
Identification of photocatalytic degradation pathways of 2-Cl-s-triazine herbicides and detection of...
20
Chemosphere, Vol. 24, No. 7, pp. 891-910, 1992 0045-6535/92 $5.00 + 0.00 Printed in Great Britain Pergamon Press plc IDENTIFICATION OF PHOTOCATALYTIC DEGRADATION PATHWAYS OF 2-CI-$-TRIAZINE HERBICIDES AND DETECTION OF THEIR DECOMPOSITION INTERMEDIATES E. Pelizzetti*, C. Minero, V. Carlin, M. Vincenti, E. Pramauro Dipartimento di Chimica Analitica, Universit~t degli Studi di Torino, Torino, Italy M. Dolci Dipartimento di Chimica Agraria, Universit~ degli Studi di Torino, Torino, Italy ABSTRACT The photocatalytic degradation of three 2-chloro-s-triazine herbicides (simazine, propazine, atrazine) over TiO 2 particles under simulated solar light has been investigated. In the early steps, the degradation progresses through competitive reactions such as oxidation of the side alkyl chain and substitution of chlorine with hydroxyl groups, the last process occurring for less than 10% of degradation. Further oxidative processes give rise to the formation of dealkylated products. Several intermediates have been detected by GC-MS and comprehensive degradation schemes are proposed. The three herbicides, hearing different alkyl groups, exhibit relevant differences in the relative role of the competitive degradation pathways. KEY WORDS : s-triazine derivatives, degradation mechanism, photodegradation, GC-MS, atrazine, simazine, propazine. 891
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Identification of photocatalytic degradation pathways of 2-Cl-s-triazine herbicides and detection of...

Chemosphere, Vol. 24, No. 7, pp. 891-910, 1992 0045-6535/92 $5.00 + 0.00 Printed in Great Britain Pergamon Press plc
IDENTIFICATION OF PHOTOCATALYTIC DEGRADATION PATHWAYS OF 2-CI-$-TRIAZINE HERBICIDES
AND DETECTION OF THEIR DECOMPOSITION INTERMEDIATES
E. Pelizzetti*, C. Minero, V. Carlin, M. Vincenti, E. Pramauro
Dipartimento di Chimica Analitica,
Universit~t degli Studi di Torino, Torino, Italy
M. Dolci
Dipartimento di Chimica Agraria,
Universit~ degli Studi di Torino, Torino, Italy
ABSTRACT
The photocatalytic degradation of three 2-chloro-s-triazine herbicides (simazine, propazine, atrazine)
over TiO 2 particles under simulated solar light has been investigated. In the early steps, the degradation
progresses through competitive reactions such as oxidation of the side alkyl chain and substitution of chlorine
with hydroxyl groups, the last process occurring for less than 10% of degradation. Further oxidative processes
give rise to the formation of dealkylated products.
Several intermediates have been detected by GC-MS and comprehensive degradation schemes are
proposed. The three herbicides, hearing different alkyl groups, exhibit relevant differences in the relative role
of the competitive degradation pathways.
KEY WORDS : s-triazine derivatives, degradation mechanism, photodegradation, GC-MS, atrazine, simazine,
propazine.
891

892
INTRODUCTION
The 2-chloro-s-triazines are amongst the most widely used herbicides. 1'2 Their degradation and
metabolic pathways have been largely investigated. Dehalogenated, dealkylated and deaminated derivatives of
the s-triazine herbicides have been isolated in different sources. 3-11
Recently, fast and efficient photocatalytic degradation has been reported for some s-triazine
herbicides. 12'13 The catalytic process can be of interest in the fate of these compounds in the soil 14 and
aquatic environments and represent a viable route for the decontamination of polluted waters. 15'16
The present paper reports on the degradation pathways and the identification of some intermediates
occurring in the photocatalytic degradation of the three widely used 2-chloro-s-triazines reported in Chart I.
Particular efforts have been devoted to the study of early stages of the process, since the latter part of the
degradation scheme has been already established for atrazine. 13 Emphasis is also given to the comparison of
the mechanisms and decomposition rates for differently alkylated compounds.
Chart I. Structural formulas, trivial and chemical names of 2-chloro-s-triazines under study.
Structural formula:
c4
R1 R=
Symbol R1 R2 Trivial name Chemical name
1,3,5-trlazlne-2,4-dlamlne- S C~-Is CL=~'Is S l m a z l n e -6-chloro-N,N'-diethyl
1,3,5-trlazlne-2,4-dlamlne- P CH(CI-kj)z CH(CI-~j) 2 Propazine -6-chloro-N~l'-(1-methylethyl)
1,3,5-trlazlne-2,4-dlamlne- A ~ CH(CI-~2 Atrazlne -6-chloro-N-ethyI-N'-(1-methylethyl)
EXPERIMENTAL SECTION
Materials and Reaeents.
Standard herbicide compounds (atrazine 99.5%, simazine 99.9%, propazine 99%) supplied by Istituto
Superiore della Sanit~ (Rome), and cyanuric acid (Aldrich 98%) were used as received. Stock solutions (1
gL -1) of triazine herbicides were prepared in ethyl acetate; reanalysis of stock solutions showed no effect of

893
the solvent on the stability of the considered herbicides. Cyanuric acid (compound X in Scheme V) is soluble
enough in water for preparing stock solutions (1 gL -1) in bidistiiled water.
Compounds III, V, VI, VIII, XVIII (see Schemes) were synthesized as described previously; 12 compounds
I, XIII, XXI were obtained by hydrolysis of S, P, A respectively, according to the literature. 17
Acetonitrile and methanol (Lichrosolv Merck) and didodecyldimethylammonium bromide (Kodak) were
used as received. Other reagents used were at least analytical grade.
All degradation experiments were carried out by using TiO 2 Degussa P25 grade as photocatalyst. In
order to avoid a possible interference from ions adsorbed on the photocatalyst in the analytical determinations,
the TiO 2 powder was irradiated and washed several times with bidistilled water until no signal due to chloride,
sulphate or sodium ions could be detected by ion chromatography (Cl- < 20 and Na + < 100 part per billion
H20 in the presence of TiO 2 5 gL-I in H20).
Irradiation ext~erim¢~.
Experiments were carried out in Pyrex glass cells on 5 mL of an aqueous suspension containing the
needed amount of semiconductor powder and substrate, by irradiating with a 1500 Watt Xenon lamp
(CO.FO.MEGRA, Milan) equipped with a 340 cutoff filter and simulating AMI solar light. The cells were
described elsewhere. 13 The total photonic flux in both cells, as measured by ferrioxalate actinometry and
corrected for the fraction of light absorbed and the quantum yield of the actinometer 18 in the range 340-545
nm, was found 5.8x10 -5 einstein min -1 cm -2.
Substrates were dissolved in water at ppm levels, under the solubility limit, 19 by putting the required
volume of the stock solution in a volumetric flask, evaporating the solvent at room temperature under nitrogen
stream and then by adding water till to the required volume. The suspensions were stirred overnight.
Analytical determinations of substrate concentration in these solutions assured that quantitative transfer was
achieved.
All the irradiation experiments were carried out at 0.5 gL-1 of TiO 2.
Analytical determinations.
The HPLC determinations were carried out with a Hitachi model L6200 pump with UV detection (model
L4200), equipped with a bonded phase octadecylsilica column (LiChrospher C18 Merck, 125 mm long, 4 mm
I.D., 5 /~m packing). The primary degradation of triazine herbicides was followed by direct injection of the
filtered (0.45 /~m) irradiated sample; UV detection was performed at 240 nm; mobile phase was 65% methanol
/ 35% water at 1 mL rain -1 and retention times were: simazine (compound S in Chart I) 2.9 rain, propazine
(compound P) 6.0 min and atrazine (compound A) 4.0 min.
For the analysis of samples irradiated for short times (range of minutes, see figures 3,4,6,7 and 9), an
extraction in ethyl acetate was carried out before GC-MS analysis. 1 /.~1 of sample was injected splitlass (30 s)
into a SE-52 capillary column (30 m, 0.32 mm). The other end of the column was inserted directly into the
ion source via a heated transfer line. The oven temperature was programmed as follows: isothermal at 60"C
for 1 rain; from 60"C to 300°C at 10*C/min; isothermal at 300°C for 10 min.
