
Homochiral Selectivity in RNA Synthesis: Montmorillonite-catalyzed Quaternary Reactions of D,...
24
PREBIOTIC CHEMISTRY Homochiral Selectivity in RNA Synthesis: Montmorillonite-catalyzed Quaternary Reactions of D, L-Purine with D, L- Pyrimidine Nucleotides Prakash C. Joshi & Michael F. Aldersley & James P. Ferris Received: 2 June 2010 / Accepted: 17 June 2010 / Published online: 20 August 2010 # Springer Science+Business Media B.V. 2010 Abstract Selective adsorption of D, L-ImpA with D, L-ImpU on the platelets of montmorillonite demonstrates an important reaction pathway for the origin of homochirality in RNA synthesis. Our earlier studies have shown that the individual reactions of D, L-ImpA or D, L-ImpU on montmorillonite catalyst produced oligomers which were only partially inhibited by the incorporation of both D- and L-enantiomers. Homochirality in these reactions was largely due to the formation of cyclic dimers that cannot elongate. We investigated the quaternary reactions of D, L-ImpA with D, L-ImpU on montmorillonite. The chain length of these oligomers increased from 9-mer to 11-mer as observed by HPLC, with a concominant increase in the yield of linear dimers and higher oligomers in the reactions involving D, L-ImpA with D, L-ImpU as compared to the similar reactions carried out with D-enantiomers only. The formation of cyclic dimers of U was completely inhibited in the quaternary reactions. The yield of cyclic dimers of A was reduced from 60% to 10% within the dimer fraction. 12 linear dimers and 3 cyclic dimers were isolated and characterized from the quaternary reaction. The homochirality and regioselectivity of dimers were 64.1% and 71.7%, respectively. Their sequence selectivity was shown by the formation of purine-pyrimidine (54–59%) linkages, followed by purine-purine (29–32%) linkages and pyrimidine-pyrimidine (9–13%) linkages. Of the 16 trimers detected, 10 were homochiral with an overall homochirality of 73–76%. In view of the greater homochirality, sequence- and regio- selectivity, the quaternary reactions on montmorillonite demonstrate an unexpectedly favorable route for the prebiotic synthesis of homochiral RNA compared with the separate reactions of enantiomeric activated mononucleotides. Keywords RNA world . Homochirality . Prebiotic chemistry . Montmorillonite . Catalysis Orig Life Evol Biosph (2011) 41:213–236 DOI 10.1007/s11084-010-9222-1 Electronic supplementary material The online version of this article (doi:10.1007/s11084-010-9222-1) contains supplementary material, which is available to authorized users. P. C. Joshi : M. F. Aldersley : J. P. Ferris (*) Rensselaer Polytechnic Institute, Department of Chemistry & Chemical Biology, The New York Center for Astrobiology, Troy, NY 12180, USA e-mail: [email protected]
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Homochiral Selectivity in RNA Synthesis: Montmorillonite-catalyzed Quaternary Reactions of D,...

PREBIOTIC CHEMISTRY
Homochiral Selectivity in RNA Synthesis:Montmorillonite-catalyzed Quaternary Reactions of D,L-Purine with D, L- Pyrimidine Nucleotides
Prakash C. Joshi & Michael F. Aldersley &
James P. Ferris
Received: 2 June 2010 /Accepted: 17 June 2010 /Published online: 20 August 2010# Springer Science+Business Media B.V. 2010
Abstract Selective adsorption of D, L-ImpA with D, L-ImpU on the platelets ofmontmorillonite demonstrates an important reaction pathway for the origin of homochiralityin RNA synthesis. Our earlier studies have shown that the individual reactions of D, L-ImpA orD, L-ImpU onmontmorillonite catalyst produced oligomers which were only partially inhibitedby the incorporation of both D- and L-enantiomers. Homochirality in these reactions waslargely due to the formation of cyclic dimers that cannot elongate. We investigated thequaternary reactions of D, L-ImpAwith D, L-ImpU on montmorillonite. The chain length ofthese oligomers increased from 9-mer to 11-mer as observed by HPLC, with a concominantincrease in the yield of linear dimers and higher oligomers in the reactions involvingD, L-ImpAwith D, L-ImpU as compared to the similar reactions carried out with D-enantiomers only. Theformation of cyclic dimers of U was completely inhibited in the quaternary reactions. The yieldof cyclic dimers of Awas reduced from 60% to 10%within the dimer fraction. 12 linear dimersand 3 cyclic dimers were isolated and characterized from the quaternary reaction. Thehomochirality and regioselectivity of dimers were 64.1% and 71.7%, respectively. Theirsequence selectivity was shown by the formation of purine-pyrimidine (54–59%) linkages,followed by purine-purine (29–32%) linkages and pyrimidine-pyrimidine (9–13%) linkages.Of the 16 trimers detected, 10 were homochiral with an overall homochirality of 73–76%. Inview of the greater homochirality, sequence- and regio- selectivity, the quaternaryreactions on montmorillonite demonstrate an unexpectedly favorable route for theprebiotic synthesis of homochiral RNA compared with the separate reactions ofenantiomeric activated mononucleotides.
Keywords RNAworld . Homochirality . Prebiotic chemistry . Montmorillonite . Catalysis
Orig Life Evol Biosph (2011) 41:213–236DOI 10.1007/s11084-010-9222-1
Electronic supplementary material The online version of this article (doi:10.1007/s11084-010-9222-1)contains supplementary material, which is available to authorized users.
P. C. Joshi :M. F. Aldersley : J. P. Ferris (*)Rensselaer Polytechnic Institute,Department of Chemistry & Chemical Biology,The New York Center for Astrobiology, Troy, NY 12180, USAe-mail: [email protected]

Introduction
RNA is proposed to be an important biopolymer in the early life on Earth where it providedboth catalysis and the ability to store genetic information (Cech et al. 1981; Guerrier-Takadaet al. 1983; Gilbert 1986). The “RNA World” hypothesis for the origin of life proposes thatRNA formed first and the DNA-protein world evolved from it (Woese 1967; Crick 1968;Orgel 1968, 2000). Most hypotheses of the origins of biological organization have suggestedthat RNAs with length in the range of 30–50 nucleotides are needed to initiate catalysis thatmakes a genetic system viable (Szostak and Ellington 1993; Joyce and Orgel 1999). An RNAconsisting of triple stem-loop structure containing 40–50 nucleotides offers a reasonable hopeof functioning as a replicase ribozyme (Szostak and Ellington 1993; Joyce and Orgel 2006).
Previous studies from our laboratory have demonstrated the montmorillonite-catalyzedformation of RNA oligomers from activated mononucleotides (Ferris 2002; Miyakawa andFerris 2003; Huang and Ferris 2006; Zagorevskii et al. 2006; Joshi et al. 2007). Oligomersof chain lengths up to 9-mers, 30-mers and 50-mers were detected by HPLC (Joshi et al.2007), MALDI mass spectra (Zagorevskii et al. 2006) and gel electrophoresis (Huang andFerris 2006), respectively. The individual D, L-ImpA and D, L-ImpU (Fig. 1) gave anaverage of 60% and 58.5% yields respectively, of cyclic dimers in the dimer fractions. Thetrimer fractions have homochiralities of 40% and 10.9%, respectively. The longestoligomers, as determined by HPLC analysis, were 7 mers from D, L-ImpA and 6 mersfrom D, L-ImpU (Joshi et al. 2007). The high yields of cyclic dimers, the lowhomochirality, and the short oligomers formed indicate that this is not an encouragingapproach to the formation of RNA oligomers that may have catalytic activity (Joshi et al.2007). This conclusion prompted the investigation of the products formed from thequaternary mixture of D, L-ImpAwith D, L-ImpU on montmorillonite. We did this becauseif complementary activated nucleotides were present on the Primitive Earth, e.g. D, L-ImpAand D, L-ImpU, they would be present as racemic mixtures (Fig. 1).
Materials and Methods
General Adenosine, A3′p, pA, A2′pA, A3′pA, A2′pU, A3′pU, anhydrous NaClO4, bacterialalkaline phosphatase (APH), 2, 2′-dithiodipyridine, imidazole, NaH2PO4, phosphorusoxychloride (POCl3), ribonuclease T2 (RNase T2), uridine, pU, U
3′p, U2′pA, U3′pA, U2′pU,U3′pU, triethyl phosphate (TEP), trifluoroacetic acid (TFA), triphenylphosphine, triethylamine(TEA) and Trizma base (TRIS) were obtained from Sigma. L-adenosine and L-uridine werepurchased from ChemGenes Corporation. Sodium pyrophosphate, perchloric acid andtriethyl phosphate (TEP) were purchased from Aldrich. N, N-Dimethylformamide(DMF), dimethyl sulfoxide (DMSO), acetonitrile (CH3CN), methanol (CH3OH), etherand acetone were obtained from Mallinckrodt. Sodium chloride and magnesium chloridewere procured from J.T. Baker. HPLC grade NaClO4 and enzyme grade ammonium acetatewere purchased from Fisher. Volclay SPV-200 was a gift from the American Colloid Co.,Arlington Heights, IL. The Na+ form of homoionic Volclay montmorillonite was prepared bytitration to pH 7. All montmorillonite-catalyzed reactions, synthesis and control studies wereperformed in a solution containing 0.2 M sodium chloride and 0.075 M magnesium chloride.
HPLC analysis was performed on a Hitachi L-6200A intelligent pump system equippedwith a Hitachi L-4000 UV detector operating at 260 nm. The products as anions wereseparated on a Dionex DNAPac®-100 (4.0×250 mm) analytical anion exchange columnfrom Dionex Corporation, Sunnyvale, CA, using a gradient of 0–0.4 M NaClO4 with 2 mM
214 P.C. Joshi et al.

