
gpu accelerated quantum chemistry
192
GPU ACCELERATED QUANTUM CHEMISTRY A DISSERTATION SUBMITTED TO THE DEPARTMENT OF CHEMISTRY AND THE COMMITTEE ON GRADUATE STUDIES OF STANFORD UNIVERSITY IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY NATHAN LUEHR February 2015
-
Upload
khangminh22 -
Category
Documents
-
view
1 -
download
0
Transcript of gpu accelerated quantum chemistry

GPU ACCELERATED QUANTUM CHEMISTRY
A DISSERTATION
SUBMITTED TO THE DEPARTMENT OF CHEMISTRY
AND THE COMMITTEE ON GRADUATE STUDIES
OF STANFORD UNIVERSITY
IN PARTIAL FULFILLMENT OF THE REQUIREMENTS
FOR THE DEGREE OF
DOCTOR OF PHILOSOPHY
NATHAN LUEHR
February 2015

http://creativecommons.org/licenses/by-nc/3.0/us/
This dissertation is online at: http://purl.stanford.edu/hb803mt5913
© 2015 by Nathan Luehr. All Rights Reserved.
Re-distributed by Stanford University under license with the author.
This work is licensed under a Creative Commons Attribution-Noncommercial 3.0 United States License.
ii

I certify that I have read this dissertation and that, in my opinion, it is fully adequatein scope and quality as a dissertation for the degree of Doctor of Philosophy.
Todd Martinez, Primary Adviser
I certify that I have read this dissertation and that, in my opinion, it is fully adequatein scope and quality as a dissertation for the degree of Doctor of Philosophy.
Hans Andersen
I certify that I have read this dissertation and that, in my opinion, it is fully adequatein scope and quality as a dissertation for the degree of Doctor of Philosophy.
Vijay Pande
Approved for the Stanford University Committee on Graduate Studies.
Patricia J. Gumport, Vice Provost for Graduate Education
This signature page was generated electronically upon submission of this dissertation in electronic format. An original signed hard copy of the signature page is on file inUniversity Archives.
iii

iv

v
ABSTRACT
This dissertation develops techniques to accelerate quantum chemistry calculations
using commodity graphical processing units (GPUs). As both the principle bottleneck
in finite basis calculations and a highly parallel task, the evaluation of Gaussian
integrals is a prime target for GPU acceleration. Methods to tailor quantum chemistry
algorithms from the bottom up to take maximum advantage of massively parallel
processors are described. Special attention is taken to make maximum use of
performance features typical of modern GPUs, such as high single precision
performance. After developing an efficient integral direct self-consistent field (SCF)
procedure for GPUs that is an order of magnitude faster than typical CPU codes, the
same machinery is extended to the configuration interaction singles (CIS) and time-
dependent density functional theory (TDDFT) methods. Finally, this machinery is
applied to molecular dynamics (MD) calculations. To extend the time scale accessible
to MD calculations of large systems, an ab initio multiple time steps (MTS) approach
is developed. For small systems, up to a few dozen atoms, an interactive interface
enabling a virtual molecular modeling kit complete with realistic ab initio forces is
developed.

vi
ACKNOWLEDGEMENTS
I thank my advisor Dr. Todd Martínez for his guidance, advice, and patience
over the past six years of work. Working in his lab has provided tremendous
professional and personal growth and has been much more fun than graduate school
has any right to be. I am also indebted to my predecessor, Ivan Ufimtsev, who began
the GPU effort and gently brought me up to speed. I have also had the advantage of
working with Dr. Tom Markland, and Dr. Christine Isborn on various projects and
have benefited tremendously in learning from them. I thank my father, Dr. Craig
Luehr, for his life-long encouragement to pursue interesting questions, and his
patience in reviewing even the least interesting sections of this dissertation. Finally, I
thank my wife, Tracy, for her unfailing encouragement and support in pursuing this
work.

vii
TABLE OF CONTENTS
CHAPTER TITLE PAGE
Title Page i
Abstract v
Acknowledgements vi
Table of Contents vii
List of Tables viii
List of Illustrations ix
Introduction 1
1 Background and Review 7
2 Integral-Direct Fock Construction on GPUs 33
3 Dynamic Precision ERI Evaluation 53
4 DFT Exchange-Correlation Evaluation on GPUs 71
5 Excited State Electronic Structure on GPUs: CIS and TDDFT 91
6 Multiple Time Step Integrators for Ab Initio MD 115
7 Interactive Ab Initio Molecular Dynamics 141
Bibliography 165

viii
LIST OF TABLES
TABLE TITLE PAGE
1.1 Runtime Comparison for GPU ERI Algorithms 27
3.1 RHF Energies Computed in Single and Double Precision 60
3.2 Double and Dynamic Precision Final RHF Energies 64
3.3 Runtime Comparison of Dynamic and Full Double Precision 66
4.1 Comparison of Becke Grid Calculations by Precision 78
4.2 Performance of CPU and GPU Becke Weight Calculations 79
4.3 Timing Breakdown: CPU and GPU Grid Generation for BPTI 80
5.1 Accuracy and Performance of GPU CIS Algorithm 103
5.2 TD-BLYP AX Build Timings for Various Quadrature Grids 106
5.3 Properties for First Bright State of Several Dendrimers 109
6.1 Performance of MTS and Velocity Verlet Integrators 134
7.1 Small Molecule TeraChem Optimization Improvements 157
7.2 Wall Time per MD Time Step for Various Small Molecules 160

ix
LIST OF ILLUSTRATIONS
FIGURE TITLE PAGE
1.1 1 Block – 1 Contracted ERI Mapping 23
1.2 1 Thread – 1 Contracted ERI Mapping 25
1.3 1 Thread – 1 Primitive ERI Mapping 26
1.4 ERI Grid Sorted by Angular Momentum 30
2.1 GPU J-Engine Algorithm 38
2.2 Organization of Coulomb ERIs by Schwarz Bound 42
2.3 GPU K-Engine Algorithm 48
3.1 Arrangement of Double and Single Precision Coulomb ERIs 59
3.2 Geometries Used for Benchmarking Mixed Precision 60
3.3 Mixed Precision Error versus Precision Threshold 61
3.4 Additional Test Systems for Dynamic Precision 65
3.5 Fock Construction Speedups by Precision 66
4.1 Pseudo-code of Serial Becke Weight Calculation 75
4.2 Benchmark Molecules for Becke Weight Kernels 79
4.3 Linear Scaling of CPU and GPU Becke Weight Kernels 80
4.4 One-Dimensional and Three-Dimensional SCF Test Systems 84
4.5 First SCF Iteration Timing Breakdown 85
4.6 Parallel Efficiency for SCF Calculations on Multiple GPUs 87
4.7 TeraChem vs. GAMESS: SCF Performance for Water Clusters 89
5.1 Geometries of Four Generations of Oligothiophene Dendrimers 99

x
5.2 Additional Systems to Benchmark Excited State Calculations 99
5.3 CIS Convergence Using Single and Double Precision 101
5.4 Timing Breakdown for Construction of AX Vectors on GPU 104
5.5 TDDFT First Excitation Energy versus System Size 108
6.1 H2O/OH- Dissociation Curves for CASE and RHF 124
6.2 Total Energy for 21ps MTS-LJFRAG Simulation 125
6.3 Energy Drift for Ab Initio MTS Integrators 127
6.4 Power Spectra: Velocity Verlet and MTS Integrators 129
6.5 Power Spectra: MTS Integrators with Various Model Forces 130
6.6 Power Spectra: CASE Verlet and CASE MTS Integrators 131
6.7 Energy Conservation of 21ps MTS-CASE Trajectory 133
7.1 Schematic of Interactive MD Communication 145
7.2 Histogram of Step Times for Interactive and Batch MD 148
7.3 Schematic of Visualized and Simulated Systems 152
7.4 Multi-GPU Parallelization Strategies 155
7.5 Total Energy Curve for AI-IMD Simulation of HCl 159
7.6 Geometries for AI-IMD Benchmark Calculations 160
7.7 Snapshots of an Interactive Simulation 161
7.8 Interactive Proton Transfer in Imidazole 162

1
INTRODUCTION
For the field of quantum chemistry, the circumvention of computational
bottlenecks is a key concern. After all, the non-relativistic Schrödinger equation in
principle describes the chemistry of a great many important organic and biological
systems.1 In practice, however, the generality and accuracy of this equation can only
be accessed at a tremendous computational cost that scales factorially with the size of
the system. Many sophisticated ab initio approximations2 have been developed to
reduce the required effort to a polynomial function of the system’s size while retaining
the general applicability of the Schrödinger equation along with an absolute accuracy
in the computed energy of at least 0.5 kcal/mol, which is ~kBT at 300K and thus the
threshold at which energies become chemically relevant.
The history of quantum chemistry has developed through a series of
algorithmic developments. However, the impact of computer hardware has been
equally important. Because each algorithm was developed to run on a particular
machine, the performance characteristics of each computer shaped the algorithms that
were developed. For example, the expression of correlated wavefunction methods
exclusively in terms of dense linear algebra operations is arguably a direct result of the
efficiency of BLAS on traditional processors. As clock speeds and serial CPU
performance ramped up in the 90’s and early 2000’s, processor architectures were
heavily consolidated until only a few remained, most notably Intel’s x86. Given the
favorable cost and performance of CPUs, it is not surprising that quantum chemistry
methods have extensively targeted the CPU.

2
However, in recent years the CPU’s serial performance has essentially
stagnated. As a result, alternative architectures are gaining traction for many
workloads. Multi-core CPUs have become commonplace. Massively parallel
streaming architectures designed for use in graphics processing units (GPUs) provide a
more extreme contrast with traditional serial processors. Today’s widening landscape
of processor designs raises the question of what shape quantum chemistry methods
will take as they move beyond the CPU.
In the following chapters we seek to answer this question by tailoring various
quantum chemistry algorithms to GPUs. This is a particularly attractive architecture
both because key computational bottlenecks in quantum chemistry map extremely
well onto massively parallel GPU processors and because low-cost high-performance
hardware is readily available and continuously improved. The introduction of the
Compute Unified Device Architecture3 (CUDA) as an extension to the C language
also greatly simplifies GPU programming, making it easily accessible for scientific
programming. The methods described in the following chapters form the foundation of
the TeraChem quantum chemistry program, which was designed from the ground up
to make optimal use of GPU hardware.4,5
The importance of this work is at least threefold. First, the methods presented
here are important in themselves because they dramatically accelerate certain quantum
chemistry calculations and make previously difficult calculations routine.6-9 Second, as
finite size constraints become dominant in hardware design, machines will become
increasingly parallel. As a result, many features and limitations that exist on modern
GPUs will become ubiquitous on high-performance architectures of the future, making

3
our work highly transferable. Third, the discussion of how to map quantum chemistry
calculations onto computer hardware goes beyond mere code optimization, since there
is a kind of natural selection at play in method development. If certain operations can
be accelerated, then methods that exploit these operations may gain advantages and be
chosen for future development. The present work follows this pattern. For example,
after introducing and optimizing the Coulomb and exchange operators for use in self-
consistent field (SCF) calculations10,11 in chapters 2-4, these same operations are
exploited to accelerate excited state methods6 in chapter 5.
At the same time that traditional processors are reaching their performance
limits, it is becoming much cheaper to fabricate custom architectures. The present
work focuses on optimizing quantum chemistry methods for a particular alternative
architecture. Perhaps in the future the inverse process will be feasible, and processors
will be tailored as much to quantum chemistry as vice versa. The series of ANTON
machines that are designed for classical MD are perhaps early examples of this
trend.12-18 The less successful GRAPE-DR architecture is similar,19 but perhaps shows
how much must be gleaned from the study of existing architectures before efficient
custom hardware can be designed for quantum chemistry.
The following chapters are organized as follows. Chapter one gives a brief
introduction to quantum chemistry and introduces the electron repulsion integrals
(ERIs) which represent an important computational bottleneck. We also review the
McMurchie-Davidson20 approach that can be used to evaluate these integrals as well
as early work to evaluate ERIs on GPU processors.21,22 Chapter two covers the
efficient implementation of Coulomb and exchange operators in TeraChem.11,23

4
Chapter three introduces dynamic precision, which is an important technique to tailor
integral evaluation to GPUs that provide much more single than double precision
performance.10 Chapter four discusses the implementation of density functional theory
(DFT) exchange-correlation potentials on GPUs.24,25 In chapter five we extend our
Coulomb and exchange operators to excited state configuration interaction singles
(CIS) and linear response time-dependent DFT (TDDFT).6 Finally, we consider
methods that leverage GPU quantum chemistry to extend the reach of ab initio
molecular dynamics (AIMD). In chapter six we discuss the use of multiple time step
(MTS) integrators to accelerate AIMD in large systems.8 And in chapter seven we turn
to accelerating calculations on small systems and introduce an interactive quantum
chemistry interface built on real-time AIMD.
REFERENCES
(1) Dirac, P. A. M. P R Soc Lond a-Conta 1929, 123, 714.
(2) Helgaker, T.; Jørgensen, P.; Olsen, J. Molecular electronic-structure theory; Wiley: New York, 2000.
(3) Schwegler, E.; Challacombe, M.; HeadGordon, M. J. Chem. Phys. 1997, 106, 9708.
(4) PetaChem, L.; Vol. 2010.
(5) Ufimtsev, I. S.; Martinez, T. J. J. Chem. Theo. Comp. 2009, 5, 2619.
(6) Isborn, C. M.; Luehr, N.; Ufimtsev, I. S.; Martinez, T. J. J. Chem. Theo. Comp. 2011, 7, 1814.
(7) Kulik, H. J.; Luehr, N.; Ufimtsev, I. S.; Martinez, T. J. J. Phys. Chem. B 2012, 116, 12501.
(8) Luehr, N.; Markland, T. E.; Martinez, T. J. J. Chem. Phys. 2014, 140, 084116.

5
(9) Ufimtsev, I. S.; Luehr, N.; Martinez, T. J. J. Phys. Chem. Lett. 2011, 2, 1789.
(10) Luehr, N.; Ufimtsev, I. S.; Martinez, T. J. J. Chem. Theo. Comp. 2011, 7, 949.
(11) Ufimtsev, I. S.; Martinez, T. J. J. Chem. Theo. Comp. 2009, 5, 1004.
(12) Grossman, J. P.; Kuskin, J. S.; Bank, J. A.; Theobald, M.; Dror, R. O.; Ierardi, D. J.; Larson, R. H.; Ben Schafer, U.; Towles, B.; Young, C.; Shaw, D. E. Acm Sigplan Notices 2013, 48, 549.
(13) Grossman, J. P.; Towles, B.; Bank, J. A.; Shaw, D. E. Des Aut Con 2013.
(14) Kuskin, J. S.; Young, C.; Grossman, J. P.; Batson, B.; Deneroff, M. M.; Dror, R. O.; Shaw, D. E. Int S High Perf Comp 2008, 315.
(15) Larson, R. H.; Salmon, J. K.; Dror, R. O.; Deneroff, M. M.; Young, C.; Grossman, J. P.; Shan, Y. B.; Klepeis, J. L.; Shaw, D. E. Int S High Perf Comp 2008, 303.
(16) Shaw, D. E.; Deneroff, M. M.; Dror, R. O.; Kuskin, J. S.; Larson, R. H.; Salmon, J. K.; Young, C.; Batson, B.; Bowers, K. J.; Chao, J. C.; Eastwood, M. P.; Gagliardo, J.; Grossman, J. P.; Ho, C. R.; Ierardi, D. J.; Kolossvary, I.; Klepeis, J. L.; Layman, T.; McLeavey, C.; Moraes, M. A.; Mueller, R.; Priest, E. C.; Shan, Y. B.; Spengler, J.; Theobald, M.; Towles, B.; Wang, S. C. Conf Proc Int Symp C 2007, 1.
(17) Shaw, D. E.; Dror, R. O.; Salmon, J. K.; Grossman, J. P.; Mackenzie, K. M.; Bank, J. A.; Young, C.; Deneroff, M. M.; Batson, B.; Bowers, K. J.; Chow, E.; Eastwood, M. P.; Ierardi, D. J.; Klepeis, J. L.; Kuskin, J. S.; Larson, R. H.; Lindorff-Larsen, K.; Maragakis, P.; Moraes, M. A.; Piana, S.; Shan, Y. B.; Towles, B. Proceedings of the Conference on High Performance Computing Networking, Storage and Analysis 2009.
(18) Towles, B.; Grossman, J. P.; Greskamp, B.; Shaw, D. E. Conf Proc Int Symp C 2014, 1.
(19) Makino, J.; Hiraki, K.; Inaba, M. 2007 Acm/Ieee Sc07 Conference 2010, 548.
(20) McMurchie, L. E.; Davidson, E. R. J. Comp. Phys. 1978, 26, 218.
(21) Yasuda, K. J. Comp. Chem. 2008, 29, 334.
(22) Ufimtsev, I. S.; Martinez, T. J. J. Chem. Theo. Comp. 2008, 4, 222.
(23) Titov, A. V.; Ufimtsev, I. S.; Luehr, N.; Martinez, T. J. J. Chem. Theo. Comp. 2013, 9, 213.

6
(24) Hwu, W.-m. GPU computing gems; Elsevier: Amsterdam ; Burlington, MA, 2011.
(25) Yasuda, K. J. Chem. Theo. Comp. 2008, 4, 1230.

7
CHAPTER ONE
BACKGROUND AND REVIEW
In this chapter we provide a brief review of the Hartree-Fock (HF) and Density
Functional Theory (DFT) quantum chemistry methods within atom-centered Gaussian
basis sets. We focus particularly on the evaluation of electron repulsion integrals
(ERIs) because these play an important role in the following chapters. The
performance of Self-consistent field (SCF) methods such as HF and DFT depends on
two principal bottlenecks. The first is the evaluation of ERIs. Formally, for a basis set
containing N functions, a total of O(N4) ERIs must be evaluated. In the limit of large
systems, efficient screening of negligibly small ERIs can reduce this number to O(N2)
or, for certain insulating systems, even O(N).1-5 The second bottleneck is the update of
the orbitals/density between SCF iterations. This is traditionally performed by
diagonalizing the N-by-N Fock matrix to obtain its eigenvectors and eigenvalues.
Eigensolvers applied to dense matrices run with a complexity of O(N3). However,
using sparse matrix algebra it is possible, again in asymptotically large systems, to
achieve O(N) scaling for this step as well.6 Thus, formal asymptotic analysis is of
limited use since the dominant bottleneck results from prefactors rather than scaling
exponents. Empirically, for systems up to at least 10,000 basis functions, integral
evaluation dominates the SCF runtime, and thus the following chapters focus
primarily on the GPU acceleration of ERI evaluation.
Numerous ERI evaluation schemes have been developed for use in traditional
CPU codes. For very high angular momentum, Rys quadrature methods7 may provide

8
an advantage on GPUs due to their smaller memory footprint.8-10 For low angular
momentum basis functions, however, the Rys and simpler McMurchie-Davidson11
approaches provide comparable performance, and the simplicity of the latter is
preferred here.
QUANTUM CHEMISTRY REVIEW
Full derivations of the HF and DFT methods as well as in-depth descriptions of
various ERI evaluation algorithms can be found elsewhere.12-14 Here we provide a
brief background in order to put the subsequent chapters in context. Unless otherwise
noted, we assume a spin-restricted wavefunction anstatz and atomic units.
Self-Consistent Field Equations in Gaussian Basis Sets
A primitive Gaussian function is defined as follows.
! i
!r( ) = Ni
!rx " xi( )ni !ry " yi( )li !rz " zi( )mi e"# i!r"!Ri
2
(1.1)
Here !r is the three-dimensional electronic coordinate,
!Ri = xi , yi , zi( ) is the
primitive’s Cartesian center (usually coinciding with the location of an atom), !i is an
exponent determining the spatial extent of the function, and Ni is a normalization
constant chosen so that the following holds.
! i!r( )! i
!r( )d!r = 1"#
#
$ (1.2)
The nonnegative integers, ni, li, and mi, fix the function’s angular momentum in the
Cartesian x-, y-, and z-directions. Their sum, "i = ni + li + mi, gives the primitive’s
total angular momentum. Functions with !i = 0,1,2 are termed, s-, p-, and d-functions

9
respectively. The set of !i +1( ) !i + 2( ) / 2 primitive functions that differ only by the
distribution of !i into n, l, and m is referred to as a primitive shell.
! I = ! i
!r( ) |!Ri =
!RI ," i =" I ,#i = #I{ } (1.3)
We use the lower case indices i, j, k, and l to refer to primitive functions and the
capital letters I, J, K, and L for primitive shells.
In order to more closely approximate solutions to the atomic Schrödinger
equation, several primitive functions (all sharing a common center, Rµ , and angular
momenta, nµ , lµ , and mµ ) are combined together into contracted basis shells using
fixed contraction weights, ci.
!µ (r) = ci" i (r)i#µ$ (1.4)
Here a segmented basis is assumed in which each primitive contributes to a single
basis function. Greek indices are used for contracted AO quantities. The notation
i !µ specifies that the primitive ! i (r) belongs to the AO contraction !µ (r) . These
contracted functions are termed atomic orbitals (AOs) in analogy to the Hydrogen
atom’s one-electron orbitals which they resemble and form the basis in which the
Schrödinger equation will be solved.
The AOs are further combined by linear contraction into molecular orbitals
(MOs), each of which represents a one-particle spatial probability distribution for an
electron in the multi-atom system.
! i!r( ) = Cµi"µ
!r( )µ
N
# (1.5)

10
The MO coefficients, Cµi , are free parameters, and their determination is the primary
objective of the SCF procedure. In order to describe an n-electron system, the one-
electron MOs are combined with spin functions in a Slater determinant.
! !x1,!x2,...,
!xn( ) = 1n!
" 1(!x1) " 2 (
!x1) " " n (!x1)
" 1(!x2 ) " 2 (
!x2 ) " " n (!x2 )
# # $ #" 1(!xn ) " 2 (
!xn ) " " n (!xn )
(1.6)
For the spin-restricted case in which two electrons occupy each spatial orbital, the spin
orbitals (depending on both spatial and spin electronic degrees of freedom) can be
defined as follows (where !k is the spin degree of freedom for the kth electron):
! 2n"1(!xk ) = #n (
!rk )$ (% k )! 2n (
!xk ) = #n (!rk )&(% k )
(1.7)
The energy of the wavefunction, ! , representing n electrons in a system
containing A fixed atomic nuclei (each with charge Za and located at position !Ra ) is
derived from the expectation value of the electronic Hamiltonian, H :
H =Za!ri !!Raa
A
" !#i
2
2+ 1
21!ri !!rjj$i
"%
&''
(
)**i
n
" (1.8)
�ERHF =
!�H�!
! != 2 " i h" i +
i
n/2
# 2 " i" i�" j" j( )i, j
n/2
# $ " i" j�" i" j( )i, j
n/2
# (1.9)
Here the MOs are assumed, without loss of generality, to be orthonormal.
!i ! j = " ij (1.10)

11
The one-electron core Hamiltonian operator, h , accounts for electron-nuclear
attraction and electron kinetic energy,
h(!r ) =
Za!r !!Raa
A
" ! #2
2 (1.11)
and the two-electron repulsion integrals (ERIs) account for pairwise repulsive
interactions between electrons.
�! i! j�!k! l( ) = d 3!r1 d 3!r2""
! i* !r1( )! j
!r1( )!k* !r2( )! l
!r2( )!r1 #!r2
(1.12)
For Kohn-Sham DFT, a similar energy expression is obtained by using the
determinant to describe non-interacting pseudo-particles whose total density matches
the ground state electron density.15
!(!r ) = 2 " i(
!r )2
i
n/2
# (1.13)
In this case, components of the Hartree-Fock energy provide good approximations for
the DFT kinetic energy and classical electron repulsion. An additional density-
dependent exchange-correlation functional, EXC !"# $% , corrects for the relatively small
energetic effects of electron exchange and correlation as well as errors from
approximating the kinetic energy as that of the Kohn-Sham determinant.
�EDFT = 2 ! i h! i +
i
n/2
" 2 ! i! i�! j! j( )i, j
n/2
" + EXC[#] (1.14)
Given the exact exchange-correlation functional, EXC !"# $% , equation 12 would provide
the exact ground state energy. Unfortunately, the exact functional is not known in any
computationally feasible form. In practice a variety of approximate functionals are

12
often employed. For simplicity, we focus on the remarkably successful class of
generalized gradient approximation (GGA) functionals. These take the form of an
integral over a local xc-kernel that depends only on the total density and its gradient.
EXC[!]= fxc(!(r), "!(r)
2 )d!r# (1.15)
To calculate the HF or DFT ground state electronic configuration, we vary the MO
coefficients, Cµi , to minimize ERHF or EDFT under the constraint of equation 8 that the
MOs remain orthonormal. Functional variation ultimately results in the following
conditions on the MO coefficients.
F(P)C = !SC (1.16)
Here P is the density matrix represented in the AO basis;
Pµ! = Cµi
i
n
" C!i* (1.17)
! is a diagonal matrix of MO energies (formally, this matrix is the set of Lagrange
multipliers enforcing the constraint that all the molecular orbital remain orthogonal,
i.e. equation 8); S is the AO overlap matrix;
Sµ! = "µ "! (1.18)
and F(P) is the non-linear Fock operator, defined slightly differently for HF and DFT
as follows.
�Fµ!
HF (P) = hµ! + P"#"#
N
$ 2 µ!�#"( )% µ"�!#( )&' () (1.19)
�Fµ!
DFT (P) = hµ! + 2 µ!�"#( )P#"#"
N
$ +Vµ!XC (1.20)
Here h is the core Hamiltonian from equation 9 in the AO basis,

13
hµ! = "µ
Za!r1 #!Raa
A
$ #%1
2"! (1.21)
and the two electron ERIs are defined in the AO basis as follows.
�
µ!�"#( ) = d 3!r1 d 3!r2$$%µ!r1( )%! !r1( )%"
!r2( )%# !r2( )!r1 &!r2
= ci c jck cll'#(
k'"(
j'!(
i'µ( d 3!r1 d 3!r2$$
) i!r1( )) j
!r1( )) k!r2( )) l
!r2( )!r1 &!r2
= ci c jck cll'#(
k'"(
j'!(
i'µ( ij�kl*+ ,-
(1.22)
Note that round braces refer to ERIs involving contracted basis functions while square
braces refer to primitive ERIs. Finally, for DFT, VXC is determined by functional
differentiation of the exchange-correlation energy expression.
Vµ!
XC = "µ
# EXC
#$"! (1.23)
Because the HF and DFT Fock operators are non-linear, equation (1.16) cannot
be solved in closed form. Instead an iterative approach is used. Starting from some
guess for the density matrix, P, the Fock matrix is constructed and then diagonalized
to obtain a matrix of approximate MO orbitals, C. The MO coefficients are then used
to construct an improved guess for the density matrix using equation (1.17), and the
process is repeated until F and P converge to stable values.
Evaluating Electron Repulsion Integrals
Having described the basic SCF working equations we turn now to the
evaluation of primitive ERIs.

14
�
ij�kl!" #$ = d 3!r1 d 3!r2%%& i!r1( )& j
!r1( )& k!r2( )& l
!r2( )!r1 '!r2
(1.24)
Efficient evaluation of the coulomb integrals within Gaussian basis sets begins by
invoking the Gaussian product theorem (GPT) of equation (1.25). This allows a pair of
Gaussian functions at different centers to be rewritten as a combined Gaussian
function centered at a point, !P , between the original centers.16
e!" i!r!!Ri( )2e!" j
!r!!Rj( )2 = Kije
!#ij!r!!Pij( )2
#ij =" i +" j
Kij = e!" i" j
" i+" j
!Ri!!Rj( )2
!Pij =
" i
!Ri +" j
!Rj
" i +" j
(1.25)
Applying equation (1.25) separately to the bra, ! i!r1( )! j
!r1( ) , and ket, ! k!r2( )! l
!r2( )
primitive pairs of equation (1.24), results in two charge distributions, ! ij and !kl and
reduces the four center ERI to a simpler two-center problem.
�
ij�kl!" #$ = % ij�%kl!" #$ (1.26)
The pair distributions, ! ij , can be factored into x-, y-, and z-components, which will
greatly simplify the problem.
! ij = Ni N j Kij
x! ijy! ij
z! ij (1.27)
The x-component of the bra distribution is shown below, the other terms being
analogous.
x! ij (x1) = x1 " xi( )ni x1 " x j( )nj e
"#ij x1"Xij( )2 (1.28)

15
Following McMurchie and Davidson we expand the pair distributions of
equation (1.28) exactly in a basis of Hermite Gaussians, {!t}.11
x! ij (x1) = x Et
nin j"tx (x1)
t
ni+nj
# (1.29)
!t
x (x1) = ""Xij
#
$%
&
'(
t
e)*ij x1)Xij( )2
(1.30)
Again, analogous expressions expand the y- and z-components. The expansion
coefficients, x Et
ij
are calculated from simple recurrence relations given below.11
x Etmn = 0, where t < 0 or t > m+ n
x Etm+1,n = 1
2 px Et!1
mn + X Pij Ri
x Etmn + (t +1) x Et+1
mn
x Etm,n+1 = 1
2 px Et!1
mn + X Pij Rj
x Etmn + (t +1) x Et+1
mn
(1.31)
Here XPQ is shorthand for Px – Qx. The pair distribution from equation (1.27) is then
written as follows.
! ij (r1) = Ni N j Kij Etuv
ij
v
mi+mj
" #tuv (r1)u
li+l j
"t
ni+nj
" (1.32)
Etuvij = x Et
nin j y Eulil j z Ev
mimj
(1.33)
!tuv (r1) = !tx (x1)!u
y ( y1)!vz (z1) (1.34)
And the overall integral from equation (1.24) is expanded as follows.
[ij | kl]= Ni N j Nk Nl Kij Kkl Etuv
ij E !t !u vkl
!v
mk+ml
" Vtuv!t !u !v
!u
lk+ll
"!t
nk+nl
"v
mi+mj
"u
li+l j
"t
ni+nj
" (1.35)

16
Here Vtuv!t !u !v represent Hermite Coulomb integrals, which are defined through equations
(1.30) and (1.34) as partial derivatives of an s-function Coulomb integral.
Vtuv!t !u !v = ["tuv |" !t !u !v ]
= (#1) !t + !u + !v $$Xij
%
&'
(
)*
t+ !t$$Yij
%
&'
(
)*
u+ !u$
$Zij
%
&'
(
)*
v+ !ve#+ij
!r1#!Pij( )2e#+kl
!r2#!Pkl( )2
!r1 #!r2
,, d!r1d!r2
(1.36)
To evaluate Vtuv!t !u !v , the simple Coulomb integral on the right in equation (1.36)
is first expressed in terms of the Boys function, Fn.
e!"ij!r1!!Pij( )2e!"kl
!r2!!Pkl( )2
!r1 !!r2
## d!r1d!r2 =
2$ 5/2
"ij"kl "ij +"kl
F0
"ij"kl
"ij +"kl
Pij ! Pkl( )2%
&'
(
)*
(1.37)
Fn (x) = t 2ne! xt
2
dt0
1
" (1.38)
In practice the Boys function is computed using an interpolation table and downward
recursion.14 Next we define the auxiliary functions, Rtuvn , as follows.
Rtuv
n = !!Xij
"
#$
%
&'
t!!Yij
"
#$
%
&'
u!
!Zij
"
#$
%
&'
v
(2)ij)kl
)ij +)kl
"
#$
%
&'
n
Fn
)ij)kl
)ij +)kl
!Pij (!Pkl( )2"
#$
%
&' (1.39)
Noting that
Rtuv
0 = !!Xij
"
#$
%
&'
t!!Yij
"
#$
%
&'
u!
!Zij
"
#$
%
&'
v
F0
(ij(kl
(ij +(kl
!Pij )!Pkl( )2"
#$
%
&' (1.40)
Equation (1.36) now becomes the following.
Vtuv!t !u !v = ("1) !t + !u + !v 2# 5/2
$ij$kl $ij +$kl
Rt+ !t ,u+ !u ,v+ !v0 $ij$kl
$ij +$kl
,!Pij "!Pkl( )2%
&'
(
)* (1.41)

17
The utility of the auxiliaries, Rtuvn , is that they can be efficiently computed from the
Boys function starting at R000n using the following recurrence relations.
Rt+1,u ,vn = tRt!1,u ,v
n+1 + Xij ! Xkl( )Rt ,u ,vn+1
Rt ,u+1,vn = uRt ,u!1,v
n+1 + Yij !Ykl( )Rt ,u ,vn+1
Rt ,u ,v+1n = vRt ,u ,v!1
n+1 + Zij ! Zkl( )Rt ,u ,vn+1
(1.42)
This brings us to the final expression for evaluating the ERI of equation (1.24).
[ij | kl]= Nij Nkl Etuvij E !t !u !v
kl
!v
mk+ml
" (#1) !t + !u + !v
$ij +$kl
Rt+ !t ,u+ !u ,v+ !v0
!u
lk+ll
"!t
nk+nl
"v
mi+mj
"u
li+l j
"t
ni+nj
" (1.43)
Nij =
Ni N j Kij 2! 5/4
"ij (1.44)
Here Nij is a convenience factor that combines all scalar factors for the bra (or ket) pair
distribution. In practice, the AO contraction coefficients from equation (1.22), cicj in
the case of the bra, are also included in this factor.
For s-functions, each quartet of primitive shells, [ij|kl], generates a single
integral. For shells with higher angular momentum a shell quartet generates multiple
integrals, since each primitive shell contains multiple functions. For example, each
shell quartet with the momentum pattern [sp|sd] will generate 18 functions (since there
are three functions in the p-shell, six in the d-shell and one in each s-shell. These 18
integrals, however, involve the same set of auxiliary integrals, Rtuv0 , and Hermite
contraction coefficients, xEtmn . As a result, it is advantageous to generate the
intermediates once, and then evaluate equation (1.43) repeatedly, once for each
integral in the shell quartet.

18
Screening Negligible Integrals
The Fock contributions in equations (1.19) and (1.20) nominally involve
contributions from N4 ERIs. However, several strategies are routinely used to avoid
calculating most of these integrals. First, the ERIs possess eight-fold symmetry so that
[ij|kl] = [kl|ij] = [ij|lk]. The point group symmetry of the molecule is sometimes also
used to eliminate even more redundant integrals. However, large systems rarely
possess such symmetry, so this approach is not applicable to the present work.
Many of the remaining integrals are so small that they can be neglected
without affecting the computed molecular properties. Because each AO basis function
is localized in space, a pair distribution, !ij , will approach zero exponentially as the
distance between primitive functions increases. Thus, an AO ERI, �µ!�"#( ) , will be
non-negligible only if µ is centered near ! and ! is near ! . For large systems, this
reduces the number of integrals to a more manageable N2. In order to efficiently
identify significant ERIs, a Cauchy-Schwarz inequality can be applied to either
contracted or primitive integrals.17
�
ij�kl!" #$ % ij�ij!" #$1/2
kl�kl!" #$1/2
(1.45)
For primitive integrals, this Schwarz bound is easily computed, because in
[ij|ij] integrals the bra and ket pair distributions share a common center greatly
simplifying the integral expressions. Thus, by checking the integral bound for each
shell quartet, it is possible to avoid computing many small integrals all together.
Another advantage of the Schwarz bound is that it can be decomposed into bra and ket

19
parts, and thus quantities of [ij|ij]1/2 can be computed once and stored with each pair
distribution rather than being recomputed for every shell quartet.
INTRODUCTION TO CUDA AND GPU PROGRAMMING
Each GPU is a massively parallel device, containing thousands of execution
cores. However, the performance of these processors results not only from the raw
width of execution units, but also from a hierarchy of parallelism that forms the
foundation of the hardware architecture and is ingeniously exposed to the programmer
through the CUDA programming model.18 Developers must understand and respect
these hierarchical boundaries if their programs are to run efficiently on GPUs.
At the lowest level, the CUDA programmer writes a small procedure – called a
kernel in “CUDA-speak” – that is to be executed by tens of thousands of individual
threads in parallel. Although each CUDA thread is logically autonomous, the
hardware does not execute each thread independently. Instead, instructions are
scheduled for groups of 32 threads, called warps, in single-instruction-multiple-thread
(SIMT) fashion. Every thread in a warp executes the same instruction stream, with
threads masked to null operations (no-ops) for instructions in which they do not
participate.
Warps are grouped into larger blocks of up to 1024 threads. Blocks are
assigned to local groups of execution units called streaming multiprocessors (SMs).
The SM provides hardware-based intra-block synchronization methods and a small
on-chip shared memory often used for intra-block communication. CUDA blocks can
be indexed in 1, 2, or 3 dimensions at the convenience of the programmer.

20
At the highest level, blocks are organized into a CUDA grid. As with blocks,
the grid can be up to 3 dimensional. In general, the grid contains many more blocks
and threads than the GPU has physical execution units. When a grid is launched, a
hardware scheduler streams CUDA blocks onto the processors. By breaking a task into
fine-grained units of work, the GPU can be kept constantly busy, maximizing
throughput performance.
In CUDA the memory is also structured hierarchically. The host memory
usually provides the largest space, but can only be accessed through the PCIe data bus
which suffers from latencies on the order of several thousand instruction cycles. The
GPU’s main (global) memory provides several gigabytes of high-bandwidth memory
capable of more than 250 GB/s of sustained throughput. In order to enable this
bandwidth, global memory accesses incur long latencies, on the order of 500 clock
cycles. Global memory operations are handled in parallel mirroring the SIMT warp
design. The large width of the memory controller allows simultaneous memory access
by all threads of a warp as long as those threads target contiguous memory locations.
Low-latency, on-chip memory is also available. Most usefully, each block can use up
to 64 KB of shared memory for intra-block communication, and each thread may use
up to 255 local registers in which to store intermediate results.
Consideration of the hardware design suggests the following basic strategies
for maximizing the performance of GPU kernels.
1) Launch many threads, ideally one to two orders of magnitude more threads than
the GPU has execution cores. For example, a Tesla K20 with 2496 cores may not
reach peak performance until at least O(105) threads are launched. Having

21
thousands of processors will only be an advantage if they are all saturated with
work. All threads are hardware scheduled, making them very lightweight to create,
unlike host threads. Also, the streaming model ensures that the GPU will not
execute more threads than it can efficiently schedule. Thus oversubscription will
not throttle performance. Context switches are also instantaneous, and this is
beneficial because they allow a processor to stay busy when it might otherwise be
stalled, for example, waiting for a memory transaction to complete.
2) Keep each thread as simple as possible. Threads with smaller shared-memory and
register footprints can be packed more densely onto each SM. This allows the
schedulers to hide execution and memory latencies by increasing the chance that a
ready-to-execute warp will be available on any given clock cycle.
3) Decouple your algorithm to be as data parallel as possible. Synchronization
between threads always reduces the effective concurrency available to the GPU
schedulers and should be minimized. For example, it is often better to re-compute
intermediate quantities rather than build shared caches, sometimes even when the
intermediates require hundreds of cycles to compute.
4) Maintain regular memory access patterns. On the CPU this is done temporally
within a single thread, on the GPU it is more important to do it locally among
threads in a warp.
5) Maintain uniform control flow within a warp. Because of the SIMT execution
paradigm, all threads in the warp effectively execute every instruction needed by
any thread in the warp. Pre-organizing work by expected code-path can eliminate
divergent control flow within each warp and improve performance.

