
Green fluorescent protein for in situ synthesis of highly uniform Au nanoparticlesand monitoring...
16
Journal of Colloid and Interface Science 326 (2008) 129–137 Contents lists available at ScienceDirect Journal of Colloid and Interface Science www.elsevier.com/locate/jcis Green fluorescent protein for in situ synthesis of highly uniform Au nanoparticles and monitoring protein denaturation Pallab Sanpui a , Shivendra B. Pandey b , Siddhartha Sankar Ghosh a,b,∗ , Arun Chattopadhyay a,c,∗ a Centre for Nanotechnology, Indian Institute of Technology Guwahati, Guwahati 781039, India b Department of Biotechnology, Indian Institute of Technology Guwahati, Guwahati 781039, India c Department of Chemistry, Indian Institute of Technology Guwahati, Guwahati 781039, India article info abstract Article history: Received 31 March 2008 Accepted 7 July 2008 Available online 17 July 2008 Keywords: Green fluorescent protein Gold nanoparticles Green synthesis Protein denaturation Purified recombinant green fluorescent protein (GFP) expressed in E. coli was used for single-step synthesis of gold nanoparticles (Au NPs) with extraordinary size specificity in aqueous medium. The fluorescence of GFP offered a probe for concomitant changes in the protein during the course of synthesis, in addition to the monitoring of the time-dependent formation of Au NPs by the surface plasmon resonance. Reaction of AuCl − 4 with the protein produced spherical Au NPs having diameters ranging from 5–70 nm. Remarkably, addition of 1.0 × 10 −5 M AgNO 3 in the medium produced uniform spherical Au NPs with particle diameter of 2.2 ± 0.5 nm. Fluorescence spectroscopic measurements suggest that during synthesis of Au NPs in absence of AgNO 3 , partial denaturation of the protein occurred resulting in the lowering of fluorescence intensity. On the other hand, when the NPs were synthesized in the presence of AgNO 3 complete denaturation of the protein with complete loss of fluorescence could be observed, which was further confirmed by native and sodium dodecyl sulfate (SDS) polyacrylamide gel electrophoresis (PAGE). However, use of AgNO 3 only resulted neither in the formation of NPs nor had any significant effect on the fluorescence of GFP. © 2008 Elsevier Inc. All rights reserved. 1. Introduction Synthesis of metal nanoparticles (NPs) with predefined size and shape is fundamentally of great importance in diverse applications such as data storage [1], imaging [2,3], catalysis [4,5], biodiag- nostics [6], miniaturization of electronic components and in the creation of functional nanostructures [7] assembled in a ‘bottom- up’ approach exploiting the size and shape dependent physico- chemical and optoelectronic properties [8,9] of these nanoscale entities. At present, a vast array of different semiconductor and metal nanoparticles can be synthesized by wet chemical methods— aqueous-phase as well as organic-phase synthesis—with fair con- trol on the size and shape of the synthesized nanoparticles [10]. However, use of stringent reaction conditions and hazardous chem- icals in the syntheses of these NPs by chemical methods not only pose problems for large-scale synthesis but also make them less attractive for biological applications. On the other hand, “green- er” methods to synthesize NPs employing biological systems have * Corresponding authors at: Centre for Nanotechnology, Indian Institute of Tech- nology Guwahati, Guwahati 781039, India. Faxes: +91 361 2582249, +91 361 2690762. E-mail addresses: [email protected] (S.S. Ghosh), [email protected] (A. Chattopadhyay). mainly centered on the microbial syntheses of nanoscale materi- als [11–16]. The microbial syntheses, however, offer less flexibility over the control of particle sizes and shapes. On the other hand, although there are some reports of synthesizing metal NPs using biological macromolecules [17–21], synthesis of uniform and well defined metal NPs of less than 5 nm, suitable for biological ap- plications, by biomolecules such as purified protein still remains a challenge. It is important to appreciate that while synthesis of small NPs using biomolecules is of great value, understanding the mechanism involved and the fate of the NPs as well as that of the biomolecules following synthesis are equally important. This assumes greater significance especially for the development of hierarchically as- sembled functional nano-structured devices and in the evaluation of the biocompatibility associated with these materials [22]. The changes in structural as well as functional stability of specific pro- teins adsorbed on macroscopic surface although have already been reported [23], the study of effects of the physicochemical proper- ties of the surface at the nanoscale on protein integrity has been started only recently [24–28] and is far from being exhaustive. In- terestingly, the primary focus in the biosynthesis of NPs has been on the product NPs, their properties and occasionally the proper- ties of the composite system. There is no report on the accom- panying changes in the bio-precursor as the synthesis of NPs takes 0021-9797/$ – see front matter © 2008 Elsevier Inc. All rights reserved. doi:10.1016/j.jcis.2008.07.015
Transcript of Green fluorescent protein for in situ synthesis of highly uniform Au nanoparticlesand monitoring...

Journal of Colloid and Interface Science 326 (2008) 129–137
Contents lists available at ScienceDirect
Journal of Colloid and Interface Science
www.elsevier.com/locate/jcis
Green fluorescent protein for in situ synthesis of highly uniform Au nanoparticlesand monitoring protein denaturation
Pallab Sanpui a, Shivendra B. Pandey b, Siddhartha Sankar Ghosh a,b,∗, Arun Chattopadhyay a,c,∗a Centre for Nanotechnology, Indian Institute of Technology Guwahati, Guwahati 781039, Indiab Department of Biotechnology, Indian Institute of Technology Guwahati, Guwahati 781039, Indiac Department of Chemistry, Indian Institute of Technology Guwahati, Guwahati 781039, India
a r t i c l e i n f o a b s t r a c t
Article history:Received 31 March 2008Accepted 7 July 2008Available online 17 July 2008
Keywords:Green fluorescent proteinGold nanoparticlesGreen synthesisProtein denaturation
Purified recombinant green fluorescent protein (GFP) expressed in E. coli was used for single-stepsynthesis of gold nanoparticles (Au NPs) with extraordinary size specificity in aqueous medium. Thefluorescence of GFP offered a probe for concomitant changes in the protein during the course of synthesis,in addition to the monitoring of the time-dependent formation of Au NPs by the surface plasmonresonance. Reaction of AuCl−4 with the protein produced spherical Au NPs having diameters ranging from5–70 nm. Remarkably, addition of 1.0 × 10−5 M AgNO3 in the medium produced uniform spherical AuNPs with particle diameter of 2.2±0.5 nm. Fluorescence spectroscopic measurements suggest that duringsynthesis of Au NPs in absence of AgNO3, partial denaturation of the protein occurred resulting in thelowering of fluorescence intensity. On the other hand, when the NPs were synthesized in the presence ofAgNO3 complete denaturation of the protein with complete loss of fluorescence could be observed, whichwas further confirmed by native and sodium dodecyl sulfate (SDS) polyacrylamide gel electrophoresis(PAGE). However, use of AgNO3 only resulted neither in the formation of NPs nor had any significanteffect on the fluorescence of GFP.
© 2008 Elsevier Inc. All rights reserved.
1. Introduction
Synthesis of metal nanoparticles (NPs) with predefined size andshape is fundamentally of great importance in diverse applicationssuch as data storage [1], imaging [2,3], catalysis [4,5], biodiag-nostics [6], miniaturization of electronic components and in thecreation of functional nanostructures [7] assembled in a ‘bottom-up’ approach exploiting the size and shape dependent physico-chemical and optoelectronic properties [8,9] of these nanoscaleentities. At present, a vast array of different semiconductor andmetal nanoparticles can be synthesized by wet chemical methods—aqueous-phase as well as organic-phase synthesis—with fair con-trol on the size and shape of the synthesized nanoparticles [10].However, use of stringent reaction conditions and hazardous chem-icals in the syntheses of these NPs by chemical methods not onlypose problems for large-scale synthesis but also make them lessattractive for biological applications. On the other hand, “green-er” methods to synthesize NPs employing biological systems have
* Corresponding authors at: Centre for Nanotechnology, Indian Institute of Tech-nology Guwahati, Guwahati 781039, India. Faxes: +91 361 2582249, +91 3612690762.
E-mail addresses: [email protected] (S.S. Ghosh), [email protected](A. Chattopadhyay).
mainly centered on the microbial syntheses of nanoscale materi-als [11–16]. The microbial syntheses, however, offer less flexibilityover the control of particle sizes and shapes. On the other hand,although there are some reports of synthesizing metal NPs usingbiological macromolecules [17–21], synthesis of uniform and welldefined metal NPs of less than 5 nm, suitable for biological ap-plications, by biomolecules such as purified protein still remains achallenge.
It is important to appreciate that while synthesis of small NPsusing biomolecules is of great value, understanding the mechanisminvolved and the fate of the NPs as well as that of the biomoleculesfollowing synthesis are equally important. This assumes greatersignificance especially for the development of hierarchically as-sembled functional nano-structured devices and in the evaluationof the biocompatibility associated with these materials [22]. Thechanges in structural as well as functional stability of specific pro-teins adsorbed on macroscopic surface although have already beenreported [23], the study of effects of the physicochemical proper-ties of the surface at the nanoscale on protein integrity has beenstarted only recently [24–28] and is far from being exhaustive. In-terestingly, the primary focus in the biosynthesis of NPs has beenon the product NPs, their properties and occasionally the proper-ties of the composite system. There is no report on the accom-panying changes in the bio-precursor as the synthesis of NPs takes
0021-9797/$ – see front matter © 2008 Elsevier Inc. All rights reserved.doi:10.1016/j.jcis.2008.07.015

130 P. Sanpui et al. / Journal of Colloid and Interface Science 326 (2008) 129–137
place. This is possibly due to easy surface plasmon resonance (SPR)based optical probe being available for metal such as Au NPs andlack of such probes for biosystems such as individual proteins andDNA.
