
Little Bite, Big Disease: Recognizing and Managing Tickborne ...
Upload
greenvillemed-scCategory
view
0download
0
Pergamon
Molecular Immunology, Vol. 32, No. 16, 1183-l 195. 1995 pp. Copyright 0 1995 Elsevier Science Ltd
Printed in Great Britain. All rights reserved 0161~5890(95)00099-2 0161-5890/95 $9.50 + 0.00
IN V1TRO AND IN I/IV0 INHIBITION OF COMPLEMENT ACTIVITY BY A SINGLE-CHAIN Fv FRAGMENT
RECOGNIZING HUMAN C5
MARK J. EVANS,*lj SCOTT A. ROLLINS,? DENNIS W. WOLFF,? RUSSELL P. ROTHER,* ALLEN J. NORIN,$ DENISE M. THERRIEN,$
GAL0 A. GRIJALVA,$ JOHN P. MUELLER,* STEVEN H. NYE,$ STEPHEN P. SQUINTO* and JAMES A. WILKINSS
*Department of Molecular Development, Alexion Pharmaceuticals, 25 Science Park, New Haven, CT 06511, U.S.A.; TDepartment of Immunobiology, Alexion Pharmaceuticals, 25 Science Park, New
Haven, CT 06511, U.S.A.; iDepartment of Process Development, Alexion Pharmaceuticals, 2.5 Science Park, New Haven, CT 06511, U.S.A.; SDepartments of Medicine, Anatomy and Cell Biology
and Surgery, State University of New York, Health Science Center, Brooklyn, NY 11203, U.S.A.
(Received 23 Murch 1995; accepted in revisedform 30 June 1995)
Abstract-Complement activation has been implicated in the pathogenesis of several human diseases. Recently, a monoclonal antibody (N19-8) that recognizes the human complement protein CS has been shown to effectively block the cleavage of C5 into C5a and CSb, thereby blocking terminal complement activation. In this study, a recombinant N19-8 s&v antibody fragment was constructed from the N19-8 variable regions, and produced in both mammalian and bacterial cells. The Nl9-8 scFv bound human C5 and was as potent as the N19-8 monoclonal antibody at inhibiting human CSb-9-mediated hemolysis of chicken erythrocytes. In contrast. the N19-8 scFv only partially retained the ability of the N19-8 monoclonal antibody to inhibit C5a generation. To investigate the ability of the Nl9-8 scFv to inhibit complement-mediated tissue damage, complement-dependent myocardial injury was induced in isolated mouse hearts by perfusion with Krebs-Henseleit buffer containing 6% human plasma. The perfused hearts sustained extensive deposition of human C3 and C5b-9, resulting in increased coronary artery perfusion pressure, end-diastolic pressure, and a decrease in heart rate until the hearts ceased beating approximately 10min after the addition of plasma. Hearts treated with human plasma supplemented with either the Nl9-8 monoclonal antibody or the N 19-S scFv did not show any detectable changes in cardiac performance for at least 1 hr following the addition of plasma. Hearts treated with human plasma alone showed extensive deposition of C3 and C5b-9, while hearts treated with human plasma containing the Nl9-8 scFv showed extensive deposition of C3, but no detectable deposition of C5b-9. Administration of a 100mg bolus dose of N19-8 scFv to rhesus monkeys inhibited the serum hemolytic activity by at least 50% for up to 2 hr. Pharmacokinetic analysis of Nl9-8 scFv serum levels suggested a two-compartment model with a T,jz~ of 27min. Together. these data suggest the recombinant Nl9-8 scFv is a potent inhibitor of the terminal complement cascade and may have potential in vivo applications where short duration inhibition of terminal complement activity is desirable.
Key words: complement, C5, single chain antibody, scFv, rhesus monkey.
INTRODUCTION body-independent, triggered by the binding of com-
The complement system consists of a diverse group of plement component C3b directly to a surface. Activation
serum proteins which interact to provide many of the of either the classical or alternative pathway generates
effector functions necessary for the elimination of cellular C3 convertase, which cleaves C3 into C3a and C3b, and
and viral pathogens (Liszewski and Atkinson, 1993). C5 convertase, which cleaves C5 into CSa and C5b. C3a
Complement activation can proceed through two distinct and C5a both function as anaphylatoxins, with C5a
enzymatic cascades. The classical pathway is activated by additionally being a potent chemotactic factor for neu-
the binding of the complement protein Cl to the Fc trophils (Gerard and Gerard, 1994). C3b and its degra-
regions of antibodies aggregated by multiple antigenic dation product, iC3b, act as opsonins by binding to
determinants. In contrast, the alternative pathway is anti- complement receptor type 1 (CRI) and complement receptor type 3 (CR3) present on neutrophils and mac- rophages (Liszewski and Atkinson, 1993). C3b- and
TAuthor to whom correspondence should be addressed. iC3b-mediated phagocytosis is believed to be a major 1183

1184 M. J. EVANS et al.
defense mechanism against bacterial or fungal infections (reviewed in Morgan, 1990). Finally, C5b initiates for- mation of the membrane attack complex consisting of the terminal complement components C5b, C6, C7, C8 and C9, which results in the osmotic damage of sus- ceptible target cells.
The complement system’s ability to inflict damage is not intrinsically limited to foreign cells, but can be directed against the host’s own cells. To prevent auto- logous complement-mediated damage, mammalian cells express several regulatory proteins that block comple- ment activation (reviewed in Lachmann, 1991). These regulatory proteins act at multiple points in the com- plement cascade by inactivating Cl (Cl inhibitor), accelerating the inactivation of the C3 and C5 convertases (complement receptor type 1, membrane cofactor protein and decay accelerating factor), or preventing formation of the membrane attack complex (CD59).
Recently the importance of complement activation in the pathogenesis of several diseases has been recognized. Although dysfunction of the complement system is not the etiologic basis of these diseases, the resultant acti- vation of complement contributes significantly to the clinical pathology. One group of diseases in which com- plement activation plays an important role is the auto- immune diseases, which are frequently correlated with the formation of complement-activating autoantibodies. For example, in myasthenia gravis, autoantibodies directed against the acetylcholine receptor result in the deposition of membrane attack complexes at the neuro- muscular junction (Engel and Arahata, 1987). Mice gen- etically deficient in C5 are resistant to development of experimental autoimmune myasthenia gravis (Christa- doss, 1988). Similarly, treatment of rats with an Fab directed against complement component C6 inhibits the development of experimental autoimmune myasthenia gravis (Biesecker and Gomez, 1989) suggesting that inhi- bition of the complement cascade at C5 or later may provide clinical benefit. Complement has also been sug- gested to play an important role in several other chronic conditions such as multiple sclerosis and rheumatoid arthritis (reviewed in Morgan, 1994).
In addition to these chronic conditions, complement activation is also believed to be an important component of several acute conditions. In reperfusion injury associ- ated with myocardial infarction, complement activation results in cardiac tissue damage by multiple mechanisms (reviewed in Homeister and Lucchesi, 1994). First, C5b- 9 deposition has been demonstrated to occur in human myocardial infarcts (Schafer et al., 1986; Hugo et al., 1990). These membrane attack complexes can result in diverse alterations in the cardiac myocyte physiology due to the influx of ions, particularly calcium ions, and can ultimately result in osmotic lysis. Secondly, C5a serves both as a chemoattractant for neutrophils and as a neutrophil activator, causing the release of proteases and the generation of toxic oxygen metabolites which further damage the myocardium. Thirdly, iC3b, a proteolytic fragment of cell surface deposited C3b, can mediate neutrophil binding to cardiac tissue through a high
affinity interaction with neutrophil CD1 1 b/CD1 8. This contributes significantly to reperfusion injury patho- genesis, since inhibition of neutrophil adherance to endo- thelial cells by either monoclonal antibodies against CD1 1 b or the compound NPC 15669 significantly reduces myocardial injury in animal models (Simpson er al., 1988; Curtis et al., 1993). The inhibition of com- plement activity by pretreatment of rats with a soluble form of complement receptor 1 (sCRl), which blocks all of the above mechanisms, reduces myocardial infarct size up to 44% (Weisman et al., 1990). Administration of sCR1 just prior to reperfusion results in a more modest 25% reduction in infarct size (Smith et al., 1993).
In cardiopulmonary bypass (CPB), complement acti- vation occurs during the extracorporeal circulation of the blood (Moore et al., 1988; Svennevig et al., 1993) and is believed to contribute significantly to the CPB-induced systemic inflammatory reaction. Recent studies in a closed loop CPB circuit have shown that an anti-C5 mon- oclonal antibody (Nl9-8) can block the appearance of several markers of platelet and neutrophil activation associated with complement mediated systemic inflam- mation (Rinder ef al., 1995). In vivo studies using pigs have demonstrated that CPB-induced lung injury can be significantly ameliorated by complement inhibition using sCR1 (Moat et al., 1992; Gillinov et cd., 1993). These findings suggest treatment of patients undergoing CPB with a complement inhibitor may produce significant clinical benefits.
Monoclonal antibodies (mAb) capable of blocking the function of several of the terminal complement proteins have been identified (Wurzner et al., 1991). However, there are several potential limitations for using murine mAbs as complement inhibitors in humans. First, the Fc portion of the antibody might itself bind to Cl and acti- vate complement. Second, the large size of an antibody results in a slow rate of tissue penetration compared to smaller antibody fragments (Cove11 et al., 1986). Finally, repeated administration of murine antibodies routinely results in a strong human immune response to the murine antibody (Miller et al., 1983; Sears et al., 1984; Khazaeli et a/., 1988). This immunogenicity in humans is greatly reduced by the construction of chimeric antibodies con- sisting of murine variable regions fused to human con- stant regions (LoBuglio et al., 1989; Moreland et al.,
1993; Weiden et al., 1993), suggesting murine variable regions alone may not be highly immunogenic in humans.
