
Vibrio cholerae O1 lineages driving cholera outbreaks during seventh cholera pandemic in Ghana

Cancer Immunol Immunother (2010) 59:737–746
DOI 10.1007/s00262-009-0794-4ORIGINAL ARTICLE
Initial characterization of an immunotoxin constructed from domains II and III of cholera exotoxin
Robert Sarnovsky · Tara Tendler · Matheusz Makowski · Maureen Kiley · Antonella Antignani · Roberta Traini · Jingli Zhang · RaYt Hassan · David J. FitzGerald
Received: 18 September 2009 / Accepted: 30 October 2009 / Published online: 29 November 2009© Springer-Verlag 2009
Abstract Immunotoxins are antibody–toxin fusion pro-teins under development as cancer therapeutics. In earlyclinical trials, immunotoxins constructed with domains IIand III of Pseudomonas exotoxin (termed PE38), have pro-duced a high rate of complete remissions in Hairy Cell Leu-kemia and objective responses in other malignancies.Cholera exotoxin (also known as cholix toxin) has a verysimilar three-dimensional structure to Pseudomonas exo-toxin (PE) and when domains II and III of each are com-pared at the primary sequence level, they are 36% identicaland 50% similar. Here we report on the construction andactivity of an immunotoxin made with domains II and III ofcholera exotoxin (here termed CET40). In cell viabilityassays, the CET40 immunotoxin was equipotent to tenfoldless active compared to a PE-based immunotoxin madewith the same single-chain Fv. A major limitation of toxin-based immunotoxins is the development of neutralizingantibodies to the toxin portion of the immunotoxin.Because of structure and sequence similarities, we evalu-ated a CET40 immunotoxin for the presence of PE-relatedepitopes. In western blots, three-of-three anti-PE antibodypreparations failed to react with the CET40 immunotoxin.More importantly, in neutralization studies neither these
antibodies nor those from patients with neutralizing titers toPE38, neutralized the CET40-immunotoxin. We proposethat the use of modular components such as antibody Fvsand toxin domains will allow a greater Xexibility in howthese agents are designed and deployed including thesequential administration of a second immunotoxin afterpatients have developed neutralizing antibodies to the Wrst.
Keywords Exotoxin · Cholix · Immunotoxin · Neutralization · Antibody
Introduction
Antibody-based therapies of human cancer have becomeWrst-line treatments in certain settings. By way of example:Her2-positive breast cancer patients are treated with Her-ceptin [1] while individuals with certain B-cell malignan-cies receive Rituxan [2]. These antibodies are given eitheralone or in combination with chemotherapy. The potentialbeneWt of using antibody-based therapy is an eVective treat-ment with low side eVects. However, when the administra-tion of an unmodiWed antibody is not eVective, severaloptions are available to make the antibody a ‘cytotoxic’agent [3]. Radionuclides, small molecular weight drugs(including prodrugs), enzymes, homing partners (such asbispeciWc antibodies), and protein toxins have each been“attached” to tumor-binding antibodies as adjuncts toincrease their eVectiveness [4–12]. Each type of modiWedantibody has beneWts and limitations [3, 13].
Recombinant immunotoxins are antibody-based thera-peutics composed of Fv fragments fused with protein toxins[11, 14–16]. The protein toxins are usually derived frombacterial or plant cytotoxic proteins and act enzymaticallywithin the cytosol of mammalian cells. Advantages of
Electronic supplementary material The online version of this article (doi:10.1007/s00262-009-0794-4) contains supplementary material, which is available to authorized users.
R. Sarnovsky · T. Tendler · M. Makowski · M. Kiley · A. Antignani · R. Traini · J. Zhang · R. Hassan · D. J. FitzGerald (&)Laboratory of Molecular Biology, CCR, NCI, NIH, HHS, 37 Convent Dr, Room 5124, Bethesda, MD 20892, USAe-mail: [email protected]
123

738 Cancer Immunol Immunother (2010) 59:737–746
toxin-based agents relate to their potency, lack of muta-genic activity, and the fact that cancer cells rarely exhibittoxin resistance. The main disadvantage is immunogenicity[17–24]. Typically, patients make neutralizing antibodieswithin weeks of receiving their Wrst course of treatment.
Pseudomonas exotoxin has been investigated for anumber of years as a cytotoxic partner with antibody frag-ments in the development of anticancer immunotoxins[11, 16]. Typically, a 38-kDa fragment of PE, encompassingdomains II and III of the parental toxin, is fused geneticallyto either a single-chain Fv (scFv) or disulWde stabilized Fv(dsFv). With the exception of treating individuals withB-cell malignancies [25], most immunotoxin trials havebeen limited to one or sometimes two cycles of therapybecause patients make neutralizing antibodies, usuallywithin 3 weeks of initiating treatment [23, 26]. Potentialstrategies to make such toxin-based agents less immuno-genic include the co-administration of immunosuppressiveagents [27–29] and the re-engineering of the parentmolecule to remove major epitopes [21, 24]. While theco-administration of immunosuppressive agents is simplein concept, it is diYcult to accomplish in Phase I/II trialsdue to the confounding problem of mixing two agentswhere the properties of one, in this case the immunotoxin,are not well understood. The prospect of engineering a bac-terial toxin to render it non-immunogenic is also challeng-ing. However, by removing the most potent antigenicepitopes, it may be possible to administer several cycles oftherapy before a neutralizing response develops [21]. Thereplacement of the toxin portion of the immunotoxin with aclosely related but immunologically distinct ‘molecularcousin’ may allow for a third approach. This strategyshould work best in situations where structural similaritiesare close enough to allow for domain swapping and the useof a ‘modular replacement strategy’.
Here, we report on the development of a novel recombi-nant immunotoxin constructed from domains II and III ofan exotoxin from Vibrio cholerae (also termed cholix toxin[30]) that shares »36% identity and 50% similarity with thecomparable domains of Pseudomonas exotoxin.
To construct this immunotoxin, a synthetic gene encod-ing amino acids 270 to 634 of cholera exotoxin (CET) wascombined with the single-chain Fv antibody (HB21scFv)directed to the human transferrin receptor. HB21-CET40was potently toxic for a number of human cancer celllines. And despite a high level of structural and sequencesimilarity between PE40 and CET40, anti-PE antibodiesdid not recognize or neutralize the CET40 immunotoxin.Thus, it is now possible to develop a distinct anti-cancertherapeutic platform centered on CET-based immuno-toxins that potentially can be administered as a Wrst-linetherapeutic agent or to individuals with prior exposure toPE-based immunotoxins.
