Induction of interleukin-10 in the T helper type 17 effector population by the G protein coupled...
14
Induction of interleukin-10 in the T helper type 17 effector popula- tion by the G protein coupled estrogen receptor (GPER) agonist G-1 Introduction CD4 + helper T lymphocytes orchestrate adaptive immune responses to invading pathogens, and are critical to the pathogenesis of numerous disease processes, including autoimmunity and cancer. They are an attractive drug target because of their central role in immunity, and their implication in a wide variety of diseases. There are several distinct lineages of CD4 + helper T cells, each specialized in enhancing specific branches of the immune system. The original paradigm described by Mossman and Coff- mann 1 divided CD4 + helper T lymphocytes into the T helper type 1 (Th1) and Th2 populations, with Th1 cells producing interferon-c (IFN-c) and coordinating cellular immunity responses and Th2 cells secreting humoral immunity mediators such as interleukin-4 (IL-4), IL-5 and IL-13. In 2005, the Th1–Th2 paradigm was expanded as the Th17 population emerged as a third class of helper/effector T cell. Th17 cells are characterized by expression of the transcription factor RORct, 2,3 and secrete pro-inflammatory cytokines including IL-21 4 and IL-17A/F. These cells are important to controlling infec- tions by extracellular pathogens, but also appear to play a deleterious role in human health by contributing to the pathogenesis of numerous autoimmune diseases. 5 In mice, Th17 differentiation depends on transforming growth Ryan L. Brunsing 1 and Eric R. Prossnitz 1,2 1 Department of Cell Biology and Physiology, University of New Mexico Health Science Cen- ter, Albuquerque, NM, and 2 UNM Cancer Center, University of New Mexico Health Science Center, Albuquerque, NM, USA doi:10.1111/j.1365-2567.2011.03471.x Received 28 February 2011; revised 16 May 2011; accepted 3 June 2011. Correspondence: E. R. Prossnitz, Department of Cell Biology & Physiology, University of New Mexico, Albuquerque, NM 87131, USA. Email: [email protected] Senior author: Eric R. Prossnitz Summary Interleukin-10 (IL-10) is a potent suppressor of the immune system, com- monly produced by CD4 + T cells to limit ongoing inflammatory responses minimizing host damage. Many autoimmune diseases are marked by large populations of activated CD4 + T cells within the setting of chronic inflammation; therefore, drugs capable of inducing IL-10 production in CD4 + T cells would be of great therapeutic value. Previous reports have shown that the small molecule G-1, an agonist of the membrane-bound G-protein-coupled estrogen receptor GPER, attenuates disease in an ani- mal model of autoimmune encephalomyelitis. However, the direct effects of G-1 on CD4 + T-cell populations remain unknown. Using ex vivo cul- tures of purified CD4 + T cells, we show that G-1 elicits IL-10 expression in T helper type 17 (Th17) -polarized cells, increasing the number of IL- 10 + and IL-10 + IL-17A + cells via de novo induction of IL-10. T-cell cul- tures differentiated in the presence of G-1 secreted threefold more IL-10, with no change in IL-17A, tumour necrosis factor-a, or interferon-c. Moreover, inhibition of extracellular signal-regulated kinase (but not p38 or Jun N-terminal kinase) signalling blocked the response, while analysis of Foxp3 and RORct expression demonstrated increased numbers of IL- 10 + cells in both the Th17 (RORct + ) and Foxp3 + RORct + hybrid T-cell compartments. Our findings translated in vivo as systemic treatment of male mice with G-1 led to increased IL-10 secretion from splenocytes fol- lowing T-cell receptor cross-linking. These results demonstrate that G-1 acts directly on CD4 + T cells, and to our knowledge provide the first example of a synthetic small molecule capable of eliciting IL-10 expression in Th17 or hybrid T-cell populations. Keywords: estrogen receptor; G-protein-coupled estrogen receptor 1; GPR30; hybrid T cell; interleukin-10; T helper type 17; regulatory T cells Ó 2011 The Authors. Immunology Ó 2011 Blackwell Publishing Ltd, Immunology, 134, 93–106 93 IMMUNOLOGY ORIGINAL ARTICLE
Transcript of Induction of interleukin-10 in the T helper type 17 effector population by the G protein coupled...

Induction of interleukin-10 in the T helper type 17 effector popula-tion by the G protein coupled estrogen receptor (GPER) agonist G-1
Introduction
CD4+ helper T lymphocytes orchestrate adaptive immune
responses to invading pathogens, and are critical to the
pathogenesis of numerous disease processes, including
autoimmunity and cancer. They are an attractive drug
target because of their central role in immunity, and their
implication in a wide variety of diseases. There are several
distinct lineages of CD4+ helper T cells, each specialized
in enhancing specific branches of the immune system.
The original paradigm described by Mossman and Coff-
mann1 divided CD4+ helper T lymphocytes into the T
helper type 1 (Th1) and Th2 populations, with Th1 cells
producing interferon-c (IFN-c) and coordinating cellular
immunity responses and Th2 cells secreting humoral
immunity mediators such as interleukin-4 (IL-4), IL-5
and IL-13. In 2005, the Th1–Th2 paradigm was expanded
as the Th17 population emerged as a third class of
helper/effector T cell. Th17 cells are characterized by
expression of the transcription factor RORct,2,3 and
secrete pro-inflammatory cytokines including IL-214 and
IL-17A/F. These cells are important to controlling infec-
tions by extracellular pathogens, but also appear to play a
deleterious role in human health by contributing to the
pathogenesis of numerous autoimmune diseases.5 In mice,
Th17 differentiation depends on transforming growth
Ryan L. Brunsing1 and Eric R.
Prossnitz1,2
1Department of Cell Biology and Physiology,
University of New Mexico Health Science Cen-
ter, Albuquerque, NM, and 2UNM Cancer
Center, University of New Mexico Health
Science Center, Albuquerque, NM, USA
doi:10.1111/j.1365-2567.2011.03471.x
Received 28 February 2011; revised 16 May
2011; accepted 3 June 2011.
Correspondence: E. R. Prossnitz, Department
of Cell Biology & Physiology, University of
New Mexico, Albuquerque, NM 87131, USA.
Email: [email protected]
Senior author: Eric R. Prossnitz
Summary
Interleukin-10 (IL-10) is a potent suppressor of the immune system, com-
monly produced by CD4+ T cells to limit ongoing inflammatory responses
minimizing host damage. Many autoimmune diseases are marked by large
populations of activated CD4+ T cells within the setting of chronic
inflammation; therefore, drugs capable of inducing IL-10 production in
CD4+ T cells would be of great therapeutic value. Previous reports have
shown that the small molecule G-1, an agonist of the membrane-bound
G-protein-coupled estrogen receptor GPER, attenuates disease in an ani-
mal model of autoimmune encephalomyelitis. However, the direct effects
of G-1 on CD4+ T-cell populations remain unknown. Using ex vivo cul-
tures of purified CD4+ T cells, we show that G-1 elicits IL-10 expression
in T helper type 17 (Th17) -polarized cells, increasing the number of IL-
10+ and IL-10+ IL-17A+ cells via de novo induction of IL-10. T-cell cul-
tures differentiated in the presence of G-1 secreted threefold more IL-10,
with no change in IL-17A, tumour necrosis factor-a, or interferon-c.
Moreover, inhibition of extracellular signal-regulated kinase (but not p38
or Jun N-terminal kinase) signalling blocked the response, while analysis
of Foxp3 and RORct expression demonstrated increased numbers of IL-
10+ cells in both the Th17 (RORct+) and Foxp3+ RORct+ hybrid T-cell
compartments. Our findings translated in vivo as systemic treatment of
male mice with G-1 led to increased IL-10 secretion from splenocytes fol-
lowing T-cell receptor cross-linking. These results demonstrate that G-1
acts directly on CD4+ T cells, and to our knowledge provide the first
example of a synthetic small molecule capable of eliciting IL-10 expression
in Th17 or hybrid T-cell populations.
Keywords: estrogen receptor; G-protein-coupled estrogen receptor 1;
GPR30; hybrid T cell; interleukin-10; T helper type 17; regulatory T cells
� 2011 The Authors. Immunology � 2011 Blackwell Publishing Ltd, Immunology, 134, 93–106 93
I M M U N O L O G Y O R I G I N A L A R T I C L E

factor-b (TGF-b) and IL-6- or IL-21-mediated signal
transducer and activator of transcription 3 (STAT3) acti-
vation,5 while IL-23 signalling plays a critical role in sta-
bilizing the Th17 phenotype.6 Although Th1, Th2 and
Th17 effector T cells coordinate a robust and diverse arse-
nal of adaptive immune responses necessary for the main-
tenance of human health, mechanisms of restraint must
limit effector responses to protect the host from immune-
mediated damage.
