
Involvement of G-protein βγ subunits on the influence of inhibitory α 2-autoreceptors on the...
10
Involvement of G-protein bg subunits on the influence of inhibitory a 2 -autoreceptors on the angiotensin AT 1 -receptor modulation of noradrenaline release in the rat vas deferens Carlos Talaia a , Glo ´ria Queiroz a, * , Helder Pinheiro a , Daniel Moura b , Jorge Gonc ¸alves a a Laboratory of Pharmacology, CEQOFFUP, Faculty of Pharmacy, University of Porto, Rua Anı ´bal Cunha, 164, 4050-047 Porto, Portugal b Institute of Pharmacology and Therapeutics, Faculty of Medicine, University of Porto, Alameda Hernani Monteiro, 4200-319 Porto, Portugal Received 9 March 2006; received in revised form 12 June 2006; accepted 7 July 2006 Available online 7 September 2006 Abstract The influence of a 2 -autoreceptors on the facilitation of [ 3 H]-noradrenaline release mediated by angiotensin II was studied in prostatic portions of rat vas deferens preincubated with [ 3 H]-noradrenaline. Angiotensin II enhanced tritium overflow evoked by trains of 100 pulses at 8 Hz, an effect that was attenuated by the AT 1 -receptor antagonist losartan (0.3–1 mM), at concentrations suggesting the involvement of the AT 1B subtype. The effect of angiotensin II was also attenuated by inhibition of phospholipase C (PLC) and protein kinase C (PKC) indicating that prejunctional AT 1 -receptors are coupled to the PLC–PKC pathway. Angiotensin II (0.3–100 nM) enhanced tritium overflow more markedly, up to 64%, under conditions that favor a 2 -autoinhibition, observed when stimulation consisted of 100 pulses at 8 Hz, than under poor a 2 -autoinhibition conditions, only up to 14%, observed when a 2 -adrenoceptors were blocked with yohimbine (1 mM) or when stimulation consisted of 20 pulses at 50 Hz. Activation of PKC with 12-myristate 13-acetate (PMA, 0.1–3 mM) also enhanced tritium overflow more markedly under strong a 2 -autoinhibition conditions. Inhibition of G i/o -proteins with pertussis toxin (8 mg/ml) or blockade of Gbg subunits with the anti-bg peptide MPS-Phos (30 mM) attenuated the effects of angiotensin II and PMA. The results indicate that activation of AT 1 -receptors coupled to the PLC–PKC pathway enhances noradrenaline release, an effect that is markedly favoured by an ongoing activation of a 2 -autoreceptors. Interaction between a 2 -adrenoceptors and AT 1 -receptors seems to involve the bg subunits released from the G i/o -proteins coupled to a 2 -adrenoceptors and protein kinase C activated by AT 1 -receptors. # 2006 Elsevier Ltd. All rights reserved. Keywords: Noradrenaline release; AT 1 -receptors; Protein kinase C; G i/o -coupled receptors; Gbg subunits; Receptor crosstalk Release of noradrenaline from sympathetic nerve terminals may be modulated (facilitated or inhibited) by activation of presynaptic receptors; the activation of one type of receptor may modify the effect mediated by another (Boehm and Kubista, 2002), a mutual influence that may involve receptors coupled to different signalling pathways. Facilitation of noradrenaline release mediated by b 2 - adrenoceptors, which are coupled to the G s –adenylyl cyclase (AC)–protein kinase A (PKA) pathway, may be attenuated by simultaneous activation of G i/o -protein coupled receptors (Majewski and Rand, 1981; Johnston and Majewski, 1986; Apparsundaram and Eikenburg, 1995; Queiroz et al., 2003) whereas facilitation mediated by bradykinin B 2 receptors, which are coupled to the G q/11 -phospholipase C (PLC)–protein kinase C (PKC) pathway, seems to require always the simultaneous activation of presynaptic G i/o -protein coupled www.elsevier.com/locate/neuint Neurochemistry International 49 (2006) 698–707 Abbreviations: AC, adenylyl cyclase; Rp-cAMPS, Rp-adenosine-3 0 ,5 0 -cyc- lic-monophosphorothioate triethylammonium; 9-CP-Ade, 9-(cyclopentyl)-ade- nine; DPCPX, 8-cyclopentyl-1,3-dipropylxanthine; PTX, pertussis toxin; PMA, phorbol 12-myristate 13-acetate; 4-M-PMA, phorbol-12-myristate-13-acetate 4-O-methyl ether; PKC, protein kinase C; PKA, protein kinase A; PLC, phospholipase C; Ro 32-0432, bisindolylmaleimide XI; TEA, tetraethylamo- nium; U-73122, 1-[6-[((17b)-3-methoxyestra-1,3,5[10]-trien-17-yl)amino]- hexyl]-1H-pyrrole-2,5-dione; U-73343, 1-[6-[((17b)-3-methoxyestra- 1,3,5[10]-trien-17-yl)amino]hexyl]-2,5-pyrrolidinedione; Brimonidine, 5- bromo-N-(2-imidazolin-2-yl)-6-quinoxalinamine * Corresponding author. Tel.: +351 22 2078 970; fax: +351 22 2078 969. E-mail address: [email protected] (G. Queiroz). 0197-0186/$ – see front matter # 2006 Elsevier Ltd. All rights reserved. doi:10.1016/j.neuint.2006.07.002
-
Upload
independent -
Category
Documents
-
view
2 -
download
0
Transcript of Involvement of G-protein βγ subunits on the influence of inhibitory α 2-autoreceptors on the...

www.elsevier.com/locate/neuint
Neurochemistry International 49 (2006) 698–707
Involvement of G-protein bg subunits on the influence of inhibitory
a2-autoreceptors on the angiotensin AT1-receptor modulation of
noradrenaline release in the rat vas deferens
Carlos Talaia a, Gloria Queiroz a,*, Helder Pinheiro a,Daniel Moura b, Jorge Goncalves a
a Laboratory of Pharmacology, CEQOFFUP, Faculty of Pharmacy, University of Porto,
Rua Anıbal Cunha, 164, 4050-047 Porto, Portugalb Institute of Pharmacology and Therapeutics, Faculty of Medicine, University of Porto,
Alameda Hernani Monteiro, 4200-319 Porto, Portugal
Received 9 March 2006; received in revised form 12 June 2006; accepted 7 July 2006
Available online 7 September 2006
Abstract
The influence of a2-autoreceptors on the facilitation of [3H]-noradrenaline release mediated by angiotensin II was studied in prostatic portions
of rat vas deferens preincubated with [3H]-noradrenaline. Angiotensin II enhanced tritium overflow evoked by trains of 100 pulses at 8 Hz, an effect
that was attenuated by the AT1-receptor antagonist losartan (0.3–1 mM), at concentrations suggesting the involvement of the AT1B subtype. The
effect of angiotensin II was also attenuated by inhibition of phospholipase C (PLC) and protein kinase C (PKC) indicating that prejunctional
AT1-receptors are coupled to the PLC–PKC pathway.
Angiotensin II (0.3–100 nM) enhanced tritium overflow more markedly, up to 64%, under conditions that favor a2-autoinhibition, observed
when stimulation consisted of 100 pulses at 8 Hz, than under poor a2-autoinhibition conditions, only up to 14%, observed when a2-adrenoceptors
were blocked with yohimbine (1 mM) or when stimulation consisted of 20 pulses at 50 Hz. Activation of PKC with 12-myristate 13-acetate (PMA,
0.1–3 mM) also enhanced tritium overflow more markedly under strong a2-autoinhibition conditions. Inhibition of Gi/o-proteins with pertussis
toxin (8 mg/ml) or blockade of Gbg subunits with the anti-bg peptide MPS-Phos (30 mM) attenuated the effects of angiotensin II and PMA.
The results indicate that activation of AT1-receptors coupled to the PLC–PKC pathway enhances noradrenaline release, an effect that is
markedly favoured by an ongoing activation of a2-autoreceptors. Interaction between a2-adrenoceptors and AT1-receptors seems to involve the bg
subunits released from the Gi/o-proteins coupled to a2-adrenoceptors and protein kinase C activated by AT1-receptors.
