
The Control Reproduction Number and Case-underreporting of Visceral Leishmaniasis in Bihar, India
Upload
independentCategory
view
3download
0
ANTIMICROBIAL AGENTS AND CHEMOTHERAPY, Feb. 2011, p. 659–666 Vol. 55, No. 20066-4804/11/$12.00 doi:10.1128/AAC.00436-10Copyright © 2011, American Society for Microbiology. All Rights Reserved.
In Silico Screening, Structure-Activity Relationship, and BiologicEvaluation of Selective Pteridine Reductase Inhibitors
Targeting Visceral Leishmaniasis�†Jaspreet Kaur,1 Pranav Kumar,1 Sargam Tyagi,3 Richa Pathak,2 Sanjay Batra,2
Prashant Singh,4 and Neeloo Singh1*Drug Target Discovery and Development Division1 and Medicinal and Process Chemistry Division,2 Central Drug Research Institute,
Chattar Manzil Palace, CSIR, P.O. Box No. 173, Lucknow 226001, India; Connexious Life Sciences, Bangalore,India3; and Department of Chemistry, Dayanand Anglo-Vedic (PG) College, Dehradun 248001, India4
Received 1 April 2010/Returned for modification 5 July 2010/Accepted 19 November 2010
In this study we utilized the concept of rational drug design to identify novel compounds with optimalselectivity, efficacy and safety, which would bind to the target enzyme pteridine reductase 1 (PTR1) inLeishmania parasites. Twelve compounds afforded from Baylis-Hillman chemistry were docked by using theQUANTUM program into the active site of Leishmania donovani PTR1 homology model. The biological activityfor these compounds was estimated in green fluorescent protein-transfected L. donovani promastigotes, and themost potential analogue was further investigated in intracellular amastigotes. Structure-activity relationshipbased on homology model drawn on our recombinant enzyme was substantiated by recombinant enzymeinhibition assay and growth of the cell culture. Flow cytometry results indicated that 7-(4-chlorobenzyl)-3-methyl-4-(4-trifluoromethyl-phenyl)-3,4,6,7,8,9-hexahydro-pyrimido[1,2-a]pyrimidin-2-one (compound 7) was10 times more active on L. donovani amastigotes (50% inhibitory concentration [IC50] � 3 �M) than onpromastigotes (IC50 � 29 �M). Compound 7 exhibited a Ki value of 0.72 �M in a recombinant enzymeinhibition assay. We discovered that novel pyrimido[1,2-a]pyrimidin-2-one systems generated from the allylamines afforded from the Baylis-Hillman acetates could have potential as a valuable pharmacological toolagainst the neglected disease visceral leishmaniasis.
Visceral leishmaniasis (VL or kala-azar) is the most devas-tating form of leishmaniasis and is caused by the invasion ofthe reticuloendothelial system (spleen, liver, and bone mar-row) by the hemoflagellate protozoan parasite Leishmania do-novani. Approximately 500,000 new cases of VL arise annuallyworldwide. More than 90% of VL cases occur in India, Bang-ladesh, Nepal, Sudan, Brazil, and Ethiopia (4). In India, highincidence has been reported from the states of Bihar, Assam,West Bengal, and Eastern Uttar Pradesh, where the incidenceof resistance and relapse is increasing. In the absence of asuitable vaccine, chemotherapy remains the only tool to treatthis parasitic infection. Chemotherapy for Leishmania includespentavalent antimonials, amphotericin B, miltefosine, andparomomycin. Disappointingly, however, all clinical agents inuse suffer from side effects which include toxicity, resistance,partial effectiveness, and high cost (17, 24). The present situ-ation, therefore, necessitates the need for discovery and devel-opment of safe, effective, and affordable drugs against VL.
The biochemical pathways present in trypanosomatidsand absent from their mammalian host provide excellentunique targets for rational drug design. The sequencing of
Leishmania genome has led to the postgenomic era for drugdiscovery. With the whole-genome sequencing of L. dono-vani clinical isolates under way in our laboratory (http://www.leishmaniaresearchsociety.org), the molecular-target-driven approach to antileishmanial drug discovery will befurther strengthened. The Leishmania enzyme pteridine re-ductase 1 (PTR1) is one such validated drug target (18). Themain function of PTR1 is the reduction of biopterin, but it alsoreduces folates and provides metabolic bypass to dihydrofolatereductase-thymidylate synthase (DHFR-TS) enzyme of Leish-mania parasite. Therefore, an inhibitor with good activity tar-geting both enzymes viz. DHFR-TS and PTR1 is a rational wayof inhibiting the folate pathway (19). Furthermore, there isrecycling of reduced pterins in Leishmania via the pterin-4�-carbinolamine dehydratase (PCD). Therefore, this regenera-tion pathway of reduced pterins (15) can also be a target forintervention.
