
(I) Synthesis of a macrocyclic complex praeseodymium and (II ...
78
Rochester Institute of Technology Rochester Institute of Technology RIT Scholar Works RIT Scholar Works Theses 8-14-1993 Organometallics: (I) Synthesis of a macrocyclic complex Organometallics: (I) Synthesis of a macrocyclic complex praeseodymium and (II ) effect of wilkinson's catalyst on the praeseodymium and (II ) effect of wilkinson's catalyst on the cleavage of cyclohexene oxide by borane cleavage of cyclohexene oxide by borane Carolyn Ruth Follow this and additional works at: https://scholarworks.rit.edu/theses Recommended Citation Recommended Citation Ruth, Carolyn, "Organometallics: (I) Synthesis of a macrocyclic complex praeseodymium and (II ) effect of wilkinson's catalyst on the cleavage of cyclohexene oxide by borane" (1993). Thesis. Rochester Institute of Technology. Accessed from This Thesis is brought to you for free and open access by RIT Scholar Works. It has been accepted for inclusion in Theses by an authorized administrator of RIT Scholar Works. For more information, please contact [email protected].
-
Upload
khangminh22 -
Category
Documents
-
view
3 -
download
0
Transcript of (I) Synthesis of a macrocyclic complex praeseodymium and (II ...

Rochester Institute of Technology Rochester Institute of Technology
RIT Scholar Works RIT Scholar Works
Theses
8-14-1993
Organometallics: (I) Synthesis of a macrocyclic complex Organometallics: (I) Synthesis of a macrocyclic complex
praeseodymium and (II ) effect of wilkinson's catalyst on the praeseodymium and (II ) effect of wilkinson's catalyst on the
cleavage of cyclohexene oxide by borane cleavage of cyclohexene oxide by borane
Carolyn Ruth
Follow this and additional works at: https://scholarworks.rit.edu/theses
Recommended Citation Recommended Citation Ruth, Carolyn, "Organometallics: (I) Synthesis of a macrocyclic complex praeseodymium and (II ) effect of wilkinson's catalyst on the cleavage of cyclohexene oxide by borane" (1993). Thesis. Rochester Institute of Technology. Accessed from
This Thesis is brought to you for free and open access by RIT Scholar Works. It has been accepted for inclusion in Theses by an authorized administrator of RIT Scholar Works. For more information, please contact [email protected].

ORGANOMETALLlCS: (I) SYNTHESIS OF A MACROCYCLIC COMPLEXOF PRAESEODYMIUM and (II) EFFECT OFWILKINSON'S CATALYST ON THE CLEAVAGE OFCYCLOHEXENE OXIDE BY BORANE
CAROLYN RUTH
AUGUST, 1993
THESIS
SUBMITTED IN PARTIAL FULFILLMENT OF THEREQUIREMENTS FOR THE DEGREE OF MASTER OF SCIENCE
APPROVED:
Terence C. MorrillProject Advisor
Department Head
Rochester Institute of TechnologyRochester, New York 14623
Department of Chemistry

PERMISSION GRANTED
Title of Thesis: ORGANOMETALLlCS: (I) SYNTHESIS OF A
MACROCYCLIC COMPLEX OF PRAESEODYMIUM and (II) EFFECT OF
WILKINSON'S CATALYST ON THE CLEAVAGE OF CYCLOHEXENE OXIDE
BY BORANE
I, C AtZo LyAi RLlT H_. hereby grant permission to
the Wallace Memorial Library of the Rochester Institute of Technology to
reproduce my thesis in whole or in part. Any reproduction will not be for
commercial use or profit.
Signature of Author

ACKNOWLEDGMENTS
The author would like to thank the Department of Chemistry at
Rochester Institute of Technology for the support of this work as well as Dr.
T.C. Morrill, research advisor, and Drs. R. Gilman, K. Turner, and W. Hallows
for serving as members of the research advisory committee.

ABSTRACT
(I) The template synthesis and characterization of the complex of
praeseodymium acetate from 2,6-diacetylpyridine and ortho-
phenylenediamine was attempted. The attempted synthesis produced a
green precipitate with a high melting point and poor solubility in organic
solvents, unlikely characteristics for compound 1 seen on the
page. This project was set aside due to the apparent failure to produce the
desired compound.
(II) The effect ofWilkinson's catalyst on the treatment of cyclohexene
oxide with borane-THF at72 for 5 1/2 hours was also investigated.
Oxidation of the organoborane formed by both the catalyzed and
uncatalyzed reactions produced cyclohexanol. Another significant product
was one which, according to GC-MS, had a molecular weight of 172. This
product was not completely identified. Studies of the progress of both the
catalyzed and uncatalyzed reactions were conducted using TLC. TLC
showed no evidence of the starting material, cyclohexene oxide, after 30
minutes of reflux. No change in the product mixture measured by TLC
occurred after 30 minutes for the catalyzed reaction and after 60 minutes for
the uncatalyzed reaction, indicating that the reactions were probably
complete at that point.

TABLE OF CONTENTS
SECTION PAGE
I. INTRODUCTION 1
II. PART ONE: HISTORICAL REVIEW 3
III. PART TWO: HISTORICAL REVIEW 9
IV. EXPERIMENTAL SECTION: Instrumentation and Chemicals 19
V. PART ONE: EXPERIMENTAL SECTION 21
VI. PART TWO: EXPERIMENTAL SECTION
A. Catalyzed Reaction 24
B. Uncatalyzed Reaction 30
C. Reaction Study Using TLC 35
VII. PART ONE: RESULTS AND DISCUSSION 41
VIII. PART TWO: RESULTS AND DISCUSSION 45
IX. REFERENCES 53
X. APPENDIX: IR, GC, GC-MS, and NMR Spectra 56
iii

INTRODUCTION
This thesis deals with two different research projects, both of which
are described below.
Part One describes the first research project: the attempted
synthesis and characterization of the praeseodymium (III) macrocyclic
complex (1) derived from 2,6-diacetylpyridine and orf/70-phenylene diamine.
2 OAc
CI-
n H20
(1)
Part Two describes the second research project: a study of the
hydroboration of cyclohexene oxide (2 ) and the effect of Wilkinsons catalyst
1

on this reaction. The products formed and the effect of the catalyst upon the
progress of the reaction were examined.
+ BHyTHFcatalyst
THF
Ethanol
NaOH
H202
(2)
no catalyst
+ BH3THF ==>3
THF
Ethanol
NaOH
H202
(2)

PART ONE: HISTORICAL REVIEW
Nuclear magnetic resonance is the most useful technique for organic
structure determination. 1H NMR, which is frequently used, reveals the
number of nonequivalent protons present in a compound, the number of
neighboring protons, and finally the total number of each type of proton. As
with any spectrometric technique, resolution is a major concern, where
resolution is the separation between 1H NMR signals. One method to
improve resolution is to increase the strength of the instrument magnetic
field in order to separate overlapping chemical shifts of nonequivalent
protons.
Solvent induced shifts have also proven to be useful in the
simplification of NMR spectra as demonstrated by Hinckley in 1969 (1). In
addition, paramagnetic ions were known to electrostatically interact with
electron rich substrates to cause NMR signal shifts as demonstrated by
Sanders and Williams in 1970 (2). As a result, the field of lanthanide shift
reagents expanded greatly during the next decade or so.
As the name suggests, LSRs (lanthanide shift reagents) employ
lanthanide metals to enhance the nonequivalence of nuclei to produce, in
many cases, first-order proton NMR spectra. LSRs require three basic
characteristics. First, the lanthanide metal cations must be paramagnetic.
Secondly, these LSRs must function as Lewis-acids: the Lewis-acidic site is
clearly the metal cation and these coordinate with the Lewis-base sites on
the organic substrate. Third, the LSRs should be soluble in nonpolar
organic solvents commonly used for NMR analysis. The NMR solvent
should be a nonpolar organic compound that is less Lewis-basic than the

substrate in order to avoid competition between the substrate and solvent for
the LSR (3).
In Figure 1 below, a proton NMR spectrum of 0.1 M toluene in
deuterated chloroform shows the effect of the binuclear shift reagents used
for spectrum (b) when compared with spectrum (a). The binuclear shift
reagents have peaks of their own, of course, but fortunately these peaks do
not appear in the same region as the toluene peaks. In Figure 2, the
structure of the shift reagents are shown (4).
"B Hc
Pr
7 6 5 4 3 2 1 Oppm
Proton NMR spectrum of 01 M toluene in CDCIj with (a) no shift reagent.
ndfb/0 2M PrfJFodi, and 0.2 M Ag?od)
o<CH>>
oQuu'CF,CF,CF,
oQuu.'CF,CF,CF,
Figure 1 Figure 2.
Paramagnetic ions exert a strong magnetic field of their own which
has the capability of inducing shifts and thus spreading out NMR spectra (5).
In order for this secondary magnetic field to be influential, the nucleus of the
substrate must be in the vicinity of the cation. This is achieved due to the
equilibria established in the following equation:
4

LSR + S = LSR-S where S = organic substrate
Crown ethers, macrocylic ethers of ethylene glycol, have been used
to encapsulate lanthanide metal cations. Employing crown ethers with the
appropriate cavity size made it possible for the metal cation to form
coordinate covalent bonds to the oxygen atoms in the ether. At the same
time, the hydrocarbon exterior around the metal cation made solubility in
nonpolar solvents possible. DeCann characterized the 15-crown-5
praeseodymium (III) nitrate complex and attempted to employ it as an NMR
shift reagent (5). The complex, however, was only soluble in polar solvents
and dissociated in solution. It was not any more effective as an NMR shift
reagent than praeseodymium (III) nitrate alone.
Nitrogen-containing macrocycles were shown to have some
advantages over crown ethers in 1983 by Hiroshi and Tsukube (6). The
hexaaza and tetraaza nitrogen containing macrocycles, shown in Figure 3,
were selective for ammonium cations over potassium cations as opposed to
a typical crown ether which was less selective among cations. The host
cation also bound more tightly to the nitrogen atoms of the polyamines than
to the oxygen atoms of the polyethers. This enabled the complex to be more
stable in solution.
c. . ; c 3ft.CM.fth
ft.CM2fth
Figure 3.