Electron impact (EI) and chemical ionization (CI) mass spectra were obtained using a Finnigan-MAT
8400 double-focusing reverse geometry mass spectrometer, interfaced to a Varian 3400 gas chromatograph and
controlled by a SuperIncos data system. The EI spectra were recorded at a source temperature of 200°C,
setting the electron energy to 70 eV and the accelerating voltage to 3 kV. The magnet was scanned from 29 to
800 Da in 0.8 s (1.0 s/cycle) and the resolution was set to m/6m = 1000 (5% valley).

894
In CI experiments, isobutane was used as the reagent gas at a pressure of 0.3 mBar. The mass spectra of
positive ions were recorded. The electron energy was set to 100 eV and the scanned mass range was 60-1000
IDa.
The extraction in organic solvents of compounds formed after long irradiation times and therefore the
gas chromatographic-mass spectrometric analysis, is prevented by their large hydrophilicity. Ammeline
(compound VI), ammelide ( compound VIII) and (iso)cyanuric acid (compound X) (see Scheme V) were
detected by ion interaction chromatography with the immobilized reagent technique (IIIR). The reverse phase
column was loaded with didodecyldimethylammonium bromide (DDDMAB) by fluxing 30 mL of a lxl0 -2 M
DDDMAB solution in acetonitrile/water 25%/75% at 1 mL min -1 and washing with 30 ml of water. A 5x10 -3
M phosphate buffer solution at pH 7.7 was used as eluant at 1 mL rain -1. The pH and the concentration of
the buffer used as eluant were optimized to give reliable analytical conditions. The complete elution of
bromide ion from the column required a conditioning time of about an hour and is evidenced at 220 nm by a
baseline decrease followed by further stabilization. The retention times were 2.0, 2.9, 5.0 and 6.4 minutes for
solvent front, ammeline (VI), ammelide (VIII) and cyanuric acid (X), respectively (UV detection at 220 nm).
The formation of chloride ions was monitored by suppressed ion chromatography, using a Biotronik IC
5000 apparatus equipped with a BTIAN separation column (20 cm long, 4 mm I.D., Biotronik) and an alkaline
buffer eluant containing K2CO 3 (3 raM) and KOH (1.5 mM) at the flow rate of 1.5 mL min -1.
RESULTS
StoichiQmetrv.
In the previous report 12, the final products originating from the photocatalytic degradation of s-triazine
derivatives have already been shown. At long irradiation times, the process leads to the formation of cyanuric
acid (X), as the major final product, and stoichiometric chloride for 2-chloro-s-triazines together with nitrate
corresponding to the two amino groups in 4 and 6 positions of the triazine ring.
Intermediate identifications.
Compounds I, III, V, VI, VIII, X and XVIII have been identified as intermediates through HPLC
analysis of the slurry at different irradiation times by comparison with the retention times of the pure
products under the same conditions.
Other sample constituents, arising from photodecomposition of the various starting materials, were
identified from their EI mass spectra by (i) comparison with pure standards, when available; (ii) comparison
with published spectra either from databases or research papers and (iii) careful interpretation based on the
fragment ions formed in the EI induced decomposition.
Further information can be obtained by the CI mass spectra 20,21 In these experiments, the base peak
corresponded to the protonated molecular ion for all the substances investigated, allowing an easy
determination of the molecular weight both for major constituents and trace compound. This was particularly
useful for molecules such as XIV and XV where extensive fragmentation prevents the detection of a
significant abundance of molecular ion in the EI mass spectrum.
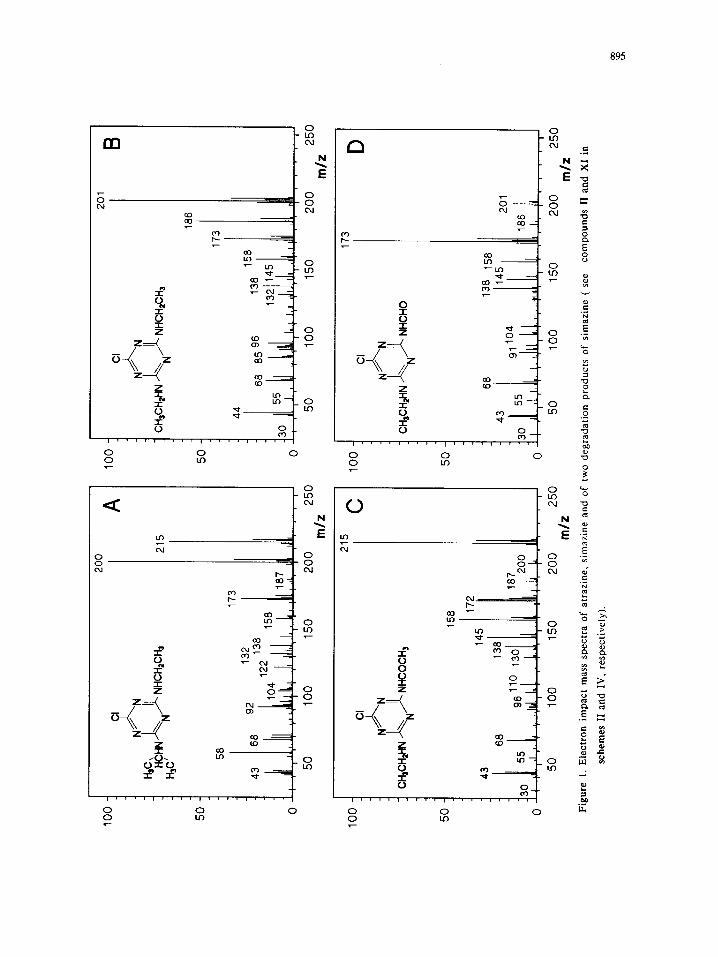
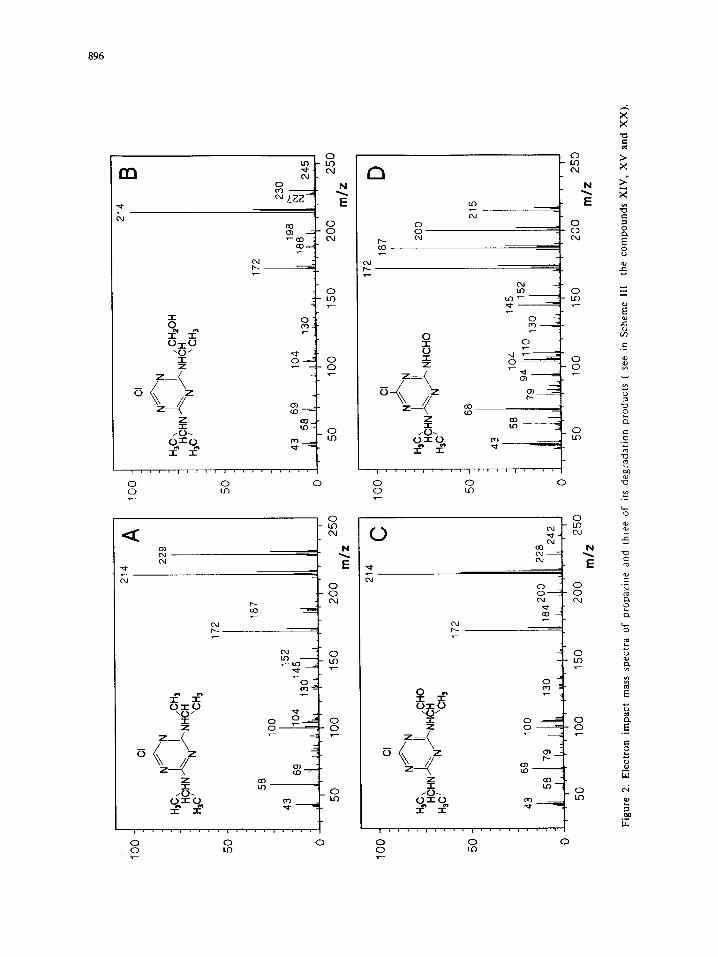
In Figures 1 and 2 the EI mass spectra are depicted for starting materials and various
photodecomposition products identified in the reaction mixtures. These spectra show that the fragmentation of
most molecular ions mainly occurS by competitive ruptures on the substituents of the triazine ring.
