
Copper-catalyzed transformations of carboradicals generated ...
Upload
khangminh22Category
view
2download
0
Indoles and Indolines – Palladium-Catalyzed Synthesis and Functionalizations
by Aromatization and Dearomatization
by
Nicolas Zeidan
A thesis submitted in conformity with the requirements for the degree of Doctor of Philosophy
Department of Chemistry University of Toronto
© Copyright by Nicolas Zeidan 2020

ii
Indoles and Indolines –
Palladium-Catalyzed Synthesis and Functionalizations
by Aromatization and Dearomatization
Nicolas Zeidan
Doctor of Philosophy
Department of Chemistry
University of Toronto
2020
Abstract
The Lautens group has developed many late transition-metal catalyzed protocols for the synthesis
of various heterocycles. The focus of this work is palladium-catalysis for the synthesis and
functionalization of indoles and indolines. This thesis is divided into three chapters concerning the
synthesis of indoles using the Fang-Lautens indole synthesis, the dearomatization of indole by a
migratory insertion strategy, and an electrophilic dearomatization of indoles with N-
fluorobenzenesulfonimide.
Chapter 1 describes the synthesis of 2-cyanoindoles using the Fang-Lautens indole synthesis. The
reaction utilizes vinyl gem-dibromo anilines to access diverse free- and N-capped 2-cyanoindoles
in good yield. An investigation of the mechanism and a one-pot procedure to access the 2-tetrazole
containing indole is presented.
Chapter 2 focuses on the palladium-catalyzed dearomatization of indoles by migratory insertion
to access chiral 2,3-functionalized indolines in good to excellent yields. Two research goals are
presented. First, a protocol for a diastereoselective arylation/direct arylation of indoles is
described, producing complex indolines from benzoyl-indoles and activated C–H bond containing

iii
aryl and heteroaryl compounds. The role of a copper additive is explored in preserving dr in the
products. Secondly, an asymmetric arylation/borylation of indoles is presented. The synthesis of
privileged 3,3’-diaryl phosphoramidite ligands as well as a preactivated mixed-boron reagent
containing an sp2-“sp3” bond is described.
Chapter 3 focuses on an electrophilic dearomatization of indoles using an electrophilic source of
fluoride in a “push-pull mechanism”. The scalable reaction produces activated indolines bearing
an exocyclic double bond on the 2-position. The amphoteric nature of these indolines is explored
and various palladium, rhodium, and copper reactions are described for the formation of new C–
H, C–B, C–C (sp3 and sp2), C–N, C–O, C–P, and C–S bonds.

iv
Acknowledgments
When I started graduate school in 2015, I did not expect to make so many great friends and
connections. Now that I have finished, I can truly say that this degree would not have been possible
without the support and guidance of so many great mentors, friends, colleagues, and comrades.
Mark Lautens, thank you for your thoughtful guidance over the years. I really appreciate that you
accepted me into your group when I came with a minimal organic chemistry background. The
more I progressed and learnt, the more I appreciated just how vast your knowledge in the field is.
I hope one day I can be as meticulous with my chemistry and management as you. To my
supervisor in Germany, Armido Studer, thank you for letting me join your group. I appreciated
expanding my knowledge in a different research pursuit. Mark Taylor, I really appreciated all the
thoughtful questions and comments at our annual meetings. Sophie Rousseaux, j’aimerais
sincèrement vous remercier pour tout votre appui et vos conseils. Merci pour m’avoir encourager
à poursuivre les études supérieures. J’apprécie infiniment que tu étais toujours disponible pour
converser durant mes années comme étudiant et je vais chérir nos discussions intellectuelles
ensemble.
To my lab colleagues, there are just too many to thank. If your name is not mentioned, I hope you
know that there are just too many memories to recall on a single page. Thank you to the original
mentors in the group. Dave Petrone, thank you for yelling just enough. Christine Le, thank you for
the Boss way you got us into the Bruker party. Zafar Qureshi, thank you for the great snowboarding
memories. Hyung Yoon, I could always count on getting your honest advice. Alvin Jang, thank
you for the friendship amidst the chemistry. Andy Yen, thank you for taking the bullet on so many
things, seeing as though we were so close in time. Thank you Heather Lam and Andrew Whyte
for your fierce and friendly competition and motivation. There is something special about being in
the same year as someone, and I will always cherish that. Thank you to all the students afterwards
for being excellent and continuing the Lautens ways. I will never forget our camping trips,
snowboarding trips, and interesting spring formals.
Many thanks to all the students whom I worked with over the years, there are just too many to
name. I am very grateful to have met so many international students and look forward to visiting

v
the Europeans when I make it across the pond. Christian Breuers, thanks for letting me crash on
your couch. Randy Sanichar, thanks for the great fishing and rum. Christian Dank, zaufst du dich
heute weider so an? Veil ich mochte. Tamara Beisel, for encouraging me to do so much, I hope
one day I can be as good of a chemist (and person) as you.
Thank you to my mother Katia Zeidan, and Father Joseph Zeidan for all your support over the
years. Thank you for all the late-night drives back home. Thank you to all my siblings (all of them,
Salam), for the late-night drinks and shiii. Finally, thank you to the boneyard boys, and sometimes
the gulag girls for all the great drops and George Dubyas.
Last and most importantly, to my partner, Milena Aitken, we made it :) Thank you very much for
your patience, every day, and every night. Being in school at the same time was not ideal but it
was fun. I am so proud that you are now a successful PA. I look forward to getting married,
spending time in Europe with you, and the rest of our lives together. Thank you for always pushing
me to do my best and holding down the fort.

vi
Table of Contents
Acknowledgments.......................................................................................................................... iv
Table of Contents ........................................................................................................................... vi
List of Tables ................................................................................................................................. ix
List of Schemes ................................................................................................................................x
List of Appendices ....................................................................................................................... xvi
List of Publications ..................................................................................................................... xvii
Abbreviations ............................................................................................................................. xviii
Chapter 1 ........................................................................................................................................1
Palladium-Catalyzed Synthesis of 2-Substituted Indoles from Vinyl gem-Dibromo
Containing Anilines ....................................................................................................................1
1.1 Introduction ..........................................................................................................................1
1.1.1 Ubiquity of Indoles ..................................................................................................1
1.1.2 Nucleophilicity of Indoles........................................................................................3
1.1.3 Synthesis of Indoles .................................................................................................3
1.1.4 Fang-Lautens Indole Synthesis ..............................................................................11
1.2 Research Goal ....................................................................................................................18
1.2.1 Motivation ..............................................................................................................18
1.2.2 Contributions..........................................................................................................20
1.2.3 Results and Discussion ..........................................................................................20
1.3 Chapter Summary ..............................................................................................................28
1.4 Experimental ......................................................................................................................29
1.4.1 General Considerations ..........................................................................................29
1.4.2 Synthesis of Starting Materials ..............................................................................29
1.4.3 Palladium-Catalyzed Synthesis of 2-Cyanoindoles ...............................................31

vii
1.4.4 Derivatization of 2-Cyanoindole............................................................................38
1.4.5 Substrates of Mechanistic Studies .........................................................................39
Chapter 2 ......................................................................................................................................39
Migratory Insertion Strategy for Indole Dearomatization ........................................................40
2.1 Introduction ........................................................................................................................40
2.1.1 Elementary View on Aromatization ......................................................................40
2.1.2 Strategies for Benzene Dearomatization ................................................................41
2.1.3 Strategies for Indole Dearomatization ...................................................................44
2.2 Research Goal 1 – Palladium-Catalyzed Dearomative Arylation/Heteroarylation of
Indoles ................................................................................................................................59
2.2.1 Motivation ..............................................................................................................59
2.2.2 Contributions..........................................................................................................59
2.2.3 Results and Discussion ..........................................................................................60
2.3 Research Goal 2 – Arylation/Borylation of Indoles ..........................................................69
2.3.1 Motivation ..............................................................................................................69
2.3.2 Contributions..........................................................................................................70
2.3.3 Results and Discussion ..........................................................................................71
2.3.4 Section Conclusion ................................................................................................82
2.4 Chapter Summary ..............................................................................................................83
2.5 Experimental ......................................................................................................................83
2.5.1 General Considerations ..........................................................................................83
2.5.2 Synthesis of Starting Materials ..............................................................................84
2.5.3 Research Goal 1 – Dearomative Palladium-Catalyzed
Arylation/Heteroarylation ......................................................................................89
2.5.4 Research Goal 2 – Dearomative Palladium-Catalyzed Arylation/Borylation .....102
Chapter 3 ....................................................................................................................................117
Dearomatization of Indoles with NFSI and Further Aromative Functionalizations ...............117

viii
3.1 Introduction ......................................................................................................................117
3.1.1 Importance of Fluorine in Biology, Medicine, Materials ....................................117
3.1.2 Strategies of Fluorine Incorporation ....................................................................118
3.1.3 Electrophilic Dearomatization of Indoles – Push-Pull Mechanism .....................124
3.2 Research Goal 3 – Synthesis and Reactions of 3,3-Difluoro-2-exo-Methylidene
Indolines ...........................................................................................................................127
3.2.1 Motivation ............................................................................................................127
3.2.2 Contributions........................................................................................................128
3.2.3 Results and Discussion ........................................................................................129
3.3 Chapter Summary ............................................................................................................136
3.4 Experimental ....................................................................................................................136
3.4.1 General Considerations ........................................................................................136
3.4.2 Synthesis of Starting Materials ............................................................................137
3.4.3 Synthesis of 3,3-Difluoro-2-exo-Methylidene Indolines .....................................138
3.4.4 Reactions of 3,3-Difluoro-2-exo-Methylidene Indolines .....................................142
General Conclusions and Outlook ..........................................................................................149
Appendix A – NMRs from Chapter 1 ..........................................................................................150
Appendix B – NMRs from Chapter 2 ..........................................................................................190
Appendix C – HPLC Chromatograms from Chapter 2 ................................................................281
Appendix D – NMRs from Chapter 3 ..........................................................................................288
Appendix E – X-Ray Crystallographic data ................................................................................330

ix
List of Tables
Table 1 Various coupling partners investigated for the synthesis of 2-substituted indoles .......... 13
Table 2 Optimized reaction conditions and effects of reaction parameters .................................. 22
Table 3 Substrate scope for the synthesis of 2-cyanoindoles ....................................................... 25
Table 4 Effects of reaction parameters in the dearomative arylation/heteroarylation of indolesa 61
Table 5 Investigating the substrate scope with respect to the indolea ........................................... 63
Table 6 Investigating the substrate scope with respect to the activated arenesa ........................... 66
Table 7 Epimerization studies and probing of the effects of CuII chloridea ................................. 68
Table 8 Optimization for the enantioselective aryl/borylationa .................................................... 76
Table 9 Ligand screen with the optimized conditionsa ................................................................. 78
Table 10 Examining the scope of the aryl/borylation of indoles .................................................. 81
Table 11 Effects of the reaction parameters in the dearomative fluorination of indolesa ........... 130
Table 12 Examining the scope of the indole fluorination with NFSIa ........................................ 132

x
List of Schemes
Scheme 1 Abundant indoles in biology, pharmaceuticals, and clandestine sources. ..................... 2
Scheme 2 General reactivity of indoles towards electrophiles (SEAr) ........................................... 3
Scheme 3 General reactivity of 3-substituted indoles towards electrophiles (SEAr) ...................... 3
Scheme 4 General reaction scheme for the Fischer indole synthesis ............................................. 4
Scheme 5 Three classic methods for the synthesis of indoles from nitroarenes. Leimgruber–
Batcho, Bartoli, and Reissert indole syntheses. .............................................................................. 5
Scheme 6 Buchwald modification of the Fischer indole synthesis................................................. 7
Scheme 7 Mori-Ban indole synthesis using Pd0 and Hegedus indole synthesis using PdII ............ 7
Scheme 8 Larock indole synthesis and important features of the reaction ..................................... 8
Scheme 9 Intramolecular -arylation of arylimines for the synthesis of 2,3-disubstituted ............ 9
Scheme 10 Synthesis of indoles by an unexpected interception by nucleopalladation ................ 10
Scheme 11 An aqueous Pd0-catalyzed arylation cyclization to 2-arylindoles .............................. 10
Scheme 12 Enantioselective variation of the Cacchi reaction by the use of privileged ligands ... 11
Scheme 13 General scheme for the Fang-Lautens indole synthesis ............................................. 11
Scheme 14 Initial report of indole synthesis using vinylic-gem-dibromides ................................ 12
Scheme 15 The first general method for the Fang-Lautens indole synthesis ............................... 13
Scheme 16 Two possible pathways. (top) kinetically favored E–selective oxidative addition,
arylation, amination. (bottom) Z–selective oxidative addition, amination, arylation ................... 14
Scheme 17 Reaction of proposed intermediates from both pathways to study second step ......... 15
Scheme 18 Proposal for a directed- Z-selective oxidative addition .............................................. 15

xi
Scheme 19 Vinylidene intermediate allowing for an E- to Z- isomerization of the correct oxidative
addition metal-complex. (top) Negatively charged complex. (bottom) Positively charged complex
....................................................................................................................................................... 16
Scheme 20 Transition-state for the Pd-facilitated E- to Z- isomerization .................................... 16
Scheme 21 Productive use of the reversible oxidative addition for the synthesis of 2-bromoindoles
....................................................................................................................................................... 17
Scheme 22 Proposed reaction: Fang-Lautens indole synthesis for the synthesis of 2-cyanoindoles
....................................................................................................................................................... 18
Scheme 23 General Pd-catalyzed cyanation of aryl-(pseudo)halides ........................................... 18
Scheme 24 Examples of biologically interesting compounds which feature 2-cyanoindole derived
core ................................................................................................................................................ 19
Scheme 25 Synthesis of 2-cyanoindoles by C–H activation ........................................................ 19
Scheme 26 Ramirez Olefination for the synthesis of gem-dibromides ......................................... 20
Scheme 27 Ramirez Olefination for the synthesis of gem-dichlorides ......................................... 21
Scheme 28 Selective tin reduction of the nitro moiety to the aniline ........................................... 21
Scheme 29 Reactivity of chloro- and bromo- containing substrates ............................................ 26
Scheme 30 Single-pot transformation of the gem-dibromides to indoles containing a tetrazole
moiety at the 2-position by amination/cyanation/azide-cyclization ............................................. 26
Scheme 31 Subjecting 1.48, a potential intermediate, to the standard reaction conditions .......... 27
Scheme 32 Subjecting 1.62, a potential intermediate, to the standard reaction conditions .......... 27
Scheme 33 Proposed mechanism for the synthesis of 2-cyanoindoles ......................................... 28
Scheme 34 Example of aromatic system vs. anti-aromatic system .............................................. 40
Scheme 35 Simple method for drawing the MO-diagram of benzene -bonds ........................... 40

xii
Scheme 36 Benefits of dearomatization. After dearomatization, -bonds react as simple olefins in
future functionalizations ............................................................................................................... 41
Scheme 37 Birch Reduction products for EDG and EWG substituted aryl-rings ........................ 41
Scheme 38 Mechanism of the Birch Reduction ............................................................................ 42
Scheme 39 Trapping of the final anion intermediate of the Birch Reduction and its use in
Palladium-chemistry as a formal source of HCN ......................................................................... 42
Scheme 40 Modern methods of dearomatization with metal catalysts or oxidants ...................... 43
Scheme 41 Cycloaddition of arenophile, MTAD, with arenes ..................................................... 43
Scheme 42 Typical functionalizations carried out on dearomatized intermediates 2.9 ................ 44
Scheme 43 Palladium-catalyzed Tsuji-Trost type reactivity of intermediate 2.8 ......................... 44
Scheme 44 Push-pull dearomatization of indoles ......................................................................... 45
Scheme 45 Mizoroki-Heck and Interrupted-Heck reactivity ........................................................ 46
Scheme 46 Migratory insertion strategy and interrupted-Heck reactivity .................................... 46
Scheme 47 Intramolecular Mizoroki-Heck Reaction for an efficient route to fused indolines .... 47
Scheme 48 Mechanism proposed for the intramolecular Mizoroki-Heck reaction of N-(2-
halobenzoyl)indoles ...................................................................................................................... 48
Scheme 49 Domino Larock annulation/dearomative Heck reaction of N-(2-iodoaryl)benzamides
....................................................................................................................................................... 49
Scheme 50 Total synthesis of (+)-Hinckdentine A using a key enantioselective dearomative Heck
reaction .......................................................................................................................................... 50
Scheme 51 Reaction conditions reported by Jia for the enantioselective dearomative Heck reaction
of pyrroles ..................................................................................................................................... 50
Scheme 52 Enantioselective reductive Heck-reaction of indoles ................................................. 51

xiii
Scheme 53 Palladium-catalyzed dearomatization of indoles by arylation/borylation then
protodeborylation, or arylation/hydride reduction ........................................................................ 52
Scheme 54 (a) Pd-catalyzed dearomative Heck reaction of indoles by a C-2 tethered arylamide.
(b) Extended chain benzamide dearomatization for dihydrobenzoquinolone formation .............. 53
Scheme 55 Palladium-catalyzed dearomative bisfunctionalization of indoles ............................. 54
Scheme 56 Pd-catalyzed diastereoselective indole 1,2-difunctionalization by a dearomatization
strategy .......................................................................................................................................... 55
Scheme 57 Palladium-catalyzed dearomative bisfunctionalization of indoles. All three examples
are highly diastereoselective ......................................................................................................... 56
Scheme 58 First Pd-catalyzed enantioselective dearomatization by difunctionalization of indoles
via arylation/alkynylation sequence .............................................................................................. 57
Scheme 59 Palladium-catalyzed diastereoselective trapping of the dearomatized benzylic
intermediate with various nucleophiles ........................................................................................ 58
Scheme 60 General reaction studied in research goal 1 ............................................................... 59
Scheme 61 Synthesis of N-benzoyl indoles .................................................................................. 60
Scheme 62 Convergence of diastereomers by the alkylation of 2.63 ........................................... 67
Scheme 63 Dearomative aryl/borylation with palladium ............................................................. 69
Scheme 64 There is a plethora of precedence for the chemical transformation of the C–B bond to
various other atoms ....................................................................................................................... 69
Scheme 65 Miyaura-Borylation of aryl-halides and domino methods ......................................... 70
Scheme 66 Initial racemic aryl/borylation of indoles ................................................................... 71
Scheme 67 Protodeborylation of 2.65a in the presence of inorganic base ................................... 71
Scheme 68 Phosphoramidite ligands and their reaction condition requirements ......................... 72

xiv
Scheme 69 B2Pin2 pre-activation for transmetallation with CsOPiv ............................................ 72
Scheme 70 Copper-catalyzed borylation developed by Santos and coworkers ............................ 73
Scheme 71 Mixed-boron transmetallation and expected byproduct neutralization ...................... 73
Scheme 72 Synthesis of mixed-boron reagent .............................................................................. 74
Scheme 73 General strategy for the synthesis of 3,3’-substituted BINOLs ................................. 74
Scheme 74 O-P-N coupling for the synthesis of phosphoramidite ligands .................................. 75
Scheme 75 Oxidation of the boron-containing product 2.65a ...................................................... 82
Scheme 76 Further oxidation with PCC to the benzylic ketone ................................................... 82
Scheme 77 Fluoro-derivative of cortisone .................................................................................. 117
Scheme 78 The synthesis of anhydrous TBAF by SNAr ............................................................ 119
Scheme 79 The fluorination of chloro- and nitro-arenes with anhydrous NFSI at room temperature
..................................................................................................................................................... 119
Scheme 80 Common N–F reagents............................................................................................. 120
Scheme 81 The synthesis of aromatic and heteroaromatic fluorides by the reaction of Grignard
reagents with NFSI ..................................................................................................................... 121
Scheme 82 Fluorination of aryl-BF3K salts with KF and an internal oxidant ............................ 121
Scheme 83 Palladium-catalyzed C–F bond formation via Pd0/PdII and PdII/PdIV cycles ........... 122
Scheme 84 In situ active catalyst formation by arylation of the charged ligand ........................ 123
Scheme 85 Palladium(II)-catalyzed direct fluorination of C–H bonds ...................................... 123
Scheme 86 Dearomatization of indoles by a push-pull mechanism ........................................... 124
Scheme 87 Dearomative allylation of indoles ............................................................................ 125

xv
Scheme 88 Intramolecular palladium-catalyzed dearomatization of indoles using 3-tethered aryl-
halides as internal electrophiles .................................................................................................. 125
Scheme 89 Fluorine or nitrogen transfer using NFSI ................................................................. 126
Scheme 90 The use of Selectfluor for fluorination and trapping with oxygen nucleophiles ...... 126
Scheme 91 Enantioselective fluorination with NFSI, and trapping with O/N-based nucleophiles
tethered at the 3-position, forming multi-cyclic products ........................................................... 127
Scheme 92 Dearomative fluorination of 2-methylindole ............................................................ 128
Scheme 93 Asymmetric allylic fluoroalkylation/trifluoromethylation ....................................... 128
Scheme 94 Medium-scale synthesis of 3.38a ............................................................................. 131
Scheme 95 Amphoteric properties of 3.38a ................................................................................ 133
Scheme 96 Palladium-catalyzed allylic functionalizationsa ....................................................... 134
Scheme 97 Cu- and Rh- addition/-fluoride elimination ........................................................... 136

xvi
List of Appendices
Appendix A – NMRs from Chapter 1 ..........................................................................................150
Appendix B – NMRs from Chapter 2 ..........................................................................................190
Appendix C – HPLC Chromatograms from Chapter 2 ................................................................281
Appendix D – NMRs from Chapter 3 ..........................................................................................288
Appendix E – X-Ray Crystallographic data ................................................................................330

List of Publications
1. “Synthesis and Reactions of 3,3-Difluoro-2-exo-Methylidene Indolines” Zeidan, N.;
Zambri, M.; Unger, S.; Dank, C.; Torelli, A.; Mirabi, B.; Lautens. M. Org. Lett. 2020, 22,
3688.
2. “Migratory Insertion Strategies for Dearomatization” Zeidan, N.; Lautens, M. Synthesis
2019, 51, 4137.
3. “Pd-catalyzed dearomative arylborylation of indoles” Shen, C.*; Zeidan, N.*; Wu, Q.;
Breuers, C. B. J.; Liu, R.-R.; Jia, Y.-X.; Lautens, M. Chem. Sci. 2019, 10, 3118.
4. “Palladium-Catalyzed Arylation/Heteroarylation of Indoles: Access to 2,3-Functionalized
Indolines” Zeidan, N.; Beisel, T.; Ross, R.; Lautens, M. Org. Lett. 2018, 20, 7332.
5. “Palladium-Catalyzed Synthesis of 2-Cyanoindoles from 2-gem-Dihalovinylanilines”
Zeidan, N.; Bognar, S.; Lee, S.; Lautens, M. Org. Lett. 2017, 19, 5058.
6. “Pd(0)-Catalyzed Dearomative Diarylation of Indoles” Petrone, D. A.; Kondo, M.;
Zeidan, N.; Lautens, M. Chem. Eur. J. 2016, 22, 5684.
7. “Dearomative Indole Bisfunctionalization via a Diastereoselective Palladium-Catalyzed
Arylcyanation” Petrone, D. A.; Yen, A.; Zeidan, N.; Lautens, M. Org. Lett. 2015, 17, 4838.

Abbreviations
Ac acetyl
aq aqueous
Ar aryl
atm atmosphere
ATR attenuated total reflectance
BINAP 1,1-binaphthalene-2,2-diyl)bis(diphenylphosphine)
bipy 2,2’-bipyridyl
Bn benzyl
Boc tert-butoxycarbonyl
BrettPhos 2-(Dicyclohexylphosphino)3,6-dimethoxy-2,4,6-triisopropyl-1,1-biphenyl
i-Bu isobutyl
n-Bu n-butyl
t-Bu tert-butyl
Bz benzoyl
°C degree Celsius
Cat catechol
cat. catalyst
Cbz carboxybenzoyl

xix
COD 1,5-cyclooctadiene
Conc. concentrated
Conv. conversion
Cy cyclohexyl
d day
DART Direct analysis in real time
DCE 1,2-dichloroethane
DCM dichloromethane
dba dibenzylideneacetone
DG directing group
DMA dimethylacetamide
DMF dimethylformamide
DMSO dimethyl sulfoxide
dppe 1,2-bis(diphenylphosphino)ethane
DTBPF 1,1-bis(di-tert-butylphosphino)ferrocene
DTPF 1,1’-bis(di-o-tolylphosphino)ferrocene
dr diastereomeric ratio
EDG electron donating group
ee enantiomeric excess
equiv molar equivalent

xx
er enantiomeric ratio
ESI electrospray ionization
Et ethyl
EtOAc ethyl acetate
EWG electron withdrawing group
g gram
h hour
HPLC high performance liquid chromatography
HRMS high resolution mass spectrometry
IR infrared
L generic ligand
LG leaving group
Me methyl
MeCN acetonitrile
min minute
mg milligram
mL milliliter
mmol millimole
mp melting point
MOM methoxymethyl

xxi
MS molecular sieves
MTAD N-methyl-1,2,4-triazoline-3,5-dione
ND not determined
NFSI N-fluorobenzenesulfonimide
MO molecular orbital
Nu nucleophile
NMR nuclear magnetic resonance
OA oxidative addition
OTf trifluoromethanesulfonate
PDC pyridinium dichromate
Ph Phenyl
Pin pinacolato
Piv pivaloyl
PMP para-methoxyphenyl
i-Pr isopropyl
Pyr pyridine
quant. quantitative
R alkyl chain
RE reductive elimination
Rf retardation factor

xxii
rt room temperature
sat. saturated
SEAr electrophilic aromatic substitution
SM starting material
SNAr nucleophilic aromatic substitution
TBAF tetrabutylammonium fluoride
Temp temperature
THF tetrahydrofuran
TLC thin layer chromatography
TMS trimethylsilyl
Tol para-tolyl
Ts para-toluenesulfonyl
X halide
Xantphos 4,5-Bis(diphenylphosphino)-9,9-dimethylxanthene

1
Chapter 1
Palladium-Catalyzed Synthesis of 2-Substituted Indoles from Vinyl gem-Dibromo Containing Anilines
1.1 Introduction
1.1.1 Ubiquity of Indoles
To describe indoles (1.1) as an important class of heterocycle would be insufficient. Indoles are
ubiquitous; they appear in elementary undergraduate courses teaching the Fischer indole synthesis
as well as advanced topics in modern chemistry describing the various methods by which to
prepare indoles from metal-catalyzed transformations. It is no surprise that the Wikipedia page for
the simple term “indole” features 8 subgroups of detailed information,1 or that a search for the
term “indole” yields 1850 results of related topics.2
The structure of the essential amino acid tryptophan (1.2) features an indole core (Scheme 1). This
amino acid is linked to essential growth in children, as a precursor to serotonin (1.3), as well as a
precursor to many of the alkaloids in bacteria and plants.3 Due to its high bioactivity, the indole
core has been featured in the structures of many pharmaceuticals such as rizatriptan (Maxalt 1.4
by Merck), a medication for treating migraines, and tadalafil (Cialis 1.5 by Eli Lilly), a medication
used to treat erectile disfunction.4
1 https://en.wikipedia.org/wiki/Indole [cited 2019/10/30]
2 https://en.wikipedia.org/w/index.php?search=indole&title=Special%3ASearch&fulltext=1&ns0=1 [cited
2019/10/30] 3 Kaushik, N. K.; Kaushik, N.; Attri, P.; Kumar, N.; Kim, C. H.; Verma, A. K.; Choi, E. H. Molecules 2013, 18, 6620.
4 CPS [Internet]. Ottawa (ON): Canadian Pharmacists Association; c2016 [cited 2019/10/30]. Available from:
http://www.e-cps.ca or http://www.myrxtx.ca.

2
Scheme 1 Abundant indoles in biology, pharmaceuticals, and clandestine sources.
The capacity of indolic compounds to stimulate central nervous activity has led to the
popularization of many illicit drugs, as well as spiritual medicines with diverse hallucinogenic
effects. In recent times, many Canadians will travel to South America to experience the effects of
“Ayahuasca”, a traditional brew containing a cocktail of active indoles such as N,N-
dimethyltryptamine (1.6, DMT), providing an intense and relatively short period of
hallucinations.5 Another popular chemical enjoyed by a segment of the population has been those
of psilocybin, 1.7, an active ingredient in certain fungi which users have reported euphoric changes
in their perspective of space and time.6
5 O’Brien, C. CTVNEWS [cited 2019/10/30] Montreal conference will put spotlight on hallucinogenic ayahuasca.
Available from: https://www.ctvnews.ca/health/montreal-conference-will-put-spotlight-on-hallucinogenic-
ayahuasca-1.4633061 6 Passie, T.; Seifert, J.; Schneider, U.; Emrich, H. M. Addict. Biol. 2002, 7, 357.

3
1.1.2 Nucleophilicity of Indoles
Indoles are electron rich heteroaromatics due to the contribution of the nitrogen lone pair into the
aromatic system and hence react by SEAr. Most importantly, indoles react nucleophilically at the
C3–carbon, as opposed to the C2–carbon to avoid dearomatizing the backbone 6-membered ring
(Scheme 2).
Scheme 2 General reactivity of indoles towards electrophiles (SEAr)
This reactivity is so pronounced that even C3-substituted indoles will react in the same way, and
thereafter shift the more stabilized group to the C2 position to rearomatize the system (Scheme 3).
Scheme 3 General reactivity of 3-substituted indoles towards electrophiles (SEAr)
1.1.3 Synthesis of Indoles
1.1.3.1 Classic Synthesis
Of all the methods for indole synthesis, the best understood and extensively utilized is the Fischer
indole synthesis, discovered by Professor Emil Fischer.7 The reaction utilizes cheap and accessible
arylhydrazines 1.8, and aliphatic aldehydes and ketones 1.9, in a condensation in the presence of
an acid catalyst (Scheme 4). The intermediate shown undergoes a [3,3]-sigmatropic rearrangement
to furnish 2,3-difunctionalized indoles 1.10. This method quickly provides access to a library of
7(a) Fischer, E.; Jourdan, F. Berichte der deutschen chemischen Gesellschaft 1883, 16, 2241. (b) Fischer, E.; Hess, O.
Berichte der deutschen chemischen Gesellschaft 1884, 17, 559.

4
indoles quickly however, it is typically limited to ketones which have only one type of -protons.
In most cases, the reaction suffers when sterically congested arylhydrazines are used.
Scheme 4 General reaction scheme for the Fischer indole synthesis
An alternate approach for the synthesis of indoles has been from nitroarenes 1.11 since they contain
many of the atoms necessary for indoles, including the nitrogen atom masked in a higher oxidation
state. Additionally, the activated nature of such arenes allows for simple manipulations (Scheme
5, top). The Leimgruber–Batcho synthesis exploits the reduced pKa of the benzylic protons of an
ortho substituent to deprotonate and subsequently condense the benzyl-anion onto the
dimethylacetal of DMF, thus incorporating all atoms necessary for an indole (1.12).8 A general
reduction of the nitro substituent to the aniline allows for a condensation and aromatization of the
system.
8 Batcho, A. D.; Leimgruber, W. Org. Synth. 1985, 63, 214.

5
Scheme 5 Three classic methods for the synthesis of indoles from nitroarenes. Leimgruber–
Batcho, Bartoli, and Reissert indole syntheses.
Similarly, the Reissert indole synthesis also traps a benzylic anion but instead with dialkyl oxalate
(1.13). Following reduction of the nitro group, indole-2-carboxylates (1.14) are obtained in good
yield (Scheme 5, middle).9 The Bartoli Indole synthesis utilizes vinyl Grignard reagents to partially
reduce nitroarenes to the O-vinylhydroxylamines which undergo a [3,3]-sigmatropic
rearrangement to the corresponding 7-substituted indoles (Scheme 5, bottom, 1.16).10 There are
many other classic indole synthesis in the literature including: Bischler–Möhlau indole synthesis
9 Reissert, A. Berichte der deutschen chemischen Gesellschaft 1897, 30, 1030.
10 Bartoli, G.; Palmieri, G.; Bosco, M.; Dalpozzo, R. Tet. Lett. 1989, 30, 2129.

6
(reaction of -bromoarylketones and anilines),11 Cadogan-Sundberg indole synthesis (phosphine
reduction of nitroarenes, cyclization onto adjacent vinyl groups),12 Fukuyama indole synthesis
(AIBN/Bu3SnH mediated radical cyclization of o-isocyanostyrenes),13 Gassman indole synthesis
(reaction of anilines with -thioether ketones),14 Hemetsberger-Knittel indole synthesis (the use
of nitrenes from azide decomposition for indole synthesis),15 Madelung indole synthesis (similar
benzylic deprotonation however on N-acetylanilines then cyclization onto the acetyl group),16
Nenitzescu indole synthesis (reaction of benzoquinone and -aminocrotonicesters, delivers 5-
hydroxyindole derivatives).17
1.1.3.2 Palladium-Catalyzed Indole Synthesis
The past half-century of novel indole synthesis reported in the literature has been dominated by
metal-catalyzed methods, largely due to the discovery of palladium catalyzed approaches.18 One
strategy published by the Buchwald group involved the synthesis of an arylhydrazine surrogate for
a Fischer indole synthesis using a Buchwald-Hartwig amination (
Scheme 6).19
11
(a) Möhlau, R. Ber. 1881, 14, 171. (b) Bischler, A.; Fireman, P. Ber. 1893, 26, 1346. 12
(a) Bunyan, P.J.; Cadogan, J.I.G. Proc. Chem. Soc, 1962, 78. (b) Sundberg, R.J. J. Org. Chem. 1965, 30, 3604. 13
Tokuyama, H.; Yamashita, T.; Reding, M. T.; Kaburagi, Y.; Fukuyama, T. J. Am. Chem. Soc. 1999, 121, 3791. 14
Gassman, P. G.; Van Bergen, T. J.; Gruetzmacher, G. J. Am. Chem. Soc. 1973, 95, 6508. 15
Hemetsberger, H.; Knittel, D. Monatshefte für Chemie. 1972, 103, 194. 16
Madelung, W. Berichte der deutschen chemischen Gesellschaft 1912, 45, 1128. 17
Nenitzescu, C. Bull. Soc. Chim. Romania 1929, 11, 37. 18
(a) Vicente, R. Org. Biomol. Chem. 2011, 9, 6469. (b) Mancuso, R.; Dalpozzo, R. Catalysts 2018, 8, 458. (c)
Molnar, A. Chem. Rev. 2011, 111, 2251. 19
(a) Wagaw, S.; Yang, B. H.; Buchwald, S. L. J. Am. Chem. Soc. 1998, 120, 6621. (b) Wagaw, S.; Yang, B. H.;
Buchwald, S. L. J. Am. Chem. Soc. 1999, 121, 10251.

7
Scheme 6 Buchwald modification of the Fischer indole synthesis
The diarylhydrazine 1.18 is hydrolyzed, condenses onto a ketone, then undergoes the [3,3]-
sigmatropic rearrangement to deliver the desired indole product (1.19).
Other notable methods of palladium catalyzed indole synthesis involve the use of olefin reactivity
with Pd0 (Mizoroki-Heck-like-reactivity) or PdII (Wacker-like-reactivity), popularized by Mori
and Ban,20 and Hegedus21 respectively (Scheme 7).
Scheme 7 Mori-Ban indole synthesis using Pd0 and Hegedus indole synthesis using PdII
20
(a) Mori, M.; Ban, Y. Tetrahedron Lett. 1976, 17, 1803. (b) Mori, M.; Chiba, K.; Ban, Y. Tetrahedron Lett. 1977,
18, 1037. (c) Ban, Y.; Wakamatsu, T.; Mori, M. Heterocycles 1977, 6, 1711. 21
Hegedus, L. S.; Allen, G. F.; Waterman, E. L. J. Am. Chem. Soc. 1976, 98, 2674.

8
In the Mori-Ban indole synthesis, ortho-halo substrates 1.23 are used as an entry point for Pd0
oxidative addition (1.24), 5-exo-trig migratory insertion (1.25), -hydride elimination, and
rearomatization generates indole products 1.1 (for an in-depth view at the Mizoroki Heck Reaction,
see section 2.1.3.2 on page 45). The Hegedus approach involves the use of PdII activation of the
olefin 1.21 towards nucleophilic attack of the aniline nitrogen, -hydride elimination, and
isomerization generates the indole products 1.1. Reoxidation of the Pd0 catalyst is necessary for
turnover (typically achieved with CuII or benzoquinone).
One of the most widely used methods is the Larock indole synthesis discovered in 1991 by Richard
C. Larock (Scheme 8).22 The power of the reaction comes from its generality and use of simple
starting materials. An anionic palladium mechanism is proposed since the use of chloride is
required (potentially generating [Pd0-Cl]– species). The reaction tolerates a number of chloride
sources, bases, and internal alkyne coupling partners.
Scheme 8 Larock indole synthesis and important features of the reaction
Ortho-haloanilines, 1.26, are used in a reaction that involves the oxidative insertion of a Pd0
catalyst into the C–X bond, and a regioselective migratory insertion into internal alkynes
generating the 6-membered palladacycle 1.27. The regioselectivity is steric driven; the larger
22
Larock, R. C.; Yum, E. K. J. Am. Chem. Soc. 1991, 113, 6689.

9
group is placed at the carbon destined to become the C–2 position due to the clashing of the new
C-C bond during the carbopalladation step.23
The area of palladium-catalyzed indole synthesis continues to grow and shown below are some
highlights of the recent literature of such reactions. Nolan and coworkers reported a convenient
synthesis of free indoles and diverse azaindoles using an -arylation strategy of arylimines 1.29
(Scheme 9).24
Scheme 9 Intramolecular -arylation of arylimines for the synthesis of 2,3-disubstituted
The authors use an electron-rich and sterically hindered NHC-palladium catalyst to access various
heterocycles in good to excellent yields. The ability of the catalyst to tolerate azaindoles is
interesting and unusual, demonstrating the power of this reaction.
In their attempt to explore an intramolecular Heck reaction of 1.31 to generate large-ring
benzo[1,4]heterocycles, Gharpure observed a competitive nucleopalladation/retro-oxa-Michael
generating N-alkylindoles (Scheme 10).25
23
Larock, R. C.; Yum, E. K.; Refvik, M. D. J. Org. Chem. 1998, 63, 7652. 24
Marelli, E.; Corpet, M.; Minenkov, Y.; Neyyappadath, R. M.; Bismuto, A.; Buccolini, G.; Curcio, M.; Cavallo, L.;
Nolan, S. P. ACS catalysis 2016, 6, 2930. 25
Gharpure, S. J.; Anuradha, D. Org. Lett. 2017, 19, 6136.

10
Scheme 10 Synthesis of indoles by an unexpected interception by nucleopalladation
Rather than undergoing a 7-exo-trig carbopalladation, the PdII oxidative addition complex 1.32
undergoes a nucleopalladation/reductive elimination. The oxazole intermediate formed then
undergoes a ring opening by retro-oxa-Michael.
Scheme 11 An aqueous Pd0-catalyzed arylation cyclization to 2-arylindoles
An interesting aqueous PdII-catalyzed synthesis of indoles is reported by Cheng and Chen (Scheme
11).26 The utility of the reaction come from its use of a reliable arylation of simple aryl
acetonitriles, providing the atom-blueprint for a cyclization of the adjacent aniline to deliver 2-
arylindoles (1.36).
26
Yu, S.; Hu, K.; Gong, J.; Qi, L.; Zhu, J.; Zhang, Y.; Cheng, T.; Chen, J. Org. Biomol. Chem. 2017, 15, 4300.
11
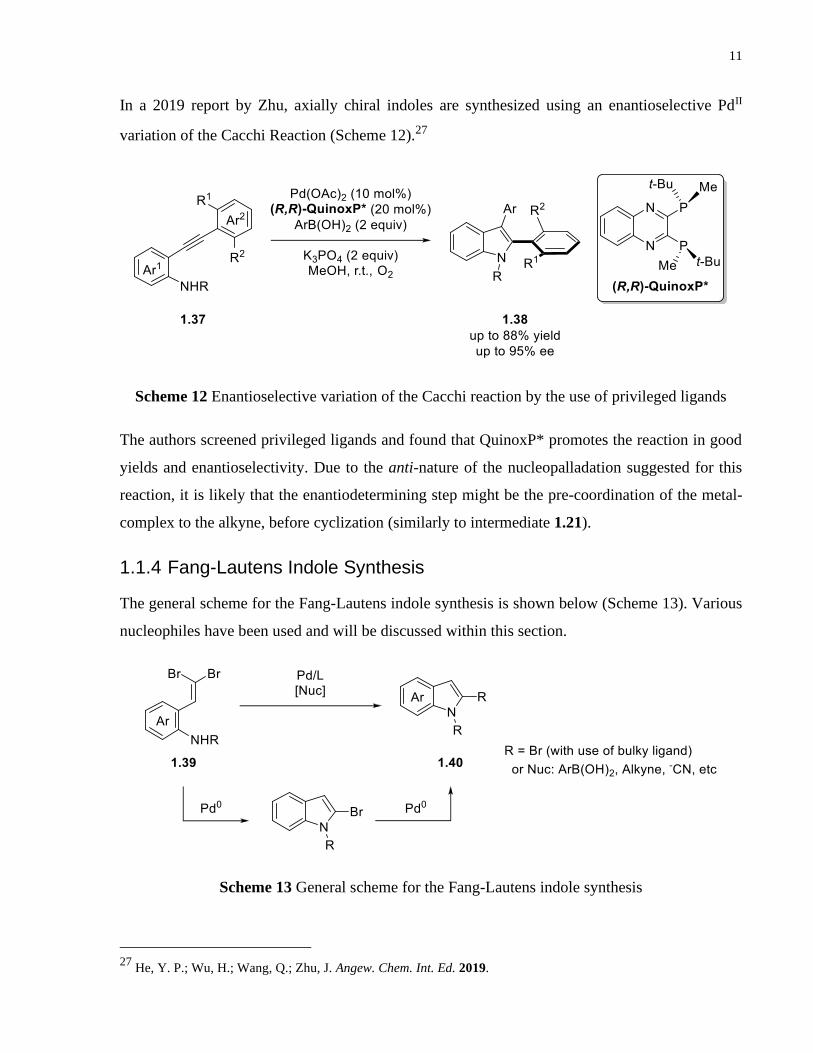
In a 2019 report by Zhu, axially chiral indoles are synthesized using an enantioselective PdII
variation of the Cacchi Reaction (Scheme 12).27
Scheme 12 Enantioselective variation of the Cacchi reaction by the use of privileged ligands
The authors screened privileged ligands and found that QuinoxP* promotes the reaction in good
yields and enantioselectivity. Due to the anti-nature of the nucleopalladation suggested for this
reaction, it is likely that the enantiodetermining step might be the pre-coordination of the metal-
complex to the alkyne, before cyclization (similarly to intermediate 1.21).
1.1.4 Fang-Lautens Indole Synthesis
The general scheme for the Fang-Lautens indole synthesis is shown below (Scheme 13). Various
nucleophiles have been used and will be discussed within this section.
Scheme 13 General scheme for the Fang-Lautens indole synthesis
27
He, Y. P.; Wu, H.; Wang, Q.; Zhu, J. Angew. Chem. Int. Ed. 2019.

12
1.1.4.1 The Discovery of the Reaction
Near the turn of the century, the usefulness of gem-dibromo compounds was already becoming
apparent,28 though it wasn’t until 2004 that Bisseret and coworkers reported two examples of a
palladium-catalyzed tandem reaction of ortho-vinylic-gem-dibromoanilines with nucleophiles,
generating substituted indoles (Scheme 14).29
Scheme 14 Initial report of indole synthesis using vinylic-gem-dibromides
The first reported reaction used diethylphosphite for the synthesis of diethyl indole-2-phosphonate
(top) and the second uses para-methoxyphenylboronic acid for a Suzuki-Miyaura trap, forming 2-
arylindole (bottom). Lautens and Fang independently reported a general version of the indole
synthesis, capable of tolerating various electronic and steric perturbations on the anilines, as well
as any aryl, heteroaryl, vinyl, and alkyl boronic acids (Scheme 15).30 The authors attribute the
excellent reactivity of the catalyst system to SPhos, a bulky and electron rich ligand novel at the
time.31
28
(a) Zapata, A. J.; Rúiz, J. J. Organomet. Chem. 1994, 479, c6. (b) Shen, W.; Thomas, S. A. Org. Lett. 2000, 2,
2857. 29
Thielges, S.; Meddah, E.; Bisseret, P.; Eustache, J. Tetrahedron Lett. 2004, 45, 907. 30
(a) Fang, Y. Q.; Lautens, M. Org. Lett. 2005, 7, 3549. (b) Fang, Y.-Q.; Lautens, M. J. Org. Chem. 2008, 73, 538. 31
Walker, S. D.; Barder, T. E.; Martinelli, J. R.; Buchwald, S. L. Angew. Chem. Int. Ed. 2004, 43, 1871.

13
Scheme 15 The first general method for the Fang-Lautens indole synthesis
Over the next decade, many groups investigated other possible coupling partners for the modular
synthesis of 2-substituted indoles using these substrates (Table 1). Intermolecular and
intramolecular Heck and Sonogashira traps were developed shortly after the first report.32 Alper
and coworkers as well as Pontikis and Florent developed protocols for the carbonylation and
trapping of indoles.33 Lan and coworkers developed an interesting direct heteroarylation using
activated heterocycles, generating bi-heteroaryl products.34
Table 1 Various coupling partners investigated for the synthesis of 2-substituted indoles
Coupling partner Product Group
Lautens (2006)
Lautens (2007)
Alper (2008)
32
Fayol, A.; Fang, Y. Q.; Lautens, M. Org. Lett. 2006, 8, 4203. (b) Nagamochi, M.; Fang, Y. Q.; Lautens, M. Org.
Lett. 2007, 9, 2955. 33
(a) Vieira, T. O.; Meaney, L. A.; Shi, Y. L.; Alper, H. Org. Lett. 2008, 10, 4899. (b) Arthuis, M.; Pontikis, R.;
Florent, J. C. Org. Lett. 2009, 11, 4608. 34
Qin, X.; Cong, X.; Zhao, D.; You, J.; Lan, J. Chem. Commun. 2011, 47, 5611.

14
Pontikis and Florent (2009)
Lan (2011)
1.1.4.2 The Mechanistic Conundrum
Bisseret and coworkers made an important remark about the mechanism of the reaction; the
oxidative addition of the E- C–Br bond is kinetically favored (1.45).29 This selective oxidative
addition has also been explored by others.35 This leads to their proposed mechanism that the
arylation should occur first, generating intermediate 1.46, which then undergoes a cycloamination,
generating 2-aryl indoles (Scheme 16, top, 1.36).
Scheme 16 Two possible pathways. (top) kinetically favored E–selective oxidative addition,
arylation, amination. (bottom) Z–selective oxidative addition, amination, arylation
Lautens and coworkers synthesized the two possible intermediates 1.46 and 1.48, and subjected
them to the reaction conditions to assess their role in the reaction (Scheme 17).30 It was found that
substrate 1.46 produced small amounts of double arylated product 1.49, which was never observed
35
Zapata, A. J.; Rúiz, J. J. Organomet. Chem. 1994, 479, c6.

15
in any other studies, casting doubt on the possibility of the arylation/amination mechanism.
Intermediate 1.48 however produced exclusively the desired indole 1.36 in excellent yield.
Scheme 17 Reaction of proposed intermediates from both pathways to study second step
Various mechanisms have been proposed for this apparent formal Z-selective oxidative addition.
It is possible the selectivity of the oxidative addition is influenced by the ortho-heteroatom in a
directed oxidative addition however, in this case, it is unlikely the soft, electron-rich Pd0–complex
would coordinate to the hard-donor (Scheme 18).36
Scheme 18 Proposal for a directed- Z-selective oxidative addition
Other possibilities discussed include a charge-separated metal vinyl carbene intermediate (Scheme
19), by either donation of the aniline (top) or a stabilized benzylic anion (bottom).37
36
(a) Pearson, R. G. J. Chem. Ed. 1987, 64, 561. (b) Finn, M. G.; Sharpless, K. B. J. Am. Chem. Soc. 1991, 113, 113.
(c) Schrock, R. R. Angew. Chem. Int. Ed. 2006, 45, 3748. (d) Tudor, R.; Ashley, M. Platin. Met. Rev. 2007, 51, 116.
(e) Deangelis, A.; Colacot, T. J. in RSC Catalysis Series, Vol. 2015 (ed. T. J. Colacot), The Royal Society of Chemistry,
Cambridge, 2015, pp. 20–90. 37
(a) Brady, K. A.; Nile, T. A. J. Organomet. Chem. 1991, 206, 299. (b) Zargarian, D.; Alper, H.

16
Scheme 19 Metal vinyl carbene intermediate allowing for an E- to Z- isomerization of the
correct oxidative addition metal-complex. (top) Negatively charged complex. (bottom) Positively
charged complex
Recent computational work on Pd-assisted E- to Z- isomerization on vinyl-iodides, supported by
experimental evidence, suggest that the isomerization may not be via a defined metal carbene
intermediate (Scheme 20).38
Scheme 20 Transition-state for the Pd-facilitated E- to Z- isomerization
The transition state is described by three important characteristics: the Pd–C bond is twisted
slightly out of plane, the Pd–C bond is shortened slightly, the C=C bond is lengthened slightly.
The changes in bond lengths however were very minute. The authors concluded, both
computationally and experimentally, that the sequence of E-selective oxidative
Organometallics, 1991, 10, 2914. (c) Amatore, C.; Bensalem, S.; Ghalem, S.; Jutand, A. Organometallics 2004,
689, 4642. (d) Tanke, R. S.; Crabtree, R. H. J. Am. Chem. Soc. 1990, 112, 7984. (e) Chung, L. W.; Wu, Y.-D.; Trost,
B. M.; Ball, Z. T. J. Am. Chem. Soc. 2003, 125, 11578. 38
Petrone, D. A.; Franzoni, I.; Ye, J.; Rodriguez, J. F.; Poblador-Bahamonde, A. I.; Lautens, M. J. Am. Chem. Soc.
2017, 139, 3546.

17
addition/isomerization was kinetically favoured compared to Z-selective oxidative addition. In a
separate computational study by Lautens and Schoenebeck, a palladium-facilitated Cis to Trans
isomerization was found to have only minor charge build up in the transition state. The lack of
bond length change as well as charge build up suggest that a long-lived metal vinyl carbene species
is not an intermediate in these isomerizations.39
1.1.4.3 Reversible Oxidative Addition
Oxidative addition has historically been considered an irreversible process when it comes to aryl-
halides and palladium catalysts. Hartwig and coworkers published a series of mechanistic
investigations into this process, and it was found that the use of excess bulky ligand favoured a
reductive elimination of the C–X bond.40 From this observation, a practical use of the reversible
oxidative addition was demonstrated by Lautens and Newman in the synthesis of 2-bromoindoles
by means of the Fang-Lautens indole synthesis using bulky phosphine ligands (Scheme 21)002E41
Scheme 21 Productive use of the reversible oxidative addition for the synthesis of
2-bromoindoles
39
Sperger, T.; Le, C. M.; Lautens, M.; Schoenebeck, F. Chem. Sci. 2017, 8, 2914. 40
(a) Roy, A. H.; Hartwig, J. F. J. Am. Chem. Soc. 2001, 123, 1232. (b) Roy, A. H.; Hartwig, J. F. J. Am. Chem. Soc.
2003, 125, 13944. (c) Roy, A. H.; Hartwig, J. F. Organometallics 2004, 23, 1533. 41
(a) Newman, S. G.; Aureggi, V.; Bryan, C. S.; Lautens, M. Chem. Commun. 2009, 5236. (b) Newman, S. G.;
Lautens, M. J. Am. Chem. Soc. 2010, 132, 11416.

18
The authors were able to run an intramolecular Buchwald-Hartwig amination even in the presence
of a C–I bond, as well as in the presence of product 1.51 which could sequester the catalyst as 1.52
or 1.53.
1.2 Research Goal
1.2.1 Motivation
The first research goal of this thesis was to develop a protocol for the synthesis of 2-cyanoindoles
using the Fang-Lautens indole synthesis,42 as the products would possess a versatile handle for
future transformations (Scheme 22).
Scheme 22 Proposed reaction: Fang-Lautens indole synthesis for the synthesis of 2-cyanoindoles
Metal-catalyzed cyanation of aromatic and heteroaromatic compounds has been an important
research objective, as the nitrile functionality serves as a versatile synthetic handles for functional
group manipulation.43 The d10 transition-metals have been the most commonly used catalysts for
cyanation by cross-coupling; C–X bonds could be transformed to C–CN bonds by means of
oxidative addition and trapping with a source of [CN]– (Scheme 23).
Scheme 23 General Pd-catalyzed cyanation of aryl-(pseudo)halides
Ionic sources of cyanide such as NaCN, KCN, and Zn(CN)2 require polar aprotic solvents. The
biggest challenge faced with palladium-catalyzed cyanation reactions is catalyst poisoning by the
irreversible formation of inactive complexes. This is typically the result of binding an excess
42
Zeidan, N.; Bognar, S.; Lee, S.; Lautens, M. Org. Lett. 2017, 19, 5058. 43
(a) Cohen, D. T.; Buchwald, S. L. Org. Lett. 2015, 17, 202. (b) Torborg, C.; Beller, M. Adv. Synth. Catal. 2009,
351, 3027. (c) Anbarasan, P.; Schareina, T.; Beller, M. Chem. Soc. Rev. 2011, 40, 5049.

19
amount of cyanide on the metal-center. This is further discussed in the context of the Fang-Lautens
indoles synthesis below.
Derivatives of 2-cyanoindoles exhibit high bioactivity and can be identified in many natural
products and pharmaceutical agents (Scheme 24).44
Scheme 24 Examples of biologically interesting compounds which feature 2-cyanoindole
derived core
Some recent methods for the synthesis of 2-cyanoindoles have utilized Rh,45 Pd,46 Cu,47 Co,48 or
Mn/Zn49 as catalysts to activate the C–H bond in the 2-position of indoles for cyanation (Scheme
25). These methods allow the direct functionalization of indoles however, the use of specific
directing groups, usually pyrimidine, are removed under harsh basic reflux.
Scheme 25 Synthesis of 2-cyanoindoles by C–H activation
44
(a) Almagro, L.; Fernandez-Perez, F.; Pedreno, M. A. Molecules 2015, 20, 2973. (b) Borza, I.; Kolok, S.; Galgoczy,
K.; Gere, A.; Horvath, C.; Farkas, S.; Greiner, I.; Domany, G. Bioorg. Med. Chem. Lett. 2007, 17, 406. 45
(a) Mishra, N. K.; Jeong, T.; Sharma, S.; Shin, Y.; Han, S.; Park, J.; Oh, J. S.; Kwak, J. H.; Jung, Y. H.; Kim, I. S.
Adv. Synth. Catal. 2015, 357, 1293. (b) Chaitanya, M.; Anbarasan, P. J. Org. Chem. 2015, 80, 3695. 46
Xu, S.; Huang, X.; Hong, X.; Xu, B. Org. Lett. 2012, 14, 4614. 47
Kou, X.; Zhao, M.; Qiao, X.; Zhu, Y.; Tong, X.; Shen, Z. Chem. - Eur. J. 2013, 19, 16880. 48
Li, J.; Ackermann, L. Angew. Chem., Int. Ed. 2015, 54, 3635. 49
Liu, W.; Richter, S. C.; Mei, R.; Feldt, M.; Ackermann, L. Chem. - Eur. J. 2016, 22, 17958.