Methods
Construction of HB21-CET40
A synthetic gene fragment (sequence provided in Fig. 1a)encoding putative domains II and III of Cholera exotoxinwas produced (at Blue Heron Biotechnology) with HindIIIand EcoRI restriction sites Xanking the gene, and providedas a pUC19 plasmid. An expression vector (pRB2506)encoding the single-chain Fv fragment of the HB21 anti-body fused with PE40 (HB21-PE40) was provided by Rich-ard Beers and Ira Pastan. The vector DNA was digestedwith the appropriate restriction endonucleases, separatedvia 0.9% agarose gel electrophoresis and the fragments gelpuriWed using a Qiaquick gel extraction kit. Ligations ofDNA fragments were performed using an approximate(3:1) insert to vector molar ratio with T4 DNA ligase(New England Biolabs) in 1£ ligation buVer, at 37°C for1.5 or 16 h at 16°C. Clones were screened by diagnosticrestriction digests and positive clones conWrmed byDNA sequencing (Johns Hopkins Sequencing Facility,Baltimore, MD).
Expression of HB21-CET40
The backbone of the expression vector has an inducible T7promoter and carries a gene encoding chloramphenicolresistance. Expression of the single-chain immunotoxinwas carried out in BL21-Star (DE3) E. coli cells (Invitro-gen) grown at 37°C in baZed Fernbach Xasks at 275 rpm.Cells were grown in Superbroth (KD Medical) supple-mented with chloramphenicol at 25 �g/ml (Sigma) and‘Overnight Express’ additives (Novagen). This mediumwas inoculated with freshly transformed cells and grownovernight (»17 h). Final culture OD600’s were »5–6.Cells were harvested by centrifugation at 4,000£g for10 min in a Sorvall 3B centrifuge. Cell pellets were storedfrozen at ¡80°C.
Preparation of inclusion bodies
Frozen cells (up to 10,000 OD units) were resuspended in25 ml of Tris–EDTA (TE 50/20-mM/mM, pH 8.0) and dis-persed using a tissuemizer. Cells were then lysed with theaddition of chicken egg white lysozyme (Sigma) to a Wnalconcentration of 200 �g/ml for 1 h at RT. Lysed cells wereincubated further for 30 min with the addition of 3.3 ml of5.0 M NaCl and 3.3 ml of 25% Triton X-100. Inclusionbodies (IB) were then recovered in the pellet following cen-trifugation for 45 min at 15,000£g (Sorvall SS-34 rotor).The pellet was resuspended in 25 ml TE 50/20, 1% vol/volTriton X-100, dispersed using a tissuemizer and centrifugedas above three more times. To remove the detergent, the
123

Cancer Immunol Immunother (2010) 59:737–746 739
inclusion bodies were washed four times in TE 50/20. TheIB pellet preparation was stored frozen at ¡80°C.
Solubilization of IB’s
IBs were solubilized initially in 6 M Guanidine–HCl, 0.1 MTris–HCl, 2 mM EDTA at pH 8.0. After 1–4 h, dith-ioerythretol (DTE) was added to a Wnal concentration of65 mM (10 mg/ml) and solubilization allowed to proceedon a rocking platform overnight at RT.
Renaturation refolding
Solubilized immunotoxin was centrifuged to remove non-soluble material and the supernatant diluted (»1:100 vol/vol) into a refolding buVer: 0.1 M Tris, 0.5 M L-Arginine–HCl, 2 mM EDTA, 0.9 mM GSSG, pH 8.0 at 10°C. After24 h, additional GSSG, (9 mM Wnal), was added for another
24 h. The refolded protein was then dialyzed against 20 mMTris–HCl,100 mM Urea pH 8.0.
Anion exchange chromatography
The post dialysis immunotoxin preparation was adjusted to2 l with deionized water and batch-adsorbed onto 50 ml ofQ-sepharose. The resin was recovered on a 2-l-Buchnerfunnel and washed with four volumes of 20 mM Tris–HCl,1 mM EDTA, pH 8.0 and then eluted using the samebuVer supplemented with 0.1, 0.35, and 0.5 M NaCl. Theimmunotoxin eluting with 0.35 M NaCl was retained foradditional chromatography. The retained material wasdiluted in low salt buVer (buVer A, 20 mM Tris, pH 8.0)and pumped onto Mono-Q 5/5 column at 1 ml/min. Proteinwas eluted from the resin with a linear gradient 0–100% ofbuVer B (20 mM Tris pH 8.0, 1.0 M NaCl), over 30 ml, col-lecting 1 ml fractions. Peak fractions were concentrated
Fig. 1 CET40 immunotoxin. a Shown is the DNA and protein sequence of the immunotoxin HB21-CET40. The initiating methionine is followed by the variable portion of the heavy chain, a glycine-serine linker, the variable portion of the light chain, a short connector sequence (including the HindIII site), and amino acids 270–634 of cholera exotoxin. b Sequence alignment via ‘ClustalX’ (2.09) analysis. Shown in descending order are amino acids 270–634 of cholix toxin [30], amino acids 270–634 of CET (corresponding to domains II and III from the exotoxin gene of Vibrio chol-erae strain 1587), and Wnally domains II and III of exotoxin A, PE40. Key common features include the location of a furin cleavage site, an NAD binding glutamic acid, and a KDEL-like motif at the C-terminus. Color-ing was per the default settings for ClustalX as deWned by ‘colprot.xml’ (colprot.par). For alignment: “*” = fully conserved; “:” = conserved within a ‘strong’ group; and “.” = conserved within a ‘weak’ group
123

740 Cancer Immunol Immunother (2010) 59:737–746
using an Amicon Ultra (10,000 MWCO) concentrator(Amicon) to a volume ·1 ml for gel Wltration.
Gel Wltration chromatography
The concentrated sample was loaded onto a TSKgelG3000-SWxl column (Tosoh Bioscience) at 0.5 ml/min,using PBS, pH 7.4, as the mobile phase. Fractions of 0.5 mlwere collected and analyzed by SDS-PAGE (Supp Fig. 1).
Cell lines
KB3-1, A549, DLD-1, Raji, 293TT (from C. Buck NCI),HUT102, and L929 cells were grown in RPMI-1640 orDMEM and 10% fetal bovine serum supplemented withpenicillin, streptomycin, glutamine, and pyruvate.