100 50
100 50
OI
N'~N
H,C\ L
II
HCHN ~. ~
NHCH2CH3
H3C /
N
58
i3
68
9i10
4 13
2 12
2 13
8 15
8
,. [
.,,,iJ,
., IL,
I, ~
,,.. ]..
l,,,
d,~7
I
' '
' '
I '
' '
' I
' '
' '
50
1 O0
150
200
215
A
200
250
m/z
Cl
N.-~-.N
c.
.c,..
, \,J
-,,co
c,.
215
C
158
145
172
I ]
IL '8,7
200
,~[ ,.
,,, I
!, ,,.
.,d.,
L. ! ,
.. !,.
J, ]J.
,..IL
:glL j,.k
!,
....
50
100
150
200
250
m/z
0 0
100
-
50-
CI
186
173
44
138
158
/
/
.l. ,.
IJ 68
85 9
6 la2
/14s
I d,
,o
? .... L
t,LJ.
.,,.,,
,J,J
,I,,,L
,Ihl
I '
' '
' I
' '
' '
I '
' '
'
50
100
150
100
-
.~oI
B
' '
' '
I
200
250
m/z
50-
Cl
N-'~L-N
c.
.c...
. \.J
- ,,c
.o
68
138
158
3,0 ji
l ~!
,.,i,,
. 91
104
[li5
,
,Ih, I
,,L ....
.... ,,,,,,
h IJ I
,
, I
' '
' '
I '
' '
' I
50
100
150
Fig
ure
1. E
lect
ron
impa
ct m
ass
spec
tra
of a
traz
ine,
sim
azin
e an
d of
tw
o de
grad
atio
n pr
oduc
ts o
f si
maz
ine
( se
e
sche
mes
II
and
IV,
resp
ecti
vely
).
173
D
201
1~6
I L
','
, ,
, ,
, I
200
250
m/z
co
mp
ou
nd
s II
and
XI
in
O0
896
m
.ca:
cO
t'M
O "O
I- O
o-to \0 / -r 0 --: Z ~ -
Z cO_ - r ~ . ) .
/ ( . ) ~
- r 1 -
O - lid
~ J
O -O
O LO
i , , , , i , , , , t ; , , , i , , , ,
O O O If)
0
Ol-O "-(~/ I Z
Z I-
-~- "I-
0 0
co
i o
_ _ E
o o
04
~oZ_
0 .~ 0 ~------ 0
o~
~O
O
i , , , , , , , , , i , , , , , , , , ,
O O O LO
O
121
cO
N
to E
o o O O tXl
~ . . O
o _ N
~ ~ - ~ ~ E
o ~ r ~
o - r o
i - r " ~ - ~ ~ 0 Z 0 0
o
Z
o " tO ~
o o o O U'~ r
O LID
00 c , J ~ t~M
o ° _ _
o
olo
I Z
z %
Z 1 -
o :
0 0
cO
i , , , , i , , , , i . . , , , ' '
o o o o
O t/3 Od
N
E
O O
O
O O
O
× X
>
>-
0 Q.
E 8
.E
g
..E
O c~
E
E
(-i

897
AU mass spectra display several common ions (m/z= 104, 110, 130, 145, etc.), which correspond to
fragments retaining the triazine moiety. Obviously, these peaks provide no information on the
photochemi~lly modified substituents of the ring.
In contrast, other spectral features (fragment ions and neutral losses) point out that some specific groups
are included in the molecular structure. For example, the peak at m/zffi 58 Da is observed only when an
isopropylamino- group is linked to the triazine. Note that the isobaric acetylamino- group does not produce
the ion m/zffi 58 (Fig. IC). Likewise, the presence of an ethylamino- group in the structure is often evidenced
by a rather abundant ion at m/zffi 44 Da (Fig. IB, IC, ID and Fig. 9 in Ref.13).
The mass spectra of triazines substituted with a formylamino- group show an intense [M-28] + peak,
corresponding to the facile loss of a CO molecule with rearrangement of a hydrogen atom; consequently, the
molecular ion signal is rather weak (Fig. ID, 2D). The cleavage of the amide bond in acetylamino-substituted
rings is less favorable. Thus, the molecular ion is abundant and alternative fragmentations compete with the
expected losses of the acetyl radical ([M-43] +) and ketene ([M-42]+). The latter peaks originate from either
the homolytic cleavage of the amide bond or a four- or six-membered rearrangement (Fig. IC and Fig. 7 in
Ref. 13). It is noteworthy that the two processes occur to a similar extent in both examples considered.
Scheme I. Proposed reaction pathways of isopropyl degradation.
,,01% OH /OH3 02. ~ -NH-?/'OH3CI-~ "-NH-CH,,cI_I 3 " -NH-C LCI_I3 OH~ O'-OH
-NH-OH:;: 02 /CH2-O-OH l, -NH-OH\oHa H.zO 1 / -NH-CO-CHa
ehydration
-NH-CH#I'I'zOH 0~2 ,,CliO 0 2 .COOH 01"t3 -NH-OH,,c ~ ~-NH-CH\cHa ~I -C02
-NH-Ct-~-C~ -NH-OHO ' ~
As shown in Scheme I, a possible preliminary step of the photocatalyzed decomposition of an isopropyl
substituent consists in the oxidation of one methyl group to the corresponding alcohol or aldehyde (see
compounds XIV and XV, respectively). The resulting products fragment extensively upon EI excitation by
loss of the hydroxymethyl- or formyl- radical, respectively. Cleavage of the C-C bond leads to an extremely
898
stable ion, in which the charge is delocalized on the adjacent nitrogen and the conjugated aromatic ring (Fig.
2B, 2C). Once it is ionized, XV promptly releases the formyl group, giving the fragment ion m/z 214 Da
(Fig. 2C). The same fragment is also produced from compound XIV by the loss of a -CH2OH radical, while
the molecular ion can be barely detected (Fig. 2B). In this spectrum, the occurrence of the [M-I8] + ion (m/z
227), corresponding to the neutral loss of a water molecule, is a further indication that a hydroxyl group is
present in the structure.
The products found in samples and their relative abundance are fully consistent with the
photodegradation mechanisms proposed, viz. no product expected based on the photodecomposition pathway is
actually missing, and the product distribution observed clearly correspond to the relative favorableness of
competitive degradations. For example, in the decomposition of atrazine the identified products originate from
either the characteristic oxidation of the ethyl or the isopropyl group. The same oxidation processes have been
previously recognized for the symmetrical structures of simazine and propazine. The abundance of the
corresponding products suggests that the isopropyl group is more readily oxidized than the ethyl. In fact, III is
much more abundant than XVIII, although XVII and XX are produced in larger amounts than XXII and
XXIII.
Deeradation kinetics.
The evolution of the concentration of initial compounds, identified intermediates and the final product
(cyanuric acid) was followed as a function of the irradiation time. The results are displayed in Figures 3-10.
LO O X
( -
N eU
E . I
GO
15 25
10
5
0 I I I I
0 10 20 30 40 i
50
A
60
Irradiation time (min)
O O X
o
r - o ~
N
E CO
I i 1
c-
O £3. E O O
20
15
10
5
0
B
(~\\comp.(lll) I \ i "e I -V1
I / \ I / \ I / \
I [~] \\
1 / ' ,~...comp.(I) \\ " '~, ........ \
. . . . . . .