Herein, we report the use of purified recombinant green fluo-rescent protein (GFP), expressed in E. coli bacteria, for single-stepsynthesis of Au NPs with extraordinary size specificity in aque-ous medium. We observed that in the presence of AuCl−4 alonethe proteins produced spherical Au NPs of 5–70 nm, whereas uni-form spherical particles smaller than 5 nm were formed in theadditional presence of AgNO3. Also, fluorescence from the proteinwas used as the probe for concomitant changes in the protein dur-ing the course of synthesis. This is in addition to the SPR probeto monitor the time-dependent formation of Au NPs. The integrityof protein’s three-dimensional structure was also followed by na-tive and sodium dodecyl sulfate polyacrylamide gel electrophoresis(SDS–PAGE).
2. Materials and methods
2.1. Growth media and chemicals
Silver nitrate (AgNO3), sodium chloride (NaCl) and dimethylsulfoxide (DMSO) were purchased from Merck India Ltd. HAuCl4solution (17%, w/v, in dilute HCl), 2,2′-dithiobis(5-nitropyridine)(DTNP) and other high purity molecular biology grade chemicalsand reagents for native and SDS–PAGE were obtained from Sigma–Aldrich Chemical Corporation. E. coli growth medium Luria–Bertani(LB) was purchased from HiMedia, India. Milli-Q grade water (Mil-lipore, USA) was used in all the experiments.
2.2. Isolation and purification of GFP
GFP (wild type) was isolated and subsequently purified fromovernight grown culture of GFP expressing E. coli. This was basedon the modification of methods reported by other workers [29,30].The procedure related to the generation of recombinant green flu-orescent protein (GFP) expressing E. coli (DH5α) cell has been de-scribed elsewhere [31]. After purification of GFP, excess salts fromthe protein sample were removed by dialysis. The dialyzed sam-ple was subsequently lyophilized and the resulting purified GFP inpowder form was stored at −20 ◦C for further use.
2.3. Synthesis of Au nanoparticles by GFP
The stock solution of GFP was prepared in 0.02 M Tris–HCl,pH 8.0, buffer containing 0.15 M NaCl and 5 mM EDTA. In a typicalreaction, 14 µgmL−1 GFP was added to 5 mL reaction medium con-taining 1.0×10−4 M HAuCl4 and incubated at 37 ◦C for 4 days. Thesame buffer, used for making the GFP stock solution, was also usedas the reaction medium. Additional reactions were also carried outin the presence of different concentration of AgNO3, keeping allother conditions the same as above. Appropriate control samples,namely sample containing only HAuCl4 in reaction buffer and sam-ple containing only GFP in reaction buffer, were also incubated insame conditions. A complementary set of experiments was alsocarried out where GFP were incubated with different concentra-tions of AgNO3 in the same reaction conditions (in absence ofHAuCl4) as above to check the formation of Ag NPs by GFP.
2.4. UV–visible and fluorescence spectroscopic measurements
The formation of Au NPs was followed by monitoring the UV–vis spectra of the samples taken at regular time intervals in aCary 100 UV–visible spectrophotometer (Varian Inc.). The fluores-cence of GFP in all the samples were also measured at regular
intervals by recording the fluorescence emission spectra of GFP(λemission = 509 nm) using excitation at 395 nm with a FluoroMax-3 (HORIBA Jobin Yvon) fluorescence spectrophotometer. Thefluorescence emission spectra of GFP with λemission = 505 nm andλexcitation = 475 nm for each sample were also recorded. The mea-surements were carried out under ambient conditions.
2.5. X-ray diffraction (XRD) measurements
As prepared solution of Au NPs was spread on glass slide andsubsequently air-dried under ambient conditions for XRD mea-surements. The measurements were performed using a Bruker D8ADVANCE (Bruker AXS Inc.) X-ray powder diffractometer usingCuKα (λ = 1.54 Å) source.
2.6. Transmission electron microscopic (TEM) analysis
5 µL of as prepared Au NPs solutions from different sampleswere drop cast on carbon coated copper TEM grids followed byair-drying. The grids were then analyzed by a JEOL 2100 UHR-TEMinstrument operating at an accelerating voltage of 120 KeV.
2.7. Protein gel electrophoresis
After incubation of GFP with HAuCl4, both in presence and inabsence of AgNO3, aliquots of the different samples were taken at90 h and subjected to native as well as SDS–PAGE. Protein profileswere visualized by staining the gel using silver staining method aswell as under UV transilluminator before silver staining in case ofnative PAGE only.
2.8. Blocking of free thiol groups of GFP
The free thiol groups of cysteine residues of GFP were modi-fied by previously described method [18]. Briefly, 10.3 µL of 5 mMDTNP in DMSO was added to 35 µL of 2 mgmL−1 GFP. The fi-nal reaction volume was made up to 5 mL with 0.02 M Tris–HCl(pH 8.0), 0.15 M NaCl and 5 mM EDTA so as to give a final GFPconcentration of 14 µgmL−1. After overnight incubation at 37 ◦Cwith gentle stirring, the reaction mixture turned to a pale yel-low colored solution. The modification of free thiol groups wasfollowed spectrophotometrically with a Cary 100 UV–visible spec-trophotometer (Varian Inc.). The solution was then incubated withHAuCl4 (final concentration of 1.0×10−4 M), both in presence andabsence of 1.0 × 10−5 M AgNO3. Appropriate control experimentswere also carried out with unmodified GFP in presence of DMSO.
3. Results and discussion
Following incubation of GFP in pH 8.0 buffer solution alongwith HAuCl4 (1.0 × 10−4 M) at 37 ◦C, the color of the solution be-came purple by 8 h indicating the formation of Au NPs. UV–visspectra of the solution (Fig. 1a), comprised of gradually increasingabsorption peak at ca. 532 nm with an additional absorption bandat ca. 680 nm. The maximum absorption was reached by 90 hof incubation. The peak at 532 nm is characteristic of transverseplasmon resonance of Au NPs, whereas the peak at 680 nm is char-acteristic of longitudinal plasmon resonance of either Au nanorodsor triangular or hexagonal shaped Au NPs [16,32]. When the samereaction was carried out in presence of 1.0 × 10−5 M AgNO3, thesolution became light pink in 10 h, which further turned to intensepink color after 48 h of incubation. The time dependent UV–visiblespectra of the solution (Fig. 1b) showed consistent increase in theabsorbance at 532 nm till 90 h of incubation. There was no sec-ond peak at higher wavelength as was observed with the sampleprepared from HAuCl4 and in absence of AgNO3. In both cases,
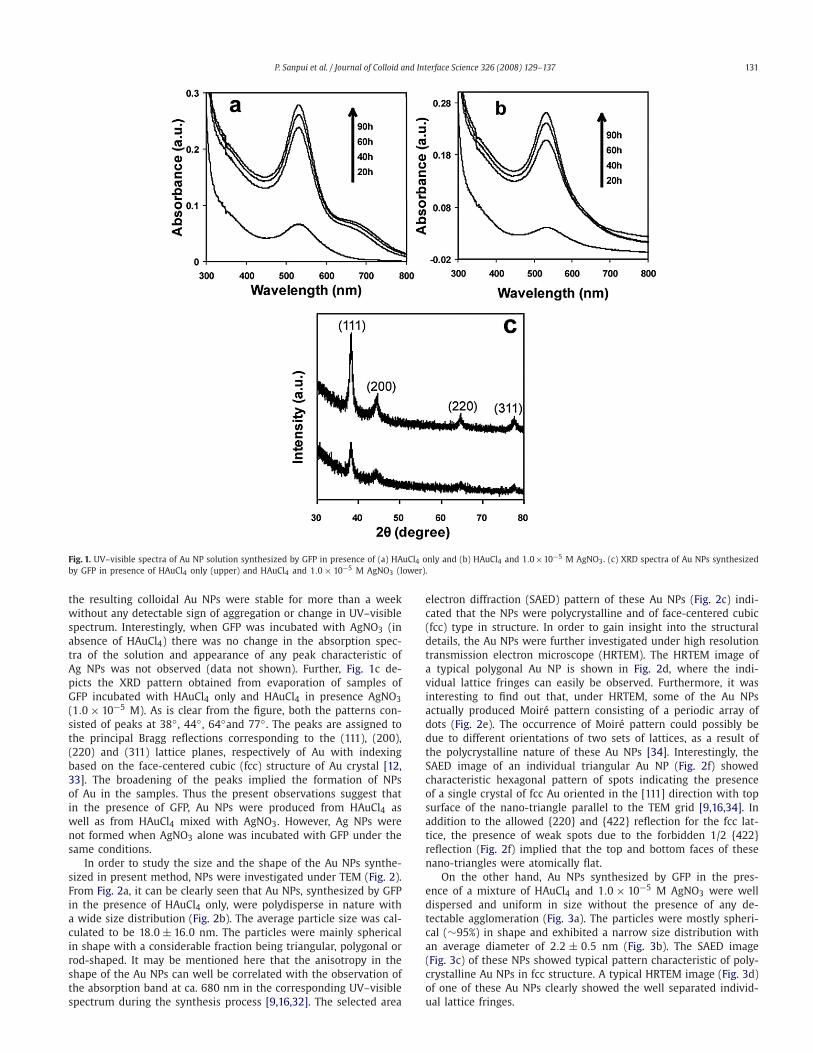
P. Sanpui et al. / Journal of Colloid and Interface Science 326 (2008) 129–137 131
Fig. 1. UV–visible spectra of Au NP solution synthesized by GFP in presence of (a) HAuCl4 only and (b) HAuCl4 and 1.0×10−5 M AgNO3. (c) XRD spectra of Au NPs synthesizedby GFP in presence of HAuCl4 only (upper) and HAuCl4 and 1.0× 10−5 M AgNO3 (lower).