Single chain antibodies contain the light chain variable region and the heavy chain variable region fused into a single tandem polypeptide maintaining the binding speci- ficity of the parental monoclonal antibody (Bird et al.,
1988; Huston et al., 1988). Binding affinities of scFv mol- ecules are frequently similar to the parental monoclonal antibody (Pantoliano et al., 1991; Kortt et al., 1994), since the folded structure of an scFv is nearly identical to the variable region structure of the parental antibody (Zdanov et al., 1994). Although an scFv possesses no effector functions, it may retain the ability to block the function of its target protein. scFv antibodies have an extremely rapid rate of tissue penetration (Yokota et al.,

Single-chain Fv fragment recognizing human C5 1185
1992). Finally, scFv antibodies can be readily produced in large quantities using E. coli (Pantoliano et al., 1991; Kortt et al., 1994). Together these characteristics suggest a potential therapeutic role for complement-inhibitory scFv antibody fragments.
The monoclonal antibody N 19-8 binds human C5 and prevents its cleavage by either the classical or alternative pathway C5 convertases (Wurzner et al., 1991). This anti- body thus blocks C5a generation and membrane attack complex formation, but does not block formation of C3a and C3b. Previously we have shown that Fabs produced from this antibody retain these properties, indicating an intact N19-8 antibody structure is not required for com- plement inhibition (Evans et a(., 1995). Here we have utilized the variable regions of the N19-8 mAb to con- struct a single chain antibody directed against human C5 and investigated the ability of this N19-8 scFv to inhibit complement activation in vitro and in vivo.
MATERIALS AND METHODS
Cloning qf‘N19-8 variable region genes
Cloning of the N19-8 variable regions was performed using a set of commercially available primers (Mouse Ig- Primer Set, Novagen, Madison, WI). Total RNA was isolated from N19-8 hybridoma cells using the acid/ guanidinium thiocyanate technique (Chomczynski and Sacchi, 1987). For first strand cDNA synthesis, 1Opg total RNA were denatured at 65’C for 5 min, chilled on ice, and added to a 100~1 reaction containing 1OmM Tris-HCl, pH 8.3, 50 mM KCl, 1.5 mM MgCl,, 10 mM dithiothreitol, 250 PM each dNTP, 20 units AMV reverse transcriptase (Seikagaku America, Rockville, MD), and 10 pmol of the appropriate 3’ primer (as described in the Ig-Prime Kit protocol). After incubation at 37°C for 1 hr, 5,ul of the cDNA synthesis reaction were added to a 100~1 PCR reaction containing: 10 mM Tris-HCl, pH 9.0 at 25”C, 50mM KCl, 1.5mM MgCl,, 0.1% (w/v) gelatin, 1.0% (v/v) Triton X-100, 200pM each dNTP, 2.5 U AmpliTuq DNA polymerase (Perkin-Elmer, Nor- walk, CT) and 25pmol of the appropriate 5’ and 3’ primers. The reaction conditions were 1 min at 95”C, 1 min at 42°C and 1 min at 72°C for 30 cycles, followed by a final extension at 72’C for 10 min. The resulting PCR products having the expected size (approximately 450 bp) were cloned into the vector pCRI1 (Invitrogen, San Diego, CA) using a T/A cloning kit (Invitrogen). DNA sequence analysis of cloned DNA fragments was performed by the dideoxy chain-termination method using double-stranded plasmid DNA as a template. Unique heavy chain and light chain variable regions were isolated by this procedure. Clones obtained from inde- pendent PCR reactions were sequenced to detect any mutations introduced during the PCR reactions.
The N19-8 scFv was constructed by using overlapping PCR with the following primers: UDEC366 CGGAATTCCATGGCCAATATTGTGCTGACCCA ATCTCC; UDEC367 CCCCCGCCACCCGACCC GCCACCTCCGGTCCGTTTTATTTCCAGCTTGG;
UDEC368 GGCGGGTCGGGTGGCGGGGGATCG GGTGGCGGAGGGTCGGACGTCAAGCTCGTGG AGTCTGGGGG; UDEC369 CCCGGATCCTACT CGAGTGAGGAGACGGTGACCGTGG.
The PCR reaction conditions were 1 x buffer (10 mM Tris-HCl, pH 8.3 (at 25°C) 50 mM KCl, 1.5 mM MgCl,, 200 PM each dNTP) containing 20 ng N 19-8 VH plasmid and 20 ng N19-8 VL plasmid as templates, 50 pmol each UDEC366 and UDEC369, and 0.5 pmol each UDEC367 and UDEC368. The reaction was subjected to 30 ampli- fication cycles, each cycle consisting of 1 min at 95°C 1 min at 5O’C, and 1 min at 72”C, followed by a final 10 min incubation at 72°C. The resulting PCR products having the expected size (approximately 780 bp) were cloned into the vector pCRI1.
Production andpurzjication of N19-8 scFz
For bacterial expression of N19-8 scFv, a fragment containing the correct N19-8 scFv sequence was sub- cloned into the bacteriophage T7 promoter vector PET- 14b (Novagen, Madison, WI) using NcoI and BamHI sites flanking the N19-8 scFv. The subsequent plasmid was used to transform E. coli strain ME2. This strain is a phage resistant derivative of the E. cofi strain W3110 lysogenized with IDE3 using a 1DE3 lysogenization kit (Novagen). This prophage contains the gene for the phage T7 polymerase behind the E. coli lac UV5 pro- moter (Studier et al.. 1990). The resulting strain express- ing N19-8 scFv was grown at 37°C in 2 1 Applikon glass vessel fermentors containing Terrific Broth [1.2% (w/v) bacto-tryptone, 2.4% (w/v) bacto-yeast extract, 0.4% (v/v) glycerol, 90mM KP04. pH 7.01 supplemented with 100 pg/ml ampicillin. The production of recombinant scFv was induced by the addition of 1 mM isopropylthio- B-D-galactoside when the OD,, of the culture reached 10. After an additional 3 hr incubation at 37°C the cells were harvested by centrifugation and the cell pellets stored at - 80°C. Cells were resuspended in 1 mM EDTA, pH 5.0 and lysed using a microfluidizer M 11 O-T (Microfluidics Corp., Newton, MA). An insoluble frac- tion was prepared by centrifugation of the lysate. N 19-8 scFv was solubilized by resuspension in 20 mM Tris, 8 M urea, pH 8.0 using a Tekmar homogenizer. The resulting suspension was incubated with stirring for 1 hr at 4°C and treated with the flocculating agent BPA-1000 (4 /d/ml, Tosohaas, Montgomeryville, PA) to remove DNA and contaminating acidic proteins. Insoluble material was removed by centrifugation at 11 ,OOOg for 25 min. After filtration through a 0.45 ,um Whatman polycap AS filter unit. 50 nM CuSO, was added and the N19-8 scFv was allowed to refold at 4°C for approximately 18 hr. The refolding mixture was exchanged into 20mM Tris-HCl, pH 8.0 by ultrafiltration and diafiltration using a spiral wound ultrafiltration cartridge (Amicon). Purification of properly refolded N19-8 scFv was achieved using Q sepharose (Pharmacia, Piscataway, NJ) chromatography in 20mM Tris-HCl, pH 8.0, followed by S-200 (Phar- macia) chromatography in phosphate-buffered saline (PBS).

1186 M. J. EVANS et al.
For mammalian cell production of the Nl9-8 scFv, the Nl9-8 light chain secretory leader sequence was cloned in front of the Nl9-8 scFv. This cassette was then cloned into the expression plasmid pAPEX-3P and stable 293- EBNA transfectants were selected using 1 pg/ml puro- mycin as previously described (Evans et al., 1995). Trans- fected cells were grown to confluence and refed serum- free EX-CELL 300 medium (JRH Biosciences, Lenexa, KS) supplemented with 1 pg/ml puromycin every 3 or 4 days. The conditioned medium was collected and con- centrated 20-fold using a stirred cell concentrator (Amicon, Beverly, MA) fitted with a YMIO membrane. The concentrated medium was adjusted to 20 mM Tris- HCI, 500mM NaCl, 5 mM imidazole, pH 8.0 (binding buffer) and incubated with Ni*+-nitriloacetic acid resin (Qiagen, Chastsworth, CA) at 4°C for 2 hr. The resin was pelleted, washed twice with binding buffer, and poured into a column. The column was washed with 25 column volumes binding buffer, 25 column volumes wash buffer (20 mM Tris-HCI, 500 mM NaCl, 25 mM imidazole, pH 8.0), and eluted with three column volumes elution buffer (20 mM Tris-HCI, 500mM NaCl, 1 M imidazole, pH 8.0). The eluted protein was further concentrated IO-fold using a Centriprep-10 (Amicon), dialysed into PBS, and stored at 4°C.