Tris at pH 8. The analysis of dimers and trimers was performed on a reverse phase AlltimaC-18, 5 μ (4.6 mm×250 mm) column (Alltech) using a gradient of 0.02 M NaH2PO4 with0.2% TFA (pH 2.5) and 30% CH3CN in water with 0.2% TFA (pH 2.5). The analysis of theisomers of U was also performed on a reverse phase Jupiter C-18, 4 μ (4.6 mm × 250 mm)column (Phenomenex) and a μBondapak C-18 (3.9 mm×300 mm) column (Waters) byusing a gradient of 0.005 M NaH2PO4 with 5% CH3OH, pH 3.5 and 0.01 M NaH2PO4 with40% CH3OH, pH 4. Large scale separations and purification of D- and L- mononucleotidesand selected dimers were carried out on a preparative Alltima C-18, 5 μ column (10 mm ×300 mm) isocratically by using TFA (0.075%) and CH3CN (2%) in H2O (pH 2.3).Electrospray mass spectra were obtained using either (1) an Agilent 1100 series LC/MSD intrap system; samples were introduced into the ion source using a syringe pump at a flowrate of ~7 μL/min or using an auto sampler, or (2) a Thermoelectron LTQ XL Orbitrap massspectrometer, fitted with an Agilent 1200 nanoflow HPLC system; the column was an AgilentTechnologies Zorbax Eclipse SB-C18 5 μm (150 mm×0.5 mm) at a flow rate of 20 μL/min.
Enzymatic hydrolysis of reaction products An alkaline phosphatase solution (0.5 units,500 μL) was adjusted to pH 8 and incubated for 1 h at 37°C to remove the 5′-terminalphosphate groups. RNase T2 hydrolysis of RNA oligomers was performed in 100 μL at 37°Cand pH 4 using two units of the enzyme for 1–4 h.
Phosphorylation of L-nucleosides L-nucleosides of adenine and uracil were phosphorylatedat the 5′-position (Ikemoto et al 1995). A mixture of L-nucleoside (30 mmol) and water(15 mmol) in TEP (1.0 mL) was treated with POCl3 (60 mmol) at 0°C for 5 h (L-adenosine)or 25 h (L-uridine). The reaction was terminated by adding the reaction mixture drop wiseinto 1.5 mL ice cold water and continuing stirring for an additional 1 h. Progress of thereaction was monitored by HPLC by withdrawing 2 μL samples at intervals, diluting to10 mL with water and analyzing a 10 μL aliquot on an Alltima reverse phase column. The
N
NN
N
NH2
O
OHOH
OPN
O-
O
N
NH
O
ONO
OHOH
OPN
O-
O
N
D-ImpU
D-ImpA
N
N N
N
NH2
O
OH OH
O P N
O-
O
N
HN
O
O NO
OH OH
O P N
O-
O
N
L-ImpU
L-ImpA
Fig. 1 Enantiomeric pairs of activated nucleotide anions: D- and L-ImpA, D- and L-ImpU
Homochiral Selectivity in RNA Synthesis 215

crude yields of L-AMP and L-UMP at the completion of the reactions were 90±5% and75±5%, respectively. TEP was removed from the reaction mixture by extraction with ether(3×5 mL). A portion of mononucleotide (below 10%) retained in the ether extract wasrecovered quantitatively by extraction with H2O (2×1 mL). The L-mononucleotides werepurified on a preparative Alltima column (purity >99.5%).
Preparation of activated nucleotides A mixture of 5′-mononucleotide (free acid, 0.16 mmol)and imidazole (2.2 mmol) was dissolved in DMF (2 mL) and DMSO (2 mL) in a 50 mL flaskand the solvent was evaporated to near dryness at a reduced pressure. The evaporation wasrepeated twice with DMF (2×2 mL) to remove residual water. The residue was dissolved inDMF (2 mL) and DMSO (2 mL) and stirred with 2, 2′-dithiodipyridine (0.64 mmol), triphenylphosphine (0.46 mmol) and TEA (0.65 mmol) for 3 h. The resulting product was recoveredfrom the clear yellow reaction mixture as a precipitate by adding the reaction mixture drop wiseto a solution of anhydrous sodium perchlorate (0.35 g) in a mixture of ether (30 mL), acetone(20 mL) and TEA (2 mL) with stirring. The stirring was continued for 2 h (to ensure completeformation of Na+ salt) and a white, flocculent solid was precipitated which was allowed tosettle (15 min). The supernatant was decanted and the remaining reaction mixture wascentrifuged. The resulting white pellet was washed twice with a mixture of ether (50 mL) andacetone (50 mL) and then with ether (2×50 mL) and dried overnight in a vacuum desiccator.Their purities were determined by reverse phase HPLC (purity >99.5%).
Synthesis of Dimers
Synthesis of linear dimers of A The D, D-ApA and D, L-ApA were synthesized for use asauthentic standards for the identification of linear dimers and also as starting materials for thesynthesis of trimers. For example: D, D-A2′pA and D, D-A3′pAwere prepared by the addition of15 mM D-adenosine (200 μL) to 20 mg montmorillonite and incubating the mixture at 24°Cfor 1 h. 10 mM D-ImpA (200 μL) was added to the mixture and the reaction was allowed tostand for 48 h at 24°C. The D, D-A2′pA and D, D-A3′pAwere purified on a preparative reversephase HPLC column, eluting in that order. The products were identified by hydrolysis withRNase T2 and APH and identification of the hydrolysis products by HPLC on a reverse phaseAlltima C-18 column. Similarly the D, L-A2′pA and D, L-A3′pAwere prepared by the additionof 15 mM D-adenosine (200 μL) to 20 mg montmorillonite and incubating the mixture for 1 h.10 mML-ImpA (200 μL) was added to the mixture and the reaction was allowed to stand for48 h at 24°C to yield D, L-A2′pA and D, L-A3′pA which were purified on a preparativereverse phase HPLC column, eluting in that order. The structures were confirmed by enzymatichydrolysis and electrospray HPLC/MS where the dimers were separated on a reverse phaseAlltima C-18 column using a gradient of 0–40% of 0.2% formic acid and 0.2% formic acid in30% CH3CN. Calculated for ApA (C20H25N10O10P, m/z=596.1493). Found for each isomer:nominal masses: 597 (M+1) and 595 (M-1).
Synthesis of linear dimers of U These products were synthesized for use as authenticstandards for the identification of linear dimers and as starting materials for the synthesis oftrimers. For example D, L-UpUs, were prepared by the addition of 15 mM D-uridine(200 μL) to 20 mg montmorillonite and allowing the mixture to stand at 24°C for 1 h.10 mML-ImpU (200 μL) was added to the mixture and was allowed to stand at 24°C for48 h. D, L-U2′pU and D, L-U3′pU were purified using a preparative reverse phase HPLCcolumn, eluting in that order. Their structures were determined by hydrolysis with RNase
216 P.C. Joshi et al.

T2 and APH and identification of the hydrolysis products by comparison with authenticstandards on a reverse phase Alltima C-18 column. The structures were confirmed byelectrospray HPLC/MS. Calculated for UpU (C18H23N4O14P) m/z=550.0948. Found foreach isomer: nominal masses: 551 (M+1) and 549 (M-1).
Synthesis of linear dimers containing both A and U These were synthesized for use asauthentic standards for the identification of linear dimers and as starting materials for the synthesisof ApUpA and ApUpU trimers. For example, D, D-ApUs were prepared by the addition 15 mMD-adenosine (200 μL) to 20mgmontmorillonite and allowing themixture to stand at 24°C for 1 h.10 mMD-ImpU (200 μL) was added to the mixture and was allowed to stand for 48 h at 24°C. D,D-A2′pU and D, D-A3′pU were purified on a preparative reverse phase HPLC column, eluting inthat order. Their structures were determined by hydrolysis with RNase T2 and APH andidentification of the hydrolysis products by HPLC on a reverse phase Alltima C-18 column. D,L-A2′pU and D, L-A3′pU were prepared by the addition 15 mM D-adenosine (200 μL) to 20 mgmontmorillonite and allowing the mixture to stand at 24°C for 1 h. 10 mM L-ImpU (200 μL)was added to the mixture and was allowed to stand for 48 h at 24°C. The D, L-A2′pU and D, L-A3′pU were purified on a preparative reverse phase HPLC column eluting in that order. D, D-and D, L- isomers of UpAwere prepared by the addition of 15 mM D-uridine (200 μL) to 20 mgmontmorillonite and allowing the mixture to stand at 24°C for 1 h. 10 mM D-ImpA or L-ImpA(200 μL each) were added to the mixture and was allowed to stand for 48 h at 24°C. D, D-U2′pA, D, D-U3′pA, D, L-U2′pA and D, L-U3′pA were purified on a preparative reverse phaseHPLC column. The structures were confirmed by electrospray HPLC/MS. Calculated for ApUor UpA (C19H24N7O12P), m/z=573.1220. Found for each isomer: nominal masses: 574 (M+1)and 572 (M-1).
Synthesis of Trimers
Synthesis of linear trimers of A D, D, D-A3′pA2′pA and D, D, D- A3′pA3′pA weresynthesized by adding 10 mM D, D-A3′pA (84 μL) to montmorillonite (4.2 mg). The mixturewas allowed to stand at 24°C for 30 min. 84 μL of 1 mM D-ImpAwas added to the reactionmixture and allowed to react for 5 hr at 24°C. The trimers were separated as a single peak onan ion exchange column in 2% yield. The D, D, D-A3′pA2′pA and D, D, D-A3′pA3′pAisomers were separated in that order on a Jupiter reverse phase column. D, D, D-A2′pA2′pAand D, D, D-A2′pA3′pAwere synthesized by adding 0.2 mM D, D-A2′pA (100 μL) to 2.0 mgmontmorillonite. The mixture was allowed to stand at 24°C for 1 h. 100 μL of 1.0 mM D-ImpA was added to the reaction mixture and allowed to react for 48 h at 24°C. The trimerswere separated as a single peak on an ion exchange column in 2% yield. The D, D, D-A2′pA2′pA and D, D, D-A2′pA3′pA isomers were separated in that order on an Alltima reversephase column.
D, D, L-A3′pA2′pA and D, D, L- A3′pA3′pA were synthesized by adding 10 mM D, D-A3′pA (84 μL) to 4.2 mg montmorillonite. The mixture was allowed to stand at 24°C for30 min. 84 μL of 1 mML-ImpAwas added to the reaction mixture and allowed to react for5 hr at 24°C. The trimers were separated as a single peak from an ion exchange column in1% yield. D, D, L-A3′pA2′pA and D, D, L-A3′pA3′pA isomers were separated in that orderon a Jupiter reverse phase column. D, D, L-A2′pA2′pA and D, D, L-A2′pA3′pA weresynthesized by adding 0.2 mM D, D-A2′pA (100 μL) to 2.0 mg montmorillonite. Themixture was allowed to stand at 24°C for 1 h. 100 μL of 1.0 mML-ImpA was added to thereaction mixture and allowed to react for 48 h at 24°C. The trimers were separated as a
Homochiral Selectivity in RNA Synthesis 217