Cytotoxicity assay
The WST-1 (Roche) was used to assess cytotoxicity. Cellswere seeded in 96-well plates at 5 £ 103 per well. After24 h, immunotoxins or immunotoxin-antibody mixtureswere added to cells for a further 48 h. Dye-containingmedia was removed and replaced with a 10% vol/vol ofWST-1 reagent in dye-free RPMI-1640 growth media.Absorbance measurements were made at 450 nm at 30 and60 min. Replicates of Wve were used for each data point andall experiments were conducted independently at leasttwice. Cycloheximide (Sigma) was added at 10 �g/ml as apositive control in all experiments. For competition experi-ments with excess antibody, DLD-1 cells were pretreatedwith 10 ug/ml of HB21 for 30 min and then HB21-CET40was added to a Wnal concentration of either 10 or 1 ng/ml.
Anti-PE antibodies
Several anti-PE antibody preparations were evaluated. Amouse monoclonal antibody, termed M40-1, was originallydescribed as a neutralizing antibody that also recognizedPE via western blots [31]. Two rabbit polyclonal antibodypreparations that reacted with PE via western blot and neu-tralized the toxin were also employed. One of these was alab reagent originally generated to formaldehyde-treatednative PE. The other was purchased from Sigma (P2318).We also used human sera from patients treated with theimmunotoxin SS1P-PE38 (see below).
Antibodies to CET40
To make antibodies reactive for CET40, a rabbit was hyper-immunized with an enzymatically inactive form of CET40(E581A) fused to HB21scFv. Immunizations and antiseraproduction were carried out at Convance. Because these sera
contained antibodies to both the scFv and CET40, westernblots were conducted on full-length PE and CET proteins.Both PE and CET were expressed in E. coli and puriWedusing the same protocol used to prepared HB21-CET40.
Western blots
Immunotoxin proteins, either HB21-PE40 or HB21-CET40(30 ng) were separated via SDS-PAGE (8–16% gradient),transferred to PVDF membranes and probed with anti-PEantibodies. CET and PE was similarly separated and trans-ferred to PVDF membranes. Either donkey anti-mouseIgG-HRP or donkey anti-rabbit IgG-HRP (Jackson, Immu-noresearch) was used to detect the primary antibodies.Reactive bands were detected by ECL and visualized onAmersham HyperWlm.
Neutralization assay
Rabbit or mouse antibodies were diluted 1:100 and mixedwith either 5 or 1 ng/ml of either HB21-PE40 or HB21-CET40 for 1 h at room temperature. At the end of the incu-bation the immunotoxin–antibody mixture was diluted 1:1with media over cells. Cells were incubated with immuno-toxin and antibodies for 48 h and then evaluated for viabil-ity using the WST-1 assay.
Human sera (from four individuals) were obtained withinformed consent before and after treatment with the PE38immunotoxin, SS1P, which is directed to the surface anti-gen mesothelin [26]. Immunotoxin treatment was at thedose level of 45 �g/kg for each the four individuals [26].The post treatment sample was documented as having neu-tralizing titers to PE38. The immunotoxin at 5 or 1 ng/mlwas mixed with a 1:100 dilution of patient sera and incu-bated for 1 h at room temperature. After this incubation,50 �l was added to each well giving a Wnal immunotoxinconcentration of 2.5 and 0.5 ng/ml, respectively, and a Wnalserum dilution of 1:200.
Results
Construction and initial characterization of a CET40 immunotoxin
A single-chain immunotoxin was constructed from a cDNAencoding the Fv portion of the HB21 antibody recognizingthe human transferrin receptor [32] and a synthetic geneencoding domains II and III of cholera exotoxin1 (here
1 (for results sections) Amino acids 270–634 of CET encompassdomains II, III and a small sub-domain termed, Ib. For simplicity,domain Ib is not routinely mentioned (also see “Discussion”).
123
Cancer Immunol Immunother (2010) 59:737–746 741
called CET) which is very similar to the toxin named ‘cho-lix toxin’ (accession number; AY876053) by Jorgensenet al. [30]. The synthetic gene, corresponding to aminoacids 270–634 of CET (the annotated DNA and proteinsequences are provided in Fig. 1a) was derived from thesequenced genome of V. cholerae strain1587 (accessionnumber for CET is ZP_01950668) and diVers from cholixtoxin in domains II and III by ten amino acids. Figure 1bshows a clustalX sequence alignment of domains II and IIIof cholix toxin (V. cholerae strain TP [33]), CET (V. chol-erae strain1587) and PE40. Key features of each toxininclude a consensus furin cleavage site (with strong conser-vation on the N-terminal site of the scissile bond and weakconservation on the C-terminal side), a conserved glutamicacid marking the NAD-binding pocket and a C-terminal aKDEL-like sequence followed by a terminal lysine. Alsothe four half cysteines are completely conserved as are sev-eral stretches of residues within domain III (from residues187–336 in Fig. 1b).
Expression of HB21-CET40 was driven by a T7 pro-moter and accomplished via growth in ‘autoinductionmedia’ under Cm selection (see “Methods”). After an over-night culture, the insoluble protein was recovered in inclu-sion bodies and puriWed according to our standardlaboratory protocol (see “Methods” and [34, 35]). BrieXy,inclusion bodies were solubilized with 6 M guanidine and areducing agent, refolded into a redox shuZing buVer, andpuriWed using anion exchange and gel Wltration chromatog-
raphy. An SDS-PAGE analysis of gel Wltration fractionsrevealed that »20% of HB21-CET40 eluted as a monomer(Supp Fig. 1). Fractions 28 and 29 were used for experi-ments presented below.