Hemolytic assay
Nl9-8 mAb or N19-8 scFv were preincubated with 5% human serum in 0.1 ml Verona1 buffered saline containing 1% gelatin (GVBS+‘) for 30min at room temperature. Chicken erythrocytes (Lampire Biological, Pipersville, PA) were washed four times with GVBS++ and re- suspended at 5 x 107cells/ml in GVBS++ containing 1 pg/ml anti-chicken erythrocyte IgG (Intercell Tech- nologies, Hopewell, NJ). After incubation at 4°C for 15 min the cells were washed twice with GVBS+ + and 3 x IO6 cells were added to the preincubated human serum. After further incubation for 30min at 37°C. the cells were pelleted by centrifugation at 10,OOOg for 2 min and the supernatant was assayed for hemoglobin release by spectrophotometry (Aal,) and for C5a content by ELISA as previously described (Wurzner et al., 1991). Hemolytic assays of serum obtained from rhesus mon- keys were performed identically except that the rhesus serum was diluted to 2.5% final concentration.
Isovolemic mouse Langendorff hearts
Hearts were harvested from ether-anesthetized female mice and immediately placed in ice cold Krebs-Henseleit buffer (KHB; 118 mM NaCl, 4.7 mM KCI, 25 mM NaHCO,, 1.2mM MgS04, 2.5 mM CaCl,, 1.2mM KH2P04, 11 mM glucose, 0.5 mM EDTA and 10 mM HEPES, pH 7.4). A blunted 20gauge needle dripping cold KHB was inserted into the aorta, advanced toward the coronary artery ostia and tied in place. The heart was then continuously flushed with ice cold KHB at a rate of approximately 1 ml/min while a water-filled polyethylene balloon, heat-formed at the end of PE-IO tubing, was
inserted into the left ventricle via the left atria1 appendage and tied securely in place.
The instrumented heart was transferred to a recir- culating perfusion apparatus. The heart chamber and buffer reservoir were submerged in the bath of a Lauda circulator set at 37°C. Buffer was drawn from the res- ervoir with a Manostat peristaltic pump, and passed through a gauze filter, a 37°C heat exchanger, and a bubble trap before entering the heart. Perfusate effluent was funneled to the base of the heart chamber for periodic sampling, and was returned to the buffer reservoir with a second Manostat peristaltic pump. Buffer in the reservoir was continuously superfused with 95% 0,/5O/, CO2 at approximately 5 cm water positive pressure.
During a 1 O-20 min setup/stabilization period each heart was perfused with 30 ml KHB at a rate of 3-4 ml per min, with the exact rate adjusted to yield an initial perfusion pressure of approximately 65 mmHg. Similarly, the left ventricular balloon volume was adjusted with water to establish an initial end diastolic balloon pressure of 12-I 5 mmHg. At t =O. 2 ml normal human plasma (preincubated with N19-8 mAb or N19-8 scFv at 4°C as indicated) was added to the buffer reservoir. Perfusion pressure and left ventricular pressure were monitored with Microswitch piezoresistive pressure transducers (156PClSGWL) positioned at heart height. The trans- ducers were connected to MB38 strain gauge modules on a MB-01 signal conditioning board, with the output routed to a DAS-1602 A/D card (Keithley Metrabyte) in a 66 MHz 486 VESA PC clone. Data were acquired using an adaptation of Labtech Notebook software. Left ven- tricular balloon pressure was sampled at 96 Hz for calcu- lation of heart rate. Perfusion pressure, left ventricular end diastolic and systolic pressures, and heart rate were saved to disk at 1 Hz for subsequent display and analysis. Data were collected from hearts for up to 1 hr after the addition of serum.
Immunohistochemistry
At the conclusion of the heart perfusion, a coronal slice from the middle of the heart was embedded in O.C.T. compound (Miles, Elkhart, IN) and frozen on dry ice. Five micron sections were transferred to poly-L-lysine coated slides and fixed with acetone. After air drying, the slides were rinsed once with PBS containing 2% FCS (PBS/FCS) and incubated for 30min at room tem- perature in PBS containing 10% FCS. The slides were rinsed with PBS/FCS, incubated with mouse anti-human C5b-9 (clone HCC5b.l; IBL Research Products Corp.) for 1 hr at room temperature, rinsed again, and incubated in PBS/FCS containing rhodamine-labeled goat anti- mouse IgG (AMAC) for 30min. The slides were again rinsed, and incubated in PBS/FCS containing fluorescein isothiocyanate-labeled goat anti-human C3 (Cappel Research Products, Durham, NC). After a final rinse, the slides were mounted and labelled sections were photo- graphed with a Nikon Diaphot inverted fluorescence microscope.

Single-chain Fv fragment recognizing human C.5 1187
Rhesus pharmacokinetics
Three rhesus monkeys (Maccaca mufatta) weighing 12- 16 kg were administered either N19-8 scFv or N19-8 mAb. In the first monkey, anesthesia was induced by the intravenous administration of 20 mg/kg sodium pen- tothal via a left forearm vein and maintained using 4% halothane. Nl9-8 scFv (100 mg in 40 ml PBS) was infused intravenously over approximately 2 min, and blood sam- ples were drawn through a right femoral artery line immediately prior to infusion and at 1, 3, 5, 10, 15, 30, 60 and 120min following infusion. A second monkey was anesthetized using intermittent boluses of sodium pentothal and received 100 mg N19-8 scFv in 100 ml PBS administered intravenously over 2 min. Venous blood samples were obtained immediately prior to infusion and at 15, 30, 60 and 120min post-infusion. Additional venous blood samples were obtained at 4,8,24 and 48 hr after infusion using ketamine to sedate the monkey. The third monkey was treated identically to the second monkey, except the infusion consisted of 100mg N19-8 mAb in 85ml PBS. The collected blood samples were allowed to clot and the serum was stored at - 80°C.
Immunoblot analysis was performed using a rabbit polyclonal antibody (ALP21) directed against bacterially produced N 19-8 scFv followed by chemiluminescence detection (ECL Western Blotting Detection reagents, Amersham, Arlington Heights, IL). Rhesus serum levels of N19-8 scFv were quantitated by a sandwich ELISA procedure. Briefly. 96-well PolySorp plates (Nunc, Nap- erville, IL) were coated with 2 pg/ml human C5 in 0.1 M carbonate, pH 9.6, blocked with blocking buffer (PBS containing 1% bovine serum albumin, 0.05% Tween-20), and washed several times with PBS + 0.5% Tween-20. Rhesus serum diluted in blocking buffer was added and allowed to bind at 37°C for 60 min. The plates were again washed and incubated with ALP21 in blocking buffer at 37°C for 60 min, washed, and incubated with peroxidase- conjugated goat anti-rabbit antibody. Following a final washing, bound Nl9-8 scFv was detected using o-phen- ylenediamine dihydrochloride as a substrate.
RESULTS
Construction andproduction qf the N19-8 scFv
The variable regions of the monoclonal antibody N19- 8 were cloned by PCR amplification using a family of degenerate 5’ primers coupled with 3’ primers specific for murine constant regions. Amino acid sequence of the amino terminus of the N19-8 light and heavy chains indicated that the appropriate variable regions had been cloned. These variable regions were used to construct a single chain Fv molecule containing the N 19-8 light chain variable region joined to the N 19-8 heavy chain variable region by a 17 amino acid linker (Fig. 1). Six histidine residues were added at the carboxyl terminus of the scFv to facilitate nickel affinity purification of the scFv as previously described (Lindner et al., 1992).
To determine if the N19-8 scFv retained the ability to bind human C5, the N19-8 scFv was initially produced
in a mammalian secretion system. The N19-8 scFv was directed to the secretory pathway by placing the Nl9-8 light chain leader sequence at the amino terminus of the N19-8 scFv. This version of the N19-8 scFv was cloned into the mammalian expression vector pAPEX-3P (Evans et al., 1995) and transfected into 293-EBNA cells. The secreted N19-8 scFv was purified from conditioned serum-free medium by utilizing nickel affinity chro- matography. SDS-PAGE analysis of the purified material revealed a single major band of the predicted molecular weight (27.6 kDa) for the processed N19-8 scFv (Fig. 2, lanes 2 and 5). The N19-8 scFv migrated slightly faster under nonreducing conditions (lane 2) as compared to reducing conditions (lane 5), indicating that scFv contained disulfide bonds resulting in a more com- pact structure. To verify that the mammalian-produced N19-8 scFv retained the ability to bind to C5, human C5 was incubated with plate-coated N19-8 scFv and bound human C5 was detected using a second monoclonal anti- body (Fig. 3). Wells coated with albumin or with N19-8 scFv alone produced a basal absorbance Ievei. Albumin coated wells incubated with human CS showed similar low values, whereas N19-8 scFv-coated wells incubated with human C5 were strongly positive, indicating that Nl9-8 scFv retained the ability to bind human C5.
