
Perceived Parental Protectiveness Promotes Positive Friend Influence
Upload
independentCategory
view
1download
0
JNK1 But Not JNK2 Promotes the Development ofSteatohepatitis in Mice
Jorn M. Schattenberg,1,2 Rajat Singh,1,2 Yongjun Wang,1,2 Jay H. Lefkowitch,3 Raina M. Rigoli,1,2
Philipp E. Scherer4 and Mark J. Czaja1,2
Nonalcoholic fatty liver disease (NAFLD) is characterized by hepatic steatosis and varying de-grees of necroinflammation. Although chronic oxidative stress, inflammatory cytokines, andinsulin resistance have been implicated in the pathogenesis of NAFLD, the mechanisms thatunderlie the initiation and progression of this disease remain unknown. c-Jun N-terminal kinase(JNK) is activated by oxidants and cytokines and regulates hepatocellular injury and insulinresistance, suggesting that this kinase may mediate the development of steatohepatitis. Thepresence and function of JNK activation were therefore examined in the murine methionine- andcholine-deficient (MCD) diet model of steatohepatitis. Activation of hepatic JNK, c-Jun, andAP-1 signaling occurred in parallel with the development of steatohepatitis in MCD diet–fedmice. Investigations in jnk1 and jnk2 knockout mice demonstrated that jnk1, but not jnk2, wascritical for MCD diet–induced JNK activation. JNK promoted the development of steatohepa-titis as MCD diet–fed jnk1 null mice had significantly reduced levels of hepatic triglycerideaccumulation, inflammation, lipid peroxidation, liver injury, and apoptosis compared withwild-type and jnk2 �/� mice. Ablation of jnk1 led to an increase in serum adiponectin but hadno effect on serum levels of tumor necrosis factor-�. In conclusion, JNK1 is responsible forJNK activation that promotes the development of steatohepatitis in the MCD diet model.These findings also provide additional support for the critical mechanistic involvementof JNK1 overactivation in conditions associated with insulin resistance and the metabolicsyndrome. Supplementary material for this article can be found on the HEPATOLOGY website(http://interscience.wiley.com/jpages/0270-9139/suppmat/index.html). (HEPATOLOGY 2006;43:163-172.)
The most common liver disease in Westerncountries is nonalcoholic fatty liver disease(NAFLD), which encompasses a spectrum
from simple steatosis to steatosis combined with varying
degrees of necroinflammation and fibrosis.1,2 The initialhepatic lipid accumulation and subsequent progression tocellular injury and inflammation termed nonalcoholicsteatohepatitis (NASH) are thought to occur through dis-tinct mechanisms.3 NAFLD is frequently associated withobesity, dyslipidemia, and insulin resistance, a group ofdisorders that constitute the metabolic syndrome.4 Al-though these conditions may predispose individuals tothe development of NAFLD, the molecular mechanismsthat underlie hepatic fat accumulation and trigger thehepatocyte injury and cell death of NASH are unknown.
There is considerable interest in delineating the factorsthat initiate the onset of necroinflammation because theexistence of steatosis by itself is insufficient to causechronic liver disease. A factor that may trigger the pro-gression to actual cell injury in the setting of a fatty liver isoxidant stress from increased expression of the prooxidantcytochrome P450 isoform 2E1 (CYP2E1),5,6 or the gen-eration of excessive reactive oxygen species from mito-chondrial dysfunction.7,8 In addition, modulators of theinflammatory and immune responses, such as adiponec-tin and tumor necrosis factor-� (TNF), may regulate stea-
Abbreviations: NAFLD, nonalcoholic fatty liver disease; NASH, nonalcoholicsteatohepatitis; CYP2E1, cytochrome P450 2E1; TNF-�, tumor necrosis factor-�;JNK, c-Jun N-terminal kinase; MCD diet, methionine- and choline-deficient diet;RT-PCR, reverse transcription-polymerase chain reaction; MDA, malondialde-hyde; ALT, alanine aminotransferase; TUNEL, terminal deoxynucleotide trans-ferase-mediated deoxyuridine triphosphate nick-end labeling; AP-1, activatorprotein-1; EMSA, electrophoretic mobility shift assay; ICAM-1, intracellular adhe-sion molecule-1; MCP-1, monocyte chemoattractant protein-1.
From the 1Department of Medicine, 2Marion Bessin Liver Research Center, and4Department of Cell Biology, Albert Einstein College of Medicine, Bronx, NY; and the3Department of Pathology, Columbia University Medical Center, New York, NY.
Received July 13, 2005; accepted October 18, 2005.Supported in part by National Institutes of Health Grants DK61498 (M.J.C.)
and DK55758 (P.E.S.).Address reprint requests to: Mark J. Czaja, M.D., Marion Bessin Liver Research
Center, Albert Einstein College of Medicine, 1300 Morris Park Avenue, Bronx, NY10461. E-mail: [email protected]; fax 718-430-8975.
Copyright © 2005 by the American Association for the Study of Liver Diseases.Published online in Wiley InterScience (www.interscience.wiley.com).DOI 10.1002/hep.20999Potential conflict of interest: Nothing to report.
163

totic liver injury.9 TNF promotes insulin resistance andbecomes a hepatotoxin when hepatocytes are sensitized toits cytotoxicity by certain factors, including overexpres-sion of cytochrome P450 2E1.10 Adiponectin may reduceliver steatosis and/or injury because of its antilipogenic,anti-inflammatory, and insulin-sensitizing proper-ties.11-13 Serum adiponectin levels are decreased in obesityand insulin-resistant states and correlate inversely withnecroinflammatory activity in human NAFLD.14
Intracellular signaling pathways are likely to be criticalregulators of both the hepatic accumulation of lipid andthe hepatocyte’s injury response in the setting of steatosis.Recently activation of the mitogen-activated protein ki-nase c-Jun N-terminal kinase (JNK) has been implicatedin the development of obesity and insulin resistance,15
two prominent risk factors for NAFLD that may act topromote steatosis. Other investigations have implicatedJNK activation in the regulation of liver injury.16 JNK isencoded for by three genes, each of which is alternativelyspliced to yield � and � forms of both a p54 and p46protein.17 In most cells, including hepatocytes, only twoof the genes, jnk1 and jnk2, are expressed.17 JNK expres-sion has been implicated in hepatocyte injury mediated byTNF,18,19 ischemia reperfusion,20 hepatitis virus,21 andbile acids.22 The mechanism(s) of this effect is unknown,although it is clear that, whereas transient JNK activationmay be beneficial to the hepatocyte, sustained JNK activ-ity triggers cell death.18 Although the differential effects ofjnk1 and jnk2 generated isoforms in liver injury are largelyunknown, recent studies in cultured hepatocytes havedemonstrated that jnk1 promotes, and jnk2 attenuateshepatocyte injury from toxic bile acids.22 Whether JNKactivation occurs in vivo in chronic liver injuries such asNASH is unknown. We have previously demonstratedthat in vitro chronic hepatocyte overexpression ofCYP2E1 as occurs in NASH causes sustained JNK acti-vation that promotes insulin resistance.10,23 These find-ings combined suggest that JNK activation may occur inNAFLD and modulate the pathophysiology of this dis-ease.