# 2006 Elsevier Ltd. All rights reserved.
Keywords: Noradrenaline release; AT1-receptors; Protein kinase C; Gi/o-coupled receptors; Gbg subunits; Receptor crosstalk
Release of noradrenaline from sympathetic nerve terminals
may be modulated (facilitated or inhibited) by activation of
Abbreviations: AC, adenylyl cyclase; Rp-cAMPS, Rp-adenosine-30,50-cyc-
lic-monophosphorothioate triethylammonium; 9-CP-Ade, 9-(cyclopentyl)-ade-
nine; DPCPX, 8-cyclopentyl-1,3-dipropylxanthine; PTX, pertussis toxin; PMA,
phorbol 12-myristate 13-acetate; 4-M-PMA, phorbol-12-myristate-13-acetate
4-O-methyl ether; PKC, protein kinase C; PKA, protein kinase A; PLC,
phospholipase C; Ro 32-0432, bisindolylmaleimide XI; TEA, tetraethylamo-
nium; U-73122, 1-[6-[((17b)-3-methoxyestra-1,3,5[10]-trien-17-yl)amino]-
hexyl]-1H-pyrrole-2,5-dione; U-73343, 1-[6-[((17b)-3-methoxyestra-
1,3,5[10]-trien-17-yl)amino]hexyl]-2,5-pyrrolidinedione; Brimonidine, 5-
bromo-N-(2-imidazolin-2-yl)-6-quinoxalinamine
* Corresponding author. Tel.: +351 22 2078 970; fax: +351 22 2078 969.
E-mail address: [email protected] (G. Queiroz).
0197-0186/$ – see front matter # 2006 Elsevier Ltd. All rights reserved.
doi:10.1016/j.neuint.2006.07.002
presynaptic receptors; the activation of one type of receptor
may modify the effect mediated by another (Boehm and
Kubista, 2002), a mutual influence that may involve receptors
coupled to different signalling pathways.
Facilitation of noradrenaline release mediated by b2-
adrenoceptors, which are coupled to the Gs–adenylyl cyclase
(AC)–protein kinase A (PKA) pathway, may be attenuated by
simultaneous activation of Gi/o-protein coupled receptors
(Majewski and Rand, 1981; Johnston and Majewski, 1986;
Apparsundaram and Eikenburg, 1995; Queiroz et al., 2003)
whereas facilitation mediated by bradykinin B2 receptors,
which are coupled to the Gq/11-phospholipase C (PLC)–protein
kinase C (PKC) pathway, seems to require always the
simultaneous activation of presynaptic Gi/o-protein coupled

C. Talaia et al. / Neurochemistry International 49 (2006) 698–707 699
Fig. 1. Experimental protocol. Tissues were incubated with [3H]-noradrenaline
([3H]NA; 0.1 mM), transferred to superfusion chambers and superfused with
medium containing the drugs indicated in the Section 1.2. In some experiments,
before incubation with [3H]NA, tissues were incubated with PTX (8 mg/ml) or
the anti-bg peptide (30 mM, MPS-Phos) or the respective solvents, which were
tested in the same experiment, in parallel control tissues. Up to five trains of 100
pulses at 8 Hz or 20 pulses at 50 Hz (S0–S4) were applied, every 30 min. S0 was
applied at t = 30 min of the superfusion but was not used for determination of
tritium overflow (not shown). Superfusate samples were collected at 5 min
intervals, from t = 55 min onwards. Ordinates represent tritium outflow as
fraction of the tissue tritium content at the onset of the respective collection
period. The area of the peaks represents tritium overflow evoked by electrical
stimulation (S1–S4). Agonists were added to the superfusion medium as
indicated. Values represent the fractional rate of tritium outflow from sol-
vent-treated tissues stimulated with trains 100 pulses at 8 Hz.
receptors (Cox et al., 2000; Trendelenburg et al., 2003; Mota
and Guimaraes, 2003). However, inconsistent effects have been
published on the influence of Gi/o-protein coupled receptors on
the effects of other Gq/11-PLC–PKC coupled receptors, such as
the angiotensin AT1-receptors. Angiotensin II mediated
facilitation of noradrenaline release was reported to be
enhanced (rabbit heart: Starke and Schumann, 1972; guinea
pig atria: Brasch et al., 1995; mouse tissues: Cox et al., 2000;
Trendelenburg et al., 2003; hearts of newborn rats: Mota and
Guimaraes, 2003), attenuated (rabbit pulmonary arteries: Costa
and Majewski, 1988) or not influenced (rat-tail artery; Mota
et al., 2000) by activation Gi/o-coupled receptors, such as the
a2-adrenoceptors.
Interaction between presynaptic receptors, although may not
be a general rule, may represent an important mechanism of fine
regulation of transmitter release under physiological conditions
and may also have important pathophysiological implications.
For example, in spontaneous hypertensive rats, hypertension
may be caused by an augmented crosstalk between receptors
activated by vasoactive peptides, such as angiotensin or
vasopressin, and a2-adrenoceptors in the renal microcirculation
(Jackson et al., 2001; Gao et al., 2003). The mechanisms
involved in such interactions are not fully elucidated, but some
evidences indicate it may occur at some step of the signalling
pathways to which the receptors are coupled (Starke, 2001).
The vas deferens has been widely used as a model to study
the postganglionic sympathetic transmission and its regulation.
In this tissue, noradrenaline release is modulated by several
presynaptic autoreceptors such as a2- and P2-receptors (von
Kugelgen et al., 1993) and by heteroreceptors such as the b2-
adrenoceptors, activated by blood born adrenaline and
adenosine A1-, A2A- and A2B-receptors, activated by locally
formed adenosine (Goncalves et al., 1996; Queiroz et al., 2003,
2004). Some of these receptors have been shown to interact. For
example, facilitation of noradrenaline release mediated by
adenosine A2-receptors, but not that mediated by b2-
adrenoceptors, is enhanced by tonic activation of Gi/o-protein
coupled receptors (Queiroz et al., 2003; Talaia et al., 2005). In
the rat vas deferens, angiotensin II also modulates sympathetic
transmission, through activation of AT1-receptors (Cox et al.,
1995; Sum et al., 1996). However, the contribuition of a
prejunctional modulation of noradrenaline release and the
influence of other prejunctional receptors on a putative release
facilitatory effect of angiotensin II in this tissue have not been
investigated. The present study aimed to investigate the effect
of angiotensin II on noradrenaline release, in the prostatic
portion of rat vas deferens, and whether the effect is influenced
and how it is influenced by activation of presynaptic
Gi/o-protein coupled receptors (i.e. a2-adrenoceptors).
1. Experimental procedures
1.1. Materials and solutions
The following drugs were used: levo-[ring-2,5,6-3H]-noradrenaline
([3H]NA), specific activity 53.0 Ci mmol�1 was from DuPont NEN (Garal,
Lisboa, Portugal); desipramine hydrochloride (DMI), 8-cyclopentyl-1,3-dipro-
pylxanthine (DPCPX), yohimbine hydrochloride, angiotensin II human,
5-bromo-N-(2-imidazolin-2-yl)-6-quinoxalinamine (brimonidine), phorbol
12-myristate 13-acetate (PMA), phorbol-12-myristate-13-acetate 4-O-methyl
ether (4-M-PMA), bisindolylmaleimide XI hydrochloride (Ro 32-0432), 9-
cyclopentyladenine (9-CP-Ade), Rp-adenosine-30,50-cyclic-monophosphor-
othioate triethylammonium (Rp-cAMPS), 1-(6-(((17b)-3-methoxyestra-1,3,5
(10)-trien-17-yl)amino)hexyl)-1H-pyrrole-2,5-dione (U-73122), 1-(6-(((17b)-
3-methoxyestra-1,3,5(10)-trien-17-yl)amino)hexyl)-2,5-pyrrolidinedione (U-
73343), pertussis toxin (PTX) and tetraethylamonium (TEA) were from Sigma
(Sintra, Portugal); losartan was from (Merck Portuguesa, Lisbon, Portugal) and
the anti-bg peptide AAVALLPAVLLALLAVTDQLGEDFFAVDLEAFL-QEF-
GLLPEKE (MPS-Phos) was from AnaSpec, Inc. (San Jose, USA). Stock
solutions of drugs were prepared with DMSO or distilled water and kept at
�20 8C. Solutions of drugs were prepared from aliquots of stock solutions that
were diluted in buffer immediately before use. The final concentration of
DMSO was 0.02%, being tested in parallel control tissue preparations.
1.2. Experimental protocol
Adult male Wistar rats (250–300 g; IBMC, Porto, Portugal) were used.
Animal handling and experiments were conducted according to the guidelines
of the European Communities Council Directive (86/609/EEC). Animals were
killed by cervical dislocation and exsanguination. Prostatic portions of vas
deferens were dissected out, cleaned of connective tissue and divided in portions
of 13 � 5 mg weight. The procedures used to label tissue preparations with
[3H]NA and to evaluate changes on electrically evoked tritium overflow as an
indicator of changes on neuronal noradrenaline release have been previously
described (e.g. Queiroz et al., 2004). Tissue preparations were incubated in 2 ml
of Krebs solution containing [3H]NA (0.1 mM; specific activity of
53.0 Ci mmol�1) for 40 min at 37 8C. In experiments with the anti-bg peptide
MPS-Phos or PTX, incubation conditions were adapted according to the
literature (Lai et al., 1983; von Kugelgen et al., 1993; Chang et al., 2000)
and tissues were incubated with anti-bg peptide MPS-Phos (30 mM) or PTX
(8 mg/ml) for 120 min and in the last 40 min period [3H]NA was also present
(Fig. 1). PTX was pre-activated by incubation with dithiothreitol (DTT) for

C. Talaia et al. / Neurochemistry International 49 (2006) 698–707700
30 min at 37 8C (Kaslow et al., 1987). Control tissues, tested in parallel (in the
same experiment), were incubated with the respective solvents (PBS for anti-bg
peptide and 5 mM DDT for PTX).