single peak on an ion exchange column in 2% yield. The D, D, L-A2′pA2′pA and D, D, L-A2′pA3′pA isomers were separated in that order on an Alltima reverse phase column.
D, L, D-A3′pA2′pA and D, L, D-A3′pA3′pA were prepared by adding 1 mM D, L-A3′pA(143 μL) to 7.15 mg montmorillonite. The mixture was allowed to stand at 24°C for 1 h.143 μL of 1 mM D-ImpA was added to the reaction mixture and allowed to react for 18 h at24°C. The trimers were separated as a single peak from an ion exchange column in 14% yield.D, L, D-A3′pA2′pA and D, L, D-A3′pA3′pA isomers were separated in that order on a Jupiterreverse phase column. The D, L, D-A2′pA2′pA and D, L, D- A2′pA3′pA were synthesized byadding 0.2 mM D, L-A2′pA (100 μL) to 2.0 mg montmorillonite. The mixture was allowed tostand at 24°C for 1 h. 100 μL of 1.0 mM D-ImpA was added to the reaction mixture andallowed to react for 48 h at 24°C. The trimers were separated as a single peak from an ionexchange column in 2% yield. The D, L, D-A2′pA2′pA and D, L, D-A2′pA3′pA isomers wereseparated in that order on an Alltima reverse phase column.
D, L, L-A3′pA2′pA and D, L, L-A3′pA3′pA were synthesized by adding 1 mM D, L-A3′pA (143 μL) to 7.15 mg montmorillonite. The mixture was allowed to stand at 24°C for1 h. 143 μL of 1 mML-ImpA was added to the reaction mixture and allowed to react for18 h at 24°C. The trimers were separated as a single peak from an ion exchange column in22% yield. The D, L, L-A3′pA2′pA and D, L, L-A3′pA3′pA isomers were eluted in that orderon a Jupiter reverse phase column. D, L, L-A2′pA2′pA and D, L, L-A2′pA3′pA weresynthesized by adding 0.2 mM D, L-A2′pA (100 μL) to 2.0 mg montmorillonite. Themixture was allowed to stand at 24°C for 1 h. 100 μL of 1.0 mML-ImpA was added to thereaction mixture and allowed to react for 48 h at 24°C. The trimers were separated as asingle peak from an ion exchange column in 2% yield. The D, L, L-A2′pA2′pA and D, L, L-A2′pA3′pA isomers were eluted in that order on an Alltima reverse phase column.
The products were identified by hydrolysis with RNase T2 and APH and byidentification of hydrolysis products by HPLC on a reverse phase Alltima column. Thestructure of trimers were confirmed by electrospray HPLC/MS where the trimers wereseparated on a reverse phase Alltima column using a gradient of 0–40% 0.2% formic acidand 0.2% formic acid in 30% CH3CN. Calculated for ApApA (C30H37N15O16P2), m/z=925.2018. Found for each isomer: nominal mass: 926 (M+1) and 924 (M-1).
Synthesis of linear trimers of U D, D, D-U2′pU2′pU and D, D, D-U2′pU3′pU weresynthesized by adding to 250 μL 1 mM D, D-U2′pU (250 μL) to 25 mg montmorillonite.The mixture was allowed to stand at 24°C for 30 min. 250 μL of 5 mM D-ImpU was addedto the reaction mixture and allowed to react for 72 hr at 24°C. The trimers were separated asa single peak on an ion exchange column in 8.7% yield. The D, D, D-U2′pU2′pU and D, D,D-U2′pU3′pU isomers were eluted in that order from a μBondapak reverse phase column.The D, D, L-U2′pU2′pU and D, D, L-U2′pU3′pU were synthesized by adding 1 mM D, D-U2′pU (250 μL) to 25 mg montmorillonite. The mixture was allowed to stand at 24°C for30 min. 250 μL of 5 mML-ImpU was added to the reaction mixture and allowed to reactfor 72 h at 24°C. The trimers were separated as a single peak from an ion exchange columnin 8.4% yield. The D, D, L-U2′pU2′pU and D, D, L- U2′pU3′pU isomers were eluted in thatorder on a μBondapak reverse phase column.
The D, D, D-U3′pU2′pU and D, D, D-U3′pU3′pU were synthesized by adding 1 mM D,D-U3′pU (250 μL) to 25 mg montmorillonite. The mixture was allowed to stand at 24°C for30 min. 250 μL of 5 mM D-ImpU was added to the reaction mixture and allowed to reactfor 72 h at 24°C. The trimers were separated as a single peak from an ion exchange columnin 10.5% yield. The D, D, D-U3′pU2′pU and D, D, D-U3′pU3′pU isomers were separated inthat order on a μBondapak reverse phase column.
218 P.C. Joshi et al.

The D, D, L-U3′pU2′pU and D, D, L-U3′pU3′pU were synthesized by adding 1 mM D, D-U3′pU (250 μL) to 25 mg montmorillonite. The mixture was allowed to stand at 24°C for30 min. 250 μL of 5 mML-ImpU was added to the reaction mixture and allowed to react for72 h at 24°C. The trimers were eluted as a single peak from an ion exchange column in 12%yield. D, D, L-U3′pU2′pU and D, D, L-U3′pU3′pU isomers were eluted in that order on aμBondapak reverse phase column.
D, L, D-U2′pU2′pU and D, L, D-U2′pU3′pU were synthesized by adding 2.5 mM D, L-U2′pU (169 μL) to 16.9 mg montmorillonite. The mixture was allowed to stand at 24°C for1 h. 169 μL of 5 mM D-ImpU was added to the reaction mixture and allowed to react for96 h at 24°C. The trimers were eluted as a single peak from an ion exchange column in23% yield. D, L, D-U2′pU2′pU and D, L, D-U2′pU3′pU isomers were separated in that orderon a μBondapak reverse phase column.
D, L, L-U2′pU2′pU and D, L, L-U2′pU3′pU were synthesized by adding 2.5 mM D, L-U2′pU(169 μL) to 16.9 mg montmorillonite. The mixture was allowed to stand at 24°C for 1 h. 169 μLof 5 mML-ImpU was added to the reaction mixture and allowed to react for 96 h at 24°C. Thetrimers were eluted from an ion exchange column in 21% yield. D, L, L-U2′pU2′pU and D, L, L-U2′pU3′pU isomers were eluted in that order on a μBondapak reverse phase column.
D, L, D-U3′pU2′pU and D, L, D-U3′pU3′pU were synthesized by adding 1 mM D, L-U3′pU(66 μL) to 6.6 mg montmorillonite. The mixture was allowed to stand at 24°C for 1 h. 66 μL of5 mM D-ImpU was added to the reaction mixture and allowed to react for 96 h at 24°C. Thetrimers were separated from an ion exchange column in 19% yield. D, L, D-U3′pU2′pU and D,L, D-U3′pU3′pU isomers were eluted in that order on a μBondapak reverse phase column.
D, L, L-U3′pU2′pU and D, L, L-U3′pU3′pU were synthesized by adding 1 mM D, L-U3′pU (66 μL) to 6.6 mg montmorillonite and the mixture was allowed to stand at 24°C for1 h. 66 μL of 5 mML-ImpU was added to the reaction mixture and allowed to react for96 h at 24°C. The trimers were separated from an ion exchange column in 18% yield. D, L,L-U3′pU2′pU and D, L, L-U3′pU3′pU isomers were eluted in that order on a μBondapakreverse phase column. The products were identified by hydrolysis with RNase T2 and APHand by identification of hydrolysis products by HPLC on a reverse phase Alltima column.The structure of trimers were confirmed by electrospray HPLC/MS where the trimers wereeluted on a reverse phase Alltima column using a gradient of 0–40% 0.2% formic acid and0.2% formic acid in 30% CH3CN. Calculated for UpUpU (C27H34N6O22P2), m/z=856.1202. Found for each isomer: nominal mass: 857 (M+1) and 855 (M-1).
Synthesis of linear trimers containing both A and U D, D, D-A3′pA2′pU and D, D, D-A3′pA3′pU were synthesized by adding 0.2 mM D, D-A3′pA (100 μL) to 2.0 mg montmorilloniteand the mixture was allowed to stand at 24°C for 60 min. 100 μL of 1.0 mM D-ImpU wasadded to the reaction mixture and allowed to react for 48 hr at 24°C. The trimers were separatedas a single peak from an ion exchange column in 2% yield. D, D, D-A3′pA2′pU and D, D, D-A3′pA3′pU were eluted in that order on an Alltima reverse phase column.
The D, D, L-A3′pA2′pU and D, D, L-A3′pA3′pU were synthesized by adding 0.2 mM D,D-A3′pA (250 μL) to 2.0 mg montmorillonite and the mixture was allowed to stand at 24°Cfor 1 h. 100 μL of 1.0 mML-ImpU was added to the reaction mixture and allowed to reactfor 48 h at 24°C. The trimers were separated as a single peak from an ion exchange columnin 2% yield. D, D, L-A3′pA2′pU and D, D, L-A3′pA3′pU isomers were eluted in that orderon an Alltima reverse phase column.
D, D, D-A2′pA2′pU and D, D, D-A2′pA3′pU were synthesized by adding 0.2 mM D, D-A2′pA (100 μL) to 2.0 mg montmorillonite and the mixture was allowed to stand at 24°Cfor 1 h. 100 μL of 1.0 mM D-ImpU was added to the reaction mixture and allowed to react
Homochiral Selectivity in RNA Synthesis 219

for 48 h at 24°C. The trimers were eluted as a single peak from an ion exchange column in2% yield. D, D, D-A2′pA2′pU and D, D, D-A2′pA3′pU isomers were eluted in that order onan Alltima reverse phase column.
D, D, L-A2′pA2′pU and D, D, L-A2′pA3′pU were synthesized by adding 0.2 mM D, D-A2′pA (100 μL) to 2.0 mg montmorillonite and the mixture was allowed to stand at 24°Cfor 1 h. 100 μL of 1.0 mML-ImpU was added to the reaction mixture and allowed to reactfor 48 h at 24°C. The trimers were eluted as a single peak from an ion exchange column in2% yield. D, D, L-A2′pA2′pU and D, D, L-A2′pA3′pU isomers were eluted in that order onan Alltima reverse phase column.
D, D, D-A3′pU2′pU and D, D, D-A3′pU3′pU were synthesized by adding 0.2 mM D, D-A3′pU (100 μL) to 2.0 mg montmorillonite and the mixture was allowed to stand at 24°Cfor 1 h. 100 μL of 1.0 mM D-ImpU was added to the reaction mixture and allowed to reactfor 48 h at 24°C. The trimers were eluted as a single peak from an ion exchange column in2% yield. D, D, D-A3′pU2′pU and D, D, D-A3′pU3′pU isomers were separated in that orderon an Alltima reverse phase column.
D, D, L-A3′pU2′pU and D, D, L-A3′pU3′pU were synthesized by adding 0.2 mM D, D-A3′pU (100 μL) to 2.0 mg montmorillonite and the mixture was allowed to stand at 24°Cfor 1 h. 100 μL of 1.0 mML-ImpU was added to the reaction mixture and allowed to reactfor 48 h at 24°C. The trimers were eluted as a single peak from an ion exchange column in2% yield. D, D, L-A3′pU2′pU and D, D, L-A3′pU3′pU isomers were separated in that orderon an Alltima reverse phase column.
D, D, D-A2′pU2′pU and D, D, D-A2′pU3′pU were synthesized by adding 0.2 mM D, D-A2′pU (100 μL) to 2.0 mg montmorillonite and the mixture was allowed to stand at 24°Cfor 1 h. 100 μL of 1.0 mMD-ImpU was added to the reaction mixture and allowed to react for48 h at 24°C. The trimers were eluted as a single peak from an ion exchange column in 2%yield. The D, D, D-A2′pU2′pU and D, D, D-A2′pU3′pU isomers were eluted in that order on anAlltima reverse phase column.
D, D, L-A2′pU2′pU and D, D, L-A2′pU3′pU were synthesized by adding 0.2 mM D, D-A2′pU (100 μL) to 2.0 mg montmorillonite and the mixture was allowed to stand at 24°Cfor 1 h. 100 μL of 1.0 mML-ImpU was added to the reaction mixture and allowed to reactat 24°C for 48 h. The trimers were eluted as a single peak from an ion exchange column in2% yield. D, D, L-A2′pU2′pU and D, D, L-A2′pU3′pU isomers were separated in that orderon an Alltima reverse phase column.
D, D, D-A3′pU2′pA and D, D, D-A3′pU3′pA were synthesized by adding 0.2 mM D, D-A3′pU (100 μL) to 2.0 mg montmorillonite and the mixture was allowed to stand at 24°Cfor 1 h. 100 μL of 1.0 mM D-ImpAwas added to the reaction mixture and allowed to reactfor 48 h at 24°C. The trimers were eluted as a single peak from an ion exchange column in2% yield. D, D, D-A3′pU2′pA and D, D, D-A3′pU3′pA isomers were separated in that orderon an Alltima reverse phase column.
D, D, L-A3′pU2′pA and D, D, L- A3′pU3′pA were synthesized by adding 0.2 mM D, D-A3′pU (100 μL) to 2.0 mg montmorillonite and the mixture was allowed to stand at 24°Cfor 1 h. 100 μL of 1.0 mML-ImpA was added to the reaction mixture and allowed to reactfor 48 h at 24°C. The trimers were eluted as a single peak from an ion exchange column in2% yield. D, D, L-A3′pU2′pA and D, D, L-A3′pU3′pA isomers were eluted in that order onan Alltima reverse phase column.
The D, D, D-A2′pU2′pA and D, D, D-A2′pU3′pA were synthesized by adding 0.2 mM D,D-A2′pU (100 μL) to 2.0 mg montmorillonite and the mixture was allowed to stand at 24°Cfor 1 h. 100 μL of 1.0 mM D-ImpAwas added to the reaction mixture and allowed to reactat 24°C for 48 h. The trimers were eluted as a single peak from an ion exchange column in
220 P.C. Joshi et al.