20
The extension of the Fang-Lautens indole synthesis for cyanation would be a valuable addition to
the literature as it can both generate the indole core and add functionality in a single step without
the need for a directing group.
1.2.2 Contributions
The results presented were obtained in collaboration with Sabine Bognar (visiting master’s student
from Germany) and Sophia Lee (undergraduate students from the University of Toronto). I
conceived the idea, directed the project, and executed most of the experiments. Sophia Lee assisted
in the optimization of the reaction and substrate synthesis. Sabine Bognar focused on synthesizing
substrates and assisted on exploring the scope of the reaction. Specific contributions are listed
within the text as well as within the experimental section.
1.2.3 Results and Discussion
1.2.3.1 Starting Material Synthesis
The synthesis of the gem-dihaloanilines is straightforward and is typically achieved in high yields
using the Ramirez Olefination (Scheme 26).50 The gem-dichlorides are also accessed in the same
way using chloroform as the surrogate of CCl2 (Scheme 27).
Scheme 26 Ramirez Olefination for the synthesis of gem-dibromides
50
Desai, N. B.; McKelvie, N.; Ramirez, F. J. Am. Chem. Soc. 1962, 84, 1745.

21
Scheme 27 Ramirez Olefination for the synthesis of gem-dichlorides
One improvement to the Ramirez-olefination was published by the Lautens group, which applies
the Horner-Wadsworth-Emmons modification by using P(Oi-Pr)3.51 This allows for a simplified
purification of the products by aqueous workup, and avoiding the tedious purification step from
the usual triphenylphosphine oxide byproduct.
The aniline is then accessed by reduction of the nitro group, most commonly achieved on large-
scale by SnCl2 in EtOH (Scheme 28). Other important methods used for the nitro-selective
reduction (leaving the gem-dibromide intact), have been Fe0–reductions, with FeCl3 catalyst, and
vanadium-doped platinum-catalyzed hydrogenation.
Scheme 28 Selective tin reduction of the nitro moiety to the aniline
1.2.3.2 Optimization
We began our studies with conditions similar to those previously reported41b and the use of
Zn(CN)2 as the cyanide source. The use of the bulky ligand allows both the monitoring of the 2-
bromoindole intermediate (since this ligand allows for reversible oxidative addition into 1.48) as
well as minimizing over coordination of cyanide ion (vide infra). After screening reaction
parameters, we found Pd(t-Bu3P)2 (5 mol %), Zn(CN)2 (0.55 equiv), Zn(TFA)2 (10 mol %) and
K3PO4 (2 equiv) in PhMe–DMA (3:1) at 110 °C for 18 h at 0.5 mmol scale to be our optimal
conditions for the preparation of 1.61 in 74% yield (Table 2, entry 1).
51
Lautens, M.; Fang, Y.-Q.; Lifchits, O. Synlett 2008, 2008, 413.

22
Table 2 Optimized reaction conditions and effects of reaction parameters
Entry Change to standard condition Conv (%) 1.61 (%) 1.48 (%)
1 None Full 74 –
2 Double the scale Full 73 –
3 Dichloride instead of dibromide Full 78 –
4 No Zn(TFA)2 92 55 5
5 Zn dust instead of Zn(TFA)2 85a 58a –
6b Zn(OAc)2 instead of Zn(TFA)2 Fullc 42 –
7b No DMA Full 15 70
8b PhMe–DMA (1:1) 46 11 –
aAverage for 3 runs, 40–70% yield. bYield determined by 1H NMR from crude using 1,3,5-trimethoxybenzene as
internal standard. cLarge amounts of 3 isolated (42%).
The reaction was run on 1 mmol scale with no change in yield (Table 2, entry 2). The use of the
gem-dichloride is possible, delivering the product in comparable yield (Table 2, entry 3). When no
additive was used, the reaction was irreproducible and failed to fully consume all of the starting
material (Table 2, entry 4). Grushin and Macgregor have recently described the modes of

23
Pd-catalyst deactivation during cyanation by presence of air, water, amines, and excess cyanide.52
Limited reports on the use of Zn(0) and Zn(OAc)2 to improve catalytic turnover are available.53
When Zn(0) was used, improved conversion was observed but the reaction remained
irreproducible with variance in yields up to 30% in parallel reactions (Table 2, entry 5). The use
of Zn(OAc)2 was detrimental to the reaction, and large amounts of a bromoalkyne 1.62 was
produced presumably by base-promoted hydrodebromination (Table 2, entry 6). The
trifluoroacetate variant was used to reduce the basicity of the acetate counter-ion to prevent the
elimination from occurring. Zn(TFA)2 led to full consumption of the starting material and the
highest yield consistently. Although the role of the additive is unclear, it may facilitate
transmetallation, thus partially regulating the amount of free cyanide in solution.
The solvent ratio was carefully selected to accommodate the opposing polarity requirements for
cyclization and cyanation as they necessitate the use of non-polar and polar conditions,
respectively. In the absence of DMA as a co-solvent, full consumption of the starting material is
achieved however, cyanation is retarded and 1.48 is the major product (Table 2, entry 7). The
bromoindole is a likely intermediate in the reaction (vide infra) suggesting the cyanation is slower.
We reason the lower reactivity to be the result of poor Zn(CN)2 solubility in PhMe.52 In contrast,
poor conversions were observed in a 1:1 solvent ratio (Table 2, entry 8). Only 1.61 was observed
suggesting cyanation of 1.48 occurred rapidly but with poor catalytic turnover. A 3:1 solvent ratio
was found to be ideal for a consecutive cyclization/cyanation. We believe this may also moderate
the amount of Zn(CN)2 present in solution throughout the reaction, hence slowing the formation
of catalytically inert, coordinatively saturated palladium complexes. DMA proved better than other
polar solvents such as THF or MeCN due to its high boiling point. Reactions with DMF yielded
greater amounts of 1.62 as a byproduct, presumably by solvent decomposition at high
temperatures.54
52
Erhardt, S.; Grushin, V. V.; Kilpatrick, A. H.; Macgregor, S. A.; Marshall, W. J.; Roe, D. C. J. Am. Chem. Soc.
2008, 130, 4828. 53
(a) Ramnauth, J.; Bhardwaj, N.; Renton, P.; Rakhit, S.; Maddaford, S. P. Synlett 2003, 2237. (b) Chidambaram, R.
Tetrahedron Lett. 2004, 45, 1441. 54
Petrone, D. A.; Yen, A.; Zeidan, N.; Lautens, M. Org. Lett. 2015, 17, 4838.

24
1.2.3.3 Exploring the Substrate Scope
With optimized conditions in hand, we proceeded to investigate the scope of the transformation
(Scheme 3). Extending the aromatic system to the naphthyl ring delivered product 1.61b with no
change in yield (74%). Alkyl substitution adjacent to the aniline or the vinyl dibromide delivered
products 1.61c and 1.61d in 71% and 69% yield respectively. Alkyl and benzyl groups on the
aniline nitrogen provided the N-caped indoles in similar yield (1.61e-g, 72%, 72%, 77%), even
when a sterically demanding isopropyl group was used. Placing an electron-donating OMe at the
para position relative to the aniline gave an improved yield (1.61h, 77%), presumably by
increasing the nucleophilicity of the nitrogen.
In contrast, an electron-withdrawing substituent para to the aniline, as in 1.61i and 1.61j bearing
CO2Me and F necessitated slightly higher catalyst loading and higher temperature to achieve full
consumption, delivering products in 48% and 67%, respectively. An opposite trend was found
when studying electronic-effects at the position para relative to the olefin. While the methoxy
substituent required forcing conditions to deliver product 1.61k in 68% yield, the methyl ester
proceeded smoothly to afford product 1.61l in 75%. Fluorine substitution led to the desired product
1.61m albeit in reduced yield (61%). Having similar electronic properties to 1.41h, reaction of
1.41n proceeded smoothly to 1.61n in slightly improved yield (77%). Interestingly, the highly
electron-rich nature of the scaffold bearing the dioxolane ring was more challenging and provided
1.61o albeit in reduced yields (53%). The reaction selectively underwent the desired reaction with
a chlorine substituent para- to the nitrogen (1.61p), leaving a tangible, heteroaryl C–X bond for
future derivatization (Scheme 29, top).

25
Table 3 Substrate scope for the synthesis of 2-cyanoindoles
1.61b (74%)
1.61c (71%)a
1.61d (69%)
1.61e (72%)a
1.61f (72%)
1.61g (77%)
1.61h (77%)
1.61i (48%)a,b
1.61j (67%)a,b
1.61k (68%)
1.61l (75%)a
1.61m (61%)
1.61n (77%)
1.61o (53%)b
1.61p (70%)
1.61q (61%)b,c
aReaction, workup, and/or purification was run by Sabine Bognar. bPd(t-Bu3P)2 (7.5 mol%), Zn(TFA)2 (15 mol%),
120 °C. cFrom 4-bromo-2-(2,2-dibromovinyl)-aniline using Zn(CN)2 (2.2 equiv).
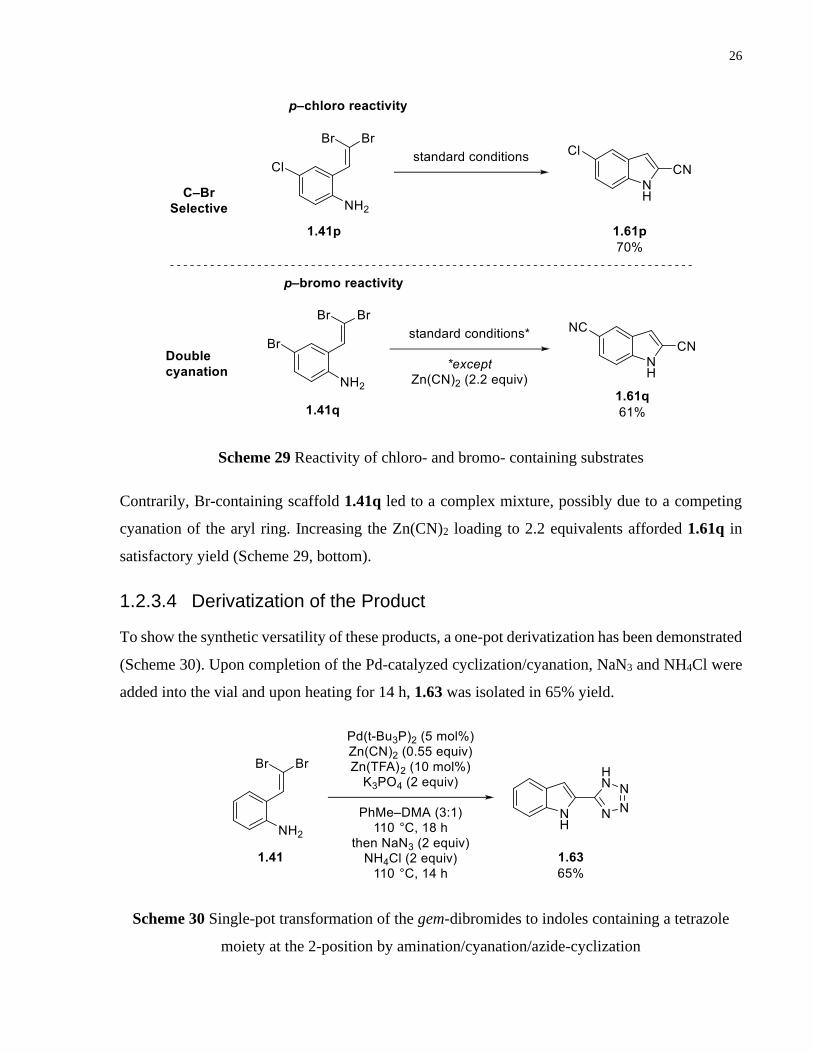
26
Scheme 29 Reactivity of chloro- and bromo- containing substrates
Contrarily, Br-containing scaffold 1.41q led to a complex mixture, possibly due to a competing
cyanation of the aryl ring. Increasing the Zn(CN)2 loading to 2.2 equivalents afforded 1.61q in
satisfactory yield (Scheme 29, bottom).
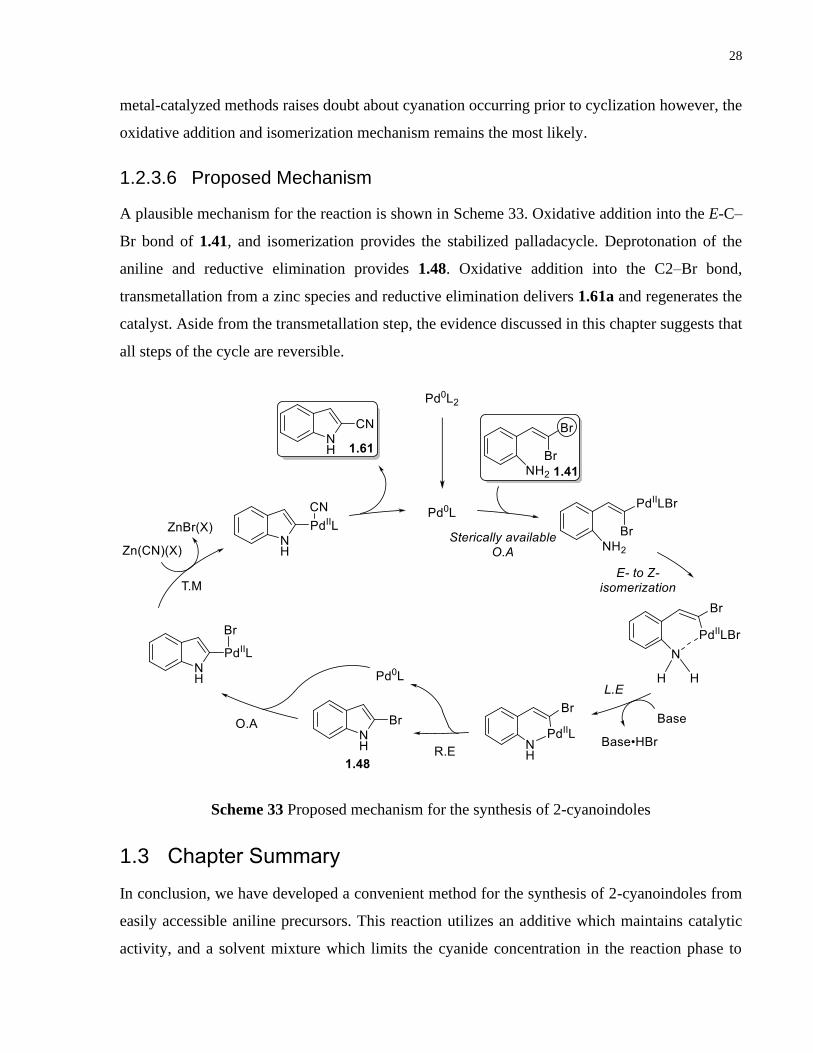
1.2.3.4 Derivatization of the Product
To show the synthetic versatility of these products, a one-pot derivatization has been demonstrated
(Scheme 30). Upon completion of the Pd-catalyzed cyclization/cyanation, NaN3 and NH4Cl were
added into the vial and upon heating for 14 h, 1.63 was isolated in 65% yield.
Scheme 30 Single-pot transformation of the gem-dibromides to indoles containing a tetrazole
moiety at the 2-position by amination/cyanation/azide-cyclization

27
1.2.3.5 Mechanistic Investigation
To further evaluate the mechanism of the transformation and gain insight on the roles of the
observed species, 1.48 and 1.62 were synthesized. Subjecting 1.48 to the standard conditions
delivered the desired product in 93% yield (Scheme 31).
Scheme 31 Subjecting 1.48, a potential intermediate, to the standard reaction conditions
This result supports the proposal that the reaction proceeds via the formation of 1.48. In contrast,
when 1.62 was subjected to the same conditions, 60% of unreacted material was recovered with
no other compounds identified (Scheme 32). This result suggests 1.62 is part of a non-productive
decomposition pathway leading to loss in mass-balance under these conditions.
Scheme 32 Subjecting 1.62, a potential intermediate, to the standard reaction conditions
We sought to investigate the order of events; the possibility of cyanation occurring prior to
cyclization. Although we have never observed these products in reactions carried out to partial
conversions, this sequence remains a plausible pathway since oxidative addition of a catalyst
across the C–(E)-Br bond would give the kinetically favored product. Selective coupling of the C–
(E)-Br with boronic acids is known,30 however, any attempts55 to cyanate by selective cross-
coupling led to full recovery of unreacted starting material. The difficulty of the cyanation using
55
Catalyst such as Pd, Ni and Cu were used with varying ligands and Zn(CN)2 or KCN as the cyanide source. Along
with 1a, gem-vinyldihalides with ortho-nitro functionality and dimethyl-protected aniline was used. All reactions led
to recovery of starting material.
28
metal-catalyzed methods raises doubt about cyanation occurring prior to cyclization however, the
oxidative addition and isomerization mechanism remains the most likely.
1.2.3.6 Proposed Mechanism
A plausible mechanism for the reaction is shown in Scheme 33. Oxidative addition into the E-C–
Br bond of 1.41, and isomerization provides the stabilized palladacycle. Deprotonation of the
aniline and reductive elimination provides 1.48. Oxidative addition into the C2–Br bond,
transmetallation from a zinc species and reductive elimination delivers 1.61a and regenerates the
catalyst. Aside from the transmetallation step, the evidence discussed in this chapter suggests that
all steps of the cycle are reversible.
Scheme 33 Proposed mechanism for the synthesis of 2-cyanoindoles
1.3 Chapter Summary
In conclusion, we have developed a convenient method for the synthesis of 2-cyanoindoles from
easily accessible aniline precursors. This reaction utilizes an additive which maintains catalytic
activity, and a solvent mixture which limits the cyanide concentration in the reaction phase to

29
prevent catalyst poisoning. Finally, the versatility of the products was demonstrated by extending
the protocol to a one-pot transformation of the nitrile to the tetrazole.
1.4 Experimental
1.4.1 General Considerations
Unless otherwise stated all reactions were run under an atmosphere of argon in oven or flame dried
glassware. Catalytic reactions were run in 3-dram vials with open-top caps fitted with a Teflon
septum. Reactions were monitored by thin-layer chromatography (TLC) on EMD Silica Gel 60
F254 plates and visualized under UV light or by immersion in potassium permanganate (KMnO4)
stain. Flash column chromatography was performed on Silicycle 40-60 m silica gel and in the
case of 2-cyanoindoles, the silica was suspended in 5% NEt3 in hexanes before loading the column.
PhMe was distilled over CaH. DMA was purchased from Sigma-Aldrich in a Sure/Seal bottle and
used without further purification. Pd(t-Bu3P)2 was purchased from Johnson Matthey. All reagents
and organic building blocks were purchased from commercial supplier (Sigma Aldrich, Strem,
Alfa Aesar, TCI, Combi-blocks) and used without further purification.
1H and 13C NMR spectra were obtained on the Agilent DD2 500 equipped with a 5mm Xsens Cold
Probe. The spectra were internally referenced to the solvent peak. 19F NMR spectra were obtained
on the Varian Mercury 400 or 500 operating at 470 or 564 MHz. Measurements were taken at 296
K and chemical shifts are reported in parts per million (ppm). Data is reported in the following
format: chemical shift ( ppm), multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m =
multiplet), coupling constant (Hz), integration. Melting points were measured on a Fischer-Johns
Melting Point Apparatus and are uncorrected. High resolution mass spectra (HRMS) were obtained
on a Micromass 70S-250 spectrometer (EI) or an ABI/Sciex QStar Mass Spectrometer (ESI) or a
JEOL AccuTOF model JMS-T1000LC mass spectrometer equipped with an IONICS® Direct
Analysis in real Time (DART) ion source.
1.4.2 Synthesis of Starting Materials
1.4.2.1 Synthetic Remarks
Compounds 1.41a–1.41d, 1.41f, 1.41g, 1.41i, 1.41j, 1.41l, and 1.41m were synthesized according
to literature procedure.30 Compound 1.41e was synthesized according to literature procedure.32b

30
Compound 1.41p and 1.41o were synthesized according to literature procedure.56 Compound
1.41n was synthesized according to literature procedure.57 Compound 1.41q was synthesized
according to literature procedure.41b
1.4.2.2 General Procedure for the Synthesis of 2-gem-Dihaloanilines
To a solution of nitrobenzaldehyde (9.07 g, 60 mmol, 1 equiv) and CBr4 (29.85 g, 90 mmol, 1.5
equiv) in DCM (300 ml, 0.2 M with respect to the aldehyde) at 0 °C was added a solution of PPh3
(47.21 g, 180 mmol, 3 equiv) in DCM (200 ml, 0.9 M) dropwise over a 30-minute period,
maintaining the temperature under 5 °C. The reaction was stirred for 30 min before warming to r.t.
and stirred for an additional 30 min. Consumption of the starting material was monitored by TLC.
The reaction was filtered through a short pad of silica gel, eluting with 10% EtOAc in hexanes
until all product was collected as monitored by TLC to reduce the amount of triphenylphosphine
oxide present. The crude was concentrated to an oil, taken up into EtOH (200 ml, 0.3 M) and was
added SnCl2⸳H2O (67.7 g, 300 mmol, 5 equiv). The reaction was refluxed at 90 °C for 45 min,
cooled, and was basified using K2CO3 to pH 10. The aqueous layer was extracted 5 times with
EtOAc, and the organic layer was washed with H2O, brine and dried over Na2SO4. The product
was purified by silica gel flash chromatography.
1.4.2.3 Characterization Data for New Compounds
2-(2,2-dibromovinyl)-4-methoxyaniline (1.41h) – The compound was
synthesized according to General Procedure 1.4.2.2 using 5.4 mmol of the
aldehyde. The product was purified by flash chromatography using hexanes–
EtOAc (5:1, v:v) as the mobile phase and was isolated as an off-white solid
(901.1 mg, 2.935 mmol, 76%, mp = 66–67 °C). 1H NMR (500 MHz, CDCl3) δ 7.35 (s, 1H), 6.91
(d, J = 2.9 Hz, 1H), 6.78 (dd, J = 8.7, 2.9 Hz, 1H), 6.67 (d, J = 8.6 Hz, 1H), 3.76 (s, 3H). 13C NMR
56
Gupta, S.; Koley, D.; Ravikumar, K.; Kundu, B. J. Org. Chem. 2013, 78, 8624. 57
Yuen, J.; Fang, Y. -Q.; Lautens, M. Org. Lett. 2006, 8, 653.

31
(125 MHz, CDCl3) δ 152.4, 137.1, 133.9, 122.8, 117.4, 116.2, 113.8, 92.7, 55.8. IR (thin film, cm-
1) 3439, 3360, 2996, 2947, 2907, 2830, 1609, 1497, 1464, 1427. HRMS (DART, M+1) Calculated
for C9H10Br2NO 305.9129, found 305.9126.
2-(2,2-dibromovinyl)-5-methoxyaniline (1.41k) – The compound was
synthesized according to General Procedure 1.4.2.2 using 3.84 mmol of the
aldehyde. The product was purified by flash chromatography using hexanes–
EtOAc (9:1, v:v) as the mobile phase and was isolated as an off-white solid
(949 mg, 3.09 mmol, 58%, mp = 59–60 °C). 1H NMR (500 MHz, CDCl3) δ 7.44 – 6.99 (m, 2H),
6.37 (d, J = 8.7 Hz, 1H), 6.21 (s, 1H), 3.97 – 3.55 (m, 5H). 13C NMR (125 MHz, CDCl3) δ 160.6,
145.1, 133.3, 130.1, 114.4, 104.1, 100.6, 91.0, 54.9. IR (thin film, cm-1) 3464, 3377, 3207, 2997,
2959, 2936, 2835, 1612, 1566, 1462. HRMS (DART, M+1) Calculated for C9H10Br2NO 305.9129,
found 305.9127.
1.4.3 Palladium-Catalyzed Synthesis of 2-Cyanoindoles
1.4.3.1 General Procedure for the Substrate Scope
To an oven dried 3-dram vial, substrate (0.5 mmol, 1 equiv), Zn(CN)2 (32.3 mg, 0.275 mmol, 0.55
equiv), Zn(TFA)2 (14.6 mg, 0.05 mmol, 10 mol %), and K3PO4 (212.3 mg, 1 mmol, 2 equiv) were
added. After the vial was purged with Ar for 5 min, 1 ml of PhMe and 1 ml of DMA was added.
To a second oven dried vial, Pd(t-Bu3P)2 (12.8 mg, 0.025 mmol, 5 mol %) was added and purged
with Ar for 5 min. The catalyst was dissolved in 2 ml of PhMe and the solution was transferred to
the substrate mixture. The reaction was stirred at 1000 rpm and 110 °C for 18 h. The reaction
contents were transferred into a separatory funnel, diluted in EtOAc, and the organic phase was
washed multiple times with water and brine. The organic layer was concentrated, and the product
was purified by silica gel flash chromatography. It was necessary to let the silica stand in 5% NEt3
in hexanes before loading the column to achieve better separation and prevent minor
decomposition.

32
1.4.3.2 Procedure for 1 mmol Scale Reaction
To an oven dried vial, substrate (1 mmol, 1 equiv), Zn(CN)2 (64.6 mg, 0.55 mmol, 0.55 equiv),
Zn(TFA)2 (29.1 mg, 0.1 mmol, 10 mol %), and K3PO4 (424.5 mg, 2 mmol, 2 equiv) were added.
After the vial was purged with Ar for 5 min, 2 ml of PhMe and 2 ml of DMA was added. To a
second oven dried vial, Pd(t-Bu3P)2 (25.6 mg, 0.05 mmol, 5 mol %) was added and purged with
Ar for 5 min. The catalyst was dissolved in 4 ml of PhMe and the solution was transferred to the
substrate mixture. The reaction was stirred at 1000 rpm and 110 °C for 18 h. The reaction contents
were transferred into a separatory funnel, diluted in EtOAc, and the organic phase was washed
multiple times with water and brine. The organic layer was concentrated, and the product was
purified by silica gel flash chromatography. It was necessary to let the silica stand in 5% NEt3 in
hexanes before loading the column to achieve better separation and prevent minor decomposition.
1.4.3.3 Characterization Data for New Compounds
1H-indole-2-carbonitrile (1.61a) – The compound was synthesized according
to General Procedure 1.4.3.1. The product was purified by flash
chromatography using hexanes–EtOAc–DCM (17:2:1, v:v:v) as the mobile
phase and was isolated as a white solid (0.5 mmol scale: 52.6 mg, 0.37 mmol, 74%, 1 mmol scale:
103.8 mg, 0.73 mmol 73%, mp = 94–95 °C). 1H NMR (500 MHz, CDCl3) δ 8.88 (s, 1H), 7.68 (dd,
J = 8.2, 1.0 Hz, 1H), 7.50 – 7.34 (m, 2H), 7.25 – 7.16 (m, 2H). 13C NMR (125 MHz, CDCl3) δ
137.1, 126.4, 126.3, 122.2, 121.8, 114.6, 114.5, 111.9, 106.2. IR (thin film, cm-1) 3131, 3078,
3051, 2974, 2926, 2236, 1653, 1558, 1522, 1410, 1348. HRMS (DART, M+1) Calculated for
C9H7N2 143.0609, found 143.0607.
1H-benzo[g]indole-2-carbonitrile (1.61b) – The compound was
synthesized according to General Procedure 1.4.3.1. The product was
purified by flash column chromatography using hexanes–EtOAc–DCM
(17:2:1, v:v:v) as the mobile phase and was isolated as an off-white solid (71.0 mg, 0.37 mmol,
74%, mp = 237–240 °C). 1H NMR (500 MHz, DMSO-d6) δ 13.28 (s, 1H), 8.39 (ddt, J = 8.2, 1.4,
0.8 Hz, 1H), 7.98 (ddt, J = 8.0, 1.3, 0.5 Hz, 1H), 7.69 (d, J = 8.7 Hz, 1H), 7.64 (ddd, J = 8.2, 7.0,
1.3 Hz, 1H), 7.60 – 7.53 (m, 2H), 7.48 (s, 1H). 13C NMR (125 MHz, DMSO-d6) δ 133.0, 131.4,
128.6, 126.3, 125.9, 122.3, 122.2, 121.5, 121.2, 120.5, 114.9, 114.4, 103.8. 3316, 3080, 3051,

33
3001, 2214, 1535, 1450. IR (thin film, cm-1) 3316, 3080, 3051, 3001, 2214, 1535, 1450. HRMS
(DART, M+1) Calculated for C13H9N2 193.0766, found 193.0770.
7-methyl-1H-indole-2-carbonitrile (1.61c) – The compound was synthesized
according to General Procedure 1.4.3.1. The data for this compound was
acquired and organized with the assistance of Sabine Bognar. The product was
purified by flash column chromatography using hexanes–EtOAc–DCM (18:1:1, v:v:v) as the
mobile phase and was isolated as an off-white solid (55.3 mg, 0.354 mmol, 71%, mp = 151–152
°C). 1H NMR (500 MHz, CDCl3) δ 8.65 (s, 1H), 7.53 (ddt, J = 8.0, 1.5, 0.7 Hz, 1H), 7.22 (d, J =
2.1 Hz, 1H), 7.19 (dt, J = 7.1, 1.0 Hz, 1H), 7.14 (dd, J = 7.9, 7.1 Hz, 1H), 2.52 (s, 3H). 13C NMR
(125 MHz, CDCl3) δ 136.9, 126.6, 126.0, 122.2, 121.2, 119.9, 115.0, 114.6, 106.0, 16.7. IR (thin
film, cm-1) 3288, 3063, 2964, 2915, 2857, 2227, 1617, 1526, 1432, 1363. HRMS (DART, M+18)
Calculated for C10H12N3 174.1031, found 174.1031.
4-methyl-1H-indole-2-carbonitrile (1.61d) – The compound was synthesized
according to General Procedure 1.4.3.1. The product was purified by flash
column chromatography using hexanes–EtOAc–DCM (18:1:1, v:v:v) as the
mobile phase and was isolated as an off-white solid (54.0 mg, 0.346 mmol,
69%, mp = 83–84 °C). 1H NMR (500 MHz, CDCl3) δ 8.92 (s, 1H), 7.35 – 7.20 (m, 3H), 7.01 (dt,
J = 6.8, 1.1 Hz, 1H), 2.56 (s, 3H). 13C NMR (125 MHz, CDCl3) δ 136.8, 131.8, 126.4, 126.4,
121.5, 114.5, 113.0, 109.3, 105.4, 18.6. IR (thin film, cm-1) 3480, 3326, 3131, 3063, 2918, 2854,
2226, 1615, 1518, 1473. HRMS (DART, M+1) Calculated for C10H9N2 157.0766, found
157.0771.
1-methyl-1H-indole-2-carbonitrile (1.61e) – The compound was synthesized
according to General Procedure 1.4.3.1. The data for this compound was
acquired and organized with the assistance of Sabine Bognar. The product was
purified by flash column chromatography using hexanes–EtOAc–DCM (40:1:1, v:v:v) as the
mobile phase and was isolated as an off-white solid (55.8 mg, 0.36 mmol, 72%). Spectral data
match literature reports.58 1H NMR (500 MHz, CDCl3) δ 7.67 (dt, J = 8.1, 1.0 Hz, 1H), 7.41 (ddd,
J = 8.5, 6.8, 1.1 Hz, 1H), 7.36 (dq, J = 8.5, 1.0 Hz, 1H), 7.21 (ddd, J = 8.1, 6.8, 1.1 Hz, 1H), 7.16
58
Ushijima, S.; Moriyama, K. Tetrahedron, 2011, 67, 958.

34
(d, J = 0.9 Hz, 1H), 3.91 (s, 3H). 13C NMR (125 MHz, CDCl3) δ 138.0, 126.1, 125.8, 122.3, 121.3,
113.6, 112.6, 110.2, 110.1, 31.5.
1-isopropyl-1H-indole-2-carbonitrile (1.61f) – The compound was
synthesized according to General Procedure 1.4.3.1. The product was purified
by flash column chromatography using hexanes–EtOAc–DCM (40:1:1, v:v:v)
as the mobile phase and was isolated as a colorless oil (66.3 mg, 0.36 mmol, 72 %). 1H NMR (500
MHz, CDCl3) δ 7.66 (ddd, J = 8.1, 0.9 Hz, 1H), 7.52 – 7.45 (m, 1H), 7.37 (ddd, J = 8.6, 7.0, 1.2
Hz, 1H), 7.22 – 7.15 (m, 2H), 4.92 (m, J = 7.0 Hz, 1H), 1.71 (d, J = 7.0 Hz, 6H). 13C NMR (125
MHz, CDCl3) δ 136.5, 126.5, 125.4, 122.4, 121.1, 114.5, 114.2, 111.0, 107.8, 49.4, 22.0. IR (thin
film, cm-1) 3117, 3053, 2980, 2936, 2882, 2222, 1612, 1514, 1447. HRMS (DART, M+1)
Calculated for C12H13N2 185.1079, found 185.1076.
1-benzyl-1H-indole-2-carbonitrile (1.61g) – The compound was synthesized
according to General Procedure 1.4.3.1. The product was purified by flash
column chromatography using hexanes–MTBE (40:1, v:v:v to 20:1, v:v:v) as
the mobile phase and was isolated as a colorless oil (89.7 mg, 0.386 mmol, 77 %). 1H NMR (500
MHz, CDCl3) δ 7.70 (dt, J = 8.1, 1.0 Hz, 1H), 7.39 – 7.27 (m, 5H), 7.25 – 7.16 (m, 4H), 5.48 (s,
2H). 13C NMR (125 MHz, CDCl3) δ 137.6, 136.1, 129.0, 128.2, 126.9, 126.5, 126.1, 122.5, 121.7,
113.8, 113.6, 110.8, 110.1, 49.1. IR (thin film, cm-1) 3119, 3063, 3032, 2926, 2220, 1616, 1603,
1518, 1452. HRMS (DART, M+1) Calculated for C16H13N2 233.1079, found 233.1078.
5-methoxy-1H-indole-2-carbonitrile (1.61h) – The compound was
synthesized according to General Procedure 1.4.3.1. The product was
purified by flash column chromatography using hexanes–EtOAc–NEt3
(17:2:1, v:v:v) as the mobile phase and was isolated as a white solid (65.9 mg, 0.38 mmol, 77%,
mp = 146–147 °C). 1H NMR (500 MHz, CDCl3) δ 8.53 (s, 1H), 7.34 – 7.28 (m, 1H), 7.11 (d, J =
1.0 Hz, 1H), 7.08 – 7.03 (m, 2H), 3.85 (s, 3H). 13C NMR (125 MHz, CDCl3) δ 155.4, 132.1, 126.7,
118.0, 114.2, 113.9, 112.6, 106.4, 101.9, 55.7. IR (thin film, cm-1) 3287, 3121, 2955, 2940, 2835,
2241, 1450, 1362, 1173. HRMS (DART, M+1) Calculated for C10H9N2O 173.0715, found
173.0717.

35
methyl 2-cyano-1H-indole-5-carboxylate (1.61i) – The compound was synthesized with the
following modification to General Procedure 1.4.3.1: Pd(t-Bu3P)2
(19.2 mg, 0.0376 mmol, 7.5 mol %), Zn(TFA)2 (21.9 mg, 0.075, 15
mol %), at 120 °C. The data for this compound was acquired and
organized with the assistance of Sabine Bognar. The product was purified by flash column
chromatography using hexanes–EtOAc–DCM (17:2:1, v:v:v) as the mobile phase and was isolated
as a white solid (48 mg, 0.240 mmol, 48%, decomp at 230 °C). 1H NMR (500 MHz, DMSO-d6)
δ 12.74 (s, 1H), 8.38 (dd, J = 1.6, 0.7 Hz, 1H), 7.91 (dd, J = 8.7, 1.7 Hz, 1H), 7.55 (dt, J = 8.8, 0.9
Hz, 1H), 7.53 (d, J = 0.9 Hz, 1H), 3.86 (s, 3H). 13C NMR (125 MHz, DMSO-d6) δ 166.5, 139.2,
125.7, 125.5, 124.6, 122.5, 114.7, 114.0, 112.5, 107.7, 52.0. IR (thin film, cm-1) 3234, 3161, 2958,
2926, 2854, 2228, 1683, 1614, 1428. HRMS (DART, M+1) Calculated for C11H9N2O2 201.0664,
found 201.0666.
5-fluoro-1H-indole-2-carbonitrile (1.61j) – The compound was
synthesized with the following modification to General Procedure 1.4.3.1:
Pd(t-Bu3P)2 (19.2 mg, 0.0376 mmol, 7.5 mol %), Zn(TFA)2 (21.9 mg, 0.075,
15 mol %), at 120 °C. The data for this compound was acquired and organized with the assistance
of Sabine Bognar. The product was purified by flash column chromatography using hexanes–
EtOAc–DCM (18:1:1, v:v:v) as the mobile phase and was isolated as an off-white solid (53.6 mg,
0.335 mmol, 67%, mp = 111–112 °C). 1H NMR (500 MHz, CDCl3) δ 9.02 (s, 1H), 7.37 (ddt, J =
9.0, 4.3, 0.8 Hz, 1H), 7.31 (ddd, J = 9.0, 2.5, 0.6 Hz, 1H), 7.19 – 7.11 (m, 2H). 13C NMR (125
MHz, CDCl3) δ 158.7 (d, J = 238.2 Hz), 133.6, 126.6 (d, J = 10.8 Hz), 115.6 (d, J = 27.1 Hz),
114.3 (d, J = 5.4 Hz), 114.1, 113.0 (d, J = 9.5 Hz), 107.7, 106.5 (d, J = 23.9 Hz). 19F NMR (564
MHz, CDCl3) δ -121.44 (m). IR (thin film, cm-1) 3295, 3090, 2991, 2917, 2227, 1635, 1578, 1520,
1492. HRMS (DART, M+1) Calculated for C9H6FN2 161.0515, found 161.0518.
6-methoxy-1H-indole-2-carbonitrile (1.61k) – The compound was
synthesized according to General Procedure 1.4.3.1. The product was
purified by flash column chromatography using hexanes–EtOAc–DCM (17:2:1, v:v:v) as the
mobile phase and was isolated as a white solid (58.7 mg, 0.34 mmol, 68%, mp = 98–99 °C). 1H
NMR (500 MHz, CDCl3) δ 8.67 (s, 1H), 7.52 (d, J = 8.8 Hz, 1H), 7.14 (s, 1H), 6.88 (dd, J = 8.8,
2.2 Hz, 1H), 6.82 (s, 1H), 3.86 (s, 3H). 13C NMR (125 MHz, CDCl3) δ 159.5, 138.0, 122.8, 120.4,
114.7, 114.7, 113.3, 104.6, 93.6, 55.5. IR (thin film, cm-1) 3358, 3011, 2965, 2932, 2218, 1628,

36
1582, 1510, 1448, 1400. HRMS (DART, M+1) Calculated for C10H9N2O 173.0715, found
173.0713.
methyl 2-cyano-1H-indole-6-carboxylate (1.61l) – The compound
was synthesized according to General Procedure 1.4.3.1. The data for
this compound was acquired and organized with the assistance of
Sabine Bognar. The product was purified by flash column chromatography using hexanes–EtOAc–
DCM (17:2:1, v:v:v) as the mobile phase and was isolated as an off-white solid (75.2 mg, 0.376
mmol, 75%, decomp at 216 °C). 1H NMR (500 MHz, DMSO-d6) δ 8.07 (dt, J = 1.6, 0.9 Hz, 1H),
7.78 (dd, J = 8.5, 0.8 Hz, 1H), 7.71 (dd, J = 8.5, 1.5 Hz, 1H), 7.44 (d, J = 0.9 Hz, 1H), 3.87 (s,
3H). 13C NMR (125 MHz, DMSO-d6) δ 166.5, 136.2, 129.1, 126.4, 121.9, 121.0, 114.2, 114.0,
113.0, 109.1, 52.2. IR (thin film, cm-1) 3279, 2361, 2330, 2226, 1692, 1559, 1534, 1560, 1441.
HRMS (DART, M+1) Calculated for C11H9N2O2 201.0664, found 201.0666.
6-fluoro-1H-indole-2-carbonitrile (1.61m) – The compound was
synthesized according to General Procedure 1.4.3.1. The product was
purified by flash column chromatography using hexanes–PhMe (3:1, v:v) as
the mobile phase and was isolated as a white solid (48.6 mg, 0.303 mmol, 61%, mp = 139–140
°C). 1H NMR (500 MHz, CDCl3) δ 8.62 (s, 1H), 7.62 (ddt, J = 8.8, 5.3, 0.7 Hz, 1H), 7.19 (dd, J
= 2.2, 1.0 Hz, 1H), 7.10 (dddd, J = 9.1, 2.3, 1.0, 0.6 Hz, 1H), 7.00 (ddd, J = 9.4, 8.8, 2.2 Hz, 1H).
13C NMR (125 MHz, CDCl3) δ 162.3 (d, J = 244.4 Hz), 137.0 (d, J = 13.0 Hz), 123.3 (d, J = 10.4
Hz), 122.8, 114.6 (d, J = 1.2 Hz), 113.9 (d, J = 2.4 Hz), 111.5 (d, J = 25.3 Hz), 106.7, 97.9 (d, J =
26.7 Hz). 19F NMR (470 MHz, CDCl3) δ -114.31 (m). IR (thin film, cm-1) 3277, 3140, 3072, 2928,
2857, 2229, 1627, 1590, 1522, 1450. HRMS (DART, M+1) Calculated for C9H6FN2 161.0515,
found 161.0516.
7-methoxy-1H-indole-2-carbonitrile (1.61n) – The compound was
synthesized according to General Procedure 1.4.3.1. The product was purified
by flash column chromatography using hexanes–EtOAc–DCM (17:2:1, v:v:v)
as the mobile phase and was isolated as a white solid (66.6 mg, 0.39 mmol, 77%, mp = 125–126
°C). 1H NMR (500 MHz, CDCl3) δ 8.85 (s, 1H), 7.25 (d, J = 8.0 Hz, 1H), 7.16 (d, J = 2.2 Hz, 1H),
7.13 (dd, J = 8.1, 7.7 Hz, 1H), 6.78 (d, J = 7.7 Hz, 1H), 3.98 (s, 3H). 13C NMR (125 MHz, CDCl3)
δ 146.1, 128.2, 127.3, 122.2, 114.4, 114.1, 114.1, 105.9, 104.8, 55.5. IR (thin film, cm-1) 3343,

37
3080, 3007, 2963, 2938, 2911, 2839, 2228 1628, 1584, 1530, 1447. HRMS (DART, M+1)
Calculated for C10H9N2O 173.0715, found 173.0716.
5H-[1,3]dioxolo[4,5-f]indole-6-carbonitrile (1.61o) – The compound
was synthesized with the following modification to General Procedure
1.4.3.1: Pd(t-Bu3P)2 (19.2 mg, 0.0376 mmol, 7.5 mol %), Zn(TFA)2 (21.9
mg, 0.075, 15 mol %), at 120 °C. The product was purified by flash column chromatography using
hexanes–EtOAc–DCM (40:5:2, v:v:v) as the mobile phase and was isolated as a white solid (48.9
mg, 0.26 mmol, 53%, mp = 210–211 °C). 1H NMR (500 MHz, DMSO-d6) δ 12.20 (s, 1H), 7.16
(s, 1H), 7.06 (s, 1H), 6.94 (s, 1H), 6.02 (s, 2H). 13C NMR (125 MHz, DMSO-d6) δ 147.9, 144.1,
132.9, 120.1, 114.9, 113.3, 103.7, 101.1, 98.8, 92.0. IR (thin film, cm-1) 3311, 3125, 3009, 2917,
2209, 1528, 1499, 1475. HRMS (DART, M+18) Calculated for C10H10N3O2 204.0773, found
204.0772.
5-chloro-1H-indole-2-carbonitrile (1.61p) – The compound was
synthesized according to General Procedure 1.4.3.1. The product was
purified by flash column chromatography using hexanes–EtOAc–DCM
(18:1:1, v:v:v) as the mobile phase and was isolated as an off-white solid (61.5 mg, 0.35 mmol,
70%, decomp at 144 °C). 1H NMR (500 MHz, CDCl3) δ 8.68 (s, 1H), 7.66 (dt, J = 1.7, 0.9 Hz,
1H), 7.36 – 7.34 (m, 2H), 7.14 (dd, J = 2.1, 0.7 Hz, 1H). 13C NMR (125 MHz, CDCl3) δ 135.1,
127.7, 127.1, 126.9, 121.3, 113.8, 113.5, 112.8, 107.6. IR (thin film, cm-1) 3310, 3115, 3069,
2963, 2924, 2232, 1518, 1483, 1414, 1346. HRMS (DART, M+1) Calculated for C9H6ClN2
177.0220, found 177.0219.
1H-indole-2,5-dicarbonitrile (1.61q) – The compound was synthesized
with the following modification to General Procedure 1.4.3.1: Zn(CN)2
(129.2 mg, 1.1 mmol, 2.2 equiv), Pd(t-Bu3P)2 (19.2 mg, 0.0376 mmol,
7.5 mol %), Zn(TFA)2 (21.9 mg, 0.075, 15 mol %), at 120 °C. The product was purified by flash
column chromatography using hexanes–EtOAc–DCM (8:1:1, v:v:v) as the mobile phase and was
isolated as a white solid (51.0 mg, 0.305 mmol, 61%, mp = 227–228 °C). 1H NMR (500 MHz,
DMSO-d6) δ 8.26 (dd, J = 1.5, 0.9 Hz, 1H), 7.71 – 7.57 (m, 2H), 7.50 (d, J = 0.8 Hz, 1H). 13C
NMR (125 MHz, DMSO-d6) δ 138.4, 128.1, 127.5, 125.5, 119.7, 113.9, 113.8, 113.7, 113.7,

38
108.6, 103.4. IR (thin film, cm-1) 3250, 3130, 3054, 2991, 2928, 2857, 2227, 1618, 1476. HRMS
(DART, M+H) Calculated for C10H6N3 168.0562, found 168.0564.
1.4.4 Derivatization of 2-Cyanoindole
2-(1H-tetrazol-5-yl)-1H-indole (1.63) – To an oven dried 3-dram vial, 1.41 (138.5 mg, 0.5 mmol,
1 equiv), Zn(CN)2 (32.3 mg, 0.275 mmol, 0.55 equiv), Zn(TFA)2 (14.6 mg, 0.05 mmol, 10 mol
%), and K3PO4 (212.3 mg, 1 mmol, 2 equiv) were added. After the vial was purged with Ar for 5
min, 1 ml of PhMe and 1 ml of DMA was added. To a second oven dried vial, Pd(t-Bu3P)2 (12.8
mg, 0.025 mmol, 5 mol %) was added and purged with Ar for 5 min. The catalyst was dissolved
in 2 ml of PhMe and the solution was transferred to the substrate mixture. The reaction was stirred
at 1000 rpm and 110 °C for 18 h. Once the vial had cooled to rt, NaN3 (65 mg, 1 mmol, 2 equiv)
and NH4Cl (53.5 mg, 1 mmol, 2 equiv) were added and the reaction was reheated to 110 ºC and
stirred at 1000 rpm for 14 h. The reaction contents were transferred into a separatory funnel, diluted
in EtOAc, and the organic phase was washed multiple times with water and brine. The organic
layer was concentrated, and the product was purified by silica gel flash chromatography
using DCM–MeOH–AcOH (100:1:1, v:v:v) as the mobile phase and was isolated as a white solid
(60.2 mg, 0.325 mmol, 65%). Spectral data was in accordance to reported data.59 1H NMR (500
MHz, DMSO-d6) δ 12.16 (s, 1H), 7.67 (d, J = 7.9 Hz, 1H), 7.51 (dd, J = 8.2, 1.0 Hz, 1H), 7.22
(ddd, J = 8.2, 7.0, 1.2 Hz, 1H), 7.19 (dd, J = 2.2, 0.9 Hz, 1H), 7.08 (ddd, J = 8.0, 7.0, 1.0 Hz, 1H).
13C NMR (125 MHz, DMSO-d6) δ 150.3, 137.5, 127.5, 123.6, 122.1, 121.2, 120.2, 112.3, 103.7.
59
Kou, X.; Zhao, M.; Qiao, X.; Zhu, Y.; Tong, X.; Shen, Z. Chem. - Eur. J. 2013, 19, 16880.

39
1.4.5 Substrates of Mechanistic Studies
2-bromo-1H-indole (1.48) – was synthesized according to literature procedure
and the spectral data matched reported value.41b 1H NMR (500 MHz, CDCl3) δ
8.03 (s, 1H), 7.56 (ddt, J = 7.8, 1.5, 0.8 Hz, 1H), 7.29 (dq, J = 8.1, 0.9 Hz, 1H),
7.20 (ddd, J = 8.2, 7.1, 1.3 Hz, 1H), 7.15 (ddd, J = 7.8, 7.1, 1.1 Hz, 1H), 6.56 (dd, J = 2.1, 0.9 Hz,
1H). 13C NMR (125 MHz, CDCl3) δ 136.4, 128.7, 122.2, 120.5, 119.6, 110.3, 108.7, 104.8.
Upon treatment of 1.48 to general procedure 1.4.3.1, 1.61 was isolated (66.1 mg, 0.465 mmol,
93%) by flash column chromatography using hexanes–EtOAc–DCM (17:2:1, v:v:v). as the mobile
phase.
2-(bromoethynyl)aniline (1.62) – was synthesized according to literature
procedure and the spectral data matched reported value.60 1H NMR (500 MHz,
DMSO-d6) δ 7.15 (ddd, J = 7.7, 1.6, 0.5 Hz, 1H), 7.06 (ddd, J = 8.2, 7.2, 1.6 Hz,
1H), 6.69 (ddd, J = 8.2, 1.2, 0.5 Hz, 1H), 6.48 (ddd, J = 7.7, 7.2, 1.2 Hz, 1H), 5.42 (s, 2H). 13C
NMR (125 MHz, DMSO-d6) δ 150.3, 132.2, 129.9, 115.7, 114.0, 105.1, 77.4, 55.8.
Upon treatment of 1.62 to general procedure 1.4.3.1, 60% of 1.62 was recovered.
Chapter 2
60
Kunzer, A. R.; Wendt, M. D. Tetrahedron Lett. 2011, 52, 1815.

40
Migratory Insertion Strategy for Indole Dearomatization
2.1 Introduction
2.1.1 Elementary View on Aromatization
Aromaticity refers to the extra level of stability that planar rings with conjugated -bonds exhibit
when they obey Huckel’s rule of 4n+2 electrons (Scheme 34, left).61 Importantly, unsaturated rings
which contain 4n electrons have reduced stability. They are said to be anti-aromatic when planar,
and will twist to avoid overlap between their -bond (Scheme 34, right).
Scheme 34 Example of aromatic system vs. anti-aromatic system
The increase in stability could be described by molecular orbital theory; aromatic compounds have
all bonding orbitals filled with paired electrons. For simple rings, drawing MO-diagrams could be
done by placing the apex of a ring towards the bottom and considering all points of the ring as
energy levels (Scheme 35).
Scheme 35 Simple method for drawing the MO-diagram of benzene -bonds
61
(a) E, Hückel, “Grundzüge der Theorie ungesáttigter und aroma tischer Verbindungen,” Verlag Chemie, Berlin,
1938, 71. (b) von E. Doering, W.; Detert, F. L. J. Am. Chem. Soc. 2002, 73, 876.

41
2.1.2 Strategies for Benzene Dearomatization
The dearomatization of aromatic compounds has been of long-standing interest as a way to access
otherwise inaccessible products. The appeal of this strategy in retrosynthesis lies in the possibility
of a difunctionalization as well as further manipulations of the remaining -systems, which now
lack resonance stabilization energy (Scheme 36).
Scheme 36 Benefits of dearomatization. After dearomatization, -bonds react as simple olefins
in future functionalizations
2.1.2.1 Classic Reactions: Birch Reduction
In the mid 1940’s, Arthur Birch reported on arguably the simplest method for the reduction of
benzene to the cyclohexadiene (Scheme 37).62 The reaction only requires cheap alkali-metals
dissolved in liquid ammonia, and alcohol, and has predictable selectivity.
Scheme 37 Birch Reduction products for EDG and EWG substituted aryl-rings
The selectivity for arenes with EDG typically proceeds to give the most substituted 1,4-diene
whilst arenes with electron-withdrawing substituents typically give the unsubstituted 1,4-diene.
The reaction proceeds by a single-electron reduction by the solvated electride [Na(NH3)6]+[e-]
62
(a) Birch, A. J. J. Chem. Soc. 1944, 430. For a collection of the work of A. J. Birch, see Tetrahedron 1988, 44, V.

42
formed in situ as a strong reducing agent capable of dearomatizing the system and is characterized
by a bright blue color (Scheme 38).63
Scheme 38 Mechanism of the Birch Reduction
Additionally, the final anion formed in the reaction can be trapped with electrophiles, providing
functionalized dienes. The Studer group has dubbed these dienes “pro-aromatic”; upon releasing
an equivalent of HCN, they gain 33–36 kcal of energy (Scheme 39, 2.5).64
Scheme 39 Trapping of the final anion intermediate of the Birch Reduction and its use in
Palladium-chemistry as a formal source of HCN
2.1.2.2 Modern Reactions: MTAD
For many years, the development of new benzene dearomatization chemistry has been largely
dominated by phenol/aniline dearomatization (Scheme 40). The dearomatization of phenols and
63
For a case of isolated electrides, see: (a) Dye, J. L. Science 1990, 247, 663. For reports examining the mechanism
of the Birch Reduction, see: (b) Krapcho, A. P.; Bothner, A. A. J. Am. Chem. Soc. 1959, 81, 3658. (c) Wooster, C. B.;
Godfrey, K. L. J. Am. Chem. Soc. 1937, 59, 596. (d) Birch, A. J. Q. Rev., Chem. Soc. 1950, 4, 69. 64
Bhunia, A.; Bergander, K.; Studer, A. J. Am. Chem. Soc. 2018, 140, 16353.