Initial attempts to produce the N19-8 scFv protein in E. coli using a periplasm secretion system resulted in low yields of properly processed N19-8 scFv (data not shown). Therefore, large-scale production of the N19-8 scFv was performed using an E. co/i intracellular expression system. E. coli strain ME2 was transformed with the expression plasmid PET-I 4b containing the N19- 8 scFv under control of a bacteriophage T7 RNA poly- merase promoter. A single purified clone expressing N 19- 8 scFv was grown at 37°C in a 2 1 Applikon glass vessel fermentor to an OD,, of approximately 10 and T7 RNA polymerase expression was induced by the addition of isopropyl+D-galactoside. Three hours following induc- tion, the cells were harvested and inclusion bodies were prepared by cell lysis and centrifugation. Nl9-8 scFv was solubilized in 8 M urea, allowed to refold at 4°C for 24 hr, and diafiltered into 20mM Tris, pH 8.0. SDS-PAGE analysis of the resulting material under reducing con- ditions revealed a single major band of the predicted molecular weight (27.7 kDa), which comigrated with N19-8 scFv produced in 293-EBNA cells. In contrast, under nonreducing conditions the refolded bacterial N 19-8 scFv preparation contained several strong bands. only one of which comigrated with the N19-8 scFv pro- duced in 293-EBNA cells. We reasoned that properly refolded scFv should comigrate under nonreducing conditions with the mammalian produced N19-8 scFv. This species of N19-8 scFv was purified by Q Sepharose chromatography followed by S-200 chromatography. The resulting purified material contained a single pre- dominant band which comigrated under either reducing or non-reducing conditions with the N19-8 scFv pro- duced in 293-EBNA cells (Fig. 2). Approximately 300 mg total N 19-8 scFv and 25 mg purified, refolded N 19-8 scFv were obtained from 80 g of starting cell paste.

1188 M. J. EVANS et al.
atg gee m a
AAT ATT GTG CTG ACC CAA TCT CCA GCT TCT TTG GCT GTG TCT CTA GGG CAG AGG NIVLTQSPASLAVSLGQR
GCC ACC A T
ATA TCC TGC AGA GCC AGT GAA AGT GTT GAT AGT TAT GAC AAT AGT TTT ATG CAC I S C(R A S E S V D S Y D N S F M HI
CDR-Ll
TGG TAC w Y
CAG CAG AAA CCA GGA CAG CCA CCC AI!& CTC CTC ATC TTT CT-l' GCA Tee AAC CTA Q Q K P G Q P P K L L I FIL A S N L
CDR-L2
GAATCT GGG GTC CCT GCC AGG TTC AGT GGC AGT GGG TCT AGG ACA GAC TTC ACC CTC ACC EGVPARFSGSGSRTDFTLT
ATT GAT CCT GTG GAG GCT GAT GAT GCT GCA ACC TAT TAC TGT CAG CAA AAT AAT GAG GTT
IDPVEADDAATYYCIQQNNEV CDR-L3
CCG AAC ACG TTC GGA GGG GGG ACC AAG CTG GAA ATA AAA egg act gga ggt ggc ggg tcg PNTlFGGGTKLEIKrtggggs
ggt ggc ggg gga tcg ggt ggc gga ggg tcg GAC GTC AAG CTC GTG GAG TCT GGG GGA GAC
4 g g g s g g g g s DVKLVESGGD
TTA GTG AAG CTT GGA GGG TCC CTG AAA CTC TCC TGT GCA GCC TCT GGA TTC ACC TTC AGT LVKLGGSLKLSCAASGFT F(S
AGC TAT TAT ATG TCT TGG GTT CGC CAG ATT TCA GAG AAG AGG CTG GAG TTG GTC GCA GCC S Y Y M S[W V R Q I S E K R L E L V AIA
CDR-Hl ATT AAT AGT AAT GGT GAT AGC ACC TAC TAT CCA GAC ACT GTG AAG GGC CGA TI’C ACC ATC
INSNGDSTYYPDTVIKGRFTI CDR-H2
TCC AGA GAC AAT GCC AAG AGC ACC CTG GAT CT’G CAA ATG AGC AGT Cl-G AAG TCT GAG GAC SRDNAKSTLDLQMSSLKSED
ACA GCC TTG TAT TTC TGT GTA AGA GAG ACT TAT TAC TAC GGG ATT AGT CCC GTC TTC GAT TALYFCVIRETYYYGISPVFD
CDR-H3
GTC TGG GGC ACA GGG ACC ACG GTC Act GTC Tee TCA ctc gag cat cat cat cat cat ca= XWGTGTTVTVSSlehhhhhh
Fig. 1. Nucleotide and predicted amino acid sequence of the N19-8 scFv. Upper case letters denote the light and heavy variable regions obtained from the hybridoma N19-8, with lower case letters denoting the linker used to join the light and heavy variable regions together and the histidine tag at the carboxyl end of the single chain. Amino acid sequences obtained from the N19-8 mAb are underlined, and the CDR sequences according to the hypervariable definition are boxed. This sequence
has been submitted to the GenBank database with the accession number L43067.
inhibition of human complement activity by Nl9-8 scFv
The ability of the bacterial Nl9-8 scFv to inhibit human complement activity was tested in a hemolytic assay. In this assay, chicken erythrocytes sensitized with rabbit antibody were lysed by human serum due to the activation of the classical complement pathway by the rabbit antibody. Addition of N19-8 mAb to the human serum inhibited erythrocyte hemolysis by approximately 80% when the N19-8 mAb was present in four-fold or greater molar excess, as compared to the human serum C5 concentration (Fig. 4). An identical inhibition curve was obtained with the N19-8 scFv, demonstrating that recombinant Nl9-8 scFv maintained the ability to block the formation of the membrane attack complex. Further,
the near identity of the mAb and scFv inhibition curves suggested the Nl9-8 mAb and the N19-8 scFv had similar affinities for human C5.
The ability of the Nl9-8 scFv to inhibit the generation of C5a was assayed by using chicken erythrocytes sen- sitized with rabbit antibody. Supernatants from the hem- olytic reactions were assayed by a sandwich ELBA to determine the amount of C5a generated during the incu- bation of the human serum with the erythrocytes. Addition of Nl9-8 mAb at a four-fold molar excess to the human serum C5 resulted in a 70% inhibition of C5a generation, while a 16-fold molar excess resulted in a 90% inhibition (Fig. 5), values nearly identical to those previously reported (Wurzner et al., 1991). In contrast, the Nl9-8 scFv inhibited C5a generation by only approxi-

Single-chain Fv fragment recognizing human C5 1189
31 -
21.5 - 14.5 - 6.5 -
1 2 3 4 5 6 Fig. 2. SDS-polyacrylamide gel of purified N 19-8 scFv. N 19-8 scFv was secreted by mammalian 293-EBNA cells into serum-
free medium and purifed using nickel affinity chromatograpy. Nl9-8 scFv produced by bacterial ME2 cells was extracted from inclusion bodies, refolded, and purified using Q Sepharose anion exchange chromatography followed by Sephacryl S-200 size-exclusion chromatography. 2Opg of purified N19-8 scFv obtained from mammalian cells (lanes 2 and 5) or from bacterial
cells (lanes 3 and 6) was subjected to SDS-polyacrylamide gel electrophoresis under nonreducing (lanes 1,2.3) or reducing (lanes 4.5.6) conditions and stained with Coomassie R-250.
MW, molecular weight markers.
mately 40% at either a four- or 16-fold molar excess. N19-8 scFv produced in 293-EBNA cells gave similar results (data not shown), indicating the diminished ability of the Nl9-8 scFv to inhibit C5a generation was not specific to the bacterially-produced material. Thus the Nl9-8 scFv completely retained the parental antibody’s ability to block membrane attack complex formation, but only partially retained the ability to block C5 cleavage.
human
Albumin N19-8 SCFV
Fig. 3. Binding of N19-8 scFv to human C5. 96well plates coated with either albumin or Nl9-8 scFv were incubated with (0) or without (D) 0.5pg/ml human CS. Bound C5 was detected using a second monoclonal antibody which binds to human C5 at an epitope distinct from the N19-8 mAb. followed by peroxidase-conjugated goat anti-mouse IgG. Quantitation was performed by monitoring hydrolysis of o-phenylene-
diamine dihydrochloride at 490 nm.
E 60-
$ 5 40- P
0 1 I I I I.111, I I I I1111, 8 n I I “q
0.1 1 10 100 Molar Ratio (inhibitor Binding SitesHuman C6)
Fig. 4. Inhibition of lysis of chicken erythrocytes by N19-8 scFv. Five per cent human serum in GVBS++ was preincubated
for 30 min at room temperature with varying amounts of either the Nl9-8 mAb or the Nl9-8 scFv. Chicken erythrocytes pre-
coated with anti-chicken erythrocyte rabbit antibody were added and the incubation was continued for 30min at 31 C.
The degree of complement-mediated erythrocyte lysis was quantified by determination of hemoglobin release (with hem- oglobin release in the absence of inhibitor defined as 100%) and is plotted as a function of the ratio of the molar concentration of inhibitor C5 binding sites to the human C5 molar concentration
present in the assay (assuming a single antibody molecule can bind to and block the function of two molecules of C5 sim- ultaneously). Data are the average of two independent exper- iments, with each value determined in duplicate in each
experiment.
Protection of murine hearts from human complement bt, Nl9-8 SCFU
To assess the ability of the Nl9-8 scFv to protect an organ from human complement attack, complement- dependent myocardial injury was induced in an isolated mouse heart by perfusion with Krebs-Henseleit buffer containing 6% human plasma. Homeister et al. have previously demonstrated that in a similar system in which
n N19-8 mAb
[3 N19-6 SCFV
No Inhibitor
0.26 4 16
Molar Ratio (Inhibitor Binding Sites:Human C5)
Fig. 5. C5a generation in the presence of Nl9-8 scFv. Hemolytic assays using 10% human serum and the indicated ratio of inhibitor to human C5 were performed as described in Fig. 3. The supernatants were assayed for human C5a content by a sandwich ELISA. A representative experiment of several per- formed is shown, with 12.5 ng/ml human CSa generated in the
absence of inhibitor in this experiment.