22
These strategies have well known analogues for CPU programming; however,
the performance penalty resulting from their violation is usually much more severe in
the case of the GPU. The tiny size of GPU caches relative to the large number of in-
flight threads defeats any possibility of cushioning the performance impact of non-
ideal programming patterns. In such cases, the task of optimization goes far beyond
simple FLOP minimization, and the programmer must consider tradeoffs from each of
the above considerations on his design.
GPU ERI EVALUATION
Parallelization Strategies
The primary challenge in implementing ERI routines on GPUs is deciding how
to map the integrals onto the GPUs execution units. Because each ERI can be
computed independently, there are many possible ways to decompose the work into
CUDA grids and blocks. For simplicity we will ignore the screening of negligible
integrals until the next chapter, and consider a simplified calculation in which each
AO is formed by a contraction of s-functions only. In this case, equation (1.43)
simplifies tremendously to the following expression.
[ij | kl]=Nij Nkl
!ij +!kl
F0
!ij!kl
!ij +!kl
Pij " Pkl( )2#
$%
&
'( (1.46)
Since we are now interested in integrals over contracted AO functions, we note that
the pair prefactors, Nij, now include the AO contraction coefficients.
A convenient way to organize ERI evaluation is to expand unique pairs of
atomic orbitals, !µ!" µ #"{ } , into a vector of dimension N(N-1)/2. The outer product

23
of this vector with itself then produces a matrix whose elements are quartets,
!µ!" ,!#!$ µ %" ,# %${ } , each representing a (bra|ket) contracted AO integral. This is
illustrated by the blue square in figure 1. Due to (bra|ket) = (ket|bra) symmetry among
the ERIs, only the upper triangle of the integral matrix need be computed. Clearly ERI
evaluation is embarrassingly parallel at the level of AOs. However, each AO integral
in the grid can include contributions from many primitive integrals. In order to
parallelize the calculation over the more finely grained primitives, a degree of
coordination must be introduced among threads. Here we review three broadly
representative decomposition schemes10,19 that will guide our work in later chapters.
Figure 1: Schematic of 1 Block – 1 Contracted Integral (1B1CI) mapping. Cyan squares on left represent contracted ERIs each mapped to the labeled CUDA block of 64 threads. Orange squares show mapping of primitive ERIs to CUDA threads (green and blue boxes, colored according to CUDA warp) for two representative integrals, the first a “contraction” over a single primitive ERI and the second involving 34=81 primitive contributions.
The first strategy assigns a CUDA block to evaluate each contracted AO ERI
and maps a 2-dimensional CUDA grid onto the 2D ERI grid. The threads within each
block work together to compute a contracted integral in parallel. This approach is
termed the one block – one contracted integral (1B1CI) scheme. It is illustrated in
figure 1. Each cyan square represents a contracted integral. The CUDA block
responsible for each contracted ERI is labeled within the square. Lower triangular
block(2, 0)
|11)
(11|
|12) |13) |22) |23) |33)
(12|
(13|
(22|
(23|
(33|
block(0, 0)
block(3, 3)
block(0, 5)
idle blocks
(0)Block (2, 0), Integral (11|13)
[22|33](1)
[idle](31)[idle]
(63)[idle]
(32)[idle]
(0)Block (3, 3), Integral (22|22)
[11|11](16)
[12|32](31)
[21|22](63)
[32|11](32)
[21|23][32|12] [33|33]
(17)[12|33][idle] [idle] [idle] [idle]

24
blocks, labeled idle in Figure 1, would compute redundant integrals due to
�bra�ket( ) = ket�bra( ) symmetry. These blocks exit immediately and, because of the
GPUs efficient thread scheduling, contribute negligibly to the overall execution time.
Each CUDA block is made up of 64 worker threads arranged in a single dimension.
Blocks are represented by orange rectangles in Figure 1. The primitive integrals are
mapped cyclically onto the threads, and each thread collects a partial sum in an on-
chip register. The first thread computes and accumulates integrals 1, 65, etc. while the
second thread handles integrals 2, 66, etc. After all primitive integrals have been
evaluated, a block level reduction produces the final contracted integral.
Two cases deserving particular consideration are illustrated in Figure 1. The
upper thread block shows what happens for very short contractions, in the extreme
case, a single primitive. Since there is only one primitive to compute, all threads other
than the first will sit idle. A similar situation arises in the second example. Here an
ERI is calculated over four AOs each with contraction length 3 for a total of 81
primitive integrals. In this case, none of the 64 threads are completely idle. However,
some load imbalance is still present, since the first 17 threads compute a second
integral while the remainder of the warp, threads 18-31, execute unproductive no-op
instructions. It should be noted that threads 32-63 do not perform wasted instructions
because the entire warp skips the second integral evaluation. Thus, “idle” CUDA
threads do not always map to idle execution units. Finally, as contractions lengthen,
load imbalance between threads in a block will become negligible in terms of the
runtime, making the 1B1CI strategy increasingly efficient.

25
Figure 2: Schematic of 1 Thread – 1 Contracted Integral (1T1CI) mapping. Cyan squares represent contracted ERIs and CUDA threads. Thread indices are shown in parentheses. Each CUDA block (red outlines) computes 16 ERIs with each thread accumulating the primitives of an independent contraction, in a local register.
A second parallelization strategy assigns entire contracted integrals to
individual CUDA threads. Since all primitives within a contraction are computed by a
single GPU thread, the sum of the final ERI can be accumulated in a local register,
avoiding the final reduction step. This coarser decomposition, which is termed the one
thread – one contracted integral (1T1CI) strategy, is illustrated in figure 2. The
contracted integrals are again represented by cyan squares, but each CUDA block,
represented by red outlines, now handles multiple contracted integrals rather than just
one. The 2-D blocks shown in Figure 2 are given dimensions 4x4 for illustrative
purposes. In practice, blocks sized at least 16x16 threads should be used. Because
threads within the same warp execute in SIMT fashion, warp divergence will result
whenever neighboring ERIs involve contractions of different lengths. To eliminate
these imbalances, the ERI grid must be pre-sorted by contraction length so that blocks
handle ERIs of uniform contraction length.
(0, 0) (0, 0)(3, 0)
(3, 3)
(1, 1)
(0, 0)
(1, 1)
(0, 3)
(0, 0) (3, 0)
(0, 3)
(12|13
)
|11)
(11|
|12) |13) |22) |23) |33)
(12|
(13|
(22|
(23|
(33| idle threads
block (0, 0)

26
Figure 3: Schematic of 1 Thread – 1 Primitive Integral (1T1PI) mapping. Cyan squares represent two-dimensional tiles of 16 x 16 primitive ERIs, each of which is assigned to a 16 x 16 CUDA block as labeled. Red lines indicate divisions between contracted ERIs. The orange box shows assignment of primitive ERIs to threads (grey squares) within a block that contains contributions to multiple contractions.
The third strategy maps each thread to a single primitive integral (1T1PI) and
ignores boundaries between primitives belonging to different AO contractions. A
second reduction step is then employed to sum the final contracted integrals from their
constituent primitive contributions. This is illustrated in Figure 3. The 1T1PI approach
provides the finest grained parallelism of the three mappings considered. It is similar
to the 1B1CI in that contracted ERIs are again broken up between multiple threads.
Here however, the primitives are distributed to CUDA blocks without considering the
contraction of which they are members. In Figure 3, cyan squares represent 2D CUDA
blocks of dimension 16x16, and red lines represent divisions between contracted
integrals. Because the block size is not an even multiple of contraction length, the
primitives computed within the same block will, in general, contribute to multiple
contracted ERIs. This approach results in perfect load balancing (for primitive
evaluation), since each thread does exactly the same amount of work. It is also notable
in that 1T1PI imposes few constraints on the ordering of primitive pairs, since they no
longer need to be grouped or interleaved by parent AO indices. However, the
|11)(1
1||12)
(12|
block(0, 0)
block(3, 3)
block(0, 5)
block(5, 5)
block(4, 1)
idle threads
Thread(0, 0)
[35|55]
Thread(7, 0)
[35|66]
Thread(0, 15)[62|55]
Thread(7, 15)[62|66]
Thread(8, 0)
[35|11]
Thread(15, 0)[35|22]
Thread(8, 15)[62|11]
Thread(15, 15)[62|22]
Block (4, 1) contributes to(11|12) and (11|13)

27
advantages of the 1T1PI scheme are greatly reduced if we also consider the
subsequent reduction step needed to produce the final contract ERIs. These reductions
involve inter-block communication and for highly contracted basis sets, can prove
more expensive than the ERI evaluation itself.
Table 1 summarizes benchmarks for each of the strategies described above.10
The example system consisted of 64 hydrogen atoms arranged in a 4x4x4 cubic lattice
with 0.74Å separating nearest neighbors. Two basis sets are considered. The 6-311G
basis represents a low contraction limit in which most (two-thirds) of the AOs include
a single primitive component. Here the 1T1PI mapping provides the best performance.
At such a minimal contraction level, very few ERIs must be accumulated between
block boundaries, minimizing required inter-block communication. The 1T1CI
method takes a close second since, for small contractions, it represents a parallel
decomposition that is only slightly coarser than the ideal 1T1PI scheme. The 1B1CI
scheme, on the other hand, takes a distant third. This is due to its poor load balancing
since, for the 6-311G basis, over 85% of the contracted ERIs involve nine or fewer
primitive ERI contributions. Thus, the vast majority of the 64 threads in each 1B1CI
CUDA block do no work.
GPU 1B1CI
GPU 1T1CI
GPU 1T1PI
CPU PQ Pre-calc
GPU-CPU Transfer GAMESS
6-311G 7.086 0.675 0.428 0.009 0.883 170.8 STO-6G 1.608 1.099 2.863 0.012 0.012 90.6
Table 1: Runtime comparison for evaluating ERIs of 64 H Atom lattice using 1B1CI, 1T1CI, and 1T1PI methods. Times are given in seconds. All GPU calculations were run on an Nvidia 8800 GTX. CPU pre-calculation records time required to build pair quantities prior to launching GPU kernels. GPU-CPU transfer provides the time required to copy the completed ERIs from device to host memory. Timings for the CPU-based GAMESS program running on an Opteron 175 CPU are included for comparison.

28
The STO-6G basis provides a sharp contrast. Here each contracted ERI
includes 64 or 1296 primitive ERI contributions. As a result for the 1T1PI scheme, the
reduction step becomes much more involved, in fact requiring more time than
primitive ERI evaluation itself. This illustrates that, for massively parallel
architectures, organizing communication is often just as important as minimizing
arithmetic instructions when optimizing performance. The 1T1CI scheme performs
similarly to the 6-311G case. The fact that all ERIs now involve uniform contraction
lengths provides a slight boost, since it requires only 60% more time to compute twice
as many primitive ERIs compared to the 6-311G basis. The 1B1CI method, improves
dramatically, as every thread of every block is now responsible for at least 20
primitive ERIs.
Finally, it should be noted that simply transferring ERIs between host and
device can take longer than the ERI evaluation itself, especially in the case of low
basis set contraction. This means that, for efficient GPU implementations, ERIs can be
re-evaluated from scratch faster than they can be fetched from host memory, and much
faster than they can be fetched from disk. This will prove an important consideration
in the next chapter.
Extension to Higher Angular Momentum
Some additional considerations are important for extension to basis functions
of higher angular momentum. For non-zero angular momentum functions, shells
contain multiple functions. As noted above, all integrals within an ERI shell depend on
the same auxiliary integrals, Rtuv0 , and Hermite contraction coefficients, xEt
mn . Thus, it
is advantageous to have each thread compute an entire shell of primitive integrals. For

29
example, a thread computing a primitive ERI of class [sp|sp] is responsible for a total
of nine integrals.
The performance of GPU kernels is quite sensitive to the register footprint of
each thread. As threads use more memory, the total number of concurrent threads
resident on each SM decreases. Fewer active threads, in turn, reduce the GPU’s ability
to hide execution latencies and lowers throughput performance. Because all threads in
a grid reserve the same register footprint, a single grid handling both low and, more
complex, high angular momentum integrals will apply the worst-case memory
requirements to all threads. To avoid this, separate kernels must be written for each
class of integral.
Specialized kernels also provide opportunities to further optimize each routine
and reduce memory usage, for example, by unrolling loops or eliminating
conditionals. This is particularly important for ERIs involving d- and higher angular
momentum functions, where loop overheads become non-trivial. For high angular
momentum integrals it is also possible to use symbolic algebra libraries to generate
unrolled kernels that are optimized for the GPU.20
Given a basis set of mixed angular momentum shells, we could naively extend
any of the decomposition strategies presented above as follows. First, build the pair
quantities as prescribed, without consideration for angular momentum class. Then
launch a series of ERI kernels, one for each momentum class, assigning a compute
unit (either block or thread depending on strategy being extended) to every ERI in the
grid. Work units assigned to ERIs that do not apply to the appropriate momentum
class could exit immediately. This strategy is illustrated for a hypothetical system

30
containing four s-shells and one p-shell in the left side of Figure 4. Each square
represents a shell quartet of ERIs, that is all ERIs resulting from combination of the
various angular momentum functions within each of the included AO shells. The
elements are colored by total angular momentum class, and a specialized kernel
evaluates elements of each color. Unfortunately, the number of integral classes
increases rapidly with the maximum angular momentum in the system. The inclusion
of d-shells would already result in the vast majority of the threads in each kernel
exiting without doing any work.
Figure 4: ERI grids colored by angular momentum class for a system containing four s-shells and one p-shell. Each square represents all ERIs for a shell quartet (a) Grid when bra and ket pairs are ordered by simple loops over shells. (b) ERI grid for same system with bra and ket pairs sorted by angular momentum, ss, then sp, then pp. Each integral class now handles a contiguous chunk of the total ERI grid.
A better approach is illustrated on the right side of Figure 4. Here we have
sorted the bra and ket pairs by the angular momenta of their constituents, ss then sp
and last pp. As a result, the ERIs of each class are localized in contiguous sub-grids,
and kernels can be dimensioned to exactly cover only the relevant integrals.
REFERENCES
(1) Challacombe, M.; Schwegler, E. J. Chem. Phys. 1997, 106, 5526.
(2) Burant, J. C.; Scuseria, G. E.; Frisch, M. J. J. Chem. Phys. 1996, 105, 8969.
11 12 13 14 15 22 23 24 25 33 34 35 44 45 55
1112
1314
1522
2324
2533
3435
4445
55
11 12 13 14 22 23 24 33 34 44 15 25 35 45 55
1112
1314
2223
2433
3444
1525
3545
55
|λσ)
(μν|
|λσ)
(μν|
(b)(a)

31
(3) Schwegler, E.; Challacombe, M. J. Chem. Phys. 1996, 105, 2726.
(4) Schwegler, E.; Challacombe, M.; HeadGordon, M. J. Chem. Phys. 1997, 106, 9708.
(5) Ochsenfeld, C.; White, C. A.; Head-Gordon, M. J. Chem. Phys. 1998, 109, 1663.
(6) Rudberg, E.; Rubensson, E. H. J Phys-Condens Mat 2011, 23.
(7) Rys, J.; Dupuis, M.; King, H. F. J. Comp. Chem. 1983, 4, 154.
(8) Yasuda, K. J. Comp. Chem. 2008, 29, 334.
(9) Asadchev, A.; Allada, V.; Felder, J.; Bode, B. M.; Gordon, M. S.; Windus, T. L. J. Chem. Theo. Comp. 2010, 6, 696.
(10) Ufimtsev, I. S.; Martinez, T. J. J. Chem. Theo. Comp. 2008, 4, 222.
(11) Mcmurchie, L. E.; Davidson, E. R. J. Comp. Phys. 1978, 26, 218.
(12) Parr, R. G.; Yang, W. Density-functional theory of atoms and molecules; Oxford University Press: Oxford, 1989.
(13) Szabo, A.; Ostlund, N. S. Modern Quantum Chemistry; McGraw Hill: New York, 1982.
(14) Helgaker, T.; Jørgensen, P.; Olsen, J. Molecular electronic-structure theory; Wiley: New York, 2000.
(15) Kohn, W.; Sham, L. J. Phys Rev 1965, 140, 1133.
(16) Boys, S. F. Proc. Roy. Soc. Lon. A 1950, 200, 542.
(17) Whitten, J. L. J. Chem. Phys. 1973, 58, 4496.
(18) NVIDIA In Design Guide; NVIDIA Corporation: docs.nvidia.com, 2013.
(19) Ufimtsev, I. S.; Martinez, T. J. Comp. Sci. Eng. 2008, 10, 26.
(20) Titov, A. V.; Ufimtsev, I. S.; Luehr, N.; Martinez, T. J. J. Chem. Theo. Comp. 2013, 9, 213.

32

33
CHAPTER TWO
INTEGRAL-DIRECT FOCK CONSTRUCTION
ON GRAPHICAL PROCESSING UNITS
Because ERIs remain constant from one SCF iteration to the next, it was once
common practice to pre-compute all numerically significant ERIs prior to the SCF. At
each iteration the two-electron Fock contributions of equation (2.1) would then be
generated from contracted ERIs, �µ!�"#( ) , stored, for example, on disk.
�Gµ! (P) = P"#
"#
N
$ 2 µ!�"#( )% µ"�!#( )&' () (2.1)
This procedure certainly minimized the floating-point operations involved in the
calculation. However, for systems containing thousands of basis functions, ERI
storage quickly becomes impractical. The integral-direct approach, pioneered by
Almlof,1 avoids the storage of ERIs by re-computing them on the fly with each
formation of the Fock matrix.
ERI evaluation represents a tremendous bottleneck in the integral-direct
approach. Thus, early implementations were careful to generate only symmetry-unique
ERIs, �µ!�"#( ) where µ !" , ! "# , and µ! " #$ . Here µ! and !" are compound
indices corresponding to the element numbers in an upper triangular matrix. Each ERI
was then combined with various density elements and scattered into multiple locations
in the Fock matrix. This reduces the number of ERIs that must be evaluated by a factor
of eight compared to a naïve implementation (based on the eightfold symmetry among
the ERIs).

34
Beyond alleviating storage capacity bottlenecks, the direct approach offers
performance advantages over conventional algorithms based on integral storage. As
observed above, ERIs can sometimes be re-calculated faster than they can be recalled
from storage (even when this storage is just across a fast PCIe bus). As advances in
instruction throughput continue to out pace those for communication bandwidths, this
balance will shift even further in favor of integral-direct algorithms. Another
advantage results from knowledge of the density matrix during Fock construction.1 By
augmenting the usual Schwarz bound with density matrix elements as follows,
�µ!�"#( )P"# $ µ!�µ!( )1/2
"#�"#( )1/2P"# (2.2)
the direct approach is able to eliminate many more integrals than is possible for pre-
computed ERIs since even quite large integrals are often multiplied by vanishing
density matrix elements.
Almlof and Ahmadi also suggested dividing the calculation of G into separate
Coulomb, J, and exchange, K, contributions, double calculating any ERIs that are
common to both.2
�Jµ! = µ!�"#( )P"#
"#
N
$ (2.3)
�Kµ! = µ"�!#( )P"#
"#
N
$ (2.4)
This division offers two primary advantages. First, for the Coulomb operator in
equation (2.3), the density elements, P!" , can be pre-contracted with the ket, !" ) .
This provides an important optimization as described later in this chapter. Second for
the exchange operator in equation (2.4) only a few non-negligible contributions need

35
to be computed. The density matrix in insulating systems with finite band gap decays
exponentially with distance.3,4 Because Gaussian basis functions are localized, the AO
density matrix remains sparse. As noted in the previous chapter, the bra, µ! , and ket,
!" , pairs are also sparse due to the locality of the Gaussian basis set. Thus, equation
(2.4) couples the bra and ket through a sparse density matrix, and few ERI
contributions survive screening in large systems. Separately calculating the exchange
term in equation (2.4) then adds few ERIs compared to the number of ERIs required
by equation (2.3) alone.
The considerations above apply perhaps even more forcefully on GPUs. As
already observed in the context of forming contracted ERIs, the GPU’s wide execution
units benefit from longer contractions of primitive ERIs. In the previous chapter, this
explained the improved performance of the 1B1CI approach for the hydrogen lattice
test case in moving from the 6-311G basis to the more highly contracted STO-6G.
Longer contractions parallelize more evenly across many cores. Expanded in terms of
primitive ERIs, the sums in Eqs. (2.3) and (2.4) include many more contributions than
any contracted ERI considered in chapter 1. Thus to simplify the parallel structure of
the Coulomb and exchange algorithms and improve GPU performance, the
construction of contracted ERIs, �µ!�"#( ) , is abandoned in the present chapter in
favor of direct construction of Coulomb and exchange matrix elements from primitive
Gaussian functions.

36
Even within the Coulomb and exchange operators, ERI symmetry must not be
taken for granted. For example, although each ERI, �µ!�"#( ) , makes multiple
contributions to the exchange matrix,
�
µ!�"#( )P!# $ Kµ" "#�µ!( )P#! $ K"µ
!µ�"#( )Pµ# $ K!" "#�!µ( )P#µ $ K"!
µ!�#"( )P!" $ Kµ# #"�µ!( )P"! $ K#µ
!µ�#"( )Pµ" $ K!# #"�!µ( )P"µ $ K#!
(2.4)
gathering disparate density matrix elements introduces irregular memory access
patterns and scattering outputs to the Fock matrix creates dependencies between
threads computing different ERIs. GPU performance is extremely sensitive to these
considerations, so that even the eight-fold reduction in work available from exploiting
the full symmetry among ERIs could be swamped by an even larger performance
slowdown resulting from fragmented memory accesses. It is helpful to start, as below,
from a naïve, but completely parallel algorithm, and then exploit ERI symmetry only
where it provides a practical benefit.
The remainder of the chapter describes the algorithm used to implement
integral-direct Coulomb and exchange operators in TeraChem. A final performance
evaluation will be delayed until the implementation of DFT exchange-correlation
terms have also been described in chapter 4.
GPU J-ENGINE
The strategies for handling ERIs developed in the previous chapter provide a
good starting point for the evaluation of the Coulomb operator in equation (2.3).5 As

37
in the previous chapter, we first consider ERIs involving only s-functions in which
each quartet of AO functions produces a single integral. This provides a clear context
in which to describe the overall structure of our approach. Later, details for evaluating
higher angular momentum functions will be provided. The first step is again to
enumerate AO pairs, !µ!" , for the bra and ket. For the moment we consider the full
lists of N2 function pairs and consider the integral matrix, I, constructed as the N2-by-
N2 product of the bra-pair column vector, µ! |( , with the ket-pair row vector, | !" ) .
Iµ! ,"# = µ! | "#( ) (2.5)
Inserting equation (2.5) into (2.3) casts the Coulomb operator as a matrix vector
product between the integral matrix and a vector of length N2 built by re-dimensioning
the usual N-by-N one-particle density matrix.
With this picture in mind, several plausible mappings to CUDA threads and
blocks suggest themselves. A simple strategy is to assign each thread to a single bra-
pair, µ! |( , and have it sweep over all kets, | !" ) , and density elements, P!" ,
accumulating the products, �µ!�"#( )P#" , to compute an independent Coulomb
element, Jµ! . This strategy is similar to the 1T1CI scheme of chapter 1 and maximizes
the independence of each thread but at the cost of rather coarse parallelism. In order to
saturate a massively parallel GPU (or ideally several GPUs) it is preferable to employ
multiple threads to compute each Jµ! . Thus a preferred approach uses 2D thread
blocks so that threads in each row stride across the integral matrix, each accumulating
a partial sum. This is shown in figure 1 for an illustrative block size of 2x2. In practice

38
a block size of 8x8 was shown to be near optimal across a range of empirical test
calculations. Once all integrals have been evaluated, a final reduction within each row
of the CUDA block produces final Jµ! elements. The reduction step adds negligibly to
the runtime regardless of primitive contraction length, because it is performed only
once per Coulomb element, rather than once per contracted ERI as in the 1B1CI case
described in the previous chapter.
Figure 1: Schematic representation of a J-Engine kernel for one angular momentum class, e.g., (ss|ss). Cyan squares represent significant ERI contributions. Sorted bra and ket vectors are represented by triangles to left and above grid. The path of a 2x2 block as it sweeps across the grid is shown in orange. The final reduction across rows of the block is illustrated within the inset to the right.
The above discussion ignored ERI symmetry for clarity. However, having
determined an efficient mapping of the Coulomb problem onto the GPU execution
units, it is important to consider what symmetries can be exploited without upsetting
the structure of the algorithm. We first note that Jµ! is symmetric. Thus it is sufficient

39
to compute its upper triangle and only the N(N+1)/2 bra pairs where µ !" need to be
considered. Similarly for ket pairs, the terms �µ!�"#( )P"# and �
µ!�"#( )P"# can be
computed together as �µ!�"#( ) P"# + P#"
1+$"#
, where the Kronecker delta is used to handle
the special case along the diagonal of the density matrix. Thus, using a slightly
transformed density, ERI symmetry again allows a reduction to ket pairs where ! "# .
If the AO shells are ordered by angular momentum, as suggested in the
previous chapter, then these symmetry reductions will also conveniently reduce the
number of specialized momentum-specific kernels that are needed. For example,
assuming a basis including s-, p-, and d-functions, the reduced pairs will include a
total of six momentum classes, ss, sp, sd, pp, pd, and dd, and require 36 specialized
Coulomb kernels, less than half of the 34 = 81 total momentum classes that might be
expected.
It is not possible to exploit the final class of ERI symmetry,
�µ!�"#( ) = "#�µ!( ) , without creating dependencies between rows of the ERI grid
and, as a result, performance sapping inter-block communication. Thus, ignoring the
screening of negligibly small integrals discussed below, the GPU J-Engine nominally
computes N4/4 integrals.
For clarity, the discussion of ERI symmetry has been carried out in terms of
contracted ERIs. However, building such contracted intermediates would require
many sums of irregular length that are difficult to parallelize efficiently across many
cores. Thus it is advantageous to construct the Coulomb operator directly from

40
primitive ERIs. Continuing with s-functions for the moment, the primitive Coulomb
matrix elements are calculated as follows.
�Jij = ci c j ij�kl!" #$ck cl P%k%l
kl& (2.6)
Here we define the AO index vector, !i , to select the contracted AO index to which
the ith primitive function belongs. (Since at present our discussion is limited to s-
functions, we ignore the structure of functions organized within shells). The
coefficients, c j , represent weights of primitive functions within AO contractions. The
final Coulomb elements are then computed in a second summation step as follows.
Jµ! = Jiji"µj"!
# (2.6)
The evaluation of equation (2.6) can be carried out as described above, except
that the bra and ket AO pairs, !µ!" , are now replaced by expanded sets of primitive
pairs, ! i! j | i "µ, j "# ,µ $#{ } . The bra prefactor from equation (1.44) is now
augmented with the contraction coefficients.
Nijbra = cicjNij (2.7)
In constructing the ket pairs, the prefactor is pre-multiplied by both the contraction
coefficients and the appropriate density matrix element.
Nklket = ckclNkl
P!k!l + P!l!k1+"!k!l
(2.8)
Along with these prefactors, the quantities !ij and !Pij from equation (1.25) and each
pair’s Schwarz contribution, Bijbra = ij | ij[ ]1/2 and Bkl
ket = kl | kl[ ]1/2 P!k!l for each bra and

41
ket pair, is transferred to the GPU. As illustrated in figure 1, a CUDA kernel then
processes the primitive ERI grid formed as the outer product of bra and ket pair arrays.
The resulting primitive Coulomb elements are returned to the host where the sum of
equation (2.6) is carried out to provide the final matrix elements.
As noted above, screening small ERIs can reduce the computational
complexity of Coulomb construction from O(N4) to O(N2). Screening is introduced in
three passes. During the initial enumeration of primitive pairs, a conservative bound is
used to remove all primitive pairs for which Bijbra < ! pair "10#15 Hartrees.a Next, when
building the bra and ket arrays, pairs are further filtered that satisfy equations (2.9) and
(2.10), respectively.
Bijbra ! " screen
maxklBklket (2.9)
Bklket ! " screen
maxijBijbra (2.10)
Here 10-12 is a typical value of ! screen .b Finally, prior to evaluating each ERI, the GPU
kernel evaluates the four-center density-weighted Schwarz bound, and the
computationally intensive ERI evaluation is skipped where the following holds.
�BijbraBkl
ket = ij�ij( )1/2 kl�kl( )1/2 P!" # $ screen (2.11)
a ! pair corresponds to the variable THREPRE in TeraChem. b ! screen corresponds to the variable THRECL in TeraChem.

42
Figure 2: Organization of ERIs for Coulomb formation. Rows and columns correspond to primitive bra and ket pairs respectively. Each ERI is colored according to the magnitude of its Schwarz bound. Data is derived from calculation on ethane molecule. Left grid is obtained by arbitrary ordering of pairs within each angular momentum class and suffers from load imbalance because large and small integrals are computed in neighboring cells. Right grid sorts bra and ket primitives by Schwarz contribution within each momentum class, providing an efficient structure for parallel evaluation.
The left half of figure 2 shows a typical distribution of Schwarz bounds for
ERIs in the primitive Coulomb grid. Merely skipping small integrals still requires
evaluating the bound of every cell in this grid. Also, because large and small integrals
are interspersed throughout the grid, CUDA warps will suffer from divergence
between threads that are evaluating an ERI and threads that are not. A more efficient
approach to screening can be achieved by sorting the final lists of bra and ket pair
quantities in descending order by Bijbra and Bkl
ket , respectively. The resulting grid,
shown on the right in figure 2, eliminates warp divergence because the significant
integrals are condensed in a contiguous region for each momentum class of ERI.
Furthermore, the bounds are now guaranteed to decrease across each row, so each
block may safely exit when the first negligible ERI is located. This eliminates the
overhead of even checking the Schwarz bound for negligible quartets.

43
For clarity, the discussion above has considered only s-functions. For integrals
involving higher angular momentum functions, it is advantageous to compute all ERIs
belonging to a shell quartet simultaneously. In order to maintain fine-grained
parallelism, it would be desirable to distribute a shell quartet among threads in a block.
However, ERI evaluation involves extensive recurrence relations such as equation
(1.42) which cannot be efficiently parallelized between many SIMT processing cores.
Thus, it is preferable to assign an independent thread to compute all ERIs within a
shell quartet. The final algorithm follows the basic pattern described for s-functions,
except that instead of function pairs, the bra and ket vectors are now built from
primitive shell pairs, ! I! J .
The quantities NIJbra , !IJ , and
!PIJ are uniform for all functions within the shell
pair and are thus organized in pair data arrays as above. The Schwarz contributions,
Bijbra/ket , are not strictly uniform for d-type functions and above. However, to maintain
rotational invariance, the Schwarz bound is computed treating both primitives as
spherical s-functions, a quantity which is uniform across the shell pair. Because each
shell pair now spans several functions, the ket bound, Bklket , must use the maximum
density element over the shell block.
BKLket = KL |KL[ ]1/2 PKLmax (2.12)
PKLmax = max
!k"!K
!l"!L
P#k#l (2.12)
Additional pair data is required to compute ERIs of non-zero angular
momentum. Since the density elements differ for each function within the shell block

44
they can no longer be included in the ket prefactor, Nijket , as in equation (2.8). The
Hermite expansion coefficients, Etuvij , from equation (1.33) are also needed for every
function pair ij in the shell pair IJ.
An important simplification both in terms of the required pair data and
computational cost of the final integral is available by inserting equation (1.43) into
equation (2.3).2
Jµ! = Nijci c ji"µj"!
# Etuvij Nklck cl
$ij +$klkl# (%1) &t + &u + &v P'k'l
E &t &u &vkl Rt+ &t ,u+ &u ,v+ &v
0
&t &u &v#
tuv#
(2.12)
Because the quantities Nkl, ck cl, and !kl are uniform for all function pairs within the
shell pair, the above sum can be segmented by primitive shell pair, KL, as follows.
Jµ! = Nijci c ji"µj"!
# Etuvij N KLcKcL
$ij +$KLKL# D %t %u %v
KL Rt+ %t ,u+ %u ,v+ %v0
%t %u %v#
tuv#
(2.12)
D !t !u !vKL = ("1) !t + !u + !v P#k#l
E !t !u !vkl
k$Kl$L
% (2.12)
Rather than separately packing McMurchie-Davidson coefficients and density
elements for every primitive function pair, combined coefficients are generated
through equation (2.12) for each pair of ket shells.
Equation (2.12) is computed in two steps in TeraChem. First the pair data
described above is copied to the GPU where the following Hermite Coulomb
contributions are computed.
Jtuvij =
Nijci c j N KLcKcL
!ij +!KLKL" D #t #u #v
KL Rt+ #t ,u+ #u ,v+ #v0
#t #u #v"
(2.12)

45
In exact analogy to the algorithm described for s-functions above, each row of CUDA
threads is assigned a bra shell pair, !I! J
, and strides across the list of kets,
accumulating all associated Jtuvij
values in separate registers. After all significant
Coulomb contribions have been computed, reductions within the rows of each block
produce the sums of equation (2.12). The Hermite Coulomb contributions are then
copied back to host memory where, in analogy to equation (2.6) the bra expansion
coefficients are used to produce the final Coulomb elements as follows.
Jµ! = Etuvij Jtuv
ij
tuv"
i#µj#!
" (2.12)
GPU K-ENGINE
The GPU-based construction of the exchange operator in equation (2.4)
follows a similar approach to that employed in the GPU J-Engine algorithm.5 Here we
highlight adjustments that are required to accommodate the increased complexity of
exchange which results from the splitting of the output index, µ! , between the bra
and ket. As a result rows of the bra-by-ket ERI grid do not contribute to a single
matrix element but scatter across an entire row of the K matrix. Additionally,
symmetry among the ERIs is more difficult to exploit for exchange since symmetric
pairs, e.g., �µ!�"#( )$ µ!�#"( ) , now contribute to multiple matrix elements. The
split of the density index, !" , between bra and ket, also precludes the pre-contraction
of density elements into the pair quantities as was done for the ket pairs in the J-
Engine above.

46
Such complications could be naïvely removed by changing the definitions of
bra and ket to so called physicist notation, where pairs include a primitive from each
of two electrons.
�µ!�"#( ) = µ" !# (2.13)
With such µ! and !" pairs, a GPU algorithm analogous to the J-Engine could easily
be developed. Unfortunately, the new definitions of bra and ket also affect the
pairwise Schwarz bound, which now becomes the following.
�µ! µ!
1/2"# "#
1/2= µµ�!!( )1/2
""�##( )1/2 (2.14)
As the distance, R , between !µ and !" increases, the quantity �µµ�!!( ) decays
slowly, as 1/ R . This should be compared to a decay of e!R2
for �µ!�µ!( ) . This
weaker bound means that essentially all N4 ERIs would need to be examined leading
to a severe performance reduction compared to the N2 Coulomb algorithm. Thus, the
scaling advantages of the µ! /!" pairing are very much worth maintaining, even at
the cost of reduced hardware efficiency.
The K-Engine begins with the usual step of enumerating AO shell pairs, !µ!" .
Because the !µ!" and !"!µ pairs contribute to different matrix elements, we neglect
symmetry and construct AO pairs for both µ ! " and µ > ! pairs. As with Coulomb
evaluation, the pairs are separated by angular momentum class, and different kernels
are tuned for each type of integral. Inclusion of µ > ! pairs requires additional pair
and kernel classes compared to the J-Engine, since, for example, kernels handling ps
pairs are distinct from those handling sp pairs.

47
Simply sorting the bra and ket primitive pairs by Schwarz bound would leave
contributions to individual K elements scattered throughout the ERI grid. In order to
avoid inter-block communication, it is necessary to localize these exchange
contributions. This is accomplished by sorting the primitive bra and ket pairs by µ -
and ! -index, respectively. Then the ERIs contributing to each element, Kµ! , form a
contiguous tile in the ERI grid. This is illustrated in figure 3. Within each segment of
µ -pairs, primitive PQs are additionally sorted by Schwarz contributions, �µ!�µ!( )1/2
,
so that significant integrals are concentrated in the top left of each µ! -tile.
Since the density index is split between bra and ket, density information cannot
be included in the primitive expansion coefficients or Schwarz contributions. Instead
additional vectors are packed for each angular momentum class, Pss , Psp , etc. The
packed density is ordered by shell pair and organized into CUDA vector types to allow
for efficient fetching. For example, each entry in the Psp block contains three
elements, spx , spy , and spz , packed into a float4 (assuming single precision) vector
type. The maximum overall density element in a shell pair, P!"
max is also pre-
computed for each shell pair, in order to minimize memory access when computing
the exchange Schwarz bound.
�!µ!"�!# !$%& '(P"$ ) !µ!"�!µ!"
%& '(1/2
!# !$�!# !$%& '(1/2
P"$
max (2.15)

48
Figure 3: Schematic of a K-Engine kernel. Bra and ket pair arrays are represented by triangles to left and above grid. The pairs are grouped by µ and ! index and then sorted by bound. The paths of four blocks are shown in orange, with the zigzag pattern illustrated by arrows in the top right. The final reduction of an exchange element within a 2x2 block is shown to the right.
To map the exchange calcuation to the GPU, a 2-D CUDA grid is employed in
which each block computes a single Kµ! element, and is thus assigned to a tile of the
primitive ERI grid. Each CUDA block passes through its tile of � µ!�!!"# $% primitive
integrals in a zigzag pattern, computing one primitive shell per thread. Ordering pairs
by bound allows a block to skip to the next row as soon as the boundary of
insignificant integrals is located. As with the J-Engine, the uniformity of ERI bounds
within each block is maximized by dimensioning square 2-D CUDA blocks. Figure 3
shows a 2x2 block for illustrative purposes, a block dimension of at least 8x8 is used
in practice. When all significant ERIs have been evaluated, a block-level reduction is

49
used to compute the final exchange matrix element. As with the J-Engine above, this
final reduction represents the only inter-thread communication required by the K-
Engine algorithm and detracts negligibly from the overall performance. The K-Engine
approach is similar to the 1B1CI ERI scheme described in chapter 1. However,
because each block is responsible for thousands of primitive ERIs, exchange
evaluation does not suffer from the load imbalance observed for the 1B1CI algorithm.
The structure of the ERI grid allows neighboring threads to fetch contiguous
elements of bra and ket data. Access to the density matrix is more erratic, because it is
not possible to order pairs within each µ!-tile simultaneously by Schwarz bound and "
or # index. As a result, neighboring threads must issue independent density fetches
that are in principle scattered across the density matrix. In practice, once screening is
applied, most significant contributions to an element, Kµ!, arise from localized "#-
blocks, where " is near µ and # is near !. Thus, CUDA textures can be used to
ameliorate the performance penalty imposed by the GPU on non-sequential density
access.
Another difficulty related to the irregular access of density elements is that the
density weighted Schwarz bounds for primitive ERIs calculated through equation
(2.15) are not strictly decreasing across each row or down each column of a µ!-tile.
As a result, the boundary between significant and negligible ERIs is not as sharply
defined as in the Coulomb case. Yet, using the density to preempt integral evaluation
in exchange is critical. This can be appreciated by comparison with the Coulomb
operator in which the density couples only to the ket pair. Since in general both the

50
density element, P!" , and Schwarz bound, �!"�!"( )1/2
, decrease as the distance r!"
increases, density weighting the Coulomb Schwarz bound has the effect of making
already small �!"�!"( )1/2
terms even smaller. In exchange, on the other hand, the
density couples the bra and ket so that small density matrix elements can reduce
otherwise large bounds and greatly reduce the number of ERIs that need to be
evaluated. In fact, for large insulator systems, we expect the total number of non-
negligible ERIs to reduce from N2 Coulomb integrals to just N exchange ERIs.
In order to incorporate the density into the ERI termination condition, the usual
exit threshold, !screen ,a is augmented by an additional guard multiplier, G=~10-3.b Each
warp of 32 threads then terminates ERI evaluation across each row of the ERI-tile
when it reaches a contiguous set of primitive ERIs where the following holds for every
thread.
�!µ!"�!µ!"#$ %&
1/2!' !(�!' !(#$ %&
1/2P"(
max< G) (2.16)
In principle, this non-rigorous exit condition could neglect significant integrals in
worst-case scenarios. However, in practice, empirical tests demonstrate that the exit
procedure produces the same result obtained when density information is not
exploited.
For SCF calculations, the density and exchange matrices are symmetric. In this
case, we need only compute the upper triangle of matrix elements. This amounts to
exploiting �µ!�"#( )$ "#�µ!( ) ERI symmetry and means that we calculate N2/2
a ! screen corresponds to the variable THREEX in TeraChem. b G corresponds to the variable KGUARD in TeraChem.