Individual preparations were then transferred to superfusion chambers
where they were held between platinum electrodes 7 mm apart, and superfused
with [3H]NA free medium at a constant flow rate of 1 ml min�1. A stimulator
(Hugo Sachs Elektronic-Type 215, March-Hungstetten, Germany), operating in
the constant current mode, was used for electrical field stimulation with square
wave pulses of 1 ms width and 50 mA current strength, yielding a voltage drop
of 20 V per chamber. The stimulation periods consisted of 100 pulses at 8 Hz or
20 pulses at 50 Hz. Up to five stimulation periods were applied. A primer
stimulation period, applied at t = 30 min (t = 0 min was the onset of super-
fusion) was not used for determination of tritium overflow; unless otherwise
indicated, subsequent stimulation periods were applied at t = 60 min (S1), t = 90
(S2), t = 120 (S3) and t = 150 (S4) min. Superfusate samples were collected at
5 min intervals, from t = 55 min onwards (Fig. 1). At the end of each experi-
ment, tritium content was determined in superfusate samples and in tissue
preparations by scintillation spectrometry (Beckman LS 6500, Beckman
Instruments, Fullerton, USA).
The solution used for incubation and superfusion contained (mM): NaCl
118.6, KCl 4.70, CaCl2 2.52, MgSO4 1.23, NaHCO3 25.0, glucose 10.0,
ascorbic acid 0.3 and dissodium EDTA 0.031; it was continuously gassed with
a mixture of 95% O2, 5% CO2 and maintained at 37 8C. Desipramine (0.4 mM;
to inhibit neuronal uptake of noradrenaline) and DPCPX (0.1 mM; to block
adenosine A1-receptors) were added at the beginning of superfusion and kept
throughout. When indicated, yohimbine (a2-adrenoceptor antagonist), U-73122
or U-73343 (phospholipase C inhibitor and its inactive analogue, respectively),
were also added at the beginning of superfusion and kept throughout. Unless
otherwise indicated, enzyme inhibitors and losartan were added to the super-
fusion medium 25 and 60 min before S2, respectively, and kept until the end of
the experiment; other drugs were added to the superfusion medium as indicated:
angiotensin II was added 10 min before Sn, PMA or its inactive analogue 4-M-
PMA were added 15 min before Sn and all were kept until the end of the
respective stimulation period. In tissues pre-incubated with the anti-bg peptide
MPS-Phos and PTX, or the respective solvents, angiotensin II, PMA or
isoprenaline, yohimbine or brimonidine were all tested in the same tissue
preparations: angiotensin II on S2, PMA on S3 and yohimbine on S4 (added
20 min before S4 and kept until the end of the experiment). In a set of these
experiments, isoprenaline instead of PMA, and brimonidine instead of yohim-
bine were assayed; they were added 6 min before S3 or before S4, respectively
and kept until the end of the stimulation period. TEA, when tested was added
25 min before S2 and kept until the end of the experiment.
Table 1
Basal tritium outflow (b1) and electrically evoked tritium overflow (S1) from contr
Drugs present Basal tritium
(b1; % of tissu
Throughout superfusion
100 pulses/8 Hz
Solvent 0.119 � 0.005
Yohimbine (1 mM) 0.152 � 0.038
20 pulses/50 Hz
Solvent 0.125 � 0.030
During incubation
Solvent (DTT; 5 mM) 0.270 � 0.056
DTT + PTX (8 mg/ml) 0.317 � 0.040
Solvent (PBS) 0.122 � 0.004
PBS + MPS-Phos (30 mM) 0.127 � 0.010
Preparations were incubated with [3H]-noradrenaline and superfused with medium c
throughout superfusion or present during the incubation). Pertussis toxin (PTX) and th
phosphate buffer saline (PBS), were present during the incubation period (see Section
(S0–S4). Presented values were obtained from control tissue preparations not expo
immediately before S1. Evoked tritium overflow was calculated by subtracting the
subsequent to the first stimulation period (S1), and was expressed as percentage of
means � S.E.M. for (n) tissue preparations. Significant differences from basal outfl* P < 0.05.
1.3. Evaluation of drug effects on evoked tritium overflow
Tritium outflow was calculated as fraction of the tissue tritium content at the
onset of the respective collection period (fractional rate of outflow, min�1). Drug
effects on basal tritium outflow were evaluated by the bn/b1 ratios and expressed
as percentage of change from the respective mean ratio obtained in the appro-
priate control; bn was the fractional rate of outflow in the 5 min period before S2–
S4 (b2–b4, respectively); b1 was the fractional rate of outflow in the 5 min period
before S1. Tritium overflow evoked by electrical stimulation was calculated by
subtracting the estimated basal outflow from the total outflow observed during
and in the 10 min period subsequent to each stimulation period and was expressed
as percentage of the total tritium present in the tissue at the onset of the
stimulation (Fig. 1). Effects of drugs added after S1 on tritium overflow were
estimated by ratios of the overflow elicited by S2–S4 (Sn) and the overflow elicited
by S1 (Sn/S1). Sn/S1 values obtained in individual experiments in which a test
compound ‘‘A’’ was added after S1 were calculated as a percentage of change
(increase or decrease) from the respective mean ratio obtained in the appropriate
control group (solvent instead of ‘‘A’’). When interaction of ‘‘A’’, added after S1,
and a drug ‘‘B’’ either added after S1 or at the beginning of superfusion was
studied, the ‘‘appropriate control’’ was a group in which B alone was used.
1.4. Presentation of data and statistical analysis
Results are expressed as means � standard errors of the mean (S.E.M.); n is
the number of tissue preparations. Statistical analysis of the effect of drugs on
basal tritium outflow and on electrically evoked tritium overflow was performed
by using the unpaired Student’s t-test or one-way analysis of variance (ANOVA)
followed by Dunnett’s multiple comparison test. P values lower than 0.05
(P < 0.05) indicate significant differences.
2. Results
2.1. General observations
The fractional rate of basal tritium outflow immediately
before S1 (b1) and the electrically evoked tritium overflow
elicited by S1 from prostatic portions of rat vas deferens in
different experimental conditions are shown in Table 1. Under
the experimental conditions used, calcium-omission from
ol preparations of the prostatic portion of rat vas deferens
outflow
e tritium min�1)
Evoked tritium overflow
(S1; % of tissue tritium)
(34) 0.210 � 0.011 (34)
(16) 0.837 � 0.064 (16)*
(8) 0.161 � 0.039 (8)*
(8)* 0.211 � 0.019 (8)
(8)* 0.285 � 0.025 (8)
(6) 0.161 � 0.012 (6)
(10) 0.174 � 0.014 (10)
ontaining DMI (0.4 mM) plus DPCPX (0.1 mM) and the drugs indicated (added
e anti-bg peptide (MPS-Phos) or the respective solvents: dithiothreitol (DTT) or
1.2). Up to five trains of 100 pulses at 8 Hz or 20 pulses at 50 Hz were applied at
sed to any drug after S1; b1 refers to tritium outflow during the 5 min period
estimated basal outflow from total outflow observed during and in the 10 min
the total tritium present in the tissue at the onset of the stimulation. Values are
ow or from tritium overflow in the presence of solvent at 100 pulses at 8 Hz.
C. Talaia et al. / Neurochemistry International 49 (2006) 698–707 701
superfusion medium abolished tritium overflow but not basal
outflow (not shown). The adenosine A1 receptor antagonist
DPCPX (0.1 mM) was present throughout superfusion in all
experiments to prevent the influence of tonic activation of
inhibitory adenosine A1 receptors.