I I I I I
0 10 20 30 40 50 60
Irradiation time (min)
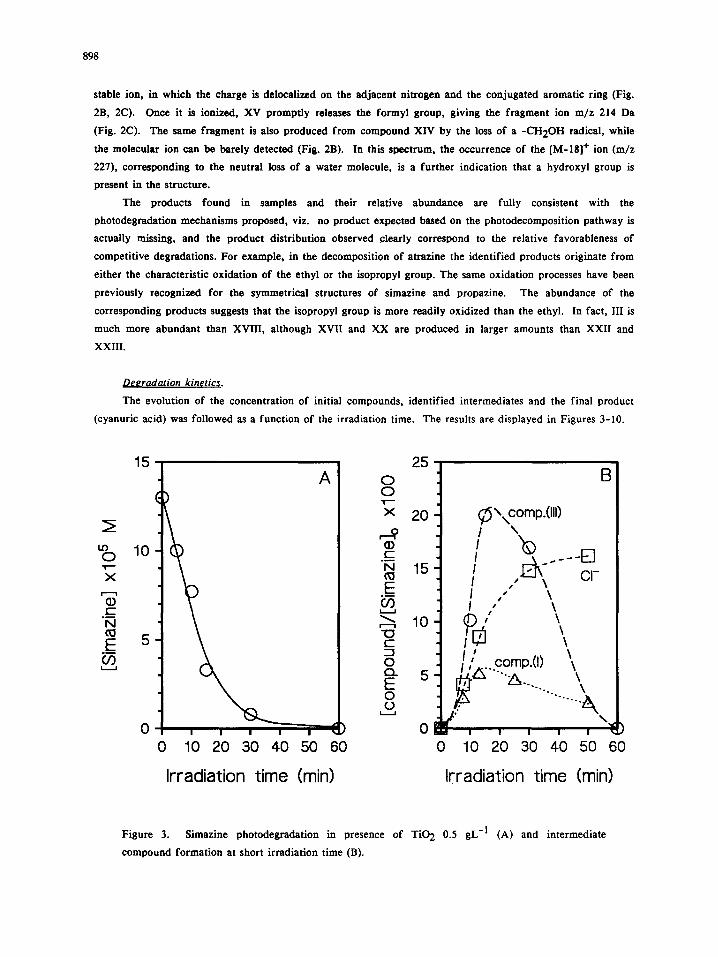
Figure 3. Simazine photodegradation in presence of TiO 2 0.5 gL -1 (A) and intermediate
compound formation at short irradiation time (B).
899
LO 0 X
-C] ¢-
o Q. E 0 0
15
10
5
0
p.(lll)
-E ]
) i
0 10 20 30
Irradiation time (min)
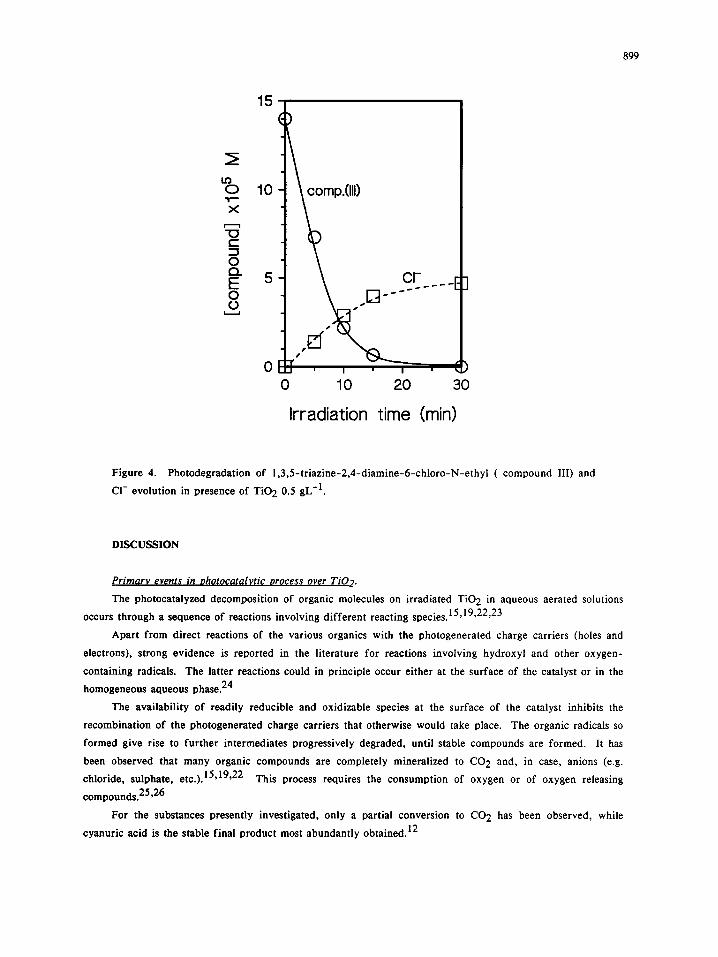
Figure 4. Photodegradation of 1,3,5-triazine-2,4-diamine-6-chloro-N-ethyl ( compound III) and
CI- evolution in presence of TiO 2 0.5 gL-1.
DISCUSSION
Primary events in Dhotocatalvtic process over TiO 2.
The photocatalyzed decomposition of organic molecules on irradiated TiO 2 in aqueous aerated solutions
occurs through a sequence of reactions involving different reacting species. 15'19'22'23
Apart from direct reactions of the various organics with the photogenerated charge carriers (holes and
electrons), strong evidence is reported in the literature for reactions involving hydroxyl and other oxygen-
containing radicals. The latter reactions could in principle occur either at the surface of the catalyst or in the
homogeneous aqueous phase. 24
The availability of readily reducible and oxidizable species at the surface of the catalyst inhibits the
recombination of the photogenerated charge carriers that otherwise would take place. The organic radicals so
formed give rise to further intermediates progressively degraded, until stable compounds are formed. It has
been observed that many organic compounds are completely mineralized to CO 2 and, in case, anions (e.g.
chloride, sulphate, etc.). 15,19,22 This process requires the consumption of oxygen or of oxygen releasing
compounds.25,26
For the substances presently investigated, only a partial conversion to CO 2 has been observed, while
cyanuric acid is the stable final product most abundantly obtained. 12
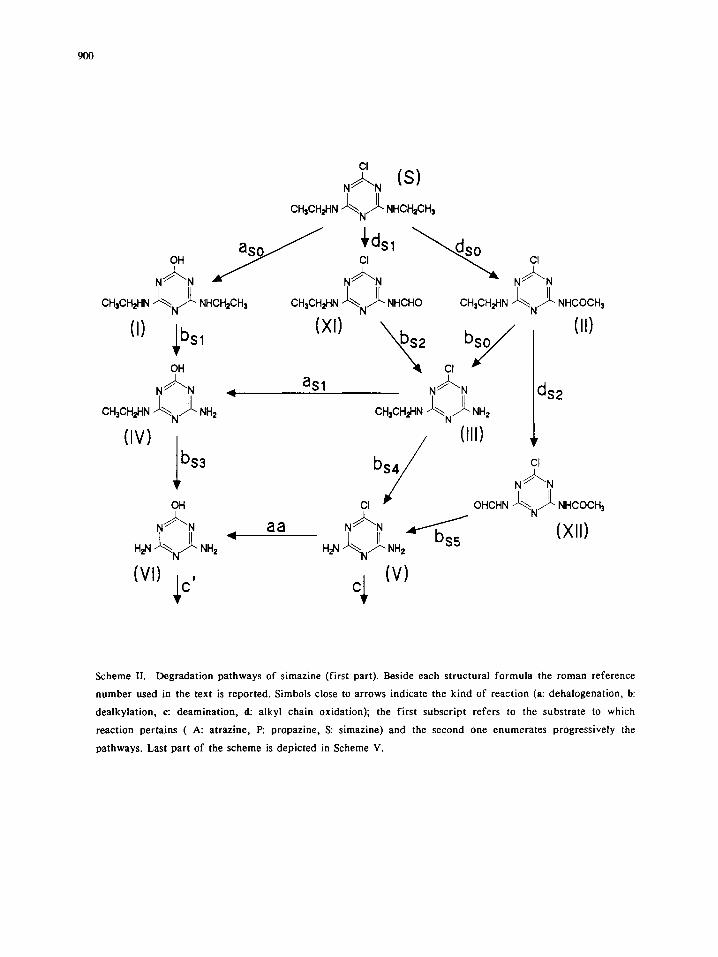
900
OH N~'~N
CHsCH~N -J~.~N ~j" NHCH,CHs
(I) ;bsl OH
N ~''N ~1 OH,CHiN -J'-~.~N..~L- NH ,
(IV) lbsa OH
(Vl) ;c'
CI
OH,CHiN ~ N ~L NHCH2CHs
CH,CHz~rtN -J'~N~L NHCHO
(Xl) "~2 c,
asl N~J'-..N
CH,OHxk-IN ~'-~N ~" NH,
a a
CI N~N
I CH,CH2HN ~.~N./~1" NHCOCH,
bso~ (it)
ds2
(ill) CI
N~N b/
Cl N~N
C~ (V)
J I OHOHN ~L~N/J~ NHOOOH 3
(xll)
Scheme II. Degradation pathways of simazine (first part). Beside each structural formula the roman reference
number used in the text is reported. Simbols close to arrows indicate the kind of reaction (a: dehalogenation, b:
dealkylation, c: deamination, d: alkyl chain oxidation); the first subscript refers to the substrate to which
reaction pertains ( A: atrazine, P: propazine, S: simazine) and the second one enumerates progressively the
pathways. Last part of the scheme is depicted in Scheme V.