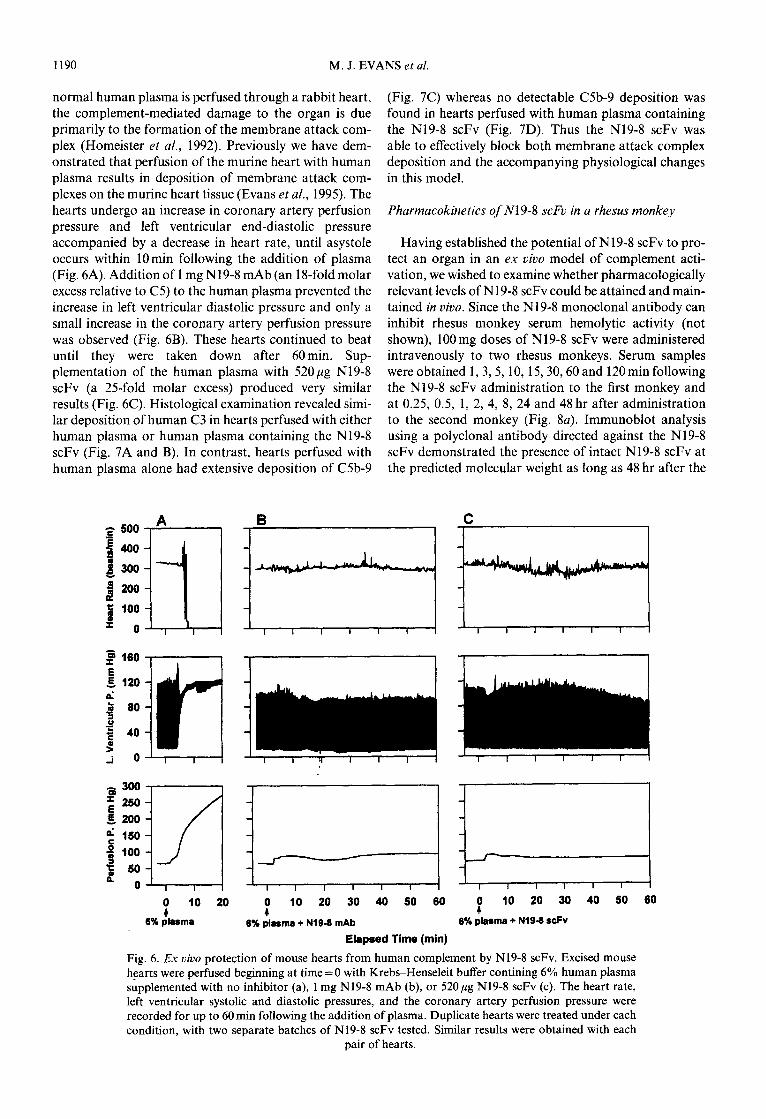
1190 M. J. EVANS et al.
normal human plasma is perfused through a rabbit heart, (Fig. 7C) whereas no detectable C5b-9 deposition was the complement-mediated damage to the organ is due found in hearts perfused with human plasma containing primarily to the formation of the membrane attack com- the N19-8 scFv (Fig. 7D). Thus the N19-8 scFv was plex (Homeister et al., 1992). Previously we have dem- able to effectively block both membrane attack complex onstrated that perfusion of the murine heart with human deposition and the accompanying physiological changes plasma results in deposition of membrane attack corn- in this model. plexes on the murine heart tissue (Evans et al., 1995). The hearts undergo an increase in coronary artery perfusion Pharmacokinetics of N19-8 scFv in a rhesus monkey pressure and left ventricular end-diastolic pressure accompanied by a decrease in heart rate, until asystole Having established the potential of N19-8 scFv to pro- occurs within 10min following the addition of plasma tect an organ in an ex vivo model of complement acti- (Fig. 6A). Addition of I mg N19-8 mAb (an 18-fold molar vation, we wished to examine whether pharmacologically excess relative to C5) to the human plasma prevented the relevant levels of N19-8 scFv could be attained and main- increase in left ventricular diastolic pressure and only a tained in vivo. Since the N19-8 monoclonal antibody can small increase in the coronary artery perfusion pressure inhibit rhesus monkey serum hemolytic activity (not was observed (Fig. 6B). These hearts continued to beat shown), 100 mg doses of N19-8 scFv were administered until they were taken down after 60min. Sup- intravenously to two rhesus monkeys. Serum samples plementation of the human plasma with 520 pg N19-8 were obtained 1,3,5, 10, 15,30,60 and 120min following scFv (a 25-fold molar excess) produced very similar the N19-8 scFv administration to the first monkey and results (Fig. 6C). Histological examination revealed simi- at 0.25, 0.5, 1, 2, 4, 8, 24 and 48 hr after administration lar deposition of human C3 in hearts perfused with either to the second monkey (Fig. 8~). Immunoblot analysis human plasma or human plasma containing the N19-8 using a polyclonal antibody directed against the N19-8 scFv (Fig. 7A and B). In contrast, hearts perfused with scFv demonstrated the presence of intact N19-8 scFv at human plasma alone had extensive deposition of C5b-9 the predicted molecular weight as long as 48 hr after the
p 500 A B
j 400
1 300
J 200
5 100
p 0 ITI:--
9 160
B 120 d
$ 60
z 2 40
f i 0
300
~~-_
s
; 260
% 200
$ 160
+ 100
3 L
50
0 Fl&l~ t 10 20 t 10 20 30 40 50 60
8% plasma 6% plasma + Nl9-9 mAb
Elapsed lime (min)
I I I I I I
P 10 20 30 40 60 60
6% plasma + NIB-9 scFv
Fig. 6. Ex vivo protection of mouse hearts from human complement by N19-8 scFv. Excised mouse hearts were perfused beginning at time = 0 with Krebs-Henseleit buffer contining 6% human plasma supplemented with no inhibitor (a), 1 mg N19-8 mAb (b), or 52Opg N19-8 scFv (c). The heart rate, left ventricular systolic and diastolic pressures, and the coronary artery perfusion pressure were recorded for up to 60 min following the addition of plasma. Duplicate hearts were treated under each condition, with two separate batches of N19-8 scFv tested. Similar results were obtained with each
pair of hearts.

Single-chain Fv fragment recognizing human C5 1191
Fig. 7. C3 and C5b-9 deposition in mouse hearts treated with 6% human plasma. Mouse hearts were perfused as described in Fig. 6 with 6% human plasma (A,C) or 6% human plasma supplemented with 52Opg N19-8 scFv (B,D) for 20 or 60min, respectively. At the conclusion of the perfusion period, coronal slices from the middle of each heart were embedded in O.C.T. compound and frozen. Panels a and c show immunohistochemical detection of C3 and CSb-9 deposition. respectively. in a mouse heart treated with 6% human plasma. Panels B and D show C3 and C5b-9 staining, respec- tively, in a mouse heart treated with human plasma containing 520 klg N19-8 scFv. Magnification is
at 200 x .
infusion, with no degradation products detected through- out the time course examined. A higher molecular weight
cross-reacting band was present in all samples including the preinfusion time point. The serum concentration of
functional N19-8 scFv present at each time point was assayed by a sandwich ELISA using human CS as the
capture molecule and is reported under the respective time points (Fig. EA). The ability of the rhesus serum to
lyse sensitized chicken erythrocytes was also determined at each time point (Fig. EB). The concentrations of N19- 8 scFv present at 15 or 30 min following administration were sufficient to inhibit rhesus serum lytic activity by 80%, while approximately 50% inhibition of lytic activity
was observed at 1 or 2 hr following administration. Assuming the concentration of C5 in rhesus serum is equivalent to that in human serum (0.4pM), a con- centration of 260 lg/ml N19-8 scFv corresponds to a 23-
fold molar excess and would be expected to effectively inhibit complement activity (Fig. 4). In contrast, a serum concentration of 20lg/ml N19-8 scFv corresponds to
only a 1.8fold molar excess and would not be predicted to inhibit complement activity (Fig. 4).
A semilogarithmic plot of the averaged values of N19-
8 scFv serum concentration obtained from the two mon- keys revealed a biphasic non-linear curve (Fig. 9). Non-
linear curve fit analysis of these results revealed a biexponential decay curve with T,,zcc of 28min (Fig. 9,
inset) and a T,,,P of 17 hr. The N 19-8 mAb was also
injected into a single rhesus monkey as a 1OOmg bolus and serum concentrations were determined at various times following this infusion (Fig. 9). The phar-
macokinetics of the N19-8 mAb were consistent with a single compartment model with a T,.? of approximately 60 hr.