Tissues were stimulated with trains of 100 pulses at 8 Hz or
20 pulses at 50 Hz. The stimulation conditions used were
selected in order to provide different levels of tritium overflow
and ongoing a2-autoinhibition (Singer, 1988). Electrical
stimulation with trains of 20 pulses at 50 Hz, elicited an
overflow of tritium which was about 67% of that elicited
by trains of 100 pulses at 8 Hz (see Table 1). The level of
ongoing a2-autoinhibition was assessed by the effect of the
a2-adrenoceptor antagonist yohimbine (1 mM) on tritium
overflow. Stimulation with trains of 100 pulses at 8 Hz
elicited an overflow of tritium that was much higher when the
a2-adrenoceptor antagonist yohimbine (1 mM) was present,
indica-ting a marked ongoing a2-autoinhibition under these
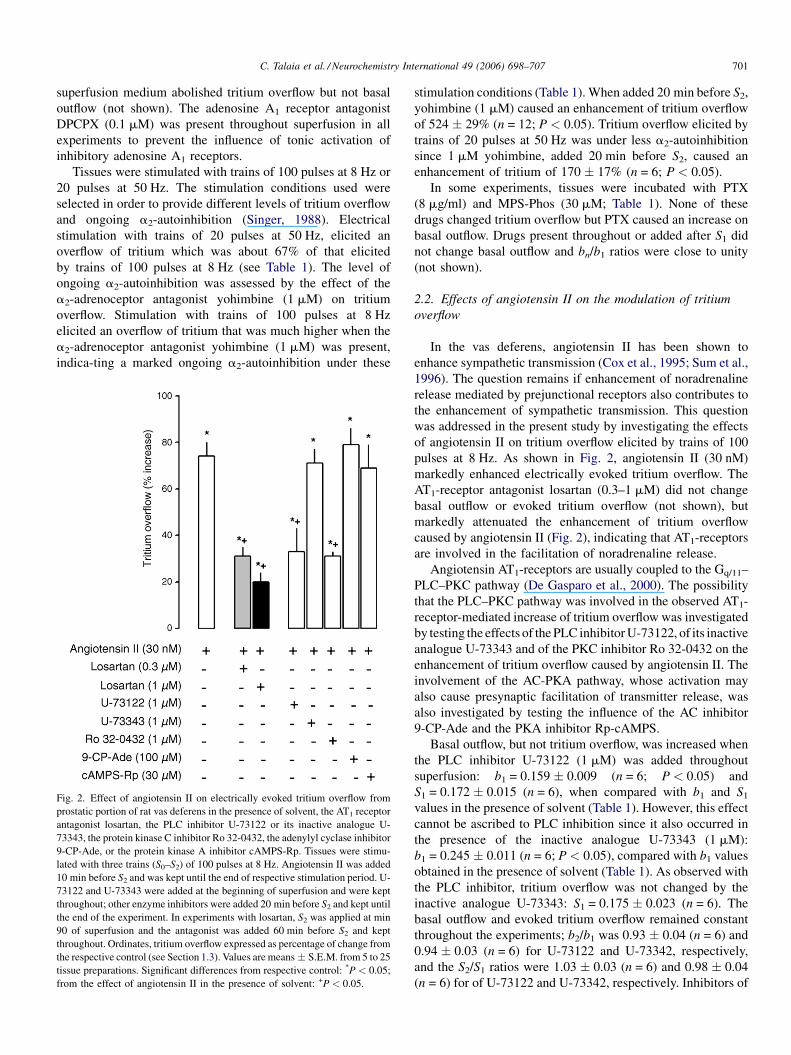
Fig. 2. Effect of angiotensin II on electrically evoked tritium overflow from
prostatic portion of rat vas deferens in the presence of solvent, the AT1 receptor
antagonist losartan, the PLC inhibitor U-73122 or its inactive analogue U-
73343, the protein kinase C inhibitor Ro 32-0432, the adenylyl cyclase inhibitor
9-CP-Ade, or the protein kinase A inhibitor cAMPS-Rp. Tissues were stimu-
lated with three trains (S0–S2) of 100 pulses at 8 Hz. Angiotensin II was added
10 min before S2 and was kept until the end of respective stimulation period. U-
73122 and U-73343 were added at the beginning of superfusion and were kept
throughout; other enzyme inhibitors were added 20 min before S2 and kept until
the end of the experiment. In experiments with losartan, S2 was applied at min
90 of superfusion and the antagonist was added 60 min before S2 and kept
throughout. Ordinates, tritium overflow expressed as percentage of change from
the respective control (see Section 1.3). Values are means � S.E.M. from 5 to 25
tissue preparations. Significant differences from respective control: *P < 0.05;
from the effect of angiotensin II in the presence of solvent: +P < 0.05.
stimulation conditions (Table 1). When added 20 min before S2,
yohimbine (1 mM) caused an enhancement of tritium overflow
of 524 � 29% (n = 12; P < 0.05). Tritium overflow elicited by
trains of 20 pulses at 50 Hz was under less a2-autoinhibition
since 1 mM yohimbine, added 20 min before S2, caused an
enhancement of tritium of 170 � 17% (n = 6; P < 0.05).
In some experiments, tissues were incubated with PTX
(8 mg/ml) and MPS-Phos (30 mM; Table 1). None of these
drugs changed tritium overflow but PTX caused an increase on
basal outflow. Drugs present throughout or added after S1 did
not change basal outflow and bn/b1 ratios were close to unity
(not shown).
2.2. Effects of angiotensin II on the modulation of tritium
overflow
In the vas deferens, angiotensin II has been shown to
enhance sympathetic transmission (Cox et al., 1995; Sum et al.,
1996). The question remains if enhancement of noradrenaline
release mediated by prejunctional receptors also contributes to
the enhancement of sympathetic transmission. This question
was addressed in the present study by investigating the effects
of angiotensin II on tritium overflow elicited by trains of 100
pulses at 8 Hz. As shown in Fig. 2, angiotensin II (30 nM)
markedly enhanced electrically evoked tritium overflow. The
AT1-receptor antagonist losartan (0.3–1 mM) did not change
basal outflow or evoked tritium overflow (not shown), but
markedly attenuated the enhancement of tritium overflow
caused by angiotensin II (Fig. 2), indicating that AT1-receptors
are involved in the facilitation of noradrenaline release.
Angiotensin AT1-receptors are usually coupled to the Gq/11–
PLC–PKC pathway (De Gasparo et al., 2000). The possibility
that the PLC–PKC pathway was involved in the observed AT1-
receptor-mediated increase of tritium overflow was investigated
by testing the effects of the PLC inhibitor U-73122, of its inactive
analogue U-73343 and of the PKC inhibitor Ro 32-0432 on the
enhancement of tritium overflow caused by angiotensin II. The
involvement of the AC-PKA pathway, whose activation may
also cause presynaptic facilitation of transmitter release, was
also investigated by testing the influence of the AC inhibitor
9-CP-Ade and the PKA inhibitor Rp-cAMPS.
Basal outflow, but not tritium overflow, was increased when
the PLC inhibitor U-73122 (1 mM) was added throughout
superfusion: b1 = 0.159 � 0.009 (n = 6; P < 0.05) and
S1 = 0.172 � 0.015 (n = 6), when compared with b1 and S1
values in the presence of solvent (Table 1). However, this effect
cannot be ascribed to PLC inhibition since it also occurred in
the presence of the inactive analogue U-73343 (1 mM):
b1 = 0.245 � 0.011 (n = 6; P < 0.05), compared with b1 values
obtained in the presence of solvent (Table 1). As observed with
the PLC inhibitor, tritium overflow was not changed by the
inactive analogue U-73343: S1 = 0.175 � 0.023 (n = 6). The
basal outflow and evoked tritium overflow remained constant
throughout the experiments; b2/b1 was 0.93 � 0.04 (n = 6) and
0.94 � 0.03 (n = 6) for U-73122 and U-73342, respectively,
and the S2/S1 ratios were 1.03 � 0.03 (n = 6) and 0.98 � 0.04
(n = 6) for of U-73122 and U-73342, respectively. Inhibitors of
C. Talaia et al. / Neurochemistry International 49 (2006) 698–707702
PKC, AC and PKA, when tested alone, did not change basal
outflow or tritium overflow (not shown).
Enhancement of tritium overflow elicited by angiotensin II
(30 nM) was markedly attenuated by the PLC inhibitor U-
73122 (1 mM) and by the PKC inhibitor Ro 32-0432 (1 mM)
and was not changed by U-73343 (1 mM; Fig. 2). Inhibition of
the AC-PKA pathway with the AC inhibitor 9-CP-Ade
(100 mM) or with the PKA inhibitor Rp-cAMPS (30 mM)
did not change the enhancement of tritium overflow caused by
angiotensin II (30 nM; Fig. 2).