To examine JNK involvement in NAFLD, studieswere performed in a methionine-and choline-deficient(MCD) diet–induced model of steatohepatitis that mim-ics some of the features of human NAFLD, including thedevelopment of steatohepatitis, CYP2E1 overexpression,increased lipid peroxidation, and hepatic insulin resis-tance.23,24 Our studies demonstrate that sustained JNKactivation occurs with the development of steatohepatitisin this model. Additional studies in jnk1 and jnk2 knock-out mice indicate that the jnk1 but not jnk2 gene is criticalfor hepatic JNK activity in vivo. Ablation of jnk1 alsosignificantly attenuated the development of steatohepati-
tis. These findings define a critical mechanistic functionfor jnk1 in experimental steatohepatitis.
Materials and Methods
Animal Model. Male and female wild-type C57BL/6,jnk1 �/�25 and jnk2 �/�26 mice (Jackson Laboratory,Bar Harbor, ME) were maintained under 12-hour light/dark cycles with unlimited access to food and water. Ge-notypes were confirmed by PCR with establishedprimers.25,26 Both knockout mice had been backcrossedonto a C57BL/6 background for more than six genera-tions. Mice were fed the MCD diet or a correspondingcontrol diet, supplemented with 300 mg DL-methionineand 200 mg choline per 100 g of diet (ICN BiomedicalsInc., Costa Mesa, CA). To accustom the animals to thehigh sucrose diet, 6-week-old animals were initiallyplaced on the control diet for 2 weeks. Animals were thenrandomly assigned to an additional 4 weeks of either con-trol diet or MCD diet. At weekly intervals, serum wasobtained by retro-orbital bleed and the mice were sacri-ficed for the removal of liver tissue. All studies were ap-proved by the Animal Care and Use Committee of theAlbert Einstein College of Medicine and followed theNational Institutes of Health guidelines on the care anduse of animals.
Protein Isolation and Western Blotting. See Sup-plementary Materials and Methods at the HEPATOLOGY
Web site (http://interscience.wiley.com/jpages/0270-9139/suppmat/index.html).
JNK Activity Assay. See Supplementary Materials andMethods at the HEPATOLOGY website (http://interscience.wiley.com/jpages/0270-9139/suppmat/index.html).
Electrophoretic Mobility Shift Assay (EMSA). SeeSupplementary Materials and Methods at the HEPATOL-OGY website (http://interscience.wiley.com/jpages/0270-9139/suppmat/index.html).
Histological Analysis. Liver specimens were fixed in10% neutral formalin and 6-�m sections stained withhematoxylin and eosin. Tissue sections were examined ina blinded fashion by a single pathologist and graded forsteatosis and inflammation. The degree of steatosis ineach specimen was determined by assessing the overallpercentage of liver parenchyma containing lipid vacuoleswith 0 � none, 1 � mild (�30%), 2 � moderate (30%-60%), and 3 � marked (�60%). Inflammation wasgraded by the presence or absence of inflammatory cellswith 0 � absent, 1 � minimal or focal occasional singleclusters of inflammatory cells present in a few microscopicfields, 2 � mild inflammation, 3 � moderate inflamma-tion, and 4 � marked inflammation.
164 SCHATTENBERG ET AL. HEPATOLOGY, January 2006

Hepatic Triglyceride Content. The triglyceride con-tent of liver tissue was determined following an extractionin a 2:1 chloroform:methanol mixture containing 0.05%sulfuric acid at �20°C for 16 hours. Triglyceride mea-surements were performed using a commercial kit accord-ing to the manufacturer’s instructions (RocheDiagnostics, Indianapolis, IN).
Reverse Transcription-Polymerase Chain Reaction(RT-PCR). See Supplementary Materials and Methodsat the HEPATOLOGY website (http://interscience.wiley.com/jpages/0270-9139/suppmat/index.html).
Lipid Peroxidation Assay. As a reflection of hepaticlevels of lipid peroxidation, malondialdehyde (MDA) lev-els were determined by colorimetric assay (EMD Bio-sciences, La Jolla, CA). Fifty mg of liver tissue werehomogenized in 20 mmol/L Tris-HCl, pH 7.4, and 500�mol/L 3,5-Di-tert-butyl-4-hydroxytoluene, and the col-orimetric reaction carried out accordingly to the manu-facturer’s instructions.
Serum Assays. Commercial kits were used to measureserum alanine aminotransferase (ALT) (TECO Diagnos-tics, Anaheim, CA), adiponectin (Linco Research Inc., St.Charles, MO), and TNF (BD Biosciences, San Diego,CA) levels. Adiponectin measurements were performed atthe same time of day in all experiments.
Terminal Deoxynucleotide Transferase-MediatedDeoxyuridine Triphosphate Nick End-Labeling Assay.The numbers of apoptotic cells in liver sections were de-termined by terminal deoxynucleotide transferase-medi-ated deoxyuridine triphosphate nick-end labeling(TUNEL) assay with a commercial kit (Promega, Madi-son, WI). Tissue sections were prepared by deparaffina-tion in xylene followed by gradual rehydration withdecreasing concentrations of ethanol. The TUNEL assaywas then performed according to the manufacturer’s in-structions. Under light microscopy the numbers ofTUNEL positive cells were counted per high-poweredfield (400� magnification) with 20 random fieldscounted per liver.
Statistical Analysis. All numerical results are ex-pressed as mean �SE and represent data from a minimumof three independent experiments. Calculations weremade with Sigma Plot 2000 (SPSS Science, Chicago, IL).Western and RT-PCR signals were quantitated using aFluorChem densitometer (Alpha Innotech, San Leo-nardo, CA).
Results
MCD Diet–Induced Steatohepatitis Is AssociatedWith Activation of JNK, c-Jun, and AP-1. The mito-gen-activated protein kinase JNK is a critical modulator
of the liver’s response to injurious stress.16 JNK also reg-ulates the development of obesity and insulin resistance,15
the two main risk factors for NAFLD. These facts suggestthat JNK may modulate the development of steatohepa-titis. To assess whether hepatic JNK activation occurs insteatohepatitis, JNK activity was examined in C57BL/6mice placed on a high carbohydrate diet lacking methio-nine and choline (MCD diet) for 4 weeks. As previouslyreported,23,24,27 animals fed this diet developed steatosis,inflammation and hepatocellular injury within 1 weekthat was sustained over the 4-week course of the experi-ments (data not shown). JNK activity was compared be-tween control diet and MCD diet–fed animals by an invitro kinase assay employing c-Jun as the substrate. JNKactivity, as reflected by levels of phospho-c-Jun on immu-noblots, was increased 7 fold within 1 week of the start ofthe MCD diet and remained elevated over the 4-weekcourse of the experiment (Fig. 1A). Levels of total c-Junwere equivalent among samples, confirming the equalloading of reaction product (Fig. 1A). JNK activationtherefore occurred early in the development of MCD di-et–induced steatohepatitis and was sustained once the dis-ease was established.