A major breakthrough in elucidating the mechanisms
of adaptive immune regulation emerged with the identifi-
cation of an array of regulatory T (Treg) -cell popula-
tions. The best defined class of Treg cells expresses the
forkhead transcription factor Foxp3 and suppresses
numerous animal models of autoimmune disease,7
whereas loss of Foxp3 function in humans and mice pre-
cipitates a fatal multi-organ autoimmune condition
marked by the inability to control T-cell responses.8,9 The
T reg cells function to dampen immune responses
through a variety of approaches, including contact-medi-
ated inhibition, secretion of perforin and granzyme A/B,
sequestration of key growth factors such as IL-2, and
secretion of suppressive cytokines including TGF-b, IL-10
and IL-35.7 Interleukin-10 in particular plays an impor-
tant role in immune homeostasis, both in mice10 and
humans,11 suggesting that it has several non-redundant
roles in regulating inflammatory responses. Many cell
types in addition to Foxp3+ cells12 can produce IL-10,
most notably several lineages of CD4+ T cells,13 including
Th1,14–16 Th214,17 and Th1718–20 cells, as well as various
types of Treg cells.21 In a feed-forward mechanism, IL-10
can drive its own expression through the induction of an
IL-10-producing Treg-cell population termed Tr1
cells.22,23 Conversely, IL-10 can also be induced indepen-
dently of IL-10 signalling in both Foxp3+ and Foxp3)
Treg-cell populations.24 Given its potent anti-inflamma-
tory effects, various strategies are being explored to target
IL-10 for therapeutic intervention.25
The intimate interplay between the critical factors in
development of Treg and Th17 cells, along with the dual
reliance on TGF-b signalling for their differentiation,26
has led to conceptualization of a Treg–Th17 axis. From a
therapeutics perspective, the identification of drugs
that promote pro-inflammatory or anti-inflammatory
responses by influencing differentiation along this axis
has gained momentum as examples of T-cell plasticity
continue to be characterized,27 in particular within the
Treg-cell and Th17-cell populations.28 Moreover, several
reports have characterized ‘hybrid’ T-cell populations
where Foxp3 is expressed in various effector T-cell popu-
lations,29 and IL-10 can be produced by Th1, Th2 and
Th17 cells.12 These results suggest that it may be possible
to treat disease by shifting the balance along the Treg–
Th17 axis in situ during ongoing immune responses. For
example, one mechanism to dampen inflammation would
be to induce IL-10 expression within Th17 cells partici-
pating in pathological inflammation. To that end, target-
ing non-cytokine signalling pathways may be a viable
option. For example, ATP,30 sphinogosine-1-phosphate31
and vitamin D32 can modulate Th17 development,
whereas antigen-presenting cell (APC)-derived indolamine
2,3-dioxygenase33 and retinoic acid34 can promote Treg-
cell populations, highlighting the importance of non-cyto-
kine signalling pathways to this paradigm.
Estrogen is a well-documented modulator of immune
function in humans and mice, capable of increasing the
expression of Foxp335 and IL-10.36 These effects translate
to human disease wherein patients with multiple sclerosis
experience a decrease in symptoms during pregnancy,37
and to murine models of autoimmune disease where
estrogen inhibits development of and reverses experimen-
tal autoimmune encephalomyelitis (EAE),38 an animal
model of multiple sclerosis. Although the effects of estro-
gen are presumed to be mediated by the classical estrogen
receptors, ERa and ERb, recent studies have pointed to
the newly described G protein-coupled estrogen receptor
GPR30/GPER as contributing to many of these responses.
We and others have recently shown that, like estradiol
(E2), the GPER-selective agonist G-1 can attenuate
EAE.38,39 In the current work we show that G-1 can evoke
IL-10 expression and secretion from CD4+ T cells differ-
entiated under Th17-polarizing conditions. G-1-mediated
IL-10 expression was blocked by the GPER-directed
antagonist G15,40 and was dependent on extracellular sig-
nal-regulated kinase (ERK) signalling, consistent with
known mechanisms of IL-10 production within effector
T-cell populations.12 Analysis of IL-17A, Foxp3 and
RORct expression demonstrated that these responses
occurred in cells expressing both IL-17A and RORct, as
well as in a population of Foxp3+ RORct+ hybrid T cells.
Taken together, our results demonstrate a novel immuno-
modulatory property for G-1. In addition, these data sug-
gest that the family of GPER-directed small molecules
may serve as model compounds for a new class of T-cell-
targeted pharmaceuticals in the treatment of autoimmune
disease and cancer.
Materials and methods
Mice
Male (7–11 weeks old) C57BL/6 and Foxp3egfp mice were
used for this study. Mice were purchased from Jackson
Laboratory (Bar Harbor, ME), and subsequently housed,
bred and cared for according to the institutional guide-
lines in the Animal Resource Facility at the University of
New Mexico. Foxp3-IRES-GFP (Foxp3egfp) transgenic
mice, which contain egfp under the control of an internal
ribosomal entry site (IRES) inserted downstream of the
foxp3 coding region, have been previously described.41
94 � 2011 The Authors. Immunology � 2011 Blackwell Publishing Ltd, Immunology, 134, 93–106
R. L. Brunsing and E. R. Prossnitz

Purification of T-cell populations
T cells were obtained from single cell suspensions follow-
ing homogenization of spleens and lymph nodes by
mechanical disruption and passage through a 70-lm
nylon filter. Suspensions were stained with anti-CD4,
anti-CD62 ligand (CD62L) and anti-CD44 antibodies
(Biolegend, San Diego, CA). Enriched populations of
CD4+ CD62Lhi and CD4+ CD44lo CD62Lhi naive T cells
were collected by flow cytometric cell sorting on a MoFlo
cell sorter (Cytomation, Carpinteria, CA). Purity was reg-
ularly > 96%. In most cases, experiments were repeated
with both types of sorted naive T cells, and no differences
were noted. Alternatively, CD4+ cells were collected from
the single cell suspensions by magnetic bead sorting,
using CD4 microbeads (Miltenyi, Bergisch Gladbach,
Germany) and positive selection on an AutoMACS (Milt-
enyi). This yielded populations with a purity > 90%.
Culture conditions
All experiments and cell purification were carried out in
RPMI-1640 medium supplemented with fetal bovine serum
(FBS), penicillin/streptomycin, L-glutamine, HEPES,
sodium pyruvate and 2-mercaptoethanol. Phenol red-free
buffers and charcoal-stripped FBS were used to minimize
exposure to estrogens or phyto/xenoestrogens that could
have confounded our results. Cells were stimulated in cul-
ture with soluble anti-CD3e (1�0 lg/ml) and anti-CD28
(2�5 lg/ml) antibodies (Biolegend), and supplemented
with various combinations of TGF-b (0�5–10 ng/ml), IL-6
(20 ng/ml) and IL-23 (20 ng/ml) as described (Biolegend
and eBiosciences, San Diego, CA). G-1 and DMSO were
added concurrently with the stimulatory antibodies and
cytokines. Non-polarizing conditions (Th0) contained no
exogenous cytokines. Th17 conditions contained TGF-
b + IL-6 ± IL-23. Experiments were carried out using 96-
well plates with 2 · 105 cells per well (106 cells/ml). For
experiments using GPER and mitogen-activated protein
(MAP) kinase inhibitors, cells were pre-incubated for 60–
90 min with 25 lM PD98059 [MAP kinase kinase (MEK)
inhibitor], 250 nM Jun N-terminal kinase (JNK) II inhibi-
tor, 100 nM SB203580 (p38 inhibitor), or 500 nM G15
(GPER antagonist,40 provided by Dr Jeffrey Arterburn at
New Mexico State University) where indicated, before the
addition of stimulatory antibodies or cytokines. All com-
pounds used in the study were dissolved in DMSO. All cul-
tures were incubated at 37� (+ 5% CO2).
Intracellular cytokine staining and analysis
Following 4 days in culture, cells were washed with med-
ium and ‘rested’ for 60–90 min at 37� (+ 5% CO2). Cul-
tures were then treated with PMA (50 ng/ml) and
ionomycin (500 ng/ml) for 4–5 hr in the presence of Bre-
feldin A (Biolegend) followed by fixation in Fixation Buf-
fer (Biolegend). Samples were then washed and stained
for intracellular proteins in Permeabilization Wash buffer
(Biolegend) for 2 hr at room temperature, and washed
with excess Permeabilization Wash buffer for 15 min at
room temperature before centrifugation and analysis.
Immediately after staining, data were collected on a FAC-
Scalibur (Becton Dickinson, Franklin Lakes, NJ). Data
analysis was performed using FLOWJO software (TreeStar,
Ashland, OR). Antibodies for staining included anti-IL-
10-allophycocyanin, anti-IL-10-phycoerythrin, anti-IL-
17A-phycoerythrin, and IL-17A-peridinin chlorophyll
protein and anti-IFN-c-allophycocyanin all from Bioleg-
end, as well as anti-RORct-phycoerythrin from eBio-
sciences.
Proliferation studies
For analysis of proliferation, freshly sorted T cells were
stained with 2�5 lM eFluor670 according to the manufac-
turer’s protocols (eBiosciences). Cells were then cultured,
stained and analysed as indicated above. Geometric mean
fluorescence intensity (GMFI) of eFluor670 was deter-
mined using FLOWJO software (TreeStar), and unstimu-
lated controls were used to differentiate between
proliferating and non-proliferating cells.
Luminex multiplex assays
Following 4 days in culture, T cells were washed with cold
medium to remove any cytokines in solution, resus-
pended in fresh medium, and counted. For cultures of
splenocytes, single cells suspensions were obtained follow-
ing homogenization of spleens by mechanical disruption
and passage through a 70-lm nylon filter. Cells were then
plated in a 96-well plate with 2 · 105 cells per well
(106 cells/ml), allowed to incubate for 60–90 min at 37�(+5% CO2), and re-stimulated with soluble anti-CD3e(2�5 lg/ml) antibody. Following the indicated stimulation
time, culture medium was collected and spun down to
remove any residual cells. The concentration of IL-4, IL-
6, IL-10, IL-17A, IFN-c and tumour necrosis factor-a(TNF-a) in the cell-free culture medium was analysed
using custom bead arrays from Millipore, and quantified
on a Luminex 100 system (Austin, TX) with the Luminex
XY plate handling platform. Assays were performed
according to the manufacturer’s protocols. Duplicate wells
were assayed for each sample, and data are representative
of the average median value for each sample. Analysis was
performed using IS 2.3 software (Luminex).
In vivo treatment with G-1
A vehicle consisting of 90% emulsion solution
(PBS + 0�9% Tween-20 + 0�9% BSA) and 10% ethanol
� 2011 The Authors. Immunology � 2011 Blackwell Publishing Ltd, Immunology, 134, 93–106 95
IL-10 induction by GPER agonist G-1

was used. For delivery of compounds, E2 or G-1 was dis-
solved in ethanol and added at appropriate concentrations
such that 100 ll per animal per injection was used. The
compound was added to each injection as part of the
10% ethanol found in the vehicle, so it was diluted such
that < 10 ll per animal per injection was required. Injec-
tions were administered in the afternoon, and to limit
stress from the long series of injections inherent in this
study, animals were sedated using isofluorane before
injection. Compound was delivered subcutaneously on
the dorsum adjacent to the hind limb, and the side of the
injection was alternated every 2 days.