43
anilines fall within two categories; nucleophilic dearomatizations with external electrophilic
species (such as metal-allyl complexes or aryl halides) or oxidative electrophilic dearomatizations
with external nucleophiles and oxidants (such as hypervalent iodine species).65
Scheme 40 Modern methods of dearomatization with metal catalysts or oxidants
Recent research on arene dearomatization led by the Sarlah group has focused on the development
of reagents which rapidly undergo cycloadditions with arenes, labeled arenophiles.66 Reagents
such as N-methyl-1,2,4-triazoline-3,5-dione (MTAD, 2.7) participate in photochemically induced
cycloadditions, forming dearomatized intermediates (2.8) bearing isolated olefins capable of future
functionalizations (Scheme 41).67
Scheme 41 Cycloaddition of arenophile, MTAD, with arenes
Their method offers an expediated route to densely substituted unsaturated ring systems. Once the
olefin has undergone functionalization, the authors have demonstrated methods for removing the
MTAD moiety. For example, a retro-[4+2] yields the conjugated diene, or reduction to access the
65
Wu, W. T.; Zhang, L.; You, S. L. Chem. Soc. Rev. 2016, 45, 1570. 66
Wertjes, W. C.; Southgate, E. H.; Sarlah, D. Chem. Soc. Rev. 2018, 47, 7996. 67
Southgate, E. H.; Pospech, J.; Fu, J.; Holycross, D. R.; Sarlah, D. Nat. Chem. 2016, 8, 922.
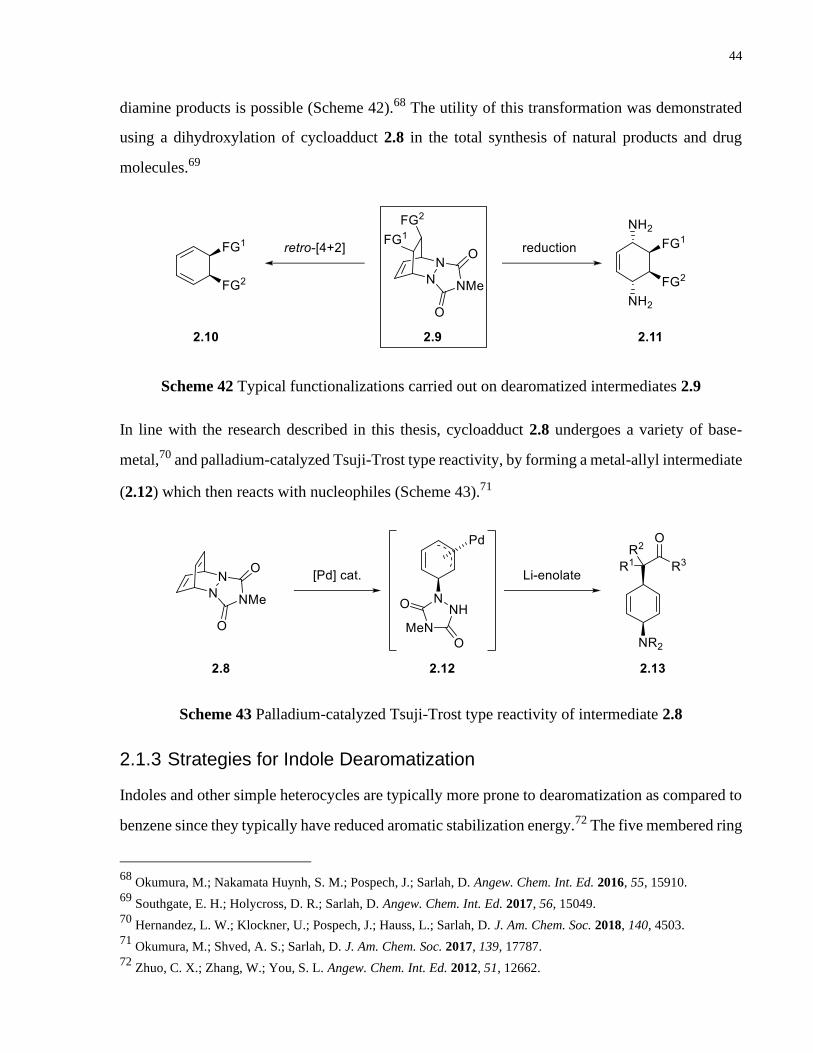
44
diamine products is possible (Scheme 42).68 The utility of this transformation was demonstrated
using a dihydroxylation of cycloadduct 2.8 in the total synthesis of natural products and drug
molecules.69
Scheme 42 Typical functionalizations carried out on dearomatized intermediates 2.9
In line with the research described in this thesis, cycloadduct 2.8 undergoes a variety of base-
metal,70 and palladium-catalyzed Tsuji-Trost type reactivity, by forming a metal-allyl intermediate
(2.12) which then reacts with nucleophiles (Scheme 43).71
Scheme 43 Palladium-catalyzed Tsuji-Trost type reactivity of intermediate 2.8
2.1.3 Strategies for Indole Dearomatization
Indoles and other simple heterocycles are typically more prone to dearomatization as compared to
benzene since they typically have reduced aromatic stabilization energy.72 The five membered ring
68
Okumura, M.; Nakamata Huynh, S. M.; Pospech, J.; Sarlah, D. Angew. Chem. Int. Ed. 2016, 55, 15910. 69
Southgate, E. H.; Holycross, D. R.; Sarlah, D. Angew. Chem. Int. Ed. 2017, 56, 15049. 70
Hernandez, L. W.; Klockner, U.; Pospech, J.; Hauss, L.; Sarlah, D. J. Am. Chem. Soc. 2018, 140, 4503. 71
Okumura, M.; Shved, A. S.; Sarlah, D. J. Am. Chem. Soc. 2017, 139, 17787. 72
Zhuo, C. X.; Zhang, W.; You, S. L. Angew. Chem. Int. Ed. 2012, 51, 12662.

45
of the indole can be dearomatized relatively easily since the 6-membered ring remains intact (vide
supra, Scheme 3, page 3). This strategy offers two important benefits: milder reaction conditions
allows for stereoselective methods, and 2,3-substituted indolines are of particular interest as they
are present in countless natural products.73
2.1.3.1 Push-Pull Dearomatization
Indole dearomatization has been largely dominated by a push-pull approach; a nucleophilic attack
of the C3–position forms an intermediate iminium ion (2.15) which is trapped by a nucleophile
(Scheme 44, 2.16).74 This topic will be the main focus of chapter 3 (see section 3.1.3 on page 124).
Scheme 44 Push-pull dearomatization of indoles
2.1.3.2 Migratory Insertion Strategy – An Umpolung Approach
One strategy for chemical dearomatization, which has been explored in recent years, is transition-
metal-catalyzed reactions which feature a migratory insertion as the dearomative step.75 The key-
step leading to dearomatized products relies on an interrupted-Heck reaction (Scheme 45).76 The
mechanism of the Mizoroki-Heck Reaction involves oxidative addition of C–X bond, syn 1,2-
migratory insertion into an alkene, and -hydride elimination (Scheme 45, Prod A). The
Interrupted Heck Reaction involves trapping of the -bound palladium intermediate with a
nucleophile by reductive elimination (Scheme 45, Prod B).
73
Boal, B. W.; Schammel, A. W.; Garg, N. K. Org. Lett. 2009, 11, 3458. 74
Roche, S. P.; Youte Tendoung, J.-J.; Tréguier, B. Tetrahedron 2015, 71, 3549. 75
Zeidan, N.; Lautens, M. Synthesis 2019, 51, 4137. 76
Thornbury, R. T.; Saini, V.; Fernandes, T. A.; Santiago, C. B.; Talbot, E. P. A.; Sigman, M. S.; McKenna, J. M.;
Toste, F. D. Chem. Sci. 2017, 8, 2890.
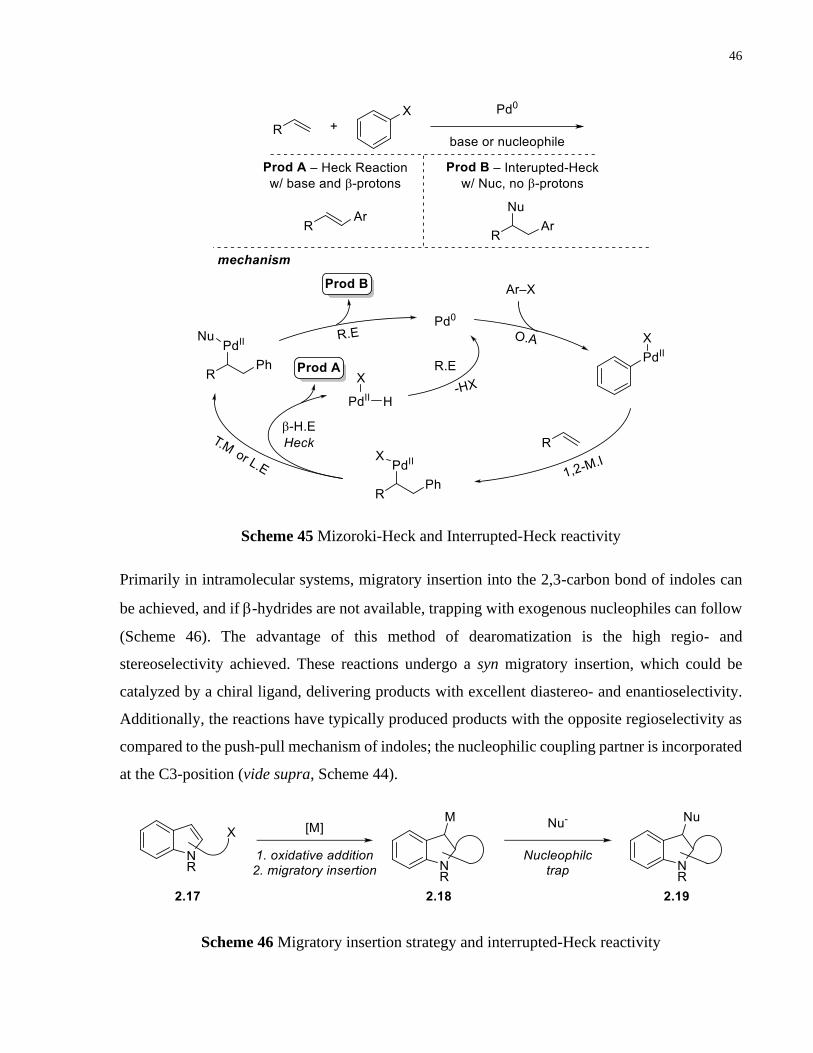
46
Scheme 45 Mizoroki-Heck and Interrupted-Heck reactivity
Primarily in intramolecular systems, migratory insertion into the 2,3-carbon bond of indoles can
be achieved, and if -hydrides are not available, trapping with exogenous nucleophiles can follow
(Scheme 46). The advantage of this method of dearomatization is the high regio- and
stereoselectivity achieved. These reactions undergo a syn migratory insertion, which could be
catalyzed by a chiral ligand, delivering products with excellent diastereo- and enantioselectivity.
Additionally, the reactions have typically produced products with the opposite regioselectivity as
compared to the push-pull mechanism of indoles; the nucleophilic coupling partner is incorporated
at the C3-position (vide supra, Scheme 44).
Scheme 46 Migratory insertion strategy and interrupted-Heck reactivity

47
2.1.3.2.1 Dearomative Monofunctionalization of Indoles
The first report of a Pd-catalyzed dearomative Heck-reaction on indoles was by Yao and Wu in
2012 (Scheme 47).77 The authors used 2,3-disubstituted-N-(2-halobenzoyl)indoles (2.20) to
accomplish the intramolecular dearomatization, delivering indolines (selected examples: 2.21–
2.23) bearing an exo-cyclic double bond in excellent yields. These substrates had been used
previously by Grigg78 and coworkers (vide infra, Scheme 55).
Scheme 47 Intramolecular Mizoroki-Heck Reaction for an efficient route to fused indolines
The reaction was found to be very efficient, utilizing only 2 mol% Pd(OAc)2 and in nearly all
cases, very high yields were achieved. A Pd0-PdII mechanism was proposed, and the active catalyst
is likely formed by the reduction of PdII by DMF (Scheme 48).79
77
Zhao, L.; Li, Z.; Chang, L.; Xu, J.; Yao, H.; Wu, X. Org. Lett. 2012, 14, 2066. 78
Brown, S.; Clarkson, S.; Grigg, R.; Thomas, W. A.; Sridharan, V.; Wilson, D. M. Tetrahedron 2001, 57, 1347. 79
Molina de la Torre, J. A.; Espinet, P.; Albéniz, A. C. Organometallics 2013, 32, 5428.

48
Scheme 48 Mechanism proposed for the intramolecular Mizoroki-Heck reaction of N-(2-
halobenzoyl)indoles
Oxidative addition into the substrate C–X bond results in a PdII complex (2.24). Following a Ag-
mediated ligand exchange (2.25), carbopalladation across the C2-C3 bond of the indole furnishes
the dearomatized intermediate with a chiral -bound benzylic Pd species (2.26). A -hydride
elimination furnishes the indoline product (2.21), and the catalyst is regenerated by reductive
elimination.
A domino approach was discovered by Jia by taking aryl-amides 2.27 and subjecting them to a
Larock indole synthesis/dearomatization sequence (Scheme 49).80 The N-(2-
bromobenzoyl)indoles 2.20 are formed in situ by Larock annulation and thereby undergo a
dearomative Heck reaction using dppf as the bidentate phosphine ligand.
80
Liang, R. X.; Xu, D. Y.; Yang, F. M.; Jia, Y. X. Chem. Commun. 2019, 55, 7711.

49
Scheme 49 Domino Larock annulation/dearomative Heck reaction of N-(2-iodoaryl)benzamides
A target-focused example of this reaction was demonstrated by Kitamura and Fukuyama who
reported a total synthesis of (+)-Hinckdentine A (2.31), utilizing an enantioselective dearomative
Heck reaction as the key step (Scheme 50).81 Until then, studies on the biological activity of this
compound were hindered due to a shortage in supply. The catalytic dearomatization of 2.29 could
be scaled up to 10 g, delivering 2.30 in near quantitative yield and excellent er, which could be
recrystallized to >99:1 er. Importantly, the authors utilized a chiral and bulky monodentate
phosphoramidite ligand L1 which would set the stage for future enantioselective
difunctionalizations (vide infra, section 2.1.3.2.2).
81
Douki, K.; Ono, H.; Taniguchi, T.; Shimokawa, J.; Kitamura, M.; Fukuyama, T. J. Am. Chem. Soc. 2016, 138,
14578.

50
Scheme 50 Total synthesis of (+)-Hinckdentine A using a key enantioselective dearomative
Heck reaction
A reliable highly enantioselective variation of the reaction was reported by Yao and Wu, shown
in Scheme 47, as well as the enantioselective dearomatization of pyrrole was achieved
simultaneously by Jia82 and You83 in 2018, using (S)-SEGPHOS as the ligand (Scheme 51).
Scheme 51 Reaction conditions reported by Jia for the enantioselective dearomative Heck
reaction of pyrroles
82
Li, X.; Zhou, B.; Yang, R. Z.; Yang, F. M.; Liang, R. X.; Liu, R. R.; Jia, Y. X. J. Am. Chem. Soc. 2018, 140, 13945. 83
Yang, P.; You, S. L. Org. Lett. 2018, 20, 7684.

51
The authors report a broad scope for both the indolines and dihydropyrrole products (2.33).
Notably, the aryl-triflate analogue of 2.20 was necessary to access the indoline products; possibly
allowing bidentate ligation via a cationic palladium mechanism with the non-coordinating
counterion.
In 2015, Jia reported the first enantioselective dearomative reductive Heck-reaction on this class
of substrates (Scheme 52).84
Scheme 52 Enantioselective reductive Heck-reaction of indoles
Excellent yields and enantioselectivity were observed, and the reaction conditions were quite
flexible in that a wide range of solvents, and bases were tolerated. Notably, a simple chiral
bidentate phosphine like BINAP was sufficient to deliver products (2.35) with high
enantioenrichment. The scope of the reaction was later extended to tolerate vinyl-halides by using
indoles bearing N-(2-bromocyclohexene) moieties.85
A second class of substrates worth mentioning arises by tethering the aryl-halide to the C-2 or C-
3 position of the heterocycle. This was first demonstrated by Ma and Xu in 2016 by tethering a
halo-aniline via an amide bond at the C-2 position of indoles (Scheme 53).86
84
Shen, C.; Liu, R. R.; Fan, R. J.; Li, Y. L.; Xu, T. F.; Gao, J. R.; Jia, Y. X. J. Am. Chem. Soc. 2015, 137, 4936. 85
Liang, R.-X.; Yang, R.-Z.; Liu, R.-R.; Jia, Y.-X. Org. Chem. Front. 2018, 5, 1840. 86
Wei, F.; Wei, L.; Zhou, L.; Tung, C.-H.; Ma, Y.; Xu, Z. Asian J. Org. Chem. 2016, 5, 971.

52
Scheme 53 Palladium-catalyzed dearomatization of indoles by arylation/borylation then
protodeborylation, or arylation/hydride reduction
Complex spirocyclic indolines/oxindoles (2.37) are accessed in high yield by means of
intramolecular arylation by migratory insertion, generating intermediate 2.38. The benzylic-
palladium intermediate is then either trapped with B2Pin2 and further protodeborylated, or directly
trapped with a strong hydridic source.
The enantioselective variant, as well as an extended scope was later developed in 2018 by Jia and
coworkers (Scheme 54).87
87
(a) Li, X.; Zhou, B.; Yang, R. Z.; Yang, F. M.; Liang, R. X.; Liu, R. R.; Jia, Y. X. J. Am. Chem. Soc. 2018, 140,
13945. (b) Douki, K.; Shimokawa, J.; Kitamura, M. Org. Biomol. Chem. 2019, 17, 1727.
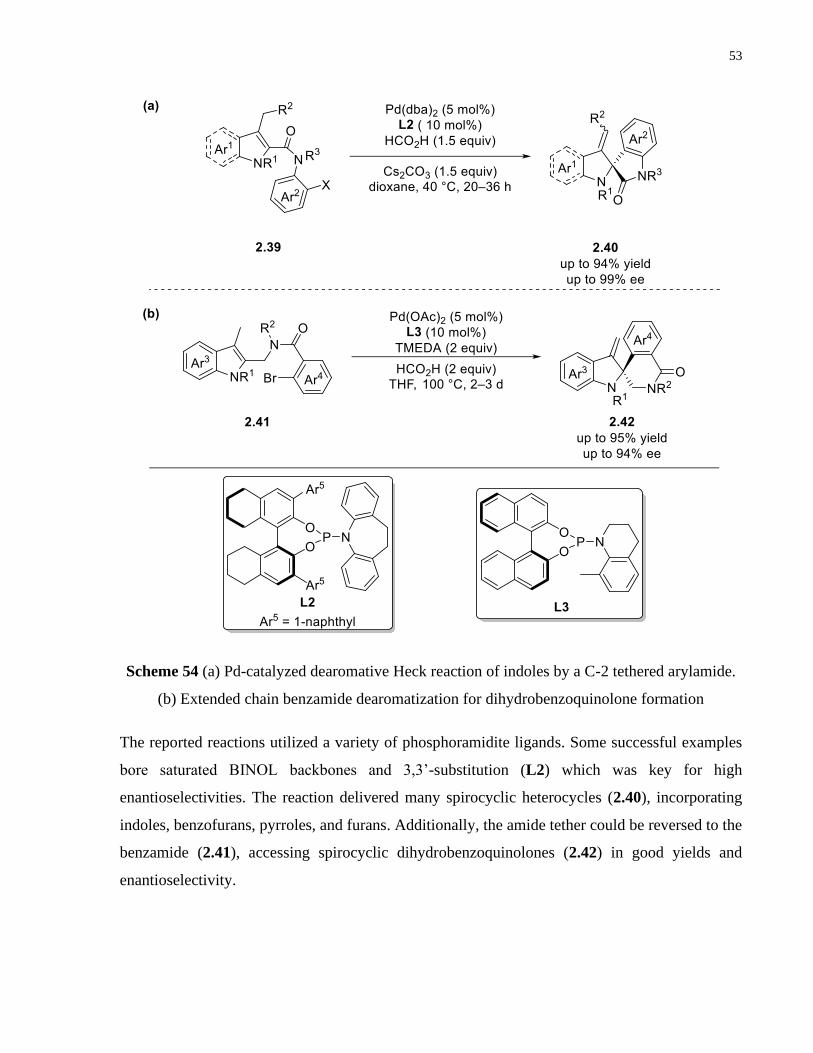
53
Scheme 54 (a) Pd-catalyzed dearomative Heck reaction of indoles by a C-2 tethered arylamide.
(b) Extended chain benzamide dearomatization for dihydrobenzoquinolone formation
The reported reactions utilized a variety of phosphoramidite ligands. Some successful examples
bore saturated BINOL backbones and 3,3’-substitution (L2) which was key for high
enantioselectivities. The reaction delivered many spirocyclic heterocycles (2.40), incorporating
indoles, benzofurans, pyrroles, and furans. Additionally, the amide tether could be reversed to the
benzamide (2.41), accessing spirocyclic dihydrobenzoquinolones (2.42) in good yields and
enantioselectivity.

54
2.1.3.2.2 Dearomative Difunctionalizations – Interrupted Heck Reaction
The seminal work on the palladium-catalyzed dearomative difunctionalization of indoles via an
interrupted Heck mechanism by Grigg was reported in 2001 (Scheme 55).78 The N-benzoyl indoles
(2.43) reported were previously explored in radical cyclizations.88 Grigg describes the “queuing
process” for what is now commonly referred to as a domino process. Furthermore, the authors
describe the idea of “relay switch” species such as carbon monoxide which switches the cascade
between intramolecular to intermolecular.
Scheme 55 Palladium-catalyzed dearomative bisfunctionalization of indoles
Only one example was reported, and it arose by a sequence of arylation and
carbonylation/heteroarylation (2.44), with the major byproduct being from the direct
carbonylation/heteroarylation (2.45). Grigg concluded that the low yield was due to the poor
reactivity of the secondary alkyl-Pd-intermediate which is sterically encumbered within the bowl
shape of the molecule (intermediate 2.46).
This reactivity wasn’t further investigated until studies by the Lautens group in 2015, where we
reported a Pd-catalyzed diastereoselective bisfunctionalization of these types of indoles with
Zn(CN)2 as the trapping nucleophile (Scheme 56).54
88
Zhang, W.; Pugh, G. Tetrahedron Lett. 1999, 40, 7591.

55
Scheme 56 Pd-catalyzed diastereoselective indole 1,2-difunctionalization by a dearomatization
strategy
The reaction utilizes DtBPF as the ligand and proceeds to give exclusively the product of syn-
addition (2.47) in excellent yields. Some control studies confirmed the hypothesis by Grigg,
namely that the product of syn-migratory insertion is the kinetic product. The nitrile nucleophile
reacts on the concave face and only epimerizes when subjected to base.
Following this report, several groups expanded the capacity of the benzylic Pd-intermediate to be
intercepted with various nucleophiles (Scheme 57). The following examples were all published
within 2016. In the first example, the benzylic-Pd intermediate is trapped with boroxines in a
Suzuki-like mechanism (2.48).89 Jing and Liang90 as well as Jia91 reported a decarboxylative
alkynylation (2.49), and Jia described the direct alkynylation (2.50).92
89
(a) Petrone, D. A.; Kondo, M.; Zeidan, N.; Lautens, M. Chem. Eur. J. 2016, 22, 5684. 90
(b) Chen, S.; Wu, X. X.; Wang, J.; Hao, X. H.; Xia, Y.; Shen, Y.; Jing, H.; Liang, Y. M. Org. Lett. 2016, 18, 4016. 91
(c) Wang, Y.; Liu, R.; Gao, J.; Jia, Y. Chin. J. Org. Chem. 2017, 37, 691-697. 92
(d) Liu, R. R.; Xu, T. F.; Wang, Y. G.; Xiang, B.; Gao, J. R.; Jia, Y. X. Chem. Commun. 2016, 52, 13664.

56
Scheme 57 Palladium-catalyzed dearomative bisfunctionalization of indoles. All three examples
are highly diastereoselective
All three of the examples were highly diastereoselective but racemic when using bulky
monodentate phosphine ligands. Although the reductive Heck published by Jia one year earlier
was enantioselective using BINAP, and tolerated a wide variety of bases and solvents (vide supra,
Scheme 52), these three reactions were reported to give poor or no enantioselectivities. There is
currently no consensus on the reasons for the lack of induction, particularly since the
enantiodetermining step is proposed to precede trapping of the nucleophile. One possibility is that
transmetallation might occur prior to addition to the aryl -bond, and thus the ligands on the metal
center are different.

57
It was not until 2017 that Jia published the first diastereo- and enantioselective dearomative
bisfunctionalization of these N-tethered indoles via an arylation/alkynylation sequence (Scheme
58).93
Scheme 58 First Pd-catalyzed enantioselective dearomatization by difunctionalization of indoles
via arylation/alkynylation sequence
The authors solved the enantioselectivity issue by utilizing a chiral, monodentate, yet bulky ligand
(L4). The ligand is similar to the phosphoramidite used by Kitamura and Fukuyama (vide supra,
Scheme 50) however it was made bulkier by installing 3,3’-aryl rings on the BINOL backbone.
An MTBE–THF mixture was found to be the optimal solvent mixture; MTBE was necessary for
high enantioselectivity and THF was necessary for improved reactivity.
In recent years, many palladium-catalyzed difunctionalization-dearomatization of indoles have
been published and although they are typically diastereoselective, it remains a challenge to access
these scaffolds in high enantioselectivities (Scheme 59).
93
Liu, R. R.; Wang, Y. G.; Li, Y. L.; Huang, B. B.; Liang, R. X.; Jia, Y. X. Angew. Chem. Int. Ed. 2017, 56, 7475.

58
Scheme 59 Palladium-catalyzed diastereoselective trapping of the dearomatized benzylic
intermediate with various nucleophiles
These reactions include the arylation-phosphorylation94 (2.52) and arylation-vinylation95 (2.54)
by Jia, and the arylation-carbonylation96 (2.53) by Wu. In all cases, the products are highly
functionalized indolines, and most are equipped with readily modifiable groups. Jia could access
the vinyl products 2.54 with modest enantioselectivity. The limitation of the enantioselective
conditions for the anion-capture chemistry is that MTBE and Et2O are currently the only two
94
Weng, J.-Q.; Xing, L.-L.; Hou, W.-R.; Liang, R.-X.; Jia, Y.-X. Org. Chem. Front. 2019, 6, 1577. 95
Liang, R.-X.; Wang, K.; Wu, Q.; Sheng, W.-J.; Jia, Y.-X. Organometallics 2019, 38, 3927. 96
Wang, H.; Wu, X.-F. Org. Lett. 2019, 21, 5264.

59
solvents which provide high ee using the phosphoramidite ligands. In the 4 cases above, MTBE
was not a suitable solvent for reactivity. This incompatibility seems to be the biggest restriction
and hence it would be desirable to develop ligands which could tolerate a broader range of solvents
for the enantioselective dearomative bisfunctionalization of indoles.
2.2 Research Goal 1 – Palladium-Catalyzed Dearomative Arylation/Heteroarylation of Indoles
2.2.1 Motivation
The first research goal related to this topic was to develop a palladium-catalyzed dearomative
bisfunctionalization of indoles by means of a migratory insertion, and direct C–H arylation with
activated heterocycles (Scheme 60).97
Scheme 60 General reaction studied in research goal 1
This method would allow for the rapid construction of a large library of complex chiral
heterocycles from simple starting materials. Additionally, direct C–H arylation would be
advantageous as it would remove the need for pre-functionalized coupling partners.
2.2.2 Contributions
The results presented were obtained in collaboration with Dr. Tamara Beisel (postdoc from
Germany) and Rachel Ross (graduate student in the Lautens group). I conceived the idea, directed
the project, and executed most of the experiments. Dr. Tamara Beisel assisted in the optimization
of the reaction and the scope of the heteroaryl coupling partner. Rachel Ross focused on exploring
97
Zeidan, N.; Beisel, T.; Ross, R.; Lautens, M. Org. Lett. 2018, 20, 7332.

60
the scope of the reaction with respect to the indoles. Specific contributions are listed within the
text as well as within the experimental section.
2.2.3 Results and Discussion
2.2.3.1 Starting Material Synthesis
Starting materials are synthesized by benzoylation of the corresponding 2-functionalized indoles
(Scheme 61). The indoles which are not commercially available could be synthesized by the Fang-
Lautens indole synthesis (See section 1.1.4 on page 11).30
Scheme 61 Synthesis of N-benzoyl indoles
Starting materials (2.34) were synthesized by deprotonating 2-functionalzied indoles (2.57), and
reacting them with derivatives of 2-halobenzoyl chlorides (see section 2.5.2). Although the yields
of this method are generally low, the cheap cost of materials and scalability of the reaction make
it ideal for scope studies.
2.2.3.2 Optimization
We began our studies with benzothiazole (BTA) as the coupling partner based on the reactivity of
the C–H bond and for its potentially useful therapeutic effects.98 After screening the reaction
parameters, we found that 0.2 mmol of 2.34-Br with 5 mol % Pd(t-Bu3P)2, 8 equiv of BTA, 2
equiv of Cs2CO3, 40 mol % CuCl2 in toluene at 120 °C for 15 h provided the dearomatized product
2.58a in quantitative yield as a single diastereomer, as confirmed by single-crystal X-ray
analysis.99 To assess the importance of the reaction parameters, we altered the conditions and
evaluated the impact (Table 4).
98
Kamal, A.; Syed, M. A. H.; Mohammed, S. M. Expert Opin. Ther. Pat. 2015, 25, 335. 99
CCDC deposition number: 1858836.

61
Table 4 Effects of reaction parameters in the dearomative arylation/heteroarylation of indolesa
Entry Change to standard condition 2.58a (%)b drc
1 None >99 >20:1
2 1.2 equiv of Cs2CO3 67 >20:1
3 4 equiv of BTA 83 >20:1
4 No CuCl2 60 3.2:1
5 CuCl instead of CuCl2 62 5.6:1
6b PivOH instead of CuCl2 74 4.6:1
7b Added 10 L H2O (2.8 equiv) 97 >20:1
8 1 g scale instead of 0.2 mmol scale 88 >20:1
aReactions run on 0.2 mmol scale. bYield of 2.58a determined by NMR using trimethoxybenzene as an internal
standard. cDiastereomeric ratio determined by NMR. BTA = benzothiazole
Importantly, other aryl phosphine, alkyl phosphine, and Buchwald ligands were not efficient in
catalyzing this reaction. Lowering the number equivalents of base (Table 4, entry 2) or BTA (Table
4,entry 3) resulted in a slightly diminished yield, with no impact on dr. Omitting CuCl2 from the
reaction (Table 4, entry 4) resulted in diminished yields as well as lower dr, suggesting that the
additive influences the rate or longevity of the catalyst as well as affects epimerization. Notably,
CuCl (Table 4, entry 5) as well as various Cu(I or II), Zn(II), Fe(III), and Yb(III) salts did not

62
improve the yield or dr. Pivalic acid, a common additive in C–H activation,100 improves the yield,
but at the expense of poor diastereoselectivity (Table 4, entry 6). Finally, we found that the reaction
tolerates a modest amount of water without catalyst deactivation (Table 4, entry 7). Gratifyingly,
we were able to scale the reaction up to 1 g with minimal loss in yield (Table 4, entry 8).
2.2.3.3 Exploring the Scope of the Reaction
With the optimized conditions in hand, we investigated the scope with respect to the indole (Table
5). Product 2.58a could be accessed on a 1 g scale, or by using the aryl-iodide or aryl-chloride
variants of 2.34 in good yield with excellent dr.
Relative stereochemistry was assigned by analogy to 2.58a. An EWG para- to the benzoyl linker
(2.58b) or an EDG para- to the halide (2.58c) were tolerated; however, the opposite electronic
pattern provided products 2.58d and 2.58e with diminished yields. Substitution at the 5-position
of the indole provided products 2.58f and 2.58g, albeit in reduced stereoselectivity. However,
excellent dr was observed for the difluoro product 2.58h. A longer alkyl chain at the 2-position, as
in the case of 2.58i or protected amine 2.58j, was tolerated. Additionally, Weinreb amide 2.58k,
and a variety of 2-aryl indoles (2.58l –2.58o), all participated in the reaction. Finally, vinyl halide
2.34p was a compatible substrate in the diarylation reaction providing product 2.58p in good yield.
Slow hydrolysis was observed for electron deficient starting materials providing 2-methylindole
as a side product. In all other cases, only product was observed by crude NMR suggesting other
decomposition pathways are operative.
100
Lafrance, M.; Fagnou, K. J. Am. Chem. Soc. 2006, 128, 16496.

63
Table 5 Investigating the substrate scope with respect to the indolea
2.58a
from 2.34-Br
(>99%, >20:1 dr)
2.58a
from 2.34-I
(65%, >20:1 dr)
2.58a
from 2.34-Cl
(48%, >20:1 dr)b
2.58b
(94%, >20:1 dr)c
2.58c
(77%, >20:1 dr)b,d
2.58d
(46%, >20:1 dr)d
2.58e
(90%, >20:1 dr)d

64
2.58f
(95%, 9.6:1 dr)
2.58g
(71%, 10.8:1 dr)d
2.58h
(52%, >20:1 dr)
2.58i
(90%, >20:1 dr)d
2.58j
(78%, >20:1 dr)d
2.58k
(63%, >20:1 dr)b,d
2.58l
(90%, >20:1 dr)d
2.58m
(73%, >20:1 dr)e,d
2.58n
(88%, >20:1 dr)d
2.58o
(87%, >20:1 dr)d

65
2.58p
(78%, >20:1 dr)d
aUnless otherwise noted, reactions run on a 0.2 mmol scale. bReaction run using 10 mol % catalyst cReaction run at
100 °C. dReactions were run by Rachel Ross. eReaction run on 0.1 mmol scale.
Next, we examined the scope with respect to the C–H partner (Table 6). Pyridine-N-oxide was an
excellent coupling partner, providing product 2.59 in excellent yield and selectivity as compared
to the standard reaction. Although not a heterocycle, pentafluorobenzene was sufficiently electron
deficient towards C-H activation to undergo the reaction and delivered product 2.60, in good yield
and excellent selectivity albeit requiring a slight increase in catalyst loading. No regioselectivity
was observed with 4-methylthiazole, providing both C-2 and C-5 arylation products in 62% (2.61)
and 31% (2.62) yields, respectively, however, they could be easily separated by column
chromatography. Indole and benzofuran did not have sufficiently low energy of activation to
undergo the desired reaction.101
101
Gorelsky, S. I.; Lapointe, D.; Fagnou, K. J. Am. Chem. Soc. 2008, 130, 10848.
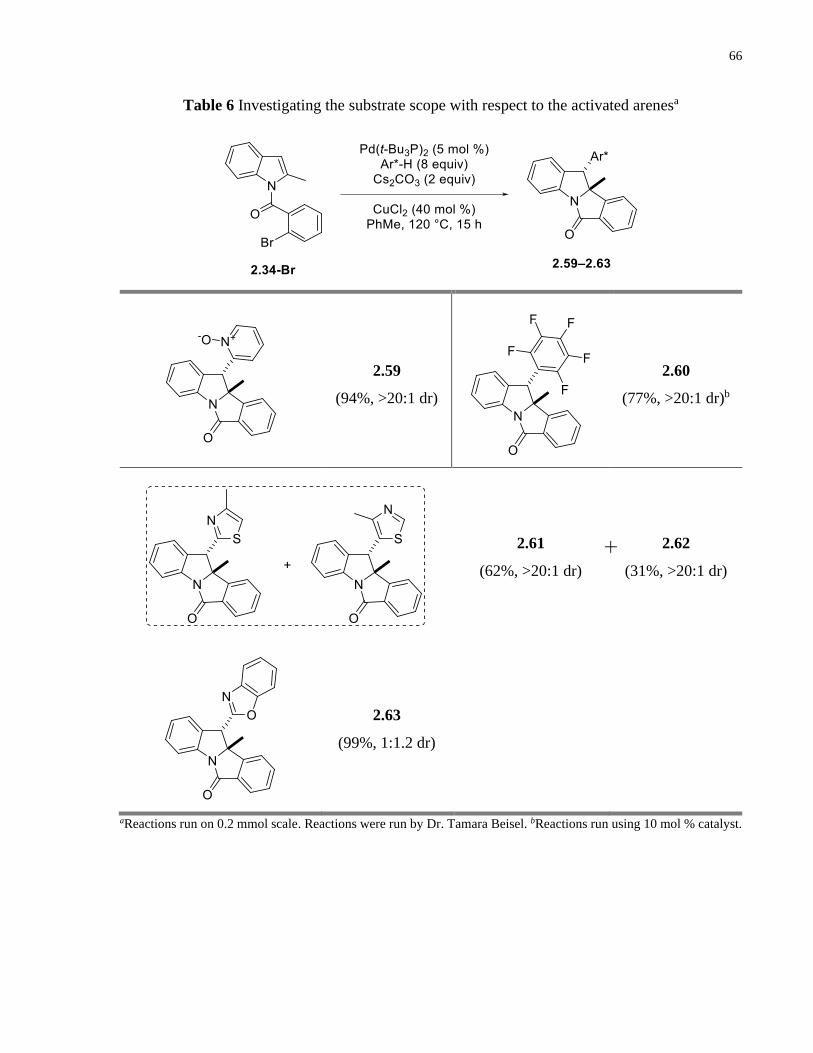
66
Table 6 Investigating the substrate scope with respect to the activated arenesa
2.59
(94%, >20:1 dr)
2.60
(77%, >20:1 dr)b
2.61
(62%, >20:1 dr)
2.62
(31%, >20:1 dr)
2.63
(99%, 1:1.2 dr)
aReactions run on 0.2 mmol scale. Reactions were run by Dr. Tamara Beisel. bReactions run using 10 mol % catalyst.

67
Under the standard conditions, benzoxazole provided the dearomatized product (2.63) in
quantitative yield, albeit with poor selectivity. The diastereomers converged to a single
diastereomer by alkylating the acidic position to give two fully substituted contiguous centers
(Scheme 62, 2.64).102
Scheme 62 Convergence of diastereomers by the alkylation of 2.63
Indoles, benzofurans, and benzothiophenes were not tolerated in the reaction and only trace
product could be observed.
2.2.3.4 Investigation of the Role of Copper in the Reaction
We also investigated the role of copperII chloride on the reaction as both the yield and the dr of the
product increased when used (vide supra, section 2.2.3.2 on page 60). We subjected a sample of
the product to Cs2CO3 in toluene at 120 °C for 15 hours (Table 7, entry 1). Although full recovery
of the product was seen, the dr eroded to 2.4:1. The addition of 40 mol% copper gave a similar
outcome (Table 7, entry 2). However, in the presence of 2 equiv of copper, full recovery of the
material was achieved with no erosion in dr (Table 7, entry 3). We attribute the need for 2 equiv
to the relative concentrations of copper and the product relative to base during the progress of the
reaction. That is, during the standard reaction, as the concentration of product increases, the
concentration of base decreases, and hence, a smaller amount of copper is sufficient to maintain
high dr values. The possibility of a diastereromeric resolution by copper was investigated (Table
102
Only one isomer was observed under these reaction conditions. Alkylation proposed to occur stereoselectively
from the convex face due to the rigid structure of the molecule (see ref Error! Bookmark not defined. on page 76).

68
7, entry 4), but no clarity was achieved. However, there was no change in dr suggesting
epimerization is suppressed in both diastereomers.
Table 7 Epimerization studies and probing of the effects of CuII chloridea
Entry Change to standard condition 2.58a+2.58a’ (%)b drc
1 None 98 2.4:1
2 CuCl2 (40 mol%) 79 2.3:1
3 CuCl2 (2 equiv) 97 >20:1
4 CuCl2 (2 equiv) and
sample of 2.58a used was 4:1 dr 89 4:1
aReactions run on 0.1 mmol scale. bYield of 2.58a determined by NMR using trimethoxybenzene as an internal
standard. cDiastereomeric ratio determined by NMR.
2.2.3.5 Conclusion of Research Goal 1
In conclusion, we have developed a palladium-catalyzed diastereoselective dearomatization of
indoles by a 1,2-arylation/direct heteroarylation sequence. Functionalized indolines are accessed
in good to excellent yields and selectivity. The role of copperII chloride was investigated and was
found to prevent epimerization of the easily epimerizable dibenzylic center, as well as improve
conversions.

69
2.3 Research Goal 2 – Arylation/Borylation of Indoles
2.3.1 Motivation
The second research goal of the chapter was to develop a palladium-catalyzed dearomative
aryl/borylation of indoles. Enantioenriched indolines which possess chiral C–B bonds are useful
for future transformations (Scheme 63).103
Scheme 63 Dearomative aryl/borylation with palladium
Boron containing molecules serve as important building blocks: the C–B bond can be used as a
handle for forging new carbon−carbon or carbon−heteroatom bonds (Scheme 64).104 This can been
achieved by metal-catalyzed, radical, oxidative, and reductive methods.
Scheme 64 There is a plethora of precedence for the chemical transformation of the C–B bond to
various other atoms
Palladium-catalysis has been used as a reliable method of borylation of aryl-halides (Scheme 65,
top).105 The domino Heck-borylation reaction, employing boron reagents as nucleophiles, is an
103 Shen, C.; Zeidan, N.; Wu, Q.; Breuers, C. B. J.; Liu, R.-R.; Jia, Y.-X.; Lautens, M. Chem. Sci. 2019, 10, 3118.
104 (a) Pelter, A.; Smith K.; Brown, H. C. Borane Reagents; Academic Press: London, 1988; (b) Miyaura, N.; Suzuki,
A. Chem. Rev. 1995, 95, 2457. (c) Davison, M.; Hughes, A. K.; Marder T. B.; Wade, K. Contemporary Boron
Chemistry; RSC: Cambridge, U.K., 2000. (d) Boronic Acids: Preparation and Applications in Organic Synthesis
Medicine and Materials, 2nd edn., (Ed.: Hall, D. G.), Wiley-VCH, Weinheim, 2011. 105 (a) Ishiyama, T.; Murata, M.; Miyaura, N. J. Org. Chem. 1995, 60, 7508. (b) Takagi, J.; Takahashi, K.; Ishiyama,
T.; Miyaura, N. J. Am. Chem. Soc. 2002, 124, 8001.

70
efficient method to synthesize Csp3-based organoboron compounds (Scheme 65, middle and
bottom).106
Scheme 65 Miyaura-Borylation of aryl-halides and domino methods
It is worthwhile to extend this methodology for the synthesis of structurally diverse, chiral
organoboron heterocycles by dearomatization. At the time of project planning, there had been
numerous studies on forming C–H and C–C bond by dearomative migratory insertion strategy
however, no C–heteroatom bond forming reactions had been explored.
2.3.2 Contributions
The results presented were obtained in collaboration with Christian Breuers (visiting master’s
student from Germany). I conceived the idea, directed the project, and executed most of the
experiments. Christian Breuers synthesized many of the ligands and assisted in the optimization.
Specific contributions are listed within the text as well as within the experimental section.
106 (a) Vachhani, D. D.; Butani, H. H.; Sharma, N.; Bhoya, U. C.; Shah, A. K.; Van der Eycken, E. V. Chem. Commun.
2015, 51, 14862. (b) Lautens, M.; Yoon, H.; Jang, Y. Synthesis 2016, 48, 1483.

71
2.3.3 Results and Discussion
2.3.3.1 Starting Material Synthesis and Initial Screening
Starting materials were accessed by the same chemical sequence as described in Research Goal 1
(see section 2.2.3.1) however, the aryl-chlorides were used as they produced higher er in the
enantioselective variant, as described below. In our initial screening using previously encountered
conditions for borylation, we were able to achieve a good yield of 2.65a with the presence of
protodeborylated product 2.66 (Scheme 66). At this point, we joined a collaborative effort for the
development of this protocol with the group of Yi-Xia Jia and they took lead in optimizing the
racemic protocol.103
Scheme 66 Initial racemic aryl/borylation of indoles
We continued our studies with the development of the enantioselective variant. Our chief concern
was that 2.65a underwent protodeborylation when subjected to inorganic bases for prolonged
reaction times at high temperatures (Scheme 67).
Scheme 67 Protodeborylation of 2.65a in the presence of inorganic base

72
Furthermore, the only class of ligands which was successful in delivering enantioenriched products
was phosphoramidites107 however, the reaction conditions needed for their success were
incompatible with our optimized conditions thus far (Scheme 68).93
Scheme 68 Phosphoramidite ligands and their reaction condition requirements
The conditions for borylation, protodeborylation, and ligand compatibility conflicted. It was clear
that the development of an enantioselective protocol would rely on the elimination of the inorganic
base in the reaction.
2.3.3.2 Mixed–Boron Reagent
Classic borylation methods rely on the use of B2Pin2 (2.67) and an inorganic base, typically
CsOPiv, KOAc, NaOt-Bu, etc. for transmetallation to a PdII complex formed in situ from oxidative
addition (Scheme 69).108
Scheme 69 B2Pin2 pre-activation for transmetallation with CsOPiv
The coordination of the base to a boron atom allows for an activated sp2-sp3 bond, where the sp2-
boron center could be transferred onto a palladium complex. The Santos group reported a copper-
107 Teichert, J. F.; Feringa, B. L. Angew. Chem. Int. Ed. 2010, 49, 2486. 108 Chow, W. K.; Yuen, O. Y.; Choy, P. Y.; So, C. M.; Lau, C. P.; Wong, W. T.; Kwong, F. Y. RSC Advances 2013,
3, 12518.

73
catalyzed hydroboration of ,-unsaturated conjugated compounds using a pre-activated mixed-
sp2-sp3 boron reagent (Scheme 70, 2.69).109
Scheme 70 Copper-catalyzed borylation developed by Santos and coworkers
The reagent allows the reaction to occur without the need of exogenous inorganic base. The
transmetallation could be explained by a coordination of the amine (Scheme 71).
Scheme 71 Mixed-boron transmetallation and expected byproduct neutralization
To our knowledge, this reagent had not been used with palladium catalysis. The acidic byproduct
produced did not affect the published copper reaction109 however, we suspected this might inhibit
the palladium catalysts as a H-PdII-Cl species. We could however neutralize the reaction with
organic base to avoid protodeborylation of the oxyphilic borylated products. The synthesis of this
mixed-boron reagent, 2.69, is simple and can be done by mixing commercially available
diolamine (2.70) with B2Pin2 (2.67) under conditions which precipitate the polar product, as it is
formed (Scheme 72).
109 (a) Gao, M.; Thorpe, S. B.; Santos, W. L. Org. Lett. 2009, 11, 3478. (b) Thorpe, S. B.; Guo, X.; Santos, W. L.
Chem. Commun. 2011, 47, 424. (c) Gao, M.; Thorpe, S. B.; Kleeberg, C.; Slebodnick, C.; Marder, T. B.; Santos, W.
L. J. Org. Chem. 2011, 76, 3997.

74
Scheme 72 Synthesis of mixed-boron reagent
2.3.3.3 Ligand Synthesis
Phosphoramidites are a class of privileged ligands discovered and popularized by Feringa and
Alexakis.110 Many ortho-functionalized BINOL derivatives have been synthesized in the
literature, mainly by ortho-lithiation/bromination, Suzuki-Miyaura coupling (Scheme 73).111
Scheme 73 General strategy for the synthesis of 3,3’-substituted BINOLs
This divergent protocol allowed for a large library of ligands to be synthesized rapidly. The
commonly employed method for O-P-N coupling requires the use of PCl3 as an electrophile with
various amines (Scheme 74).112 A work-up with DCM and pentanes allows for the isolation of the
ligands in high yields without the need for column chromatography (see section 2.5.4.2.5).
110 (a) de Vries, A. H. M.; Meetsma, A.; Feringa, B. L. Angew. Chem. Int. Ed. 1996, 35, 2374. (b) Alexakis, A.; Rosset,
S.; Allamand, J.; March, S.; Guillen, F.; Benhaim, C. Synlett 2001, 9, 1375. 111 Wu, T. R.; Shen, L.; Chong, J. M. Org. Lett. 2004, 6, 2701. 112 Smith, C. R.; RajanBabu, T. V. Org. Lett. 2008, 10, 1657.

75
Scheme 74 O-P-N coupling for the synthesis of phosphoramidite ligands
2.3.3.4 Optimization of the Enantioselective Reaction
The enantioselective Heck/borylation reaction of 2.34 was then investigated using
phosphoramidite L5 as the chiral ligand (Table 8). It was necessary to change solvents from DCE
the MTBE since the former was not efficient in the enantioselective variant (Table 8, entry 1 and
2). Although the bromo- and iodo- variations of the substrates delivered product in higher yields
than the aryl-chloride, it was evident that the smaller halide improved the enantioselectivities
(Table 8, entries 3–4). In the case of the aryl-chloride, there was no reaction using B2Pin2 (Table
8, entry 5). We employed an organic base to neutralize the by-product of the boron-reagent and
the conversion improved to 50% while maintaining the ee (Table 8, entry 6).
Preliminary screening suggested that increasing the steric bulk of the ligand improved the yield of
the reaction (Table 8, entry 7) and therefore further optimization studies were carried out with
ligand L6 (Table 8, entries 8–11). Other amines (Table 8, entry 8) or lowering the temperature
(Table 8, entry 9) were not effective. By increasing ligand and reagent loading, the product was
delivered in 74% yield and 94% ee (Table 8, entry 10 and 11). The absolute stereochemistry of
2.65a was assigned by single-crystal X-ray analysis.113
113 CCDC deposition number: 1855293

76
Table 8 Optimization for the enantioselective aryl/borylationa
Entry X Additive Changes to condition yield (%) er (%)
1 Br None None 73 82:18
2 Br None DCE as solvent 34 60:40
3 I None None 40 75:25
4 Cl None None 15 94:6
5 Cl K2CO3 (2 equiv) B2Pin2 (2 equiv) NR –
6 Cl NEt3 (3 equiv) None 50 94:6
7 Cl NEt3 (3 equiv) Using L6 65 94:6
––––––––––Entries 8–11 using ligand L.6 (N-Cy, p-OMe) ––––––––––
8 Cl i-Pr2NEt (3 equiv) None 27 –
9 Cl NEt3 (3 equiv) 80 °C NR –
10 Cl NEt3 (3 equiv) 10 mol% ligand 68 95.5:4.5
11 Cl NEt3 (5 equiv) 10 mol% ligand
3 equiv 2.69 74 97:3
aIsolated yield. er was determined by chiral HPLC. NR = no reaction.

77
With optimized conditions in hand, we studied the effect of ligand substitution on the reaction
using the conditions in Table 8, entry 11 (Table 9). Many ligands, including BINAP, Prophos,
Binapine, and the parent Josiphos or Walphos produced trace or no product in the reaction.
To our surprise, 3,3’-unsubstituted ligands produced trace or no product (Table 9, L7–L9). We
confirmed that the cyclohexylamine ligand L6 was superior to the initially used ligand (Table 9,
L5–L6). Sterically smaller alkyl groups on the nitrogen were worse in catalyzing the reaction
(Table 9, L10). However, attempting to add steric bulk, while maintaining the electronic properties
of the 3,3’-anisole did not improve the reaction (Table 9, L11–L12). Increasing the electron
density in the aryl rings did not improve the reaction (Table 9, L13). We examined the effects of
increasing the steric bulk at the para position while slightly reducing the electronic perturbations,
and although the yield improved, the enantioselectivity slightly diminished (Table 9, L14).
Investigating electron-withdrawing substitutions also did not further improve the selectivity
however, the sterically congested 3,5-CF3-aryl substituted ligand increased the yield as compared
to the anisole ligand (Table 9, L15–L17).
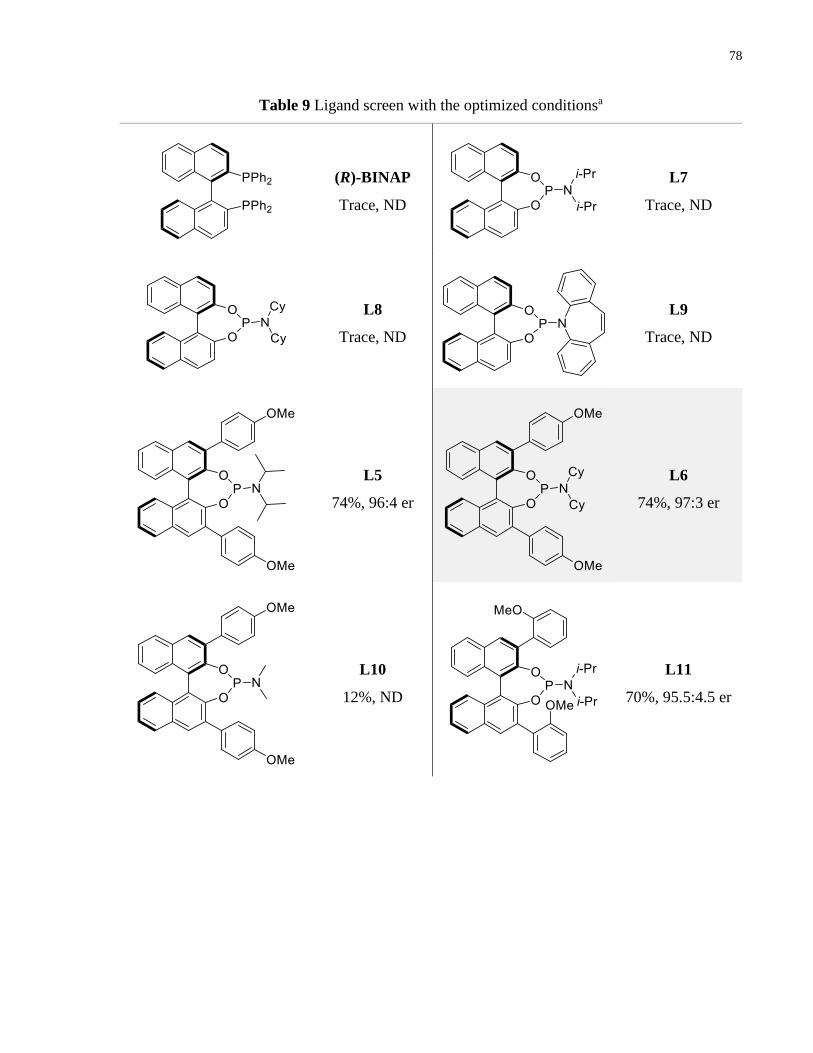
78
Table 9 Ligand screen with the optimized conditionsa
(R)-BINAP
Trace, ND
L7
Trace, ND
L8
Trace, ND
L9
Trace, ND
L5
74%, 96:4 er
L6
74%, 97:3 er
L10
12%, ND
L11
70%, 95.5:4.5 er

79
L12
71%, 90.5:9.5
er
L13
34%, 93:7 er
L14
80%, 93.5:6.5
er
L15
52%, 89:11 er
L16
23%, 92.5:7.5
er
L17
80%, 81.5:18.5
er
aYields determined by NMR using trimethoxybenzene as an internal standard. The er was determined by chiral HPLC.

80
2.3.3.5 Exploring the Substrate Scope
We then examined the scope of the dearomative Heck/borylation on various aryl-chloride
substrates (Table 10). Absolute stereochemistry was assigned by analogy to 2.65a. Sterically
hindered aryl-halide (2.65b), as well as substrates with functionalities para to the amide tether
(2.65c, 2.65d) provided products in diminished enantioselectivities. In contrast, substitution para
to the chloride (2.65e) or on the indole moiety provided 2.65f -2.65g in moderate yields and
excellent enantioselectivities. Aryl functionality at the C2-position of the indole provided 2.65h
and 2.65i in moderate and good yields with excellent enantioselectivities. Finally, a heterocycle
containing scaffold 2.65j was accessed in good yield albeit in a diminished enantioselectivity.