51
ERIs (in the absence of screening). This is four times more ERIs than are generated by
traditional CPU codes that take full advantage of the eight-fold ERI symmetry.
However, comparisons by instruction count are too simplistic when analyzing the
performance of massively parallel architectures. In the present case, freeing the code
of inter-block dependencies boosts GPU performance more than enough to
compensate for a four-fold work increase.
Because the AO density matrix elements, Pµ! , in insulating systems rapidly
decays to zero with increasing distance, rµ! , it is possible to pre-screen entire
exchange elements, Kµ! , for which the sum in equation (2.4) will be negligibly small.
A rigorous bound on each exchange matrix element can be evaluated using the
Schwarz inequality as follows.6
�Kµ! = µ"�!#( )P"#
"#
N
$ % µ"�µ"( )1/2P"# !#�!#( )1/2
"#
N
$ (2.17)
Or casting the AO Schwarz bounds in matrix form, �Qµ! = µ!�µ!( )1/2
:
Kµ! " (QPQ)µ! (2.18)
Whenever !Rµ is far from
!R! , P!" will be zero for all !" near !µ and !" near !" . In
practice, it is possible to impose a simple distance cutoff so that Kµ! is approximated
as follows.
�
Kµ! =µ"�!#( )P"#
"#
N
$ if rµ! < RMASK
0 otherwise
%
&'
('
(2.19)

52
The mask condition is trivially pre-computed for each element, Kµ! , and packed in a
bit mask, 1 indicating that the exchange element should be evaluated and 0 indicating
that it should be skipped. Each block of CUDA threads checks its mask at the
beginning of the exchange kernel, and blocks that are assigned a zero bit exit
immediately. As will be shown for practical calculations in chapter 4, this simple
distance mask accelerates the K-Engine algorithm to scale essentially linearly with the
size of the system without introducing book keeping overhead7-10 that could easily
reduce the K-Engine’s overall efficiency.
REFERENCES
(1) Almlof, J.; Faegri, K.; Korsell, K. J. Comp. Chem. 1982, 3, 385.
(2) Ahmadi, G. R.; Almlof, J. Chem. Phys. Lett. 1995, 246, 364.
(3) Kohn, W. Phys Rev 1959, 115, 809.
(4) des Cloizeaux, J. Phys Rev 1964, 135, A685.
(5) Ufimtsev, I. S.; Martinez, T. J. J. Chem. Theo. Comp. 2009, 5, 1004.
(6) Kussmann, J.; Ochsenfeld, C. J. Chem. Phys. 2013, 138.
(7) Burant, J. C.; Scuseria, G. E.; Frisch, M. J. J. Chem. Phys. 1996, 105, 8969.
(8) Ochsenfeld, C.; White, C. A.; Head-Gordon, M. J. Chem. Phys. 1998, 109, 1663.
(9) Schwegler, E.; Challacombe, M. J. Chem. Phys. 1999, 111, 6223.
(10) Neese, F.; Wennmohs, F.; Hansen, A.; Becker, U. Chem. Phys. 2009, 356, 98.

! "#!
CHAPTER THREE
DYNAMIC PRECISION ERI EVALUATIONa
$%&'!()*+*,-../!01'*+,10!-'!2(,'341)!5*01(!+-41!6-)07-)1!6-51!'6(7,!
8)141,0(3'! 9(81,8*-.! :()! 4-,/! 8-';'! *,! 2(4938-8*(,-.! 2614*'8)/<! *,2.30*,+!
1.128)(,*2! '8)3283)1! 861()/<=>=?! !"# $%$&$'# 4(.123.-)! 0/,-4*2'<==! -,0! 149*)*2-.!
:()21>:*1.0! @-'10! 4(.123.-)! 0/,-4*2'AB<=C>=D! E61! 141)+1,21! (:! 861! F&GH!
0151.(941,8! :)-417();! :)(4! IJKGKH! 6-'! '*49.*:*10! 861! )193)9('*,+! (:! 86*'!
6-)07-)1! :()! '2*1,8*:*2! 2(4938*,+<="! 2(49-)10! 8(! 1-)./! 1::()8'! (,! '*4*.-)!
-)26*81283)1'!86-8!6-0!)1.*10!(,!1L9.*2*8./!)12-'8*,+!'2*1,8*:*2!-.+()*864'!*,!81)4'!
(:! .(7! .151.! +)-96*2'! *,'8)328*(,'AB<=M! I151)861.1''<! @12-3'1! $%&'! 6-51! @11,!
2-)1:3../! 01'*+,10! :()! 4-L*434! 91):()4-,21! *,! '912*:*2! +)-96*2'! 9)(21''*,+!
8-';'<! *8! *'! '8*..! *49()8-,8! 761,! -0-98*,+! '2*1,8*:*2! 2(01'! 8(! )3,! (,! $%&'! 8(!
)1'9128! '912*-.*N10! 6-)07-)1! 2(,'8)-*,8'! '326! -'!414()/! -221''! 9-881),'! -,0!
,(,>3,*:()4! 1::*2*1,2/! (:! :.(-8*,+! 9(*,8! -)*86418*2! *,! 0*::1)1,8! 9)12*'*(,A! K8! *'!
3,.*;1./!86-8!861'1!.*4*8-8*(,'!7*..!151)!@1!:3../!1.*4*,-810!'*,21!861/!9)(5*01!861!
:(3,0-8*(,! (:! 861! $%&'! 2(4938-8*(,-.! 9)(71''A! $%&! 6-)07-)1! 1L21.'! *,!
4-''*51./!9-)-..1.! :.(-8*,+>9(*,8! 2(4938-8*(,! *,!9-)8!@12-3'1! *8! *'! +((0!-8! .*88.1!
1.'1A!
E61! F(3.(4@! -,0! OL26-,+1! -.+()*864'! 0151.(910! *,! 26-981)! C! 6-51<! (:!
2(3)'1<!-.)1-0/!@11,!2-)1:3../!01'*+,10!8(!4-L*4*N1!:*,1>+)-*,10!9-)-..1.*'4!-,0!
!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!-!H0-9810!7*86!91)4*''*(,!:)(4!IA!P316)<!KAQA!&:*48'15<!-,0!EARA!S-)8*,1N<!()#*+,-)#.+,')#*'-/0&A!!"##<!T<!UDU>U"DA!F(9/)*+68!VC??UW!H41)*2-,!F614*2-.!Q(2*18/A!

!"D!
)1+3.-)! 414()/! -221''! 9-881),'A! E61! 9)1'1,8! 26-981)! 1L81,0'! 86-8! 7();! 8(!
2(,'*01)!861!(98*4-.!3'1!(:!:.(-8*,+>9(*,8!9)12*'*(,!*,!861!F(3.(4@!-,0!OL26-,+1!
(91)-8()'A!
OXK!5-.31'!'9-,!-!7*01!0/,-4*2!)-,+1!:)(4!Y=?#!0(7,!8(!Y=?>=?!Z-)8)11'!
@1.(7!76*26!OXK'!2-,!@1!,1+.12810!76*.1!4-*,8-*,*,+!2614*2-.!-223)-2/A!Q*,+.1!
9)12*'*(,!2-,,(8!-223)-81./!-22343.-81!,34@1)'!-2)(''!86*'!7*01!0/,-4*2!)-,+1!
(:!86*)811,!012*4-.!()01)'A!E63'<!1-)./!'2*1,8*:*2!9)(+)-4'!)1.*10!(,!8)*2;'!'326!-'!
2(491,'-810! '344-8*(,! *,! ()01)! 8(! 4-*,8-*,! 9)12*'*(,! 03)*,+! .-)+1!
2-.23.-8*(,'A=T![*86!861!-051,8!(:!6-)07-)1>@-'10!0([email protected]!9)12*'*(,!*,'8)328*(,'!
:()! 2(44(0*8/! F%&'! *,! 861! =UB?'<! *8! @12-41! '8-,0-)0! 9)-28*21! 8(! 3'1! 0([email protected]!
9)12*'*(,! 3,*:()4./! :()! -..! :.(-8*,+>9(*,8! (91)-8*(,'! *,! \3-,834! 2614*'8)/!
2-.23.-8*(,'A! K,! :-28<! :()! 5-)*(3'! -)26*81283)-.! -,0! -.+()*864*2! )1-'(,'<! '*,+.1!
9)12*'*(,! -)*86418*2! (::1)10! (,./! -! '.*+68! 91):()4-,21! -05-,8-+1! (51)! 0([email protected]!
9)12*'*(,A!!
P*;1!1-)./!F%&'<! 861! :*)'8!F&GH>1,[email protected]!$%&'!6-0!,(!'399()8! :()!0([email protected]!
9)12*'*(,! -)*86418*2<! 014-,0*,+! 2-)1! *,! 861*)! 3'1! :()! \3-,834! 2614*'8)/!
-99.*2-8*(,'A! F(,51,*1,8./! 861! .-81'8! $%&'! :3../! '399()8! 0([email protected]! 9)12*'*(,!
-)*86418*2!7*86!91):()4-,21!*,!861!)-,+1!(:!'151)-.!63,0)10!$]P^%Q<!'391)*()!8(!
86-8!(::1)10!@/!F%&'A!I151)861.1''<!151,!861!.-81'8!$%&'!2(,8*,31!8(!9)(5*01!39!
8(! #CL! 4()1! 91):()4-,21! 8(! '*,+.1! 9)12*'*(,! (91)-8*(,'A! E6*'! 0*'9-)*8/! '814'!
:)(4! 861!6-)07-)1_'!910*+)11! *,!+)-96*2'<!761)1! 861)1! *'! .*88.1!,110! :()!0([email protected]!
9)12*'*(,!-223)-2/!-,0!861!,121''-)/!*,2)1-'1!*,!2*)23*8)/!*'!0*::*23.8!8(!`3'8*:/A!^,!
-,/! 9)(21''()<! '*,+.1! 9)12*'*(,! 1L6*@*8'! :3)861)! 91):()4-,21! -05-,8-+1'! -'! -!

! ""!
)1'3.8! (:! *8'! '4-..1)! 414()/! :((89)*,8<! 76*26! )10321'! 0-8-! @-,07*086!
)1\3*)141,8'C!-,0!*,2)1-'1'!861!,34@1)!(:!5-.31'!86-8!2-,!@1!2-2610!*,!(,>26*9!
)1+*'81)'A! E63'<! :()! 4-L*434! 91):()4-,21! (,! $%&'<! *8! )14-*,'! *49()8-,8! 8(!
:-5()!'*,+.1!9)12*'*(,!-)*86418*2!-'!4326!-'!9(''*@.1A!
! E([email protected],21!$%&!91):()4-,21!7*86!2614*2-.!-223)-2/<!\3-,834!2614*'8)/!
*49.141,8-8*(,'! 6-51! -0(9810! 4*L10! 9)12*'*(,! -99)(-261'! *,! 76*26! 0([email protected]!
9)12*'*(,! (91)-8*(,'! -)1! -0010! '9-)*,+./! 8(! -,! (861)7*'1! '*,+.1! 9)12*'*(,!
2-.23.-8*(,A! S-8)*L! 43.8*9.*2-8*(,! *,! 861! 2(,81L8! (:! )1'(.38*(,>(:>861>*01,8*8/!
Sa..1)>%.1''18! 91)83)@-8*(,! 861()/! 6-'! @11,! '6(7,! 8(! 9)(5*01! -223)-81!4*L10!
9)12*'*(,!)1'3.8'<!151,!761,!861!4-`()*8/!(:!(91)-8*(,'!-)1!2-))*10!(38!*,!'*,+.1!
9)12*'*(,AD<M! Q*,+.1! 9)12*'*(,! OXK! 15-.3-8*(,! 6-'! @11,! '3221'':3../! -3+41,810!
7*86!0([email protected]!9)12*'*(,!-22343.-8*(,!*,8(!861!4-8)*L!1.141,8'!(:!861!F(3.(4@!-,0!
1L26-,+1! (91)-8()'A#<"! bG([email protected]! 9)12*'*(,! -22343.-8*(,c! '*49./! 41-,'! 86-8! 861!
OXK'!-)1!15-.3-810!*,!'*,+.1!9)12*'*(,!@38!-!0([email protected]!9)12*'*(,!5-)*[email protected]! *'!3'10!8(!
-22343.-81!861!9)(0328'!(:!01,'*8/!4-8)*L!1.141,8'!-,0!OXK'!76*26!4-;1!39!861!
:*,-.!(91)-8()! V1A+A!F(3.(4@!()!1L26-,+1WA! ! ]()!1L-49.1<! 861!F(3.(4@!(91)-8()!
2-,!@1!2(,'8)32810!-'d!!
Jµ!64+ = P"#
32 µ! | "#( )32 (3.1)
761)1!861!'391)'2)*98'!*,0*2-81!861!,34@1)!(:!@*8'!(:!9)12*'*(,!3'10!8(!2(49381!
[email protected]!81)4!-,0!861!OXK'!-)1!+*51,!-'d!
µ! | "#( ) = $µ (r1)$! (r1)$" (r2 )$# (r2 )r1 % r2
& dr1dr2 (3.2)

!"M!
F(4938*,+!-!:17!(:!861!.-)+1'8!OXK'!*,!:3..!0([email protected]!9)12*'*(,!6-'!-.'(!@11,!'6(7,"!
8(!*49)(51!-223)-2/!2(49-)10!8(!2-.23.-8*(,'!3'*,+!(,./!'*,+.1!9)12*'*(,!:()!-..!
OXK'A! K,2)141,8-.! 2(,'8)328*(,! (:! 861! ](2;! 4-8)*L=B! 2-,! -.'(! *49)(51! 861!
-223)-2/!(:! '*,+.1!9)12*'*(,!OXK!15-.3-8*(,A#!]*,-../<! *8!6-'!@11,!'3++1'810<! 86-8!
:3..! '*,+.1!9)12*'*(,!2-,!@1!'-:1./!149.(/10! *,! 861!1-).*1'8!QF]! *81)-8*(,'A"!Q326!
'8)-81+*1'!6-51!9)(51,!1::128*51! *,! *49)(5*,+!4*L10!9)12*'*(,! )1'3.8'! :()!4-,/!
2-.23.-8*(,'! )19()810! 8(!0-81A!Z(7151)<!,(! '/'814-8*2! '830/!(:!4*L10!9)12*'*(,!
OXK!15-.3-8*(,!6-'!@11,!3,01)8-;1,A!K,!:-28<!861!9)12*'*(,!238(::'!43'8!@1!26('1,!
2-)1:3../!:()!1-26!4(.123.-)!'/'814!'830*10!8(!+3-)-,811!86-8!861!-@'(.381!1))()!
*'!7*86*,!-!8(.1)[email protected]!)-,+1A!
K,! 861! 9)1'1,8! 26-981)<! 71! *,8)(0321! -! '/'814-8*2! 4186(0! (:! @(86!
2(,8)(..*,+!1))()!-,0!4*,*4*N*,+!0([email protected]!9)12*'*(,!(91)-8*(,'!*,!4*L10!9)12*'*(,!
](2;!4-8)*L!2(,'8)328*(,A![1!@1+*,!@/!01'2)*@*,+!-!4*L10!9)12*'*(,!'26141<! *,!
76*26! -..! *,81+)-.'! -)1! 2(493810! (,! 861!$%&<!7*86! .-)+1!OXK'! 2-.23.-810! *,! :3..!
0([email protected]! 9)12*'*(,! -,0! '4-..! *,81+)-.'! *,! '*,+.1! 9)12*'*(,! 7*86! 0([email protected]! 9)12*'*(,!
-22343.-8*(,A! [1! '6(7! 86-8! 861! )1.-8*51! 1))()! *,! '326! 2-.23.-8*(,'! *'! 71..!
@16-510! :()! -,! -))-/! (:! '/'814'! -,0! 2-,! @1! 2(,8)(..10! 8(! 9)(5*01! -,! 1::128*51!
9)12*'*(,! -,0!91):()4-,21! *,81)410*-81!@18711,! 86-8! (:! '*,+.1! -,0! :3..! 0([email protected]!
9)12*'*(,A! K,!()01)! 8(! :3)861)!012)1-'1! 861!,34@1)!(:!0([email protected]!9)12*'*(,! *,81+)-.!
15-.3-8*(,'<!71!'3++1'8!-!,17!4186(0!86-8!71!81)4!12%!-$3#/4,3$5$'%! *,!76*26!
861! 1::128*51! 9)12*'*(,! *'! -0`3'810! 0/,-4*2-../! @18711,! QF]! *81)-8*(,'A! K,! 86*'!
7-/!861!4*,*434!,34@1)!(:!0([email protected]!9)12*'*(,!*,81+)-.'!2-,!@1!3'10!8(!(@8-*,!-,/!

! "T!
01'*)10!.151.!(:!-223)-2/A!]*,-../<!71!9)1'1,8!91):()4-,21!-,0!-223)-2/!)1'3.8'!8(!
@1,264-);!861!0/,-4*2!9)12*'*(,!-99)(-26!-'!*49.141,810!*,!E1)-F614A=U!!
MIXED PRECISION IMPLEMENTATION
E61! 4-+,*8301! (:! -,! OXK! *'! 2(44(,./! @(3,010! 3'*,+! 861! Q267-)N!
*,1\3-.*8/AC?! K,! 0*)128! QF]! 2(01'<=B! 861! @(3,0! 2-,! @1! :3)861)! )103210! 3'*,+!
1.141,8'!(:!861!01,'*8/!4-8)*L!-'d!
µ! | "#( )P"# $ µ! | µ!( )1/2 "# | "#( )1/2 P"# (3.3)
e12-3'1! (,./! -@'(.381! -223)-2/! *'! )1\3*)10! *,! 2614*2-.! -99.*2-8*(,'<! f-'30-!
*,8)(03210!-! 238(::! (,! 861!01,'*8/>71*+6810!Q267-)N!@(3,0! 8(!+)(39!OXK'! *,8(!
87(!@-8261'A"!E6('1!76('1!@(3,0!:1..!@1.(7!861!238(::!71)1!2-.23.-810!*,!'*,+.1!
9)12*'*(,! (,! 861! $%&A! K,81+)-.'! 76('1! @(3,0! 7-'! .-)+1)! 86-,! 861! 238(::! 71)1!
15-.3-810!*,!0([email protected]!9)12*'*(,!(,!861!F%&A!
Jµ!64+ =
P"#32 µ! | "#( )32 if P"# µ! | µ!( ) "# | "#( ) $ % prec
P"#64 µ! | "#( )64 if P"# µ! | µ!( ) "# | "#( ) > % prec
&'(
)(
(3.4)
H'! 6-'! @11,! 9)15*(3'./! ,(810<=! 7*86! 861! -051,8! (:! )(@3'8! 0([email protected]! 9)12*'*(,!
'399()8!(,!861!$%&!*8! *'!,(!.(,+1)!,121''-)/!8(!15-.3-81!'(41!OXK'!(,!861!F%&A!
[1!6-51!*49.141,810!-!4*L10!9)12*'*(,!](2;!4-8)*L!15-.3-8*(,!'26141!'*4*.-)!
8(! 86-8! '3++1'810!@/!f-'30-A! K,'81-0! (:! 3'*,+! 861!F%&<! 6(7151)<!71!0151.(910!
0([email protected]!9)12*'*(,!-,-.(+31'!(:!(3)!9)15*(3'./!)19()810!'*,+.1!9)12*'*(,!F(3.(4@!
-,0! 1L26-,+1! )(38*,1'A#! K49.141,8-8*(,! 018-*.'! -)1! 01'2)*@10! *,! 861! 9)15*(3'!
26-981)A!

!"B!
K,! ()01)! 8(!4-;1! 861!4('8! (:! 861!$%&_'!414()/! @-,07*086<! 861! 0([email protected]!
-,0!'*,+.1!9)12*'*(,!*,81+)-.'!-)1!6-,0.10!*,!-!87(>9-''!-.+()*864A!H'!9)15*(3'./!
01'2)*@10<! (3)! OXK! -.+()*864! (91)-81'! 0*)128./! (,! 861! @)-! -,0! ;18! 9)*4*8*51!
$-3''*-,!9-*)'<!76*26!-)1!'()810!@/!012)1-'*,+!Q267-)N!@(3,0A!K,!861!:*)'8!9-''<!
0-8-!:()!861!.-)+1'8!9)*4*8*51!9-*)'!*'!9-2;10!*,8(!0([email protected]!9)12*'*(,!-))-/'!-,0!-,/!
OXK! 76('1! @(3,0! *'! +)1-81)! 86-,! 861! 9)12*'*(,! 86)1'6(.0! *'! 2-.23.-810! 3'*,+!
0([email protected]! 9)12*'*(,! $%&! ;1),1.'A! K,! 861! '12(,0! 9-''<! '4-..1)! 9)*4*8*51! 9-*)'! -)1!
-0010!8(!861!@)-!-,0!;18!0-8-<!76*26!*'!)1-''[email protected]!*,8(!'*,+.1!9)12*'*(,!-))-/'!
-,0!9)(21''10!@/!'*,+.1!9)12*'*(,!;1),1.'A!
e12-3'1! 861! :(3)>*,01L<! 01,'*8/>71*+6810! Q267-)N! @(3,0! *'! 2(493810!
(,./!7*86*,!861!$%&!;1),1.'<!'(41!039.*2-8*(,!(223)'!@18711,!861!'18'!(:!'*,+.1!
-,0!0([email protected]!9)12*'*(,!9)*4*8*51!9-*)'<!-,0!1-26!;1),1.!43'8! :*.81)!(38! *,0*5*03-.!
OXK'!76('1! @(3,0! :-..'! (38'*01! (:! 861! )1.15-,8! )-,+1A! Q*,21! -'! @1:()1! 861! 9-*)!
\3-,8*8*1'! -)1! ()01)10! @/! Q267-)N! @(3,0<! 861! '*,+.1! -,0! 0([email protected]! 9)12*'*(,!
*,81+)-.'!)1'*01!*,!2(,8*+3(3'!@.(2;'!-'!'6(7,!*,!:*+3)1!=A!E6*'!4*,*4*N1'!7-)9!
0*51)+1,21!(,!861!$%&<!'*,21!,1*+6@()*,+!86)1-0'!7*..!*,!+1,1)-.!';*9!()!2(49381!
*,81+)-.'! 8(+1861)A![61,!:*.81)*,+<! *8! *'!1''1,8*-.! 86-8!@(86!861!'*,+.1!-,0!0([email protected]!
9)12*'*(,! ;1),1.'! 6-,0.1! 861! @(3,0'! *01,8*2-../A! ^861)7*'1<! 861! 0*::1)1,8!
)(3,0*,+!@16-5*()!1L6*@*810!@/!'*,+.1!-,0!0([email protected]!9)12*'*(,!-)*86418*2!7*..!2-3'1!
*,81+)-.'! 2.('1! 8(! 861! @(3,0! 8(! @1! ';*9910! ()! 0([email protected]! 2(3,810A! K,! (3)!
*49.141,8-8*(,<!861!0([email protected]!9)12*'*(,!;1),1.'!2-'8!861!@(3,0!\3-,8*8*1'!8(!'*,+.1!
9)12*'*(,! @1:()1! 0181)4*,*,+! 761861)! 861! -''(2*-810! *,81+)-.'! -,0! 861*)!
2(,8)*@38*(,'!7*..!@1!15-.3-810A!

! "U!
Figure 1: Organization of double and single precision workloads within Coulomb ERI grids. Rows and columns correspond to primitive bra and ket pairs. On left each ERI is colored according to the magnitude of its Schwarz bound. On right ERIs are colored by required precision. Yellow ERIs require double precision while those in green may be evaluated in single precision. Blue ERIs are neglected entirely.
MIXED PRECISION ACCURACY
E61!4(.123.1'!'6(7,!*,!:*+3)1!C!71)1!26('1,!-'!)19)1'1,8-8*51!81'8!2-'1'!
8(! '830/! 861!-223)-2*1'!(:! '151)-.!4*L10!9)12*'*(,! 86)1'6(.0'A!$1(418)*1'!71)1!
9)19-)10! -8! @(86! -,!(98*4*N10!XZ]gM>#=$!4*,*434!-,0!-!0*'8()810! +1(418)/!
(@8-*,10! @/! 91):()4*,+! IJE! 0/,-4*2'! -8! YC???hA! E61! 0([email protected]! 9)12*'*(,! $%&!
*49.141,8-8*(,! 7-'! :*)'8! @1,264-);10! -+-*,'8! $HSOQQC=! 3'*,+! 861! 01:-3.8!
$HSOQQ!2(,51)+1,21!-,0! 87(>1.128)(,! 86)1'6(.0'A!E61! )1'3.8*,+! :*,-.! 1,1)+*1'<!
'6(7,! *,! [email protected]! =<! *,0*2-81! +((0! -+)1141,8A! Q7*826*,+! 8(! '*,+.1! 9)12*'*(,! OXK'!
01+)-01'! 861! )1'3.8! @/! #>"! ()01)'! (:! 4-+,*8301A! Z(7151)<! *8! '6(3.0! @1!
1496-'*N10!86-8!:()!4(.123.1'!2(,8-*,*,+!-'!4-,/!-'!(,1!63,0)10!-8(4'<!'*,+.1!
9)12*'*(,!9)(5*01'!-01\3-81!)1'3.8'!i!861!-@'(.381!1))()!(:!861!1,1)+/!2(493810!
7*86!'*,+.1!9)12*'*(,!OXK'! *'!71..!@1.(7!=!;2-.g4(.!151,! :()!I13)(;*,*,!H!7*86!
="T!-8(4'A!!

!M?!
Figure 2: Molecular geometries used to benchmark the correlation between precision cutoff and the effective precision of the final energy. Optimized geometries (shown here) were used in addition to distorted nonequilibrium geometries prepared by carrying out RHF/STO-3G NVT dynamics at 2000 K (1000 K for Ascorbic Acid).
! H'2()@*2!H2*0! P-28('1! F/-,(@-281)*-.!E(L*,!$HSOQQ! >MB?A"BCBUDT! >=CBUAMMMMC"?! >CDU=AC?"BBU#!E1)-F614!G%! >MB?A"BCBUDT! >=CBUAMMMMC"?! >CDU=AC?"BBU?!E1)-F614!Q%! >MB?A"BCB?T=! >=CBUAMMMDCMM! >CDU=AC?"#MM?!! ! ! !! I13)(;*,*,!H! "LM!I-,(83@1! !$HSOQQ! >D?BUAMBB#TT?! >=#TU?A=D="=T=! !E1)-F614!G%! >D?BUAMBB#TMC! >=#TU?A=D="=TM! !E1)-F614!Q%! >D?BUAMBTUBCD! >=#TU?A=#BUUBT! !Table 1: RHF/6-31G final energies in Hartrees compared between GAMESS (set at default convergence and two-electron thresholds), our GPU accelerated TeraChem code performing all calculations in double precision (TeraChem DP), and TeraChem using single precision for ERIs with double precision accumulation into the Fock matrix elements (TeraChem SP). Distorted nonequilibrium geometries from RHF/STO-3G NVT dynamics at 2000K (1000K for Ascorbic Acid) were used.
I1L8!71!15-.3-810!861!4*L10!9)12*'*(,!-99)(-26!@/!5-)/*,+!861!9)12*'*(,!
86)1'6(.0! @18711,! =?>U! Z-)8)11'! V,1-)./! -..! *,81+)-.'! -)1! 15-.3-810! *,! 0([email protected]!
9)12*'*(,W! -,0! =A?! Z-)8)11'! V1''1,8*-../! -..! *,81+)-.'! 3'1! '*,+.1! 9)12*'*(,WA!
I1+.*+*@./!'4-..!OXK'!71)1!'2)11,10!-22()0*,+!8(!861!01,'*8/!71*+6810!Q267-)N!
3991)!@(3,0!(:!1\3-8*(,!V#A#W!3'*,+!-!2(,'1)5-8*51!86)1'6(.0!8(!1,'3)1!86-8!-,/!

! M=!
0*::1)1,21'! *,! 861! :*,-.! 1,1)+/! -@(51! Y=?>T! Z-)8)11'! 7(3.0! @1! 0(4*,-810! @/!
4*L10! 9)12*'*(,! 1))()'A! E61! -51)-+1! )1.-8*51! 1,1)+/! 0*::1)1,21! @18711,! :3..!
0([email protected]! -,0!4*L10!9)12*'*(,! QF]! 1,1)+*1'! :()! 861! 81,! 81'8! +1(418)*1'!01'2)*@10!
-@(51! *'! 9.(8810! -'! -! :3,28*(,! (:! 9)12*'*(,! 86)1'6(.0! *,! :*+3)1! #A! H.86(3+6! 861!
-@'(.381! 1))()! *,2)1-'1'! -.(,+! 7*86! 861! 8(8-.! 1,1)+/! (:! 861! '/'814'<! 861!
)1.-8*(,'6*9!@18711,!861!9)12*'*(,!238(::!-,0!)1.-8*51!1))()!*'!)(3+6./!.*,1-)!-,0!
*'! *,0191,01,8! (:! '/'814! '*N1! -,0! @-'*'! '18A! E63'<! 1-26! 86)1'6(.0! 2-,! @1!
-''(2*-810!7*86!-,!1::128*51!)1.-8*51!1))()!*,!861!1,1)+/A!
!
Figure 3. Average relative precision error in final RHF energies versus the precision threshold. Both minimized and distorted non-equilibrium geometries for the molecules in figure 2 are included in averages. Error bars represent two standard deviations above the mean. The black line represents the empirical error bound given of equation (3.5).
&'*,+!:*+3)1!#!*8!*'!9(''*@.1!8(!149*)*2-../!9)1>'1.128!-!9)12*'*(,!86)1'6(.0!
2())1'9(,0*,+!8(!-,/!-223)-2/!)1\3*)141,8!3'*,+!(,./!861!1'8*4-810!8(8-.!1,1)+/!
V8(! 2-.23.-81! )1.-8*51! 1))()WA! H! )1-'(,-@./! 2(,'1)5-8*51! 149*)*2-.! @(3,0! (,! 861!
9)12*'*(,! 1))()! *'! +*51,! *,! 861! :(..(7*,+! :()43.-! -,0! 9.(8810! :()! )1:1)1,21! *,!
:*+3)1!#A!

!MC!
Err Thre( ) = 2.0 !10"6Thre0.7 (3.5)
e/!*,51)8*,+!1\3-8*(,!V#A"W<!71!2(3.0!'1.128!-,!-99)(9)*-81!9)12*'*(,!86)1'6(.0!-8!
861! '8-)8! (:! 861! QF]! 9)(2103)1! -,0! 3'1! 861! 4*,*434! -..([email protected]! 1::128*51!
9)12*'*(,A!Z(7151)<! 86*'!7(3.0! )1\3*)1! 1-)./! *81)-8*(,'!76('1!01,'*8/!4-8)*21'!
-)1!6*+6./!-99)(L*4-81!8(!3'1!861!:3..! .151.!(:!9)12*'*(,!,11010!-8!2(,51)+1,21A!
E6*'! *'! 1'912*-../!7-'81:3.! *,! 51)/! .-)+1! '/'814'! '*,21! 861!-223)-2/! )1\3*)10!-8!
2(,51)+1,21! ,1-)'! 86-8! (:! :3..! 0([email protected]! 9)12*'*(,A! E(! :3)861)! )10321! 861! 3'1! (:!
0([email protected]!9)12*'*(,!*,!1-)./!*81)-8*(,'!76*.1!'8*..!-26*15*,+!861!)1\3*)10!-223)-2/!-8!
2(,51)+1,21<!71!*,8)(0321!-!0/,-4*2!9)12*'*(,!-99)(-26<!01'2)*@10!@1.(7A!
DYNAMIC PRECISION IMPLEMENTATION
E61!1''1,21!(:!861!0/,-4*2!9)12*'*(,!-99)(-26!*'!8(!3'1!1\3-8*(,!V#A"W!8(!
'1.128! -! 0*::1)1,8! 86)1'6(.0! :()! 1-26! *81)-8*(,! (:! 861! QF]! 9)(2103)1A! O-)./!
*81)-8*(,'!6-51!@11,!'6(7,!8(!8(.1)-81!)1.-8*51./!.-)+1!1))()'!*,!861!](2;!4-8)*L!
7*86(38! 6-491)*,+! 2(,51)+1,21A#<CC![1! 8-;1! 861!4-L*434!1.141,8! (:! 861!GKKQ!
1))()! 5128()C#! :)(4! 861! 9)15*(3'! *81)-8*(,! -'! -!418)*2! (:! 86*'! 8(.1)-,21<! -,0! -8!
1-26! *81)-8*(,! 71! '1.128! -! 86)1'6(.0! 9)(5*0*,+! 9)12*'*(,! '-:1./! @1.(7! 861! GKKQ!
1))()A! E6*'! 1,'3)1'! 86-8! 861! 9)12*'*(,! 1))()! *'! -! '4-..! 2(,8)*@38()! 8(! 861! 8(8-.!
1))()A!e/!)1032*,+!861!9)12*'*(,!86)1'6(.0!+)-03-../!-'!2(,51)+1,21!9)(+)1''1'<!
*8! *'! 9(''*@.1! 8(! -99)(-26! :3..! 0([email protected]! 9)12*'*(,! )1'3.8'! 76*.1! 4*,*4*N*,+! 861!
,34@1)!(:!-283-.!0([email protected]!9)12*'*(,!(91)-8*(,'A!
E(!*49)(51!91):()4-,21<!(3)!QF]!2(01!3'1'!-,!*81)-8*51!390-81!-99)(-26!
V-.'(!;,(7,!-'!*,2)141,8-.!](2;!4-8)*L!:()4-8*(,W!8(!@3*.0!39!861!](2;!4-8)*L!

! M#!
(51)! 861! 2(3)'1! (:! 861! QF]! 9)(2103)1A=B! E61! 390-81! -99)(-26! 012(49('1'! 861!
](2;!4-8)*L!-'!!
Fi+1 Pi+1( ) = Fi Pi( ) + F Pi+1 ! Pi( ) , (3.6)
'(!86-8!(,./!861!.-'8!81)4!,110'!8(!@1!2-.23.-810!*,!1-26!QF]!*81)-8*(,A!Z1)1!Pi !-,0!
Fi Pi( ) !-)1! 861! 01,'*8/! -,0! ](2;! 4-8)*21'! -8! 861! $>86! QF]! *81)-8*(,A! e12-3'1!
26-,+1'!*,!861!01,'*8/!4-8)*L!@12(41!51)/!'4-..!,1-)!2(,51)+1,21<!861!*81)-8*51!
](2;! -99)(-26! -..(7'! 4-,/! -00*8*(,-.! *,81+)-.'! 8(! @1! '2)11,10A! E/9*2-../! 86*'!
9)(5*01'! -,! (51)-..! '911039! @18711,! CL! -,0! #L! (51)! 861! 2(,51,8*(,-.! QF]!
-99)(-26A! Z(7151)<! 861! ,-j51! *49.141,8-8*(,! (:! 0/,-4*2! 9)12*'*(,! 01'2)*@10!
-@(51! 2-3'1'! 861! *81)-8*51! ](2;! 4186(0! 8(! 2(,51)+1! *,2())128./<! @12-3'1! 1-26!
390-81!(:!861!](2;!4-8)*L!2-,!,(8!2())128!:()!861!9)12*'*(,!1))()!(:!861!9)15*(3'!
'819A!
X-861)! 86-,! -@-,0(,*,+! 861! *81)-8*51! ](2;! -.+()*864! -.8(+1861)<! 71!
*,8)(0321!861!:(..(7*,+!-0`3'841,8A*L!*'!)12-.23.-810!:)(4!
'2)-826A!e18711,! 86)1'6(.0! )10328*(,'<! 861! :-'81)! *81)-8*51!](2;!390-81! '26141!
2-,!@1!'-:1./!149.(/10A!
RESULTS
E(! @1,264-);! (3)! 0/,-4*2! 9)12*'*(,! -99)(-26<! 71! 91):()410! XZ]!
1,1)+/! 2-.23.-8*(,'! (,! 861! 81'8! +1(418)*1'! 9)1'1,810! -@(51<! -'! 71..! -'! '(41!

!MD!
.-)+1)!'/'814'!'6(7,!*,!:*+3)[email protected]!C!014(,'8)-81'!861!-223)-2/!9)(5*010!@/!
(3)! 0/,-4*2! 9)12*'*(,! -99)(-26A! K,! 1-26! 2-.23.-8*(,<! 861! 0/,-4*2! 9)12*'*(,!
4186(0!*'!'3221'':3.!*,!)19)(032*,+!861!:3..!0([email protected]!9)12*'*(,!)1'3.8'!8(!7*86*,!861!
2(,51)+1,21! 2)*81)*-A! ]3)861)4()1<! 861! ,34@1)! (:! QF]! *81)-8*(,'! )1\3*)10! 8(!
)1-26! 2(,51)+1,21! V-.'(! '6(7,! *,! [email protected]! CW! *'! 1''1,8*-../! *01,8*2-.! @18711,!
0/,-4*2! -,0! 0([email protected]! 9)12*'*(,A! ]*,-../<! 861! QF]! 1,1)+/! 0*::1)1,21! @18711,!
0/,-4*2! -,0! 0([email protected]! 9)12*'*(,! )14-*,'! :-*)./! 2(,'8-,8! (51)! 861! )-,+1! (:! 81'8!
'/'814'<!*,0*2-8*,+!86-8!(3)!149*)*2-.!1))()!@(3,0!*'!)1-'(,-@./!2-.*@)-810A!
Double Precision Dynamic Precision Precision Error
Conv Thres Final Energy Iter Final Energy Iter
Ascorbic Acid (Minimum)
-680.6986413210 16 -680.6986413153 16 5.70E-09 10-7
-680.6986413213 12 -680.6986413898 12 6.85E-08 10-5
Ascorbic Acid (1000K)
-680.5828947151 17 -680.5828947066 17 8.50E-09 10-7
-680.5828947060 12 -680.5828947665 12 6.05E-08 10-5
Lactose (Minimum)
-1290.0883460632 14 -1290.0883460414 14 2.18E-08 10-7
-1290.0883460086 10 -1290.0883459660 10 4.26E-08 10-5
Lactose (2000K)
-1289.6666249592 15 -1289.6666249614 15 2.20E-09 10-7
-1289.6666249365 11 -1289.6666248603 11 7.62E-08 10-5
Cyano Toxin (Minimum)
-2492.3971992758 19 -2492.3971992873 19 1.15E-08 10-7
-2492.3971992730 13 -2492.3971985116 13 7.61E-07 10-5
Cyano Toxin (2000K)
-2491.2058890235 21 -2491.2058890017 21 2.18E-08 10-7
-2491.2058889916 13 -2491.2058886707 13 3.21E-07 10-5
Neurokinin A (Minimum)
-4091.3672645555 19 -4091.3672645944 20 3.89E-08 10-7
-4091.3672645489 14 -4091.3672644494 14 9.95E-08 10-5
Neurokinin A (2000K)
-4089.6883762179 21 -4089.6883761946 21 2.33E-08 10-7
-4089.6883760772 15 -4089.6883758130 15 2.64E-07 10-5
Nanotube (Minimum)
-13793.7293925221 24 -13793.7293925323 23 1.02E-08 10-7
-13793.7293924922 15 -13793.7293928287 15 3.37E-07 10-5
Nanotube (2000K)
-13790.1415175662 29 -13790.1415175584 27 7.80E-09 10-7
-13790.1415175332 18 -13790.1415191026 18 1.57E-06 10-5
Crambin
-17996.6562925538 18 -17996.6562926036 18 4.98E-08 10-7
-17996.6562925535 12 -17996.6562927894 12 2.36E-07 10-5
Ubiquitin
-29616.4426376594 24 -29616.4426376596 24 2.00E-10 10-7
-29616.4426376302 18 -29616.4426376655 18 3.53E-08 10-5
T-Cadherin EC1
-36975.6726049407 21 -36975.6726049394 21 1.30E-09 10-7
-36975.6726049265 16 -36975.6726048777 16 4.88E-08 10-5
Ribonuclease A
-50813.1471248227 19 -50813.1471248179 19 4.80E-09 10-7
-50813.1471247051 12 -50813.1471250247 12 3.20E-07 10-5
Table 2. Comparison of double and dynamic precision final RHF/6-31G energies (listed in Hartree). Precision error is taken as the absolute difference between double and dynamic precision energies. The number of SCF iterations required to reach convergence is listed (Iter) as well as the threshold used to converge the maximum element of the DIIS error matrix.

! M"!
!
Figure 4. Additional molecules used to test the dynamic precision algorithm.
[email protected]! #! '344-)*N1'! 861! 91):()4-,21! (:! (3)! -.+()*864! (,! 87(! $%&!
9.-8:()4'A! ^,! 861! (.01)! E1'.-! F=?M?! $%&<! 0/,-4*2! 9)12*'*(,! -221.1)-81'! 861!
2(,'8)328*(,!(:!861!](2;!4-8)*L!@/!39!8(!DL!(51)!:3..!0([email protected]!9)12*'*(,A!E61!E1'.-!
FC?"?!*,2.301'!-!+)1-81)!9)(9()8*(,!(:!0([email protected]!9)12*'*(,!3,*8'<!-,0!-'!-!)1'3.8!861!
91):()4-,21! 4-)+*,! @18711,! 0([email protected]! -,0! '*,+.1! 9)12*'*(,! -)*86418*2! *'!
,-))(710A! Z(7151)<! 151,! 61)1! 0/,-4*2! 9)12*'*(,! ](2;! 4-8)*L! 2(,'8)328*(,! *'!
@18711,!CL!-,0!#L!:-'81)!86-,!86-8!91):()410!*,!:3..!0([email protected]!9)12*'*(,A!]*+3)1!"!
2(49-)1'!](2;!4-8)*L!2(,'8)328*(,!3'*,+!0/,-4*2<!4*L10<!-,0!'*,+.1!9)12*'*(,!8(!
:3..! 0([email protected]! 9)12*'*(,! 2-.23.-8*(,'! (,! E1'.-! F=?M?! $%&'A! Z1)1!-$6,1# /4,3$5$'%!
)1:1)'!8(!'8-8*2-../!:*L*,+!861!9)12*'*(,!86)1'6(.0!:()!861!1,8*)1!QF]!9)(2103)1!-8!
861!5-.31!9)1'2)*@10!@/!*,51)8*,+!1\3-8*(,!V#A"WA!G/,-4*2!9)12*'*(,!2(,'*'81,8./!