We studied PTR1 (GenBank accession no. AY547305) froman L. donovani clinical isolate as a potential drug target. Basedon a three-dimensional model drawn on recombinant PTR1isolated from a clinical isolate of L. donovani in our laboratory(12), we carried out molecular modeling and docking studieswith annulated-pyrimidinone systems afforded from Baylis-Hillman chemistry. These scaffolds incorporate the guanidinegroup in a rigid framework. Several guanidine-based com-pounds are known to display antiparasitic activity, which isbelieved to be via their interactions with the folate pathways. Ithas been shown that aromatic adducts of Baylis-Hillman reac-tion were shown to be active against malarial parasites (6, 14)and Leishmania parasites (1, 5), which prompted us to evaluate
* Corresponding author. Mailing address: Drug Target Discoveryand Development Division, Central Drug Research Institute, ChattarManzil Palace, P.O. Box No. 173, Lucknow 226001, India. Phone:91-941-5002065. Fax: 91-522-22623405. E-mail: [email protected].
† Supplemental material for this article may be found at http://aac.asm.org/.
� Published ahead of print on 29 November 2010.
659

the selective activity of the aromatic Baylis-Hillman derivatives(20) presented in Fig. 1 against L. donovani, the main causalagent of VL. In the present study, 12 Baylis-Hillman deriva-tives were investigated against an in silico target, followed by invitro enzymatic and cell-based assay, resulting in the identifi-cation of a potent inhibitor of PTR1.
MATERIALS AND METHODS
Macrophage culture. The J774A.1 mouse (BALB/c) macrophage cell line wasobtained from National Centre for Cell Science (Pune, India) and used as acellular host for the in vitro intracellular test of antileishmanial activity againstamastigotes. The cells were maintained in RPMI 1640 medium (Gibco-BRL)adjusted to contain 2 g of sodium bicarbonate/liter, 6 g of HEPES/liter, 10%
FIG. 1. Chemical structures of test compounds. The structures of the 12 Baylis-Hillman derivatives from the in-house chemical library ofCentral Drug Research Institute, Lucknow, India, are depicted. Compounds 4, 5, 7, 8, 9, and 10 were the hexahydro pyrimido pyrimidinones, andcompounds 1, 2, 3, 6, 11, and 12 were the substituted allyl amines.
660 KAUR ET AL. ANTIMICROB. AGENTS CHEMOTHER.

(vol/vol) heat-inactivated fetal bovine serum (HI-FBS; Gibco, Germany), and100 U penicillin and 100 �g of streptomycin/ml at 37°C in a humidified atmo-sphere of 95% air and 5% CO2.
Routine L. donovani parasite culture and counting. Green fluorescent protein(GFP)-transfected L. donovani cells were prepared (23) and cultured in medium199 (pH 7.2) (Sigma) supplemented with Hanks salts, 2.05 mM L-glutamine, 12mM HEPES buffer (Sigma), 10% (vol/vol) HI-FBS, 100 U of penicillin/ml, 100�g of streptomycin/ml and additionally in the presence of 150 �g of Geneticinsulfate (G418)/ml. The cells were grown in vented T25 tissue culture flasksand maintained at 25°C. Promastigote cultures were initiated at 106 parasitesper ml and subcultured every 3 to 4 days. Parasite counts were performed induplicate by using a hemocytometer and particle counter (Beckman Coulter,Fullerton, CA).
Computational procedure. The screening process was initiated with the de-velopment of homology model of L. donovani PTR1 (LdPTR1). For homologyor comparative modeling we used restrained based modeling implemented in theMODELER program (21) interfaced with InsightII 2000.1 (Accelrys) using L.major PTR1 (LmPTR1) (sharing 91% sequence identity with LdPTR1) crystalstructure as a template (PDB code 1E7W) (8). The detailed procedure ofsequence analysis, comparative protein modeling, and refinement of the modelhas been reported recently (12). The accuracy of predictions by comparativemodeling depends on the degree of sequence similarity. Molecular docking ofthe compounds in the active site of this LdPTR1 homology model was performedusing the modern docking engine QUANTUM 3.3 software (www.q-lead.com/backup/QUANTUM_Docking_Software.pdf) as described previously (10).