As further evidence of JNK activation, levels of active,phosphorylated JNK were examined in MCD diet–fedanimals by Western blot analysis. Increased levels of phos-phorylated p54 and p46 JNK were detected from 1 to 4weeks after the start of the MCD diet by immunoblotting(Fig. 1B). A slight increase in JNK phosphorylation wasalso observed in control diet–fed animals after 2 weeksdespite their lack of increased JNK activity. Still phospho-JNK levels in MCD diet–fed mice were always at least2-fold greater than those in control diet–fed animals (Fig.1B). Levels of total JNK were unchanged over time byeither diet (Fig. 1B).
Downstream effects of JNK activation are partially me-diated through phosphorylation and activation of c-Jun.In parallel with the increases in JNK activity and phos-phorylation, levels of phosphorylated c-Jun were in-creased 3 to 6 fold in animals fed MCD diet over the4-week time course. c-Jun expression is regulated tran-scriptionally as well as by posttranslational phosphoryla-tion, and levels of total c-Jun protein increased 3 to 5 foldin animals fed MCD diet over 4 weeks (Fig. 1B). Controldiet–fed animals also had a slight increase in expression oftotal c-Jun that did not reach statistical significance (Fig.1B).
c-Jun is a critical subunit of the transcription factorAP-1 (activator protein-1). Elevated levels of total andactivated c-Jun may therefore lead to increased AP-1 ac-tivation in MCD diet–fed mice. Analysis of levels of AP-1
HEPATOLOGY, Vol. 43, No. 1, 2006 SCHATTENBERG ET AL. 165
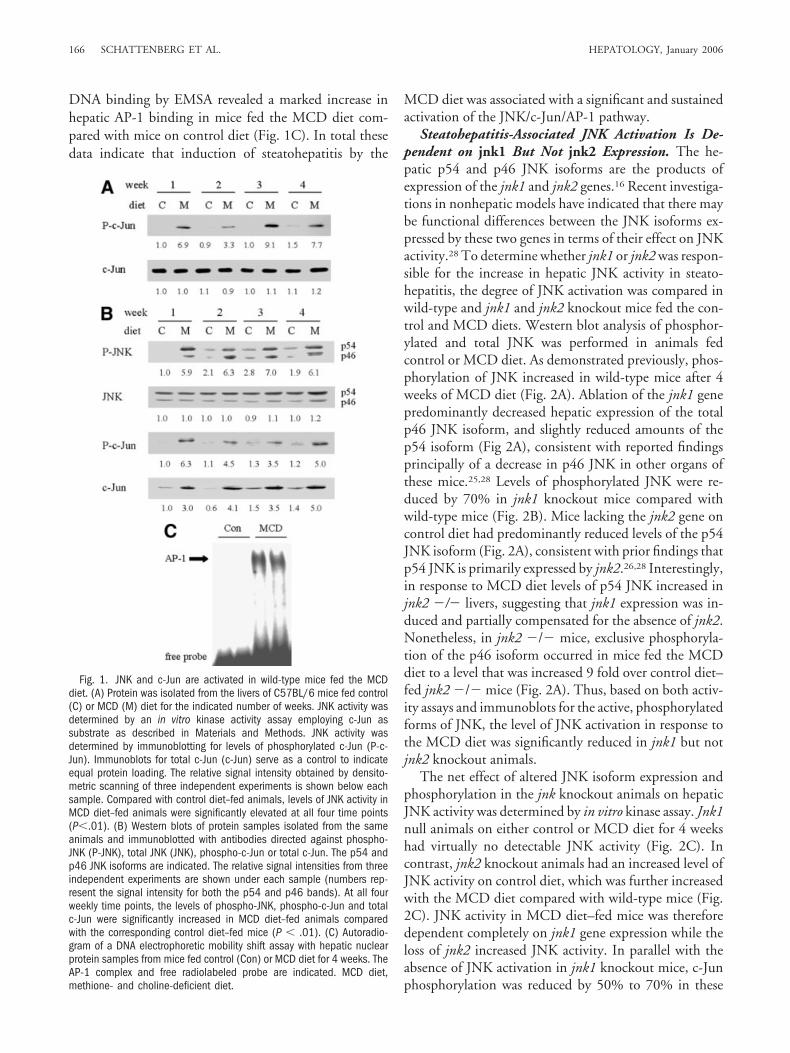
DNA binding by EMSA revealed a marked increase inhepatic AP-1 binding in mice fed the MCD diet com-pared with mice on control diet (Fig. 1C). In total thesedata indicate that induction of steatohepatitis by the
MCD diet was associated with a significant and sustainedactivation of the JNK/c-Jun/AP-1 pathway.
Steatohepatitis-Associated JNK Activation Is De-pendent on jnk1 But Not jnk2 Expression. The he-patic p54 and p46 JNK isoforms are the products ofexpression of the jnk1 and jnk2 genes.16 Recent investiga-tions in nonhepatic models have indicated that there maybe functional differences between the JNK isoforms ex-pressed by these two genes in terms of their effect on JNKactivity.28 To determine whether jnk1 or jnk2 was respon-sible for the increase in hepatic JNK activity in steato-hepatitis, the degree of JNK activation was compared inwild-type and jnk1 and jnk2 knockout mice fed the con-trol and MCD diets. Western blot analysis of phosphor-ylated and total JNK was performed in animals fedcontrol or MCD diet. As demonstrated previously, phos-phorylation of JNK increased in wild-type mice after 4weeks of MCD diet (Fig. 2A). Ablation of the jnk1 genepredominantly decreased hepatic expression of the totalp46 JNK isoform, and slightly reduced amounts of thep54 isoform (Fig 2A), consistent with reported findingsprincipally of a decrease in p46 JNK in other organs ofthese mice.25,28 Levels of phosphorylated JNK were re-duced by 70% in jnk1 knockout mice compared withwild-type mice (Fig. 2B). Mice lacking the jnk2 gene oncontrol diet had predominantly reduced levels of the p54JNK isoform (Fig. 2A), consistent with prior findings thatp54 JNK is primarily expressed by jnk2.26,28 Interestingly,in response to MCD diet levels of p54 JNK increased injnk2 �/� livers, suggesting that jnk1 expression was in-duced and partially compensated for the absence of jnk2.Nonetheless, in jnk2 �/� mice, exclusive phosphoryla-tion of the p46 isoform occurred in mice fed the MCDdiet to a level that was increased 9 fold over control diet–fed jnk2 �/� mice (Fig. 2A). Thus, based on both activ-ity assays and immunoblots for the active, phosphorylatedforms of JNK, the level of JNK activation in response tothe MCD diet was significantly reduced in jnk1 but notjnk2 knockout animals.