!MM!
(3891):()4'! 861! '*49.1)! 4*L10! 9)12*'*(,! '26141! 01'9*81! )1\3*)*,+! 91)*(0*2!
)1@3*.0'! (:! 861! ](2;! 4-8)*LA! S()1! *49()8-,8./<! 0/,-4*2! 9)12*'*(,! 2(,'*'81,8./!
9)(5*01'! @18711,! T?! -,0! B?k! (:! 861! 91):()4-,21! (:! '*,+.1! 9)12*'*(,! 76*.1!
9)(5*0*,+! )1'3.8'! 2(49-)[email protected]! 8(! :3..!0([email protected]!9)12*'*(,<! -,0! 86*'!9-881),! )14-*,'!
*,8-28!151,!:()!861!.-)+1'8!'/'814'A!
Nvidia Tesla C1060 Nvidia Tesla C2050 Dynamic
Runtime Fock Speedup
Total Speedup
Dynamic Runtime
Fock Speedup
Total Speedup
Ascorbic Acid 2.23 sec 4.0 3.4 2.93 sec 2.0 1.4 Lactose 9.70 sec 4.1 3.8 8.41 sec 2.2 1.9 Cyano Toxin 87.66 sec 4.2 4.0 68.44 sec 2.4 2.3 Neurokinin A 197.91 sec 3.8 3.7 149.76 sec 2.3 2.3 Nanotube 1716.88 sec 3.0 2.7 1155.58 sec 3.0 2.6 Crambin 1104.22 sec 2.9 2.5 762.09 sec 2.1 1.8 Ubiquitin 11833.58 sec 2.8 2.5 7517.68 sec 2.3 2.0 T-Cadherin EC1 17408.21 sec 2.7 2.4 10781.42 sec 2.3 1.9 Ribonuclease A 21869.37 sec 2.7 2.3 --- --- --- Table 3. Runtime comparison between dynamic and full double precision for RHF/6-31G single point energy calculations converged to a maximum DIIS error of 10-5 a.u. Calculations were run on a dual Intel Xeon X5570 platform with 72 gigabytes of ram. Smaller systems (from ascorbic acid to neurokinin) utilized a single GPU, while 8 GPUs operated in parallel for the larger systems. The speedups for Fock matrix construction (coulomb and exchange operator evaluation, including all data packing and host-GPU transfers) are listed along with the speedup of the entire energy evaluation. The Tesla C2050 could not treat Ribonuclease A at the RHF/6-31G level due to memory constraints.
Figure 5. Speedups for the construction of the Fock matrix accumulated over all SCF iterations using single, dynamic, and mixed precision relative to full double precision performance. Calculations were run on Nvidia Tesla C1060 GPUs and were converged to a DIIS error of 10-7 a.u. Mixed precision calculations used a static precision threshold chosen for each system to give an absolute accuracy of 10-7 Hartree. A single GPU was employed for the smaller systems (from 20 to 200 atoms), while the larger systems utilized 8 GPUs in parallel. Single precision failed to converge for ubiquitin.

! MT!
CONCLUSION
[1!6-51!014(,'8)-810!86-8!@/!0/,-4*2-../!-0`3'8*,+!861!)-8*(!(:!*,81+)-.'!
2-.23.-810!*,!'*,+.1!-,0!0([email protected]!9)12*'*(,!(,!861!$%&!*8!*'!9(''*@.1!8(!4*,*4*N1!861!
,34@1)! (:! 0([email protected]! 9)12*'*(,! -)*86418*2! (91)-8*(,'! *,! 2(,'8)328*,+! 861! ](2;!
4-8)*L!76*.1! '8*..! '/'814-8*2-../! 2(,8)(..*,+! 861! 1))()A! OL9.(*8*,+! 86*'! :.1L*@*.*8/!
71!6-51!23'8(4*N10!(3)!](2;!4-8)*L!)(38*,1'!:()!4-L*434!91):()4-,21!(,!861!
$%&A!^3)!0/,-4*2!9)12*'*(,!*49.141,8-8*(,!*'[email protected]!8(!-26*151!*,!1L21''!(:!T?k!
(:!'*,+.1!9)12*'*(,_'!91):()4-,21!76*.1!4-*,8-*,*,+!-223)-2/!2(49-)[email protected]!8(!:3..!
0([email protected]!9)12*'*(,A!
]()! 1L8)141./! .-)+1! '/'814'<! 861! )1\3*)10! )1.-8*51! -223)-2/! 4-/! 71..!
1L81,0! @1/(,0! 861! 2-9-2*8/! (:! 0([email protected]! 9)12*'*(,ACD! K,! 86*'! .*4*8! 861! -99)(-26!
(38.*,10! -@(51! 7*..! -+-*,! 9)(51! 3'1:3.! *,! '/'814-8*2-../! *49)(5*,+! 0([email protected]!
9)12*'*(,! 7*86! -! 4*,*434! (:! 6*+61)! 9)12*'*(,! -)*86418*2! (91)-8*(,'A! H! 4()1!
2(49)161,'*51! 43.8*>9)12*'*(,! '8)-81+/! 2-,! @1! 1-'*./! 1,5*'*(,10<! :()! 1L-49.1!
3'*,+! '*,+.1<! 0([email protected]! -,0! \3-0)39.1! 9)12*'*(,! 15-.3-8*(,! (:! 0*::1)1,8! OXK'<!
-22()0*,+! 8(! 861*)! 4-+,*8301A! ]3)861)4()1<! 861! '-41! 0/,-4*2-.! 9)12*'*(,!
-99)(-26!2-,!@1!-99.*10!8(!861!2-.23.-8*(,!(:!861!1L26-,+1>2())1.-8*(,!(91)-8()!
*,!01,'*8/!:3,28*(,-.!861()/!-,0!'*4*.-)!91):()4-,21!+-*,'!7*..!@1!(@8-*,10A!
REFERENCES
(1) Ufimtsev, I. S.; Martinez, T. J. J. Chem. Theo. Comp. 2008, 4, 222.
(2) Ufimtsev, I. S.; Martinez, T. J. Comp. Sci. Eng. 2008, 10, 26.
(3) Ufimtsev, I. S.; Martinez, T. J. J. Chem. Theo. Comp. 2009, 5, 1004.

!MB!
(4) Vogt, L.; Olivares-Amaya, R.; Kermes, S.; Shao, Y.; Amador-Bedolla, C.; Aspuru-Guzik, A. J. Phys. Chem. A 2008, 112, 2049.
(5) Yasuda, K. J. Comp. Chem. 2008, 29, 334.
(6) Olivares-Amaya, R.; Watson, M. A.; Edgar, R. G.; Vogt, L.; Shao, Y. H.; Aspuru-Guzik, A. J. Chem. Theo. Comp. 2010, 6, 135.
(7) Asadchev, A.; Allada, V.; Felder, J.; Bode, B. M.; Gordon, M. S.; Windus, T. L. J. Chem. Theo. Comp. 2010, 6, 696.
(8) Anderson, J. A.; Lorenz, C. D.; Travesset, A. J. Comp. Phys. 2008, 227, 5342.
(9) Genovese, L.; Ospici, M.; Deutsch, T.; Mehaut, J.-F.; Neelov, A.; Goedecker, S. J. Chem. Phys. 2009, 131, 034103.
(10) Yasuda, K. J. Chem. Theo. Comp. 2008, 4, 1230.
(11) Ufimtsev, I. S.; Martinez, T. J. J. Chem. Theo. Comp. 2009, 5, 2619.
(12) Friedrichs, M. S.; Eastman, P.; Vaidyanathan, V.; Houston, M.; Legrand, S.; Beberg, A. L.; Ensign, D. L.; Bruns, C. M.; Pande, V. S. J. Comp. Chem. 2009, 30, 864.
(13) Harvey, M. J.; Giupponi, G.; DeFabiritiis, G. J. Chem. Theo. Comp. 2009, 5, 1632.
(14) Stone, J. E.; Phillips, J. C.; Freddolino, P. L.; Hardy, D. J.; Trabuco, L. G.; Schulten, K. J. Comp. Chem. 2007, 28, 2618.
(15) Kirk, D. B.; Hwu, W. W. Programming Massively Parallel Processors: A Hands-On Approach; Morgan Kauffman Burlington, MA, 2010.
(16) Levine, B.; Martinez, T. J. Abst. Pap. Amer. Chem. Soc. 2003, 226, U426.
(17) Kahan, W. Comm. ACM 1965, 8, 40.
(18) Almlof, J.; Faegri, K.; Korsell, K. J. Comp. Chem. 1982, 3, 385.
(19) Luehr, N.; Ufimtsev, I. S.; Martinez, T. J. J. Chem. Theo. Comp. 2011, 7, 949.
(20) Whitten, J. L. J. Chem. Phys. 1973, 58, 4496.
(21) Schmidt, M. W.; Baldridge, K. K.; Boatz, J. A.; Elbert, S. T.; Gordon, M. S.; Jensen, J. H.; Koseki, S.; Matsunaga, N.; Nguyen, K. A.; Su, S. J.; Windus, T. L.; Dupuis, M.; Montgomery, J. A. J. Comp. Chem. 1993, 14, 1347.
(22) Rudberg, E.; Rubensson, E. H.; Salek, P. J. Chem. Theo. Comp. 2009, 5, 80.

! MU!
(23) Pulay, P. J. Comp. Chem. 1982, 3, 556.
(24) Takashima, H.; Kitamura, K.; Tanabe, K.; Nagashima, U. J. Comp. Chem. 1999, 20, 443.
!
!
!
!
!
!
!
!
!
!
!
!
!
!
!
!
!
!
!
!

!T?!
!

71
CHAPTER FOUR
DFT EXCHANGE-CORRELATION EVALUATION ON GPUS
The exchange-correlation potential is the final ingredient for a complete GPU
accelerated SCF program. Although not as intensive as the Coulomb and exchange
procedures, the evaluation of the exchange-correlation potential represents a second
major bottleneck for DFT calculations. We limit our consideration here to the common
class of generalized gradient approximation (GGA) functionals. In the general spin-
polarized case, where !" # !$ , equations (1.15) and (1.23) for the exchange-
correlation energy and potential become the following.1,2
EXC = fxc(!" (
!r ),!# (!r ),$ "" (
!r ),$ "# (!r ),$ ## (
!r ))d 3!r% (4.1)
Vµ!
XC" = # fxc#$"
%µ%! + 2 # fxc#& ""
'$" +# fxc#& "(
'$(
)
*+,
-./'(%µ%! )
0
122
3
455
6 d 3!r (4.2)
Where an analogous expression to equation (4.2) is used for the beta-spin Vµ!XC" , and
! represents the gradient invariants as follows.
! " #" (!r ) = $%" (
!r ) &$% #" (!r ) (4.3)
Unlike the Gaussian ERIs used to construct the Coulomb and exchange
operators, typical xc-kernel functions, fxc , are not amenable to analytical integration.
Instead, real-space numerical quadrature is employed to evaluate equations (4.1) and
(4.2). The quadrature grid itself proves to be non-trivial, and we consider it first before
discussing the evaluation of the exchange-correlation potential more properly.

72
QUADRATURE GRID GENERATIONa
Because molecular potentials and charge densities exhibit discontinuous cusps
at each nucleus, accurate numerical integration requires very dense quadrature grids at
least in these local regions. Thus efficient quadrature schemes introduce spherical
grids centered at each atom with integration points becoming more diffuse at larger
radial distances. For example, combining Euler-Maclaurin3 radial and Lebedev4
angular quadratures for each atom-centered grid allows integration around an atom
centered at !a as follows,
f (!r )d 3!r! " EiLj
j# f (!a + !rij )
i# = $p f (
!a + !rp )p# (4.4)
where Ei and Lj represent radial and angular weights respectively, p is a combined
ij index and ! p = Ei Lj . Each atomic grid independently integrates over all !3 but is
most accurate in the region of its own nucleus. In forming molecular grids, quadrature
points originating from different atoms must be weighted to normalize the total sum
over all atoms and to ensure that each atomic quadrature dominates in the region
around its nucleus. An elegant scheme introduced by Becke5 accomplishes this by
defining a spatial function, wa (!r ) , for each atomic quadrature, a , that gives the non-
negative weight assigned to that quadrature in the region of !r . Double counting is
avoided by constraining the weight function so that the following holds for all !r .
wa (!r )a! = 1 (4.5)
a Adapted from GPU Computing Gems, Emerald Edition, N. Luehr, I. Ufimtsev, T. Martinez, “Dynamical Quadrature Grids, Applications in Density Functional Calculations,” 35-42, Copyright (2011), with permission from Elsevier.

73
Constructing wa (!r ) so that it is near unity in the region of the ath nucleus also causes
each atomic quadrature to dominate in its most accurate region. The final quadrature is
then evaluated as follows.
f (!r )d 3!r! " wa (
!Ra +!rp )#p f (
!Ra +!rp )
p$
a$ = wap#p f (
!rap )p$
a$ (4.6)
Because the quadrature grid is now slaved to the molecular geometry, it must
provide well-defined gradients with respect to nuclear motion.6 This is easily
accomplished by ensuring that wa (!r ) is differentiable. Thus, Becke proposed the
following weighting scheme.5
wnp =Pn(!rnp )
Pm(!rnp )m! (4.7)
Pa (!r ) = s(µab)
b!a" (4.8)
µab(!r ) =
!Ra !!r !!Rb !!r
!Ra !
!Rb
=!ra !!rb!
Rab
(4.9)
s(µ) = 1
21! g(µ)( )
(4.10)
g(µ) = p( p( p(µ))) (4.11)
p(µ) = 3
2µ ! 1
2µ3 (4.12)
As is easily verified from the equations above, the effort to compute the Becke
weight at each grid point is determined by the denominator of equation (4.7) and
scales as O(N2) with the number of atoms in the system. Since the total number of grid
points also increases linearly with number of atoms, the total effort to compute the

74
quadrature grid is O(N3). Although the prefactor is modest if compared to Coulomb
and Exchange evaluation, generating the quadrature grid still becomes the dominant
bottleneck for calculations on large systems that are now routinely handled by the
methods described in chapters 2 and 3.
In order to reduce the formal scaling of grid generation, Stratmann and
coworkers replaced Becke’s switching function, s(µ) defined in equation (4.10), with
the following piecewise definition, where the parameter a is given the empirically
determined value of 0.64.7
s(µ) =
1 !1" µ " !a121! z(µ)( ) !a " µ " +a
0 +a " µ " +1
#
$%%
&%%
(4.13)
z(µ) = 116
35(µ / a)! 35(µ / a)3 + 21(µ / a)5 ! 5(µ / a)7"# $% (4.14)
Equation (4.13) drives the Becke weight to zero significantly faster for distant
quadrature points than equation (4.10). This allows a distance cutoff to be introduced
so that the sum and product of equations (4.7) and (4.8) need only consider a small set
of atoms in the neighborhood of !rnp . For large systems, the number of significant
neighbors becomes constant leading to linear scaling.
In our implementation, the GPU is used to evaluate equation (4.7) for each grid
point since this step dominates the runtime of the grid setup procedure. Before the
kernel is called, the spherical grids are expanded around each atom by table lookup,
the points are sorted into spatial bins, and lists of neighbor atoms are generated for
each bin. These steps are nearly identical for CPU and GPU implementations. The

75
table lookup and point sorting are already very fast on the CPU, and no GPU
implementation is planned. Building atom lists for each bin is more time consuming
and may benefit from further GPU acceleration in the future, but is not a focus of the
present chapter.
The serial implementation of Becke weight generation falls naturally into the
triple loop pseudo-coded in figure 1. Since the denominator includes the numerator as
one of its terms, we focus on calculating the former, setting aside the numerator in a
local variable when we come upon it.
Figure 1: Pseudo-code listing of Becke Kernel as a serial triple loop.
In order to accelerate this algorithm on the GPU, we first needed to locate a
fine-grained yet independent task to serve as the basis for our device threads. These
arise naturally by dividing loop iterations in the serial implementation into CUDA
blocks and threads. Our attempts at optimization fell into two categories. The finest
level of parallelism was to have each block cooperate to produce the weight for a
single point (block-per-point). In this scheme CUDA threads were arranged into 2D
01 for each point in point_array 02 P_a = P_sum = 0.0f 03 list = point.atom_list 04 for each atomA in list 05 Ra = dist(atomA, point) 06 P_tmp = 1.0f 07 for each atomB in list 08 Rb = dist(atomB, point) 09 rec_Rab = rdist(atomA, atomB) 10 mu = (Ra-Rb) * rec_Rab 11 P_tmp *= S(mu) 12 if(P_tmp < P_THRE) 13 break 14 P_sum += P_tmp 15 if(atomA == point.center_atom) 16 P_a = P_tmp 17 point.weight = point.spherical_weight * P_a/P_sum

76
blocks. The y-dimension specified the atomA index, and the x-dimension specified the
atomB index (following notation from figure 1). The second optimization scheme split
the outermost loop and assigned an independent thread to each weight (thread-per-
point).
In addition to greater parallelism, the block-per-point arrangement offered an
additional caching advantage. Rather than re-calculating each atom-to-point distance
(needed at lines 5 and 8), these values were cached in shared memory avoiding
roughly nine floating-point operations in the inner loop.
Unfortunately, this boost was more than offset by the cost of significant block
level cooperation. First of all, lines 11 and 14 required block-level reductions in the x
and y dimensions respectively. Secondly, because the number of members in each
atom list varies from as few as 2 to more than 80 atoms, any chosen block dimension
was necessarily suboptimal for many bins. Finally, the early exit condition at line 12
proved problematic. Since s(µ) falls between 0.0 and 1.0, each iteration of the atomB
loop can only reduce the value of P_tmp. Once it has been effectively reduced to zero,
we can safely exit the loop. As a result, calculating multiple “iterations” of the loop in
parallel required wasted terms to be calculated before the early exit was reached by all
threads of a warp.
On the other hand, the coarser parallelism of the thread-per-point did not
reduce its occupancy on the GPU. In the optimized kernel each thread required a
modest 29 registers and no shared memory. Thus, reasonable occupancies of 16 warps
per SM were obtained (using a Tesla C1060 GPU). Since a usual calculation requires

77
millions of quadrature points (and thus threads), there was also plenty of work
available to saturate the GPU many execution cores.
The thread-per-point implementation was, however, highly sensitive to
branching within a warp. Because each warp is executed in SIMD fashion,
neighboring threads can drag each other through non-essential instructions. This can
happen in two ways, 1) a warp contains points from bins with varying neighbor list
lengths or 2) threads in a warp exit at different iterations of the inner loop. Since many
bins have only a few points and thus warps often span multiple bins, case one can
become an important factor. Fortunately, it is easily mitigated by presorting the bins
such that neighboring warps will have similar loop limits.
The second case is more problematic and limited initial versions of the kernel
to only 132 GFLOPS. This was only a third of the Tesla C1060’s single issue
potential. Removing the early exit and forcing the threads to complete all inner loop
iterations significantly improved the GPU’s floating-point throughput to 260
GFLOPS, 84% of the theoretical single-issue peak. However, the computation time
also increased, as the total amount of work more than tripled for our test geometry. To
minimize branching, the points within each bin were further sorted such that nearest
neighbors were executed in nearby threads. In this way, each thread in a warp behaved
as similarly as possible under the branch conditions in the code. This adjustment
ultimately provided a modest performance increase to 187 GFLOPS of sustained
performance, about 60% of the single issue peak. However, thread divergence remains
a significant barrier to greater efficiency.

78
After the bins and points have been sorted on the host, the data is moved to the
GPU. We copy each point’s Cartesian coordinates, spherical weights (!p in equation
(4.4)), central atom index, and bin index to GPU global memory arrays. The atomic
coordinates for each bin list are copied to the GPU in bin-major order. This order
allows threads across a bin barrier to better coalesce their reads, which is necessary
since threads working on different bins often share a warp. Once the data is arranged
on the GPU, the kernel is launched, calculates the Becke weight and combines it with
the spherical weight in place. Finally, the weights are copied back to the host and
stored for later use in the DFT calculation.
Precision Number of Electrons Difference from Full Double Precision
Kernel Execution Time
Single 1365.9964166131 3.5x10-6 306.85 (ms) Mixed 1365.9964131574 3.9x10-8 307.68 (ms) Double 1365.9964131181 N/A 3076.76 (ms) Table 1: Comparison of single, mixed, and full double precision calculation of total charge for Olestra, a molecule with 1366 electrons.
Because the GPU is up to an order of magnitude faster using single rather than
double precision arithmetic, our kernel was designed to minimize double precision
operations. Double precision is most important when accumulating small numbers into
larger ones or when taking differences of nearly equal numbers. With this in mind we
were able to improve our single precision results using only a handful of double
precision operations. Specifically, the accumulation at line 14 and the final arithmetic
of line 17 in figure 1 were carried out in double precision. This had essentially no
impact on performance, but improved correlation with the full double precision results
by more than an order of magnitude.

79
The mixed precision GPU code can be compared to a CPU implementation of
the same algorithm. To level the playing field, the CPU code uses the same mixed
precision scheme developed for the GPU. However, the CPU implementation was not
parallelized to multiple cores. The reference timings were taken using an Intel Xeon
X5570 at 2.93 GHz and an nVidia Tesla C1060 GPU. Quadrature grids were
generated for a representative set of test geometries shown in figure 2. These ranged in
size from about 100 to nearly 900 atoms.
Figure 2: Test Molecules
Molecule Atoms (Avg. List)
Points GPU Kernel Time (ms)
CPU Kernel Time (ms)
Speedup
Taxol 110 (19) 607828 89.1 12145.3 136X Valinomycin 168 (25) 925137 212.0 26132.2 123X Olestra 453 (18) 2553521 262.9 30373.6 116X BPTI 875 (39) 4785813 2330.3 364950.8 157X Table 2: Performance comparison between CPU and GPU Becke weight calculation. Sorting and GPU data transfers are not included. Along with the total number of atoms in each molecule, the mean length of the neighbor list for that molecule is reported in parentheses. Times are reported in milliseconds.
Table 2 compares the CPU and GPU treatments of the Becke weight
evaluation (code corresponding to figure 1). The observed scaling is not strictly linear
with system size. This is because the test molecules exhibit varying atomic densities.
When the atoms are more densely packed, more neighbors appear in the inner loops.
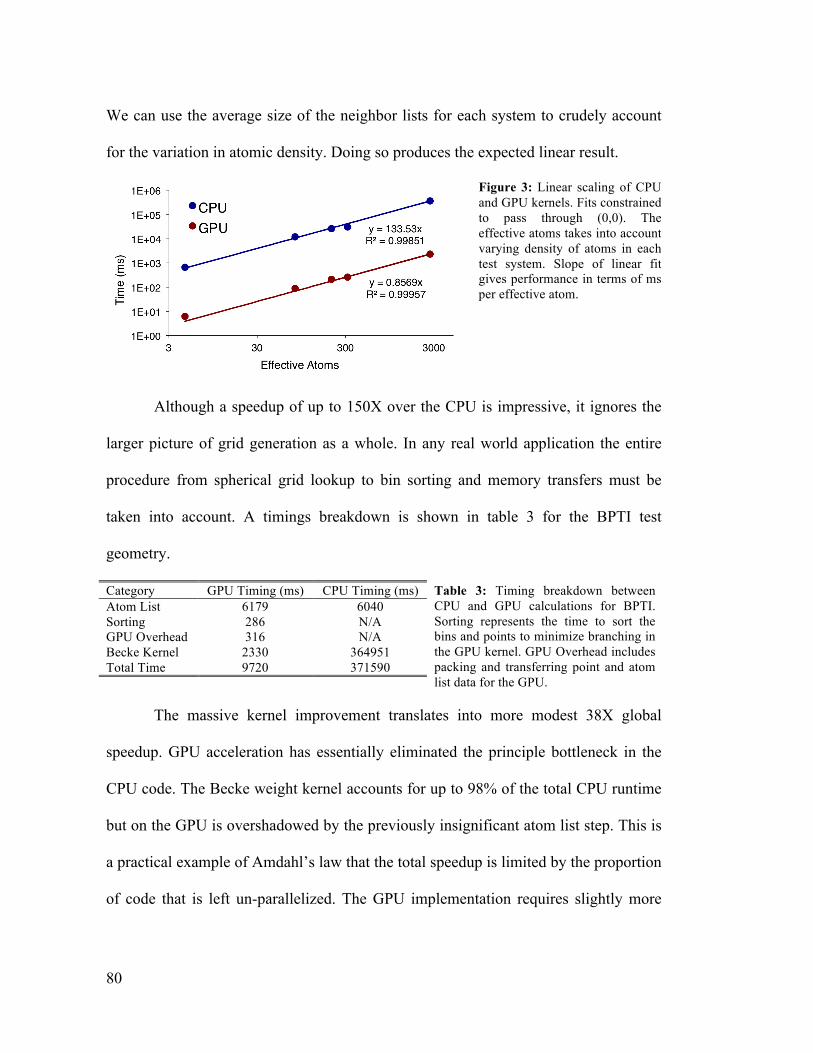
80
We can use the average size of the neighbor lists for each system to crudely account
for the variation in atomic density. Doing so produces the expected linear result.
Figure 3: Linear scaling of CPU and GPU kernels. Fits constrained to pass through (0,0). The effective atoms takes into account varying density of atoms in each test system. Slope of linear fit gives performance in terms of ms per effective atom.
Although a speedup of up to 150X over the CPU is impressive, it ignores the
larger picture of grid generation as a whole. In any real world application the entire
procedure from spherical grid lookup to bin sorting and memory transfers must be
taken into account. A timings breakdown is shown in table 3 for the BPTI test
geometry.
Category GPU Timing (ms) CPU Timing (ms) Table 3: Timing breakdown between CPU and GPU calculations for BPTI. Sorting represents the time to sort the bins and points to minimize branching in the GPU kernel. GPU Overhead includes packing and transferring point and atom list data for the GPU.
Atom List 6179 6040 Sorting 286 N/A GPU Overhead 316 N/A Becke Kernel 2330 364951 Total Time 9720 371590
The massive kernel improvement translates into more modest 38X global
speedup. GPU acceleration has essentially eliminated the principle bottleneck in the
CPU code. The Becke weight kernel accounts for up to 98% of the total CPU runtime
but on the GPU is overshadowed by the previously insignificant atom list step. This is
a practical example of Amdahl’s law that the total speedup is limited by the proportion
of code that is left un-parallelized. The GPU implementation requires slightly more

81
time to generate the atom lists due to the additional sorting by length required to
minimize warp divergence on the GPU.
The final weights for many individual quadrature points are negligibly small.
These are removed from the final grid, and the Cartesian coordinates and weights of
all remaining points are stored for use in later integration.
wap!p "# i!rap "
!ri (4.15)
EXCHANGE-CORRELATION POTENTIAL EVALUATION
With the numerical quadrature grid in hand, we now consider the evaluation of
the exchange-correlation potential, Vxc in equation (4.2). Following Yasuda’s GPU
implementation,2 the calculation of Vxc is broken down into three parts. First, the
electronic density and its gradient are evaluated at each grid point. Second, the DFT
functional is evaluated at each grid point. And third, the functional values are summed
to produce the final matrix elements.
As with the ERI kernels considered previously, the calculation is segmented by
the angular momenta of the basis functions. For GGA functionals, however, the
exchange-correlation potential involves only one-electron terms, so that kernels deal
with pairs rather than quartets of basis shells. Thus, for a basis limited to s- , p-, and d-
functions, only six kernel classes are needed, ss, sp, sd, pp, pd, and dd. Threads
assigned to higher angular momenta shells again compute terms for all primitive
functions within the shell pair.

82
The alpha-spin electronic density and density gradient at each grid point, !ri ,
are calculated from the one particle density matrix P! as follows (with analogous
expressions for the beta spin case).
!" (!ri ) = Pµ#
" $µ (!ri )$# (!ri )µ#% (4.16)
!"# (!ri ) = 2 Pµ$
# %µ (!ri )!%$ (!ri )µ$& (4.17)
To compute these quantities, each CUDA thread is assigned a unique grid point and
loops over significant primitive shell pairs. As a result of the exponential decay of the
basis functions, !µ , the sum for each grid point need only include a few significant
AOs. Significant AO primitive pairs are spatially binned by the center of charge, !Pij in
equation (1.25), and sorted by increasing exponent. Each thread loops over all bins,
but exits each bin as soon as the first negligible point-pair interaction is reached. The
quadrature points are similarly binned to minimize warp divergence by ensuring that
neighboring threads share similar sets of significant basis functions.
Next, the density at each quadrature point is copied to host memory and used
to evaluate the xc-kernel function and its various derivatives on the grid. Specifically,
the following values are computed for each grid point.
ai =! i fxc "# (!ri ),"$ (
!ri ),% ## (!ri ),% #$ (
!ri ),% $$ (!ri )( )
bi =! i
& fxc!ri( )
&"#
ci =! i 2& fxc
!ri( )&% ##
'"# +& fxc
!ri( )&% #$
'"$
(
)*+
,-
(4.18)

83
This step has a small computational cost and can be performed on the host without
degrading performance. This is desirable as a programming convenience since
implementing various density functionals can be performed without editing CUDA
kernels, and because the host provides robust and efficient support for various
transcendental functions needed by some functionals. During this step, the functional
values, ai , are also summed to evaluate the total exchange-correlation energy per
equation (4.1).
The final step is to construct AO matrix elements for the exchange-correlation
potential. This task is again performed on the GPU. For all non-negligible primitive
pairs, !K! L , the pair quantities KKL ,
!PKL , and !KL from equation (1.25) are packed
into arrays and transferred to device memory. The grid coordinates, !ri , are spatially
binned and transferred to the GPU with corresponding values bi and ci . A CUDA
kernel then performs the summation,
!Vµ! = "µ ("ri )
bi2"! ("ri )+ ci#"! (
"ri )$%&
'()i
*
= ckcll+!*
k+µ* , k (
"ri )bi2, l ("ri )+ ci#, l (
"ri )$%&
'()i
* (4.19)
and the final matrix elements are computed as follows.
Vµ!XC = !Vµ! + !V!µ (4.20)
This calculation can be envisioned as a 2D matrix in which each row
corresponds to a primitive shell pair, !K! L , and each column is associated with a
single grid point. The CUDA grid is configured as a 1-D grid of 2-D blocks, similar to
the GPU J-Engine described in chapter 2. A stack of blocks spanning the primitive

84
pairs sweeps across the matrix. The points are spatially binned so that entire bins can
be skipped whenever a negligible point-primitive pair combination is encountered. As
in the Coulomb algorithm, each row of threads across a CUDA block cooperates to
produce a primitive matrix element, !Vkl . These are copied to the host where the final
summation into AO matrix elements takes place.
SCF PERFORMANCE EVAULATION
Having completed our discussion of GPU acceleration of SCF calculations we
illustrate the application of our work to practical calculations. All GPU calculations
described below were carried out using the TeraChem quantum chemistry package. To
elucidate the computational scaling of our GPU implementation with increasing
system size, we consider two types of systems. First, linear alkene chains ranging in
length between 25 and 707 carbon atoms provide a benchmark at the low-dimensional
limit. Second, cubic water clusters containing between 10 and 988 molecules provide
a dense, three-dimensional test system that is more representative of interesting
condensed phase systems. Examples of each system type are shown in figure 4.
Figure 4: One-dimensional alkene and three dimensional water-cube test systems. Alkene lengths vary from 24 to 706 carbon atoms and water cubes range from 10 to nearly 850 water molecules. A uniform density is used for all water boxes.
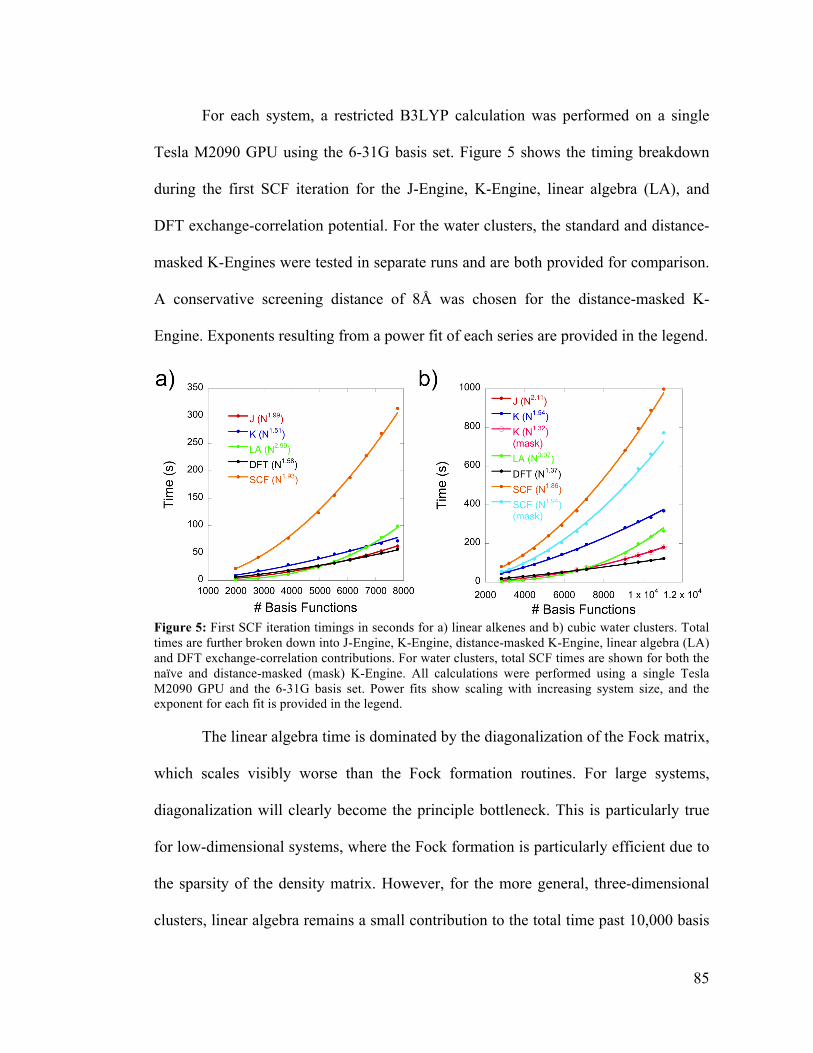
85
For each system, a restricted B3LYP calculation was performed on a single
Tesla M2090 GPU using the 6-31G basis set. Figure 5 shows the timing breakdown
during the first SCF iteration for the J-Engine, K-Engine, linear algebra (LA), and
DFT exchange-correlation potential. For the water clusters, the standard and distance-
masked K-Engines were tested in separate runs and are both provided for comparison.
A conservative screening distance of 8Å was chosen for the distance-masked K-
Engine. Exponents resulting from a power fit of each series are provided in the legend.
Figure 5: First SCF iteration timings in seconds for a) linear alkenes and b) cubic water clusters. Total times are further broken down into J-Engine, K-Engine, distance-masked K-Engine, linear algebra (LA) and DFT exchange-correlation contributions. For water clusters, total SCF times are shown for both the naïve and distance-masked (mask) K-Engine. All calculations were performed using a single Tesla M2090 GPU and the 6-31G basis set. Power fits show scaling with increasing system size, and the exponent for each fit is provided in the legend.
The linear algebra time is dominated by the diagonalization of the Fock matrix,
which scales visibly worse than the Fock formation routines. For large systems,
diagonalization will clearly become the principle bottleneck. This is particularly true
for low-dimensional systems, where the Fock formation is particularly efficient due to
the sparsity of the density matrix. However, for the more general, three-dimensional
clusters, linear algebra remains a small contribution to the total time past 10,000 basis

86
functions. If the more efficient masked K-Engine can be employed, the crossover
point, at which linear algebra becomes dominant, shifts to about 8,000 basis functions.
Although the time per basis function is lower for the alkene test series, the
overall scaling is similar for both test series. This behavior is good evidence that we
have penetrated the asymptotic regime of large systems, where the dimensionality of
the physical system should impact the prefactor rather than the exponent. That we
easily reach this crossover point on GPUs is itself noteworthy. Even more noteworthy
is the fact that the K-Engine exhibits sub-quadratic scaling without imposing any of
the assumptions or book-keeping mechanisms such as neighbor lists common among
linear scaling exchange algorithms.8-11 The further imposition of a simple distance
mask on exchange elements provides an effective linear scaling approach for
insulating systems. The scaling of the SCF with the masked K-Engine further
exemplifies the impact of the linear algebra computation on the overall efficiency. The
N3 scaling of LA becomes significantly more dominant in large systems. The validity
of the distance-based exchange screening approximation was confirmed by comparing
the final converged absolute SCF energies of each water cluster calculated with and
without screening. The absolute total electronic energy is accurate to better than 0.02
kcal/mol in all systems considered, which is well within chemical accuracy.
Our GPU J-Engine scales quadratically with system size, which comports well
with our scaling analysis in chapter 2. Further acceleration would require modification
of the long-ranged Coulomb potential, for example by factorization into a multipole
expansion.12 Extrapolating beyond figure 5, this may become necessary for water
clusters beyond 12,000 basis functions, when the J-Engine finally becomes the
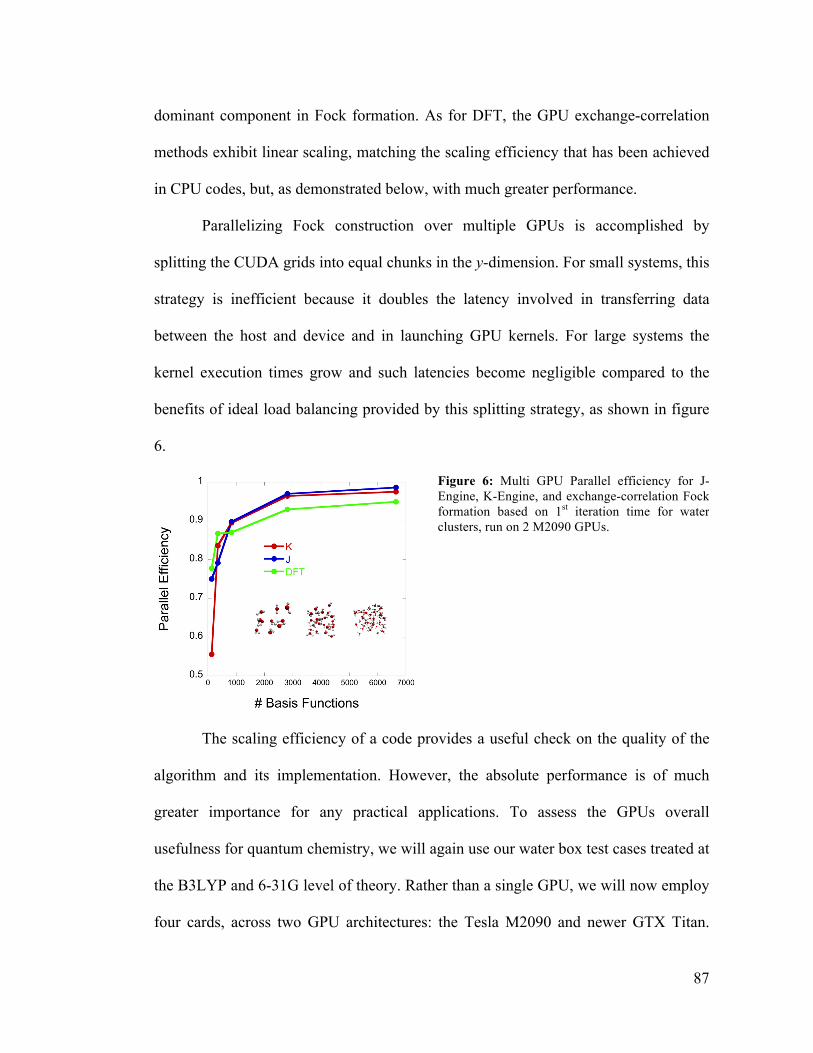
87
dominant component in Fock formation. As for DFT, the GPU exchange-correlation
methods exhibit linear scaling, matching the scaling efficiency that has been achieved
in CPU codes, but, as demonstrated below, with much greater performance.
Parallelizing Fock construction over multiple GPUs is accomplished by
splitting the CUDA grids into equal chunks in the y-dimension. For small systems, this
strategy is inefficient because it doubles the latency involved in transferring data
between the host and device and in launching GPU kernels. For large systems the
kernel execution times grow and such latencies become negligible compared to the
benefits of ideal load balancing provided by this splitting strategy, as shown in figure
6.
Figure 6: Multi GPU Parallel efficiency for J-Engine, K-Engine, and exchange-correlation Fock formation based on 1st iteration time for water clusters, run on 2 M2090 GPUs.
The scaling efficiency of a code provides a useful check on the quality of the
algorithm and its implementation. However, the absolute performance is of much
greater importance for any practical applications. To assess the GPUs overall
usefulness for quantum chemistry, we will again use our water box test cases treated at
the B3LYP and 6-31G level of theory. Rather than a single GPU, we will now employ
four cards, across two GPU architectures: the Tesla M2090 and newer GTX Titan.