the resulting colloidal Au NPs were stable for more than a weekwithout any detectable sign of aggregation or change in UV–visiblespectrum. Interestingly, when GFP was incubated with AgNO3 (inabsence of HAuCl4) there was no change in the absorption spec-tra of the solution and appearance of any peak characteristic ofAg NPs was not observed (data not shown). Further, Fig. 1c de-picts the XRD pattern obtained from evaporation of samples ofGFP incubated with HAuCl4 only and HAuCl4 in presence AgNO3(1.0 × 10−5 M). As is clear from the figure, both the patterns con-sisted of peaks at 38◦ , 44◦ , 64◦and 77◦ . The peaks are assigned tothe principal Bragg reflections corresponding to the (111), (200),(220) and (311) lattice planes, respectively of Au with indexingbased on the face-centered cubic (fcc) structure of Au crystal [12,33]. The broadening of the peaks implied the formation of NPsof Au in the samples. Thus the present observations suggest thatin the presence of GFP, Au NPs were produced from HAuCl4 aswell as from HAuCl4 mixed with AgNO3. However, Ag NPs werenot formed when AgNO3 alone was incubated with GFP under thesame conditions.
In order to study the size and the shape of the Au NPs synthe-sized in present method, NPs were investigated under TEM (Fig. 2).From Fig. 2a, it can be clearly seen that Au NPs, synthesized by GFPin the presence of HAuCl4 only, were polydisperse in nature witha wide size distribution (Fig. 2b). The average particle size was cal-culated to be 18.0 ± 16.0 nm. The particles were mainly sphericalin shape with a considerable fraction being triangular, polygonal orrod-shaped. It may be mentioned here that the anisotropy in theshape of the Au NPs can well be correlated with the observation ofthe absorption band at ca. 680 nm in the corresponding UV–visiblespectrum during the synthesis process [9,16,32]. The selected area
electron diffraction (SAED) pattern of these Au NPs (Fig. 2c) indi-cated that the NPs were polycrystalline and of face-centered cubic(fcc) type in structure. In order to gain insight into the structuraldetails, the Au NPs were further investigated under high resolutiontransmission electron microscope (HRTEM). The HRTEM image ofa typical polygonal Au NP is shown in Fig. 2d, where the indi-vidual lattice fringes can easily be observed. Furthermore, it wasinteresting to find out that, under HRTEM, some of the Au NPsactually produced Moiré pattern consisting of a periodic array ofdots (Fig. 2e). The occurrence of Moiré pattern could possibly bedue to different orientations of two sets of lattices, as a result ofthe polycrystalline nature of these Au NPs [34]. Interestingly, theSAED image of an individual triangular Au NP (Fig. 2f) showedcharacteristic hexagonal pattern of spots indicating the presenceof a single crystal of fcc Au oriented in the [111] direction with topsurface of the nano-triangle parallel to the TEM grid [9,16,34]. Inaddition to the allowed {220} and {422} reflection for the fcc lat-tice, the presence of weak spots due to the forbidden 1/2 {422}reflection (Fig. 2f) implied that the top and bottom faces of thesenano-triangles were atomically flat.
On the other hand, Au NPs synthesized by GFP in the pres-ence of a mixture of HAuCl4 and 1.0 × 10−5 M AgNO3 were welldispersed and uniform in size without the presence of any de-tectable agglomeration (Fig. 3a). The particles were mostly spheri-cal (∼95%) in shape and exhibited a narrow size distribution withan average diameter of 2.2 ± 0.5 nm (Fig. 3b). The SAED image(Fig. 3c) of these NPs showed typical pattern characteristic of poly-crystalline Au NPs in fcc structure. A typical HRTEM image (Fig. 3d)of one of these Au NPs clearly showed the well separated individ-ual lattice fringes.
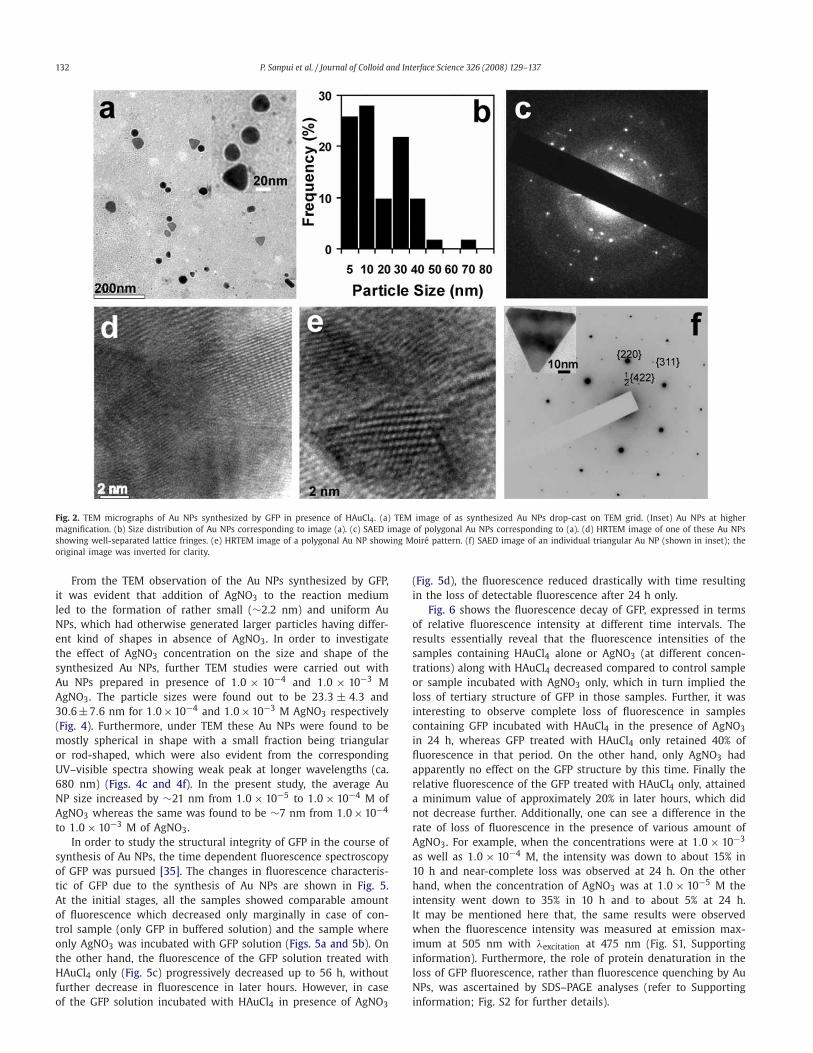
132 P. Sanpui et al. / Journal of Colloid and Interface Science 326 (2008) 129–137
Fig. 2. TEM micrographs of Au NPs synthesized by GFP in presence of HAuCl4. (a) TEM image of as synthesized Au NPs drop-cast on TEM grid. (Inset) Au NPs at highermagnification. (b) Size distribution of Au NPs corresponding to image (a). (c) SAED image of polygonal Au NPs corresponding to (a). (d) HRTEM image of one of these Au NPsshowing well-separated lattice fringes. (e) HRTEM image of a polygonal Au NP showing Moiré pattern. (f) SAED image of an individual triangular Au NP (shown in inset); theoriginal image was inverted for clarity.
From the TEM observation of the Au NPs synthesized by GFP,it was evident that addition of AgNO3 to the reaction mediumled to the formation of rather small (∼2.2 nm) and uniform AuNPs, which had otherwise generated larger particles having differ-ent kind of shapes in absence of AgNO3. In order to investigatethe effect of AgNO3 concentration on the size and shape of thesynthesized Au NPs, further TEM studies were carried out withAu NPs prepared in presence of 1.0 × 10−4 and 1.0 × 10−3 MAgNO3. The particle sizes were found out to be 23.3 ± 4.3 and30.6±7.6 nm for 1.0×10−4 and 1.0×10−3 M AgNO3 respectively(Fig. 4). Furthermore, under TEM these Au NPs were found to bemostly spherical in shape with a small fraction being triangularor rod-shaped, which were also evident from the correspondingUV–visible spectra showing weak peak at longer wavelengths (ca.680 nm) (Figs. 4c and 4f). In the present study, the average AuNP size increased by ∼21 nm from 1.0 × 10−5 to 1.0 × 10−4 M ofAgNO3 whereas the same was found to be ∼7 nm from 1.0×10−4
to 1.0 × 10−3 M of AgNO3.In order to study the structural integrity of GFP in the course of
synthesis of Au NPs, the time dependent fluorescence spectroscopyof GFP was pursued [35]. The changes in fluorescence characteris-tic of GFP due to the synthesis of Au NPs are shown in Fig. 5.At the initial stages, all the samples showed comparable amountof fluorescence which decreased only marginally in case of con-trol sample (only GFP in buffered solution) and the sample whereonly AgNO3 was incubated with GFP solution (Figs. 5a and 5b). Onthe other hand, the fluorescence of the GFP solution treated withHAuCl4 only (Fig. 5c) progressively decreased up to 56 h, withoutfurther decrease in fluorescence in later hours. However, in caseof the GFP solution incubated with HAuCl4 in presence of AgNO3
(Fig. 5d), the fluorescence reduced drastically with time resultingin the loss of detectable fluorescence after 24 h only.