Flow cytometer-based growth inhibition assay. L. donovani promastigotes andamastigotes of a clinical isolate expressing GFP were used for the in vitroevaluation of compounds for antileishmanial activity. The test was performed byflow cytometry as described previously (23). Briefly, log-phase promastigotes(2 � 106 cells/ml) in medium 199 supplemented with 10% HI-FBS were seededin 48-well tissue culture plates (Greiner Bio-One, Germany). The samples werecoincubated with different concentrations (6, 12.5, 25, 50, and 100 �g/ml) of 12compounds. Untreated cells served as a control. All compounds investigatedduring the present study were generated in-house by the methodology reportedearlier (Fig. 1) (20). The stock solutions (10 mM) were prepared in dimethylsulfoxide (DMSO) and stored at 4°C. After incubation at 26°C for 48 h, theparasites were washed and suspended in phosphate-buffered saline (PBS; pH7.2), followed by flow cytometry analysis (Becton Dickinson FACSCalibur, serialno. E1136). J774A.1 cells, a mouse (BALB/c) macrophage cell line (105 cells/well), in six-well culture plates (Nunc, Illinois) were infected with GFP-trans-fected promastigotes at a parasite/macrophage ratio of 10:1, followed by incu-bation at 37°C in 5% CO2 for 0 to 18 h. At 18 h after the parasites had enteredthe macrophages, free parasites were eliminated, and macrophages were treatedwith various concentrations (0.25, 0.50, 1.0, 1.5, and 2 �g/ml) of 7-(4-chloroben-zyl)-3-methyl-4-(4-trifluoromethyl-phenyl)-3,4,6,7,8,9-hexahydro-pyrimido[1,2-a]pyrimidin-2-one (compound 7), the most promising compound. The final drugconcentration was 2 �g/ml (4.59 �M) with a DMSO concentration of 0.20% inthe sixth assay well. At 48 h after treatment, infected macrophages were har-vested by washing them with PBS (pH 7.2) containing 3 mM EDTA and 10 mMglucose to easily dislodge the cells without using trypsin-EDTA and then ana-lyzed by using a flow cytometer equipped with a 15-mV, 488-nm, air-cooledargon ion laser using forward scatter (FSC) versus side scatter (SSC) to gate themacrophage population and an FL1 histogram to quantify the fluorescence of theviable cells. Intracellular amastigotes were detected and sorted according to theirrelative fluorescence intensities of GFP inside amastigotes compared to those ofuninfected cells. At least 10,000 cells were acquired for each analysis and ana-lyzed by using CellQuest Pro software. The infection rate of macrophages wasmeasured by putting markers in histograms for uninfected and infected cultures.Miltefosine was used as the reference drug because it is clinically used in theregions of Bihar (India) where leishmaniasis is endemic. Experiments wereperformed in triplicate. In vitro antileishmanial activity was expressed as theconcentration inhibiting parasite growth by 50% (i.e., the IC50 � the standarddeviation [SD]) after 48 h of incubation. The percent inhibition for each drugconcentration calculated as follows: [(the mean fluorescence intensity [MFI]without drug � the MFI with drug)/(the MFI without drug)] � 100. The IC50 wasobtained by plotting a graph of the percentage of inhibition at different concen-trations of all of the events using Origin 6.1 version software.
In vitro cytotoxicity against the macrophages. The J774A.1 cell line was incu-bated in a 96-well plate containing 104 cells/well. The plates were incubatedovernight in a CO2 incubator, with a supply of 5% CO2 at 37°C. Compound 7 atdifferent concentrations (2 to 50 �g/ml) was dispensed in triplicate, and threewells were left as control wells. The plates were incubated for 72 h, and an MTT
[3-(4,5-dimethylthiazole-2-yl)-2-5-diphenyl tetrazolium bromide] assay was per-formed to assess cell proliferation or viability (16).
Target protein expression and in vitro inhibition assay of recombinant enzyme(LdPTR1). The recombinant enzyme LdPTR1 used for the enzyme inhibitionstudies was purified based on its N-terminal His6 tag by affinity chromatographywith a Ni2�-IDA Hi-Trap chelating Sepharose column in AKTAprime Plus (GEHealthcare) (13). Reductase activity (LdPTR1) was assayed in a 96-well plate byusing a FLUOstarX Galaxy from BMG Lab Technologies, and the volume ofeach reaction was 300 �l. The assays were initiated by the addition of enzyme att � 0, and the rate were measured for 15-s intervals for 3 min. NADPH (SRLMumbai, India) oxidation was monitored at 340 nm. The inhibition studies forLdPTR1 were performed at 303 K in the presence of NADPH (100 �M) andseveral fixed concentrations (0 to 120 �M) of substrate biopterin (catalog no.B2517, Sigma) at pH 4.8 in 20 mM sodium acetate buffer (catalog no. S7899;Sigma). Since pteridines exhibit absorbance changes when reduced, the extinc-tion coefficient 7,230 M�1 cm�1 was used for the coupled oxidation/reduction ofNADPH/biopterin (2). The kinetic parameters Km and Vmax for the pteridinesubstrates were evaluated by fitting the Michaelis-Menten equation by nonlinearregression. For inhibition studies, recombinant LdPTR1 was incubated withcompound 7, and NADPH and the reaction initiated with the substrate bio-pterin. Lineweaver-Burk plots were used to determine the mode of inhibition byinhibitor, examining ligand competition under conditions where [inhibitor] ��[enzyme] and [biopterin].