Results
G-1 elicits IL-10 production in CD4+ T cells underTh17 polarizing conditions
To investigate the direct effects of G-1 on CD4+ T cells,
we chose to use purified cultures of naive T cells activated
by polyclonal stimulation with anti-CD3e and anti-CD28
antibody. This eliminated secondary effects caused by the
activity of G-1 on APCs within the culture. Furthermore,
primary cells from male mice were used throughout the
study to avoid potential confounding effects of either; (i)
varying estrogen levels in female mice, or (ii) the inflam-
matory effects of ovariectomy. We have also determined
that CD4+ CD44loCD62Lhi naive T-cell and CD4+ Foxp3+
T-reg cell populations express the G-1 target GPER (R. L.
Brunsing and E. R. Prossnitz, manuscript in preparation).
Given that G-1 can protect mice from EAE38,39 and
the importance of the the Th17 lineage to this model,3
we began by determining the effects of G-1 on naive
T-cell differentiation under Th17-polarizing conditions
(TGF-b/IL-6 ± IL-23). Hence, naive T cells from 7- to
11-week-old male C57BL/6 mice were collected by FACS
and stimulated for 4 days ex vivo, supplemented with
combinations of TGF-b, IL-6 and IL-23. Following
4 days of stimulation, cells were analysed for expression
of IFN-c, IL-17A and IL-10 by intracellular cytokine
staining. Expression of IL-10 was present exclusively in
cultures treated with IL-6 (Fig. 1a), consistent with pre-
vious findings using ex vivo culture systems where treat-
ment with TGF-b alone blocks IL-10 expression in
differentiating CD4+ T cells,13 and efficient induction of
IL-10 secretion from effector T-cell populations requires
one of the STAT activating cytokines (for example,
IL-6).19 As expected, IL-17A expression was also largely
dependent on Th17-polarizing conditions (i.e. treatment
with both TGF-b and IL-6; Fig. 1a), although a small
number of IL-17A+ cells was observed in the TGF-b-
treated cultures (data not shown), and was enhanced by
the addition of IL-23. G-1 treatment resulted in an
increase in the percentage of IL-10+ cells within Th 17
cell-polarized cultures (Fig. 1b), including within cultures
supplemented with IL-23 (Fig. 1c), which is known to be
important in stabilizing the phenotype of Th17 popula-
tions.6 This G-1-mediated IL-10 expression was specific
as no increase in the prevalence IL-17A+ cells was
observed in either of the Th17-polarizing conditions
(Fig. 1b,c). In addition, G-1 treatment had no effect on
IFN-c expression in cultures stimulated with CD3/28
alone (Fig. 1d); however, few IFN-c+ cells were detected
in the other culture conditions tested (see Supplementary
material, Fig. S1).
To determine whether the induction of IL-10+ cells
translated into a specific increase in the secretion of IL-10
from G-1-treated cultures, naive T cells were collected
and stimulated as above, in the presence of TGF-b and
IL-6. After 4 days of differentiation, DMSO-treated and
G-1-treated cells were collected, washed with medium to
remove any cytokines released over the course of differen-
tiation, and re-plated at 106 cells/ml. Cells were then
re-stimulated with anti-CD3e antibody for 24 hr, after
which culture medium was analysed for the presence of
newly secreted IL-6, IL-10, IL-17A, TNF-a and IFN-c by
Luminex multiplex assay. Cells differentiated in the pres-
ence of G-1 produced approximately threefold more IL-10
than control cultures (Fig. 2a), consistent with our obser-
vation that G-1 induced an IL-10-producing population.
No difference in the secretion of IL-6, IL-17A, TNF-a or
IFN-c was detected (Fig. 2b–e), again suggesting that G-1
was specifically driving the production of the anti-inflam-
matory cytokine IL-10, and not pro-inflammatory media-
tors such as TNF-a and IFN-c. Taken together, these data
show that G-1 can specifically drive IL-10 expression
within, and secretion from, CD4+ T-cell populations.
Induction of an IL-10+ IL-17A+ and IL-10+ IL-17A)
population by G-1
As G-1-induced IL-10 expression was dependent on
Th17-polarizing conditions, we sought to determine the
relationship between G-1-induced IL-10+ cells and those
expressing the characteristic Th17 cytokine IL-17A.
Hence, naive T cells were again collected by FACS and
polyclonally stimulated in the presence of TGF-b and
IL-6. Cells were cultured with increasing doses of G-1
(1–500 nM) and analysed for IL-17A and IL-10 by intra-
cellular cytokine staining (Fig. 3a). Our data reveal a
dose-dependent increase in IL-10+ IL-17A) (Fig. 3a,b)
and IL-10+ IL-17A+ cells (Fig. 3a,c) within G-1-treated
cultures. A similar trend was observed under IL-23 polar-
izing conditions (Fig. 1a and data not shown). In addi-
tion, G-1-mediated IL-10 expression was blocked by the
recently described GPER antagonist G15.40 The induction
of a population of IL-10+ IL-17A+ cells suggests that G-1
can elicit IL-10 expression within cells that have differen-
tiated to the Th17 lineage. Taken together, these data
show that G-1 can elicit IL-10 production within the
96 � 2011 The Authors. Immunology � 2011 Blackwell Publishing Ltd, Immunology, 134, 93–106
R. L. Brunsing and E. R. Prossnitz

Th17 compartment, a response that is blocked by the
GPER-selective antagonist G15.
ERK signalling is critical for G-1-mediated IL-10expression
Interleukin-10 production within Th populations has
been shown to be dependent on signalling through extra-
cellular signal-regulated kinases ERK1/2,12,13 one of three
MAP kinase cascades, the others comprising JNK1/2 and
p38. GPER has been shown to activate the ERK pathway,
although predominantly in cancer cells.42 To test whether
G-1-mediated induction of IL-10 was dependent on MAP
kinase signalling, naive T cells were treated with either
PD98059, an inhibitor of the ERK pathway, SB203580,
an inhibitor of the p38 pathway, or the JNK II inhibitor,
and stimulated under Th17-polarizing conditions as
before. Consistent with other published reports,13 we
found that inhibition of p38 had no effect on IL-10
expression in Th17-polarized cells. Similarly, JNK signal-
ling appeared not to be required for G-1-mediated
induction of IL-10 (Fig. 4a). In contrast, there was no
difference in the percentage of IL-10+ cells observed
between control and G-1-treated cultures when cells were
cultured with the ERK inhibitor PD98059 (Fig. 4a,b),
consistent with a role for ERK signalling specifically in
DM
SO
IL-1
0
IL-1
0
IL-1
0
IL-1
0
IL-1
0IL
-10
IL-1
0
IL-1
0
IL-1
0
IL-1
0
IL-1
0
G-1
IL-17A IL-17A IL-17A IL-17A IL-17A
IL-17A IL-17AIL-17AIL-17AIL-17A
IL-17A
0·057
0·095
1·25 0·096
0·36
1·31 0·11
0·12
1·08 0·37 4·82 0·067
0·61
21·2 9·12
5·12
19·8 9·27
9·470·56
0·11
0·57
16·4 6·08
7·11
CD3/28
TGF-β1
IL-6
IL-23
1·27 3·82 14·5 6·4
11·5
+ +
+
+ +
+
+
+
+
+
+
+
–
–
–
–
– –
–
–
(a)
TGF-β /IL-6/IL-23
IFNγ +
TGF-β /IL-6
2·5
2·0
1·5
1·0
0·5
0·0IL-10+ IL-17A+
2·5
2·0
1·5
1·0
0·5
0·0IL-10+ IL-17A+
DMSODMSOG-1G-1
NS
NS
2·5
2·0
1·5
1·0
0·5
0·0
CD3/28 alone
NS
DMSO
G-1
Rel
ativ
e %
Rel
ativ
e %
Rel
ativ
e %
*
***
(b) (c) (d)
Figure 1. The G-protein-coupled estrogen receptor (GPER) -directed agonist G-1 induces interleukin-10 (IL-10) production from CD4+ T cells.
CD4+ CD44lo CD62Lhi naive CD4+ T cells were collected by FACS and cultured for 4 days ex vivo with various combinations of transforming
growth factor-b (TGF-b), IL-6 and IL-23, and supplemented with 100 nm G-1 or vehicle (DMSO, control). Cells were subsequently stained for
intracellular IFN-c, IL-17A and IL-10, then analysed by flow cytometry. (a) Representative plots from the various conditions showing intracellular
IL-17A and IL-10. (b) Quantification of data from five to seven independent experiments showing relative number of total IL-10+ cells and total
IL-17A+ cells in cultures treated with TGF-b + IL-6. (c) Quantification of data from four to seven independent experiments showing relative
prevalence of total IL-10+ cells and total IL-17A+ cells in cultures treated with TGF-b + IL-6 + IL-23. (d) Quantification of IFN-c+ cells in cul-
tures stimulated with anti-CD3/28 in non-polarizing conditions (i.e. without the addition of any cytokines). P-values determined by Student’s t-
test; *P < 0�05; ***P < 0�0005. Error bars = SEM; NS, not significant.
� 2011 The Authors. Immunology � 2011 Blackwell Publishing Ltd, Immunology, 134, 93–106 97
IL-10 induction by GPER agonist G-1

G-1-mediated IL-10 induction. These data suggest that
G-1 mediates IL-10 expression by activating ERK signal-
ling in CD4+ T cells.