88
Notably, a 1533 atom single-point energy calculation on a cubic water cluster required
only 2.47 hours for the entire SCF procedure, and ab initio dynamics, requiring
thousands of single point evaluations are feasible for systems containing thousands of
basis functions.
As a point of comparison, the same calculations were carried out using the
CPU implementation available in the GAMESS program.13 Parameters such as
integral neglect thresholds and SCF convergence criteria were matched as closely as
possible to the previous GPU calculations. The CPU calculation was also parallelized
over all 8 CPU cores available in our dual Xeon X5680 3.33GHz server. Figure 7
shows the speedup of the GPU implementation relative to GAMESS for the first SCF
iteration including both Fock construction and diagonalization, the latter of which is
performed in both codes on the CPU. Performance is similar between the two GPU
models on small structures for both alkenes and water clusters. However, GTX Titan
exhibits a larger speedup relative to GAMESS above 100 orbitals and reaches 400x at
the largest systems examined. During Fock construction, the CPU cores are left idle in
our implementation. It is also possible to reserve some work for the CPU,14 but given
the performance advantage of our GPU implementation, this would offer a rather
small performance improvement.

89
Figure 7: Total SCF time of TeraChem on 8 CPUs and 4 GPUs, relative to GAMESS on 8 CPUs for water clusters.
REFERENCES
(1) Pople, J. A.; Gill, P. M. W.; Johnson, B. G. Chem. Phys. Lett. 1992, 199, 557.
(2) Yasuda, K. J. Chem. Theo. Comp. 2008, 4, 1230.
(3) Murray, C. W.; Handy, N. C.; Laming, G. J. Mol. Phys. 1993, 78, 997.
(4) Lebedev, V. I.; Laikov, D. N. Dokl Akad Nauk+ 1999, 366, 741.
(5) Becke, A. D. J. Chem. Phys. 1988, 88, 2547.
(6) Johnson, B. G.; Gill, P. M. W.; Pople, J. A. J. Chem. Phys. 1993, 98, 5612.
(7) Stratmann, R. E.; Scuseria, G. E.; Frisch, M. J. Chem. Phys. Lett. 1996, 257, 213.
(8) Burant, J. C.; Scuseria, G. E.; Frisch, M. J. J. Chem. Phys. 1996, 105, 8969.
(9) Ochsenfeld, C.; White, C. A.; Head-Gordon, M. J. Chem. Phys. 1998, 109, 1663.
(10) Schwegler, E.; Challacombe, M. J. Chem. Phys. 1999, 111, 6223.
(11) Neese, F.; Wennmohs, F.; Hansen, A.; Becker, U. Chem. Phys. 2009, 356, 98.
(12) White, C. A.; Head-Gordon, M. J. Chem. Phys. 1994, 101, 6593.
(13) Schmidt, M. W.; Baldridge, K. K.; Boatz, J. A.; Elbert, S. T.; Gordon, M. S.; Jensen, J. H.; Koseki, S.; Matsunaga, N.; Nguyen, K. A.; Su, S. J.; Windus, T. L.; Dupuis, M.; Montgomery, J. A. J. Comp. Chem. 1993, 14, 1347.

90
(14) Asadchev, A.; Gordon, M. S. J. Chem. Theo. Comp. 2012, 8, 4166.

91
CHAPTER FIVE
EXCITED STATE ELECTRONIC STRUCTURE
ON GPUS: CIS AND TDDFTa
In the previous chapters we described GPU algorithms to accelerate the
construction of the Fock matrix for use in SCF calculations. We now demonstrate how
those efficient Fock methods can be applied more widely to dramatically increase the
performance of post-SCF calculations such as single excitation configuration
interaction (CIS),1 time-dependent Hartree-Fock (TDHF) and linear response time-
dependent density functional theory (TDDFT).2-6 These single-reference methods are
widely used for ab initio calculations of electronic excited states of large molecules
(more than 50 atoms, thousands of basis functions) because they are computationally
efficient and straightforward to apply.7-9 Although highly correlated and/or multi-
reference methods such as multireference configuration interaction (MRCI10),
multireference perturbation theory (MRMP11 and CASPT212), and equation-of-motion
coupled cluster methods (SAC-CI13 and EOM-CC14,15) allow for more reliably
accurate treatment of excited states, including those with double excitation character,
these are far too computationally demanding for large molecules.
CIS/TDHF is essentially the excited state corollary of the ground state Hartree-
Fock method, and thus similarly suffers from a lack of electron correlation. Because
a Adapted from C.M. Isborn, N. Luehr, I.S. Ufimtsev, and T.J. Martinez, J. Chem. Theo. Comput. 2011, 7, 1814-1823. This is an unofficial adaptation of an article that appeared in an ACS publication. ACS has not endorsed the content of this adaptation or the context of its use.

92
of this, CIS/TDHF excitation energies are consistently overestimated, often by ~1eV.8
The TDDFT method includes dynamic correlation through the exchange correlation
functional, but standard non-hybrid TDDFT exchange-correlation functionals tend to
underestimate excitation energies, particularly for Rydberg and charge transfer states.5
The problem in charge-transfer excitation energies is due to the lack of the correct 1/r
Coulombic attraction between the separated charges of the excited electron and hole.16
Charge transfer excitation energies are generally improved with hybrid functionals and
also with range separated functionals that separate the exchange portion of the DFT
functional into long and short range contributions.17-21 Neither CIS nor TDDFT (with
present-day functionals) properly include the effects of dispersion, but promising
results have been obtained with an empirical correction to standard DFT
functionals,22,23 and there are continued efforts to include dispersion directly in the
exchange-correlation functional.24,25 Both the CIS and TDDFTa single reference
methods lack double excitations and are unable to model conical intersections or
excitations in molecules that have multi-reference character.26,27 In spite of these
limitations, the CIS and TDDFT methods can be generally expected to reproduce
trends for one-electron valence excitations, which are a majority of the transitions of
photochemical interest. TDDFT using hybrid density functionals, which include some
percentage of Hartree-Fock (HF) exact exchange, have been particularly successful in
modeling the optical absorption of large molecules. Furthermore, the development of
new DFT functionals and methods is an avid area of research, with many new
a By TDDFT we here refer to the adiabatic linear response formalism with presently available functionals.

93
functionals introduced each year. Thus it is likely that the quality of the results
available from TDDFT will continue to improve. A summary of the accuracy currently
available for vertical excitation energies is available in a recent study by Jacquemin et
al. which compares TDDFT results using 29 functionals for ~500 molecules.28
Although CIS and TDDFT are the most tractable methods for treating excited
states of large molecules, their computational cost still prevents application to many
systems of photochemical interest. Thus, there is considerable interest in extending the
capabilities of CIS/TDDFT to even larger molecules, beyond hundreds of atoms.
Quantum mechanics/molecular mechanics (QM/MM) schemes provide a way
to model the environment of a photophysically interesting molecule by treating the
molecule with QM and the surrounding environment with MM force fields.29-33
However, it is difficult to know when the MM approximations break down and when a
fully QM approach is necessary. With fast, large scale CIS/TDDFT calculations, all
residues of a photoactive protein could be treated quantum mechanically to explore the
origin of spectral tuning, for example. Explicit effects of solvent-chromophore
interactions, including hydrogen bonding, charge transfer, and polarization, could be
fully included at the ab initio level in order to model solvatochromic shifts.
GPUs provide a promising route to large-scale CIS and TDDFT calculations.
GPUs have been applied to achieve speed-ups of 1-2 orders of magnitude in ground
state electronic structure,34-36 ab initio molecular dynamics37 and empirical force field-
based molecular dynamics calculations.38-41 In this chapter we extend the
implementation of GPU quantum chemistry beyond the SCF methods36,42 described in
previous chapters to the calculation of excited electronic states. The performance

94
provided by GPU hardware for evaluating ERIs allows full QM treatment of the
excited states of very large systems – both large chromophores and chromophores in
which the environment plays a critical role and should be treated with QM. In this
chapter we present the results of implementing CIS and TDDFT within the Tamm-
Dancoff approximation using GPUs to drastically accelerate the bottlenecks of two-
electron integral evaluation, density functional quadrature, and matrix multiplication.
This results in CIS calculations over 200x faster than those achieved running on a
comparable CPU platform. Benchmark CIS/TDDFT timings are presented for a
variety of systems.
CIS/TDDFT IMPLEMENTATION USING GPUS
The linear response formalism of TDHF and TDDFT has been thoroughly
presented in review articles.4,7,8,43 Only the equations relevant for this work are
presented here. The TDHF/TDDFT working equation is
A BB A
!"#
$%&
XY
!"#
$%&=' 1 0
0 (1!"#
$%&
XY
!"#
$%&
, (5.1)
where for TDHF (neglecting spin indices for simplicity)
Aai,bj = ! ij!ab "a # " i( ) + ia | jb( ) # ij | ab( ) , (5.2)
Bai,bj = ia | bj( )! ib | aj( ) , (5.3)
and for TDDFT
Aai,bj = ! ij!ab "a # " i( ) + ia | jb( ) + ij | fxc | ab( ), (5.4)
Bai,bj = ia |bj( ) + ib | fxc | aj( ) . (5.5)
The ERIs are defined as usual.

95
ia | jb( ) =! i(r1)!a (r1)! j (r2)!b (r2)
r1 " r2## dr1dr2 (5.6)
Within the adiabatic approximation of DFT, the exact time-dependent exchange-
correlation potential is approximated using the time-independent ground state
exchange-correlation functional Exc[!] as follows.
ia | fxc | jb( ) = ! i(r1)!a (r1)" 2Exc
"#(r1)"#(r2)$$ ! j (r2)!b (r2)dr1dr2 (5.7)
The i,j and a,b indices represent occupied and virtual MOs, respectively, in the
Hartree-Fock/Kohn-Sham (KS) ground state determinant.
Setting the B matrix to zero within TDHF results in the CIS equation, while in
TDDFT this same neglect yields the equation known as the Tamm-Dancoff
approximation (TDA):
AX =!X . (5.8)
In part because DFT virtual orbitals provide a better starting approximation to the
excited state than HF orbitals, the neglect of the B matrix within TDA is known to
accurately reproduce full TDDFT results for non-hybrid DFT functionals.7,8,44
Furthermore, previous work has shown that a large contribution from the B matrix in
TDDFT (and to a lesser extent also in TDHF) is often indicative of a poor description
of the ground state, either due to singlet-triplet instabilities or multi-reference
character.45 Thus, if there is substantial deviation between the full TDDFT and TDA-
TDDFT excitation energies, the TDA results will often be more accurate.
A standard iterative Davidson algorithm46 has been implemented to solve the
CIS/TDA-TDDFT equations. As each AX matrix-vector product is formed, the

96
required two-electron integrals are calculated over primitive basis functions within the
atomic orbital (AO) basis directly on the GPU. Within CIS, the AX matrix-vector
product is calculated as follows.
ACISX( )bj = ! ij!ab "a # " i( ) + ia | jb( ) # (ij | ab)[ ]Xiaia$ (5.9)
ia | jb( ) ! (ij | ab)[ ]Xiaia" = Cµj
µ#" C#bFµ# (5.10)
Fµ! = T"#"#$ µ! |"#( ) % µ" |!#( ){ } (5.11)
T!" = Xiaia# C!iC"a (5.12)
Here C!i are the SCF MO coefficients of the HF/KS determinant, and T!" is a non-
symmetric transition density matrix. For very small systems there is no performance
advantage with GPU computation of the matrix multiplication steps in equations
(5.10) and (5.12), in which case we compute these quantities on the CPU. For larger
systems, the matrix-vector multiplications are performed on the GPU using dgemm
calls to the CUBLAS library, a CUDA BLAS implementation.47
Equation (5.11) represents the dominant computational bottleneck of this
integral direct CIS algorithm. Fortunately, this equation is equivalent to the Coulomb
and exchange matrix builds described in chapters 2 – 4 except that the non-symmetric
transition density, T, replaces the symmetric one-particle density matrix, P. Thus, the
portion of the F matrix from the product of T!" with the first integral in equation (5.11)
is computed with the GPU J-engine algorithm. The portion of the F matrix from the
product of T!" with the second integral in equation (5.11) is computed with the K-
engine algorithm. The coulomb matrix remains symmetric even with a non-symmetric

97
transition density matrix. Thus, it is possible to continue to eliminate redundant µ# "
#µ bra and !" " "! ket pairs as in the SCF routine as long as the sum of transpose
density elements is used as follows.
Jµ! = J!µ = µ! | "#( ) T"# + 1$%"#( )T#"( )
"&#'
(5.13)
The excited state exchange matrix is not symmetric. We must thus calculate both the
upper and lower triangle contributions with separate calls to the ground state GPU K-
engine routines. Since the SCF K-engine already ignores µ# " #µ bra and !" " "!
ket symmetries, the result is that, ignoring screening, all O(N4) ERIs are computed for
the excited state K contribution, while the J term requires the same O(N4/4) ERIs
computed in the ground state.
Compared to the J-engine, the K-engine suffers from a second disadvantage for
both ground and excited state calculations. Unlike the J-matrix GPU implementation,
the K-matrix algorithm cannot map the density matrix elements onto the ket integral
data, since the density index now spans both bra and ket indices. This leads to two
negative consequences. First, each thread must load an independent density matrix
element which results in slower non-coalesced GPU memory accesses. Second, the
sparsity of the density cannot be included in the pre-sorting of ket pairs. Thus, the
integral bounds are not guaranteed to strictly decrease and a mix of significant and
insignificant ERIs must be computed. As a result of these drawbacks, the exchange
algorithm is less efficient on the GPU and actually takes longer to calculate than its
Coulomb counterpart even though density screening should in principle leave many
fewer significant ERI contributions in the case of exchange. In practice a K/J timing

98
ratio for ground state SCF calculations is observed to be between 3 and 5 for systems
studied below.
Evaluation of the derivative of the exchange-correlation potential needed for
TDDFT excited states7 is performed using the same numerical quadrature as the
ground state exchange-correlation potential. We again use a Becke type quadrature
scheme48 with Lebedev angular49 and Euler-Maclaurin radial50 quadrature grids. The
same basic approach described in the previous chapter is again employed to compute
the exchange-correlation matrix elements of equation (5.7).35,51 The expensive steps
are evaluating the electron density/gradient at the grid quadrature points to
numerically evaluate the necessary functionals and summing the values on the grid to
assemble the matrix elements of the exchange-correlation potential. For the excited
state calculations, we generate the second functional derivative of the exchange
correlation functional only once, saving its value at each quadrature point in memory.
Then, for each Davidson iteration, the appropriate integrals are evaluated, paired with
the saved functional derivative values and summed into matrix elements using GPU
kernels analogous to the ground state case detailed in the previous chapter.
RESULTS AND DISCUSSION
We evaluate the performance of our GPU-based CIS/TDDFT algorithm on a
variety of test systems: B3PPQ – 6,6’-bis(2-(1-triphenyl)-4-phenylquinoline - an
oligoquinoline recently synthesized and characterized by the Jenekhe group for use in
OLED devices52 and characterized theoretically by Tao and Tretiak;53 four generations
of oligothiophene dendrimers that are being studied for their interesting photophysical
properties54-56; the entire photoactive yellow protein (PYP)57 solvated by TIP3P58

99
water molecules; and deprotonated trans-thiophenyl-p-coumarate, an analogue of the
PYP chromophore59 that takes into account the covalent cysteine linkage, solvated
with an increasing number of QM waters.
Figure 1. Structures, number of atoms, and basis functions (fns) using the 6-31G basis set for four generations of oligothiophene dendrimers, S1-S4. Carbon atoms are orange, sulfur atoms are yellow.
Figure 2. Structures, number of atoms, and basis functions (fns) for the 6-31G basis for benchmark systems photoactive yellow protein (PYP), the solvated PYP chromophore, and oligoquinoline B3PPQ. For PYP, carbon, nitrogen, oxygen and sulfur atoms are green, blue, red, and yellow, respectively. For the other molecules, atom coloration is as given in figure 1, with additional red and blue coloration for oxygen and nitrogen atoms, respectively.
Benchmark structures are shown in figures 1 and 2 along with the number of
atoms and basis functions for a 6-31G basis set. For the solvated PYP chromophore,
only three structures are shown in figure 2, but benchmark calculations are presented

100
for 15 systems with increasing solvation, starting from the chromophore in vacuum
and adding water molecules up to a 16 Ångstrom solvation shell, which corresponds to
900 water molecules.
For our benchmark TDDFT calculations, we use the generalized gradient
approximation with Becke’s exchange functional60 combined with the Lee, Yang, and
Parr correlation functional61 (BLYP). During the SCF procedure for the ground state
wavefunction, we use two different DFT grids. A sparse grid of ~1000 grid
points/atom is used to converge the wave function until the DIIS error reaches a value
of 0.01, followed by a more dense grid of ~3000 grid points/atom until the ground
state wave function is fully converged. This denser grid is also used for the excited
state TDDFT timings reported herein, unless otherwise noted. The Coulomb and
exchange integral screening thresholds are set to 1x10-11 atomic units. Coulomb and
exchange integrals with products of the density element and Schwarz bound below the
integral screening threshold are not computed, and exchange evaluation is terminated
when the products of density element and Schwarz bound fall below this value times a
guard factor of 0.001. The timings reported were obtained using a dual quad-core Intel
Xeon X5570 platform with 72 GB RAM and eight Tesla C1060 GPUs.
All CPU operations are performed in full double precision arithmetic,
including one-electron integral evaluation, integral accumulation, and diagonalization
of the subspace matrix of A. Calculations carried out on the GPU (Coulomb and
exchange operator construction and DFT quadrature) use mixed precision unless
otherwise noted. The mixed precision integral evaluation is a hybrid of 32-bit and 64-
bit arithmetic. In this case, integrals with Schwarz bounds larger than 0.001 a.u. are

101
computed in full double precision, and all others are computed in single precision with
double precision accumulation into the final matrix elements. To study the effects of
using single precision on excited state calculations, we have run the same CIS
calculations using both single and double precision integral evaluation for many of our
benchmark systems.
Figure 3. Plot of single and double precision convergence behavior for the 1st CIS/6-31G excited state of five benchmark systems. A typical convergence threshold of 10-5 in the residual norm is indicated with a straight black line. Convergence behavior is generally identical for single and double precision integration until very small residual values well below the convergence threshold. Some calculations do require double precision for convergence. One such example is shown here for a snapshot of the PYP chromophore (PYPc) with 94 waters.
In general we find that mixed (and often even single) precision arithmetic on
the GPU is more than adequate for CIS/TDDFT, with convergence being achieved in
as many or fewer iterations than is required for the identical convergence criterion for
GAMESS. In most cases we find that the convergence behavior is nearly identical for
single and double precision until the residual vector is quite small. Figure 3 shows the
typical single and double precision convergence behavior as represented by the CIS
residual vector norm convergence for B3PPQ, the 1st and 3rd generations of
oligothiophene dendrimers S1 and S3, and a snapshot of the PYP chromophore
surrounded by 14 waters. A common convergence criterion on the residual norm is

102
shown with a straight black line at 10-5 a.u. Note that for the examples in figure 3, we
are not using mixed precision – all two electron integrals on the GPU are done in
single precision. This is therefore an extreme example (other calculations detailed in
this paper used mixed precision where large integrals and quadrature contributions are
calculated in double precision) and serves to show that CIS and TDDFT are generally
quite robust, irrespective of the precision used in the calculation. Nevertheless, a few
problematic cases have been found in which single precision integral evaluation is not
adequate and where double precision is needed to achieve convergence.a During the
course of hundreds of CIS calculations performed on snapshots of the dynamics of the
PYP chromophore solvated by various numbers of water molecules, a small number
(<1%) of cases yield ill-conditioned Davidson convergence when single precision is
used for the GPU-computed ERIs and quadrature contributions. For illustration, the
single and double precision convergence behavior for one of these rare cases, here the
PYP chromophore with 94 waters, is shown in figure 3. In practice, this is not a
problem since one can always switch to double precision and this can be done
automatically when convergence problems are detected.
Timings and excitation energies for some of the test systems are given in table
1 and compared to the GAMESS quantum chemistry package version 12 Jan 2009
(R3). The GAMESS timings are obtained using the same Intel Xeon X5570 eight-core
machine as for the GPU calculations (where GAMESS is running in parallel over all
eight cores). The numerical accuracy of the excitation energies for mixed precision
a Of course, if the convergence threshold was made sufficiently small, all calculations would require double (or better) precision throughout.

103
GPU integral evaluation is excellent for all systems studied; the largest discrepancy
between GAMESS and our GPU implementation is less than 0.0001eV. Speedups are
given for both the total SCF time and CIS computation time, with a large increase in
performance times obtained using the GPU for both ground and excited state methods.
The speedups increase as system size increases, with SCF speedups outperforming
CIS speedups. For the largest system compared with GAMESS, which is the 29 atom
chromophore of PYP surrounded by 487 QM water molecules, the speedup is well
over 400# for SCF and 200# for CIS.
molecule (atoms; basis functions) CIS timings (s) Speedup DES0/S1 (au) GPU GAMESS SCF CIS GPU GAMESS
B3PPQ oligoquinoline (112; 700) 38.6 371.5 15 10 0.1729276 0.1729293 S2 oligothiophene dendrimer (128; 958) 117.5 755.9 15 6 0.1511511 0.1511509 PYP chromophore + 101 waters (332; 1501) 164.8 3032.7 52 18 0.1338027 0.1338021 PYP chromophore + 146 waters (467; 2086) 286.7 8654.9 90 30 0.1337468 0.1337463 PYP chromophore + 192 waters (605; 2684) 431.5 20546.8 133 48 0.1338623 0.1338617 PYP chromophore + 261 waters (812; 3581) 715.4 57800.5 212 81 0.1339657 0.1339651 PYP chromophore + 397 waters (1220; 5349) 1459.3 243975.7 353 167 0.1341203 0.1341196 PYP chromophore + 487 waters (1490; 6519) 2408.1 562606.6 421 234 0.1343182 0.1343174 Table 1. Accuracy and performance of the CIS algorithm on a dual Intel Xeon X5570 (eight CPU cores) with 72 GB RAM. GPU Calculations use eight Tesla C1060 GPU cards.
The dominant computational parts in building the CIS/TDDFT AX vector can
be divided into Coulomb matrix, exchange matrix and DFT contributions. Figure 4
plots the CPU+GPU time consumed by each of these three contributions (both CPU
and GPU times are included here, although the CPU time is a very small fraction of
the total). J and K timings are taken from an average of the ten initial guess AX builds
for a CIS calculation, and the DFT timings are from an average of the initial guess AX
builds for a TD-BLYP calculation. The initial guess transition densities are very sparse
and thus this test highlights the differing efficiency of screening and thresholding in

104
these three contributions. The J-timings for CIS and BLYP are similar, and only those
for CIS are reported. Power law fits are shown as solid lines and demonstrate near-
linear scaling behavior of all three contributions to the AX build. The coulomb matrix
and DFT quadrature steps are closest to linear scaling, with observed scaling of N1.2
and N1.1, respectively, where N is the number of basis functions. The exchange
contribution scales as N1.6. These empirical scaling data demonstrate that with proper
sorting and integral screening, the AX build in CIS and TDDFT scales much better
than quadratic, with no loss of accuracy in excitation energies.
Figure 4. Contributions to the time for building an initial AX vector in CIS and TD-BLYP. Ten initial X vectors are created based on MO energy gaps, and the timing reported is the average time for building AX for those ten vectors. The timings are obtained on a dual Intel Xeon X5570 platform with eight Tesla C1060 GPUs. Data (symbols) are fit to power law (solid line, fitting parameters in inset). Fewer points are included for the TD-BLYP timings because the SCF procedure does not convergea for the solvated PYP chromophore with a large number of waters or for the full PYP protein.
a This is due to the well-known problem with non-hybrid DFT functionals having erroneously low-lying charge transfer states that can prevent SCF convergence.
y=7.3!10-5 x1.2 R=0.97 y=5.0!10-5 x1.6 R=0.99 y=4.9!10-4 x1.04 R=0.97

105
Of the three integral contributions (Coulomb, exchange, and DFT quadrature),
the computation of the exhcange matrix is clearly the bottleneck. This is due to the
three issues with exchange computation previously discussed: 1) the J-engine
algorithm takes full advantage of density sparsity because of efficient density
screening that is not possible for our K-engine implementation, 2) exchange kernels
access the density in memory non-contiguously, and 3) exchange lacks the µ! " !µ
and #" " "# symmetry. It is useful to compare the time required to calculate the
exhcange contribution to the first ground-state SCF iteration (which is the most
expensive iteration due to the use of Fock matrix updating62) and to the AX vector
build for CIS (or TDDFT). We find that the exchange contribution is on average 1.5x
slower in CIS/TDDFT compared to ground state SCF. One might have expected the
excited state computation to be 2x slower because of the two K-engine calls, but the
algorithm is able to exploit the greater sparsity of the transition density matrix
(compared to the ground state density matrix).
Due to efficient screening of ERI contributions to the Coulomb matrix, the J-
engine similarly exploits the increased sparseness of the transition density, and
therefore is faster than the ground state 1st iteration J-engine calculation. In the current
implementation the Coulomb evaluation profits more from transition density sparsity
than that for exchange since it scales better with system size (N1.2 vs N1.6).
As can be seen in figure 4, the DFT integration usually takes more time than
the Coulomb contribution, in spite of the fact that DFT integration scales more nearly
linearly with system size. This is because of the larger prefactor for DFT integration,
which is related to the density of the quadrature grids used. It has previously been

106
noted63 that very sparse grids can be more than adequate for TDDFT. We further
support this claim with the data presented in table 2, where we compare the lowest
excitation energies and average TD-BLYP integration times for the initial guess
vectors for six different grids on two of the test systems. For both molecules, the
excitation energies from the sparsest grid agree well with those of the more dense
grids, but with a substantial reduction in integration time, suggesting that a change to
an ultra sparse grid for the TDDFT portion of the calculation could result in
considerable time savings with little to no loss of accuracy. The TD-BLYP values
computed with NWChem64 using the default ‘medium’ grid are also given to show the
accuracy of our implementation. The small (<0.001eV) differences in excitation
energies between our GPU-based TD-BLYP and NWChem are likely due to slightly
differing ground state densities, which differ in energy by 0.000008 a.u. for the
chromophore and 0.0019 a.u. for the S2 dendrimer.
PYP chromophore (29 atoms) Table 2. TD-BLYP timings (average time for the DFT quadrature in one AX build for the initial 10 AX vectors) and first excitation energies using increasingly dense quadrature grids. For comparison, NWChem excitation energies are also given using the default ‘medium’ grid. Number of points/atom refers to the pruned grid for TeraChem and the unpruned grid for NWChem. NWChem was run on a different architecture, so timings are not directly comparable.
grid points points/atom time (s) $E (au) 0 29497 1017 0.06 0.08516162 1 81461 2809 0.11 0.08516510 2 182872 6305 0.22 0.08516472 3 330208 11386 0.38 0.08516266 4 841347 29011 0.91 0.08516267 5 2126775 73337 2.30 0.08516268
NWChem/medium
21655 n/a 0.08516691 S2 dendrimer (128 atoms) Grid points points/atom time (s) $E (au)
0 141684 1106 0.28 0.08394399 1 382576 2988 0.51 0.08394430 2 848918 6632 0.98 0.08394427 3 1506502 11769 1.64 0.08394433 4 3770640 29458 3.72 0.08394431 5 9472331 74002 9.24 0.08394431
NWChem/medium
25061 n/a 0.08395211

107
GPU-accelerated CIS and TDDFT methods can calculate excited states of
much larger molecules than can currently be studied with previously existing ab initio
methods. For the well-behaved valence transitions in the PYP systems, CIS
convergence requires very few Davidson iterations. The total wall time (SCF+CIS)
required to calculate the 1st CIS/6-31G excited state of the entire PYP protein (10,869
basis functions) is less than 7 hours, with ~5.5 hours devoted to the SCF procedure,
and ~1.5 hours to the CIS procedure. Most (1.2 hours) of the CIS time is spent
computing exchange contributions. We can thus treat the protein with full QM and
study how mutation within PYP will affect the absorbance. For any meaningful
comparison with the experimental absorption energy of PYP at 2.78 eV,59 many
configurations need to be taken into account. For this single configuration, the CIS
excitation energy of 3.69 eV is much higher than the experimental value, as expected
with CIS. The TD-B3LYP bright state (S5) is closer to the experimental value, but still
too high at 3.33 eV.
Solvatochromic studies in explicit water are problematic for standard DFT
methods, including hybrid functionals, due to the well-known difficulty in treating
charge transfer excitations.16,65 In calculating the timings for the first excited state of
the PYP chromophore with increasing numbers of waters, we found that the energy of
the CIS first excited state quickly leveled off and stabilized, while that for TD-BLYP
and TD-B3LYP generally decreased to nonsensical values, at which point the ground
state SCF convergence was also problematic. This behavior of the first excitation
energies for the PYP chromophore with increasing numbers of waters is shown in
figure 5 for CIS, TD-BLYP, and TD-B3LYP. While the 20% HF exchange in the

108
hybrid TD-B3LYP method does improve the excitation energies over TD-BLYP, the
energies are clearly incorrect for both methods, and a higher level of theory or a range-
corrected functional19,21 is certainly necessary for studying excitations involving
explicit QM waters.
Figure 5. The first excitation energy (eV) of the PYP chromophore with increasing numbers of surrounding water molecules. Both TD-BLYP and TD-B3LYP exhibit spurious low-lying charge transfer states. The geometry is taken from a single AMBER dynamics snapshot.
The recent theoretical work by Badaeva et al. examining the one and two
photon absorbance of oligothiophene dendrimers was limited to results for the first
three generations S1-S3, even though experimental results were available for S4.54-56
In table 3, we compare our GPU accelerated results on the first bright excited state
(oscillator strength > 1.0) using TD-B3LYP within the TDA to the full TD-B3LYP
and experimental results. Results within the TDA are comparable to those from full
TD-B3LYP, for both energies and transition dipole moments. Our results for S4 show
the continuing trend of decreasing excitation energy and increasing transition dipole
moment with increasing dendrimer generation.

109
Exp GPU TD-B3LYP CPU TD-B3LYP $E $E µge $E µge
S1 3.25 3.13 9.2 3.00 8.0 S2 3.25 2.98 10.6 2.92 10.1 S3 3.20 2.93 12.0 2.89 11.9 S4 3.19 2.83 13.3
Table 3. Experimental and calculated vertical transition energies (eV) and transition dipole moments (D) for the lowest energy bright state. Experimental results were taken from Refs. 54 and 56. GPU accelerated TD-B3LYP was computed within the Tamm-Dancoff approximation. TD-B3LYP results taken from Ref. 56.
CONCLUSION
We have implemented ab initio CIS and TDDFT calculations using GPUs,
allowing full QM calculation of the excited states of large systems. The numerical
accuracy of the excitation energies is shown to be excellent using mixed precision
integral evaluation. A small percentage of cases require full double precision
integration. For these occasional issues, we can easily switch to full double precision
to achieve the desired convergence. The ability to use lower precision in much of the
CIS and TDDFT calculation is reminiscent of the ability to use coarse grids when
calculating correlation energies, as shown previously for pseudospectral methods.63,66-
69 Recently, it has also been shown70 that single precision can be adequate for
computing correlation energies with Cholesky decomposition methods which are
closely related to pseudospectral methods.71 Both quadrature and precision errors
generally behave as relative errors, while chemical accuracy is an absolute standard
(often taken to be ~1 kcal/mol). Thus, coarser grids and/or lower precision can be
safely used when the quantity being evaluated is itself small (and therefore less
relative accuracy is required), as is the case for correlation energies and/or excitation
energies.

110
For some of the smaller benchmark systems, we present speedups as compared
to the GAMESS quantum chemistry package running over 8 processor cores. The
speedups obtained for CIS and TDDFT calculations range from 6x to 234x, with
increasing speedups with increasing system size.
The increased size of the molecules that can be treated using our GPU-based
algorithms exposes some failings of DFT and TDDFT. Specifically, the charge-
transfer problem16 of TDDFT and the delocalization problem72 of DFT both seem to
become more severe as the molecules become larger, especially for the case of
hydrated chromophores with large numbers of surrounding quantum mechanical water
molecules.
REFERENCES
(1) Foresman, J. B.; Head-Gordon, M.; Pople, J. A.; Frisch, M. J. The Journal of Physical Chemistry 1992, 96, 135.
(2) Runge, E.; Gross, E. K. U. Physical Review Letters 1984, 52, 997.
(3) Gross, E. K. U.; Kohn, W. Physical Review Letters 1985, 55, 2850.
(4) Casida, M. E. In Recent Advances in Density Functional Methods; Chong, D. P., Ed.; World Scientific: Singapore, 1995.
(5) Casida, M. E.; Jamorski, C.; Casida, K. C.; Salahub, D. R. The Journal of Chemical Physics 1998, 108, 4439.
(6) Appel, H.; Gross, E. K. U.; Burke, K. Physical Review Letters 2003, 90, 043005.
(7) Hirata, S.; Head-Gordon, M.; Bartlett, R. J. The Journal of Chemical Physics 1999, 111, 10774.
(8) Dreuw, A.; Head-Gordon, M. Chem. Rev. 2005, 105, 4009.
(9) Burke, K.; Werschnik, J.; Gross, E. K. U. The Journal of Chemical Physics 2005, 123, 062206.

111
(10) Dallos, M.; Lischka, H.; Shepard, R.; Yarkony, D. R.; Szalay, P. G. J. Chem. Phys. 2004, 120, 7330.
(11) Kobayashi, Y.; Nakano, H.; Hirao, K. Chem. Phys. Lett. 2001, 336, 529.
(12) Roos, B. O. Acc. Chem. Res. 1999, 32, 137.
(13) Tokita, Y.; Nakatsuji, H. J. Phys. Chem. B 1997, 101, 3281.
(14) Krylov, A. I. Ann. Rev. Phys. Chem. 2008, 59, 433.
(15) Stanton, J. F.; Bartlett, R. J. J. Chem. Phys. 1993, 98, 7029.
(16) Dreuw, A.; Weisman, J. L.; Head-Gordon, M. The Journal of Chemical Physics 2003, 119, 2943.
(17) Iikura, H.; Tsuneda, T.; Yanai, T.; Hirao, K. The Journal of Chemical Physics 2001, 115, 3540.
(18) Heyd, J.; Scuseria, G. E.; Ernzerhof, M. The Journal of Chemical Physics 2003, 118, 8207.
(19) Tawada, Y.; Tsuneda, T.; Yanagisawa, S.; Yanai, T.; Hirao, K. The Journal of Chemical Physics 2004, 120, 8425.
(20) Rohrdanz, M. A.; Herbert, J. M. The Journal of Chemical Physics 2008, 129, 034107.
(21) Rohrdanz, M. A.; Martins, K. M.; Herbert, J. M. The Journal of Chemical Physics 2009, 130, 054112.
(22) Grimme, S. J. Comp. Chem. 2004, 25, 1463.
(23) Grimme, S. J. Comp. Chem. 2006, 27, 1787.
(24) Vydrov, O. A.; Van Voorhis, T. J. Chem. Phys. 2010, 132, 164113.
(25) Dion, M.; Rydberg, H.; Schroder, E.; Langreth, D. C.; Lundqvist, B. I. Phys. Rev. Lett. 2004, 92, 246401.
(26) Maitra, N. T.; Zhang, F.; Cave, R. J.; Burke, K. The Journal of Chemical Physics 2004, 120, 5932.
(27) Levine, B. G.; Ko, C.; Quenneville, J.; Martinez, T. J. Mol. Phys. 2006, 104, 1039.
(28) Jacquemin, D.; Wathelet, V.; Perpeate, E. A.; Adamo, C. J. Chem. Theo. Comp. 2009, 5, 2420.

112
(29) Warshel, A.; Levitt, M. J. Mol. Biol. 1976, 103, 227.
(30) Virshup, A. M.; Punwong, C.; Pogorelov, T. V.; Lindquist, B. A.; Ko, C.; Martinez, T. J. J. Phys. Chem. B 2009, 113, 3280.
(31) Ruckenbauer, M.; Barbatti, M.; Muller, T.; Lischka, H. J. Phys. Chem. A 2010, 114, 6757.
(32) Polli, D.; Altoe, P.; Weingart, O.; Spillane, K. M.; Manzoni, C.; Brida, D.; Tomasello, G.; Orlandi, G.; Kukura, P.; Mathies, R. A.; Garavelli, M.; Cerullo, G. Nature 2010, 467, 440.
(33) Schafer, L.; Groenhof, G.; Boggio-Pasqua, M.; Robb, M. A.; Grubmuller, H. PLoS Comp. Bio. 2008, 4, e1000034.
(34) Vogt, L.; Olivares-Amaya, R.; Kermes, S.; Shao, Y.; Amador-Bedolla, C.; Aspuru-Guzik, A. The Journal of Physical Chemistry A 2008, 112, 2049.
(35) Yasuda, K. J. Chem. Theo. Comp. 2008, 4, 1230.
(36) Ufimtsev, I. S.; Martinez, T. J. J. Chem. Theo. Comp. 2009, 5, 1004.
(37) Ufimtsev, I. S.; Martinez, T. J. J. Chem. Theo. Comp. 2009, 5, 2619.
(38) Stone, J. E.; Phillips, J. C.; Freddolino, P. L.; Hardy, D. J.; Trabuco, L. G.; Schulten, K. J. Comp. Chem. 2007, 28, 2618.
(39) Anderson, J. A.; Lorenz, C. D.; Travesset, A. J. Comp. Phys. 2008, 227, 5342.
(40) Liu, W.; Schmidt, B.; Voss, G.; Muller-Wittig, W. Comp. Phys. Comm. 2008, 179, 634.
(41) Friedrichs, M. S.; Eastman, P.; Vaidyanathan, V.; Houston, M.; Legrand, S.; Beberg, A. L.; Ensign, D. L.; Bruns, C. M.; Pande, V. S. J. Comp. Chem. 2009, 30, 864.
(42) Ufimtsev, I. S.; Martinez, T. J. J. Chem. Theo. Comp. 2008, 4, 222.
(43) Grabo, T.; Petersilka, M.; Gross, E. K. U. Journal of Molecular Structure: THEOCHEM 2000, 501-502, 353.
(44) Hirata, S.; Head-Gordon, M. Chem. Phys. Lett. 1999, 314, 291.
(45) Cordova, F.; Doriol, L. J.; Ipatov, A.; Casida, M. E.; Filippi, C.; Vela, A. J. Chem. Phys. 2007, 127, 164111.
(46) Davidson, E. R. J. Comp. Phys. 1975, 17, 87.