Cytotoxic activity of the HB21-CET40
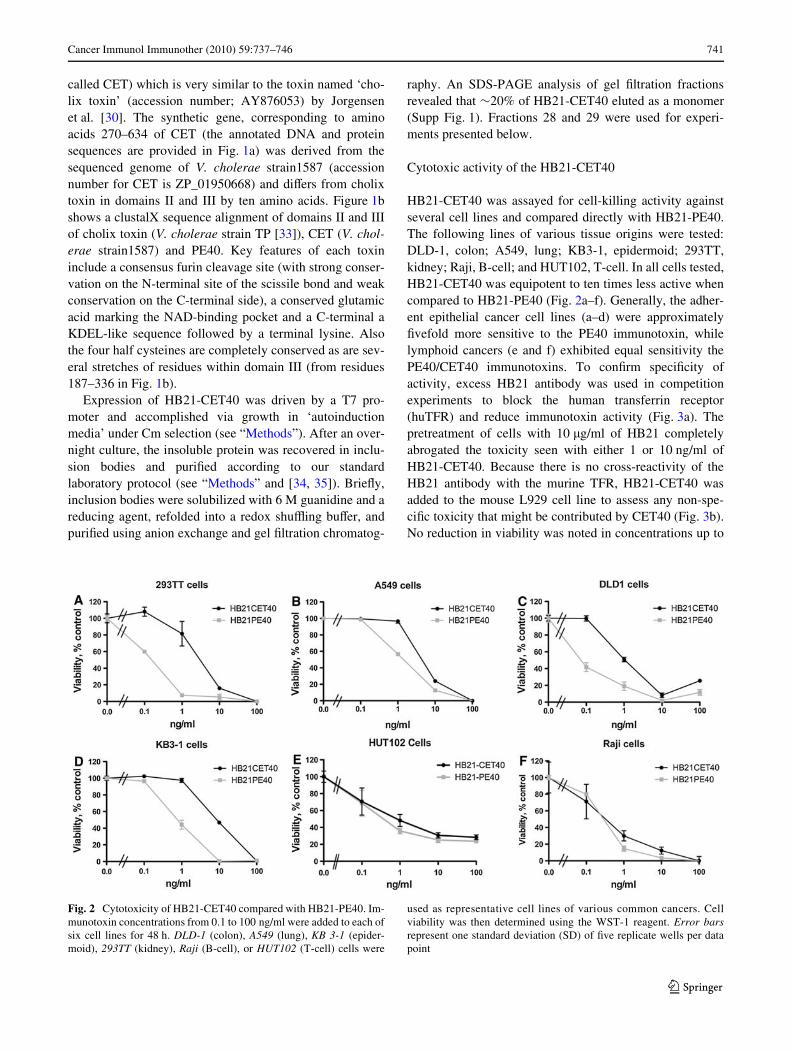
HB21-CET40 was assayed for cell-killing activity againstseveral cell lines and compared directly with HB21-PE40.The following lines of various tissue origins were tested:DLD-1, colon; A549, lung; KB3-1, epidermoid; 293TT,kidney; Raji, B-cell; and HUT102, T-cell. In all cells tested,HB21-CET40 was equipotent to ten times less active whencompared to HB21-PE40 (Fig. 2a–f). Generally, the adher-ent epithelial cancer cell lines (a–d) were approximatelyWvefold more sensitive to the PE40 immunotoxin, whilelymphoid cancers (e and f) exhibited equal sensitivity thePE40/CET40 immunotoxins. To conWrm speciWcity ofactivity, excess HB21 antibody was used in competitionexperiments to block the human transferrin receptor(huTFR) and reduce immunotoxin activity (Fig. 3a). Thepretreatment of cells with 10 �g/ml of HB21 completelyabrogated the toxicity seen with either 1 or 10 ng/ml ofHB21-CET40. Because there is no cross-reactivity of theHB21 antibody with the murine TFR, HB21-CET40 wasadded to the mouse L929 cell line to assess any non-spe-ciWc toxicity that might be contributed by CET40 (Fig. 3b).No reduction in viability was noted in concentrations up to
Fig. 2 Cytotoxicity of HB21-CET40 compared with HB21-PE40. Im-munotoxin concentrations from 0.1 to 100 ng/ml were added to each ofsix cell lines for 48 h. DLD-1 (colon), A549 (lung), KB 3-1 (epider-moid), 293TT (kidney), Raji (B-cell), or HUT102 (T-cell) cells were
used as representative cell lines of various common cancers. Cellviability was then determined using the WST-1 reagent. Error barsrepresent one standard deviation (SD) of Wve replicate wells per datapoint
123
742 Cancer Immunol Immunother (2010) 59:737–746
100 ng/ml. From the data above, we conclude that CET40can be used to construct potent and antigen-speciWc recom-binant immunotoxins.
Cross-reactivity of anti-PE antibodies
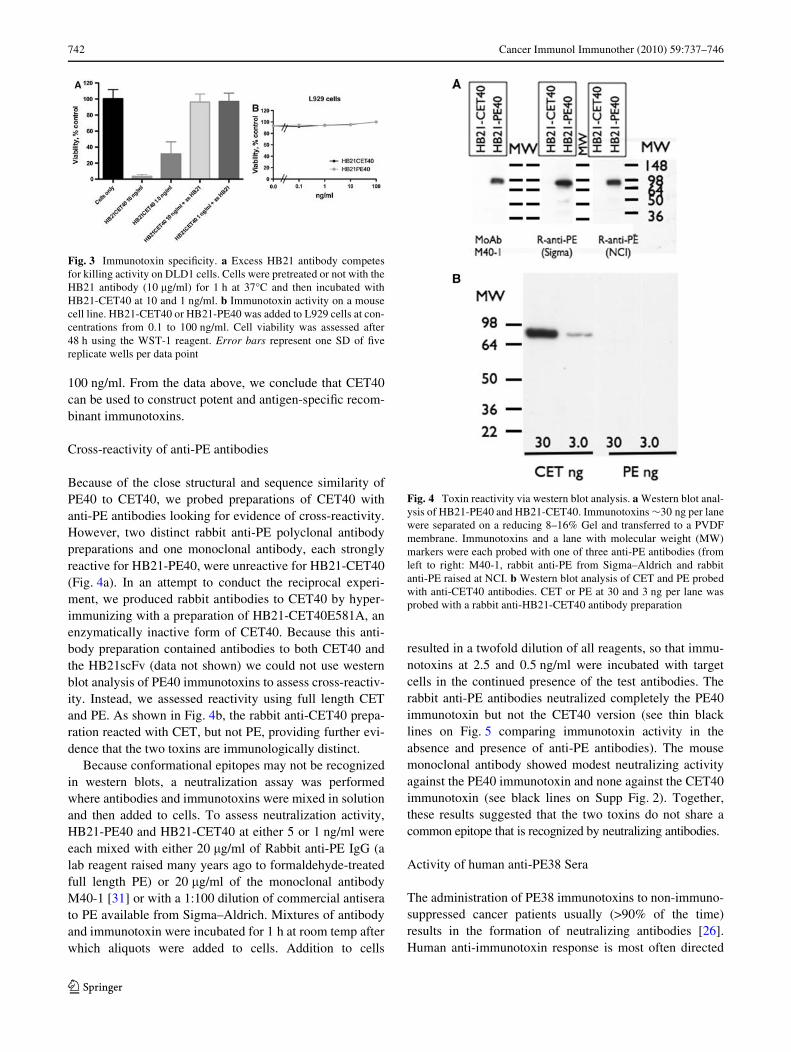
Because of the close structural and sequence similarity ofPE40 to CET40, we probed preparations of CET40 withanti-PE antibodies looking for evidence of cross-reactivity.However, two distinct rabbit anti-PE polyclonal antibodypreparations and one monoclonal antibody, each stronglyreactive for HB21-PE40, were unreactive for HB21-CET40(Fig. 4a). In an attempt to conduct the reciprocal experi-ment, we produced rabbit antibodies to CET40 by hyper-immunizing with a preparation of HB21-CET40E581A, anenzymatically inactive form of CET40. Because this anti-body preparation contained antibodies to both CET40 andthe HB21scFv (data not shown) we could not use westernblot analysis of PE40 immunotoxins to assess cross-reactiv-ity. Instead, we assessed reactivity using full length CETand PE. As shown in Fig. 4b, the rabbit anti-CET40 prepa-ration reacted with CET, but not PE, providing further evi-dence that the two toxins are immunologically distinct.