The net effect of altered JNK isoform expression andphosphorylation in the jnk knockout animals on hepaticJNK activity was determined by in vitro kinase assay. Jnk1null animals on either control or MCD diet for 4 weekshad virtually no detectable JNK activity (Fig. 2C). Incontrast, jnk2 knockout animals had an increased level ofJNK activity on control diet, which was further increasedwith the MCD diet compared with wild-type mice (Fig.2C). JNK activity in MCD diet–fed mice was thereforedependent completely on jnk1 gene expression while theloss of jnk2 increased JNK activity. In parallel with theabsence of JNK activation in jnk1 knockout mice, c-Junphosphorylation was reduced by 50% to 70% in these
Fig. 1. JNK and c-Jun are activated in wild-type mice fed the MCDdiet. (A) Protein was isolated from the livers of C57BL/6 mice fed control(C) or MCD (M) diet for the indicated number of weeks. JNK activity wasdetermined by an in vitro kinase activity assay employing c-Jun assubstrate as described in Materials and Methods. JNK activity wasdetermined by immunoblotting for levels of phosphorylated c-Jun (P-c-Jun). Immunoblots for total c-Jun (c-Jun) serve as a control to indicateequal protein loading. The relative signal intensity obtained by densito-metric scanning of three independent experiments is shown below eachsample. Compared with control diet–fed animals, levels of JNK activity inMCD diet–fed animals were significantly elevated at all four time points(P�.01). (B) Western blots of protein samples isolated from the sameanimals and immunoblotted with antibodies directed against phospho-JNK (P-JNK), total JNK (JNK), phospho-c-Jun or total c-Jun. The p54 andp46 JNK isoforms are indicated. The relative signal intensities from threeindependent experiments are shown under each sample (numbers rep-resent the signal intensity for both the p54 and p46 bands). At all fourweekly time points, the levels of phospho-JNK, phospho-c-Jun and totalc-Jun were significantly increased in MCD diet–fed animals comparedwith the corresponding control diet–fed mice (P � .01). (C) Autoradio-gram of a DNA electrophoretic mobility shift assay with hepatic nuclearprotein samples from mice fed control (Con) or MCD diet for 4 weeks. TheAP-1 complex and free radiolabeled probe are indicated. MCD diet,methione- and choline-deficient diet.
166 SCHATTENBERG ET AL. HEPATOLOGY, January 2006

animals on either diet compared with wild-type mice (Fig.2A). Levels of total c-Jun were also decreased in controldiet and MCD diet–fed jnk1 �/� mice (Fig. 2A).
JNK affects the function of it downstream effector c-Jun by phosphorylating sites that increase the AP-1 tran-scriptional activity of c-Jun. Levels of c-Jun gene
expression are regulated by AP-1 and therefore also JNKdependent. The effect of jnk1 ablation on the MCD diet–induced increase in AP-1 DNA binding was examined.AP-1 DNA binding was increased in all three types ofmice fed the MCD diet (Fig. 2D). However, levels ofAP-1 DNA binding were decreased 30% in jnk1 �/�animals as compared with wild-type and jnk2 �/� mice.
MCD Diet–fed jnk1 �/� Mice Have ImprovedHistology. The marked decrease in JNK activation inMCD diet–fed jnk1 null mice made these animals anappropriate model for a study of the effects of the absenceof JNK activity on the development of steatohepatitis.Wild-type, jnk1 �/� and jnk2 �/� mice all gainedweight on control diet over the 4-week course of the ex-periment, although the increase in jnk2 null mice wassignificantly greater (Table 1). All three mouse strains lostsimilar amounts of weight on the MCD diet (Table 1).The ratio of liver weight to body weight, a determinant ofhepatomegaly, was significantly greater in wild-type andjnk2 �/� mice than in jnk1 �/� mice fed the MCD diet(Table 1).
Histological analysis revealed minimal steatosis and noinflammation in all three types of mice fed the control diet(Fig. 3A-C). MCD diet–fed mice developed macrove-sicular and microvesicular steatosis and inflammatory ag-gregates of lymphocytes and neutrophils (Fig. 3D-F).However, jnk1 �/� mice on the MCD diet developedmarkedly reduced hepatic steatosis and inflammation(Fig. 3E). This impression was confirmed by blinded his-tological grading of liver sections. The grade of steatosiswas approximately 2-fold greater in wild-type and jnk2knockout mice than in jnk1 null mice (Fig. 4A). Jnk1�/� mice had a statistically significant decrease in steato-sis on control diet as well. Similarly the inflammationgrade in jnk1 knockout mice was reduced 45% compared
4™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™™Fig. 2. Jnk1 �/� but not jnk2 �/� mice have decreased JNK,
c-Jun and AP-1 activation. (A) Western blots of hepatic protein fromwild-type C57BL/6 (WT), jnk1 �/� and jnk2 �/� mice fed the control(C) or MCD (M) diet for 4 weeks. Samples were immunoblotted withantibodies for the p54 and p46 isoforms of phosphorylated (P-JNK) andtotal (JNK) JNK, and phosphorylated (P-c-Jun) and total (c-Jun) c-Jun.The relative signal intensity obtained by densitometric scanning of threeindependent experiments is shown below each sample. (B) Densitomet-ric values for p54 and p46 phospho-JNK among the different groups fromWestern blot analysis of three independent experiments (*P�.02 com-pared with MCD diet–fed wild-type mice). (C) A JNK in vitro kinase assaywas performed on the same samples as in panel A. Levels of JNK activityare reflected in the relative amounts of phosphorylated c-Jun while totalc-Jun levels indicate equivalent sample loading. The relative signalintensity obtained by densitometric scanning of three independent ex-periments is shown below each sample. (D) Autoradiogram of a DNAEMSA for AP-1 with hepatic nuclear protein samples from the identicalmice. The AP-1 complex and free radiolabeled probe are indicated. MCD,methione and choline deficient.
HEPATOLOGY, Vol. 43, No. 1, 2006 SCHATTENBERG ET AL. 167

with that in wild-type and jnk2 knockout mice (Fig. 4B).Thus, although wild-type and jnk1 and jnk2 knockoutmice all developed steatosis and inflammation on theMCD diet, the degree of steatosis and inflammation wassignificantly reduced in animals lacking jnk1.
Triglyceride Content, Proinflammatory Gene Ex-pression and Levels of Lipid Peroxidation Are De-creased in jnk1 Knockout Mice Fed the MCD Diet.To further validate the histological differences in steatosisobserved among the three mouse strains on MCD diet,hepatic triglyceride content was determined. Triglyceridecontent among the three strains of mice fed the controldiet was equivalent (Fig. 5A). A 5-fold increase in hepatictriglyceride content occurred after 4 weeks of MCD dietin wild-type and jnk2 knockout mice (Fig. 5A). In con-trast, consistent with the histological grading, the increasein hepatic triglyceride content in jnk1 knockout mice wassignificantly decreased by 60% compared with the levelsin wild-type and jnk2 null mice (Fig. 5A).