DISCUSSION
The best characterized complement inhibitor under development as a pharmacological agent is a recom- binant soluble form of CR1 (sCR1) which has been dem-
onstrated to partially block the development of disease manifestations in several disease models including post- ischemic myocardial damage, skeletal muscle ischemia.
and burn injury (Weisman ei ai., 1990; Mulligan et al., 1992; Pemberton et al., 1993). However, sCR1 has several disadvantages as a complement inhibitor. First, sCR1 is
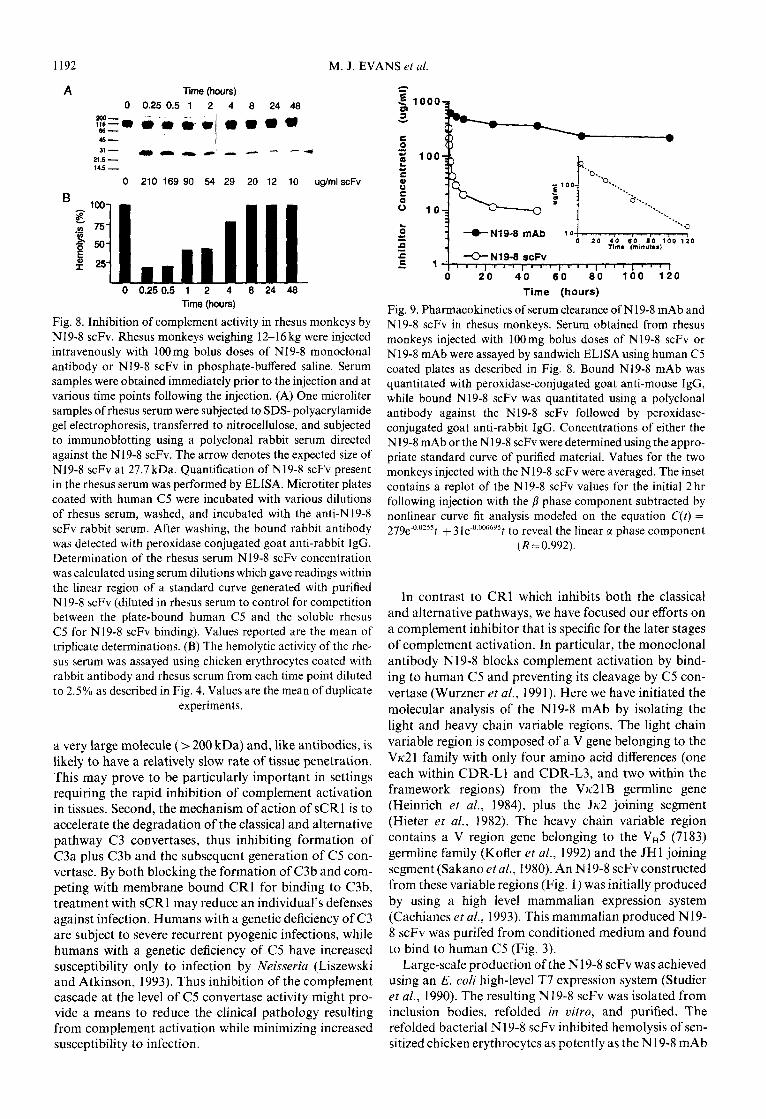
1192 M. J. EVANS et al.
A Time (hours) 0 0.25 0.5 1 2 4 0 2446
pvr,'&&-" l)l)aD(L
45- 31 - r)rr-crr---~
21.5 - 1+5-
0 210 169 90 54 29 20 12 10 ug/ml scFv
0 0.25 0.5 1 2 4 0 24 4% lime (hours) Time (hours)
Fig. 8. Inhibition of complement activity in rhesus monkeys by N19-8 scFv. Rhesus monkeys weighing 12-16 kg were injected intravenously with 100mg bolus doses of N19-8 monoclonal antibody or N19-8 scFv in phosphate-buffered saline. Serum samples were obtained immediately prior to the injection and at various time points following the injection. (A) One microliter samples of rhesus serum were subjected to SDS-polyacrylamide gel electrophoresis, transferred to nitrocellulose, and subjected to immunoblotting using a polyclonal rabbit serum directed against the N19-8 scFv. The arrow denotes the expected size of N19-8 scFv at 27.7 kDa. Quantification of N19-8 scFv present in the rhesus serum was performed by ELISA. Microtiter plates coated with human C5 were incubated with various dilutions of rhesus serum, washed, and incubated with the anti-N19-8 scFv rabbit serum. After washing, the bound rabbit antibody was detected with peroxidase conjugated goat anti-rabbit IgG. Determination of the rhesus serum N19-8 scFv concentration was calculated using serum dilutions which gave readings within the linear region of a standard curve generated with purified N19-8 scFv (diluted in rhesus serum to control for competition between the plate-bound human C5 and the soluble rhesus C5 for N 19-8 scFv binding). Values reported are the mean of triplicate determinations. (B) The hemolytic activity of the rhe- sus serum was assayed using chicken erythrocytes coated with rabbit antibody and rhesus serum from each time point diluted to 2.5% as described in Fig. 4. Values are the mean of duplicate
experiments.
a very large molecule (> 200 kDa) and, like antibodies, is likely to have a relatively slow rate of tissue penetration. This may prove to be particularly important in settings requiring the rapid inhibition of complement activation in tissues. Second, the mechanism of action of sCR1 is to accelerate the degradation of the classical and alternative pathway C3 convertases, thus inhibiting formation of C3a plus C3b and the subsequent generation of CS con- vertase. By both blocking the formation of C3b and com- peting with membrane bound CR1 for binding to C3b, treatment with sCR1 may reduce an individual’s defenses against infection. Humans with a genetic deficiency of C3 are subject to severe recurrent pyogenic infections, while humans with a genetic deficiency of C5 have increased susceptibility only to infection by Neisseria (Liszewski and Atkinson, 1993). Thus inhibition of the complement cascade at the level of C5 convertase activity might pro- vide a means to reduce the clinical pathology resulting from complement activation while minimizing increased susceptibility to infection.
4
0 20 40 60 00 100 120
Fig. 9. Pharmacokinetics of serum clearance of N19-8 mAb and N19-8 scFv in rhesus monkeys. Serum obtained from rhesus monkeys injected with 100mg bolus doses of N19-8 scFv or N19-8 mAb were assayed by sandwich ELISA using human C5 coated plates as described in Fig. 8. Bound Nl9-8 mAb was quantitated with peroxidase-conjugated goat anti-mouse IgG, while bound N19-8 scFv was quantitated using a polyclonal antibody against the N19-8 scFv followed by peroxidase- conjugated goat anti-rabbit IgG. Concentrations of either the N19-8 mAb or the N19-8 scFv were determined using the appro- priate standard curve of purified material. Values for the two monkeys injected with the N19-8 scFv were averaged. The inset contains a replot of the N19-8 scFv values for the initial 2 hr following injection with the p phase component subtracted by nonlinear curve fit analysis modeled on the equation C(t) = 279e-O_O??St + 3 ~e-0.000695 t to reveal the linear a phase component
(R = 0.992).
In contrast to CR1 which inhibits both the classical and alternative pathways, we have focused our efforts on a complement inhibitor that is specific for the later stages of complement activation. In particular, the monoclonal antibody N19-8 blocks complement activation by bind- ing to human C5 and preventing its cleavage by C5 con- vertase (Wurzner et al., 199 1). Here we have initiated the molecular analysis of the N19-8 mAb by isolating the light and heavy chain variable regions. The light chain variable region is composed of a V gene belonging to the Vlc21 family with only four amino acid differences (one each within CDR-Ll and CDR-L3, and two within the framework regions) from the Vk-21B germline gene (Heinrich et al., 1984). plus the Jlc2 joining segment (Hieter et al., 1982). The heavy chain variable region contains a V region gene belonging to the Vn5 (7183) germline family (Kofler et al., 1992) and the JHl joining segment (Sakano et al., 1980). An N 19-8 scFv constructed from these variable regions (Fig. 1) was initially produced by using a high level mammalian expression system (Cachianes et al., 1993). This mammalian produced N19- 8 scFv was purifed from conditioned medium and found to bind to human C5 (Fig. 3).
Large-scale production of the N 19-8 scFv was achieved using an E. coli high-level T7 expression system (Studier et al., 1990). The resulting N19-8 scFv was isolated from inclusion bodies, refolded in vitro. and purified. The refolded bacterial N 19-8 scFv inhibited hemolysis of sen- sitized chicken erythrocytes as potently as the N19-8 mAb

Single-chain Fv fragment recognizing human C5 1193
(Fig. 4). Both molecules inhibited hemolytic activity by 50% when present at approximately a three-fold molar excess of inhibitor relative to human CS. In contrast, a significant difference between the N19-8 mAb and the N19-8 scFv was apparent in their ability to inhibit C5a generation (Fig. 5). The N19-8 mAb inhibited C5a gen- eration by 70% when present at a four-fold molar excess, and 90% when present at a 16-fold molar excess. The Nl9-8 scFv inhibited C5a generation by only 40% at either a four- or 16-fold molar excess. The hemolytic assay measures generation of the membrane attack com- plex by classical pathway generation of C5b and can be inhibited by antibodies to other components of the membrane attack complex (Wurzner et al., 1991). In con- trast the C5a generation assay measures the ability of the C5 convertase to bind and cleave C5. Previously we have found that Fabs derived from Nl9-8 blocked C5a gener- ation as effectively as the parental antibody (Evans et al., 1995). The N19-8 mAb epitope is located on the CS/? chain (data not shown), while the cleavage of C5 into C5a and C5b occurs on the C50: chain. The N19-8 mAb and Fab may thus not bind to the precise regions of C5 recognized by the classical pathway C5 convertase (C4b2a3b) or the alternative pathway C5 convertase (C3bBb3b), but rather they may block interaction of the convertases with C5 through a steric hindrance mech- anism. The size of the scFv (28 kDa) may be too small to efficiently block cleavage of C5 by either C5 convertase. Alternatively, the N19-8 scFv may inhibit only one of the two C5 convertases. However, the Nl9-8 scFv must remain bound to C5b and block its interaction with C6 since the scFv prevented formation of the membrane attack complex as effectively as the mAb. Finally, these properties of the Nl9-8 scFv are specific for the Nl9-8 epitope, as we have recently created an scFv which recog- nizes a different epitope on C5 and is capable of very effectively blocking C5a generation and hemolytic activity (M.J.E. and S.A.R., unpublished observations).