The ERK pathway is known to be a potent activator of
cell proliferation. To determine if G-1-mediated increases
in IL-10 were the result of increased proliferation of cells
expressing IL-10 rather than induction of IL-10 de novo,
naive T cells were stained with the proliferation dye eFlu-
or670 before stimulation in culture. We were unable to
detect any significant difference in the proportion of
dividing cells following G-1 treatment. The observation
that G-1-treated cultures demonstrate attenuated dilution
of the eFluor dye compared with the DMSO-treated cul-
tures (Fig. 5) indicates that the increase in IL-10+ cells
following G-1 treatment is not the result of an increase in
cell proliferation, and in fact shows that proliferating cells
are going through fewer divisions when treated with G-1,
perhaps because of the action of IL-10. In addition, the
dramatic increase in the number of non-dividing cells
expressing IL-10 in G-1-treated cultures (as indicated in
the upper right quadrant in Fig. 5b) suggests that G-1
can specifically drive expression of IL-10 independent of
cell division. Taken together, these data show that G-1
stimulates de novo IL-10 expression within differentiating
Th17 cells through direct action on T cells via an ERK-
dependent mechanism.
IL-10 induction occurs within a hybrid T-cellpopulation
An emerging paradigm in T-cell biology is the induction
of ‘hybrid’ T-cell populations that express one of the
canonical effector T-cell transcription factors (for example
T-bet from the Th1 lineage) as well as Foxp3.29 These
cells appear to play a role in the regulation of specific
types of inflammatory responses, where the expression of
Foxp3 imparts a suppressive phenotype, and the expres-
sion of the lineage-specific factor such as T-bet leads to a
repertoire of gene products (e.g. chemokine receptors)
that allow for targeting to sites of inflammation. Presum-
ably, this provides a mechanism for the recruitment of
regulatory T cells to sites on ongoing inflammatory
responses. To investigate the expression of Foxp3 together
with RORct, naive T cells were collected from Foxp3egfp
transgenic mice.41 Cells were stimulated for 4 days in the
presence of TGF-b and IL-6 with or without G-1 added
to the culture. Following differentiation, IL-10, IL-17A,
RORct and Foxp3 were analysed by intracellular cytokine
staining or detection of endogenous GFP expression
by flow cytometry. G-1 was equally effective at inducing
IL-10 production within Foxp3) RORct+ Th17 cells as in
Foxp3+ RORct+ hybrid T cells (Fig. 6). The Th17 (i.e.
RORct+) subset yielded an increase in both IL-10+ IL-
17A+ and IL-10+ IL-17A) cells, while only IL-10+ IL-17A)
cells were detected in the hybrid T-cell population. In fact
no IL-17A+ cells were present in the Foxp3+ population
(data not shown). These data demonstrate the ability of
G-1 to induce IL-10 within the recently described hybrid
Th17 population in addition to conventional (Fox-
p3) RORct+) Th17 cells.
In vivo treatment with G-1 leads to increased IL-10secretion from splenocytes
Our results show that treatment of naive T cells with G-1
in culture can lead to increased IL-10 expression and
secretion. To determine if these findings translated
in vivo, wild-type mice were injected subcutaneously with
G-1 for 7 consecutive days, after which isolated spleno-
cytes were stimulated in culture with anti-CD3e and anti-
CD28 antibodies. Samples of supernatant were collected
24, 48 and 72 hr after stimulation and analysed for
10
5
0DMSO G-1
G-1
G-1
G-1
NS
6
4
2
0DMSO
DMSODMSO
DMSO
G-1
NS NS
NS
200
200
200
150
100
50
00
0
600
400
400
300
100
**
IL-1
0 (n
g/m
l)IL
-6 (
pg/m
l)
IL-1
7A (
ng/m
l)
IFN
- γ (
pg/m
l)
TN
F- α
(pg
/ml)
(a) (b)
(d)(c)
(e)
Figure 2. Cytokine secretion following ex vivo treatment with G-1.
CD4+ CD62Lhi naive CD4+ T cells were collected by FACS and cul-
tured for 4 days ex vivo with anti-CD3/28 + interleukin-6 (IL-
6) + transforming growth factor-b (TGF-b) in the presence of
100 nm G-1 (black bars) or DMSO (white bars). Cells were washed
on day 4 and re-stimulated with anti-CD3e alone. Culture medium
was collected after 24 hr and analysed for the presence of secreted
(a) IL-10, (b) IL-17A, (c) interferon-c (IFN-c), (d) tumour necrosis
factor-a (TNF-a), and (e) IL-6 by Luminex multiplex assay. Data are
the means from three independent experiments done in triplicate.
P-values determined by Student’s t-test; *P < 0�02. Error bars =
SEM; NS, not significant.
98 � 2011 The Authors. Immunology � 2011 Blackwell Publishing Ltd, Immunology, 134, 93–106
R. L. Brunsing and E. R. Prossnitz

secreted IL-6, IL-10, IL-17A, IFN-c and TNF-a by Lumin-
ex multiplex assay. No trends were observed for any of
the analytes following 24 hr of stimulation (Fig. 7). As
postulated, following 72 hr of stimulation cells from the
G-1 treated mice produced significantly more IL-10
(Fig. 7a), in agreement with our results with cultured
DM
SO
G-1
IL-1
0IL
-10
IL-1
0IL
-10
IL-1
0IL
-10
IL-1
0IL
-10
15·2 3·29
4·95
15·7 3·74
5·01
14·5 3·9
5·39 6·18
5·0117·4 32·6 10·6
3·9
39·9 12·6
2·87
5·73
4·9417·94·42
5·77
17·2
IL-17A IL-17A IL-17A IL-17A
IL-17AIL-17AIL-17AIL-17A
G-1
(a)
50
40
30
20
10
01 10 100 500
15
10
5
01 10 100 500
G-1 (nM)
DMSO
G-12·0
1·5
1·0
0·5
0·0+ G15
% o
f tot
al p
op.
% o
f tot
al p
op.
***
***
IL-10+IL-17A– IL-10+IL-17A+
Rel
ativ
e %
IL-1
0+
G-1 (nM)
*
*
TGF-β/IL-6
G-1
DMSODMSO
G-1
**
*
(b)(c)
(d)
Figure 3. The G-protein-coupled estrogen receptor (GPER) -directed agonist G-1 induces interleukin-10 (IL-10) production in IL-17A+ cells.
CD4+ CD44lo CD62Lhi or CD4+ CD62Lhi naive CD4+ T cells were collected by FACS and cultured for 4 days ex vivo with anti-CD3/28 + IL-
6 + transforming growth factor-b (TGF-b). Increasing doses of G-1 (1–500 nm, black bars) or equivalent amounts of vehicle (DMSO, white bars)
were added, or cultures were pre-treated with the GPER antagonist G15. Cells were subsequently stained for intracellular IL-17A and IL-10, and anal-
ysed by flow cytometry. (a) Representative plots from the various conditions showing intracellular IL-17A and IL-10. (b–c) Quantification of data
from one of two independent experiments showing the percentage of cells that are (b) IL-10+ IL-17A) and (c) IL-10+ IL-17A+ for the given condi-
tions. (d) Summary of data from two independent experiments showing that the GPER-directed antagonist G15 (500 nm) can block G-1 (100 nm)-
mediated IL-10 induction. P-values determined by Student’s t-test; *P < 0�01; **P < 0�001; ***P < 0�0001. Error bars = SD (b,c) or SEM (d).
� 2011 The Authors. Immunology � 2011 Blackwell Publishing Ltd, Immunology, 134, 93–106 99
IL-10 induction by GPER agonist G-1
naive T cells. Moreover, there was a statistically significant
difference between the time–course of IL-10 secretion for
the cells from G-1-treated mice compared with those
from vehicle-treated animals, as determined by analysis of
variance (Fig. 7a).
Some unexpected results where obtained as well. We
observed that G-1-treated splenocytes demonstrated a sta-
tistically significant increase in the secretion of IL-17A at
48 hr (Fig. 7b). This differed from our findings in Fig. 1,
where no change in the prevalence IL-17A+ cells was
observed, and Fig. 2 where naive T cells cultured with
G-1 produced similar levels of IL-17A compared with
control cells. Additionally, splenocytes from G-1-treated
mice produced decreased levels of IFN-c relative to those
that were treated with vehicle alone (Fig. 7c), suggesting
that in addition to driving production of IL-10 and IL-
17A, G-1 may act systemically to reduce the levels of
IFN-c. This result differed from those shown in Figs 1
and 2 as well, where no changes in IFN-c expression were
noted. These observations probably reflect the complex
nature of the in vivo environment, with secondary effects
resulting from activity on other immune populations. No
changes in the secretion of TNF-a (Fig. 7d) or IL-6
(Fig. 7e) were detected, in agreement with our findings
from Fig. 2. Collectively, these data suggest that pharma-
cological stimulation of GPER in vivo leads to an increase
in the production of the cytokines IL-10 and IL-17A, and
decreased production of the pro-inflammatory cytokine
IFN-c following T-cell activation, yielding an overall anti-
inflammatory environment.