The field of lanthanide metal/nitrogen containing macrocyclic
complexes and their potential as proton NMR shift reagents has been
investigated by many researchers. In 1979, Alan Hart and co-workers
illustrated that the template condensation of 2,6 diacetylpyridine with
ethylenediamine gives the 18-member hexaaza macrocyclic complexes of
the type [M(N03 )3 L] where M=La or Ce and L= 2,7,1 3,1 8,-tetramethyl-
3,6,1 4,1 7,23,24-hexaazatricyclo [1 7.3.1 8-12] tetracosa-1(23),2,6,8,10, 12
(24), 1 3,1 7,1 9,21 -decaene (7). The complexes are kinetically stable in water
with respect to dissociation of the macrocycle. The Ce complex in particular
has proven to be a potential aqueous NMR shift reagent.
L.M. Vallarino and co-workers illustrated in 1984 that the 18-member
hexaaza-macrocyclic ligand having the formula C22H26 N6- can De frmed
via the templating effect, used by Hart, with any one of the lanthanide ions
(excluding radioactive Pm) along with the condensation of 2,6- diacetyl
pyridine and 1,2-diaminoethane (8):
rNHj H2N
^0.1 M N.C1 r
N... j y
Nn 2
OAc'
S Ln(OAc)j J"
. ! JG"
O O
^-v
nHjO
U Pr. Od. Eu. U

Research involving lanthanide metal/nitrogen containing macrocyclic
complexes and their potential as proton NMR shift reagents has continued at
Rochester Institute of Technology under the direction of T.C. Morrill and his
research students. Mary DiSano, during 1988-89, synthesized a hexaaza
macrocyclic ligand coordinated to Pr (3+) as a potential lanthanide shift
reagent (9). Nitrate, carbonate, picrate, and chloroacetate counteranions
were employed in order to put on anions that readily dissociate compared to
the original praseodymium acetate template complex. Unfortunately, a
successful lanthanide shift reagent was not synthesized. Insolubility of the
complexes in suitable NMR reagents, difficulties in purification of the
complexes, and failure of the complexes to induce meaningful NMR shifts in
the spectrum of 1- heptanol were some of the problems encountered.
During 1990, Eileen George, an undergraduate student at RIT,
attempted the reaction seen below (10). In this reaction, the acetate and
chloride anions on a hexaaza macromolecule are changed to
tetraphenylborate anions with the hope that the tetraphenylborate ions
would increase solubility in nonpolar NMR solvents and because of their
bulkiness would coordinate to the metal cation less strongly. The inhibited
coordination to the cation would, theoretically, allow the cation to coordinate
to basic sites in organic substrates and thus improve the resolution of the
NMR spectra of those substrates. This project was only partly successful
with respect to the attempt to change the anions on the complex to
tetraphenylborate anions. Analysis by IR and NMR showed that only one
tetraphenylborate anion had been substituted for the acetate and chloride
anions and not three as are indicated in the reaction below:
N.i N ^N-,
I3:- NN 3B(CH)
f '"i*:1+ 12nA'
+ xsNaB(CH) ? > J' ' *
^y.- ^y.n H O

In the summer of 1 991,Dr. Robert Bryant from the University of
Rochester suggested that a lanthanide macromolecule made with four
aromatic rings instead of two would make a complex that was more planar
and hence would make a better contrast agent for MRI (11). From that
suggestion came this thesis project: the synthesis of a hexaaza
macromolecule (1 ) from 2,6 - diacetylpyridine, ortfto-phenylenediamine, and
praeseodymium acetate using the template synthesis method successfully
employed by A. Hart (7) and L. Vallarino (8), and then, if the synthesis was
successful, to test it as a NMR shift reagent:
-A-a
NH2NaCl
+ Pr(OAc)3NH, CH3OH
2 OAc
a-
nH20
(1)

PART TWO: HISTORICAL REVIEW
The synthetic procedure which can lead to net anti-Markovnikov
hydration of an alkene is hydroboration-oxidation. It was developed by H.C.
Brown and his coworkers at Purdue University in the 1950's as part of a
broad program designed to demonstrate how boron-containing reagents
could be applied to organic chemical synthesis (12).
Diborane, B2H5, is a colorless gas which can dissolve in aprotic
solvents like tetrahydrofuran (THF). These solvents form"loose"
Lewis acid-
base complexes with borane, BH3 (13):
H
H* A /\
h'\\* 2
(_J' Mq
THF
Hydroboration is a reaction in which borane (BH3), or alternatively,
RBH2 or R2BH compounds add to a carbon-carbon bond. New carbon-
hydrogen and carbon-boron bonds are created. An alkylborane is formed
from an alkene in a hydroboration reaction. The B-H bond always adds syn
across a C=C bond (12):
R
0^
H
H BH3:THF
R h H rh
THF LI

The mechanism in Figure 4 below shows how the B-H unit adds to the
pi bond in an alkene (14). Because the pi bond is electron rich and borane
is electron poor, it is reasonable to formulate an initial Lewis acid-base
complex requiring the participation of the empty p orbital on BH3, as in the
borane-THF complex. Subsequently, one of the hydrogens is transferred by
means of a four-center transition state to one of the alkene carbons, while
the boron shifts to the other without any additional intermediates. The
stereochemistry is syn. All three B-H bonds are reactive in this way.
Hydroboration is not only stereospecific but also regioselective. The boron
binds to the less-hindered (less-substituted) carbon.
Q-0
Emprj p orfriul
Mechanism of Hydroboration:
-Q G
cc-
H-B H
H>UXH
H
Boraiw-ilkent compkv
HjBX
Figure 4.
10

Following hydroboration, the organoborane is oxidized by treatment
with hydrogen peroxide in aqueous base. This is the oxidation stage of the
sequence; hydrogen peroxide is the oxidizing agent, and the organoborane
is converted to an alcohol (12):
VJUbh, H202
Cr^ OH
EtOH
O
The mechanism for the oxidation stage is (15):
H202 + 'OH >HOO"
+ H20
\/R
B-R + "OOH > [ B V ] >
/ /0N
0(0H
"OH + B-OR
/
H20
B-O-R > B-OH +ROH
' Hydrolysis
B-OH contains two more B-H bonds, so the above hydroboration-
oxidation process can take plaGe two more times.
Additional advantages of a hydroboration-oxidation synthesis of
alcohols are simplicity of procedure, relatively mild reaction conditions (for
11

example, temperature), high overall yields, and absence of skeletal
rearrangements (15).
In 1985, a paper was published by Mannig and Noth (16) on catalytic
hydroboration using Wilkinson's catalyst and a relatively unreactive
hydroborating reagent: 1,3,2-benzodioxabarole (catecholborane).
Catecholborane reacts with alkynes and alkenes, but only at elevated
temperatures:
aII B-H ? R-CC-H
*
Z"0^IL / 70 C H B-0
2 hn.O
catecholborane
C^O RR" R h
R"
Bx \-Sf Vh ?
sc-c'
- y-4{o
L0/ R'
- lOOj R^\
catecholborane
In the presence of Wilkinson catalyst, [chlorotris(triphenyl phosphine)
rhodium (I) ] (3), seen in Figure 5, these reactions proceeded rapidly (25
minutes) at room temperature (20).
/Rh
PCPb3)
(Pb3)P<':P(Pbj)
(3)
Figure 5.
12

Moreover, when molecules such as 5-hexene-2-one, which contain
both a double bond and a carbonyl group, were reacted with catechol
borane in the presence of Wilkinson's catalyst, the carbon-carbon double
bond was completely reduced and the keto carbonyl was not reduced. See
reaction below. Therefore, Wilkinson's catalyst activated the double bond
and not the carbonyl (16).
CO- -*--co
(3)
0^
The complex mechanism for the result above was proposed by
Mannig and Noth in 1985 and can be represented by a catalytic loop (17).
See Figure 6. Wilkinson's catalyst is a 16 electron complex. In solution,
Wilkinson's catalyst dissociates with one of its triphenylphosphine ligands
and after oxidative addition of the B-H bond from catecholborane (and
possibly complexation with a solvent molecule), Wilkinson's catalyst
becomes a stable 18 electron complex. See (c) below. This complex then
binds with an olefin present in solution. The next step of the reaction
involves insertion of the double bond into the Rh-H bond. Once this is
complete, irreversible reductive elimination of the organoborane returns
Wilkinson's catalyst back to a 16 electron complex which can undergo
reaction again.
13

L-PPh,
reductive
elimination
RnljCI WlllunMO I CJtJiyS,
t
RhL,a
Ch<
c-ci
u C /
***** V. /\ J.-i _Xolefin
':i'
W Nl bntf,n9migration
Figure 6.
Wilkinson's catalyst was first used in 1966 for the homogeneous
hydrogenation of double and triple bonds (18). In fact, the hydroboration
mechanism above is merely a modification of the homogeneous
hydrogenation mechanism.
KR'"
(3)
H2,25
C
Ethanol
RR"
*R'"
R' HR
H
Before Wilkinson's catalyst, hydrogenations were performed
heterogeneously with metals such as platinum or palladium (19). These
reactions were often performed at high temperatures and pressures, and the
yields were often low. Wilkinson's catalyst hydrogenations can be
14

performed at room temperature and standard pressure, with the time of
reaction being short and the yields high.
One interesting point about the ability of Wilkinson's catalyst to
activate double and triple bonds is the possibility of activating other pi
systems, or in the case of cyclopropane rings,"pseudo"
pi systems.
The cyclopropane molecule has attracted a great deal of interest
because of its highly strained bond angles and its unusual chemistry, which
is in several respects similar to that of alkenes. Walsh (20) proposed an
orbital model for bonding in three-membered rings designed to account for
this similarity. Because the H-C-H bond angle in cyclopropane is close to
1 20 (21), Walsh chose sp2 hybridization for the carbon, with one hybrid
being used for each C-H bond and the third pointing directly into the center
of the ring. See (a) in Figure 7 below. The unhybridized p orbital then lies
in the ring plane. See (b) in Figure 7 below (22).
<
(bi
Figure 7.
15