Fig. 6 shows the fluorescence decay of GFP, expressed in termsof relative fluorescence intensity at different time intervals. Theresults essentially reveal that the fluorescence intensities of thesamples containing HAuCl4 alone or AgNO3 (at different concen-trations) along with HAuCl4 decreased compared to control sampleor sample incubated with AgNO3 only, which in turn implied theloss of tertiary structure of GFP in those samples. Further, it wasinteresting to observe complete loss of fluorescence in samplescontaining GFP incubated with HAuCl4 in the presence of AgNO3
in 24 h, whereas GFP treated with HAuCl4 only retained 40% offluorescence in that period. On the other hand, only AgNO3 hadapparently no effect on the GFP structure by this time. Finally therelative fluorescence of the GFP treated with HAuCl4 only, attaineda minimum value of approximately 20% in later hours, which didnot decrease further. Additionally, one can see a difference in therate of loss of fluorescence in the presence of various amount ofAgNO3. For example, when the concentrations were at 1.0 × 10−3
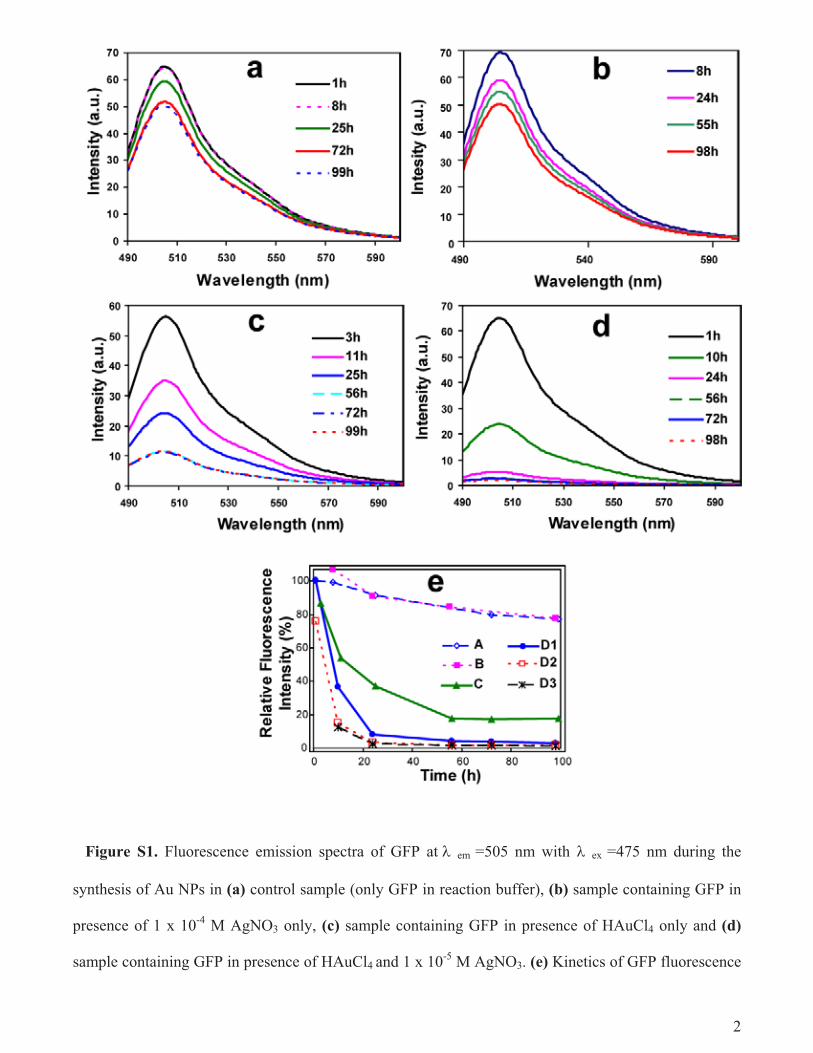
as well as 1.0 × 10−4 M, the intensity was down to about 15% in10 h and near-complete loss was observed at 24 h. On the otherhand, when the concentration of AgNO3 was at 1.0 × 10−5 M theintensity went down to 35% in 10 h and to about 5% at 24 h.It may be mentioned here that, the same results were observedwhen the fluorescence intensity was measured at emission max-imum at 505 nm with λexcitation at 475 nm (Fig. S1, Supportinginformation). Furthermore, the role of protein denaturation in theloss of GFP fluorescence, rather than fluorescence quenching by AuNPs, was ascertained by SDS–PAGE analyses (refer to Supportinginformation; Fig. S2 for further details).
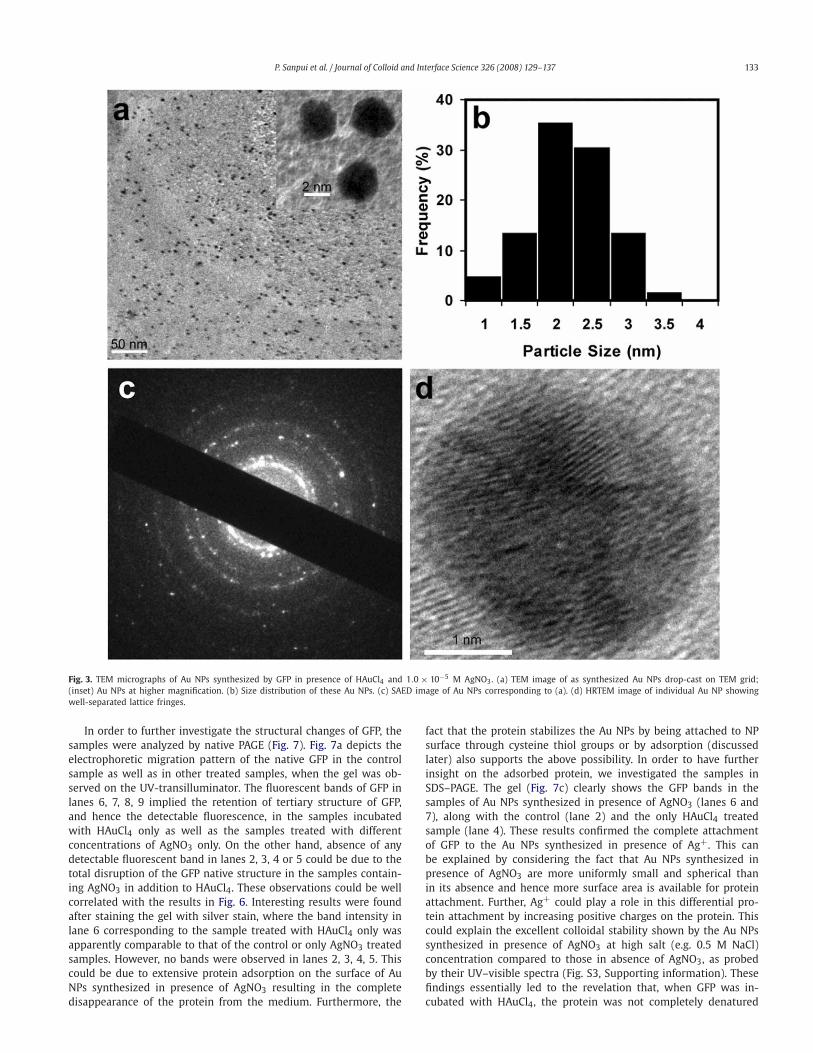
P. Sanpui et al. / Journal of Colloid and Interface Science 326 (2008) 129–137 133
Fig. 3. TEM micrographs of Au NPs synthesized by GFP in presence of HAuCl4 and 1.0 × 10−5 M AgNO3. (a) TEM image of as synthesized Au NPs drop-cast on TEM grid;(inset) Au NPs at higher magnification. (b) Size distribution of these Au NPs. (c) SAED image of Au NPs corresponding to (a). (d) HRTEM image of individual Au NP showingwell-separated lattice fringes.