The decreased histological evidence of inflammation injnk1 �/� mice was further supported by RT-PCR anal-ysis of mRNA expression of two proinflammatory genes,ICAM-1 and MCP-1. Both genes have previously beendemonstrated to be upregulated in parallel with the de-velopment of MCD diet–induced steatohepatitis.29 Sim-ilar to findings of Leclercq et al.,29 the MCD diet resultedin marked increases in ICAM-1 (intracellular adhesion
molecule-1) and MCP-1 (monocyte chemoattractantprotein-1) mRNA expression in wild-type mice (Fig. 5B).Both genes were upregulated in jnk1 �/� mice but thelevels of ICAM-1 and MCP-1 mRNA in these mice weresignificantly decreased by 48% and 65%, respectively,compared with wild-type mice (Fig. 5C). Levels of bothmRNAs were decreased in jnk2 �/� mice compared withwild-type mice but the changes were not statistically sig-nificant.
MCD diet–induced experimental steatohepatitis andhuman NASH are associated with increased oxidativestress.7,24 The generation of excessive reactive oxygen spe-cies leads to the nonenzymatic formation of lipid hy-droperoxides that cause cellular injury through amodification of cellular macromolecules. To additionallyassess the function of jnk1 and jnk2 expression in steato-hepatitis, hepatic levels of MDA, a product of lipid per-oxidation, and serum ALT, a marker of hepatocellularinjury, were determined in MCD diet–fed mice. MDAlevels increased 2 fold in wild-type and jnk2 knockoutmice on MCD diet compared with control diet (Fig. 5D).However, the increase in MDA levels in jnk1 �/� micewas significantly decreased by 25% compared with wild-type and jnk2 knockout mice (Fig. 5D). MDA levels didnot differ significantly among control diet–fed animals.
Liver Injury and Cell Death Are Decreased in theAbsence of jnk1. To examine whether reductions in ste-
Table 1. Change in Body Weights and Liver-to-Body Weight Ratios of Wild-Type (WT), jnk1 �/� and jnk2 �/� Mice Fed aControl (Con) or Methionine- and Choline-Deficient (MCD) Diet for 4 Weeks*
WT jnk1 �/� jnk2 �/�
Con MCD Con MCD Con MCD
Change in body weight (g) �2.7 � 0.7 �5.7 � 0.6 �3.8 � 0.8 �5.1 � 0.5 �6.7 � 1.5‡ �4.2 � 0.5Liver:body (%) 4.7 � 0.3 5.3 � 0.3 4.2 � 0.5 4.1 � 0.2§ 4.2 � 0.1 5.1 � 0.3
*Change in body weight is expressed as the gain (�) or loss (�) in weight in grams over the 4 weeks of diet administration. Liver weight is expressed as a percentageof body weight.
‡There is a statistically significant difference in this value from wild-type mice on control diet or §from wild-type and jnk2 �/� mice on MCD diet (all P � .02).
Fig. 3. Jnk1 �/� mice fed the MCD diethave improved histology compared with wild-type and jnk2 �/� mice. Hematoxylin andeosin stained sections of wild-type (WT), jnk1�/� and jnk2 �/� livers after 4 weeks ofcontrol (Con) or MCD diet. Findings are notablefor a reduced degree of steatosis and inflam-mation in the liver from the jnk1 �/� MCDdiet–fed mouse compared with those from theWT and jnk2 �/� mice. MCD diet, methione-and choline-deficient diet.
168 SCHATTENBERG ET AL. HEPATOLOGY, January 2006

atosis, inflammation, and oxidative stress translated into adecrease in hepatocyte injury, levels of serum ALT andhepatic apoptosis were compared in the three types ofmice. In accordance with previously published observa-tions,24,27 ALT levels increased 15 fold in MCD diet–fedwild-type mice (Fig. 6A). While jnk2 �/� mice devel-
oped a similar increase in serum ALT as wild-type mice,this increase was significantly reduced by 40% in jnk1knockout mice (Fig. 6A).
It has been suggested that hepatocyte injury in steato-hepatitis results in hepatocyte apoptosis.30 Consistentwith this concept, the number of TUNEL-positive cells
Fig. 4. The livers of MCD diet–fed jnk1 null mice have decreasedgrades of steatosis and inflammation. The degree of steatosis (A) andinflammation (B) was graded in a blinded fashion using the scalesdescribed in Materials and Methods in wild-type (WT), jnk1 �/ � andjnk2 �/� mice. Results are from 4 independent experiments and eachdata point represents a minimum of 4 mice for control diet and 10 micefor MCD diet (*P � .01 for steatosis and P � .03 for inflammationcompared with wild-type or jnk2 �/� mice). MCD diet, methione- andcholine-deficient diet.
Fig. 5. MCD diet–fed jnk1 �/� mice have decreased levels of hepatic triglycerides, proinflammatory gene expression and MDA. (A) Levels ofhepatic triglyceride content for wild-type (WT), jnk1 �/�, and jnk2 �/� mice fed the control or MCD diet for 4 weeks. Data are from 4 independentexperiments and represent a total of 5 control diet and 7 MCD diet–fed animals (*P � .02 compared with wild-type or jnk2 �/� mice). (B)Representative RT-PCR results for ICAM-1, MCP-1 and �-actin in the three types of mice that were fed control (C) or MCD (M) diet for 4 weeks. (C)Densitometry scanning of signal intensities of RT-PCR results from 3 independent experiments. ICAM-1 and MCP-1 expression levels were normalizedto the �-actin signal from the same samples (*P � .01 compared with wild-type mice). (D) Levels of MDA in WT, jnk1 �/� and jnk2 �/ � micefed the control or MCD diet for 4 weeks. Data are from 4 independent experiments and represent 5 control diet and 7 MCD diet–fed animals (*P �.02 compared with wild-type or jnk2 �/� mice). MCD diet, methione- and choline-deficient diet.
Fig. 6. Ablation of jnk1 decreases liver injury and apoptosis. (A)Levels of serum ALT in wild-type (WT), jnk1 �/� and jnk2 �/� micefed control or MCD diet for 4 weeks. Data are from 4 independentexperiments and represent 4 control diet and 7 MCD diet–fed animals (*P � .02 compared with wild-type or jnk2 �/� mice). (B) Numbers ofapoptotic cells per high-powered field (HPF) by TUNEL assay for the threetypes of mice fed 4 weeks of diet. Results are from 3 independentexperiments (*P � .001 compared with wild-type or jnk2 �/� mice).MCD diet, methione- and choline-deficient diet.
HEPATOLOGY, Vol. 43, No. 1, 2006 SCHATTENBERG ET AL. 169
increased 10 to 20 fold in the livers of MCD diet–fedwild-type and jnk2 �/� mice (Fig. 6B). In contrast, thenumber of apoptotic cells in MCD diet–fed jnk1 �/�mice was only 27% of that in wild-type animals (Fig. 6B).Thus, the absence of jnk1 ultimately led to a significantdecrease in hepatocellular injury and death.