2.3. Effects of angiotensin AT1-receptors under different
levels of ongoing a2-autoinhibition
In several sympathetically innervated tissues, the level of
ongoing activation of a2-autoreceptors has been shown to
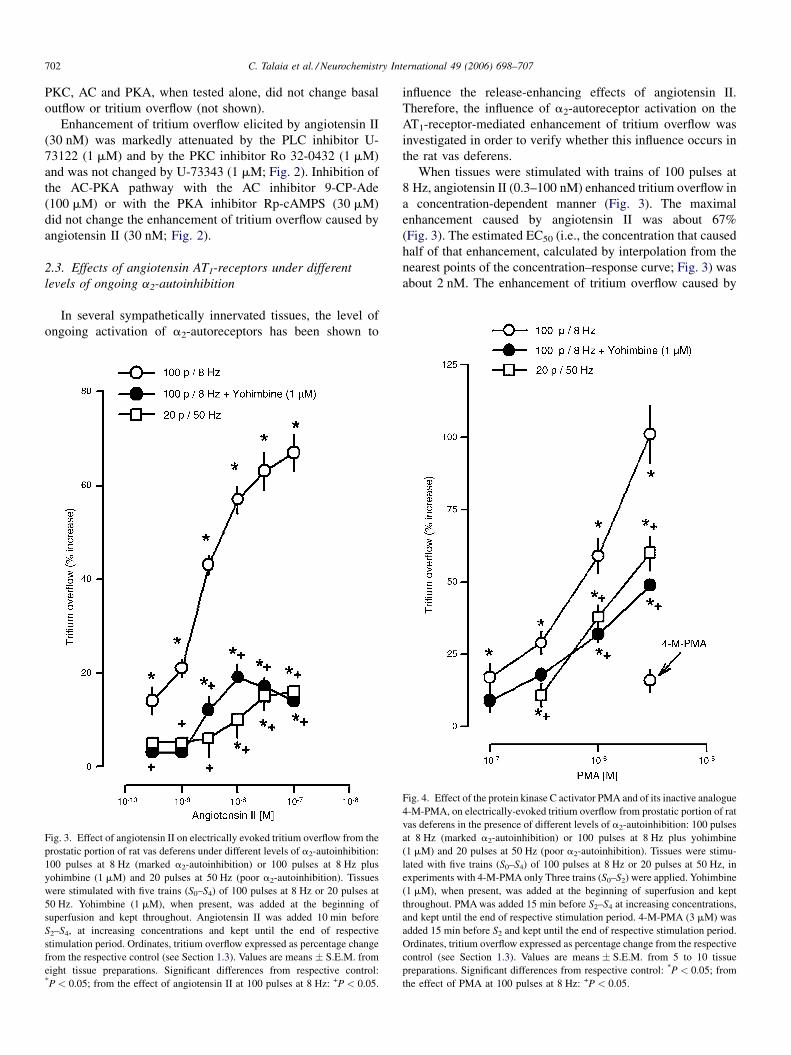
Fig. 3. Effect of angiotensin II on electrically evoked tritium overflow from the
prostatic portion of rat vas deferens under different levels of a2-autoinhibition:
100 pulses at 8 Hz (marked a2-autoinhibition) or 100 pulses at 8 Hz plus
yohimbine (1 mM) and 20 pulses at 50 Hz (poor a2-autoinhibition). Tissues
were stimulated with five trains (S0–S4) of 100 pulses at 8 Hz or 20 pulses at
50 Hz. Yohimbine (1 mM), when present, was added at the beginning of
superfusion and kept throughout. Angiotensin II was added 10 min before
S2–S4, at increasing concentrations and kept until the end of respective
stimulation period. Ordinates, tritium overflow expressed as percentage change
from the respective control (see Section 1.3). Values are means � S.E.M. from
eight tissue preparations. Significant differences from respective control:*P < 0.05; from the effect of angiotensin II at 100 pulses at 8 Hz: +P < 0.05.
influence the release-enhancing effects of angiotensin II.
Therefore, the influence of a2-autoreceptor activation on the
AT1-receptor-mediated enhancement of tritium overflow was
investigated in order to verify whether this influence occurs in
the rat vas deferens.
When tissues were stimulated with trains of 100 pulses at
8 Hz, angiotensin II (0.3–100 nM) enhanced tritium overflow in
a concentration-dependent manner (Fig. 3). The maximal
enhancement caused by angiotensin II was about 67%
(Fig. 3). The estimated EC50 (i.e., the concentration that caused
half of that enhancement, calculated by interpolation from the
nearest points of the concentration–response curve; Fig. 3) was
about 2 nM. The enhancement of tritium overflow caused by
Fig. 4. Effect of the protein kinase C activator PMA and of its inactive analogue
4-M-PMA, on electrically-evoked tritium overflow from prostatic portion of rat
vas deferens in the presence of different levels of a2-autoinhibition: 100 pulses
at 8 Hz (marked a2-autoinhibition) or 100 pulses at 8 Hz plus yohimbine
(1 mM) and 20 pulses at 50 Hz (poor a2-autoinhibition). Tissues were stimu-
lated with five trains (S0–S4) of 100 pulses at 8 Hz or 20 pulses at 50 Hz, in
experiments with 4-M-PMA only Three trains (S0–S2) were applied. Yohimbine
(1 mM), when present, was added at the beginning of superfusion and kept
throughout. PMA was added 15 min before S2–S4 at increasing concentrations,
and kept until the end of respective stimulation period. 4-M-PMA (3 mM) was
added 15 min before S2 and kept until the end of respective stimulation period.
Ordinates, tritium overflow expressed as percentage change from the respective
control (see Section 1.3). Values are means � S.E.M. from 5 to 10 tissue
preparations. Significant differences from respective control: *P < 0.05; from
the effect of PMA at 100 pulses at 8 Hz: +P < 0.05.

C. Talaia et al. / Neurochemistry International 49 (2006) 698–707 703
angiotensin II was attenuated in the presence of the a2-
adrenoceptor-antagonist yohimbine (1 mM) and when tissues
were stimulated under conditions that also led to poor ongoing
a2-autoinhibition (20 pulses at 50 Hz, Fig. 3), indicating that
effects mediated by prejunctional angiotensin AT1-receptors on
tritium overflow are also influenced by the ongoing activation of
a2-autoreceptors, and the existence of an interaction between
AT1-receptors and a2-adrenoceptors in this tissue.
2.4. Mechanism of interaction between angiotensin AT1
and a2-adrenoceptors
In order to elucidate the molecular mechanisms of the
interaction between AT1-receptors and a2-adrenoceptors, the
effect of several drugs that interfere with the signalling pathways
to which these receptors are coupled was tested. The effects of
two compounds that disrupt the Gi/o-proteins, PTX and the anti
bg-peptide MPS-Phos, were investigated on the modulation of
tritium overflow mediated by a2-adrenoceptors and on the
effects of angiotensin II and PMA, which activate the AT1-
transduction pathway at the receptor or PKC level, respectively.
The results obtained indicate that prejunctional AT1-
receptores are coupled to the PLC–PKC pathway. Therefore,
the influence of a direct activator of PKC on tritium overflow,
evoked under different levels of ongoing a2-autoinhibition (see
Section 2.1) was tested. The PKCactivator PMA (0.1–3 mM), but
not its inactive analogue 4-M-PMA, enhanced, in a concentra-
tion-dependent manner, tritium overflow evoked by 100 pulses at
8 Hz (Fig. 4). The effect of PMA, like that caused by angiotensin
II, was attenuated by blockade of a2-adrenoceptors with 1 mM
yohimbine or when tissues were stimulated under conditions
Fig. 5. Effect of the a2-adrenoceptor agonist brimonidine (A) and the a2-adrenocep
prostatic portions of rat vas deferens incubated with PTX or the anti-bg peptide MPS
up to five trains (S0–S4) of 100 pulses at 8 Hz. Angiotensin II was added 10 min befo
results on Fig. 6); brimonidine or yohimbine were added 6 and 20 min before S4, re
Ordinates, tritium overflow expressed as percentage change from the respective contr
Significant differences from respective control: *P < 0.05; from the effect of yohimbi+P < 0.05.
that also led to poor ongoing a2-autoinhibition (20 pulses at
50 Hz; Fig. 4).
Incubation of the tissues with PTX (8 mg/ml), at a concen-
tration that hasbeen shown to uncouple Gi/o-proteins in this tissue
(Lai et al., 1983; von Kugelgen et al., 1993) prevented the
decrease on tritium overflow caused by the a2-adrenoceptor
agonist brimonidine (1 mM; Fig. 5A). A similar effect was
observed when tissues were incubated with the anti-bg peptide
MPS-Phos, an inhibitor of Gbg subunits (Chang et al., 2000; Orr
et al., 2002), at a concentration shown to prevent effects mediated
by Gbg subunits (30 mM; Chang et al., 2000). Furthermore,
yohimbine (1 mM) enhanced tritium overflow by preventing
ongoing activation of a2-autoreceptors (Fig. 5B) but this effect
was much smaller in tissues that were previously incubated with
PTX and MPS-Phos (Fig. 5B). Taken together, these results
indicate that, under these experimental conditions, PTX and
MPS-Phos are inhibiting the pathway to which the a2-adre-
noceptors are coupled.