113
(47) White, C. A.; Head-Gordon, M. J. Chem. Phys. 1994, 101, 6593.
(48) Becke, A. D. The Journal of Chemical Physics 1988, 88, 2547.
(49) Lebedev, V. I.; Laikov, D. N. Dokl. Akad. Nauk 1999, 366, 741.
(50) Murray, C. W.; Handy, N. C.; Laming, G. J. Mol. Phys. 1993, 78, 997.
(51) Brown, P.; Woods, C.; McIntosh-Smith, S.; Manby, F. R. J. Chem. Theo. Comp. 2008, 4, 1620.
(52) Hancock, J. M.; Gifford, A. P.; Tonzola, C. J.; Jenekhe, S. A. The Journal of Physical Chemistry C 2007, 111, 6875.
(53) Tao, J.; Tretiak, S. J. Chem. Theo. Comp. 2009, 5, 866.
(54) Ramakrishna, G.; Bhaskar, A.; Bauerle, P.; Goodson, T. The Journal of Physical Chemistry A 2007, 112, 2018.
(55) Harpham, M. R.; Suzer, O.; Ma, C.-Q.; Bauerle, P.; Goodson, T. J. Am. Chem. Soc. 2009, 131, 973.
(56) Badaeva, E.; Harpham, M. R.; Guda, R.; Suzer, O.; Ma, C.-Q.; Bauerle, P.; Goodson, T.; Tretiak, S. J. Phys. Chem. B 2010, 114, 15808.
(57) Yamaguchi, S.; Kamikubo, H.; Kurihara, K.; Kuroki, R.; Niimura, N.; Shimizu, N.; Yamazaki, Y.; Kataoka, M. Proceedings of the National Academy of Sciences 2009, 106, 440.
(58) Jorgensen, W. L.; Chandrasekhar, J.; Madura, J. D.; Impey, R. W.; Klein, M. L. The Journal of Chemical Physics 1983, 79, 926.
(59) Nielsen, I. B.; Boye-Peronne, S.; El Ghazaly, M. O. A.; Kristensen, M. B.; Brondsted Nielsen, S.; Andersen, L. H. Biophysical Journal 2005, 89, 2597.
(60) Becke, A. D. Physical Review A 1988, 38, 3098.
(61) Lee, C.; Yang, W.; Parr, R. G. Phys. Rev. B 1988, 37, 785.
(62) Almlof, J.; Faegri, K.; Korsell, K. J. Comp. Chem. 1982, 3, 385.
(63) Ko, C.; Malick, D. K.; Braden, D. A.; Friesner, R. A.; Martinez, T. J. J. Chem. Phys. 2008, 128, 104103.
(64) Valiev, M.; Bylaska, E. J.; Govind, N.; Kowalski, K.; Straatsma, T. P.; VanDam, H. J. J.; Wang, D.; Nieplocha, J.; Apra, E.; Windus, T. L.; deJong, W. A. Comp. Phys. Comm. 2010, 181, 1477.

114
(65) Grimme, S.; Parac, M. ChemPhysChem 2003, 4, 292.
(66) Martinez, T. J.; Carter, E. A. J. Chem. Phys. 1993, 98, 7081.
(67) Martinez, T. J.; Carter, E. A. J. Chem. Phys. 1994, 100, 3631.
(68) Martinez, T. J.; Carter, E. A. J. Chem. Phys. 1995, 102, 7564.
(69) Martinez, T. J.; Mehta, A.; Carter, E. A. J. Chem. Phys. 1992, 97, 1876.
(70) Vysotskiy, V. P.; Cederbaum, L. S. J. Chem. Theo. Comp. 2010, Articles ASAP; DOI: 10.1021/ct100533u.
(71) Martinez, T. J.; Carter, E. A. In Modern Electronic Structure Theory, Part II; Yarkony, D. R., Ed.; World Scientific: Singapore, 1995, p 1132.
(72) Cohen, A. J.; Mori-Sanchez, P.; Yang, W. Science 2008, 321, 792.

115
CHAPTER SIX
MULTIPLE TIME STEP INTEGRATORS
FOR AB INITIO MOLECULAR DYNAMICSa
The GPU-accelerated electronic structure methods developed in previous
chapters provide an excellent foundation for ab initio molecular dynamics (AIMD)
simulations. For ab initio dynamics, the calculation of the electronic structure at each
time step is the dominant bottleneck, and GPU acceleration does little to alter this
balance. However, with GPUs, AIMD can be routinely applied to very large systems,
containing up to one thousand atoms. As is well understood from classical dynamics
simulations, such large systems include a mix of fast and slow degrees of freedom.
The fastest modes determine the maximum acceptable time step, while the slower
modes typically represent the motions of scientific interest. In the present chapter we
discuss the application of multiple time step (MTS) integrators to extend the
simulation time scale accessible to large AIMD simulations.
MTS integration techniques1-7 are standard tools used to increase the
computational efficiency of molecular dynamics calculations based on empirical force
fields. These MTS schemes exploit the fact that the forces in chemical systems can
typically be split into fast-varying and slow-varying parts. This splitting is then
leveraged to integrate the slow-varying parts with a longer time step and the fast-
varying parts with a shorter time step. Since the “slow forces”, such as the long-range
a Adapted with permission from N. Luehr, T.E. Markland, and T.J. Martinez, J. Chem. Phys. 2014, 140, 084116. Copyright 2014, AIP Publishing LLC.

116
electrostatic interactions, are typically more computationally expensive to evaluate
than the “fast forces”, such as covalent bond stretches, the ability to evaluate them less
often affords significant speed-ups. For empirical potentials this separation is often
straightforward as the Hamiltonian is typically written as a sum of terms such as van
der Waals, bond stretching, torsional, and electrostatic interactions which can be easily
assigned to the “slow” or “fast” part.
In contrast, ab initio molecular dynamics (AIMD) schemes compute the
potential energy surface on which the nuclei evolve by solving the electronic
Schrodinger equation at each time step. This introduces significant flexibility,
allowing for bond rearrangement (difficult with empirical force fields that generally
assume a prescribed bonding topology),8 electron transfer,9 and transitions between
electronic states.10,11 The most straightforward AIMD approach is the Born-
Oppenheimer scheme (BOMD).12-14 In BOMD, the electronic degrees of freedom are
assumed to relax adiabatically at each nuclear geometry, and an electronic structure
problem is solved fully self-consistently at each time step. An advantage of this
approach is that dynamics always occur on a Born-Oppenheimer potential energy
surface. In contrast, other AIMD methods employ an extended Lagrangian scheme
(Car-Parrinello or CPMD),15,16 where new fictitious degrees of freedom corresponding
to the coefficients in the electronic wavefunction are integrated simultaneously with
the nuclear motion. The CPMD method avoids iteration to self-consistency in the
solution of the electronic wavefunction at each time step, at the expense of introducing
electronic time-scales that are faster than those of atomic motion and thus
necessitating smaller time steps for CPMD than would be possible using BOMD.

117
MTS schemes have been developed to mitigate the computational cost of integrating
fast electronic degrees of freedom in CPMD.17-19 These exploit the time-scale
separation between the fictitious electronic and nuclear degrees of freedom and allow
the outer (nuclear) time step to approach the BOMD limit.
Applying MTS schemes to decompose nuclear motions in AIMD methods
presents a much greater challenge since ab initio forces do not naturally separate into
fast-varying and slow-varying components. Thus, for a long time it has appeared that
MTS schemes could not be applied straightforwardly to BOMD (or, equivalently, to
the nuclear degrees of freedom in CPMD). Recent work has shown that this
conclusion is too harsh, demonstrating that one can treat different components of the
electronic structure problem (specifically, the Hartree-Fock and Moller-Plesset
contributions) with different time steps in an MTS scheme.20 This approach leverages
the well-known fact that the dynamic electron correlation correction to the Hartree-
Fock potential energy surface varies slowly with molecular geometry.
In this chapter, we demonstrate two ways of splitting the electronic
Hamiltonian that enable AIMD calculations to exploit MTS integrators. The first of
these relies on a fragment decomposition of the Hamiltonian. Such fragment
decompositions have been previously proposed to accelerate electronic structure
computations for large molecular systems.21-30 In those cases, the energy expression in
terms of fragments is viewed as an approximation to the true potential energy surface,
and neglected interactions (e.g., relating to charge transfer between fragments) are
rarely quantified or corrected. In contrast, our scheme uses the fragment
decomposition only as an intermediary representation during inner time steps with

118
corrections included at outer steps. As such, the dynamics occurs on the potential
surface without any fragment approximations.
The second MTS scheme we introduce exploits a splitting of the Coulomb
operator in the electronic Hamiltonian. This is closely related to the Coulomb-
Attenuated Schrödinger Equation (CASE) approximation that has been proposed to
accelerate electronic structure calculations.31,32 Again, the advantage of our approach
is that unlike the CASE approximation, which entirely neglects long-range
electrostatic effects, our scheme yields results which are equivalent to those obtained
in a calculation employing the usual Coulomb operator while simultaneously allowing
for the computational speedups afforded by the CASE approach.
THEORY
Trotter factorization of the Liouville operator provides a systematic approach
to derive symplectic time-reversible molecular dynamics integrators for systems
containing many time-scales.3 We begin by briefly reviewing this formalism in order
to highlight its desirable properties for the AIMD force splitting schemes presented
below. The classical Liouville operator for n degrees of freedom with coordinates, xk,
and conjugate momenta, pk is
iL = !xk
!!xk
+ fk
!!pk
"
#$
%
&'
k
n
( (6.1)
where fk is the force acting on the kth degree of freedom. The classical propagator,
eiLT , exactly evolves the system by a time period T from an initial phase space point

119
!(t) = x(t),p(t){ } at time t to its destination at time t + T via the operation
! t +T( ) = eiLT! t( ) .
For a multidimensional system with a general choice of interactions, this
operation cannot be performed analytically for the full propagator. The Trotter
factorization method solves this problem by splitting the Liouville operator,3
iL = iLx + iLp = !xk
!!xkk
n
" + fk
!!pkk
n
" (6.2)
and applying the symmetric Suzuki-Trotter formula.33,34
ea+b ! ea/2 M eb/ M ea/2 M( )M
+O 1M 2
"#$
%&' (6.3)
Thus one obtains
!(t +"T ) = eiLp"T /2eiLx"T eiLp"T /2!(t) (6.4)
where !T = T / M is the time step. This expression becomes exact as M ! " i.e. !T
! 0. In practice one uses a finite time step, which is sufficiently small to well
represent the fastest varying force in the system. Regardless of the time step, the
family of Trotter factorized integrators retains important symplectic and time-
reversible properties.3,35
The above Trotter factorized integrator is identical to the traditional Velocity
Verlet scheme.36 The operator eiLp!T
can be shown to perform the operations of a
momentum shift through a time interval #T:3
x(t),p(t){ }! x(t), p(t) + "T f (x(t)){ } (6.5)

120
while eiLx!T performs a coordinate shift:
x(t),p(t){ }! x(t)+"Tp(t) / m, p(t){ } (6.6)
where m is the vector of masses for the degrees of freedom.
The strength of the Trotter factorization, however, is that it allows much more
general decompositions of the Liouville operator. We now consider the case where the
total force on each degree of freedom can be separated into fast and slow components.
fi = fiF + fi
S (6.7)
Splitting the momentum shift operator, the Liouville operator can now be decomposed
into three terms.
iL = iLx + iLp
F + iLpS = !xk
!!xkk
n
" + fkF !!pkk
n
" + fkS !!pkk
n
" (6.8)
Proceeding as above with Suzuki-Trotter expansion, one obtains the following MTS
integrator.3
!(t + "T ) = eiLp
S "T /2 eiLpF#T /2eiLx#T eiLp
F#T /2( )N
eiLpS "T /2!(t)
(6.9)
Here, !T = N"T is the outer time step, and the bracketed term evolves the positions
and momenta of the system by the smaller inner time step, !T , under the action of
only the fast varying forces. The advantage of the MTS scheme is that the outer time
step can be chosen with respect to the fastest slow force and evaluated 1/Nth as often
compared to traditional Verlet integrators. Since force evaluation is the dominant
computational step in AIMD calculations, the MTS approach can in principle provide

121
an N-fold speedup over traditional integrators, as long as the appropriate
decomposition into slow and fast forces can be found.
In BOMD both long- and short-range atomic forces depend on nonlinear SCF
equations. Thus, a strict algebraic force decomposition of the type commonly used in
classical MTS integrators is not possible. Fortunately, the RESPA strategy is flexible
enough to allow a broad range of numerical decompositions. We introduce the
following, almost trivial, scheme.
iL = !xk
!!xkk
n
" + Fkmod !
!pkk
n
" + FkAI # Fk
mod( ) !!pkk
n
" = iLx + iLpF + iLp
S
(6.10)
Here FkAI
is the full ab initio force acting on the kth degree of freedom while Fkmod is
an approximate model force intended to capture the short-range behavior of FkAI .
Assuming the model force is smooth and numerically well behaved, the resulting ab
initio MTS integrators evolve dynamics on the same potential energy surface as
traditional BOMD approaches. Of course, a poor choice of the model force could
leave short-range, high-frequency components in the difference force, FkAI ! Fk
mod ,
limiting the outer time step and defeating any speedup. However, the same logic also
works in reverse. We are free to relax an exact model for the short-range force until
short-range discrepancies appear in FkAI ! Fk
mod with the same timescale as the fastest
long-range forces. In other words, the outer time step retains some ability to correct
errors in our short-range model force. In practice we will show that sufficiently
accurate model forces are easily computed, so it is the latter line of reasoning that is
most relevant.

122
Our first approach to model the short-range ab initio force is to split an
extended system into small independent fragments. A very general approach would
require a method to fragment macro-molecules across covalent bonds as well as
automatically distribute electrons among fragments. For simplicity, the present work
focuses on systems where these refinements are not needed, in particular, large water
clusters. In the method we christen MTS-FRAG, the model force is constructed as a
sum of independent ab initio gradient calculations on each water molecule in vacuo.
Because all molecular calculations require equal computational effort, the work to
compute the model force scales linearly with the size of the system (i.e., number of
fragments). On the other hand, the global ab initio gradient needed in the outer time
step requires computational effort that is at least quadratic in the system size [for
conventional implementations of Hartree-Fock (HF) or density functional theory
(DFT)]. Thus, for large systems, the short-range force calculation becomes essentially
free and we expect the overall speedup to increase linearly with the size of the outer
MTS time step. The same analysis applies to other ab initio methods such as
perturbation theory and coupled cluster. In these cases, the scaling of effort with
respect to molecular size is often considerably steeper, which will make our MTS-
FRAG scheme even more efficient compared to an implementation where the ab initio
force is evaluated using HF or DFT.
As presented above, the fragment approach completely ignores all
intermolecular interactions during the inner time steps. It is illustrative to also consider
a simple refinement, in which we add Lennard-Jones and Coulomb terms between all
W water molecules to the model force.

123
Fk
LJ = !k
ArOmOn
12 " CrOmOn
6 +qiq j
rijj#n$
i#m$
%
&''
(
)**n=m+1
W
$m=1
W
$ (6.11)
We denote an MTS method using this Lennard-Jones augmented fragment model
force as MTS-LJFRAG. In the present work, we used empirical parameters (A, C, and
charges qk) directly from the TIP3P water model without modification.37 Evaluation of
this term adds negligible effort to the more expensive ab initio fragment calculations
but allows atoms to avoid strongly repulsive regions near neighboring molecules
during inner integration steps. Polarization and charge transfer effects are still
neglected, but these are expected to vary on longer time scales. Further refinement of
these fragment models is no doubt possible. However, we introduce these coarse
approximations here in order to explore the ability of the outer time step to correct for
small but significant errors in the short-range model force.
The primary limitation of the fragment approaches described above is that the
atomic decomposition into fragments cannot be adjusted during the course of a
simulation without destroying the reversibility and energy conservation of the
integrator. This rules out arbitrary bond rearrangements, one of the key features we
would like to preserve from traditional AIMD. As an alternative we consider the
MTS-CASE scheme, where the model force is obtained from an electronic structure
calculation with a truncated (i.e. short-range) Coulomb operator. This is accomplished
with the following substitution in the electronic Hamiltonian (and also in the Coulomb
interaction between the nuclear point charges).
1r! erfc("r)
r (6.12)

124
Here ! is a constant parameter with units of inverse distance that determines the
range at which the Coulomb interaction is effectively quenched. Because the CASE
model force does not require an explicit molecular decomposition, it should, in
principle, have no difficulty with bond rearrangements. However, the accuracy with
which the CASE approximation can describe distorted transition geometries is a
serious concern. Figure 1 shows potential energy scans at the restricted Hartree-Fock
(RHF) and CASE levels of theory for the pictured dissociation of a hydroxide ion and
water molecule. While CASE accurately reproduces the RHF minimum-energy
oxygen-oxygen distance, the binding energy is catastrophically underestimated. Such
artifacts imply that physically relevant trajectories cannot be obtained from the CASE
potential surface. However, in MTS-CASE the outer time step corrects for the
difference between the CASE potential and the full RHF surface. Hence the MTS-
CASE trajectory evolves on the full potential surface and so should accurately
describe transition geometries.
Figure 1: Dissociation curves for an H2O/OH- cluster at the CASE and RHF levels of theory using the 6-31G basis set. Potential curves are generated from optimized geometries at constrained oxygen-oxygen distances. The energy for each curve is taken relative to its minimum value. The equilibrium bond distances are very similar. However, CASE severely under-estimates the binding energy, and includes an unphysical kink near 5 Angstroms.

125
RESULTS AND DISCUSSION
Figure 2: Energy conservation for 21ps simulation of an (H2O)57 water cluster using the MTS-LJFRAG integrator with outer and inner time steps of 2.5 and 0.5fs respectively. The simulation was run at the RHF/3-21G level of theory in the NVE ensemble after 5ps NVT equilibration at 350K. The cluster was confined by a spherical boundary chosen to lead to a density of 1g/mL. The blue curve shows total energy in kcal/mol shifted by +2.72x106 kcal/mol. The cyan line is a least-squares fit of total energy to show drift. Slope is 1.39x10-2 kcal/mol/ps for all degrees of freedom, i.e. 2.74x10-5 kcal/mol/ps/dof. The red curve shows total kinetic energy for scale.
In order to test these approaches, we implemented the ab initio MTS methods
described above in a development version of TeraChem.38 We first simulated the
dynamics of an (H2O)57 cluster at the RHF/3-21G level of theory. In all calculations,
the cluster was confined to a density of 1g/mL by applying a spherical quadratic
repulsive potential to oxygen atoms beyond the sphere’s radius. The barrier potential
was included in the inner time step of the MTS trajectories due to its negligible

126
computational cost compared to the ab initio force evaluation. The system was first
equilibrated for 5 ps using a standard Velocity Verlet36 integrator with the Bussi-
Parinello thermostat.39 During equilibration we used an MD time step of 1.0 fs, the
target temperature was 350K, and the thermostat time constant was 100 fs.
After equilibration, we collected a series of 21ps micro-canonical (NVE)
trajectories using a range of time steps for each of the MTS-FRAG, MTS-LJFRAG,
and MTS-CASE integrators outlined above. All simulations were started from
identical initial conditions. Baseline dynamics using the standard Velocity Verlet
integrator were collected with time steps between 0.5 and 1.5fs, since higher values
lead to unstable trajectories. Ab initio MTS trajectories used an inner time step of
0.5fs, with outer time steps ranging from 1.5 to 3.0fs. For the MTS-CASE integrator,
two screening ranges were tested: $=0.33 Bohr-1, which corresponds to an effective
screening distance of only 1.6 Å, and $=0.18 Bohr-1, which corresponds to 2.9Å.
The drift in total energy over the course of a simulation provides a test for the
quality of an integrator. An energy curve for an MTS-LJFRAG simulation using an
outer time step of 2.5fs is shown in figure 2. The drift was extracted by performing a
linear fit of the total energy over the 21ps trajectory and is almost unnoticeable on the
scale of the natural kinetic energy fluctuations of the system. The extracted slope is
2.74x10-5 kcal/mol/ps/dof, i.e. during the entire 21ps trajectory 5.75x10-4 kcal/mol of
energy is added to each degree of freedom due to inaccuracies in the integration of the
equations of motion. To put this in perspective, this is equivalent to a 0.289 K rise in
temperature of the system over that time. Current AIMD trajectories are generally
limited to around 100ps and hence even on this time-scale the increase in temperature
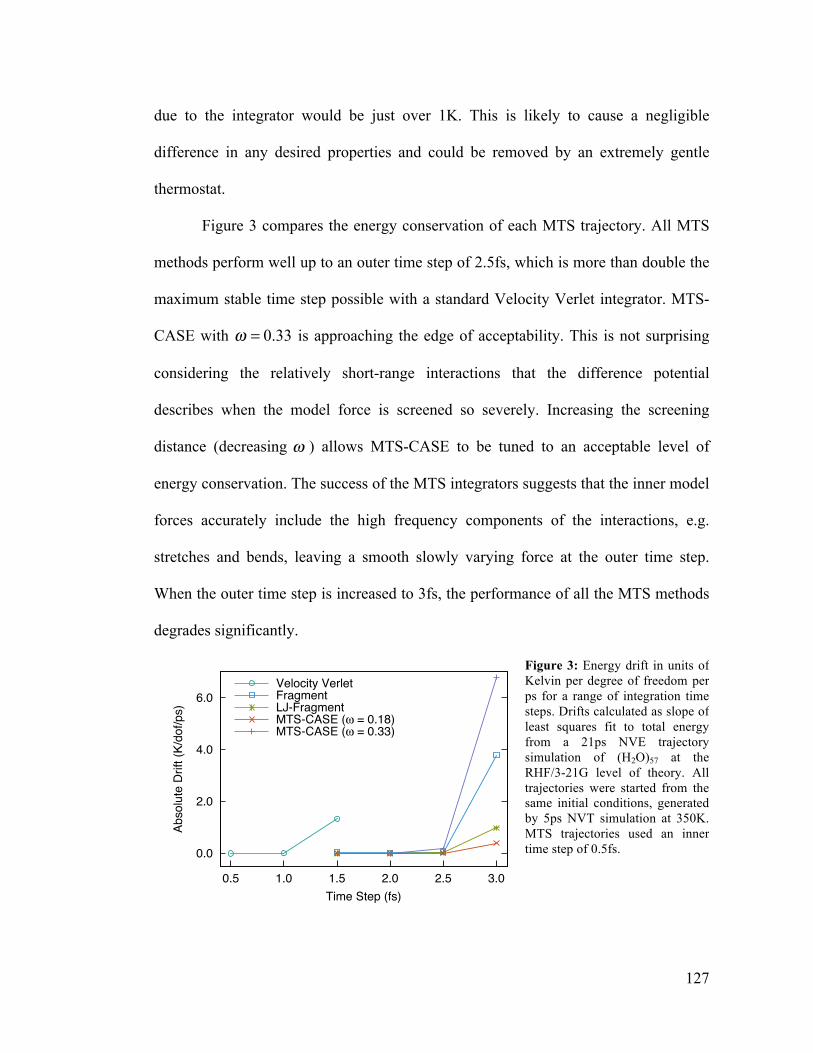
127
due to the integrator would be just over 1K. This is likely to cause a negligible
difference in any desired properties and could be removed by an extremely gentle
thermostat.
Figure 3 compares the energy conservation of each MTS trajectory. All MTS
methods perform well up to an outer time step of 2.5fs, which is more than double the
maximum stable time step possible with a standard Velocity Verlet integrator. MTS-
CASE with ! = 0.33 is approaching the edge of acceptability. This is not surprising
considering the relatively short-range interactions that the difference potential
describes when the model force is screened so severely. Increasing the screening
distance (decreasing ! ) allows MTS-CASE to be tuned to an acceptable level of
energy conservation. The success of the MTS integrators suggests that the inner model
forces accurately include the high frequency components of the interactions, e.g.
stretches and bends, leaving a smooth slowly varying force at the outer time step.
When the outer time step is increased to 3fs, the performance of all the MTS methods
degrades significantly.
Figure 3: Energy drift in units of Kelvin per degree of freedom per ps for a range of integration time steps. Drifts calculated as slope of least squares fit to total energy from a 21ps NVE trajectory simulation of (H2O)57 at the RHF/3-21G level of theory. All trajectories were started from the same initial conditions, generated by 5ps NVT simulation at 350K. MTS trajectories used an inner time step of 0.5fs. 0.0
2.0
4.0
6.0
0.5 1.0 1.5 2.0 2.5 3.0
Abso
lute
Drif
t (K/
dof/p
s)
Time Step (fs)
Velocity VerletFragmentLJ-FragmentMTS-CASE (! = 0.18)MTS-CASE (! = 0.33)

128
The failure of all methods with a 3fs outer time step is not surprising since
MTS integrators such as RESPA are known to suffer from non-linear resonance
instabilities.40,41 These instabilities, which arise due to interactions between the fast
forces and long time steps, mean that the fastest forces in the system limit how
infrequently the slowest interactions must be calculated at the outer time step. In the
case of our system the fastest modes, due to OH stretches of the dangling hydrogen
atoms at the surface of the cluster, oscillate at 4000 cm-1 (figure 4), corresponding to a
time period of %=8.5 fs. For a harmonic oscillator one can show that the maximum
stable outer time step is given by &Tmax = %/' which yields a resonance limit on the
time step of 2.7fs.42,43 This matches our observations precisely, and suggests that the
present limitations of our method do not stem from inadequacy of the model
potentials. It has been shown that resonance instabilities can be effectively mitigated
using specially designed Langevin thermostats.41,44 The application of these
techniques here is beyond the scope of the present study, but should increase the outer
time step by a further factor of 2-4 fold and thus further improve the computational
speedups reported here.

129
Figure 4: Power spectrum comparison between Velocity Verlet with 0.5fs time step (red), Velocity Verlet with 1.0fs time step (blue) and MTS-LJFRAG integrator with 2.5 and 0.5fs outer and inner time steps respectively (green). Spectra are based on 21ps NVE trajectories at the RHF/3-21G level of theory. System consists of 57 water molecules confined by a spherical boundary to a density of 1g/mL.
While energy conservation provides a test of the stability of MTS methods one
would also like to ensure that more subtle properties of the system remain unchanged.
For example, figure 4 shows the power spectrum of the system using 0.5fs and 1.0fs
time steps with a traditional Velocity Verlet integrator. Although in figure 3 we found
these both to provide acceptable energy conservation, the 1.0fs Verlet integrator shows
a clear frequency shift at high frequencies compared to the 0.5fs Verlet and MTS
integrators. Hence, although using standard Velocity Verlet may be stable, one should
take care regarding the properties obtained when large time-steps are employed.
0
0.1
0.2
0.3
0.4
0 1000 2000 3000 4000
Rel
ativ
e Po
wer
Wavenumber (cm-1)
Verlet 0.5fsVerlet 1.0fs
LJ-FRAG

130
Figure 5: Power spectrum comparison between standard Velocity Verlet (red), and MTS trajectories. Upper box compares simple fragment (blue), and Lennard-Jones augmented fragment (green) methods. Lower box compares MTS-CASE integration using two values of omega, 0.33 (blue) and 0.18 (green), which represent 1.6 and 2.9Å cutoffs of the Coulomb potential respectively. Spectra are based on 21ps NVE trajectories at the RHF/3-21G level of theory. System consists of 57 water molecules confined by a spherical boundary to a density of 1g/mL. MTS integrators use 2.5fs and 0.5fs for outer and inner time steps respectively. Verlet integrator uses 0.5fs time step.
Figure 5 shows the power spectra from the MTS trajectories using a 2.5fs
outer time step and 0.5fs inner time step compared with that obtained using standard
Velocity Verlet with a 0.5fs time step. In all cases the agreement is very good, with the
peak positions being very well reproduced. Remarkably all MTS methods with a 2.5fs
outer time step capture the power spectrum better than a Verlet integrator using a 1.0fs
time step. Peak intensities show greater variation, though this is likely due to the large
statistical error bars on power spectra obtained from a single 21ps trajectory.
It is clear from figure 5 that the empirically fit terms in the LJFRAG model
improve on the simpler FRAG approach. This suggests that the unmodified TIP3P
0
0.2
0.4R
elat
ive
Pow
erVerlet 0.5fs
FRAGLJ-FRAG
0
0.2
0.4
0 1000 2000 3000 4000Wavenumber (cm-1)
Verlet 0.5fsCASE (0.33)CASE (0.18)

131
force field is capturing some of the relevant high frequency intermolecular forces,
such as those from hydrogen bonds that are not included by the simpler monomer
fragment approach. Among the CASE spectra, the $=0.18 model shows only marginal
improvement over the coarser $=0.33 cutoff. Both outperform the simple fragment
approach, and are roughly equivalent to the Lennard-Jones fragment model.
Figure 6: Power spectra resulting from Velocity Verlet integrated dynamics on the CASE (blue) and RHF (red) potential energy surfaces compared with MTS-CASE (green) integration. The CASE approximation uses a cutoff of 0.33 Bohr-1 (1.6 Å) for both the Verlet and MTS integrators. Verlet-RHF and MTS-CASE spectra are based on 21ps NVE trajectories using the 3-21G basis set. Verlet-CASE spectrum is based on a shorter 14ps trajectory at the same level of theory. The system consists of 57 water molecules confined by a spherical boundary to a density of 1g/mL. Outer and inner timesteps for the MTS integrator are 2.5fs and 0.5fs respectively. Verlet integrators use 0.5fs time steps.
It is also instructive to compare MTS-CASE dynamics with that obtained by
traditional (Velocity Verlet) integration on the CASE potential surface. Figure 6
compares the power spectrum obtained from a 14ps simulation of dynamics on the
CASE (!=0.33) potential energy surface using a 0.5fs Velocity Verlet integrator to
that obtained from 0.5fs (RHF) Verlet and 2.5fs/0.5fs MTS-CASE (!=0.33). The
Verlet-CASE power spectrum is in pronounced disagreement with that obtained from
dynamics on the RHF surface. This is not surprising given the major quantitative
0
0.1
0.2
0.3
0.4
0.5
0 1000 2000 3000 4000
Rel
ativ
e Po
wer
Wavenumber (cm-1)
Verlet-RHFVerlet-CASE (!=0.33)MTS-CASE (!=0.33)

132
differences between the RHF and CASE potential surfaces shown in figure 1. It is
notable that the differences are most pronounced at the highest frequencies since one
might expect the O-H stretch to be relatively well preserved within CASE, given that
the Coulomb operator is unmodified at short ranges. However, this peak shows much
less structure in the CASE spectra and is significantly red-shifted from 4000 to
3600cm-1. Given these significant differences, it is remarkable that the MTS-CASE
trajectory gives a power spectrum close to that obtained on the RHF surface despite
using the CASE potential as the model for high-frequency force updates.
Thus far we have only considered water clusters, which do not undergo
covalent bond rearrangements on the simulated time-scale. However, one of the main
advantages of AIMD simulation is the ability for the system to undergo spontaneous
covalent bond breaking and formation during the simulation. Hence to evaluate the
ability of our MTS-CASE approach to describe bond rearrangements, we simulated
the proton transfer dynamics of a hydroxide ion solved in a cluster of 64 water
molecules. The system was equilibrated using the same procedure and boundary
conditions as the (H2O)57 cluster described above. A 21ps microcanonical simulation
was run at the RHF/3-21G level of theory using the MTS-CASE ($=0.33) integrator.
Figure 7 shows a 2ps window of the total trajectory. The energy drift of 1.54x10-4
kcal/mol/ps/dof (0.077K/ps/dof) was calculated by a linear fit to the entire 21ps
trajectory. For each frame the hydroxide oxygen atom was determined by assigning
each hydrogen atom to the nearest oxygen atom and then reporting the index of the
oxygen atom with a single assigned hydrogen (red line in figure 7).

133
Figure 7: Energy conservation of MTS-CASE integrator. Plot shows 2ps window from a longer 21ps trajectory. The NVE simulation was run at the RHF/3-21G level of theory after 5ps of NVT calibration to 350K. The inner and outer time steps were 0.5 and 2.5fs. The Coulomb attenuation parameter was 0.33 Bohr-1. The system was made up of a hydroxide ion solvated by 64 water molecules and was confined by a spherical barrier to a density of 1g/mL. A representative snapshot shows a proton transition between two oxygen atoms highlighted in blue. The blue curve shows the total energy in kcal/mol shifted by -3.10x106 kcal/mol. The green line gives a least squares fit of the total energy curve for the entire 21ps trajectory. Its slope records a drift of 8.7x10-2 kcal/mol/ps for the entire system or 1.5x10-4 kcal/mol/ps/dof. The red line indicates the index of the hydroxide oxygen atom for each time step. This was determined by assigning each hydrogen atom to its nearest oxygen neighbor, and then reporting the oxygen with a single assigned hydrogen. The magenta curve shows the total kinetic energy for scale.
As shown in the upper panel of figure 7, a proton oscillates primarily between
two oxygen atoms (shown in blue in the lower panel). During the entire 21ps
trajectory we observed over 500 proton transfer events. However, the total energy drift
is comparable to that reported above for the nonreactive (H2O)57 cluster. This is
remarkable on two counts. First, it demonstrates that the MTS scheme is applicable
even when bonds are being formed and broken – a difficult task for empirical force
fields and also for the fragment-based MTS-FRAG and MTS-LJFRAG schemes

134
described above. Second, the CASE approach we are using here is the most
aggressively screened of the two we have considered. With a screening factor of
$=0.33 bohr-1, the Coulomb interaction between any two charged particles is already
being attenuated at a distance of 1.6Å. Hence, in the inner time step, the proton barely
interacts with any electronic orbitals other than those on the two nearest heavy atoms.
All other interactions are corrected in the outer time step, which demonstrates
remarkable robustness by its ability to maintain the energy conservation observed
above.
Integrator Time Step (fs) Time/Step (sec) Steps/Day fs/day Speedup Velocity Verlet 0.5 430 201 100 1.0 Velocity Verlet 1.0 422 204 204 2.0 MTS-LJFrag 1.5 460 188 282 2.8 MTS-LJFrag 2.0 471 183 367 3.7 MTS-LJFrag 2.5 485 178 446 4.4 Table 1: Performance of MTS Lennard-Jones fragment compared to standard Velocity Verlet integrator. The system consists of 120 water molecules confined by a spherical boundary to a density of 1g/mL. The simulation was run at the RHF/6-31G level of theory using a single NVIDIA Tesla C2070 GPU. Step sizes for MTS integrator refer to the outer time step; 0.5fs inner time steps were used throughout. Timings are averaged over 100 MD steps and are in units of wall-time seconds per outer step. Speedups are computed by comparison to the 0.5fs Velocity Verlet integrator.
Finally, we consider the computational efficiency of our ab initio MTS
approach based on our initial implementation in TeraChem. Table 1 summarizes the
performance of the Lennard-Jones fragment MTS scheme relative to a standard
Velocity Verlet integrator with a time step of 0.5fs. Even though the Velocity Verlet
integrator is stable with a 1fs time step (figure 3), such a large time step noticeably
alters the dynamics, as demonstrated by the shift of the high frequency peak in the
power spectrum (figure 4). Hence, Velocity Verlet with a 0.5fs time step is the
appropriate comparison. These calculations were carried out with a larger (H2O)120
cluster treated at the RHF/6-31G level of theory and confined to a density of 1g/mL by

135
a spherical boundary. All calculations used a single Tesla C2070 GPU. Because the
outer time step still requires a full SCF gradient evaluation, the best that can be
achieved is a 5x speedup, if we compare an MTS integrator with a 2.5fs outer time
step to a standard Velocity Verlet method with a time step of 0.5fs. We are able to
achieve up to 4.4x speedup, which is over 88% efficient.
As with the fragment models, the CASE approximation leads to linear scaling
computational effort in evaluating the inner time steps.32 This is now due to improved
screening of electron repulsion integrals afforded by a short-range coulomb operator.
The present experimental code does not yet exploit the improved CASE screening.
However, previous work has shown that CASE at the screening levels employed here
offers significant computational speed-ups.32
CONCLUSION
In this paper we have demonstrated new approaches that allow MTS
integrators to be applied generally to AIMD calculations. We exploited the ability of
the outer MTS time steps to correct low frequency modeling errors within the inner
time steps. Thus we were able to employ drastically simplified short-range
approximations to the ab initio forces in the inner time steps. Despite these
computational savings, the resulting methods remain robust, exhibiting symplecticity
and time-reversibility and providing excellent energy conservation and even improved
dynamical properties compared to Verlet integrators with moderately large time steps.
As with single-step integrators our ab initio MTS methods can be systematically
improved by reducing the time steps or, for MTS-CASE, also by increasing the
screening distance.

136
The model forces used here are inspired by linear scaling approximations.
However, while linear scaling methods attempt to globally capture all significant
interactions, MTS model forces need only represent the highest frequencies within the
system, and even here low frequency differences between the model and ab initio
systems can be tolerated. This means that much looser thresholds can be employed in
the case of MTS model forces. Thus, even for systems too small to reach the crossover
point for traditional linear scaling methods, our MTS scheme can provide a significant
speedup. For larger systems where linear scaling approaches are applicable, the MTS
approaches introduced here can still be applied, since its looser thresholds should
allow the model forces to be computed more cheaply than the more accurate linear-
scaling gradient.
Other model forces are obviously possible. For example, our use of the TIP3P
force field suggests a completely empirical model force. However, the benefits of such
models are limited by the cost of the global gradient calculation in the outer time step.
In the present case, reducing the computational effort of the inner time step would at
best yield a 13% speedup over the fragment approaches described above. Thus, future
work should focus on extending the outer time step and on reducing the cost of the
global gradient evaluation. For example, employing existing Langevin thermostats44 to
remove spurious resonance effects should allow the outer time step to be extended to
10 or even 20 fs and provide greater performance gains.
REFERENCES
(1) Tuckerman, M. E.; Berne, B. J. The Journal of Chemical Physics 1991, 95, 8362.

137
(2) Tuckerman, M. E.; Berne, B. J.; Martyna, G. J. The Journal of Chemical Physics 1991, 94, 6811.
(3) Tuckerman, M. E.; Berne, B. J.; Martyna, G. J. The Journal of Chemical Physics 1992, 97, 1990.
(4) Tuckerman, M. E.; Berne, B. J.; Rossi, A. The Journal of Chemical Physics 1991, 94, 1465.
(5) Tuckerman, M. E.; Martyna, G. J.; Berne, B. J. The Journal of Chemical Physics 1990, 93, 1287.
(6) Grubmuller, H.; Heller, H.; Windemuth, A.; Schulten, K. Mol. Sim. 1991, 6, 121.
(7) Streett, W. B.; Tildesley, D. J.; Saville, G. Mol. Phys. 1978, 35, 639.
(8) Carloni, P.; Rothlisberger, U.; Parrinello, M. Acc. Chem. Res. 2002, 35, 455.
(9) VandeVondele, J.; Sulpizi, M.; Sprik, M. Ang. Chem. Int. Ed. 2006, 45, 1936.
(10) Virshup, A. M.; Punwong, C.; Pogorelov, T. V.; Lindquist, B. A.; Ko, C.; Martinez, T. J. J. Phys. Chem. B 2009, 113, 3280.
(11) Ben-Nun, M.; Martinez, T. J. Adv. Chem. Phys. 2002, 121, 439.
(12) Barnett, R. N.; Landman, U.; Nitzan, A.; Rajagopal, G. J. Chem. Phys. 1991, 94, 608.
(13) Leforestier, C. J. Chem. Phys. 1978, 68, 4406.
(14) Payne, M. C.; Teter, M. P.; Allan, D. C.; Arias, T. A.; Joannopoulos, J. D. Rev. Mod. Phys. 1992, 64, 1045.
(15) Car, R.; Parrinello, M. Phys. Rev. Lett. 1985, 55, 2471.
(16) Tuckerman, M. E.; Ungar, P. J.; von Rosenvinge, T.; Klein, M. L. J. Phys. Chem. 1996, 100, 12878.
(17) Hartke, B.; Gibson, D. A.; Carter, E. A. Int. J. Quant. Chem. 1993, 45, 59.
(18) Gibson, D. A.; Carter, E. A. J. Phys. Chem. 1993, 97, 13429.
(19) Tuckerman, M. E.; Parrinello, M. J. Chem. Phys. 1994, 101, 1316.
(20) Steele, R. P. J. Chem. Phys. 2013, 139, 011102.
(21) Yang, W. T. Physical Review Letters 1991, 66, 1438.