901
Simazine Degradation.
The major photocatalytic degradation paths of simazine are depicted in Scheme II and V, as deduced
from chromatographic analysis and mass spectrometric data. As mentioned in the previous section, N-
ethylammelide (originating from IV) and 2-chloro-4-hydroxy-6-ethylamino-l ,3,5-tr iazine (originating from
III) are present only in traces and not shown in the scheme. These compounds together with other
dechlorinated derivatives were also found in the treatment of atrazine with UV/ozonation. 27,28
From the comparison of Figure 3A and 3B, it can be deduced that path as0 accounts for ca. 10% of
simazine disappearance (after 15 minutes ca. 75% of simazine is degraded, but compound I and chloride ions
reach the stoichiometric molar ratio of ca. 0.06 and 0.10, respectively). Figure 3B shows also the concentration
variation as a function of time for compound III, which is the most relevant intermediate after 5 rain of
irradiation, but decreases rapidly at longer irradiation times. Compound III can originate from deacetylation
of compound II and through the formation of XI and subsequent oxidation/decarboxylation. Compound III
disappears even faster than simazine as shown in Fig. 4, but in this case dehalogenation route seems to play a
more important role (note that after 15 minutes, when the concentration of III is reduced at ca. 5% of the
initial one, chloride ions are ca. 30% of stoichiometric).
~ t ~ t
o 75 viii A o 40 I~ B
6O ~ ..A.. / ,
~ / ,,"'..... v ~,, ~,~ ~- /,~ ~--..... ',,, 020 ,~, -"... D
~ot :/v / x I ~ : A I x '
0 5 12 16 0 5 14 16
Irradiation time (hours) Irradiation time (hours)
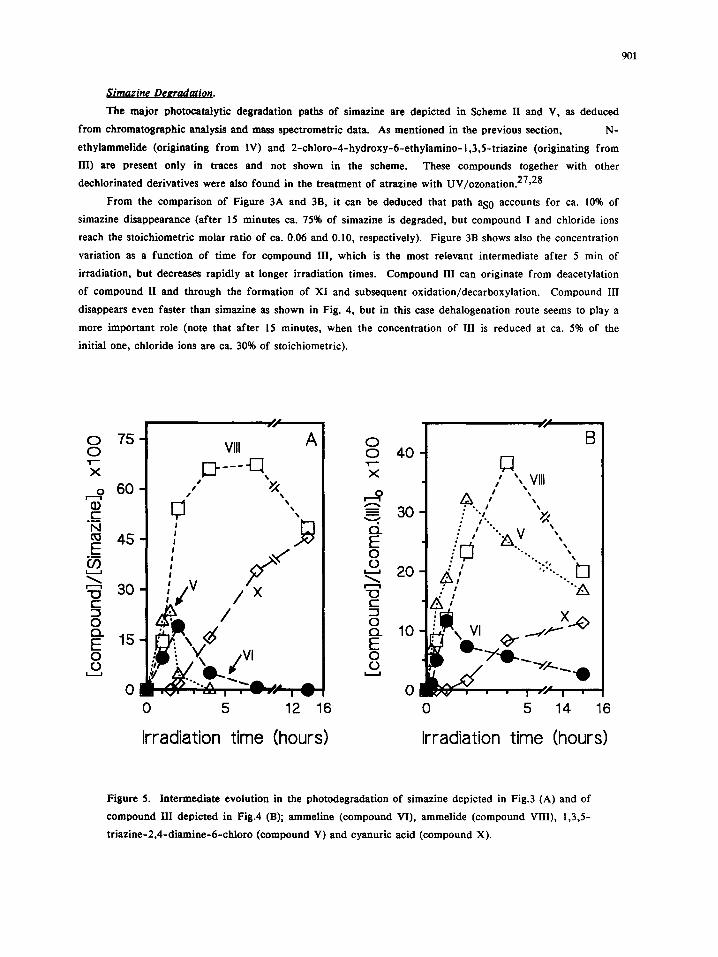
Figure 5. Intermediate evolution in the photodegradation of simazine depicted in Fig.3 (A) and of
compound III depicted in Fig.4 (B); ammeline (compound VI), ammelide (compound VIII), 1,3,5-
tr iazine-2,4-diamine-6-chloro (compound V) and cyanuric acid (compound X).

902
Then, the decomposition at the early stages apparently proceeds through a competition between the
chlorine substitution (at position 6) by a hydroxyl group (a wall-known process in the photocatalytic
transformation of other haloaromatic compounds) 15,16,22 and the oxidation of the ethylamino groups at the 2
and 4 positions of the 1,3,5-triazine ring.
The mechanism of the latter process, on the basLs of the product identified (II and, in a lesser extent,
XI), can be attributed to the reaction of a hydroxyl radical on the methylene carbon of the ethyl group. The
free radical formed thereafter can react with molecular oxygen, as follows:
"OH
-NH-CH 2-cH 3 > -NH-CH-CH 3
0 - 0 • O-OH
o2 I I > -NH-CH-CH 3 > -NH-CH-CH 3
the hydroperoxide can either dehydrate or rearrange, yielding II and XI, respectively. Further oxidation steps
can cause carboxylic group formation. The subsequent photocatalytic decarboxylation (also a well-known
process) leads to dealkylated compounds. 19'23'29'30
The onset and decrease of intermediates V, VI (ammeline), VIII (ammelide) and the final product X
(cyanuric acid) is depicted in Fig. 5A and 5B for the degradation of simazine and compound III, respectively.
As expected, the general trend is similar for simazine and III, being compound V the intermediate appearing
first and ammelide the intermediate that reaches the highest concentration. Interestingly, compound XII
(although detected in low concentration) suggests that V can originate also through the sequence ds0 + ds2 +
dss. Since it has been shown that path aa (giving ammeline from V) is negligible (essentially only traces of
ammelide are observed in the photocatalytic degradation of V and the formation of cyanuric acid goes mainly
through path c + c"' + aa'), 13 ammeline formation in simazine degradation (up to the maximum value of ca.
15%) has to occur mainly through the sequence as0 + bSl + bs3 from simazine and aSl + bs3 from III. The
final slow formation of cyanuric acid from ammelide and V has been reported. 13
Several intermediates present in Scheme II have been identified in previous studies on s-triazine
herbicides transformations. The main differences arise from the relative importance of the different pathways.
While soil transformation occurs mainly through substitution of chlorine with a hydroxyl group, 3
microbial degradation takes place essentially through dealkylation paths. 7 Metabolic degradation observed in
chicken goes in both directions. 5
The action of chemical oxidizing agents such as ozone 27'28 and Fenton's reagent 31 (ferrous sulfate -
hydrogen peroxide) similarly lead to extensive dealkylation together with partial hydroxy substitution of the
chlorine atom.
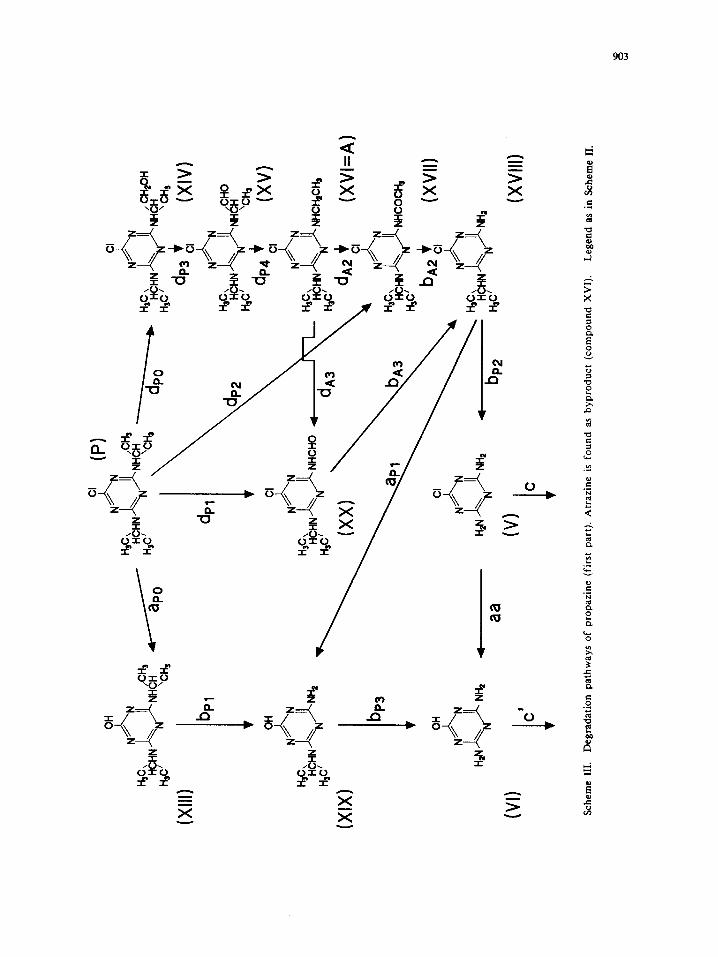
ProDazine Deeradalion.