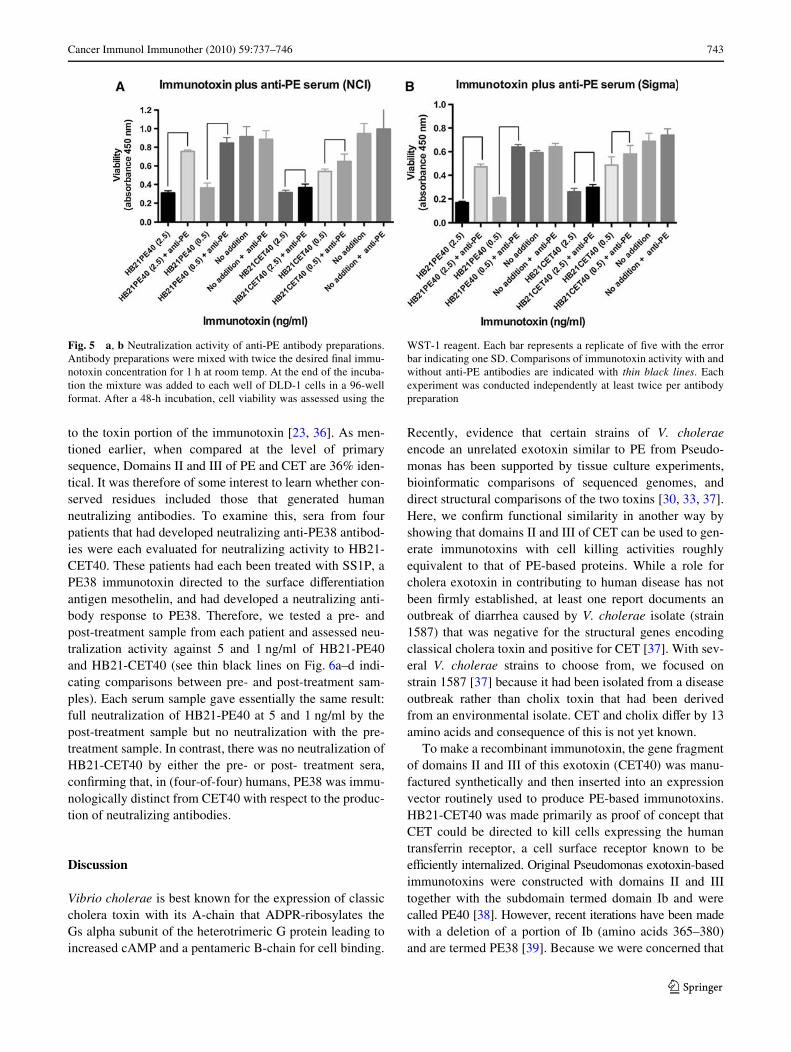
Because conformational epitopes may not be recognizedin western blots, a neutralization assay was performedwhere antibodies and immunotoxins were mixed in solutionand then added to cells. To assess neutralization activity,HB21-PE40 and HB21-CET40 at either 5 or 1 ng/ml wereeach mixed with either 20 �g/ml of Rabbit anti-PE IgG (alab reagent raised many years ago to formaldehyde-treatedfull length PE) or 20 �g/ml of the monoclonal antibodyM40-1 [31] or with a 1:100 dilution of commercial antiserato PE available from Sigma–Aldrich. Mixtures of antibodyand immunotoxin were incubated for 1 h at room temp afterwhich aliquots were added to cells. Addition to cells
resulted in a twofold dilution of all reagents, so that immu-notoxins at 2.5 and 0.5 ng/ml were incubated with targetcells in the continued presence of the test antibodies. Therabbit anti-PE antibodies neutralized completely the PE40immunotoxin but not the CET40 version (see thin blacklines on Fig. 5 comparing immunotoxin activity in theabsence and presence of anti-PE antibodies). The mousemonoclonal antibody showed modest neutralizing activityagainst the PE40 immunotoxin and none against the CET40immunotoxin (see black lines on Supp Fig. 2). Together,these results suggested that the two toxins do not share acommon epitope that is recognized by neutralizing antibodies.