81
Table 10 Examining the scope of the aryl/borylation of indoles
2.65a
74%, 97:3
2.65b
59%, 89.5:10.5
2.65c
88%, 84:16
2.65d
65%, 84:16
2.65e
66%, 95.5:4.5
2.65f
59%, 95.5:4.5
2.65g
65%, 96.5:3.5
2.65h
73%, 97:3
2.65i
40%, 97:3
2.65j
81%, 91:9

82
2.3.3.6 Derivatization of the Products
We investigated the modular versatility of the C–B bond. Oxidation of compound 2.65a with
perborate provided the chiral alcohol 2.77 (Scheme 75).114
Scheme 75 Oxidation of the boron-containing product 2.65a
Additionally, the alcohol could be further oxidized to the ketone 2.78 with no loss in enantiomeric
excess (Scheme 76).115
Scheme 76 Further oxidation with PCC to the benzylic ketone
2.3.4 Section Conclusion
In conclusion, we have developed a dearomative difunctionalization of indoles through a
palladium-catalyzed intramolecular aryl/borylation reaction. Indolines possessing vicinal
borylated tertiary and tetrasubstituted stereocenters were accessed in good yields and excellent
diastereoselectivities. The asymmetric variant of this reaction was explored with a new BINOL-
based chiral phosphoramidite ligand and moderate to excellent enantioselectivities were obtained
114
Kubota, K.; Hayama, K.; Iwamoto, H.; Ito, H. Angew. Chem. Int. Ed. 2015, 54, 8809. 115
For more derivatizations of the C–B bond, see work by Jia in ref 103.

83
(up to 94% ee). Transformations of the benzylic boron to the alcohol and ketone were presented
to show the synthetic versatility of the products.
2.4 Chapter Summary
This chapter has explored the developments of dearomative reactions of indoles by migratory
insertion and nucleophilic trapping. The regioselectivity of the reaction is complementary to
typical indole dearomatization by electrophiles. Currently, these nucleophilic trapping reactions
are known asymmetrically with phosphoramidite ligands, which are typically expensive. I expect
that future research would aim to find cheaper routes to the enantioenriched products by the
development of new catalysts.
2.5 Experimental
2.5.1 General Considerations
Reactions were monitored by thin-layer chromatography (TLC) on EMD Silica Gel 60 F254 plates
and visualized under UV light or by immersion in iodine on silica stain. Flash column
chromatography was performed on Silicycle 40-60 m silica gel. PhMe was distilled over CaH.
Pd(t-Bu3P)2 was purchased from Johnson Matthey. All reagents and organic building blocks were
purchased from commercial supplier (Sigma Aldrich, Strem, Alfa Aesar, TCI, Combi-blocks) and
used without further purification.
1H and 13C NMR spectra were obtained on the Agilent DD2 500 equipped with a 5mm Xsens Cold
Probe. The spectra were internally referenced to the solvent peak. 19F NMR spectra were obtained
on the Varian Mercury 400 or 500 operating at 470 or 564 MHz. Measurements were taken at 296
K and chemical shifts are reported in parts per million (ppm). Data is reported in the following
format: chemical shift ( ppm), multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m =
multiplet), coupling constant (Hz), integration. Melting points were measured on a Fisher-Johns
Melting Point Apparatus and are uncorrected. High resolution mass spectra (HRMS) were obtained
on a Micromass 70S-250 spectrometer (EI) or an ABI/Sciex QStar Mass Spectrometer (ESI) or a
JEOL AccuTOF model JMS-T1000LC mass spectrometer equipped with an IONICS® Direct
Analysis in real Time (DART) ion source. Chiral HPLC analysis was performed on an Agilent HP
1100 or HP 1200 series modular system, operated by ChemStation LC 3D software. Column
specification and conditions described when used.

84
2.5.2 Synthesis of Starting Materials
2.5.2.1 Synthetic Remarks
Starting materials 2.34 were synthesized according to literature procedure.54,89 All other starting
materials were synthesized by a slight variation shown below.
2.5.2.2 General Procedure for the Synthesis of N-(2-halobenzoyl)indoles
To a suspension of benzoic acid derivatives (1.2 –2 equiv) in DCM (1 M) and a few drops of DMF
at 0 °C, was added oxalyl chloride (1.2–2 equiv). Stirring ceased when the reaction stopped
evolving gas and a homogeneous solution was observed. The solvent was evaporated, and the acyl-
chloride formed was redissolved in THF (1 M). In a second flask, a solution of indole (1–50 mmol,
1 equiv) in THF (1 M), using an ice water bath as a heat sink, was added NaH (60% dispersion in
mineral oil, 1.2 equiv) and was stirred at room temperature for 10–30 minutes (noticeable dark
colour change upon deprotonation). The solution of acyl-chloride was added to the deprotonated
indole over 1 minute and the reaction was stirred for 2 hours (noticeable light color change). The
reaction was quenched with NH4Cl(aq) (Caution: exothermic and vigorous reaction with left over
NaH). The aqueous layer was extracted with EtOAc, and the organic layer was washed with
NaHCO3(aq) and brine. After evaporation, the crude mixture was columned on silica gel using
relatively non-polar conditions containing triethylamine to remove all traces of indole which
typically elute rapidly (typically seen as a pink–red color upon standing of pure material).

85
2.5.2.3 Characterization Data for New Compounds
(2-iodophenyl)(2-methyl-1H-indol-1-yl)methanone – The compound
was synthesized according to General Procedure 2.5.2.2. The product was
purified by flash column chromatography using pentanes–EtOAc (100:1,
v:v) as the mobile phase and was isolated as a white solid (1.7 g, 4.6 mmol,
46%). 1H NMR (300 MHz, CDCl3) δ 2.26 (d, J = 1.2 Hz, 3H), 6.42 (p, J = 1.1 Hz, 1H), 7.10 (ddd,
J = 8.5, 7.3, 1.4 Hz, 1H), 7.16 – 7.31 (m, 3H), 7.42 – 7.55 (m, 3H), 7.93 (dd, J = 8.0, 1.1 Hz, 1H).
13C NMR (125 MHz, CDCl3) δ 16.6, 93.2, 110.5, 115.1, 119.9, 123.7, 123.8, 128.7, 129.2, 130.1,
132.0, 137.0, 137.5, 140.0, 142.3, 169.2. IR (thin film, cm-1) 3053, 2964, 2924, 2856, 1682, 1595,
1574, 1455, 1308. HRMS (DART, M+H) Calc’d for C16H13INO 362.0042, found 362.0030.
(2-bromo-4-methoxyphenyl)(2-methyl-1H-indol-1-yl)methanone
– The compound was synthesized according to General Procedure
2.5.2.2. The product was purified by flash column chromatography
using pentanes–toluene–DCM (2:2:1, v:v:v) as the mobile phase and
was isolated as a white solid (1.04 g, 3.0 mmol, 60%, mp = 57–62 °C). 1H NMR (500 MHz,
CDCl3) δ 2.30 (d, J = 1.2 Hz, 3H), 3.89 (s, 3H), 6.40 (p, J = 1.1 Hz, 1H), 6.96 (dd, J = 8.6, 2.5 Hz,
1H), 7.09 (ddd, J = 8.4, 7.2, 1.3 Hz, 1H), 7.16 – 7.20 (m, 2H), 7.24 – 7.29 (m, 1H), 7.40 – 7.46
(m, 2H). 13C NMR (125 MHz, CDCl3) δ 16.1, 55.8, 109.7, 113.8, 114.5, 118.8, 119.7, 121.6,
123.2, 123.4, 129.8, 130.4, 131.0, 136.8, 137.4, 161.9, 167.6. IR (thin film, cm-1) 3074, 2970,
2929, 2841, 1702, 1610, 1594, 1482, 1466, 1375, 1289, 1321, 1217, 1133, 1025, 752, 732, 696.
HRMS (DART, M+H) Calc’d for C24H19N2O2S 399.11672, found 399.11646.
1-(2-bromobenzoyl)-N-methoxy-N-methyl-1H-indole-2-
carboxamide – The compound was synthesized according to General
Procedure 2.5.2.2. The product was purified by flash column
chromatography using pentanes–EtOAc (5:1, v:v) as the mobile phase
and was isolated as a white solid (658.3 mg, 1.7 mmol, 85%, mp = 72–
76 °C). 1H NMR (300 MHz, CDCl3) δ 3.08 (s, 4H), 3.50 (s, 3H), 7.01 (s, 1H), 7.24 – 7.32 (m,
2H), 7.36 – 7.46 (m, 3H), 7.50 – 7.56 (m, 1H), 7.59 – 7.65 (m, 1H), 7.65 – 7.70 (m, 1H). 13C NMR
(125 MHz, CDCl3) δ 33.4, 61.4, 113.1, 114.8, 121.3, 122.0, 124.0, 126.4, 127.4, 128.7, 130.8,
132.5, 132.5, 133.7, 136.6, 136.6, 163.1, 166.7. IR (thin film, cm-1) 3060, 2985, 2947, 1694, 1653,

86
1457, 1420, 1366, 1332, 1312, 958, 754, 692, 625. HRMS (DART, M+H) Calc’d for
C18H15BrN2O3 387.0344, found 387.0343.
(2-chlorophenyl)(2-methyl-1H-indol-1-yl)methanone – The compound
was synthesized according to General Procedure 2.5.2.2. The product was
purified on silica by flash chromatography, eluting with pentanes to
pentanes–NEt3 (40:1, v:v). The product was isolated as a white solid (60% yield, mp = 58–59 °C).
1H NMR (500 MHz, CDCl3) δ 7.52–7.46 (m, 4H), 7.45–7.39 (m, 2H), 7.23 (td, J = 7.5, 1.0 Hz,
1H), 7.14 (ddd, J = 8.5, 7.3, 1.3 Hz, 1H), 6.42 (p, J = 1.1 Hz, 1H), 2.27 (d, J = 1.3 Hz, 3H); 13C
NMR (125 MHz, CDCl3) δ 166.9, 137.2, 136.8, 136.4, 132.0, 131.7, 130.3, 129.9, 129.2, 127.4,
123.7, 123.6, 119.8, 114.9, 110.4, 16.2. HRMS (DART, M+H) calculated for C16H13ClNO
270.0686, found 270.0685.
(2-chloro-3-methylphenyl)(2-methyl-1H-indol-1-yl)methanone –
The compound was synthesized according to General Procedure 2.5.2.2.
The crude reaction mixture was purified on silica by flash
chromatography, eluting with pentanes to pentanes–EtOAc (50:1, v:v). The product was isolated
as a white solid (47% yield, mp = 85–86 °C). 1H NMR (500 MHz, CDCl3) δ 7.46–7.38 (m, 3H),
7.32–7.30 (m, 2H), 7.20 (td, J = 7.5, 1.0 Hz, 1H), 7.11 (ddd, J = 8.5, 7.3, 1.3 Hz, 1H), 6.40 (t, J =
1.0 Hz, 1H), 2.45 (d, J = 0.7 Hz, 3H), 2.24 (d, J = 1.2 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ
167.3, 137.8, 137.3, 136.8, 136.8, 133.0, 131.4, 129.9, 127.0, 126.5, 123.7, 123.5, 119.7, 115.0,
110.3, 20.2, 16.2. HRMS (DART, M+H) calculated for C17H15ClNO 284.0842, found 284.0849.
(2-chloro-4-methoxyphenyl)(2-methyl-1H-indol-1-yl)methanone
– The compound was synthesized according to General Procedure
2.5.2.2. The crude reaction mixture was purified on silica by flash
chromatography. Two columns were required, first eluting with pentanes–EtOAc (10:1, v:v), then
pentanes–DCM–toluene (2:2:1, v:v:v). The product was isolated as a white solid (31% yield, mp
= 105–106 °C). 1H NMR (500 MHz, CDCl3) δ 7.46–7.42 (m, 2H), 7.30–7.24 (m, 1H), 7.20–7.16
(m, 1H), 7.09 (ddd, J = 8.5, 7.2, 1.3 Hz, 1H), 7.00 (d, J = 2.4 Hz, 1H), 6.92 (dd, J = 8.6, 2.5 Hz,
1H), 6.41–6.39 (m, 1H), 3.88 (s, 3H), 2.31 (d, J = 1.2 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ
166.9, 162.2, 137.4, 136.8, 133.5, 131.1, 129.8, 128.3, 123.4, 123.2, 119.7, 115.7, 114.4, 113.3,
109.7, 55.8, 16.0. HRMS (DART, M+H) calculated for C17H15ClNO2 300.0791, found 300.0790.

87
(2-chloro-4-fluorophenyl)(2-methyl-1H-indol-1-yl)methanone – The compound was
synthesized according to General Procedure 2.5.2.2. The crude reaction
mixture was purified on silica by flash chromatography, eluting with
pentanes to pentanes–Et2O (100:1, v:v). The product was isolated as a
white solid (35% yield, mp = 51–52 °C). 1H NMR (500 MHz, CDCl3) δ 7.51 (dd, J = 8.6, 5.8 Hz,
1H), 7.46 (ddd, J = 7.7, 1.3, 0.7 Hz, 1H), 7.32 (dq, J = 8.3, 0.9 Hz, 1H), 7.27–7.19 (m, 2H), 7.17–
7.11 (m, 2H), 6.42 (t, J = 1.1 Hz, 1H), 2.28 (d, J = 1.2 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ
166.2, 163.8 (d, J = 255.6 Hz), 137.2, 136.8, 133.5 (d, J = 10.8 Hz), 132.8 (d, J = 3.8 Hz), 131.2
(d, J = 9.4 Hz), 130.0, 123.8, 123.8, 120.0, 118.1 (d, J = 24.9 Hz), 115.1 (d, J = 21.8 Hz), 114.7,
110.5, 16.3. 19F NMR (377 MHz, CDCl3) δ -105.88 (q, J = 7.9 Hz). HRMS (DART, M+H)
calculated for C16H12ClFNO 288.0591, found 288.0590.
(2-chloro-5-fluorophenyl)(2-methyl-1H-indol-1-yl)methanone – The
compound was synthesized according to General Procedure 2.5.2.2. The
crude reaction mixture was purified on silica by flash chromatography,
eluting with pentanes to pentanes–Et2O, (100:1, v:v). The product was
isolated as a white solid (36% yield, mp = 73–75 °C). 1H NMR (500 MHz, CDCl3) δ 7.48–7.43
(m, 2H), 7.42–7.39 (m, 1H), 7.25–7.19 (m, 3H), 7.14 (ddd, J = 8.5, 7.3, 1.4 Hz, 1H), 6.43 (t, J =
1.2 Hz, 1H), 2.28 (d, J = 1.2 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 165.4 (d, J = 2.0 Hz), 161.2
(d, J = 250.3 Hz), 137.7 (d, J = 7.2 Hz), 136.9, 136.6, 131.9 (d, J = 8.0 Hz), 129.9, 126.7 (d, J =
3.6 Hz), 123.9, 123.8, 119.9, 119.1 (d, J = 22.8 Hz), 116.3 (d, J = 24.7 Hz), 114.7, 110.8, 16.2. 19F
NMR (377 MHz, CDCl3) δ -112.96 (td, J = 8.0, 4.6 Hz). HRMS (DART, M+H) calculated for
C16H12ClFNO 288.0591, found 288.0584.
(2-chlorophenyl)(5-fluoro-2-methyl-1H-indol-1-yl)methanone –
The compound was synthesized according to General Procedure
2.5.2.2. The crude reaction mixture was purified on silica by flash
chromatography, eluting with pentanes to pentanes–Et2O (100:1, v:v). The product was isolated
as a white solid (47% yield, mp = 84–85 °C). 1H NMR (500 MHz, CDCl3) δ 7.56–7.38 (m, 5H),
7.10 (dd, J = 8.6, 2.6 Hz, 1H), 6.85 (td, J = 9.1, 2.6 Hz, 1H), 6.36 (p, J = 1.2 Hz, 1H), 2.19 (d, J =
1.2 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 166.8, 159.8 (d, J = 240.0 Hz), 138.9, 136.3, 133.2
(d, J = 1.4 Hz), 132.2, 131.8, 131.1 (d, J = 10.0 Hz), 130.5, 129.3, 127.6, 116.1 (d, J = 9.1 Hz),
111.3 (d, J = 24.7 Hz), 110.2 (d, J = 3.7 Hz), 105.6 (d, J = 23.8 Hz), 16.3. 19F NMR (377 MHz,

88
CDCl3) δ -119.68 (td, J = 8.6, 8.2, 4.6 Hz). HRMS (DART, M+H) calculated for C16H12ClFNO
288.0591, found 288.0596.
(2-chlorophenyl)(5-methoxy-2-methyl-1H-indol-1-yl)methanone
– The compound was synthesized according to General Procedure
2.5.2.2. The crude reaction mixture was purified on silica by flash
chromatography, eluting with pentanes to pentanes–NEt3 (20:1, v:v). The product was isolated as
a white solid (41% yield, mp = 54–55 °C). 1H NMR (500 MHz, CDCl3) δ 7.51–7.46 (m, 3H),
7.43–7.39 (m, 1H), 7.31 (d, J = 9.0 Hz, 1H), 6.92 (d, J = 2.4 Hz, 1H), 6.72 (dd, J = 9.0, 2.6 Hz,
1H), 6.33 (dt, J = 1.9, 0.9 Hz, 1H), 3.82 (s, 3H), 2.20 (d, J = 1.2 Hz, 3H). 13C NMR (125 MHz,
CDCl3) δ 166.5, 156.4, 137.9, 136.4, 131.8, 131.6, 131.4, 130.9, 130.2, 129.1, 127.3, 115.7, 111.6,
110.4, 102.9, 55.6, 16.2. HRMS (DART, M+H) calculated for C17H15ClNO2 300.0791, found
300.0784.
(2-chlorophenyl)(2-phenyl-1H-indol-1-yl)methanone – The compound
was synthesized according to General Procedure 2.5.2.2. The crude
reaction mixture was purified on silica by flash chromatography, eluting
with pentanes to pentanes–Et2O (100:1, v:v). The product was isolated as
a white solid (46% yield, mp = 86–87 °C). 1H NMR (500 MHz, CDCl3) δ 8.14 (ddt, J = 8.1, 1.4,
0.8 Hz, 1H), 7.61 (ddd, J = 7.3, 1.6, 0.7 Hz, 1H), 7.40–7.29 (m, 2H), 7.28–7.25 (m, 2H), 7.21 (ddd,
J = 7.6, 1.6, 0.6 Hz, 1H), 7.16–7.08 (m, 5H), 7.03 (ddd, J = 7.7, 6.7, 1.9 Hz, 1H), 6.66 (d, J = 0.7
Hz, 1H). 13C NMR (125 MHz, CDCl3) δ 167.4, 140.5, 137.8, 135.6, 132.8, 132.5, 131.8, 130.6,
129.9, 129.5, 128.8, 127.7, 127.7, 126.3, 125.0, 124.0, 120.6, 115.2, 111.4. HRMS (DART, M+H)
calculated for C21H15ClNO 332.0842, found 332.0844.
(2-chlorophenyl)(2-(4-fluorophenyl)-1H-indol-1-yl)methanone – The
compound was synthesized according to General Procedure 2.5.2.2. The
crude reaction mixture was purified on silica by flash chromatography,
eluting with pentanes to pentanes–MTBE (50:1, v:v). The product was
isolated as a white solid (21% yield, mp = 94–96 °C). 1H NMR (500 MHz,
CDCl3) δ 8.11 (ddt, J = 8.2, 1.4, 0.8 Hz, 1H), 7.60 (ddd, J = 7.2, 1.7, 0.7
Hz, 1H), 7.39–7.31 (m, 2H), 7.25–7.21 (m, 3H), 7.18 (ddd, J = 8.1, 7.2, 1.7 Hz, 1H), 7.13 (ddd, J
= 8.2, 1.4, 0.5 Hz, 1H), 7.08 (td, J = 7.5, 1.3 Hz, 1H), 6.85–6.80 (m, 2H), 6.64 (d, J = 0.8 Hz, 1H).

89
13C NMR (125 MHz, CDCl3) δ 167.3, 162.1 (d, J = 248.2 Hz), 139.3, 137.6, 135.5, 132.3, 131.9,
130.6 (d, J = 7.6 Hz), 130.5, 130.0, 129.3, 128.9 (d, J = 3.6 Hz), 126.5, 125.2, 124.1, 120.6, 115.2,
114.8 (d, J = 21.9 Hz), 111.6. 19F NMR (377 MHz, CDCl3) δ -113.45 (ddd, J = 13.9, 9.0, 5.6 Hz).
HRMS (DART, M+H) calculated for C21H14ClFNO 350.0748, found 350.0743.
(2-chloropyridin-3-yl)(2-methyl-1H-indol-1-yl)methanone – The
compound was synthesized according to General Procedure 2.5.2.2. The
crude reaction mixture was purified on silica by flash chromatography,
eluting with pentanes–EtOAc (10:1 to 5:1, v:v). The product was isolated as a white solid (38%
yield, mp = 66–67 °C). 1H NMR (500 MHz, CDCl3) δ 8.60 (dd, J = 4.9, 2.0 Hz, 1H), 7.85 (dd, J
= 7.6, 2.0 Hz, 1H), 7.48–7.41 (m, 3H), 7.23 (td, J = 7.5, 1.0 Hz, 1H), 7.15 (ddd, J = 8.6, 7.3, 1.3
Hz, 1H), 6.44 (p, J = 1.2 Hz, 1H), 2.23 (d, J = 1.2 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 165.2,
151.5, 148.0, 138.0, 136.7, 136.6, 132.9, 130.0, 124.1, 124.0, 122.7, 120.0, 114.8, 111.1, 16.5.
HRMS (DART, M+H) calculated for C15H12ClN2O 271.0638, found 271.0630.
2.5.3 Research Goal 1 – Dearomative Palladium-Catalyzed Arylation/Heteroarylation
2.5.3.1 General Procedure for the Arylation/Heteroarylation
In an oven-dried 2-dram vial equipped with an open-top septum cap purging with an argon balloon
was added substrate 2.34 (0.2 mmol), Pd(t-Bu3P)2 (5 mol %), Cs2CO3 (2 equiv), CuCl2 (40 mol
%), and the heterocycle (8 equiv) if it is a solid at room temperature. After a 10-minute purge,
PhMe (2 mL, 0.1 M) and, in the cases where a liquid heterocycle was used, the heterocycle (8
equiv), in that order. The balloon was removed, and the reaction was heated to 120 °C, for 15 h. It
was necessary to stir the reaction at 1000–1400 rpm to avoid clumping of the base. Upon
completion, the reaction was filtered through a silica plug, concentrated, and columned via silica
gel chromatography.

90
2.5.3.2 Characterization Data for New Compounds
(±)-11-(benzo[d]thiazol-2-yl)-10b-methyl-10b,11-dihydro-6H-
isoindolo[2,1-a]indol-6-one (2.58a) – Was synthesized according to
General Procedure 2.5.3.1. The product was purified by flash column
chromatography using pentanes–EtOAc (9:1 to 7:3, v:v) as the mobile phase
and was isolated as an off-white (73.7 mg, 0.2 mmol, >99%, mp = 223–226
°C). 1H NMR (500 MHz, CDCl3) δ 7.52 – 7.45 (m, 2H), 7.38 (dt, J = 7.6,
1.0 Hz, 1H), 7.34 (ddd, J = 8.3, 7.1, 1.2 Hz, 2H), 7.29 (td, J = 7.5, 1.3 Hz, 1H), 7.24 (td, J = 7.5,
1.1 Hz, 1H), 7.18 (td, J = 7.7, 1.2 Hz, 2H), 5.02 (d, J = 0.6 Hz, 1H), 1.85 (s, 3H). 13C NMR (125
MHz, CDCl3) δ 171.0, 168.4, 152.3, 147.3, 139.3, 136.3, 135.5, 133.0, 132.5, 130.2, 128.9, 127.2,
126.0, 125.3, 125.0, 124.6, 123.6, 122.9, 121.8, 117.8, 75.7, 55.5, 28.3. IR (thin film, cm-1) 3080,
3051, 2980, 2926, 1694, 1603, 1486, 1466, 1357, 1312, 904, 758. HRMS (DART, M+H) Calc’d
for C23H17N2OS 369.1062, found 369.1067.
Syn-diastereomer: 1H NMR (500 MHz, CDCl3) δ 8.16 (ddd, J = 8.2, 1.2, 0.6 Hz, 1H), 7.97 (ddd,
J = 8.0, 1.3, 0.6 Hz, 1H), 7.92 (dt, J = 7.6, 1.0 Hz, 1H), 7.81 (dt, J = 7.7, 0.9 Hz, 1H), 7.76 (dt, J =
7.9, 0.8 Hz, 1H), 7.69 (td, J = 7.5, 1.2 Hz, 1H), 7.60 – 7.52 (m, 2H), 7.51 – 7.42 (m, 3H), 7.21 (td,
J = 7.6, 1.1 Hz, 1H), 5.22 (t, J = 1.2 Hz, 1H), 1.46 (s, 3H). 13C NMR (125 MHz, CDCl3) δ 167.8,
167.7, 153.6, 150.6, 140.0, 135.8, 135.1, 133.4, 132.5, 129.6, 129.2, 126.6, 126.5, 125.7, 125.2,
125.1, 123.4, 123.2, 121.8, 117.8, 75.3, 55.0, 23.6. HRMS (DART, M+H) Calc’d for C23H17N2OS
369.1062, found 369.1062.
Gram-scale: Reaction was run according to General Procedure 2.5.3.1 however, on a scale of 1 g
(3.2 mmol of 1a) in a 100 mL Schlenk bomb (producing 1.04 g of product).
(±)-11-(benzo[d]thiazol-2-yl)-9-fluoro-10b-methyl-10b,11-dihydro-
6H-isoindolo[2,1-a]indol-6-one (2.58b) – Was synthesized according to
General Procedure 2.5.3.1 however using 100 °C reaction temperature.
The product was purified by flash column chromatography using
pentanes–EtOAc (20:1 to 7:3, v:v) as the mobile phase and was isolated
as an off-white (72.65 mg, 0.188 mmol, 94%, mp = 210–214 °C). 1H NMR (500 MHz, CDCl3) δ
7.93 – 7.88 (m, 1H), 7.86 (dd, J = 7.9, 1.1 Hz, 1H), 7.69 (dd, J = 8.4, 5.0 Hz, 1H), 7.51 (td, J = 7.8,
1.1 Hz, 2H), 7.40 – 7.33 (m, 2H), 7.24 – 7.17 (m, 2H), 7.10 (dd, J = 8.2, 2.3 Hz, 1H), 6.94 (td, J =

91
8.7, 2.1 Hz, 1H), 5.01 (s, 1H), 1.84 (s, 3H). 13C NMR (125 MHz, CDCl3) δ 170.6, 167.3, 165.5
(d, J = 254.0 Hz), 152.5, 150.0 (d, J = 9.8 Hz), 139.3, 135.9, 135.4, 130.3, 129.1 (d, J = 2.0 Hz),
127.2, 126.7 (d, J = 9.8 Hz), 126.2, 125.4, 125.2, 123.1, 121.8, 117.8, 116.8 (d, J = 23.6 Hz), 111.2
(d, J = 24.2 Hz), 75.3 (d, J = 2.5 Hz), 55.4, 28.2. 19F NMR (377 MHz, CDCl3) δ -104.95 (td, J =
8.8, 5.1 Hz). IR (thin film, cm-1) 3064, 2972, 2928, 2856, 1713, 1628, 1602, 1479, 1354, 1192.
HRMS (DART, M+H) Calc’d for C23H16FN2OS 387.0967, found 387.0976.
(±)-11-(benzo[d]thiazol-2-yl)-9-methoxy-10b-methyl-10b,11-
dihydro-6H-isoindolo[2,1-a]indol-6-one (2.58c) – The compound
was synthesized according to General Procedure 2.5.3.1 using 10 mol
% catalyst loading. This reaction was run by Rachel Ross. The product
was purified by flash column chromatography using pentanes–EtOAc
(9:1 to 7:3, v:v) as the mobile phase and was isolated as an orange
solid (61.1 mg, 0.15 mmol, 77%, mp = 57–62 °C). 1H NMR (500 MHz, CDCl3) δ 1.84 (s, 3H),
3.69 (s, 3H), 4.99 – 5.02 (m, 1H), 6.75 (dd, J = 8.4, 2.2 Hz, 1H), 6.84 (dd, J = 2.2, 0.5 Hz, 1H),
7.16 (td, J = 7.5, 1.1 Hz, 1H), 7.21 (ddd, J = 8.3, 7.2, 1.2 Hz, 1H), 7.32 – 7.40 (m, 2H), 7.46 – 7.52
(m, 2H), 7.61 (dd, J = 8.4, 0.5 Hz, 1H), 7.85 (ddt, J = 7.9, 1.1, 0.5 Hz, 1H), 7.90 (ddd, J = 8.2, 1.1,
0.6 Hz, 1H). 13C NMR (125 MHz, CDCl3) δ 171.3, 168.4, 163.4, 152.3, 150.1, 139.8, 135.7, 130.2,
127.1, 126.1, 126.0, 125.4, 125.0, 122.8, 121.9, 117.6, 116.4, 107.8, 75.2, 55.8, 55.5, 28.7. IR
(thin film, cm-1) 3074, 2970, 2929, 2841, 1702, 1610, 1594, 1482, 1466, 1375, 1289, 1321, 1217,
1133, 1025, 752, 732, 696. HRMS (DART, M+H) Calc’d for C24H19N2O2S 399.1167, found
399.1165.
(±)-11-(benzo[d]thiazol-2-yl)-10b-methyl-8-(trifluoromethyl)-
10b,11-dihydro-6H-isoindolo[2,1-a]indol-6-one (2.58d) – The
compound was synthesized according to General Procedure 2.5.3.1. This
reaction was run by Rachel Ross. The product was purified by flash
column chromatography using pentanes–EtOAc (9:1 to 7:3, v:v) as the
mobile phase and was isolated as a brown solid (40.0 mg, 0.09 mmol,
46%, mp = 123–128 °C). 1H NMR (500 MHz, CDCl3) δ 1.86 (s, 3H), 5.06 (d, J = 0.6 Hz, 1H),
7.19 – 7.24 (m, 2H), 7.37 (ddd, J = 8.4, 7.3, 1.2 Hz, 2H), 7.49 – 7.58 (m, 4H), 7.88 (tt, J = 8.2, 0.8
Hz, 2H), 7.99 (dt, J = 1.6, 0.8 Hz, 1H). 13C NMR (125 MHz, CDCl3) δ 28.3, 55.3, 75.9, 117.9,
121.9, 122.0 (q, J = 3.9 Hz), 123.0, 123.6 (q, J = 272.7 Hz), 124.3, 125.3, 125.8, 126.3, 127.3,

92
129.2 (q, J = 3.6 Hz), 130.4, 131.6 (q, J = 33.1 Hz), 134.0, 135.3, 136.0, 138.9, 150.2 – 151.0 (m),
152.4, 166.6, 170.3. 19F NMR (376 MHz, CDCl3) δ -62.53. IR (thin film, cm-1) 3064, 2972, 2926,
2852, 1706, 1630, 1605, 1479, 1435, 1322, 1117, 753. HRMS (DART, M+H) Calc’d for
C24H16F3N2OS 437.0935, found 437.0934.
(±)-11-(benzo[d]thiazol-2-yl)-8-methoxy-10b-methyl-10b,11-
dihydro-6H-isoindolo[2,1-a]indol-6-one (2.58e) – The compound was
synthesized according to General Procedure 2.5.3.1. This reaction was
run by Rachel Ross. The product was purified by flash column
chromatography using pentanes–EtOAc (9:1 to 7:3, v:v) as the mobile
phase and was isolated as an orange solid (67.4 mg, 0.17 mmol, 85%
(+5% diastereomer), mp = 150–155 °C). 1H NMR (500 MHz, CDCl3) δ 1.82 (s, 3H), 3.73 (s, 3H),
4.98 – 5.00 (m, 1H), 6.85 (dd, J = 8.4, 2.5 Hz, 1H), 7.15 – 7.23 (m, 3H), 7.25 – 7.29 (m, 1H), 7.33
– 7.38 (m, 2H), 7.46 – 7.53 (m, 2H), 7.88 (ddt, J = 10.0, 7.9, 0.8 Hz, 2H). 13C NMR (125 MHz,
CDCl3) δ 28.4, 55.5, 55.7, 75.4, 107.1, 117.8, 120.9, 121.8, 123.0, 124.4, 125.0, 125.3, 126.0,
127.2, 130.2, 134.5, 135.6, 136.5, 139.3, 139.7, 152.4, 160.3, 168.3, 171.3. IR (thin film, cm-1)
2929, 1702, 1610, 1478, 1434, 1357, 1321, 1285, 1145, 1053, 1013, 832, 776, 760, 732. HRMS
(DART, M+H) Calc’d for C24H19N2O2S 399.1167, found 399.1153.
(±)-11-(benzo[d]thiazol-2-yl)-2-methoxy-10b-methyl-10b,11-
dihydro-6H-isoindolo[2,1-a]indol-6-one (2.58f) – The compound was
synthesized according to General Procedure 2.5.3.1. The product was
purified by flash column chromatography using pentanes–EtOAc (9:1
to 7:3, v:v) as the mobile phase and was isolated as a white solid (68.54
mg, 0.172 mmol, 86% (+9% diastereomer), mp = 171–175 °C). 1H
NMR (500 MHz, CDCl3) δ 7.88 (ddd, J = 8.3, 1.2, 0.6 Hz, 1H), 7.78 (dd, J = 8.7, 0.5 Hz, 1H),
7.70 (ddd, J = 7.4, 1.3, 0.7 Hz, 1H), 7.48 (ddd, J = 8.0, 1.2, 0.7 Hz, 1H), 7.38 – 7.32 (m, 2H), 7.28
(td, J = 7.5, 1.3 Hz, 1H), 7.24 (td, J = 7.4, 1.2 Hz, 1H), 7.19 (ddd, J = 8.2, 7.2, 1.2 Hz, 1H), 7.02
(ddd, J = 8.7, 2.6, 0.4 Hz, 1H), 6.90 (dt, J = 2.6, 0.5 Hz, 1H), 4.97 (d, J = 0.6 Hz, 1H), 3.77 (s, 3H),
1.84 (s, 3H). 13C NMR (125 MHz, CDCl3) δ 170.8, 168.3, 157.7, 152.3, 147.2, 137.7, 135.5, 133.3,
132.8, 132.3, 128.9, 126.0, 125.0, 124.5, 123.5, 122.9, 121.8, 118.5, 115.5, 112.8, 76.1, 55.8, 55.8,
28.1. IR (thin film, cm-1) 3004, 2966, 2943, 2836, 1718, 1700, 1489, 1362, 1279. HRMS (DART,
M+H) Calc’d for C24H19N2O2S 399.1167, found 399.1160.
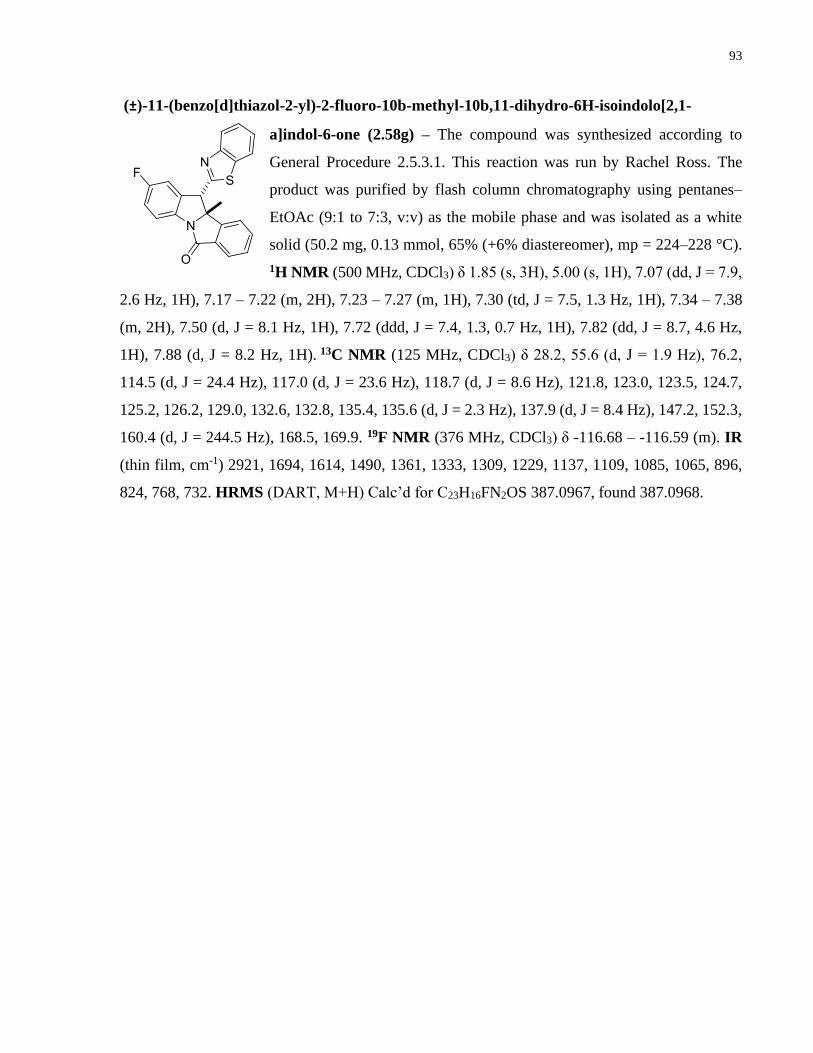
93
(±)-11-(benzo[d]thiazol-2-yl)-2-fluoro-10b-methyl-10b,11-dihydro-6H-isoindolo[2,1-
a]indol-6-one (2.58g) – The compound was synthesized according to
General Procedure 2.5.3.1. This reaction was run by Rachel Ross. The
product was purified by flash column chromatography using pentanes–
EtOAc (9:1 to 7:3, v:v) as the mobile phase and was isolated as a white
solid (50.2 mg, 0.13 mmol, 65% (+6% diastereomer), mp = 224–228 °C).
1H NMR (500 MHz, CDCl3) δ 1.85 (s, 3H), 5.00 (s, 1H), 7.07 (dd, J = 7.9,
2.6 Hz, 1H), 7.17 – 7.22 (m, 2H), 7.23 – 7.27 (m, 1H), 7.30 (td, J = 7.5, 1.3 Hz, 1H), 7.34 – 7.38
(m, 2H), 7.50 (d, J = 8.1 Hz, 1H), 7.72 (ddd, J = 7.4, 1.3, 0.7 Hz, 1H), 7.82 (dd, J = 8.7, 4.6 Hz,
1H), 7.88 (d, J = 8.2 Hz, 1H). 13C NMR (125 MHz, CDCl3) δ 28.2, 55.6 (d, J = 1.9 Hz), 76.2,
114.5 (d, J = 24.4 Hz), 117.0 (d, J = 23.6 Hz), 118.7 (d, J = 8.6 Hz), 121.8, 123.0, 123.5, 124.7,
125.2, 126.2, 129.0, 132.6, 132.8, 135.4, 135.6 (d, J = 2.3 Hz), 137.9 (d, J = 8.4 Hz), 147.2, 152.3,
160.4 (d, J = 244.5 Hz), 168.5, 169.9. 19F NMR (376 MHz, CDCl3) δ -116.68 – -116.59 (m). IR
(thin film, cm-1) 2921, 1694, 1614, 1490, 1361, 1333, 1309, 1229, 1137, 1109, 1085, 1065, 896,
824, 768, 732. HRMS (DART, M+H) Calc’d for C23H16FN2OS 387.0967, found 387.0968.

94
(±)-11-(benzo[d]thiazol-2-yl)-2,9-difluoro-10b-methyl-10b,11-
dihydro-6H-isoindolo[2,1-a]indol-6-one (2.58h) – The compound
was synthesized according to General Procedure 2.5.3.1. The product
was purified by flash column chromatography using pentanes–EtOAc
(20:1 to 7:3, v:v) as the mobile phase and was isolated as a white solid
(42.06 mg, 0.104 mmol, 52%, mp = 178–182 °C). 1H NMR (500 MHz,
CDCl3) δ 7.94 (s, 1H), 7.80 (dd, J = 8.7, 4.6 Hz, 1H), 7.69 (dd, J = 8.4, 4.9 Hz, 1H), 7.60 (d, J =
34.1 Hz, 1H), 7.40 (d, J = 7.0 Hz, 1H), 7.25 – 7.16 (m, 2H), 7.14 – 7.02 (m, 2H), 6.94 (t, J = 8.6
Hz, 1H), 5.01 (s, 1H), 1.85 (s, 3H). 13C NMR (125 MHz, CDCl3) δ 167.4, 166.5114., 164.5, 161.4,
159.5, 149.8 (d, J = 9.7 Hz), 135.5 (d, J = 2.3 Hz), 128.9 (d, J = 2.1 Hz), 126.8 (d, J = 10.0 Hz),
126.3, 125.4, 121.9, 118.7 (d, J = 8.6 Hz), 117.2, 117.1 (d, J = 2.6 Hz), 116.9, 114.6 (d, J = 24.4
Hz), 111.2 (d, J = 24.3 Hz), 55.5. IR (thin film, cm-1) 3069, 2971, 2854, 1714, 1623, 1598, 1485,
1191. HRMS (DART, M+H) Calc’d for C23H15F2N2OS 405.0873, found 405.0872.
(±)-11-(benzo[d]thiazol-2-yl)-10b-ethyl-10b,11-dihydro-6H-
isoindolo[2,1-a]indol-6-one (2.58i) – The compound was synthesized
according to General Procedure 2.5.3.1. This reaction was run by Rachel
Ross. The product was purified by flash column chromatography using
pentanes–EtOAc (9:1 to 7:3, v:v) as the mobile phase and was isolated as a
white solid (68.6 mg, 0.18 mmol, 90%, mp = 229–231 °C). 1H NMR (500
MHz, CDCl3) δ 0.69 (t, J = 7.3 Hz, 3H), 2.25 (qd, J = 7.1, 3.3 Hz, 2H), 5.04 (s, 1H), 7.14 – 7.21
(m, 2H), 7.22 – 7.30 (m, 2H), 7.30 – 7.37 (m, 3H), 7.48 (td, J = 7.7, 1.1 Hz, 2H), 7.72 (dd, J = 7.3,
1.3 Hz, 1H), 7.82 – 7.93 (m, 2H). 13C NMR (125 MHz, CDCl3) δ 8.0, 33.5, 55.1, 79.2, 117.7,
121.8, 122.9, 123.6, 124.4, 125.0, 125.2, 126.0, 127.0, 128.9, 130.1, 132.4, 134.0, 135.6, 136.7,
139.7, 145.7, 152.4, 169.0, 171.2. IR (thin film, cm-1) 3051, 3037, 2984, 2973, 2934, 2876, 1689,
1600, 1516, 1480, 1459, 1365, 1106, 753. HRMS (DART, M+H) Calc’d for C24H19N2OS
383.1218, found 383.1227.

95
(±)-2-(3-(11-(benzo[d]thiazol-2-yl)-6-oxo-6H-isoindolo[2,1-
a]indol-10b(11H)-yl)propyl)isoindoline-1,3-dione (2.58j) –
The compound was synthesized according to General Procedure
2.5.3.1. This reaction was run by Rachel Ross. The product was
purified by flash column chromatography using pentanes–EtOAc
(9:1 to 1:1, v:v) as the mobile phase and was isolated as an orange
solid (84.6 mg, 0.16 mmol, 78%, mp = 205–208 °C). 1H NMR (500 MHz, CDCl3) δ 1.18 – 1.31
(m, 1H), 1.66 (ddtd, J = 13.5, 11.6, 6.8, 4.9 Hz, 1H), 2.21 – 2.35 (m, 2H), 3.56 (t, J = 7.0 Hz, 2H),
5.02 (s, 1H), 7.17 (tdd, J = 7.6, 4.8, 1.1 Hz, 2H), 7.19 – 7.24 (m, 2H), 7.24 – 7.28 (m, 1H), 7.30 –
7.35 (m, 2H), 7.44 – 7.49 (m, 2H), 7.67 – 7.72 (m, 3H), 7.79 – 7.81 (m, 2H), 7.85 (ddt, J = 9.6,
8.0, 0.8 Hz, 2H). 13C NMR (125 MHz, CDCl3) δ 23.0, 37.7, 37.7, 55.3, 78.3, 117.8, 121.7, 122.9,
123.4, 123.6, 124.6, 125.0, 125.4, 126.0, 127.1, 129.1, 130.2, 132.1, 132.7, 133.6, 134.1, 135.6,
136.3, 139.7, 145.5, 152.3, 168.3, 169.1, 170.9. IR (thin film, cm-1) 3061, 2929, 2857, 1710, 1606,
1486, 1466, 1405, 1362, 1301, 1125, 760, 720, 692, 672. HRMS (DART, M+H) Calc’d for
C33H24N3O3S 542.1538, found 542.1530.
(±)-11-(benzo[d]thiazol-2-yl)-N-methoxy-N-methyl-6-oxo-6H-
isoindolo[2,1-a]indole-10b(11H)-carboxamide (2.58k) – The
compound was synthesized according to General Procedure 2.5.3.1 using
10 mol % catalyst loading. This reaction was run by Rachel Ross. The
product was purified by flash column chromatography using pentanes–
EtOAc (9:1 to 7:3, v:v) as the mobile phase and was isolated as a yellow
solid (56.0 mg, 0.13 mmol, 63%, mp = 194–198 °C). 1H NMR (400 MHz, CDCl3) δ 3.10 (s, 3H),
3.42 (s, 3H), 6.40 (s, 1H), 7.17 (tt, J = 7.6, 1.4 Hz, 2H), 7.23 – 7.34 (m, 3H), 7.35 (dd, J = 7.6, 1.1
Hz, 1H), 7.40 – 7.52 (m, 3H), 7.69 – 7.73 (m, 1H), 7.85 (dd, J = 8.1, 5.9 Hz, 2H). 13C NMR (125
MHz, CDCl3) δ 34.5, 51.7, 61.3, 81.5, 117.0, 121.6, 123.2, 124.1, 124.2, 124.9, 125.6, 125.9,
126.8, 129.5, 130.0, 132.6, 134.2, 135.4, 136.5, 139.9, 141.5, 152.8, 168.9, 169.6, 170.0. IR (thin
film, cm-1) 3068, 2976, 2935, 1715, 1653, 1486, 1470, 1357, 1307, 758, 733. HRMS (DART,
M+H) Calc’d for C25H20N3O3S 442.1225, found 442.1219.

96
(±)-11-(benzo[d]thiazol-2-yl)-10b-phenyl-10b,11-dihydro-6H-
isoindolo[2,1-a]indol-6-one (2.58l) – The compound was synthesized
according to General Procedure 2.5.3.1. This reaction was run by Rachel
Ross. The product was purified by flash column chromatography using
pentanes–EtOAc (9:1 to 8:2, v:v) as the mobile phase and was isolated as a
white solid (77.8 mg, 0.18 mmol, 90%, mp = 190–195 °C). 1H NMR (500
MHz, CDCl3) δ 5.66 (s, 1H), 7.12 – 7.30 (m, 6H), 7.38 (qd, J = 6.6, 1.3 Hz, 3H), 7.50 (td, J = 7.7,
1.1 Hz, 2H), 7.53 (d, J = 8.0 Hz, 1H), 7.67 – 7.69 (m, 1H), 7.84 – 7.88 (m, 2H), 7.97 (d, J = 8.1
Hz, 1H), 8.00 (dd, J = 8.0, 1.0 Hz, 1H). 13C NMR (125 MHz, CDCl3) δ 57.4, 80.8, 117.6, 121.8,
123.1, 124.6, 124.6, 125.1, 125.3, 125.6, 126.1, 126.9, 128.4, 128.8, 129.2, 130.3, 132.3, 132.6,
135.8, 136.0, 140.0, 142.8, 146.8, 152.7, 169.0, 171.0. IR (thin film, cm-1) 3070, 3046, 3029, 1734,
1709, 1604, 1479, 1459, 1348, 1304, 1140, 748. HRMS (DART, M+H) Calc’d for C28H19N2OS
431.1218, found 431.1227.
(±)-11-(benzo[d]thiazol-2-yl)-10b-(4-methoxyphenyl)-10b,11-
dihydro-6H-isoindolo[2,1-a]indol-6-one (2.58m) – The compound
was synthesized according to General Procedure 2.5.3.1 on 0.1 mmol
scale. This reaction was run by Rachel Ross. The product was purified
by flash column chromatography using pentanes–EtOAc (9:1 to 8:2,
v:v) as the mobile phase and was isolated as a white solid (33.7 mg,
0.07 mmol, 73%, mp = 240–245 °C). 1H NMR (500 MHz, CDCl3) δ 3.75 (s, 3H), 5.61 (d, J = 0.6
Hz, 1H), 6.85 – 6.92 (m, 2H), 7.15 (dtd, J = 8.6, 7.5, 1.1 Hz, 2H), 7.18 – 7.23 (m, 2H), 7.25 – 7.28
(m, 1H), 7.38 (ddd, J = 8.3, 7.2, 1.2 Hz, 1H), 7.46 – 7.53 (m, 3H), 7.67 (ddd, J = 7.4, 1.3, 0.7 Hz,
1H), 7.71 – 7.78 (m, 2H), 7.95 (ddd, J = 8.2, 1.2, 0.7 Hz, 1H), 7.98 (ddt, J = 7.9, 1.1, 0.5 Hz, 1H).
13C NMR (125 MHz, CDCl3) δ 55.4, 57.3, 80.5, 114.5, 117.6, 121.8, 123.0, 124.5, 124.5, 125.1,
125.6, 126.1, 126.5, 126.9, 128.7, 130.2, 132.3, 132.6, 134.6, 135.6, 136.2, 139.9, 147.2, 152.5,
159.6, 169.0, 171.0. IR (thin film, cm-1) 3072, 2935, 2839, 1715, 1607, 1511, 1478, 1466, 1306,
1258, 1183, 1037, 754, 729. HRMS (DART, M+H) Calc’d for C29H21N2OS 461.1324, found
461.1324.

97
(±)-11-(benzo[d]thiazol-2-yl)-10b-(4-(trifluoromethyl)phenyl)-
10b,11-dihydro-6H-isoindolo[2,1-a]indol-6-one (2.58n) – The
compound was synthesized according to General Procedure 2.5.3.1.
This reaction was run by Rachel Ross. The product was purified by flash
column chromatography using pentanes–EtOAc (9:1 to 8:2, v:v) as the
mobile phase and was isolated as a white solid (87.5 mg, 0.18 mmol,
88%, mp = 210–214 °C). 1H NMR (500 MHz, CDCl3) δ 5.62 (s, 1H), 7.14 – 7.29 (m, 5H), 7.40
(t, J = 7.4 Hz, 1H), 7.48 – 7.56 (m, 3H), 7.64 (d, J = 8.0 Hz, 2H), 7.70 (dd, J = 7.4, 1.4 Hz, 1H),
7.95 – 8.03 (m, 4H). 13C NMR (125 MHz, CDCl3) δ 57.30, 80.51, 117.68, 121.87, 123.12, 123.91
(q, J = 272.2 Hz), 124.56, 124.80, 125.23, 125.79, 125.87, 126.22 (q, J = 3.8 Hz), 126.96, 127.21,
129.21, 130.51, 130.71 (q, J = 32.7 Hz), 132.27, 132.84, 135.45, 135.86, 139.81, 145.89, 146.90,
152.74, 168.87, 170.54. 19F NMR (376 MHz, CDCl3) δ -62.70. IR (thin film, cm-1) 3078, 2925,
1723, 1606, 1486, 1470, 1325, 1305, 1161, 1117, 1069, 1025, 844, 824, 752, 732, 692, 612. HRMS
(DART, M+H) Calc’d for C29H18F3N2OS 499.1092, found 499.1090.
(±)-11-(benzo[d]thiazol-2-yl)-8-methyl-10b-phenyl-10b,11-dihydro-
6H-isoindolo[2,1-a]indol-6-one (2.58o) – The compound was synthesized
according to General Procedure 2.5.3.1. This reaction was run by Rachel
Ross. The product was purified by flash column chromatography using
pentanes–EtOAc (9:1 to 8:2, v:v) as the mobile phase and was isolated as a
white solid (73.8 mg, 0.17 mmol, 87%, mp = 88–93 °C). 1H NMR (500
MHz, CDCl3) δ 2.19 (s, 3H), 5.63 (s, 1H), 7.01 (ddd, J = 8.0, 1.6, 0.7 Hz, 1H), 7.13 (td, J = 7.5,
1.1 Hz, 1H), 7.21 – 7.29 (m, 3H), 7.33 – 7.42 (m, 4H), 7.46 – 7.51 (m, 2H), 7.54 (ddd, J = 7.9, 1.2,
0.6 Hz, 1H), 7.81 – 7.85 (m, 2H), 7.96 – 8.01 (m, 2H). 13C NMR (125 MHz, CDCl3) δ 21.3, 57.3,
80.6, 117.5, 121.9, 123.1, 124.2, 124.8, 125.0, 125.2, 125.5, 126.1, 126.9, 128.2, 129.1, 130.2,
132.5, 133.8, 135.8, 136.2, 139.0, 140.0, 143.2, 144.2, 152.6, 169.1, 171.1. IR (thin film, cm-1)
3066, 2925, 2853, 1710, 1602, 1486, 1466, 1357, 1309, 1241, 1137, 760, 732, 708, 628. HRMS
(DART, M+H) Calc’d for C29H21N2OS 445.1375, found 445.1379.