Predicted drug-like properties of Baylis-Hillman derivatives as growth inhib-itors. L. donovani growth inhibitors were further examined for desirable drug-like properties using ADME Boxes v4.0 software (Pharma Algorithms, Toronto,Ontario, Canada) (9). In brief, this algorithm predicted human adsorption andmetabolism bioavailability for new compounds using a combination of bothprobabilistic and mechanistic methods. A bioavailable compound was defined asone that should satisfy the following criteria: dissolve in the stomach or intestineunder variable pH, withstand acid hydrolysis at a pH of �2, permeate theintestinal membrane by passive or active transport, withstand P-glycoproteinefflux in concert with metabolic enzymes in intestine, and withstand first-passmetabolism in the liver. Based on predictions, oral bioavailability was classifiedas follows: poor, �30%; moderate, 30 to 70%; and good, 70%. ADME Boxessoftware was also used to predict the toxicities of the compounds (i.e., the AMESin vitro test for gene mutation in bacteria, hERG [hERG is an outwardly di-rected, i.e., repolarizing, cardiac potassium current that is voltage and timedependent; that is, it is activated or “turns on” with depolarization and deacti-vates or “turns off” on repolarization], skin irritation, the 50% lethal dose inmice, and Cyp450 inhibition). For genotoxicity, we calculated the probability thata compound would register as positive in an Ames mutagenicity screening test,while hERG in silico assessment was calculated as the probability of a compoundbeing an hERG channel inhibitor at clinically relevant concentrations. Toxicitypredictions have an associated reliability index (RI), defined as follows: RI � 0.3,not reliable; RI � 0.3 to 0.5, borderline reliability; RI � 0.5 to 0.75, moderatereliability; and RI � 0.75, high reliability (9).
Statistical analysis. The data are presented as means � the SD. The statisticalsignificance of differences in percentages between treated and untreated wasanalyzed by one-way analysis of variance using GraphPad Prism software.
RESULTS
Molecular docking of Baylis-Hillman derivatives. To ex-plore their probable binding modes, molecular docking of the12 compounds (Fig. 1) was performed into the active site ofpreviously reported LdPTR1 homology model (12) by usingQUANTUM 3.3 software. They were also superimposed in theligand-binding pocket of LdPTR1. The superimpositionshowed almost similar spatial position (Fig. 2). Compounds 1,2, 3, 6, 11, and 12 were substituted allyl amines precursors forthe synthesis of hexahydro pyrimido pyrimidinones 4, 5, 7, 8, 9,and 10. The docking results indicated that compound 7 mayhave the highest potential since it has minimum predicted IC50
of 2.98 � 10�6 mol/liter with minimum binding free energy of�32.16 kJ/mol (Fig. 3A; see also Table S1 in the supplementalmaterial). Compound 7 was also docked into the active siteof Trypanosoma cruzi DHFR-TS (TcDHFR-TS) (http://www.pdb.org/pdb/explore/explore.do?structureId�2H2Q), human
VOL. 55, 2011 PTERIDINE REDUCTASE INHIBITORS AS ANTILEISHMANIALS 661

DHFR (hDHFR) (structure taken from 1drf doi:10.2210/pdb1drf/pdb), and Toxoplasma gondii PCD (TgPCD) since nocrystal structure was available in Leishmania (Fig. 3B, C, andD). Compound 7 showed predicted IC50s of 2.99 � 10�5,9.84 � 10�4, and 7.34 � 10�5 mol/liter with binding freeenergies of �26.33, �17.5, and �24.06 kJ/mol for TcDHFR-TS, hDHFR, and TgPCD, respectively (see Table S2 in thesupplemental material).
In vitro efficacy of Baylis-Hillman derivatives against L. do-novani promastigotes and intracellular amastigotes. The com-pounds were incubated with GFP-expressing promastigotes,for 48 h before flow cytometry analysis. The IC50s for com-pounds 4, 5, 7, 8, 9, and 10 were determined to be 31, 59, 29,120, 68, and 72 �M, respectively, and the IC50s for compounds1, 3, 11, and 12 were determined to be 125, 131, 150 and 121�M, respectively. Compounds 2 and 6 were found to be inac-tive in a cell-based assay. Among the hexahydro pyrimido pyri-midinone derivatives, compound 7 has maximum activity withIC50 of 29 �M. So compound 7 was selected for further studyin amastigotes. Promastigotes expressing GFP were used toinfect J774A.1 cells. Our Leishmania promastigote transfec-tants proliferated and were infective to macrophages, resulting
FIG. 2. Superimposition of compounds and shape of the ligand-binding pocket. (A) All 12 Baylis-Hillman derivatives were superim-posed. The 12 compounds are displayed as stick models in differentcolors (compound 1, red; compound 2, orange; compound 3, green;compound 4, marine blue; compound 5, light pink; compound 6, pur-ple blue; compound 7, dark pink; compound 8, olive; compound 9,yellow; compound 10, light pink; compound 11, brown; compound 12,cyan). (B) The shape of the ligand-binding pocket is represented by thecolored mesh that overlays the superimposition of all 12 compounds inthe pocket.
FIG. 3. Predicted binding models for compound 7 (green) in the binding site of LdPTR1 (blue) (A), compound 7 (blue) in the binding site ofTcDHFR-TS (red) (B), compound 7 (blue) in the binding site of hDHFR (red) (C), and compound 7 (blue) in the binding site of TgPCD (red) (D).