The enhancement of tritium overflow caused by angiotensin II
(30 nM) and PMA (1 mM), but not that caused by the b-
adrenoceptor agonist isoprenaline, was markedly attenuated in
tissues treated with PTX (8 mg/ml; Fig. 6). Isoprenalinewas used
asnegative control, in this set of experiments because it facilitates
noradrenaline release by activation of b2-adrenoceptors that are
not coupled to Gq-proteins and the effect they mediate is not
influenced by ongoing activation of a2-adrenoceptors (Talaia
et al., 2005).
The anti-bg peptide MPS-Phos (30 mM) also attenuated
the enhancement of tritium overflow caused by angiotensin II
(30 nM) or PMA (1 mM; Fig. 6). However, MPS-Phos failed
to change the enhancement of tritium overflow caused by the
tor antagonist yohimbine (B), on electrically evoked tritium overflow from the
-Phos or the respective solvents (see Section 1.2). Tissues were stimulated with
re S2, PMA or isoprenaline were added 15 and 6 min before S3, respectively (see
spectively, and all were kept up to the end of the respective stimulation period.
ol (see Section 1.3). Values are means � S.E.M. from 6 to 12 tissue preparations.
ne in tissues incubated the respective solvents of PTX and of the anti-bg peptide,

C. Talaia et al. / Neurochemistry International 49 (2006) 698–707704
Fig. 6. Effects of angiotensin II, PMA, isoprenaline and TEA, on electrically
evoked tritium overflow from prostatic portion of rat vas deferens incubated
with PTX or anti-bg peptide (anti-bg) or the respective solvents (see Section
1.2). Tissues were stimulated with up to five trains (S0–S4) of 100 pulses at 8 Hz.
Angiotensin II was added 10 min before S2, PMA or isoprenaline were added 15
and 6 min before S3, respectively, and all were kept up to the end of the
respective stimulation period. TEA was added 20 min before S2 and kept
throughout. Only in experiments with TEA, yohimbine (1 mM) was present
throughout superfusion. Ordinates, tritium overflow expressed as percentage of
change from the respective control (see Section 1.3). Values are mean-
s � S.E.M. from 5 to 8 tissue preparations. Significant differences from
respective control (solvent): *P < 0.05; from the effect of angiotensin II,
PMA, isoprenaline and TEA in tissues incubated with the respective solvents
of PTX and of the anti-bg peptide, +P < 0.05.
non-selective K+-channel blocker TEA (1 mM; Fig. 6),
excluding the possibility that MPS-Phos attenuates the effects
of any drug that enhances tritium overflow.
3. Discussion
The electrically evoked tritium overflow from tissue
preparations pre-incubated with [3H]NA has been shown to
reflect action potential-evoked neuronal release of noradrena-
line (Starke, 1977). Therefore, changes in tritium overflow
elicited by drugs, from the prostatic portions of rat vas deferens
pre-incubated with [3H]NA, were assumed to reflect changes in
neuronal release of noradrenaline, as observed in previous
studies (Queiroz et al., 2004).
In the rat vas deferens, angiotensin II facilitates the
purinergic component of sympathetic transmission through
activation of AT1-receptors (Cox et al., 1995; Sum et al., 1996).
The present study shows that angiotensin II also facilitates
noradrenaline release, an effect that may contribute to the
overall facilitation of sympathetic transmission. This facilita-
tion was blocked by the selective AT1-receptor antagonist
losartan (Duncia et al., 1992), although concentrations much
higher than those needed to antagonise the constrictor response
to angiotensin II were needed. In our experiments the blockade
of the prejunctional angiotensin II effect was obtained only
after incubation for 60 min with 0.3 mM losartan. This agrees
with the suggestion that prejunctional angiotensin AT1-
receptors belong to the AT1B subtype, thus requiring higher
concentrations of losartan to be blocked (Pinheiro et al., 2002;
Nap et al., 2004; Guimaraes and Pinheiro, 2005).
Angiotensin AT1-receptors are generally coupled to the
Gq/11–PLC–PKC pathway, the same transduction mechanism
activated by AT1-receptors that mediate a facilitation of
noradrenaline release in the rat vas deferens. This conclusion is
supported by the observation that the effect of angiotensin II
was attenuated by inhibition of PLC with U-73122 and by
inhibition of PKC with Ro 32-0432. Both compounds have
been used at concentrations shown to selectively inhibit PLC
and PKC, respectively (Wirkner et al., 2000; Wilkinson et al.,
1993). The possibility that prejunctional angiotensin AT1-
receptors might also be coupled to the AC-PKA pathway was
excluded since the effect of angiotensin II was not changed by
inhibition of AC or PKA, with 9-CP-Ade and Rp-cAMPS,
respectively.
In the rat vas deferens, facilitation of noradrenaline by
adenosine A2A-receptors, which are coupled to the PLC–PKC
pathway and by adenosine A2B-receptors, which are coupled to
the AC-PKA pathway requires, or at least is much amplified by
activation of the release inhibitory a2-adrenoceptors (Queiroz
et al., 2003; Talaia et al., 2005). The possibility that activation
a2-autoreceptors may also influence the facilitation of nora-
drenaline mediated by angiotensin AT1-receptors was investi-
gated by studying the effects of angiotensin II on noradrenaline
release elicited under different conditions of ongoing a2-
autoinhibition. The level of a2-autoinhibition was assessed by
the enhancement of noradrenaline release caused by blockade of
a2-adrenoceptors with yohimbine, tested at 1 mM, a concentra-
tion that is more than 10-fold its pA2 value (Bylund and Ray-
Prenger, 1989). When tissues were stimulated with trains of 100
pulses at 8 Hz, noradrenaline release was under more marked
influence of a2-autoreceptors than when tissues were stimulated
with trains of 20 pulses at 50 Hz, since yohimbine enhanced
more markedly noradrenaline release evoked by trains of 100
pulses at 8 Hz than by trains of 20 pulses at 50 Hz. Stimulation
with brief trains of pulses applied at high frequency (20 pulses at
50 Hz), does not favour activation of a2-autoreceptors (Singer,
1988; Starke, 2001). Poor ongoing a2-autoinhibition conditions
were also created by stimulating the tissues with trains of 100
pulses at 8 Hz in the presence of the a2-adrenoceptor antagonist
yohimbine (1 mM), at a concentration that, in the experimental
conditions used, caused maximal facilitation of noradrenaline
release.
Angiotensin II enhanced noradrenaline release more
markedly when tissues were stimulated with trains of 100
pulses at 8 Hz (strong ongoing a2-autoinhibition conditions),
than when a2-adrenoceptors were blocked with yohimbine, or
when release was evoked by trains of 20 pulses at 50 Hz (both
conditions lead to poor ongoing a2-autoinhibition). The
possibility that noradrenaline release was already maximal
when a2-adrenoceptors were blocked, preventing further

C. Talaia et al. / Neurochemistry International 49 (2006) 698–707 705
increases from being observed, has been excluded because the
b-adrenoceptor-agonist isoprenaline caused a facilitation of
noradrenaline of the same magnitude with or without a2-
adrenoceptor blockade (Talaia et al., 2005). Furthermore, two
different conditions that lead to poor a2-autoinhibition also lead
to an attenuation of the angiotensin II effect, indicating that in
this tissue, AT1-receptors and a2-adrenoceptors interact in a
way that angiotensin AT1-receptor-mediated facilitation of
noradrenaline release requires, or is amplified, by an ongoing
activation of a2-adrenoceptors. A similar interaction has been
previously described in other sympathetically innervated
tissues (rabbit heart; Starke and Schumann, 1972; guinea pig
atria; Brasch et al., 1995; mouse atria; Cox et al., 2000; mouse
tissues; Trendelenburg et al., 2003; rat heart; Mota and
Guimaraes, 2003). However it does not seem to be a general
phenomena since in some tissues facilitation of noradrenaline
release by angiotensin II is not influenced (rat tail artery; Mota
et al., 2000) or is attenuated (rabbit pulmonary artery; Costa and
Majewski, 1988) by activation of a2-autoreceptors; therefore
the understanding of the molecular mechanism behind this
interaction may contribute to explain this variability.
Angiotensin AT1-receptors are coupled to the Gq/11–PLC–
PKC pathway. Facilitation of noradrenaline release caused by
direct activation of PKC, like facilitation caused by angiotensin
II, was smaller under poor a2-autoinhibition conditions (100
pulses at 8 Hz plus yohimbine or 20 pulses at 50 Hz). Evidence
that enhancement of tritium overflow caused by PMA is
mediated by PKC was provided by a previous observation that
the PKC inhibitor Ro 32-0432 prevents the release enhancing
effect of PMA (Queiroz et al., 2004). Since interaction is also
observed when the Gq/11–PLC–PKC pathway is activated at
PKC level, the interaction between the a2-adrenoceptors and
angiotensin AT1-receptor-signalling pathways must occur
mainly at steps downstream PKC activation. However,
involvement of other steps located upstream PKC activation
cannot be excluded since blockade of a2-adrenoceptors or
inactivation of Gi/o-proteins with PTX attenuated more
markedly the facilitation of noradrenaline release caused by
angiotensin II than that caused by direct activation of PKC.