Walsh proposed that these atomic orbitals interact in two sets. The
three sp2 hybrids pointing toward the ring center overlap and form three
molecular orbitals, one bonding and two antibonding (22). These orbitals
are shown in (a) of Figure 8 below. The bonding member of the set holds
two electrons which are delocalized in the region inside the ring. The three
p orbitals interact through overlap that is between the end-on type
characteristic of sigma bonds and the side-on type characteristic of pi bonds
to form the three delocalized orbitals shown below in (b) of Figure 8. This
time two are bonding and one is antibonding. Each of the bonding pi-type
orbitals has one node, but the net effect is bonding.
7 Vi
<.*)
Figure 8.
The Walsh model makes clear the pi nature of cyclopropane and also
indicates that significant electron density will be concentrated outside the
equilateral triangle defined by the internuclear lines (22). The possibility
thus exists that hydroborating reagents activated by Wilkinson's catalyst
could facilitate the ring opening reaction of a cyclopropane ring.
Research in the area of using Wilkinson's catalyst to activate
cyclopropane rings in a hydroboration reaction has been carried on at
16

Rochester Institute of Technology under the direction of Dr. T.C. Morrill. One
of his students, Kevin Gillman, worked with quadricyclene (tetracyclo
[3.2.O.2-704'6
] heptane) (23). Quadricyclene contains about 96 kcal/mole
of strain energy due to its restricted polycyclic nature. When Gillman
allowed quadricyclene to react with catecholborane and Wilkinson's
catalyst, the product was notricyclanol in 9.9% yield:
1. (PPhj^RiiCl
Catecbolborme
THF ,5C
2. H202 , OH
quadricyclene notricyclanol (9.9%)
Donna Chen, in the same laboratories, attempted the catalyzed
hydroboration of norcarane (bicyclo[4.1.0.]heptane) using BH3-THF. GC-MS
analysis showed that only the starting material, norcarane, appeared in the
ether extract (24):
o + BH3:THF
l.(PPh3)3Rha
Reflux
ZHjOj.OH
norcarane
Cyclohexene oxide, (7-oxabicyclo[4.1.0]heptane), (2) seen below is
very similar to norcarane except that it contains a 3-membered heterocyclic
ring. The oxygen atom has unshared pairs of electrons to which the rhodium
17
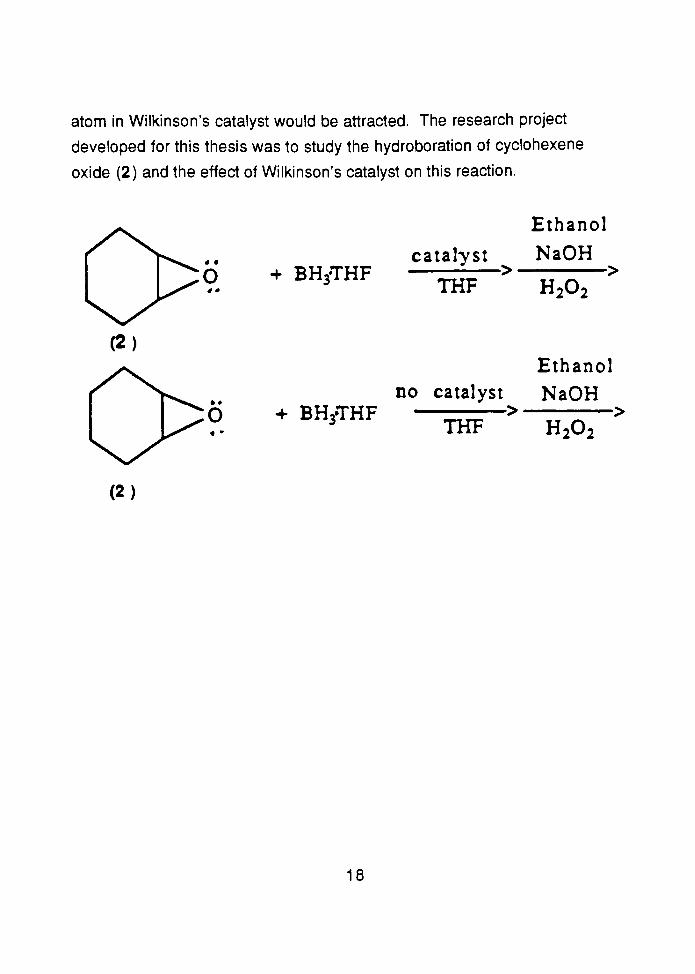
atom in Wilkinson's catalyst would be attracted. The research project
developed for this thesis was to study the hydroboration of cyclohexene
oxide (2) and the effect of Wilkinson's catalyst on this reaction.
+ BH3THF
catalyst
THF
no catalyst
+ BH3THF=7^r>
3THF
Ethanol
NaOH
H202
Ethanol
NaOH
H202
(2)
18

EXPERIMENTAL SECTION
Instrumentation and Chemicals:
GC Spectra were obtained with a Hewlett Packard 5890
Series II Chromatograph. The Helium flow rate was 1.3 mL/min. A
J&W Durabond-5 capillary column was used that is relatively
nonpolar and composed of 95% dimethyl-(5%)-diphenyl-
polysiloxane.
GC Parameters Used: Initial temperature =80 (2 minutes)
Ramp: 10/minute (7 minutes)
Final temperature =150 (6 minutes)
Injection Port =225
Detector =225
GC-Mass Spectroscopy was performed on a Hewlett Packard
5995 Spectrometer which was equipped with a J&W Durabond-1
column composed of 100% dimethyl-polysiloxane (nonpolar). The
parameters used were the same as those listed for the gas
chromatograph above.
Infrared Spectra were obtained with a Perkin Elmer 1760X
FT-IR Spectrometer and also with a Perkin Elmer 681
Spectrophotometer. The solid samples in Part One were analyzed
as KBr pellets and the liquids obtained in Part Two were
dissolved in deuterated chloroform and analyzed between salt
cells.
19

The reagents and solvents used were purchased from
Aldrich Chemical Company, Inc., Fisher Scientific, Inc., and J.T.
Baker, Inc. No further purification of these chemicals was done.
Dry nitrogen was prepared by passing reagent grade nitrogen
from Linde Air Products through a Fieser's solution (25) followed
by passage through concentrated H2S04 and finally through
anhydrous KOH.
Proton NMR spectra were obtained on a Bruker 200 MHz,
courtesy of Fisons Pharmaceutical Company. Deuterated
chloroform was used as solvent.
TLC was done using silica gel glass plates, Si-250F,
obtained from J.T. Baker, Inc. The plates were developed with a
3:2 pentane:ethyl ether mixture and then sprayed with a developer
and heated on a hot plate. The development spray (26) was
composed of 0.5 mL p-anisaldehyde in 9 mL 95% ethanol
containing 0.5 mL H2S04 and a few drops of glacial acetic acid.
Liquid chromatographic (LC) columns were made using
fluorescent silica gel of pore size 60 obtained from E. Merck,
Germany.
All mobile phase solutions for TLC and for liquid
chromatographic columns were prepared in volume/volume ratios.
Melting points were determined with a Mel-temp apparatus
from Lab Specialties Inc. and required no correction as per
calibration by benzoic acid.
The thermogravimetric analysis was obtained from a
Perkin-Elmer TGS-2 instrument heated at a rate of 20
degrees/min. under nitrogen gas at a flow rate of 55 mL/min.
20

PART ONE: EXPERIMENTAL SECTION
Attempted Template Synthesis of the Praeseodymium (III)Macrocyclic Complex derived from 2,6-Diacetylpyridine and
Orf/70-phenylenediamine
c -a
NH2NaCl
2 ^JL *L^ +2 B J + Pr(OAc)3
NH2CH3OH
O
; %KN^^^2 OAc
-
a-
nH20
(1)
Praeseodymium acetate hydrate, 0.3720 g (1 mmol), was mixed with
anhydrous methanol and swirled into a 250 mL round-bottom flask. Then
0.3264 g (2 mmol) of 2,6 - diacetylpyridine dissolved in 20 mL methanol and
0.2163 g (2 mmol) of o/t/70-phenylenediamine dissolved in 5 mL methanol
were both poured into the flask. To this mixture was added 0.0292 g NaCl in
5 mL methanol.
The mixture refluxed at65 for 5 hours. The color gradually changed over a
period of 3 hours from cloudy green to cloudy yellow. The reaction mixture
was allowed to cool to room temperature. A pale green precipitate
21

separated from the yellow reaction liquid and this was removed by filtration
through filter paper. The precipitate had a mass of 0.2536 g after being
dried in a 1 00 oven for one hour.
An IR spectrum of the green precipitate showed a strong broad band
between 3200 and 3400 cm"1
(OH) and two strong peaks between 1375 and
1575 cm-1 (COO"). There was also a weak narrow band at 1600 cm-1 (C=N).
The precipitate was slightly soluble in water and DMSO; it was insoluble in
benzene, chloroform, pyridine, ethyl acetate, carbon disulfide, and acetone.
While attempting to determine a melting point, the precipitate seemed to
shrink, looked slightly wet and became a darker green at 160. Between
220and 260, the green solid stuck to the sides of the melting point tube,
while a greenish liquid rested on the bottom of the tube. At 280, the
material turned brown (decomposed).
The green precipitate was placed in a crucible for a combustion test. It
burned slowly and completely charred.
The precipitate tested negative for the presence of chloride ion. The
alcoholic silver nitrate test produced no precipitate and the Beilstein test
showed an orange flame.
Thermogravimetric analysis suggested that 4.9% of the green precipitate
was water.
Tests to determine purity with TLC were unsatisfactory due to the difficulty of
dissolving the precipitate in organic solvents.
This reaction was also carried out in acetonitrile and in dioxane as solvents.
Although some of the results obtained from product analysis were
22

unsatisfactory (especially solubility), complete characterization of the
products was not done.
This research project was set aside in October, 1992. The reasons for this
decision are discussed in Part One of the Results and Discussion section.
23