98
(±)-11-(benzo[d]thiazol-2-yl)-10b-methyl-7,8,9,10,10b,11-hexahydro-
6H-isoindolo[2,1-a]indol-6-one (2.58p) – The compound was synthesized
according to General Procedure 2.5.3.1. This reaction was run by Rachel
Ross. The product was purified by flash column chromatography using
pentanes–EtOAc (9:1, v:v) as the mobile phase and was isolated as an orange
solid (57.9 mg, 0.16 mmol, 78%, mp = 206–209 °C). 1H NMR (500 MHz,
CDCl3) δ 1.06 (ddtt, J = 12.7, 9.2, 6.4, 3.6 Hz, 1H), 1.19 – 1.28 (m, 1H), 1.41 – 1.48 (m, 1H), 1.52
(dtdd, J = 13.2, 7.8, 5.3, 2.6 Hz, 1H), 1.64 (s, 3H), 1.93 (dddt, J = 17.7, 8.0, 5.3, 2.4 Hz, 1H), 2.08
– 2.22 (m, 3H), 4.76 (s, 1H), 7.12 (td, J = 7.5, 1.1 Hz, 1H), 7.25 – 7.33 (m, 2H), 7.44 (tt, J = 7.7,
1.4 Hz, 2H), 7.69 (dt, J = 8.1, 2.1 Hz, 2H), 7.97 (d, J = 8.2 Hz, 1H). 13C NMR (125 MHz, CDCl3)
δ 20.4, 21.5, 21.8, 23.2, 25.0, 54.7, 77.1, 117.7, 121.9, 123.0, 124.9, 125.2, 126.2, 127.1, 130.1,
133.8, 135.8, 136.0, 139.9, 152.5, 158.9, 171.5, 172.4. IR (thin film, cm-1) 3067, 3037, 2958, 2931,
2859, 1688, 1601, 1511 1479, 1459, 1432, 1332, 1306, 1127, 1101, 772. HRMS (DART, M+H)
Calc’d for C23H21N2OS 373.1375, found 373.1376.
(±)-2-(10b-methyl-6-oxo-10b,11-dihydro-6H-isoindolo[2,1-a]indol-11-
yl)pyridine 1-oxide (2.59) – The compound was synthesized according to
General Procedure 2.5.3.1. This reaction was run by Dr. Tamara Beisel. The
product was purified by flash column chromatography using DCM–MeOH
(200:1 to 50:1, v:v) as the mobile phase and was isolated as a white solid
(47.94 mg, 0.146 mmol, 73%, mp = 164–166 °C). 1H NMR (500 MHz, CDCl3) δ 8.10 (dd, J =
6.5, 1.2 Hz, 1H), 7.91 (dt, J = 7.7, 0.9 Hz, 1H), 7.83 (dt, J = 8.1, 0.7 Hz, 1H), 7.66 (ddd, J = 7.5,
1.2, 0.7 Hz, 1H), 7.49 – 7.45 (m, 1H), 7.38 (td, J = 7.6, 1.2 Hz, 1H), 7.29 (td, J = 7.5, 1.0 Hz, 1H),
7.21 – 7.18 (m, 2H), 6.85 (ddd, J = 7.6, 6.4, 2.0 Hz, 1H), 6.72 (td, J = 7.8, 1.2 Hz, 1H), 6.00 (dd, J
= 7.9, 2.1 Hz, 1H), 5.58 (s, 1H), 1.91 (s, 3H). 13C NMR (125 MHz, CDCl3) δ 168.7, 151.6, 147.9,
140.4, 139.0, 135.5, 132.8, 132.4, 129.7, 129.0, 126.8, 125.5, 125.5, 125.5, 124.2, 123.9, 123.3,
117.8, 117.5, 49.2, 28.1. IR (thin film, cm-1) 3083, 2971, 2925, 2245, 1722, 1694, 1486, 1430,
1361, 906. HRMS (DART, M+H) Calc’d for C21H17N2O2 329.1290, found 329.1287.

99
(±)-10b-methyl-11-(perfluorophenyl)-10b,11-dihydro-6H-
isoindolo[2,1-a]indol-6-one (2.60) – The compound was synthesized
according to General Procedure 2.5.3.1 using 10 mol % catalyst loading.
This reaction was run by Dr. Tamara Beisel. The product was purified by
flash column chromatography using pentanes–EtOAc (20:1, v:v) as the
mobile phase and was isolated as a white solid (61.7 mg, 0.15 mmol, 77%,
mp = 191–196 °C). 1H NMR (400 MHz, CDCl3) δ 1.80 (s, 3H), 4.78 (s, 1H), 7.06 – 7.10 (m, 1H),
7.11 – 7.18 (m, 2H), 7.39 – 7.46 (m, 3H), 7.79 (d, J = 7.9 Hz, 1H), 7.82 – 7.87 (m, 1H). 13C NMR
(125 MHz, CDCl3) δ 29.2, 45.7, 74.8, 115.0, 117.4, 121.8, 125.1, 125.4, 125.9, 129.3, 129.6, 132.8,
132.9, 132.9, 134.2, 140.7, 140.7, 147.8, 168.6, 168.7. 19F NMR (376 MHz, CDCl3) δ -162.12 (td,
J = 21.6, 8.1 Hz), -160.76 (td, J = 21.7, 8.0 Hz), -154.31 (t, J = 21.1 Hz), -144.58 (ddd, J = 22.6,
7.9, 3.6 Hz), -139.78 (dd, J = 22.4, 8.3 Hz). IR (thin film, cm-1) 3047, 2972, 2930, 2868, 1707,
1528, 1503, 1488, 1466, 1357, 1337, 1308, 1133, 1000, 979, 762, 683. HRMS (DART, M+H)
Calc’d for C22H13F5NO 402.0917, found 402.0922.
(±)-10b-methyl-11-(4-methylthiazol-2-yl)-10b,11-dihydro-6H-
isoindolo[2,1-a]indol-6-one (2.61) – The compound was synthesized
according to General Procedure 2.5.3.1. This reaction was run by Dr. Tamara
Beisel. The product was purified by flash column chromatography using
pentanes–EtOAc (4:1 to 7:3, v:v) as the mobile phase and was isolated as a
white solid (41.22 mg, 0.124 mmol, 62%, mp = 144–149°C). 1H NMR (500
MHz, CDCl3) δ 7.83 (ddd, J = 7.9, 1.1, 0.5 Hz, 1H), 7.71 (dt, J = 7.6, 1.0 Hz, 1H), 7.47 (td, J =
7.8, 1.3 Hz, 1H), 7.43 – 7.37 (m, 2H), 7.36 – 7.31 (m, 2H), 7.17 (td, J = 7.5, 1.1 Hz, 1H), 6.40 (p,
J = 1.0 Hz, 1H), 4.89 (s, 1H), 2.25 (d, J = 1.1 Hz, 3H), 1.79 (s, 3H). 13C NMR (125 MHz, CDCl3)
δ 169.7, 168.5, 152.0, 147.7, 139.4, 136.7, 133.0, 132.1, 130.0, 128.7, 127.0, 125.2, 124.4, 123.9,
117.7, 114.6, 75.8, 54.8, 27.9, 17.0. IR (thin film, cm-1) 3098, 3039, 2968, 2924, 2858, 1721, 1699,
1529, 1483, 1361, 1131. HRMS (DART, M+H) Calc’d for C20H16N2OS 333.1062, found
333.1055.

100
(±)-10b-methyl-11-(4-methylthiazol-5-yl)-10b,11-dihydro-6H-isoindolo[2,1-a]indol-6-one
(2.62) – The compound was synthesized according to General Procedure
2.5.3.1. This reaction was run by Dr. Tamara Beisel. The product was purified
by flash column chromatography using pentanes–EtOAc (4:1 to 7:3, v:v) as
the mobile phase and was isolated as a white solid (20.61 mg, 0.062 mmol,
31%, mp = 171-175 °C). 1H NMR (500 MHz, CDCl3) δ 8.20 (s, 1H), 7.83 –
7.79 (m, 1H), 7.76 (dt, J = 7.5, 1.0 Hz, 1H), 7.42 (dtd, J = 13.5, 7.6, 1.3 Hz, 2H), 7.35 (td, J = 7.5,
1.1 Hz, 1H), 7.21 (ddt, J = 7.7, 1.4, 0.7 Hz, 1H), 7.17 – 7.12 (m, 2H), 4.68 (s, 1H), 2.54 (s, 3H),
1.78 (s, 3H). 13C NMR (125 MHz, CDCl3) δ 168.4, 151.5, 147.8, 147.7, 138.8, 138.4, 133.2, 132.9,
132.7, 129.6, 128.9, 126.5, 125.3, 124.8, 122.3, 117.7, 75.9, 48.9, 27.9, 15.8. IR (thin film, cm-1)
3079, 2969, 2925, 2856, 1702. 1603, 1478, 1463, 1357, 1308, 756. HRMS (DART, M+H) Calc’d
for C20H16N2OS 333.1062, found 333.1058.
(±)-11-(benzo[d]oxazol-2-yl)-10b-methyl-10b,11-dihydro-6H-
isoindolo[2,1-a]indol-6-one (2.63) – The compound was synthesized
according to General Procedure 2.5.3.1. This reaction was run by Dr.
Tamara Beisel. The product was purified by flash column chromatography
using pentanes–EtOAc (9:1, v:v) as the mobile phase and two white solids
were isolated corresponding to the anti-isomer and syn-isomer.
Anti-diastereomer: 31.72 mg, 0.09, 45%, mp = 239-244 °C. 1H NMR (500 MHz, CDCl3) δ 7.86
(dt, J = 7.9, 0.8 Hz, 1H), 7.81 – 7.76 (m, 1H), 7.51 (d, J = 7.8 Hz, 1H), 7.47 (td, J = 7.8, 1.2 Hz,
1H), 7.33 – 7.26 (m, 4H), 7.14 (tdd, J = 7.9, 3.4, 1.2 Hz, 2H), 7.08 (ddd, J = 8.3, 7.2, 1.1 Hz, 1H),
7.02 (d, J = 8.2 Hz, 1H), 4.82 (s, 1H), 1.84 (s, 3H). 13C NMR (125 MHz, CDCl3) δ 168.6, 163.4,
150.7, 147.4, 140.3, 140.0, 133.7, 132.9, 132.7, 130.0, 129.0, 126.5, 125.1, 125.0, 124.8, 124.2,
122.4, 119.9, 118.0, 110.5, 74.8, 51.0, 28.2. IR (thin film, cm-1) 3053, 2970, 2931, 2922, 2854,
1708, 1615, 1570, 1480, 1353, 755. HRMS (DART, M+H) Calc’d for C23H17N2O2 353.1290,
found 353.1285.
Syn-diastereomer: 38.44 mg, 0.109 mmol, 54%, mp = 194-196 °C. 1H NMR (500 MHz, CDCl3)
δ 7.93 (d, J = 7.6 Hz, 1H), 7.87 – 7.84 (m, 1H), 7.82 (d, J = 7.6 Hz, 1H), 7.76 (d, J = 7.4 Hz, 1H),
7.71 (td, J = 7.5, 1.1 Hz, 1H), 7.68 – 7.65 (m, 1H), 7.59 – 7.53 (m, 2H), 7.48 – 7.40 (m, 3H), 7.25
– 7.21 (m, 1H), 5.00 (s, 1H), 1.47 (s, 3H). 13C NMR (125 MHz, CDCl3) δ 167.6, 163.2, 151.0,

101
150.1, 141.2, 139.6, 133.6, 133.4, 132.6, 129.4, 129.4, 127.0, 125.7, 125.3, 125.2, 124.9, 123.0,
120.6, 117.7, 110.8, 75.0, 50.6, 23.3. IR (thin film, cm-1) 3080, 3050, 2974, 2929, 2856, 1706,
1606, 1569, 1479, 1354, 750. HRMS (DART, M+H) Calc’d for C23H17N2O2 353.1290, found
353.1296.
2.5.3.3 Alkylation of 2.63
(±)-tert-butyl 2-(11-(benzo[d]oxazol-2-yl)-10b-methyl-6-oxo-10b,11-dihydro-6H-
isoindolo[2,1-a]indol-11-yl)acetate (2.64) – I designed the experiment and the reaction was run
by Rachel Ross. In a dried vial under argon was added 2.63 (70.5 mg, 0.2 mmol, 1 equiv, 1:1.2 dr)
and DMF (2 mL, 0.1 M) and cooled in an ice bath. In the following order, NaH (4.8 mg, 0.24
mmol, 1.2 equiv), and TBAI (7.4 mg, 0.04 mmol, 20 mol %) were added. The alkyl bromide (30
µL, 0.4 mmol, 2 equiv) was added quickly. The reaction was stirred for 4 hours, quenched with
NH4Cl, and extracted with EtOAc. The organic layer was washed thoroughly with water, brine,
and dried over MgSO4, before pumping off the solvent and purification by flash column
chromatography using pentanes–EtOAc (9:1 to 5:1, v:v) as the mobile phase and was isolated as
a white solid (60.7 mg, 0.13 mmol, 65%, mp = 179-184 °C). 1H NMR (400 MHz, CDCl3) δ 1.12
(s, 9H), 1.28 (s, 3H), 1.82 (d, J = 15.5 Hz, 1H), 2.95 (d, J = 15.5 Hz, 1H), 7.23 – 7.29 (m, 1H),
7.38 – 7.44 (m, 2H), 7.47 (td, J = 7.7, 1.3 Hz, 1H), 7.61 (td, J = 7.5, 1.0 Hz, 1H), 7.64 – 7.70 (m,
1H), 7.74 (td, J = 7.5, 1.2 Hz, 1H), 7.77 – 7.81 (m, 1H), 7.82 – 7.87 (m, 2H), 7.90 (dt, J = 7.7, 0.9
Hz, 1H), 7.96 (dt, J = 7.6, 1.0 Hz, 1H). 13C NMR (125 MHz, CDCl3) δ 23.8, 27.9, 43.9, 54.6, 77.8,
81.5, 110.9, 117.5, 120.6, 123.8, 124.8, 124.9, 125.1, 125.3, 128.9, 129.7, 129.9, 133.1, 133.7,
136.0, 137.8, 141.4, 146.8, 150.8, 165.6, 166.6, 168.3. IR (thin film, cm-1) 3072, 2980, 2931, 2872,
1736, 1707, 1482, 1457, 1370, 1341, 1154, 1137, 758, 737. HRMS (DART, M+H) Calc’d for
C29H27N2O4 467.1971, found 467.1976.
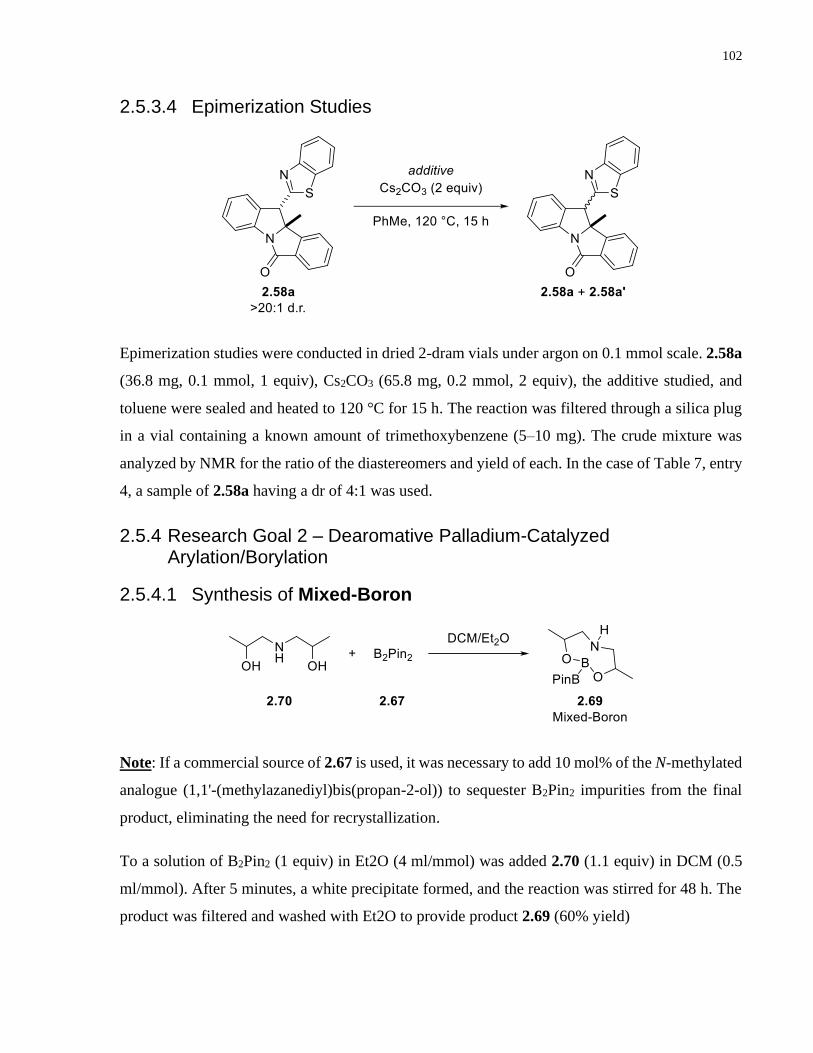
102
2.5.3.4 Epimerization Studies
Epimerization studies were conducted in dried 2-dram vials under argon on 0.1 mmol scale. 2.58a
(36.8 mg, 0.1 mmol, 1 equiv), Cs2CO3 (65.8 mg, 0.2 mmol, 2 equiv), the additive studied, and
toluene were sealed and heated to 120 °C for 15 h. The reaction was filtered through a silica plug
in a vial containing a known amount of trimethoxybenzene (5–10 mg). The crude mixture was
analyzed by NMR for the ratio of the diastereomers and yield of each. In the case of Table 7, entry
4, a sample of 2.58a having a dr of 4:1 was used.
2.5.4 Research Goal 2 – Dearomative Palladium-Catalyzed Arylation/Borylation
2.5.4.1 Synthesis of Mixed-Boron
Note: If a commercial source of 2.67 is used, it was necessary to add 10 mol% of the N-methylated
analogue (1,1'-(methylazanediyl)bis(propan-2-ol)) to sequester B2Pin2 impurities from the final
product, eliminating the need for recrystallization.
To a solution of B2Pin2 (1 equiv) in Et2O (4 ml/mmol) was added 2.70 (1.1 equiv) in DCM (0.5
ml/mmol). After 5 minutes, a white precipitate formed, and the reaction was stirred for 48 h. The
product was filtered and washed with Et2O to provide product 2.69 (60% yield)

103
2.5.4.2 Synthesis of Phosphoramidite Ligands
Synthesis of Phosphoramidite ligands was performed by first building 3,3’-substitued BINOL
derivatives according to literature procedure (summarized below),111 then coupling to PCl3 using
a slight variation to literature protocols.112 Spectral data for known compounds could be obtained
free of charge by following the citation above.
2.5.4.2.1 MOM-Protection of BINOL
To NaH (60% in mineral oil, 2.2 equiv) in THF (0.5 M) at 0 °C was slowly added a solution of
BINOL (2.79, 1 equiv) in THF (0.67 M). The reaction was stirred for 1 h, then was warmed to
room temperature for 15 minutes. The reaction was cooled back down to 0 °C, and chloromethyl
methyl ether (2.2 equiv) was added dropwise. The reaction was stirred at room temperature for 5
hours, after which was quenched with aqueous ammonium chloride, extracted with ethyl acetate
and the organic layer was concentrated. The product was columned in hexanes–ethyl acetate
(10:1).
2.5.4.2.2 Ortho-Bromination of BINOL
To a solution of mom-protected BINOL (2.71) in Et2O (17 mL/mmol) was added n-BuLi (3 equiv)
dropwise. The reaction was stirred for 3 h, then THF (11 mL/mmol) was added and the reaction

104
mixture was further stirred for an additional hour. After cooling the flask in an ice water bath,
dibromotetrachloroethane (3 equiv) was added quickly in one portion. The reaction was stirred at
room temperature for 5 hours, after which was quenched with aqueous ammonium chloride,
extracted with Et2O and the organic layer was concentrated. The product was columned in
hexanes–ethyl acetate (10:1).
2.5.4.2.3 Suzuki-Miyaura Cross-Coupling
To 3,3’-dibromo binaphthyl 2.72 (1 equiv), Pd(PPh3)4 (10 mol%) in DME (6.7 mL/mmol) in a
round bottom flask at room temperature under argon was added an arylboronic acid (3.5 equiv)
and 2 M aqueous Na2CO3 (5 equiv). The mixture was stirred at reflux for 10 h, cooled, passed
through a Celite pad, and concentrate. The residue was redissolved in DCM, washed with NH4Cl,
water, brine and concentrated. The products were purified typically by column chromatography
using hexanes–ethyl acetate (10:1).
2.5.4.2.4 Acidic Solid Support Deprotection of MOM-Protecting Group
A mixture of mom-protected BINOL derivatives (2.80, 1 equiv) and Amberlyst 15 resin (100 w%)
in THF–MeOH (1:1) was heated to reflux for 15 h. The reaction was cooled to room temperature,
filtered through a Celite pad, and concentrated. The products were purified typically by column
chromatography using hexanes–ethyl acetate (10:1).

105
2.5.4.2.5 General Procedure for the Synthesis of Phosphoramidite Ligands
To a flame dried round bottom flask cooled under a flow of argon was added DCM (0.1 M with
respect to BINOL derivative), and PCl3 (1 equiv). The flask was cooled to 0 °C, and NEt3 (5 equiv)
was added slowly. The reaction was warmed to room temperature. The dialkylamine of choice
(2.74, 1 equiv) was added dropwise, and the reaction was stirred for 5 h. The BINOL derivative
(2.73, 1 equiv) was added in one portion through the top and the reaction was stirred for an
additional 12 h. The reaction was filtered through a silica pad carefully, eluting with a mixture of
hexanes–DCM (20% to 60%). It is crucial that the ligand is not purified using EtOAc since there
are byproducts that coelute.
2.5.4.2.6 Characterization Data for Ligands
N,N-diisopropyldinaphtho[2,1-d:1',2'-f][1,3,2]dioxaphosphepin-4-
amine – The ligand was synthesized according to General Procedure
2.5.4.2.5 and was isolated as a white foam which could be crushed into
a crystalline solid. The reaction was run by Christian Breuers. 1H NMR
(600 MHz, CDCl3) δ 7.98 (d, J = 8.7 Hz, 1H), 7.95 – 7.91 (m, 3H), 7.54
(dd, J = 8.7, 0.9 Hz, 1H), 7.48 (d, J = 8.7 Hz, 1H), 7.44 (dd, J = 8.7, 1.0 Hz, 1H), 7.43 – 7.40 (m,
2H), 7.35 (dd, J = 8.6, 1.1 Hz, 1H), 7.28 (ddd, J = 8.3, 6.7, 1.3 Hz, 1H), 7.26 – 7.23 (m, 1H), 3.42
(dhept, J = 10.7, 6.8 Hz, 2H), 1.26 (d, J = 6.8 Hz, 6H), 1.22 (d, J = 6.7 Hz, 6H). 13C NMR (125

106
MHz, CDCl3) δ 150.6, 150.5, 150.3, 133.0, 132.9, 132.9, 132.8, 131.4, 131.4, 130.6, 130.6, 130.4,
130.3, 130.3, 129.5, 129.5, 128.4, 128.3, 128.3, 127.3, 127.2, 126.0, 126.0, 125.9, 124.7, 124.4,
124.2, 124.1, 122.6, 122.5, 122.5, 122.0, 122.0, 45.0, 44.9, 24.7, 24.6. 31P NMR (243 MHz,
CDCl3) δ 151.73.
N,N-dicyclohexyldinaphtho[2,1-d:1',2'-f][1,3,2]dioxaphosphepin-4-
amine – The ligand was synthesized according to General Procedure
2.5.4.2.5 and was isolated as a white foam which could be crushed into a
crystalline solid. The reaction was run by Christian Breuers. 1H NMR
(500 MHz, CDCl3) δ 7.96 (d, J = 8.8 Hz, 1H), 7.93 – 7.87 (m, 3H), 7.52 (dd, J = 8.7, 0.9 Hz, 1H),
7.45 (d, J = 8.8 Hz, 1H), 7.44 – 7.38 (m, 4H), 7.33 (dd, J = 8.6, 1.1 Hz, 1H), 7.29 – 7.21 (m, 2H),
2.82 (dtt, J = 15.3, 11.9, 3.6 Hz, 2H), 1.98 – 1.42 (m, 14H), 1.08 – 0.81 (m, 6H). 13C NMR (126
MHz, CDCl3) δ 150.6, 150.5, 150.3, 133.0, 133.0, 132.8, 132.8, 131.4, 131.4, 130.6, 130.4, 130.3,
130.2, 130.2, 129.4, 129.4, 128.4, 128.2, 127.2, 127.1, 127.0, 126.0, 125.9, 124.7, 124.4, 124.2,
124.2, 122.6, 122.5, 122.3, 122.0, 122.0, 54.3, 54.2, 35.3, 26.6, 26.5, 25.6. 31P NMR (243 MHz,
CDCl3) δ 151.92.
5-(dinaphtho[2,1-d:1',2'-f][1,3,2]dioxaphosphepin-4-yl)-5H-
dibenzo[b,f]azepine – The ligand was synthesized according to
General Procedure 2.5.4.2.5 and was isolated as a white foam which
could be crushed into a crystalline solid. 1H NMR (600 MHz, CDCl3)
δ 8.02 (d, J = 8.8 Hz, 1H), 7.95 – 7.92 (m, 1H), 7.82 – 7.77 (m, 1H),
7.67 (dd, J = 8.7, 0.8 Hz, 1H), 7.49 – 7.45 (m, 1H), 7.42 (ddd, J = 8.1, 6.6, 1.3 Hz, 1H), 7.39 (ddd,
J = 8.0, 6.6, 1.2 Hz, 1H), 7.34 (dd, J = 8.5, 1.1 Hz, 1H), 7.28 (ddd, J = 6.9, 6.0, 1.7 Hz, 2H), 7.25
– 7.20 (m, 6H), 7.16 (dd, J = 7.2, 2.0 Hz, 1H), 7.02 (d, J = 11.5 Hz, 1H), 6.99 – 6.94 (m, 2H), 6.91
(d, J = 8.7 Hz, 1H), 6.57 (td, J = 7.6, 1.6 Hz, 1H). 13C NMR (151 MHz, CDCl3) δ 150.1, 150.0,
148.8, 148.8, 143.1, 143.0, 142.6, 136.6, 136.5, 135.3, 133.0, 133.0, 132.3, 132.3, 131.6, 131.5,
131.5, 130.4, 130.3, 129.3, 129.2, 129.2, 129.1, 129.1, 129.0, 128.7, 128.5, 128.4, 128.0, 127.2,
126.9, 126.8, 126.8, 126.3, 126.2, 125.8, 125.0, 124.4, 124.4, 124.3, 122.3, 122.2, 121.6, 121.3,
121.2. 31P NMR (243 MHz, CDCl3) δ 137.93.
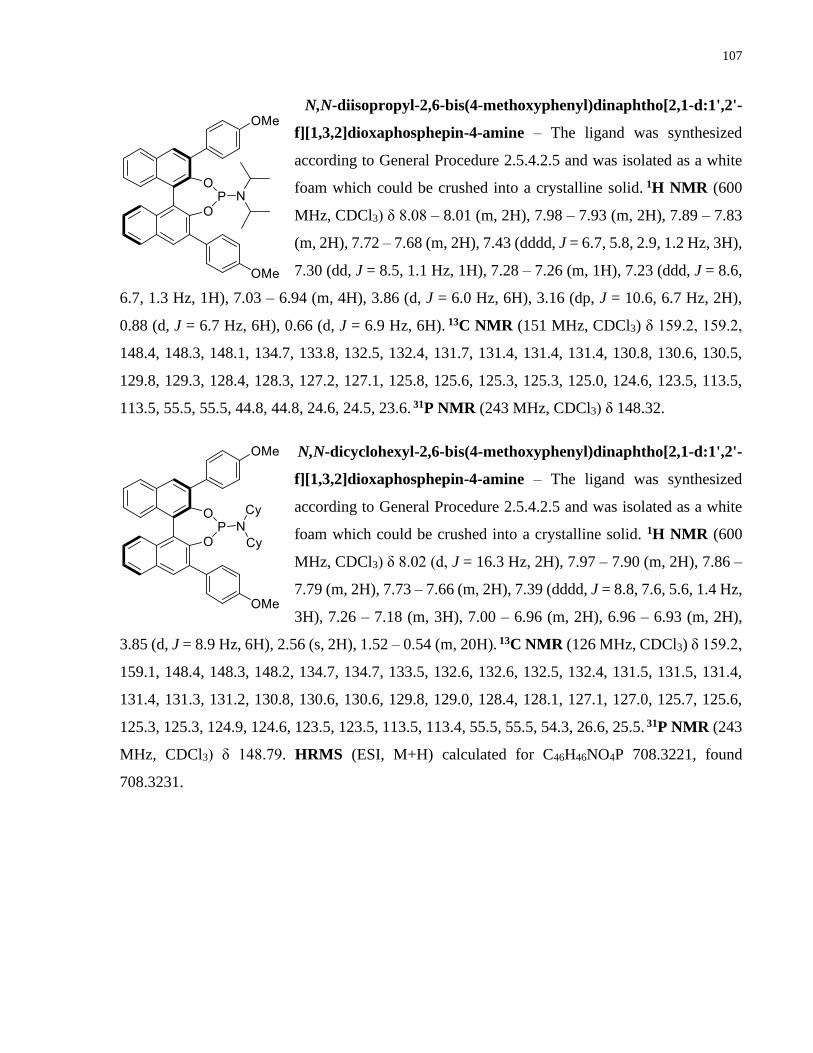
107
N,N-diisopropyl-2,6-bis(4-methoxyphenyl)dinaphtho[2,1-d:1',2'-
f][1,3,2]dioxaphosphepin-4-amine – The ligand was synthesized
according to General Procedure 2.5.4.2.5 and was isolated as a white
foam which could be crushed into a crystalline solid. 1H NMR (600
MHz, CDCl3) δ 8.08 – 8.01 (m, 2H), 7.98 – 7.93 (m, 2H), 7.89 – 7.83
(m, 2H), 7.72 – 7.68 (m, 2H), 7.43 (dddd, J = 6.7, 5.8, 2.9, 1.2 Hz, 3H),
7.30 (dd, J = 8.5, 1.1 Hz, 1H), 7.28 – 7.26 (m, 1H), 7.23 (ddd, J = 8.6,
6.7, 1.3 Hz, 1H), 7.03 – 6.94 (m, 4H), 3.86 (d, J = 6.0 Hz, 6H), 3.16 (dp, J = 10.6, 6.7 Hz, 2H),
0.88 (d, J = 6.7 Hz, 6H), 0.66 (d, J = 6.9 Hz, 6H). 13C NMR (151 MHz, CDCl3) δ 159.2, 159.2,
148.4, 148.3, 148.1, 134.7, 133.8, 132.5, 132.4, 131.7, 131.4, 131.4, 131.4, 130.8, 130.6, 130.5,
129.8, 129.3, 128.4, 128.3, 127.2, 127.1, 125.8, 125.6, 125.3, 125.3, 125.0, 124.6, 123.5, 113.5,
113.5, 55.5, 55.5, 44.8, 44.8, 24.6, 24.5, 23.6. 31P NMR (243 MHz, CDCl3) δ 148.32.
N,N-dicyclohexyl-2,6-bis(4-methoxyphenyl)dinaphtho[2,1-d:1',2'-
f][1,3,2]dioxaphosphepin-4-amine – The ligand was synthesized
according to General Procedure 2.5.4.2.5 and was isolated as a white
foam which could be crushed into a crystalline solid. 1H NMR (600
MHz, CDCl3) δ 8.02 (d, J = 16.3 Hz, 2H), 7.97 – 7.90 (m, 2H), 7.86 –
7.79 (m, 2H), 7.73 – 7.66 (m, 2H), 7.39 (dddd, J = 8.8, 7.6, 5.6, 1.4 Hz,
3H), 7.26 – 7.18 (m, 3H), 7.00 – 6.96 (m, 2H), 6.96 – 6.93 (m, 2H),
3.85 (d, J = 8.9 Hz, 6H), 2.56 (s, 2H), 1.52 – 0.54 (m, 20H). 13C NMR (126 MHz, CDCl3) δ 159.2,
159.1, 148.4, 148.3, 148.2, 134.7, 134.7, 133.5, 132.6, 132.6, 132.5, 132.4, 131.5, 131.5, 131.4,
131.4, 131.3, 131.2, 130.8, 130.6, 130.6, 129.8, 129.0, 128.4, 128.1, 127.1, 127.0, 125.7, 125.6,
125.3, 125.3, 124.9, 124.6, 123.5, 123.5, 113.5, 113.4, 55.5, 55.5, 54.3, 26.6, 25.5. 31P NMR (243
MHz, CDCl3) δ 148.79. HRMS (ESI, M+H) calculated for C46H46NO4P 708.3221, found
708.3231.

108
N,N-diisopropyl-2,6-bis(2-methoxyphenyl)dinaphtho[2,1-d:1',2'-
f][1,3,2]dioxaphosphepin-4-amine – The ligand was synthesized
according to General Procedure 2.5.4.2.5 and was isolated as a white
foam which could be crushed into a crystalline solid. 1H NMR (600
MHz, CDCl3) δ 8.13 (s, 1H), 8.00 (s, 1H), 7.98 – 7.91 (m, 2H), 7.67 (dd,
J = 7.6, 1.8 Hz, 1H), 7.57 – 7.50 (m, 1H), 7.51 – 7.23 (m, 10H), 7.11 –
6.95 (m, 5H), 3.77 (d, J = 25.2 Hz, 6H), 3.16 (dp, J = 10.6, 6.7 Hz, 2H),
0.74 (d, J = 6.7 Hz, 6H), 0.65 (d, J = 6.9 Hz, 6H). 13C NMR (151 MHz, CDCl3) δ 157.5, 157.4,
149.4, 149.3, 148.9, 133.2, 133.0, 133.0, 132.8, 132.2, 131.4, 131.3, 131.1, 130.6, 129.9, 129.2,
129.0, 128.7, 128.4, 128.3, 127.5, 127.4, 127.4, 125.6, 125.4, 125.1, 124.7, 124.7, 124.6, 124.1,
121.9, 120.2, 120.1, 120.0, 111.1, 110.4, 55.8, 55.6, 55.5, 44.7, 44.6, 24.3, 24.3, 23.5, 23.5, 18.5,
13.7, 1.2. 31P NMR (162 MHz, CDCl3) δ 147.10. HRMS (ESI, M+1) calculated for C40H39NO4P
628.2611, found 628.2611.
N,N-diisopropyl-2,6-bis(3-methoxyphenyl)dinaphtho[2,1-d:1',2'-
f][1,3,2]dioxaphosphepin-4-amine – The ligand was synthesized
according to General Procedure 2.5.4.2.5 and was isolated as a white
foam which could be crushed into a crystalline solid. 1H NMR (600
MHz, CDCl3) δ 8.13 (d, J = 0.7 Hz, 1H), 8.07 (s, 1H), 8.02 – 7.92 (m,
2H), 7.53 (dd, J = 2.6, 1.6 Hz, 1H), 7.49 (ddd, J = 7.7, 1.6, 1.0 Hz, 1H),
7.48 – 7.43 (m, 3H), 7.39 – 7.25 (m, 8H), 6.95 (ddd, J = 7.8, 2.6, 1.5
Hz, 1H), 6.92 (ddd, J = 8.3, 2.6, 1.0 Hz, 1H), 3.85 (d, J = 15.8 Hz, 6H), 3.16 (dp, J = 10.8, 6.8 Hz,
2H), 0.87 (d, J = 6.7 Hz, 6H), 0.66 (d, J = 6.9 Hz, 6H). 13C NMR (151 MHz, CDCl3) δ 159.4,
159.3, 148.3, 148.3, 148.0, 139.5, 139.4, 135.2, 135.2, 134.0, 132.7, 132.7, 131.2, 130.5, 130.3,
130.0, 128.9, 128.9, 128.5, 128.5, 127.2, 127.1, 126.0, 125.9, 125.3, 125.1, 124.7, 123.4, 123.4,
123.0, 122.9, 122.9, 115.8, 115.7, 115.7, 113.8, 113.3, 55.5, 55.4, 44.9, 44.8, 24.5, 24.4, 23.6. 31P
NMR (162 MHz, CDCl3) δ 148.96. HRMS (ESI, M+1) calculated for C40H39NO4P 628.2611,
found 628.2609.
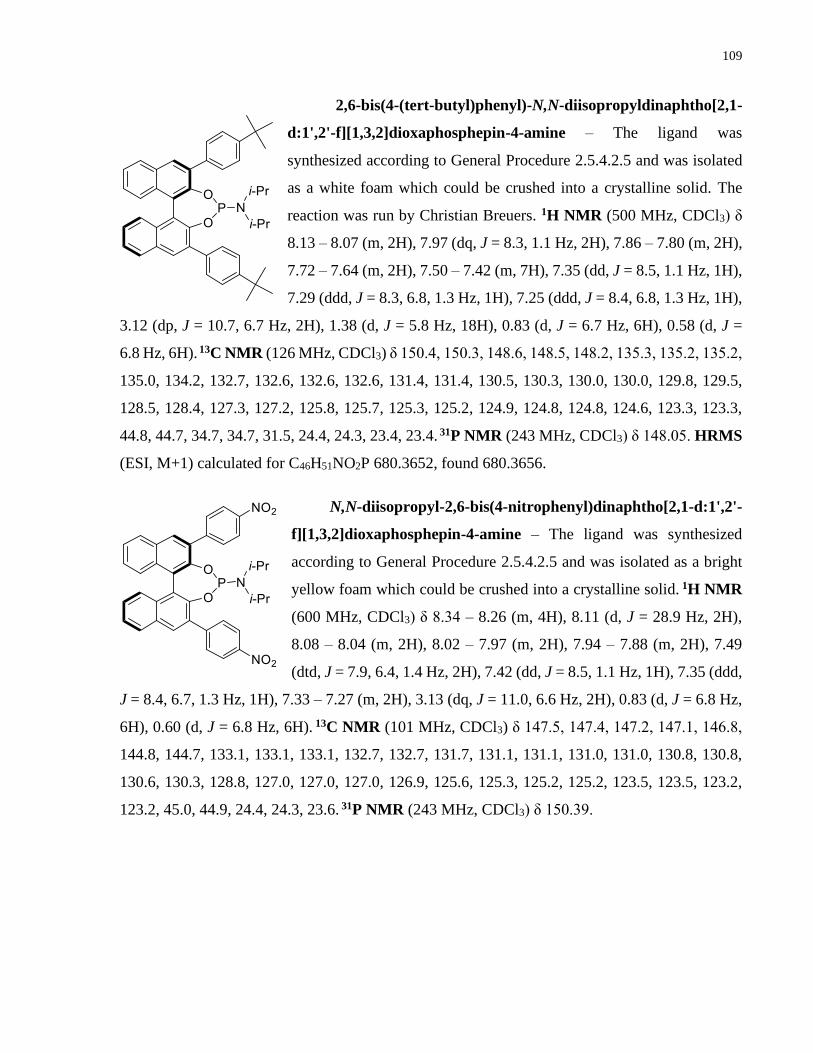
109
2,6-bis(4-(tert-butyl)phenyl)-N,N-diisopropyldinaphtho[2,1-
d:1',2'-f][1,3,2]dioxaphosphepin-4-amine – The ligand was
synthesized according to General Procedure 2.5.4.2.5 and was isolated
as a white foam which could be crushed into a crystalline solid. The
reaction was run by Christian Breuers. 1H NMR (500 MHz, CDCl3) δ
8.13 – 8.07 (m, 2H), 7.97 (dq, J = 8.3, 1.1 Hz, 2H), 7.86 – 7.80 (m, 2H),
7.72 – 7.64 (m, 2H), 7.50 – 7.42 (m, 7H), 7.35 (dd, J = 8.5, 1.1 Hz, 1H),
7.29 (ddd, J = 8.3, 6.8, 1.3 Hz, 1H), 7.25 (ddd, J = 8.4, 6.8, 1.3 Hz, 1H),
3.12 (dp, J = 10.7, 6.7 Hz, 2H), 1.38 (d, J = 5.8 Hz, 18H), 0.83 (d, J = 6.7 Hz, 6H), 0.58 (d, J =
6.8 Hz, 6H). 13C NMR (126 MHz, CDCl3) δ 150.4, 150.3, 148.6, 148.5, 148.2, 135.3, 135.2, 135.2,
135.0, 134.2, 132.7, 132.6, 132.6, 132.6, 131.4, 131.4, 130.5, 130.3, 130.0, 130.0, 129.8, 129.5,
128.5, 128.4, 127.3, 127.2, 125.8, 125.7, 125.3, 125.2, 124.9, 124.8, 124.8, 124.6, 123.3, 123.3,
44.8, 44.7, 34.7, 34.7, 31.5, 24.4, 24.3, 23.4, 23.4. 31P NMR (243 MHz, CDCl3) δ 148.05. HRMS
(ESI, M+1) calculated for C46H51NO2P 680.3652, found 680.3656.
N,N-diisopropyl-2,6-bis(4-nitrophenyl)dinaphtho[2,1-d:1',2'-
f][1,3,2]dioxaphosphepin-4-amine – The ligand was synthesized
according to General Procedure 2.5.4.2.5 and was isolated as a bright
yellow foam which could be crushed into a crystalline solid. 1H NMR
(600 MHz, CDCl3) δ 8.34 – 8.26 (m, 4H), 8.11 (d, J = 28.9 Hz, 2H),
8.08 – 8.04 (m, 2H), 8.02 – 7.97 (m, 2H), 7.94 – 7.88 (m, 2H), 7.49
(dtd, J = 7.9, 6.4, 1.4 Hz, 2H), 7.42 (dd, J = 8.5, 1.1 Hz, 1H), 7.35 (ddd,
J = 8.4, 6.7, 1.3 Hz, 1H), 7.33 – 7.27 (m, 2H), 3.13 (dq, J = 11.0, 6.6 Hz, 2H), 0.83 (d, J = 6.8 Hz,
6H), 0.60 (d, J = 6.8 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 147.5, 147.4, 147.2, 147.1, 146.8,
144.8, 144.7, 133.1, 133.1, 133.1, 132.7, 132.7, 131.7, 131.1, 131.1, 131.0, 131.0, 130.8, 130.8,
130.6, 130.3, 128.8, 127.0, 127.0, 127.0, 126.9, 125.6, 125.3, 125.2, 125.2, 123.5, 123.5, 123.2,
123.2, 45.0, 44.9, 24.4, 24.3, 23.6. 31P NMR (243 MHz, CDCl3) δ 150.39.
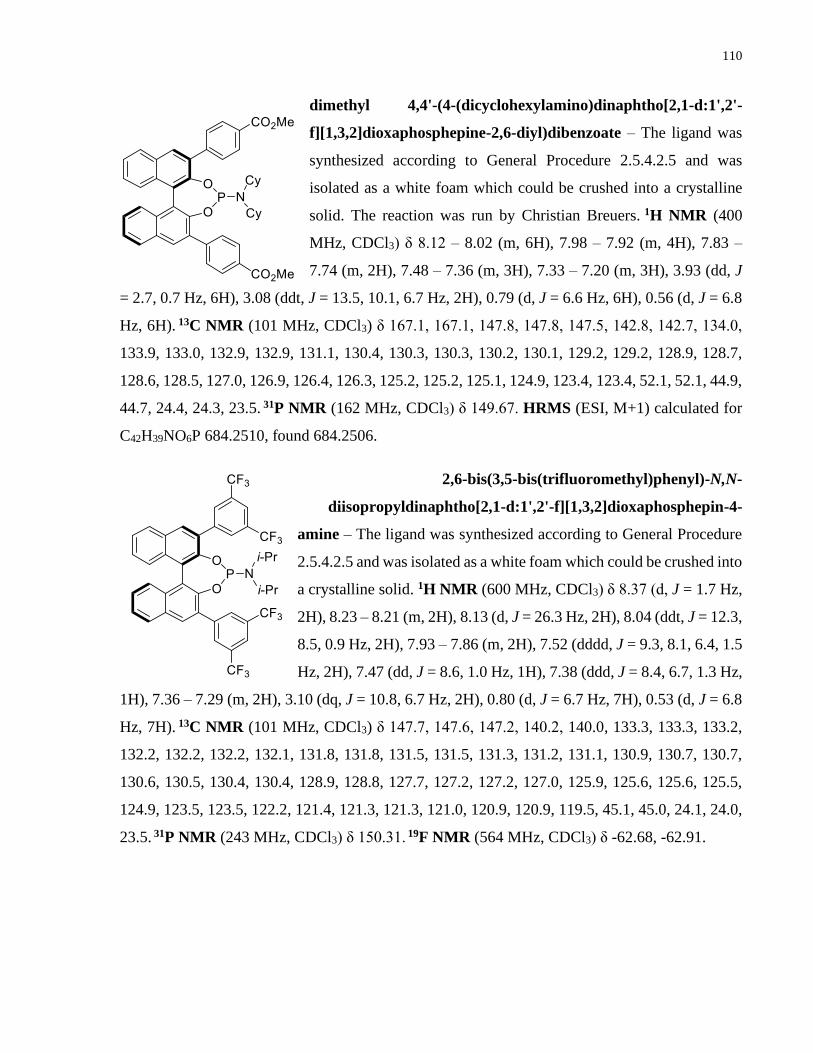
110
dimethyl 4,4'-(4-(dicyclohexylamino)dinaphtho[2,1-d:1',2'-
f][1,3,2]dioxaphosphepine-2,6-diyl)dibenzoate – The ligand was
synthesized according to General Procedure 2.5.4.2.5 and was
isolated as a white foam which could be crushed into a crystalline
solid. The reaction was run by Christian Breuers. 1H NMR (400
MHz, CDCl3) δ 8.12 – 8.02 (m, 6H), 7.98 – 7.92 (m, 4H), 7.83 –
7.74 (m, 2H), 7.48 – 7.36 (m, 3H), 7.33 – 7.20 (m, 3H), 3.93 (dd, J
= 2.7, 0.7 Hz, 6H), 3.08 (ddt, J = 13.5, 10.1, 6.7 Hz, 2H), 0.79 (d, J = 6.6 Hz, 6H), 0.56 (d, J = 6.8
Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 167.1, 167.1, 147.8, 147.8, 147.5, 142.8, 142.7, 134.0,
133.9, 133.0, 132.9, 132.9, 131.1, 130.4, 130.3, 130.3, 130.2, 130.1, 129.2, 129.2, 128.9, 128.7,
128.6, 128.5, 127.0, 126.9, 126.4, 126.3, 125.2, 125.2, 125.1, 124.9, 123.4, 123.4, 52.1, 52.1, 44.9,
44.7, 24.4, 24.3, 23.5. 31P NMR (162 MHz, CDCl3) δ 149.67. HRMS (ESI, M+1) calculated for
C42H39NO6P 684.2510, found 684.2506.
2,6-bis(3,5-bis(trifluoromethyl)phenyl)-N,N-
diisopropyldinaphtho[2,1-d:1',2'-f][1,3,2]dioxaphosphepin-4-
amine – The ligand was synthesized according to General Procedure
2.5.4.2.5 and was isolated as a white foam which could be crushed into
a crystalline solid. 1H NMR (600 MHz, CDCl3) δ 8.37 (d, J = 1.7 Hz,
2H), 8.23 – 8.21 (m, 2H), 8.13 (d, J = 26.3 Hz, 2H), 8.04 (ddt, J = 12.3,
8.5, 0.9 Hz, 2H), 7.93 – 7.86 (m, 2H), 7.52 (dddd, J = 9.3, 8.1, 6.4, 1.5
Hz, 2H), 7.47 (dd, J = 8.6, 1.0 Hz, 1H), 7.38 (ddd, J = 8.4, 6.7, 1.3 Hz,
1H), 7.36 – 7.29 (m, 2H), 3.10 (dq, J = 10.8, 6.7 Hz, 2H), 0.80 (d, J = 6.7 Hz, 7H), 0.53 (d, J = 6.8
Hz, 7H). 13C NMR (101 MHz, CDCl3) δ 147.7, 147.6, 147.2, 140.2, 140.0, 133.3, 133.3, 133.2,
132.2, 132.2, 132.2, 132.1, 131.8, 131.8, 131.5, 131.5, 131.3, 131.2, 131.1, 130.9, 130.7, 130.7,
130.6, 130.5, 130.4, 130.4, 128.9, 128.8, 127.7, 127.2, 127.2, 127.0, 125.9, 125.6, 125.6, 125.5,
124.9, 123.5, 123.5, 122.2, 121.4, 121.3, 121.3, 121.0, 120.9, 120.9, 119.5, 45.1, 45.0, 24.1, 24.0,
23.5. 31P NMR (243 MHz, CDCl3) δ 150.31. 19F NMR (564 MHz, CDCl3) δ -62.68, -62.91.

111
2.5.4.3 General Procedure for the Aryl/Borylation
In a dry and purged 1D vial, Pd(dba)2 (5 mol%) and L6 (10 mol%) were pre-stirred in 2 mL of
MTBE at 40 °C for 15 minutes. The catalyst solution was transferred (washing with 2 mL of
MTBE, total 4 mL) into a Schlenk tube containing the corresponding aryl-chloride (0.2 mmol) and
Mixed-Boron (3 equiv). NEt3 (5 equiv) was added and the reaction was sealed and heated to 100
°C for 18 h. The reaction was filtered through silica and purified by column chromatography.
2.5.4.4 Characterization Data for New Compounds
10b-methyl-11-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-10b,11-
dihydro-6H-isoindolo[2,1-a]indol-6-one (2.65a) – The compound was
synthesized according to General Procedure 2.5.4.3 and was purified by silica
flash chromatography, eluting with pentanes–EtOAc–DCM (10:1:1, v:v:v).
Product isolated as a white solid (53.5 mg, 0.148 mmol, 74%, 97:3 er, mp = 193-195 °C). 1H NMR
(500 MHz, CDCl3) δ 7.86 (d, J = 7.5 Hz, 1H), 7.68 (d, J = 7.5 Hz, 1H), 7.56 (td, J = 7.5, 1.0 Hz,
1H), 7.51 – 7.44 (m, 2H), 7.27 (t, J = 7.5 Hz, 1H), 7.14 (d, J = 7.0 Hz, 1H), 7.06 (td, J = 7.5, 1.0
Hz, 1H), 2.92 (s, 1H), 1.66 (s, 3H), 0.84 (s, 6H), 0.68 (s, 6H). 13C NMR (125 MHz, CDCl3) δ
168.1, 150.0, 138.8, 138.6, 133.6, 132.0, 128.4, 127.1, 124.58, 124.56, 124.4, 122.6, 117.4, 83.4,
73.9, 28.6, 24.3, 23.8. HRMS (ESI, M+1) Calculated for C22H25BNO3 362.1922, found 362.1921.
[α]D20 = +90.7 (c 1.0, CHCl3). HPLC [Daicel Chiralpak IA column (25 cm × 0.46 cm), n-hexane–
i-PrOH (90:10, v:v), 0.75 mL/min, tR(major) = 9.9 min, tR(minor) = 7.8 min].
10,10b-dimethyl-11-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-
10b,11-dihydro-6H-isoindolo[2,1-a]indol-6-one (2.65b) – The compound
was synthesized according to General Procedure 2.5.4.3 and was purified by
silica flash chromatography, eluting with pentanes–EtOAc–DCM (10:1:1,

112
v:v:v). Product isolated as a white solid (44.4 mg, 0.118 mmol, 59%, 89.5:10.5 er, mp = 161–162
°C). 1H NMR (500 MHz, CDCl3) δ 7.71–7.66 (m, 2H), 7.35 (t, J = 7.4 Hz, 1H), 7.30 (dt, J = 7.5,
1.0 Hz, 1H), 7.25 (td, J = 7.7, 1.4 Hz, 1H), 7.14 (ddt, J = 7.5, 1.3, 0.7 Hz, 1H), 7.04 (td, J = 7.5,
1.1 Hz, 1H), 2.98 (s, 1H), 2.49 (s, 3H), 1.68 (s, 3H), 0.82 (s, 6H), 0.67 (s, 6H). 13C NMR (125
MHz, CDCl3) δ 167.5, 148.0, 138.6, 138.1, 134.1, 133.8, 132.6, 128.6, 127.2, 124.5, 124.3, 122.3,
117.2, 83.4, 74.2, 26.4, 24.3, 23.8, 18.7. HRMS (DART, M+1) Calculated for C23H27BNO3
376.2079, found 376.2046. [α]D20 = +125.2 (c 1.0, CHCl3). HPLC [Daicel Chiralpak IA column
(25 cm × 0.46 cm), n-hexane–i-PrOH (90:10, v:v), 0.75 mL/min, tR(major) = 8.7 min, tR(minor) = 7.6
min.
9-methoxy-10b-methyl-11-(4,4,5,5-tetramethyl-1,3,2-
dioxaborolan-2-yl)-10b,11-dihydro-6H-isoindolo[2,1-a]indol-6-
one (2.65c) – The compound was synthesized according to General
Procedure 2.5.4.3 and was purified by silica flash chromatography,
eluting with pentanes–EtOAc–DCM (10:1:1, v:v:v). Product isolated as a white solid (69.2 mg,
0.177 mmol, 88%, 84:16 er, mp = 243-245 °C). 1H NMR (500 MHz, CDCl3) δ 7.67 (d, J = 7.5
Hz, 1H), 7.38 (d, J = 8.5 Hz, 1H), 7.34 (d, J = 2.5 Hz, 1H), 7.26 (td, J = 7.5, 1.0 Hz, 1H), 7.15 –
7.11 (m, 2H), 7.06 (td, J = 7.5, 1.0 Hz, 1H), 3.88 (s, 3H), 2.89 (s, 1H), 1.64 (s, 3H), 0.86 (s, 6H),
0.72 (s, 6H). 13C NMR (125 MHz, CDCl3) δ 168.0, 160.4, 142.4, 139.0, 138.6, 135.0, 127.1, 124.6,
124.4, 123.5, 120.2, 117.3, 107.2, 83.4, 73.7, 55.8, 28.7, 24.5, 23.9. HRMS (ESI, M+1) Calculated
for C23H27BNO4 392.2028, found 392.2044. [α]D20 = +172.4 (c 1.0, CHCl3). HPLC [Daicel
Chiralpak IA column (25 cm × 0.46 cm), n-hexane–i-PrOH (90:10, v:v), 0.75 mL/min, tR(major) =
13.5 min, tR(minor) = 10.8 min.
(10bR,11S)-9-fluoro-10b-methyl-11-(4,4,5,5-tetramethyl-1,3,2-
dioxaborolan-2-yl)-10b,11-dihydro-6H-isoindolo[2,1-a]indol-6-one
(2.65d) – The compound was synthesized according to General
Procedure 2.5.4.3 and was purified by silica flash chromatography,
eluting with pentanes–EtOAc–DCM (10:1:1, v:v:v). Product isolated as a white solid (49.0 mg,
0.129 mmol, 65%, 84:16 er, mp = 152–154 °C). 1H NMR (500 MHz, CDCl3) δ 7.82 (ddd, J = 8.0,
5.1, 0.8 Hz, 1H), 7.64 (dd, J = 7.8, 1.0 Hz, 1H), 7.25 (td, J = 7.6, 1.3 Hz, 1H), 7.17–7.12 (m, 3H),
7.05 (td, J = 7.5, 1.1 Hz, 1H), 2.90 (s, 1H), 1.64 (s, 3H), 0.86 (s, 6H), 0.73 (s, 6H). 13C NMR (125
MHz, CDCl3) δ 167.1, 165.5 (d, J = 252.5 Hz), 152.8 (d, J = 9.3 Hz), 138.6, 138.1, 129.5 (d, J =

113
2.2 Hz), 127.2, 126.7 (d, J = 9.6 Hz), 124.7, 124.5, 117.3, 115.9 (d, J = 23.4 Hz), 110.1 (d, J =
23.8 Hz), 83.6, 73.5 (d, J = 2.6 Hz), 28.6, 24.4, 23.9. 19F NMR (377 MHz, CDCl3) δ -106.75 (dt,
J = 9.0, 4.5 Hz). HRMS (DART, M+1) Calculated for C22H24BFNO3 380.1833, found 380.1832.
[α]D20 = +87.3 (c 1.0, CHCl3). HPLC [Daicel Chiralpak IA column (25 cm × 0.46 cm), n-hexane–
i-PrOH (90:10, v:v), 0.75 mL/min, tR(major) = 10.7 min, tR(minor) = 8.5 min.
(10bR,11S)-10-fluoro-10b-methyl-11-(4,4,5,5-tetramethyl-1,3,2-
dioxaborolan-2-yl)-10b,11-dihydro-6H-isoindolo[2,1-a]indol-6-one
(2.65e) – The compound was synthesized according to General Procedure
2.5.4.3 and was purified by silica flash chromatography, eluting with
pentanes–EtOAc–DCM (10:1:1, v:v:v). Product isolated as a white solid
(50.3 mg, 0.133 mmol, 66%, 95.5:4.5 er, mp = 170–172 °C). 1H NMR (500 MHz, CDCl3) δ 7.67–
7.63 (m, 1H), 7.51 (dd, J = 7.7, 2.5 Hz, 1H), 7.43 (dd, J = 8.3, 4.4 Hz, 1H), 7.28–7.23 (m, 3H),
7.14 (ddt, J = 7.4, 1.3, 0.7 Hz, 1H), 7.06 (td, J = 7.5, 1.1 Hz, 1H), 2.90 (s, 1H), 1.64 (s, 3H), 0.85
(s, 6H), 0.72 (s, 6H). 13C NMR (125 MHz, CDCl3) δ 166.7 (d, J = 3.3 Hz), 163.1 (d, J = 247.7
Hz), 145.6 (d, J = 2.6 Hz), 138.6, 138.34, 135.8 (d, J = 8.4 Hz), 127.2, 124.7, 124.7, 124.0 (d, J =
8.4 Hz), 119.2 (d, J = 23.5 Hz), 117.4, 111.2 (d, J = 23.2 Hz), 83.5, 73.7, 28.6, 24.4, 23.9. 19F
NMR (377 MHz, CDCl3) δ -112.89 – -113.09 (m). HRMS (DART, M+1) Calculated for
C22H24BFNO3 380.1833, found 380.1836. [α]D20 = +72.7 (c 1.0, CHCl3). HPLC [Daicel Chiralpak
IA column (25 cm × 0.46 cm), n-hexane–i-PrOH (90:10, v:v), 0.75 mL/min, tR(major) = 8.6 min,
tR(minor) = 6.8 min.
(10bR,11S)-2-fluoro-10b-methyl-11-(4,4,5,5-tetramethyl-1,3,2-
dioxaborolan-2-yl)-10b,11-dihydro-6H-isoindolo[2,1-a]indol-6-one
(2.65f) – The compound was synthesized according to General Procedure
2.5.4.3 and was purified by silica flash chromatography, eluting with
pentanes–EtOAc–DCM (10:1:1, v:v:v). Product isolated as a white solid (44.6 mg, 0.118 mmol,
59%, 95.5:4.5 er, mp = 193–194 °C). 1H NMR (500 MHz, CDCl3) δ 7.84 (dt, J = 7.5, 1.0 Hz, 1H),
7.61–7.53 (m, 2H), 7.49–7.43 (m, 2H), 6.97–6.91 (m, 1H), 6.85 (ddd, J = 8.3, 2.5, 0.8 Hz, 1H),
2.89 (s, 1H), 1.65 (s, 3H), 0.83 (s, 6H), 0.67 (s, 6H). 13C NMR (125 MHz, CDCl3) δ 168.1, 160.2
(d, J = 242.2 Hz), 149.7, 140.9 (d, J = 8.5 Hz), 134.8 (d, J = 2.2 Hz), 133.4, 132.1, 128.6, 124.7,
122.7, 117.9 (d, J = 9.0 Hz), 113.5 (d, J = 23.5 Hz), 112.2 (d, J = 24.3 Hz), 83.7, 74.5, 28.5, 24.4,
23.9. 19F NMR (377 MHz, CDCl3) δ -118.53 – -118.71 (m). HRMS (DART, M+1) Calculated for

114
C22H24BFNO3 380.1833, found 380.1837. [α]D20 = +86.0 (c 1.0, CHCl3). HPLC [Daicel Chiralpak
IA column (25 cm × 0.46 cm), n-hexane–i-PrOH (90:10, v:v), 0.75 mL/min, tR(major) = 10.1 min,
tR(minor) = 8.4 min.
(10bR,11S)-2-methoxy-10b-methyl-11-(4,4,5,5-tetramethyl-1,3,2-
dioxaborolan-2-yl)-10b,11-dihydro-6H-isoindolo[2,1-a]indol-6-one
(2.65g) – The compound was synthesized according to General
Procedure 2.5.4.3 and was purified by silica flash chromatography,
eluting with pentanes–EtOAc–DCM (10:1:1, v:v:v). Product isolated as a white solid (50.7 mg,
0.129 mmol, 65%, 96.5:3.5 er, mp = 189–191 °C). 1H NMR (500 MHz, CDCl3) δ 7.83 (dt, J =
7.5, 1.0 Hz, 1H), 7.57 (d, J = 8.5 Hz, 1H), 7.53 (td, J = 7.4, 1.2 Hz, 1H), 7.48–7.42 (m, 2H), 6.77
(ddd, J = 8.5, 2.6, 0.5 Hz, 1H), 6.71 s(dd, J = 2.6, 0.8 Hz, 1H), 3.78 (s, 3H), 2.86 (s, 1H), 1.64 (s,
3H), 0.83 (s, 6H), 0.66 (s, 6H). 13C NMR (125 MHz, CDCl3) δ 168.0, 157.0, 149.9, 140.5, 133.8,
132.4, 131.8, 128.4, 124.5, 122.5, 117.7, 111.6, 111.3, 83.5, 74.3, 55.6, 28.5, 24.4, 23.9. HRMS
(DART, M+1) Calculated for C23H27BNO4 392.2033, found 392.2028. [α]D20 = +28.6 (c 1.0,
CHCl3). HPLC [Daicel Chiralpak IA column (25 cm × 0.46 cm), n-hexane–i-PrOH (90:10, v:v),
0.75 mL/min, tR(major) = 16.2 min, tR(minor) = 10.8 min.
10b-phenyl-11-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-10b,11-
dihydro-6H-isoindolo[2,1-a]indol-6-one (2.65h) – The compound was
synthesized according to General Procedure 2.5.4.3 and was purified by silica
flash chromatography, eluting with pentanes–EtOAc–DCM (10:1:1, v:v:v).
Product isolated as a white solid (61.8 mg, 0.146 mmol, 73%, 97:3 er, mp = 180–183 °C). 1H
NMR (500 MHz, CDCl3) δ 7.85 (d, J = 7.5 Hz, 1H), 7.78 (d, J = 8.0 Hz, 1H), 7.66–7.63 (m, 2H),
7.57 (d, J = 7.5 Hz, 1H), 7.49 (td, J = 7.5, 1.0 Hz, 1H), 7.41 (td, J = 7.5, 1.0 Hz, 1H), 7.31 – 7.28
(m, 2H), 7.27–7.23 (m, 1H), 7.21 (t, J = 7.5 Hz, 1H), 7.06 (d, J = 7.5 Hz, 1H), 7.02 (td, J = 7.5,
1.0 Hz, 1H), 3.62 (s, 1H), 0.89 (s, 6H), 0.73 (s, 6H). 13C NMR (125 MHz, CDCl3) δ 168.7, 149.3,
144.2, 139.1, 138.6, 133.1, 132.3, 128.6, 128.4, 127.6, 127.2, 124.8, 124.7, 124.6, 124.2, 123.7,
117.3, 83.7, 79.0, 24.4, 23.9. HRMS (ESI, M+1) Calculated for C27H27BNO3 424.2079, found
424.2049. [α]D20 = +219.1 (c 1.0, CHCl3). HPLC [Daicel Chiralpak IA column (25 cm × 0.46 cm),
n-hexane–i-PrOH (90:10, v:v), 0.75 mL/min, tR(major) = 11.1 min, tR(minor) = 12.9 min.