Discussion
It is known that CD4+ T cells play a critical role in the
pathogenesis of many of the most prominent diseases of
the Western world, including cancer, autoimmunity and
infectious diseases. The cytokine IL-10 is a potent sup-
pressor of immune responses, capable of acting on a mul-
titude of cell types to dampen inflammatory responses to
and limit host damage by infection and autoimmune dis-
ease. In this study, we demonstrated that the GPER-direc-
ted agonist G-1 can drive IL-10 production from Th17-
(a) DMSOG-1
NS3
2
1
0
DM
SO
(b)
IL-1
0IL
-10
IL-17A
IL-10+IL-17A+
2·0
1·5
1·0
0·5
0·0Control PD98059
DMSOG-1
DMSOG-1
DMSOG-1
Control PD98059Control PD98059
2·0
1·5
1·0
0·5
0·0
2·0
1·5
1·0
0·5
0·0
Rel
ativ
e %
(c) IL-10+IL-17A–
NS
IL-10–IL-17A+
Rel
ativ
e %
Rel
ativ
e %
G-1
25·5 5·22
6·29
38 10·4 27·6 15
IL-1
0IL
-10
4·44 11
12·5
14·326·3PD98059No inhibitor
Rel
ativ
e %
IL-1
0+
TGF-β /IL6 + PD + JNK inh + SB
IL-17A
IL-17A IL-17A
*****
**
**
***
***
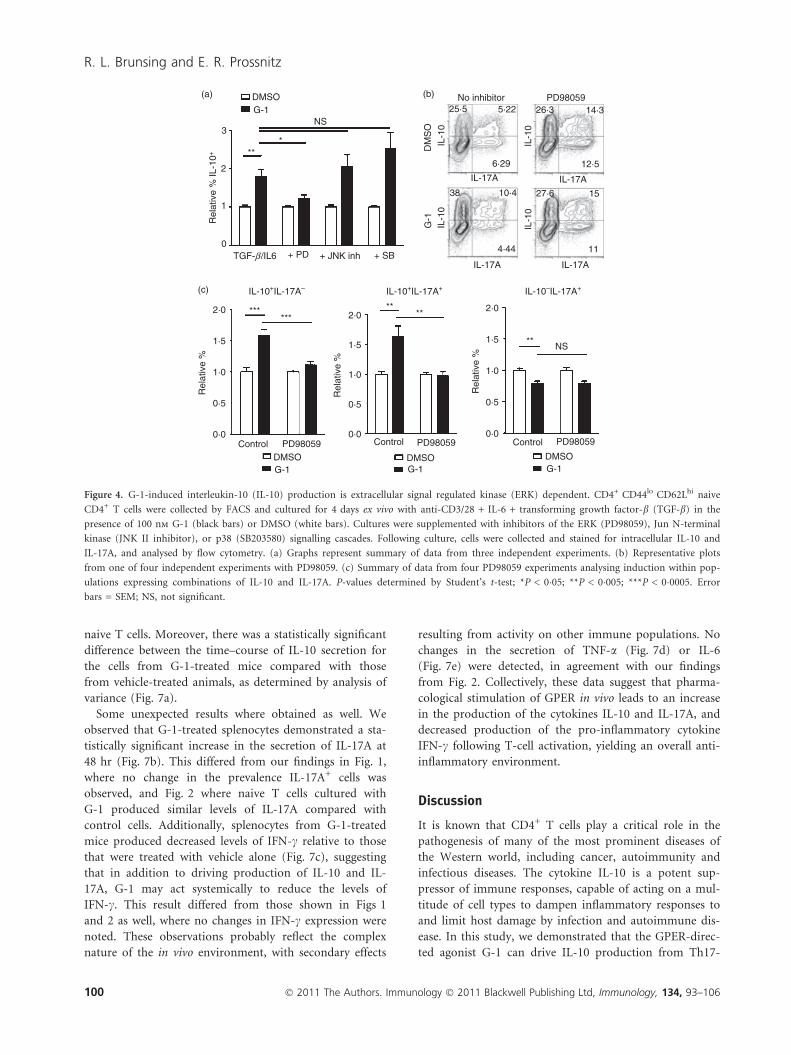
Figure 4. G-1-induced interleukin-10 (IL-10) production is extracellular signal regulated kinase (ERK) dependent. CD4+ CD44lo CD62Lhi naive
CD4+ T cells were collected by FACS and cultured for 4 days ex vivo with anti-CD3/28 + IL-6 + transforming growth factor-b (TGF-b) in the
presence of 100 nm G-1 (black bars) or DMSO (white bars). Cultures were supplemented with inhibitors of the ERK (PD98059), Jun N-terminal
kinase (JNK II inhibitor), or p38 (SB203580) signalling cascades. Following culture, cells were collected and stained for intracellular IL-10 and
IL-17A, and analysed by flow cytometry. (a) Graphs represent summary of data from three independent experiments. (b) Representative plots
from one of four independent experiments with PD98059. (c) Summary of data from four PD98059 experiments analysing induction within pop-
ulations expressing combinations of IL-10 and IL-17A. P-values determined by Student’s t-test; *P < 0�05; **P < 0�005; ***P < 0�0005. Error
bars = SEM; NS, not significant.
100 � 2011 The Authors. Immunology � 2011 Blackwell Publishing Ltd, Immunology, 134, 93–106
R. L. Brunsing and E. R. Prossnitz

polarized CD4+ T-cell populations. We observed an
increase in the number of cells expressing IL-10 within,
and increased IL-10 secretion from, the G-1-treated
cultures. This response was not the result of global
changes in cytokine production as G-1 had no effect on
the expression of IL-17A under Th17-polarizing condi-
tions, or in the induction of IFN-c in non-polarizing
(Th0) conditions. We also observed no significant change
in the secretion of IL-6, IL-17A, TNF-a or IFN-c from G-
1-treated cultures, demonstrating high selectivity for the
mechanism of G-1-mediated IL-10 induction.
We did occasionally detect fewer cells in G-1-treated
cultures relative to those treated with DMSO (RLB and
ERP, unpublished observation), but this was not a con-
sistent finding. This observation may reflect variability in
the temporal dynamics of IL-10 induction between
different experiments or G-1-mediated induction of reg-
ulatory T-cell populations. As noted above, we observed
a slight but significant decrease in proliferation of G-1-
treated cultures (Fig. 5). Additionally, we noted a small
but significant increase in expression of the apoptotic/
cell death marker Annexin V in G-1-treated cultures
(see Supplementary material, Fig. S2). Either of these
effects may be contributing to a decrease in cell number
in G-1-treated cultures. Further studies will be required
to determine whether these findings reflect the direct
effects of G-1 on cellular proliferation and viability, or
are secondary to the induction of regulatory T-cell
populations.
Systemic delivery of G-1 drove IL-10 production from
splenocytes following T-cell activation in culture. It is
notable that this effect does not require overt in vivo anti-
gen recognition. This result may reflect that G-1-mediated
signalling in naive T cells leads to an alteration in their
resting state, perhaps through transcriptional mechanisms.
Another possibility is that there is carryover of G-1 dur-
ing purification of splenocytes before culture, where anti-
gen presentation is mimicked using stimulatory
antibodies, or that the effects are the result of the low lev-
els of T-cell activation inherent in naive mice. Along
those lines, we have consistently found a small population
of memory cells within the spleen of untreated mice, sug-
gesting low levels of immune activation in ‘naive’ animals
(data not shown). It is also possible that pre-existing
memory T cells are responsible for G-1’s effect in this set-
ting, as G-1 can drive IL-10 secretion from this popula-
tion (unpublished observation). In agreement with our
observations from cultured T cells (Fig. 2), systemic
administration of G-1 had no effect on IL-6 or TNF-asecretion. Conversely, we did detect increased secretion of
IL-17A following in vivo treatment with G-1, while also
observing a decrease in the production of IFN-c. These
differences from results with purified T-cell cultures may
reflect the effects of G-1 on other immune populations
following in vivo treatment. Such populations may also be
contributing to the observed IL-10 secretion, directly or
indirectly. Another possibility includes G-1-mediated
IL-10 production during the week-long injections of G-1,
leading to inactivation of splenic APCs and a decrease in
the secretion of Th1-polarizing cytokines like IL-12, and
hence to lower IFN-c production.
Th17 cells are localized in high numbers to sites of
autoimmune inflammation. Our data suggest that it may
be possible to induce IL-10 in situ where large numbers
of Th17 cells persist, through systemic treatment with
G-1. The feasibility of this therapeutic approach is suggested
by experiments in which IL-10+ Th17 cells differentiated
with TGF-b and IL-6 alone inhibited the development of
EAE following adoptive transfer of neuropeptide-reactive
Th17 cells.19 This effect was dependent on IL-10 produc-
tion19 and suggests that such cells can inhibit fully differ-
entiated pathogenic T-cell populations through the
secretion of IL-10 in situ, as would likely be required in
the case of a viable therapeutic intervention based on the
results of our study. While our finding that systemic G-1
could increase IL-17A secretion from murine splenocytes
warrants further attention, it must be noted that IL-17A
has been shown to exhibit immunosuppressive properties
in several settings, including in the development of ath-
erosclerosis43–45 and the induction of T-cell-mediated
colitis.46 Moreover, the IL-10+ Th17 cells discussed above
that were shown to exhibit bystander suppressive effects
in EAE also produced high levels of IL-17A.19 Conse-
quently, the induction of IL-17A is reconcilable with its
(a) DMSO G-1
NS1·0
0·8
0·6
0·4
0·2
0·0% Proliferating ∝ 1/(MFI)
IL-1
0
15·8
64·9 18·7
0·61
DMSO
G-1
20·2 3·37
52·7 23·7
eFluor670
eFluor670
(b) 104
104
103
103
102
102
101
101
100
IL-1
0
104
103
102
101
100
100
104
103
10210
110
0
A.U
.
**
Figure 5. G-1 effects are not dependent on proliferation.
CD4+ CD62Lhi naive CD4+ T cells were collected by FACS and
stained with the proliferation dye eFluor670 before culture. Follow-
ing differentiation for 4 days ex vivo in culture with anti-CD3/
28 + interleukin-6 (IL-6) + transforming growth factor-b (TGF-b)
in the presence of 100 nm G-1 (black bars) or DMSO (white bars),
cells were stained for intracellular IL-10. (a) Percentage of cells
proliferating and the inverse of geometric mean fluorescence inten-
sity (GMFI), a measure of total proliferation. (b) Sample plots show-
ing IL-10 expression and eFluor670 staining. The upper right
quadrant shows cells expressing IL-10 without evidence of prolifera-
tion. Data show one of two independent experiments. P-values
determined by Student’s t-test; **P < 0�02. Error bars = SD; NS, not
significant, A.U., arbitrary units.