138
(22) Yang, W. T. Physical Review A 1991, 44, 7823.
(23) Gordon, M. S.; Freitag, M.; Bandyopadhyay, P.; Jensen, J. H.; Kairys, V.; Stevens, W. J. J. Phys. Chem. A 2001, 105, 293.
(24) Gordon, M. S.; Smith, Q. A.; Xu, P.; Slipchenko, L. V. Ann. Rev. Phys. Chem. 2013, 64, 553.
(25) He, X.; Zhang, J. Z. H. J. Chem. Phys. 2006, 124, 184703.
(26) Steinmann, C.; Fedorov, D. G.; Jensen, J. H. PLOS One 2013, 8, e60602.
(27) Xie, W.; Orozco, M.; Truhlar, D. G.; Gao, J. J. Chem. Theo. Comp. 2009, 5, 459.
(28) Fedorov, D. G.; Nagata, T.; Kitaura, K. Phys. Chem. Chem. Phys. 2012, 14, 7562.
(29) Pruitt, S. R.; Addicoat, M. A.; Collins, M. A.; Gordon, M. S. Phys. Chem. Chem. Phys. 2012, 14, 7752.
(30) Collins, M. A. Phys. Chem. Chem. Phys. 2012, 14, 7744.
(31) Adamson, R. D.; Dombroski, J. P.; Gill, P. M. W. Chem. Phys. Lett. 1996, 254, 329.
(32) Adamson, R. D.; Dombroski, J. P.; Gill, P. M. W. J. Comp. Chem. 1999, 20, 921.
(33) Suzuki, M. Commun Math Phys 1976, 51, 183.
(34) Trotter, H. F. Proc. Amer. Math. Soc. 1959, 10, 545.
(35) Sanz-Serna, J. M.; Calvo, M. P. Numerical Hamiltonian Problems; Chapman and Hall: London, 1994.
(36) Swope, W. C.; Andersen, H. C.; Berens, P. H.; Wilson, K. R. J. Chem. Phys. 1982, 76, 637.
(37) Jorgensen, W. L.; Chandrasekhar, J.; Madura, J. D.; Impey, R. W.; Klein, M. L. The Journal of Chemical Physics 1983, 79, 926.
(38) Ufimtsev, I. S.; Martinez, T. J. J. Chem. Theo. Comp. 2009, 5, 2619.
(39) Bussi, G.; Donadio, D.; Parrinello, M. J. Chem. Phys. 2007, 126, 014101.
(40) Biesiadecki, J. J.; Skeel, R. D. J. Comp. Phys. 1993, 109, 318.

139
(41) Ma, Q.; Izaguirre, J. A.; Skeel, R. D. Siam J Sci Comput 2003, 24, 1951.
(42) Barth, E.; Schlick, T. J. Chem. Phys. 1998, 109, 1633.
(43) Han, G. W.; Deng, Y. F.; Glimm, J.; Martyna, G. Comp. Phys. Comm. 2007, 176, 271.
(44) Morrone, J. A.; Markland, T. E.; Ceriotti, M.; Berne, B. J. J. Chem. Phys. 2011, 134, 014103.

140

141
CHAPTER SEVEN
INTERACTIVE AB INITIO MOLECULAR DYNAMICS
The previous chapter was focused on extending AIMD to large systems, where
long time scales are important. In this chapter we explore the impact of accelerated
AIMD applied to small systems containing up to a few dozen atoms. In this regime,
the steady advance of computers could soon transform the basic models used to
understand and describe chemistry. In terms of quantitative models in general,
researchers no longer seek human comprehensible, closed-form equations such as the
ideal gas law. Instead algorithmic models that are evaluated through intensive
computer simulations have become the norm. As a few examples, ab initio electronic
structure, molecular dynamics (MD), and minimum energy reaction path optimizers
are now standard tools for describing chemical systems.
Despite their often-impressive accuracy, quantitative models sometimes
provide surprisingly little scientific insight. For example, individual MD trajectories
are chaotic and can be just as inscrutable to human understanding as the physical
experiment they seek to elucidate. Qualitative cartoon-like models, it seems, are
essential to inspire human imagination and satisfy our curiosity. As an illustration,
consider the pervasive use of primitive ball-and-stick type molecular models in
chemistry. The success of these physical models lies as much in their ability to capture
human imagination and support interesting geometric questions as it does in their
inherent realism. Useful models must be both accurate and playful.

142
Fortunately, computers are capable of much more than crunching numbers for
quantitative models. With the development of computer graphics and the explosion of
immersive gaming technologies, computers also provide a powerful platform for
human interaction. Starting with works by Johnson1 and Levinthal2 in the 1960’s,
molecular viewers were developed first to visualize and manipulate x-ray structures
and later to visualize the results of MD simulations as molecular movies. The next
goal was to allow researchers to interact with realistic physical simulations in real time
as they ran. Along these lines, the Sculpt project provided an interactive geometry
optimizer that included a user-defined spring force in a modified steepest descent
optimizer.3 By furnishing the molecular potential from a classical force field further
simplified with rigid bonds and strict distance cutoffs for non-bonded interactions,
Sculpt could achieve real-time calculation rates for protein systems containing up to
eighty residues, which was certainly impressive at the time.
Later work replaced Sculpt’s geometry optimizer with a MD kernel.4,5 Rather
than being limited to minimum energy structures, the user could then probe dynamical
behavior of protein systems, watching the dynamics trajectory unfold in real time and
insert arbitrary spring forces to steer the dynamics in any direction of interest. Force-
feedback devices have also been used to control molecular tugs.6 These allow users to
feel as well as see the molecular interactions and increase the precision of user control.
Of course, arbitrary user interaction is a (sometimes large) source of energy flow into
the simulation. Aggressive thermostats are necessary to reduce this heating.4 As a
consequence, the results of interactive dynamics are not immediately applicable, for
example, in calculating statistical properties. However, for small forces the trajectories

143
will explore phase space regions that are still relevant to dynamics on standard
ensembles, and thus offer many qualitative mechanistic insights.
Classical force fields suffer from two disadvantages that hamper their
application to general-purpose chemical modeling. First they are empirically tuned,
and as a result are valid only in a finite region of configuration space. This is
particularly problematic for interactive simulations, where the user is free to pull the
system into highly distorted configurations. The second, more important disadvantage
is that covalent bonds modeled by classical springs cannot be rearranged during the
simulation. Ab initio forces, calculated from first principles electronic structure theory,
do not suffer from these disadvantages and provide ideal potentials for use with
interactive dynamics. However, due to prohibitive computational costs, it remains
difficult to evaluate ab initio forces at the fast rate necessary to support real-time
dynamics.
Recently, a divide-and-conquer implementation of the semi-empirical atom
superposition and electron delocalization molecular orbital (ASED-MO) model has
been developed to support real-time energy minimization and MD in the SAMSON
program.7,8 SAMSON offers impressive performance, able to perform real-time
calculations on systems containing up to thousands of atoms. User interaction is
implemented by alternating between “user action steps” in which the user moves or
inserts atoms, and standard optimization or MD steps. In order to keep the
computational complexity manageable, SAMSON applies several approximations
beyond those inherent in ASED-MO. The global system is split into overlapping sub-
problems that are solved independently in parallel. Also for large systems, distant

144
atoms are frozen so that only forces for atoms in the region of user interaction need to
be calculated in each step.
Several previous attempts at full ab initio interactive dynamics have been
reported. In early work, Marti and Reiher avoided on the fly electronic structure
calculations by using an interpolated potential surface.9 The potential surface is pre-
calculated over some relevant configuration space. Then during dynamics, the force at
each time step is obtained from a simple moving least-squares calculation. However, it
is difficult to predict a priori what regions of configuration space will be visited. A
partial solution is to periodically add additional interpolation points where and when
higher accuracy is desired.10 However, the number of needed interpolation points
grows exponentially with the dimensionality of the system. Thus, for non-trivial
systems, it is essential to evaluate the ab initio gradient on the fly. Recently the
feasibility of such calculations has been tested using standard packages and tools.
Combining the Turbomole DFT package, minimal basis sets, effective core potentials
and a quad core processor, Haag and Reiher achieve update rates on the order of a
second for systems containing up to 8 atoms.11
In the following we present the results of our own implementation of
interactive ab initio dynamics. By using the GPU accelerated TeraChem12 code and
carefully streamlining the calculation, interactive simulations are possible for systems
up to a few dozen atoms. The final result is a virtual molecular modeling kit that
combines intuitive human interaction with the accuracy and generality of an ab initio
quantum mechanical description.

145
METHOD
The ab initio interactive MD (AI-IMD) system described below is based on the
interactive MD (IMD) interface that was previously developed to enable interactive
steered molecular dynamics in the context of classical force fields.6 A high level
overview of the original scheme is shown in figure 1. Molecular visualization and
management of the haptic (or “touch”) interface is handled by VMD.13 Along with the
current molecular geometry, VMD displays a pointer that the user controls through a
3D haptic device (shown in figure 5). Using the pointer, the user can select and tug an
atom feeling the generated force through feedback to the haptic device. VMD also
sends the haptic forces to a separate MD program, in our case TeraChem,12 where they
are included with the usual ab initio gradient in the following haptic-augmented force.
F (R,t) = !" Eqm (R)+ Fhap (t) (7.1)
After integrating the system forward in time, TeraChem returns updated coordinates
for display in VMD.
Figure 1: Schematic representation of the IMD interface previously developed for classical MD calculations. VMD is responsible for visualization while TeraChem performs AIMD calculations in real time.
A major advantage of the IMD scheme is that it uses spring forces to cushion
the user’s interaction with the system, rather than raw position updates. This allows
the user to add weak biases that do not totally disrupt the initial momentum of the
TeraChemVMD MD Coords
Haptic Forces
Forc
e Fe
edba
ckPo
inte
r Pos
ition
MD Coords
Pointer Position
3D Display
Haptic Device

146
system. It also avoids severe discontinuities that would overthrow the numerical
stability of standard MD integrators. As a result, the system’s dynamics and energy are
always well-defined, albeit for a time-dependent Hamiltonian, and the magnitude of
haptic perturbations can in principle be accurately measured and controlled.
Communication across the IMD interface is asynchronous. In VMD the render
loop does not wait for updated coordinates between draws and force updates are sent
to TeraChem continuously as they happen rather than waiting for the next MD time-
step. Similarly, TeraChem does not wait to receive haptic force updates between time
steps. This scheme was designed to minimize communication latencies.6
Asynchronous communication also logically decouples the software components and
allows each to operate in terms of generic streams of coordinates and simplifies the
process of adapting the system from classical to ab initio MD as detailed below.
Due to the considerations above, the IMD approach provides a robust starting
point for AI-IMD. However, several adjustments to the classical IMD approach are
needed to accommodate ab initio calculations. These are detailed in the following
sections.
Simulation Rate
A primary benefit of interactive modeling is that molecular motions, as well as
static structures, can be intuitively represented and manipulated. Thus it is critical to
maintain a sensation of smooth motion. To achieve this, past research has targeted
simulation rates of at least ten or twenty MD steps per second.4,5,8 Such update rates
are comparable to video frame rates and certainly result in smooth motion. At present,
however, quantum chemistry calculations requiring less than fifty milliseconds are

147
possible only for trivially small systems. In order to reach larger molecules and basis
sets, it is important to decouple graphical updates from the underlying MD time steps.
Ultimately, the necessary simulation rate is dictated not by graphics considerations,
but by the time scale of the motion being studied. The goal of interactive MD is to
shift the movement of atoms to the time scale of seconds on which humans naturally
perceive motion. For molecular vibrations, this requires simulating a few
femtoseconds of molecular trajectory per second of wall time. Assuming a 1 fs time
step, each gradient evaluation is then allowed at least 200ms to execute. Experiments
show that up to a full second between ab initio gradient evaluations is workable,
though the resulting dynamics become increasingly sluggish.
In addition to high performance, an interactive interface requires a uniform
simulation rate. Each second of displayed trajectory must correspond to a standard
interval of simulated time. This is critical both to convey a visual sensation of smooth
motion as well as to provide consistent haptic physics. For example, the effective
power input of a given haptic input will increase with the simulation rate, since the
same duration of pull in wall time translates into longer pulls in simulated time.6
Problematically, the effort needed to evaluate the ab initio gradient varies widely
depending on the molecular coordinates. For many geometries, such as those near
equilibrium, the SCF equations can be converged in just a few iterations by using
guess orbitals from previous MD steps. For strongly distorted geometries, however,
hundreds of SCF iterations are sometimes required. Even worse, the SCF calculation
may diverge causing the entire calculation to abort. Since users tend to drive the
system away from equilibrium, difficult to converge geometries are more common in

148
interactive MD than in dynamics run on traditional ensembles (see figure 2). To
handle these distorted geometries, we employ the very robust ADIIS+DIIS
convergence accelerator.14,15 However, for well-behaved geometries, ADIIS+DIIS was
found on average to require more iterations than DIIS alone. Thus, the best approach
is to converge with standard DIIS for up to ~20 iterations, and switch over to
ADIIS+DIIS only where DIIS fails.
Figure 2: Histogram of wall-times for 1000 steps of MD run with user interaction (red) and without haptic input (blue). The system was the uncharged imidaloze molecule pictured. The simulation used the unrestricted Hartree-Fock electronic structure method with the 3-21G basis. Each step of the SCF was converged to 2.0e-5 in the maximum element of the commutator SDF-FDS.
In order to establish a fixed simulation rate we consider the variance in timings
for individual MD steps illustrated for a particular system in figure 2. Noting that the
large variance in MD step timings is primarily the result of a few outliers, the target
wall time for a simulation step, Twall, can be set far below the worst-case gradient
evaluation time. For the vast majority of steps, the ab initio gradient then completes
before Twall has elapsed and the MD integrator must pause briefly to allow the display
to keep pace. For pathologically long MD steps, such as those requiring many ADIIS
iterations, the displayed trajectory is frozen and haptic input ignored until the MD step
completes.

149
Integrating the haptic force
A key strength of AI-IMD is that it allows users to make and break bonds
between atoms. However, this level of control requires haptic forces that are stronger
than the bonding interactions between atoms. The situation in classical IMD is very
different. Classical force fields in general cannot handle bond reorganization. Thus,
IMD was originally conceived to control only weak non-bonded interactions. In
practice, the inclusion of this strong haptic force in equation (7.1) can produce
noticeable energetic artifacts in the MD simulation. For example, an unbound atom
vibrating around a fixed haptic pointer might visibly gain amplitude with each
oscillation. Such non-conservative dynamics result in rapid heating of the system that
quickly swamps the user’s control. The cause is simply that the MD time step that
would appropriately integrate a closed system is too long to accurately handle the
stronger haptic forces. An obvious solution is to use a shorter time step. However,
reducing the time step would adversely slow the simulation rate, severely reducing the
size of systems that can be modeled interactively. As the system becomes more
sluggish, the user will also tend to increase the haptic force, exacerbating the problem.
A more elegant solution is to use a multiple time step integrator, such as
reversible RESPA,16 to separate the haptic forces from the ab initio interactions. The
weaker ab initio forces can then use a longer MD time step and be evaluated less
frequently (in wall time) than the stronger haptic forces. Between each ab initio
update, l sub-steps are used to accurately integrate the haptic force as follows.

150
Vi(n+1/2,0) !Vi
(n,l ) +Ai(n) "t2
l #
Vi(n+1/2,m+1/2) !Vi
(n+1/2,m ) +Fihap (tn,m )2mi
$ t
Xi(n,m+1) !Xi
(n,m ) +Vi(n+1/2,m+1/2)$ t
Vi(n+1/2,m+1) !Vi
(n+1/2,m+1/2) +Fihap (tn,m+1)2mi
$ t
%
&
'''
(
'''
Xi(n+1,0) !Xi
(n,l )
Ai(n+1) ! )*iE
qm (Xi(n+1,0) )
mi
Vi(n+1,0) !Vi
(n+1/2,l ) +Ai(n+1) "t
2
(7.2)
Here !t is the MD time step between ab initio force updates while the inner
time step, "t=!t/l, governs the interval between haptic force evaluations. A is the
acceleration due only to the ab initio forces, while Fhap (tn,m ) is the haptic force vector
evaluated at time tn,m = n!t +m" t . Compared to the computational costs of an
electronic structure calculation, it is trivial to integrate the haptic force in each sub-
step. Thus, the MTS scheme runs at the full speed of the simpler velocity Verlet
algorithm.
A second difficulty arises when incorporating strong haptic forces in MD. As
shown in figure 1 above, in the classical IMD scheme, VMD is responsible for
calculating the haptic force based on the currently displayed positions of the targeted
atom and haptic pointer. However, due to communication latencies and the time
required for VMD to complete each graphical update, the forces received by
TeraChem lag the simulated trajectory by at least 7ms. Importantly, this lag shifts the
forces in resonance with the system’s nuclear motion. The result is again uncontrolled

151
heating of the haptic vibrational mode. The solution is to modify the IMD scheme so
that haptic positions, rather than pulling forces, are sent to TeraChem. TeraChem can
then calculate the haptic forces at the correct instantaneous molecular geometry.
Decoupling display from simulation
Having developed a robust MD integrator for interactive simulations, we now
consider how to best display molecular motion to the user. In order to maintain the
visual sensation of smooth motion, the displayed coordinates must be updated with a
minimum frequency of ~20Hz. Assuming, as above, that it requires several hundred
milliseconds of wall time to evaluate each MD step, multiple visual frames must be
generated for each simulated MD step. To accomplish this we distinguish between the
simulated system which consists of the usual coordinates, velocities, and accelerations
at each time step, i, and a separate display system, !X(t) , which is continuous in time
and closely follows the simulated system, !X(i!t) " Xi .
By separating the simulation and display problems, the numerical integrity of
the overall model can be maintained while maximizing interactivity. Robust MD
integrators guarantee the numerical stability of the simulation and provide well-
defined physical properties, such as potential and kinetic energies, at each MD time
step. At the same time, the displayed system is free to compromise accuracy for
additional responsiveness. This is advantageous because the tolerances of human
perception are much more forgiving than those required for stable numerical
integration.

152
Figure 3: Schematic of simulated and visualized systems. Visualized frames (right boxes) lag the simulated coordinates (left boxes) by one ab initio gradient evaluation. The visualized coordinates are interpolated between MD time steps, but the simulated and visualized coordinates match after each step. The visualized haptic position is read from the device at each visual frame, but is sampled by the simulation once per MD step. The l MTS substeps of equation 7.2 calculate the haptic forces from a common haptic position.
Consider a simplified case in which the simulated trajectory is integrated using
the velocity Verlet integrator.17 In each MD step, the simulated system is propagated
t femtoseconds forward in simulation time as follows.
Step 1: X(n+1) = X(n) +V(n)!t + 12A(n)!t 2
Step 2: Ai(n+1) = "#iE
ai (X(n+1) )+ Fihap ((n +1)!t)
mi
Step 3: V(n+1) = V(n) + A(n) +A(n+1)
2!t
(7.3)
Here the evaluation of the ab initio gradient dominates the wall time required to
compute each MD step, Twall. Latency between haptic inputs and the system’s
response would be minimized by immediately displaying each coordinate vector,
X(n+1), as it is calculated in step 1. However, in this case displaying further motion
VisualizationTeraChemSimulation
Wal
l Tim
e
Update Xn-1 → Xn
Calculate
-∇E(Xn) → Fn
Sample hn-1
Update Xn → Xn+1
Calculate
-∇E(Xn+1) → Fn+1
Sample hn
Update Xn → Xn+1
Sample hn
Hap
tic P
ositi
on

153
during the time consuming calculation of A(n+1) in step 2 would require extrapolation
forward in time. In practice such extrapolation leads to noticeable artifacts as the
simulated and displayed coordinates diverge. An alternative is to buffer the displayed
trajectory by one MD step. Thus X(n) is displayed as X(n+1) is calculated in step 1, and
the display can then interpolate toward a known X(n+1) during the succeeding gradient
evaluation. In this way, smooth motion is achieved. The distinction between simulated
and visualized systems is illustrated in figure 3.
A variety of interpolation schemes are possible for !X . For example, linear
interpolation would give the following trajectory.
!X(nTwall + t) = X
(n) + X(n+1) !X(n)
Twallt (7.4)
Here t is given in wall time. While equation (7.4) provides continuous molecular
coordinates, the visualized velocity jumps discontinuously by
! !V = A(n+1) !t 2
Twall (7.5)
after each MD step. These velocity jumps can be reduced to
! !V = A
(n) "A(n"1)
2!t 2
Twall (7.6)
by continuously accelerating the display coordinates over the interpolated path as
follows.
!X(nTwall + t) = X
(n) +V(n) !tTwall
t + 12A(n) !t 2
Twall2 t 2 (7.7)

154
This trajectory is again continuous in coordinate space, and additionally, in the special
case of a constant acceleration, the velocity is also continuous between MD steps.
Since for MD simulations, molecular forces change gradually from step to step,
equation (7.7) is sufficient to reduce velocity discontinuities below the threshold of
human perception.
The simple approach of equation (7.7) is easily extended to the more
complicated MTS integrator developed above. For example, the displayed trajectory
can be defined piecewise between sub steps as follows.
!X n +ml( )Twall + t( ) = X(n,m ) +V(n,m ) !t
Twallt + 12A(n,m ) !t 2
Twall2 t 2 (7.8)
However, since the ab initio gradient “kicks” the velocity only at the outer time step,
equation (7.8) corresponds to a linear interpolation in the ab initio forces similar to
equation (7.4). Amortizing the ab initio acceleration over the time step, while applying
each haptic acceleration over its own sub-step produces a smoother trajectory.
!X n +ml( )Twall + t( ) = X(n,0) +V(n,0) !t
Twallt + 12A(n) !t 2
Twall2 t 2 (7.9)
As a result of this smoothing, the simulated and displayed systems do not
match at each sub-step. However, by applying the same haptic forces calculated for
the simulated system to the visual system, the systems will match to within machine
precision at the outer time steps. To avoid any buildup of round-off error, the display
coordinates are re-synced to the simulated system after each step.

155
In the present implementation, TeraChem is responsible for calculating both
the simulated and displayed trajectories. At intervals of about 10ms, a!communication
thread evaluates !X(t) and sends the updated coordinates to VMD.
Optimizing TeraChem for small systems
TeraChem was originally designed to handle large systems such as
proteins.18,19 To enable these huge calculations, the ERIs contributing to the Coulomb
and exchange operators are computed using GPUs. To maximize performance,
TeraChem uses custom unrolled procedures for each angular momentum class of ERIs
(e.g. one function handles Coulomb (ss|ss) type contributions, another (ss|sp), and so
on).20 For large systems, near perfect load balancing is achieved by distributing each
type of ERI across all GPUs in a computer. This is shown in the top frame of figure 4.
However, for small systems, there is not enough parallel work in an ERI batch to
saturate even one, much less as many as eight GPUs. Thus new strategy was
implemented as illustrated in the lower frame of figure 4.
Figure 4: Multi-GPU parallelization strategies. (A) Static load balancing ideal for large systems, where each class of integrals (e.g. sssp) provides enough work to saturate all GPUs. (B) Dynamic scheduling in which each GPU computes independent integral classes, allowing small jobs to saturate multiple GPUs with work.
Here each ERI class runs on a single GPU, and different GPUs run different types of
ERI kernels in parallel. The throughput of each GPU was further improved by using
CUDA streams to enable simultaneous execution of multiple kernels on each GPU.
(A)
(B)Queue 3 ssss sssp sssd ssps ... dddd
GPU3Queue 2 ssss sssp sssd ssps ... dddd
GPU2Queue 1 ssss sssp sssd ssps ... dddd
GPU1
ssss sssp sssd ssps sspp sspdGPU1
...GPU3
GPU2
ddddUnified Queue

156
Dynamic load balancing was enabled through Intel Cilk Plus work-stealing queues.
Together these schemes significantly increase the parallelism exposed by a small
calculation.
When handling small systems, it is also essential to minimize execution
latencies that may not have been apparent in larger calculations. In GPU code,
communication across the PCIe bus is a common cause of latency. This was
minimized by using asynchronous CUDA streams which are provided as part of the
CUDA runtime. CUDA streams reduce latencies for host-GPU communication and
enable memory transfers to overlap kernel execution. Another important cause of
latency is the allocation and release of GPU memory. While memory allocation is an
expensive operation on most architectures, allocations are particularly costly on the
GPU, triggering synchronization across devices and even serializing the execution of
kernels run on separate GPUs. To eliminate these costs, large blocks of memory are
pre-allocated from each GPU at the start of the calculation. Individual CPU threads
request these pre-allocated blocks as needed. A mutual exclusion lock is used to
guarantee thread safety during the assignment of GPU memory blocks. Once assigned,
the memory block is the sole property of a single thread and can be used without
further synchronization. Extending this system, blocks of page-locked host memory
are also pre-allocated and distributed along with the GPU memory blocks. By
assembling GPU input data in page-locked host memory, the driver can avoid staging
transfers through an internal buffer, further improving latency.
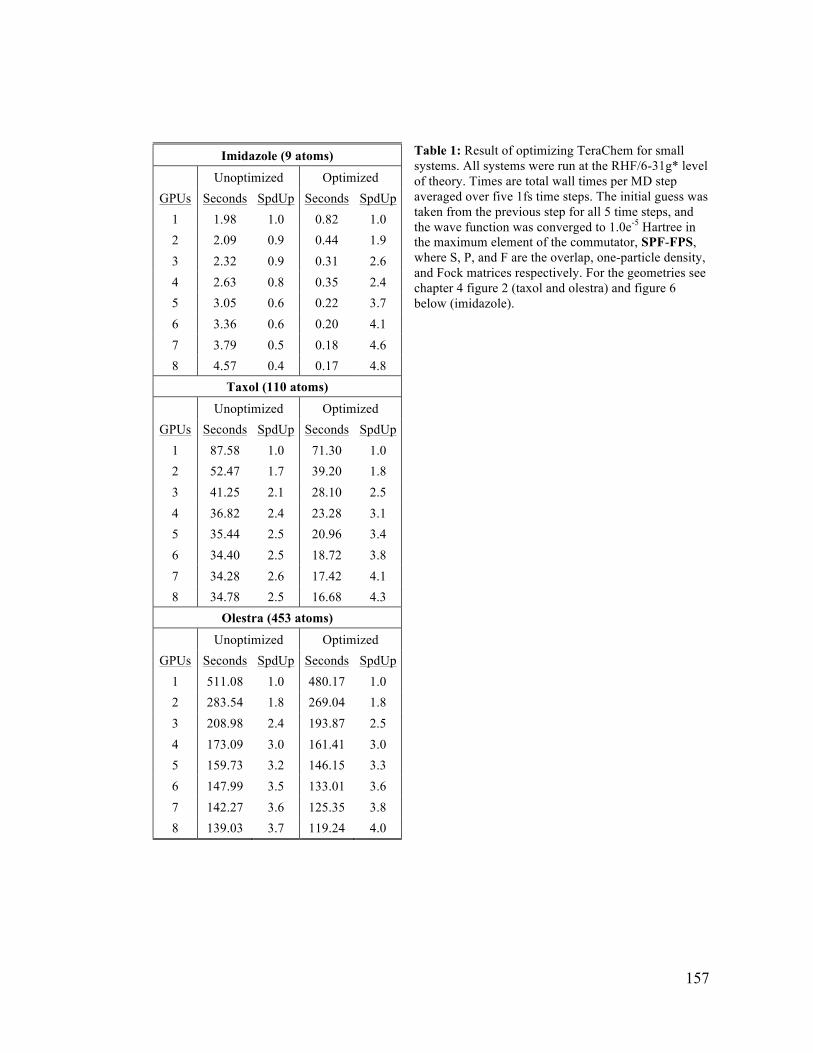
157
Table 1: Result of optimizing TeraChem for small systems. All systems were run at the RHF/6-31g* level of theory. Times are total wall times per MD step averaged over five 1fs time steps. The initial guess was taken from the previous step for all 5 time steps, and the wave function was converged to 1.0e-5 Hartree in the maximum element of the commutator, SPF-FPS, where S, P, and F are the overlap, one-particle density, and Fock matrices respectively. For the geometries see chapter 4 figure 2 (taxol and olestra) and figure 6 below (imidazole).
Imidazole (9 atoms)
Unoptimized Optimized GPUs Seconds SpdUp Seconds SpdUp
1 1.98 1.0 0.82 1.0 2 2.09 0.9 0.44 1.9 3 2.32 0.9 0.31 2.6 4 2.63 0.8 0.35 2.4 5 3.05 0.6 0.22 3.7 6 3.36 0.6 0.20 4.1 7 3.79 0.5 0.18 4.6 8 4.57 0.4 0.17 4.8
Taxol (110 atoms)
Unoptimized Optimized GPUs Seconds SpdUp Seconds SpdUp
1 87.58 1.0 71.30 1.0 2 52.47 1.7 39.20 1.8 3 41.25 2.1 28.10 2.5 4 36.82 2.4 23.28 3.1 5 35.44 2.5 20.96 3.4 6 34.40 2.5 18.72 3.8 7 34.28 2.6 17.42 4.1 8 34.78 2.5 16.68 4.3
Olestra (453 atoms)
Unoptimized Optimized GPUs Seconds SpdUp Seconds SpdUp
1 511.08 1.0 480.17 1.0 2 283.54 1.8 269.04 1.8 3 208.98 2.4 193.87 2.5 4 173.09 3.0 161.41 3.0 5 159.73 3.2 146.15 3.3 6 147.99 3.5 133.01 3.6 7 142.27 3.6 125.35 3.8 8 139.03 3.7 119.24 4.0

158
ASSESSMENT
In order to benchmark the numerical stability of the AI-IMD integrator, we
first consider the trivial diatomic system, HCl. Because the user does work through the
haptic forces, interactive dynamics will not in general conserve the total energy of the
system. Energy is conserved, however, in the special case of a fixed pulling position
since the spring force can be construed as a function of only the molecular geometry,
and the Hamiltonian becomes time independent. Indeed, in this case, the resulting
dynamics simply evolves on a force-modified potential energy surface.21 Using this
special case, the AI-IMD integrator is validated as follows. First the HCl molecule is
minimized at the RHF/6-31G** level of theory. The resulting geometry is aligned
along the y-axis with the chlorine atom located at the origin and the hydrogen at
y=1.265 Angstroms. The test system also includes a haptic spring connecting the
hydrogen atom to a fixed pulling point at y=1.35 Angstroms.
Starting from the above initial geometry, AI-IMD calculations were run using
varying haptic spring constants and MTS sub-steps. All calculations used a fixed time
step of 1.0fs to integrate the ab initio forces. For smaller haptic force constants, below
0.7 Hartree/Bohr2, the haptic-induced motions occur on timescales comparable to ab
initio forces encountered in near-equilibrium MD. Thus, a simple velocity Verlet
integrator conserves energy about as well as the MTS approach developed above. At
larger forces, however, velocity Verlet becomes increasingly unstable. Figure 5
demonstrates the stability of our AI-IMD integrator for a spring constant of 2.6
Hartree/Bohr2. Here the velocity Verlet integrator rapidly diverges, while an MTS
integrator using 5 sub-steps remains stable. Although overall energy drifts increase at

159
higher forces, the MTS approach does not exhibit wild divergence even at a force
constant of 4.0 Hartree/Bohr2. This is important because explosive divergence is a
much greater obstacle to controlling the system than is a slow energy drift. In general,
the motion of the haptic tugging point will itself induce heating so that a thermostat is
already required to counter the slow accumulation of energy during long simulations.
Figure 5: Total energy for IMD simulation of HCl with fixed tugging point. HCl was aligned with y-axis, with Cl intially at the origin and H at y=1.265 Angstoms. The haptic force pulled toward y=1.35 Angstroms with a force constant of 2.6 Hartree/Bohr2. A standard veloctiy Verlet integrator (red) suffers from wild energy oscillations and diverges before reaching 500fs. The AI-IMD MTS integrator (blue) used 5 sub-steps per ab initio force evaluation and shows no instability throughout the entire 5ps trajectory (of which 1ps is shown).
We turn next to more interesting systems that better illustrate the potential of
AI-IMD. As currently implemented in TeraChem, AI-IMD can be applied to systems
containing up to a few dozen atoms using double-zeta basis sets at the SCF level of
theory. For smaller systems polarization functions can also be employed. AI-IMD is
thus well suited to treat reactions of small organic molecules. Table 2 shows the
average wall time per MD step for simulations on a variety of representative
molecules. Here the average is calculated from short 250 step non-interactive
trajectories using the spin-restricted Hartree-Fock method and various basis sets. The
initial geometries were optimized at the Hartree-Fock/3-21G level of theory and are
shown in figure 4. At each MD step, the wave function was converged to 1.0e-4
Hartree in the maximum element of the commutator, SPF-FPS, where S, P, and F are
0
50
100
150
200
250
300
350
0 0.2 0.4 0.6 0.8 1
Tota
l Ene
rgy
- E0
(kca
l/mol
)
Time (ps)
velocity VerletMTS, 5 sub-steps

160
the overlap, one-particle density, and Fock matrices respectively. The initial density
for each SCF is obtained from the converged density of the previous MD step. The
Coulomb and exchange operators are computed predominantly using single precision
floating-point operations, with double precision used to accumulate each contribution
into the Fock matrix. This scheme has been shown to be accurate for systems up to
100 atoms.22
Molecule (atoms) STO-3G 3-21G 6-31G** Table 2: Wall time per MD time step. Times listed in ms and averaged over 250 steps of AIMD at RHF level of theory. Initial geometries are shown in figure 6. The MD time step was 1.0fs. Simulations used 8 Nvidia GTX970 GPUs in parallel.
HCl (2) 3.16 3.84 39.52 CH3F+OH- (7) 11.60 15.40 92.24 Imidazole (9) 21.64 36.00 157.28 Caffeine (24) 101.60 195.56 717.88 Quinoline (30) 112.72 201.68 858.84 Spiropyran (40) 246.88 401.84 2492.20
Figure 6: Initial geometries for benchmark calculations listed in table 2. All structures represent minima at the RHF/3-21G level of theory.
Figure 7 illustrates a typical interactive simulation. Here the quinoline system
introduced above is interactively modeled using RHF and the STO-3G basis set. Each
ab initio gradient is allowed 200 ms of wall time. The MTS integrator uses 10 sub-
steps and the force constant of the haptic spring is set to 0.15 Hartree/Bohr2.
Throughout the simulation, the coordinates of the three ammonia nitrogen atoms are

161
frozen to avoid their diffusion into the vacuum. This system has been previously used
to experimentally study long distance proton transfer.23 In a similar spirit, we use a
haptic tug to remove a proton from the central ammonia and form an ammonium ion
on the right as shown in panels (B) and (C) of figure 7. The system then spontaneously
responds by transferring three additional protons and lengthening the central C-C bond
between the quinoline rings to form 7-ketoquinoline as shown in panel (D).
Figure 7: Snapshots of interactive simulation of 7-hydroxyquinoline with three ammonia molecules. (A) The simulation is started from the geometry shown in figure 4. The cartesian coordinates of the ammonia nitrogen atoms are fixed to avoid diffusion. (B) Force is applied to transfer a proton from the central ammonia to form an ammonium ion on the right (C). Subsequently, three protons spontaneously transfer ultimately converting 7-hydroxyquinoline to a tuatomeric 7-ketoquinoline (D).
We find force-feedback through the haptic device to be an important feature in
AI-IMD for several reasons. First, it improves user control by providing a cue to
pulling distance. Even with a 3D display, it is sometimes difficult to estimate exact
positions within the simulated system. Since the force increases as the spring is

162
stretched, the feedback force helps the user determine where and how hard they are
pulling on the system. Second, feedback can provide vector field information that is
not easily represented visually. For example, in simulating the imidazole molecule
shown in figure 8, the N-H bonds can in general be bent much more easily than
stretched. In attempting to transfer the N-H proton between nitrogen atoms, the user
feels that a bending motion is easier and naturally moves the proton along a realistic
path. It the user tries to pull the atom into a forbidden region, here the middle of the !-
bonding structure of the aromatic ring, the proton resists and instead follows the path
shown around the perimeter. It is difficult to represent the field of such repulsions
visually. Thus, a force-feedback interface provides a unique and useful perspective.
Figure 8: Interactive proton transfer in imidazole molecule. The haptic pointer is attached to a proton originally on the far side of the molecule (translucent atom) and used to pull it across to the nearer nitrogen atom. The path of the haptic pointer is shown in green while that of the transfering proton is colored in white.
CONCLUSION
Interactive models have a long history in chemical research. The venerable ball
and stick model, invented more than a century ago still plays an important role in
shaping our understanding of Chemistry. In that same spirit, interactive ab initio
calculations represent a new synthesis of intuitive human interfaces with accurate
numerical methods. AI-IMD can already be applied to systems containing up to a few
dozen atoms at the Hartree-Fock level of theory. And, as computers and algorithms

163
continue to improve, the scope of on-the-fly calculations will continue to widen. Work
is already in progress to extend our work here to higher-level DFT methods and to
provide on-the-fly orbital display. These features present no technical challenges
beyond what has already been accomplished for Hartree-Fock calculations above.
The integration methods presented here provide robust energy conservation for
strong haptic forces as long as the ab initio forces are similar to those encountered in
equilibrium MD. Of course, the user is free to slam the system into much higher-
energy configurations where this assumption simply does not hold. More research is
needed to develop graceful ways to conserve the system’s energy in such cases, for
example by switching to an empirical description which can be rapidly evaluated at a
much shorter time step than is possible for the ab initio forces. Such developments
would greatly improve the resilience of AI-IMD simulations particularly for non-
expert users. The final test of any model is whether it provides fertile ground in which
researchers can formulate and test imaginative scientific questions. This explains the
longevity of existing interactive models in chemistry and surely recommends AI-IMD
as an area deserving much future research.
REFERENCES
(1) Johnson, C. K. OR TEP: A FORTRAN Thermal-Ellipsoid Plot Program for Crystal Structure Illustrations, Oak Ridge National Laboratory, 1965.
(2) Levinthal, C. Sci Am 1966, 214, 42.
(3) Surles, M. C.; Richardson, J. S.; Richardson, D. C.; Brooks, F. P. Protein Sci 1994, 3, 198.
(4) Leech, J.; Prins, J. F.; Hermans, J. Ieee Comput Sci Eng 1996, 3, 38.

164
(5) Prins, J. F.; Hermans, J.; Mann, G.; Nyland, L. S.; Simons, M. Future Gener Comp Sy 1999, 15, 485.
(6) Stone, J. E.; Gullingsrud, J.; Schulten, K. ACM Symposium on 3D Graphics 2001, 191.
(7) Bosson, M.; Grudinin, S.; Redon, S. J. Comp. Chem. 2013, 34, 492.
(8) Bosson, M.; Richard, C.; Plet, A.; Grudinin, S.; Redon, S. J. Comp. Chem. 2012, 33, 779.
(9) Marti, K. H.; Reiher, M. J. Comp. Chem. 2009, 30, 2010.
(10) Haag, M. P.; Marti, K. H.; Reiher, M. Chemphyschem 2011, 12, 3204.
(11) Haag, M. P.; Reiher, M. Int J Quantum Chem 2013, 113, 8.
(12) Ufimtsev, I. S.; Martinez, T. J. J. Chem. Theo. Comp. 2009, 5, 2619.
(13) Humphrey, W.; Dalke, A.; Schulten, K. J Mol Graph Model 1996, 14, 33.
(14) Hu, X. Q.; Yang, W. T. J. Chem. Phys. 2010, 132.
(15) Pulay, P. J. Comp. Chem. 1982, 3, 556.
(16) Tuckerman, M.; Berne, B. J.; Martyna, G. J. J. Chem. Phys. 1992, 97, 1990.
(17) Swope, W. C.; Andersen, H. C.; Berens, P. H.; Wilson, K. R. J. Chem. Phys. 1982, 76, 637.
(18) Kulik, H. J.; Luehr, N.; Ufimtsev, I. S.; Martinez, T. J. J. Phys. Chem. B 2012, 116, 12501.
(19) Ufimtsev, I. S.; Luehr, N.; Martinez, T. J. J. Phys. Chem. Lett. 2011, 2, 1789.
(20) Ufimtsev, I. S.; Martinez, T. J. J. Chem. Theo. Comp. 2008, 4, 222.
(21) Ong, M. T.; Leiding, J.; Tao, H. L.; Virshup, A. M.; Martinez, T. J. J. Am. Chem. Soc. 2009, 131, 6377.
(22) Luehr, N.; Ufimtsev, I. S.; Martinez, T. J. J. Chem. Theo. Comp. 2011, 7, 949.
(23) Tanner, C.; Manca, C.; Leutwyler, S. Chimia 2004, 58, 234.