The ability of N19-8 scFv to protect an organ from complement-mediated damage was assessed with a hang- ing heart model in which isolated mouse hearts were perfused with Krebs-Henseleit buffer containing 6% human plasma. In rabbit hearts the resulting organ dam- age is dependent on C8, suggesting that membrane attack complex formation is the major mechanism of tissue dam- age in this model (Homeister et al., 1992). The membrane bound complement inhibitors decay accelerating factor, membrane cofactor protein, and CD59 are all restricted in their action to complement of the homologous species (Lachmann, 1991), so the murine inhibitors present on the murine vascular endothelium are unable to inhibit the formation of the membrane attack complex by human complement. This model thus serves as a stringent test of the ability of a soluble molecule to inhibit membrane attack complex formation. Functional changes in the cardiac status of the murine heart resulting from acti- vation of the human plasma complement included increased end-diastolic pressure, increased coronary per- fusion pressure. and reduced heart rate until the hearts ceased beating approximately 10 min after the addition
of human plasma (Fig. 6A). Extensive deposition of C3 and C5b-9 was found in the heart tissue (Fig. 7). When the human plasma was supplemented with either the N19-8 mAb (Fig. 6B) or the N19-8 scFv (Fig. 6C), the mouse hearts continued to function with no detectable decrease in heart rate, increase in left ventricular diastolic pressure, or increase in coronary artery perfusion pres- sure for at least 60 min. C3 deposition was evident in the hearts treated with human plasma containing the N19-8 scFv. As expected, no CSb-9 deposition was detected in these hearts (Fig. 7).
The ability of Nl9-8 scFv and Nl9-8 mAb to inhibit complement in vivo was assessed in rhesus monkeys. Rhe- sus serum hemolytic activity was inhibited by greater than 50% for up to 2 hr following the administration of a 100 mg dose of Nl9-8 scFv (Fig. 8) and for at least 72 hr following the administration of a 100 mg dose of N19-8 mAb. Pharmacokinetic analysis suggested a two- phase serum clearance curve, with a T,!*c~ of 28 min and T&3 of 17 hr (Fig. 9). Another scFv, the CC49 scFv, has been injected into rhesus monkeys (Milenic et al., 1991) and was found to have an extremely rapid plasma clear- ance rate (TiIZ~ of 3.9min and T&I of 4.2 hr). A sig- nificant difference between the CC49 scFv and the Nl9- 8 scFv is their target antigen, as the CC49 scFv binds to a cell surface protein whereas the N19-8 scFv binds to a serum protein. Further, the clearance of scFv molecules is believed to occur predominantly by renal clearance and several protein parameters including size, hydrophobicity and charge can influence the rate of renal clearance. Although scFv fragments have been reported to dimerize (Griffiths et al., 1993; Kortt et al., 1994), size exclusion chromatagraphy suggested that the N19-8 scFv was pre- dominantly in a monomeric form (not shown) and the extended half-life relative to the CC49 scFv was unlikely to be due to dimerization. The T,!2a value obtained for the N19-8 scFv is similar to that of factor D, a similarly sized (25 kDa) component of the alternative pathway, which has a fractional catabolic rate of 60%/hr (Pascual et al., 1988).
The N19-8 scFv concentrations achieved in the rhesus serum may be pharmacologically efficacious in settings such as reperfusion injury and CPB. The scFv levels achieved in the rhesus serum (approximately a 23-fold molar excess relative to C5) are similar to the levels used in the hanging heart model (a 25fold molar excess). The degree of complement activation in this model may greatly exceed that occuring in the presence of human membrane-bound complement inhibitors and we have not determined the minimal amount of N19-8 scFv which can protect in this model. Extrapolation of the rhesus monkey data suggests short-term inhibition of com- plement activity in humans would require a dose of approximately 500mg, within the range of antibody doses given previously to patients with no adverse effects (Khazaeli et al., 1988). Additionally, the recent devel- opment of scFv antibodies with higher affinity for human C5 may reduce this dose (M.J.E. and S.A.R, unpublished observations).
The period of time necessary for complement inhi-

1194 M. J. EVANS et al.
bition in cardiopulmonary bypass is well defined, essen- tially lasting throughout the time of extracorporeal recirculation until the administration of protamine, gen- erally about 2-4 hr. In contrast, the time period necessary for complement inhibition in cardiac reperfusion injury is less clear. Furthermore, it is likely that inhibition of complement within the cardiac tissue will be important in reperfusion injury, suggesting an advantage for com- plement inhibition using scFv antibodies. It is, however, unclear whether blockade of the complement cascade at the level of C5 can reduce reperfusion injury as achieved by blockade at the level of C3 convertase (Weisman et al., 1990; Smith et al., 1993). Animal studies will be required to determine if the anti-C5 scFv can significantly reduce reperfusion injury in viuo.
Complement-mediated tissue damage also occurs in several autoimmune disorders including rheumatoid arthritis, myasthenia gravis, and immune complex neph- ritis (Morgan, 1994). Treatment of these diseases will require prolonged inhibition of complement activity. Monoclonal antibodies typically have significantly shor- ter half lives in humans than do human antibodies, and result in formation of human antibodies directed against the murine antibody (Khazaeli et al., 1994). The cloning of the N19-8 variable regions will allow the production of a humanized antibody for further in vivo analysis of the potential therapeutic utility of complement inhibitors in multiple disease settings.
Acknowledgements--We would like to thank Nicholas Markel for E. coli fermentation, Tad Thomas for guidance with puri- fication of the bacterial N19-8 scFv, and Jeff Burkwit, Amy Chodera, Stephanie DeCesare, Michelle Giannoni, Sandra Hartman and Eva Setter for technical assistance.
REFERENCES
Biesecker G. and Gomez C. (1989) Inhibition of acute passive transfer experimental autoimmune myasthenia gravis with Fab antibody to complement C6. J. Immun. 142,2654-2659.
Bird R. E., Hardman K. D., Jacobson J. W., Johnson S., Kauf- man B. M., Lee S.-M., Lee T., Pope S. H., Riordan G. S. and Whitlow M. (1988) Single chain antigen-binding proteins. Science 242,423426.
Cachianes G., Ho C., Weber R. F., Williams S. R., Goeddel D. V. and Leung D. W. (1993) Epstein-Barr virus-derived vec- tors for transient and stable expression of recombinant proteins. BioTechniques l&255-259.
Christadoss P. (1988) C5 gene influences the development of murine myasthenia gravis. J. Zmmun. 140, 2589-2592.
Chomczynski P. and Sacchi N. (1987) Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol- chloroform extraction. Analvt. Biochem. 162, 156159.
Cove11 D. G., Barbet J., Holton 0. D., Black C. D. V., Parker R. J. and Weinstein J. N. (1986) Pharmacokinetics of monoclonal immunoglobulin G,, F(ab’),, and Fab’ in mice. Cancer Res. 46,3969-3978.
Curtis W. E., Gillinov A. M., Wilson I. C., Bator J. M., Burch R. M., Cameron D. E. and Gardner T. J. (1993) Inhibition of neutrophil adhesion reduces myocardial infarct size. Ann. thorac. Surg. 56, 106991073
Engel A. G. and Arahata K. (1987) The membrane attack
complex of complement at the endplate in myasthenia gravis. Ann. N. Y. Acad. Sci. 505, 326332.
Evans M. J., Hartman S. L., Wolff D. W., Rollins S. A. and Squint0 S. P. (1995) Rapid expression of an anti-human C5 chimeiric Fab utilizing a vector that replicates in COS and 293 cells. J. immunol. Meth. 184, 123-138.
Gerard C. and Gerard N. P. (1994) C5a anaphyloatoxin and its seven transmembrane-segment receptor. A. Rev. Immun. 12,775-808.
Gillinov, A. M., DeValeria P. A., Winkelstein J. A., Wilson I., Curtis W. E., Shaw D., Yeh C. G., Rudolph A. R., Baum- gartner W. A., Herskowitz A. and Cameron D. E. (1993) Complement inhibition with soluble complement receptor type 1 in cardiopulmonary bypass. Ann. thorac. Surg. 55, 619624.
Griffiths A. D., Malmqvist M., Marks J. D., Bye J. M., Emble- ton M. J., McCafferty J., Baier M., Holliger K. P., Gorick B. D., Hughes-Jones N. C., Hoogenboom H. R. and Winter G. (1993) Human anti-self antibodies with high specificity from phage display libraries. EMBO J. 12, 725-734.
Heinrich G., Traunecker A. and Tonegawa S. (1984) Somatic mutation creates diversity in the major group of mouse immu- noglobulin IC light chains. J. exp. Med. 159,417435.
Hieter P. A., Maize1 J. V. and Leder P. (1982) Evolution of human immunoglobulin IC J region genes. J. bioi. Chem. 257, 1516-1522.
Homeister J. W. and Lucchesi B. R. (1994) Complement acti- vation and inhibition in myocardial ischemia and reperfusion injury. A. Rev. pharmac. Toxicol. 34, 1740.
Homeister J. W., Satoh P. and Lucchesi B. R. (1992) Effects of complement activation in the isolated heart. Role of the terminal complement components. Circ. Res. 71, 303-3 19.
Hugo F., Hamdoch T., Mathey D., Schafer H. and Bhakdi S. (1990) Quantitative measurement of SC5b-9 and C5b-9(m) in infarcted areas of human myocardium. Clin. exp. Zmmun. 81, 132-136.