In order to further investigate the structural changes of GFP, thesamples were analyzed by native PAGE (Fig. 7). Fig. 7a depicts theelectrophoretic migration pattern of the native GFP in the controlsample as well as in other treated samples, when the gel was ob-served on the UV-transilluminator. The fluorescent bands of GFP inlanes 6, 7, 8, 9 implied the retention of tertiary structure of GFP,and hence the detectable fluorescence, in the samples incubatedwith HAuCl4 only as well as the samples treated with differentconcentrations of AgNO3 only. On the other hand, absence of anydetectable fluorescent band in lanes 2, 3, 4 or 5 could be due to thetotal disruption of the GFP native structure in the samples contain-ing AgNO3 in addition to HAuCl4. These observations could be wellcorrelated with the results in Fig. 6. Interesting results were foundafter staining the gel with silver stain, where the band intensity inlane 6 corresponding to the sample treated with HAuCl4 only wasapparently comparable to that of the control or only AgNO3 treatedsamples. However, no bands were observed in lanes 2, 3, 4, 5. Thiscould be due to extensive protein adsorption on the surface of AuNPs synthesized in presence of AgNO3 resulting in the completedisappearance of the protein from the medium. Furthermore, the
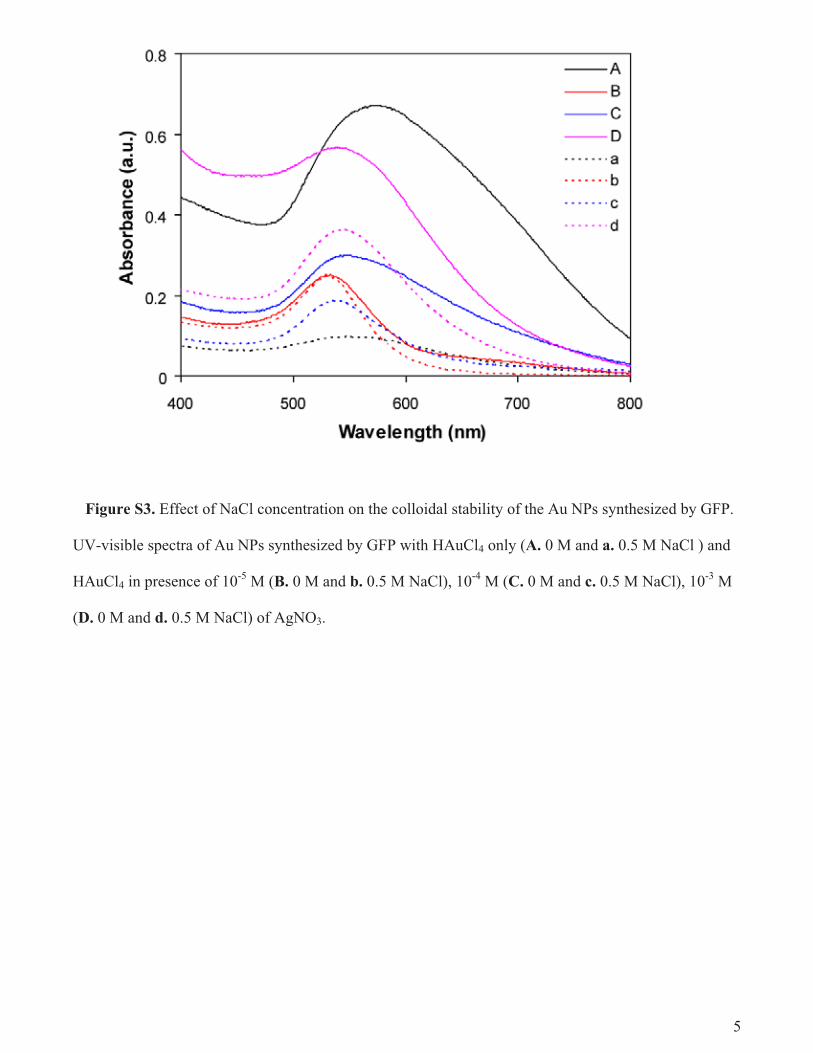
fact that the protein stabilizes the Au NPs by being attached to NPsurface through cysteine thiol groups or by adsorption (discussedlater) also supports the above possibility. In order to have furtherinsight on the adsorbed protein, we investigated the samples inSDS–PAGE. The gel (Fig. 7c) clearly shows the GFP bands in thesamples of Au NPs synthesized in presence of AgNO3 (lanes 6 and7), along with the control (lane 2) and the only HAuCl4 treatedsample (lane 4). These results confirmed the complete attachmentof GFP to the Au NPs synthesized in presence of Ag+. This canbe explained by considering the fact that Au NPs synthesized inpresence of AgNO3 are more uniformly small and spherical thanin its absence and hence more surface area is available for proteinattachment. Further, Ag+ could play a role in this differential pro-tein attachment by increasing positive charges on the protein. Thiscould explain the excellent colloidal stability shown by the Au NPssynthesized in presence of AgNO3 at high salt (e.g. 0.5 M NaCl)concentration compared to those in absence of AgNO3, as probedby their UV–visible spectra (Fig. S3, Supporting information). Thesefindings essentially led to the revelation that, when GFP was in-cubated with HAuCl4, the protein was not completely denatured
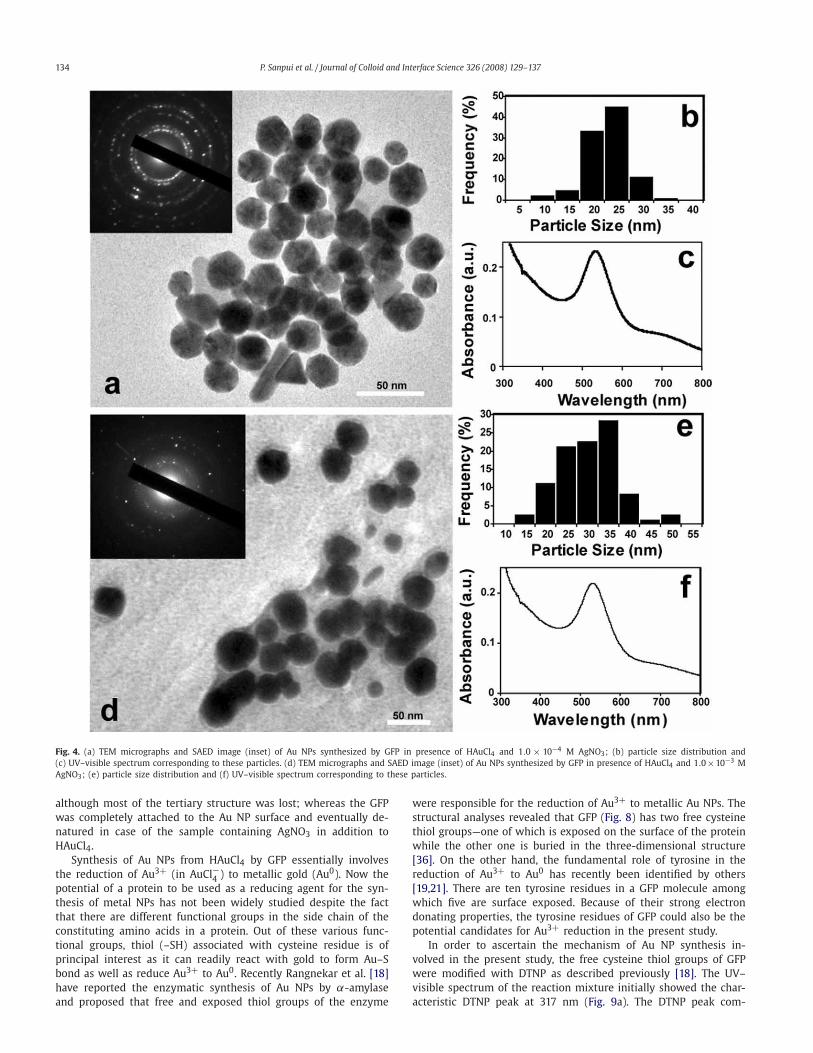
134 P. Sanpui et al. / Journal of Colloid and Interface Science 326 (2008) 129–137
Fig. 4. (a) TEM micrographs and SAED image (inset) of Au NPs synthesized by GFP in presence of HAuCl4 and 1.0 × 10−4 M AgNO3; (b) particle size distribution and(c) UV–visible spectrum corresponding to these particles. (d) TEM micrographs and SAED image (inset) of Au NPs synthesized by GFP in presence of HAuCl4 and 1.0×10−3 MAgNO3; (e) particle size distribution and (f) UV–visible spectrum corresponding to these particles.
although most of the tertiary structure was lost; whereas the GFPwas completely attached to the Au NP surface and eventually de-natured in case of the sample containing AgNO3 in addition toHAuCl4.
Synthesis of Au NPs from HAuCl4 by GFP essentially involvesthe reduction of Au3+ (in AuCl−4 ) to metallic gold (Au0). Now thepotential of a protein to be used as a reducing agent for the syn-thesis of metal NPs has not been widely studied despite the factthat there are different functional groups in the side chain of theconstituting amino acids in a protein. Out of these various func-tional groups, thiol (–SH) associated with cysteine residue is ofprincipal interest as it can readily react with gold to form Au–Sbond as well as reduce Au3+ to Au0. Recently Rangnekar et al. [18]have reported the enzymatic synthesis of Au NPs by α-amylaseand proposed that free and exposed thiol groups of the enzyme
were responsible for the reduction of Au3+ to metallic Au NPs. Thestructural analyses revealed that GFP (Fig. 8) has two free cysteinethiol groups—one of which is exposed on the surface of the proteinwhile the other one is buried in the three-dimensional structure[36]. On the other hand, the fundamental role of tyrosine in thereduction of Au3+ to Au0 has recently been identified by others[19,21]. There are ten tyrosine residues in a GFP molecule amongwhich five are surface exposed. Because of their strong electrondonating properties, the tyrosine residues of GFP could also be thepotential candidates for Au3+ reduction in the present study.