� 2011 The Authors. Immunology � 2011 Blackwell Publishing Ltd, Immunology, 134, 93–106 101
IL-10 induction by GPER agonist G-1

ability to attenuate EAE, despite the established impor-
tance of Th17 cells to EAE induction,3,47,48 and the fact
that systemic neutralization of IL-17A/F attenuates clinical
symptoms in this model.49 However, there is also clear
evidence that IL-17A can contribute to pathogenic
inflammation.5 Future studies aimed at determining the
context in which G-1 or any related compounds elicit
critical Th17 factors like IL-17A/F, IL-21, IL-22, IL-23
and the aryl hydrocarbon receptor will be critical to
determining the setting(s) in which G-1 has therapeutic
potential.
The observation of G-1-induced IL-17A secretion may
offer some insight into autoimmune pathophysiology.
There is a longstanding debate about how the apparent
immunosuppressive activities of E2 can be reconciled with
the higher prevalence of autoimmune disease in women.
It is possible that E2-mediated activation of GPER may
drive increased IL-17A production under specific circum-
stances, and that this contributes to augmented autoim-
mune pathogenesis in women. Future studies aimed at
investigating this possibility should be directed at delin-
eating the specific conditions in which GPER activation
leads to IL-17A, and perhaps IL-17F, production. It
would be interesting to correlate these findings with stud-
ies investigating the expression of ERa, ERb and GPER,
which may vary over time. An explanation for the sexual
dimorphism in the prevalence of autoimmune disease
may reside in identifying a setting where GPER plays a
predominant role in estrogen signalling, perhaps as the
result of down-regulation of ERa and ERb within specific
cell populations, under conditions where GPER activation
leads to production of IL-17A or even IL-17F.
If these properties can be definitively described, there is
also the possibility that G-1 may serve a role in T-cell-
Isotype control “Hybrid” T cells
RORgtRORgt
IL-10+IL-17A–
Th17 cells
DMSO
G-1
Rel
ativ
e %
IL-1
0+IL
-17A
–
Rel
ativ
e %
IL-1
0+IL
-17A
–
Rel
ativ
e %
IL-1
0+IL
-17A
–
Rel
ativ
e %
IL-1
0–IL
-17A
+
Rel
ativ
e %
IL-1
0–IL
-17A
+
Rel
ativ
e %
IL-1
0+IL
-17A
+
Rel
ativ
e %
IL-1
0+IL
-17A
+
Total pop Th17 cells Hybrid T cells
NSNS
Th17 cellsTotal popTh17 cellsTotal pop
DMSO G-1 DMSO G-1
0 0 0 0
1 1 1 1
2 2 2 2
3 *** ***
0
1
2
3
0
1
2
3
0
1
2
3
3 33
IL-10+IL-17A+IL-10–IL-17A+
0·027
0·65
Fox
p3
Fox
p3
****** *
(a)
(b)
(c) (d)
Figure 6. G-1 induces interleukin-10 (IL-10) production within the hybrid T-cell population. CD4+ CD62Lhi naive CD4+ T cells were collected
by FACS from Foxp3egfp mice and cultured for 4 days ex vivo with anti-CD3/28 + IL-6 + transforming growth factor-b (TGF-b) in the presence
of 100 nm G-1 (black bars) or DMSO (white bars). Cells were collected and stained for intracelluar IL-10, IL-17A, and RORct, and analysed by
flow cytometry. Cells that were Foxp3+ RORct+ were designated as hybrid T cells, whereas those that were Foxp3) RORct+ were designated as T
helper type 17 (Th17) cells. (a) Gating logic to determine hybrid T-cell and Th17 populations. (b–d) Graphs represent summary of data from
three independent experiments showing the relative percentage of (b) IL-10+ IL-17A), (c) IL-10+ IL-17A+, and (d) IL-10) IL-17A+ populations.
P-values determined by Student’s t-test; *P < 0�05; **P < 0�005; ***P < 0�0005. Error bars = SEM; NS, not significant.
102 � 2011 The Authors. Immunology � 2011 Blackwell Publishing Ltd, Immunology, 134, 93–106
R. L. Brunsing and E. R. Prossnitz

based tumour vaccine strategies. Evidence suggests that
polarization of tumour-specific T-cells towards a Th17
phenotype before adoptive transfer can enhance tumour
eradication.50 G-1 or a related compound may serve as a
cost-effective and safe alternative to recombinant cyto-
kines during T-cell culture, or even as a systemic adjuvant
treatment to help stabilize the cells following adoptive
transfer, especially given the fact that we observed
increased IL-17A production following in vivo G-1 treat-
ments. Moreover, further delineating the role of GPER in
polarization along the Treg–Th17 axis, may uncover other
pharmacological approaches, such as the use of G15, that
can elicit anti-tumour responses by driving conversion of
Treg cells into Th17 populations. This strategy was vali-
dated in principle through the use of indoleamine 2,3-
dioxygenase inhibitors in the B16 melanoma model.33
Our findings also suggest that GPER-mediated induc-
tion of IL-10 may play a role in estrogen’s ability to
suppress autoimmune diseases. Two previous reports
demonstrated that G-1 can suppress EAE.38,39 In one
study, the authors found that G-1’s protective effects cor-
related with increased programmed death-1 (PD-1)
expression on Foxp3+ Treg cells, and were dependent on
PD-1 expression as PD-1 knockout mice were not pro-
tected from disease by G-1.38 Notably, the authors also
observed increased IL-10 production from G-1-treated
splenocytes collected from diseased animals compared
with placebo controls, an effect lost in the PD-1 knockout
mice.38 This correlates well with our results in Fig. 7, as
we observed increased IL-10 production from splenocytes
of G-1-treated mice. Notably, IL-10 production in CD4+ T
cells can inhibit the development of EAE,18 a disease
whose pathogenesis is dependent on RORct expression.3
The fact that we demonstrated G-1 leads to an increase in
IL-10 within RORct+ cells, and that IL-10 induction
occurs even in the presence of IL-23, leads to the hypothe-
sis that G-1 suppressed EAE through the induction of
IL-10 production from RORct+ cells specifically within the
600
4
IL-1
7A (
ng/m
l)
IL-1
0 (p
g/m
l)IL
-6 (
pg/m
l)IF
N-γ
(ng
/ml)
TN
F-α
(pg
/ml)
3
2
1
0
400
400
300
200
100
500
200
0
0
0
0
20
40
60
80 Vehicle (n = 3)
G-1 (n = 5)Vehicle (n = 3)G-1 (n = 5)
Vehicle (n = 3)G-1 (n = 5)
Vehicle (n = 3)G-1 (n = 5)
Vehicle (n = 3)
***
*
*
G-1 (n = 5)
10
2-way ANOVA: P = 0·05
2-way ANOVA: P = 0·05
2-way ANOVA: P = 0·09
20
30
24
24
48
48
Time in culture (hr) Time in culture (hr)
Time in culture (hr)Time in culture (hr)
Time in culture (hr)
72 24 48 72
24 48 7272
24 48 72
(a)(b)
(d)(c)
(e)
Figure 7. Cytokine secretion following in vivo treatment with G-1. Seven- to eleven-week-old male wild-type C57BL/6 mice were injected with G-1
(5 lg/day) or vehicle for 7 consecutive days. One day following the last injection, splenocytes were collected and cultured in the presence of anti-
CD3e (1�0 lg/ml) and anti-CD28 (2�5 lg/ml) antibody. Culture medium was collected after 24, 48, and 72 hr and analysed for the presence of
secreted (a) IL-10, (b) IL-17A, (c) interferon-c (IFN-c), (d) tumour necrosis factor-a (TNF-a) and (e) interleukin-6 (IL-6) by Luminex multiplex
assay. Graphs are mean data with three to five mice per group. P-values comparing G-1 and vehicle at a given time-point were determined by Stu-
dent’s t-test; *P � 0�05, **P � 0�01. P-values comparing G-1 and vehicle curves were determined by two-way anova as indicated. Error bars = SEM.
� 2011 The Authors. Immunology � 2011 Blackwell Publishing Ltd, Immunology, 134, 93–106 103
IL-10 induction by GPER agonist G-1

central nervous system via a PD-1-dependent mechanism.
It has also been recently shown that estrogen can protect
mice from EAE in a Foxp3-indpendent manner.51 Again
an increase in IL-10 was noted, though it is not known
what cells were responsible for this effect. Additionally,
other studies have shown that: (i) E2 can increase IL-10
production in vivo in a GPER-dependent manner,36 and
(ii) the in vitro suppressive activity of Treg cells from PD-
1 knockout mice was enhanced following in vivo treatment
with E2, without changing the expression levels of Foxp3.52
One hypothesis to explain these results may be that E2 sig-
nalling through classical estrogen receptors substitutes for
PD-1-mediated signalling in the induction of IL-10 from
effector populations when E2 is used in lieu of G-1. Fur-
ther studies using conditional knockouts of IL-10 within
the CD4+ compartment, and analysis of GPER, ERa, and
ERb signalling in Foxp3+ and Foxp3) populations, includ-
ing the specific requirement of PD-1 expression, will be
needed to definitively address these questions.
G-1 has been characterized as a selective agonist for the
G protein-coupled estrogen receptor GPER,53 a recently
identified non-classical member of the estrogen receptor
family.54 Consistent with this mechanism of action, G-1-
mediated IL-10 expression was inhibited by the addition
of the GPER-directed antagonist G15.40 Our results are
also supported by observations that G-1-mediated inhibi-
tion of EAE is dependent on GPER expression.38
Although small molecules can be subject to off-target
activity, it is unlikely that both G-1 and G15 would exhi-
bit off-target profiles that mimic their established activi-
ties towards GPER. Nevertheless, further investigation
into the G-1 target(s) in T cells is warranted. Although
GPER)/) mice exist, they exhibit higher levels of apopto-
sis in double-negative thymocytes and lack the E2-medi-
ated increase in this population following systemic
estrogen administration.55 Therefore studies aimed at ver-
ifying GPER as the target of G-1 within the T-cell popula-
tion will need to employ inducible knockout strategies or
retroviral RNAi targeting of GPER to avoid the con-
founding effects of aberrant thymic T-cell development
observed in GPER)/) mice.