165
BIBLIOGRAPHY
Adamson, R. D., J. P. Dombroski, and P. M. W. Gill. "Chemistry without Coulomb Tails." Chemical Physics Letters 254.5-6 (1996): 329-36.
---. "Efficient Calculation of Short-Range Coulomb Energies." J. Comp. Chem. 20 (1999): 921-27.
Ahmadi, G. R., and J. Almlof. "The Coulomb Operator in a Gaussian Product Basis." Chemical Physics Letters 246.4-5 (1995): 364-70.
Almlof, J., K. Faegri, and K. Korsell. "Principles for a Direct Scf Approach to Lcao-Mo Ab Initio Calculations." Journal of Computational Chemistry 3.3 (1982): 385-99.
Anderson, Joshua A., Chris D. Lorenz, and A. Travesset. "General Purpose Molecular Dynamics Simulations Fully Implemented on Graphics Processing Units." Journal of Computational Physics 227.10 (2008): 5342-59.
Anglada, E., J. Junquera, and J. M. Soler. "Efficient Mixed-Force First-Principles Molecular Dynamics." Physical Review E 68.5 (2003): 055701.
Appel, H., E. K. U. Gross, and K. Burke. "Excitations in Time-Dependent Density-Functional Theory." Physical Review Letters 90.4 (2003): 043005.
Asadchev, A., et al. "Uncontracted Rys Quadrature Implementation of up to G Functions on Graphical Processing Units." Journal of Chemical Theory and Computation 6.3 (2010): 696-704.
Asadchev, A., and M. S. Gordon. "Mixed-Precision Evaluation of Two-Electron Integrals by Rys Quadrature." Computer Physics Communications 183.8 (2012): 1563-67.
---. "New Multithreaded Hybrid Cpu/Gpu Approach to Hartree-Fock." Journal of Chemical Theory and Computation 8.11 (2012): 4166-76.
Badaeva, E., et al. "Excited-State Structure of Oligothiophene Dendrimers: Computational and Experimental Study." J. Phys. Chem. B 114 (2010): 15808-17.

166
Barnett, R. N., et al. "Born-Oppenheimer Dynamics Using Density Functional Theory - Equilibruium and Fragmentation of Small Sodium Clusters." J. Chem. Phys. 94 (1991): 608-16.
Barth, E., and T. Schlick. "Extrapolation Versus Impulse in Multiple-Timestepping Schemes. Ii. Linear Analysis and Applications to Newtonian and Langevin Dynamics." Journal of Chemical Physics 109.5 (1998): 1633-42.
Becke, A. D. "A Multicenter Numerical-Integration Scheme for Polyatomic-Molecules." Journal of Chemical Physics 88.4 (1988): 2547-53.
---. "A Multicenter Numerical Integration Scheme for Polyatomic Molecules." The Journal of Chemical Physics 88.4 (1988): 2547-53.
---. "Density-Functional Exchange-Energy Approximation with Correct Asymptotic Behavior." Physical Review A 38.6 (1988): 3098.
Ben-Nun, M., and T. J. Martinez. "Ab Initio Quantum Molecular Dynamics." Adv. Chem. Phys. 121 (2002): 439-512.
Bhaskaran-Nair, K., et al. "Noniterative Multireference Coupled Cluster Methods on Heterogeneous Cpu-Gpu Systems." Journal of Chemical Theory and Computation 9.4 (2013): 1949-57.
Biesiadecki, J. J., and R. D. Skeel. "Dangers of Multiple Time-Step Methods." Journal of Computational Physics 109.2 (1993): 318-28.
Bosson, M., S. Grudinin, and S. Redon. "Block-Adaptive Quantum Mechanics: An Adaptive Divide-and-Conquer Approach to Interactive Quantum Chemistry." Journal of Computational Chemistry 34.6 (2013): 492-504.
Bosson, M., et al. "Interactive Quantum Chemistry: A Divide-and-Conquer Ased-Mo Method." Journal of Computational Chemistry 33.7 (2012): 779-90.
Boys, S. F. "Electronic Wave Functions. 1. A General Method of Calculation for the Stationary States of Any Molecular System." Proceedings of the Royal Society of London Series a-Mathematical and Physical Sciences 200.1063 (1950): 542-54.
Brown, P., et al. "Massively Multicore Parallelization of Kohn-Sham Theory." J. Chem. Theo. Comp. 4 (2008): 1620-26.
Burant, J. C., G. E. Scuseria, and M. J. Frisch. "A Linear Scaling Method for Hartree-Fock Exchange Calculations of Large Molecules." Journal of Chemical Physics 105.19 (1996): 8969-72.

167
Burke, Kieron, Jan Werschnik, and E. K. U. Gross. "Time-Dependent Density Functional Theory: Past, Present, and Future." The Journal of Chemical Physics 123.6 (2005): 062206-9.
Bussi, G., D. Donadio, and M. Parrinello. "Canonical Sampling through Velocity Rescaling." Journal of Chemical Physics 126.1 (2007): 014101.
Car, Roberto, and Michele Parrinello. "Unified Approach for Molecular Dynamics and Density-Functional Theory." Phys. Rev. Lett. 55 (1985): 2471-74.
Carloni, P., U. Rothlisberger, and M. Parrinello. "The Role and Perspective of Ab Initio Molecular Dynamics in the Study of Biological Systems." Acc. Chem. Res. 35 (2002): 455-64.
Case, David A., et al. "The Amber Biomolecular Simulation Programs." Journal of Computational Chemistry 26.16 (2005): 1668-88.
Casida, Mark E. "Time Dependent Density Functional Response Theory for Molecules." Recent Advances in Density Functional Methods. Ed. Chong, D. P. Singapore: World Scientific, 1995.
Casida, Mark E., et al. "Molecular Excitation Energies to High-Lying Bound States from Time-Dependent Density-Functional Response Theory: Characterization and Correction of the Time-Dependent Local Density Approximation Ionization Threshold." The Journal of Chemical Physics 108.11 (1998): 4439-49.
Challacombe, M., and E. Schwegler. "Linear Scaling Computation of the Fock Matrix." Journal of Chemical Physics 106.13 (1997): 5526-36.
Cohen, A. J., P. Mori-Sanchez, and W. Yang. "Insights into Current Limitations of Density Functional Theory." Science 321 (2008): 792-94.
Collins, M. A. "Systematic Fragmentation of Large Molecules by Annihilation." Phys. Chem. Chem. Phys. 14 (2012): 7744-51.
Cordova, F., et al. "Troubleshooting Time-Dependent Density Functional Theory for Photochemical Applications: Oxirane." J. Chem. Phys. 127 (2007): 164111.
Dallos, M., et al. "Analytic Evaluation of Nonadiabatic Coupling Terms at the Mr-Ci Level. Ii. Minima on the Crossing Seam: Formaldehyde and the Photodimerization of Ethylene." J. Chem. Phys. 120 (2004): 7330-39.
Davidson, E. R. "Iterative Calculation of a Few of Lowest Eigenvalues and Corresponding Eigenvectors of Large Real-Symmetric Matrices." Journal of Computational Physics 17.1 (1975): 87-94.

168
Davidson, Ernest R. "The Iterative Calculation of a Few of the Lowest Eigenvalues and Corresponding Eigenvectors of Large Real-Symmetric Matrices." Journal of Computational Physics 17.1 (1975): 87-94.
Daw, M. S. "Model for Energetics of Solids Based on the Density-Matrix." Physical Review B 47.16 (1993): 10895-98.
DePrince, A. E., and J. R. Hammond. "Coupled Cluster Theory on Graphics Processing Units I. The Coupled Cluster Doubles Method." Journal of Chemical Theory and Computation 7.5 (2011): 1287-95.
DePrince, A. E., et al. "Density-Fitted Singles and Doubles Coupled Cluster on Graphics Processing Units." Molecular Physics 112 (2014): 844-52.
Deraedt, H., and B. Deraedt. "Applications of the Generalized Trotter Formula." Physical Review A 28.6 (1983): 3575-80.
des Cloizeaux, J. "Energy Bands + Projection Operators in Crystal - Analytic + Asymptotic Properties." Physical Review 135.3A (1964): A685-+.
Devadoss, C., P. Bharathi, and J. S. Moore. "Energy Transfer in Dendritic Macromolecules: Molecular Size Effects and the Role of an Energy Gradient." Journal of the American Chemical Society 118.40 (1996): 9635-44.
Dion, M., et al. "Van Der Waals Density Functional for General Geometries." Phys. Rev. Lett. 92 (2004): 246401.
Dirac, P. A. M. "Quantum Mechanics of Many-Electron Systems." Proceedings of the Royal Society of London Series a-Containing Papers of a Mathematical and Physical Character 123.792 (1929): 714-33.
Dreuw, A., and M. Head-Gordon. "Single-Reference Ab Initio Methods for the Calculation of Excited States of Large Molecules." Chemical Reviews 105.11 (2005): 4009-37.
Dreuw, Andreas, Jennifer L. Weisman, and Martin Head-Gordon. "Long-Range Charge-Transfer Excited States in Time-Dependent Density Functional Theory Require Non-Local Exchange." The Journal of Chemical Physics 119.6 (2003): 2943-46.
Fedorov, D. G., T. Nagata, and K. Kitaura. "Exploring Chemistry with the Fragment Molecular Orbital Method." Phys. Chem. Chem. Phys. 14 (2012): 7562-77.
Foresman, James B., et al. "Toward a Systematic Molecular Orbital Theory for Excited States." The Journal of Physical Chemistry 96.1 (1992): 135-49.

169
Friedrichs, Mark S., et al. "Accelerating Molecular Dynamic Simulation on Graphics Processing Units." Journal of Computational Chemistry 30.6 (2009): 864-72.
Genovese, L., et al. "Density Functional Theory Claculation on Many-Cores Hybrid Cpu-Gpu Architectures." J. Chem. Phys. 131 (2009): 034103.
Gibson, D. A. , and E. A. Carter. "Time-Reversible Multiple Time Scale Ab Initio Molecular Dynamics." J. Phys. Chem. 97 (1993): 13429-34.
Gordon, M. S., et al. "The Effective Fragment Potential Method: A Qm-Based Mm Approach to Modeling Environmental Effects in Chemistry." J. Phys. Chem. A 105 (2001): 293-307.
Gordon, M. S., et al. "Accurate First Principles Model Potentials for Intermolecular Interactions." Ann. Rev. Phys. Chem. 64 (2013): 553-78.
Grabo, T., M. Petersilka, and E. K. U. Gross. "Molecular Excitation Energies from Time-Dependent Density Functional Theory." Journal of Molecular Structure: THEOCHEM 501-502 (2000): 353-67.
Grimme, Stefan. "Accurate Description of Van Der Waals Complexes by Density Functional Theory Including Empirical Corrections." Journal of Computational Chemistry 25.12 (2004): 1463-73.
---. "Semiempirical Gga-Type Density Functional Constructed with a Long-Range Dispersion Correction." Journal of Computational Chemistry 27.15 (2006): 1787-99.
Grimme, S., and M. Parac. "Substantial Errors from Time-Dependent Density Functional Theory for the Calculation of Excited States of Large Pi Systems." ChemPhysChem 4 (2003): 292-95.
Gross, E. K. U., and Walter Kohn. "Local Density-Functional Theory of Frequency-Dependent Linear Response." Physical Review Letters 55.26 (1985): 2850.
Grossman, J. P., et al. "Hardware Support for Fine-Grained Event-Driven Computation in Anton 2." Acm Sigplan Notices 48.4 (2013): 549-60.
Grossman, J. P., et al. "The Role of Cascade, a Cycle-Based Simulation Infrastructure, in Designing the Anton Special-Purpose Supercomputers." 2013 50th Acm / Edac / Ieee Design Automation Conference (Dac) (2013).
Grubmuller, H., et al. "Generalized Verlet Algorithm for Efficient Molecular Dynamics Simulations with Long-Range Interactions." Mol. Sim. 6 (1991): 121-42.

170
Guidon, M., et al. "Ab Initio Molecular Dynamics Using Hybrid Density Functionals." Journal of Chemical Physics 128.21 (2008): 214104.
Haag, M. P., K. H. Marti, and M. Reiher. "Generation of Potential Energy Surfaces in High Dimensions and Their Haptic Exploration." Chemphyschem 12.17 (2011): 3204-13.
Haag, M. P., and M. Reiher. "Real-Time Quantum Chemistry." International Journal of Quantum Chemistry 113.1-2 (2013): 8-20.
Han, G. W., et al. "Error and Timing Analysis of Multiple Time-Step Integration Methods for Molecular Dynamics." Computer Physics Communications 176.4 (2007): 271-91.
Hancock, Jessica M., et al. "High-Efficiency Electroluminescence from New Blue-Emitting Oligoquinolines Bearing Pyrenyl or Triphenyl Endgroups." The Journal of Physical Chemistry C 111.18 (2007): 6875-82.
Harpham, Michael R., et al. "Thiophene Dendrimers as Entangled Photon Sensor Materials." Journal of the American Chemical Society 131.3 (2009): 973-79.
Hartke, B. , D. A. Gibson, and E. A. Carter. "Multiple Time Scale Hartree-Fock Molecular Dynamics." Int. J. Quant. Chem. 45 (1993): 59-70.
Harvey, M. J., G. Giupponi, and G. DeFabiritiis. "Acemd: Accelerating Biomolecular Dynamics in the Microsecond Time Scale." J. Chem. Theo. Comp. 5 (2009): 1632-39.
He, X., and J. Z. H. Zhang. "The Generalized Molecular Fractionation with Conjugate Caps/Molecular Mechanics Method for Direct Calculation of Protein Energy." J. Chem. Phys. 124 (2006): 184703.
Helgaker, Trygve, Poul Jørgensen, and Jeppe Olsen. Molecular Electronic-Structure Theory. New York: Wiley, 2000.
Heyd, Jochen, Gustavo E. Scuseria, and Matthias Ernzerhof. "Hybrid Functionals Based on a Screened Coulomb Potential." The Journal of Chemical Physics 118.18 (2003): 8207-15.
Hirata, So, and Martin Head-Gordon. "Time-Dependent Density Functional Theory within the Tamm-Dancoff Approximation." Chemical Physics Letters 314.3-4 (1999): 291-99.

171
Hirata, So, Martin Head-Gordon, and Rodney J. Bartlett. "Configuration Interaction Singles, Time-Dependent Hartree--Fock, and Time-Dependent Density Functional Theory for the Electronic Excited States of Extended Systems." The Journal of Chemical Physics 111.24 (1999): 10774-86.
Hu, X. Q., and W. T. Yang. "Accelerating Self-Consistent Field Convergence with the Augmented Roothaan-Hall Energy Function." Journal of Chemical Physics 132.5 (2010).
Humphrey, W., A. Dalke, and K. Schulten. "Vmd: Visual Molecular Dynamics." Journal of Molecular Graphics & Modelling 14.1 (1996): 33-38.
Hwu, Wen-mei. Gpu Computing Gems. Amsterdam ; Burlington, MA: Elsevier, 2011.
Iikura, Hisayoshi, et al. "A Long-Range Correction Scheme for Generalized-Gradient-Approximation Exchange Functionals." The Journal of Chemical Physics 115.8 (2001): 3540-44.
Isborn, C. M., et al. "Excited-State Electronic Structure with Configuration Interaction Singles and Tamm-Dancoff Time-Dependent Density Functional Theory on Graphical Processing Units." Journal of Chemical Theory and Computation 7.6 (2011): 1814-23.
Jacquemin, Denis, et al. "Extensive Td-Dft Benchmark: Singlet-Excited States of Organic Molecules." Journal of Chemical Theory and Computation 5.9 (2009): 2420-35.
Johnson, B. G., P. M. W. Gill, and J. A. Pople. "The Performance of a Family of Density Functional Methods." Journal of Chemical Physics 98.7 (1993): 5612-26.
Johnson, Carroll K. Or Tep: A Fortran Thermal-Ellipsoid Plot Program for Crystal Structure Illustrations. Oak Ridge, TN: Oak Ridge National Laboratory, 1965.
Jorgensen, William L., et al. "Comparison of Simple Potential Functions for Simulating Liquid Water." The Journal of Chemical Physics 79.2 (1983): 926-35.
Kahan, W. "Further Remarks on Reducing Truncation Errors." Communications of the Acm 8.1 (1965): 40-&.
Kirk, D. B., and W. W. Hwu. Programming Massively Parallel Processors: A Hands-on Approach. Burlington, MA: Morgan Kauffman 2010.

172
Ko, C., et al. "Pseudospectral Time-Dependent Density Functional Theory." J. Chem. Phys. 128 (2008): 104103.
Kobayashi, Y., H. Nakano, and K. Hirao. "Mutlireference Moller-Plesset Perturbation Theory Using Spin-Dependent Orbital Energies." Chem. Phys. Lett. 336 (2001): 529-35.
Kohn, W. "Analytic Properties of Bloch Waves and Wannier Functions." Physical Review 115.4 (1959): 809-21.
Kohn, W., and L. J. Sham. "Self-Consistent Equations Including Exchange and Correlation Effects." Physical Review 140.4A (1965): 1133-&.
Krylov, A. I. "Equation-of-Motion Coupled Cluster Methods for Open-Shell Adn Electronically Excited Species: The Hitchiker's Guide to Fock Space." Ann. Rev. Phys. Chem. 59 (2008): 433-62.
Kulik, H. J., et al. "Ab Initio Quantum Chemistry for Protein Structures." Journal of Physical Chemistry B 116.41 (2012): 12501-09.
Kuskin, J. S., et al. "Incorporating Flexibility in Anton, a Specialized Machine for Molecular Dynamics Simulation." 2008 Ieee 14th International Symposium on High Peformance Computer Architecture (2008): 315-26.
Kussmann, J., and C. Ochsenfeld. "Pre-Selective Screening for Matrix Elements in Linear-Scaling Exact Exchange Calculations." Journal of Chemical Physics 138.13 (2013).
Langlois, J.-M., et al. "Rule-Based Trial Wave Functions for Generalized Valence Bond Theory." J. Phys. Chem. 98 (1994): 13498-505.
Larson, R. H., et al. "High-Throughput Pairwise Point Interactions in Anton, a Specialized Machine for Molecular Dynamics Simulation." 2008 Ieee 14th International Symposium on High Peformance Computer Architecture (2008): 303-14.
Lebedev, V. I., and D. N. Laikov. "Quadrature Formula for the Sphere of 131-Th Algebraic Order of Accuracy." Doklady Akademii Nauk 366.6 (1999): 741-45.
---. "Quadrature Formula for the Spehre of 131-Th Algebraic Order of Accuracy." Dokl. Akad. Nauk 366 (1999): 741-45.
Lee, Chengteh, Weitao Yang, and Robert G. Parr. "Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density." Physical Review B 37.2 (1988): 785.

173
Leech, J., J. F. Prins, and J. Hermans. "Smd: Visual Steering of Molecular Dynamics for Protein Design." Ieee Computational Science & Engineering 3.4 (1996): 38-45.
Leforestier, C. "Classical Trajectories Using the Full Ab Initio Potential Energy Surface H-+Ch4 -> Ch4 + H-." J. Chem. Phys. 68 (1978): 4406-10.
Levine, B., and T. J. Martinez. "Hijacking the Playstation2 for Computational Chemistry." Abst. Pap. Amer. Chem. Soc. 226 (2003): U426.
Levine, Benjamin G., et al. "Conical Intersections and Double Excitations in Time-Dependent Density Functional Theory." Molecular Physics 104.5-7 (2006): 1039-51.
Levinthal, C. "Molecular Model-Building by Computer." Scientific American 214.6 (1966): 42-&.
Li, X. H., V. E. Teige, and S. S. Iyengar. "Can the Four-Coordinated, Penta-Valent Oxygen in Hydroxide Water Clusters Be Detected through Experimental Vibrational Spectroscopy?" Journal of Physical Chemistry A 111.22 (2007): 4815-20.
Li, X. P., R. W. Nunes, and D. Vanderbilt. "Density-Matrix Electronic-Structure Method with Linear System-Size Scaling." Physical Review B 47.16 (1993): 10891-94.
Liu, Weiguo, et al. "Accelerating Molecular Dynamics Simulations Using Graphics Processing Units with Cuda." Comp. Phys. Comm. 179.9 (2008): 634-41.
Luehr, Nathan, Thomas E. Markland, and Todd J. Martinez. "Multiple Time Step Integrators in Ab Initio Molecular Dynamics." Journal of Chemical Physics 140 (2014): 084116.
Luehr, N., I. S. Ufimtsev, and T. J. Martinez. "Dynamic Precision for Electron Repulsion Integral Evaluation on Graphical Processing Units (Gpus)." Journal of Chemical Theory and Computation 7.4 (2011): 949-54.
Ma, Q., J. A. Izaguirre, and R. D. Skeel. "Verlet-I/R-Respa/Impulse Is Limited by Nonlinear Instabilities." Siam Journal on Scientific Computing 24.6 (2003): 1951-73.
Ma, W. J., et al. "Gpu-Based Implementations of the Noniterative Regularized-Ccsd(T) Corrections: Applications to Strongly Correlated Systems." Journal of Chemical Theory and Computation 7.5 (2011): 1316-27.

174
Ma, Z. H., and M. E. Tuckerman. "On the Connection between Proton Transport, Structural Diffusion, and Reorientation of the Hydrated Hydroxide Ion as a Function of Temperature." Chemical Physics Letters 511.4-6 (2011): 177-82.
Maitra, Neepa T., et al. "Double Excitations within Time-Dependent Density Functional Theory Linear Response." The Journal of Chemical Physics 120.13 (2004): 5932-37.
Makino, J., K. Hiraki, and M. Inaba. "Grape-Dr: 2-Pflops Massively-Parallel Computer with 512-Core, 512-Gflops Processor Chips for Scientific Computing." 2007 Acm/Ieee Sc07 Conference (2010): 548-58.
Marti, K. H., and M. Reiher. "Haptic Quantum Chemistry." Journal of Computational Chemistry 30.13 (2009): 2010-20.
Martinez, T. J., and E. A. Carter. "Pseudospectral Double-Excitation Configuration Interaction." J. Chem. Phys. 98 (1993): 7081-85.
---. "Pseudospectral Moller-Plesset Perturbation Theory through Third Order." J. Chem. Phys. 100 (1994): 3631-38.
---. "Pseudospectral Methods Applied to the Electron Correlation Problem." Modern Electronic Structure Theory, Part Ii. Ed. Yarkony, D. R. Singapore: World Scientific, 1995. 1132-65.
---. "Pseudospectral Multi-Reference Single and Double-Excitation Configuration Interaction." J. Chem. Phys. 102 (1995): 7564-72.
Martinez, T. J., A. Mehta, and E. A. Carter. "Pseudospectral Full Configuration Interaction." J. Chem. Phys. 97 (1992): 1876-80.
Marx, D., A. Chandra, and M. E. Tuckerman. "Aqueous Basic Solutions: Hydroxide Solvation, Structural Diffusion, and Comparison to the Hydrated Proton." Chemical Reviews 110.4 (2010): 2174-216.
Mcmurchie, L. E., and E. R. Davidson. "One-Electron and 2-Electron Integrals over Cartesian Gaussian Functions." Journal of Computational Physics 26.2 (1978): 218-31.
McMurchie, Larry E., and Ernest R. Davidson. "One- and Two-Electron Integrals over Cartesian Gaussian Functions." Journal of Computational Physics 26.2 (1978): 218-31.

175
Minary, P., M. E. Tuckerman, and G. J. Martyna. "Long Time Molecular Dynamics for Enhanced Conformational Sampling in Biomolecular Systems." Physical Review Letters 93.15 (2004): 150201.
Morrone, J. A., et al. "Efficient Multiple Time Scale Molecular Dynamics: Using Colored Noise Thermostats to Stabilize Resonances." Journal of Chemical Physics 134.1 (2011): 014103.
Murray, C. W. , N. C. Handy, and G. J. Laming. "Quadrature Schemes for Integrals of Density Functional Theory." Mol. Phys. 78 (1993): 997.
Neese, F., et al. "Efficient, Approximate and Parallel Hartree-Fock and Hybrid Dft Calculations. A 'Chain-of-Spheres' Algorithm for the Hartree-Fock Exchange." Chemical Physics 356.1-3 (2009): 98-109.
Nielsen, I. B., et al. "Absorption Spectra of Photoactive Yellow Protein Chromophores in Vacuum." Biophysical Journal 89.4 (2005): 2597-604.
NVIDIA. Cuda C Programming Guide. 2013. Design Guide. March 7, 2014.
Ochsenfeld, C., C. A. White, and M. Head-Gordon. "Linear and Sublinear Scaling Formation of Hartree-Fock-Type Exchange Matrices." Journal of Chemical Physics 109.5 (1998): 1663-69.
Olivares-Amaya, R., et al. "Accelerating Correlated Quantum Chemistry Calculations Using Graphical Processing Units and a Mixed Precision Matrix Multiplication Library." Journal of Chemical Theory and Computation 6.1 (2010): 135-44.
Ong, M. T., et al. "First Principles Dynamics and Minimum Energy Pathways for Mechanochemical Ring Opening of Cyclobutene." Journal of the American Chemical Society 131.18 (2009): 6377-+.
Parr, Robert G., and Weitao Yang. Density-Functional Theory of Atoms and Molecules. International Series of Monographs on Chemistry. Oxford: Oxford University Press, 1989.
Payne, M. C., et al. "Iterative Minimization Techniques for Ab Initio Total Energy Calculations - Molecular Dynamics and Conjugate Gradients." Rev. Mod. Phys. 64 (1992): 1045-97.
PetaChem, LLC. "Http://Www.Petachem.Com." Web. November 30, 2010 2010.
Polli, D., et al. "Conical Intersection Dynamics of the Primary Photoisomerization Event in Vision." Nature 467 (2010): 440-43.

176
Pople, J. A., P. M. W. Gill, and B. G. Johnson. "Kohn-Sham Density-Functional Theory within a Finite Basis Set." Chemical Physics Letters 199.6 (1992): 557-60.
Prins, J. F., et al. "A Virtual Environment for Steered Molecular Dynamics." Future Generation Computer Systems 15.4 (1999): 485-95.
Pruitt, S. R., et al. "The Fragment Molecular Orbintal and Systematic Molecular Fragmentation Methods Applied to Water Clusters." Phys. Chem. Chem. Phys. 14 (2012): 7752-64.
Pulay, P. "Improved Scf Convergence Acceleration." Journal of Computational Chemistry 3.4 (1982): 556-60.
Ramakrishna, Guda, et al. "Oligothiophene Dendrimers as New Building Blocks for Optical Applications†." The Journal of Physical Chemistry A 112.10 (2007): 2018-26.
Reza Ahmadi, G., and Jan Almlof. "The Coulomb Operator in a Gaussian Product Basis." Chemical Physics Letters 246.4-5 (1995): 364-70.
Rohrdanz, Mary A., and John M. Herbert. "Simultaneous Benchmarking of Ground- and Excited-State Properties with Long-Range-Corrected Density Functional Theory." The Journal of Chemical Physics 129.3 (2008): 034107-9.
Rohrdanz, Mary A., Katie M. Martins, and John M. Herbert. "A Long-Range-Corrected Density Functional That Performs Well for Both Ground-State Properties and Time-Dependent Density Functional Theory Excitation Energies, Including Charge-Transfer Excited States." The Journal of Chemical Physics 130.5 (2009): 054112-8.
Roos, B. O. "Theoretical Studies of Electronically Excited States of Molecular Systems Using Multiconfigurational Perturbation Theory." Acc. Chem. Res. 32 (1999): 137-44.
Rubensson, E. H., E. Rudberg, and P. Salek. "Density Matrix Purification with Rigorous Error Control." Journal of Chemical Physics 128.7 (2008).
Ruckenbauer, M., et al. "Nonadiabatic Excited-State Dynamics with Hybrid Ab Initio Quantum-Mechanical/Molecular-Mechanical Methods: Solcavtion of the Pentadieniminium Cation in Apolar Media." J. Phys. Chem. A 114 (2010): 6757-65.

177
Rudberg, E., and E. H. Rubensson. "Assessment of Density Matrix Methods for Linear Scaling Electronic Structure Calculations." Journal of Physics-Condensed Matter 23.7 (2011).
Rudberg, E., E. H. Rubensson, and P. Salek. "Automatic Selection of Integral Thresholds by Extrapolation in Coulomb and Exchange Matrix Constructions." Journal of Chemical Theory and Computation 5.1 (2009): 80-85.
Runge, Erich, and E. K. U. Gross. "Density-Functional Theory for Time-Dependent Systems." Physical Review Letters 52.12 (1984): 997.
Rys, J., M. Dupuis, and H. F. King. "Computation of Electron Repulsion Integrals Using the Rys Quadrature Method." Journal of Computational Chemistry 4.2 (1983): 154-57.
Sanz-Serna, J. M., and M. P. Calvo. Numerical Hamiltonian Problems. London: Chapman and Hall, 1994.
Schafer, L., et al. "Chromophore Protonation State Controls Photoswitching of the Fluoroprotein Asfp595." PLoS Comp. Bio. 4 (2008): e1000034.
Schmidt, M. W., et al. "General Atomic and Molecular Electronic-Structure System." Journal of Computational Chemistry 14.11 (1993): 1347-63.
Schwegler, E., and M. Challacombe. "Linear Scaling Computation of the Hartree-Fock Exchange Matrix." Journal of Chemical Physics 105.7 (1996): 2726-34.
---. "Linear Scaling Computation of the Fock Matrix. Iv. Multipole Accelerated Formation of the Exchange Matrix." Journal of Chemical Physics 111.14 (1999): 6223-29.
---. "Linear Scaling Computation of the Fock Matrix. Iii. Formation of the Exchange Matrix with Permutational Symmetry." Theoretical Chemistry Accounts 104.5 (2000): 344-49.
Schwegler, E., M. Challacombe, and M. HeadGordon. "Linear Scaling Computation of the Fock Matrix. 2. Rigorous Bounds on Exchange Integrals and Incremental Fock Build." Journal of Chemical Physics 106.23 (1997): 9708-17.
Shaw, D. E., et al. "Anton, a Special-Purpose Machine for Molecular Dynamics Simulation." Isca'07: 34th Annual International Symposium on Computer Architecture, Conference Proceedings (2007): 1-12.

178
Shaw, D. E., et al. "Millisecond-Scale Molecular Dynamics Simulations on Anton." Proceedings of the Conference on High Performance Computing Networking, Storage and Analysis (2009).
Stanton, J. F. , and R. J. Bartlett. "The Equation of Motion Coupled Cluster Method. A Systematic Biorthogonal Approach to Molecular Excitation Energies, Transition Probabilities, and Excited Satte Properties." J. Chem. Phys. 98 (1993): 7029-39.
Steele, R. P. . "Communication: Multiple-Timestep Ab Initio Molecular Dynamics with Electron Correlation." J. Chem. Phys. 139 (2013): 011102.
Steinmann, C., D. G. Fedorov, and J. H. Jensen. "Mapping Enzymatic Catalysis Using the Effective Fragment Molecular Orbital Method: Towards All Ab Initio Biochemistry." PLOS One 8 (2013): e60602.
Stone, John E., Justin Gullingsrud, and Klaus Schulten. "A System for Interactive Molecular Dynamics Simulation." ACM Symposium on 3D Graphics (2001): 191.
Stone, John E., et al. "Accelerating Molecular Modeling Applications with Graphics Processors." Journal of Computational Chemistry 28.16 (2007): 2618-40.
Strain, M. C., G. E. Scuseria, and M. J. Frisch. "Achieving Linear Scaling for the Electronic Quantum Coulomb Problem." Science 271.5245 (1996): 51-53.
Stratmann, R. E., G. E. Scuseria, and M. J. Frisch. "Achieving Linear Scaling in Exchange-Correlation Density Functional Quadratures." Chemical Physics Letters 257.3-4 (1996): 213-23.
Streett, W. B., D. J. Tildesley, and G. Saville. "Multiple Time-Step Methods in Molecular Dynamics." Mol. Phys. 35 (1978): 639-48.
Surles, M. C., et al. "Sculpting Proteins Interactively - Continual Energy Minimization Embedded in a Graphical Modeling System." Protein Science 3.2 (1994): 198-210.
Suzuki, M. "Generalized Trotters Formula and Systematic Approximants of Exponential Operators and Inner Derivations with Applications to Many-Body Problems." Communications in Mathematical Physics 51.2 (1976): 183-90.
Swope, W. C., et al. "A Computer-Simulation Method for the Calculation of Equilibrium-Constants for the Formation of Physical Clusters of Molecules - Application to Small Water Clusters." Journal of Chemical Physics 76.1 (1982): 637-49.

179
Szabo, Attila, and Neil S. Ostlund. Modern Quantum Chemistry. New York: McGraw Hill, 1982.
Takashima, H., et al. "Is Large-Scale Ab Initio Hartree-Fock Calculation Chemically Accurate? Toward Improved Calculation of Biological Molecule Properties." Journal of Computational Chemistry 20.4 (1999): 443-54.
Tanner, C., C. Manca, and S. Leutwyler. "7-Hydroxyquinoline Center Dot(Nh3)(3): A Model for Excited State H-Atom Transfer Along an Ammonia Wire." Chimia 58.4 (2004): 234-36.
Tao, Jianmin, and Sergei Tretiak. "Optical Absorptions of New Blue-Light Emitting Oligoquinolines Bearing Pyrenyl and Triphenyl Endgroups Investigated with Time-Dependent Density Functional Theory." Journal of Chemical Theory and Computation 5.4 (2009): 866-72.
Tawada, Yoshihiro, et al. "A Long-Range-Corrected Time-Dependent Density Functional Theory." The Journal of Chemical Physics 120.18 (2004): 8425-33.
Titov, A. V., et al. "Generating Efficient Quantum Chemistry Codes for Novel Architectures." Journal of Chemical Theory and Computation 9.1 (2013): 213-21.
Tokita, Y., and H. Nakatsuji. "Ground and Excited States of Hemoglobin Co and Horesradish Peroxidase Co: Sac/Sac-Ci Study." J. Phys. Chem. B 101 (1997): 3281-89.
Towles, B., et al. "Unifying on-Chip and Inter-Node Switching within the Anton 2 Network." 2014 Acm/Ieee 41st Annual International Symposium on Computer Architecture (Isca) (2014): 1-12.
Trotter, H. F. "On the Product of Semi-Groups of Operators." Proc. Amer. Math. Soc. 10 (1959): 545-51.
Tuckerman, M., B. J. Berne, and G. J. Martyna. "Reversible Multiple Time Scale Molecular-Dynamics." Journal of Chemical Physics 97.3 (1992): 1990-2001.
Tuckerman, Mark E, and Bruce J Berne. "Molecular Dynamics in Systems with Multiple Time Scales: Systems with Stiff and Soft Degrees of Freedom and with Short and Long Range Forces." The Journal of Chemical Physics 95 (1991): 8362-64.
Tuckerman, Mark E, Bruce J Berne, and G J Martyna. "Molecular Dynamics Algorithm for Multiple Time Scales: Systems with Long Range Forces." The Journal of Chemical Physics 94 (1991): 6811-15.

180
---. "Reversible Multiple Time Scale Molecular Dynamics." The Journal of Chemical Physics 97 (1992): 1990-2001.
Tuckerman, Mark E, Bruce J Berne, and A Rossi. "Molecular Dynamics Algorithm for Multiple Time Scales: Systems with Disparate Masses." The Journal of Chemical Physics 94 (1991): 1465-69.
Tuckerman, M. E., A. Chandra, and D. Marx. "Structure and Dynamics of Oh-(Aq)." Accounts of Chemical Research 39.2 (2006): 151-58.
Tuckerman, Mark E, G J Martyna, and Bruce J Berne. "Molecular Dynamics Algorithm for Condensed Systems with Multiple Time Scales." The Journal of Chemical Physics 93 (1990): 1287-91.
Tuckerman, M. E., D. Marx, and M. Parrinello. "The Nature and Transport Mechanism of Hydrated Hydroxide Ions in Aqueous Solution." Nature 417.6892 (2002): 925-29.
Tuckerman, M. E., and Michele Parrinello. "Integrating the Car-Parrinello Equations. Ii. Multiple Time-Scale Techniques." J. Chem. Phys. 101 (1994): 1316-29.
Tuckerman, M. E., et al. "Ab Initio Molecular Dynamics Simulations." J. Phys. Chem. 100 (1996): 12878-87.
Ufimtsev, I. S., N. Luehr, and T. J. Martinez. "Charge Transfer and Polarization in Solvated Proteins from Ab Initio Molecular Dynamics." Journal of Physical Chemistry Letters 2.14 (2011): 1789-93.
Ufimtsev, I. S., and T. J. Martinez. "Quantum Chemistry on Graphical Processing Units. 1. Strategies for Two-Electron Integral Evaluation." Journal of Chemical Theory and Computation 4.2 (2008): 222-31.
---. "Graphical Processing Units for Quantum Chemistry." Computing in Science & Engineering 10.6 (2008): 26-34.
---. "Quantum Chemistry on Graphical Processing Units. 3. Analytical Energy Gradients, Geometry Optimization, and First Principles Molecular Dynamics." Journal of Chemical Theory and Computation 5.10 (2009): 2619-28.
---. "Quantum Chemistry on Graphical Processing Units. 2. Direct Self-Consistent-Field Implementation." Journal of Chemical Theory and Computation 5.4 (2009): 1004-15.
Valiev, M., et al. "Nwchem: A Comprehensive and Scalable Open-Source Solution for Large Scale Molecular Simulations." Comp. Phys. Comm. 181 (2010): 1477.

181
VandeVondele, J., M. Sulpizi, and M. Sprik. "From Solvent Fluctuations to Quantitative Redox Properties of Quinones in Methanol and Acetonitrile." Ang. Chem. Int. Ed. 45 (2006): 1936-38.
Virshup, A. M., et al. "Photodynamics in Complex Enviroments: Ab Initio Multiple Spawning Quantum Mechanical/Molecular Mechanical Dynamics." J. Phys. Chem. B 113 (2009): 3280-91.
---. "Photodynamics in Complex Environments: Ab Initio Multiple Spawning Quantum Mechanical/Molecular Mechanical Dynamics." J. Phys. Chem. B 113 (2009): 3280-91.
Vogt, L., et al. "Accelerating Resolution-of-the-Identity Second-Order Moller-Plesset Quantum Chemistry Calculations with Graphical Processing Units." Journal of Physical Chemistry A 112.10 (2008): 2049-57.
Vogt, Leslie, et al. "Accelerating Resolution-of-the-Identity Second-Order Moller‚Àíplesset Quantum Chemistry Calculations with Graphical Processing Units‚Ć." The Journal of Physical Chemistry A 112.10 (2008): 2049-57.
Vydrov, O. A., and T. Van Voorhis. "Implementation and Assessment of a Simple Nonlocal Van Der Waals Density Functional." J. Chem. Phys. 132 (2010): 164113.
Vysotskiy, V. P., and L. S. Cederbaum. "Accurate Quantum Chemistry in Single Precision Arithmetic: Correlation Energy." J. Chem. Theo. Comp. Articles ASAP; DOI: 10.1021/ct100533u (2010).
Warshel, A., and M. Levitt. "Theoretical Studies of Enzymatic Reactions: Dielectric, Electrostatic and Steric Stabilization of the Carbenium Ion in the Reaction of Lysozyme." J. Mol. Biol. 103 (1976): 227-49.
Watson, M. A., et al. "Accelerating Correlated Quantum Chemistry Calculations Using Graphical Processing Units." Computing in Science & Engineering 12.4 (2010): 40-50.
White, C. A., and M. Head-Gordon. "Derivation and Efficient Implementation of the Fast Multipole Method." Journal of Chemical Physics 101.8 (1994): 6593-605.
Whitten, J. L. "Coulombic Potential-Energy Integrals and Approximations." Journal of Chemical Physics 58.10 (1973): 4496-501.
---. "Coulombic Potential Energy Integrals and Approximations." The Journal of Chemical Physics 58.10 (1973): 4496-501.

182
Xie, W., et al. "X-Pol Potential: An Electronic Structure Based Force Field for Molecular Dynamics Simulation of a Solvated Protein in Water." J. Chem. Theo. Comp. 5 (2009): 459-67.
Yamaguchi, Shigeo, et al. "Low-Barrier Hydrogen Bond in Photoactive Yellow Protein." Proceedings of the National Academy of Sciences 106.2 (2009): 440-44.
Yang, W. T. "Direct Calculation of Electron-Density in Density-Functional Theory." Physical Review Letters 66.11 (1991): 1438-41.
---. "Direct Calculation of Electron-Density in Density-Functional Theory - Implementation for Benzene and a Tetrapeptide." Physical Review A 44.11 (1991): 7823-26.
Yasuda, K. "Accelerating Density Functional Calculations with Graphics Processing Unit." Journal of Chemical Theory and Computation 4.8 (2008): 1230-36.
---. "Two-Electron Integral Evaluation on the Graphics Processor Unit." Journal of Computational Chemistry 29.3 (2008): 334-42.




