Scheme III represents the degradation routes for photocatalytic degradation of propazine. Although the
scheme is similar to that for simazine (propyl instead of ethyl substituents at the amino groups), some
qualitative and quantitative differences can be observed in the plots of intermediate concentrations versus
time.
Compounds XIV and XV (Scheme III), that are amongst the possible compounds formed through
oxidation of the propyl group, can be detected after short irradiation times (5 and 10 minutes). After the
same irradiation period compound XVIII is the major intermediate detected.
OH J"'N
I~C.
, ~.
~ II
/C
H3
HCHN
"~. ~
NH
CH
1.130
/ N
\C
H~
(XIII)
lbp
1
OH
N-'2"L.N
H3
C~
I HC
HN'J~
.~ .~
NH
, H=
C ~
N
(XlX
) lbe
3
OH
N~',,,
N H~
~ NH
=
(Vl)
$c'
.~.
(P)
o,
H3C,
, 'J
' H
,,Cl.~
N ,
,,'~',,.
, N
....
x
d. v
H,,C
/ N
, "C
HI
I \
d,,c,, Ix,vl
dp 1
~ ,, ~
L"N
~_
s
H3C'
, ~,
~ I[
/Clio
I
\ap2
HCHN
-"~.
.~NH
CH
'i, \"
H,c
' ",
"c~
o ~
dp4 ~
(X
V)
N/~
"N
\ " "
~N
H,c'
,"
. dA
3 \
~c,'_
?
-(xx)
~
'-
\ a.,
,.2 c',
(XV,
=A)
\ ~c
"J"-N
b
,t aa
H~
I'~N.
~"NH
, =
bp 2
~HN
~'~N
H, (
XVll)
(v) ~
c (xv
ltt)
Sch
eme
III.
Deg
rada
tion
pat
hway
s of
pro
pazi
ne (
firs
t pa
rt).
Atr
azin
e is
fou
nd a
s by
prod
uct
(com
poun
d X
VI)
. L
egen
d as
in
Sch
eme
II.
O '1=-"
X r - ' - i
r " N
Q . O
El_ I,--,..I
Other interesting compounds detected at non-negligible concentration arc XVII and XX, present also in
the atrazine degradation (see Scheme IV). It is worth noting that also atrazine is likely a propazine
degradation side-product, present in traces. Different pathways (some of which not depicted in the scheme)
can be responsible of the presence of these derivatives. From the products identified above, the mechanism
depicted in Scheme III can be hypothesized; the reaction sequence proposes an initial attack of hydroxyl
radicals (or other hydrogen atom abstracting species) to the side chains to form primary radicals, which further
react with 02 and H20.
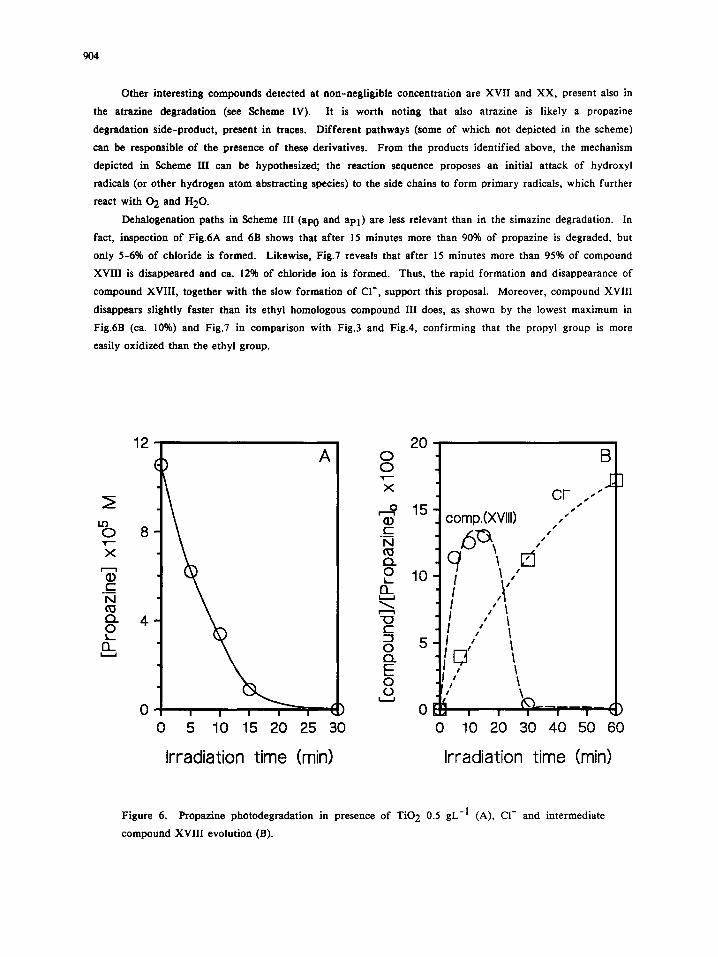
Dehalogenation paths in Scheme III (ap0 and apl ) are less relevant than in the simazine degradation. In
fact, inspection of Fig.6A and 6B shows that after 15 minutes more than 90% of propazine is degraded, but
only 5-6% of chloride is formed. Likewise, Fig.7 reveals that after 15 minutes more than 95% of compound
XVIII is disappeared and ca. 12% of chloride ion is formed. Thus, the rapid formation and disappearance of
compound XVIII, together with the slow formation of CI-, support this proposal. Moreover, compound XVIII
disappears slightly faster than its ethyl homologous compound III does, as shown by the lowest maximum in
Fig.6B (ca. 10%) and Fig.7 in comparison with Fig.3 and Fig.4, confirming that the propyl group is more
easily oxidized than the ethyl group.
4
12 20
0 I I I I I
5 10 15 20 25
A O O X
o Q.) o- N
O
13_ I , , - - I
" 0 c- "-I 0
E o 0
9O4
15
10
5
0
CF a j ~ [ ] J
s s
s J
0 30 40 50 60
c0mp.(XVlll)
! ,t"
[ , I t , I
I I !
0 10 20 30
Irradiation time (min) Irradiation time (min)
Figure 6. Propazine photodegradation in presence of TiO 2 0.5 gL -1 (A), CI- and intermediate
compound XVIII evolution (B).