Previous studies suggest that interaction between Gi/o-
protein coupled receptors and Gq11-protein coupled receptors
may occur at multiple steps of the signalling pathways, namely:
(i) enhancement of PLC activity by bg subunits released from
Gi/o-proteins (Blank et al., 1992; Banno et al., 1998; Selbie and
Hill, 1998); (ii) PKC removal of Ca2+-channel-inhibition
mediated by the bg subunits released from Gi/o-proteins
(Swartz, 1993; Hamid et al., 1999; Barrett and Rittenhouse,
2000; Lee et al., 2004); (iii) PKC-mediated desensitisation of
the Gi/o-proteins (Murthy et al., 2000) or the Gi/o-coupled
receptors (Fredholm and Lindgren, 1988; Liang et al., 1998).
The present study indicates an involvement of G-protein bg
subunits on this interaction. The anti-bg peptide MPS-Phos, a
cell permeable peptide that prevents the effects of bg subunits
(Chang et al., 2000; Orr et al., 2002) attenuated the facilitation
of noradrenaline release caused by angiotensin II. The main
source of bg subunits seem to be the Gi/o-proteins activated by
a2-adrenoceptors because MPS-Phos also attenuated the
release enhancing effects caused by direct activation of
PKC, which bypasses activation of Gq11-proteins and PLC
coupled to the angiotensin AT1-receptors. The effect of the
MPS-Phos seems to be selective for bg subunits because it did
not changed the effect of TEA that enhances noradrenaline
release by a mechanism independent of G-protein activation
(Kirpekar et al., 1976). Furthermore, bg subunits are key
players in the interaction observed because MPS-Phos did not
change the facilitation of noradrenaline release mediated by b-
adrenoceptors, which are coupled to G-proteins but do not
interact with the a2-adrenoceptors (Talaia et al., 2005).
Influence of bg subunits on the PLC–PKC signalling
pathway has been described upstream or downstream PKC
activation. Upstream PKC, a possible step at which interaction
may occur is the activation of PLC. It has been shown that bg
subunits released from Gi/o-proteins, upon activation of Gi/o-
protein-coupled receptors, can enhance activation of PLCb by
the Gaq subunits released from Gq11-coupled receptors (Blank
et al., 1992; Banno et al., 1998; Selbie and Hill, 1998)
contributing to trigger or amplify the effect mediated by
angiotensin AT1-receptors. A possible step downstream PKC
activation is the regulation of the opening-closure rate of
voltage-gated Ca2+-channels, a key-step in prejunctional
modulation of transmitter release and one of the targets of
the a2-adrenoceptor-mediated inhibition of noradrenaline
release (Hamid et al., 1999). The bg subunits, released from
Gi/o-proteins, inhibit Ca2+ influx and transmitter release, an
effect that can be attenuated by PKC (Swartz, 1993; Hamid
et al., 1999; Barrett and Rittenhouse, 2000; Lee et al., 2004) and
this may provide a mechanism by which PKC can cause a
facilitation of noradrenaline release that depends on the level of
ongoing a2-autoinhibition.
Irrespectively of the step at which interaction occurs, the
common link is the involvement of the bg subunits released by
activation of Gi/o-coupled receptors. The observation that MPS-
Phos also attenuated facilitation of noradrenaline release
caused by direct activation of PKC allows to conclude that: (i)
the main source of the bg subunits involved are released by
activation of Gi/o proteins; (ii) the interaction must involve steps
downstream PKC activation.
Modulation of neurotransmitter release by presynaptic
receptors was an interesting finding and changed the way
neuronal communication was understood. The release inhibi-
tory receptors are viewed as a logical mechanism for a neuron
to control the amount of neurotransmitter required, but the
physiological role of facilitatory receptors is still not yet very
clear. One possibility is that they can work to select the more
convenient cotransmitter combination (Goncalves et al., 1996;
Driessen et al., 1996). The interactions like those reported in the
present study may indicate another function for these receptors:
that they may represent a mechanism to control the effects of
the release inhibitory receptors, preventing excessive inhibi-
tion.
Interaction between prejunctional facilitatory and inhibitory
receptors involved in the modulation of noradrenaline release
have been described in several tissues, and also in the tissue
used in the present study. However, interactions do not follow a

C. Talaia et al. / Neurochemistry International 49 (2006) 698–707706
Fig. 7. Model proposed to explain the influence of a2-adrenoceptors on the
modulation of noradrenaline release by angiotensin AT1-receptors. Activation
of angiotensin AT1-receptors leads to activation of PLC and PKC. Activation of
a2-adrenoceptors, which are coupled to Gi/o-proteins, leads to the release of bg
subunits that may enhance activation of PLC (Blank et al., 1992; Banno et al.,
1998) or inhibit the secretory machinery (Blackmer et al., 2001) and the opening
of N-type Ca2+-channels (Delmas et al., 1999). PKC enhances noradrenaline
release by acting on secretory machinery (Shoji-Kasai et al., 2002) and/or the N-
type Ca2+-channels, removing the inhibition mediated by the bg subunits, and
thus causing desinhibition (facilitation) of transmitter release (Hamid et al.,
1999).
constant pattern. It is possible that the occurrence of
interactions between receptors depends on the cellular
organization of receptors or cluster of receptors, that have
common molecular targets, whose function they regulate, or
that share common intracellular signalling proteins (Oh and
Schnitzer, 2001; Hur and Kim, 2002; Razani et al., 2002).
In conclusion, in the prostatic portion of the rat vas deferens,
angiotensin AT1-receptor-activation facilitates noradrenaline
release and this may contribute to the enhancement of
sympathetic transmission reported in this tissue. The angio-
tensin AT1-receptor-mediated facilitation of noradrenaline
release is greatly enhanced by an ongoing activation of a2-
adrenoceptors and the mechanisms proposed to explain such
interaction involves steps mainly downstream PKC activation
and the bg subunits released upon activation of Gi/o-protein
coupled a2-adrenoceptors (Fig. 7).
Acknowledgements
This work was supported by FCT (I&D n. 226/94) and
POCI/SAU-FCT/60714/2004.
References
Apparsundaram, S., Eikenburg, D.C., 1995. Role of prejunctional beta adre-
noceptors in rat cardiac sympathetic neurotransmission. J. Pharmacol. Exp.
Ther. 272, 519–526.
Banno, Y., et al., 1998. Stimulation by G protein bg subunits of phospholipase
C beta isoforms in human platelets. Thromb. Haemost. 79, 1008–1013.
Barrett, C.F., Rittenhouse, A.R., 2000. Modulation of N-type calcium channel
activity by G-proteins and protein kinase C. J. Gen. Physiol. 115, 277–286.
Blackmer, T., et al., 2001. G protein bg subunit-mediated presynaptic inhibi-
tion: regulation of exocytotic fusion downstream of Ca2+ entry. Science 292,
293–297.
Blank, J.L., et al., 1992. Activation of cytosolic phosphoinositide phospho-
lipase C by G-protein bg subunits. J. Biol. Chem. 267, 23069–23075.
Boehm, S., Kubista, H., 2002. Fine-tuning of sympathetic transmitter release
via ionotropic and metabotropic presynaptic receptors. Pharmacol. Rev. 4,
43–99.
Brasch, H., et al., 1995. In field-stimulated guinea-pig atria an AT1-receptor
mediated increase of noradrenaline release by angiotensin II is seen only in
the presence of prejunctional autoinhibition. Adv. Exp. Med. Biol. 377,
293–298.
Bylund, D.B., Ray-Prenger, C., 1989. Alfa-2A and alfa-2B adrenergic receptor
subtypes: attenuation of cyclic AMP production in cell lines containing only
one receptor subtype. J. Pharmacol. Exp. Ther. 251, 640–644.
Chang, M., et al., 2000. Dissecting G protein-coupled receptor signaling
pathways with membrane-permeable blocking peptides. J. Biol. Chem.
275, 7021–7029.
Costa, M., Majewski, H., 1988. Facilitation of noradrenaline release from
sympathetic nerves through activation of ACTH receptors, b-adrenoceptors
and angiotensin II receptors. Br. J. Pharmacol. 95, 993–1001.
Cox, S.L., et al., 1995. Evidence for the involvement of different receptor
subtypes in the pre- and postjunctional actions of angiotensin II at rat
sympathetic neuroeffector sites. Br. J. Pharmacol. 114, 1057–1063.
Cox, S.L., et al., 2000. Enhancement of noradrenaline release by angiotensin II
and bradykinin in mouse atria: evidence for cross talk between Gq/11 protein-
and Gi/o protein-coupled receptors. Br. J. Pharmacol. 129, 1095–1102.