Our results have begun to elucidate the mechanisms by
which G-1 induces IL-10 expression and production. Addi-
tion of the MEK1 inhibitor PD98059 blocked G-1-medi-
ated IL-10 induction, whereas addition of inhibitors of the
p38 and JNK pathways was without effect. These findings
are consistent with reports that ERK signalling is necessary
for the induction of IL-10 in Th1 and Th2 cells, and con-
tributes to IL-10 expression in Th17 populations, with no
detectable difference when p38 signalling is blocked.13 Why
addition of PD98059 led to a mild increase in the number
of IL-10+ cells within control (DMSO) cultures is unclear
(Fig. 4b). This stands in contrast to the previous reports
discussed above,12,13 yet we consistently observed this
effect. Interestingly, in the work by Saraiva et al.13 blockade
of ERK signalling only led to a partial inhibition of IL-10
induction from Th17 cultures. This suggests there are two
pathways of IL-10 induction in Th17 cells, the ‘ERK-
dependent pathway’ described above, and an alternative
pathway. One hypothesis to explain the discrepancy
between our findings and previous reports would be that
this alternative pathway: (i) is inhibited by ERK signalling
(an ‘ERK-sensitive pathway’), and (ii) is the predominant
pathway for IL-10 induction in culture conditions using
charcoal-stripped FBS in lieu of normal FBS, as we have
done here. Given that ERK signalling is implicated in IL-10
expression within Th1 and Th2 cells, it will be interesting
to determine whether G-1 can drive IL-10 production
under Th1- or Th2-polarizing conditions. The lack of IL-
10 expression in unpolarized (Th0) cells is not unexpected.
Interleukin-10 production in Th populations requires
STAT activation via IL-4, IL-6, IL-12, IL-21 and IL-27.18,20
However, these cytokines are produced by APCs and dif-
ferentiated T-cell populations and are likely to be in lim-
ited supply in the pure cultures of naive T cells that we
employed. We observed that G-1 was unable to induce IL-
10 production in differentiating naive T cells without the
addition of both TGF-b and IL-6 to the culture medium,
suggesting that G-1 cannot replace any of the critical sig-
nals necessary to induce IL-10 in Th17 cells. It appears that
the function of TGF-b in Th17 development is to block the
differentiation of Th1 and Th2 cells.56 Hence our observa-
tion that G-1 treatment with IL-6 alone does not consis-
tently elicit IL-10 production despite detectable levels of
IL-10+ cells perhaps reflects a dependence on Th17 differ-
entiation. Future studies will need to address this question.
Finally, the IL-10+ IL-17A+ cells we identified appear to
be part of the autoregulatory pathway,21 as they express
RORct but not Foxp3. In fact we detected virtually no IL-
17A+ cells within the Foxp3+ population. While not com-
pletely unexpected, because Foxp3 can inhibit some of
the transcriptional activity of RORct,57 Foxp3+ IL-17A+
cells have been previously reported.58 Our observation
that G-1 induces IL-10 expression in Foxp3+ RORct+
hybrid T cells suggests that, in addition to generating IL-
10 production in populations already localized at the site
of inflammation, G-1 may also enhance the suppressive
function of Treg populations drawn in from the circula-
tion. Such a response would not be unprecedented as
T-bet-induced CXCR3 expression in Foxp3+ cells has
been shown to play a role in targeting Treg cells to sites
of Th1-type inflammation.59 If IL-10 can be stably
induced in hybrid T-cell populations following in vivo G-
1 treatment, their suppressive activity may be enhanced as
they are recruited to sites of ongoing inflammation.
Numerous attempts have been made to harness the
immunosuppressive properties of IL-10 for therapeutic
benefit, many of which have been based on the use of
biologics.25 To our knowledge, this is the first evidence that
a synthetic small molecule can shift the balance along the
104 � 2011 The Authors. Immunology � 2011 Blackwell Publishing Ltd, Immunology, 134, 93–106
R. L. Brunsing and E. R. Prossnitz

Treg–Th17 axis in favour of IL-10 production, in this case
by acting directly on T-cell populations. These data build on
previous results demonstrating that dexamethasone and
retinoic acid can elicit IL-10 from polyclonally stimulated
naive T cells when IL-4, IL-12 and IFN are neutralized.60
Also worth noting is the fact that it is becoming increas-
ingly clear that GPER probably plays a smaller role in the
majority of classical estrogen responses, such as uterine
imbibition, as compared with its better known counter-
part ERa.40 Hence G-1 may be associated with a more
tolerable adverse effect profile.
Our findings suggest that the membrane-permeable
small molecule G-1 may serve as a novel T-cell-targeted
immunosuppressive agent in settings where large popula-
tions of Th17 cells exist, for example in rheumatoid
arthritis, inflammatory bowel disease, or psoriasis. G-1
may also prove useful for in vitro generation of IL-10-
producing cells for adoptive immunotherapy. Future
studies delineating the specific signalling mechanisms and
targets of G-1 and other related compounds will be semi-
nal to the continued development of this new class of
immunoregulatory estrogenic small molecules. The selec-
tivity of G-139,53 and its attractive pharmacological prop-
erties38 make this compound a strong candidate for
pharmaceutical development, paving the way for the
development of novel T-cell targeted immunotherapeu-
tics.
Acknowledgements
This work was supported by National Institutes of Health
grants R01 CA116662, CA118743 and CA127731 (E.R.P.).
Data were generated in the Flow Cytometry Shared
Resource Center supported by the University of New
Mexico Health Sciences Center and the University of New
Mexico Cancer Center. The authors would like to thank
Drs Rick Lyons and Mary Lipscomb for insightful discus-
sions, Kristin Owens and Lori Deihl for their technical
assistance, and Dr Helen Hathaway for her expertise in
the care and use of mice.
Authorship contributions and disclosure ofconflict of interest
R.L.B. and E.R.P. designed and interpreted experiments
and wrote the manuscript. R.L.B. carried out experiments
and E.R.P. holds a U.S. patent on G-1 and G15.
References
1 Mosmann TR, Coffman RL. TH1 and TH2 cells: different patterns of lymphokine
secretion lead to different functional properties. Annu Rev Immunol 1989; 7:145–73.
2 Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, Weaver
CT. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from
the T helper type 1 and 2 lineages. Nat Immunol 2005; 6:1123–32.
3 Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, Littman
DR. The orphan nuclear receptor RORct directs the differentiation program of proin-
flammatory IL-17+ T helper cells. Cell 2006; 126:1121–33.
4 Wei L, Laurence A, Elias KM, O’Shea JJ. IL-21 is produced by Th17 cells and drives
IL-17 production in a STAT3-dependent manner. J Biol Chem 2007; 282:34605–10.
5 Torchinsky MB, Blander JM. T helper 17 cells: discovery, function, and physiological
trigger. Cell Mol Life Sci 2010; 67:1407–21.
6 McGeachy MJ, Chen Y, Tato CM et al. The interleukin 23 receptor is essential for the
terminal differentiation of interleukin 17-producing effector T helper cells in vivo. Nat
Immunol 2009; 10:314–24.
7 Vignali DA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev 2008;
8:523–32.
8 Patey-Mariaud de Serre N, Canioni D, Ganousse S, Rieux-Laucat F, Goulet O, Ruemm-
ele F, Brousse N. Digestive histopathological presentation of IPEX syndrome. Mod
Pathol 2009; 22:95–102.
9 Clark LB, Appleby MW, Brunkow ME, Wilkinson JE, Ziegler SF, Ramsdell F. Cellular
and molecular characterization of the scurfy mouse mutant. J Immunol 1999; 162:2546–
54.
10 Kuhn R, Lohler J, Rennick D, Rajewsky K, Muller W. Interleukin-10-deficient mice
develop chronic enterocolitis. Cell 1993; 75:263–74.
11 Glocker EO, Kotlarz D, Boztug K et al. Inflammatory bowel disease and mutations
affecting the interleukin-10 receptor. N Eng J Med 2009; 361:2033–45.
12 Saraiva M, O’Garra A. The regulation of IL-10 production by immune cells. Nat Rev
2010; 10:170–81.
13 Saraiva M, Christensen JR, Veldhoen M, Murphy TL, Murphy KM, O’Garra A. Inter-
leukin-10 production by Th1 cells requires interleukin-12-induced STAT4 transcription
factor and ERK MAP kinase activation by high antigen dose. Immunity 2009; 31:209–
19.
14 Del Prete G, De Carli M, Almerigogna F, Giudizi MG, Biagiotti R, Romagnani S.
Human IL-10 is produced by both type 1 helper (Th1) and type 2 helper (Th2) T cell
clones and inhibits their antigen-specific proliferation and cytokine production.
J Immunol 1993; 150:353–60.
15 Meyaard L, Hovenkamp E, Otto SA, Miedema F. IL-12-induced IL-10 production by
human T cells as a negative feedback for IL-12-induced immune responses. J Immunol
1996; 156:2776–82.
16 Jankovic D, Kullberg MC, Feng CG et al. Conventional T-bet+Foxp3– Th1 cells are the
major source of host-protective regulatory IL-10 during intracellular protozoan infec-
tion. J Exp Med 2007; 204:273–83.
17 Fiorentino DF, Bond MW, Mosmann TR. Two types of mouse T helper cell. IV. Th2
clones secrete a factor that inhibits cytokine production by Th1 clones. J Exp Med
1989; 170:2081–95.
18 Fitzgerald DC, Zhang GX, El-Behi M et al. Suppression of autoimmune inflammation
of the central nervous system by interleukin 10 secreted by interleukin 27-stimulated T
cells. Nat Immunol 2007; 8:1372–9.