2% yield. D, D, D-A2′pU2′pA and D, D, D-A2′pU3′pA isomers were eluted in that order onan Alltima reverse phase column.
D, D, L-A2′pU2′pA and D, D, L-A2′pU3′pA were synthesized by adding 0.2 mM D, D-A2′pU (100 μL) to 2.0 mg montmorillonite and the mixture was allowed to stand at 24°Cfor 1 h. 100 μL of 1.0 mML-ImpA was added to the reaction mixture and allowed to reactat 24°C for 48 h. The trimers were eluted as a single peak from an ion exchange column in2% yield. D, D, L-A2′pU2′pA and D, D, L-A2′pU3′pA isomers were eluted in that order onan Alltima reverse phase column.
The products were identified by hydrolysis with RNase T2 and APH and byidentification of hydrolysis products on a reverse phase Alltima HPLC column. Thestructures of trimers were confirmed by electrospray HPLC/MS where the trimers wereseparated on a reverse phase Alltima column using a gradient of 0–40% formic acid andformic acid (0.2%) in 30% CH3CN. Calculated for ApApU and/or ApUpA and/or UpApA(C29H36N12O18P2): m/z=902.1746. Found for each isomer 903.1 (M+1) and 901.09 (M-1).Calculated for ApUpU and/or UpApU and/or UpUpA (C28H35N9O20P2), m/z=879.1474.Found for each isomer: nominal mass: 880 (M+1) and 878 (M-1).
Montmorillonite-catalyzed oligomerization of a racemic mixture of D, L-ImpA and D, L-ImpU Stock solutions of D-ImpA, L-ImpA, D-ImpU and L-ImpU (15 mM) were prepared in a0.075 M magnesium chloride and 0.2 M sodium chloride reagent. The ImpA and ImpUconcentrations were determined by quantitative HPLC analysis of 10-5 M samples on a reversephase column. The D, L- mixtures (0.15–15 mM) were prepared by mixing the correct volumeof each solution calculated on the basis of these analyses. 200-μL racemic mixture was addedto 10 mg Na+-montmorillonite, the suspension was vortexed and allowed to stand at 24°C for aperiod of 2 h, 18 h or 3 days. The supernatant was collected from reaction mixtures containingmontmorillonite by centrifugation at 14,000 rpm for 8 min. The reaction products weredesorbed from montmorillonite by extracting four times (2 x1 h, overnight and 1×1 h) with200 μL of 30 % CH3CN in 0.1 M NaCl elution reagent. Each extract was collected bycentrifugation and combined with the supernatant to give 0.9 mL of combined extracts. Thecombined extract was diluted to 1.0 mL and filtered (Alltima 0.45 μm nylon syringe filter),adjusted to pH 4 with 1 M HClO4 and incubated at 37°C for 4 h to cleave any unreactedimidazole group from the activated nucleotides. Each of the dimer, trimer and tetramer fractionswas separated from the combined extracts on the ion exchange column as a cluster of peaks.Products were hydrolyzed by APH to cleave the 5′-terminal phosphate group prior to analysison a reverse phase column.
Reaction and analysis in the absence of minerals Control solutions of D, L-ImpA and D, L-ImpU mixtures were prepared in a 0.075 M magnesium chloride and 0.2 M sodium chloridereagent (200 μL) without montmorillonite. The mixtures were allowed to stand at 24°C for3 days. At the end of reaction time, the mixture was diluted to 1.0 mL with the 30% CH3CN in0.1 M NaCl reagent, adjusted to pH 4 with 1 M HClO4 and incubated at 37°C for 4 h to cleaveany unreacted imidazole group from the activated nucleotides. The samples were furtherhydrolyzed by APH before analysis on an Alltima reverse phase column.
Molecular modeling of activated nucelotides After an initial minimum energy structure hadbeen determined using molecular mechanics, as found in Spartan ′06 (by Wavefunction,Inc., Irvine, CA), the minimum energy of the molecule was determined using the Hartree-Fock 3-21G or 6-31G* basis sets. The electrostatic potential maps and dipole momentswere then calculated. The diagrams in Fig. 8 have areas of negative potential colored red
Homochiral Selectivity in RNA Synthesis 221

and positive regions blue, as per convention. The dipole moments for the activatednucleotide anions, ImpA and ImpU, together with the corresponding zwitterionic species,are listed in Table 6.
Results and Discussion
Unless otherwise stated, the concentration of activated nucleotide solutions is that of the totalactivated nucleotide present in eqimolar mixtures. In our study of the reactions of a 15 mMequimolar quaternary mixture of the activated monomers, D, L-ImpA and D, L-ImpU, weobserved byHPLC that the chain length of the oligomers was 11mers with this quaternary system(Reaction 4a, Table 1). The homochirality of dimer was 64.1% (Table 2). The homochirality oftrimers increased to 74.2% (Table 4) compared with 38±3% with D, L-ImpA (Joshi et al. 2007)or 10±1% with D, L-ImpU (Joshi et al. 2007) when reacted alone. In the dimer fraction, thecyclic dimers were formed in 30.5±6.5% yield compared with 59.0±1.0% with D, L-ImpA orD, L-ImpU reacted alone with montmorillonite. The lower yields of cyclic dimers and higherhomochirality of trimers suggests that it may be possible to selectively elongate these oligomersand preserve homochirality (Ferris 2002; Ferris et al. 1996).
Formation of oligomers The montmorillonite-catalyzed reaction of a 15 mM D-ImpA withD-ImpU mixture for 3 days produced oligomers as long as 9-mers as determined by HPLCanalysis on an ion exchange column (Reaction 3, Table 1 and Fig. 2a). The chain length ofoligomers increased to 11-mer in the montmorillonite-catalyzed reaction of the 15 mMequimolar quaternary mixture of D, L-ImpA and D, L-ImpU (Reaction 4a, Table 1 andFig. 2b). These findings differed significantly from the results obtained in Reactions 1 and 2where there were much higher yields of cyclic dimers (Joshi et al. 2007). In the quaternary
Table 1 Percent yield of oligomers formed by the montmorillonite-catalyzed reactions of (1) D, L-ImpA, (2)D, L-ImpU, (3) D-ImpA with D-ImpU and (4a, 4b, 4c) D, L-ImpA with D, L-ImpU
Reaction# 1 2 3 4a 4b 4c
Time 18 h 18 h 3 day 3 day 18 h 18 h
Molarity 15 mM 15 mM 15 mM 15 mM 1.2 mM 0.6 mM
Chain length
I 51.5±4.3 63.3±2.23 21.9±1.2 30.3±0.8 65.5±1.7 79.6±1.10
II 37.5±2.4 26.6±1.21 54.7±1.2 43.3±3.0 28.1±1.1 17.5±0.2
III 7.7±0.1 6.6±0.30 13.8±0.5 14.5±0.8 5.2±0.6 2.9±0.40
IV 2.40±0.02 2.51±0.05 5.3±0.6 5.8±1.0 1.3±0.10
V 0.64±0.04 0.92±0.10 2.44±0.4 3.00±0.3
VI 0.12±0.02 0.06±0.01 1.11±0.2 1.50±0.01
VII 0.02±0.00 0.51±0.01 0.70±0.00
VIII 0.23±0.01 0.43±0.00
IX Ta 0.20±0.00
X 0.11±0.00
XI Ta
a Trace
222 P.C. Joshi et al.

Table 2 Dimers formed by the reactions of D, L-ImpA + D, L-ImpU on montmorillonitea. Proportions ofeach dimer, homochirality and regioselectivity
Dimers 1.2 mM ImpA + ImpU 0.6 mM ImpA + ImpU
2 h 18 h (4b) 2 h 18 h (4c)
Cyclic dimers
D, D & L, L-c-A3′pU3′p 26.3 19.7 23.3 15.2
D, L & L, D-c-A3′pA3′p 6.7 3.5 4.4 3.3
D, D & L, L-c-A3′pA3′p 4.4 3.3 6.9 5.1
Linear dimers
D, D & L, L-U2′pU 4.9 1.6 3.2 2.1
D, L & L, D-U2′pU 4.1 3.9 6.0 5.4
D, D & L, L-U3′pU + D, D & L, L-A2′pU 8.3 8.1 9.5 10.2
D, L & L, D-U3′pU 3.1 2.5 2.7 3.8
D, L & L, D-A2′pU 7.2 9.4 10.0 9.7
D, D & L, L-A2′pA 3.3 3.9 3.0 2.9
D, D & L, L-A3′pU 11.3 19.3 12.1 20.5
D, L & L, D-A3′pU 6.6 8.7 5.4 8.1
D, L & L, D-A2′pA 4.0 4.6 3.5 2.7
D, D & L, L-A3′pA 5.6 6.8 6.3 6.8
D, L & L, D-A3′pA 4.2 4.7 3.7 4.2
Homochirality (%)b 64.1 62.6 64.3 62.8
Regioselectivity (%)c 72.5 72.6 69.6 72.1
a The oligomerization reactions were carried out in an MgCl2 (0.075 M) + NaCl (0.2 M) reagentb Overall homochirality was 63.5±0.8%c Regioselectivity refers to the 3′, 5′-phosphodiester bonds. The overall regioselectivity was 71.7±1.4%
(b) (a)
5 10 15 20 25 30 Time, minutes
5 10 15 20 25 30 Time, minutes
Fig. 2 Anion exchange HPLC analysis of the montmorillonite catalyzed reactions of 15 mM D-ImpA withD-ImpU (2a, Reaction 3) and 15 mM D, L-ImpA with D, L-ImpU (2b, Reaction 4a). Oligomer length isindicated by the numbers within each HPLC trace
Homochiral Selectivity in RNA Synthesis 223