Activity of human anti-PE38 Sera
The administration of PE38 immunotoxins to non-immuno-suppressed cancer patients usually (>90% of the time)results in the formation of neutralizing antibodies [26].Human anti-immunotoxin response is most often directed
Fig. 3 Immunotoxin speciWcity. a Excess HB21 antibody competesfor killing activity on DLD1 cells. Cells were pretreated or not with theHB21 antibody (10 �g/ml) for 1 h at 37°C and then incubated withHB21-CET40 at 10 and 1 ng/ml. b Immunotoxin activity on a mousecell line. HB21-CET40 or HB21-PE40 was added to L929 cells at con-centrations from 0.1 to 100 ng/ml. Cell viability was assessed after48 h using the WST-1 reagent. Error bars represent one SD of Wvereplicate wells per data point
Fig. 4 Toxin reactivity via western blot analysis. a Western blot anal-ysis of HB21-PE40 and HB21-CET40. Immunotoxins »30 ng per lanewere separated on a reducing 8–16% Gel and transferred to a PVDFmembrane. Immunotoxins and a lane with molecular weight (MW)markers were each probed with one of three anti-PE antibodies (fromleft to right: M40-1, rabbit anti-PE from Sigma–Aldrich and rabbitanti-PE raised at NCI. b Western blot analysis of CET and PE probedwith anti-CET40 antibodies. CET or PE at 30 and 3 ng per lane wasprobed with a rabbit anti-HB21-CET40 antibody preparation
123
Cancer Immunol Immunother (2010) 59:737–746 743
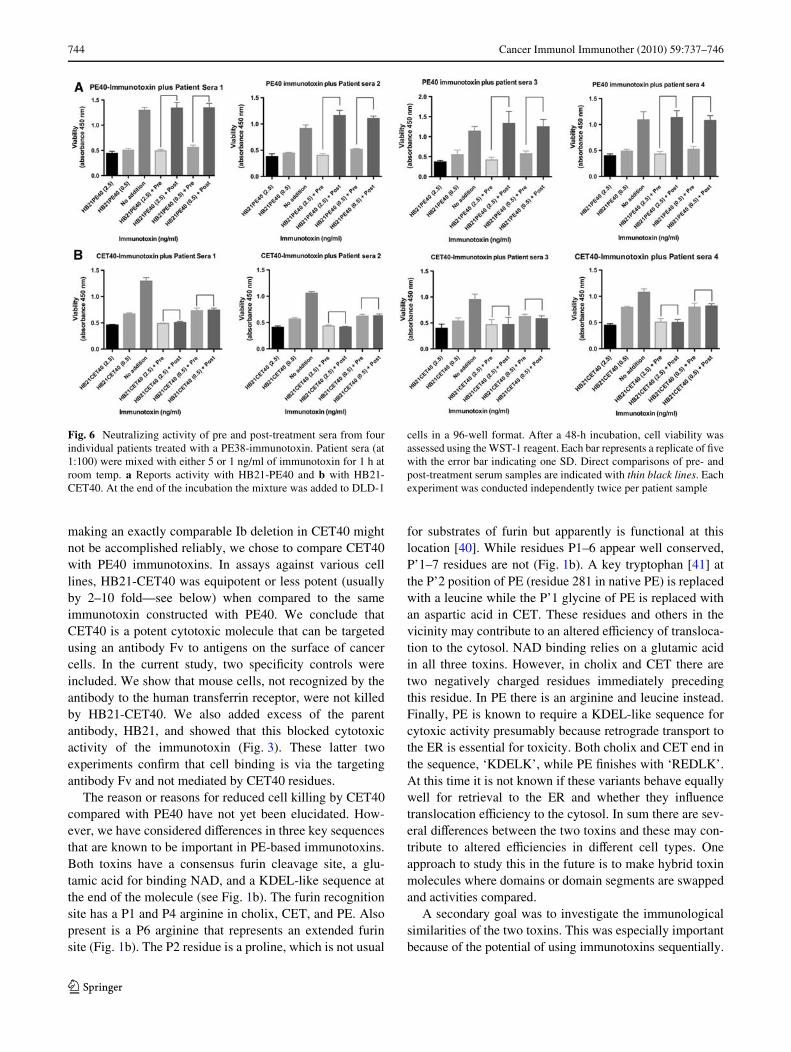
to the toxin portion of the immunotoxin [23, 36]. As men-tioned earlier, when compared at the level of primarysequence, Domains II and III of PE and CET are 36% iden-tical. It was therefore of some interest to learn whether con-served residues included those that generated humanneutralizing antibodies. To examine this, sera from fourpatients that had developed neutralizing anti-PE38 antibod-ies were each evaluated for neutralizing activity to HB21-CET40. These patients had each been treated with SS1P, aPE38 immunotoxin directed to the surface diVerentiationantigen mesothelin, and had developed a neutralizing anti-body response to PE38. Therefore, we tested a pre- andpost-treatment sample from each patient and assessed neu-tralization activity against 5 and 1 ng/ml of HB21-PE40and HB21-CET40 (see thin black lines on Fig. 6a–d indi-cating comparisons between pre- and post-treatment sam-ples). Each serum sample gave essentially the same result:full neutralization of HB21-PE40 at 5 and 1 ng/ml by thepost-treatment sample but no neutralization with the pre-treatment sample. In contrast, there was no neutralization ofHB21-CET40 by either the pre- or post- treatment sera,conWrming that, in (four-of-four) humans, PE38 was immu-nologically distinct from CET40 with respect to the produc-tion of neutralizing antibodies.
Discussion
Vibrio cholerae is best known for the expression of classiccholera toxin with its A-chain that ADPR-ribosylates theGs alpha subunit of the heterotrimeric G protein leading toincreased cAMP and a pentameric B-chain for cell binding.
Recently, evidence that certain strains of V. choleraeencode an unrelated exotoxin similar to PE from Pseudo-monas has been supported by tissue culture experiments,bioinformatic comparisons of sequenced genomes, anddirect structural comparisons of the two toxins [30, 33, 37].Here, we conWrm functional similarity in another way byshowing that domains II and III of CET can be used to gen-erate immunotoxins with cell killing activities roughlyequivalent to that of PE-based proteins. While a role forcholera exotoxin in contributing to human disease has notbeen Wrmly established, at least one report documents anoutbreak of diarrhea caused by V. cholerae isolate (strain1587) that was negative for the structural genes encodingclassical cholera toxin and positive for CET [37]. With sev-eral V. cholerae strains to choose from, we focused onstrain 1587 [37] because it had been isolated from a diseaseoutbreak rather than cholix toxin that had been derivedfrom an environmental isolate. CET and cholix diVer by 13amino acids and consequence of this is not yet known.
To make a recombinant immunotoxin, the gene fragmentof domains II and III of this exotoxin (CET40) was manu-factured synthetically and then inserted into an expressionvector routinely used to produce PE-based immunotoxins.HB21-CET40 was made primarily as proof of concept thatCET could be directed to kill cells expressing the humantransferrin receptor, a cell surface receptor known to beeYciently internalized. Original Pseudomonas exotoxin-basedimmunotoxins were constructed with domains II and IIItogether with the subdomain termed domain Ib and werecalled PE40 [38]. However, recent iterations have been madewith a deletion of a portion of Ib (amino acids 365–380)and are termed PE38 [39]. Because we were concerned that
Fig. 5 a, b Neutralization activity of anti-PE antibody preparations.Antibody preparations were mixed with twice the desired Wnal immu-notoxin concentration for 1 h at room temp. At the end of the incuba-tion the mixture was added to each well of DLD-1 cells in a 96-wellformat. After a 48-h incubation, cell viability was assessed using the
WST-1 reagent. Each bar represents a replicate of Wve with the errorbar indicating one SD. Comparisons of immunotoxin activity with andwithout anti-PE antibodies are indicated with thin black lines. Eachexperiment was conducted independently at least twice per antibodypreparation
123
744 Cancer Immunol Immunother (2010) 59:737–746
making an exactly comparable Ib deletion in CET40 mightnot be accomplished reliably, we chose to compare CET40with PE40 immunotoxins. In assays against various celllines, HB21-CET40 was equipotent or less potent (usuallyby 2–10 fold—see below) when compared to the sameimmunotoxin constructed with PE40. We conclude thatCET40 is a potent cytotoxic molecule that can be targetedusing an antibody Fv to antigens on the surface of cancercells. In the current study, two speciWcity controls wereincluded. We show that mouse cells, not recognized by theantibody to the human transferrin receptor, were not killedby HB21-CET40. We also added excess of the parentantibody, HB21, and showed that this blocked cytotoxicactivity of the immunotoxin (Fig. 3). These latter twoexperiments conWrm that cell binding is via the targetingantibody Fv and not mediated by CET40 residues.