19 McGeachy MJ, Bak-Jensen KS, Chen Y, Tato CM, Blumenschein W, McClanahan T,
Cua DJ. TGF-beta and IL-6 drive the production of IL-17 and IL-10 by T cells and
restrain TH-17 cell-mediated pathology. Nat Immunol 2007; 8:1390–7.
20 Stumhofer JS, Silver JS, Laurence A et al. Interleukins 27 and 6 induce STAT3-medi-
ated T cell production of interleukin 10. Nat Immunol 2007; 8:1363–71.
21 Maynard CL, Weaver CT. Diversity in the contribution of interleukin-10 to T-cell-med-
iated immune regulation. Immunol Rev 2008; 226:219–33.
22 Battaglia M, Gregori S, Bacchetta R, Roncarolo MG. Tr1 cells: from discovery to their
clinical application. Semin Immunol 2006; 18:120–7.
23 Roncarolo MG, Gregori S, Battaglia M, Bacchetta R, Fleischhauer K, Levings MK. Inter-
leukin-10-secreting type 1 regulatory T cells in rodents and humans. Immunol Rev
2006; 212:28–50.
24 Maynard CL, Harrington LE, Janowski KM, Oliver JR, Zindl CL, Rudensky AY, Weaver
CT. Regulatory T cells expressing interleukin 10 develop from Foxp3+ and Foxp3) pre-
cursor cells in the absence of interleukin 10. Nat Immunol 2007; 8:931–41.
25 O’Garra A, Barrat FJ, Castro AG, Vicari A, Hawrylowicz C. Strategies for use of IL-10
or its antagonists in human disease. Immunol Rev 2008; 223:114–31.
26 Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK.
Reciprocal developmental pathways for the generation of pathogenic effector TH17 and
regulatory T cells. Nature 2006; 441:235–8.
27 Bluestone JA, Mackay CR, O’Shea JJ, Stockinger B. The functional plasticity of T cell
subsets. Nat Rev 2009; 9:811–6.
28 Lee YK, Mukasa R, Hatton RD, Weaver CT. Developmental plasticity of Th17 and Treg
cells. Curr Opin Immunol 2009; 21:274–80.
29 Barnes MJ, Powrie F. Hybrid Treg cells: steel frames and plastic exteriors. Nat Immunol
2009; 10:563–4.
30 Atarashi K, Nishimura J, Shima T et al. ATP drives lamina propria TH17 cell differenti-
ation. Nature 2008; 455:808–12.
� 2011 The Authors. Immunology � 2011 Blackwell Publishing Ltd, Immunology, 134, 93–106 105
IL-10 induction by GPER agonist G-1

31 Liao JJ, Huang MC, Goetzl EJ. Cutting edge: alternative signaling of Th17 cell develop-
ment by sphingosine 1-phosphate. J Immunol 2007; 178:5425–8.
32 Colin EM, Asmawidjaja PS, van Hamburg JP, Mus AM, van Driel M, Hazes JM, van
Leeuwen JP, Lubberts E. 1,25-Dihydroxyvitamin D3 modulates Th17 polarization and
interleukin-22 expression by memory T cells from patients with early rheumatoid
arthritis. Arthritis Rheum 2010; 62:132–42.
33 Sharma MD, Hou DY, Liu Y et al. Indoleamine 2,3-dioxygenase controls conversion of
Foxp3+ Tregs to TH17-like cells in tumor-draining lymph nodes. Blood 2009;
113:6102–11.
34 Mucida D, Park Y, Kim G, Turovskaya O, Scott I, Kronenberg M, Cheroutre H. Reci-
procal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science
2007; 317:256–60.
35 Polanczyk MJ, Hopke C, Huan J, Vandenbark AA, Offner H. Enhanced FoxP3 expres-
sion and Treg cell function in pregnant and estrogen-treated mice. J Neuroimmunol
2005; 170:85–92.
36 Yates MA, Li Y, Chlebeck PJ, Offner H. GPR30, but not estrogen receptor-alpha, is
crucial in the treatment of experimental autoimmune encephalomyelitis by oral ethinyl
estradiol. BMC Immunol 2010; 11:20.
37 Confavreux C, Hutchinson M, Hours MM, Cortinovis-Tourniaire P, Moreau T. Rate of
pregnancy-related relapse in multiple sclerosis. Pregnancy in Multiple Sclerosis Group.
N Eng J Med 1998; 339:285–91.
38 Wang C, Dehghani B, Li Y, Kaler LJ, Proctor T, Vandenbark AA, Offner H. Membrane
estrogen receptor regulates experimental autoimmune encephalomyelitis through
up-regulation of programmed death 1. J Immunol 2009; 182:3294–303.
39 Blasko E, Haskell CA, Leung S et al. Beneficial role of the GPR30 agonist G-1 in an
animal model of multiple sclerosis. J Neuroimmunol 2009; 214:67–77.
40 Dennis MK, Burai R, Ramesh C et al. In vivo effects of a GPR30 antagonist. Nat Chem
Biol 2009; 5:421–7.
41 Haribhai D, Lin W, Relland LM, Truong N, Williams CB, Chatila TA. Regulatory T
cells dynamically control the primary immune response to foreign antigen. J Immunol
2007; 178:2961–72.
42 Filardo EJ, Quinn JA, Bland KI, Frackelton AR Jr. Estrogen-induced activation of Erk-1
and Erk-2 requires the G protein-coupled receptor homolog, GPR30, and occurs via
trans-activation of the epidermal growth factor receptor through release of HB-EGF.
Mol Endocrinol 2000; 14:1649–60.
43 Ait-Oufella H, Herbin O, Bouaziz JD et al. B cell depletion reduces the development of
atherosclerosis in mice. J Exp Med 2010; 207:1579–87.
44 Taleb S, Romain M, Ramkhelawon B et al. Loss of SOCS3 expression in T cells
reveals a regulatory role for interleukin-17 in atherosclerosis. J Exp Med 2009;
206:2067–77.
45 Taleb S, Tedgui A, Mallat Z. Interleukin-17: friend or foe in atherosclerosis? Curr Opin
Lipidol 2010; 21:404–8.
46 O’Connor W Jr, Kamanaka M, Booth CJ, Town T, Nakae S, Iwakura Y, Kolls JK, Flav-
ell RA. A protective function for interleukin 17A in T cell-mediated intestinal inflam-
mation. Nat Immunol 2009; 10:603–9.
47 Cua DJ, Sherlock J, Chen Y et al. Interleukin-23 rather than interleukin-12 is the criti-
cal cytokine for autoimmune inflammation of the brain. Nature 2003; 421:744–8.
48 Langrish CL, Chen Y, Blumenschein WM et al. IL-23 drives a pathogenic T cell popu-
lation that induces autoimmune inflammation. J Exp Med 2005; 201:233–40.
49 Hofstetter HH, Ibrahim SM, Koczan D, Kruse N, Weishaupt A, Toyka KV, Gold R.
Therapeutic efficacy of IL-17 neutralization in murine experimental autoimmune
encephalomyelitis. Cell Immunol 2005; 237:123–30.
50 Muranski P, Boni A, Antony PA et al. Tumor-specific Th17-polarized cells eradicate
large established melanoma. Blood 2008; 112:362–73.
51 Subramanian S, Yates M, Vandenbark AA, Offner H. Oestrogen-mediated protection of
experimental autoimmune encephalomyelitis in the absence of Foxp3+ regulatory T
cells implicates compensatory pathways including regulatory B cells. Immunology 2011;
132:340–7.
52 Polanczyk MJ, Hopke C, Vandenbark AA, Offner H. Treg suppressive activity involves
estrogen-dependent expression of programmed death-1 (PD-1). Int Immunol 2007;
19:337–43.
53 Bologa CG, Revankar CM, Young SM et al. Virtual and biomolecular screening con-
verge on a selective agonist for GPR30. Nat Chem Biol 2006; 2:207–12.
54 Prossnitz ER, Arterburn JB, Smith HO, Oprea TI, Sklar LA, Hathaway HJ. Estrogen
signaling through the transmembrane G protein-coupled receptor GPR30. Annu Rev
Physiol 2008; 70:165–90.
55 Wang C, Dehghani B, Magrisso IJ et al. GPR30 contributes to estrogen-induced thymic
atrophy. Mol Endocrinol 2008; 22:636–48.
56 Das J, Ren G, Zhang L et al. Transforming growth factor beta is dispensable for the
molecular orchestration of Th17 cell differentiation. J Exp Med 2009; 206:2407–16.
57 Zhou L, Lopes JE, Chong MM et al. TGF-beta-induced Foxp3 inhibits TH17 cell differ-
entiation by antagonizing RORct function. Nature 2008; 453:236–40.
58 Beriou G, Costantino CM, Ashley CW, Yang L, Kuchroo VK, Baecher-Allan C, Hafler
DA. IL-17-producing human peripheral regulatory T cells retain suppressive function.
Blood 2009; 113:4240–9.
59 Koch MA, Tucker-Heard G, Perdue NR, Killebrew JR, Urdahl KB, Campbell DJ. The
transcription factor T-bet controls regulatory T cell homeostasis and function during
type 1 inflammation. Nat Immunol 2009; 10:595–602.
60 Spolski R, Kim HP, Zhu W, Levy DE, Leonard WJ. IL-21 mediates suppressive effects
via its induction of IL-10. J Immunol 2009; 182:2859–67.
Supporting Information
Additional Supporting Information may be found in the
online version of this article:
Figure S1. G-1 does not alter IFNc expression.
Figure S2. G-1 increases Annexin V expression.
Please note: Wiley-Blackwell are not responsible for the
content or functionality of any supporting materials sup-
plied by the authors. Any queries (other than about miss-
ing material) should be directed to the corresponding
author for the article.
106 � 2011 The Authors. Immunology � 2011 Blackwell Publishing Ltd, Immunology, 134, 93–106
R. L. Brunsing and E. R. Prossnitz