662 KAUR ET AL. ANTIMICROB. AGENTS CHEMOTHER.
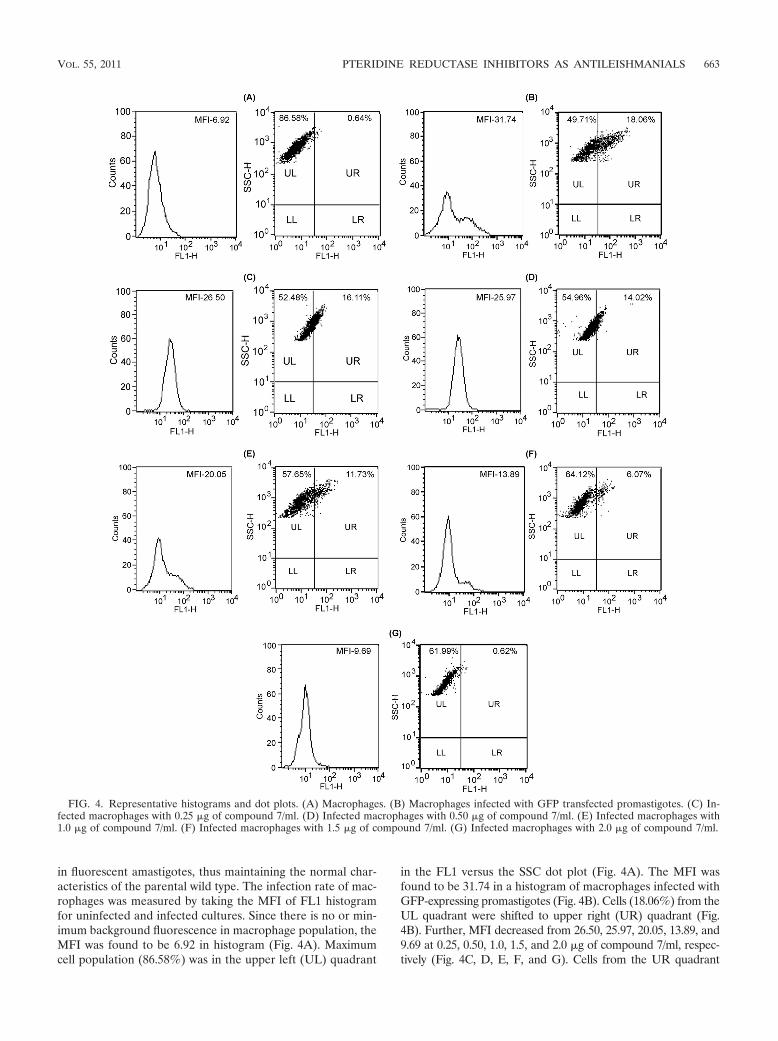
in fluorescent amastigotes, thus maintaining the normal char-acteristics of the parental wild type. The infection rate of mac-rophages was measured by taking the MFI of FL1 histogramfor uninfected and infected cultures. Since there is no or min-imum background fluorescence in macrophage population, theMFI was found to be 6.92 in histogram (Fig. 4A). Maximumcell population (86.58%) was in the upper left (UL) quadrant
in the FL1 versus the SSC dot plot (Fig. 4A). The MFI wasfound to be 31.74 in a histogram of macrophages infected withGFP-expressing promastigotes (Fig. 4B). Cells (18.06%) from theUL quadrant were shifted to upper right (UR) quadrant (Fig.4B). Further, MFI decreased from 26.50, 25.97, 20.05, 13.89, and9.69 at 0.25, 0.50, 1.0, 1.5, and 2.0 �g of compound 7/ml, respec-tively (Fig. 4C, D, E, F, and G). Cells from the UR quadrant
FIG. 4. Representative histograms and dot plots. (A) Macrophages. (B) Macrophages infected with GFP transfected promastigotes. (C) In-fected macrophages with 0.25 �g of compound 7/ml. (D) Infected macrophages with 0.50 �g of compound 7/ml. (E) Infected macrophages with1.0 �g of compound 7/ml. (F) Infected macrophages with 1.5 �g of compound 7/ml. (G) Infected macrophages with 2.0 �g of compound 7/ml.
VOL. 55, 2011 PTERIDINE REDUCTASE INHIBITORS AS ANTILEISHMANIALS 663

decreased from 16.11, 14.02, 11.73, 6.07, and 0.62% at 0.25, 0.50,1.0, 1.5, and 2.0 �g/ml of compound 7, respectively (Fig. 4C, D, E,F and G). Flow cytometric results indicated that compound 7 was10 times (IC50 � 3 �M) more active in L. donovani amastigotesthan in promastigotes (IC50 � 29 �M).
Cytotoxicity of test compounds against the J774A.1 cell line.Compound 7 was checked against the J774A.1 cells using anMTT assay to determine whether the dose used for IC50 onintramacrophage amastigotes was toxic to the cell itself. Ex-periments revealed that the 50% cell cytotoxicity concentration(CC50) value was far higher (73 �M) than the IC50 dose (3�M) for intracellular amastigotes.