Huston J. S., Levinson D., Mudgett-Hunmter M., Tai M.-S., Novotyn J., Margolies M. N., Ridge R. J., Bruccoleri R.. Haber E., Crea R. and Oppermann, H. (1988) Protein engin- eering of antibody binding sites: Recovery of specific activity in an anti-digoxin single-chain Fv analogue produced in Escherichia coli. Proc. natn. Acad. Sci. U.S.A. 85,5879-5883.
Khazaeli M. B., Conry R. M. and LoBuglio A. F. (1994) Human immune response to monoclonal antibodies. J. Immunother. 15,42-52.
Khazaeli M. B., Saleh M. N., Wheeler R. H., Huster W. J., Holden H.. Carrano R. and LoBuglio A. F. (1988) Phase I trial of multiple large doses of murine monoclonal antibody C017-1A. II Pharmacokinetics and immune response. J. natn. Cancer Inst. 80,937-942.
Kofler R., Geley S., Kofler H. and Helmberg A. (1992) Mouse variable-region gene families: complexity, polymorphism and use in non-autoimmune responses. Immun. Rev. 128, 5-2 1.
Kortt A. A., Malby R. L., Caldwell J. B., Gruen L. C., Ivanic N., Lawrence M. C., Howlett G. J., Webster R. G.. Hudson P. J. and Colman, P. M. (1994) Recombinant anti-sialidase single-chain variable fragment antibody. Characterization, formation of dimer and higher-molecular-mass multimers and the solution of the crystal structure of the single-chain variable fragment/sialidase complex. Eur. J. Biochem. 221, 151-157.
Lachmann P. J. (1991) The control ofhomologous lysis. Immun.
Today 12,312-315. Lindner P., Guth B., Wulfing C., Krebber C.. Steipe B., Muller
F. and Pluckthun A. (1992) Purification of native proteins

Single-chain Fv fragment recognizing human C5 1195
from the cytoplasm and periplasm of Escherichia coli using IMAC and histidine tails: a comparison of proteins and pro- tocols. Meth. Companion Meth. Enzvm. 4,41-56.
Liszewski M. K. and Atkinson J. P. (1993) The complement system. In Fundamental Immunology (Edited by Paul W. E.), p. 917. Raven Press, New York.
A. (1995) Blockade of C5a and C5b-9 generation inhibits leukocyte and platelet activation during extracorporeal cir- culation. J. clin. Invest. 96, 15641572.
LoBuglio A. F., Wheeler R. H., Trang J., Haynew A., Rogers K., Harvey E. B.. Sun L.. Ghrayeb J. and Khazaeli M. B. (1989) Mouse/human chimeric monoclonal antibody in man: kinetics and immune response. Proc. natn. Acad. Sci. U.S.A.
86,4220-4224.
Sakano H., Maki R., Kurosawa Y., Roeder W. and Tonegawa S. (1980) Two types of somatic recombination are necessary for the generation of complete immunoglobulin heavy-chain genes. Nature 286,676-683.
Milenic D. E., Yokota T., Filpula D. R., Finkelman M. A. J., Dodd S. W., Wood J. F.. Whitlow M., Snoy P. and Schlom J. (I 99 I ) Construction, binding properties, metabolism, and tumor targeting of a single-chain Fv derived from the pan- carcinoma monoclonal antibody CC49. Cancer Res. 51, 6363-6371.
Schafer H., Mathey D., Hugo F. and Bhakdi S. (1986) Depo- sition of the terminal C5b-9 complement complex in infarcted areas of human myocardium. J. Zmmun. 137,1945-1949.
Sears H. F., Herlyn D., Steplewski Z. and Koprowski H. (1984) Effects of monoclonal antibody immunotherapy on patients with gastrointestinal adenocarcinoma. J. biol. Response Modzjiers 3, 138-150.
Miller R. A., Oseroff A. R., Stratte P. T. and Levy R. (1983) Monoclonal antibody therapeutic trials in seven patients with T-cell lymphoma. Blood 62,988-995.
Moat N. E., Macnaughton P. D., Pallares L. C. M., Freeman A., Hibbs M. and Evans T. W. (1992) Complement inhibition may attenuate acute lung injury after cardiopulmonary bypass in pigs. Am. Rev. Respir. Dis. 145, A845.
Moreland L. W., Bucy R. P., Tilden A., Pratt P. W., LoBuglio A. F., Khazaeli M.. Everson M. P., Daddona P., Ghrayeb J., Kilgarriff C., Sanders M. E. and Koopman, W. J. (1993) Use of a chimeric monoclonal anti-CD4 antibody in patients with refractory rheumatoid arthritis. Arthrit. Rheum. 36,307- 318.
Simpson P. J., Todd R. F., Fantone J. C., Mickelson J. K., Griffin J. D. and Lucchesi B. R. (1988) Reduction of exper- imental canine myocardial reperfusion injury by a mono- clonal antibody (anti-Mel, anti-CD1 lb) that inhibits leuko- cyte adhesion. J. clin. Invest. 81, 624-629.
Smith E. F., Griswold D. E., Egan J. W., Hillegass L. M., Smith R. A. G., Hibbs M. J. and Gagnon R. C. (1993) Reduction of myocardial reperfusion injury with human soluble com- plement receptor type 1 (BRL 55730). Eur. J. Pharmac. 236,477-48 1.
Studier F. W.. Rosenberg A. H., Dunn J. J. and Dubendorff J. W. (1990) Use of T7 RNA polymerase to direct expression of cloned genes. Meth. Enzym. 185, 60-89.
Moore F. D., Warner K. G.. Assousa S., Valeri C. R. and Khuri S. F. (1987) The effects of complement activation curing cardiopulmonary bypass. Ann. Surg. 208,95-103.
Morgan B. P. (1990) Complement and infectious diseases. In Complement. Clinical Aspects and Relevance to Disease (Edited by Morgan B.P.), p. 97. Academic Press, New York.
Morgan B. P. (1994) Clinical complementology: recent progress and future trends. Eur. 1. c/in. invest. 24, 219-228.
Mulligan M. S., Yeh C. G., Rudolph A. R. and Ward P. A. (1992) Protective effects of soluble CR1 in complement-and neutrophil-mediated tissue injury. J. Immun. 148, 1479-1485.
Pantoliano M. W., Bird R. E., Johnson S., Asel E. D., Dodd S. W., Wood J. F. and Hardman. K. D. (1991) Conformational stability, folding, and ligand-binding affinity of single-chain Fv immunoglobulin fragments expressed in Escherichia coli. Biochemistry 30, 10117-10125.
Svennevig, J. L., Geiran 0. R., Karlsen H., Pedersen T., Mollnes T. E., Kongsgard U. and Froysaker T. (1993) Com- plement activation during extracorporeal circulation: in vitro comparison of Duraflo II heparin-coated and uncoated oxy- genator circuits. J. thorac. cardiovasc. Surg. 106,466-471.
Weiden P. L., Breitz H. B., Seiler C. A., Bjorn M. J., Ratliff B.A., Mallett R., Beaumier P. L.. Appelbaum J. W., Fritzberg A. R. and Salk, D. (1993) Rhenium-186-labeled chimeric antibody NR-LU-13: pharmacokinetics, biodistribution and immunogenicity relative to murine analog NR-LU-10. J. nucl. Med. 34,2111-2119.
Weisman H. F., Bartow T., Leppo M. K., Marsh H. C., Carson G. R., Concino M. F., Boyle M. P., Roux K. H., Weisfeldt M. L. and Fearon D. T. (1990) Soluble human complement receptor type I : in vivo inhibitor of complement suppressing post-ischemic myocardial inflammation and necrosis. Science 249, 146-151.
Pemberton M., Anderson G., Vetvicka V., Justus D. E. and Ross G. D. (1993) Microvascular effects of complement blockade with soluble recombinant CR1 on ischemia/ reperfusion injury of skeletal muscle. J. Immun. 150, 5104- 5113.
Wurzner R., Schulze M., Happe L., Franzke A., Bieber F. A., Oppermann M. and Gotze 0. (1991) Inhibition of terminal complement complex formation and cell lysis by monoclonal antibodies. Complement Injamm. 8, 328-340.
Yokota T., Milenic D. E., Whitlow M. and Schlom J. (1992) Rapid tumor penetration of a single-chain Fv and com- parison with other immunoglobulin forms. Cancer Res. 52, 3402-3408.
Pascual M., Steiger G., Estreicher J., Macon K., Volanakis J. Zdanov A., Li Y., Bundle D. R., Deng S.-J.. MacKenzie C. R., E. and Schifferli. J. A. (1988) Metabolism of complement Narang S. A.. Young N. M. and Cygler M. (1994) Structure factor D in renal failure. Kidney Int. 34, 529-536. of a single-chain antibody variable domain (Fv) fragment
Rinder C. S., Rinder H. M., Smith B. R., Fitch J. C. K., Smith complexed with a carbohydrate antigen at 1.7-A resolution, M. J., Tracey J. B.. Matis L. A., Squint0 S. P. and Rollins S. Proc. natn. Acad. Sci. U.S.A. 91, 6423-6427.
Copyright © 2022 FDOKUMEN








![Recognizing acute delirium as part of your routine [RADAR]](https://static.fdokumen.com/doc/165x107/632322e9887d24588e0485f5/recognizing-acute-delirium-as-part-of-your-routine-radar.jpg)











