowski, R. A., MacArthur, M. W., & Moss, D. S. (1993).PROCHECK: A program to check the stereochemical qual-ity of protein structures. Journal of Applied Crystallogra-phy, 26, 283–291.
Liang, L., Angleson, K. J., & Dores, R. M. (2013). Using thehuman melanocortin-2 receptor as a model for analyzinghormone/receptor interactions between a mammalian MC2receptor and ACTH(1-24). General and ComparativeEndocrinology, 181, 203–210.
Luger, T. A., Schwarz, H., Scholzen, K. T., Schwarz, A., &Brazoska, R. (1999). Role of epidermal cell-derived alpha-melanocyte stimulating hormone in ultraviolet light medi-ated local immunosuppresssion. Annals of the New YorkAcademy of Sciences, 885, 209–216.
Massova, I., & Kollman, P. A. (2000). Combined molecularmechanical and continuum solvent approach (MM-PBSA/GBSA) to predict ligand binding. Perspectives in DrugDiscovery and Design, 18, 113–135.
Moukhametzianov, R., Warne, T., Edwards, P. C., Serrano-Vega,M. J., Leslie, A. G. W., Tate, C. G., & Schertler, G. F. X.(2011). Two distinct conformations of helix 6 observed inantagonist-bound structures of a 1-adrenergic receptor.Proceedings of the National Academy of Sciences, 108,8228–8232.
18 M. Dieudonné and K.V. Ramesh
Dow
nloa
ded
by [
122.
167.
6.57
] at
22:
55 0
4 M
ay 2
014

Mountjoy, K. G., Robbins, L. S., Mortrud, M. T., & Cone, R. D.(1992). The cloning of a family of genes that encode themelanocortin receptors. Science, 257, 1248–1251.
Mukherjee, S., & Zhang, Y. (2011). Protein-protein complexstructure predictions by multimeric threading and templaterecombination. Structure, 19, 955–966.
Nasu, K., Hirakawa, T., Okamoto, M., Nishida, M., Kiyoshima,C., Matsumoto, H. … Narahara, H. (2011). Advancedsmall cell carcinoma of the uterine cervix treated by neoad-juvant chemotherapy with irinotecan and cisplatin followedby radical surgery. Rare Tumors, 3, e6.
Onufriev, A., Bashford, D., & Case, D. A. (2004). Exploringprotein native states and large-scale conformational changeswith a modified generalized born model. Proteins: Struc-ture, Function, and Bioinformatics, 55, 383–394.doi:10.1002/prot.20033
Padhi, A. K., Kumar, H., Vasaikar, S. V., Jayaram, B., & Gomes,J. (2012). Mechanisms of loss of functions of human angio-genin variants implicated in amyotrophic lateral sclerosis.PLoS ONE, 7, e32479.
Pastor, R. W., Brooks, B. R., & Szabo, A. (1988). An analysisof the accuracy of Langevin and molecular dynamics algo-rithms. Molecular Physics, 65, 1409–1419.
Pettersen, E. F., Goddard, T. D., Huang, C. C., Couch, G.,Greenblatt, D. M., Meng, E. C., & Ferrin, T. E. (2004).UCSF Chimera: A visualization system for exploratoryresearch and analysis. Journal of Computational Chemistry,25, 1605–1612.
Pogozheva, I. D., Chai, B.-X., Lomize, I. L., Fong, T. M.,Weinberg, D. H., Nargund, R. P., … Mulholland, M. W.(2005). Interactions of human melanocortin 4 receptor withnonpeptide and peptide agonists. Biochemistry, 44, 11329–11341.
Prabhavathi, M., Ashokkumar, K., Geetha, N., & Saradha Devi,K. M. (2011). Homology modeling and structure predictionof thioredoxin (TRX) protein in wheat. International Jour-nal of Bioscience, 1, 20–32.
Ryckaert, J. P., Ciccotti, G., & Berendsen, H. J. C. (1997).Numerical integration of the cartesian equations of motionof a system with constraints: Molecular dynamics of n-alkanes. Journal of Computational Physics, 23, 327–341.
Sandeep, S., Priyadarshini, V., Pradhan, D., Munikumar, M., &Umamaheswari, A. (2012). Docking and molecular dynam-ics simulations studies of human protein kinase catalyticsubunit alpha with antagonist. Journal of Clinical ScienceResearch, 1, 15–23.
Schiöth, H. B., Chhajlani, V., Muceniece, R., Klusa, V., &Wikberg, J. E. S. (1996). Major pharmacological distinctionof the ACTH receptor from other melanocortin receptors.Life Sciences, 59, 797–801.