115
(10bS,11S)-10b-(4-fluorophenyl)-11-(4,4,5,5-tetramethyl-1,3,2-
dioxaborolan-2-yl)-10b,11-dihydro-6H-isoindolo[2,1-a]indol-6-one
(2.65i) – The compound was synthesized according to General Procedure
2.5.4.3 and was purified by silica flash chromatography, eluting with
pentanes–EtOAc–DCM (10:1:1, v:v:v). Product isolated as a white solid
(35.0 mg, 0.0793 mmol, 40%, 97:3, mp = 163–165 °C). 1H NMR (500 MHz, CDCl3) δ 7.84 (dt, J
= 7.5, 1.0 Hz, 1H), 7.75 (dd, J = 7.8, 1.0 Hz, 1H), 7.61–7.56 (m, 2H), 7.53–7.45 (m, 2H), 7.40 (td,
J = 7.3, 1.3 Hz, 1H), 7.23 (td, J = 7.6, 1.4 Hz, 1H), 7.08–7.04 (m, 1H), 7.01 (td, J = 7.5, 1.1 Hz,
1H), 6.97–6.91 (m, 2H), 3.56 (s, 1H), 0.86 (s, 6H), 0.71 (s, 6H). 13C NMR (125 MHz, CDCl3) δ
168.6, 162.1 (d, J = 246.6 Hz), 149.2, 139.9 (d, J = 3.1 Hz), 139.0, 138.4, 133.0, 132.4, 128.5,
127.3, 126.6 (d, J = 8.1 Hz), 124.9, 124.7, 124.2, 123.6, 117.4, 115.5 (d, J = 21.6 Hz), 83.7, 78.6,
24.4, 23.9. 19F NMR (376 MHz, CDCl3) δ -114.95 – -115.03 (m). HRMS (DART, M+1)
Calculated for C27H26BFNO3 442.1990, found 442.1994. [α]D20 = +183.2 (c 1.0, CHCl3). HPLC
[Daicel Chiralpak IA column (25 cm × 0.46 cm), n-hexane–i-PrOH (90:10, v:v), 0.75 mL/min,
tR(major) = 10.5 min, tR(minor) = 13.1 min.
(11S,11aR)-11a-methyl-11-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-
yl)-11,11a-dihydro-5H-pyrido[2',3':3,4]pyrrolo[1,2-a]indol-5-one (2.65j)
– The compound was synthesized according to General Procedure 2.5.4.3 and
was purified by silica flash chromatography, eluting with pentanes–EtOAc–
DCM (10:1:1, v:v:v). Product isolated as a white solid (58.7mg, 0.162 mmol, 81%, 91:9 er, mp =
120–122 °C). 1H NMR (500 MHz, CDCl3) δ 8.72 (dd, J = 4.9, 1.6 Hz, 1H), 8.12 (dd, J = 7.7, 1.6
Hz, 1H), 7.68 (ddt, J = 7.7, 1.0, 0.5 Hz, 1H), 7.39 (dd, J = 7.7, 4.9 Hz, 1H), 7.27 (tdd, J = 7.6, 1.3,
0.6 Hz, 1H), 7.20 (ddt, J = 7.6, 1.4, 0.7 Hz, 1H), 7.08 (td, J = 7.5, 1.2 Hz, 1H), 3.01 (s, 1H), 1.71
(s, 3H), 0.86 (s, 6H), 0.71 (s, 6H). 13C NMR (125 MHz, CDCl3) δ 169.7, 166.5, 152.5, 138.4,
137.9, 132.6, 127.4, 127.3, 124.9, 124.8, 123.1, 117.5, 83.4, 74.7, 27.0, 24.5, 23.7. HRMS (DART,
M+1) Calculated for C21H24BN2O3 363.1880, found 363.1878. [α]D20 = +105.6 (c 1.0, CHCl3).
HPLC [Daicel Chiralpak IA column (25 cm × 0.46 cm), n-hexane–i-PrOH (90:10, v:v), 0.75
mL/min, tR(major) = 12.4 min, tR(minor) = 8.5 min.

116
2.5.4.5 Synthetic Transformation of Products
Oxidation of 3a to 11-hydroxy-10b-methyl-10b,11-dihydro-6H-
isoindolo[2,1-a]indol-6-one (2.77) – The oxidation was performed
according to the literature procedure.116 In a reaction vial, 2.65a (72.2 mg,
0.2 mmol) was dissolved in THF/H2O (1:1, 2 mL). NaBO3•4H2O (153.9 mg,
1.0 mmol) was then added at room temperature. After stirred for 2 h, the reaction mixture was
extracted three times with EtOAc, dried over MgSO4, and filtered. The crude mixture was purified
by flash column chromatography eluting with pentanes–EtOAc, (1:1, v:v) to afford the product
2.77 as a white solid (50.2 mg, 0.2 mmol, >99%, 96.5:3.5 er, mp = 99–101 °C). 1H NMR (500
MHz, CDCl3) δ 7.62 (td, J = 7.5, 1.0 Hz, 1H), 7.58 – 7.50 (m, 4H), 7.46 (td, J = 7.5, 1.0 Hz, 1H),
7.36 (td, J = 7.5, 1.0 Hz, 1H), 7.23 (td, J = 7.5, 1.0 Hz, 1H), 4.86 (s, 1H), 2.62 (s, 1H), 1.49 (s,
3H). 13C NMR (125 MHz, CDCl3) δ 168.6, 147.0, 139.2, 137.0, 133.1, 132.5, 130.4, 128.8, 126.7,
124.8, 124.4, 123.2, 117.8, 76.2, 76.0, 24.5. HRMS (DART, M+1) Calculated for C16H14NO2
252.1019, found 252.1022. [α]D20 = +178.4 (c 1.0, CHCl3). HPLC [Daicel Chiralpak IA column
(25 cm × 0.46 cm), n-hexane–i-PrOH (70:30, v:v), 0.8 mL/min, tR(major) = 11.1 min, tR(minor) = 6.4
min.
Oxidation of 4 to10b-methyl-6H-isoindolo[2,1-a]indole-6,11(10bH)-
dione (2.78) – In a reaction vial, alcohol 2.77 (50.3 mg, 0.2 mmol) was
dissolved in CHCl3 (2 mL). Pyridinium chlorochromate (64.7 mg, 0.6 mmol)
was then added at room temperature. And the reaction mixture was heated to
35 °C and stirred overnight. The solvent was then removed under vacuum and the residue was
purified by chromatography on silica gel, eluting with pentanes–EtOAc (5:1, v:v) to afford the
product 2.78 as a white solid (42.4 mg, 0.17 mmol, 85%, 96:4 er). Spectral data matched those
reported in the literature117: 1H NMR (500 MHz, CDCl3) δ 7.99 (d, J = 8.0 Hz, 1H), 7.87–7.82 (m,
2H), 7.80 – 7.75 (m, 2H), 7.72 (td, J = 7.5, 1.0 Hz, 1H), 7.54 (td, J = 7.5, 1.0 Hz, 1H), 7.30 (td, J
= 7.5, 1.0 Hz, 1H), 1.82 (s, 3H). [α]D20 = +578.9 (c 1.0, CHCl3). HPLC [Daicel Chiralpak IA
column (25 cm × 0.46 cm), n-hexane–i-PrOH (90:10, v:v), 0.75 mL/min, tR(major) = 11.0 min,
tR(minor) = 12.8 min.
116
Kubota, K.; Hayama, K.; Iwamoto, H.; Ito, H. Angew. Chem. Int. Ed. 2015, 54, 8809. 117
Liu, R.-R.; Wang, Y.-G.; Li, Y.-L.; Huang, B.-B.; Liang, R.-X.; Jia, Y.-X. Angew. Chem. Int. Ed. 2017, 56, 7475.

117
Chapter 3
Dearomatization of Indoles with NFSI and Further Aromative Functionalizations
3.1 Introduction
3.1.1 Importance of Fluorine in Biology, Medicine, Materials
Fluorine-containing molecules continue to account for a large portion of pharmaceutical and
agrochemical compounds due to an increase in solubility, lipophilicity, and biological stability
against oxidation as compared to the C–H bond containing analogues.118 In 1955, the first fluorine-
containing drug was approved in the United-States and by 2016, 30% of drugs contained at least
one fluorine atom.119 This drug was a derivative of cortisone with one H to F substitution, and it
was found to have a significant increase in glucocorticoid activity compared to the non-fluorinated
counterpart (Scheme 77, 3.1).120
Scheme 77 Fluoro-derivative of cortisone
Of the 31 recently approved active ingredients in fungicides, herbicides, and insecticides, 19
contain at least one fluorine atom (over 60%).121 The benefits of incorporating a fluorine extends
to other stages of active pharmaceutical ingredients (API) development. For example, the nuclei
118
(a) Zhou, Y.; Wang, J.; Gu, Z.; Wang, S.; Zhu, W.; Acena, J. L.; Soloshonok, V. A.; Izawa, K.; Liu, H., Chem.
Rev. 2016, 116, 422. (b) Fujiwara, T.; O'Hagan, D., J. Fluorine Chem. 2014, 167, 16. 119
O’Hagan, D. J. Fluorine Chem. 2010, 131, 1071. 120
Fried, J.; Sabo, E. F. J. Am. Chem. Soc. 1954, 76, 1455. 121
Beer, A. Agrow-Agribusiness Intelligence Annual Review. 2018,
https://agrow.agribusinessintelligence.infoma.com/-/media/agri/agrow/ag-market-reviews-
pdfs/supplements/agrowannualreview2018.pdf (accessed January 15, 2020).

118
can be used to track compounds in vitro or in vivo by NMR or MRI respectively.122 The 18F
radionuclide is the most commonly used radioisotope in positron emitting tomography due to its
convenient half-life (110 minutes) and favorable resolution (attributed to its low positron energy
of 635 keV).123
3.1.2 Strategies of Fluorine Incorporation
Diverse strategies have been employed for fluorine incorporation into small molecules, including
the use of catalysts, nucleophilic reagents, electrophilic reagents, and radical additions.124
3.1.2.1 Nucleophilic Fluorine Reagents
Alkali-metal fluorides are commonly used as a source of F- however, one must consider their
hygroscopicity as there are competing reactions with hydroxides. Other common reagents include
alkylammonium fluorides such as tetrabutylammonium fluoride (TBAF)125 since there is a weaker
ionic bonding energy to overcome as well as an increased solubility in organic solvents.126
Similarly, TBAF suffers from being hygroscopic and thus there is an interest in sources of “naked”
fluoride anion. Contrary to typical displacement reactions, it was found that the nucleophilicity of
alkali-metal fluorides and TBAF in tert-butyl alcohol is significantly increased.127 It is proposed
that while the solvent is capable of hydrogen bonding sufficiently to solvate fluoride anion, there
is limited coordination due to the bulkiness of the tert-butyl groups.127 DiMagno and coworkers
reported the synthesis of dry TBAF by the SNAr reaction of hexafluorobenzene (3.1) with
tetrabutylammonium cyanide (Scheme 78, 3.3).128
122
Ruiz-Cabello, J.; Barnett, B. P.; Bottomley, P. A.; Bulte, J. W., NMR Biomed. 2011, 24, 114. 123
Jacobson, O.; Kiesewetter, D. O.; Chen, X., Bioconjugate Chem. 2015, 26, 1. 124
(a) Szpera, R.; Moseley, D. F. J.; Smith, L. B.; Sterling, A. J.; Gouverneur, V., Angew. Chem. Int. Ed. 2019, 58,
14824. (b) Champagne, P. A.; Desroches, J.; Hamel, J. D.; Vandamme, M.; Paquin, J. F., Chem. Rev. 2015, 115, 9073.
(c) Campbell, M. G.; Ritter, T., Chem. Rev. 2015, 115, 612. (d) Studer, A., Angew. Chem. Int. Ed. 2012, 51, 8950. 125
Yoshida, Y.; Kimura, Y. Chem. Lett. 1988, 17, 1355. 126
Clark, J. H. Chem. Rev. 1980, 80, 429. 127
Kim, D. W.; Jeong, H. J.; Lim, S. T.; Sohn, M. H.; Katzenellenbogen, J. A.; Chi, D. Y. J. Org. Chem. 2008, 73,
957. 128
Sun, H.; DiMagno, S. G. J. Am. Chem. Soc. 2005, 127, 2050.

119
Scheme 78 The synthesis of anhydrous TBAF by SNAr
Anhydrous TBAF displayed remarkable nucleophilicity compared to commercial sources of
TBAF, and electron-deficient chloro- and nitro- arenes could be fluorinated at room temperature
in short reaction times (Scheme 79).129
Scheme 79 The fluorination of chloro- and nitro-arenes with anhydrous NFSI at room
temperature
Finally, nucleophilic fluorination has been explored using palladium catalysis. The challenges and
mechanistic peculiarities are described in section 3.1.2.3 on page 121.
3.1.2.2 Electrophilic Fluorine Reagents
For decades, various electrophilic fluorinating reagents have been well developed and widely
used.130 Due to fluorine being the most electronegative atom on the periodic table, the only obvious
electrophilic source of fluorine is F2. Since it is an extremely toxic and reactive gas, it is used in
the synthesis of many reagents with properties better suited for laboratory use.131 The most
common family of reagents are those which contain N–F bonds (Scheme 80).
129
Sun, H.; DiMagno, S. G. Angew. Chem. Int. Ed. 2006, 45, 2720. 130
Taylor, S. D.; Kotoris, C. C.; Hum, G. Tetrahedron 1999, 55, 12431. 131
Lal, G. S.; Pez, G. P.; Syvret, R. G. Chem. Rev. 1996, 96, 1737.

120
Scheme 80 Common N–F reagents
One of the most commonly used electrophilic sources of fluorine is N-fluorobenzenesulfonimide
(3.10, NFSI) due to its shelf-stability, controlled reactivity, modest cost, ability to vary its
electronic properties, and the availability of a 18F variant.132 Although the N–F bond is polarized
towards fluorine due to the difference in electronegativity, formal F+ transfer occurs by two
mechanisms. The first mechanism is proposed to occur by SN2 at the fluorine atom since the *N–
F orbital is more sterically accessible.133 The second mechanism proposed occurs by single
electron transfer. There is evidence that both pathways take place simultaneously depending on
reaction conditions.134
One of the most reliable methods for the fluorination of simple aromatic and heteroaromatic
compounds (3.12) is the reaction of Grignard reagents (3.13) with electrophilic N–F reagents
(Scheme 81).135
132
(a) Teare, H.; Robins, E. G.; Arstad, E.; Luthra, S. K.; Gouverneur, V., Chem. Commun. 2007, 2330. (b)
Buckingham, F.; Kirjavainen, A. K.; Forsback, S.; Krzyczmonik, A.; Keller, T.; Newington, I. M.; Glaser, M.; Luthra,
S. K.; Solin, O.; Gouverneur, V., Angew. Chem. Int. Ed. 2015, 54, 13366. (c) Meyer, D.; Jangra, H.; Walther, F.;
Zipse, H.; Renaud, P., Nat. Commun. 2018, 9, 4888. 133
Nyffeler, P. T.; Duron, S. G.; Burkart, M. D.; Vincent, S. P. P.; ́ Wong, C.-H. Angew. Chem. Int. Ed. 2005, 44,
192. 134
(a) Lee, K. Y.; Kochi, J. K. J. Chem. Soc., Perkin Trans. 2 1992, 1011. (b) Bockman, T. M.; Lee, K. Y.; Kochi, J.
K. J. Chem. Soc., Perkin Trans. 2 1992, 1581. (c) Vincent, S. P.; Burkart, M. D.; Tsai, C.-Y.; Zhang, Z.; Wong, C.-
H. J. Org. Chem. 1999, 64, 5264. (c) Brandt, J. R.; Lee, E.; Boursalian, G. B.; Ritter, T. Chem. Sci. 2014, 5, 169. 135
Yamada, S.; Gavryushin, A.; Knochel, P. Angew. Chem. Int. Ed. 2010, 49, 2215.

121
Scheme 81 The synthesis of aromatic and heteroaromatic fluorides by the reaction of Grignard
reagents with NFSI
The Sanford group developed an interesting fluorination with a relatively cheap source of fluoride,
KF, with a large excess of Cu(OTf)2, which served two roles (Scheme 82).136 The copper was the
mediator for the reaction between the aryl-BF3K reagents (3.15) and the fluoride anion, and
subsequently, as the internal oxidant for a productive reductive elimination.
Scheme 82 Fluorination of aryl-BF3K salts with KF and an internal oxidant
Electrophilic fluorination reactions involving palladium as a catalyst have also been explored. The
strategy for such reactions as well as the mechanistic uniqueness are described in the next section.
3.1.2.3 Pd-Catalyzed C–F Bond Formation (nucleophilic & electrophilic)
There are two strategies for fluorine incorporation using palladium-catalysts: low valent Pd0-PdII
mechanism and high valent PdII-PdIV mechanism (Scheme 83).
136
Ye, Y.; Schimler, S. D.; Hanley, P. S.; Sanford, M. S. J. Am. Chem. Soc. 2013, 135, 16292.

122
Scheme 83 Palladium-catalyzed C–F bond formation via Pd0/PdII and PdII/PdIV cycles
The transformation of aryl-halides/triflates to aryl fluorides via a Pd0-mechanism was successfully
accomplished by the Buchwald group in 2009 (Scheme 83, left).137 Until then, the reductive
elimination of Caryl–F was underexplored due to the formation of unproductive species such as
dimers as well as the competing kinetics for the reductive elimination of phosphine ligands.138 The
authors utilized a bulky monodentate phosphine ligand to force a T-shaped three-coordinate
complex (3.17), facilitating reductive elimination. Interestingly, it was found that the ligand was
functionalized in situ to form the active catalyst (Scheme 84, 3.18).137
137
(a) Lee, H. G.; Milner, P. J.; Buchwald, S. L. J. Am. Chem. Soc. 2014, 136, 3792. (b) Watson, D. A.; Su, M.;
Teverovskiy, G.; Zhang, Y.; Garcia-Fortanet, J.; Kinzel, T.; Buchwald, S. L. Science 2009, 325, 1661. (c) Sather, A.
C.; Buchwald, S. L. Acc. Chem. Res. 2016, 49, 2146. 138
Grushin, V. V. Chem. - Eur. J. 2002, 8, 1006.

123
Scheme 84 In situ active catalyst formation by arylation of the charged ligand
The second approach features a kinetically favored reductive elimination by a mechanism which
invokes a palladium(IV) species (Scheme 83, right).139 Sanford and coworkers reported the first
example of this type of reactivity in the fluorination of 2-arylpyridines (3.19) (Scheme 85).139a The
use of an electrophilic source of fluorine as an oxidant led to two possible mechanisms being
proposed: a direct substitution (nucleophilic attack of Pd–C on F+) or the oxidative addition to
high-valent palladium-complexes with a reduced barrier to reductive elimination. Future studies
involving the isolations and characterization of key PdIV-complexes supported the mechanism
which features a PdIV reductive elimination complex.139b,c
Scheme 85 Palladium(II)-catalyzed direct fluorination of C–H bonds
139
(a) Hull, K. L.; Anani, W. Q.; Sanford, M. S. J. Am. Chem. Soc. 2006, 128, 7134. (b) Ball, N. D.; Sanford, M. S.
J. Am. Chem. Soc. 2009, 131, 3796. (c) Racowski, J. M.; Gary, J. B.; Sanford, M. S. Angew. Chem. Int. Ed. 2012, 51,
3414.

124
3.1.3 Electrophilic Dearomatization of Indoles – Push-Pull Mechanism
One may take advantage of indole nucleophilicity for dearomatization. When substituted at the 3-
position, indoles typically participate in substitution reactions with electrophiles to form the 3,3-
disubstituted iminium, which in many cases can be isolated (Scheme 86, 3.22).140 These iminiums
can then be trapped with nucleophiles (hence push-pull), before (3.23a) or after migration (3.23b)
of a group to the 2-position (path a or path b).
Scheme 86 Dearomatization of indoles by a push-pull mechanism
Arguably some of the best understood electrophilic metal-complexes, allyl-palladium species,
have been used in a wide variety of reactions concerning indoles.141 Trost and coworkers reported
an asymmetric dearomative allylation of indoles (Scheme 87).142 Interestingly, the boron-based
Lewis acid was found to both increase reactivity, and influence the enantioselectivity. 3,3-
Disubstituted indoles (3.25) are isolated in good to excellent yield and selectivity.
140
Roche, S. P.; Youte Tendoung, J.-J.; Tréguier, B. Tetrahedron 2015, 71, 3549. 141
Trost, B. M. Tetrahedron 2015, 71, 5708. 142
Trost, B. M.; Quancard, J. J. Am. Chem. Soc. 2006, 128, 6314.

125
Scheme 87 Dearomative allylation of indoles
Similarly, the oxidative addition complexes of palladium and aryl-halides are considered
electrophilic as they can be trapped by various nucleophiles. By this logic, You and coworkers
developed a simple intramolecular dearomatization of indoles with aryl-bromides tethered at the
3-position (Scheme 88, 3.26).143 The authors found large amounts of 1,2-migration when the 5-
membered ring was generated however, they were able to trap the imines with a hydride source,
generating the indoline products in good yields (3.28).
Scheme 88 Intramolecular palladium-catalyzed dearomatization of indoles using 3-tethered aryl-
halides as internal electrophiles
NFSI (3.10) has been shown to be a versatile reagent, capable of fluorine or sulfonamide transfer
to heterocycles, typically by modifying the solvent (Scheme 89).144 The incorporation of fluorine
143
Wu, K. J.; Dai, L. X.; You, S. L. Org. Lett. 2012, 14, 3772. 144
(a) Sakurai, F.; Yukawa, T.; Taniguchi, T., Org. Lett. 2019, 21, 7254. (b) Wang, X. J.; Lei, B. W.; Ma, L. F.; Jiao,
H. X.; Xing, W. H.; Chen, J. M.; Li, Z. Y., Adv. Synth. Catal. 2017, 359, 4284. (c) Meanwell, M.; Nodwell, M. B.;
Martin, R. E.; Britton, R., Angew. Chem. Int. Ed. 2016, 55, 13244. (d) Liu, H. H.; Wang, Y.; Deng, G. J.; Yang, L.,
Adv. Synth. Catal. 2013, 355, 3369. (e) Lim, Y. H.; Ong, Q.; Duong, H. A.; Nguyen, T. M.; Johannes, C. W., Org.
Lett. 2012, 14, 5676. (f) Lozano, O.; Blessley, G.; Martinez del Campo, T.; Thompson, A. L.; Giuffredi, G. T.; Bettati,
M.; Walker, M.; Borman, R.; Gouverneur, V., Angew. Chem. Int. Ed. 2011, 50, 8105.

126
proved to be a useful method to rapidly access functionalized isatin-analogues (Scheme 89,
right).145
Scheme 89 Fluorine or nitrogen transfer using NFSI
The electrophilic dearomatization of indoles has been explored with various N–F reagents such as
NFSI and Selectfluor. The most common strategy involves the fluorination and subsequent
trapping of the iminium formed. Jiao and coworkers used Selectfluor and oxygen nucleophiles to
access highly substituted indolines (Scheme 90).146 Water (3.30), alcohols (3.31), and N-tethered
alcohols (3.32) were tolerated well in the reaction.
Scheme 90 The use of Selectfluor for fluorination and trapping with oxygen nucleophiles
Simultaneously and independently, the Gouverneur group developed an enantioselective
fluorination of indoles, trapping with 3-tethered alkyl alcohols, phenol, and various N-nucleophiles
145
(a) Lim, Y. H.; Ong, Q.; Duong, H. A.; Nguyen, T. M.; Johannes, C. W., Org. Lett. 2012, 14, 5676. (b) Bhrigu,
B.; Pathak, D.; Siddiqui, N.; Alam, M.; Ahsan, W., Int. J. Pharm. Sci. Drug Res. 2010, 2, 229. 146
Lin, R.; Ding, S.; Shi, Z.; Jiao, N. Org. Lett. 2011, 13, 4498.

127
(Scheme 91).147 2,3-Cis-fused indolines (3.34) bearing a fluorine atom at one of the two
stereogenic centers are isolated in good yield and selectivity. The chirality stems from the use of a
hydroquinidine derived organocatalyst ((DHQ)2PHAL).
Scheme 91 Enantioselective fluorination with NFSI, and trapping with O/N-based nucleophiles
tethered at the 3-position, forming multi-cyclic products
3.2 Research Goal 3 – Synthesis and Reactions of 3,3-Difluoro-2-exo-Methylidene Indolines
3.2.1 Motivation
The focus of this project was to develop a dearomative fluorination of 2-methylindoles, generating
3,3-difluoroindolines bearing an exocyclic double bond (Scheme 92).148 The reactivity of these
indolines was studied, mainly in reactions concerning allylic-fluorides.
147
Lozano, O.; Blessley, G.; Martinez del Campo, T.; Thompson, A. L.; Giuffredi, G. T.; Bettati, M.; Walker, M.;
Borman, R.; Gouverneur, V. Angew. Chem. Int. Ed. 2011, 50, 8105. 148
Zeidan, N.; Zambri, M.; Unger, S.; Dank, C.; Torelli, A.; Mirabi, B.; Lautens, M. Org. Lett. 2020, 22, 3688.

128
Scheme 92 Dearomative fluorination of 2-methylindole
The products generated possess allylic fluorides capable of undergoing a variety of future
derivatizations. Allylic fluorides have been long studied, and continue to be of interest due to their
greater reactivity towards certain Tsuji-Trost type reactions.149 For example, the Trost group
recently developed an asymmetric palladium-catalyzed trifluoromethylation of allylic fluorides
using chiral Trost ligands (Scheme 93).150
Scheme 93 Asymmetric allylic fluoroalkylation/trifluoromethylation
3.2.2 Contributions
The results presented were obtained in collaboration with Matthew Zambri (an undergraduate
student at the University of Toronto), Sven Unger (a visiting student from Germany), Dr. Christian
Dank (a postdoc from Austria), Bijan Mirabi (a PhD student in the Lautens group), and Alexa
Torelli (a PhD student in the Lautens group). I conceived the idea, directed the project, and
executed most of the experiments. Matthew Zambri investigated the scope of the reaction and
optimized some derivatizations. Sven Unger optimized and ran palladium-allylation reactions. The
149
Hazari, A.; Gouverneur, V.; Brown, J. M. Angew. Chem. Int. Ed. 2009, 48, 1296. 150
Trost, B. M.; Gholami, H.; Zell, D. J. Am. Chem. Soc. 2019, 141, 11446.

129
other collaborators contributed specific derivatizations based on their expertise. All contributions
are specified within the text as well as within the experimental section.
3.2.3 Results and Discussion
3.2.3.1 Starting Material Synthesis
The starting materials were synthesized by the same chemical transformation as Chapter 2 (see
section 2.5.2.2). NFSI is a commercially available and bench stable reagent.
3.2.3.2 Optimization
We began our studies with 2.2 equivalents of NFSI in MTBE at 60 °C. The reaction parameters
were varied to study their effects (Table 11). Other solvents were tolerated (Table 11, entry 1–3)
however, the reaction was best in THF (Table 11, entry 4). Notably, DCM gave no product,
possibly due to the background amidation reaction reported by Yang.151 Dropping the temperature
resulted in diminished yield (Table 11, entry 5). Concentrating the reaction increased the yield
(Table 11, entry 6) however, further increasing the concentration to 0.5 M resulted in the
immediate appearance of a black colour and decomposition ensued (Table 11, entry 7). A wide
range of additives, organic bases, inorganic bases, and acids were screened, in hopes of activating
either the substrate or NFSI, and we found that the addition of NH4Cl increased the yield to 60%
(Table 11, entry 8).
151
Liu, H. H.; Wang, Y.; Deng, G. J.; Yang, L., Adv. Synth. Catal. 2013, 355, 3369.

130
Table 11 Effects of the reaction parameters in the dearomative fluorination of indolesa
Entry Solvent Variation 3.38a (%) 3.39 (%)
1 MTBE none 32 –
2 DCM none – –
3 MeCN none 26 –
4 THF none 47 <5
5 THF 40 °C 37 15
6 THF 0.2 M 48 15
7 THF 0.5 M decomp –
8 THF 0.2 M and 1 equiv NH4Cl 60 –
aReactions run on 0.2 mmol scale and yields were determined by NMR using trifluorotoluene as an internal standard.
The fluorination of 2-methylindole (3.37a) with NFSI according to the conditions in Table 11,
entry 8 delivered exomethylidene containing indoline 3.38a as a crystalline, bench stable
product.152
3.2.3.3 Decomposition Studies and Scale-up
It was found that upon increasing the concentration or the scale of the reaction past 1 mmol, the
reaction immediately turned black and only decomposition of the starting material was observed.
For this reason, we added NFSI via a syringe pump.This allowed us to increase the initial
concentration with respect to the substrate (0.5 M) while decreasing the decomposition pathway
152
CCDC deposition number 1956222.

131
involving NFSI. The reaction mixture was filtered through a silica plug to remove the polar by-
products of NFSI. Importantly, the product was recrystalized in pentanes, removing the need for
relatively wasteful column chromatographic purification on a larger scale.
Scheme 94 Medium-scale synthesis of 3.38a
3.2.3.4 Exploring the Substrate Scope
Next, we investigated the impact of the protecting group on nitrogen as well as a few substituents
on the indole backbone (Table 12). The substrate bearing an electron-rich benzoyl group delivered
product 3.38b in comparable yield. However, electron-withdrawing groups had a negative effect
on yield, with the strong -withdrawing nitro group having the greatest impact (3.38c and 3.38d).
The opposite trend was found when introducing electronic perturbations at the 5-position of the
indole; MeO- functionalized indole (3.38e) provided the product in slightly diminished yield and
F- containing indole (3.38f) was accessed in good yield. Dimethoxy containing indoline 3.38g was
isolated in low yield, however an interesting restrictive rotation around the C–N bond was
elucidated by NMR, suggesting meta-functionalized benzoyl groups are too sterically bulky for
the reaction. Azaindole derivative produced product 3.38h in modest yield however, the product
was found to be unstable and decomposed shortly following isolation. A carbonyl moiety on the
nitrogen was necessary as only acetyl and Boc protected indoles gave the desired product (1H
NMR and 19F NMR), accompanied by inseparable byproducts. Benzyl and tosyl protected indoles
decomposed upon reaction with NFSI. Extending the carbon chain at the 2-position led to no
reactivity.
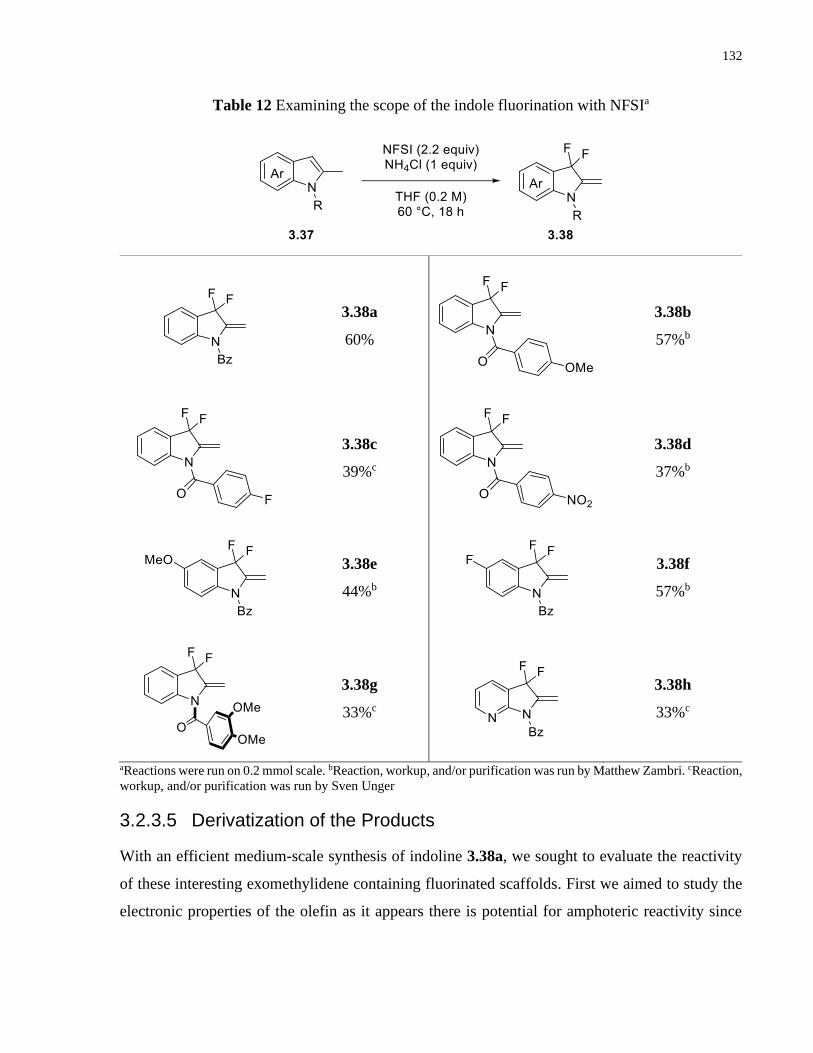
132
Table 12 Examining the scope of the indole fluorination with NFSIa
3.38a
60%
3.38b
57%b
3.38c
39%c
3.38d
37%b
3.38e
44%b
3.38f
57%b
3.38g
33%c
3.38h
33%c
aReactions were run on 0.2 mmol scale. bReaction, workup, and/or purification was run by Matthew Zambri. cReaction,
workup, and/or purification was run by Sven Unger
3.2.3.5 Derivatization of the Products
With an efficient medium-scale synthesis of indoline 3.38a, we sought to evaluate the reactivity
of these interesting exomethylidene containing fluorinated scaffolds. First we aimed to study the
electronic properties of the olefin as it appears there is potential for amphoteric reactivity since

133
there are -bond withdrawing fluorine atoms as well as weak -donation from the nitrogen atom
(Scheme 95).
Scheme 95 Amphoteric properties of 3.38a
Gratifyingly, we found that the olefin could participate in an SN2’ reaction with morpholine acting
as the nucleophile (3.40). The reaction required DMA as a solvent and no product was isolated in
toluene, in contrast with the outcome when using palladium (vide infra). Rapid and quantitative
epoxidation with DMDO was also achieved with preservation of the fluorine moiety (3.41, reaction
run by Dr. Christian Dank).
Functionalization of allylic and propargylic fluorides has been studied with various nucleophiles,
metal-catalysts, and organocatalysts.153,149 Reactions catalyzed by palladium and platinum have
been investigated in detail and the reactivity is found to be superior compared to many commonly
used oxygen-based leaving groups.153b, 153c We aimed to further elucidate the electrophilic nature
of these indolines as potential substrates in palladium-allylation chemistry (Scheme 96).
Phenylsilane, as a hydride source, delivered 3-fluoroindole 3.43 in quantitative yield.154 When 1.5
equivalents of morpholine were reacted in toluene, a mixture of mono- and di-addition products
were formed. However, upon increasing the loading of morpholine (5 equivalents), we could
153
(a) Nishimine, T.; Fukushi, K.; Shibata, N.; Taira, H.; Tokunaga, E.; Yamano, A.; Shiro, M.; Shibata, N., Angew.
Chem. Int. Ed. 2014, 53, 517. (b) Benedetto, E.; Keita, M.; Tredwell, M.; Hollingworth, C.; Brown, J. M.; Gouverneur,
V., Organometallics 2012, 31, 1408. (c) Pacheco, M. C.; Purser, S.; Gouverneur, V., Chem. Rev. 2008, 108, 1943. 154
Narumi, T.; Tomita, K.; Inokuchi, E.; Kobayashi, K.; Oishi, S.; Ohno, H.; Fujii, N., Org. Lett. 2007, 9, 3465.

134
isolate the double addition product 3.44 in 80% yield. The second addition may occur via an
oxidative addition into the heteroaryl-fluoride bond.155
Scheme 96 Palladium-catalyzed allylic functionalizationsa
aUnless otherwise stated, reactions run on a 0.1 mmol scale using [(C3H5)PdCl]2 (2.5–5 mol%) and xantphos (5–10
mol%). Reaction conditions: (a) XPhos (5 mol%) instead of xantphos, PhSiH3 (2 equiv), Et3N (1 equiv), EtOH, 50
°C, 6h. (b) morpholine (5 equiv), PhMe, 100 °C, 6 h. (c) Sodium phenylsulfinate (5 equiv), PhMe, 100 °C, 18 h, then
LiOH (2 M), 2h. (d) HP(O)(OEt)2 (2 equiv), CsF (1 equiv), PhMe, 70 °C, 18 h. (e) sodium dimethylmalonate (5 equiv),
15-crown-5 (5 equiv), DCM, r.t., 2h.
155
Amii, H.; Uneyama, K., Chem. Rev. 2009, 109, 2119.

135
In a similar manner, sulfonylation of 3.38a led to the product of double addition (3.45), which
upon treatment with LiOH led to cleavage of the labile benzoyl moiety.156 A clean phosphorylation
reaction was observed using dialkylphosphite as a nucleophile, producing 3.46 in good yield.157
Finally, using sodium dimethylmalonate as a nucleophile, according to the literature procedure,
produced 3.47 in excellent yield.153C
We also explored the viability of metal-catalyzed addition to the olefin, through a sterically
congested metal-complex 3.48 which could undergo a -fluoride elimination (Scheme 97).158 The
rhodium-catalyzed arylation/-fluoride elimination of 3.38a provided the mono-arylated product
3.49 in good yield and maintained a fluoride atom within the product [reaction run by Bijan
Mirabi]. Similarly, the copper-catalyzed borylation/-fluoride elimination provided product 3.50,
which possesses two handles for future transformations [reaction run by Alexa Torelli].
156
Eichelmann, H.; Gais, H.-J., Tetrahedron: Asymmetry 1995, 6, 643. 157
(a) Hirao, T.; Masunaga, T.; Ohshiro, Y.; Agawa, T., Synthesis 1981, 1981, 56. (b) Hong, Y.; Liu, W.; Dong, M.;
Chen, X.; Xu, T.; Tian, P.; Tong, X., Org. Lett. 2019, 21, 5742. 158
(a) Jang, Y. J.; Rose, D.; Mirabi, B.; Lautens, M., Angew. Chem. Int. Ed. 2018, 57, 16147. (b) Huang, Y.; Hayashi,
T., J. Am. Chem. Soc. 2016, 138, 12340. (c) Gao, P.; Yuan, C.; Zhao, Y.; Shi, Z., Chem 2018, 4, 2201.

136
Scheme 97 Cu- and Rh- addition/-fluoride elimination
3.3 Chapter Summary
In conclusion, we have developed an efficient and scalable protocol for the synthesis of a gem-
difluoroindoline bearing an exocyclic and allylic olefin without the need for column
chromatography. These scaffolds are versatile building blocks capable of acting in an amphoteric
sense as well as with various Pd/Cu/Rh reactions forming new C–H, C–B, C–C, C–N, C–O, C–P,
and C–S bonds.
3.4 Experimental
3.4.1 General Considerations
Metal catalyzed reactions were run under an atmosphere of argon in oven or flame dried 2-dram
vials with open-top caps fitted with a Teflon septum. Reactions were monitored by thin-layer
chromatography (TLC) on EMD Silica Gel 60 F254 plates and visualized under UV light or by
immersion in iodine on silica stain. Flash column chromatography was performed on Silicycle 40-
60 m silica gel. PhMe was distilled over CaH. All reagents and organic building blocks were
purchased from commercial supplier (Sigma Aldrich, Strem, Alfa Aesar, TCI, Combi-blocks) and
used without further purification.
1H and 13C NMR spectra were obtained on the Agilent DD2 500 equipped with a 5mm Xsens Cold
Probe. The spectra were internally referenced to the solvent peak. 19F and 31P NMR spectra were
obtained on the Varian Mercury 400 or 500 operating at 470 or 564 MHz. Measurements were
taken at 296 K and chemical shifts are reported in parts per million (ppm). Data is reported in the
following format: chemical shift ( ppm), multiplicity (s = singlet, d = doublet, t = triplet, q =
quartet, m = multiplet), coupling constant (Hz), integration. High resolution mass spectra (HRMS)
were obtained on a Micromass 70S-250 spectrometer (EI) or an ABI/Sciex QStar Mass
Spectrometer (ESI) or a JEOL AccuTOF model JMS-T1000LC mass spectrometer equipped with
an IONICS® Direct Analysis in real Time (DART) ion source.

137
3.4.2 Synthesis of Starting Materials
3.4.2.1 Synthetic Remarks
Starting materials were synthesized according to the previous protocol with using non-halogenated
benzoyl chlorides (see section 2.5.2.2 on page 84). Characterization data for known compounds
matched the literature data.159
3.4.2.2 Characterization Data for New Compounds
(2-methyl-1H-indol-1-yl)(4-nitrophenyl)methanone (3.37d) – The
compound was synthesized according to general procedure 2.5.2.2. The
product was purified by flash column chromatography using pentanes–
EtOAc–NEt3 (100:1:1 v:v) as the mobile phase and was isolated as a
yellow solid (203.6 mg, 0.7264 mmol, 14.5%). 1H NMR (400 MHz, CDCl3) δ 8.39 – 8.31 (m,
2H), 7.91 – 7.84 (m, 2H), 7.48 (dt, J = 7.8, 1.0 Hz, 1H), 7.18 (ddd, J = 7.9, 7.2, 1.0 Hz, 1H), 7.05
(ddd, J = 8.4, 7.2, 1.3 Hz, 1H), 6.95 (dq, J = 8.4, 0.9 Hz, 1H), 6.47 (p, J = 1.1 Hz, 1H), 2.41 (d, J
= 1.2 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 167.8, 150.2, 141.3, 137.7, 136.9, 130.6, 129.9,
124.1, 123.5, 123.4, 120.3, 114.4, 110.0, 16.2. IR (thin film, cm-1) 3399, 3113, 3070, 3052, 2926,
2858, 1686, 1602, 1454, 1525, 1193, 804, 708. HRMS (DART, M+H) Calculated for C16H13N2O3
281.0926, found 281.0928.
(2-methyl-1H-pyrrolo[2,3-b]pyridin-1-yl)(phenyl)methanone
(3.37g) – The compound was synthesized according to general
procedure 2.5.2.2. The product was purified by flash column
chromatography using pentanes–EtOAc–NEt3 (30:1:1 v:v) as the
mobile phase and was isolated as a white solid (2.34 g, 7.92 mmol, 72%). 1H NMR (400 MHz,
CDCl3) δ 7.45 – 7.40 (m, 1H), 7.38 (dq, J = 8.3, 0.9 Hz, 1H), 7.20 – 7.12 (m, 2H), 7.11 – 7.04 (m,
2H), 6.96 (dd, J = 7.5, 1.7 Hz, 1H), 6.36 (t, J = 1.0 Hz, 1H), 3.92 (s, 3H), 3.77 (s, 3H), 2.28 (d, J
= 1.2 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 167.7, 153.2, 146.8, 137.9, 137.2, 132.1, 129.9,
124.7, 123.5, 123.3, 120.2, 119.7, 115.1, 115.1, 109.7, 61.8, 56.1, 16.3. IR (thin film, cm-1) 2981,
2927, 2840, 1668, 1592, 1574, 1456, 1441, 1361, 984, 749. HRMS (DART, M+H) Calculated for
C18H18NO3 296.1281, found 296.1282.
159
Ramella, V.; He, Z.; Daniliuc, C. G.; Studer, A., Org. Lett. 2015, 17, 664.