reaction (4a, Table 1) the percent yield of oligomers (Table 1, Products III-XI) increasedsignificantly while the yield of dimers decreased from 54.7% to 43.3%. Errors in yieldsquoted in Tables 1, 2 and 4 are estimated from three or more experiments.
Oligomerization reactions of 1.2 mM and 0.6 mM of D, L-ImpA with D, L-ImpU(Reaction 4b and 4c, Table 1 and 2) on montmorillonite catalyst were carried out for 2 hand 18 h. Low concentrations of reactants were used to minimize the formation of higheroligomers; otherwise any differences in the rates of elongation of the dimers would result ina change of the proportion of dimeric products and similarly the higher oligomers.
Reaction 4b produced products up to tetramers when reacted for a period of 2 h and18 h, respectively. In Reaction 4c, only dimers and trimers were detected after 2 h and 18 h.The terminal 5′-phosphate of all oligomers was cleaved using alkaline phosphatase (APH)and the dephosphorylated products were separated by HPLC using an Alltima reverse phasecolumn. In the absence of the formation of oligomers of long chain length in Reaction 4band 4c, the dimers were analyzed directly on an Alltima C-18 column (Fig. 3). Theindividual linear and cyclic dimers were separated by HPLC to determine their yields(Table 2). These values were compared with the relative yields of linear and cyclic dimersobtained from Reactions 1 and 2 (Joshi et al. 2007) carried out at 0.15 mM and 0.6 mMconcentrations.
When 0.6 mM solutions of the activated mononucleotides in modifications toReactions 1 and 2 were reacted for 18 h, the cyclic and linear dimers were formed in 56:44 and 46: 54 ratios, respectively (Joshi et al. 2007). In the quaternary Reaction 4c, therelative ratio of cyclic vs. linear dimers was 24: 76. This is attributed to the differences inbinding of purine and pyrimidine mononucleotides to the catalytic sites of themontmorillonite. Previous studies established that purine nucleotides bind more stronglyto montmorillonite than do the pyrimidine nucleotides (Ferris et al. 1989; Kawamura andFerris 1999; Joshi et al. 2009). It is also known that 67% of oligomer formed from D-ImpAhas 3′, 5′-phosphodiester bonds while only 20% of those formed from D-ImpU have 3′, 5′-phosphodiester bonds (Ding et al 1996; Ferris and Ertem 1993). The difference inregioselectivity is attributed to the difference in the orientation of the activated
(1) UppU (2) D, D & L, L-c-A3 pU3 p(3) Uridine (4) D, L & L, D-c-A pA p(5) 3 , 5 -c-UMP (6) D, D & L, L-c-A pA p(7) D, D & L, L-U2 pU (8) Adenosine (9) D, L & L, D-U pU (10) D, D & L, L-U pU &
D, D & L, L-A pU(11) D, L & L, D-U pU(12) D, L & L, D-A pU(13) D, D & L, L-A pA(14) D, D & L, L-A pU(15) D, L & L, D-A pU(16) D, L & L, D-A pA(17) D, D & L, L-A pA(18) D, L & L, D-A pA10 15 20 25 30 35
Time, minutes
′ ′
′′
3′3′
3′
2′
2′
2′2′
2′
3′′
3′
3′
3′3′
3′3′
Fig. 3 Reverse phase HPLC analysis of the predominantly dimer products of the reaction of 0.6 mM D, L-ImpA with D, L-ImpU on montmorillonite for 2 h followed by alkaline phosphatase hydrolysis
224 P.C. Joshi et al.

mononucleotide when bound on montmorillonite. In quaternary reactions involving bothpurine and pyrimidine activated monomers, their binding to the platelets of montmorillonitewith different orientations prevented intramolecular cyclization. The cyclic dimer wouldalso reduce the number of catalytic sites in montmorillonite due to their relatively strongeradsorption on montmorillonite surfaces. The lower yields of cyclic dimers results in aninevitable increase in chain length of oligomers. The stronger retention of cyclic dimers onmontmorillonite was determined by monitoring the extraction profile of oligomers frommontmorillonite by the successive use of increasingly efficient reagents. Elution with 0.1 Mammonium acetate led largely to desorption of linear oligomers. Extraction of the samemontmorillonite sample with 0.1 M sodium pyrophosphate eluted moderate quantities of cyclicdimers. A final extraction of the same montmorillonite sample with 0.1 M NaCl in 30%acetonitrile completed the extraction of cyclic dimers. This last reagent is used routinely toremove adsorbed products from the montmorillonite (see Materials and Methods).
Identification of reaction products The HPLC analysis of the products of Reaction 4c(Table 1) after APH hydrolysis resulted in the detection of 18 peaks on an Alltima C-18column (Fig. 3). The major peaks (3 and 8) were identified as uridine and adenosine,respectively while peaks 1 and 5 are UppU and 3′, 5′-cyclic-UMP, respectively. Theremaining peaks are cyclic and linear dimers examples of which are found in Fig. 4.
a)
N
NN
N
NH2
O
OHO
N
N N
N
NH2
O
OH O
OP
-O O
O
PO O-
c) d)
b)
Fig. 4 Examples of linear and cyclic dimer anion structures: a) D, D-3′, 5′-ApA; b) D, D-3′, 5′-UpU; c) D,D-c-3′, 5′-ApA and d) D, D-c-3′, 5′-UpU
Homochiral Selectivity in RNA Synthesis 225

Procedures
1. Analysis of Cyclic Dimers. The structures of the cyclic dimers were confirmed byenzymatic hydrolysis of purified products with RNase T2 and APH (Scheme 1) toknown products and by electrospray HPLC/MS.
a) The D, D & L, L-c-A3′pU3′p dimers (2, Fig. 3) were partially (50%) hydrolyzed byRNase T2 to equal amounts of 3′-UMP and 3′-AMP due to cleavage of 3′,5′ bond ofthe D, D- enantiomers (Scheme 1, a). The L, L-enantiomer is not recognized by theenzyme so only 50% hydrolysis occurs. Further digestion of the RNase T2 hydrolysatewith alkaline phosphatase yielded equal amounts of uridine and adenosine while the L,L-enantiomer is again unaffected (Scheme 1, a). The nucleosides were identified byco-elution on a reverse phase HPLC column. Electrospray mass spectral analysis ofthe product 2 nominalmasses : Mþ 1 ¼ð 636;M� 1 ¼ 634Þ was consistent with theassigned structure (calculated for C19H23N7O14P2; m/z=635.0778).
b) The structures of D, L- & L, D-c-A3′pA3′p (4, Fig. 3) were determined byhydrolyses with RNase T2 followed by APH to give L, D-A3′pA (Scheme 1, b).Electrospray mass spectral analysis of the product nominal masses : Mþ 1 ¼ð659;M� 1 ¼ 657Þ was consistent with the assigned structure (calculated forC20H24N10O12P2; m/z=658.1050).
c) D, D&L, L-c-A3′pA3′p (6, Fig. 3) were partially (50%) hydrolyzed with RNase T2 to3′-AMP which was further hydrolyzed by APH to adenosine (Scheme 1, c).Electrospray mass spectral analysis of product 6 nominalmasses : Mþ 1 ¼ð659;M� 1 ¼ 657Þ was consistent with the assigned structure (calculated forC20H24N10O12P2; m/z=658.1050).
2. Analysis of homochiral linear dimers. Six of the 12 linear dimers were identified by theAPH hydrolysis of the products of Reaction 4c (Fig. 3) and the subsequent HPLC co-injection of the hydrolyzed products with authentic standards. The authentic standardswere: D, D- & L, L-U2′pU (7); D, D- & L, L-U3′pU (10); D, D- & L, L-A2′pU (10); D, D-& L, L-A2′pA (13); D, D- & L, L-A3′pU (14) and D, D- & L, L-A3′pA (17). The L, L-enantiomers of the linear dimers were synthesized by adsorbing L-adenosine or L-uridineon montmorillonite followed by treatment with L-ImpA or L-ImpU to yield L, L-A2′pA; L,L-A3′pA; L, L-U2′pU; L, L-U3′pU; L, L-A2′pU; L, L-A3′pU; L, L-U2′pA and L, L-U3′pA.
RNase T2 APH
a. D, D & L, L-c-A3 pU3 p L, L-c-A3 pU3 p + D-A3 p + D-U3 p L, L-c-A3 pU3 p + A + U (50%)
RNase T2 APH
b. D, L & L, D-c-A3 pA3 p L, D-A3 pA3 p L, D-A3 pA
RNase T2 APH
c. D, D & L, L-c-A3 pA3 p L, L-c-A3 pA3 p + D-A3 p L, L-c-A3 pA3 p + A (50%)
Key: D-A3 p = D-Adenosine-3 -monophosphate; D-U3 p = D-Uridine-3 -monophosphate; A = D-Adenosine; U = D-Uridine; APH = hydrolysis by alkaline phosphatase; RNaseT2 = hydrolysis by ribonuclease T2
Scheme 1 Enzymatic hydrolysis of cyclic dimers
226 P.C. Joshi et al.

These L, L-enantiomers of linear dimers co-eluted with the corresponding D, D-enantiomers on a C-18 reverse phase HPLC column. The L, L-enantiomers (3′, 5′) arenot hydrolyzed by RNase T2, therefore, only 50% hydrolysis of the D, D- & L, L-homochiral (3′, 5′) linear dimers were observed (Scheme 2). Neither D, D- nor L, L-2′, 5′enantiomeric linear dimers are hydrolyzed by RNase T2 (Scheme 2).
3. Analysis of heterochiral linear dimers. The D, L-enantiomers of heterochiral dimers (D,L-A2′pA; D, L-A3′pA; D, L-U2′pU; D, L-U3′pU; D, L-A2′pU; D, L-A3′pU; D, L-U2′pAand D, L-U3′pA) were synthesized in the laboratory. The authentic samples of L, D-enantiomers were synthesized by adsorbing L-adenosine or L-uridine to montmoril-lonite followed by treatment with D-ImpA or D-ImpU to yield L, D-A2′pA, L, D-A3′pA, L, D-U2′pU, L, D-U3′pU; L, D-A2′pU, L, D-A3′pU; L, D-U2′pA and L, D-U3′pA. Six of the eight dimers were identified in the montmorillonite-catalyzedquaternary reactions of D, L-ImpA + D, L-ImpU. These dimers were: D, L & L, D-
APH RNaseT2
1. D, D & L, L-pU2
pU D, D & L, L-U2
pU D, D & L, L-U2
pU
APH RNaseT2
2. D, L & L, D-pU2
pU D, L & L, D-U2
pU D, L & L, D-U2
pU
APH RNaseT2
3. D, D & L, L-pU3
pU D, D & L, L-U3
pU D-U3
p + U + L, L-U3
pU
50%
APH RNaseT2
4. D, L & L, D-pU3
pU D, L & L, D-U3
pU D-U3
p + U + L, D-U3
pU
50%
APH RNaseT2
5. D, D & L, L-pA2
pU D, D & L, L-A2
pU D, D & L, L-A2
pU
APH RNaseT2
6. D, L & L, D-pA2
pU D, L & L, D-A2
pU D, L & L, D-A2
pU
APH RNaseT2
7. D, D & L, L-pA3
pU D, D & L, L-A3
pU D-A3
p+ U + L, L-A3
pU
50%
APH RNaseT2
8. D, L & L, D-pA3
pU D, L & L, D-A3
pU D-A3
p + U + L, D-A3
pU
50%
APH RNaseT2
9. D, D & L, L-pA2
pA D, D & L, L-A2
pA D, D & L, L-A2
pA
APH RNaseT2
10. D, L & L, D-pA2
pA D, L & L, D-A2
pA D, L & L, D-A2
pA
APH RNaseT2
11. D, D & L, L-pA3
pA D, D & L, L-A3
pA D-A3
p+ A + L, L-A3
pA
50%
APH RNaseT2
12. D, L & L, D-pA3
pA D, L & L, D-A3
pA D-A3
p + A + L, D-A3
pA
50%
______________________________________________________________________________________
Key: U = D-Uridine; D-U3
p = D-Uridine-3 -monophosphate; A = D-Adenosine; D-A3
p = D-
Adenosine-3 -monophosphate; APH = hydrolysis by alkaline phosphatase; RNaseT2 = hydrolysis by
ribonuclease T2
Scheme 2 Enzymatic hydrolysis of linear dimers
Homochiral Selectivity in RNA Synthesis 227