Sciretti, D., Bruscolini, P., Pelizzola, A., Pretti, M., & Jaramillo,A. (2009). Computational protein design with side-chainconformational entropy. Proteins: Structure, Function, andBioinformatics, 74, 176–191.
Sebag, J. A., & Hinkle, P. M. (2009). Regions of melanocortin2 (MC2) receptor accessory protein necessary for dualtopology and MC2 receptor trafficking and signaling. Jour-nal of Biological Chemistry, 284, 610–618.
Seelig, S., Lindley, B. D., & Sayers, G. (1975). A newapproach to the structure-activity relationship for ACTHanalogs using isolated adrenal cortex cells. In J. G. Hard-man, & B. W. O’Malley (Eds.), Methods in enzymology,Vol. 39 (pp. 347–359). New York, NY: Academic Press.
Sheinerman, F. B., & Honig, B. (2002). On the role of electro-static interactions in the design of protein–protein inter-faces. Journal of Molecular Biology, 318, 161–177.
Sitkoff, D., Sharp, K. A., & Honig, B. (1994). Accurate calcu-lation of hydration free energies using macroscopic solventmodels. Journal of Physical Chemistry, 98, 1978–1988.
Smith, D. M. A., Straatsma, T. P., & Squier, T. C. (2012).Retention of conformational entropy upon calmodulin bind-ing to target peptides is driven by transient salt bridges.Biophysical Journal, 103, 1576–1584.
Stambulchik, E. (Producer). (1997). Xmgrace. Retrieved fromhttp://plasma-gate.weizmann.ac.il/Xmgr
Still, W. C., Tempczyk, A., & Hawley, R. C. (1990). Semiana-lytical treatment of solvation for molecular mechanics anddynamics. Journal of the American Chemical Society, 112,6127–6129.
Suzuki, I., Cone, R. D., Im, S., Nordlund, J., & Abdel-Malek,Z. A. (1996). Binding of melanotropic hormones to themelanocortin receptor MC1R on human melanocytes stimu-lates proliferation and melanogenesis. Endocrinology, 137,1627–1633.
Torsten, S., Jurgen, K., Nicolas, G., & Manuel, C. P. (2003).SWISS-MODEL: An automated protein homology-model-ing server. Nucleic Acids Research, 31, 3381–3385.
Tsigo, C. A. K., Hung, W., & Chrousos, G. P. (1993). Heredi-tary isolated glucocorticoid deficiency is associated withabnormalities of the adrenocorticotropin receptor gene.Journal of Clinical Investigation, 92, 2458–2461.
Van der Ploeg, L. H. T., Martin, W. J., Howard, A. D., Nargund,R. P., Austin, C. P., Guan, X., … MacIntyre, D. E. (2002). Arole for the melanocortin 4 receptor in sexual function. Pro-ceedings of the National Academy of Sciences, 99, 11381–11386.
Versteeg, D. H. G., Van Bergen, P., Adan, R. A. H., & DeWildt, D. J. (1998). Melanocortins and cardiovascular regu-lation. European Journal of Pharmacology, 360, 1–14.
de Vries, S. J., & Bonvin, A. M. J. J. (2011). CPORT: A con-sensus interface predictor and its performance in predic-tion-driven docking with HADDOCK. PLoS ONE, 6,e17695.
de Vries, S. J., van Dijk, M., & Bonvin, A. M. J. J. (2010).The HADDOCK web server for data-driven biomoleculardocking. Nature Protocols, 5, 883–897.
Wang, Z. X., Zhang, W., Wu, C., Lei, H., Cieplak, P., & Duan,Y. (2006). Strike a balance: Optimization of backbone tor-sion parameters of AMBER polarizable force field for sim-ulations of proteins and peptides. Journal of ComputationalChemistry, 27, 781–790.
Warrell, D. A., Timothy, M. C., Firth, J. D., & Benz, E. J.(2005). Oxford textbook of medicine, vol. 2. Oxford:Oxford University Press.
Weiser, J., Shenkin, P. S., & Still, W. C. (1999). Approximateatomic surfaces from linear combinations of pairwise over-laps (LCPO). Journal of Computational Chemistry, 20,217–230.
Wikberg, J. (1999). Melanocortin receptors: Perspectives fornovel drugs. European Journal of Pharmacology, 375,295–310.
Zhang, Y. (2008). I-TASSER server for protein 3D structureprediction. BMC bioinformatics, 9, 40–47.
Zhang, Y., DeVries, M. E., & Jeffrey, S. (2006). Structure mod-eling of all identified G protein–Coupled receptors in thehuman genome. PLoS Computational Biology, 2, e13.
Human adrenocorticotropic hormone 19
Dow
nloa
ded
by [
122.
167.
6.57
] at
22:
55 0
4 M
ay 2
014




