138
(3,4-dimethoxyphenyl)(2-methyl-1H-indol-1-yl)methanone (3.37h) – The
compound was synthesized according to general procedure 2.5.2.2. The
product was purified by flash column chromatography using hexanes–
EtOAc–DCM (20:1:1 v:v) as the mobile phase and was isolated as a white
solid (590.7 mg, 2.5 mmol, 74%).1H NMR (500 MHz, CDCl3) δ 8.03 (dd, J = 4.8, 1.6 Hz, 1H),
7.80 – 7.74 (m, 3H), 7.65 – 7.59 (m, 1H), 7.48 – 7.42 (m, 2H), 7.05 (dd, J = 7.7, 4.8 Hz, 1H), 6.39
(q, J = 1.1 Hz, 1H), 2.59 (d, J = 1.2 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 169.7, 149.7, 142.8,
139.1, 135.0, 133.4, 130.9, 128.4, 127.6, 121.6, 118.3, 104.8, 15.3. IR (thin film, cm-1) 3013, 2960,
2931, 1686, 1596, 1560, 1402, 1305, 1251, 904, 812. HRMS (DART, M+H) Calculated for
C15H13N2O 237.1022, found 237.1026.
3.4.3 Synthesis of 3,3-Difluoro-2-exo-Methylidene Indolines
3.4.3.1 General Procedure
In an oven-dried 2-dram vial equipped with a open-top septum cap purging with an argon balloon
was added substrate 3.37 (0.2 mmol), NFSI (0.44.mmol), and NH4Cl (0.2 mmol). After a 10-
minute purge, THF (1 mL, 0.2 M) was added. The balloon was removed, and the reaction was
heated to 60 °C, for 12–24 h. Upon completion, the reaction was diluted with pentanes to crash
out the polar byproducts and filtered through a silica plug, eluting with a 2:1 solvent mixture of
pentanes–Et2O. The reaction was concentrated and purified via silica gel chromatography. When
required, analytically pure material was obtained by recrystallization in pentanes.
3.4.3.2 Procedure for the 20 mmol-Scale Reaction
(3,3-difluoro-2-methyleneindolin-1-yl)(phenyl)methanone (3.38) – In a flame dried round
bottom flask equipped with an argon balloon was added substrate 3.37a (11 or 20 mmol, 1 equiv),
NH4Cl (1 equiv), and THF (0.5 M) and was stirred at 60 °C. NFSI (2.2 equiv) in THF (1 M) was
added using a syringe pump over 2 h and the reaction was further stirred for 24 h. Upon completion,
polar materials were crashed out using pentanes and the suspension was filtered over a silica plug

139
eluting with pentanes–Et2O (2:1). The product was recrystalized using pentanes and small amounts
of Et2O to afford analytically pure 3.38a (44% yield for 11 mmol and 20 mmol scales).
3.4.3.3 Characterization Data for New Compounds
(3,3-difluoro-2-methyleneindolin-1-yl)(phenyl)methanone (3.38a) – Was
synthesized according to general procedure 3.4.3.1. The product was purified
by flash column chromatography using pentanes–Et2O (50:1 to 30:1, v:v) as
the mobile phase and was isolated as a white solid (32.6 mg, 0.12 mmol,
60%). 1H NMR (400 MHz, CDCl3) δ 7.68 – 7.60 (m, 3H), 7.60 – 7.54 (m, 1H), 7.53 – 7.44 (m,
2H), 7.41 – 7.35 (m, 2H), 7.22 (ddd, J = 7.7, 4.7, 3.7 Hz, 1H), 5.43 (td, J = 3.0 Hz, 1H), 5.18 (td,
J = 3.6, 2.6 Hz, 1H). 13C NMR (125 MHz, CDCl3) δ 168.8, 142.8 (t, J = 5.8 Hz), 142.6 (t, J = 26.9
Hz), 135.2, 132.9 (t, J = 1.5 Hz), 132.2, 129.0, 128.5, 124.8 (t, J = 1.7 Hz), 124.1, 122.9 (t, J =
24.8 Hz), 118.2 (t, J = 240.6 Hz), 116.4, 102.0 (t, J = 3.3 Hz). 19F NMR (377 MHz, CDCl3) δ -
85.13. IR (thin film, cm-1) 3338, 3151, 3129, 3061, 3029, 1673, 1616, 1600, 1462, 1355, 1152,
1047, 767. HRMS (DART, M+H) Calculated for C16H12F2NO 272.0887, found 272.0892.
(3,3-difluoro-2-methyleneindolin-1-yl)(4-
methoxyphenyl)methanone (3.38b) – Was synthesized according to
general procedure 3.4.3.1. The product was purified by flash column
chromatography using pentanes–Et2O (30:1, v:v) as the mobile phase
and was isolated as a white solid (34.6 mg, 0.1148 mmol, 57%). 1H NMR (400 MHz, CDCl3) δ
7.67 – 7.58 (m, 3H), 7.42 – 7.33 (m, 2H), 7.22 – 7.16 (m, 1H), 6.99 – 6.92 (m, 2H), 5.41 (td, J =
3.2, 2.5 Hz, 1H), 5.20 (td, J = 3.6, 2.5 Hz, 1H), 3.88 (s, 3H). 13C NMR (100 MHz, CDCl3 δ 168.4,
163.0, 143.2 (t, J = 6.0 Hz), 143.0 (t, J = 26.6 Hz), 132.9 (t, J = 1.5 Hz), 131.1, 127.0, 124.4 (t, J
= 1.7 Hz), 124.0, 122.7 (t, J = 24.8 Hz), 118.3 (t, J = 239.9 Hz), 116.1, 114.2, 101.1 (t, J = 3.3 Hz),
55.6. 19F NMR (377 MHz, CDCl3) δ -85.04. IR (thin film, cm-1) 3130, 3012, 2937, 2842, 1736,
1671, 1604, 1578, 1469, 1252, 756. HRMS (DART, M+H) Calculated for C17H14NO2F2 302.0987,
found 302.0991.
(3,3-difluoro-2-methyleneindolin-1-yl)(4-fluorophenyl)methanone
(3.38c) – Was synthesized according to general procedure 3.4.3.1 with the
following variations. The reaction was run on 0.5 mmol scale. The product
contained two unknown inseparable byproducts. The yield was measured

140
by proton NMR using 10.6 mg of TMB as an internal standard (0.197 mmol, 39%). 1H NMR (400
MHz, CDCl3) δ 7.74 – 7.65 (m, 3H), 7.50 – 7.39 (m, 2H), 7.28 – 7.23 (m, 1H), 7.23 – 7.17 (m,
2H), 5.49 (td, J = 3.0 Hz, 1H), 5.21 (td, J = 3.3 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ 167.4,
164.9 (d, J = 253.8 Hz), 143.0 – 142.3 (m), 132.8 (t, J = 1.5 Hz), 131.2 (d, J = 8.9 Hz), 131.0 (d,
J = 3.5 Hz), 124.8 (t, J = 1.7 Hz), 124.0, 122.7 (t, J = 24.8 Hz), 118.1 (t, J = 240.1 Hz), 116.1 (d,
J = 8.1 Hz), 116.0, 101.7 (t, J = 3.3 Hz). 19F NMR (377 MHz, CDCl3) δ -85.39, -105.72 (td, J =
8.4, 7.9, 4.2 Hz). IR (thin film, cm-1) 3125, 3068, 3015, 2969, 3927, 1664, 1615, 1602, 1482, 1350,
1270, 1196, 1048. HRMS (DART, M+H) Calculated for C16H11NOF3 290.0787, found 290.0783.
(3,3-difluoro-2-methyleneindolin-1-yl)(4-nitrophenyl)methanone
(3.38d) – Was synthesized according to general procedure 3.4.3.1. The
product was purified by flash column chromatography using pentanes–
Et2O (50:1 to 30:1, v:v) as the mobile phase and was isolated as a
yellow solid (23.4 mg, 0.074 mmol, 37%). 1H NMR (400 MHz, CDCl3) δ 8.38 – 8.31 (m, 2H),
7.84 – 7.77 (m, 2H), 7.72 – 7.63 (m, 1H), 7.43 (d, J = 4.2 Hz, 2H), 7.28 (dd, J = 8.5, 4.8 Hz, 1H),
5.50 (td, J = 2.9 Hz, 1H), 5.14 (td, J = 3.2 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 166.2, 149.7,
142.4 (t, J = 27.3 Hz), 142.1 (t, J = 5.8 Hz), 140.7, 133.2 (t, J = 1.5 Hz), 129.6, 125.6 (t, J = 1.7
Hz), 124.3, 123.1 (t, J = 24.8 Hz), 117.9 (t, J = 240.5 Hz), 116.4, 103.0 (t, J = 3.1 Hz). 19F NMR
(377 MHz, CDCl3) δ -86.03. IR (thin film, cm-1) 3111, 2981, 1674, 1615, 1601, 1526, 1472, 1346,
1294, 762. HRMS (DART, M+H) Calculated for C16H11F2N2O3 317.0738, found 317.0743.
(3,3-difluoro-5-methoxy-2-methyleneindolin-1-
yl)(phenyl)methanone (3.38e) – Was synthesized according to general
procedure 3.4.3.1. The product was purified by flash column
chromatography using pentanes–Et2O (50:1 to 30:1, v:v) as the mobile
phase and was isolated as a white solid (26.5 mg, 0.088 mmol, 44%). 1H NMR (400 MHz, CDCl3)
δ 7.63 – 7.52 (m, 3H), 7.50 – 7.43 (m, 2H), 7.34 (dt, J = 9.1, 1.5 Hz, 1H), 7.14 (dt, J = 2.8, 1.4 Hz,
1H), 6.93 (ddt, J = 9.1, 2.6, 1.1 Hz, 1H), 5.41 (td, J = 2.9 Hz, 1H), 5.14 (td, J = 3.3 Hz, 1H), 3.83
(s, 3H). 13C NMR (125 MHz, CDCl3) δ 166.2, 149.7, 142.4 (t, J = 27.3 Hz), 142.1 (t, J = 5.8 Hz),
140.7, 133.2 (t, J = 1.5 Hz), 129.6, 125.6 (t, J = 1.7 Hz), 124.3, 123.1 (t, J = 24.8 Hz), 117.9 (t, J
= 240.5 Hz), 116.4, 103.0 (t, J = 3.1 Hz). 19F NMR (377 MHz, CDCl3) δ -85.45. IR (thin film,
cm-1) 3137, 3068, 3028, 3012, 2967, 2921, 2851, 2835, 1655, 1486, 1361, 1220, 1047. HRMS
(DART, M+H) Calculated for C17H14F2NO2 302.0993, found 302.0995.

141
phenyl(3,3,5-trifluoro-2-methyleneindolin-1-yl)methanone (3.38f) –
Was synthesized according to general procedure 3.4.3.1. The product was
purified by flash column chromatography using pentanes–Et2O (50:1, v:v)
as the mobile phase and was isolated as a white solid (32.98 mg, 0.114
mmol, 57%). 1H NMR (400 MHz, CDCl3) δ 7.66 – 7.52 (m, 3H), 7.52 –
7.43 (m, 2H), 7.37 – 7.30 (m, 1H), 7.13 (td, J = 8.9, 2.8 Hz, 1H), 5.41 (td, J = 3.1 Hz, 1H), 5.01
(td, J = 3.3 Hz, 1H). 13C NMR (125 MHz, CDCl3) ) δ 168.3, 157.1 (t, J = 1.8 Hz), 142.8 (t, J =
26.8 Hz), 136.4 (t, J = 6.0 Hz), 135.4, 131.9, 128.9, 128.3, 123.8 (t, J = 24.4 Hz), 119.5 (t, J = 1.6
Hz), 118.2 (t, J = 240.4 Hz), 117.7, 107.9, 102.1 (t, J = 3.3 Hz), 55.9. 19F NMR (377 MHz, CDCl3)
δ -85.57, -116.44 (dd, J = 7.3 Hz). IR (thin film, cm-1) 3141, 3065, 3017, 2926, 2855, 1666, 1482,
1268, 1188. HRMS (DART, M+H) Calculated for C16H11F3NO 290.0793, found 290.0790.
(3,3-difluoro-2-methyleneindolin-1-yl)(3,4-
dimethoxyphenyl)methanone (3.38g) – Was synthesized according to
general procedure 3.4.3.1. The product was purified by flash column
chromatography using pentanes–Et2O (50:1 to 30:1, v:v) as the mobile
phase and was isolated as a white solid (39.7 mg, 0.12 mmol, 33%). 1H NMR (400 MHz, CDCl3)
δ 7.62 (dt, J = 7.6, 1.0 Hz, 1H), 7.41 – 7.28 (m, 2H), 7.24 – 7.18 (m, 1H), 7.17 – 7.13 (m, 1H),
7.07 (dd, J = 8.3, 1.5 Hz, 1H), 6.95 (dd, J = 7.6, 1.5 Hz, 1H), 5.43 (d, J = 2.3 Hz, 1H), 5.34 (s,
1H), 3.90 (s, 3H), 3.80 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 166.4, 153.1, 146.0, 142.4 (t, J =
5.9 Hz), 142.1 (t, J = 26.9 Hz), 133.0 (t, J = 1.5 Hz), 131.0, 125.1 (t, J = 1.9 Hz), 125.0, 124.0,
123.2 (t, J = 24.8 Hz), 119.5, 118.1 (t, J = 239.7 Hz), 116.4, 114.9, 102.3, 61.7, 56.1. 19F NMR
(377 MHz, CDCl3) δ -82.94 (d, J = 270.8 Hz), -86.23 (d, J = 271.5 Hz). IR (thin film, cm-1) 3012,
2938, 2839, 1680, 1615, 1398, 1582, 1471, 1358, 1290, 1270, 1048, 751. HRMS (DART, M+H)
Calculated for C18H16F2NO3 332.1093, found 332.1088.
(3,3-difluoro-2-methylene-2,3-dihydro-1H-pyrrolo[2,3-b]pyridin-1-
yl)(phenyl)methanone (3.38h) – Was synthesized according to general
procedure 3.4.3.1. The product was purified by flash column chromatography
using pentanes–EtOAc (30:1, v:v) as the mobile phase and was isolated as a
white solid (18.1 mg, 0.066 mmol, 33%). 1H NMR (400 MHz, CDCl3) δ 8.14 (ddt, J = 5.0, 1.9,
1.0 Hz, 1H), 7.93 (dq, J = 7.6, 1.5 Hz, 1H), 7.68 – 7.59 (m, 2H), 7.55 (ddt, J = 7.9, 7.0, 1.3 Hz,
1H), 7.47 – 7.37 (m, 2H), 7.06 (dd, J = 7.6, 5.0 Hz, 1H), 6.27 (td, J = 4.1, 2.2 Hz, 1H), 5.63 (td, J

142
= 3.4, 2.1 Hz, 1H). 13C NMR (126 MHz, CDCl3) δ 168.8, 155.4 (t, J = 6.0 Hz), 152.0 (t, J = 1.7
Hz), 141.2 (t, J = 26.6 Hz), 135.5, 133.3, 132.2, 129.1, 128.2, 119.6 (t, J = 1.4 Hz), 116.3 (t, J =
241.0 Hz), 115.8 (t, J = 25.7 Hz), 102.2 (t, J = 3.2 Hz). 19F NMR (377 MHz, CDCl3) δ -84.64. IR
(thin film, cm-1) 3029, 2961, 2924, 2856, 1684, 1594, 1473, 1449, 1420, 1272, 1059, 902. HRMS
(DART, M+H) Calculated for C19H11F2N2O 273.0834, found 273.0835.
3.4.4 Reactions of 3,3-Difluoro-2-exo-Methylidene Indolines
3.4.4.1 Allylic Substitution with Morpholine Nucleophile (SN2’)
(3-fluoro-2-(morpholinomethyl)-1H-indol-1-yl)(phenyl)methanone (3.40) – In an oven-dried
2-dram vial equipped with an open-top cap with septum and an argon balloon was added substrate
3.38a (0.1 mmol), DMA (1 mL), and morpholine (10 equiv) in that order. The balloon was
removed, the reaction was sealed tightly, and was heated to 70 °C for 18 h. The reaction was
filtered over silica, concentrated, and columned using a pentanes–EtOAc (10:1, v:v) eluting phase
to isolated 3.40 as a white solid (28.76 mg, 0.85 mmol, 85%). 1H NMR (400 MHz, CDCl3) δ 7.77
– 7.69 (m, 2H), 7.65 – 7.54 (m, 2H), 7.54 – 7.41 (m, 3H), 7.29 – 7.19 (m, 2H), 3.54 – 3.43 (m,
6H), 2.15 – 2.06 (m, 4H). 13C NMR (101 MHz, CDCl3 δ 169.7, 147.8 (d, J = 251.8 Hz), 135.7,
134.2 (d, J = 5.3 Hz), 132.8, 129.8, 128.6, 125.3, 122.9, 119.0 (d, J = 18.3 Hz), 118.8 (d, J = 22.4
Hz), 116.9 (d, J = 2.6 Hz), 114.3 (d, J = 1.8 Hz), 67.0, 52.7, 50.6 (d, J = 2.7 Hz). 19F NMR (377
MHz, CDCl3) δ -168.89. IR (thin film, cm-1) 3058, 2962, 2854, 2814, 1694, 1454, 1411, 1343,
1320, 1116, 747. HRMS (DART, M+H) Calculated for C20H20N2O2F 339.1503, found 339.1507.
3.4.4.2 DMDO epoxidation

143
(3,3-difluorospiro[indoline-2,2'-oxiran]-1-yl)(phenyl)methanone (3.41) – This experiment was
devised by Nicolas Zeidan and executed by Dr. Christian Dank. DMDO was synthesized according
the literature procedure.160 3.3 mL of DMDO in acetone (0.066 M, 1.1 equiv) was added to 3.38a
(54.25 mg, 0.2 mmol, 1 equiv) at 0 °C. The reaction was gradually warmed to r.t. and stirred for
20 h. The clear solution was concentrated under reduced pressure to give 3.41 as a slightly yellow
oil (57.45, 0.2 mmol, >99%).1H NMR (400 MHz, CDCl3) δ 7.70 – 7.53 (m, 4H), 7.53 – 7.43 (m,
2H), 7.38 (tq, J = 7.5, 1.2 Hz, 1H), 7.24 (dd, J = 8.0, 7.1 Hz, 1H), 7.13 – 7.02 (m, 1H), 3.75 (ddd,
J = 4.6, 2.7, 2.0 Hz, 1H), 3.31 (dd, J = 4.5, 0.5 Hz, 1H). 13C NMR (126 MHz, CDCl3) δ 169.1,
143.8 (dd, J = 6.7, 5.4 Hz), 134.8, 132.8, 132.1, 128.6, 128.3, 124.6 (t, J = 1.8 Hz), 123.7, 121.3
(dd, J = 25.2, 24.7 Hz), 119.4 (dd, J = 249.9, 243.5 Hz), 117.2, 76.1 (dd, J = 41.6, 25.3 Hz), 48.2
(d, J = 4.2 Hz). 19F NMR (377 MHz, CDCl3) δ -93.82 (d, J = 259.7 Hz), -109.25 (d, J = 259.7
Hz). IR (thin film, cm-1) 3060.2, 3004.7, 2920.1, 1971.9, 1674.0, 1615.2, 1600.9, 1472.8, 1405.8,
1341.7, 1293.8, 1195.1, 1158.2, 1140.2, 1115.7, 1076.3, 948.59, 759.96, 699.23. HRMS (DART,
M+H) Calculated for C16H12NO2F2 288.0831, found 288.0830.
3.4.4.3 Pd-Catalyzed Hydro-Defluorination
(3-fluoro-2-methyl-1H-indol-1-yl)(phenyl)methanone (3.43) – In an oven-dried 2-dram vial
equipped with an open-top cap with septum and an argon balloon was added substrate 3.38a (0.1
mmol), [(C3H5)PdCl]2 (2.5 mol%), and xphos (5 mol%). The vial was purged for 10 minutes and
thereafter, EtOH (1 mL), PhSiH3 (2 equiv), and NEt3 (1 equiv) were added successively. The
balloon was removed, the reaction was sealed tightly and was heated to 50 °C for 6 h. The reaction
was filtered over silica, concentrated, and columned using a pentanes–Et2O (100:1, v:v) eluting
phase to isolated 3.43 as a white solid (25.3 mg, 0.999 mmol, >99%). 1H NMR (600 MHz, CDCl3)
δ 7.73 – 7.68 (m, 2H), 7.67 – 7.61 (m, 1H), 7.55 – 7.47 (m, 3H), 7.21 (ddd, J = 7.9, 6.8, 1.2 Hz,
1H), 7.15 – 7.07 (m, 2H), 2.31 (d, J = 2.5 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 169.6, 147.0
160
Organic Syntheses 2013, 90, 350.

144
(d, J = 248.3 Hz), 135.7, 133.5 (d, J = 5.2 Hz), 133.0, 129.7, 129.0, 124.3, 123.1, 120.4 (d, J =
18.6 Hz), 119.6 (d, J = 25.2 Hz), 116.3 (d, J = 2.6 Hz), 114.8 (d, J = 1.9 Hz), 11.1 (d, J = 1.5 Hz).
19F NMR (564 MHz, CDCl3) δ -171.22. IR (thin film, cm-1) 3058, 2964, 2927, 2854, 1687, 1645,
1600, 1456, 1347, 1316, 743. HRMS (DART, M+H) Calculated for C16H13NOF 254.0976, found
254.0977.
3.4.4.4 Pd-Catalyzed Double Amination
(3-morpholino-2-(morpholinomethyl)-1H-indol-1-yl)(phenyl)methanone (3.44) – In an oven-
dried 2-dram vial equipped with an open-top cap with septum and an argon balloon was added
substrate 3.38a (0.1 mmol), [(C3H5)PdCl]2 (2.5 mol%), and Xantphos (5 mol%). The vial was
purged for 10 minutes and thereafter, PhMe (1 mL), and morpholine (5 equiv) were added
successively. The balloon was removed, the reaction was sealed tightly, and it was heated to 100
°C for 6 h. The reaction was filtered over silica, concentrated, and columned using a pentanes–
EtOAc (10:1, v:v) eluting phase to isolate 3.44 as a white solid (32.3 mg, 0.0796 mmol, 80%). 1H
NMR (400 MHz, CDCl3) δ 7.78 – 7.70 (m, 3H), 7.63 – 7.54 (m, 1H), 7.51 – 7.43 (m, 3H), 7.21 –
7.13 (m, 2H), 3.89 – 3.83 (m, 4H), 3.51 (s, 2H), 3.46 (t, J = 4.7 Hz, 4H), 3.28 – 3.21 (m, 4H), 2.09
– 2.02 (m, 4H). 13C NMR (125 MHz, CDCl3) δ 170.0, 136.3, 135.9, 133.2, 132.7, 130.8, 129.9,
128.5, 125.7, 123.9, 122.0, 119.8, 114.1, 68.0, 67.0, 52.9, 52.8, 51.9. IR (thin film, cm-1) 2958,
2852, 1688, 1454, 1344, 1327, 1261, 1115, 748. HRMS (DART, M+H) Calculated for
C24H28N3O3 406.2125, found 406.2127.
3.4.4.5 Pd-Catalyzed Double sulfonylation

145
3-(phenylsulfonyl)-2-((phenylsulfonyl)methyl)-1H-indole (3.45) – In an oven-dried 2-dram vial
equipped with an open-top cap with septum and an argon balloon was added substrate 3.38a (0.2
mmol), [(C3H5)PdCl]2 (5 mol%), Xantphos (10 mol%), and sodium benzene sulfinate (5 equiv).
The vial was purged for 10 minutes and thereafter, PhMe (1 mL) was added. The balloon was
removed, the reaction was sealed tightly, and it was heated to 100 °C for 18 h. The reaction was
filtered over silica, concentrated, and columned using a pentanes–EtOAc (5:1 to 2:1, v:v) eluting
phase to isolate 3.45 as a white solid (40.3 mg, 0.098 mmol, 49%). 1H NMR (400 MHz, CDCl3)
δ 9.72 (s, 1H), 7.86 – 7.77 (m, 3H), 7.77 – 7.70 (m, 2H), 7.70 – 7.61 (m, 1H), 7.59 – 7.41 (m, 4H),
7.41 – 7.33 (m, 2H), 7.33 – 7.28 (m, 1H), 7.22 (ddd, J = 8.2, 7.1, 1.1 Hz, 1H), 5.25 (s, 2H). 13C
NMR (126 MHz, CDCl3) δ 142.9, 137.6, 135.5, 134.7, 132.9, 129.6, 129.6, 129.2, 128.5, 126.5,
125.0, 124.3, 123.0, 120.2, 115.4, 112.1, 52.7. IR (thin film, cm-1) 3317, 3069, 3011, 2918, 1534,
1447, 1426, 1302, 1131, 908, 733. HRMS (DART, M+H) Calculated for C21H21N2O4S2
429.09372, found 429.09327.
3.4.4.6 Pd-Catalyzed Allylic Phosphorylation
diethyl ((1-benzoyl-3-fluoro-1H-indol-2-yl)methyl)phosphonate (3.46) – In an oven-dried 2-
dram vial equipped with an open-top cap with septum and an argon balloon was added substrate
3.38a (0.1 mmol), [(C3H5)PdCl]2 (2.5 mol%), and Xantphos (5 mol%), and CsF (1 equiv). The vial
was purged for 10 minutes and thereafter, PhMe (1 mL), and diethyl phosphite (2 equiv) were
added successively. The balloon was removed, the reaction was sealed tightly, and it was heated
to 70 °C for 18 h. The reaction was filtered over silica, concentrated, and columned using a
pentanes–EtOAc (3:1 to 2:1, v:v) eluting phase to isolate 3.46 as a white solid (26.1 mg, 0.067 mmol,
67%). 1H NMR (400 MHz, CDCl3) δ 7.83 – 7.76 (m, 2H), 7.69 – 7.60 (m, 1H), 7.52 (dddd, J =
13.8, 7.7, 6.9, 1.4 Hz, 3H), 7.17 (ddd, J = 8.0, 7.2, 0.9 Hz, 1H), 7.01 (ddt, J = 8.5, 7.2, 1.3 Hz, 1H),
6.63 (dd, J = 8.6, 2.2 Hz, 1H), 4.01 (dqd, J = 8.3, 7.1, 2.5 Hz, 4H), 3.74 (dd, J = 21.0, 1.5 Hz, 2H),
1.17 (t, J = 7.1 Hz, 6H). 13C NMR (126 MHz, CDCl3) δ 169.7 (d, J = 1.1 Hz), 147.8 (dd, J = 252.0,
11.2 Hz), 135.1, 134.0 (dd, J = 5.4, 1.9 Hz), 133.3, 130.3, 128.9, 124.5 (d, J = 2.1 Hz), 122.7 (d, J

146
= 1.6 Hz), 119.3 (dd, J = 17.9, 4.0 Hz), 116.7 (dd, J = 2.3 Hz), 114.6 (dd, J = 24.5, 14.7 Hz), 114.2
(dd, J = 1.8 Hz), 62.4 (d, J = 6.5 Hz), 22.2 (dd, J = 141.0, 2.2 Hz), 16.3 (d, J = 5.9 Hz). 19F NMR
(377 MHz, CDCl3) δ -168.38 (d, J = 12.1 Hz). 31P NMR (162 MHz, CDCl3) δ 23.03 (d, J = 11.9
Hz). IR (thin film, cm-1) 3062, 2983, 2931, 3909, 1692, 1638, 1453, 1349, 1019, 853. HRMS
(DART, M+H) Calculated for C20H22FNO4P 390.1265, found 390.1266.
3.4.4.7 Pd-Catalyzed Double Alkylation
dimethyl 2-(1-benzoyl-2-(3-methoxy-2-(methoxycarbonyl)-3-oxopropyl)-1H-indol-3-
yl)malonate (3.47) – In an oven-dried 2-dram vial equipped with an open-top cap with septum
and an argon balloon was added substrate 3.38a (0.1 mmol), [(C3H5)PdCl]2 (2.5 mol%), and
Xantphos (5 mol%), sodium dimethylmalonate (5 equiv), and 15-crown-5 (5 equiv). The vial was
purged for 10 minutes and thereafter, DCM (1 mL) was added. The balloon was removed, the
reaction was sealed tightly, and it was stirred at r.t. for 2 h. The reaction was filtered over silica,
concentrated, and columned using a pentanes–EtOAc (10:1 to 5:1, v:v) eluting phase to isolate
3.47 as a white solid (46.4 mg, 0.09364 mmol, 94%). 1H NMR (500 MHz, CDCl3) δ 7.75 – 7.69
(m, 2H), 7.66 (ddt, J = 8.8, 7.3, 1.3 Hz, 1H), 7.61 (ddd, J = 8.0, 1.3, 0.7 Hz, 1H), 7.54 – 7.46 (m,
2H), 7.12 (ddd, J = 8.0, 7.2, 1.0 Hz, 1H), 6.94 (ddd, J = 8.4, 7.2, 1.2 Hz, 1H), 6.48 (dt, J = 8.5, 0.9
Hz, 1H), 5.16 (s, 1H), 4.07 (t, J = 7.6 Hz, 1H), 3.78 (s, 7H), 3.69 – 3.64 (m, 8H). 13C NMR (126
MHz, CDCl3) δ 170.0, 169.2, 168.5, 136.7, 136.0, 134.9, 133.6, 130.1, 129.0, 128.1, 123.6, 122.8,
121.3, 114.4, 114.2, 52.9, 52.9, 51.6, 49.0, 25.2. IR (thin film, cm-1) 3655, 2981, 2973, 2889, 1755,
1737, 1458, 1311, 1154. HRMS (DART, M+H) Calculated for C26H26NO9 496.1602, found 496.1597.

147
3.4.4.8 Rhodium-Catalyzed Arylation
(3-fluoro-2-(4-methoxybenzyl)-1H-indol-1-yl)(phenyl)methanone (3.49) – This experiment
was devised by Nicolas Zeidan and executed by Bijan Mirabi. In an oven-dried 2-dram vial
equipped with an open-top cap with septum and an argon balloon was added substrate 3.38a (0.2
mmol), aryl boronic acid (2.5 equiv), [Rh(COD)OH]2 (2.5 mol%), and rac-BINAP (5 mol%). The
vial was purged for 10 minutes and thereafter, dioxane (1 mL) and distilled H2O (0.1 mL) was
added. The balloon was removed, the reaction was sealed tightly, and it was stirred at 120 °C for
24 h. The reaction was cooled to room temperature and quenched with an aqueous solution of
saturated NaHCO3. The mixture was diluted with EtOAc, washed once with an aqueous solution
of NaHCO3, once with H2O, and once with brine. The organic layer was dried over magnesium
sulfate, filtered, and concentrated under reduced pressure. The crude was purified by flash column
chromatography pentanes–EtOAc (9:1, v:v) to obtain product 3.49 as a yellow film (40.25 mg,
0.112 mmol, 56%). 1H NMR (500 MHz, CDCl3) δ 7.63 – 7.57 (m, 2H), 7.55 – 7.50 (m, 2H), 7.46
– 7.40 (m, 2H), 7.22 (ddd, J = 8.0, 7.2, 0.8 Hz, 1H), 7.07 (ddd, J = 8.5, 7.2, 1.3 Hz, 1H), 7.00 –
6.93 (m, 2H), 6.84 (ddt, J = 8.5, 2.3, 0.8 Hz, 1H), 6.75 – 6.71 (m, 2H), 4.24 (d, J = 1.9 Hz, 2H),
3.73 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 169.4 (d, J = 1.2 Hz), 158.3, 147.3 (d, J = 250.0 Hz),
135.3, 133.9 (d, J = 5.4 Hz), 133.0, 130.3 (d, J = 2.0 Hz), 129.8, 129.4, 128.8, 124.5, 122.9, 122.9
(d, J = 24.0 Hz), 119.9 (d, J = 18.6 Hz), 116.7 (d, J = 2.6 Hz), 114.5 (d, J = 1.8 Hz), 113.9, 55.3,
29.2 (d, J = 1.8 Hz). 19F NMR (377 MHz, CDCl3) δ -170.14. IR (neat) 3063, 2913, 2988, 2843,
1693, 1639, 1607, 1511, 1458, 1355, 1313, 1235, 1174, 1110, 1047, 894, 755, 720, 699, 663, 638.
HRMS (DART, M+H) Calculated for C23H19NO2F 360.1394, found 360.1396.

148
3.4.4.9 Copper-Catalyzed Borylation
(3-fluoro-2-((4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)methyl)-1H-indol-1-
yl)(phenyl)methanone (3.50) – This experiment was devised by Nicolas Zeidan and executed by
Alexa Torelli. In an oven-dried 2-dram vial equipped with an open-top cap with septum and an
argon balloon was added Tetrakisacetonitrile copper hexafluorophosphate (4 mol%), bppp (6
mol%), and sodium tert-butoxide (1.5 equiv). Anhydrous MTBE (1 mL) was added to the vial and
the catalyst solution was stirred for 5 minutes. Bis(pinacolato)diboron (1.5 equiv) was added in 1
mL of anhydrous MTBE and the suspension was stirred for 5 minutes. Substrate 3.38a (0.2 mmol,
1 equiv) was dissolved in anhydrous MTBE (2 mL) and charged to the reaction vial. The reaction
was stirred for 10 h then filtered through a celite pad and concentrated. A crude NMR with TMB
as an internal standard suggested the yield is around 89%. The product was purified by flash
column chromatography using pentanes–EtOAc (6% to 10%) to isolate 3.50 as a white solid (60.6
mg, 0.1598, 80%). 1H NMR (500 MHz, Chloroform-d) δ 7.79 – 7.71 (m, 2H), 7.67 – 7.60 (m,
1H), 7.55 – 7.45 (m, 3H), 7.18 – 7.11 (m, 1H), 6.92 (ddd, J = 8.5, 7.2, 1.3 Hz, 1H), 6.47 (dd, J =
8.5, 2.2 Hz, 1H), 2.66 (d, J = 1.9 Hz, 2H), 1.13 (s, 12H). 13C NMR (126 MHz, CDCl3) δ 170.0 (d,
J = 1.3 Hz), 146.2 (d, J = 245.9 Hz), 135.7, 132.9 (d, J = 5.3 Hz), 132.8, 130.0, 128.7, 123.2, 122.6,
122.1 (d, J = 25.4 Hz), 120.6 (d, J = 18.7 Hz), 116.1 (d, J = 2.6 Hz), 114.3 (d, J = 1.8 Hz), 83.9,
24.8, 9.6. 19F NMR (377 MHz, Chloroform-d) δ -172.70. IR (thin film, cm-1) 2936, 1692, 1631,
1448, 1323, 1238, 1204, 1143, 970, 845, 767, 713, 658. HRMS (DART, M+H) Calculated for
C22H24BNO3F 380.1834, found 380.1828.

149
General Conclusions and Outlook
The synthesis of indoles and their reactivity is a flourishing field, even after many years of
important discoveries. Research in metal-catalyzed synthesis of indoles will continue to grow as
complementary methods are required for the rapid synthesis of functionalized indoles. In this
regard, base-metal catalysts will be crucial in the development of inexpensive alternatives.
Students looking to pursue methodologies for the synthesis of indoles should focus on modular
methods which yield diversified indoles at the non-traditional positions (such as 2-, 4-, 7-).
Likewise, the dearomatization of indoles, and related heterocycles, will continue to be relevant as
they allow an atom-economic route to otherwise difficult to access saturated chiral cycles.
Indolines show high bioactivity and therefore will remain to be important targets of small-molecule
API development in the foreseeable future. It is advisable for those pursuing this field to develop
new catalysts for dearomatization reactions. Additionally, research into the dearomatization of
non-heterocycles by migratory insertion is lagging, and therefore I would expect this will be the
focus of research in the nearby future. The use of custom boron regents is very underdeveloped as
well. Students looking to pursue any palladium-catalyzed functionalizations are encouraged to use
the mixed boron reagent presented in chapter 2.
Finally, the need for new C–F bond forming reactions, especially those involving interesting
molecules, is crucial. Combining the ideas of dearomatization and electrophilic fluorination is a
steppingstone towards cheaper alternatives. The use of base-metals such as cobalt and nickel for
radical incorporation of these N–F reagents will likely be an interesting avenue for chemists to
come. The stable difluoroindoline presented in chapter 3 shows a remarkable stability as well as
olefin reactivity. Students pursuing related work are encouraged to attempt enantioselective
desymmetrisation reactions using palladium, rhodium, or other metals as they have been shown to
work well.

150
Appendix A – NMRs from Chapter 1

151

152

153
154

155

156

157

158

159

160

161

162

163

164

165
166

167

168

169

170

171

172

173

174

175

176

177

178

179
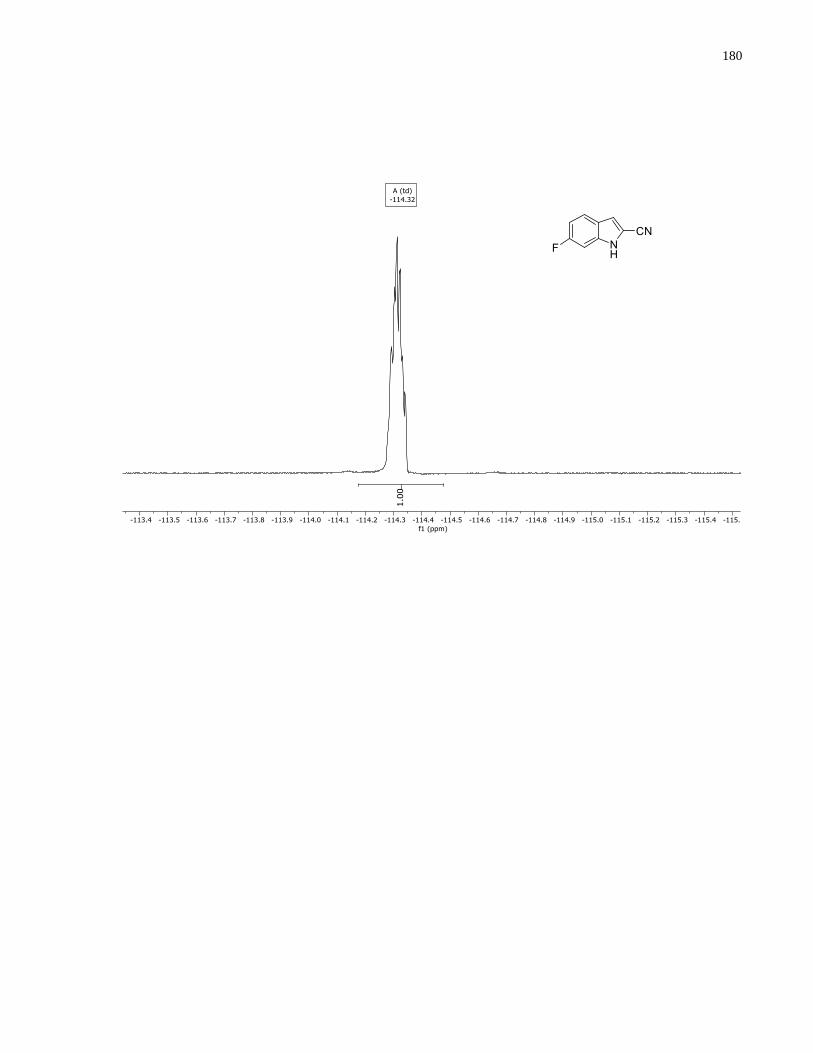
180
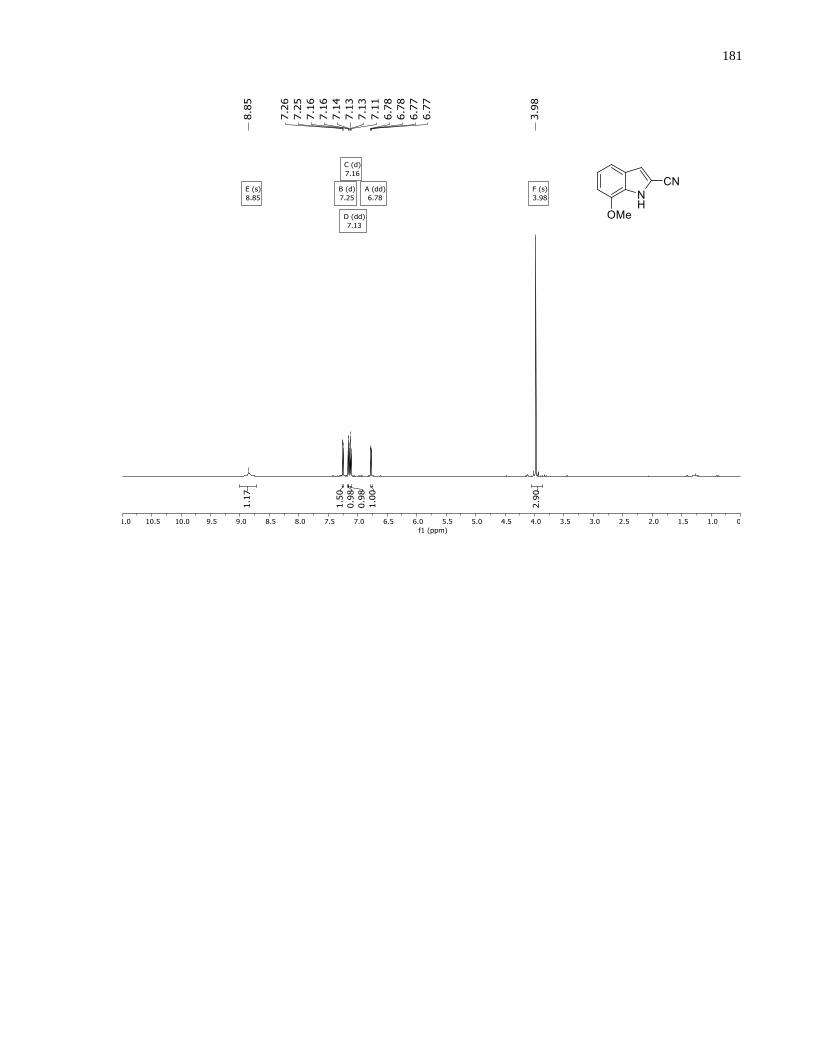
181

182
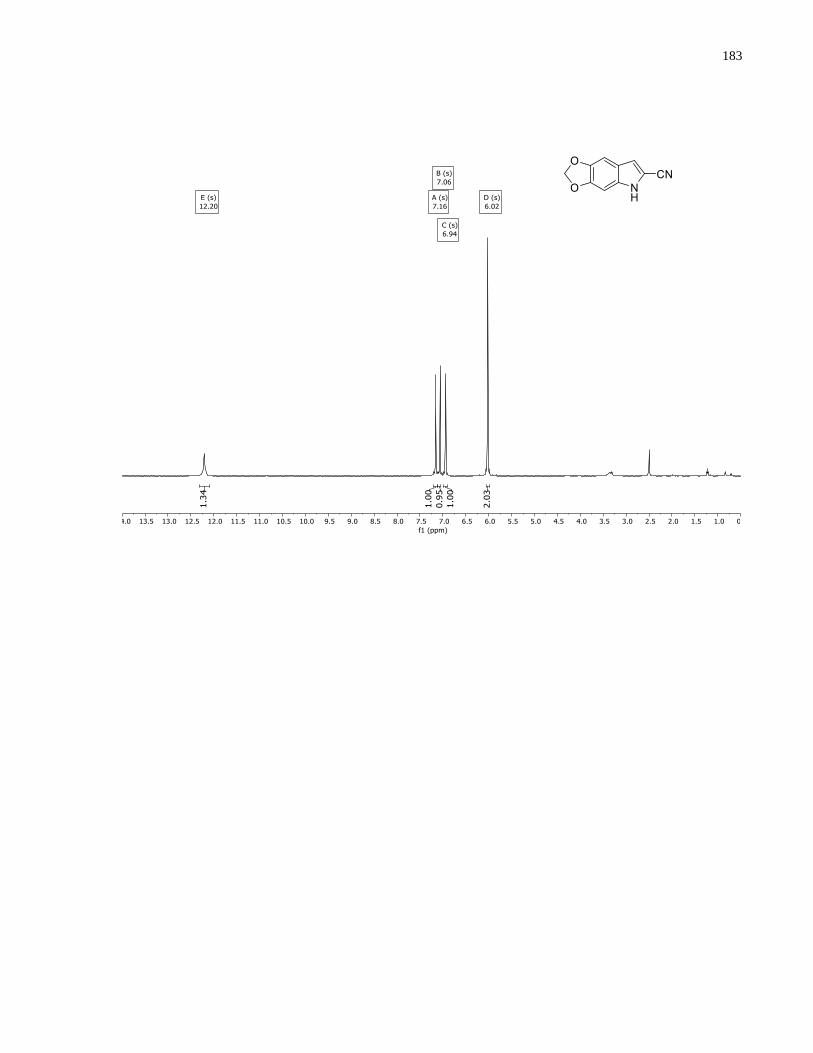
183

184
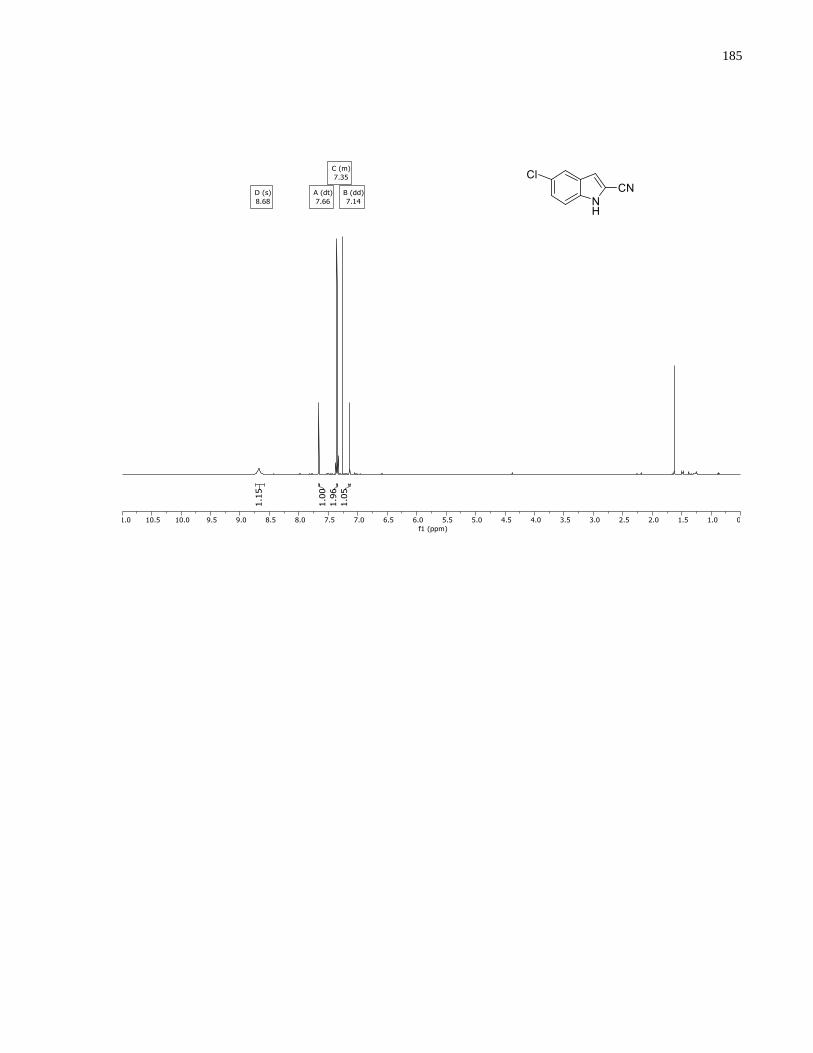
185
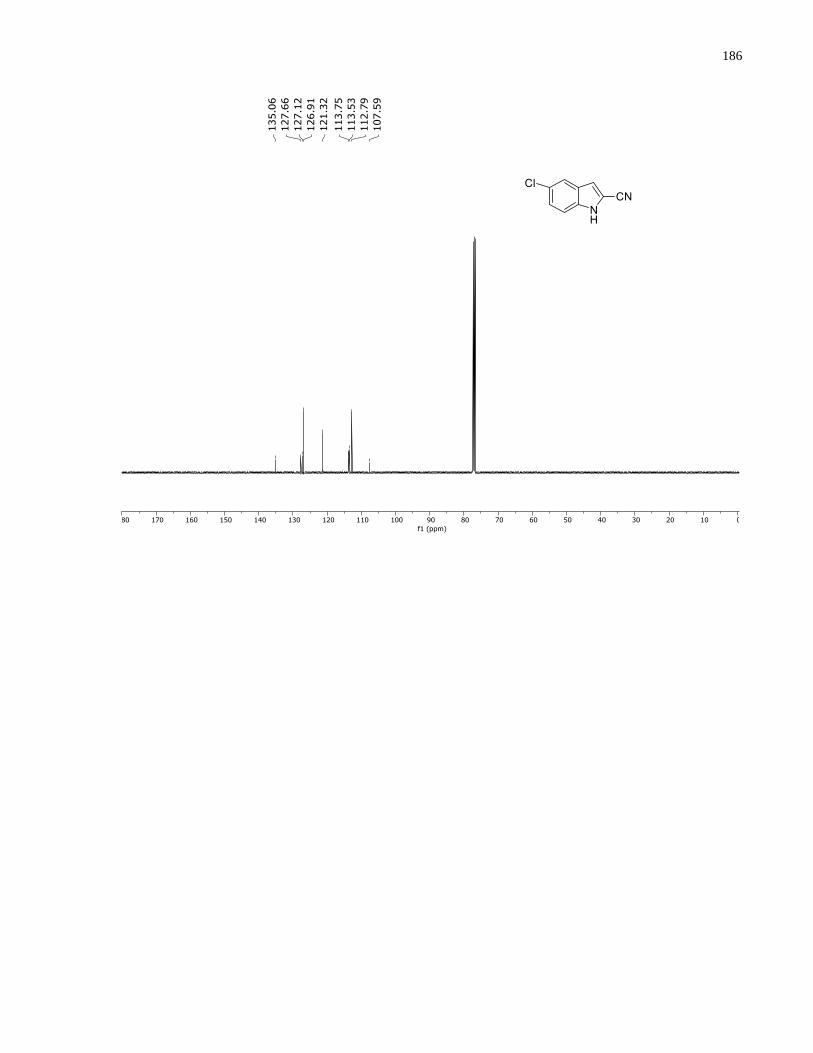
186

187

188
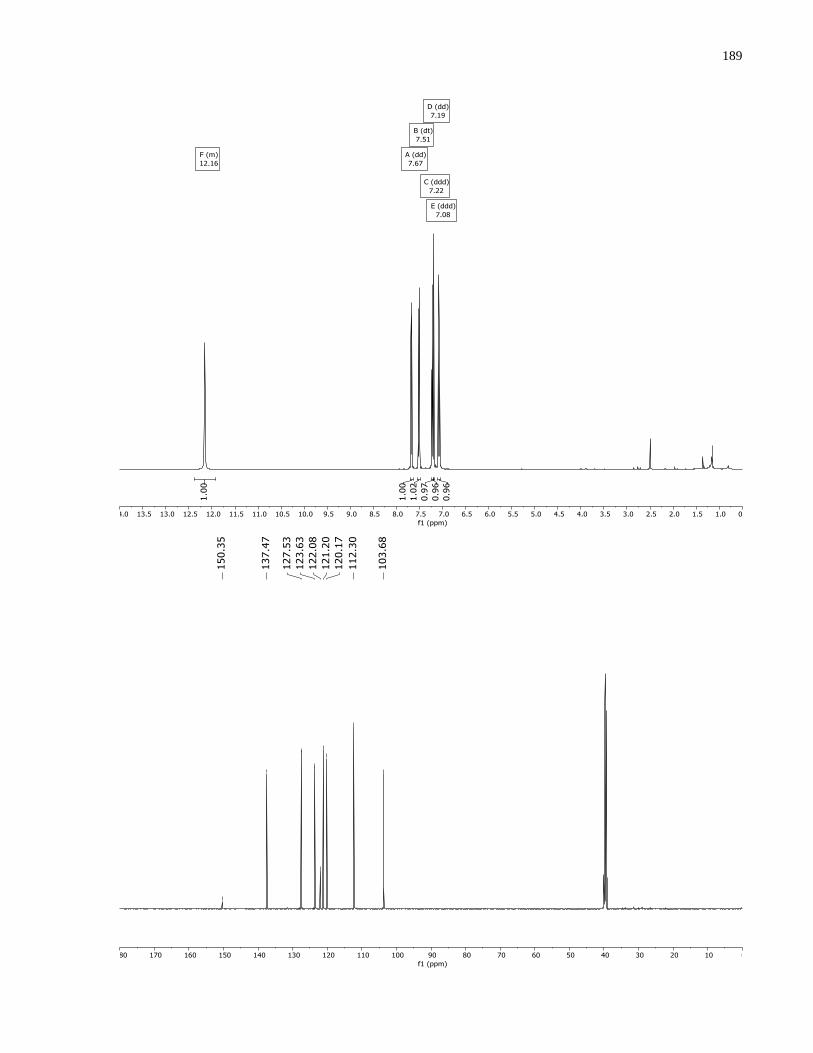
189

190
Appendix B – NMRs from Chapter 2

191

192

193

194

195

196

197

198

199
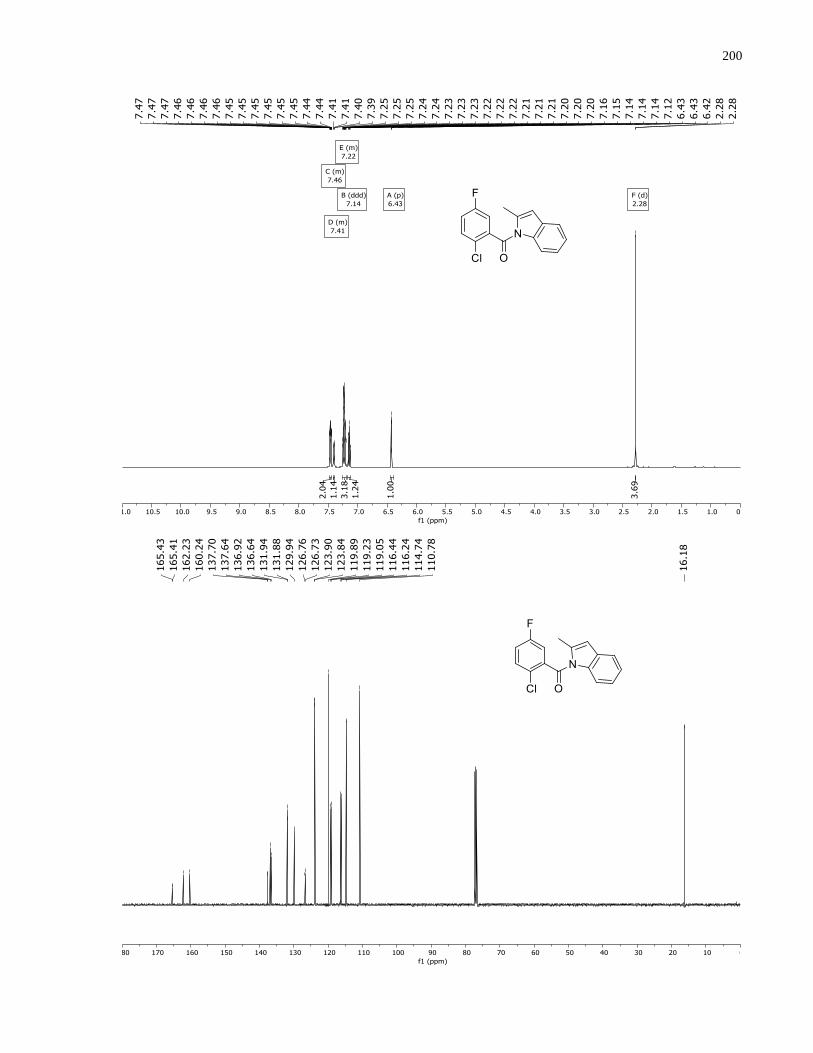
200

201

202

203

204

205

206

207

208

209
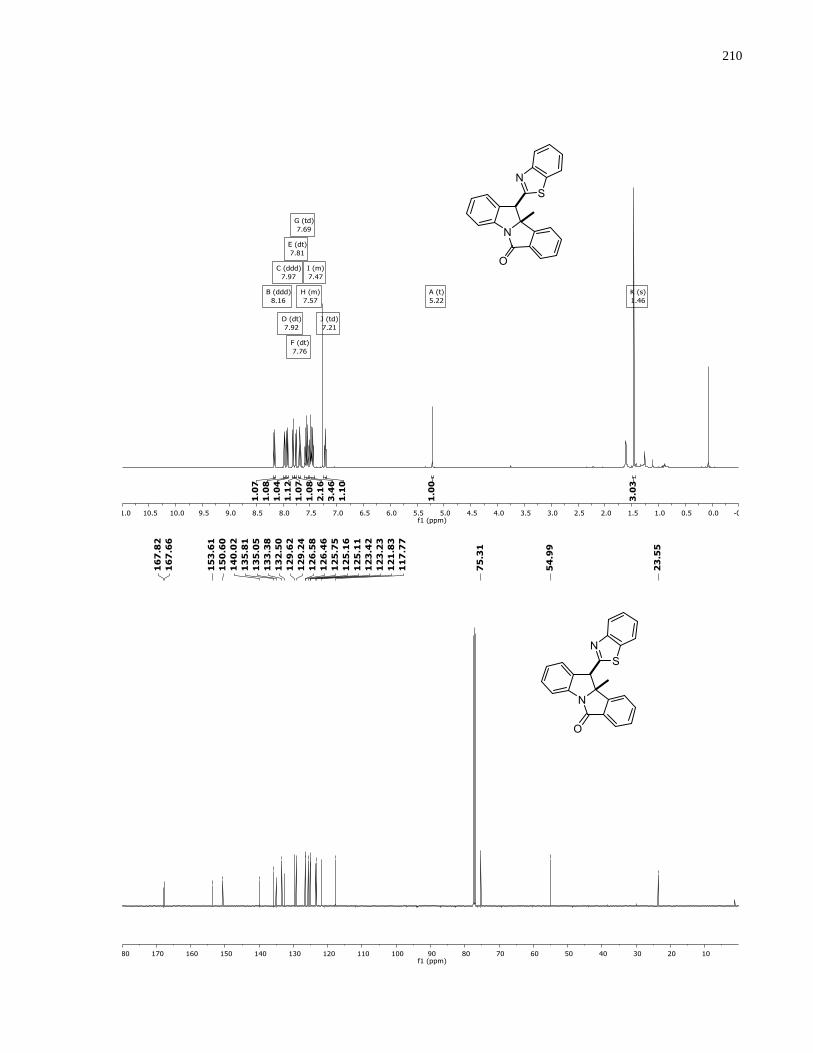
210
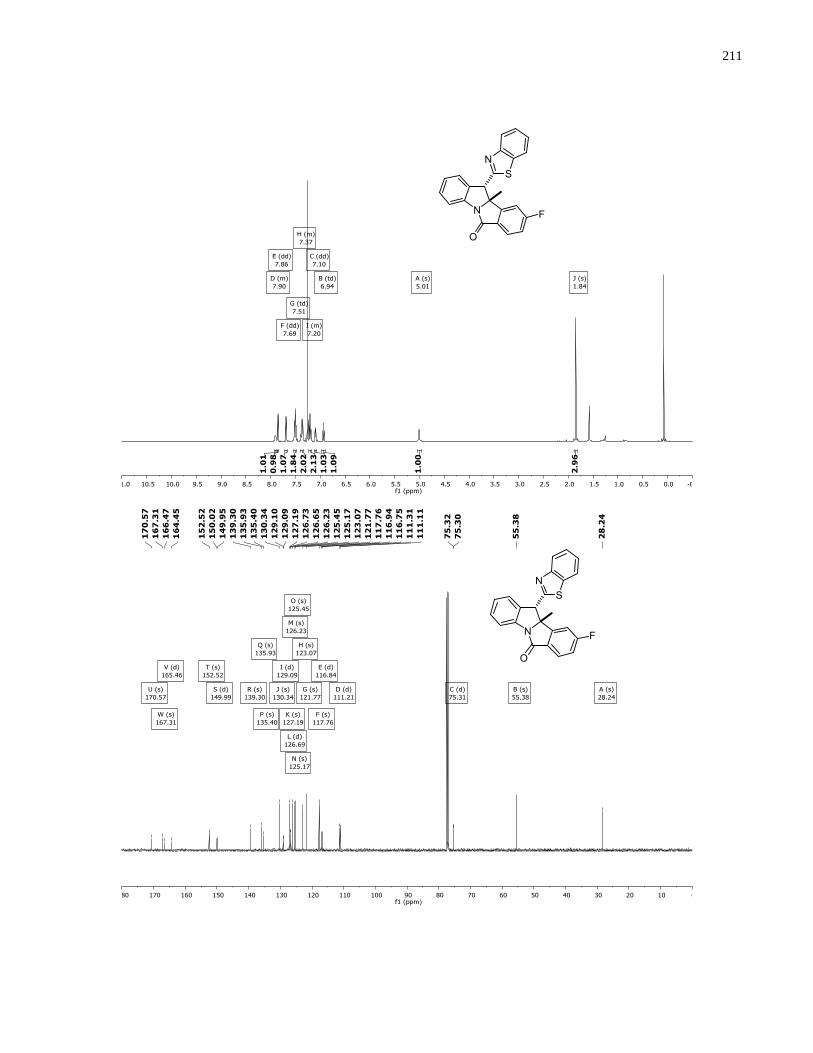
211