PART TWO: EXPERIMENTAL SECTION
Treatment of Cyclohexene Oxide with BoraneTHF Complex in
the presence of Wilkinson's Catalyst
+ BH3-THF
Ethanol
catalyst NaOH
THF H202
(2)
A 3-neck round-bottom flask was flushed by a five-minute flow of
deoxygenated and dried nitrogen gas. Then, 100 mL (0.1 mole) of 1 M
BH3-THF was injected into the flask using a syringe followed by the addition
of 0.1 g CIRh[P(C6H5)3]3, Wilkinson's catalyst. After the catalyst was
dissolved, 10.0 g (0.1 mole) of cyclohexene oxide was added. The color of
the reaction mixture changed from clear orange-brown to clear dark brown
within a half hour. A reflux was maintained at72 for 5 1/2 hours along with
continuous stirring and a nitrogen atmosphere.
The reaction flask was then placed in an ice bath until the temperature of the
reaction mixture was approximately 5. Then 40 mL of 95% ethanol,
followed by 30 mL of 3M NaOH (aq) and 40 mL of 30% hydrogen peroxide
were added with continued stirring and a pause of several minutes after
each addition. The temperature rose to20
with some fizzing after the
addition of ethanol. The mixture was allowed to cool 5-10 degrees. When
the NaOH was added, the temperature rose to approximately30 and a
24

white solid formed. The reaction mixture gradually changed to a pale yellow
color after the addition of hydrogen peroxide. It was then allowed to sit
overnight with continued stirring.
The heterogeneous mixture of brown liquid and white solid in the reaction
flask was treated with 4 X 100 mL portions of ethyl ether. The separated
ether fraction was combined with anhydrous magnesium sulfate to remove
dissolved water, filtered, and then reduced in volume to 15 mL. At this point,
a cloudy, pale yellow oil was seen.
The white precipitate which formed when 3 M NaOH (aq) was added to the
reaction mixture became tinged with a pale yellow color after sitting in the
reaction mixture overnight. It was separated from the water fraction by
filtration through filter paper followed by flushing with distilled water. The
flushing removed some of the yellow color from the precipitate. The
precipitate was air dried; the mass was 2.4 grams.
The above reaction, workup, and extraction procedure was repeated three
times. The volume of the yellow oil obtained was 20 mL the second time and
18 mL the third time. Examination of the white precipitate was done just
once in the manner described later in this section.
Flame tests were done on BH3-THF and on the yellow oil to test for the
presence of boron. The flame of BH3-THF was yellow with green edges.
The flame of the yellow oil was predominantly blue with some yellow in it.
The yellow oil produced a burning sensation on the skin. It tested positive
for peroxide using acidified sodium chromate since the color of the chromate
ion changed from yellow to blue when mixed with a sample of the yellow oil.
25

GC analysis of the pale, yellow oil showed it to be a mixture of two primary
products which were in a 5:1 ratio for trial one, 6:1 ratio for trial two, and a 5:1
ratio for trial three. The GC retention times of these two products were,
respectively, 4.2 and 10.6 minutes.
The retention times of the two products by GC-MS analysis (appendix) were
4.2 and 10.5 minutes. The molecular weight of the 4.2 minute product is 100
and the molecular weight of the 10.5 minute product is 172.
TLC of the yellow oil revealed three primary spots. The Rf values were 0.9,
0.7 and 0.2. The colors of the sprayed spots were dark blue, rose, and
brown respectively. The rose-colored spot had a small blue"cap"
on it. TLC
of reagent grade cyclohexanol was done for comparative purposes. A rose-
colored spot with an Rf value of 0.7 was observed. See drawings below.
Rose-
Brown<a <r
fy-o.2.
TLC of Unpurified Ether Extract TLC of Reagent Grade Cyclohexanol
Attempts were made using liquid chromatography to separate the various
products in the yellow oil so that the TLC spots could be associated with
specific peaks seen in the GC analysis and the mass spectra. It was found
that separating the 0.9 spot (blue) from the 0.7 spot (rose) could be achieved
fairly well by eluting the column with pure hexane. The substance which
caused the fastest blue spot on the TLC plate travelled fastest through the
26

liquid silica gel column. When the column was flushed with mixtures of
hexane and pentane in various proportions (4:1 ,3:1
,1 :1 ,
1 :3), small
amounts of the substances which caused the blue and rose spots on the
TLC plates came out together. Significant quantities of the substance
causing the rose-colored spot came out when ethyl ether was added to
pentane in the mobile phase. Mixtures of pentane and ethyl ether in various
proportions (4:1,3:1
, 2:1 ,1 :1
,1 :2, 1 :3, 1 :4) were tried. The substance
causing the rose-colored spot came out easily with even small amounts of
ethyl ether in the mobile phase, but other substance(s) causing the brown
spot on TLC plates started to come off the column with it.
The fractions coming off the liquid column which were relatively free of the
0.7 rose-colored spot were found now to contain two principal substances,
both showing blue spots on the TLC plates: Rf values of 0.8 and 0.7 were
measured. A TLC plate with only one spot was not found. See drawing
below of a fraction free of the rose-colored spot.
Faint Blue.
Dark
OorKinc.
TLC of Hexane-eluted fraction
The fractions showing the two blue spotswere subjected to GC analysis.
Only one primary peak at 10.6minutes was seen in addition to the solvent
peak. No peak at 4.2 minutes appeared. The NMR spectrum of the fractions
showing two blue spots is shown in the appendix.
27

An IR spectrum was obtained using the same sample dissolved in
deuterated chloroform that was used for the NMR spectrum just mentioned.
Strong peaks seen were: 2850-2950 cm-1 (C-H aliphatic stretch); 1450 cm-1,
1375 cm-1
(CH2, CH3 bends); 1075 cm-1 (C-0 stretch). Weak peaks
(possible contamination) seen were: 3625 cm-1 (free OH); 3500-3100 cm-1
(OH).
Fractions from the column which were rich in the substance which produced
the rose-colored spot with the Rf value of 0.7 were also subjected to GC
analysis. A large peak at 4.2 minutes was seen with these fractions.
The fractions which were rich in the substance which produced the rose-
colored spot were dissolved in deuterated chloroform and analyzed by IR.
Peaks seen were: a sharp, narrow peak at 3625 cm-1 (free OH); a broad
peak from 3550 to 3150 cm-1 (OH); 2850-2950 cm-1(C-H aliphatic stretch);
1450 cm-1, 1375 cnr1(CH2, CH3 bends); 1075 cm-1,1140cm-1 (C-0 stretch).
Since the GC-MS library file suggested that the product with the retention
time of 4.2 minutes was cyclohexanol, reagent grade cyclohexanol was
subjected to TLC, GC, IR, and GC-MS analysis on the same instruments
used for analyzing the yellow oil and the chromatographed fractions.
Reagent grade cyclohexanol produced a rose-colored spot with an R{ value
of 0.7 on the TLC plates, a peak at 4.2 minutes on the GC, and and an IR
spectrum virtually identical to the one described in the preceeding
paragraph. The mass spectra of the 4.2 minute product and of reagent
grade cyclohexanol are seen in the appendix and are also virtually identical.
The GC-MS library file had no suggestions as to the identity of the product
with the molecular weight of 172, so no comparative studies with reagent
grade samples on TLC, GC and GC-MS were done.
28

All fractions collected from the liquid chromatography column with hexane,
hexane-pentane mixtures, and pentane-ethyl ether mixtures were clear and
colorless. None of these fractions tested positive for peroxides with acidified
sodium dichromate. The yellow dichromate solution remained yellow in
color when mixed with all of the fractions just mentioned.
The white precipitate produced by the addition of 3 M NaOH (aq) to the
reaction mixture was only very slightly soluble in chloroform and benzene
and insoluble in methanol and DMSO. The IR spectrum showed a broad
peak from 3600-3100 cm-1 (OH). No other identifiable functional peaks were
seen. It had a melting point of68-69
. Immediately after melting, it
appeared to bubble within the melting point tube. When this same
precipitate was heated in an open beaker, a liquid was driven off leaving a
white solid. The melting point of this solid from which the liquid had been
driven off was above 250.
29

Treatment of Cyclohexene Oxide with BoraneTHF Complex in
the Absence of Wilkinson's Catalyst
Ethanol
no catalyst NaOH+ BH3-THF
THF HoO2^2
(2)
A 3-neck round-bottom flask was flushed by a five-minute flow of
deoxygenated and dried nitrogen gas. Then 100 mL (0.1 mole) of 1 M
BH3-THF was injected into the flask using a syringe followed by the addition
of 10.0 g (0.1 mole) of cyclohexene oxide. The reaction mixture immediately
began boiling on its own. A reflux was maintained at72 for 5 1/2 hours
along with continuous stirring and a nitrogen atmosphere. The reaction
mixture remained clear and colorless throughout the reflux.
The reaction flask was then placed in an ice bath until the temperature of
the reaction mixture was approximately 5. Then 40 mL of 95% ethanol,
followed by 30 mL of 3 M NaOH (aq) and 40 mL of 30% hydrogen peroxide
were added with continued stirring and a pause of several minutes after
each addition. The temperature rose to 20 with some fizzing after the
addition of ethanol. The mixture was allowed to cool 5-10 degrees. When
the NaOH was added, the temperature rose to approximately30 and a
white solid formed. The reaction mixture did not change color significantly
after the addition of hydrogen peroxide. It was then allowed to sit overnight
with continued stirring.
30

The heterogeneous mixture of colorless liquid and white solid in the
reaction flask were treated with 4 X 100 mL portions of ethyl ether. The
separated ether fraction was combined with anhydrous magnesium sulfate
to remove dissolved water, filtered, and then reduced in volume to 25 mL. At
this point, a pale yellow oil was observed.
The white precipitate which formed when 3 M NaOH (aq) was added to the
reaction mixture became tinged with a pale yellow color after sitting in the
reaction mixture overnight. It was separated from the water fraction by
filtration through filter paper followed by flushing with distilled water. The
flushing removed some of the yellow color from the precipitate. It was then
air dried. This precipitate melted at68
; it bubbled in the melting point tube
immediately after melting. No further examination of this white precipitate
was done.
The above reaction, workup, and extraction procedure was repeated three
times. The volume of the yellow oil obtained was 20 mL the second time
and 15 mL the third time.
Flame tests were done on BH3-THF and on the yellow oil to test for the
presence of boron. The flame of BH3-THF was yellow with green edges.
The flame of the yellow oil was predominantly blue with some yellow in it.
The yellow oil produced a burning sensation on the skin. It tested positive
for peroxide using acidified sodium chromate since the color of the chromate
ion changed from yellow to blue when mixed with a sample of the yellow oil.
GC analysis of the pale, yellow oil showed it to be a mixture of two primary
products which were in a 1 2:1 ratio for trial one, a 9:1 ratio for trial two, and a
1 2:1 ratio for trial three. The GC retention times of these two products were,
respectively, 4.2 and 10.6 minutes.
31