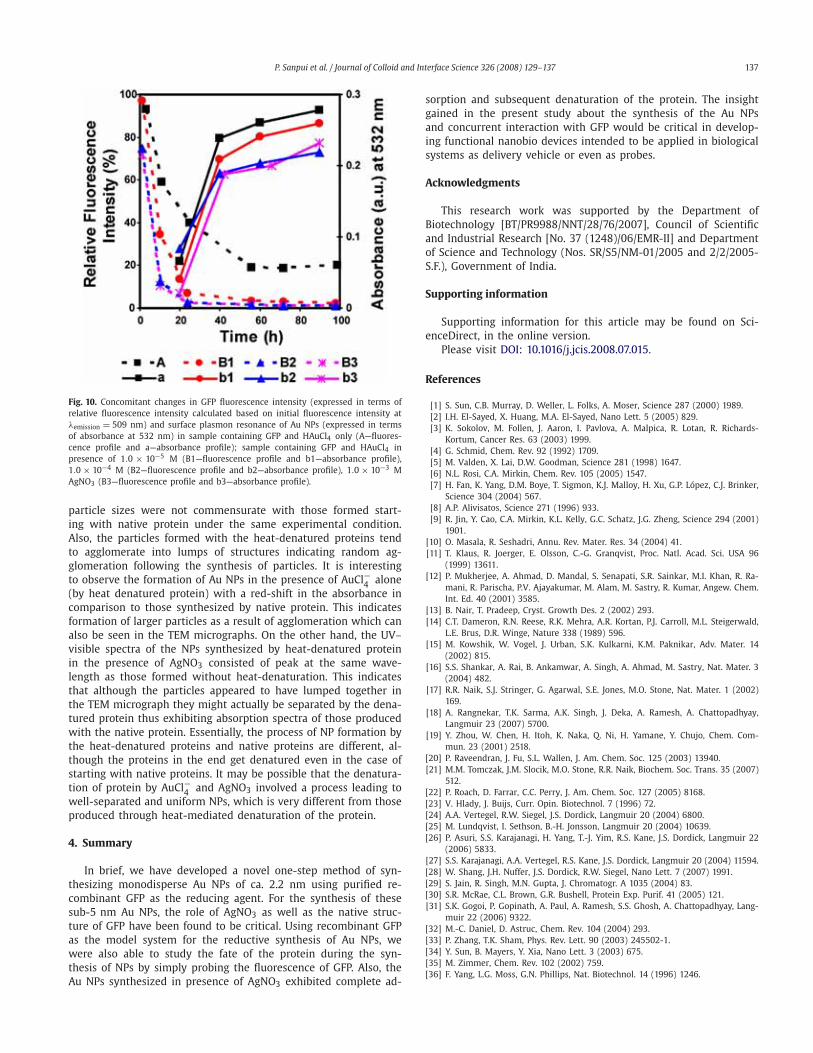
In order to ascertain the mechanism of Au NP synthesis in-volved in the present study, the free cysteine thiol groups of GFPwere modified with DTNP as described previously [18]. The UV–visible spectrum of the reaction mixture initially showed the char-acteristic DTNP peak at 317 nm (Fig. 9a). The DTNP peak com-
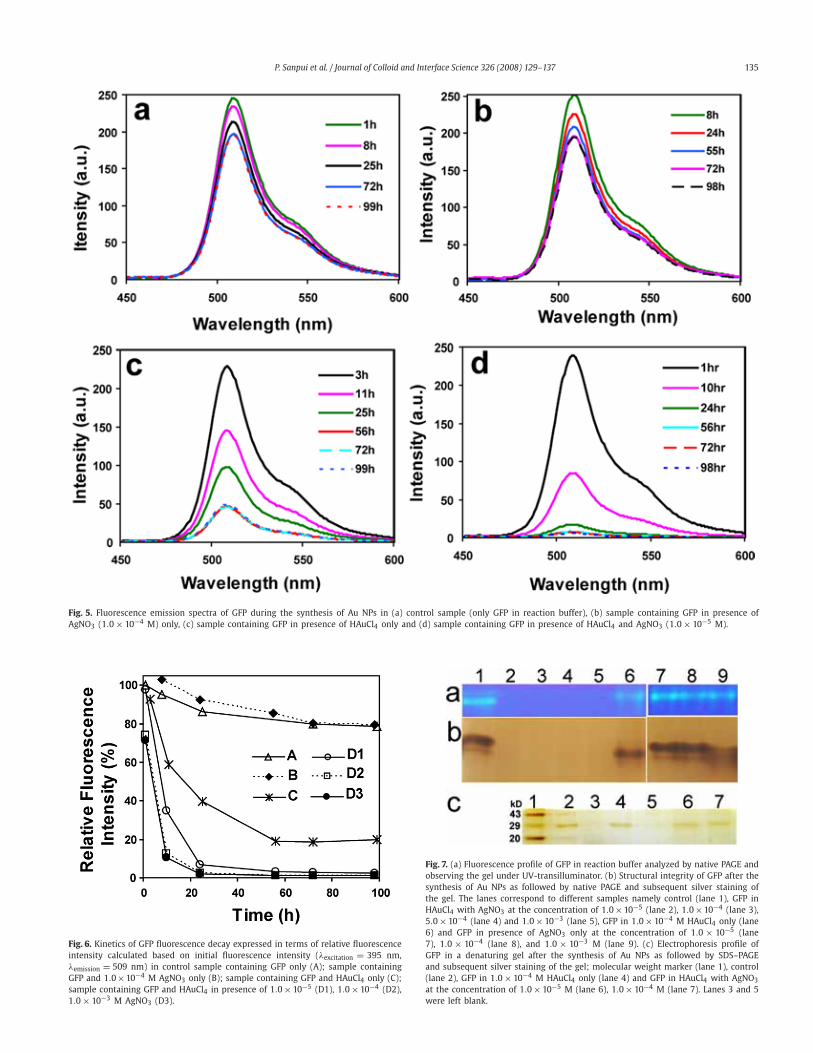
P. Sanpui et al. / Journal of Colloid and Interface Science 326 (2008) 129–137 135
Fig. 5. Fluorescence emission spectra of GFP during the synthesis of Au NPs in (a) control sample (only GFP in reaction buffer), (b) sample containing GFP in presence ofAgNO3 (1.0× 10−4 M) only, (c) sample containing GFP in presence of HAuCl4 only and (d) sample containing GFP in presence of HAuCl4 and AgNO3 (1.0× 10−5 M).
Fig. 6. Kinetics of GFP fluorescence decay expressed in terms of relative fluorescenceintensity calculated based on initial fluorescence intensity (λexcitation = 395 nm,λemission = 509 nm) in control sample containing GFP only (A); sample containingGFP and 1.0×10−4 M AgNO3 only (B); sample containing GFP and HAuCl4 only (C);sample containing GFP and HAuCl4 in presence of 1.0× 10−5 (D1), 1.0× 10−4 (D2),1.0× 10−3 M AgNO3 (D3).
Fig. 7. (a) Fluorescence profile of GFP in reaction buffer analyzed by native PAGE andobserving the gel under UV-transilluminator. (b) Structural integrity of GFP after thesynthesis of Au NPs as followed by native PAGE and subsequent silver staining ofthe gel. The lanes correspond to different samples namely control (lane 1), GFP inHAuCl4 with AgNO3 at the concentration of 1.0×10−5 (lane 2), 1.0×10−4 (lane 3),5.0× 10−4 (lane 4) and 1.0× 10−3 (lane 5), GFP in 1.0× 10−4 M HAuCl4 only (lane6) and GFP in presence of AgNO3 only at the concentration of 1.0 × 10−5 (lane7), 1.0 × 10−4 (lane 8), and 1.0 × 10−3 M (lane 9). (c) Electrophoresis profile ofGFP in a denaturing gel after the synthesis of Au NPs as followed by SDS–PAGEand subsequent silver staining of the gel; molecular weight marker (lane 1), control(lane 2), GFP in 1.0× 10−4 M HAuCl4 only (lane 4) and GFP in HAuCl4 with AgNO3
at the concentration of 1.0× 10−5 M (lane 6), 1.0× 10−4 M (lane 7). Lanes 3 and 5were left blank.
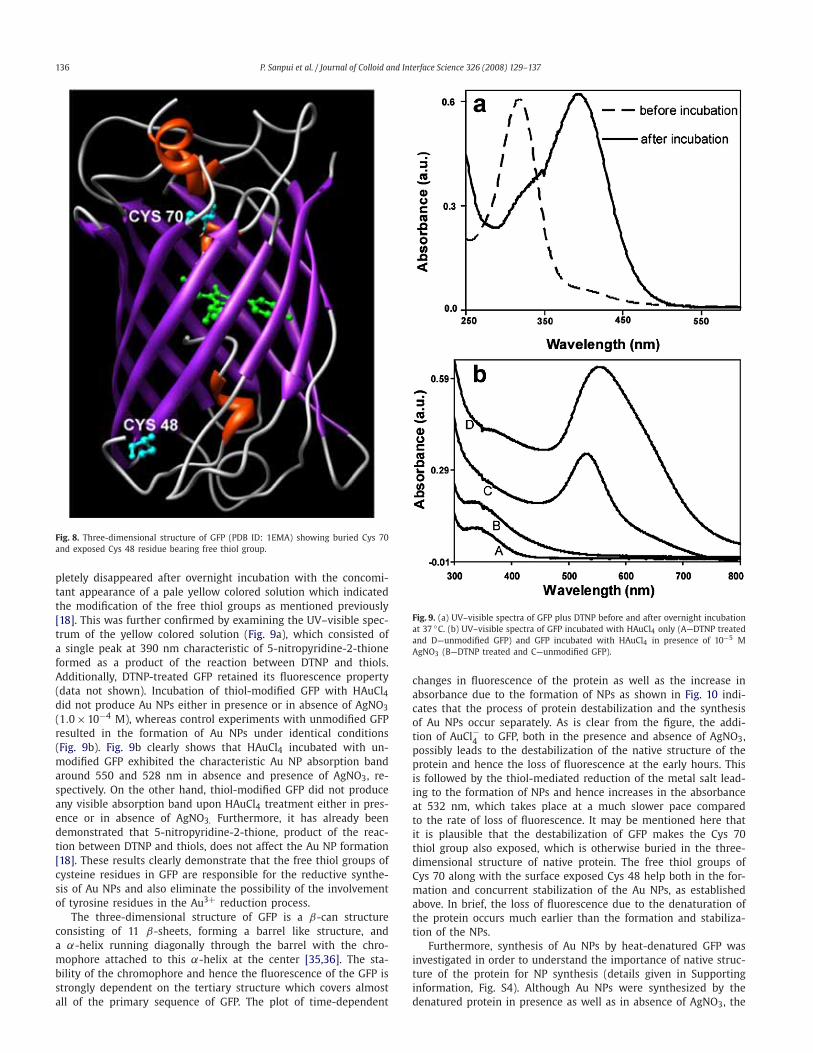
136 P. Sanpui et al. / Journal of Colloid and Interface Science 326 (2008) 129–137
Fig. 8. Three-dimensional structure of GFP (PDB ID: 1EMA) showing buried Cys 70and exposed Cys 48 residue bearing free thiol group.