De Gasparo, et al., 2000. International Union of Pharmacology. XXIII. The
angiotensin II receptors. Pharmacol. Rev. 52, 415–472.
Delmas, P., et al., 1999. bg Dimers derived from Go and Gi proteins contribute
different components of adrenergic inhibition of Ca2+ channels in rat
sympathetic neurons. J. Physiol. 518, 23–36.
Driessen, B., et al., 1996. Opposite modulation of noradrenaline and ATP
release in guinea-pig vas deferens through prejunctional beta-adrenocep-
tors: Evidence for the beta2 subtype. Naunyn-Schmied. Arch. Pharmacol.
353, 564–571.
Duncia, J.V., et al., 1992. The discovery of DuP 753, a potent orally active non-
peptide angiotensin II receptor antagonist. Med. Res. Rev. 12, 149–191.
Fredholm, B.B., Lindgren, E., 1988. Protein kinase C activation increases
noradrenaline release from rat hippocampus and modifies the inhibitory
effect of a2-adrenoceptor and adenosine A1-receptor agonists. Naunyn-
Schmied. Arch. Pharmacol. 337, 477–483.
Gao, L., et al., 2003. a2-Adrenoceptors potentiate angiotensin II- and vaso-
pressin-induced renal vasoconstriction in spontaneously hypertensive rats.
J. Pharmacol. Exp. Ther. 305, 581–586.
Goncalves, J., et al., 1996. Opposite modulation of cotransmitter release in
guinea-pig vas deferens: increase of noradrenaline and decrease of ATP
release by activation of prejunctional b-adrenoceptors. Naunyn-Schmied.
Arch. Pharmacol. 353, 192–194.
Guimaraes, S., Pinheiro, H., 2005. Functional evidence that in the cardiovas-
cular system AT1 angiotensin II receptors are AT1B prejunctionally and
AT1A postjunctionally. Cardiovasc. Res. 67, 208–215.
Hamid, J., et al., 1999. Identification of an integration center for cross talk
between protein kinase C and G protein modulation of N-type calcium
channels. J. Biol. Chem. 274, 6195–6202.
Hur, E.-M., Kim, K.-T., 2002. G protein-coupled receptor signalling and cross-
talk achieving rapidity and specificity. Cell. Signal. 14, 397–405.
Jackson, E.K., et al., 2001. Enhanced interaction between renovascular a2-
adrenoceptors and angiotensin II receptors in genetic hypertension. Hyper-
tension 38, 353–360.
Johnston, H., Majewski, H., 1986. Prejunctional b-adrenoceptors in rabbit
pulmonary artery and mouse atria: effect of a-adrenoceptor blockade and
phosphodiesterase inhibition. Br. J. Pharmacol. 87, 553–562.
Kaslow, H.R., et al., 1987. Structure-activity of the activation of pertussis toxin.
Biochemistry 26, 123–127.
Kirpekar, S.M., et al., 1976. Effect of tetraethylamonium and barium on the
release of noradrenaline from perfused cat spleen by nerve stimulation and
potassium. Naunyn-Schmied. Arch. Pharmacol. 294, 23–29.

C. Talaia et al. / Neurochemistry International 49 (2006) 698–707 707
Lai, R.-T., et al., 1983. Effect of islet-activating protein (IAP) on contractile
responses of rat vas deferens: evidence for participation of Ni (inhibitory
GTP binding regulating protein) in the a2-adrenoceptor-mediated response.
Eur. J. Pharmacol. 90, 453–456.
Lee, J.J., et al., 2004. Activation of protein kinase C antagonizes the opioid
inhibition of calcium current in rat spinal dorsal horn neurons. Brain Res.
1017, 108–119.
Liang, M., et al., 1998. Phosphorylation and functional desensitisation of the
a2A-adrenergic receptor by protein kinase C. Mol. Pharmacol. 54, 44–49.
Majewski, H., Rand, M.J., 1981. An interaction between prejunctional a-
adrenoceptors and prejunctional b-adrenoceptors. Eur. J. Pharmacol. 69,
493–498.
Mota, A., Guimaraes, S., 2003. Influence of a2-autoreceptor-stimulation on the
facilitation by angiotensin II and bradykinin of noradrenaline release.
Naunyn-Schmied. Arch. Pharmacol. 368, 443–447.
Mota, A., et al., 2000. Lack of interaction between a2-autoreceptors and
prejunctional receptors mediating a facilitatory effect on noradrenaline
release. Pharmacol. Res. 42, 383–388.
Murthy, K.S., et al., 2000. Heterologous desensitisation of response mediated
by selective PKC-dependent phosphorylation of Gi-1 and Gi-2. Am. J.
Physiol. Cell Physiol. 279, C925–C934.
Nap, A., et al., 2004. AT1-receptor blockade and sympathetic neurotransmis-
sion in cardiovascular disease. Auton. Autacoid Pharmacol. 23, 285–296.
Oh, P., Schnitzer, J.E., 2001. Segregation of heterotrimeric G proteins in cell
surface microdomains. Mol. Biol. Cell. 12, 685–698.
Orr, A.W., et al., 2002. Trombospondin stimulates focal adhesion disassembly
through Gi- and phosphoinositide 3-kinase-dependent ERK activation. J.
Biol. Chem. 277, 20453–20460.
Pinheiro, H., et al., 2002. A comparison of several AT1 angiotensin II
antagonists at pre-and postjunctional angiotensin II receptors of the rat
tail artery. Naunyn-Schmied. Arch. Pharmacol. 366, 537–542.
Queiroz, G., et al., 2004. Coupling to protein kinases A and C of adenosine A2B
receptors involved in the facilitation of noradrenaline release in the prostatic
portion of rat vas deferens. Neuropharmacology 47, 216–224.
Queiroz, G., et al., 2003. Adenosine A2A receptor-mediated facilitation of
noradrenaline release involves protein kinase C activation and attenuation of
presynaptic inhibitory receptor-mediated effects in the rat vas deferens. J.
Neurochem. 85, 740–748.
Razani, B., et al., 2002. Caveolae: from cell biology to animal physiology.
Pharmacol. Rev. 54, 431–467.
Selbie, L.A., Hill, S.J., 1998. G protein-coupled-receptor crosstalk: the fine-
tuning of multiple receptor-signaling pathways. Trends Pharmacol. Sci. 19,
87–93.
Shoji-Kasai, Y., et al., 2002. Protein kinase C-mediated translocation of
secretory vesicles to plasma membrane and enhancement of neurotrans-
mitter release from PC12 cells. Eur. J. Neurosci. 15, 1390–1394.
Singer, E.A., 1988. Transmitter release from brain slices elicited by single
pulses: a powerful method to study presynaptic mechanisms. Trends
Pharmacol. Sci. 9, 274–276.
Starke, K., 1977. Regulation of noradrenaline release by presynaptic receptor
systems. Rev. Physiol. Biochem. Pharmacol. 77, 1–124.
Starke, K., 2001. Presynaptic autoreceptors in the third decade: focus on a2-
adrenoceptors. J. Neurochem. 78, 685–693.
Starke, K., Schumann, H.J., 1972. Interactions of angiotensin, phenoxybenza-
mine and propanolol on noradrenaline release during sympathetic nerve
stimulation. Eur. J. Pharmacol. 18, 27–30.
Sum, C.-S., et al., 1996. Potentiation of purinergic transmission by angiotensin
in prostatic rat vas deferens. Br. J. Pharmacol. 118, 1523–1529.
Swartz, K.J., 1993. Modulation of Ca2+ channels by protein kinase C in rat
central and peripheral neurons: disruption of G protein-mediated inhibition.
Neuron 11, 305–320.
Talaia, C., et al., 2005. Interaction between adenosine A2B receptors and a2-
adrenoceptors on the modulation of noradrenaline release in the rat vas
deferens: possible involvement of a group 2 adenylyl cyclase isoform.
Neurochem. Int. 47, 418–429.
Trendelenburg, A.U., et al., 2003. Cross talk between presynaptic angiotensin
receptors, bradykinin receptors and a2-autoreceptors in sympathetic neu-
rons: a study in a2-adrenoceptor-deficient mice. Br. J. Pharmacol. 138,
1389–1402.
von Kugelgen, I., et al., 1993. Axon terminal P2-purinoceptors in feedback
control of sympathetic transmitter release. Neuroscience 56, 263–267.
Wilkinson, S.E., et al., 1993. Isoenzyme specificity of bisindolylmaleimides,
selective inhibitors of protein kinase C. Biochem. J. 294, 335–337.
Wirkner, K., et al., 2000. Inhibition by adenosine A2A receptors of NMDA
and AMPA currents in rat neostriatal neurons. Br. J. Pharmacol. 130,
259–269.




