The retention times of these two products by GC-MS analysis were 4.2 and
10.2 minutes respectively. The molecular weight of the 4.2 minute product is
100 and the molecular weight of the 10.2 minute product is 172. See mass
spectra in the appendix.
TLC of the yellow oil revealed three primary spots. The Rf values were 0.9,
0.7 and 0.2. The colors of the sprayed spots were dark blue, rose, and
brown respectively. The rose-colored spot had a small blue"cap"
on it. TLC
of reagent grade cyclohexanol was done for comparative purposes. A rose-
colored spot with an Rf value of 0.7 was observed. See drawings below.
TLC of Unpurified Ether Extract TLC of Reagent Grade Cyclohexanol
Attempts were made using liquid chromatography to separate the various
products in the yellow oil so that the TLC spots could be associated with
specific peaks seen in the GC analysis and the mass spectra. It was found
that separating the 0.9 spot (blue) from the 0.7 spot (rose) could be achieved
fairly well by eluting the column with pure hexane. The substance which
caused the fastest blue spot on the TLC plate travelled fastest through the
32

liquid silica gel column. When the column was flushed with mixtures of
hexane and pentane in various proportions (4:1 ,3:1,1:1,1 :3), small
amounts of the substances which caused the blue and rose spots on the
TLC plates came out together. Significant quantities of the substance
causing the rose-colored spot came out when ethyl ether was added to
pentane in the mobile phase. Mixtures of pentane and ethyl ether in
various proportions (4:1, 3:1 , 2:1 , 1 :1
,1 :2, 1 :3, 1 :4) were tried. The
substance causing the rose-colored spot came out easily with even small
amounts of ethyl ether in the mobile phase, but other substance(s) causing
the brown spot on TLC plates also came off the column with it.
The fractions coming off the liquid column which were relatively free of the
0.7 rose-colored spot were found now to contain two principal substances,
both showing blue spots on the TLC plates: Rf values of 0.8 and 0.7 were
measured. A TLC plate with only one spot was not found. See drawing
below of a fraction free of the rose-colored spot.
Faint Blux
Darkfeiu.1
OorKBUe.
TLC of Hexane-eluted fraction
The fractions showing the two blue spots were subjected to GC analysis.
Only one primary peak at 10.6 minutes was seen in addition to the solvent
peak. No peak at 4.2 minutes appeared.
33

No NMR or IR spectra of the samples showing a GC peak at 1 0.6 minutes
was obtained because of the difficulty of collecting sufficient quantities of it.
Fractions from the column which were rich in the substance which produced
the rose-colored spot were also subjected to GC analysis. A large peak at
4.2 minutes was seen with these fractions.
The fractions which were rich in the substance which produced the rose-
colored spot were dissolved in deuterated chloroform and then analyzed by
IR. Peaks seen were: a sharp, narrow peak at 3625 cm-1 (free OH); a broad
peak from 3550 to 3150 cm-1 (OH); 2850-2950 cm-1 (C-H aliphatic stretch);
1450 cm-1, 1375cm-1
(CH2> CH3 bends); 1075 cm-1, 1140cm"1 (C-0 stretch).
Since the GC-MS library file suggested that the product with the retention
time of 4.2 minutes was cyclohexanol, reagent grade cyclohexanol was
subjected to TLC, GC, IR, and GC-MS analysis on the same instruments
used for analyzing the yellow oil and the chromatographed fractions.
Reagent grade cyclohexanol produced a rose-colored spot with an Rf value
of 0.7 on the TLC plates, a peak at 4.2 minutes on the GC, and and an IR
spectrum virtually identical to the one described in the preceeding
paragraph. The mass spectra of the 4.2 minute product and of reagent
grade cyclohexanol are seen in the appendix and are also virtually identical.
The GC-MS library file had no suggestions as to the identity of the
substance with the molecular weight of 172, so no comparative studies with
reagent grade samples on TLC, GC, and GC-MS were done.
All fractions collected from the liquid chromatography column with hexane,
hexane-pentane mixtures, and pentane-ethyl ether mixtures were clear and
colorless. None of these fractions tested positive for peroxides with acidified
sodium dichromate since the yellow dichromate solution remained yellow
when mixed with all of the fractions just mentioned.
34

Reaction Study Using TLC of the Treatment of
Cyclohexene Oxide with Borane-THF in the Presence of
Wilkinson's Catalyst
+ BH3THFcatalyst
THF
Ethanol
NaOH
H202
(2)
Samples were removed from the reaction mixture with a capillary
tube at the following intervals: 1, 2, 3, 5, 10, 15, 30, 60, 120,
180, and 240 minutes. The samples were spotted on TLC plates,
developed, sprayed, heated, and examined. Samples of the solvent
and starting materials - THF, borane-THF, cyclohexene oxide
dissolved in THF, and Wilkinson's catalyst dissolved in THF - were
also spotted on the TLC plates, developed, sprayed, heated, and
compared with the collected reaction samples.
Drawings of the TLC plates on which only solvent and starting
materials were spotted are seen below.
35

THF Cyclohexene
Oxide/THF
BlueLint R^t-o
8luc C n<l
bh3/thf Wilkinson's Catalyst/THF
Line
-Blue- Li *e.^='0
"7ellU>A*4L
Spat-
36

Drawings of the TLC plates on which reaction mixture samples at
1, 5, 15, and 30 minutes were spotted are seen below. The TLC
plates of samples collected after 30 minutes were virtually
identical to the 30 minute plate. This TLC reaction study was
done only once.
1 minute 5 minutes
to
Brown.Specks
.Band
FlzjzjmSpot
-DarK Blue.
os-
U2V& Slue.
15 minutes 30 minutes
k
Blue
-ftjS*.
Likt
sr^e.
37

Reaction Study Using TLC of the Treatment of
Cyclohexene Oxide with Borane-THF in the Absence of
Wilkinson's Catalyst
Ethanol
no catalyst NaOH+ BH3THF
THF h,02^2
(2)
Samples were removed from the reaction mixture with a capillary
tube at the following intervals: 1, 2, 3, 6, 10, 15, 30, 60, 120,
180, and 240 minutes. The samples were spotted on TLC plates,
developed, sprayed, heated, and examined. Samples of the solvent
and starting materials - THF, borane-THF, and cyclohexene oxide
dissolved in THF - were also spotted on the TLC plates, developed,
sprayed, heated, and compared with the collected reaction
samples. Drawings of the solvent and starting materials are seen
in the previous section dealing with the TLC reaction study of the
Wilkinson's catalyst reaction.
Drawings of the TLC plates on which reaction mixture samples at
3, 6, 10, 30, and 60 minutes were spotted are seen below. The
TLC plates after 60 minutes were virtually identical to the 60
minute plate.
38

3 minutes 6 minutes
Fuz-zu.Spot?"
DjirlC
BlueR5C
^c+
10 minutes 30 minutes
U <^T
>Tue_
39

60 minutes
The reaction study of the treatment of cyclohexene oxide in the
absence of Wilkinson's catalyst was done twice. The appearance
of the TLC plates was virtually identical for both studies.
40

PART ONE: RESULTS AND DISCUSSION
As early as 1979, the template synthesis and properties of
macrocyclic complexes of La and Ce nitrates from 2,6-diacetylpyridine and
ethylene diamine were reported by Alan Hart and coworkers (27). The
template synthesis and properties of the complexes of several lanthanide
acetates and perchlorates using 2,6-diacetylpyridine and ethylene diamine
was demonstrated by L.M. Vallarino and coworkers in 1985 (8).
We were interested in the template synthesis and possible shift
reagent properties of a complex of praeseodymium acetate using2,6-
diacetylpyridine and orfno-phenylenediamine:
^aNH,
NH,
NaCl
+ Pr(OAc)3CH3OH
2 OAc
CI-
n H20
(D
41

The template synthesis and properties of the complexes of Ce, Pr, and
Nd nitrates from 2,6-diacetylpyridine and orffro-phenylenediamine were
reported in 1985 by Wanda Radecka-Paryzek (28 ). Professor Tom Bell of
SUNY at Stony Brook in 1989 suggested a simple lanthanide complex of the
macrocycle formed from 2,6-diacetylpyridine and orf/70-phenylenediamine
had been claimed, but he was doubtful that any bona fide complexes of this
ligand existed (29).
Since Dr. Robert Bryant from the University of Rochester in the
summer of 1991 (11) suggested that a lanthanide macromolecule made
with four aromatic rings instead of two would make a complex that was more
planar and hence would make a better MRI reagent, the decision was made
to attempt the synthesis of compound 1 using the successful synthetic
procedures employed by Hart and by Vallarino.
The green precipitate obtained from the attempted synthesis was
disappointing with respect to solubility characteristics. Since it precipitated
from the reaction solvent (methanol), NMR analysis could not be done in
methanol-d^ Furthermore, it was only slightly soluble in the polar solvents
water and DMSO, and insoluble in benzene, chloroform, pyridine, ethyl
acetate, carbon disulfide, and acetone as well as methanol. The solubility
problems made its use as a MRI reagent very limited.
Poor solubility of the green precipitate also made determination of
purity using TLC plates practically impossible.
The IR spectrum showed strong OH andCOO"
stretches. There was
an extremely weak peak at 1600cm-1 (C=N stretch) compared to the IR peak
seen on a spectrum for the complex formed using 2,6-diacetylpyridine and
ethylenediamine as done by Eileen George (10). If the synthesis had
produced 1,the C=N peak was expected to be much stronger.
The high melting point and also the negative test for chloride ion were
further indications that the product formed in our attempted template
synthesis was not 1 . This project was set aside in October, 1 992.
42