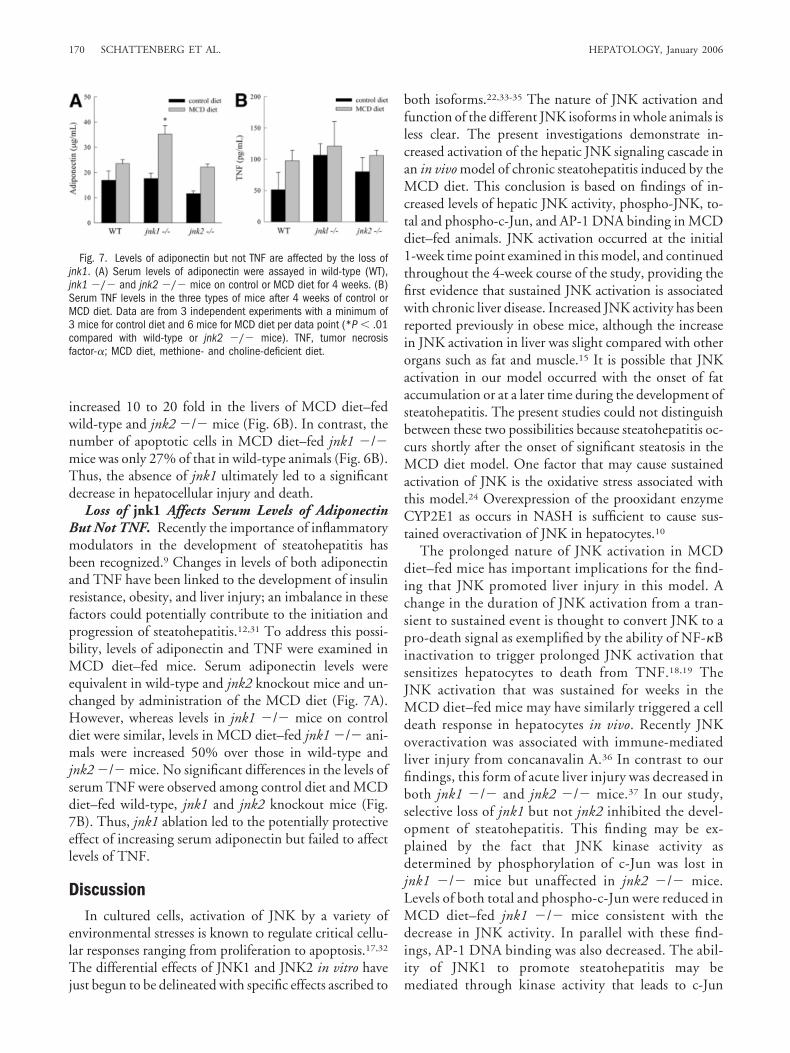
Loss of jnk1 Affects Serum Levels of AdiponectinBut Not TNF. Recently the importance of inflammatorymodulators in the development of steatohepatitis hasbeen recognized.9 Changes in levels of both adiponectinand TNF have been linked to the development of insulinresistance, obesity, and liver injury; an imbalance in thesefactors could potentially contribute to the initiation andprogression of steatohepatitis.12,31 To address this possi-bility, levels of adiponectin and TNF were examined inMCD diet–fed mice. Serum adiponectin levels wereequivalent in wild-type and jnk2 knockout mice and un-changed by administration of the MCD diet (Fig. 7A).However, whereas levels in jnk1 �/� mice on controldiet were similar, levels in MCD diet–fed jnk1 �/� ani-mals were increased 50% over those in wild-type andjnk2 �/� mice. No significant differences in the levels ofserum TNF were observed among control diet and MCDdiet–fed wild-type, jnk1 and jnk2 knockout mice (Fig.7B). Thus, jnk1 ablation led to the potentially protectiveeffect of increasing serum adiponectin but failed to affectlevels of TNF.
DiscussionIn cultured cells, activation of JNK by a variety of
environmental stresses is known to regulate critical cellu-lar responses ranging from proliferation to apoptosis.17,32
The differential effects of JNK1 and JNK2 in vitro havejust begun to be delineated with specific effects ascribed to
both isoforms.22,33-35 The nature of JNK activation andfunction of the different JNK isoforms in whole animals isless clear. The present investigations demonstrate in-creased activation of the hepatic JNK signaling cascade inan in vivo model of chronic steatohepatitis induced by theMCD diet. This conclusion is based on findings of in-creased levels of hepatic JNK activity, phospho-JNK, to-tal and phospho-c-Jun, and AP-1 DNA binding in MCDdiet–fed animals. JNK activation occurred at the initial1-week time point examined in this model, and continuedthroughout the 4-week course of the study, providing thefirst evidence that sustained JNK activation is associatedwith chronic liver disease. Increased JNK activity has beenreported previously in obese mice, although the increasein JNK activation in liver was slight compared with otherorgans such as fat and muscle.15 It is possible that JNKactivation in our model occurred with the onset of fataccumulation or at a later time during the development ofsteatohepatitis. The present studies could not distinguishbetween these two possibilities because steatohepatitis oc-curs shortly after the onset of significant steatosis in theMCD diet model. One factor that may cause sustainedactivation of JNK is the oxidative stress associated withthis model.24 Overexpression of the prooxidant enzymeCYP2E1 as occurs in NASH is sufficient to cause sus-tained overactivation of JNK in hepatocytes.10
The prolonged nature of JNK activation in MCDdiet–fed mice has important implications for the find-ing that JNK promoted liver injury in this model. Achange in the duration of JNK activation from a tran-sient to sustained event is thought to convert JNK to apro-death signal as exemplified by the ability of NF-�Binactivation to trigger prolonged JNK activation thatsensitizes hepatocytes to death from TNF.18,19 TheJNK activation that was sustained for weeks in theMCD diet–fed mice may have similarly triggered a celldeath response in hepatocytes in vivo. Recently JNKoveractivation was associated with immune-mediatedliver injury from concanavalin A.36 In contrast to ourfindings, this form of acute liver injury was decreased inboth jnk1 �/� and jnk2 �/� mice.37 In our study,selective loss of jnk1 but not jnk2 inhibited the devel-opment of steatohepatitis. This finding may be ex-plained by the fact that JNK kinase activity asdetermined by phosphorylation of c-Jun was lost injnk1 �/� mice but unaffected in jnk2 �/� mice.Levels of both total and phospho-c-Jun were reduced inMCD diet–fed jnk1 �/� mice consistent with thedecrease in JNK activity. In parallel with these find-ings, AP-1 DNA binding was also decreased. The abil-ity of JNK1 to promote steatohepatitis may bemediated through kinase activity that leads to c-Jun
Fig. 7. Levels of adiponectin but not TNF are affected by the loss ofjnk1. (A) Serum levels of adiponectin were assayed in wild-type (WT),jnk1 �/� and jnk2 �/� mice on control or MCD diet for 4 weeks. (B)Serum TNF levels in the three types of mice after 4 weeks of control orMCD diet. Data are from 3 independent experiments with a minimum of3 mice for control diet and 6 mice for MCD diet per data point (*P � .01compared with wild-type or jnk2 �/� mice). TNF, tumor necrosisfactor-�; MCD diet, methione- and choline-deficient diet.