U2′pU (9); D, L & L, D-U3′pU (11); D, L & L, D-A2′pU (12); D, L & L, D-A3′pU (15);D, L & L, D-A2′pA (16) and D, L & L, D-A3′pA (18) (Fig. 3). Two homochiral dimers,D, D & L, L-U3′pU and D, D & L, L-A2′pU co-eluted on a C-18 Alltima column (10,Fig. 3). They were identified by their behavior with RNase T2 and APH (Scheme 2).The relative yields of linear and cyclic dimers formed in the montmorillonite-catalyzedreactions of 1.2 mM and 0.6 mM D, L-ImpA with D, L-ImpU for 2 h and 18 h aresummarized in Table 2.
One of the most significant findings of this study is that no cyclic dimers of U weredetected in the quaternary reactions. Our earlier studies showed that in the montmorillonite-catalyzed reactions of D, L-ImpU (Reaction 2, Table 1), which were carried out at 0.15 mMand 0.6 mM concentrations for 18 h, 46% and 68% of the total dimer are cyclic dimers of U(Joshi et al. 2007). Among them, heterochiral dimer, D, L & L, D-c-U3′pU3′p, accounted for69% and 65% of the total of cyclic dimer, respectively. It is clear that preferentialcyclization of D- and L-enantiomers of ImpU in the binary reactions takes place because ofspecific orientations of ImpU on the platelets of montmorillonite. Addition of D, L-ImpA tothe D, L-ImpU caused changes in binding and as a result no cyclic dimers of U are formed.Another significant observation is that in the quaternary reactions the homochiral selectivityof linear dimers of U (pUpU) is increased from 39% to 63%. The importance of the absenceof cyclic dimers of U is that longer oligomers form from linear dimers.
The absence of cyclic dimers of U in the quaternary reactions was further confirmed byanalyzing the dimers on three different reverse phase columns. The dimer fractions werecollected separately from Reactions 2 and 4a of Table 1 using an ion exchange column. Theisolated dimers were then analyzed on Alltima, μBondapak and Jupiter reverse phasecolumns using various elution reagents and gradients. In each analysis no cyclic dimers ofU were detected. The absence of cyclic dimers of U was also confirmed by electrosprayHPLC/MS analysis of the total dimer fraction where no m/z corresponding toC18H22N4O16P2 (m/z=612.0506) was detected. No pUpAs were detected. On the contrary,pApUs were formed as the major dimer (Table 3). Three 3′, 5′ homochiral dimers, pU3′pU,pA3′pU and pA3′pA, emerged as the main reaction products of the dimer fraction. Theoverall homochirality and regioselectivity of dimers were 63.5±0.8% and 71.7±1.4%,respectively. The dimer outcomes are summarized in Table 3. A list of possible dimers fromthe quaternary reaction between D, L-ImpA + D, L-ImpU on montmorillonite is given inthe supplementary material (S-1).
In an uncatalyzed reaction of 15 mM D, L-ImpA + D, L-ImpU mixture carried out for3 days, only traces of dimer were detected (below 0.1%). Major reaction products wereUppU, 3′, 5′-c-UMP, AppA and 3′, 5′-c-AMP. No cyclic and linear dimers were detected inthe uncatalyzed quaternary reactions of 1.2 mM and 0.6 mM D, L-ImpA + D, L-ImpU.
Table 3 Selectivity of dimers formed in the reactions of D, L-ImpA + D, L-ImpU on montmorillonite
Dimers 1.2 mM ImpA 0.6 mM ImpA
2 h 18 h 2 h 18 h
pUpU 13.3% 9.9% 13.6% 13.2%
pUpA 0.0% 0.0% 0.0% 0.0%
pApA 32.8% 31.1% 32.4% 29.3%
pApU 53.9% 59.0% 54.0% 57.5%
228 P.C. Joshi et al.

Identification of trimers The trimers formed in Reaction 4a of Table 1 were isolated usingan ion exchange column, hydrolyzed with APH and separated on an Alltima C-18 reversephase column to give fifteen peaks (Fig. 5). Individual products were identified bycomparison with synthetic standards, selective enzymatic hydrolysis using RNase T2
followed by APH and by HPLC/MS analysis.
Preliminary electrospray HPLC/MS analysis of the entire trimer fraction analyzeddetected m/z 1006.0 (M+1) and 1004.0 (M-1) for pApApA (calculated forC30H38N15O19P3, m/z=1005.1681); m/z 983 (M+1) and 981 (M-1) for pApApU, pApUpAor pUpApA (calculated for C29H37N12O21P3, m/z=982.141) and m/z 960.0 (M+1) and958.0 (M-1) for pApUpU, pUpApU or pUpUpA (calculated for C28H36N9O23P3, m/z=959.1137). No m/z corresponding to pUpUpU (C27H35N6O25P3, m/z = 936.0865) wasdetected. Electrospray HPLC/MS analysis of 5′-terminal dephosphorylated trimers alsoconfirmed the presence of trimers with C28H35N9O20P2, m/z=879.1474 (possibly UpUpA,and/or ApUpU and/or UpApU); C29H36N12O18P2, m/z=902.1746 (possibly UpApA and/orApUpA and/or ApApU) and C30H37N15O16P2, m/z=925.2018 (ApApA). Linear trimers ofU (UpUpU; calculated for C30H37N15O16P2, m/z=856.1202) were not detected.
Further analysis of the enantiomers was carried out in three parts that includedidentification of (1) trimers containing only A, (2) trimers contain both A and U and (3)trimers containing only U.
1. Identification of trimers containing only A. Eight isomers of ApApAwere synthesized.These were D, D, D-A3′pA2′pA; D, D, D-A3′pA3′pA; D, D, L-A3′pA2′pA; D, D, L-A3′pA3′pA; D, L, D-A3′pA2′pA; D, L, D-A3′pA3′pA; D, L, L-A3′pA2′pA and D, L, L-A3′pA3′pA. Six enantiomers were detected in the trimer fraction of Reaction 4a(Table 1) as: D, D, D & L, L, L-A3′pA2′pA (9); D, L, D & L, D, L-A3′pA2′pA (11); D,D, L & L, L, D-A3′pA2′pA (12); D, D, D & L, L, L-A3′pA3′pA (13); D, D, L & L, L, D-A3′pA3′pA (14) and D, L, D & L, D, L-A3′pA3′pA (15, Fig. 5). The heterochiral trimersD, L, D & L, D, L-A3′pA2′pA (11) and D, D, L & L, L, D-A3′pA2′pA (12) were co-eluted on the C-18 Alltima column. The remaining trimers, D, L, L & L, D, D-
(1) D, D, D & L, L, L-A2 pA2 pU
(2) D, D, D & L, L, L-A3 pU pU
(3) D, D, D & L, L, L- A pU pA &
D, D, D & L, L, L-A pU pU
(4) D, D, D & L, L, L-A pA pU
(5) D, D, D & L, L, L-A pA pU
(6, 7) see text
(8) D, D, D & L, L, L-A pA pU
(9) D, D, D & L, L, L-A pA pA
(10) D, D, D & L, L, L-A pU pA
(11) D, L, D & L, D, L-A pA pA
(12) D, D, L & L, L, D-A pA pA
(13) D, D, D & L, L, L-A pA pA
(14) D, D, L & L, L, D- A pA pA
(15) D, L, D & L, D, L-A pA pA 22 24 26 28 30 32
Time, minutes
′
2′
2′
2′
2′
2′
2′
2′
′
′
3′
3′ 3′
3′
3′
3′
3′
3′ 3′
3′
3′
3′ 3′
3′
3′3′
3′
3′
Fig. 5 Reverse phase HPLC analysis of the trimers of the reaction of 15 mM D, L-ImpA + D, L-ImpU onmontmorillonite after alkaline phosphatase hydrolysis.
Homochiral Selectivity in RNA Synthesis 229