In the fall of 1992, Dr. Morrill had another opportunity to discuss these
macrocycles withTom Bell (30). Dr. Bell suggested that the properties we
had observed could be associated with the structure in Figure 9, a complex
that had been produced and verified in his laboratories. (Pr3*) was not part
of the complex Bell and his coworkers synthesized, but was probably part of
the complex we had synthesized since praeseodymium imparts a green
color to its compounds.) The poor solubility problems and high melting point
might be explained by the cross bonding between the acetyl groups.
Figure 9.
Before this project had been set aside, we also attempted to repeat
the work of Radecka-Paryzek (28) since she had claimed success in the
synthesis of the praeseodymium nitrate complex formed from 2,6-
diacetylpyridine and orf/70-phenylenediamine:
43

X-CCNH2
+ Pr(N03)3
NH,CH3OH
3
2 H20
We followed the synthetic method briefly described byRadecka-
Paryzek in the literature, which seemed virtually identical to the procedure
followed by Hart and by Vallarino, but our results were disappointing.
According to the Radecka-Paryzek paper published in 1985 (28), a yellow-
brown precipitate was obtained from the reaction above. The IR spectrum of
this precipitate included the following bands: 3450-3350 cm-1 (OH stretch);
1608 cm-1 (C=N stretch); 1595, 1568, 1455 and 1000 cm"1 (pyridine
vibrations); 1465, 1295, 1040, 823 and 743 cnr1 (coordinated nitrate ion
stretch). Her results were also verified by NMR, Mass and UV spectra.
We obtained a yellow-brown precipitate from our three attempted
syntheses of the praeseodymium nitrate macrocyclic complex seen above.
The IR spectrum showed the following absorption bands: 3350 cm-1 (N-H
stretch); a small peak at 3100cm-1 (aromatic C-H stretch); two small peaks
at 2900 and 2950cm-1 (aliphatic C-H stretch); small peak at 1700
cm-1
(C=0 stretch); peaks at 1600 and 1650 cm"1 (C=N stretches); 1495cm-1
(CH2 aliphatic bend).There was no band for the OH stretch. We were not
able to determine whether our synthetic method was exactly the same as
that of Radecka-Paryzek. No current address for her was available.
44
PART TWO: RESULTS AND DISCUSSION
The work described in Part Two of the Experimental Section
developed out of earlier studies done at RIT in which attempts were made to
open cyclopropane rings using two B-H bond sources and in some cases,
Wilkinson's catalyst.
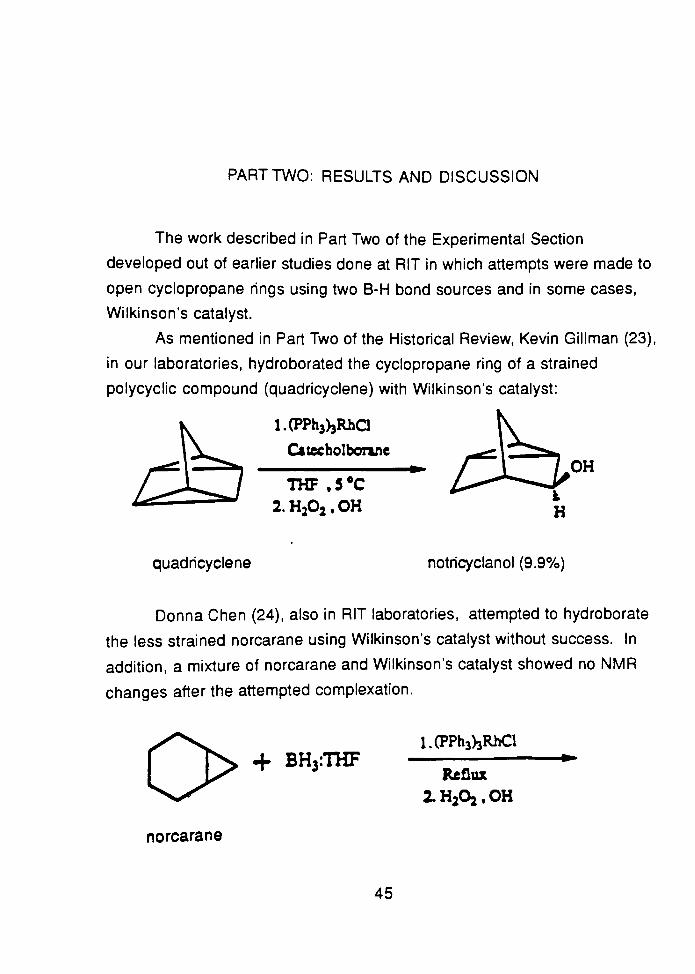
As mentioned in Part Two of the Historical Review, Kevin Gillman (23),
in our laboratories, hydroborated the cyclopropane ring of a strained
polycyclic compound (quadricyclene) with Wilkinson's catalyst:
l.(PPh3)3Rha
Catecholbonne
THF .5*C
2.H202,OH
quadricyclene notricyclanol (9.9%)
Donna Chen (24), also in RIT laboratories, attempted to hydroborate
the less strained norcarane using Wilkinson's catalyst without success. In
addition, a mixture of norcarane and Wilkinson's catalyst showed no NMR
changes after the attempted complexation.
onorcarane
-f BH3:THF
l.(PPh3)3RhCl
Reflux
2.H2Oj.OH
45

These results indicated that most three-membered rings were difficult
to cleave in spite of their pi bond character unless there was extreme ring
strain as in the case of quadricyclene. We thought that a three-membered
ring containing oxygen might be hydroborated more easily because of the
unshared pairs of electrons on the oxygen atom, thus giving the ring more
electron density. We also thought that using an elevated temperature would
facilitate this process.
H.C. Brown and coworkers found that the reduction of epoxides with
borane-THF was slow at room temperatures (31). Addition of trifluoroborane
(32) or a catalytic amount of sodium borohydride (33) to borane-THF,
however, was shown to effect ring opening faster at room temperature. We
simultaneously tested the effectiveness of an elevated temperature (72)
and of Wilkinson's catalyst in the opening of the epoxide ring of cyclohexene
oxide:
bh3-thfcatalyst
Ethanol
NaOH
THF H,0
(2)
+ BH3-THFno catalyst
THF
T->2
Ethanol
NaOH
H,02^2
(2)
If an epoxide ring is opened and an organoborane is formed, the
standard oxidation procedure by alkaline hydrolysis of the alkoxyboranes
which are formed yields alcohols (Part Two: Historical Review).
The treatment of cyclohexene oxide with borane-THF using
Wilkinson's catalyst was repeated three times with consistent results. A pale
yellow oil was obtained from the ether extract. Approximately 60% of the
46

yellow oil was one product, according to GC analysis. The rose-colored TLC
spot with an R, value of 0.7, the IR and mass spectra, and the GC retention
time of 4.2 minutes of this product matched the results found with reagent
grade cyclohexanol. Thus, approximately 60% of the ether-extracted
product mixture from the Wilkinson's catalyst reaction was cyclohexanol.
The treatment of cyclohexene oxide with borane-THF in the absence
of Wilkinson's catalyst was also repeated three times with consistent results.
A pale yellow oil was obtained from the ether extract. Approximately 70% of
the yellow oil was one product, according to GC analysis. The rose-colored
TLC spot with an Rf value of 0.7, the IR and mass spectra, and the GC
retention time of 4.2 minutes of this product also matched the results found
with reagent grade cyclohexanol. Thus, approximately 70% of the ether-
extracted product mixture from the uncatalyzed reaction was cyclohexanol.
A summary of the results of the catalyzed and uncatalyzed reactions
is shown in the Table below:
Catalyst Temp Reflux Time Primary Product % of the
of Ether Extract Ether Extract
YES 72 5.5 HOURS CYCLOHEXANOL 60%
NO 72 5.5 HOURS CYCLOHEXANOL 70%
Since the yield of cyclohexanol was higher when no catalyst was
used, it can be concluded that no catalyst is needed if the temperature is
held at 72 and that the presence of the catalyst may actually interfere
slightly with theproduction of the alcohol.
A suggested mechanism for the synthesis of cyclohexanol by means
of the hydroboration-oxidation of cyclohexene oxide is as follows:
47
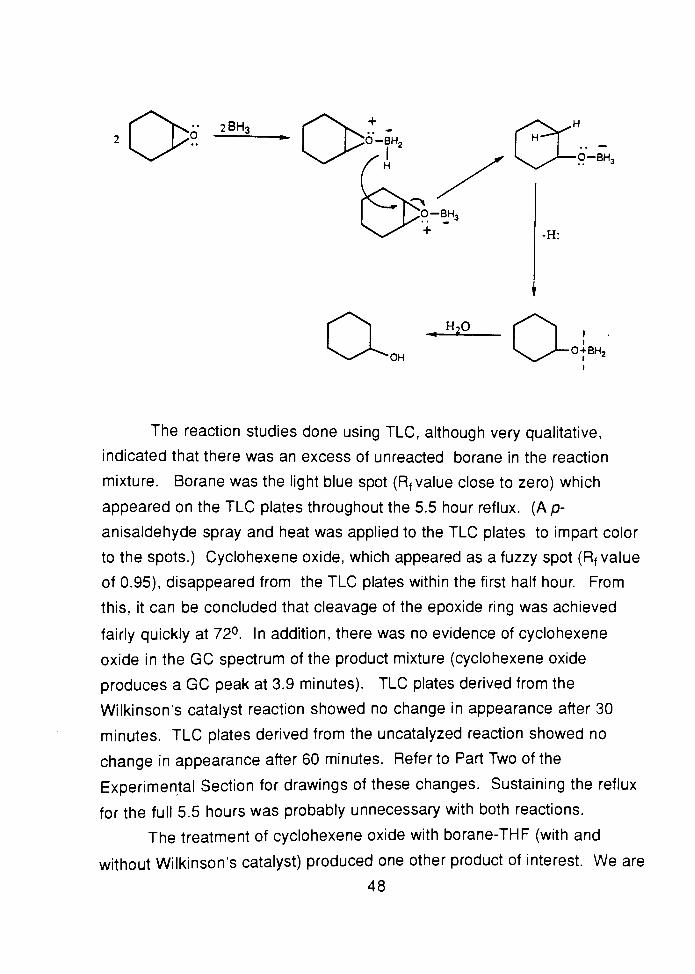
2BH,
H20
OH0+BH2
The reaction studies done using TLC, although very qualitative,
indicated that there was an excess of unreacted borane in the reaction
mixture. Borane was the light blue spot (Rf value close to zero) which
appeared on the TLC plates throughout the 5.5 hour reflux. (Ap-
anisaldehyde spray and heat was applied to the TLC plates to impart color
to the spots.) Cyclohexene oxide, which appeared as a fuzzy spot (Rf value
of 0.95), disappeared from the TLC plates within the first half hour. From
this, it can be concluded that cleavage of the epoxide ring was achieved
fairly quickly at 72. In addition, there was no evidence of cyclohexene
oxide in the GC spectrum of the product mixture (cyclohexene oxide
produces a GC peak at 3.9 minutes). TLC plates derived from the
Wilkinson's catalyst reaction showed no change in appearance after 30
minutes. TLC plates derived from the uncatalyzed reaction showed no
change in appearance after 60 minutes. Refer to Part Two of the
Experimental Section for drawings of these changes. Sustaining the reflux
for the full 5.5 hours was probably unnecessary with both reactions.
The treatment of cyclohexene oxide with borane-THF (with and
without Wilkinson's catalyst) produced one other product of interest. We are
48