170 SCHATTENBERG ET AL. HEPATOLOGY, January 2006

phosphorylation, the phosphorylation of other pro-teins,37 or an effect totally unrelated to its kinase activity.The present investigations cannot differentiate amongthese possibilities. Our studies are consistent with, but notproof of, the conclusion that the classic c-Jun kinase ac-tivity of JNK promoted steatohepatitis because only lossof the specific JNK isoform responsible for c-Jun kinaseactivity resulted in reduced disease. This finding supportsin vitro studies in non-hepatic cells demonstrating thatthe JNK1 but not JNK2 isoform is responsible for c-Junkinase activity and that loss of JNK2 may act to furtherincrease JNK activation.35 In our studies jnk2 �/� micealso had increased JNK activity although it did not trans-late into an increase in steady-state levels of phosphory-lated c-Jun. The protective effect of loss of JNK1 but notJNK2 is also consistent with in vitro hepatocyte studiesthat first demonstrated a selective protective effect ofJNK1 but not JNK2.22 A determination of whether JNK1but not JNK2 always serves as an injurious signaling path-way in the hepatocyte, or the roles of the two isoformsvary depending on the injurious stimulus, requires furtherinvestigation into other forms of in vivo liver injury.
The development of both hepatic steatosis and injurywas significantly but not completely inhibited by loss ofJNK1. The partial nature of the response indicates thateither JNK-independent mechanisms contribute to thisprocess or that JNK2 is capable of partially replacing thefunction of JNK1. JNK1 activation could serve as eitherof the two “hits” that culminate in steatohepatitis.3 First,JNK1 may promote the lipid accumulation that leads tothe initial steatosis as loss of JNK1 significantly reducedthe development of steatosis in the MCD diet model. Aprimary reduction in hepatic lipid accumulation shouldtranslate into a secondary decrease in liver injury becauselipid serves as a substrate that fuels the oxidant stress thatcauses cellular injury. JNK1 may promote the develop-ment of steatosis through its ability to induce insulin re-sistance, as MCD diet–induced steatohepatitis isassociated with JNK-dependent hepatic insulin resis-tance.23 Alternatively, JNK1 may have some as yet unde-scribed effect on lipid synthesis or oxidation. Second,JNK1 activation could act as the second “hit” in the de-velopment of steatohepatitis by acting to initiate the pro-cess of cellular injury. The JNK/c-Jun pathway mediateshepatocyte injury from the cytokine TNF and oxi-dants.10,18,19,38 TNF and oxidative stress have both beenimplicated in induction of the cellular injury ofNASH,24,31 suggesting that JNK1 activation may pro-mote hepatocellular death from these factors in this dis-ease. The fact that endoplasmic reticulum stress inducesinsulin resistance and a JNK-dependent apoptosis39,40
suggests that JNK1 may promote steatohepatitis through
activation of ER stress. However, hepatic ER stress doesnot appear to occur in the MCD diet model as there is noassociated increase in protein levels of the ER stress mark-ers BiP and GADD153 in the liver (data not shown).Finally, it is possible that JNK1 activation may act as thefirst or second hit, not through its direct effects in the liverbut through extrahepatic effects on inflammatory or im-munomodulatory factors such as cytokines or adipokines.Loss of jnk1 failed to alter serum TNF levels but didsignificantly increase adiponectin levels as previously re-ported in obese mice.12,41 Adiponectin levels are inverselycorrelated with disease activity in human NASH,14 andadiponectin administration prevents experimentalNASH.12,41 Thus, it is possible that increased adiponectinlevels in jnk1�/� mice may have inhibited the develop-ment of steatohepatitis. Against this possibility is the factthat the degree of MCD diet–induced steatohepatitis isequivalent in wild-type and adiponectin knockout ani-mals (Czaja, MJ, unpublished data). To completely ex-clude an effect of increased adiponectin levels in thismodel, however, the effects of adiponectin supplementa-tion on MCD diet–fed wild-type mice would have to beinvestigated.
These investigations demonstrate that sustained JNKactivation occurs in chronic steatohepatitis and promotesboth the steatosis and liver injury that comprise NASH.Therefore, JNK not only modulates acute states of injuryand cell death but also chronic conditions of tissue injuryas well. Further studies will have to address whether anti-JNK treatment is effective in reducing already-establishedsteatohepatitis. However, these findings together withother recent studies demonstrating the potential thera-peutic benefit of JNK inhibitors in diabetes,42 suggest thatJNK inhibition may have widespread benefit in the treat-ment of complications of the insulin resistant state.
References1. Angulo P. Nonalcoholic fatty liver disease. N Engl J Med 2002;346:1221-
1231.2. Neuschwander-Tetri BA, Caldwell SH. Nonalcoholic steatohepatitis:
summary of an AASLD Single Topic Conference. HEPATOLOGY 2003;37:1202-1219.
3. Day CP, James OF. Steatohepatitis: a tale of two “hits”? Gastroenterology1998;114:842-845.
4. Marchesini G, Bugianesi E, Forlani G, Cerrelli F, Lenzi M, Manini R, et al.Nonalcoholic fatty liver, steatohepatitis, and the metabolic syndrome.HEPATOLOGY 2003;37:917-923.
5. Chalasani N, Gorski JC, Asghar MS, Asghar A, Foresman B, Hall SD, et al.Hepatic cytochrome P450 2E1 activity in nondiabetic patients with non-alcoholic steatohepatitis. HEPATOLOGY 2003;37:544-550.
6. Weltman MD, Farrell GC, Hall P, Ingelman-Sundberg M, Liddle C.Hepatic cytochrome P450 2E1 is increased in patients with nonalcoholicsteatohepatitis. HEPATOLOGY 1998;27:128-133.
7. Sanyal AJ, Campbell-Sargent C, Mirshahi F, Rizzo WB, Contos MJ, SterlingRK, et al. Nonalcoholic steatohepatitis: association of insulin resistance andmitochondrial abnormalities. Gastroenterology 2001;120:1183-1192.
HEPATOLOGY, Vol. 43, No. 1, 2006 SCHATTENBERG ET AL. 171

8. Laurent A, Nicco C, Tran VN, Borderie D, Chereau C, Conti F, et al.Pivotal role of superoxide anion and beneficial effect of antioxidant mole-cules in murine steatohepatitis. HEPATOLOGY 2004;39:1277-1285.
9. Czaja MJ. Liver injury in the setting of steatosis: crosstalk between adipo-kine and cytokine. HEPATOLOGY 2004;40:19-22.
10. Liu H, Jones BE, Bradham C, Czaja MJ. Increased cytochrome P-450 2E1expression sensitizes hepatocytes to c-Jun-mediated cell death fromTNF-�. Am J Physiol Gastrointest Liver Physiol 2002;282:G257-G266.
11. Berg AH, Combs TP, Du X, Brownlee M, Scherer PE. The adipocyte-secreted protein Acrp30 enhances hepatic insulin action. Nat Med 2001;7:947-953.
12. Masaki T, Chiba S, Tatsukawa H, Yasuda T, Noguchi H, Seike M, et al.Adiponectin protects LPS-induced liver injury through modulation ofTNF-� in KK-Ay obese mice. HEPATOLOGY 2004;40:177-184.
13. Qi Y, Takahashi N, Hileman SM, Patel HR, Berg AH, Pajvani UB, et al.Adiponectin acts in the brain to decrease body weight. Nat Med 2004;10:524-529.