Kinetics of L. donovani PTR1. First, we optimized the en-zyme concentration for PTR1 activity using 100 �M NADPHand 100 �M biopterin in order to determine the kinetic pa-rameters such as Km, Vmax, and pH optima for the substratebiopterin. The initial velocity of reaction increases with theincrease of enzyme concentration up to 0.348 �M; at thissaturation point further increases in the enzyme concentrationhad no effect on the initial velocity of the reaction (Fig. 5A).Thus, for all of the kinetics and inhibition studies, we used a0.348 �M concentration of the enzyme. Next, we examined thepH dependence of LdPTR1 activity. A sharp peak of activitywas observed at pH 4.8 (Fig. 5B). At this optimum pH, PTR1activity with oxidized biopterin exhibited standard Michaelis-Menten kinetics (Fig. 5C). Km and Vmax values for biopterinwere derived from a Lineweaver-Burk plot (Fig. 5D). A Km of5.859 � 1.023 �M and a Vmax of 0.134 � 0.0589 �mol/min/mgwas obtained for the substrate biopterin. For NADPH, a Km of18.51 �M and a Vmax of 0.66 �mol/min/mg were obtained.Using methotrexate as inhibitor, the Ki value was found to be
1.246 �M for LdPTR1 against the substrate biopterin (Fig.5D). Inhibition studies of LdPTR1 with compound 7 yielded aKi value of 0.72 �M (Fig. 5D), and no inhibition was seen at190 �M with hDHFR (catalog no. D6566).
Predicted drug-like properties of Baylis-Hillman deriva-tives. Predicted drug-like properties of compound 7 usingADME Boxes v4.0 software established human adsorption andmetabolism bioavailability, dissolved in the stomach or intes-tine under variable pH, withstand acid hydrolysis at pH 1.8,permeate through intestinal membrane by passive transport,withstand P-glycoprotein efflux in concert with metabolic en-zymes in intestine and withstand first-pass metabolism in theliver. Based on predictions, compound 7 has more than 70%oral bioavailability.
DISCUSSION
In our continuing search for new antileishmanials (12) andin connection with our efforts to study the bioproperties ofseveral cyclic derivatives generated from the Baylis-Hillmanchemistry, we initiated an investigation of the bioactivity ofannulated pyrimidinone systems against clinical isolates of L.donovani.
We were interested in understanding the interactions ofpyrimido[1,2-a]pyrimidinone derivatives (20) with LdPTR1.Molecular docking studies of the LdPTR1 binding site re-vealed a very clear preference for the Baylis-Hillman deriva-tives. The hexahydro pyrimido pyrimidinone ring could fit wellin a hydrophobic pocket formed by residues Ala-230, Tyr-191,Tyr-194, Phe-113, Pro-224, and Leu-18. The halogen-substi-tuted phenyl rings on either side of the pyrimidine core are
FIG. 5. Kinetic parameters of L. donovani PTR1. (A) The optimum enzyme concentration for PTR1 activity was determined using 100 �MNADPH and 100 �M biopterin. (B) The pH dependence of PTR1 activity was determined using 20 mM sodium acetate buffer (pH 3.6 to 6.5).(C) The PTR1 activity was assayed with recombinant LdPTR1 using 20 mM sodium acetate (pH 4.8) for the substrate biopterin. (D) Lineweaver-Burk plot for methotrexate and compound 7 inhibition of LdPTR1. Circles indicate uninhibited enzyme, squares indicate assays in the presenceof compound 7, and triangles indicate assays in the presence of methotrexate.
664 KAUR ET AL. ANTIMICROB. AGENTS CHEMOTHER.

oriented in such a way so that the compounds attain a butter-fly-like conformation that is commonly seen in the case of HIVreverse transcriptase inhibitors (7). Because the active site ofLdPTR1 was mostly surrounded by hydrophobic side chains,hydrogen bonding interactions with compound 7 were notprominent (Fig. 3A). However, the hexahydro pyrimido pyri-midinone core of six compounds was held in the pocket by acombination of van der Waals and hydrophobic interactionswith the protein, and major hydrophobic contacts occurredbetween the hexahydro pyrimido pyrimidinone core and theside chains of Phe-113, Tyr-194, and Ala-230. As seen from ourdocking studies, Phe-113 acts as a possible basic and -donorsite and is involved in edge to face - stacking interactionwith the substituted phenyl rings. This interaction seems veryimportant for LdPTR1 inhibition and may be responsible forthe stability of the protein ligand complex (Fig. 3A). Alongwith the IC50, QUANTUM 3.3 software also calculates the freebinding energy (G bind), electrostatic and salvation energy(E es), van der Waals energies (E vdw), entropy contribution(TdS), ligand internal energy change (E tor), total charge,mass, number of flexible bonds of the ligand and RMS distancebetween the initial and final positions. The use of this softwarealso indicates the different conformers of each compound withIC50s and binding free energies. Conformer 1 of each com-pound has a minimum free binding energy. If we compared allof the compounds according to binding free energies, com-pound 7 has minimum value of �32.16 kJ/mol (see Table S1 inthe supplemental material).
The docking results were in accordance with the biologicalactivity. We tested the effect of several concentrations of theBaylis-Hillman derivatives against promastigotes. The hexahy-dro pyrimido pyrimidinone derivatives have greater biologicalactivity than substituted allyl amines. Among the hexahydropyrimido pyrimidinone derivatives, compound 7 has maximumactivity with an IC50 of 29 �M. Thus, compound 7 was selectedfor further study in amastigotes. Since, the macrophage-amas-tigote model is considered the gold standard for establishingthe drug sensitivity profile of an antileishmanial compound,promastigotes expressing GFP were used to infect a J774A.1macrophage cell line. Flow cytometric results indicated thatcompound 7 was 10 times more active on L. donovani amasti-gotes than on promastigotes. From these results, we can inferthat this compound was able to biotransform within the mac-rophages to give a more active compound.