The reason or reasons for reduced cell killing by CET40compared with PE40 have not yet been elucidated. How-ever, we have considered diVerences in three key sequencesthat are known to be important in PE-based immunotoxins.Both toxins have a consensus furin cleavage site, a glu-tamic acid for binding NAD, and a KDEL-like sequence atthe end of the molecule (see Fig. 1b). The furin recognitionsite has a P1 and P4 arginine in cholix, CET, and PE. Alsopresent is a P6 arginine that represents an extended furinsite (Fig. 1b). The P2 residue is a proline, which is not usual
for substrates of furin but apparently is functional at thislocation [40]. While residues P1–6 appear well conserved,P’1–7 residues are not (Fig. 1b). A key tryptophan [41] atthe P’2 position of PE (residue 281 in native PE) is replacedwith a leucine while the P’1 glycine of PE is replaced withan aspartic acid in CET. These residues and others in thevicinity may contribute to an altered eYciency of transloca-tion to the cytosol. NAD binding relies on a glutamic acidin all three toxins. However, in cholix and CET there aretwo negatively charged residues immediately precedingthis residue. In PE there is an arginine and leucine instead.Finally, PE is known to require a KDEL-like sequence forcytoxic activity presumably because retrograde transport tothe ER is essential for toxicity. Both cholix and CET end inthe sequence, ‘KDELK’, while PE Wnishes with ‘REDLK’.At this time it is not known if these variants behave equallywell for retrieval to the ER and whether they inXuencetranslocation eYciency to the cytosol. In sum there are sev-eral diVerences between the two toxins and these may con-tribute to altered eYciencies in diVerent cell types. Oneapproach to study this in the future is to make hybrid toxinmolecules where domains or domain segments are swappedand activities compared.
A secondary goal was to investigate the immunologicalsimilarities of the two toxins. This was especially importantbecause of the potential of using immunotoxins sequentially.
Fig. 6 Neutralizing activity of pre and post-treatment sera from fourindividual patients treated with a PE38-immunotoxin. Patient sera (at1:100) were mixed with either 5 or 1 ng/ml of immunotoxin for 1 h atroom temp. a Reports activity with HB21-PE40 and b with HB21-CET40. At the end of the incubation the mixture was added to DLD-1
cells in a 96-well format. After a 48-h incubation, cell viability wasassessed using the WST-1 reagent. Each bar represents a replicate of Wvewith the error bar indicating one SD. Direct comparisons of pre- andpost-treatment serum samples are indicated with thin black lines. Eachexperiment was conducted independently twice per patient sample
123

Cancer Immunol Immunother (2010) 59:737–746 745
Despite exhibiting segments of high sequence homologyand being very similar structurally, there appears to be verylittle immunological cross-reactivity between PE40 andCET40. None of the anti-PE antibodies reacted with theCET40 immunotoxin and, using a recently generated anti-body to a CET40 immuntoxin, we showed no cross-reactivitywith PE. Perhaps the most relevant test was the lack of across-neutralizing response by patients who had receivedPE38 immunotoxins. In four-of-four samples, post treatmentbut not pretreatment sera neutralized the cytotoxic activity ofHB21-PE40 but not the CET40 immunotoxin (Fig. 6). Thissuggests that the major neutralizing epitopes of PE38 are notshared with CET40. If this initial trend extends to the major-ity of patients treated with PE38 immunotoxins, then we candevise a strategy whereby PE38 immunotoxins are adminis-tered Wrst and then treatment is switched to a CET40 immu-notoxin using the same targeting antibody. In our experiencewith PE38 immunotoxins, patients rarely make antibodies tothe Fv fragment of the immunotoxin, so this strategy shouldallow one or two additional cycles of treatment [23].
Acknowledgments This research was supported by the IntramuralResearch Program of the NIH, National Cancer Institute, Center forCancer Research. The content of this publication does not necessarilyreXect the views or policies of the Department of Health and HumanServices, nor does mention of trade names, commercial products, ororganizations imply endorsement by the US Government.
ConXict of interest statement No conXicts are noted.