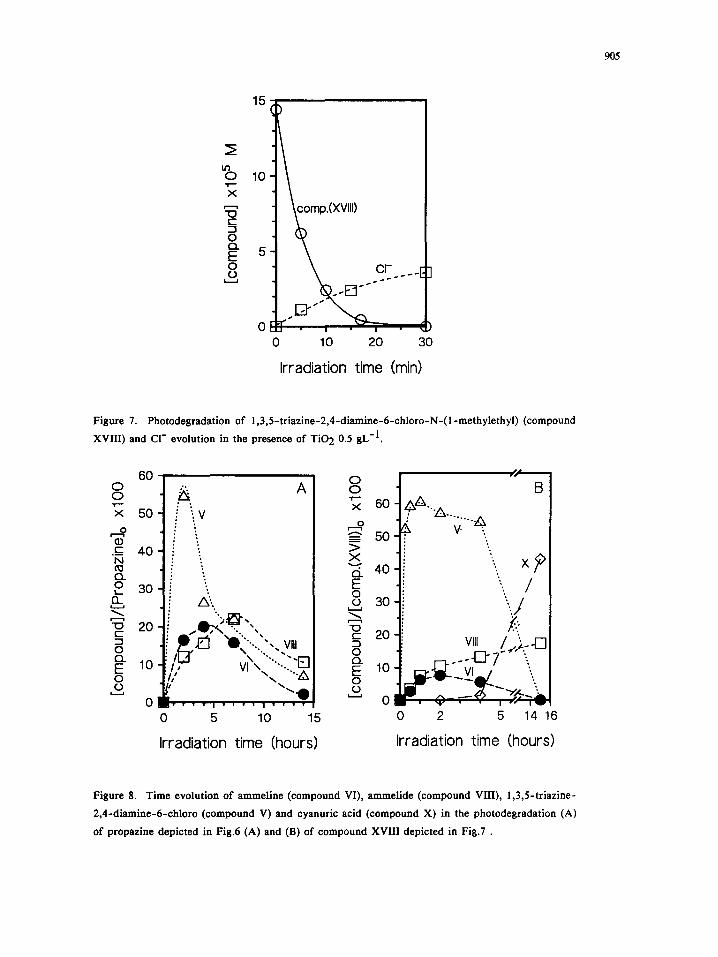
905
15
2~
O
x
"o c 0 ~L E O o
10
mp.(XVlll)
k ~ . E y o r . . . . r.
0 10 20 30
Irradiation time (min)
Figure 7. Photodegradation of 1,3,5-triazine-2,4-diamine-6-chloro-N-(l-methylethyl) (compound
XVIII) and C1- evolution in the presence of TiO 2 0.5 gL-1.
60 o A o "&
,- f,.. x 50 , V
. _ 4 0 N
O 30 .a_, X'..
20 i / "', o ='- ti ~,z~ "e~'....'-,, viii
10 ~S,I,I~ \ , ~ ..12 E v~" .. 0
, o 0 ~ , , "@
0 5 10 15
Irradiation time (hours)
Pf r r
B 0 x 60 z%.. %.
_~ 50 >
X " F £~_ 40 ;. E ., / O " o ao - j
. . - 4 3 - 7 " " oE 1° 1 J ~ " ~ - v - ' ~ . , ' 1
.o 0 I P - , - e - - ~ . . e - ~ 0 2 5 14 16
Irradiation time (hours)
Figure 8. Time evolution of ammeline (compound VI), ammelide (compound VIII), 1,3,5-triazine-
2,4-diamine-6-chloro (compound V) and cyanuric acid (compound X) in the photodegradation (A)
of propazine depicted in Fig.6 (A) and (B) of compound XVIII depicted in Fig.7 .
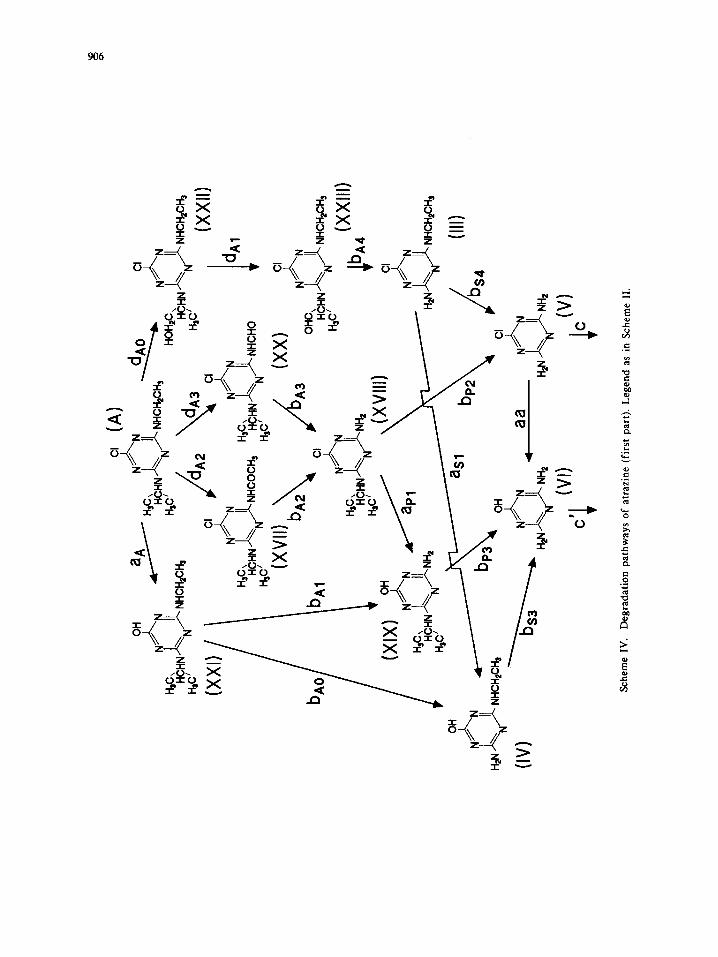
(IV)
,Q (
A)
OH
a A
N "~"
"N
dAo
cl ,-~
'-,
~'~
,N~
.~N
,=,.
c,.~
.~
, I-I:
C\ ~
II
I.I:C
/ N
HOH2
C\ -j-
jj
H:cH?
HN"~N
/~" NHC
H~X~H
: //d
~
d A
3 HH
CHN-
~N,/~
- NH
CH:CH
.. (X
Xl)
/ /
N.~N
~A
2
x~
~ I
(XX
ll)
N N
d /
\ HC
HN .-L
~. ,...~
k. NHC
O CH:
HC
HN--~
~k. N
HCHO
H:
C\
~ I
H:C\
I
A1
/ ~
H,c'
N H~
C' --
/
\ (x
v")b
\ /b
(x
x)
bAo/
\b
^,
^2~
Cl /U
A3
OHHC
~HN~
,~-.NH
CH.CH
:
/ ~
HCI-IN
~./~'N
I-12
lUA4
~
lr
H,C
~
N
I' /
(x,x
)~
~ ~x
v,,,)
/
.._
N ? "
,N ~
ap1
~ ,N ~
"N
OH ~
I~
(~HN
"L~N
"~'N
H'
"~
Hi
~l "L'~
N~ NH
Ci'IIC
H:
i~'~.
~ ~
as'
bp2~
//b
s 4
(''')
~ NH
CH2C
I'I3 CI
jl~
N/
~"N
aa
N/~L
"N
H~ ~N
~ N
H, (v
l) (v
) c'[
,~c
S
chem
e IV
. D
egra
dati
on p
athw
ays
of a
traz
ine
(fir
st p
art)
. L
egen
d as
in
Sch
eme
II.

907
l from
(Vl) OH N.J"-.N
j
C
(Vlll) OH N-"~N
HO-~.~N/~" NH,
previous
aa
schemes I
Cl N.J.~, N (V)
I C
c, (Vll) a a ' N~"-N
IC" IC '" (X) N..~ N ~, aa" N I N (IX)
- . O \ N J - O =
Scheme V. Degradation pathways for simazine, propazine and atrazine (last part). Legend as in
Scheme II.
These considerations suggest that the degradation paths occurring through compounds XVIII and XX are
predominant. Accordingly, compound V is the main intermediate detected in the degradation of both
propazine (Fig. 8A) and compound XVIII (Fig. 8B) and ammeline has been produced in lower amount in
comparison with the case of simazine. Ammeline originates from propazine through routes ap0 + bp1 + bp3
and from XVIII through apl + bp3.
Atrazine deeradalion.
The degradation of atrazine (the asymmetrically substituted compound) can be rationalized in the light of
the previously cited findings. In Scheme IV, the main degradation pathways are depicted. While compounds
XXI, XXII and XXIII are peculiar of the atrazine photodegradation, compounds XVII and XX were detected
also in the propazine decomposition. Anyhow, the chemistry of the atrazine degradation processes is the same
discussed for simazine (on the ethyl substituent) and propazine (on the isopropyl substituent).