212

213

214

215

216

217

218

219

220

221

222
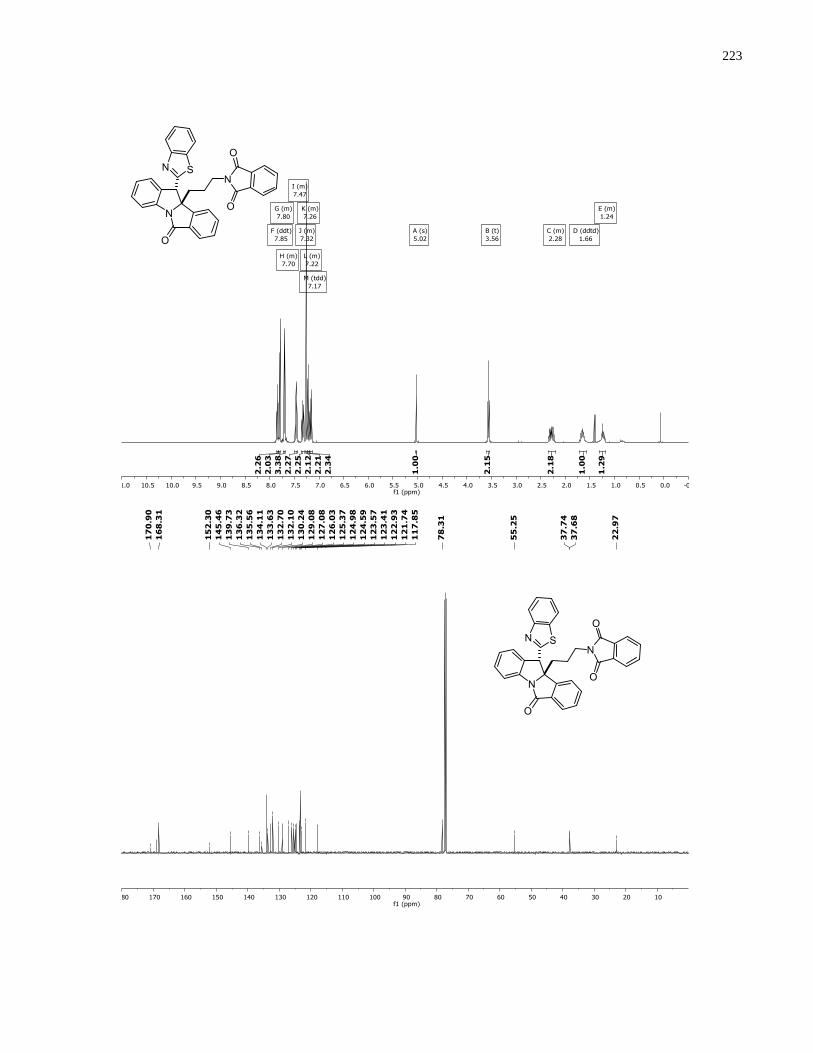
223

224

225

226

227

228

229
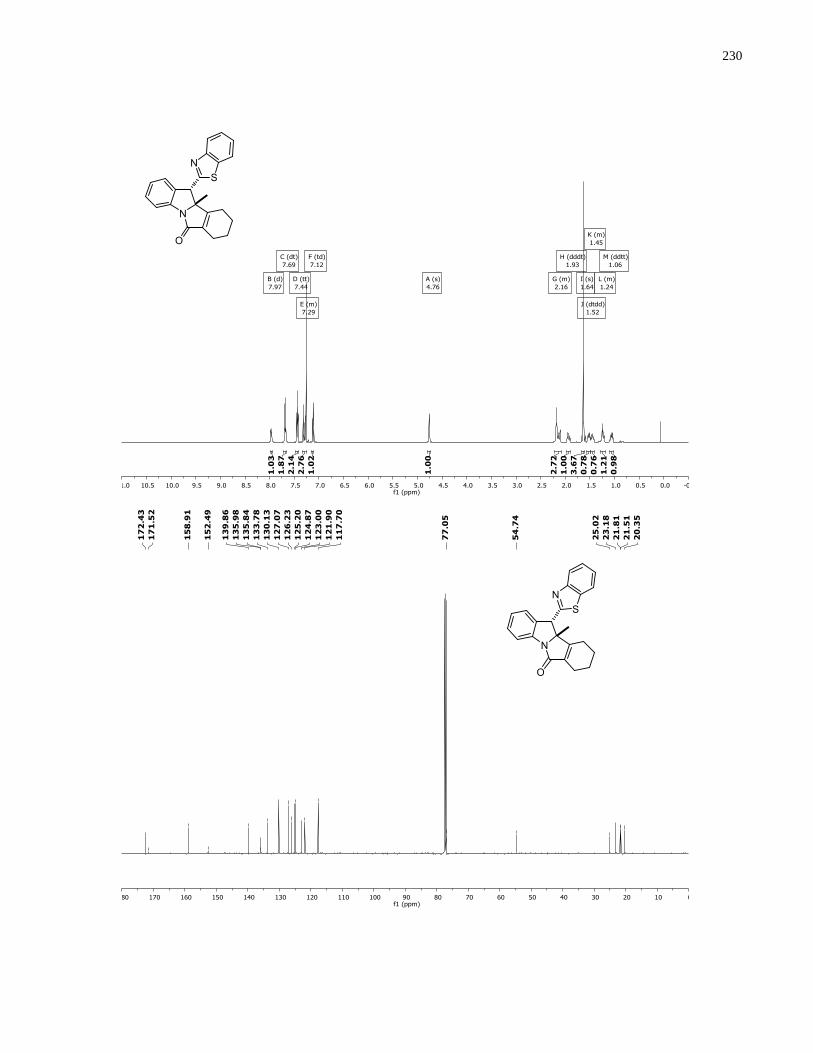
230
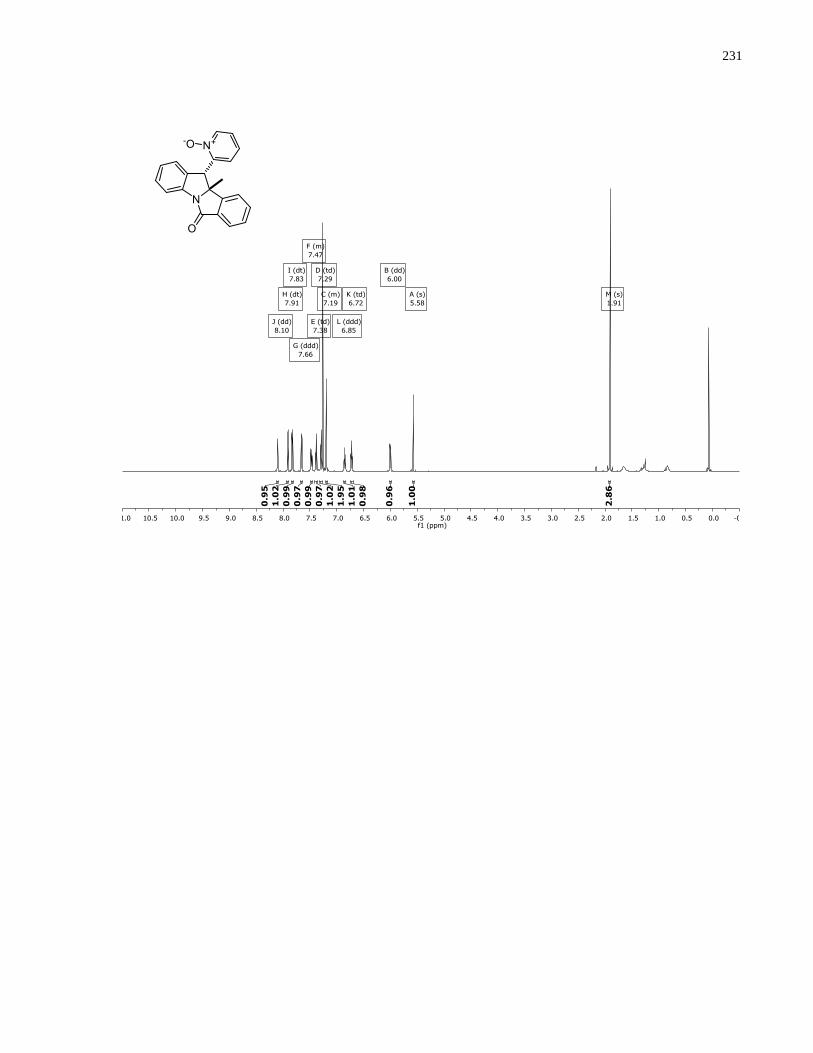
231

232

233

234

235
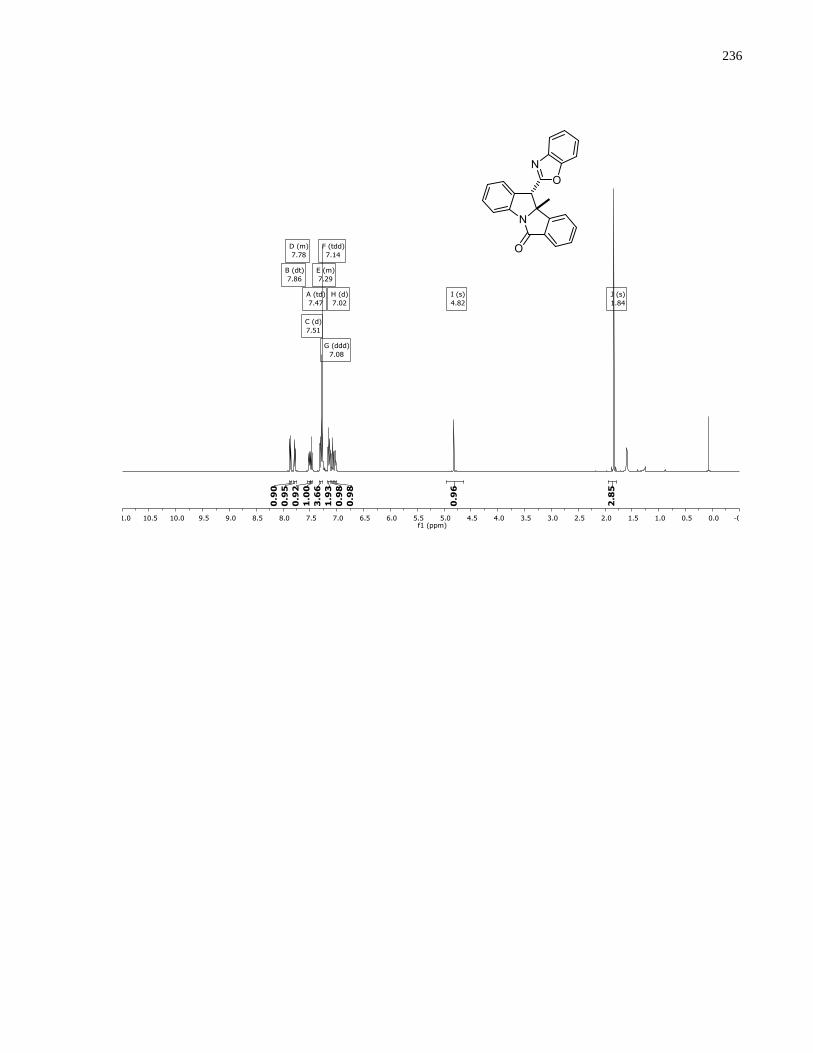
236
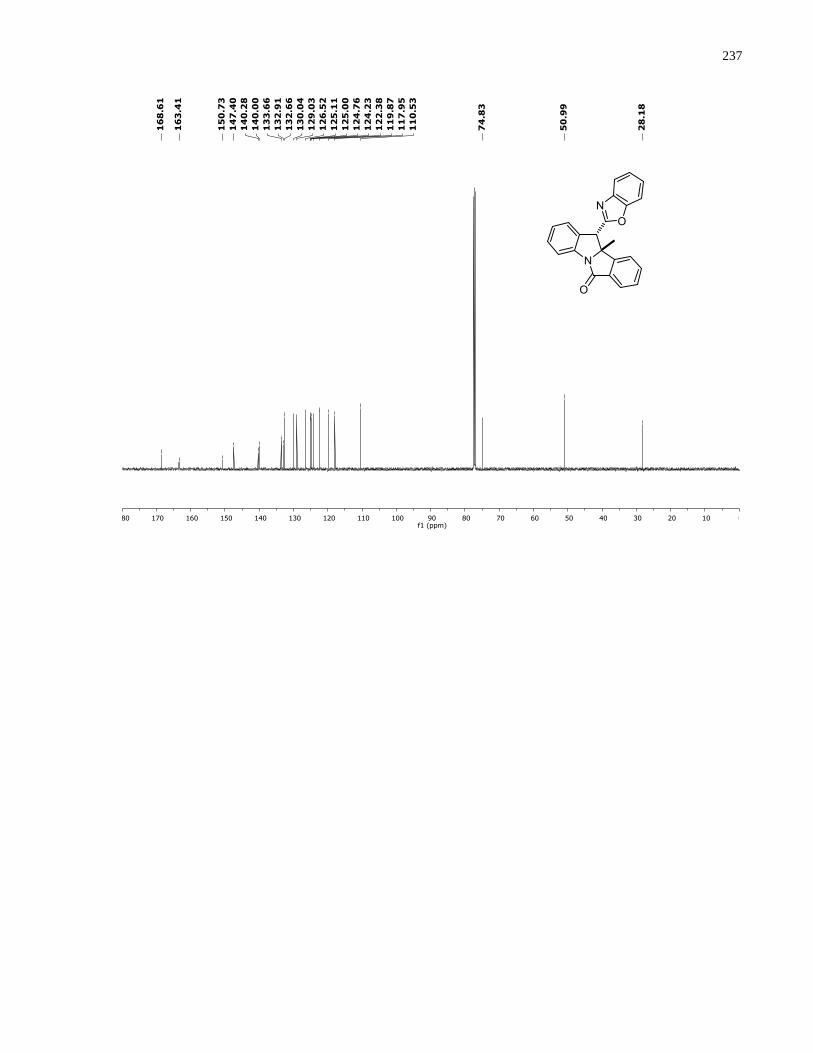
237
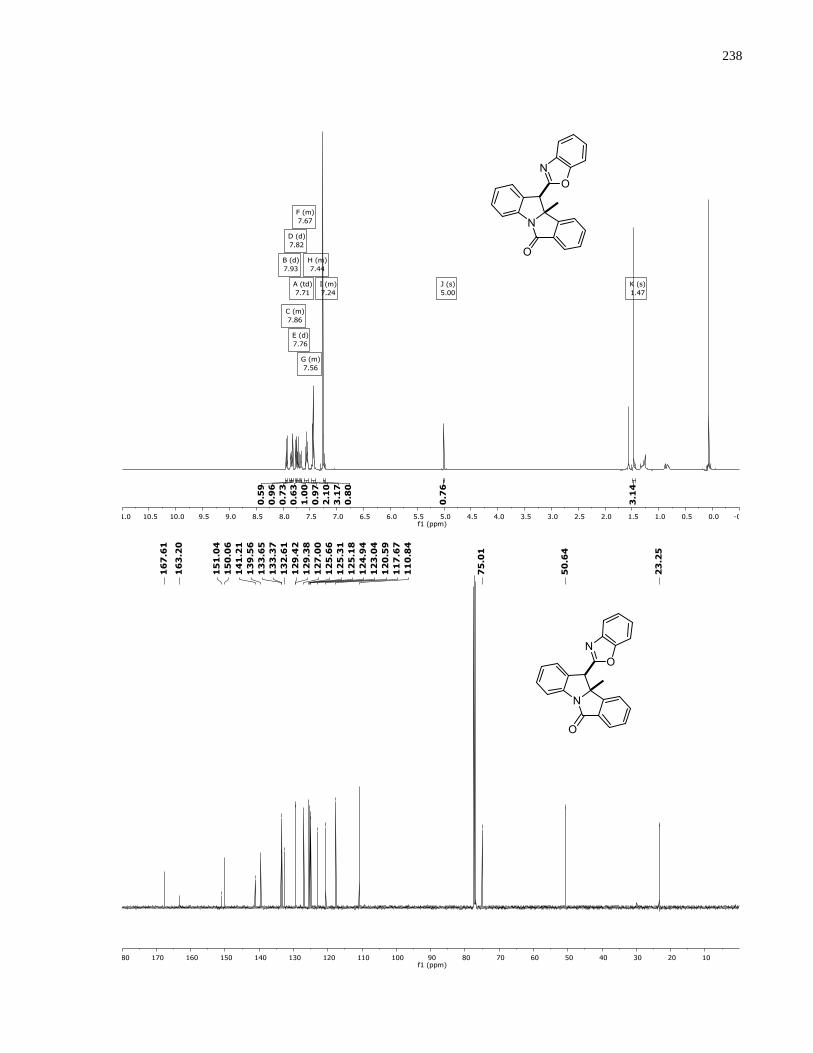
238
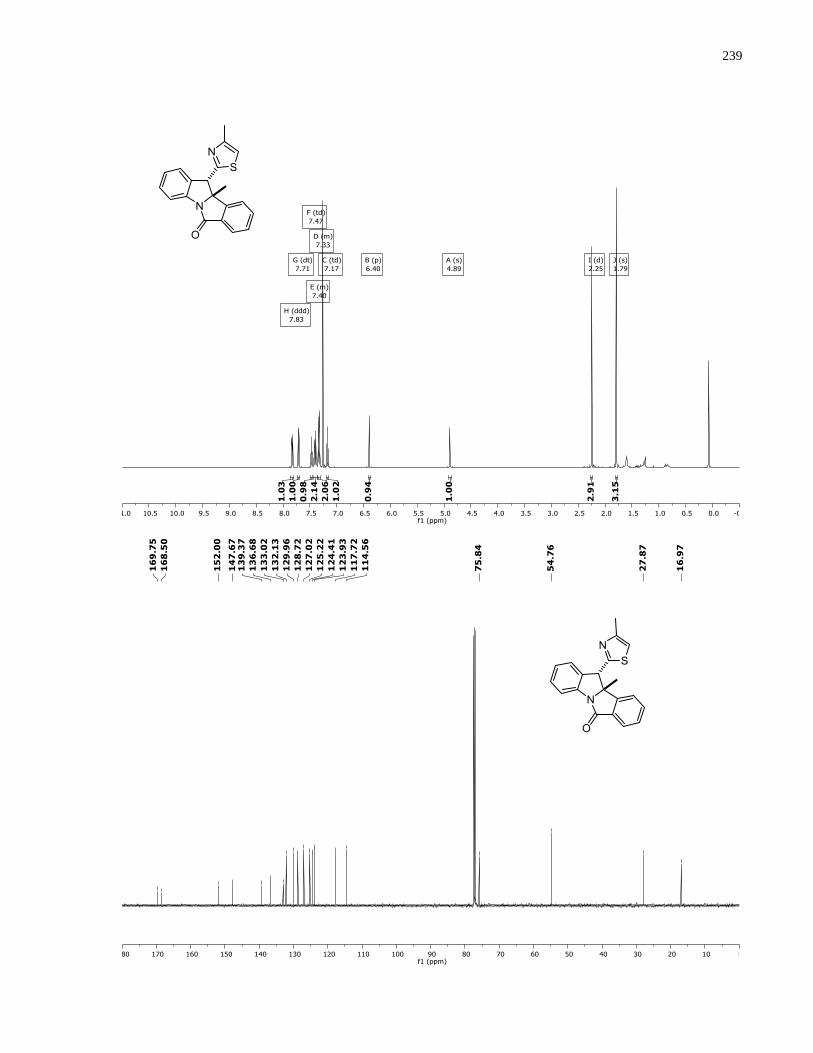
239

240

241

242

243

244

245
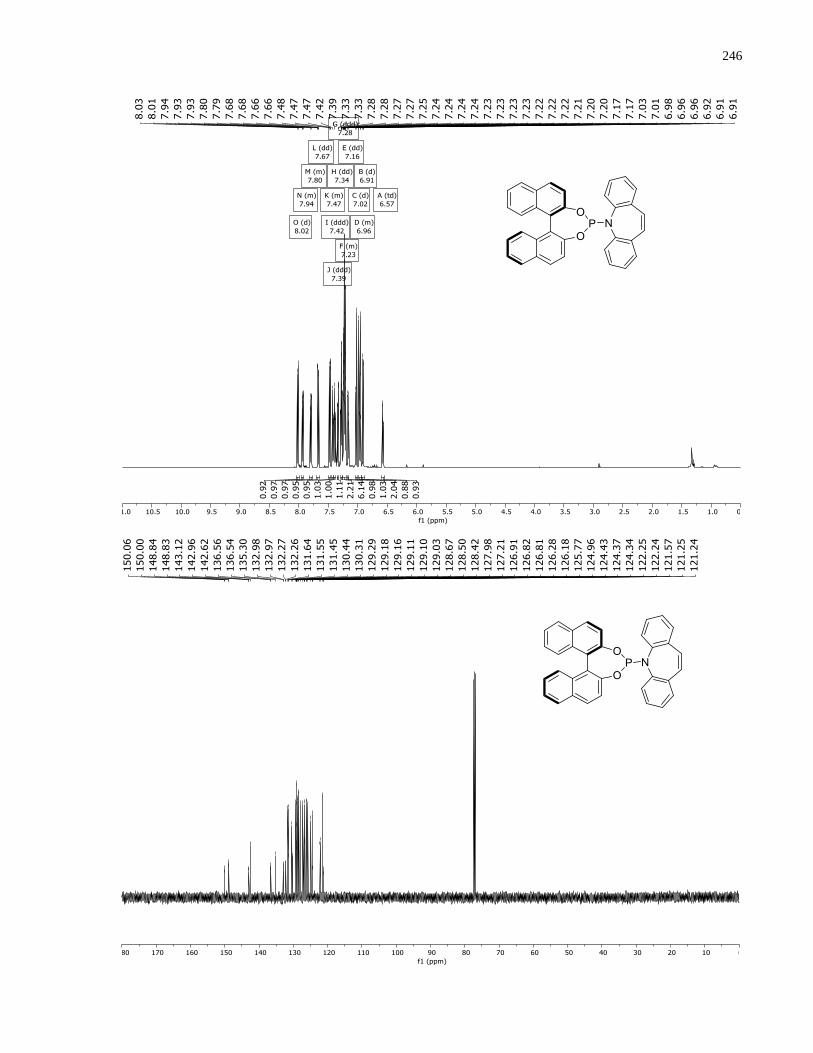
246

247

248

249

250

251

252
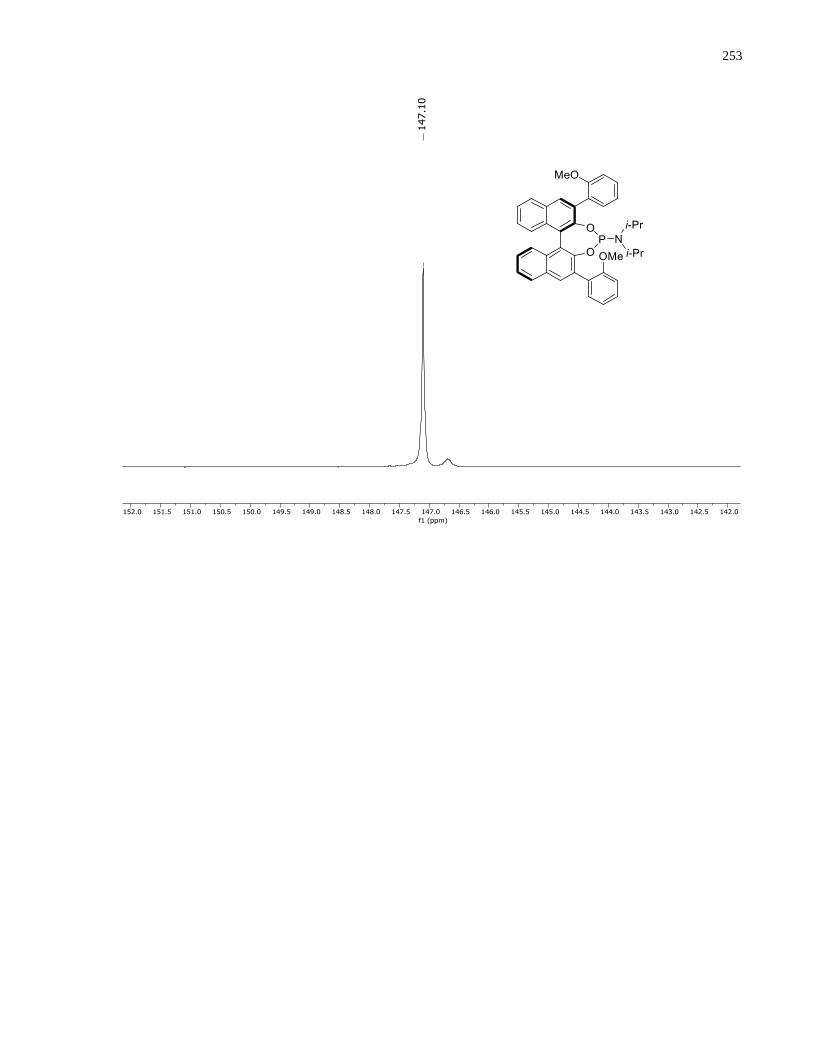
253

254

255
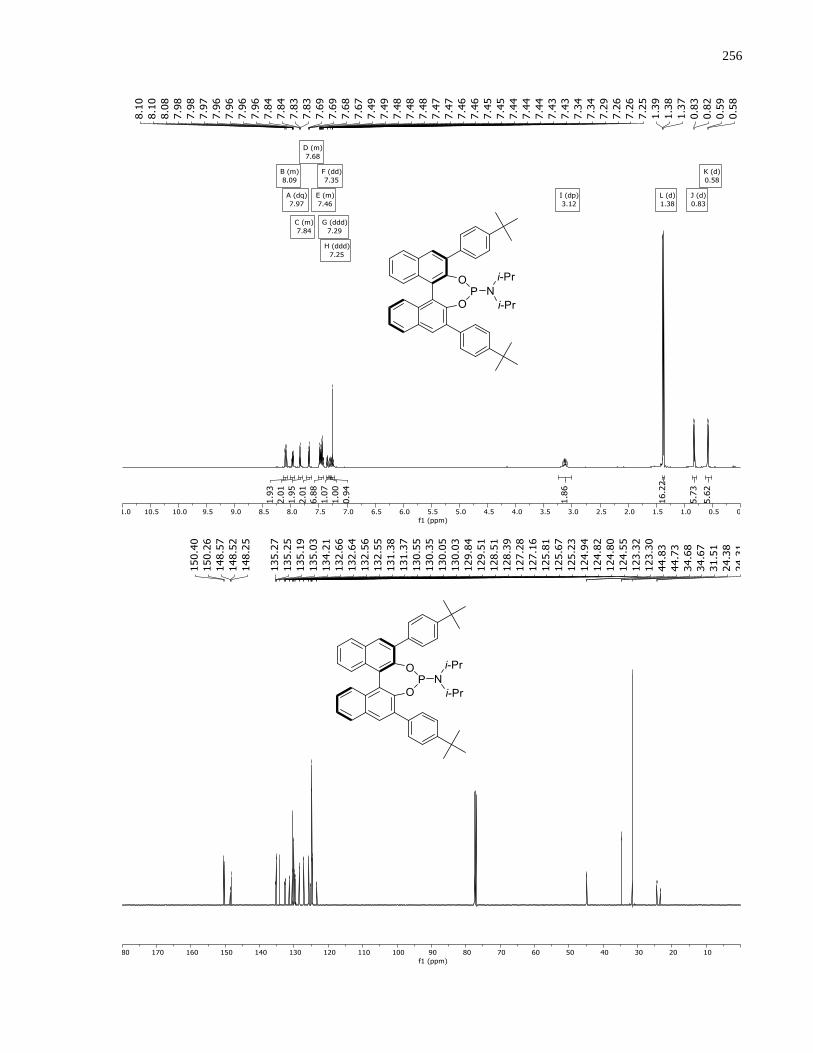
256

257

258

259

260

261

262

263
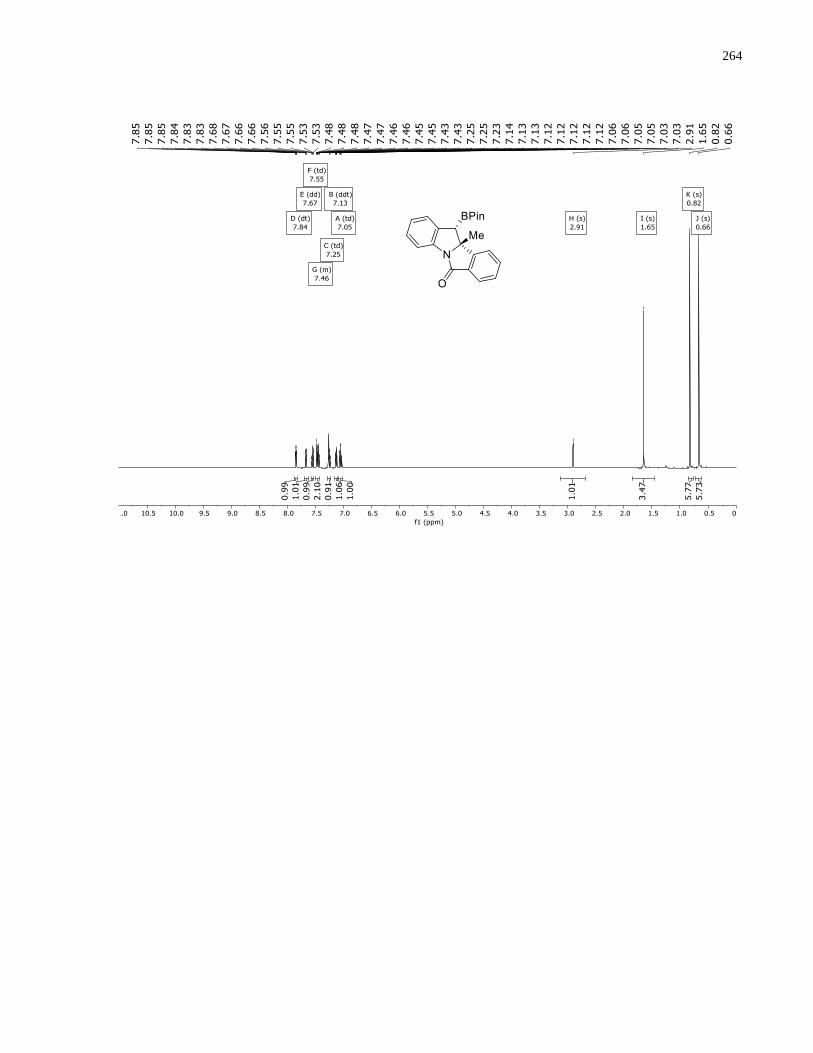
264

265

266

267

268

269

270

271

272
273
274

275

276
277

278

279

280

281
Appendix C – HPLC Chromatograms from Chapter 2

282

283
284

285

286
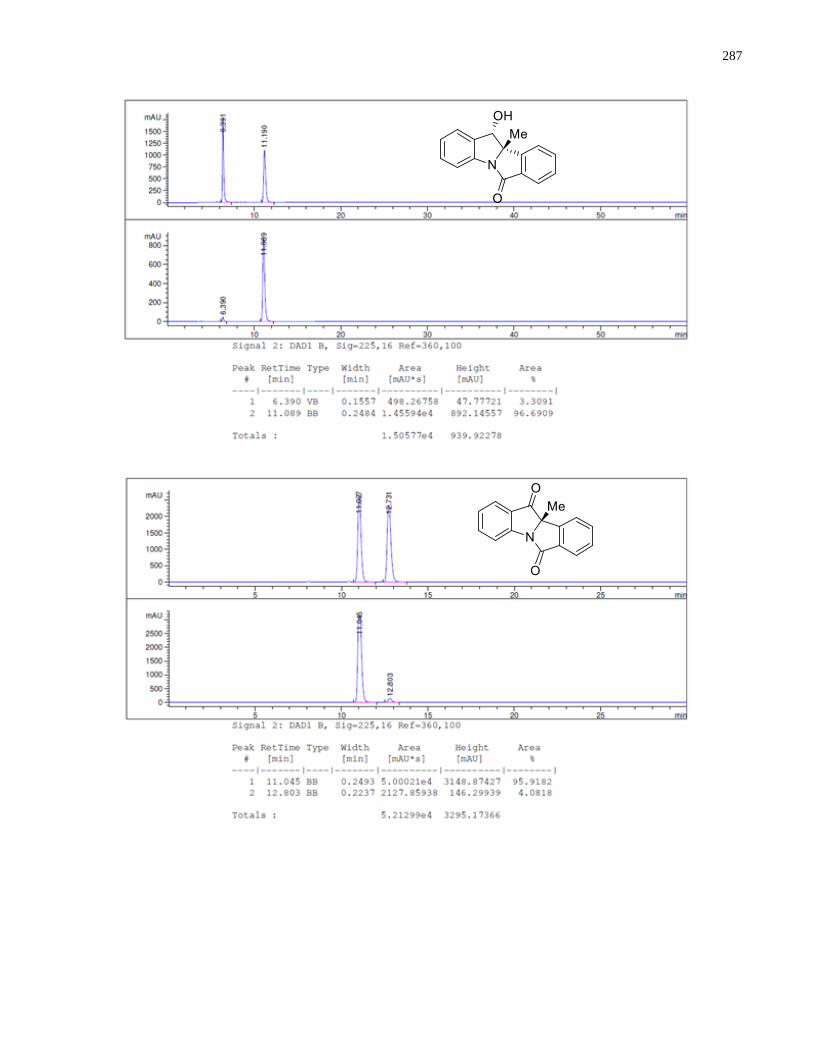
287

288
Appendix D – NMRs from Chapter 3

289

290

291
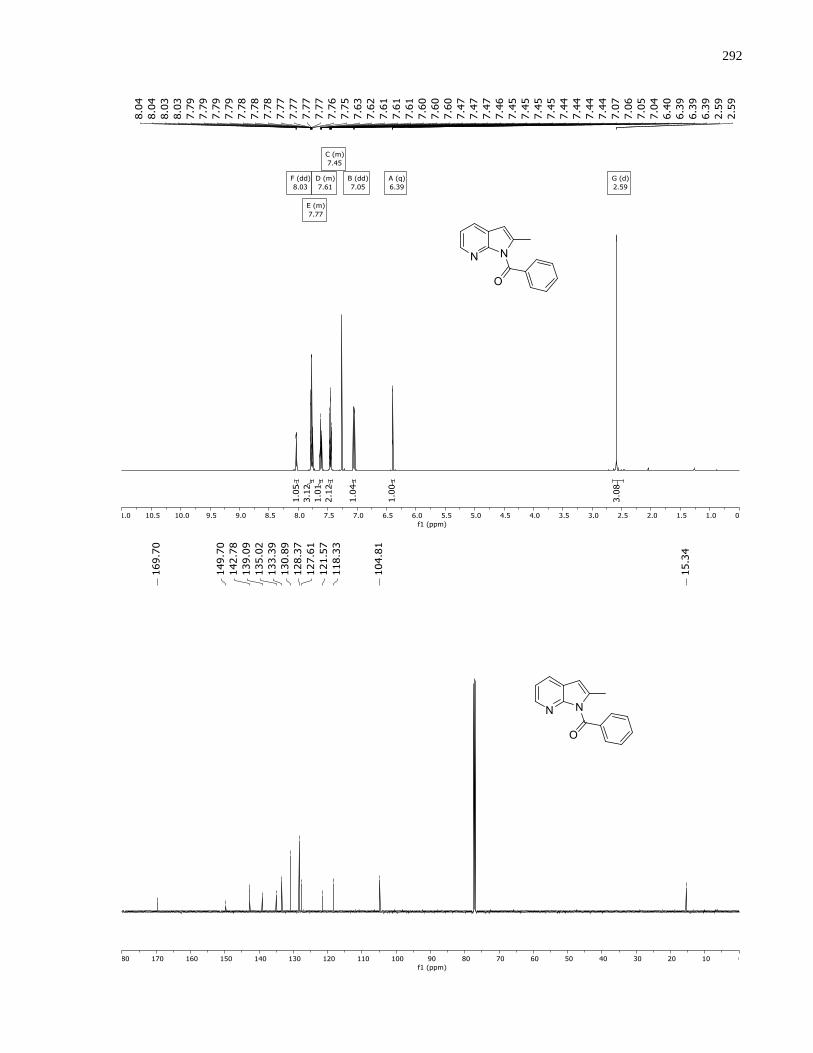
292

293
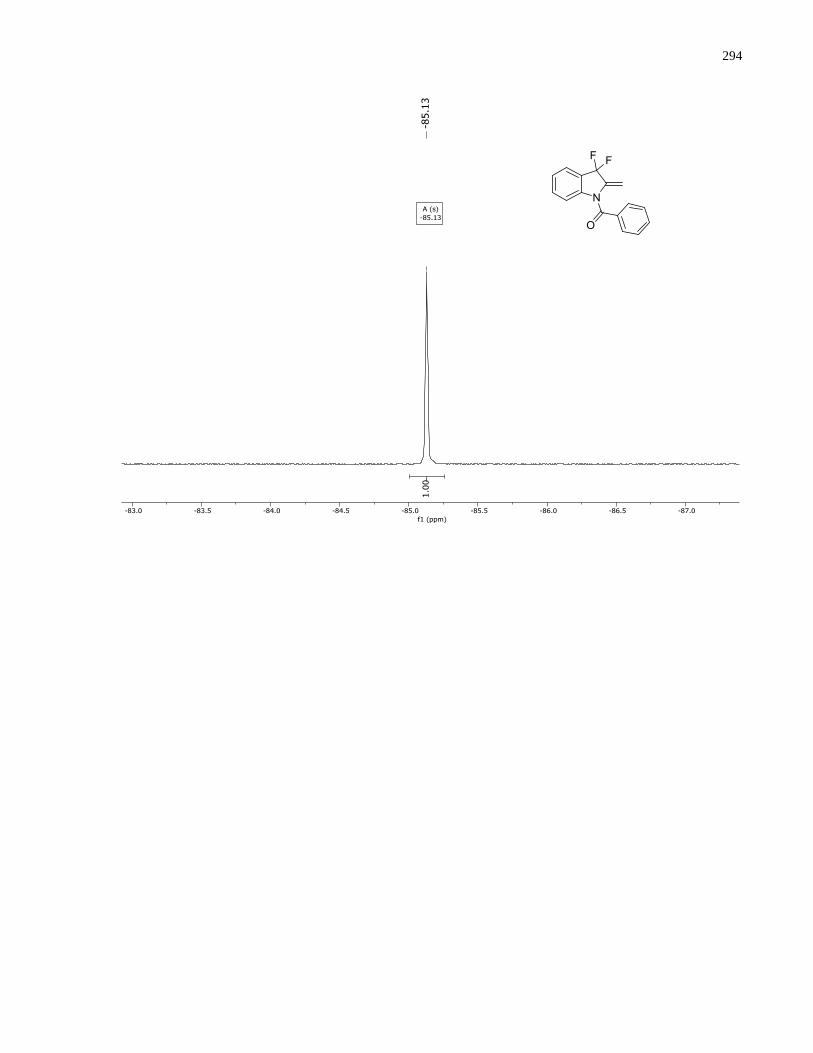
294

295

296
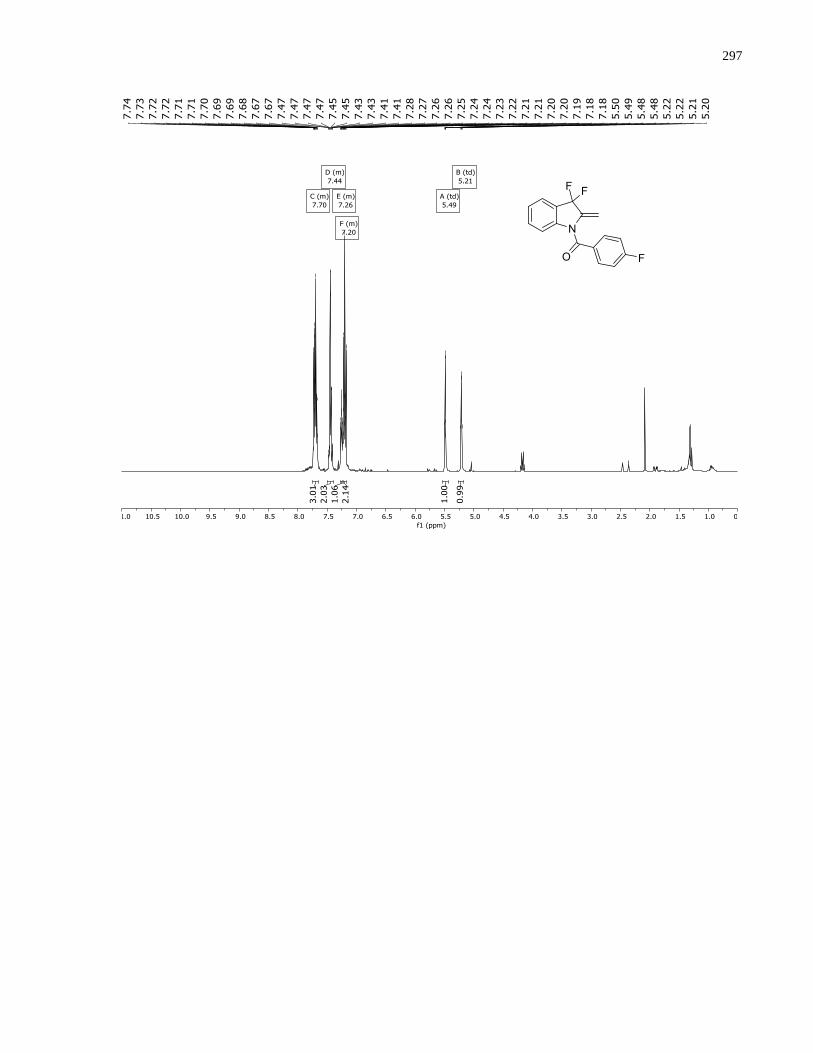
297

298

299

300
301

302
303

304

305

306

307

308

309
310

311

312

313

314
315
316
317
318
319
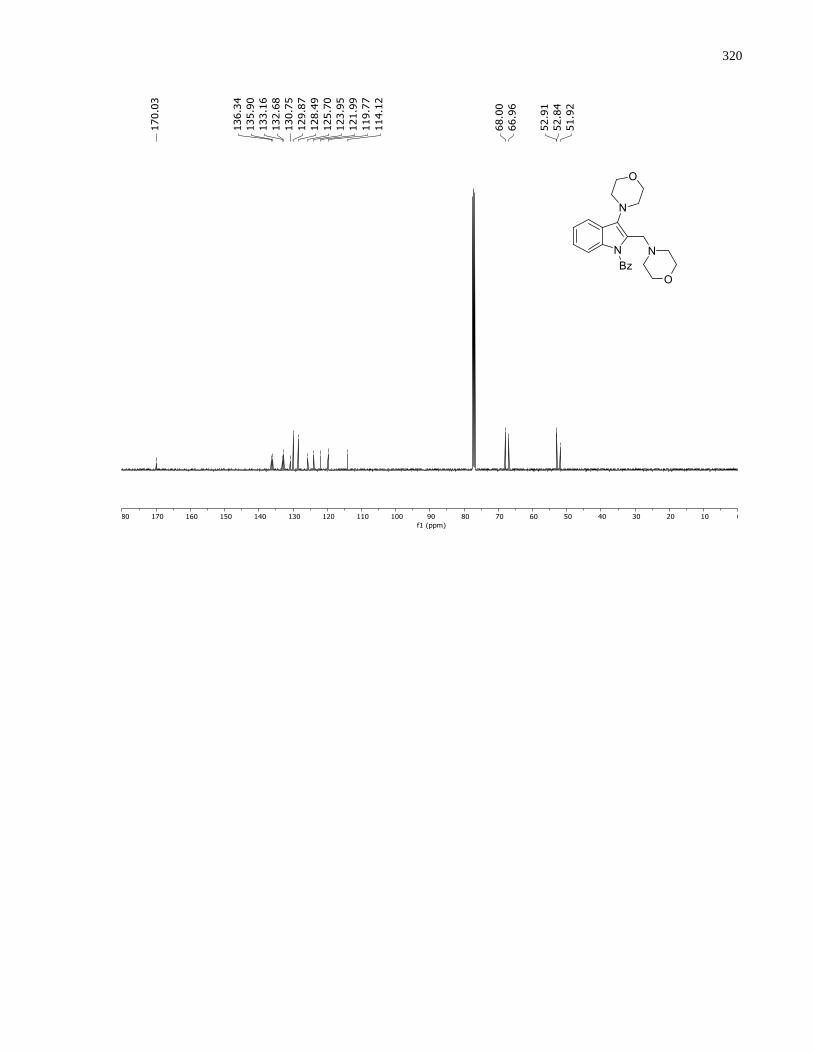
320

321

322

323

324

325

326

327

328

329

330
Appendix E – X-Ray Crystallographic data
Experiments were run by Dr. Alan Lough
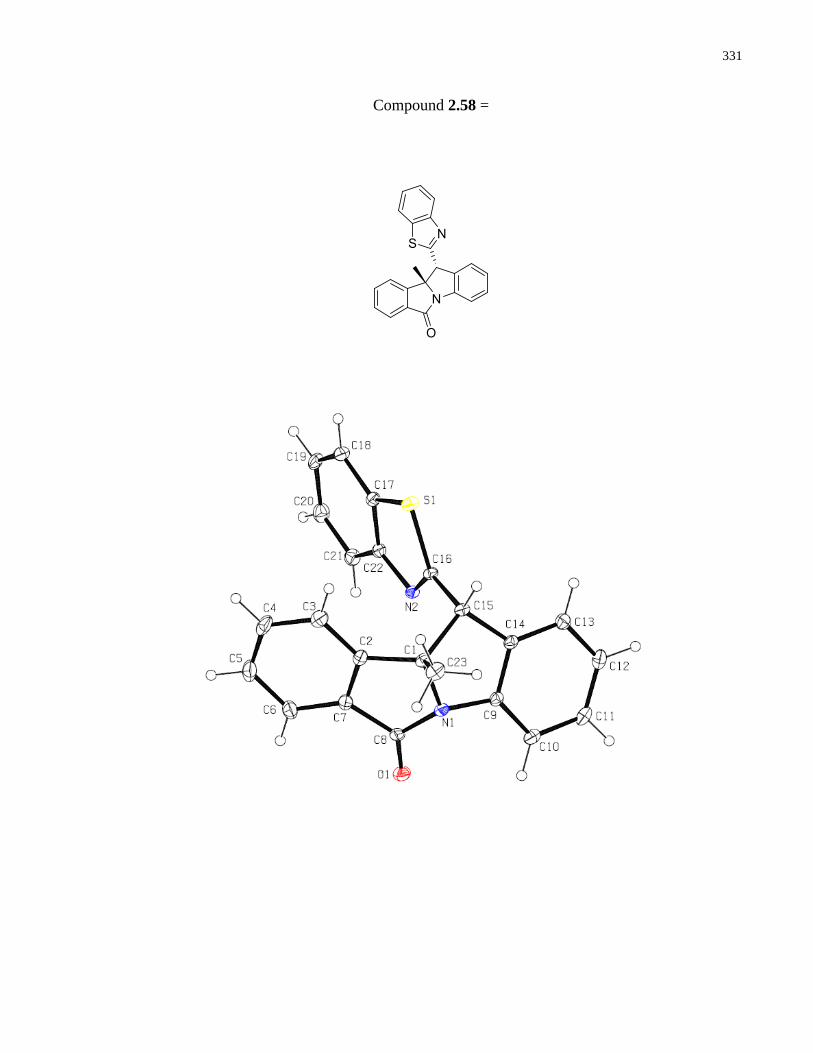
331
Compound 2.58 =

332
Table 1. Crystal data and structure refinement for d1847a_a.
Identification code d1847a_a
Empirical formula C23 H16 N2 O S
Formula weight 368.44
Temperature 150(2) K
Wavelength 0.71073 Å
Crystal system Orthorhombic
Space group Pna21
Unit cell dimensions a = 15.0523(7) Å = 90°.
b = 11.1701(6) Å = 90°.
c = 10.6235(5) Å = 90°.
Volume 1786.19(15) Å3
Z 4
Density (calculated) 1.370 Mg/m3
Absorption coefficient 0.197 mm-1
F(000) 768
Crystal size 0.180 x 0.150 x 0.060 mm3
Theta range for data collection 2.270 to 27.500°.
Index ranges -19<=h<=18, -14<=k<=14, -13<=l<=13
Reflections collected 11663
Independent reflections 4064 [R(int) = 0.0436]
Completeness to theta = 25.242° 99.6 %

333
Absorption correction Semi-empirical from equivalents
Max. and min. transmission 0.7456 and 0.7051
Refinement method Full-matrix least-squares on F2
Data / restraints / parameters 4064 / 1 / 245
Goodness-of-fit on F2 1.021
Final R indices [I>2sigma(I)] R1 = 0.0404, wR2 = 0.0855
R indices (all data) R1 = 0.0537, wR2 = 0.0911
Absolute structure parameter -0.02(4)
Extinction coefficient n/a
Largest diff. peak and hole 0.304 and -0.240 e.Å-3

334
Compound 2.65a =

335
Table 1. Crystal data and structure refinement for d1866a_a.
Identification code d1866a_a
Empirical formula C22 H24 B N O3
Formula weight 361.23
Temperature 150(2) K
Wavelength 1.54178 Å
Crystal system Monoclinic
Space group P21
Unit cell dimensions a = 9.9629(2) Å = 90°.
b = 10.4616(2) Å = 90.9230(10)°.
c = 18.5419(3) Å = 90°.
Volume 1932.33(6) Å3
Z 4
Density (calculated) 1.242 Mg/m3
Absorption coefficient 0.646 mm-1
F(000) 768
Crystal size 0.250 x 0.170 x 0.170 mm3
Theta range for data collection 2.383 to 67.563°.
Index ranges -11<=h<=11, -12<=k<=11, -22<=l<=22
Reflections collected 73419
Independent reflections 6794 [R(int) = 0.0627]

336
Completeness to theta = 67.563° 98.6 %
Absorption correction Semi-empirical from equivalents
Max. and min. transmission 0.7529 and 0.6898
Refinement method Full-matrix least-squares on F2
Data / restraints / parameters 6794 / 2087 / 947
Goodness-of-fit on F2 1.043
Final R indices [I>2sigma(I)] R1 = 0.0401, wR2 = 0.1036
R indices (all data) R1 = 0.0410, wR2 = 0.1049
Absolute structure parameter 0.01(5)
Extinction coefficient n/a
Largest diff. peak and hole 0.364 and -0.248 e.Å-3

337
Compound 3.38a

338
Table 1. Crystal data and structure refinement for d19161_a.
Identification code d19161_a
Empirical formula C16 H11 F2 N O
Formula weight 271.26
Temperature 150(2) K
Wavelength 0.71073 Å
Crystal system Monoclinic
Space group P21/n
Unit cell dimensions a = 14.5503(19) Å = 90°.
b = 5.7406(6) Å = 102.044(6)°.
c = 15.530(2) Å = 90°.
Volume 1268.6(3) Å3
Z 4
Density (calculated) 1.420 Mg/m3
Absorption coefficient 0.109 mm-1
F(000) 560
Crystal size 0.180 x 0.100 x 0.040 mm3
Theta range for data collection 1.745 to 24.997°.
Index ranges -17<=h<=17, -6<=k<=6, -18<=l<=18
Reflections collected 18687
Independent reflections 2232 [R(int) = 0.1186]
Completeness to theta = 24.997° 100.0 %

339
Absorption correction Semi-empirical from equivalents
Max. and min. transmission 0.7456 and 0.6471
Refinement method Full-matrix least-squares on F2
Data / restraints / parameters 2232 / 0 / 182
Goodness-of-fit on F2 1.011
Final R indices [I>2sigma(I)] R1 = 0.0522, wR2 = 0.0818
R indices (all data) R1 = 0.1115, wR2 = 0.0993
Extinction coefficient 0.0106(16)
Largest diff. peak and hole 0.173 and -0.194 e.Å
Copyright © 2022 FDOKUMEN















![Synthesis of a 3-(α-Styryl)benzo[ b ]-thiophene Library via Bromocyclization of Alkynes and Palladium-Catalyzed Tosylhydrazones Cross-Couplings: Evaluation as Antitubulin Agents](https://static.fdokumen.com/doc/165x107/6323817f48d448ffa006b4bd/synthesis-of-a-3-styrylbenzo-b-thiophene-library-via-bromocyclization-of.jpg)




![Stacking patterns of thieno[3,2- b ]thiophenes functionalized by sequential palladium-catalyzed Suzuki and Heck cross-coupling reactions](https://static.fdokumen.com/doc/165x107/6344b1c438eecfb33a063498/stacking-patterns-of-thieno32-b-thiophenes-functionalized-by-sequential-palladium-catalyzed.jpg)