As described elsewhere in the present study, PTR1 providesmetabolic bypass to the enzyme DHFR-TS. In order to de-velop effective chemotherapy, it will be necessary to inhibitboth of these enzymes. Given the structural overlap among thesubstrates of these two enzymes, it is conceivable that a singlecompound might be found with good efficacy against bothenzymes. DHFR-TSs from the L. major, T. brucei, and T. cruzishow a high degree of structural similarity (11, 25). Further,the structure of the three protozoan DHFR-TS and the en-zyme specificity is significantly different from that of the humanenzyme, suggesting the possibility of selective drug design.Thus, compound 7 was docked in the active sites of TcDHFR-TS, hDHFR, and TgPCD enzymes (Fig. 3B, C, and D). Com-pound 7 showed reasonable levels of selectivity for theTcDHFR-TS over the hDHFR and TgPCD (see Table S2 inthe supplemental material). These values were comparable to
the predicted IC50 of LdPTR1 and showed a greater affinity ofcompound 7 for LdPTR1.
Recombinant enzyme inhibition was performed to finallyconfirm the target specificity of compound 7 to LdPTR1. Weoptimized enzyme concentration at 0.348 �M for LdPTR1activity (Fig. 5A). Next, we examined the pH dependence ofthe LdPTR1 activity. For biopterin, the enzymatic activity in-creased when the pH of the assay buffer was decreased. Asharp peak of activity was observed at pH 4.8 (Fig. 5B), it wasalmost the same as that reported for the native and recombi-nant LmPTR1 (18). Moreover, we observed that the proteinwas stable for more than 3 h when incubated at 4°C in buffersolution from pH 3 to 9. Therefore, the lower activity observedat higher pH values could not be caused by enzyme denatur-ation but was more probably caused by the occurrence of lessfavorable conformational forms of the protein or possibly byaltered ionic forms of the substrates. At this optimum pH,PTR1 activity with oxidized biopterin exhibited standardMichaelis-Menten kinetics (Fig. 5C). Km and Vmax values forbiopterin were derived from Lineweaver-Burk plot (Fig. 5D).The properties of recombinant LdPTR1 were similar to thoseof native LmPTR1 (18), which has 91% sequence identity withthe LdPTR1, and also the active site and NADPH binding sitewas highly conserved in these two distantly related species (13).
Methotrexate is a known antifolate inhibitor of Plasmodiumfalciparum DHFR (22). Using methotrexate as the inhibitor,the Ki value was found to be 1.24 �M for LdPTR1 against thesubstrate biopterin (Fig. 5D). The Ki for methotrexate inhibi-tion was not much less at pH 4.8 against the substrate biopterinreactions performed using PTR1 (Fig. 5D). The overexpres-sion of PTR1 enzyme could reduce the folates because it pro-vides a metabolic bypass to DHFR-TS enzyme. The Ki valuefor LdPTR1 was found to be three times less than that forLmPTR1 (3). Further, the Ki values for LmDHFR andhDHFR were found to be 130 and 3.4 pM, respectively (3),suggesting that methotrexate is inhibiting PTR1 less thanDHFR. Inhibition studies of LdPTR1 with compound 7yielded a Ki value of 0.72 �M (Fig. 5D), and no inhibition wasseen at 190 �M with hDHFR.
Additional preclinical data for compound 7 in experimentalhamsters, as well as regarding the mode of cell death associ-ated with L. donovani parasites, will be examined in our futurestudies. Such a hexahydro pyrimido pyrimidinone scaffoldcould have potential as a valuable pharmacological tool againstthe disease VL.
ACKNOWLEDGMENTS
We thank A. L. Vishwakarma, SAIF Division, for flow cytometryanalysis.
Financial support was provided by the Department of Biotechnol-ogy, New Delhi, India (grant BT/PR5452/BRB/10/430/2004).
This is CDRI communication no. 7913.
REFERENCES
1. Barbosa, T. P., et al. 2009. Improved synthesis of seven aromatic Baylis-Hillman adducts (BHA): evaluation against Artemia salina Leach and Leish-mania chagasi. Eur. J. Med. Chem. 44:1726–1730.
2. Bello, A. R., B. Nare, D. Freedman, L. Hardy, and S. M. Beverley. 1994.PTR1: a reductase mediating salvage of oxidized pteridines and methotrex-ate resistance in the protozoan parasite Leishmania major. Proc. Natl. Acad.Sci. U. S. A. 91:11442–11446.
3. Cavazzuti, A., et al. 2008. Discovery of potent pteridine reductase inhibitorsto guide antiparasite drug development. Proc. Natl. Acad. Sci. U. S. A.105:1448–1453.
VOL. 55, 2011 PTERIDINE REDUCTASE INHIBITORS AS ANTILEISHMANIALS 665

4. Chappuis, F., et al. 2007. Visceral leishmaniasis: what are the needs fordiagnosis, treatment and control? Nat. Rev. Microbiol. 5:873–882.