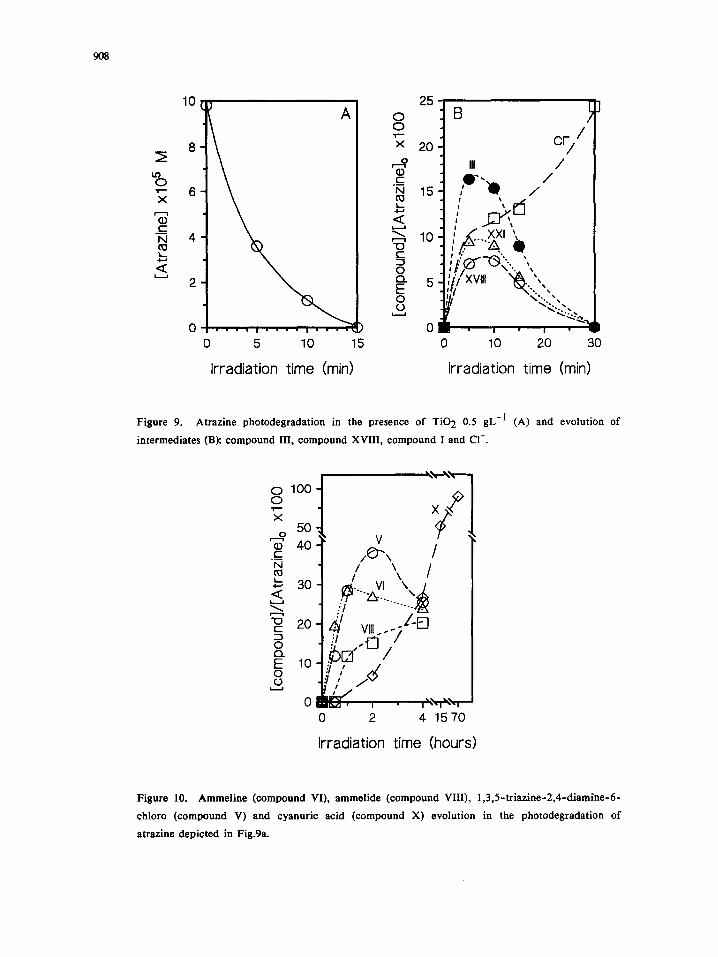
908
t t )
O x
cD E U
<
10
0
A
5 10 15
Irradiation time (min)
O O x
cD
N
< t ' - - I
E O O. E O o
25
20
15
10
B
,e'-, e jz , d
I
cl[// /
/ /
2f'-x3~... ',,
5 XVlll '~...,. " ,
0 10 20 30
Irradiation time (min)
Figure 9. Atrazine photodegradation in the presence of TiO 2 0.5 gL =1 (A) and evolution of
intermediates (B): compound III, compound XVIII, compound I and CI-.
o 100 O X < ~ > ' I " - -
X ~ 5o ~ 4o
.___ / N
~ 3o <
~ 2o E
O
E lO O o
0 .--ca , , , ' - ~ - , 0 2 4 1570
Irradiation time (hours)
Figure 10. Ammeline (compound VI), ammelide (compound VIII), 1,3,5-triazine-2,4-diamine-6-
chloro (compound V) and cyanuric acid (compound X) evolution in the photodegradation of
atrazine depicted in Fig.9a.

9O9
Dehalogenation route a A can account for ca. 8%, as from comparison of data in Fig 9B and 10. In the
meanwhile, compound III reaches higher concentrations than XVIII, as expected if the attack to the isopropyi
group is favored.
The behavior of intermediates reported in Fig. 10 shows that several paths contribute comparably to the
progress of the degradation: thus, V originates through paths bA2 (or hA3) + bp2 and hA4 + bs4; ammeline
through paths involving dehalogenation and further dealkylation of intermediates III, XVIII, IV and XIX.
CONCLUSIONS
From the present study it can be concluded that the photoeatalytic degradation of 2-ehloro-s-triazine
pesticides occurs through dehalogenation routes only in a very limited extent. Alkyl chain oxidation and
subsequent dealkylation represent the main degradation path, followed by the formation of hydroxy
derivatives, up to the final product, namely cyanuric acid.
The choice of proper degradation conditions and of the analytical procedures allows a detailed
identification of the reaction intermediates. The same compounds found in this in vitro study are likely to be
present also in other transformation routes taking place in the environment or in metabolic processes.
It is noteworthy that upon the change of the alkyl substituent in the side amino groups, viz. from ethyl-
ethyl (simazine), to propyl-propyl (propazine) and finally to the asymmetric derivative bearing an ethyl and a
propyl (atrazine), relevant variations are induced in the relative role of different pathways depicted in the
degradation schemes.
ACKNOWLEDGEMENTS
This research received generous support from Eniricerche and from EEC (Contract EV4V-0068-C(CD)).
REFERENCES
1) H.J.Sanders, Chem.Eng.News, (August 3, 1981) p.20.
2) G.L.Berg, "Farm Chemical Handbook" (Meister Publ., Willonghby, Ohio, 1985).
3) S.U.Khan, "Pesticides in the Soil Environment" (Elsevier, Amsterdam, 1980).
4) S.U.Khan and M.Schnitzer, J.Environ.Sci.Health B 13, 299 (1978).
5) T.S.Foster and S.V.Khan, J.Agric.Food Chem. 24, 566 (1976).
6) M.L.Montgomery, D.L.Botsford and V.H.Freed, J.Agric. Food Chem. 17, 1241 (1969).
7) D.D.Kaufman and P.C.Kearney, Residue Rev. 32, 235 (1970).
8) J.E.Bakke, J.D.Larson and J.Price, J.Agr.Food Chem. 20, 602 (1972).
9) D.F.Paris and D.L.Lewis, Residue Rev. 45, 95 (1973).
10) C.I.Harris, J.Agr.Food Chem. 15, 157 (1967).
11) W.E.Pereira and C.E.Rostad, Environ.Sci.Technol. 24, 1400 (1990).
12) E.Pelizzetti, C.Minero, E.Pramauro, M.Barbeni, V.Maurino and M.L.Tosato, Chim.lnd. (Milano) 69(10), 88
(1987).

910
13) E.Pelizzetti, V.Maurino, C.Minero, V.Carlin, E.Pramauro, O.Zerbinati and M.L.Tosato, Environ.Sci.Technol.
24, 1559 (1990).
14) E.Pelizzetti, V.Carlin, V.Maurino, C.Minero, M.Dolei and A.Marehesini, Soil Sci. 150, 523 (1990).
15) D.F.Ollis, E.Pelizzetti and N.Sorpone, in "Photocatalysis" N.Serpone and E.Pelizzetti Eds. (Wiley, New
York, 1989), Ch. 18.
16) D.F.OIIis, Environ.Sci.Technol. 19, 480 (1985).
17) S.J.Plust, J.R.Loehe, F.J.Feher, J.H.Benediet and H.F.Herbrandson, J.Org.Chem. 46, 3661 (1981).
18) J.Calvert and J.N.Pitts, "Photochemistry" (Wiley, New York, 1966).
19) R.W.Matthews, J.Catal. 111,264 (1988).
20) C.E.Rostad, W.E.Pereira and T.J.Leiker, Biomed.Environ.Mass Spectrom. 18, 820 (1989).
21) L.Q.Huang and M.J.I.Mattina, Biomed.Environ.Mass Spectrom. 18, 828 (1989).
22) J.C.D'Oliveira, G.AI-Sayyed and P.Pichat, Environ.Sci. Technol. 24, 990 (1990).
23) A.Chemseddine and H.P.Bohem, J.Mol.Cat. 60, 295 (1990).
24) C.S.Turehi and D.F.Ollis, J.Catal. 122, 178 (1990).
25) E.Pelizzetti, V.Carlin, C.Minero and M.Graetzel, New J.Chem., 15, 351 (1991).
26) A.Sclafani, L.Palmisano and E.Davi, New J.Chem. 14, 265 (1990).
27) P.C.Kearney, M.T.Muldoon, C.J.Somich, J.M.Ruth and D.J.Voaden, J.Agric.Food Chem. 36, 1301 (1988).
28) C.J.Somich, M.T.Muldoon and P.C.Kearney, Environ.Sci.Technol. 24, 745 (1990).
29) B.Kriutler and A.J.Bard, J.Am.Chem.Soc. 100, 5985 (1978).
30) J.M.Herrmann, M.N.Mozzanega and P.Pichat, J. Photochem. 22, 333 (1983).
31) J.R.Plimmer, P.C.Kearney and U.LKlingebill, J.Agric.Food Chem. 19, 572 (1971).
(Received in Germany 21 October 1991; accepted 8 January 1992)




