uncertain of the identity of this product, largely because of our inability to
obtain adequately pure samples of it. According to GC-MS, this product had
a M.W. = 172. It produced one of the fastest travelling blue spots on TLC
plates, had a GC retention time of 10.6 minutes, and showed strong peaks
representing an aliphatic C-H stretch, CH2 and CH3 bends, and a C-0
stretch in the IR spectrum. The IR spectrum suggests an ether. Ethers
which could possibly result from the reactions we tried are trans -1 ,2 -
diethoxycyclohexane, (4), or c/'s-1,2-diethoxycyclohexane, (5), (M.W. = 172).
See Figure 10 below.
cc ccOC2H5
OC2H5
Trans^ ,2-diethoxycyclohexane C/s-1 ,2-diethoxycyclohexane
(4) (5)
Figure 1 0.
A literature reference was found for the trans form, compound 4, of
this particular ether (34 ). No references were found for cis -1,2-diethoxy
cyclohexane in the index of Chemical Abstracts going back to1952. In the
reference for the trans form of the ether, the NMR peaks were listed as
follows: 1.15 (t,6H), 1.5-2.3 (m,8H), 3.15 (m, 2H), 3.55 (q, 4H). The NMR
spectrum was recorded using Varian EM-390 and Varian FT-80A
spectrometers.
The NMR spectrum for our product with a GC peak of 10.6 minutes
shows the following peaks: multiplets at 0.8, 1 .2, 1.6, 2.0, 2.9, 3.15, and 3.5.
This spectrum was recorded using a Bruker 200 MHz spectrometer.
Although there is agreement of this spectrumwith some of the NMR peaks
49

associated with pure trans -1,2-diethoxycyclohexane,
there are additional
peaks, not related to the trans diether, and these indicate a mixture.
This same sample, which had been eluted with pure hexane from the
LC column, had also shown two significant blue spots on the TLC plate, so
the inconclusive results of the NMR spectrum were not surprising.
Trans -1 ,2-diethoxycyclohexane is not commercially available. We
have not synthesized it from the frans-1,2-cyclohexanediol, so comparative
studies with our reaction product were not done.
We suggest the following mechanism for the synthesis of 4 from the
hydroboration-oxidation of cyclohexene oxide:
O BH,
O BH,
The white precipitate which formed when 3 M NaOH (aq) was added
to the reaction mixture had a melting point of68-69 and showed a broad
peak representing the OH functional group on the IR spectrum. The
50

recorded melting point of Na2B4O7-10H2O, sodium tetraborate
decahydrate, is 60. The recorded melting point of Na2B407-8H20, sodium
tetraborate octahydrate, is 75. The white precipitate produced by the
reactions in this thesis could be a impure mixture of these two borate
compounds and/or other borates or organoborates. Both the white
precipitate and the borate compounds just mentioned have an Rf value of
0.0 using the same stationary and mobile phases that were used for all other
TLC tests performed. The white solid and the borate compounds mentioned
above would not burn when subjected to a Fisher burner flame. The
production of a borate compound by a hydroboration-oxidation reaction is
common.
Approximately 25% of the GC peak areas from the ether extracted
products resulting from these reactions had GC retention times of three
minutes or less. No special effort was made to identify these short retention
time compounds.
The most interesting part of this research was trying to identify the
product with the molecular weight of 172, which we referred to as our
"mysterycompound."
It was noted earlier that a higher percentage of
cyclohexanol occurred in the ether extract when no Wilkinson's catalyst was
used. It should also be noted that a higher percentage of the "mystery
compound"
occurred in the ether extract when Wilkinson's catalyst was
used. Since the quantities of the yellow oil extracted by ether were
approximately the same for the catalyzed and the uncatalyzed reactions, this
means that more of the "mysterycompound"
was obtained in terms of
quantity, not only in terms of percentage of the ether extract, when
Wilkinsons catalyst was present.
We suggest that if research continues on the hydroboration of
cyclohexene oxide, larger quantities ofWilkinson's catalyst should be tried
to see if this would further improve the yield of the "mysterycompound."
Another suggestion stems from the unavailability of a commercial
51

source of compound 4. Attempts should be made to synthesize 4 in our
laboratories by using the commercially available trans-1 ,2-cyclohexanediol.
Once 4 is synthesized, GC, TLC, and GC-MS comparison studies can be
made with the hexane-eluted fractions from the LC column.
More effort also needs to be expended in the area of finding optimal
temperature and time parameters for this reaction.
In summary, the following significant information was learned from the
experimental work described in Part Two: (1) the epoxide ring of
cyclohexene oxide was completely cleaved by borane-THF at 72 in less
than 0.5 hours; (2) at 72, Wilkinson's catalyst was not essential to this
cleavage; (3) TLC plates showed that there was no cyclohexene oxide
remaining in the reaction mixture after 0.5 hours of reflux, indicating that the
extended 5.5 hour reflux time was unnecessary; (4) we observed
cyclohexanol as the primary product found in the ether-extracted product
mixture; (5) approximately 70% of the ether-extracted product mixture was
cyclohexanol in the case of the uncatalyzed reaction, while approximately
60% of the mixture was cyclohexanol in the case of the catalyzed reaction,
suggesting that the catalyst may have interfered slightly with alcohol
formation; and (6) there was some evidence that another minor product
formed was frans-1,2-diethoxycyclohexane.
52

(1
(2
(3
(4
(5
(6
(7
(8
(9
REFERENCES
Hinckley, C.C. J. Amer. Chem. Soc. 1 969, 91, 5160-5162.
Sanders, Jeremy, K.M.; Williams, D.H. J. Amer. Chem. Soc. 1 971, 93,641-645.
Sievers, R.E., Ed. "Nuclear Magnetic Resonance Shift Reagents";Academic: New York, 1973; pp. 1-2, 21-22.
Morrill, T. C. "Lanthanide Shift Reagents in Stereochemical Analysis";VCH: 1986; pp.1 59-1 60.
DeCann, Dale. MS Thesis, Rochester Institute of Technology, June,1979.
Hiroshi, Tsukube. J. Chem. Soc. Chem. Commun. 1984, 315-316.
Arif, A.M.; Gray, C.J.; Hart, F.A.; Hursthouse, M.B. J. Chem. Soc. Dalton
Trans. 19 87, 1665-1673.
DeCola, L; Smailes, D.L.; Vallarino, L.M. Inorg. Chem. 1986, 25, 1729-
1732.
DiSano, Mary. MS Thesis; Rochester Institute of Technology, Rochester,
New York, June, 1989.
(10) George, E.S. Spring Research 1989 Report; Rochester Institute of
Technology, Rochester, New York, 1989.
(11) Bryant, Robert. University of Rochester, private communication, 1991.
(12) Carey, F.A. "Organic Chemistry"; McGraw-Hill, Inc: New York, 1992; pp.
225-226.
(13) Brown, H.C. "Hydroboration"; W.A. Benjamin, Inc: New York, 1962.
53

(14) Vollhardt, K.P.C. "Organic Chemistry"; W.H. Freeman: New York, 1987;p. 478.
(15) Noland, W.E., Editor-in-Chief. "Organic Syntheses, Collective Volume6"; John Wiley & Sons: New York; pp. 923-924.
(16) Mannig, D.; Noth, H. Angew. Chem. Int. Ed. Engl. 1985, 24, 878-879.
(17) Evans, D.A.; Fu, G.C. J. Org. Chem. 1990, 55, 2280.
(18) Harmon, R.E.; Parsons, J.L.; Cooke, D.W.; Gupta, S.K.; Schoolenberg,J. J. Org. Chem. 1969, 34, 3684.
(19) Rylander, P.N. "Catalytic Hydrogenation Over Platinum Metals";Academic Press: New York, 1967.
(20) Walsh, A.D. Trans. Faraday Soc. 1949, 45, 179.
(21) Gunthard, Hs.H.; Lord, R.C.; McCubbin, F.K. J. Chem. Phys., 1956, 25,768.
(22) Lowry, T.H. "Mechanism and Theory in Organic Chemistry, Third
Edition"; Harper & Row Publishers, Inc.: New York, 1987, pp. 31-33.
(23) Gillman, Kevin. MS Thesis, Rochester Institute of Technology,
Rochester, New York, August, 1991.
(24) Morrill, T.C.; Gillman, K.; Feng, P.; Chen, D. Paper presented at
National ACS Meeting, San Francisco, California, April, 1992.
Abstract No. 289: "Hydroboration of Highly Strained Cyclopropane
Rings Promoted by WilkinsonsCatalyst."
(25) Fieser, L.F.; Fieser, M. "Reagents for Organic Reactions"; John Wiley &
Sons: New York, 1967; p. 393.
(26) Gordon, A.J. ; Ford, R.A. 'The Chemist's Companion"; John Wiley &
Sons; New York, 1972; p. 379.
(27) Backer-Dirks, J.D.J. ; Gray, C.J.; Hart, F.A.; Hursthouse, M.B.; Schoop,
B.C. Chem Commun. 1979, 774.
54