pletely disappeared after overnight incubation with the concomi-tant appearance of a pale yellow colored solution which indicatedthe modification of the free thiol groups as mentioned previously[18]. This was further confirmed by examining the UV–visible spec-trum of the yellow colored solution (Fig. 9a), which consisted ofa single peak at 390 nm characteristic of 5-nitropyridine-2-thioneformed as a product of the reaction between DTNP and thiols.Additionally, DTNP-treated GFP retained its fluorescence property(data not shown). Incubation of thiol-modified GFP with HAuCl4did not produce Au NPs either in presence or in absence of AgNO3(1.0×10−4 M), whereas control experiments with unmodified GFPresulted in the formation of Au NPs under identical conditions(Fig. 9b). Fig. 9b clearly shows that HAuCl4 incubated with un-modified GFP exhibited the characteristic Au NP absorption bandaround 550 and 528 nm in absence and presence of AgNO3, re-spectively. On the other hand, thiol-modified GFP did not produceany visible absorption band upon HAuCl4 treatment either in pres-ence or in absence of AgNO3. Furthermore, it has already beendemonstrated that 5-nitropyridine-2-thione, product of the reac-tion between DTNP and thiols, does not affect the Au NP formation[18]. These results clearly demonstrate that the free thiol groups ofcysteine residues in GFP are responsible for the reductive synthe-sis of Au NPs and also eliminate the possibility of the involvementof tyrosine residues in the Au3+ reduction process.
The three-dimensional structure of GFP is a β-can structureconsisting of 11 β-sheets, forming a barrel like structure, anda α-helix running diagonally through the barrel with the chro-mophore attached to this α-helix at the center [35,36]. The sta-bility of the chromophore and hence the fluorescence of the GFP isstrongly dependent on the tertiary structure which covers almostall of the primary sequence of GFP. The plot of time-dependent
Fig. 9. (a) UV–visible spectra of GFP plus DTNP before and after overnight incubationat 37 ◦C. (b) UV–visible spectra of GFP incubated with HAuCl4 only (A—DTNP treatedand D—unmodified GFP) and GFP incubated with HAuCl4 in presence of 10−5 MAgNO3 (B—DTNP treated and C—unmodified GFP).
changes in fluorescence of the protein as well as the increase inabsorbance due to the formation of NPs as shown in Fig. 10 indi-cates that the process of protein destabilization and the synthesisof Au NPs occur separately. As is clear from the figure, the addi-tion of AuCl−4 to GFP, both in the presence and absence of AgNO3,possibly leads to the destabilization of the native structure of theprotein and hence the loss of fluorescence at the early hours. Thisis followed by the thiol-mediated reduction of the metal salt lead-ing to the formation of NPs and hence increases in the absorbanceat 532 nm, which takes place at a much slower pace comparedto the rate of loss of fluorescence. It may be mentioned here thatit is plausible that the destabilization of GFP makes the Cys 70thiol group also exposed, which is otherwise buried in the three-dimensional structure of native protein. The free thiol groups ofCys 70 along with the surface exposed Cys 48 help both in the for-mation and concurrent stabilization of the Au NPs, as establishedabove. In brief, the loss of fluorescence due to the denaturation ofthe protein occurs much earlier than the formation and stabiliza-tion of the NPs.
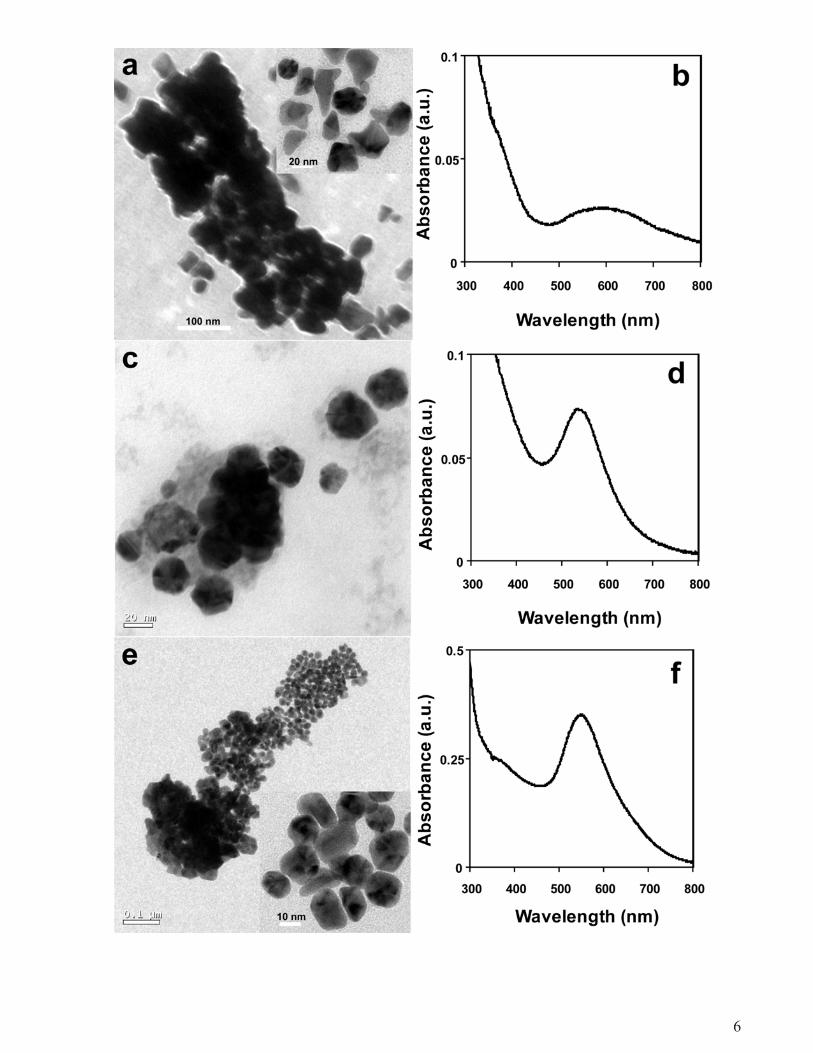
Furthermore, synthesis of Au NPs by heat-denatured GFP wasinvestigated in order to understand the importance of native struc-ture of the protein for NP synthesis (details given in Supportinginformation, Fig. S4). Although Au NPs were synthesized by thedenatured protein in presence as well as in absence of AgNO3, the
P. Sanpui et al. / Journal of Colloid and Interface Science 326 (2008) 129–137 137
Fig. 10. Concomitant changes in GFP fluorescence intensity (expressed in terms ofrelative fluorescence intensity calculated based on initial fluorescence intensity atλemission = 509 nm) and surface plasmon resonance of Au NPs (expressed in termsof absorbance at 532 nm) in sample containing GFP and HAuCl4 only (A—fluores-cence profile and a—absorbance profile); sample containing GFP and HAuCl4 inpresence of 1.0 × 10−5 M (B1—fluorescence profile and b1—absorbance profile),1.0 × 10−4 M (B2—fluorescence profile and b2—absorbance profile), 1.0 × 10−3 MAgNO3 (B3—fluorescence profile and b3—absorbance profile).
particle sizes were not commensurate with those formed start-ing with native protein under the same experimental condition.Also, the particles formed with the heat-denatured proteins tendto agglomerate into lumps of structures indicating random ag-glomeration following the synthesis of particles. It is interestingto observe the formation of Au NPs in the presence of AuCl−4 alone(by heat denatured protein) with a red-shift in the absorbance incomparison to those synthesized by native protein. This indicatesformation of larger particles as a result of agglomeration which canalso be seen in the TEM micrographs. On the other hand, the UV–visible spectra of the NPs synthesized by heat-denatured proteinin the presence of AgNO3 consisted of peak at the same wave-length as those formed without heat-denaturation. This indicatesthat although the particles appeared to have lumped together inthe TEM micrograph they might actually be separated by the dena-tured protein thus exhibiting absorption spectra of those producedwith the native protein. Essentially, the process of NP formation bythe heat-denatured proteins and native proteins are different, al-though the proteins in the end get denatured even in the case ofstarting with native proteins. It may be possible that the denatura-tion of protein by AuCl−4 and AgNO3 involved a process leading towell-separated and uniform NPs, which is very different from thoseproduced through heat-mediated denaturation of the protein.
4. Summary
In brief, we have developed a novel one-step method of syn-thesizing monodisperse Au NPs of ca. 2.2 nm using purified re-combinant GFP as the reducing agent. For the synthesis of thesesub-5 nm Au NPs, the role of AgNO3 as well as the native struc-ture of GFP have been found to be critical. Using recombinant GFPas the model system for the reductive synthesis of Au NPs, wewere also able to study the fate of the protein during the syn-thesis of NPs by simply probing the fluorescence of GFP. Also, theAu NPs synthesized in presence of AgNO3 exhibited complete ad-
sorption and subsequent denaturation of the protein. The insightgained in the present study about the synthesis of the Au NPsand concurrent interaction with GFP would be critical in develop-ing functional nanobio devices intended to be applied in biologicalsystems as delivery vehicle or even as probes.
Acknowledgments
This research work was supported by the Department ofBiotechnology [BT/PR9988/NNT/28/76/2007], Council of Scientificand Industrial Research [No. 37 (1248)/06/EMR-II] and Departmentof Science and Technology (Nos. SR/S5/NM-01/2005 and 2/2/2005-S.F.), Government of India.