A3′pA2′pA and D, L, L & L, D, D-A3′pA3′pAwere not detected in the reaction mixture.The ApApA isomers accounted for 27.4% of the total trimer yield and homochiraltrimers accounted for 66% of the total ApApA isomers. In contrast, the homochiralityof ApApA isomers in Reaction 1 of Table 1 at 0.6 mM and 1.2 mM ImpAconcentrations was only 38–42% (Joshi et al. 2007).
2. Identification of trimers containing both A and U. No dimers of the type pUpA weredetected (Table 3). The yield of pUpU was low (Table 3). Therefore, the possibility offorming pUpApA or pUpApU did not exist. Four isomers of D, D, D-ApApU (D, D,D-A2′pA2′pU; D, D, D-A2′pA3′pU; D, D, D-A3′pA2′pU and D, D, D-A3′pA3′pU) weresynthesized and used as standards. They co-eluted with the trimers 1, 4, 5 and 8,respectively (Fig. 5). Four isomers of D, D, D-ApUpAwere also synthesized (D, D, D-A3′pU2′pA; D, D, D-A3′pU3′pA; D, D, D-A2′pU2′pA and D, D, D-A2′pU3′pA). Only D,D, D-A3′pU2′pA and D, D, D-A3′pU3′pA co-eluted with trimers 3 and 10 of the trimermixture (Fig. 5). Four isomers of D, D, D-ApUpU were synthesized (D, D, D-A3′pU2′pU; D, D, D-A3′pU3′pU; D, D, D-A2′pU2′pU and D, D, D-A2′pU3′pU) and onlyD, D, D-A3′pU2′pU and D, D, D-A3′pU3′pU co-eluted with trimer 2 and 3 of the trimermixture (Fig. 5).
3. Confirmation of absence of trimers containing only U. No trimers of U were detected byHPLC by co-injection on a C-18 Alltima column with an authentic sample. These resultswere further confirmed by analyzing the same samples on a μBondapak and Jupiter C-18columns. These results were also supported by the preliminary electrospray HPLC/MSanalysis of the trimer mixture where no m/z peaks corresponding to UpUpU weredetected. This is consistent with the low yield of dimers of U (Table 3).
Identification of 6 and 7 (Fig. 5) In the course of these studies, forty-four additionalheterotrimers were synthesized for comparison by HPLC with the unidentified trimers.Their syntheses were carried out in two steps: (1) by adding D-adenosine tomontmorillonite and then reacting the mixture with an activated mononucleotide (D- orL-ImpA or ImpU) and (2) adding the individual purified dimers to montmorillonite andreacting further with an activated mononucleotide to give the trimer. These were:
D, D, D-A2′pA2′pA; D, D, D-A2′pA3′pA; D, D, L-A2′pA2′pA; D, D, L-A2′pA3′pA;D, L, D-A2′pA2′pA; D, L, D-A2′pA3′pA; D, L, L-A2′pA2′pA; D, L, L-A2′pA3′pA;D, D, L-A3′pA2′pU; D, D, L-A3′pA3′pU; D, D, L-A2′pA2′pU; D, D, L-A2′pA3′pU;D, L, D-A3′pA2′pU; D, L, D-A3′pA3′pU; D, L, D-A2′pA2′pU; D, L, D-A2′pA3′pU;D, L, L-A2′pA2′pU; D, L, L-A2′pA3′pU; D, L, L- A3′pA2′pU; D, L, L-A3′pA3′pU;D, D, L-A2′pU2′pA; D, D, L-A2′pU3′pA; D, D, L-A3′pU2′pA; D, D, L-A3′pU3′pA;D, L, D-A2′pU2′pA; D, L, D-A2′pU3′pA; D, L, D-A3′pU2′pA; D, L, D-A3′pU3′pA;D, L, L-A2′pU2′pA; D, L, L-A2′pU3′pA; D, L, L-A3′pU2′pA; D, L, L-A3′pU3′pA;D, D, L-A2′pU2′pU; D, D, L-A2′pU3′pU; D, D, L-A3′pU2′pU; D, D, L-A3′pU3′pU;D, L, D-A2′pU2′pU; D, L, D-A2′pU3′pU; D, L, D-A3′pU2′pU; D, L, D-A3′pU3′pU;D, L, L-A2′pU2′pU; D, L, L-A2′pU3′pU; D, L, L-A3′pU2′pU and D, L, L-A3′pU3′pU.
None of the above co-eluted with peaks 6 and 7 using standard HPLC analysis. Thesepeaks were also analyzed by HPLC-MS. No conclusive identification of these two peakshas been obtained to date. Since these peaks were absent in the homochiral reactions of D-ImpAwith D-ImpU on montmorillonite (Table 1, Reaction 3), the material in either of thesepeaks cannot be a homochiral trimer. Thus, in the worst possible scenario, they could be asyet unidentified heterochiral trimers. The entry for Peaks 6 and 7 in Table 4 reflects this.
230 P.C. Joshi et al.

Consequently, the minimum overall homochirality of trimers formed in the Reaction 4a is74.2±1.6%.
The effect of concentration on trimer formation in Reaction 4a The identification of trimersis based on the analysis of enantiomers isolated from Reaction 4a, which produced oligomers upto 11-mer (Table 1). For the assessment of homochirality, an ideal reaction condition will bewhere no tetramers are detected. Therefore, the montmorillonite-catalyzed quaternary reactionsof D, L-ImpA with D, L-ImpU were carried out at 4.8, 2.4, 1.2, 0.6 and 0.15 mM totalactivated mononucleotide concentrations for a period of 2 h and 18 h. The trimers werecollected from each reaction mixture on an ion exchange column. They were dephosphorylatedwith APH and analyzed on a C-18 HPLC column. All the trimers that were identified fromReaction 4a (15 mM) were also detected at 4.8 mM and 2.4 mM concentrations (Table 4) andfollowed an identical elution profile on HPLC as was observed with trimers from Reaction 4a(Fig. 5). The homochirality of trimer in 4.8 mM and 2.4 mM samples were 72.9% and 73.8%,respectively. The HPLC elution profiles of trimers from the 1.2 mM (4b) and 0.6 mM (4c)samples were also similar to Reaction 4a but a few isomers that were formed in extremelysmall quantities could not be integrated. Their quantitative yields were assessed by calculatingthe peak area manually. The homochirality of trimers in 1.2 mM and 0.6 mM reactions ofReaction 4 were found to be 74.5% and 73.2%, respectively. The 0.15 mM reaction mixture ofreactants yielded only a limited number of trimers; therefore, no data are presented. Thehomochirality from dimer and trimer are summarized in Tables 5.
Table 4 Trimers formed by the reactions of varying concentrations of D, L-ImpA + D, L-ImpU onmontmorillonitea: Proportions of each trimer and homochirality
Trimer D, L-ImpA + D, L-ImpU
15 mM 4.8 mM 2.4 mM
Homochiral
D, D, D & L, L, L-A2′pA2′pU 4.7 5.1 5.7
D, D, D & L, L, L-A2′pA3′pU 6.7 6.1 5.9
D, D, D & L, L, L-A3′pU2′pU 10.0 10.8 12.1
D, D, D & L, L, L-A3′pU2′pU & D, D, D & L, L, L-A3′pU2′pA 10.8 11.6 13.0
D, D, D & L, L, L-A3′pU3′pA 2.2 2.8 2.8
D, D, D & L, L, L-A3′pA2′pU 18.0 16.3 15.9
D, D, D & L, L, L-A3′pA3′pU 7.6 7.2 5.7
D, D, D & L, L, L-A3′pA2′pA 13.4 10.3 9.9
D, D, D & L, L, L-A3′pA3′pA 2.6 2.7 2.8
Heterochiral
D, D, L & L, L, D-A3′pA2′pA & D, L, D & L, D, L-A3′pA2′pA 4.7 3.7 3.5
D, D, L & L, L, D-A3′pA3′pA 3.2 2.8 2.6
D, L, D & L, D, L-A3′pA3′pA 0.50 0.60 0.70
Peaks 6 and 7 15.6c 20.0c 19.4c
Homochirality (%)b 76.0d 72.9d 73.8d
a The oligomerization reactions were carried out in a MgCl2 (0.075 M) + NaCl (0.2 M) reagentb Overall homochirality was a minimum of 74.2±1.6%cMaximum heterochiralitydMinimum homochirality
Homochiral Selectivity in RNA Synthesis 231
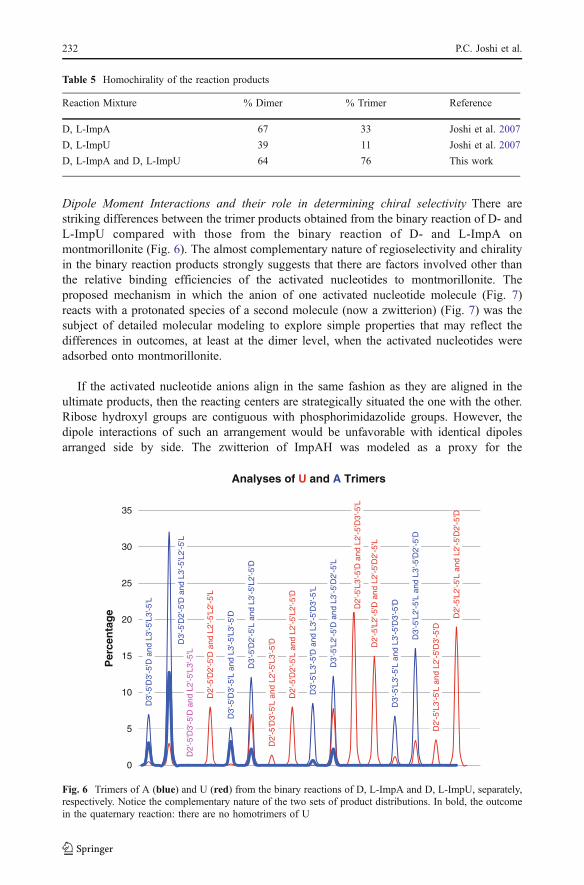
Dipole Moment Interactions and their role in determining chiral selectivity There arestriking differences between the trimer products obtained from the binary reaction of D- andL-ImpU compared with those from the binary reaction of D- and L-ImpA onmontmorillonite (Fig. 6). The almost complementary nature of regioselectivity and chiralityin the binary reaction products strongly suggests that there are factors involved other thanthe relative binding efficiencies of the activated nucleotides to montmorillonite. Theproposed mechanism in which the anion of one activated nucleotide molecule (Fig. 7)reacts with a protonated species of a second molecule (now a zwitterion) (Fig. 7) was thesubject of detailed molecular modeling to explore simple properties that may reflect thedifferences in outcomes, at least at the dimer level, when the activated nucleotides wereadsorbed onto montmorillonite.
If the activated nucleotide anions align in the same fashion as they are aligned in theultimate products, then the reacting centers are strategically situated the one with the other.Ribose hydroxyl groups are contiguous with phosphorimidazolide groups. However, thedipole interactions of such an arrangement would be unfavorable with identical dipolesarranged side by side. The zwitterion of ImpAH was modeled as a proxy for the
D3'
-5'D
3'-5
'D a
nd L
3'-5
'L3'
-5'L
D3'
-5'D
2'-5
'D a
nd L
3'-5
'L2'
-5'L
D2'
-5'D
3'-5
'D a
nd L
2'-5
'L3'
-5'L
D3'
-5'D
3'-5
'L a
nd L
3'-5
'L3'
-5'D
D3'
-5'D
2'-5
'L a
nd L
3'-5
'L2'
-5'D
D3'
-5'L
3'-5
'D a
nd L
3'-5
'D3'
-5'L
D3'
-5'L
2'-5
'D a
nd L
3'-5
'D2'
-5'L
D3'
-5'L
3'-5
'L a
nd L
3'-5
'D3'
-5'D
D3'
-5'L
2'-5
'L a
nd L
3'-5
'D2'
-5'D
D2'
-5'D
3'-5
'D a
nd L
2'-5
'L3'
-5'L
D2'
-5'D
2'-5
'D a
nd L
2'-5
'L2'
-5'L
D2'
-5'D
3'-5
'L a
nd L
2'-5
'L3'
-5'D
D2'
-5'D
2'-5
'L a
nd L
2'-5
'L2'
-5'D
D2'
-5'L
3'-5
'D a
nd L
2'-5
'D3'
-5'L
D2'
-5'L
2'-5
'D a
nd L
2'-5
'D2'
-5'L
D2'
-5'L
3'-5
'L a
nd L
2'-5
'D3'
-5'D
D2'
-5'L
2'-5
'L a
nd L
2'-5
'D2'
-5'D
0
5
10
15
20
25
30
35
Per
cen
tag
e
Analyses of U and A Trimers
Fig. 6 Trimers of A (blue) and U (red) from the binary reactions of D, L-ImpA and D, L-ImpU, separately,respectively. Notice the complementary nature of the two sets of product distributions. In bold, the outcomein the quaternary reaction: there are no homotrimers of U
Table 5 Homochirality of the reaction products
Reaction Mixture % Dimer % Trimer Reference
D, L-ImpA 67 33 Joshi et al. 2007
D, L-ImpU 39 11 Joshi et al. 2007
D, L-ImpA and D, L-ImpU 64 76 This work
232 P.C. Joshi et al.