5. De Souza, R. O. M. A., et al. 2007. High selective leishmanicidal activity of3-hydroxy-2-methylene-3-(4-bromophenyl) propanenitrile and analogouscompounds. Eur. J. Med. Chem. 42:99–102.
6. De Souza, R. O. M. A., B. A. Meireles, L. C. S. Aguiar, and M. L. A. A.Vasconcellos. 2004. Hexamethylenetetramine as a cheap and convenientalternative catalyst in the Baylis-Hillman reaction: synthesis of aromaticcompounds with anti-malarial activity. Synthesis 10:1595–1600.
7. Gehlhaar, D. K., et al. 1995. Molecular recognition of the inhibitor AG-1343by HIV-1 protease: conformationally flexible docking by evolutionary pro-gramming. Chem. Biol. 2:317–324.
8. Gourley, D. G., et al. 2001. Pteridine reductase mechanism correlates pterinmetabolism with drug resistance in trypanosomatid parasites. Nat. Struct.Biol. 8:521–525.
9. Japertas, P., R. Didziapetris, and A. Petrauskas. 2003. Fragmental methodsin the analysis of biological activities of diverse compound sets. Mini Rev.Med. Chem. 3:797–808.
10. Kaur, J., S. Sundar, and N. Singh. 2010. Molecular docking, structure-activity relationship and biologic evaluation of the anticancer drug monastrolas pteridine reductase inhibitor in Leishmania donovani clinical isolate. J.Antimicrob. Chemother. 65:1742–1748.
11. Knighton, D. R., et al. 1994. Structure of and kinetic channelling in bifunc-tional dihydrofolate reductase-thymidylate synthase. Nat. Struct. Biol. 1:186–194.
12. Kumar, P., et al. 2008. Leishmania donovani pteridine reductase 1: biochem-ical properties and structure modeling studies. Exp. Parasitol. 120:73–79.
13. Kumar, P., H. Kothari, and N. Singh. 2004. Overexpression in Escherichiacoli and purification of pteridine reductase 1 (PTR1) from a clinical isolateof Leishmania donovani. Protein Expr. Purif. 38:228–236.
14. Kundu, M. K., et al. 1999. Antimalarial activity of 3-hydroxyalkyl-2-methyl-enepropionic acid derivatives. Bioorg. Med. Chem. Lett. 9:731–736.
15. Lye, L. F., M. L. Cunningham, and S. M. Beverley. 2002. Characterization ofquinonoid-dihydropteridine reductase (QDPR) from the lower eukaryoteLeishmania major. J. Biol. Chem. 41:38245–38253.
16. Manandhar, K. D., et al. 2008. Antileishmanial activity of nano-amphoter-icin B deoxycholate. J. Antimicrob. Chemother. 62:376–380.
17. Murray, H. W., J. D. Berman, C. R. Davies, and N. G. Saravia. 2005.Advances in leishmaniasis. Lancet 366:1561–1577.
18. Nare, B., L. Hardy, and S. M. Beverley. 1997. The roles of pteridine reduc-tase 1 and dihydrofolate reductase-thymidylate synthase in pteridine metab-olism in the protozoan parasite Leishmania major. J. Biol. Chem. 272:13883–13891.
19. Nare, B., J. Luba, L. W. Hardy, and S. Beverley. 1997. New approaches toLeishmania chemotherapy: pteridine reductase 1 (PTR1) as a target andmodulator of antifolate sensitivity. Parasitology 114:S101–S110.
20. Pathak, R., and S. Batra. 2007. Expeditious synthesis of 5,6,7,8-tetrahydro-imidazo-[1,2-a]pyrimidin-2-ones and 3,4,6,7,8,9-hexahydro-pyrimido-[1,2-a]pyrimidin-2-ones. Tetrahedron 63:9448–9455.
21. Sali, A., and T. L. Blundell. 1993. Comparative protein modeling by satis-faction of spatial restraints. J. Mol. Biol. 234:779–815.
22. Shallom, S., K. Zhang, L. Jiang, and P. K. Rathod. 1999. Essential protein-protein interactions between Plasmodium falciparum thymidylate synthaseand dihydrofolate reductase domains. J. Biol. Chem. 274:37781–37786.
23. Singh, N., and A. Dube. 2004. Fluorescent Leishmania: application to anti-leishmanial drug testing. Am. J. Trop. Med. Hyg. 71:400–402.
24. Sundar, S., and M. Chatterjee. 2006. Visceral leishmaniasis–current thera-peutic modalities. Indian J. Med. Res. 123:345–352.
25. Zuccotto, F., A. C. R. Martin, R. A. Laskowski, J. M. Thornton, and I. H.Gilbert. 1998. Dihydrofolate reductase: a potential drug target in trypano-somes and leishmania. J. Comput.-Aided Mol. Des. 12:241–257.
666 KAUR ET AL. ANTIMICROB. AGENTS CHEMOTHER.
Copyright © 2022 FDOKUMEN




