Supporting information
Supporting information for this article may be found on Sci-enceDirect, in the online version.
Please visit DOI: 10.1016/j.jcis.2008.07.015.
References
[1] S. Sun, C.B. Murray, D. Weller, L. Folks, A. Moser, Science 287 (2000) 1989.[2] I.H. El-Sayed, X. Huang, M.A. El-Sayed, Nano Lett. 5 (2005) 829.[3] K. Sokolov, M. Follen, J. Aaron, I. Pavlova, A. Malpica, R. Lotan, R. Richards-
Kortum, Cancer Res. 63 (2003) 1999.[4] G. Schmid, Chem. Rev. 92 (1992) 1709.[5] M. Valden, X. Lai, D.W. Goodman, Science 281 (1998) 1647.[6] N.L. Rosi, C.A. Mirkin, Chem. Rev. 105 (2005) 1547.[7] H. Fan, K. Yang, D.M. Boye, T. Sigmon, K.J. Malloy, H. Xu, G.P. López, C.J. Brinker,
Science 304 (2004) 567.[8] A.P. Alivisatos, Science 271 (1996) 933.[9] R. Jin, Y. Cao, C.A. Mirkin, K.L. Kelly, G.C. Schatz, J.G. Zheng, Science 294 (2001)
1901.[10] O. Masala, R. Seshadri, Annu. Rev. Mater. Res. 34 (2004) 41.[11] T. Klaus, R. Joerger, E. Olsson, C.-G. Granqvist, Proc. Natl. Acad. Sci. USA 96
(1999) 13611.[12] P. Mukherjee, A. Ahmad, D. Mandal, S. Senapati, S.R. Sainkar, M.I. Khan, R. Ra-
mani, R. Parischa, P.V. Ajayakumar, M. Alam, M. Sastry, R. Kumar, Angew. Chem.Int. Ed. 40 (2001) 3585.
[13] B. Nair, T. Pradeep, Cryst. Growth Des. 2 (2002) 293.[14] C.T. Dameron, R.N. Reese, R.K. Mehra, A.R. Kortan, P.J. Carroll, M.L. Steigerwald,
L.E. Brus, D.R. Winge, Nature 338 (1989) 596.[15] M. Kowshik, W. Vogel, J. Urban, S.K. Kulkarni, K.M. Paknikar, Adv. Mater. 14
(2002) 815.[16] S.S. Shankar, A. Rai, B. Ankamwar, A. Singh, A. Ahmad, M. Sastry, Nat. Mater. 3
(2004) 482.[17] R.R. Naik, S.J. Stringer, G. Agarwal, S.E. Jones, M.O. Stone, Nat. Mater. 1 (2002)
169.[18] A. Rangnekar, T.K. Sarma, A.K. Singh, J. Deka, A. Ramesh, A. Chattopadhyay,
Langmuir 23 (2007) 5700.[19] Y. Zhou, W. Chen, H. Itoh, K. Naka, Q. Ni, H. Yamane, Y. Chujo, Chem. Com-
mun. 23 (2001) 2518.[20] P. Raveendran, J. Fu, S.L. Wallen, J. Am. Chem. Soc. 125 (2003) 13940.[21] M.M. Tomczak, J.M. Slocik, M.O. Stone, R.R. Naik, Biochem. Soc. Trans. 35 (2007)
512.[22] P. Roach, D. Farrar, C.C. Perry, J. Am. Chem. Soc. 127 (2005) 8168.[23] V. Hlady, J. Buijs, Curr. Opin. Biotechnol. 7 (1996) 72.[24] A.A. Vertegel, R.W. Siegel, J.S. Dordick, Langmuir 20 (2004) 6800.[25] M. Lundqvist, I. Sethson, B.-H. Jonsson, Langmuir 20 (2004) 10639.[26] P. Asuri, S.S. Karajanagi, H. Yang, T.-J. Yim, R.S. Kane, J.S. Dordick, Langmuir 22
(2006) 5833.[27] S.S. Karajanagi, A.A. Vertegel, R.S. Kane, J.S. Dordick, Langmuir 20 (2004) 11594.[28] W. Shang, J.H. Nuffer, J.S. Dordick, R.W. Siegel, Nano Lett. 7 (2007) 1991.[29] S. Jain, R. Singh, M.N. Gupta, J. Chromatogr. A 1035 (2004) 83.[30] S.R. McRae, C.L. Brown, G.R. Bushell, Protein Exp. Purif. 41 (2005) 121.[31] S.K. Gogoi, P. Gopinath, A. Paul, A. Ramesh, S.S. Ghosh, A. Chattopadhyay, Lang-
muir 22 (2006) 9322.[32] M.-C. Daniel, D. Astruc, Chem. Rev. 104 (2004) 293.[33] P. Zhang, T.K. Sham, Phys. Rev. Lett. 90 (2003) 245502-1.[34] Y. Sun, B. Mayers, Y. Xia, Nano Lett. 3 (2003) 675.[35] M. Zimmer, Chem. Rev. 102 (2002) 759.[36] F. Yang, L.G. Moss, G.N. Phillips, Nat. Biotechnol. 14 (1996) 1246.

1
Green fluorescent protein for in situ synthesis of highly uniform Au
nanoparticles and monitoring protein denaturation
Pallab Sanpui1, Shivendra B. Pandey
2, Siddhartha Sankar Ghosh
1, 2, 4 and Arun Chattopadhyay
1, 3, 4
1 Centre for Nanotechnology, Indian Institute of Technology Guwahati, Guwahati 781039, India
2 Department of Biotechnology, Indian Institute of Technology Guwahati, Guwahati 781039, India
3Department of Chemistry, Indian Institute of Technology Guwahati, Guwahati 781039, India
E-mail: [email protected] and [email protected]
4 Corresponding author. [email protected] (S.S.G.); [email protected] (A.C.).
Supporting Information
2
Figure S1. Fluorescence emission spectra of GFP at em =505 nm with ex =475 nm during the
synthesis of Au NPs in (a) control sample (only GFP in reaction buffer), (b) sample containing GFP in
presence of 1 x 10-4
M AgNO3 only, (c) sample containing GFP in presence of HAuCl4 only and (d)
sample containing GFP in presence of HAuCl4 and 1 x 10-5
M AgNO3. (e) Kinetics of GFP fluorescence

3
loss expressed in terms of relative fluorescence intensity calculated based on initial fluorescence
intensity ( excitation =475 nm, emission =505 nm) in control sample containing GFP only (A); sample
containing GFP and 1 x 10-4
M AgNO3 only (B); sample containing GFP and HAuCl4 only (C); sample
containing GFP and HAuCl4 in presence of 1 x 10-5
M (D1), 1 x 10-4
M (D2), 1 x 10-3
M AgNO3 (D3).
Analysis of GFP in different Au NP samples by partially denaturing SDS-PAGE:
In order to investigate whether the loss of GFP fluorescence was due to denaturation of the protein or
the quenching of fluorescence by the Au NPs, different samples were analyzed by the standard SDS-
PAGE without boiling the samples before loading (partial denaturation condition) (figure S2). When
the gel was observed under a UV-transilluminator before silver staining (Figure S2 (a)), fluorescent
bands were seen in control and Au NP sample synthesized in absence of AgNO3; whereas no
fluorescent bands were present in the AgNO3 supplemented Au NP samples. The same gel when silver-
stained, shown in figure S2 (b), revealed protein bands in AgNO3 supplemented Au NP samples along
with other samples namely control and Au NPs synthesized in absence of AgNO3. Interestingly,
additional non-fluorescent protein band can be observed in case of Au NPs synthesized in absence of
AgNO3 (figure S2(b), lane 3), which could be attributed to the denatured GFP population present in the
sample supporting fluorescence spectroscopic results (figure 5(c) and 6). Finally, the absence of any
fluorescent band in the AgNO3 supplemented Au NP samples as well as the presence of additional non-
fluorescent band in non- AgNO3 supplemented Au NP sample demonstrates that the loss of fluorescence
of GFP adsorbed on the Au NPs is due to the denaturation of the protein structure rather than quenching
of fluorescence by Au NPs.

4
Figure S2. Electrophoresis profile of GFP in a partially denaturing gel after synthesis of Au NPs in
different samples: (a) the gel under UV-transillumination and (b) subsequent silver staining of the gel;
Control (lane 2), GFP in 1.0 x 10-4
M HAuCl4 only (lane 3) and GFP in HAuCl4 with AgNO3 at the
concentration of 1.0 x 10-5
M (lane 5), 1.0 x 10-4
M (lane 6). Purified GFP reconstituted in SDS free
sample loading buffer was loaded in lane 1 as a reference. Lane 4 was left blank.
5
Figure S3. Effect of NaCl concentration on the colloidal stability of the Au NPs synthesized by GFP.
UV-visible spectra of Au NPs synthesized by GFP with HAuCl4 only (A. 0 M and a. 0.5 M NaCl ) and
HAuCl4 in presence of 10-5
M (B. 0 M and b. 0.5 M NaCl), 10-4
M (C. 0 M and c. 0.5 M NaCl), 10-3
M
(D. 0 M and d. 0.5 M NaCl) of AgNO3.
6

7
Figure S4. TEM micrographs Au NPs synthesized by heat-denatured GFP in presence of HAuCl4
only (a); HAuCl4 and 10-5
M AgNO3 (c); HAuCl4 and 10-4
M AgNO3 (e). The UV-visible spectra of
these Au NPs are shown in b, d and f, respectively.




