electrophilic species in Fig. 7, formed by the protonation of the ImpA anion by a protonfrom montmorillonite (Table 6). This has a dipole in the opposite sense to that of the anion.Therefore, the nucleotide species shown in the formation of D, D-ImpApA (Fig. 8a) havealmost directly opposing dipoles. Thus, dipole-dipole interactions encourage the reaction inFig. 7.
The interactions described also explain the homochiral nature (Joshi et al. 2007) of co-reactions involving D- and L-isomers on montmorillonite. The imidazolide anions of D-ImpA and L-ImpA will have dipoles that will be in the opposite sense but they would notreadily react since they are both negatively charged. On the other hand, pairs of reactingmolecular species with opposing dipoles can be identified. The first pair would be the D-ImpA anion and the protonated D-form as a zwitterion referred to already (Fig. 8a), togetherwith the analogous L-species as the second pair. Both would clearly react to give ahomochiral outcome. Reaction of D-ImpA anions and the L-zwitterion is disfavored sincethe dipoles are both in the same direction or the reacting centers would be far apart.Conversely, modeling reveals that the ImpU anion and the analogous zwitterion do not havedirectly opposing dipoles as seen in the formation of ImpUpU (Fig. 8b), and so the effectpresent with ImpA species is absent or at least different. If the dipoles are aligned, then thereacting centers are far apart. This is in keeping with the marked difference in homochiralityobserved with the A and U series of oligomers (Joshi et al. 2007). Other combinations ofactivated nucleotide anions/zwitterions on the catalytic montmorillonite surface leading tothe dimer products encountered are shown in Fig. 8c and d. Cyclic dimers can be readilyincluded in this analysis if two zwitterionic species align with dipoles anti parallel. Thenecessary reacting groups in these two neutral species are now adjacent for the formation ofa cyclic products such as c-D, D-ApAp (Fig. 8e) and c-D, L-ApUp (Fig. 8f), the latter beingthe major dimer product (Table 2). Preliminary results from the modeling of charges on theactivated nucleotide species are interesting. The most nucelophilic –OH group is the 3′–OHgroup of the ImpA anion. The most electrophilic phosphorus center is found in the ImpUHzw/Na+ complex. These results are in keeping with the major product dimer being pA3′pU(Supplementary Material S-1).
Table 6 Components and dipole moments (μ) in Debyes (D) used for the activated nucleotide interactions inFig. 8
HF 6-31G* D-ImpA D-ImpAH zw D-ImpU D-ImpUH zw
μ 8.9 16.0 6.1 18.5 D
Fig. 7 Proposed phosphodiesterbond formation on montmoril-lonite. XH is an undifferentiatedprotic species within the claygalleries (Joshi et al. 2009)
Homochiral Selectivity in RNA Synthesis 233

Anion Anion
Anion Anion
Zwitterion Zwitterion
a) D, D-ImpApA b) D, D-ImpUpU
Zwitterion Zwitterion
Zwitterion Zwitterion
Zwitterion Zwitterion
c) D, D-ImpApU d) D, L-ImpApU
e) c-D, D-ApAp f) c-D, L-ApUp
Fig. 8 Structures of pairings of the activated nucleotide species present (Table 6) on the catalyticmontmorillonite surfaces, showing the dipole interactions which give the final products named
234 P.C. Joshi et al.

Conclusions
It is expected that if there were nucleosides formed on the primitive Earth they would bepresent as racemic mixtures. The reaction between D, L-ImpA and D, L-ImpU wasexpected to generate a more complex mixture of products than those formed by D, L-ImpAand D, L-ImpU individually (Supplementary Material, S-2 and S-3). It was surprising todiscover that the homochirality of the dimers and trimers formed from the quaternaryreaction of D, L-ImpA and D, L-ImpU was greater than was observed from the reaction ofD, L-ImpA or D, L-ImpU alone. The homochirality of dimers was 63.5% while that of thetrimers was 74.2%. It is postulated that the montmorillonite catalyst binds the quaternarymixture of activated nucleotides on its surface so that homochiral products are favored becauseof dipole interactions especially with A. This outcome may also be due to the faster elongationof the linear D, L-mixtures of ImpA and ImpU compared with their cyclization reactions.
Catalytic montmorillonite platelets serve as selective templates for the formation of RNAoligomers. Montmorillonite catalysis differs from enzymatic catalysis (Joyce et al. 1984)where elongation is terminated if the chirality of the nucleotide to be added to the oligomerhas chirality opposite to that of the RNA template. An RNA template is a specific catalystwhile that of the montmorillonite is a selective catalyst. While the oligomer formed bymontmorillonite catalysis will have some errors in its sequence it may still be able togenerate catalytic RNAs. The specific catalysis of the RNA template will not be able togenerate RNA oligomers because synthesis is immediately terminated if a specific RNAtemplate is not available (Inoue and Orgel 1982; Joyce et al. 1984).
The yields of cyclic dimers were much lower in the quaternary reaction of a mixture of D, L-ImpAwith D, L-ImpU. No cyclic dimers of U were detected in the montmorillonite-catalyzedquaternary reactions of 0.15 mM and 0.6 mM of D, L-ImpA and D, L-ImpU whereas similarreactions carried out with the reactions of D, L-ImpU produced 46–68% of cyclic dimers (Joshiet al. 2007). Similarly, the yields of cyclic dimers of A were reduced from 56–66% to 6–11%in the quaternary reactions. Homochiral selectivity was also observed when the linear dimerselongated to trimers. If there was no selectivity in the montmorillonite-catalyzed reactions,there would have been a 25% yield of heterochiral trimers and 256 possible oligomers, eachwith a yield 0.39% (S-2). One pair of isomers, D, L, D & L, D, L-A3′pA3′pA, was identifiedat a level below the statistical level (0.78%), (Table 4), whereas all the other compounds inTable 4 showed very large differences from the statistical expectations. We detected only 16isomers which were mainly the D, D, D & L, L, L enantiomers for an average homochiralselectivity of 74.2±1.6%. This is a major increase in the homochiral selectivity from 38–43%and 10–11% observed in the reactions of D, L-ImpA or D, L-ImpU alone (Joshi et al. 2007).The higher oligomers will be predominantly homochiral if this trend continues.
Acknowledgement The authors would like to thank Dr. Dmitri V. Zagorevskii for the mass spectral analysis(supported by grant NSF CHE-0091892 for funds to purchase the Agilent 1100 HPLC system and MSD TrapInstrument) and Dr John W. Delano, University at Albany, for constructive suggestions. Volclay is a gift fromthe American Colloid Company. This research was supported by Astrobiology grant NNA09DA80A.
References
Cech TR, Zung AJ, Grabowski PJ (1981) In vitro splicing of the ribosomal RNA precursor of Tetrahymena:involvement of a guanosine nucleotide in the excision of the intervening sequence. Cell 27:487–496
Homochiral Selectivity in RNA Synthesis 235

Crick FHC (1968) The origin of the genetic code. J Mol Biol 38:367–379Ding ZP, Kawamura K, Ferris JP (1996) Oligomerization of uridine phosphorimidazolides on montmoril-
lonite: a model for the prebiotic synthesis of RNA on minerals. Orig Life Evol Biosph 26:151–171Ferris JP (2002) Montmorillonite catalysis of 30–50 mer oligonucleotides: laboratory demonstration of
potential steps in the origin of the RNA world. Orig Life Evol Biosph 32:311–332Ferris JP, Ertem G (1993) Montmorillonite catalysis of RNA oligomer formation in aqueous solution. A
model for the prebiotic formation of RNA. J Am Chem Soc 115:12270–12275Ferris JP, Ertem G, Agarwal VK (1989) The adsorption of nucleotides and polynucleotides on
montmorillonite clay. Orig Life Evol Biosph 19:153–164Ferris JP, Hill AR Jr, Liu R, Orgel LE (1996) Synthesis of long prebiotic oligomers on mineral surfaces.
Nature 381:59–61Gilbert W (1986) The RNA world. Nature 319:618Guerrier-Takada C, Gardiner K, Marsh T, Pace N, Altman S (1983) The RNA moiety of ribonuclease P is the
catalytic subunit of the enzyme. Cell 35:849–857Huang W, Ferris JP (2006) One-step, regioselective synthesis of up to 50-mers of RNA oligomers by
montmorillonite catalysis. J Am Chem Soc 128:8914–8919Ikemoto T, Haze A, Hatano H, Kitamoto Y, Ishida M, Nara K (1995) Phosphorylation of nucleosides with
phosphorus oxychloride in trialkyl phosphate. Chem Pharm Bull 43:210–215Inoue T, Orgel LE (1982) Oligomerization of (guanosine 5′-phosphor)-2-methylimidazolide on poly (C): an
RNA polymerase model. J Mol Biol 162:201–217Joshi PC, Pitsch S, Ferris JP (2007) Selectivity of montmorillonite catalyzed prebiotic reactions of D, L-
nucleotides. Orig Life Evol Biosph 37:3–26Joshi PC, Aldersley MF, Delano JW, Ferris JP (2009) Mechanism of montmorillonite catalysis in the
formation of RNA oligomers. J Am Chem Soc 131:13369–13374Joyce GF, Orgel LE (1999) Prospects for understanding the origin of the RNAworld. In: Gesteland RF, Cech
TR, Atkins JF (eds) The RNA World, 2nd edn. Cold Spring Harbor Laboratory Press, Cold SpringHarbor, pp 49–77
Joyce GF, Orgel LE (2006) Progress toward understanding the origin of the RNA world. In: Gesteland RF,Cech TR, Atkins JF (eds) The RNAWorld, 3rd edn. Cold Spring Harbor Laboratory Press, Cold SpringHarbor, pp 23–56
Joyce GF, Visser GM, van Boeckel CAA, van Boom JH, Orgel LE, van Westrenen J (1984) Chiral selectionin poly (C)-directed synthesis of oligo(G). Nature 310:602–604
Kawamura K, Ferris JP (1999) Clay catalysis of oligonucleotides formation: kinetics of the reaction of the 5′-phosphorimidazolides of nucleotides with the non-basic heterocycles uracil and hypoxanthine. Orig LifeEvol Biosph 29:563–591
Miyakawa S, Ferris JP (2003) Sequence- and regioselectivity in the montmorillonite-catalyzed synthesis ofRNA. J Am Chem Soc 125:8202–8208
Orgel LE (1968) Evolution of the genetic apparatus. J Mol Biol 38:381–393Orgel LE (2000) Perspectives: origin of life: a simple nucleic acid. Science 290:1306–1307Szostak JW, Ellington AD (1993) In vitro selection of functional RNA sequences. In: Gesteland RF, Atkins
JF (eds) The RNA World. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, pp 511–533Woese C (1967) The genetic code, the molecular basis for genetic expression. Harper and Row, New YorkZagorevskii DV, Aldersley MF, Ferris JP (2006) MALDI analysis of oligonucleotides directly from
montmorillonite. J Am Soc Mass Spectrom 17:1265–1270
236 P.C. Joshi et al.




