14. Hui JM, Hodge A, Farrell GC, Kench JG, Kriketos A, George J. Beyondinsulin resistance in NASH: TNF-� or adiponectin? HEPATOLOGY 2004;40:46-54.
15. Hirosumi J, Tuncman G, Chang L, Gorgun CZ, Uysal KT, Maeda K, et al.A central role for JNK in obesity and insulin resistance. Nature 2002;420:333-336.
16. Czaja MJ. The future of GI and liver research: editorial perspectives. III.JNK/AP-1 regulation of hepatocyte death. Am J Physiol Gastrointest LiverPhysiol 2003;284:G875-G879.
17. Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell2000;103:239-252.
18. Liu H, Lo CR, Czaja MJ. NF-�B inhibition sensitizes hepatocytes toTNF-induced apoptosis through a sustained activation of JNK and c-Jun.HEPATOLOGY 2002;35:772-778.
19. Schwabe RF, Uchinami H, Qian T, Bennett BL, Lemasters JJ, BrennerDA. Differential requirement for c-Jun NH2-terminal kinase in TNF�-and Fas-mediated apoptosis in hepatocytes. FASEB J 2004;18:720-722.
20. Uehara T, Xi Peng X, Bennett B, Satoh Y, Friedman G, Currin R, et al.c-Jun N-terminal kinase mediates hepatic injury after rat liver transplan-tation. Transplantation 2004;78:324-332.
21. Wang WH, Gregori G, Hullinger RL, Andrisani OM. Sustained activationof p38 mitogen-activated protein kinase and c-Jun N-terminal kinase path-ways by hepatitis B virus X protein mediates apoptosis via induction ofFas/FasL and tumor necrosis factor (TNF) receptor 1/TNF-� expression.Mol Cell Biol 2004;24:10352-10365.
22. Qiao L, Han SI, Fang Y, Park JS, Gupta S, Gilfor D, et al. Bile acid regulationof C/EBP�, CREB, and c-Jun function, via the extracellular signal-regulatedkinase and c-Jun NH2-terminal kinase pathways, modulates the apoptoticresponse of hepatocytes. Mol Cell Biol 2003;23:3052-3066.
23. Schattenberg JM, Wang Y, Singh R, Rigoli RM, Czaja MJ. HepatocyteCYP2E1 overexpression and steatohepatitis lead to impaired hepatic insu-lin signaling. J Biol Chem 2005;280:9887-9894.
24. Leclercq IA, Farrell GC, Field J, Bell DR, Gonzalez FJ, Robertson GR.CYP2E1 and CYP4A as microsomal catalysts of lipid peroxides in murinenonalcoholic steatohepatitis. J Clin Invest 2000; 105:1067-1075.
25. Dong C, Yang DD, Wysk M, Whitmarsh AJ, Davis RJ, Flavell RA. De-fective T cell differentiation in the absence of Jnk1. Science 1998;282:2092-2095.
26. Yang DD, Conze D, Whitmarsh AJ, Barrett T, Davis RJ, Rincon M, et al.Differentiation of CD4� T cells to Th1 cells requires MAP kinase JNK2.Immunity 1998;9:575-585.
27. Rinella ME, Green RM. The methionine-choline deficient dietary modelof steatohepatitis does not exhibit insulin resistance. J Hepatol 2004;40:47-51.
28. Sabapathy K, Hochedlinger K, Nam SY, Bauer A, Karin M, Wagner EF.Distinct roles for JNK1 and JNK2 in regulating JNK activity and c-Jun-dependent cell proliferation. Mol Cell 2004;15:713-725.
29. Leclercq IA, Farrell GC, Sempoux C, dela Pena A, Horsmans Y. Curcumininhibits NF-�B activation and reduces the severity of experimental steato-hepatitis in mice. J Hepatol 2004;41:926-934.
30. Feldstein AE, Canbay A, Angulo P, Taniai M, Burgart LJ, Lindor KD, etal. Hepatocyte apoptosis and fas expression are prominent features of hu-man nonalcoholic steatohepatitis. Gastroenterology 2003;125:437-443.
31. Lin HZ, Yang SQ, Chuckaree C, Kuhajda F, Ronnet G, Diehl AM. Met-formin reverses fatty liver disease in obese, leptin-deficient mice. Nat Med2000; 6:998-1003.
32. Karin M. Mitogen-activated protein kinase cascades as regulators of stressresponses. Ann N Y Acad Sci 1998;851:139-146.
33. Dietrich N, Thastrup J, Holmberg C, Gyrd-Hansen M, Fehrenbacher N,Lademann U, et al. JNK2 mediates TNF-induced cell death in mouseembryonic fibroblasts via regulation of both caspase and cathepsin proteasepathways. Cell Death Differ 2004;11:301-313.
34. Eminel S, Klettner A, Roemer L, Herdegen T, Waetzig V. JNK2 translo-cates to the mitochondria and mediates cytochrome c release in PC12 cellsin response to 6-hydroxydopamine. J Biol Chem 2004;279:55385-55392.
35. Liu J, Minemoto Y, Lin A. c-Jun N-terminal protein kinase 1 (JNK1), butnot JNK2, is essential for tumor necrosis factor alpha-induced c-Jun kinaseactivation and apoptosis. Mol Cell Biol 2004;24:10844-10856.
36. Maeda S, Chang L, Li ZW, Luo JL, Leffert H, Karin M. IKKbeta isrequired for prevention of apoptosis mediated by cell-bound but not bycirculating TNFalpha. Immunity 2003;19:725-737.
37. He T, Stepulak A, Holmstrom TH, Omary MB, Eriksson JE. The inter-mediate filament protein keratin 8 is a novel cytoplasmic substrate forc-Jun N-terminal kinase. J Biol Chem 2002;277:10767-10774.
38. Czaja MJ, Liu H, Wang Y. Oxidant-induced hepatocyte injury from men-adione is regulated by ERK and AP-1 signaling. HEPATOLOGY 2003;37:1405-1413.
39. Kaufman RJ. Orchestrating the unfolded protein response in health anddisease. J Clin Invest 2002;110:1389-1398.
40. Nakatani Y, Kaneto H, Kawamori D, Yoshiuchi K, Hatazaki M, MatsuokaTA, et al. Involvement of endoplasmic reticulum stress in insulin resistanceand diabetes. J Biol Chem 2005;280:847-851.
41. Xu A, Wang Y, Keshaw H, Xu LY, Lam KS, Cooper GJ. The fat-derivedhormone adiponectin alleviates alcoholic and nonalcoholic fatty liver dis-eases in mice. J Clin Invest 2003;112:91-100.
42. Kaneto H, Nakatani Y, Miyatsuka T, Kawamori D, Matsuoka TA, Mat-suhisa M, et al. Possible novel therapy for diabetes with cell-permeableJNK-inhibitory peptide. Nat Med 2004;10:1128-1132.
172 SCHATTENBERG ET AL. HEPATOLOGY, January 2006
Copyright © 2022 FDOKUMEN




