(28) Radecka-Paryzek, W. Inorg. Chim. Acta A 985, 109, L21.
(29) Bell, Tom. SUNY at Stony Brook, Letter, July, 1989.
(30) Bell, Tom. SUNY at Stony Brook, private communication, Fall, 1992.
(31) Brown, H.C.; Heim, P.; Yoon, N.M. J. Am. Chem. Soc. 1 970, 92, 1637.
(32) Brown, H.C.; Yoon, N.M. Chem. Commun. 1968, 1549.
(33) Brown, H.C.; Yoon, N.M. J. Am Chem. Soc. 1968, 90, 2686.
(34) Barluenga, J.; Alfonso-Cires, L.; Campos, P.J.; Asensio, G.
Tetrahedron 1984, 40, 2563-2568.
55

APPENDIX
56

<SBwple> <Co(wient>
Sntft Reaoent
6.302 mg( 6.302 dq)
<Reference>
92/07/22 12-47 Platinum
0.000 no <Sampllno>
25. , .1 0 sec
TG/DTA<Name>
Ruth
<Date>
c
o1-
o
-2.5
<Temp prooranlC) lC/<tn ) lnml>
1* 25.0-300 0 5 00 0.00
<6as>
nltrooen 100 0 nl/nln
q 0 ni /min
100.5
-7.5L
25 95
Rochester Institute of Technoloov
165 235
TEMP C (Heating)
TGA ANALYSIS OF PRODUCT FROM ATTEMPTED SYNTHESIS OF
PRAESEODYMIUM ACETATE MACROCYCLIC COMPLEX



IT to TYPE KIOTM *R(*I|
t.144 tu rv .It! .Ml?*
t.l4 ? vv .Ill .11743
t.tll 11*1 VP It* .11*11
2.2*7 SI229M p 41 41.42711
t.47* 1*3*2 IP ,122 .11492
t.891 *sr4i rv ,114.14*14
t.*IS 7*71 VI ,121.ItiSt
i.i4i 1*91 PV ,111 .11171
1.111 11*1 VV .111 .11919
1.1*9 sis vv .129 .11299
1.11* 78i ri .119 .l**27
.*? 11*7 ip .172 .129*1
!. *47*2I PI ,177 l.ll71
TL **E*1.I74I*I7
facto*.1.IIIIE*!!
,^
MT>2
VJaI
GC Spectrum of Cyclohexene Oxide(Dissolved In Deuterated Chloroform)

Ota*
*T ARE* TYPE yiDTH AMAX
2.124 12274272 PV .131S7.21**
2. If* 94.81* VI .21 4. 1**43
2.1*4 2*741 IV .44 .**
l.*S3 729 VI .27 .337
l.*3 17**2 PP .72 .343
4.272 114239 PI .1723S.*2*74
TOTAL AKEA-2. 132SE*7
MUL FACT0*1.?*
yS
Sov^
*yr
LtfcloUunoL-}
GC Spectrum of Cyclohexanol
(Dissolved in Deuterated Chloroform)

41141
T
1.44*
.4
l.tlj
I.I
1.114
.74
1.111
1.474
t.lSt
l.tll
>.44l
a. u*
1.111
J*
14444
*
1411
74
1*14
4411411
4ttJ
11154
urn
14444
14*1
1444
45
tan
4714111
lllllii
TTM IBTM
.1*4
v.l
v .It
7 .a*
Po.it
vv.41
.11
.14
.>
.4t
.til
vy .44
?> .tit
4.tat
rt .441
*T4, I.ttS4ttt7l '4CI01.|.t444t.tt
J>*
J>
1141
.11744
111
a*
.14447
.41141
I.iaaat
.77l|
.11711
.17111
.14741
tllll
tl7lt
.14
.till!
GC Spectrum of Mixture: Cyclohexene Oxide and Cyclohexanol
(Dissolved In Deuterated Chloroform)

.iLu
AREA*
ST AREA TYPE UIOTH AREA):
2.042 393600 PB .024 14 .22772
2. 13* 143 *P .006 .00343
2.24* 315* PP .140 . 12338
2.348 72964 PV .024 1 .74884
2.424 383680 ve .029 9 . 19624
3.268 1003 pp .044 .82404
3. 867
^"TT25B
72
2544i'02~
pp
pe
.046 .82898
...87*
-*P588293>
3.712 2393 IV .040 .06220
3.773 3023 VB .067 12044
6.23* 1707 *p. 101 04891
6.941 12109 PB .043 29823
7.818 1404 PO .296 83365
9.336 2124 vv . 112 03891
10.220 336 PV .017 eeeos
^8.536 343230
'4172138
VI . 133 13 06835_}
TOTAL AREA-
IUL FACTOR'l ,0**E4 0
*
GC SPECTRUM OF ETHER EXTRACT FROM CATALYZED REACTION

flREflM
IT n*Eft TYPE HtOTH area*
2.*27 1711077 PI .123 11.72131
.22* (221 IP .31 .1111
1.131 1131*11 PV .2( 3.32239
2.123 1*23231 VI .22 3.1*7*8
2.731 *33 IP .11 .89320
2.1*1 lit PI .111 .08*1
2.3*3 7*33 IV .11( .133*9
3.(71 19*7 VP .111 .1232*
1.1*8 (311 pp .171 .9321*
CTTZ i37i3*ee re .229 *.9i.iiT~;3.73* 12*1 V .33 .88932
3.113 11721 VI .171 .8392I
.33 379*7 PI .13 .29317
*.*** 1171 PV .1(2 ees9i
10.210 3331 pp .It .92(9*
CIS. 63* 11257*8 PI .11* s.t**i>
TOTftL ft*Ert-)l.*7(2E07
MUL FftCTO-l.l**OE**0
GC SPECTRUM OF ETHER EXTRACT FROM UNCATALYZED REACTION

DC
O
O
CX
OJ
X
o
Q
Wt-
<C
a
E-
w
Q
&u
O
z:
L>
EC
a,co

J
o
z<
X
oJ Zu cej ob Cb
o
2 Ow a
O X
< uw
a aw
Q HW <
CM DC>4 W
J HC 3
Z tij
<: a
i
ce z
< Q
CQ W
>
O OCO
Z CO
:o -
ce at-
uu
Cu
CO
a

Q
Oce
CuZ
ce>4 ooe fc.
< o
z ce?1 O
ce J
Cu x
uQ
u a
O H<c <
ce ceE- W
1 u
ce a
u
x z
H -
w
a
o >i-i
z o
3 CO
ce co
H -
u au
Cl,CO

XE-
Socogce wS
o z
&=
o
Cd
Cd
I Cu
Cd"
X U2 _.
CdCd~
z^
^ COoet-
CJCd
a,
CO
ce
co

B.BE5u
4. BE3-
2-BCSd
B
fcckn 2B Ci.779 ntn) of OftTR: MIXTURE . 0
%7 39 B3
I'l 4'
SI
X
SB 4B SB
a? =769
'/ /I \ I
"
1 1 1 i1 1 1| 1|
79 97
/
JU-
BB 7B
Hitt/Ch*PBBB 9B
TIC of ORTRsmJ fXTURL. 0
(J
c
D
S.BCB -i
4.BC6-;3.BEB-
|C
38. BEE:
a. X.BEB:
B- Jl 1 JTim* (nln.)
GC-MS of Cyclohexene Oxide
u
c
II
B
C
3
cr
Scan 313 C4.B32 111
7
4-r
of ORTR: MIXTURE. 0
B.BC3-
4.BES"
E. BE3-
Bji
9
/44
\
J.lll
34
I ,, \
B2
IBB
\1
3B 4B SB BB 7B BB 9B IBB
TIC of ORTR: MIXTURE. O
u
c
m
c
a
a
a.
S. BEB
4.BCB
3. BEB
C.BE6
I. BEB
B JLTi *> tun. )
GC-MS of Cyclohexanol

10000-j90Q&1
eooo]7000J 2?
6000-j>
S32ef 41
4S00|/
3000J20001
leaolj |0jl 1 . .Ill
Scan 338 (4.302 min) of DATFhNOWCK. D
5?
.u.
67
\ 7.
/
62/
:103j
9000
'8000
;7000
6000
"5000
[4000
[3000
100 ;2000
N^-1000
^0
MASS SPECTRUM OF PRIMARY PRODUCT IN ETHER EXTRACT
Scr 356 (4.456 -in) of DfiTfi:CYCHEXOL. DIO000 /
57
: I0CD[
9000 90on
8000 8000
7000 27 7000
8000 / BOOQ
5000
4000? 67
V /
5DDD
4000
3000 \ 71 30DO
zcoo / 100 :20QO
IOQQIi'
.11 1 , .iil! 1 ...II. . . 1 ,.\ [IBQO
MASS SPECTRUM OF BAKER-ANALYZED REAGENT CYCLOHEXANOL

in 537 (4.S&S nir.) of DR7R: NOWCK.O
B.Brs:
c B.BCSit
4. BE3:c
I t.BES:
B
B744
\34
l ,.,li.ill. ^a
te
B7
/
-..,, SI. ^
3B 4B SB 6B 7B SB BB IBB
Matt /Chargt
vS.BEB'
4. BEB
J C-BEB
C 1.BE6
Ti vie Coin.)
GC-MS OF PRIMARY PRODUCT IN ETHER EXTRACT
J 3.0C5
1.0E5
I.OC5"
JJ
C
B.OE6
r 5. 0E6
c 4.1E6
* 3.0E6
I a.ncs
c I. DEB
SO 100 1&J3
TIC of OflrRiNOpJCK. 0
k.
4 & a
Ti < (PMPl.j
4
10 12- t
GC-MS OF PRODUCT (M.W.=172) IN ETHER EXTRACT

O a
\ o o^" * *
^*
so Si ?">>ru ommtnv
8*010 >
f
*p eeeeeesaao<m<o <%
o*b
*
r in *em rm n *
xuff in
S3
^3SS feSSSSSS ssssss 3& ssssrssls




