References
1. Hudis CA (2007) Trastuzumab—mechanism of action and use inclinical practice. N Engl J Med 357:39–51
2. Cheson BD, Leonard JP (2008) Monoclonal antibody therapy forB-cell non-Hodgkin’s lymphoma. N Engl J Med 359:613–626
3. Heimann DM, Weiner LM (2007) Monoclonal antibodies in ther-apy of solid tumors. Surg Oncol Clin N Am 16:775–792, viii
4. Green DJ, Pagel JM, Pantelias A et al (2007) Pretargeted radioimmu-notherapy for B-cell lymphomas. Clin Cancer Res 13:5598s–5603s
5. Rybak SM (2008) Antibody–onconase conjugates: cytotoxicityand intracellular routing. Curr Pharm Biotechnol 9:226–230
6. Liu XY, Pop LM, Vitetta ES (2008) Engineering therapeuticmonoclonal antibodies. Immunol Rev 222:9–27
7. Singh Y, Palombo M, Sinko PJ (2008) Recent trends in targetedanticancer prodrug and conjugate design. Curr Med Chem15:1802–1826
8. Brumlik MJ, Daniel BJ, Waehler R et al (2008) Trends in immu-noconjugate and ligand-receptor based targeting development forcancer therapy. Expert Opin Drug Deliv 5:87–103
9. Carter PJ, Senter PD (2008) Antibody–drug conjugates for cancertherapy. Cancer J 14:154–169
10. Goldenberg DM, Sharkey RM (2007) Novel radiolabeled antibodyconjugates. Oncogene 26:3734–3744
11. Pastan I, Hassan R, FitzGerald DJ et al (2007) Immunotoxin treat-ment of cancer. Annu Rev Med 58:221–237
12. Kreitman RJ, Pastan I (2006) Immunotoxins in the treatment ofrefractory hairy cell leukemia. Hematol Oncol Clin North Am20:1137–1151, viii
13. Ricart AD, Tolcher AW (2007) Technology insight: cytotoxicdrug immunoconjugates for cancer therapy. Nat Clin Pract Oncol4:245–255
14. Frankel AE, Kreitman RJ, Sausville EA (2000) Targeted toxins.Clin Cancer Res 6:326–334
15. Frankel AE, Neville DM, Bugge TA et al (2003) Immunotoxintherapy of hematologic malignancies. Semin Oncol 30:545–557
16. Pastan I, Hassan R, Fitzgerald DJ et al (2006) Immunotoxin ther-apy of cancer. Nat Rev Cancer 6:559–565
17. Schnell R, Borchmann P, Staak JO et al (2003) Clinical evaluationof ricin A-chain immunotoxins in patients with Hodgkin’s lym-phoma. Ann Oncol 14:729–736
18. Schnell R, Staak O, Borchmann P (2002) A Phase I study with ananti-CD30 ricin A-chain immunotoxin (Ki-4.dgA) in patients withrefractory CD30+ Hodgkin’s and non-Hodgkin’s lymphoma. ClinCancer Res 8:1779–1786
19. Frankel AE (2004) Reducing the immune response to immunotox-in. Clin Cancer Res 10:13–15
20. Messmer D, Kipps TJ (2005) Treatment of solid tumors withimmunotoxins. Breast Cancer Res 7:184–186
21. Onda M, Beers R, Xiang L et al (2008) An immunotoxin withgreatly reduced immunogenicity by identiWcation and removal ofB cell epitopes. Proc Natl Acad Sci USA 105:11311–11316
22. Onda M, Nagata S, FitzGerald DJ et al (2006) Characterization ofthe B cell epitopes associated with a truncated form of Pseudomo-nas exotoxin (PE38) used to make immunotoxins for the treatmentof cancer patients. J Immunol 177:8822–8834
23. Posey JA, Khazaeli MB, Bookman MA et al (2002) A phase Itrial of the single-chain immunotoxin SGN-10 (BR96 sFv-PE40)in patients with advanced solid tumors. Clin Cancer Res8:3092–3099
24. Weldon JE, Xiang L, Chertov O et al (2009) A protease-resistantimmunotoxin against CD22 with greatly increased activity againstCLL and diminished animal toxicity. Blood 113(16):3792–3800
25. Kreitman RJ, Wilson WH, Bergeron K et al (2001) EYcacy of theanti-CD22 recombinant immunotoxin BL22 in chemotherapy-resistant hairy-cell leukemia. N Engl J Med 345:241–247
26. Hassan R, Bullock S, Premkumar A et al (2007) Phase I study ofSS1P, a recombinant anti-mesothelin immunotoxin given as abolus I.V. infusion to patients with mesothelin-expressingmesothelioma, ovarian, and pancreatic cancers. Clin Cancer Res13:5144–5149
27. Hassan R, Williams-Gould J, Watson T et al (2004) Pretreatmentwith rituximab does not inhibit the human immune responseagainst the immunogenic protein LMB-1. Clin Cancer Res 10:16–18
28. Knechtle SJ (2001) Treatment with immunotoxin. Philos Trans RSoc Lond B Biol Sci 356:681–689
29. Pai LH, FitzGerald DJ, Tepper M et al (1990) Inhibition of anti-body response to Pseudomonas exotoxin and an immunotoxincontaining Pseudomonas exotoxin by 15-deoxyspergualin in mice.Cancer Res 50:7750–7753
30. Jorgensen R, Purdy AE, Fieldhouse RJ et al (2008) Cholix toxin,a novel ADP-ribosylating factor from Vibrio cholerae. J BiolChem 283:10671–10678
31. Ogata M, Pastan I, FitzGerald D (1991) Analysis of Pseudomonasexotoxin activation and conformational changes by using mono-clonal antibodies as probes. Infect Immun 59:407–414
32. Batra JK, Fitzgerald DJ, Chaudhary VK et al (1991) Single-chainimmunotoxins directed at the human transferrin receptor contain-ing Pseudomonas exotoxin A or diphtheria toxin: anti-TFR(Fv)-PE40 and DT388-anti-TFR(Fv). Mol Cell Biol 11:2200–2205
33. Purdy A, Rohwer F, Edwards R et al (2005) A glimpse into theexpanded genome content of Vibrio cholerae through identiWca-tion of genes present in environmental strains. J Bacteriol187:2992–3001
123

746 Cancer Immunol Immunother (2010) 59:737–746
34. Buchner J, Brinkmann U, Pastan I (1992) Renaturation of a single-chain immunotoxin facilitated by chaperones and protein disulWdeisomerase. Biotechnology (N Y) 10:682–685
35. Buchner J, Pastan I, Brinkmann U (1992) A method for increasingthe yield of properly folded recombinant fusion proteins: single-chain immunotoxins from renaturation of bacterial inclusionbodies. Anal Biochem 205:263–270
36. Kreitman RJ, Wilson WH, White JD et al (2000) Phase I trial ofrecombinant immunotoxin anti-Tac(Fv)-PE38 (LMB-2) in patientswith hematologic malignancies. J Clin Oncol 18:1622–1636
37. Dalsgaard A, Albert MJ, Taylor DN et al (1995) Characterizationof Vibrio cgolerae non-O1 serogroups obtained from an outbreakof diarrhea in Lima, Peru. J Clin Microbiol 33:2715–2722
38. Chaudhary VK, Queen C, Junghans RP et al (1989) A recombi-nant immunotoxin consisting of two antibody variable domainsfused to Pseudomonas exotoxin. Nature 339:394–397
39. Brinkmann U, Pai LH, FitzGerald DJ et al (1991) B3(Fv)-PE38KDEL, a single-chain immunotoxin that causes completeregression of a human carcinoma in mice. Proc Natl Acad Sci USA88:8616–8620
40. Matthews DJ, Goodman LJ, Gorman CM et al (1994) A survey offurin substrate speciWcity using substrate phage display. ProteinSci 3:1197–1205
41. Zdanovsky AG, Chiron M, Pastan I et al (1993) Mechanism ofaction of Pseudomonas exotoxin. IdentiWcation of a rate-limitingstep. J Biol Chem 268:21791–21799
123
Copyright © 2022 FDOKUMEN




















